 Wenda Huang1
Wenda Huang1 Dongsheng Guo
Dongsheng Guo- 1Department of Neurosurgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Neurosurgery, Beijing TianTan Hospital, Capital Medical University, Beijing, China
Glioblastoma is the most common primary malignant tumor in the brain and has a dismal prognosis despite patients accepting standard therapies. Alternation of genes and deregulation of proteins, such as receptor tyrosine kinase, PI3K/Akt, PKC, Ras/Raf/MEK, histone deacetylases, poly (ADP-ribose) polymerase (PARP), CDK4/6, branched-chain amino acid transaminase 1 (BCAT1), and Isocitrate dehydrogenase (IDH), play pivotal roles in the pathogenesis and progression of glioma. Simultaneously, the abnormalities change the cellular biological behavior and microenvironment of tumor cells. The differences between tumor cells and normal tissue become the vulnerability of tumor, which can be taken advantage of using targeted therapies. Small molecule inhibitors, as an important part of modern treatment for cancers, have shown significant efficacy in hematologic cancers and some solid tumors. To date, in glioblastoma, there have been more than 200 clinical trials completed or ongoing in which trial designers used small molecules as monotherapy or combination regimens to correct the abnormalities. In this review, we summarize the dysfunctional molecular mechanisms and highlight the outcomes of relevant clinical trials associated with small-molecule targeted therapies. Based on the outcomes, the main findings were that small-molecule inhibitors did not bring more benefit to newly diagnosed glioblastoma, but the clinical studies involving progressive glioblastoma usually claimed “noninferiority” compared with historical results. However, as to the clinical inferiority trial, similar dosing regimens should be avoided in future clinical trials.
Introduction
Glioblastoma (GBM) is the most common primary malignant brain tumor. Based on the data from 2011 to 2015 in the United States, the average annual age-adjusted incidence of GBM is 3.21 per 100,000 population, with an overall prevalence of 9.23 per 100,000 population (1). Maximal safe surgical resection followed by radiotherapy with concomitant and adjuvant temozolomide has gradually become the standard regimen since 2005 (2). However, after standard therapy, almost every patient recurs within 6 months, and there is no effective therapy for recurrent GBM (3, 4). Although plenty of effort has been made toward studying the mechanisms of pathogenesis and progression of glioma, there was no significant change in treatment regimen and survival for GBM patients. In the past 20 years, targeted cancer therapies were promising methods, and simultaneously, various targeted drugs gradually entered clinical trials and then were approved by the FDA for cancer treatment.
Gliomas were deemed to derive from neural stem cells and, based on different classification criteria, can be generally divided into circumscribed gliomas and diffuse gliomas, or into low-grade (WHO I, II) and high-grade (WHO III, IV), or adult-type and pediatric-type (5–7). Futhermore, adult high-grade diffuse glioma affects the most people and is also the hardest to cure, predominantly including GBM (WHO IV) and anaplastic glioma (AG, WHO III, including AOD: anaplastic oligodendroglioma and AA: anaplastic astrocytoma) and accounts for approximately 80%. In 2008, Parsons et al. computed by the TCGA database and proposed three core alterations of signaling pathways in GBM: P53, retinoblastoma pathway (pRB), and receptor tyrosine kinases (RTKs) (8). Besides, mutation of phosphatase and tensin homolog deleted on chromosome ten (PTEN), neurofibromatosis (NF1), isocitrate dehydrogenase (IDH), B-Raf Proto-Oncogene (BRAF), and chromosome 1p19q co-deleted are common in glioma, even being used for molecular classification and prognosis prediction. In 2016, it was the first time the WHO Classification of CNS Tumors used molecule profiles to define the sub-types of gliomas. It is an important way to translate the abundant knowledge of the moleculer mechanisms to clinical applications (9). But regardless of IDH mutation, 1p/19q co-deletion, and H3 K27 mutation, the therapy regimen is similar and the classification method plays a limited role in guiding treatment. On the other hand, all the typical hallmarks of cancer can be detected in GBM, especially angiogenesis, tumor-promoting immune microenvironment, and reprogramming metabolism, which is gradually getting attention (10). The relative mechanisms of the hallmarks become therapeutic targets (Figure 1). Over the last two decades, since the specimen which was capable of generating tumor cells containing multiple lineage markers was reported, the concept of glioma stem cells (GSCs) has been more and more accepted by scientists (11, 12). GSCs are ascribed to a population accounting for heterogeneity in glioma mass and therapeutic resistance through self-renewal and differentiation to variant subpopulations. The elucidation of features of GSCs may be a key step in developing next-generation therapies (13). However, GSCs were not a bulk of homogeneous cells but rather a mosaic of discrete populations with distinct features (14). The complexity of GBM requires us to more deeply understand the mechanisms, in particular, the genetic and phenotypic characteristics of subclones distributed in the mass of tumor (15, 16).
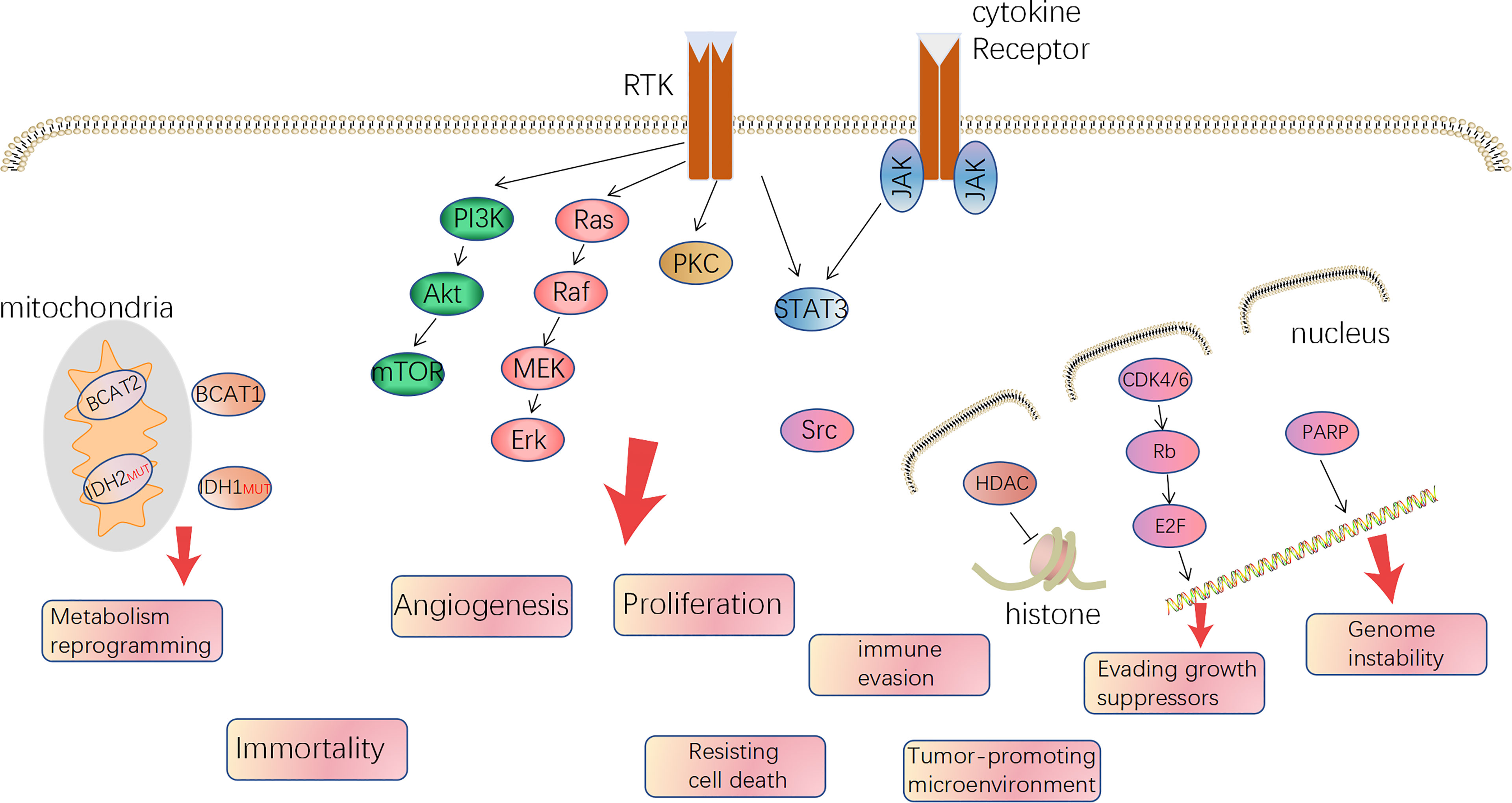
Figure 1 The major druggable targets in glioblastoma and corresponded hallmarks of tumor.
NCCN guidelines on central nervous system cancers indicate that the preferable treatment option for patients with recurrent GBM is enrollment in clinical trials due to the dismal outcomes of other therapies, including carmustine/lomustine, TMZ, radiotherapy, and bevacizumab (Bev.). There are more than 1,500 clinical trials associated with glioma completed or ongoing, among which contain hundreds of targeted therapies (https://clinicaltrials.gov/, Figure 2). Targeted therapies predominantly include two methods: small molecule and monoclonal antibodies. Simultaneously, various gene therapies such as microRNA, lncRNA, and exosome are a hotspot of research. Compared with monoclonal antibodies, small-molecule inhibitors possess several exclusive characteristics: reduced financial burden, taken orally, and more extensive druggable targets, including intracellular proteins (17). According to the targets of the small molecule inhibitors, we divided them into three main categories: tyrosine-kinase inhibitor (TKI), non-receptor tyrosine kinase (nRTK), intracellular signal transduction pathway inhibitor, and cellular biological process inhibitor. TKIs are still the most studied small-molecule inhibitors. But the outcomes of TKIs for the treatment of GBM and AG are often disappointing. An important reason for this is the complex network of intracellular signaling pathways leading to drug resistance. Besides, interference with biological processes and impact on the hallmarks of tumor growth can inhibit the growth of tumors through different mechanisms. In this review, we revisited the outcomes of recent clinical trials of adult high-grade glioma (mainly including GBM and AG) published in PubMed and the functional mechanisms of small-molecule inhibitors (Additional File 1). We hope to provide advice on the combination of drugs in future clinical trials by understanding the characteristics of different drugs.
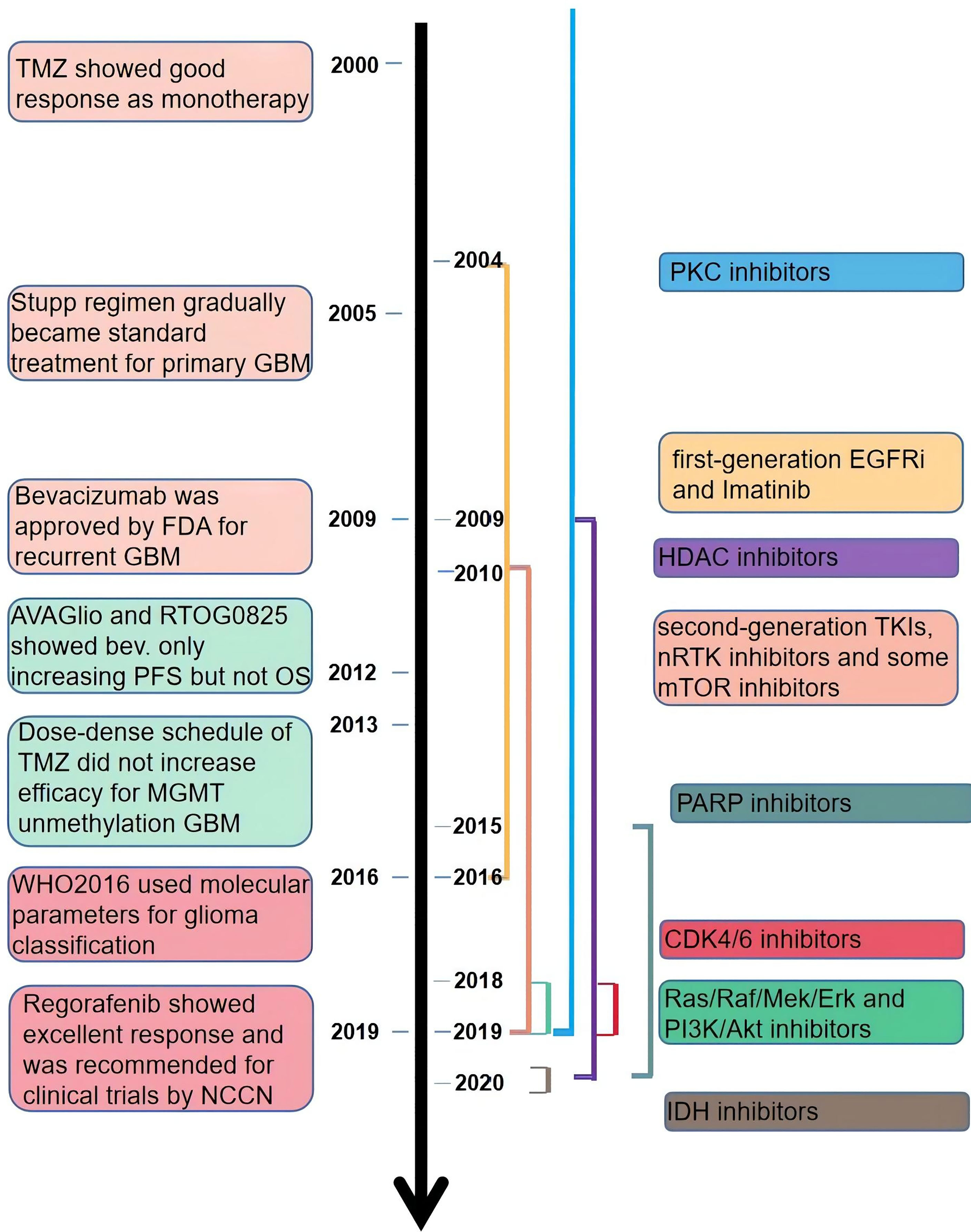
Figure 2 The time line of publicly published clinical trials of the small-molecule inhibitors.
RTK
Receptor tyrosine kinases (RTKs) and the downstream signal transduction are the most characterized networks associated with glioma pathogenesis and progression. To date, scientists have found more than 60 RTKs such as EGFR, VEGFR, MET, PDGFR, and FGFR (18, 19). Mutations in RTKs are also the most frequent alterations in GBM, namely, copy number variation, structure variation, nucleotide variation, and over-activation of the autocrine growth factor/receptor loop (18, 20). And then RTKs cause deregulation of several important pathways, namely, Ras/MAPK, PI3K/Akt/PTEN, PLC gamma/PKC, and Jak/STAT3 (21, 22). For instance, more than 50% of GBM harbor EGFR amplification and 25% cause EGFR mutation (23). EGFRvIII, a common mutant of EGFR which is continually activated even without ligand binding to the receptor, occurs after the pathogenesis of GBM to change the pattern of tumor growth and increase the intra-tumoral heterogeneity and resistance to targeted therapies (23). PDGFR, VEGFR, and FGFR are the most important RTKs associated with tumor angiogenesis (24). c-Met, also known as mesenchymal–epithelial transition factor, plays an important role in migration, therapy resistance, and vasculogenesis. In addition, Ret, c-Kit (CD177), and Flt3 (CD135) are also the targets of TKIs. The dynamic change of variant RTKs over time and space in tumor entities accounts for the complexity of GBM and therapy resistance after recurrence (3). RTKs also regulate the expression of GSC relevant transcription factors such as OLIG2, SOX2, and ZEB, which are also GSC marker molecules (25, 26). According to the chronological order, TKIs include three categories, and up to now, the most studied TKIs in glioma are predominantly first and second generation. The first-generation EGFR inhibitors, also known as type I, including erlotinib and gefitinib, can reversely bind to the ATP-binding site of EGFR to inhibit the activity of the receptor (27, 28). The second-generation inhibitors, such as afatinib, neratinib, vandetanib, and dacomitinib, irreversibly inhibit EGFR. Besides EGFR, the second-generation inhibitors can often inhibit multiple other RTKs, such as Her2, PDGFR, MET, and VEGFR (29–32). The third-generation inhibitor, Osimertinib, is used to overcome the resistance due to the T790M mutation, which is usually acquired after treatment with first-generation EGFR inhibitors. But researchers found that the acquired exon 20 C797S mutation would lead to resistance to Osimertinib within 10 months (33). So, the next-generation inhibitor, such as EAI045, is proposed to solve the problem of T790M mutation and C797S mutation inducing resistance (33). sorafenib, anlotinib, cabozantinib, cediranib, imatinib, lenvatinib, sunitinib, pazopanib, and motesanib enter clinical trials in glioma after approval by the FDA for renal cell carcinoma, lung cancer, thyroid cancer, and other solid tumors. They can target PDGFR, FGFR, C-kit, MET, and some nRTKs, which play great roles in the growth of GBM (Figure 3).
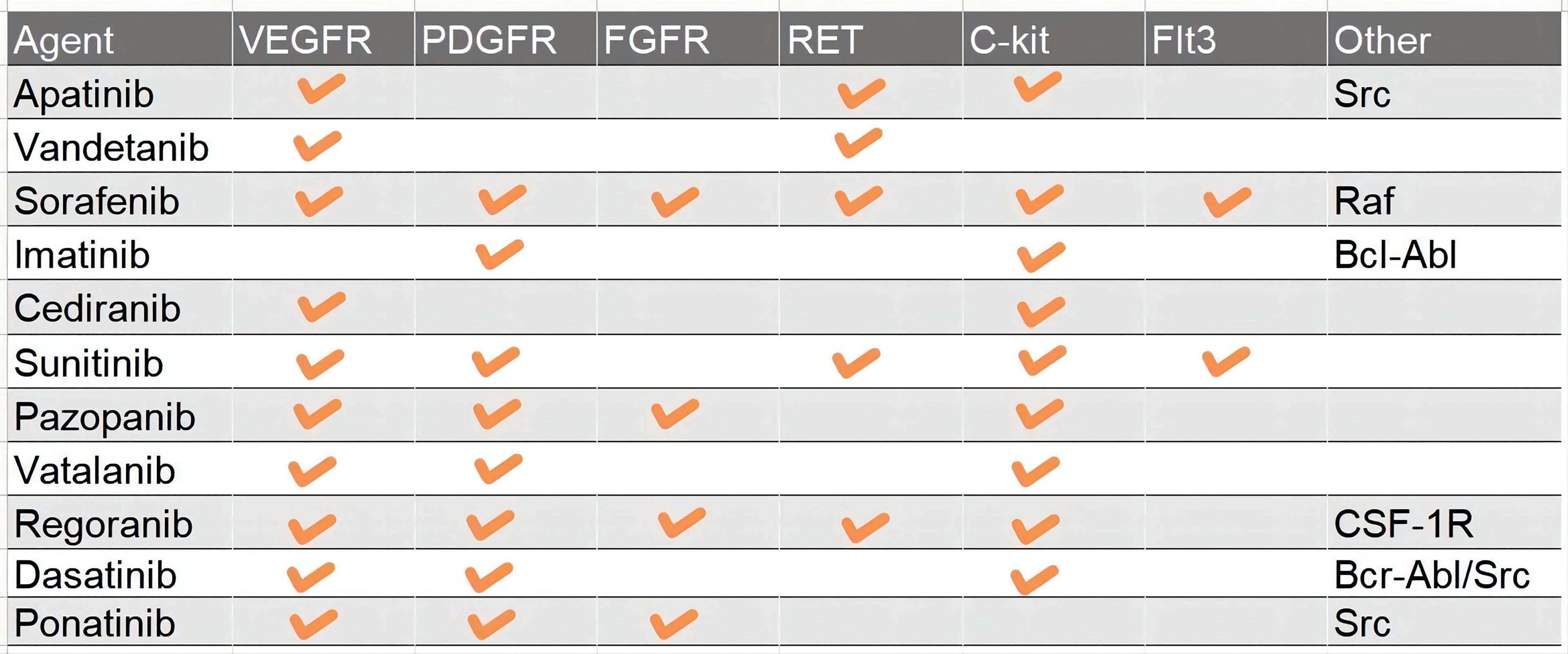
Figure 3 A table which summary the major targets of Pan-TKIs.
Until June 2021, there have been approximately 85 clinical trials of diverse TKIs to treat GBM and AG published in PubMed, including 17 studies for newly diagnosed GBM and 68 studies for progressive GBM. When comparing the results extracted from the publication with the baseline (historic results of 9 important clinical trials), there are 27 of 40 studies that show improvement in radiological response rates (ORs). These studies mainly involved patients with recurrent or progressive GBM. PFS was evaluated as mPFS in 71 studies and as PFS6 in 61 studies. There were 6 of 17 studies involving newly diagnosed GBM and 13 of 63 studies involving recurrent GBM that had an improvement in mPFS compared with baseline. OS was evaluated as mOS in 72 studies and as OS12 in 22 studies. But the most urgent problem is the only improvement of PFS while infrequently increasing the OS (Additional File 1).
Erlotinib is the most studied TKI in glioma. A phase II trial in 2014 that added Bev. and erlotinib to TMZ after completion of radiation involving 59 patients with newly diagnosed GBM showed an amazing result (mPFS: 13.5 m; mOS: 19.8 m), although the study did not reach the primary endpoint of improved OS (34). A study in 2010 using erlotinib as monotherapy for recurrent GBM had 40% PFS6 and 53% OS12 with 14 months of mOS (35). In summary, regardless of monotherapy or combined regimen, the result did not reach the expected outcome. And the improvement of ORs was partly deemed to be unauthentic because of improper imaging criteria (McDonald criteria). Combining erlotinib with other small-molecule inhibitors, such as sorafenib and sirolimus, did not play a greater role. Maybe due to the toxicity of sirolimus and its derivate temsirolimus, erlotinib combined with sirolimus decreased the PFS and OS in a phase II trial for recurrent malignant glioma (36, 37). Sorafinib is a pan-TKI, and it can also effectively inhibit the Ras pathway. Based on hypothesizing that inhibition of both EGFR and Ras would improve the survival time of patients with recurrent GBM, a phase II trial in 2013 was launched but did not reach the effective hypothesis H0 (30% increase in overall survival time compared with historical controls) (38). More clinical trial results will be helpful to clarify the efficacy of the combination of sorafenib and erlotinib. To date, the studies of erlotinib in glioma have stopped in phase II trials, and there is not enough evidence to show the potential efficacy of erlotinib.
Gefitinib is another well-characterized first-generation EGFR inhibitor. Just like erlotinib, it is more effective in cancers with mutated and overactive EGFR. Gefitinib did not show the potential to increase the OS of glioma. In 2012, a phase I/II trial involving 178 newly diagnosed GBM combining gefitinib with radiotherapy showed a disappointing result (mPFS: 4.9 m, mOS: 11.5 m) compared to baseline (38). Two important trials using gefitinib monotherapy for recurrent CNS tumor (NCT00025675, 2003) or combination with radiation for newly diagnosed GBM (NCT00052208, 2003) involving 105 and 158 patients respectively did thus far not show their results. There are no more results to evaluate the efficacy of gefitinib. As such, the outcomes of two classic first-generation EGFR inhibitors are a little disappointing. The insensitivity of GBM to erlotinib and gefitinib is partly due to the different models of EGFR overactivity in GBM cells. The two first-generation TKIs have a higher inhibiting ability of mutant EGFR such as L858R and exon 19 deletion type EGFR, which increases the kinase activity. In contrast to non-small cell lung cancer and adenocarcinoma harboring activating nucleotide mutations within the EGFR kinase domain, GBM overly activates EGFR by the increasing copy number and the substituted mutations occur in the extracellular domain (39) (Figure 4).
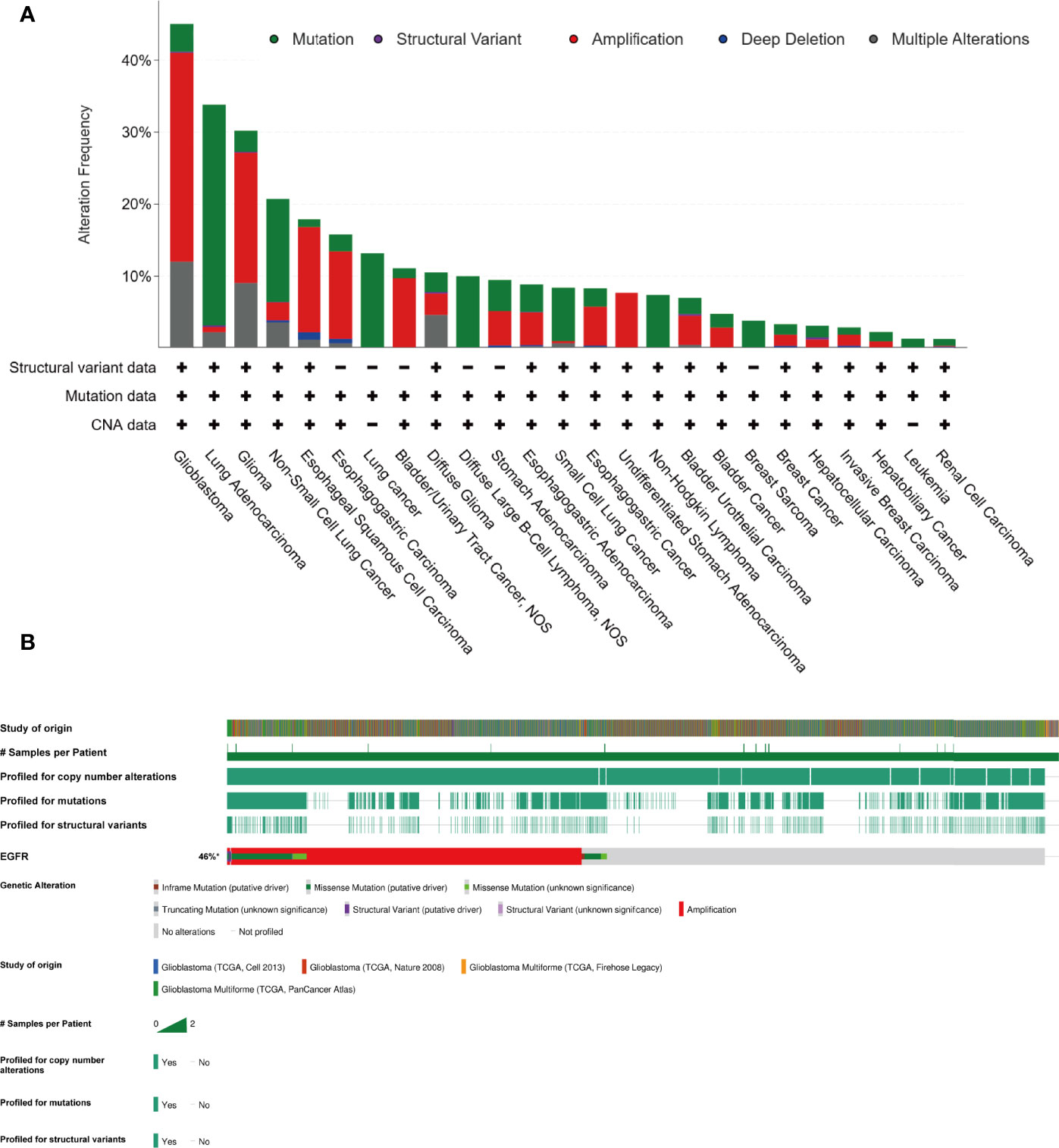
Figure 4 In contrast to lung cancer, glioma majorly harboring amplification of EGFR, instead of nucleotide mutation (cBioPortal). (A) The EGFR alteration frequency in different tumor types based on the PanCancer study “Pan-cancer analysis of whole genomes (ICGC/TCGA, Nature 2020)” in cBioportal database. (B) The collection of genetic alteration of EGFR based on four different studies in cBioportal database.
Lapatinib can selectively inhibit EGFR and HER2. Lapatinib combined with TMZ is a potential salvage option for recurrent ependymoma due to a phase II study (NCT00826241) demonstrating clinical activity with objective responses and prolonged disease control associated with disease-related symptom improvements (40). To date, three clinical trials including 74 patients did not present good results (41–43).
Afatinib is a second-generation irreversible ErbB family inhibitor, mainly used to treat cases of NSCLC that harbor mutations in the EGFR gene. Simultaneously, afatinib can effectively block EGFRvIII, a common mutant occurring in glioma (23, 44). A phase I study (NCT00977431) of afatinib combined with chemoradiotherapy demonstrated a favorable objective rate but simultaneously resulted in a high rate of serious adverse events. In 2015, a parallel-arm phase II trial (NCT00727506) showed that the addition of afatinib significantly decreased the PFS6 compared with TMZ alone (44). In the future, a pulsatile-increased dosing schedule of afatinib will be tested in glioma. It may be feasible to make a difference and reduce the adverse effects (44).
Vandetanib acts as a kinase inhibitor of several receptors, including VEGFR, EGFR, and the RET-tyrosine kinase. Because of the dual inhibition of EGFR and VEGFR2, vandetanib was supposed to play a greater role than combining erlotinib and Bev. in inhibiting angiogenesis. The prevailing rates and PFS usually showed favorable increases. A phase I/II study (NCT00441142) of parallel arm design demonstrated that adding vandetanib to TMZ and radiation can modestly increase the OS (45). These results give researchers the faith to continue their clinical studies. There are more than 7 phase I studies ongoing.
Sorafenib is a classical pan-TKI, targeting VEGFR, PDGFR, FGFR, c-Kit, Flt3, and RAF kinases, which are important in the angiogenesis of solid tumors. Pan-TKI is a trend of development of small-molecule inhibitors because of the complex crosstalk between various growth factors and compensatory mechanisms of RTKs. GBM cells can rapidly switch their signaling addiction from reliance on a single RTK to multiple RTK pathways. This may be driven by glioma stem cells (46). In general, sorafenib did not demonstrate sufficient efficacies to improve the outcome of patients with glioma. Many phase II trials (NCT00597493, NCT00544817, and NCT00621686) that have added sorafenib into the Stupp protocol or Bev. showed disappointing outcomes, even those inferior to Bev. monotherapy (47–49). In addition, sorafenib combined with erlotinib can play a synergic role by inhibiting EGFR and other RTKs in theory. But a phase II study (NCT00445588, 2013) using the combination regimen only showed an OS of 5.7 months and a PFS6 rate of 14% (38). Sorafenib combined with mTOR inhibitors such as temsirolimus did not improve the outcome in two phase I/II studies (NCT00335764, NCT00329719). Many clinical trials evaluating targeted combinatorial blockade frequently have disappointing outcomes due to overlapping toxic events, including serious diarrhea, seizure, and rash. Alternative administration schedules will probably reduce toxicity (17).
Imatinib is a specific inhibitor of several tyrosine kinase enzymes, including ABL, c-Kit, and PDGFR by occupying the tyrosine kinase active site to decrease the activity of kinases. PDGFR is deemed as a driven gene in low-grade glioma and is overexpressed in GBM. Imatinib has limited efficacy as monotherapy in GBM, and the chemotherapeutic agent hydroxyurea (HU) was found to play a synergic role when combined with imatinib due to imatinib sensitizing cancer to chemoradiotherapy and the penetrability of the blood–brain barrier (BBB) of HU (50). A multi-center phase II study (NCT00290771) involving 231 patients with progressive GBM showed a good safety profile among patients, which receiving up to 1,000 mg of imatinib and 1,000 mg of hydroxyurea. But the efficacy is not enough to induce anti-tumor (51). A phase III study (NCT00154375) involving 240 patients with TMZ-resistant GBM presented similar results of not meeting the primary study end point despite a good safety profile (52). Thus far, imatinib has not brought good news for glioma patients, just like in CML.
Cediranib is mainly a VEGFR inhibitor, and later the inhibiting c-Kit was found. Cediranib had a narrower inhibiting spectrum of RTKs than other pan-TKIs. But just because of this, cediranib has the chance to function as anti-VEGF therapy in combination with chemoradiotherapy for newly diagnosed GBM (NCT00662506 in 2008; NCT01062425 in 2010). The adverse effects of multiple TKIs are predominantly driven by the inhibition of VEGFR (17). The anti-angiogenesis and normalization of micro-vessels driven by anti-VEGF such as Bev. are deemed to sensitize patients to radiotherapy by increasing the supply of oxygen (53). The impact of angiogenesis inhibition on tumor distribution of TMZ is also a key factor considering the combination of standard therapy and anti-angiogenesis drugs (54).
Sunitinib inhibits all subtypes of PDGF-Rs and VEGFRs and c-Kit, RET, so that the safety is disquieting (55). Recently, sunitinib has mainly functioned as a monotherapy in clinical trials of CNS tumors, but the outcomes of patients were dismal. A phase II study (NCT00923117) of sunitinib to treat recurrent brain cancer has been terminated because of an unexpected result. The OS of Bev. resistant patients is 0.92 months and that of Bev. naive patients is 1.08 months. The regimen of sunitinib monotherapy caused high adverse event rates of 100% with more than 30% serious adverse events, such as skin, mucosal, and gastrointestinal adverse events (56). Bev. is a unique targeted therapy drug recently approved by the FDA for recurrent GBM. The various TKIs are considered in combination with Bev. or to be used in Bev.-resistant patients, even the VEGFR inhibitors. Continuous schedules enhance the efficacy of sunitinib. How to balance the dose plan and safety is the key factor to acquiring a satisfactory outcome.
Pazopanib limits tumor growth by targeting angiogenesis via the inhibition of VEGFR, PDGFR, c-KIT, and FGFR (57, 58). A phase II study (NCT00459381) involving 35 patients with recurrent GBM was disappointing (59). Adding chemotherapeutic drug topotecan with pazopanib did not significantly increase the OS of patients with recurrent GBM (NCT01931098) regardless of prior Bev. exposure or not. Simultaneously inhibition of EGFR and other RTKs increased the efficacy and theoretically led to a series of phase I trials to evaluate the safety and maximum toxic dose of the combination of EGFR inhibitors and pazopanib (60–62). But the accurate efficacy of GBM warrants more studies.
Vatalanib was designed for advanced cancers, especially those that have not responded to chemotherapy. It inhibits all known VEGF receptors, as well as PDGFR-beta and c-kit, but is most selective for VEGFR-2 (63). The outcomes of two phase I trials for newly diagnosed GBM were not poor compared with baseline, but the efficacy of vatalanib for treating GBM needs to be tested in more clinical trials.
Regorafenib, as a sorafenib derivative and pan-TKI of several kinases involved in tumor angiogenesis (VEGFR1–3 and TIE2), oncogenesis (KIT, RET, RAF1, and BRAF genes), the tumor microenvironment (PDGFR and FGFR), and tumor immunity (colony-stimulating factor 1 receptor), is recommended in the newest NCCN guideline for testing in future clinical trials due to a phase II parallel trial harboring a notable outcome (64, 64). In addition, regorafenib can induce autophagy arrest by the PSAT1/PRKAA autophagy-initiating pathway (65). By 2015, it had two US approvals for advanced cancers. However, the conclusion from the phase II study was defective. The outcome of regorafenib was just better than that of the control arm in the trial instead of baseline (66).
Many other TKIs are being tested in clinical trials, namely, anlotinib, cabozantinib, dovitinib, vatalanib, tandutinib, lenvatinib, and nintedanib, but so far, there are few trials with outcomes of these small-molecule inhibitors. Some TKIs have been approved by the FDA for other types of cancer and have not been widely tested in glioma. For instance, third-generation osimertinib with improved CNS penetration has broader coverage for mutated versions of EGFR.
In summary, (i) first EGFRi such as gefitinib and erlotinib, and PDGFRi imatinib may play great roles in specific patients with over-acting EGFR (especially EGFR with kinase domain mutation) and PDGFR. However, Pan-TKIs, such as sorafenib, sunitinib, and regorafenib did not show enough efficacy thus far (Table 1). (ii) Afatinib, which block EGFRvIII, a common structure mutation occurring after over-activating wild-type EGFR. The judgment of the time window of afatinib is a challenge of deeply understanding of heterogeneity in GBM temporally and regionally. (iii) VEGFR is a more important target than EGFR for GBM therapy. Anti-angiogenesis is a promising method including vandetanib, cediranib, vatalanib, and cabozantinib, just like Bev. Normalizing the immature vessels in the mass of glioma may play a supporting role in other therapies. (iv) Modest efficacy of inhibiting wild-type EGFR, effectively suppressing VEGFR, FGFR to anti-angiogenesis, along with inhibiting EGFRvIII may play an unexpected role. (v) Blindly using Pan-TKIs such as sorafenib, sunitinib, and regorafenib always did not show better efficacy and rather brought the risk of off-target effects. (vi) The side effects of TMZ and radiation are brain necrosis and myelosuppression, respectively. The side effects of TKIs are diarrhea and rash. Using TKIs and decreasing the dose of chemoradiotherapy may reduce the side effects, especially for patients over the age of 70 or patients with poor performance status to improve the life quality. (vii) The methylation status of the MGMT promoter is an important prognosis factor for TMZ therapy (Stupp regimen, MGMT methylated mOS 23.2 m, mPFS 10.5 m; MGMT unmethylated mOS 16.0 m, mPFS 7.8 m). As for MGMT unmethylated patients, TKIs may play an effective role. In the future, RTKs will still be the prime target for treatment of glioma because of their irreplaceable functions. To seriously appreciate the efficacy of various TKIs, we need more idealized randomized controlled trials just like AVAglio and RTOG 0825 for Bev.
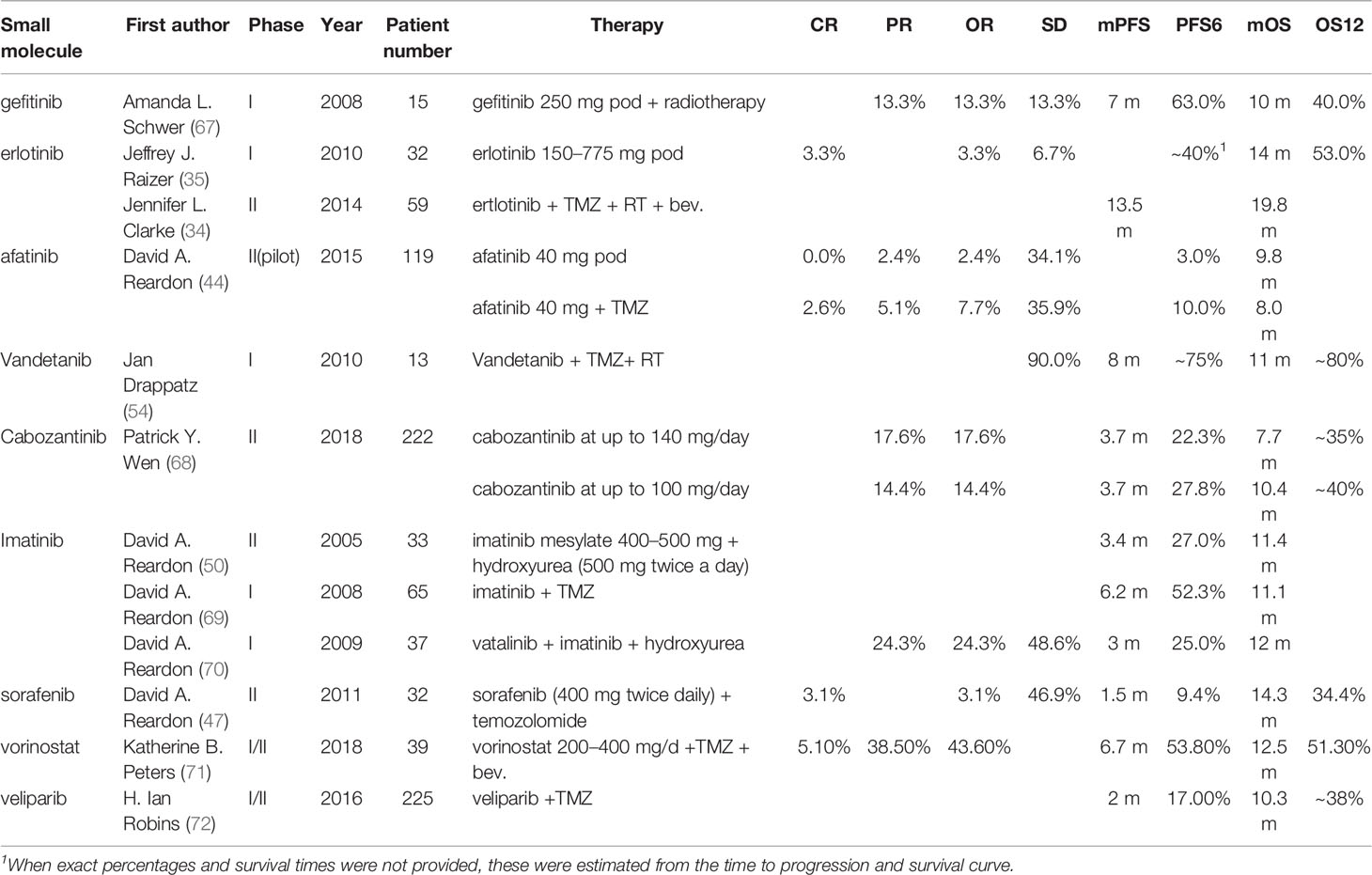
Table 1 Part of clinical studies which claimed “non-inferiority” for historical results.
nRTK and Intracellular Signal Transduction Pathways
Recently, nRTKs have gradually come into the field of view of researchers. Approximately 32 nRTKs have been identified in human cells, such as the SRC family, FAK family, and ABL family. Most of them are located in the cytoplasm, with part of them anchored on the cellular membrane by amino-terminal modification (73, 74). At first, the main function of nRTKs is associated with the immune system, such as activation of T and B cells (75). Despite lacking an RTK-like extracellular ligand-binding domain, nRTKs possess an ATP-binding site and a tyrosine kinase catalytic domain. So nRTKs can function partly as the same as transmembrane tyrosine kinases to regulate cell growth, proliferation, differentiation, adhesion, migration, and apoptosis. Beyond that, as nRTKs are located in the cytoplasm, they can bind proteins, lipids, and DNA through different domains to act broadly. Small-molecule inhibitors play a dominant role in targeted therapy for nRTKs instead of monoclonal antibodies due to their intracellular location and structure. In addition, some proteins functioning as part of the intracellular signal transduction pathway, such as Akt, MEK, and Erk, are also targets of therapy and discussed in this part.
Until June 2021, there are 48 clinical studies (20 for newly diagnosed GBM and 28 for progressive GBM) involving 9 small molecules published in PubMed. The radiological response rates are optimistic but not mTOR inhibitors compared to baseline. Thus far, the PFS and OS have not shown obvious benefits (Additional File 1).
Src
Since Francis et al. found the first proto-oncogene v-Src in the Rous sarcoma virus, 11 members of the human Src Kinase family (SFK) have gradually been discovered (76). Among these, c-Src, Yes, Fyn, Lyn, and Lck have functional roles in glioma involved in survival, proliferation, migration, angiogenesis, irradiation therapy resistance, and even stemness maintenance (77–81). Src kinase is the first characterized proto-oncogene (79, 82). The Src Kinase family thus likely functions as a traffic node of a complicated gene regulatory network initiated by membranes such as growth factor receptors, G protein-coupled receptors, and cytokine receptors (83, 84). Then, SRC-family kinases cross-talk with multiple pathways and are involved in the regulation of FAK/STAT3, Wnt/beta-catenin, caspase, cyclin/CDK, and integrin/FAK intracellularly (85–87). In GBM, the activity of Src kinase was significantly increased even though there was little missense mutation or amplification (88). Whereas mutation or loss of PTEN and EGFRvIII mutation would activate Fyn and c-Src, respectively, SRC-family kinase activity increasing is due to upstream deregulation instead of SRC per se (23, 78, 89). CD90 high-expression cells, which are identified as glioma stem cells, are sensitive to Src inhibitors (77). Moreover, Src activation and downstream multiple RTKs partially account for radiotherapy resistance. Si306, an inhibitor of c-Src, can increase the sensitivity to radiotherapy (79). To date, dasatinib and ponatinib have entered clinical trials in the area of glioma. These drugs also belong to pan-TKI.
Dasatinib is a second-generation TKI that can inhibit many nRTKs, including all types of kinases of the SRC family, such as c-Src, Lyn, Fyn, and Yes, which play important roles in the brain. Simultaneously, in a high concentration, it can impair other kinase activities, including BCR-ABL, Eph A2, c-kit, and VEGFR2, particularly PDGFR, which is altered in most diffuse intrinsic pontine gliomas (90, 91). To date, dasatinib has been evaluated along with conventional treatments or other TKIs in phase I and phase II clinical trials for GBM and diffuse pontine glioma in children and adults. The safety and tolerability of dasatinib demonstrated in the studies is disappointing, particularly when combined with lomustine in recurrent GBM and in combination with crizotinib in children with glioma (92, 93). Although theoretically anti-Src therapy can suppress radiation resistance, in a phase II trial involving 217 patients with newly diagnosed GBM, and the combination of dasatinib with concomitant radiation and TMZ did not improve PFS and OS compared with placebo (NCT00869401). Bev. and dasatinib in combination can numerically increase the PFS but not OS compared with Bev. alone (NCT00892177) (94). The above results suggest that it is effective to combine dasatinib with TKIs or various targeted drugs at the level of cellular mechanisms, such as c-MET, PDGFR, and VEGFR inhibitors (95, 96), but useless in clinical trials.
Ponatinib, the third-generation TKI for CML, targets Src and VEGFR, PDGFR, and FGFR, which are three crucial receptors associated with angiogenesis. Therefore, ponatinib is a potent anti-angiogenesis drug. It did not show a promising effect in a phase II study including 15 patients with Bev.-refractory GBM (97).
Bosutinib is a small molecule inhibitor targeting BCR-ABL and Src and is used for treating CML. In 2014, a phase II study involving only 11 patients with recurrent GBM showed a better mOS of 11.7 m compared with baseline. More clinical trials are warranted to evaluate the efficacy of bosutinib (98).
PKC
Protein kinase C is a serine/threonine kinase that includes three subgroups: the classical isoforms, novel isoforms, and atypical isoforms. Among the subgroups, there are tens of isoforms functioning differently in the survival, proliferation, adhesion, migration, and therapy resistance of glioma cells because of the diversity of phosphorylation sites, types of stimuli, and cell environment (99). The traditional signal pathway is the PLC/PIP2/IP3 pathway. But studies in various cancers, including GBM, showed that overexpression of PKC contributes to tumor pathogenesis and seems to be involved in EGFR, PI3K/Akt, Ras/MEK/MAPK, hedgehog, and TNFα signaling, which are deregulated in GBM and are deemed to be potent targets. In addition, PRKD2, a member of the PKC-activated protein kinase family, was identified as a mediator of GBM growth involving decreasing p53 and regulating the phosphorylation of retinoblastoma protein (100). RTKs, p53, and Rb, the three pathways that are commonly deregulated in GBM, crosstalk with PKC (101). Therefore, PKC inhibition is an important approach for the treatment of GBM and many trials test its efficacy. Targeting atypical PKC decreases tumor growth in EGFR inhibitor-resistant mouse models of GBM (102). PKCδ, an isoform of novel PKC, acts as a critical mediator of the maintenance of tumor stem cells through an autocrine loop with positive feedback that is driven by the PKCδ/STAT3/IL-23/JAK signaling axis (103).
Tamoxifen is a selective estrogen receptor modulator used to treat and prevent breast cancer in women (104). Strictly speaking, tamoxifen is not a classical small-molecule inhibitor. In GBM, it functions as a targeted inhibitor of PKC. Because overexpression of PKC in GBM is associated with TMZ and irradiation resistance and tamoxifen has no significant overlapping toxicities with most other drugs, clinical trials combine tamoxifen with traditional therapies for newly diagnosed GBM. Up until now, there have been no definite results for the regimen in phase II trials. A phase II trial of RTOG protocol BR-0021 combining high-dose tamoxifen with radiation did not exhibit improved effects (105). The later trials combining tamoxifen with TMZ and RT also did not have a different outcome.
Enzastaurin is a synthetic bisindolylmaleimide with potential antineoplastic activity. Enzastaurin binds to the ATP-binding site and selectively inhibits PKC β, a classical isoform involved in the induction of VEGF-stimulated neo-angiogenesis. In glioma, enzastaurin functions as an antiangiogenesis drug, entering clinical trials (106). In 2010, enzastaurin entered a phase III study as monotherapy but showed modest efficacy compared with the control arm of lomustine or baseline (107).
PI3K/Akt/mTOR
PI3K/Akt/mTOR, including the endogenous inhibitors PTEN, TSC1/2, and PHLPP, is one of the most common and most characterized malfunction pathways in glioma (108). High activity in the pathway, in general, portends a poor outcome (109). Since Powis et al. proposed that inhibition of PI3K by wortmannin could repress growth of human tumor xenografts in mice (110), there have been more than 20 types of small-molecule inhibitors entering clinical trials and more than 50 types tested in laboratories. In addition, many inhibitors can regulate the activity of PI3K/Akt to inhibit tumors, though they do not immediately target the pathway, such as oxymatrine, baicalin, and gartanin (111–113). Primarily, targeted therapy for PI3K/Akt/mTOR is mainly proposed to solve the resistance of chemoradiotherapy (114). On the one hand, the pathway is the busiest road of signal transduction that is initiated by growth factors or known as various RTKs and G protein-coupled receptors (GPCRs) on the cytoplasm membrane. On the other hand, it can crosstalk with diverse pathways intracellularly, such as Ras/Raf, Notch, STAT3, and Wnt/catenin. All of the proteins in the pathway, and upstream RTKs, and the endogenous inhibitors of the pathway are usually detected malfunctioning in glioma (114). Whereas in this pathway, every protein is an independent target, there is a combination regimen targeting more than one protein to increase efficacy. Simultaneously, PI3K has four classes and various isoforms with different catalytic-subs which play different roles. Among them, class I is dominant in cancers (114, 115). Therefore, there are pan-PI3K, isoform-selective, and dual PI3K/mTOR inhibitors. The common mutation of PIK3CA (encodes p110α, a 110 kDa catalytic subunit of Class IA) in cancer causes constitutive activation of PI3K and downstream Akt, and it is perhaps a promising target for therapy, but the mutation is not common in glioma despite 90% of GBMs harboring deregulated PI3K (116, 117). Other isoforms also impact the pathogenesis of solid tumors and hematologic malignancies. For instance, PTEN-deficient GBM largely depended on p110α for proliferation and p110β for migration (118). Recently researchers found many inhibitors can increase reactive oxygen species (ROS) to regulate the ROS-JUN-p53 loop and then decrease the level of phosphorylation and activity of PI3K/Akt/mTOR (119, 120). Inhibition of PI3K would promote the expression of the genes associated with glioma stem cells, such as SOX2, OCT4, and MSI1 (121). This is probably one of the mechanisms of resistance.
Akt, also known as PKB, includes three isoforms, and among these, Akt1 plays a major role in the PI3K/Akt pathway (122). It is not only an indispensable part of the PI3K/Akt pathway but also a serine/threonine-specific protein kinase that activates NF-κB, Bcl-2 family protein, and MDM2 to regulate multiple cellular processes (123). In addition, Akt also accepts the immediate regulation driven by other kinases (124). The drugs, which target Akt, combined with TMZ and fractional radiation, gradually enter the field of vision because chemoradiotherapy would increase the level of phosphorous Akt (125, 126).
Lastly, targeting mTOR is the most studied approach for the treatment of glioma. There are two different complexes, regulatory-associated protein mTORC1 and rapamycin insensitive mTORC2, the latter functioning in resistance to rapamycin by activating Akt in a positive feedback manner (127). 2-hydroxyglutarate (2HG) produced by IDH1/2 mutation promotes mTOR activity by depleting KDM4A and decreasing DEPTOR protein stability (128), presenting another mechanism of IDH1/2 mutation promoting the genesis of glioma and the possibility of a combination IDH inhibitor and mTOR inhibitor. Simultaneously, the pathway is a hotspot of studies on microRNA, lncRNA, and exosomes because of the extensive functions of noncoding RNA and PI3K/Akt (129–132). In addition, the downstream targets of the pathway, such as p70S6K, 4EBP1, and eIF-4E, are also druggable targets (133). However, in summary, the approach of targeting PI3K/Akt is failing in glioma clinical trials (Additional File 1). Single-target drugs are usually fed back to activate upstream and other pathways such as Erk through complex networks (124, 133). The content of phosphorous Akt, the status of PTEN, and other proteins may be prognostic factors that indicate the sensitivity to PI3K/Akt inhibitors (134, 135), but to date, the tissue analysis usually does not show that the molecular markers correlate with survival. The occurrence of heterogeneity and stem cells within the tumor adds to these difficulties (121).
PI3K
As mentioned above, there are isoform-selecting and pan-PI3K inhibitors. The former includes eganelisib, idelalisib, and alpelisib, but does not enter clinical trials in glioma. Eganelisib is a highly selective inhibitor of the enzyme PIK3C gamma (p110gamma) (136). But p110 gamma is predominantly expressed in the pancreas, skeletal muscle, liver, and heart instead of the brain (137). Idelalisib, approved by the FDA in 2014, blocks selectively P110δ, which is expressed in normal and malignant B-cells. Alpelisib is a PI3K alpha specific inhibitor for the treatment of breast cancer with a PIK3CA mutation after disease progression (138).
The drugs targeting PI3K that have entered clinical trials are mainly Pan-PI3K inhibitors, namely, taselisib, pilaralisib, buparlisib, and copanlisib. Thus far, the trials are mainly MATCH (Molecular Analysis for Therapy Choice) models because PI3K has diverse effects on the regulation of biological processes in a wide variety of human cancers (139). Taselisib is a pan-PI3K inhibitor but has the strongest activity to repress p110α (140, 141). Therefore, it is used in cancers with PIK3CA mutations, in particular uterine serous carcinomas and breast cancers, which are accompanied by hormone receptor changes because of the cross-talk between estrogen receptor (ER) and PI3K (141). The relationship between the expression of estrogen receptors and glioma expression has gained attention recently because of sex-specific differences in GBM (142). Copanlisib is predominantly against PI3K-α and PI3K-δ isoforms (143). A phase II MATCH trial involving the above two pan-PI3K inhibitors is recruiting (NCT02465060).
Pilaralisib, another pan-PI3K inhibitor, has been tested in early clinical trials. A phase I trial indicated that pilaralisib had a favorable safety profile (144), although some researchers thought that the toxicity of pan-PI3K was high (141).
Buparlisib is the most studied pan-PI3K inhibitor in glioma. It presented a promising prospect in preclinical studies. At low concentrations, buparlisib can inhibit the migration and invasion of GBM cells, and at higher concentrations and with a longer duration of drug exposure, it can promote apoptosis of cells (145, 146). It is controversial how the status of PTEN influences the efficacy of buparlisib. Koul showed that PTEN and EGFR status did not correlate with sensitivity, but mutation or wild type of p53 could change the method of death of tumor cells after treatment with buparlisib (147). But Xie et al. found that the buparlisib activity of anti-p110beta was poor and that may limit the efficacy of treating PTEN-deficient GBM (118). Buparlisib was also combined with other targeted therapies such as HSP990 (an Hsp90 inhibitor) and ABT-737 (a Bcl-2 inhibitor) because inhibiting the proteins in other pathways would cause compensatory overactivation of PI3K/Akt (148, 149). Yet, buparlisib is combined with other therapies is in phase I or phase II clinical trials (150–152). To date, the safety profile of buparlisib is deemed poor, and the regimen did not show enough efficacy to improve the outcome.
Akt
Akt inhibitors, including ipatasertib, capivasertib, MK2206, afuresertib, and perifosine, thus far are less studied in glioma. Ipatasertib is currently in phase II trials for the treatment of solid tumors. Capivasertib and afuresertib have mainly been tested in preclinical studies. MK2206 acts as an allosteric Akt inhibitor and is a highly selective inhibitor of pan-Akt, namely, of all three Akt isoforms Akt1, Akt2, and Akt3 (153). MK2206 can induce apoptosis and autophagy, increasing the efficacy of gefitinib (154). Narayan et al. showed that MK2206 at a low concentration (1 μM) reduced the phosphorylation of Thr308 and Ser473 residues of AKT in both adherent GBM cells and spheroids to sensitize them to irradiation and TMZ. At a high concentration (>5 μM), it inhibited invasion and migration of cells (155). But in PTEN-deficient cells, MK2206 could not decrease the level of phosphorylation of mTOR and S6K, the effectors of the PI3K/Akt pathway, nor induce apoptosis or autophagy.
mTOR
mTOR inhibitors are the most studied small molecules in the pathway. Now there are three generations of inhibitors and more than 70 clinical trials. Everolimus, a generation I mTOR inhibitor, is a medication used as an immunosuppressant to prevent rejection of organ transplants and in the treatment of renal cell cancer and other tumors, approved by the FDA for various conditions. Everolimus is more selective for the mTORC1 protein complex (156). It binds to its protein receptor FKBP12, which directly interacts with mTORC1 with little impact on the mTORC2 complex (156). Besides, everolimus could inhibit the production of glutathione from glutamine and glutamate and then inhibit the repair of DNA damage caused by carboplatin (157). But everolimus can lead to a hyper-activation of the kinase Akt via inhibiting the mTORC1 negative feedback loop while not inhibiting the mTORC2 positive feedback to Akt. And it has been evaluated in GBM for more than a decade with 26 trials registered on ClinicalTrials.gov (158). But a randomized phase II clinical trial RTOG 0913 (NCT01062399) presented a disappointing result: that adding everolimus into standard therapy reduced mOS by 4.7 months relative to the control arm (159). This leads us to consider that the penetration through the BBB, the toxicity of everolimus, and the activity of mTORC2 are influential but uncontrollable (158). But the next two early phase studies in pediatric patients demonstrated the manageable toxicity profile of everolimus (160, 161). On this basis, we think the prospect of mTOR inhibitors is promising. The PI3K/Akt/mTOR is a busy traffic node in cells, hence it is an important target just like RTKs and Ras/Raf/MAPK. The combination of mTOR inhibitors and targeted inhibition of Ras and PDGFR α is still a possible way to treat glioma (160, 162). The dual inhibitor of mTORC1 and mTORC2 may be a vulnerability in cancer (163). In addition, everolimus plays an irreplaceable role in the treatment of tuberous sclerosis complex and is mainly used for neurofibromatosis type 1-associated pediatric low-grade glioma and subependymal giant cell astrocytoma.
Sirolimus, also known as rapamycin and initially developed as an antifungal agent, functions as an immunosuppressive small-molecule inhibitor by inhibiting activation and sensitivity to interleukin-2 (IL-2) of T and B cells through inhibition of mTOR (164). Although it is a generation I inhibitor of mTOR like everolimus, it can also inhibit mTORC2. However, this has the adverse effect of increasing the risk of type 2 diabetes (165). The effect of sirolimus is complex, even though many studies have indicated that sirolimus can enhance mouse lifespan in a sex-specific manner (166). Temsirolimus, a derivative and prodrug of sirolimus, is converted to sirolimus (rapamycin) in vivo. But temsirolimus also shows exclusive activity on its own, and the trials in glioma mainly use temsirolimus instead of sirolimus. Outcomes of temsirolimus in clinical trials are frequently worse than those in the control arm. The combination of temsirolimus and TKIs, such as erlotinib and sorafenib, limits the dose of temsirolimus to one-tenth of MTD so as to achieve acceptable safety because of the side effects of temsirolimus. Thus, the regimen should be tested in more studies combining mTOR inhibitors and TKIs (37, 167, 168). Combining temsirolimus and perifosine, an Akt inhibitor, theoretically inhibits mTORC1 and suppresses the activity of Akt, which is activated by mTORC2 in a back-feed manner. The regimen was tolerable in a phase I study (169).
Ras/Raf/MEK/Erk
Ras/Raf/MEK/Erk is another busy traffic road in cells in addition to PI3K/Akt. Firstly, Ras is a protein superfamily of small GTPases and, in general, is responsible for cell proliferation (170). Ras is one of the most frequently altered proteins in cancer, but rarely in glioma (171). However, it is not a common target of therapy for cancer despite the many efforts contributed to the relative studies (172). Raf kinases are a family of three serine/threonine-specific protein kinases, including A-Raf, B-Raf, and C-Raf. BRAF became the focus of research recently since a large portion of human tumors carry oncogenic ‘driver’ mutations in the BRAF gene, with 11% of glioma cell lines harboring BRAF mutations (171, 173). The activating mutation in BRAF is a common change in pediatric low-grade gliomas (174). The BRAF V600E mutation inducing the constitutive activity of Raf is frequently detected in low-grade pleomorphic xanthoastrocytoma, ganglioglioma, extra-cerebellar pilocytic astrocytoma, and epithelioid GBMs instead of other types of high-grade glioma (175). But the overactivation of Ras/Raf is highly frequent in GBM. The BRAF V600E mutation is a successful target in melanoma, non-small cell lung cancer, and thyroid cancer. In high-grade gliomas, it may become a biomarker of benefit from dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) dual-targeted therapy (176). MEK is also known as MAPKK, and Erk is a classical MAPK, a classical cascade amplification pathway. The pathway connects extracellularly initiated signals from receptors on the surface of cells to the DNA in nuclei to regulate multiple biological processes. In addition, MEK and MAPK can function as kinases to immediately phosphorylate other proteins. Deregulation of the MAPK/Erk pathway is a necessary step in the malignant transformation of many cancers.
In summary, the intracellular signal transduction pathway is a significant embodiment of the complexity of biological regulation. On the one hand, every kinase can accept regulation from upstream kinase and activate downstream kinase to form a complicated network. This network is critical for maintaining the rule cell behavior. However, the overactivation of the pathway is usually due to upstream RTKs and GPCRs but not the abnormality of proteins comprising the pathway. Therefore, we suppose that it is not an ideal target for tumor treatment. Perhaps, targeting the intracellular signal transduction pathway would play an assistant role if the tumor showed significant overactivation or TKIs caused the second pathway overactivation by tissue analysis. The utilization of these inhibitors still requires biomarkers for precision medicine.
Proteins Regulating Cellular Biological Processes
In addition to the kinome, there are several proteins regulating cellular biological processes. The small molecules inhibiting the proteins are usually highly selective but difficult to target. There were 11 small molecules that were involved in 20 clinical trials that were published in PubMed (Additional File 1).
HDAC
Epigenetic changes refer to alternations affecting the expression of genes and the phenotypes of cells but not changing the sequence of DNA. It plays an important role in multiple aspects of pathogenesis and therapy resistance (177). In general, post-translational acetylation of histones modulates the structure of chromatin by relaxing the latter to promote the binding of transcription factors with motifs in genes and then increase the expression level of tumor suppressor genes (178). In addition, GSCs have a distinct pattern of epigenetic alterations, including DNA methylation and histone modifications (13). The process is controlled by the balance between the histone acetyltransferases (HATs) and histone deacetylases (HDACs). There are at least four classes of HDACs, including various isoforms (179). Different isoforms function in various ways. This indicates that HDAC inhibitors targeting multiple isoforms may be more effective (180–184). Class I and class II were studied more thoroughly, and the disruption of HDACs was observed in multiple cancers, particularly diffuse intrinsic pontine gliomas with H3K27M mutations (182, 185–187). HDAC inhibitors decondensing chromatin can prevent DNA double-strand break repair and regulate the stemness of GBM cells to induce radio sensitization (180, 188, 189). HDAC inhibitors also regulate the post-translational acetylation of proteins in the cytoplasm to influence the function of the latter, which is involved in angiogenesis, stemness, immune regulation, and ultimately inducing apoptosis. For example, many studies suggested that HDACs could downregulate the activity of p53, a classical tumor suppressor protein (185, 190, 191). HDACs block the NF-κB pathway by acetylation to inhibit resistance to TMZ and radiation (192, 193). But there was a study that indicated HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) favored the acquisition of TMZ resistance by increasing recruitment of SP1, C-JUN, NF-κB, and p300 within the relaxing MGMT promoter region (194). HDAC inhibitors repress EGFR/EGFRvIII expression inducing the expression of FOXO1—a tumor suppressor—in MYC-driven medulloblastoma cells (195, 196). HDACs also influence the polarization of microglia mediated by glioma cells (197). In addition, Nguyen et al. found that HDAC inhibitors blunted glycolysis in a c-Myc dependent manner and lowered ATP levels to elicit metabolic reprogramming to dependence on fatty acid oxidation (198). In summary, HDACs impact multiple processes through epigenetic changes. In the past few years, there have been a number of studies in which HDAC inhibitors were combined with radiation, chemotherapy, and other targeted drugs, presenting exciting efficacy in vitro and in vivo (192, 199–201). Because it is pharmacologically much simpler to inhibit an enzyme than to induce one, HDAC inhibition has gained enormous clinical interest as an anticancer strategy (177). At present, HDAC inhibitors as anti-tumor drugs are mainly used for the treatment of hematological neoplasms. Some HDAC inhibitors, such as valproic acid, have been used as anti-seizure drugs in GBM (202). To date, besides vorinostat, entinostat, Panobinostat, and VPA, many new HDAC inhibitors are still found and being studied preclinically. Interestingly, HDAC inhibitors are also being studied for their potential to induce viral HIV-1 expression in latently infected cells and disrupt latency (203).
Vorinostat, also known as SAHA, binds to the active sites of HDACs and acts as a chelator for zinc ions, which are also found in the active sites of HDACs. It acts on classes I, II, and IV of HDAC (204). In addition, SAHA could trigger autophagy in GBM stem cells through the Akt/mTOR pathway. Many clinical trials are ongoing or completed, but most are phase II or earlier. Based on preclinical results, vorinistat-combined drug with chemoradiotherapy, Bev., or bortezomib (a proteasome inhibitor) (205, 206). In a phase II north central cancer treatment group study involving 66 patients with recurrent GBM, vorinostat monotherapy showed nice tolerability but modest drug activity (207), and a phase I/II trial of vorinostat combined with traditional therapy for newly diagnosed GBM presented the same result. Because of the compromised efficacy presented in recent trials, there are additional trials registered in NIH except for a phase I trial of pembrolizumab and vorinostat combined with temozolomide for newly diagnosed GBM. Now preclinical studies focus on molecule markers such as IDH1 mutation, pChek2, and Bcl-XL, imaging parameters, which indicate sensitivity to vorinostat and other HDAC inhibitors (208–210), or to identifying some targeted inhibitors, such as melatonin, chloroquine, and gamitrinib, which can function as synergic effects with HDAC inhibitors (201, 211).
Romidepsin, also known as FK228 or Istodax, is a natural product obtained from bacteria. It acts as a prodrug, with the disulfide bond undergoing reduction within the cell to release a zinc-binding thiol. Thiol binds to a zinc atom in the binding pocket of Zn-dependent HDAC to block its activity. So, in theory, it acts on HDACs of the zinc-dependent classes I, IIa, IIb, and IV (179). Romidepsin decreases the expression of p21 to induce apoptosis (212). Wu et al. found that romidepsin increased the sensitivity to TMZ by blocking the PI3K/Akt/mTOR pathway (213). A phase I/II trial of romidepsin for adults with recurrent malignant glioma failed (214).
Panobinostat, trade name Farydak, is a hydroxamic acid and acts as a non-selective HDAC inhibitor. There are plenty of studies to research the activity in the treatment of diffuse intrinsic pontine glioma, a lethal pediatric brain cancer usually harboring mutations altering the epigenetic regulatory histone tail (H3 K27M) (215). Panobinostat can increase H3 acetylation and H3K27 trimethylation to partial rescue of the H3K27M-induced global hypotrimethylation phenotype (216). At present, Panobinostat is being tested in phase I clinical trials for diffuse intrinsic pontine glioma and GBM. Its potential of anti-angiogenesis led to combination with Bev. but failed in phase II (217).
Valproic acid is presently extensively used for epilepsy, bipolar disorder, and migraine headaches. In addition to inhibiting HADCs, it can affect GABA levels and voltage-gated sodium channels (218). Retrospective trials indicated that patients with GBM receiving valproic acid during the traditional regimen of TMZ and radiation had an improved prognosis by sensitizing to chemoradiotherapy (219–222). But Berendsen et al. proposed that epileptogenic GBM is a favorable prognostic factor regardless of using antiepileptic drugs or not (223). In 2016, a retrospective trial including 1,869 patients showed that valproic acid or levetiracetam, another anti-epileptic drug, did not improve the outcome of newly diagnosed GBM (224). To date, the activity of valproic acid in GBM is controversial. A phase II trial involving 43 patients with high-grade gliomas who received valproic acid and TMZ plus radiation indicated no increase in mOS (NCT00302159).
Belinostat, trade name Beleodaq, previously known as PXD101, is a hydroxamate-based pan-HDAC inhibitor that induces apoptosis by upregulation of p21 and through multiple pathways just like other HDAC inhibitors (225). There is currently no glioma clinical trial registered with the NIH.
CDK4/6
Deregulation of cyclin‐dependent kinase 4/6 (CDK4/6) is detected in various types of cancers, including glioma (226). Targeted inhibition of CDK4/6 has emerged as an efficient approach for treating breast cancer, with three small-molecule inhibitors approved by the FDA: ribociclib, palbociclib, and abemaciclib (227). CDK4/6 inhibitors primarily arrest the cell cycle in the G1 phase by regulating the CDK4/6–retinoblastoma (Rb)–E2F pathway to inhibit proliferation. However, as more and more studies are conducted in vitro and in vivo, the new targets of CDK4/6 inhibitors and other pathways involved in CDK4/6 have been proposed, involving cellular metabolism, autophagy, and immune evasion, despite the high selectivity of third-generation CDK4/6 inhibitors (228). The on-target and off-target mechanisms of CDK4/6 inhibitors have been reviewed by Denisa et al. (227). According to the TCGA database, the CDK4/6-Rb-E2F axis is deregulated in about 80% of GBMs, including endogenous inhibitors of CDKs, such as p16, an INK4 family protein (229). A large-scale data analysis on comprehensive genomic profiling indicated that 47.1% of brain gliomas harbored at least one cyclin alteration (230). In glioma, the activity of CDK4/6 inhibitors depends on the wild type of Rb. Mutation or deletion of Rb causes resistance to the drug. The deficiency of endogenous inhibitors, such as p16 and p18, or known as CDKN2A and CDKN2C, is a strong predictor of sensitivity to CDK4/6 inhibitors (231, 232). Besides, SHH and MYC amplification of group 3 medulloblastoma is sensitive to inhibition of CDK4/6 (233). The RTKs or growth factor pathway, p53, and CDKs/Rb/E2F pathways are the three most characterized pathways involved in the pathogenesis of glioma (234). Previous studies found that the combination of CDK4/6 and RTK inhibitors, such as mTOR inhibitors or c-Met/Trk inhibitors, could play a synergy (229, 235), possibly because the monotherapy of TKIs or CDK inhibitors would activate each other. Simultaneously, inhibition of CDK4/6 can delay radiation resistance and TMZ resistance by repressing double-strand break repair (236, 237). In general, the trials of CDK4/6 inhibitors are mainly in the early phase.
Palbociclib is the first and most studied CDK4/6 inhibitor in glioma therapy. But acquired resistance is still a problem that urgently needs to be addressed (238). On the one hand, the deregulation of the CDKs/Rb/E2F axis is a source of resistance to palbociclib. For example, gliomas with deletion or mutation of Rb are naturally insensitive to CDK4/6 inhibitors (232), but the wild-type Rb cancers would overexpress the Rb protein to gradually promote resistance to palbociclib (239). In addition, the isoforms of cyclins and CDKs, such as CDK2 and cyclin E, can play compensatory roles (227). However, a study found that palbociclib only induced reversible quiescence but not irreversible senescence in glioma stem cells (240). Thus far, palbociclib entered phase II trials for refractory or recurrent GBM, but for the moment it did not show enough efficacy to improve the outcome.
Ribociclib is used along with an aromatase inhibitor (such as letrozole). Some studies indicated the good CNS penetration of ribociclib (241). Ribociclib demonstrates a synergistic effect when combined with ALK or MEK inhibitors in the treatment of neuroblastoma (242). A phase I/II study of ribociclib following radiation therapy in children with newly diagnosed diffuse intrinsic pontine glioma indicated that the regimen was feasible (243).
Based on CDK4/6 inhibitor suppressing immune evading, the regimen of abemaciclib plus cancer immunotherapy, such as humanized antibody targeting the programmed cell death protein 1 (PD-1) receptor of lymphocytes pembrolizumab, has entered clinical trials. Thus far, there has been no result.
PARP
Poly (ADP-Ribose) polymerase (PARP) is a family of enzymes that catalyze the synthesis of linear or branched polymers of ADP-ribose (PAR) using NAD+ as substrate, and functions as radiosensitizer (244). Once PARP detects single-strand DNA breaks, it binds to DNA through the DNA-binding domain to induce structural change. Then it initiates the synthesis of a polymeric adenosine diphosphate ribose chain. The auto-PARylation of PARP on the auto-modification domain is a signal that recruits DNA ligase III (LigIII), DNA polymerase beta (polβ), and scaffolding proteins such as X-ray cross-complementing gene 1 (XRCC1) to repair the damaged DNA, a process known as base excision repair (BER) (245). Poly (ADP-ribose) glycohydrolase (PARG) degrades the PAR chain to recycle the PARP. Thus far, there are 18 members in the family, but PARP1 and PARP2 play major roles in repairing DNA, especially the first one. Beyond that, the other members perform other functions, and the PARylation of proteins as an important and ubiquitous post-translational modification involving histones and various transcription factors can also regulate multiple processes of cells, such as caspase-independent apoptosis by translating apoptosis-inducing factors into the nucleus (246–248). The high expression of PARP-1 was detected in multiple types of cancers (248). In the beginning, studies demonstrated that PARP inhibitors repressed the activity of PARP to hinder the BER system by competing with the substrate NAD+ (249). This would cause the accumulation of DNA damage of N-methylpurines (N7-methylguanine and N3-methyladenine) generated by TMZ and then eventually increase the sensitivity to TMZ and other alkylating agents. Notably, in the preclinical studies, the function of sensitization is still remarkable in MGMT-proficient and MMR-deficient glioma cells, which in general are regarded as drug-resistant cells (250–252). Higuchi et al. found that the TMZ sensitizer role of the PARP inhibitor was independent of base excision repair. There are perhaps other pathways by targeting which PARP inhibitors enhance the efficacy of chemoradiotherapy (253). For example, sustained inhibition of PARP-1 activity delays GBM recurrence by enhancing radiation-induced senescence (253). Similarly, PARP inhibitors can sensitize cells to ionizing radiation even in glioma stem cells, which can promote resistance to radiation by overexpressing PARP (254, 255), because the radiation generates single-strain DNA to exert clinical effect (256). Another important anti-tumor mechanism of PARP inhibitors is “synthetic lethality.” In some breast and ovarian cancers with BRCA mutations, meaning the deficiency of the homologous recombination repair system (HR), BER driven by PARP functions as compensation. So, the inhibition of PARP can maximize the effect of anti-tumor (257, 258). In glioma, deletion of PTEN, commonly occurring in primary GBM, can impact genomic stability by regulating the expression of RAD51, an important homologous recombination repair component (259). In in vitro, the cell lines with PTEN deficiency are more sensitive to PARP inhibitors than the wild-type PTEN cell lines (244). Inducing DNA damage is an important method to treat tumors. Except for chemotherapy and radiotherapy, topoisomerase inhibitors, topotecan and irinotecan, and inhibitors of DNA-dependent protein kinase (DNA-PK), a key enzyme involved in nonhomologous DNA end joining (NHEJ), can combine with PARP inhibitors to enhance the efficacy of antitumors (244). Recently, studies have demonstrated that IDH1/2 mutations could not only impair the HR system but also compromise the BER associated with PARP by decreasing NAD+ availability. This may be a reason for patients with IDH1/2 mutations have a better prognosis relative to IDH wild type when they receive chemoradiotherapy, and it also renders tumor cells sensitive to PARP inhibitors (260–262). Simultaneously, wild type p53, associated with cell cycle checkpoints and DNA repair, is supposed to indicate sensitivity to PARP inhibitors (263, 264). HDAC can repress DNA damage repair by epigenetic downregulation, which can play a synergistic role with PARP inhibitors (265). The inhibition of PARP can probably be used as an adjuvant therapy and there have been some clinical trials ongoing based on this theory. The loss of p53-binding protein 1 (53BP1), an antagonism factor of BRCA1, and the promoter of NHEJ can cause resistance to PARP inhibitors (266).
Olaparib, particularly in glioma harboring IDH mutation, the clinical trials were mainly carried out within recent 3 years, and there are no results thus far. OPARATIC trial, a phase I trial, demonstrated that olaparib could reliably penetrate recurrent GBM at radio sensitizing concentrations (267). Niraparib was tested in a phase II trial as a radiosensitizer (NCT04715620). Junko et al. showed that niraparib has the strongest potency in trapping PARP compared to olaparib and veliparib (268). In addition, pamiparib, talazoparib, and rucaparib which have been tested in other cancers.
In conclusion, the proteins regulating some specific cell processes, such as epigenetic changes, cell cycle, and DNA repair, may make a difference in glioma treatment. But in general, the relevant clinical trials are in their early phase. These inhibitors play functions by different mechanisms to have chance to combinate with TKIs in future clinical trials.
Conclusion
Small-molecule inhibitors did not change the survival of GBM patients in clinical trials. But at present, it is not reasonable to define small-molecule inhibitors as a failure. On the one hand, most trials involving recurrent GBM would stop therapy of small molecules after the progression of the tumor. But the mPFS of recurrent GBM is only 2 months. It causes the duration of the small molecule therapies to be less than 2 months. The short exposure duration limits the efficacy of small molecules. In addition, the efficacy of small molecules includes relieving edema, improving of neurological function, and improvement of life quality. On the other hand, anatomical characteristics of the brain restrain the delivery and efficacy of drugs. Heterogeneity within the GBM mass, and specifically mosaicism of glioma stem cells, is a critical factor in maximizing efficacy. In clinical practice, the tailored therapy associated with molecular subtyping in different regions of a tumor mass and the change of recurrence is more useful than personal therapy. The time and space obstacles could be tackled by several methods, such as new approaches to drug delivery of drugs and the scheduling of drug administration. In the future, as the finding of new targets and new technologies for drug discovery replaces the “Me-too” model, small molecules will play a more functional role in the treatment of cancer and other diseases.
Author Contributions
WH performed the selection of literature, drafted the manuscript, and prepared the figures and tables. DG designed this review, critically instructed the writing, and revised the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This project was supported by the National Natural Science Foundation of China (grant no. 81874086).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.911876/full#supplementary-material
Additional File 1 | Table S1.xlsx: A summary of outcomes in clinical studies in adult patients with glioblastoma.
Additional File 2 | Table S2.xlsx: Some clinical trials being conducted.
Abbreviations
GBM, glioblastoma multiforme; TCGA, The Cancer Genome Atlas; GSCs, glioma stem cells; NCCN, National Comprehensive Cancer Network; TMZ, Temozolomide; RT, Radiotherapy; Bev., Bevacizumab; EGFR, Epidermal growth factor receptor; OS, Overall survival; mOS, Median overall survival; PFS, Progressive free survival; mPFS, Median progressive free survival; OR, Response rate; PR, partial response; CR, complete response; SD, stable disease; RTK, Receptor tyrosine kinase; TKI, Tyrosine kinase inhibitor; nRTK, Nonreceptor tyrosine kinase; BBB, Blood–brain barrier; HDAC, Histone deacetylases; mTOR, mammalian target of rapamycin; PTEN, Phosphatase and tensin homolog; PARP, Poly (ADP-Ribose) polymerase; Grb,2Growth factor receptor-bound protein 2; GPCRs, G Protein-Coupled Receptors; NHEJ, Nonhomologous DNA end joining; HR, Homologous recombination repair system; BER, Base excision repair.
References
1. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. : CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro-Oncology (2018) 20(suppl_4):v1–v86. doi: 10.1093/neuonc/noy131
2. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of Radiotherapy With Concomitant and Adjuvant Temozolomide Versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol (2009) 10(5):459-66. doi: 10.1016/S1470-2045(09)70025-7
3. Campos B, Olsen LR, Urup T, Poulsen HS. A Comprehensive Profile of Recurrent Glioblastoma. Oncogene (2016) 35(45):5819–25. doi: 10.1038/onc.2016.85
4. Tan AC, Ashley DM, Lopez GY, Malinzak M, Friedman HS, Khasraw M. Management of Glioblastoma: State of the Art and Future Directions. CA Cancer J Clin (2020) 70(4):299–312. doi: 10.3322/caac.21613
5. Chen J, McKay RM, Parada LF. Malignant Glioma: Lessons From Genomics, Mouse Models, and Stem Cells. Cell (2012) 149(1):36–47. doi: 10.1016/j.cell.2012.03.009
6. Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, et al. Epidermal Growth Factor Receptor and Ink4a/Arf: Convergent Mechanisms Governing Terminal Differentiation and Transformation Along the Neural Stem Cell to Astrocyte Axis. Cancer Cell (2002) 1(3):269–77. doi: 10.1016/S1535-6108(02)00046-6
7. Kwon C, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, et al. Pten Haploinsufficiency Accelerates Formation of High-Grade Astrocytomas. Cancer Res (2008) 68(9):3286–94. doi: 10.1158/0008-5472.CAN-07-6867
8. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Sci (New York NY) (2008) 321(5897):1807–12. doi: 10.1126/science.1164382
9. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
10. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
11. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of Human Brain Tumour Initiating Cells. Nature (2004) 432(7015):396–401. doi: 10.1038/nature03128
12. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and Characterization of Tumorigenic, Stem-Like Neural Precursors From Human Glioblastoma. Cancer Res (2004) 64(19):7011–21. doi: 10.1158/0008-5472.CAN-04-1364
13. Mitchell K, Troike K, Silver DJ, Lathia JD. The Evolution of the Cancer Stem Cell State in Glioblastoma: Emerging Insights Into the Next Generation of Functional Interactions. Neuro-Oncology (2021) 23(2):199–213. doi: 10.1093/neuonc/noaa259
14. Merkle FT, Mirzadeh Z, Alvarez-Buylla A. Mosaic Organization of Neural Stem Cells in the Adult Brain. Sci (New York NY) (2007) 317(5836):381–4. doi: 10.1126/science.1144914
15. Reinartz R, Wang S, Kebir S, Silver DJ, Wieland A, Zheng T, et al. Functional Subclone Profiling for Prediction of Treatment-Induced Intratumor Population Shifts and Discovery of Rational Drug Combinations in Human Glioblastoma. Clin Cancer Res Off J Am Assoc Cancer Res (2017) 23(2):562–74. doi: 10.1158/1078-0432.CCR-15-2089
16. Tirosh I, Suvà ML. Tackling the Many Facets of Glioblastoma Heterogeneity. Cell Stem Cell (2020) 26:303–4. doi: 10.1016/j.stem.2020.02.005
17. Bedard PL, Hyman DM, Davids MS, Siu LL. Small Molecules, Big Impact: 20 Years of Targeted Therapy in Oncology. Lancet (Lond Eng) (2020) 395(10229):1078–88. doi: 10.1016/S0140-6736(20)30164-1
18. De Witt Hamer PC. Small Molecule Kinase Inhibitors in Glioblastoma: A Systematic Review of Clinical Studies. Neuro-Oncology (2010) 12(3):304–16. doi: 10.1093/neuonc/nop068
19. Appert-Collin A, Hubert P, Crémel G, Bennasroune A. Role of ErbB Receptors in Cancer Cell Migration and Invasion. Front Pharmacol (2015) 6:283. doi: 10.3389/fphar.2015.00283
20. Kapoor GS, O'Rourke DM. Receptor Tyrosine Kinase Signaling In Gliomagenesis: Pathobiology And Therapeutic Approaches. Cancer Biol Ther (2003) 2(4):330–42. doi: 10.4161/cbt.2.4.507
21. Tilak M, Holborn J, New LA, Lalonde J, Jones N. Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiforme. Int J Mol Sci (2021) 22(4):1831. doi: 10.3390/ijms22041831
22. Runkle KB, Kharbanda A, Stypulkowski E, Cao X, Wang W, Garcia BA, et al. Inhibition of DHHC20-Mediated EGFR Palmitoylation Creates a Dependence on EGFR Signaling. Mol Cell (2016) 62(3):385–96. doi: 10.1016/j.molcel.2016.04.003
23. Eskilsson E, Rosland GV, Talasila KM, Knappskog S, Keunen O, Sottoriva A, et al. EGFRvIII Mutations can Emerge as Late and Heterogenous Events in Glioblastoma Development and Promote Angiogenesis Through Src Activation. Neuro-Oncology (2016) 18(12):1644–55. doi: 10.1093/neuonc/now113
24. Orpana A, Salven P. Angiogenic and Lymphangiogenic Molecules in Hematological Malignancies. Leuk Lymphoma (2002) 43(2):219–24. doi: 10.1080/10428190290005964
25. Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, VandenBerg S, et al. PDGFR Alpha-Positive B Cells are Neural Stem Cells in the Adult SVZ That Form Glioma-Like Growths in Response to Increased PDGF Signaling. Neuron (2006) 51:187–99. doi: 10.1016/j.neuron.2006.06.012.32
26. Jimenez-Pascual A, Hale JS, Kordowski A, Pugh J, Silver DJ, Bayik D, et al. ADAMDEC1 Maintains a Growth Factor Signaling Loop in Cancer Stem Cells. Cancer Discov (2019) 9(11):1574–89. doi: 10.1158/2159-8290.CD-18-1308
27. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of non-Small-Cell Lung Cancer to Gefitinib. N Engl J Med (2004) 350(21):2129–39. doi: 10.1056/NEJMoa040938
28. Raymond E, Faivre S, Armand JP. Epidermal Growth Factor Receptor Tyrosine Kinase as a Target for Anticancer Therapy. Drugs (2000) 60 Suppl 1:15–23, 41-42. doi: 10.2165/00003495-200060001-00002
29. Galanti D, Inno A, La Vecchia M, Borsellino N, Incorvaia L, Russo A, et al. Current Treatment Options for HER2-Positive Breast Cancer Patients With Brain. Crit Rev Oncol Hematol (2021) 161:103329. doi: 10.1016/j.critrevonc.2021.103329
30. Maarof NNN, Alsalahi A, Abdulmalek E, Fakurazi S, Tejo BA, Abdul Rahman MB. Efficacy of Afatinib in the Treatment of Patients With Non-Small Cell Lung Cancer. Cancers (2021) 13(4):688. doi: 10.3390/cancers13040688
31. Galanti D, Inno A, La Vecchia M, Borsellino N, Incorvaia L, Russo A, et al. Current Treatment Options for HER2-Positive Breast Cancer Patients With Brain Metastases. Crit Rev Oncol Hematol (2021) 161:103329. doi: 10.1016/j.critrevonc.2021.103329
32. Fallahi P, Ferrari SM, Galdiero MR, Varricchi G, Elia G, Ragusa F, et al. Molecular Targets of Tyrosine Kinase Inhibitors in Thyroid Cancer. Semin Cancer Biol (2020) 79:180–96. doi: 10.1016/j.semcancer.2020.11.013
33. Wang S, Song Y, Liu D. EAI045: The Fourth-Generation EGFR Inhibitor Overcoming T790M and C797S Resistance. Cancer Lett (2017) 385:51–4. doi: 10.1016/j.canlet.2016.11.008
34. Clarke JL, Molinaro AM, Phillips JJ, Butowski NA, Chang SM, Perry A, et al. A Single-Institution Phase II Trial of Radiation, Temozolomide, Erlotinib, and Bevacizumab for Initial Treatment of Glioblastoma. Neuro Oncol (2014) 16:984–90. doi: 10.1093/neuonc/nou029
35. Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, et al. A Phase I Trial of Erlotinib in Patients With Nonprogressive Glioblastoma Multiforme Postradiation Therapy, and Recurrent Malignant Gliomas and Meningiomas. Neuro-Oncology (2010) 12(1):87–94. doi: 10.1093/neuonc/nop017
36. Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JEN, et al. Phase 2 Trial of Erlotinib Plus Sirolimus in Adults With Recurrent Glioblastoma. J Neurooncol (2010) 96:219–30. doi: 10.1007/s11060-009-9950-0
37. Wen PY, Chang SM, Lamborn KR, Kuhn JG, Norden AD, Cloughesy TF, et al. Phase I/II Study of Erlotinib and Temsirolimus for Patients With Recurrent Malignant Gliomas: North American Brain Tumor Consortium Trial 04-02. Neuro Oncol (2014) 16(4):567–78. doi: 10.1093/neuonc/not247
38. Peereboom DM, Ahluwalia MS, Ye X, Supko JG, Hilderbrand SL, Phuphanich S, et al. NABTT 0502: A Phase II and Pharmacokinetic Study of Erlotinib and Sorafenib for Patients With Progressive or Recurrent Glioblastoma Multiforme. Neuro Oncol (2013) 15:490–6. doi: 10.1093/neuonc/nos322
39. Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Sci (New York NY) (2004) 305(5687):1163–7. doi: 10.1126/science.1101637
40. Gilbert MR, Yuan Y, Wu J, Mendoza T, Vera E, Omuro A, et al. A Phase II Study of Dose-Dense Temozolomide and Lapatinib for Recurrent Low-Grade and Anaplastic Supratentorial, Infratentorial, and Spinal Cord Ependymoma. Neuro Oncol (2021) 23:468–77. doi: 10.1093/neuonc/noaa240
41. Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, et al. A Phase I/II Trial of GW572016 (Lapatinib) in Recurrent Glioblastoma Multiforme: Clinical Outcomes, Pharmacokinetics and Molecular Correlation. Cancer Chemother Pharmacol (2010) 65:353–61. doi: 10.1007/s00280-009-1041-6
42. Karavasilis V, Kotoula V, Pentheroudakis G, Televantou D, Lambaki S, Chrisafi S, et al. A Phase I Study of Temozolomide and Lapatinib Combination in Patients With Recurrent High-Grade Gliomas. J Neurol (2013) 260:1469–80. doi: 10.1007/s00415-012-6812-z
43. Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, et al. A Phase I/II Trial of Pazopanib in Combination With Lapatinib in Adult Patients With Relapsed Malignant Glioma. Clin Cancer Res (2013) 19:900–8. doi: 10.1158/1078-0432.CCR-12-1707
44. Reardon DA, Nabors LB, Mason WP, Perry JR, Shapiro W, Kavan P, et al. Phase I/randomized Phase II Study of Afatinib, an Irreversible ErbB Family Blocker, With or Without Protracted Temozolomide in Adults With Recurrent Glioblastoma. Neuro Oncol (2015) 17:430–9. doi: 10.1093/neuonc/nou160.48
45. Lee EQ, Kaley TJ, Duda DG, Schiff D, Lassman AB, Wong ET, et al. A Multicenter, Phase II, Randomized, Noncomparative Clinical Trial of Radiation and Temozolomide With or Without Vandetanib in Newly Diagnosed Glioblastoma Patients. Clin Cancer Res (2015) 21:3610–8. doi: 10.1158/1078-0432.CCR-14-3220
46. Hamed HA, Tavallai S, Grant S, Poklepovic A, Dent P. Sorafenib/regorafenib and Lapatinib Interact to Kill CNS Tumor Cells. J Cell Physiol (2015) 230(1):131–9. doi: 10.1002/jcp.24689
47. Reardon DA, Vredenburgh JJ, Desjardins A, Peters K, Gururangan S, Sampson JH, et al. Effect of CYP3A-Inducing Anti-Epileptics on Sorafenib Exposure: Results of a Phase II Study of Sorafenib Plus Daily Temozolomide in Adults With Recurrent Glioblastoma. J Neurooncol (2011) 101:57–66. doi: 10.1007/s11060-010-0217-6
48. Hainsworth JD, Ervin T, Friedman E, Priego V, Murphy PB, Clark BL. Lamar RE: Concurrent Radiotherapy and Temozolomide Followed by Temozolomide and Sorafenib in the First-Line Treatment of Patients With Glioblastoma Multiforme. Cancer (2010) 116:3663–9. doi: 10.1002/cncr.25275
49. Galanis E, Anderson SK, Lafky JM, Uhm JH, Giannini C, Kumar SK, et al. Phase II Study of Bevacizumab in Combination With Sorafenib in Recurrent Glioblastoma (N0776): A North Central Cancer Treatment Group Trial. Clin Cancer Res (2013) 19:4816–23. doi: 10.1158/1078-0432.CCR-13-0708
50. Reardon DA, Egorin MJ, Quinn JA, Rich JN, Gururangan S, Vredenburgh JJ, et al. Phase II Study of Imatinib Mesylate Plus Hydroxyurea in Adults With Recurrent Glioblastoma Multiforme. J Clin Oncol (2005) 23(36):9359–68. doi: 10.1200/JCO.2005.03.2185
51. Reardon DA, Dresemann G, Taillibert S, Campone M, van den Bent M, Clement P, et al. Multicentre Phase II Studies Evaluating Imatinib Plus Hydroxyurea in Patients With Progressive Glioblastoma. Br J Cancer (2009) 101(12):1995–2004. doi: 10.1038/sj.bjc.6605411
52. Dresemann G, Weller M, Rosenthal MA, Wedding U, Wagner W, Engel E, et al. Et Al: Imatinib in Combination With Hydroxyurea Versus Hydroxyurea Alone as Oral Therapy in Patients With Progressive Pretreated Glioblastoma Resistant to Standard Dose Temozolomide. J Neurooncol (2010) 96(3):393–402. doi: 10.1007/s11060-009-9976-3
53. Jain RK. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Sci (New York NY) (2005) 307(5706):58–62. doi: 10.1126/science.1104819
54. Drappatz J, Norden AD, Wong ET, Doherty LM, Lafrankie DC, Ciampa A, et al. Phase I Study of Vandetanib With Radiotherapy and Temozolomide for Newly Diagnosed Glioblastoma. Int J Radiat Oncol Biol Phys (2010) 78:85–90. doi: 10.1016/j.ijrobp.2009.07.1741
55. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and Safety of Sunitinib in Patients With Advanced Gastrointestinal Stromal Tumour After Failure of Imatinib: A Randomised Controlled Trial. Lancet (Lond Engl) (2006) 368(9544):1329–38. doi: 10.1016/S0140-6736(06)69446-4
56. Kreisl TN, Smith P, Sul J, Salgado C, Iwamoto FM, Shih JH, et al. Continuous Daily Sunitinib for Recurrent Glioblastoma. J Neurooncol (2013) 111:41–8. doi: 10.1007/s11060-012-0988-z
57. Zivi A, Cerbone L, Recine F, Sternberg CN. Safety and Tolerability of Pazopanib in the Treatment of Renal Cell Carcinoma. Expert Opin Drug Saf (2012) 11:851–9. doi: 10.1517/14740338.2012.712108
58. Verweij J, Sleijfer S. Pazopanib, a New Therapy for Metastatic Soft Tissue Sarcoma. Expert Opin Pharmaco (2013) 14(7):929–35. doi: 10.1517/14656566.2013.780030
59. Iwamoto FM, Lamborn KR, Robins HI, Mehta MP, Chang SM, Butowski NA, et al. Phase II Trial of Pazopanib (GW786034), an Oral Multi-Targeted Angiogenesis Inhibitor, for Adults With Recurrent Glioblastoma (North American Brain Tumor Consortium Study 06-02). Neuro Oncol (2010) 12:855–61. doi: 10.1093/neuonc/noq025
60. de Jonge MJA, Hamberg P, Verweij J, Savage S, Suttle AB, Hodge J, et al. Phase I and Pharmacokinetic Study of Pazopanib and Lapatinib Combination Therapy in Patients With Advanced Solid Tumors. Invest New Drugs (2013) 31:751–9. doi: 10.1007/s10637-012-9885-8
61. Inada-Inoue M, Ando Y, Kawada K, Mitsuma A, Sawaki M, Yokoyama T, et al. Phase 1 Study of Pazopanib Alone or Combined With Lapatinib in Japanese Patients With Solid Tumors. Cancer Chemother Pharmacol (2014) 73:673–83. doi: 10.1007/s00280-014-2374-3
62. Henary H, George GC, Wheler J, Naing A, Piha-Paul S, Fu S, et al. A Phase 1 Study of Intermittently Administered Pazopanib in Combination With Continuous Daily Dosing of Lapatinib in Patients With Solid Tumors. Cancer Chemother Pharmacol (2015) 76:597–603. doi: 10.1007/s00280-015-2824-6
63. Los M, Roodhart JM, Voest EE. Target Practice: Lessons From Phase III Trials With Bevacizumab and Vatalanib in the Treatment of Advanced Colorectal Cancer. Oncologist (2007) 12(4):443–50. doi: 10.1634/theoncologist.12-4-443
64. Lombardi G, De Salvo GL, Brandes AA, Eoli M, Rudà R, Faedi M, et al. Regorafenib Compared With Lomustine in Patients With Relapsed Glioblastoma (REGOMA): A Multicentre, Open-Label, Randomised, Controlled, Phase 2 Trial. Lancet Oncol (2019) 20:110–9. doi: 10.1016/S1470-2045(18)30675-2
65. Jiang J, Zhang L, Chen H, Lei Y, Zhang T, Wang Y, et al. Regorafenib Induces Lethal Autophagy Arrest by Stabilizing PSAT1 in Glioblastoma. Autophagy (2020) 16(1):106–22. doi: 10.1080/15548627.2019.1598752
66. Stupp R. Drug Development for Glioma: Are We Repeating the Same Mistakes? Lancet Oncol (2019) 20:10–2. doi: 10.1016/S1470-2045(18)30827-1
67. Schwer AL, Damek DM, Kavanagh BD, Gaspar LE, Lillehei K, Stuhr K, et al. A Phase I Dose-Escalation Study of Fractionated Stereotactic Radiosurgery in Combination With Gefitinib in Patients With Recurrent Malignant Gliomas. Int J Radiat Oncol Biol Phys (2008) 70:993–1001. doi: 10.1016/j.ijrobp.2007.07.2382
68. Wen PY, Drappatz J, de Groot J, Prados MD, Reardon DA, Schiff D, et al. Phase II Study of Cabozantinib in Patients With Progressive Glioblastoma: Subset Analysis of Patients Naive to Antiangiogenic Therapy. Neuro Oncol (2018) 20:249–58. doi: 10.1093/neuonc/nox154
69. Reardon DA, Desjardins A, Vredenburgh JJ, Sathornsumetee S, Rich JN, Quinn JA, et al. Safety and Pharmacokinetics of Dose-Intensive Imatinib Mesylate Plus Temozolomide: Phase 1 Trial in Adults With Malignant Glioma. Neuro Oncol (2008) 10:330–40. doi: 10.1215/15228517-2008-003
70. Reardon DA, Egorin MJ, Desjardins A, Vredenburgh JJ, Beumer JH, Lagattuta TF, et al. Phase I Pharmacokinetic Study of the Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitor Vatalanib (PTK787) Plus Imatinib and Hydroxyurea for Malignant Glioma. Cancer (2009) 115:2188–98. doi: 10.1002/cncr.24213
71. Peters KB, Lipp ES, Miller E, Herndon JEN, McSherry F, Desjardins A, et al. Phase I/II Trial of Vorinostat, Bevacizumab, and Daily Temozolomide for Recurrent Malignant Gliomas. J Neurooncol (2018) 137:349–56. doi: 10.1007/s11060-017-2724-1
72. Robins HI, Zhang P, Gilbert MR, Chakravarti A, de Groot JF, Grimm SA, et al. A Randomized Phase I/II Study of ABT-888 in Combination With Temozolomide in Recurrent Temozolomide Resistant Glioblastoma: An NRG Oncology RTOG Group Study. J Neurooncol (2016) 126:309–16. doi: 10.1007/s11060-015-1966-z
73. Siveen KS, Prabhu KS, Achkar IW, Kuttikrishnan S, Shyam S, Khan AQ, et al. Role of Non Receptor Tyrosine Kinases in Hematological Malignances and its Targeting by Natural Products. Mol Cancer (2018) 17(1):31. doi: 10.1186/s12943-018-0788-y
74. Neet K, Hunter T. Vertebrate non-Receptor Protein-Tyrosine Kinase Families. Genes Cells Devoted Mol Cell Mech (1996) 1(2):147–69. doi: 10.1046/j.1365-2443.1996.d01-234.x
75. Weiss A, Littman DR. Signal Transduction by Lymphocyte Antigen Receptors. Cell (1994) 76(2):263–74. doi: 10.1016/0092-8674(94)90334-4
76. Roskoski R. Src Protein-Tyrosine Kinase Structure, Mechanism, and Small Molecule Inhibitors. Pharmacol Res (2015) 94:9–25. doi: 10.1016/j.phrs.2015.01.003
77. Avril T, Etcheverry A, Pineau R, Obacz J, Jegou G, Jouan F, et al. CD90 Expression Controls Migration and Predicts Dasatinib Response in Glioblastoma. Clin Cancer Res (2017) 23(23):7360–74. doi: 10.1158/1078-0432.CCR-17-1549
78. Li X, Tao Z, Wang H, Deng Z, Zhou Y, Du Z. Dual Inhibition of Src and PLK1 Regulate Stemness and Induce Apoptosis Through Notch1-SOX2 Signaling in EGFRvIII Positive Glioma Stem Cells (GSCs). Exp Cell Res (2020) 396(1):112261. doi: 10.1016/j.yexcr.2020.112261
79. Ahluwalia MS, de Groot J, Liu WM, Gladson CL. Targeting SRC in Glioblastoma Tumors and Brain Metastases: Rationale and Preclinical Studies. Cancer Lett (2010) 298(2):139–49. doi: 10.1016/j.canlet.2010.08.014
80. Cammarata FP, Torrisi F, Forte GI, Minafra L, Bravatà V, Pisciotta P, et al. Proton Therapy and Src Family Kinase Inhibitor Combined Treatments on U87 Human Glioblastoma Multiforme Cell Line. Int J Mol Sci (2019) 20(19):4745. doi: 10.3390/ijms20194745
81. Torrisi F, Vicario N, Spitale FM, Cammarata FP, Minafra L, Salvatorelli L, et al. The Role of Hypoxia and SRC Tyrosine Kinase in Glioblastoma Invasiveness and Radioresistance. Cancers (2020) 12(10):2860. doi: 10.3390/cancers12102860
82. Han X, Zhang W, Yang X, Wheeler CG, Langford CP, Wu L, et al. Et Al: The Role of Src Family Kinases in Growth and Migration of Glioma Stem Cells. Int J Oncol (2014) 45(1):302–10. doi: 10.3892/ijo.2014.2432
83. Eom H, Kaushik N, Yoo K, Shim J, Kwon M, Choi M, et al. MerTK Mediates STAT3-KRAS/SRC-Signaling Axis for Glioma Stem Cell Maintenance. Artif Cells Nanomed Biotechnol (2018) 46(sup2):87–95. doi: 10.1080/21691401.2018.1452022
84. Lan T, Wang H, Zhang Z, Zhang M, Qu Y, Zhao Z, et al. Downregulation of β-Arrestin 1 Suppresses Glioblastoma Cell Malignant Progression Vis Inhibition of Src Signaling. Exp Cell Res (2017) 357(1):51–8. doi: 10.1016/j.yexcr.2017.04.023
85. Wei J, Ma L, Li C, Pierson CR, Finlay JL, Lin J. Targeting Upstream Kinases of STAT3 in Human Medulloblastoma Cells. Curr Cancer Drug Targets (2019) 19(7):571–82. doi: 10.2174/1568009618666181016165604
86. Bunda S, Heir P, Li A, Mamatjan Y, Zadeh G, Aldape K. C-Src Phosphorylates and Inhibits the Function of the CIC Tumor Suppressor Protein. Mol Cancer Res (2020) 18(5):774–86. doi: 10.1158/1541-7786.MCR-18-1370
87. Villarroel A, Del Valle-Pérez B, Fuertes G, Curto J, Ontiveros N, Garcia de Herreros A, et al. Src and Fyn Define a New Signaling Cascade Activated by Canonical and non-Canonical Wnt Ligands and Required for Gene Transcription and Cell Invasion. Cell Mol Life Sci CMLS (2020) 77(5):919–35. doi: 10.1007/s00018-019-03221-2
88. Wick W, Weller M, Weiler M, Batchelor T, Yung AWK, Platten M. Pathway Inhibition: Emerging Molecular Targets for Treating Glioblastoma. Neuro-Oncology (2011) 13(6):566–79. doi: 10.1093/neuonc/nor039
89. Dey N, Crosswell HE, De P, Parsons R, Peng Q, Su JD, et al. The Protein Phosphatase Activity of PTEN Regulates SRC Family Kinases and Controls Glioma Migration. Cancer Res (2008) 68(6):1862–71. doi: 10.1158/0008-5472.CAN-07-1182
90. Brattås MK, Reikvam H, Tvedt THA, Bruserud Ø. Dasatinib as an Investigational Drug for the Treatment of Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia in Adults. Expert Opin Inv Drug (2019) 28(5):411–20. doi: 10.1080/13543784.2019.1597052
91. Schiff D, Sarkaria J. Dasatinib in Recurrent Glioblastoma: Failure as a Teacher. Neuro-Oncology (2015) 17(7):910–1. doi: 10.1093/neuonc/nov086
92. Broniscer A, Jia S, Mandrell B, Hamideh D, Huang J, Onar-Thomas A, et al. Phase 1 Trial, Pharmacokinetics, and Pharmacodynamics of Dasatinib Combined With Crizotinib in Children With Recurrent or Progressive High-Grade and Diffuse Intrinsic Pontine Glioma. Pediatr Blood Cancer (2018) 65(7):e27035. doi: 10.1002/pbc.27035
93. Franceschi E, Stupp R, van den Bent MJ, van Herpen C, Laigle Donadey F, Gorlia T, et al. EORTC 26083 Phase I/II Trial of Dasatinib in Combination With CCNU in Patients With Recurrent Glioblastoma. Neuro Oncol (2012) 14:1503–10. doi: 10.1093/neuonc/nos256
94. Galanis E, Anderson SK, Twohy EL, Carrero XW, Dixon JG, Tran DD, et al. A Phase 1 and Randomized, Placebo-Controlled Phase 2 Trial of Bevacizumab Plus Dasatinib in Patients With Recurrent Glioblastoma: Alliance/North Central Cancer Treatment Group N0872. Cancer-Am Cancer Soc (2019) 125(21):3790–800. doi: 10.1002/cncr.32340
95. Greish K, Jasim A, Parayath N, Abdelghany S, Alkhateeb A, Taurin S, et al. Micellar Formulations of Crizotinib and Dasatinib in the Management of Glioblastoma Multiforme. J Drug Target (2018) 26(8):692–708. doi: 10.1080/1061186X.2017.1419357
96. Truffaux N, Philippe C, Paulsson J, Andreiuolo F, Guerrini-Rousseau L, Cornilleau G, et al. Preclinical Evaluation of Dasatinib Alone and in Combination With Cabozantinib for the Treatment of Diffuse Intrinsic Pontine Glioma. Neuro-Oncology (2015) 17(7):953–64. doi: 10.1093/neuonc/nou330
97. Lee EQ, Muzikansky A, Duda DG, Gaffey S, Dietrich J, Nayak L, et al. Phase II Trial of Ponatinib in Patients With Bevacizumab-Refractory Glioblastoma. Cancer Med-US (2019) 8(13):5988–94. doi: 10.1002/cam4.2505
98. Taylor JW, Dietrich J, Gerstner ER, Norden AD, Rinne ML, Cahill DP, et al. Phase 2 Study of Bosutinib, a Src Inhibitor, in Adults With Recurrent Glioblastoma. J Neurooncol (2015) 121:557–63. doi: 10.1007/s11060-014-1667-z
99. Anderson JC, Duarte CW, Welaya K, Rohrbach TD, Bredel M, Yang ES, et al. Kinomic Exploration of Temozolomide and Radiation Resistance in Glioblastoma Multiforme Xenolines. Radiother Oncol (2014) 111(3):468–74. doi: 10.1016/j.radonc.2014.04.010
100. Bernhart E, Damm S, Heffeter P, Wintersperger A, Asslaber M, Frank S, et al. Silencing of Protein Kinase D2 Induces Glioma Cell Senescence via P53-Dependent and -Independent Pathways. Neuro Oncol (2014) 16(7):933–45. doi: 10.1093/neuonc/not303
101. Funato K, Hayashi T, Echizen K, Negishi L, Shimizu N, Koyama-Nasu R, et al. SIRT2-Mediated Inactivation of P73 is Required for Glioblastoma Tumorigenicity. EMBO Rep (2018) 19(11):e45587. doi: 10.15252/embr.201745587
102. Kusne Y, Carrera-Silva EA, Perry AS, Rushing EJ, Mandell EK, Dietrich JD, et al. Targeting aPKC Disables Oncogenic Signaling by Both the EGFR and the Proinflammatory Cytokine Tnfα in Glioblastoma. Sci Signal (2014) 7(338):a75. doi: 10.1126/scisignal.2005196
103. Kim RK, Suh Y, Hwang E, Yoo KC, Choi KS, An S, et al. Pkcδ Maintains Phenotypes of Tumor Initiating Cells Through Cytokine-Mediated Autocrine Loop With Positive Feedback. Oncogene (2015) 34(46):5749–59. doi: 10.1038/onc.2015.29
104. Gierach GL, Curtis RE, Pfeiffer RM, Mullooly M, Ntowe EA, Hoover RN, et al. Association of Adjuvant Tamoxifen and Aromatase Inhibitor Therapy With Contralateral Breast Cancer Risk Among US Women With Breast Cancer in a General Community Setting JAMA Oncol (2017) 3(2):186–93. doi: 10.1001/jamaoncol.2016.3340
105. Robins HI, Won M, Seiferheld WF, Schultz CJ, Choucair AK, Brachman DG, et al. Phase 2 Trial of Radiation Plus High-Dose Tamoxifen for Glioblastoma Multiforme: RTOG Protocol BR-0021. Neuro Oncol (2006) 8:47–52. doi: 10.1215/S1522851705000311
106. Seystahl K, Weller M. Is There a World Beyond Bevacizumab in Targeting Angiogenesis in Glioblastoma? Expert Opin Investig Drugs (2012) 21(5):605–17. doi: 10.1517/13543784.2012.670219
107. Wick W, Puduvalli VK, Chamberlain MC, van den Bent MJ, Carpentier AF, Cher LM, et al. Phase III Study of Enzastaurin Compared With Lomustine in the Treatment of Recurrent Intracranial Glioblastoma. J Clin Oncol (2010) 28:1168–74. doi: 10.1200/JCO.2009.23.2595
108. Zhao H, Wang J, Shao W, Wu C, Chen Z, To ST, et al. Recent Advances in the Use of PI3K Inhibitors for Glioblastoma Multiforme: Current Preclinical and Clinical Development. Mol Cancer (2017) 16(1):100. doi: 10.1186/s12943-017-0670-3
109. Liu M, Lin Y, Zhang XC, Tan YH, Yao YL, Tan J, et al. Et Al: Phosphorylated mTOR and YAP Serve as Prognostic Markers and Therapeutic Targets in Gliomas. Lab Invest (2017) 97(11):1354–63. doi: 10.1038/labinvest.2017.70
110. Powis G, Berggren M, Gallegos A, Frew T, Hill S, Kozikowski A, et al. Advances With Phospholipid Signalling as a Target for Anticancer Drug Development. Acta Biochim Pol (1995) 42(4):395–403. doi: 10.18388/abp.1995_4893
111. Dai Z, Wang L, Wang X, Zhao B, Zhao W, Bhardwaj SS, et al. Oxymatrine Induces Cell Cycle Arrest and Apoptosis and Suppresses the Invasion of Human Glioblastoma Cells Through the EGFR/PI3K/Akt/mTOR Signaling Pathway and STAT3. Oncol Rep (2018) 40(2):867–76. doi: 10.3892/or.2018.6512
112. Zhu Y, Fang J, Wang H, Fei M, Tang T, Liu K, et al. Baicalin Suppresses Proliferation, Migration, and Invasion in Human Glioblastoma Cells via Ca(2+)-Dependent Pathway. Drug Des Devel Ther (2018) 12:3247–61. doi: 10.2147/DDDT.S176403
113. Luo M, Liu Q, He M, Yu Z, Pi R, Li M, et al. Gartanin Induces Cell Cycle Arrest and Autophagy and Suppresses Migration Involving PI3K/Akt/mTOR and MAPK Signalling Pathway in Human Glioma Cells. J Cell Mol Med (2017) 21(1):46–57. doi: 10.1111/jcmm.12937
114. Burris HAR. Overcoming Acquired Resistance to Anticancer Therapy: Focus on the PI3K/AKT/mTOR Pathway. Cancer Chemoth Pharm (2013) 71(4):829–42. doi: 10.1007/s00280-012-2043-3
115. Cantley LC. The Phosphoinositide 3-Kinase Pathway. Sci (New York NY) (2002) 296(5573):1655–7. doi: 10.1126/science.296.5573.1655
116. Gymnopoulos M, Elsliger M, Vogt PK. Rare Cancer-Specific Mutations in PIK3CA Show Gain of Function. P Natl Acad Sci USA (2007) 104(13):5569–74. doi: 10.1073/pnas.0701005104
117. Netland IA, Førde HE, Sleire L, Leiss L, Rahman MA, Skeie BS, et al. Treatment With the PI3K Inhibitor Buparlisib (NVP-BKM120) Suppresses the Growth of Established Patient-Derived GBM Xenografts and Prolongs Survival in Nude Rats. J Neurooncol (2016) 129(1):57–66. doi: 10.1007/s11060-016-2158-1
118. Xie S, Ni J, McFaline-Figueroa JR, Wang Y, Bronson RT, Ligon KL, et al. Divergent Roles of PI3K Isoforms in PTEN-Deficient Glioblastomas. Cell Rep (2020) 32(13):108196. doi: 10.1016/j.celrep.2020.108196
119. Lin CJ, Chen TL, Tseng YY, Wu GJ, Hsieh MH, Lin YW, et al. Honokiol Induces Autophagic Cell Death in Malignant Glioma Through Reactive Oxygen Species-Mediated Regulation of the P53/PI3K/Akt/mTOR Signaling Pathway. Toxicol Appl Pharmacol (2016) 304:59–69. doi: 10.1016/j.taap.2016.05.018
120. Zhang L, Wang H, Ding K, Xu J. FTY720 Induces Autophagy-Related Apoptosis and Necroptosis in Human Glioblastoma Cells. Toxicol Lett (2015) 236(1):43–59. doi: 10.1016/j.toxlet.2015.04.015
121. Jones NM, Rowe MR, Shepherd PR, McConnell MJ. Targeted Inhibition of Dominant PI3-Kinase Catalytic Isoforms Increase Expression of Stem Cell Genes in Glioblastoma Cancer Stem Cell Models. Int J Oncol (2016) 49(1):207–16. doi: 10.3892/ijo.2016.3510
122. Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, et al. The mTOR-Regulated Phosphoproteome Reveals a Mechanism of Mtorc1-Mediated Inhibition of Growth Factor Signaling. Sci (New York NY) (2011) 332(6035):1317–22. doi: 10.1126/science.1199498
123. Song G, Ouyang G, Bao S. The Activation of Akt/PKB Signaling Pathway and Cell Survival. J Cell Mol Med (2005) 9(1):59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x
124. Wang W, Shen T, Dong B, Creighton CJ, Meng Y, Zhou W, et al. MAPK4 Overexpression Promotes Tumor Progression via Noncanonical Activation of AKT/mTOR Signaling. J Clin Invest (2019) 129(3):1015–29. doi: 10.1172/JCI97712
125. Yu Z, Xie G, Zhou G, Cheng Y, Zhang G, Yao G, et al. NVP-BEZ235, a Novel Dual PI3K-mTOR Inhibitor Displays Anti-Glioma Activity and Reduces Chemoresistance to Temozolomide in Human Glioma Cells. Cancer Lett (2015) 367(1):58–68. doi: 10.1016/j.canlet.2015.07.007
126. Mehta M, Khan A, Danish S, Haffty BG, Sabaawy HE. Radiosensitization of Primary Human Glioblastoma Stem-Like Cells With Low-Dose AKT Inhibition. Mol Cancer Ther (2015) 14(5):1171–80. doi: 10.1158/1535-7163.MCT-14-0708
127. Duzgun Z, Eroglu Z, Biray Avci C. Role of mTOR in Glioblastoma. GENE (2016) 575(2 Pt 1):187–90. doi: 10.1016/j.gene.2015.08.060
128. Carbonneau M, GL M, Lalonde ME, Germain MA, Motorina A, Guiot MC, et al. The Oncometabolite 2-Hydroxyglutarate Activates the mTOR Signalling Pathway. Nat Commun (2016) 7:12700. doi: 10.1038/ncomms12700
129. Li Y, Ma X, Wang Y, Li G. miR-489 Inhibits Proliferation, Cell Cycle Progression and Induces Apoptosis of Glioma Cells via Targeting SPIN1-Mediated PI3K/AKT Pathway. BioMed Pharmacother (2017) 93:435–43. doi: 10.1016/j.biopha.2017.06.058
130. Nan Y, Guo H, Guo L, Wang L, Ren B, Yu K, et al. MiRNA-451 Inhibits Glioma Cell Proliferation and Invasion Through the mTOR/HIF-1α/VEGF Signaling Pathway by Targeting Cab39. Hum Gene Ther Clin Dev (2018) 29(3):156–66. doi: 10.1089/humc.2018.133
131. Xu DH, Chi GN, Zhao CH, Li DY. Long Noncoding RNA MEG3 Inhibits Proliferation and Migration But Induces Autophagy by Regulation of Sirt7 and PI3K/AKT/mTOR Pathway in Glioma Cells. J Cell Biochem (2018). doi: 10.1002/jcb.28026
132. Yao J, Wang Z, Cheng Y, Ma C, Zhong Y, Xiao Y, et al. M2 Macrophage-Derived Exosomal microRNAs Inhibit Cell Migration and Invasion in Gliomas Through PI3K/AKT/mTOR Signaling Pathway. J Transl Med (2021) 19(1):99. doi: 10.1186/s12967-021-02766-w
133. Fan QW, Nicolaides TP, Weiss WA. Inhibiting 4EBP1 in Glioblastoma. Clin Cancer Res (2018) 24(1):14–21. doi: 10.1158/1078-0432.CCR-17-0042
134. von Achenbach C, Weller M, Kaulich K, Gramatzki D, Zacher A, Fabbro D, et al. Synergistic Growth Inhibition Mediated by Dual PI3K/mTOR Pathway Targeting and Genetic or Direct Pharmacological AKT Inhibition in Human Glioblastoma Models. J Neurochem (2020) 153(4):510–24. doi: 10.1111/jnc.14899
135. Gao Y, Li L, Zheng H, Zhou C, Chen X, Hao B, et al. KIF3C is Associated With Favorable Prognosis in Glioma Patients and may be Regulated by PI3K/AKT/mTOR Pathway. J Neurooncol (2020) 146(3):513–21. doi: 10.1007/s11060-020-03399-7
136. Zhu J, Li K, Yu L, Chen Y, Cai Y, Jin J, et al. Targeting Phosphatidylinositol 3-Kinase Gamma (PI3Kγ): Discovery and Development of its Selective Inhibitors. Med Res Rev (2021) 41(3):1599–621. doi: 10.1002/med.21770
137. Stoyanov B, Volinia S, Hanck T, Rubio I, Loubtchenkov M, Malek D, et al. Cloning and Characterization of a G Protein-Activated Human Phosphoinositide-3 Kinase. Sci (New York NY) (1995) 269(5224):690–3. doi: 10.1126/science.7624799
138. André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med (2019) 380:1929–40. doi: 10.1056/NEJMoa1813904
139. Burger MT, Pecchi S, Wagman A, Ni ZJ, Knapp M, Hendrickson T, et al. Identification of NVP-BKM120 as a Potent, Selective, Orally Bioavailable Class I PI3 Kinase Inhibitor for Treating Cancer. ACS Med Chem Lett (2011) 2(10):774–9. doi: 10.1021/ml200156t
140. Ndubaku CO, Heffron TP, Staben ST, Baumgardner M, Blaquiere N, Bradley E, et al. Discovery of 2-{3-[2-(1-Isopropyl-3-Methyl-1H-1,2–4-Triazol-5-Yl)-5,6-Dihydrobenzo[F]Imidazo[1,2-D][1,4]Oxazepin-9-Yl]-1H-Pyrazol-1-Yl}-2-Methylpropanamide (GDC-0032): A β-Sparing Phosphoinositide 3-Kinase Inhibitor With High Unbound Exposure and Robust in Vivo Antitumor Activity. J Med Chem (2013) 56(11):4597–610. doi: 10.1021/jm4003632
141. Rodon J, Tabernero J. Improving the Armamentarium of PI3K Inhibitors With Isoform-Selective Agents: A New Light in the Darkness. Cancer Discov (2017) 7(7):666-9. doi: 10.1158/2159-8290.CD-17-0500
142. Carrano A, Juarez JJ, Incontri D, Ibarra A, Guerrero Cazares H. Sex-Specific Differences in Glioblastoma. Cells-Basel (2021) 10(7):1783. doi: 10.3390/cells10071783
143. Munoz J, Follows GA, Nastoupil LJ. Copanlisib for the Treatment of Malignant Lymphoma: Clinical Experience and Future Perspectives. Target Oncol (2021) 16(3):295–308. doi: 10.1007/s11523-021-00802-9
144. Edelman G, Rodon J, Lager J, Castell C, Jiang J, Van Allen EM, et al. Phase I Trial of a Tablet Formulation of Pilaralisib, a Pan-Class I PI3K Inhibitor, in Patients With Advanced Solid Tumors. Oncologist (2018) 23:401–38. doi: 10.1634/theoncologist.2017-0691
145. Foster KA, Jane EP, Premkumar DR, Morales A, Pollack IF. NVP-BKM120 Potentiates Apoptosis in Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Resistant Glioma Cell Lines via Upregulation of Noxa and Death Receptor 5. Int J Oncol (2015) 47(2):506–16. doi: 10.3892/ijo.2015.3035
146. Speranza MC, Nowicki MO, Behera P, Cho CF, Chiocca EA, Lawler SE. BKM-120 (Buparlisib): A Phosphatidyl-Inositol-3 Kinase Inhibitor With Anti-Invasive Properties in Glioblastoma. Sci Rep (2016) 6:20189. doi: 10.1038/srep20189
147. Koul D, Fu J, Shen R, LaFortune TA, Wang S, Tiao N, et al. Antitumor Activity of NVP-BKM120–a Selective Pan Class I PI3 Kinase Inhibitor Showed Differential Forms of Cell Death Based on P53 Status of Glioma Cells. Clin Cancer Res (2012) 18(1):184–95. doi: 10.1158/1078-0432.CCR-11-1558
148. Wachsberger PR, Lawrence YR, Liu Y, Rice B, Feo N, Leiby B, et al. Hsp90 Inhibition Enhances PI-3 Kinase Inhibition and Radiosensitivity in Glioblastoma. J Cancer Res Clin Oncol (2014) 140(4):573–82. doi: 10.1007/s00432-014-1594-6
149. Jane EP, Premkumar DR, Morales A, Foster KA, Pollack IF. Inhibition of Phosphatidylinositol 3-Kinase/AKT Signaling by NVP-BKM120 Promotes ABT-737-Induced Toxicity in a Caspase-Dependent Manner Through Mitochondrial Dysfunction and DNA Damage Response in Established and Primary Cultured Glioblastoma Cells. J Pharmacol Exp Ther (2014) 350(1):22–35. doi: 10.1124/jpet.114.212910
150. Rosenthal M, Clement PM, Campone M, Gil-Gil MJ, DeGroot J, Chinot O, et al. Buparlisib Plus Carboplatin or Lomustine in Patients With Recurrent Glioblastoma: A Phase Ib/II, Open-Label, Multicentre, Randomised Study. ESMO Open (2020) 5(4):e000672. doi: 10.1136/esmoopen-2020-000672
151. van den Bent M, Azaro A, De Vos F, Sepulveda J, Yung W, Wen PY, et al. A Phase Ib/II, Open-Label, Multicenter Study of INC280 (Capmatinib) Alone and in Combination With Buparlisib (BKM120) in Adult Patients With Recurrent Glioblastoma. J Neurooncol (2020) 146(1):79–89. doi: 10.1007/s11060-019-03337-2
152. Hainsworth JD, Becker KP, Mekhail T, Chowdhary SA, Eakle JF, Wright D, et al. Phase I/II Study of Bevacizumab With BKM120, an Oral PI3K Inhibitor, in Patients With Refractory Solid Tumors (Phase I) and Relapsed/Refractory Glioblastoma (Phase II). J Neurooncol (2019) 144(2):303–11. doi: 10.1007/s11060-019-03227-7
153. Cheng Y, Ren X, Zhang Y, Patel R, Sharma A, Wu H, et al. eEF-2 Kinase Dictates Cross-Talk Between Autophagy and Apoptosis Induced by Akt Inhibition, Thereby Modulating Cytotoxicity of Novel Akt Inhibitor MK-2206. Cancer Res (2011) 71(7):2654–63. doi: 10.1158/0008-5472.CAN-10-2889
154. Cheng Y, Zhang Y, Zhang L, Ren X, Huber-Keener KJ, Liu X, et al. MK-2206, a Novel Allosteric Inhibitor of Akt, Synergizes With Gefitinib Against Malignant Glioma via Modulating Both Autophagy and Apoptosis. Mol Cancer Ther (2012) 11(1):154–64. doi: 10.1158/1535-7163.MCT-11-0606
155. Narayan RS, Fedrigo CA, Brands E, Dik R, Stalpers LJ, Baumert BG, et al. The Allosteric AKT Inhibitor MK2206 Shows a Synergistic Interaction With Chemotherapy and Radiotherapy in Glioblastoma Spheroid Cultures. BMC Cancer (2017) 17(1):204. doi: 10.1186/s12885-017-3193-9
156. Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, et al. Alternative Rapamycin Treatment Regimens Mitigate the Impact of Rapamycin on Glucose Homeostasis and the Immune System. Aging Cell (2016) 15(1):28–38. doi: 10.1111/acel.12405
157. Poore B, Yuan M, Arnold A, Price A, Alt J, Rubens JA, et al. Inhibition of Mtorc1 in Pediatric Low-Grade Glioma Depletes Glutathione and Therapeutically Synergizes With Carboplatin. Neuro Oncol (2019) 21(2):252–63. doi: 10.1093/neuonc/noy150
158. Babak S, Mason WP. mTOR Inhibition in Glioblastoma: Requiem for a Dream? Neuro Oncol (2018) 20(5):584–5. doi: 10.1093/neuonc/noy034
159. Chinnaiyan P, Won M, Wen PY, Rojiani AM, Werner-Wasik M, Shih HA, et al. A Randomized Phase II Study of Everolimus in Combination With Chemoradiation in Newly Diagnosed Glioblastoma: Results of NRG Oncology RTOG 0913. Neuro Oncol (2018) 20(5):666–73. doi: 10.1093/neuonc/nox209
160. Miklja Z, Yadav VN, Cartaxo RT, Siada R, Thomas CC, Cummings JR, et al. Everolimus Improves the Efficacy of Dasatinib in Pdgfrα-Driven Glioma. J Clin Invest (2020) 130(10):5313–25. doi: 10.1172/JCI133310
161. Cacchione A, Lodi M, Carai A, Miele E, Tartaglia M, Megaro G, et al. Upfront Treatment With mTOR Inhibitor Everolimus in Pediatric Low-Grade Gliomas: A Single-Center Experience. Int J Cancer (2020). doi: 10.1002/ijc.33438
162. Failly M, Korur S, Egler V, Boulay JL, Lino MM, Imber R, et al. Combination of Sublethal Concentrations of Epidermal Growth Factor Receptor Inhibitor and Microtubule Stabilizer Induces Apoptosis of Glioblastoma Cells. Mol Cancer Ther (2007) 6(2):773–81. doi: 10.1158/1535-7163.MCT-06-0566
163. Flannery PC, DeSisto JA, Amani V, Venkataraman S, Lemma RT, Prince EW, et al. Preclinical Analysis of MTOR Complex 1/2 Inhibition in Diffuse Intrinsic Pontine Glioma. Oncol Rep (2018) 39(2):455–64. doi: 10.3892/or.2017.6122
164. Mukherjee S, Mukherjee U. A Comprehensive Review of Immunosuppression Used for Liver Transplantation. J Transplant (2009) 2009:701464. doi: 10.1155/2009/701464
165. Johnston O, Rose CL, Webster AC, Gill JS. Sirolimus Is Associated With New-Onset Diabetes in Kidney Transplant Recipients. J Am Soc Nephrol JASN (2008) 19(7):1411–8. doi: 10.1681/ASN.2007111202
166. Arriola Apelo SI, Lamming DW. Rapamycin: An InhibiTOR of Aging Emerges From the Soil of Easter Island. J Gerontol Ser A Biol Sci Med Sci (2016) 71(7):841–9. doi: 10.1093/gerona/glw090
167. Lee EQ, Kuhn J, Lamborn KR, Abrey L, DeAngelis LM, Lieberman F, et al. Phase I/II Study of Sorafenib in Combination With Temsirolimus for Recurrent Glioblastoma or Gliosarcoma: North American Brain Tumor Consortium Study 05-02. Neuro Oncol (2012) 14(12):1511–8. doi: 10.1093/neuonc/nos264
168. Schiff D, Jaeckle KA, Anderson SK, Galanis E, Giannini C, Buckner JC, et al. Phase 1/2 Trial of Temsirolimus and Sorafenib in the Treatment of Patients With Recurrent Glioblastoma: North Central Cancer Treatment Group Study/Alliance N0572. Cancer-Am Cancer Soc (2018) 124(7):1455–63. doi: 10.1002/cncr.31219
169. Kaley TJ, Panageas KS, Pentsova EI, Mellinghoff IK, Nolan C, Gavrilovic I, et al. Phase I Clinical Trial of Temsirolimus and Perifosine for Recurrent Glioblastoma. Ann Clin Transl Neurol (2020) 7(4):429–36. doi: 10.1002/acn3.51009
170. Wennerberg K, Rossman KL, Der CJ. The Ras Superfamily at a Glance. J Cell Sci (2005) 118(Pt 5):843–6. doi: 10.1242/jcs.01660
171. Basto D, Trovisco V, Lopes JM, Martins A, Pardal F, Soares P, et al. Mutation Analysis of B-RAF Gene in Human Gliomas. Acta Neuropathol (2005) 109:207–10. doi: 10.1007/s00401-004-0936-x
172. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-Targeted Therapies. Nat Rev Drug Discov (2021). doi: 10.1038/s41573-021-00220-6
173. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF Gene in Human Cancer. Nature (2002) 417(6892):949–54. doi: 10.1038/nature00766
174. Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ, et al. Activating Mutations in BRAF Characterize a Spectrum of Pediatric Low-Grade Gliomas. Neuro Oncol (2010) 12:621–30. doi: 10.1093/neuonc/noq007
175. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E Mutation in 1,320 Nervous System Tumors Reveals High Mutation Frequencies in Pleomorphic Xanthoastrocytoma, Ganglioglioma and Extra-Cerebellar Pilocytic Astrocytoma. Acta Neuropathol (2011) 121(3):397–405. doi: 10.1007/s00401-011-0802-6
176. Johanns TM, Ferguson CJ, Grierson PM, Dahiya S, Ansstas G. Rapid Clinical and Radiographic Response With Combined Dabrafenib and Trametinib in Adults With BRAF-Mutated High-Grade Glioma. J Natl Compr Canc Netw (2018) 16(1):4–10. doi: 10.6004/jnccn.2017.7032
177. Shabason JE, Tofilon PJ, Camphausen K. Grand Rounds at the National Institutes of Health: HDAC Inhibitors as Radiation Modifiers, From Bench to Clinic. J Cell Mol Med (2011) 15(12):2735–44. doi: 10.1111/j.1582-4934.2011.01296.x
178. Wei L, Hong S, Yoon Y, Hwang SN, Park JC, Zhang Z, et al. Early Prediction of Response to Vorinostat in an Orthotopic Rat Glioma Model. NMR BioMed (2012) 25(9):1104–11. doi: 10.1002/nbm.2776
179. Eyüpoglu IY, Savaskan NE. Epigenetics in Brain Tumors: HDACs Take Center Stage. Curr Neuropharmacol (2016) 14(1):48–54. doi: 10.2174/1570159X13666151030162457
180. Marampon F, Megiorni F, Camero S, Crescioli C, McDowell HP, Sferra R, et al. HDAC4 and HDAC6 Sustain DNA Double Strand Break Repair and Stem-Like Phenotype by Promoting Radioresistance in Glioblastoma Cells. Cancer Lett (2017) 397:1–11. doi: 10.1016/j.canlet.2017.03.028
181. Zhang Z, Wang Y, Chen J, Tan Q, Xie C, Li C, et al. Silencing of Histone Deacetylase 2 Suppresses Malignancy for Proliferation, Migration, and Invasion of Glioblastoma Cells and Enhances Temozolomide Sensitivity. Cancer Chemother Pharmacol (2016) 78(6):1289–96. doi: 10.1007/s00280-016-3188-2
182. Ecker J, Oehme I, Mazitschek R, Korshunov A, Kool M, Hielscher T, et al. Targeting Class I Histone Deacetylase 2 in MYC Amplified Group 3 Medulloblastoma. Acta Neuropathol Commun (2015) 3:22. doi: 10.1186/s40478-015-0201-7
183. Pak E, MacKenzie EL, Zhao X, Pazyra-Murphy MF, Park P, Wu L, et al. A Large-Scale Drug Screen Identifies Selective Inhibitors of Class I HDACs as a Potential Therapeutic Option for SHH Medulloblastoma. Neuro Oncol (2019) 21(9):1150–63. doi: 10.1093/neuonc/noz089
184. Yang WB, Hsu CC, Hsu TI, Liou JP, Chang KY, Chen PY, et al. Increased Activation of HDAC1/2/6 and Sp1 Underlies Therapeutic Resistance and Tumor Growth in Glioblastoma. Neuro Oncol (2020) 22(10):1439–51. doi: 10.1093/neuonc/noaa103
185. Marks PA, Richon VM, Miller T, Kelly WK. Histone Deacetylase Inhibitors. Adv Cancer Res (2004) 91:137–68. doi: 10.1016/S0065-230X(04)91004-4
186. Anastas JN, Zee BM, Kalin JH, Kim M, Guo R, Alexandrescu S, et al. Re-Programing Chromatin With a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell (2019) 36(5):528–44. doi: 10.1016/j.ccell.2019.09.005
187. Meel MH, de Gooijer MC, Metselaar DS, Sewing A, Zwaan K, Waranecki P, et al. Combined Therapy of AXL and HDAC Inhibition Reverses Mesenchymal Transition in Diffuse Intrinsic Pontine Glioma. Clin Cancer Res (2020) 26(13):3319–32. doi: 10.1158/1078-0432.CCR-19-3538
188. Arrizabalaga O, Moreno-Cugnon L, Auzmendi-Iriarte J, Aldaz P, Ibanez DCI, Garros-Regulez L, et al. High Expression of MKP1/DUSP1 Counteracts Glioma Stem Cell Activity and Mediates HDAC Inhibitor Response. Oncogenesis (2017) 6(12):401. doi: 10.1038/s41389-017-0003-9
189. Marampon F, Leoni F, Mancini A, Pietrantoni I, Codenotti S, Ferella L, et al. Histone Deacetylase Inhibitor ITF2357 (Givinostat) Reverts Transformed Phenotype and Counteracts Stemness in In Vitro and In Vivo Models of Human Glioblastoma. J Cancer Res Clin Oncol (2019) 145(2):393–409. doi: 10.1007/s00432-018-2800-8
190. Singh MM, Johnson B, Venkatarayan A, Flores ER, Zhang J, Su X, et al. Preclinical Activity of Combined HDAC and KDM1A Inhibition in Glioblastoma. Neuro Oncol (2015) 17(11):1463–73. doi: 10.1093/neuonc/nov041
191. Buyandelger B, Bar EE, Hung KS, Chen RM, Chiang YH, Liou JP, et al. Histone Deacetylase Inhibitor MPT0B291 Suppresses Glioma Growth In Vitro and In Vivo Partially Through Acetylation of P53. Int J Biol Sci (2020) 16(16):3184–99. doi: 10.7150/ijbs.45505
192. Pal S, Kozono D, Yang X, Fendler W, Fitts W, Ni J, et al. Dual HDAC and PI3K Inhibition Abrogates Nfκb- and FOXM1-Mediated DNA Damage Response to Radiosensitize Pediatric High-Grade Gliomas. Cancer Res (2018) 78(14):4007–21. doi: 10.1158/0008-5472.CAN-17-3691
193. Li ZY, Li QZ, Chen L, Chen BD, Wang B, Zhang XJ, et al. Histone Deacetylase Inhibitor RGFP109 Overcomes Temozolomide Resistance by Blocking NF-κb-Dependent Transcription in Glioblastoma Cell Lines. Neurochem Res (2016) 41(12):3192–205. doi: 10.1007/s11064-016-2043-5
194. Kitange GJ, Mladek AC, Carlson BL, Schroeder MA, Pokorny JL, Cen L, et al. Inhibition of Histone Deacetylation Potentiates the Evolution of Acquired Temozolomide Resistance Linked to MGMT Upregulation in Glioblastoma Xenografts. Clin Cancer Res (2012) 18(15):4070–9. doi: 10.1158/1078-0432.CCR-12-0560
195. Liffers K, Kolbe K, Westphal M, Lamszus K, Schulte A. Histone Deacetylase Inhibitors Resensitize EGFR/EGFRvIII-Overexpressing, Erlotinib-Resistant Glioblastoma Cells to Tyrosine Kinase Inhibition. Target Oncol (2016) 11(1):29–40. doi: 10.1007/s11523-015-0372-y
196. Pei Y, Liu KW, Wang J, Garancher A, Tao R, Esparza LA, et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell (2016) 29(3):311–23. doi: 10.1016/j.ccell.2016.02.011
197. Maleszewska M, Steranka A, Smiech M, Kaza B, Pilanc P, Dabrowski M, et al. Sequential Changes in Histone Modifications Shape Transcriptional Responses Underlying Microglia Polarization by Glioma. GLIA (2021) 69(1):109–23. doi: 10.1002/glia.23887
198. Nguyen T, Zhang Y, Shang E, Shu C, Torrini C, Zhao J, et al. HDAC Inhibitors Elicit Metabolic Reprogramming by Targeting Super-Enhancers in Glioblastoma Models. J Clin Invest (2020) 130(7):3699–716. doi: 10.1172/JCI129049
199. Festuccia C, Mancini A, Colapietro A, Gravina GL, Vitale F, Marampon F, et al. The First-in-Class Alkylating Deacetylase Inhibitor Molecule Tinostamustine Shows Antitumor Effects and is Synergistic With Radiotherapy in Preclinical Models of Glioblastoma. J Hematol Oncol (2018) 11(1):32. doi: 10.1186/s13045-018-0576-6
200. Zhang Y, Ishida CT, Ishida W, Lo SL, Zhao J, Shu C, et al. Combined HDAC and Bromodomain Protein Inhibition Reprograms Tumor Cell Metabolism and Elicits Synthetic Lethality in Glioblastoma. Clin Cancer Res (2018) 24(16):3941–54. doi: 10.1158/1078-0432.CCR-18-0260
201. Gonçalves RM, Agnes JP, Delgobo M, de Souza PO, Thomé MP, Heimfarth L, et al. Late Autophagy Inhibitor Chloroquine Improves Efficacy of the Histone Deacetylase Inhibitor SAHA and Temozolomide in Gliomas. Biochem Pharmacol (2019) 163:440–50. doi: 10.1016/j.bcp.2019.03.015
202. Watanabe S, Kuwabara Y, Suehiro S, Yamashita D, Tanaka M, Tanaka A, et al. Valproic Acid Reduces Hair Loss and Improves Survival in Patients Receiving Temozolomide-Based Radiation Therapy for High-Grade Glioma. Eur J Clin Pharmacol (2017) 73(3):357–63. doi: 10.1007/s00228-016-2167-1
203. Rasmussen TA, Schmeltz Søgaard O, Brinkmann C, Wightman F, Lewin SR, Melchjorsen J, et al. Comparison of HDAC Inhibitors in Clinical Development: Effect on HIV Production in Latently Infected Cells and T-Cell Activation. Hum Vacc Immunother (2013) 9(5):993–1001. doi: 10.4161/hv.23800
204. Richon V. Cancer Biology: Mechanism of Antitumour Action of Vorinostat (Suberoylanilide Hydroxamic Acid), a Novel Histone Deacetylase Inhibitor. Br J Cancer (2021) L6:p53–90. doi: 10.1038/sj.bjc.6603463
205. Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, et al. Phase II Trial of Vorinostat in Combination With Bortezomib in Recurrent Glioblastoma: A North Central Cancer Treatment Group Study. Neuro Oncol (2012) 14(2):215–21. doi: 10.1093/neuonc/nor198
206. Chinnaiyan P, Chowdhary S, Potthast L, Prabhu A, Tsai YY, Sarcar B, et al. Phase I Trial of Vorinostat Combined With Bevacizumab and CPT-11 in Recurrent Glioblastoma. Neuro Oncol (2012) 14(1):93–100. doi: 10.1093/neuonc/nor187
207. Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, et al. Phase II Trial of Vorinostat in Recurrent Glioblastoma Multiforme: A North Central Cancer Treatment Group Study. J Clin Oncol (2009) 27(12):2052–8. doi: 10.1200/JCO.2008.19.0694
208. Tosi U, Kommidi H, Adeuyan O, Guo H, Maachani UB, Chen N, et al. PET, Image-Guided HDAC Inhibition of Pediatric Diffuse Midline Glioma Improves Survival in Murine Models. Sci Adv (2020) 6(30):b4105. doi: 10.1126/sciadv.abb4105
209. Kim GH, Choi SY, Oh TI, Kan SY, Kang H, Lee S, et al. IDH1(R132H) Causes Resistance to HDAC Inhibitors by Increasing NANOG in Glioblastoma Cells. Int J Mol Sci (2019) 20(11):2679. doi: 10.3390/ijms20112679
210. Pont LM, Naipal K, Kloezeman JJ, Venkatesan S, van den Bent M, van Gent DC, et al. DNA Damage Response and Anti-Apoptotic Proteins Predict Radiosensitization Efficacy of HDAC Inhibitors SAHA and LBH589 in Patient-Derived Glioblastoma Cells. Cancer Lett (2015) 356(2 Pt B):525–35. doi: 10.1016/j.canlet.2014.09.049
211. Sung GJ, Kim SH, Kwak S, Park SH, Song JH, Jung JH, et al. Inhibition of TFEB Oligomerization by Co-Treatment of Melatonin With Vorinostat Promotes the Therapeutic Sensitivity in Glioblastoma and Glioma Stem Cells. J Pineal Res (2019) 66(3):e12556. doi: 10.1111/jpi.12556
212. Sawa H, Murakami H, Kumagai M, Nakasato M, Yamauchi S, Matsuyama N, et al. Histone Deacetylase Inhibitor, FK228, Induces Apoptosis and Suppresses Cell Proliferation of Human Glioblastoma Cells In Vitro and In Vivo. Acta Neuropathol (2004) 107(6):523–31. doi: 10.1007/s00401-004-0841-3
213. Wu Y, Dong L, Bao S, Wang M, Yun Y, Zhu R. FK228 Augmented Temozolomide Sensitivity in Human Glioma Cells by Blocking PI3K/AKT/mTOR Signal Pathways. BioMed Pharmacother (2016) 84:462–9. doi: 10.1016/j.biopha.2016.09.051
214. Iwamoto FM, Lamborn KR, Kuhn JG, Wen PY, Yung WK, Gilbert MR, et al. A Phase I/II Trial of the Histone Deacetylase Inhibitor Romidepsin for Adults With Recurrent Malignant Glioma: North American Brain Tumor Consortium Study 03-03. Neuro Oncol (2011) 13(5):509–16. doi: 10.1093/neuonc/nor017
215. Vitanza NA, Biery MC, Myers C, Ferguson E, Zheng Y, Girard EJ, et al. Optimal Therapeutic Targeting by HDAC Inhibition in Biopsy-Derived Treatment-Naïve Diffuse Midline Glioma Models. Neuro Oncol (2021) 23(3):376–86. doi: 10.1093/neuonc/noaa249
216. Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, et al. Functionally Defined Therapeutic Targets in Diffuse Intrinsic Pontine Glioma. Nat Med (2015) 21(6):555–9. doi: 10.1038/nm.3855
217. Lee EQ, Reardon DA, Schiff D, Drappatz J, Muzikansky A, Grimm SA, et al. Phase II Study of Panobinostat in Combination With Bevacizumab for Recurrent Glioblastoma and Anaplastic Glioma. Neuro Oncol (2015) 17(6):862–7. doi: 10.1093/neuonc/nou350
218. Ghodke-Puranik Y, Thorn CF, Lamba JK, Leeder JS, Song W, Birnbaum AK, et al. Valproic Acid Pathway: Pharmacokinetics and Pharmacodynamics. Pharmacogenet Genom (2013) 23(4):236–41. doi: 10.1097/FPC.0b013e32835ea0b2
219. Barker CA, Bishop AJ, Chang M, Beal K, Chan TA. Valproic Acid Use During Radiation Therapy for Glioblastoma Associated With Improved Survival. Int J Radiat Oncol Biol Phys (2013) 86(3):504–9. doi: 10.1016/j.ijrobp.2013.02.012
220. Redjal N, Reinshagen C, Le A, Walcott BP, McDonnell E, Dietrich J, et al. Valproic Acid, Compared to Other Antiepileptic Drugs, is Associated With Improved Overall and Progression-Free Survival in Glioblastoma But Worse Outcome in Grade II/III Gliomas Treated With Temozolomide. J Neurooncol (2016) 127(3):505–14. doi: 10.1007/s11060-016-2054-8
221. Weller M, Gorlia T, Cairncross JG, van den Bent MJ, Mason W, Belanger K, et al. Prolonged Survival With Valproic Acid Use in the EORTC/NCIC Temozolomide Trial for Glioblastoma. Neurology (2011) 77(12):1156–64. doi: 10.1212/WNL.0b013e31822f02e1
222. Kerkhof M, Dielemans JC, van Breemen MS, Zwinkels H, Walchenbach R, Taphoorn MJ, et al. Effect of Valproic Acid on Seizure Control and on Survival in Patients With Glioblastoma Multiforme. Neuro Oncol (2013) 15(7):961–7. doi: 10.1093/neuonc/not057
223. Berendsen S, Varkila M, Kroonen J, Seute T, Snijders TJ, Kauw F, et al. Prognostic Relevance of Epilepsy at Presentation in Glioblastoma Patients. Neuro Oncol (2016) 18(5):700–6. doi: 10.1093/neuonc/nov238
224. Happold C, Gorlia T, Chinot O, Gilbert MR, Nabors LB, Wick W, et al. Does Valproic Acid or Levetiracetam Improve Survival in Glioblastoma? A Pooled Analysis of Prospective Clinical Trials in Newly Diagnosed Glioblastoma. J Clin Oncol (2016) 34(7):731–9. doi: 10.1200/JCO.2015.63.6563
225. Kusaczuk M, Krętowski R, Stypułkowska A, Cechowska-Pasko M. Molecular and Cellular Effects of a Novel Hydroxamate-Based HDAC Inhibitor - Belinostat - in Glioblastoma Cell Lines: A Preliminary Report. Invest New Drug (2016) 34(5):552–64. doi: 10.1007/s10637-016-0372-5
226. Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, et al. Pharmacologic Inhibition of Cyclin-Dependent Kinases 4 and 6 Arrests the Growth of Glioblastoma Multiforme Intracranial Xenografts. Cancer Res (2010) 70(8):3228–38. doi: 10.1158/0008-5472.CAN-09-4559
227. Hendrychová D, Jorda R, Kryštof V. How Selective are Clinical CDK4/6 Inhibitors? Med Res Rev (2021) 41(3):1578–98. doi: 10.1002/med.21769
228. Yuan K, Wang X, Dong H, Min W, Hao H, Yang P. Selective Inhibition of CDK4/6: A Safe and Effective Strategy for Developing Anticancer Drugs. Acta Pharm Sin B (2021) 11(1):30–54. doi: 10.1016/j.apsb.2020.05.001
229. Olmez I, Brenneman B, Xiao A, Serbulea V, Benamar M, Zhang Y, et al. Combined CDK4/6 and mTOR Inhibition Is Synergistic Against Glioblastoma via Multiple Mechanisms. Clin Cancer Res (2017) 23(22):6958–68. doi: 10.1158/1078-0432.CCR-17-0803
230. Jardim DL, Millis SZ, Ross JS, Woo MS, Ali SM, Kurzrock R. Cyclin Pathway Genomic Alterations Across 190,247 Solid Tumors: Leveraging Large-Scale Data to Inform Therapeutic Directions. Oncologist (2021) 26(1):e78–89. doi: 10.1634/theoncologist.2020-0509
231. Wiedemeyer WR, Dunn IF, Quayle SN, Zhang J, Chheda MG, Dunn GP, et al. Pattern of Retinoblastoma Pathway Inactivation Dictates Response to CDK4/6 Inhibition in GBM. Proc Natl Acad Sci USA (2010) 107(25):11501–6. doi: 10.1073/pnas.1001613107
232. Cen L, Carlson BL, Schroeder MA, Ostrem JL, Kitange GJ, Mladek AC, et al. P16-Cdk4-Rb Axis Controls Sensitivity to a Cyclin-Dependent Kinase Inhibitor PD0332991 in Glioblastoma Xenograft Cells. Neuro Oncol (2012) 14(7):870–81. doi: 10.1093/neuonc/nos114
233. Cook SM, Genovesi LA, Nakamoto MW, Davis MJ, Knobluagh SE, Ji P, et al. Inhibition of CDK4/6 by Palbociclib Significantly Extends Survival in Medulloblastoma Patient-Derived Xenograft Mouse Models. Clin Cancer Res (2017) 23(19):5802–13. doi: 10.1158/1078-0432.CCR-16-2943
234. Liu S, Tang Y, Yuan X, Yuan D, Liu J, Li B, et al. Inhibition of Rb and mTOR Signaling Associates With Synergistic Anticancer Effect of Palbociclib and Erlotinib in Glioblastoma Cells. Invest New Drugs (2018) 36(6):961–9. doi: 10.1007/s10637-018-0575-z
235. Olmez I, Zhang Y, Manigat L, Benamar M, Brenneman B, Nakano I, et al. Combined C-Met/Trk Inhibition Overcomes Resistance to CDK4/6 Inhibitors in Glioblastoma. Cancer Res (2018) 78(15):4360–9. doi: 10.1158/0008-5472.CAN-17-3124
236. Cao Y, Li X, Kong S, Shang S, Qi Y. CDK4/6 Inhibition Suppresses Tumour Growth and Enhances the Effect of Temozolomide In Glioma Cells. J Cell Mol Med (2020) 24(9):5135–45. doi: 10.1111/jcmm.15156
237. Hashizume R, Zhang A, Mueller S, Prados MD, Lulla RR, Goldman S, et al. Inhibition of DNA Damage Repair by the CDK4/6 Inhibitor Palbociclib Delays Irradiated Intracranial Atypical Teratoid Rhabdoid Tumor and Glioblastoma Xenograft Regrowth. Neuro Oncol (2016) 18(11):1519–28. doi: 10.1093/neuonc/now106
238. Portman N, Alexandrou S, Carson E, Wang S, Lim E, Caldon CE. Overcoming CDK4/6 Inhibitor Resistance in ER-Positive Breast Cancer. Endocr-relat Cancer (2019) 26(1):R15–30. doi: 10.1530/ERC-18-0317
239. Guarducci C, Bonechi M, Boccalini G, Benelli M, Risi E, Di Leo A, et al. Mechanisms of Resistance to CDK4/6 Inhibitors in Breast Cancer and Potential Biomarkers of Response. Breast Care (Basel Switzerland) (2017) 12(5):304–8. doi: 10.1159/000484167
240. Morris-Hanon O, Marazita MC, Romorini L, Isaja L, Fernandez-Espinosa DD, Sevlever GE, et al. Palbociclib Effectively Halts Proliferation But Fails to Induce Senescence in Patient-Derived Glioma Stem Cells. Mol Neurobiol (2019) 56(11):7810–21. doi: 10.1007/s12035-019-1633-z
241. Tien AC, Li J, Bao X, Derogatis A, Kim S, Mehta S, et al. A Phase 0 Trial of Ribociclib in Recurrent Glioblastoma Patients Incorporating a Tumor Pharmacodynamic- and Pharmacokinetic-Guided Expansion Cohort. Clin Cancer Res (2019) 25(19):5777–86. doi: 10.1158/1078-0432.CCR-19-0133
242. Wood AC, Krytska K, Ryles HT, Infarinato NR, Sano R, Hansel TD, et al. Dual ALK and CDK4/6 Inhibition Demonstrates Synergy Against Neuroblastoma. Clin Cancer Res (2017) 23(11):2856–68. doi: 10.1158/1078-0432.CCR-16-1114
243. DeWire M, Fuller C, Hummel TR, Chow L, Salloum R, de Blank P, et al. A Phase I/II Study of Ribociclib Following Radiation Therapy in Children With Newly Diagnosed Diffuse Intrinsic Pontine Glioma (DIPG). J Neurooncol (2020) 149(3):511–22. doi: 10.1007/s11060-020-03641-2
244. Leonetti C, Biroccio A, Graziani G, Tentori L. Targeted Therapy for Brain Tumours: Role of PARP Inhibitors. Curr Cancer Drug (2012) 12(3):218–36. doi: 10.2174/156800912799277403
245. Isabelle M, Moreel X, Gagné J, Rouleau M, Ethier C, Gagné P, et al. Investigation of PARP-1, PARP-2, and PARG Interactomes by Affinity-Purification Mass Spectrometry. Proteome Sci (2010) 8:22. doi: 10.1186/1477-5956-8-22
246. Schreiber V, Dantzer F, Ame J, de Murcia G. Poly(ADP-Ribose): Novel Functions for an Old Molecule. Nat Rev Mol Cell Biol (2006) 7(7):517–28. doi: 10.1038/nrm1963
247. Smith SJ, Long A, Barrow JH, Macarthur DC, Coyle B, Grundy RG. Pediatric High-Grade Glioma: Identification of Poly(ADP-Ribose) Polymerase as a Potential Therapeutic Target. Neuro Oncol (2011) 13(11):1171–7. doi: 10.1093/neuonc/nor115
248. Galia A, Calogero AE, Condorelli R, Fraggetta F, La Corte A, Ridolfo F, et al. PARP-1 Protein Expression in Glioblastoma Multiforme. Eur J Histochem (2012) 56(1):e9. doi: 10.4081/ejh.2012.e9
249. Scalia M, Satriano C, Greca R, Stella AM, Rizzarelli E, Spina-Purrello V. PARP-1 Inhibitors DPQ and PJ-34 Negatively Modulate Proinflammatory Commitment of Human Glioblastoma Cells. Neurochem Res (2013) 38(1):50–8. doi: 10.1007/s11064-012-0887-x
250. Tentori L, Leonetti C, Scarsella M, D'Amati G, Vergati M, Portarena I, et al. Systemic Administration of GPI 15427, a Novel Poly(ADP-Ribose) Polymerase-1 Inhibitor, Increases the Antitumor Activity of Temozolomide Against Intracranial Melanoma, Glioma, Lymphoma. Clin Cancer Res (2003) 9:5370–9.
251. Tentori L, Portarena I, Torino F, Scerrati M, Navarra P, Graziani G. Poly(ADP-Ribose) Polymerase Inhibitor Increases Growth Inhibition and Reduces G(2)/M Cell Accumulation Induced by Temozolomide in Malignant Glioma Cells. GLIA (2002) 40(1):44–54. doi: 10.1002/glia.10113
252. Daniel RA, Rozanska AL, Mulligan EA, Drew Y, Thomas HD, Castelbuono DJ, et al. Central Nervous System Penetration and Enhancement of Temozolomide Activity in Childhood Medulloblastoma Models by Poly(ADP-Ribose) Polymerase Inhibitor AG-014699. Br J Cancer (2010) 103(10):1588–96. doi: 10.1038/sj.bjc.6605946
253. Higuchi F, Nagashima H, Ning J, Koerner M, Wakimoto H, Cahill DP. Restoration of Temozolomide Sensitivity by PARP Inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin Cancer Res (2020) 26(7):1690–9. doi: 10.1158/1078-0432.CCR-19-2000
254. Venere M, Hamerlik P, Wu Q, Rasmussen RD, Song LA, Vasanji A, et al. Therapeutic Targeting of Constitutive PARP Activation Compromises Stem Cell Phenotype and Survival of Glioblastoma-Initiating Cells. Cell Death Differ (2014) 21(2):258–69. doi: 10.1038/cdd.2013.136
255. Lugli N, Kamileri I, Halazonetis TD. PARP Inhibitors and IR Join Forces to Strike Glioblastoma-Initiating Cells. Cell Death Differ (2014) 21(2):192–3. doi: 10.1038/cdd.2013.172
256. Dungey FA, Löser DA, Chalmers AJ. Replication-Dependent Radiosensitization of Human Glioma Cells by Inhibition of Poly(ADP-Ribose) Polymerase: Mechanisms and Therapeutic Potential. Int J Radiat Oncol Biol Phys (2008) 72(4):1188–97. doi: 10.1016/j.ijrobp.2008.07.031
257. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific Killing of BRCA2-Deficient Tumours With Inhibitors of Poly(ADP-Ribose) Polymerase. Nature (2005) 434(7035):913–7. doi: 10.1038/nature03443
258. Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature (2005) 434(7035):917–21. doi: 10.1038/nature03445
259. McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM, et al. PTEN Loss Compromises Homologous Recombination Repair in Astrocytes: Implications for Glioblastoma Therapy With Temozolomide or Poly(ADP-Ribose) Polymerase Inhibitors. Cancer Res (2010) 70(13):5457–64. doi: 10.1158/0008-5472.CAN-09-4295
260. Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, et al. 2-Hydroxyglutarate Produced by Neomorphic IDH Mutations Suppresses Homologous Recombination and Induces PARP Inhibitor Sensitivity. Sci Transl Med (2017) 9(375):eaal2463. doi: 10.1126/scitranslmed.aal2463
261. Lu Y, Kwintkiewicz J, Liu Y, Tech K, Frady LN, Su YT, et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res (2017) 77(7):1709–18. doi: 10.1158/0008-5472.CAN-16-2773
262. IDH-Mutant Tumors Vulnerable to PARP Inhibition. Cancer Discov (2017) 7(4):F4. doi: 10.1158/2159-8290.CD-NB2017-026
263. Sabbatino F, Fusciello C, Somma D, Pacelli R, Poudel R, Pepin D, et al. Effect of P53 Activity on the Sensitivity of Human Glioblastoma Cells to PARP-1 Inhibitor in Combination With Topoisomerase I Inhibitor or Radiation. Cytom A (2014) 85(11):953–61. doi: 10.1002/cyto.a.22563
264. Sizemore ST, Mohammad R, Sizemore GM, Nowsheen S, Yu H, Ostrowski MC, et al. Synthetic Lethality of PARP Inhibition and Ionizing Radiation is P53-Dependent. Mol Cancer Res (2018) 16(7):1092–102. doi: 10.1158/1541-7786.MCR-18-0106
265. Rasmussen RD, Gajjar MK, Jensen KE, Hamerlik P. Enhanced Efficacy of Combined HDAC and PARP Targeting in Glioblastoma. Mol Oncol (2016) 10(5):751–63. doi: 10.1016/j.molonc.2015.12.014
266. Wang YT, Yuan B, Chen HD, Xu L, Tian YN, Zhang A, et al. Acquired Resistance of Phosphatase and Tensin Homolog-Deficient Cells to Poly(ADP-Ribose) Polymerase Inhibitor and Ara-C Mediated by 53BP1 Loss and SAMHD1 Overexpression. Cancer Sci (2018) 109(3):821–31. doi: 10.1111/cas.13477
267. Hanna C, Kurian KM, Williams K, Watts C, Jackson A, Carruthers R, et al. Pharmacokinetics, Safety, and Tolerability of Olaparib and Temozolomide for Recurrent Glioblastoma: Results of the Phase I OPARATIC Trial. Neuro Oncol (2020) 22(12):1840–50. doi: 10.1093/neuonc/noaa104
Keywords: glioblastoma, small molecule inhibitor, molecular mechanism, TKI—tyrosine kinase inhibitor, clinical trial
Citation: Huang W, Hao Z, Mao F and Guo D (2022) Small Molecule Inhibitors in Adult High-Grade Glioma: From the Past to the Future. Front. Oncol. 12:911876. doi: 10.3389/fonc.2022.911876
Received: 03 April 2022; Accepted: 13 May 2022;
Published: 17 June 2022.
Edited by:
Dinesh Thotala, Washington University in St. Louis, United StatesReviewed by:
Raghupathy Vengoji, University of Nebraska Medical Center, United StatesHonglin Jin, Huazhong Agricultural University, China
Copyright © 2022 Huang, Hao, Mao and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongsheng Guo, VGpndW9kb25nc2hlbmdAMTYzLmNvbQ==; Feng Mao, Mjk3NjIwMDU4OEBxcS5jb20=