 Anne Cuypers
Anne Cuypers Anh-Co Khanh Truong
Anh-Co Khanh Truong Lisa M. Becker1
Lisa M. Becker1 Paula Saavedra-García
Paula Saavedra-García Peter Carmeliet
Peter Carmeliet- 1Laboratory of Angiogenesis and Vascular Metabolism, Center for Cancer Biology (CCB), Vlaams Instituut voor Biotechnologie (VIB) and Department of Oncology, Leuven Cancer Institute (LKI), KU Leuven, Leuven, Belgium
- 2Laboratory of Angiogenesis and Vascular Heterogeneity, Department of Biomedicine, Aarhus University, Aarhus, Denmark
- 3Center for Biotechnology, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates
Tumor vessel co-option (VCO) is a non-angiogenic vascularization mechanism that is a possible cause of resistance to anti-angiogenic therapy (AAT). Multiple tumors are hypothesized to primarily rely on growth factor signaling-induced sprouting angiogenesis, which is often inhibited during AAT. During VCO however, tumors invade healthy tissues by hijacking pre-existing blood vessels of the host organ to secure their blood and nutrient supply. Although VCO has been described in the context of AAT resistance, the molecular mechanisms underlying this process and the profile and characteristics of co-opted vascular cell types (endothelial cells (ECs) and pericytes) remain poorly understood, resulting in the lack of therapeutic strategies to inhibit VCO (and to overcome AAT resistance). In the past few years, novel next-generation technologies (such as single-cell RNA sequencing) have emerged and revolutionized the way of analyzing and understanding cancer biology. While most studies utilizing single-cell RNA sequencing with focus on cancer vascularization have centered around ECs during sprouting angiogenesis, we propose that this and other novel technologies can be used in future investigations to shed light on tumor EC biology during VCO. In this review, we summarize the molecular mechanisms driving VCO known to date and introduce the models used to study this phenomenon to date. We highlight VCO studies that recently emerged using sequencing approaches and propose how these and other novel state-of-the-art methods can be used in the future to further explore ECs and other cell types in the VCO process and to identify potential vulnerabilities in tumors relying on VCO. A better understanding of VCO by using novel approaches could provide new answers to the many open questions, and thus pave the way to develop new strategies to control and target tumor vascularization.
Introduction
Sprouting angiogenesis is often regarded as the most significant mechanism of tumor vascularization and thus became the main target for anti-angiogenic therapy (AAT) (1). However, tumors are also able to ensure their blood and nutrient supply by means of alternative vascularization mechanisms, such as vessel splitting (intussusceptive angiogenesis), vascular mimicry, and vessel co-option (VCO) (2–5). Vessel co-option has first been observed by Francesco Pezzella in his pioneering work published in the mid 90s (2, 3), where he described an “alveolar or putative nonangiogenic” vascularization pattern. Interestingly, besides the acknowledgement of alternative vascularization patterns, in the 2011 new addition of the landmark review on the “Hallmarks of cancer: The new generation” (6), tumor vascularization was still described as “induction of angiogenesis”. Only this year, Doug Hanahan acknowledged in the updated “The Hallmarks of cancer: New dimensions” publication the importance of alternative modes of tumor vascularization and updated the hallmark to “inducing or assessing vasculature” (7). Thus, besides being discovered for almost three decades, only now the importance of VCO is being realized more completely by the scientific community. It is therefore not surprising that only little is known about this complex process.
During sprouting angiogenesis, vessel growth occurs by proliferation and migration of endothelial cells (ECs) from preexisting vessels; during VCO, there is no new blood vessel formation but instead, cancer cells hijack pre-existing blood vessels to grow and to invade the healthy tissue (8, 9) (Figure 1). A main feature of co-opting cancer cells is an increased motility and invasion to grow along pre-existing vessels (8, 11, 12). Often, the surrounding cancer cells compress the co-opted vessel, which can generate a hypoxic tumor core (13, 14) (Figure 1).
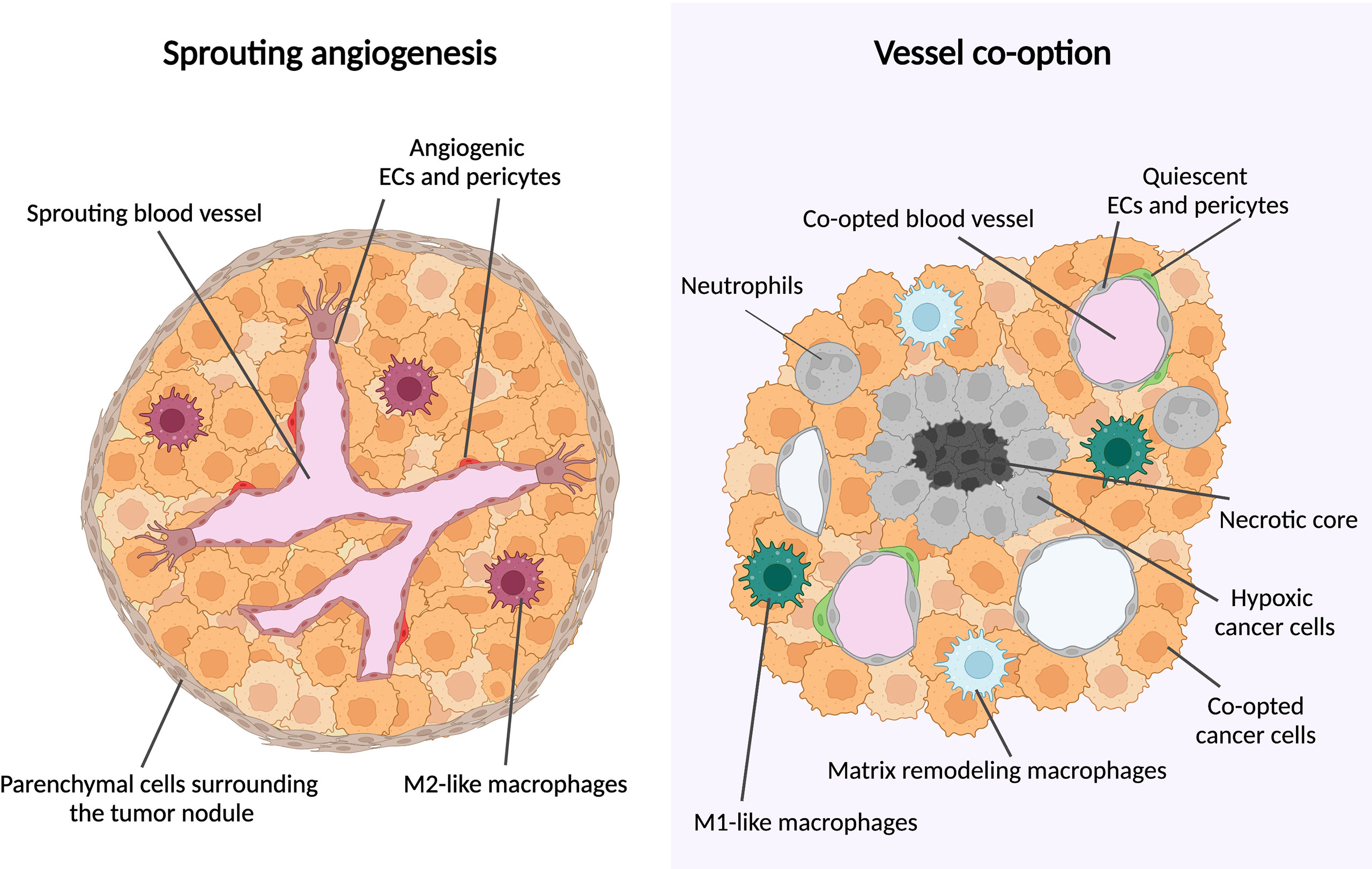
Figure 1 Metastatic tumor growth via vessel co-option versus sprouting angiogenesis. Schematic overview of metastatic tumor growth by vessel co-option versus sprouting angiogenesis. Growth via vessel co-option (left panel): During vessel co-option, cancer cells co-opt the healthy lung structures in an irregular and infiltrative manner, resulting in a tumor with a necrotic core. Cell types thus far associated with vessel co-option are indicated (neutrophils, M1-like macrophages, matrix-remodeling macrophages). Growth via sprouting angiogenesis (right panel): Metastases growing mainly via sprouting angiogenesis are characterized by a globular shape, excluding healthy alveolar cells. New blood vessel formation is achieved by proliferation and migration of ECs out of pre-existing blood vessels. M2-like macrophages are enriched in metastases growing via sprouting angiogenesis. This figure is adapted from (10).
VCO is largely understudied when compared to sprouting angiogenesis, however, it is potentially of great interest with regards to cancer therapy, as many studies suggest VCO to present a resistance mechanism against AAT (9, 15–17). Furthermore, VCO is known to be associated with worse prognosis and is frequently occurring both in primary tumors as well as in metastases (8, 9). The most common organs, in which tumors are described to use non-angiogenic mechanisms such as VCO are the liver, lung and brain, both for primary tumors, or metastases occurring in these organs (8, 9) (Figure 2). It was also hypothesized that VCO is involved in the formation of clear cell renal cell carcinoma (RCC) (18). Future investigations will further uncover the relationship between VCO and clear cell RCC.
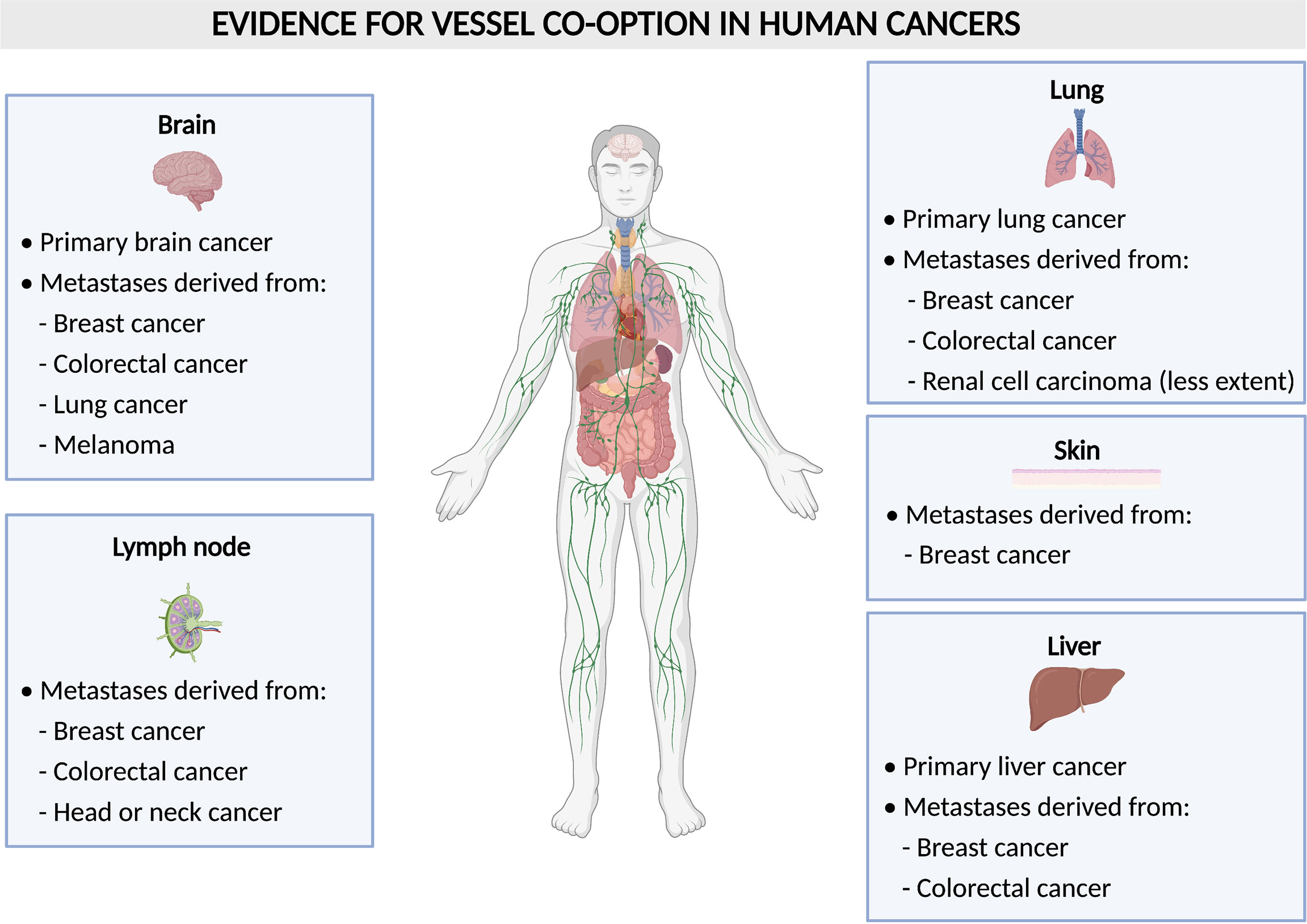
Figure 2 Vessel co-option in human tumors. Schematic overview of human tumors (per organ) with evidence for vessel co-option.
So far, approaches to study VCO are mainly focused on light microscopy and histopathological analysis of the growth pattern (15). Compared to angiogenesis, the molecular mechanisms of VCO are largely unknown and to-date, there are only a handful of papers available offering any mechanistic insights. That is the main reason why, thus far, no therapeutic strategies exist to inhibit VCO. Therefore, novel experimental strategies are warranted to gather detailed insights on this process, that can ultimately lay the foundation for development of therapeutics. Sequencing technologies, for instance, have revolutionized our understanding of cancer, and might be interesting to also study VCO-dependent tumors.
In this review, we highlight what is known to date about the models that can be used to study VCO, the underlying molecular mechanisms that drive VCO and their relevance in AAT resistance. Furthermore, we focus on the potential therapeutic strategies to target tumor VCO, based on newly gained insights into co-opted cell types. Lastly, we call attention to recently emerged approaches such as single-cell RNA sequencing and spatial transcriptomics and propose how these can be used in the future to further explore VCO (8, 17, 19, 20).
Models demonstrating vessel co-option
Evidence for vessel co-option in human cancers
During VCO, cancer cells migrate and hijack pre-existing blood vessels of the host organ in order to grow and to invade the surrounding tissue (Figure 1). This implies that cancer cells create their vascular supply in a non-angiogenic manner. Tumor VCO has first been described in lung cancer as an alveolar non-angiogenic growth pattern (3). Often, VCO in the lungs occurs in metastatic lesions (e.g. lung metastasis from renal cell carcinoma (15), but it can also arise in primary lung cancer (15, 21). In fact, co-optive or alveolar growth patterns have been observed in about half of the non-small cell lung cancers and in a third of small-cell lung cancers (22, 23). In addition to the occurrence in lung cancer, metastases in the lungs often grow via VCO. Here, metastases from colorectal and breast cancer have most often been associated with VCO, while lung metastases from RCC grow less frequently via VCO (9, 15). Notably, RCC has been known as one of the most angiogenic cancer types (24), thus it is very intriguing that the growth of RCC-derived metastases can partly rely on VCO, instead of angiogenesis.
Besides the discovery in lung cancer, co-option in tumors and metastasis has also been observed in many other organs including brain, liver, lymph node, colorectal and skin in patients (8, 25) (Figure 2). Gliomas are tumors in which VCO has been described the most (Figure 2). In human primary high-grade gliomas, cancer cells may surround brain capillaries or replace pericytes and astrocytes by adhering to the abluminal surface to co-opt pre-existing vessels which is referred to as ‘perivascular cuffing’ (25, 26). In primary low-grade gliomas, cancer cells co-opt vessels by infiltration of the parenchyma (27). In rat gliomas, it has been shown that tumors can grow by co-opting the pre-existing vasculature of the host. Upon co-option, the host vasculature does not use angiogenesis at first to support tumor growth but instead disintegrates, and thus creates an avascular tumor and impairs tumor growth. However, the remaining tumor recovers via robust angiogenesis by healthy vessels at the margins of the tumor (28).
In addition to primary brain tumors, VCO has also been identified in metastatic lesions in the brain, originating from distinct primary tumors. VCO was frequently observed in biopsy samples from patients with brain metastasis (from breast, melanoma, colorectal, and lung cancer) (29–31) (Figure 2). Moreover, assessment of brain metastasis from malignant melanoma revealed low vascular endothelial growth factor-A (VEGF-A) expression in the parenchyma, which led to an infiltrative phenotype, in which the pre-existing vasculature of the brain was co-opted. When the human melanoma cell line Mel57 was modulated to express recombinant VEGF-A-165, the metastases exhibited a fast infiltrative and expansion growth pattern with a marked central necrotic core and the co-opted peritumoral and intratumoral blood vessels were dilated causing an increase in vessel permeability (32).
In primary and metastatic liver tumors, cancer cells co-opt sinusoidal vessels and incorporate them in the tumor microenvironment, a process referred to as ‘replacement pattern’ (25, 33) (Figure 2). Here, the metastases grow without or with only minimal angiogenesis. In one study, for instance, liver specimens were resected from untreated tumors and examined by histology for their growth pattern. During replacement growth, tumors retain the basic architecture and cancer cells are well-differentiated and irregularly distributed, and the portal tracts are included in the tumor tissue (33). In addition to lesions from liver tumors themselves, metastases originating from different primary tumors are often found in the liver (Figure 2). Here, breast cancer liver metastases grow 90% via VCO, while there is less evidence of co-optive growth in colorectal carcinoma liver metastasis (16, 25, 34, 35). In lymph nodes, on the other hand, metastatic cancer cells derived from colorectal cancer, breast cancer, head or neck cancer can co-opt pre-existing vessels (25, 36–38) (Figure 2). Breast cancer metastases tend to grow in several organs via VCO, as for instance, VCO is also observed in more than 50% of metastatic cancer cells in breast cancer skin metastasis (39) (Figure 2). Lastly, melanoma cells have also been observed to co-opt blood vessels by cohesive migration on the abluminal surface in brain metastasis (32, 40–42) (Figure 2).
Animal models to study vessel co-option
Multiple animal models have been described to study VCO including intracarotid, intracardiac, intravenous and orthotopic transplantation in mice, but also zebrafish and chick chorioallantoic membrane (CAM) models (Table 1 summarizes the models demonstrating VCO per tumor type) (25). Importantly, the tumor vasculature is dependent on the anatomical site where the cell lines are injected (15). For instance, in several models, metastatic lesions from a particular tumor type use angiogenesis in one organ, but VCO in a different organ (15). Therefore, a limitation in vitro is that it is not possible to discriminate whether a cell line is classified for angiogenic or non-angiogenic tumor growth. In Table 1, we highlight known models, using different cell lines, that have been used to investigate the underlying mechanisms of tumor VCO.
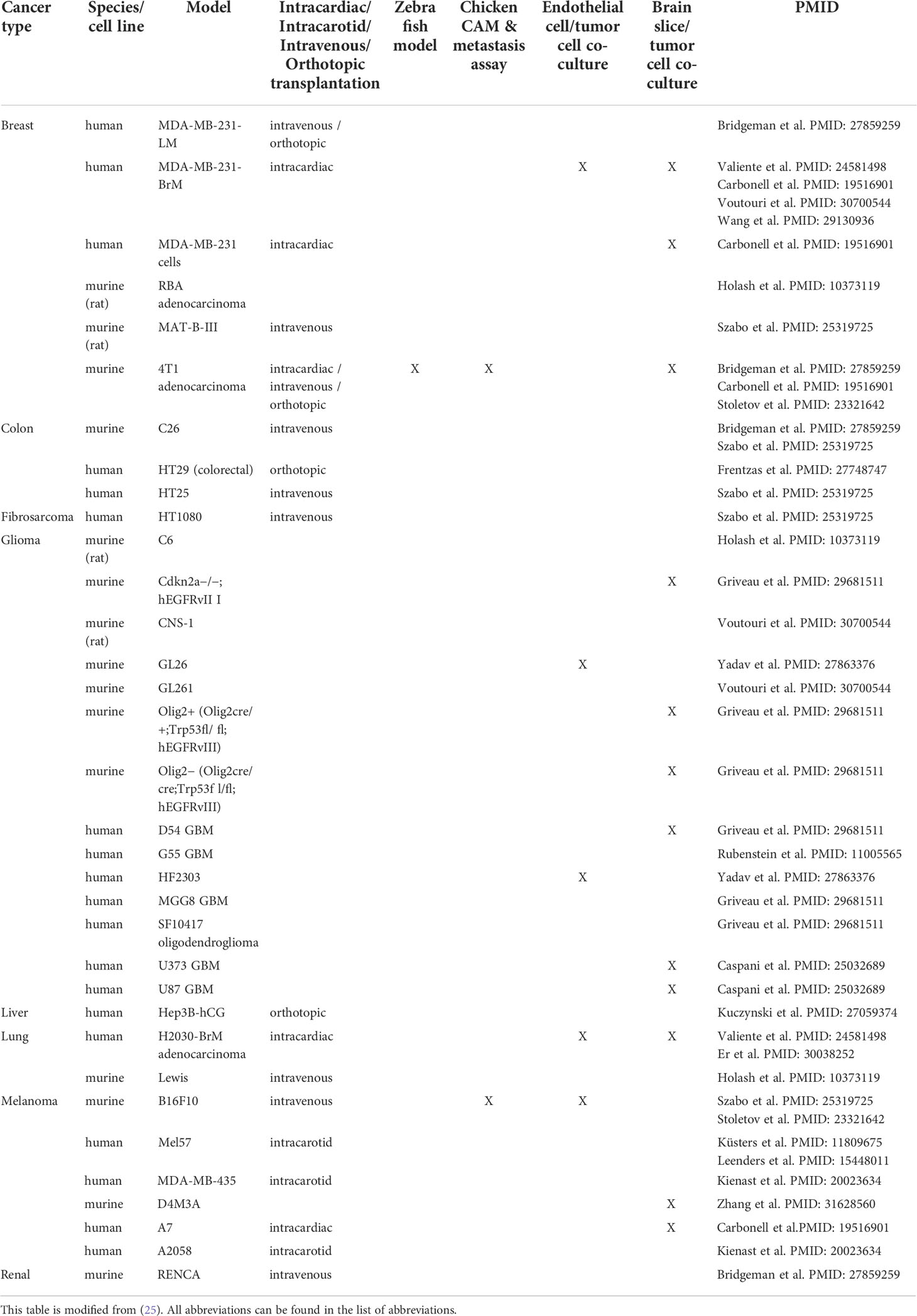
Table 1 Summary of models demonstrating tumor vessel co-option per tumor type.
Intracardiac injection of cancer cells occurs via the left ventricle of the heart in anesthetized mice and allows the cells to circulate in the body before reaching the microvasculature of the liver and lungs (20, 25, 43–45). This transplantation approach is mainly used to model brain and bone metastasis. Using this method, researchers discovered that plasminogen activator-inhibitory serpins can stimulate the survival of cancer cells (20, 25). In addition, by comparing transcriptomic signatures of brain metastatic subpopulation variants isolated from breast and lung cancer cell lines, they reported that L1CAM in brain metastases mediates VCO (20). In another study, the role of long noncoding RNAs in brain metastasis of breast cancer was studied after intracardiac injection of MDA-MB-231-Br cells and a specific long noncoding RNA (Lnc-BM) was documented to be essential for VCO in the brain (45).
Upon intracarotid injection, cancer cells are directly injected into the internal carotid artery of anesthetized mice to create experimental brain metastases. With this method, infiltrative co-optive growth patterns are visible after injection of human melanoma cell lines (M14, Mel57, 530) (32). VEGF-A regulates the progression of brain metastases of melanoma without inducing angiogenesis but by using the pre-existing vasculature (32). Melanoma brain metastases upon intracarotid transplantation also grew via VCO when mice were treated with AAT (46). Intracarotid injections of melanoma cells (A2058 and MDA-MB-435 human melanoma cells) led to perivascular growth using VCO in the resulting brain metastases, whereas injection of lung cancer cells with the same method caused angiogenic brain metastases, as assessed by multiphoton laser scanning microscopy (42).
Intravenous injection of cancer cells occurs via the jugular vein or tail vein and is mainly used to study lung metastasis. Jugular vein injection of mice with Lewis lung carcinoma cells causes lung tumors capable of co-opting the microvasculature (28). Also, experimental lung metastases generated after intravenous injection of several human and murine cancer cell lines (MAT-B-III rat mammary carcinoma, C26 murine colon carcinoma, HT25 human colon carcinoma, HT1080 human fibrosarcoma and B16 murine melanoma) showed that these metastases grow via VCO of the pulmonary vessels (47). Furthermore, when generating lung metastases models using murine renal adenocarcinoma cells (RENCA) and murine colon carcinoma cells (C26), VCO appeared to be a resistance mechanism to AAT (15). Tail vein injection of MDA-MB-231-LM cells (lung metastatic subpopulation) also causes cancer cells to spread perivascularly in the lung, mediated by neural cell adhesion molecule L1 (L1CAM) (44).
Orthotopic transplantation of cancer cells is performed by injecting them into specific anatomical sites and is commonly used to investigate the role of the microenvironment in tumorigenesis and metastasis (25, 48). Spontaneously developing metastases after orthotopic injection better recapitulate interactions between the tumor and the host as well as the specific characteristics of the whole metastatic process. In contrast to other methods, it takes more time to develop such metastases in mice. The perivascular growth of brain metastases was verified also in spontaneous metastases model of primary 4T1 mammary tumors (43). Moreover, MDA-MB-231-LM or MDA-MB-231LM2-4 cells co-opt alveolar capillaries when forming lung metastases after orthotopic mammary fat pad injections (15, 44). Injection of human HT29 colorectal cancer cells into a mouse liver generated advanced liver metastases growing via VCO. In this model, the importance of cancer cell motility during the co-opting process was documented (16). Moreover, orthotopic injection of Hep3B-hCG cells (human hepatocellular carcinoma) into mouse livers showed that VCO is used as an acquired resistance mechanism to AAT (49).
Zebrafish embryos are utilized to create metastases by injection of cancer cells into the cardinal vein (25, 50). Using these embryos helps to visualize early processes of brain colonization, like VCO and extravasation. Using this model, HMLE mammary cancer cells and 4T1 mammary cancer cells (murine) were shown to co-opt brain arteries, with a primary role of the connexin gap junction protein Cx43 in brain colonization and metastatic extravasation (50).
CAM in vivo models can also be used to explore how tumors grow and how they spread along blood vessels. A small hole is created on the top of the fertilized egg to allow the membrane to detach from the shell, followed by the placement of a matrigel containing a cancer cell mixture onto the CAM. After inoculation, spreading and growth of the cancer cells along the blood vessels is visualized by imaging. After a few incubation days, organs such as the liver or the brain can be isolated for imaging. In a study exploiting the chick embryo model, HMLE mammary carcinoma cells, B16 melanoma cells and 4T1 mammary cancer cells co-opt the blood vessels and several gap junction proteins (Cx43, Cx26) were found to play an important role during brain colonization and VCO (50).
Despite the limitations pointed out above, several in vitro models exist to study the cellular and molecular mechanisms of VCO by cancer cells. For instance, EC - cancer cell co-cultures have been used to study the interaction between blood vessels and cancer cells (25, 40, 45). ECs can be stimulated to create capillary-like structures (but these are not perfused like in vivo blood vessels) and can be monitored how cancer cells migrate along the vessels by time-lapse imaging (25). Another model that can be used in vitro is brain slice - cancer cell co-culture (11, 20, 25, 26, 43, 45, 51). This model, which has been used to study molecular and cellular processes in the microenvironment of the brain, is generated by cutting brain slices with a vibratome. Brain slices are then cultured onto porous membrane inserts in the brain slice medium and incubated for a short time. Afterwards, cancer cells are pipetted onto the brain slice surface and the interaction between brain blood vessels and cancer cells can be visualized by using fluorescence microscopy (25).
This method was used in a study, in which the authors plated human and murine cancer cells (A7, K1735M2, MDA231BR, MDA-MB-231, 4T1 cells) onto murine brain slices and observed that cancer cells spread rapidly along the blood vasculature (43). Human U87 and U373 glioblastoma multiforme (GBM) cells that were seeded onto brain slices were also shown to co-opt the pre-existing vasculature (26). When culturing human D54 GBM cells onto brain slices, bradykinin appeared to be critical for the perivascular migration of brain cancer cells (11). Moreover, when low and highly invasive breast cancer cells (MDA231 vs. MDA231-BrM) and lung adenocarcinoma cells (H2030 vs. H2030-BrM) were seeded onto brain slices, low-invasive brain parental cancer cells succumbed via serpin- and plasmin-dependent pathways, whereas brain invasive BrM cells moved towards and disseminated over brain capillaries (20). Brain slices were also used to investigate the roles of Wnt7 in glioma cells during VCO. The authors discovered that Olig2+ (Wnt7 wildtype) glioma cells used VCO, while Wnt7 null glioma cells did not migrate along blood vessels via VCO (52). Furthermore, co-option was arrested when Wnt signaling was inhibited in SF10417 human oligodendroglioma cells and Cdkn2a-/-;hEGFRvIII glioma progenitor cells. In addition, murine D4M3A melanoma cells (established from the metastatic melanoma Tyr::CreER;BrafCA;Ptenlox/lox mouse model (53)) plated onto brain slices from immunocompetent VE-Cadherin reporter mice (Cdh5-CreERT2;ZSGreenloxp/stop/loxp) mice, only migrated along the pre-existing vasculature of the brain (25, 52). Overall, while extremely helpful in studying VCO mechanisms, one should consider that the microenvironment and culture conditions from in vitro models are different from the in vivo microenvironment, therefore the mentioned results have to be verified with in vivo models.
Together, these in vivo and in vitro models have proven their value in unraveling possible VCO molecular mechanisms and their response to therapies. However, there are still limitations associated when using these models. To give some examples, for the intracardiac transplantation for instance, the efficiency of metastasis generated is low and dependent on the cell line used (25). A limitation of the intracarotid approach is that the carotid artery is ligated, potentially causing injury which might influence VCO (25). For orthotopic transplantation, a limitation is that it takes a long time to create metastases compared to the other methods described (25).
Molecular mechanisms of vessel co-option and vessel co-option as resistance mechanism to anti-angiogenic therapy
Cancer cell – EC interactions
As defined, VCO relies on an interaction between cancer cells and the host vasculature, directly with ECs or indirectly via pericytes. Therefore, understanding these cell-cell interactions is key to understand cancer cell invasion via VCO (Figure 3). The phenotypic features of non-angiogenic cancer cells include reduced adhesion to each other and to the extracellular matrix (ECM), together with increased motility and invasion (8, 11, 12). To date, only a few proteins and mechanisms are proposed for their involvement in the formation and progression of tumors relying on VCO.
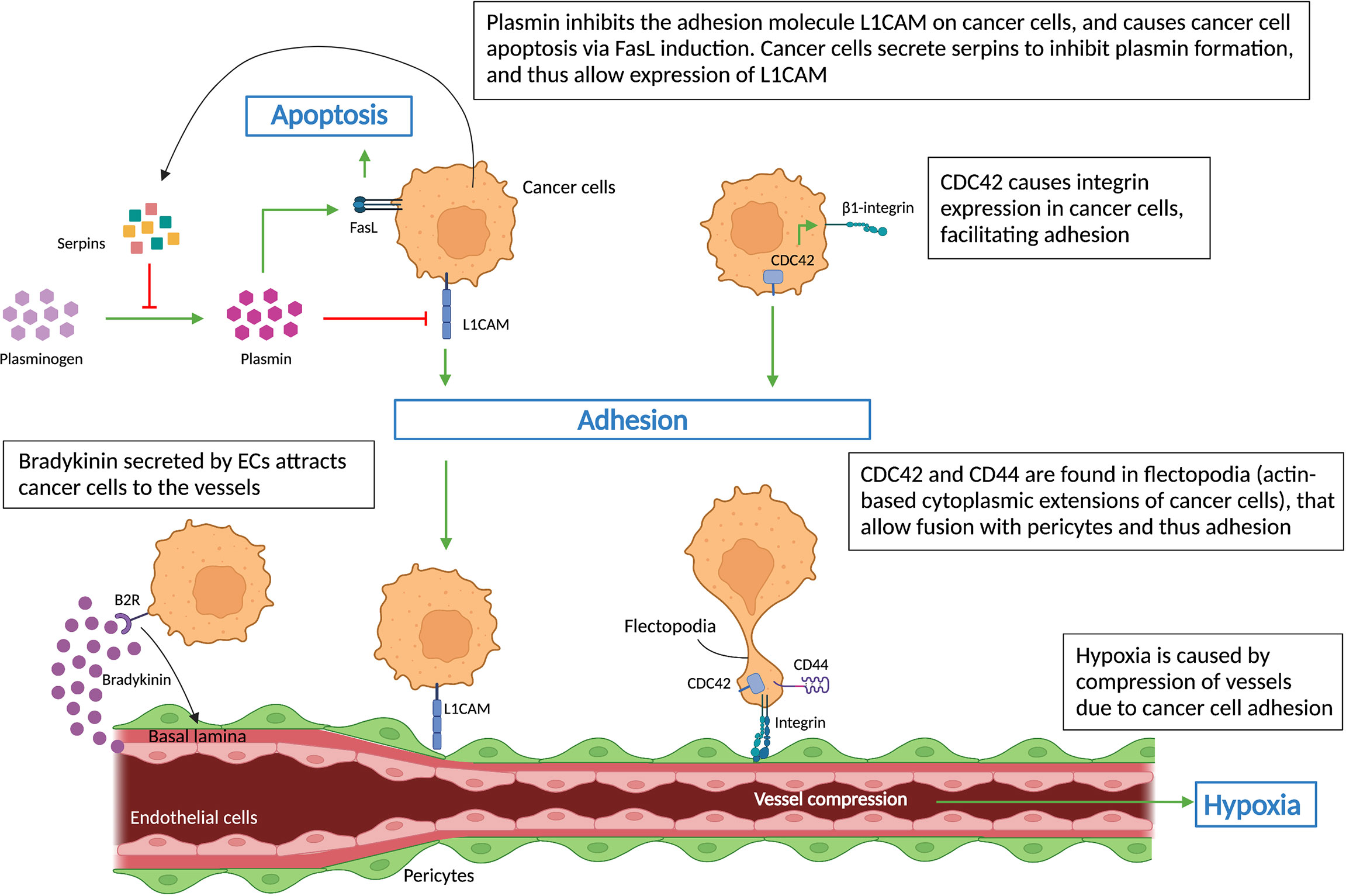
Figure 3 Cell-cell interactions during vessel co-option. Schematic graph showing key regulators (L1CAM, CDC42, CD44, integrins, serpins) of non-angiogenic, co-opted cancer cells, which cause cancer cells’ adhesion to vessels – a hallmark of vessel co-option. Cancer cells’ attachment to the pre-existing vessels leads to vessel compression, which in turn results in hypoxia. All abbreviations can be found in the list of abbreviations.
Bradykinin, the ligand of the B2 receptor (B2R), a G-protein coupled receptor, has been strongly associated with directing invading glioma cells toward blood vessels (11). B2R is highly expressed on glioma cells isolated from patient biopsies and causes their migration towards pre-existing blood vessels via a bradykinin gradient released by blood vessels (Figure 3), as demonstrated in rat brain slices (54). Inhibition of bradykinin in turn reduced the numbers of cancer cells associated with blood vessels (54).
Tumors and metastases relying on VCO are often characterized by a large hypoxic core (54), suggesting that hypoxia-driven mechanisms may impact VCO. For instance, hypoxia enhances prolyl-4-hydroxylase α1 (P4HA1) expression, which can convert bradykinin to hydroxyprolyl-bradykinin, as shown in human pancreatic cancer (55). In this study, circulating hydroxyprolyl-bradykinin/bradykinin ratios measured in patient plasma samples mirrored local tissue hypoxia regulated by hypoxia-inducible factor 1 (HIF1α). In fact, while further studies are required, these results may propose circulating hydroxyprolyl-bradykinin/bradykinin ratios as indicators of tumor hypoxia, patient prognosis and treatment responses (55).
Highly invasive, motile cancer cells are another feature of co-opted tumors and metastases. Cancer cell migration/invasion can be affected by components of the plasminogen-plasmin system (56). In an animal model of brain metastasis, L1CAM was identified in metastatic cells, where it assists their adherence to brain capillaries. The study demonstrated that cancer cells also expressed serpins, including neuroserpin and serpin B2, which inhibits stromal cell-derived plasmin – an L1CAM inactivator, and inducer of cancer cell death via FasL signaling (Figure 3). Thus inhibition of plasmin by serpins promotes their adhesion to brain vessels via L1CAM, reduces tumor apoptosis and induces VCO and metastasis (20) (Figure 3).
Cancer cells, after moving along pre-existing vessels, adhere to the basement membrane of ECs or pericytes in order to exploit VCO (57). Therefore, the interaction between cancer cells and vascular cells is a determining factor for the formation of co-opted lesions. Integrins are ECM adhesion molecules that mediate not only cell-cell interactions but also cell-ECM interactions, which are involved in metastasis and drug resistance (58, 59). β1 integrin levels on metastatic brain cells regulate their interaction with different components of the basal lamina from brain capillaries such as fibronectin, laminin, vitronectin, and collagen I and IV (60). In breast cancer, β4 integrin plays a similar role in the cancer cell – ECM interaction (43, 60, 61). Moreover, β1 integrin is also the main target of cell division control protein 42 homolog (CDC42), a member of the Rho GTPase family associated with actin-dependent cytoplasm extension regulation (Figure 3), which was shown to mediate cancer cell - EC interactions (62). Interestingly, novel treatments using small molecules and miRNAs to inhibit the abnormal expression of CDC42 may slow down the metastasis process (62, 63). In addition, CDC42, together with cell adhesion molecule CD44, were found to be crucial for the motility of cancer cells as they are highly concentrated in the flectopodia, the actin-based cytoplasmic extensions on the cancer cell membrane that impact the contractile activity of pericytes (Figure 3). Cancer cells fuse with pericytes via flectopodia creating a hybrid cell that expands the tumor margin, thus promoting VCO and tumor-related hypoxia by vessel constriction (26, 64) (Figure 3).
Signaling pathways leading to vessel co-option
A theoretical model postulates that, to establish VCO, cancer cells first invade the tissue parenchyma, displace existing healthy tissue cells, and then interact with the vascular cells (65). These cancer cell-EC interactions during VCO trigger molecular alterations in both cell types that are crucial for the motility, adhesion and invasion of cancer cells to pre-existing vessels (Figure 4). Here, we highlight signaling pathways in co-opted cancer cells and co-opting ECs that have been associated with VCO.
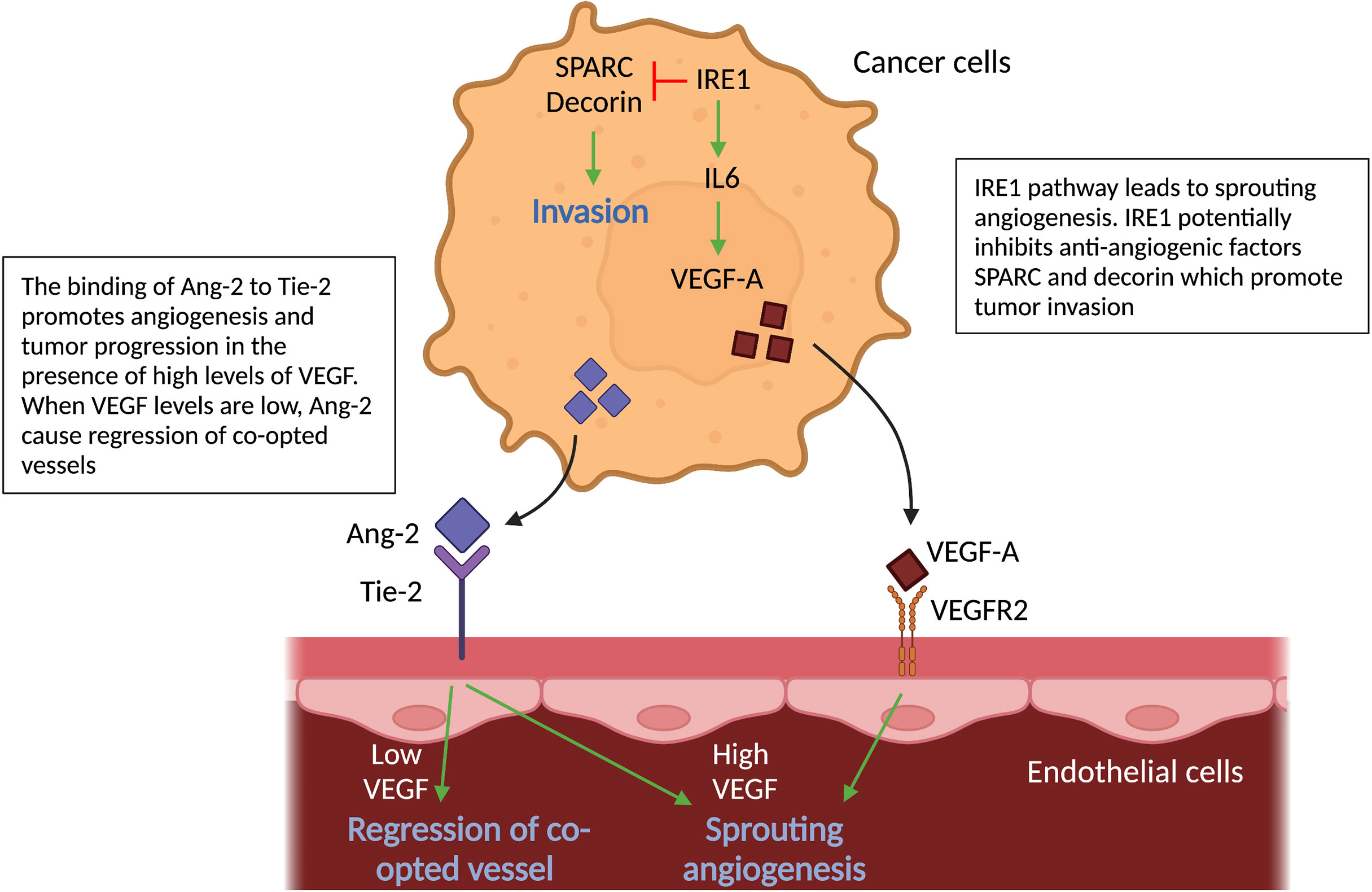
Figure 4 Molecular pathways in cancer cells and vascular cells driving angiogenesis. Schematic graph showing the known pathways associated with angiogenesis, the inhibition of which has been associated with vessel co-option. Ang2-Tie signaling: Signaling of Ang-2 through its receptor Tie-2 can cause sprouting angiogenesis if VEGF levels in the tumor microenvironment are high. If VEGF levels are low, Ang-2- Tie-2 signaling leads to regression of co-opted vessels. IRE1 signaling: IRE1 in cancer cells promote VEGF production, which can induce sprouting angiogenesis. On the other hand, it is indicated, that IRE1 impedes cancer cell invasion via inhibition of anti-angiogenic factors, such as SPARC and Decorin. All abbreviations can be found in the list of abbreviations.
The vascular growth factor angiopoietin-2 (Ang-2), which can be secreted by cancer cells (amongst others), is known to cause regression of co-opted blood vessels (58). Ang-2 is a ligand of the endothelial tyrosine kinase-receptor (Tie-2) (66). Binding of Ang-2 to its Tie-2 receptor results in a loss of vascular integrity and an increase in vascular permeability (66). Furthermore, in the presence of high levels of VEGF, Ang-2 leads to EC proliferation and triggers sprouting angiogenesis; in contrast, when VEGF levels are low, Ang-2 causes loss of vascular structures with marked regression of co-opted vessels. Ang-2 therefore may inhibit VCO in tumors (67–70) (Figure 4).
Moreover, inositol-requiring enzyme 1 (IRE1), an endoplasmic reticulum transmembrane stress sensor and a pivotal mediator of the unfolded protein response, plays a role in VCO (71). Using the CAM model and a murine orthotopic brain transplant model, reduced expression of IRE1 in glioma cells downregulates pro-angiogenic factors (VEGF-A; Interleukin-6 (IL-6)) and upregulates anti-angiogenic factors (such as SPARC and Decorin) linked to mesenchymal differentiation, tumor invasion and VCO (71). Thus, one may suppose IRE1 inhibits these anti-angiogenic factors (Figure 4). In addition, ectopic expression of IL-6 in IRE1-deficient tumors restores the angiogenic phenotype and neutralizes VCO, however it does not reverse cancer cell infiltration (71) (Figure 4).
Several signaling pathways may underlie the migratory, invasive characteristics of co-opted cancer cells. As invasive cancer cells are regularly associated with an epithelial-to-mesenchymal transition (EMT)-like phenotype, it comes to no surprise that some of the proposed mechanisms are related to EMT in cancer cells. During EMT, cancer cells lose their epithelial features and acquire mesenchymal features, thereby becoming less adherent to each other and more migratory (72). In the following, we list downstream signaling in cancer cells related to a migratory EMT-like phenotype and VCO (Figure 5).
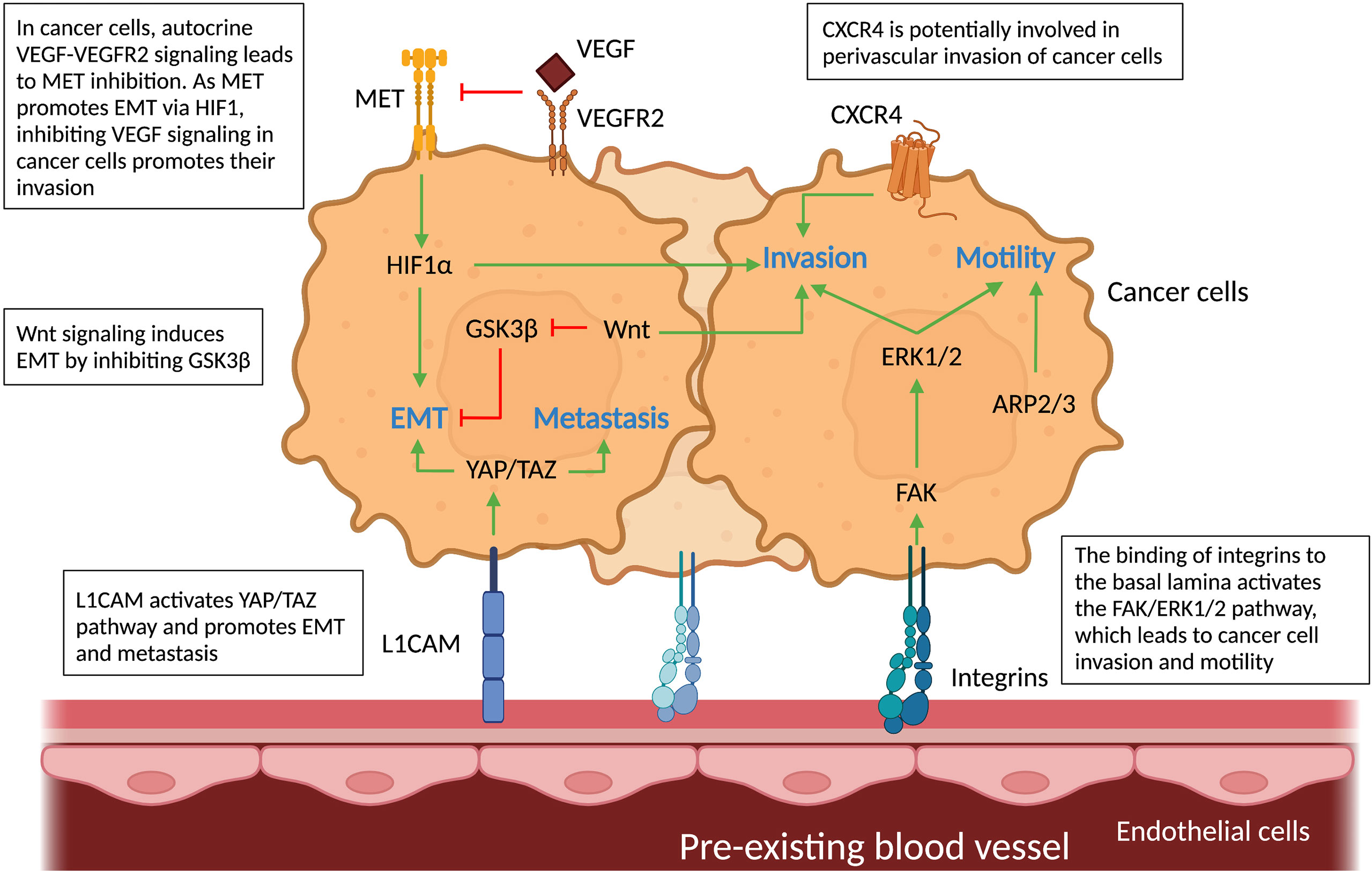
Figure 5 Molecular pathways in cancer cells and vascular cells driving vessel co-option. Schematic graph showing the known pathways associated with vessel co-option (VCO) and how they relate to each other creating a complex network. Top: Cancer cells: The pathways involved in VCO in cancer cells and their roles are shown: metastasis (YAP/TAZ), invasion (HIF1α, FAK-ERK1/2, Wnt, CXCR4), motility (FAK-ERK1/2, ARP2/3) and EMT (GSK3β, MET, YAP/TAZ). Bottom: Tumor vessels: Binding of cancer cells to tumor vessels via integrins and L1CAM can result in motile and invasive cancer cell phenotypes. All abbreviations can be found in the list of abbreviations.
First, as explained in the previous section, integrins play a key role in cell-cell interaction, but they also activate downstream pathways related to VCO. For instance, the interaction between cancer cells and ECs through β1 integrin leads to the activation of focal adhesion kinase (FAK) and extracellular signal-regulated kinase (ERK) 1/2 in co-opting cancer cells. FAK, a tyrosine kinase highly expressed in GBM cells, has been associated with cell motility and cancer cell invasion – features of tumor VCO (43, 73) (Figure 5). Whether the induction of FAK via cancer cell-EC interaction indeed leads to VCO remains to be further validated. Second, CXC-chemokine receptor 4 (CXCR4) signaling is potentially critical during VCO (Figure 5), as this pathway is involved in perivascular invasion of brain-metastasizing cancer cells through stimulation by brain EC-derived CXC-chemokine ligand 12 (CXCL12) (25, 74). In fact, CXCR4 was found highly expressed on metastatic glioblastoma cells relying on VCO (74).
A third possible signaling pathway is the Yes-associated protein (YAP) – transcriptional co-activator with PDZ-binding motif (TAZ) pathway (75–77). YAP and TAZ are primary sensors that play a crucial role in various aspects of cancer progression including promoting an EMT-like cancer cell phenotype and metastasis (75–77). Interestingly, L1CAM, known for its roles in VCO, activates the mechanotransduction effector YAP (44). It is moreover known that L1CAM-mediated pericyte-like spreading plays a major role in the initiation of metastasis (44). Further studies are required to directly link YAP-TAZ signaling in cancer cells to L1CAM1-dependent VCO. Fourth, the characteristic hypoxia of co-opted tumors generates high levels of HIF1α, a known inducer of the EMT transcription factor Zinc finger E-box binding homeobox 2 (ZEB2) that suppresses ephrinB2, thereby enhancing tumor invasiveness (78) (Figure 5). Lastly, canonical Wnt signaling in non-angiogenic cancer cells can induce an EMT-like phenotype by inhibition of the glycogen synthase kinase 3β (GSK3β), which leads to increased Snail stability (8, 79–81). Importantly, a subtype of Olig2+ oligodendrocyte-like glioma cells was shown to upregulate Wnt7 expression and promote invasion of cancer cells via co-option of existing brain vessels, linking Wnt7 signaling to VCO (52) (Figure 5).
Vessel co-option as resistance mechanism to anti-angiogenic therapy
A link between AAT resistance and VCO has been demonstrated in different types of tumors such as brain, colorectal, breast, renal and liver cancer (9, 15, 46, 49, 82, 83). For example, treatment with Bevacizumab (an anti-angiogenic drug that targets VEGF-A), used to treat kidney, colon, rectum, lung, or breast tumors, and some brain tumors, causes a metabolic shift toward glycolysis making the remaining glioblastoma cell populations less dependent on angiogenesis and promoting the VCO typical pro-invasive phenotype in a subset of GBM patients (84, 85). Thus, drivers of these invasive (EMT-like) cancer cell phenotypes upon AAT are likely associated with VCO. For instance, in an in vivo liver metastasis model, AAT induces the actin-related protein 2/3 complex (ARP2/3), which promotes cancer cells motility and VCO (16) (Figure 5). Another pathway that might be involved in VCO is the hepatocyte growth factor (HGF) signaling mediated through binding to the tyrosine kinase receptor MET. VEGF inhibition is accompanied by activation of MET, which is correlated with poor prognosis and resistance to therapy (82, 86–92). An EMT phenotype could be induced in GBM cells upon genetic or pharmacologic VEGF inhibition in an HGF-MET dependent manner (85) (Figure 5). Importantly, invasion and metastasis were induced by inhibiting VEGF signaling pathways, which could be impeded by concurrent MET inhibition (85, 89). Furthermore, VEGF inhibition, in addition to increasing MET levels, leads to enrichment of hypoxia-associated markers (including HIF1α (Figure 5), carbonic anhydrase 9 (CA9) and GLUT1), as well as downregulation of epithelial adherens junction proteins T-cadherin and E-cadherin, thus creating more mesenchymal, invasive cancer cells (85).
More functional studies are required to identify the underlying drivers of VCO. It is important to note that many of the described signaling pathways are only loosely linked, or even only hypothesized to be involved in the VCO process, and experimental evidence providing direct links to tumor VCO is yet to be imparted. Notably, the roles of EMT in VCO are not yet fully understood, and the existing links need further strengthening (65, 72). Moreover, while VEGF inhibition has been often connected to tumor VCO, VEGF is also known to downregulate cancer cell invasion via enhanced recruitment of the protein tyrosine phosphatase 1B (PTP1B, which can directly dephosphorylate MET and VEGFR2), thereby suppressing cancer cell migration (85). Nonetheless, these initial insights are a promising start to better characterize the cellular and molecular mechanisms of VCO.
Potential therapeutic anti-tumor vessel co-option strategies
As mentioned above, there is accumulating evidence linking VCO to resistance to AAT. In fact, one may also speculate whether the failure of several phase III clinical trials for various anti-angiogenic drugs (93) could be (partly) caused by tumors switching to VCO as alternative mode of vascularization. Here, cancer cells may potentially reside along existing vessels for survival and switch into a state of temporary dormancy. While these intriguing questions have yet to be investigated, targeting of VCO may add a significant contribution in developing novel anti-cancer therapies, in particular when combined with AAT. To develop potential strategies for VCO inhibition, it is important to understand which cell types in the tumor microenvironment can be targeted, as well as how we could target those cells (Figure 6). Here, we are proposing some potential targets and approaches for VCO inhibition, many of which are based on the mechanisms suggested in the previous section.
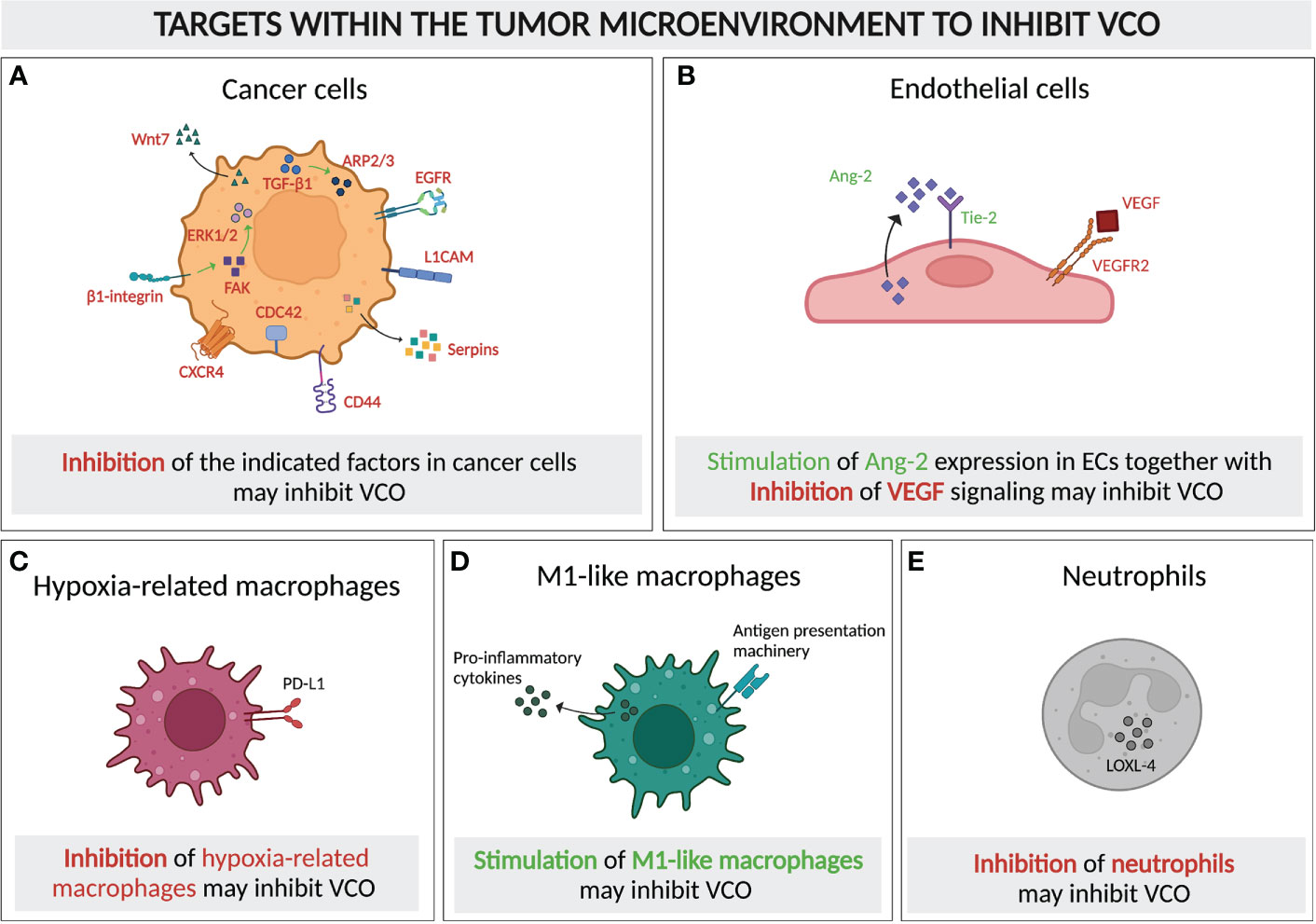
Figure 6 Molecular and cellular targeting to inhibit vessel co-option. Schematic graph showing the cellular components of a typical VCO-related tumor microenvironment and their potential as targets to inhibit VCO, either by inhibiting (A–C) or promoting the presence and/or signaling of these cells. Targets for inhibition: (A) Tumor cells with signaling pathways and molecules for potential targeting highlighted (TGF-β1, ARP2/3, EGFR, L1CAM, Serpins, CD44, CDC42, CXCR4, β1-integrin, FAK, ERK1/2, Wnt7), (C) Hypoxia-related macrophages (PD-L1 as potential target) and (E) Neutrophils (LOXL-4 presenting a potential target) Targets for dual inhibition and stimulation: (B) ECs could potentially dually targeted: VEGF signaling could be inhibited, while simultaneously promoting Ang-2 signaling. = Targets for stimulation: (D) M1-like macrophages should be promoted. All abbreviations can be found in the list of abbreviations. All figures were generated with Biorender.com.
Targeting cancer cells
As the majority of studies on VCO has centered around molecular mechanisms of co-opted cancer cells, targeting those cells to achieve VCO inhibition has attracted attention, in particular focusing on motility or adhesion pathways. The invasion of cancer cells to the perivascular environment relies on their motility, in which motility-involving molecules such as ARP2/3, L1CAM, serpins, CD44, CDC42, CXCR4, epidermal growth factor receptor (EGFR), as well as Wnt7 signaling, are critical players (16, 20, 25, 26, 43, 52, 64, 73, 74, 94–96) (Figure 6A). Knockout of a subunit of ARP2/3 suppressed VCO in a preclinical model of advanced colorectal cancer lung metastasis (CRCLM) (16). The inhibition of VCO was demonstrated by a decrease in cancer cell migration and a replacement histopathological growth pattern (HGP), where VCO is exploited to ensure the blood supply (16, 34, 97). Moreover, the upregulation of ARP2/3 in VCO-dependent cancer cells relies on runt-related transcription factor-1 (RUNX1), which is expressed upon the induction of transforming growth factor beta-1 (TGF-β1) (98, 99) (Figure 6A). In addition, L1CAM and serpins, the adhesion-assisting molecules expressed by metastatic cells, are other potential targets due to their cruciality in the formation of vessel co-opted metastases (20) (Figure 6A). Another interesting approach could be to suppress CD44 and CDC42, which might impair VCO by impeding the cancer cell-pericyte fusion (Figure 6A). This double inhibition has also been shown to facilitate pericyte conversion into a phagocytic/macrophage-like phenotype, favoring an innate immune response against the tumor (26, 64). Intriguingly, targeting CXCR4, a crucial molecule for brain microvasculature invasion by glioblastoma cells, suppresses tumor invasion and renders tumors sensitive to radiation therapy (25, 74), thus presenting another potential intriguing target (Figure 6A).
As few studies investigated the roles of different molecules in VCO upon AAT, combinatorial treatment approaches, integrating AAT with blockade of VCO can be considered. For instance, AAT via VEGFR2 blockade reduced intracerebral glioblastoma growth, but caused an increase in tumor migration with a VCO pattern. Interestingly, this increase in cancer cell migration could be inhibited by combined treatment with VEGFR2 and EGFR antibodies, as was demonstrated by a decreased migration of glioblastoma cells in an in vitro system (94, 95). Furthermore, Wnt7, which is critical for VCO in Olig2+ glioma cells, showed an increased expression in glioma preclinical models and patients after anti-VEGF treatment. Use of Wnt7 inhibitors was efficient to prevent vessel contact of glioma cells, thus limiting the metastasis of vessel co-opted cancer cells (52) (Figure 6A). The β1-integrin subunit mediates the adhesion of metastatic cancer cells to the vascular basement membrane of brain blood vessels. Blockade of this subunit in cancer cells could prevent their adhesion to the membrane of vascular cells, thus attenuating the establishment and growth of metastasis (43) (Figure 6A). Moreover, inhibition of ERK1/2 and FAK, the downstream molecules of β1-integrin, were associated with a beneficial outcome when combined with AAT (43, 73, 96, 100–102) (Figure 6A).
Targeting the tumor microenvironment
Tumors not only contain proliferating cancer cells, but comprise other cell types such as vascular cells, immune cells and a variety of connective-tissue cells, which make up the tumor microenvironment (103, 104). To date, molecular targeting of cancer cells to overcome VCO by preventing their mobility and adhesion towards the vasculature has caught considerable attention. However, strategies targeting other non-tumoral cells could be further exploited, in particular with the outlook on single-cell RNA studies on VCO in the future (as outlined in the next section), which may yield additional insights into the vessel co-opted tumor microenvironment.
Once tumors commit to VCO, an effective way of preventing tumor progression would be to target the existing tumor vasculature. In vessel co-opted tumors, Ang-2 is overexpressed by mature ECs (67). Ang-2, together with VEGF, promotes angiogenesis and tumor progression (67, 105). However, in the absence of VEGF, overexpression of Ang-2 leads to EC apoptosis, vessel destabilization and vessel loss, thus generating a hypoxic core which induces cancer cell apoptosis (106, 107). Therefore, the combination of anti-VEGF together with enhancing the endothelial Ang-2 expression might inhibit the progression of cancer cells along the existing vasculature (Figures 4, 6B).
While additional targeting of the tumor vasculature could be considered a potential approach to overcome VCO (upon AAT), a recent single-cell study investigating VCO in preclinical models discovered that both co-opted ECs and pericytes display gene expression signatures highly similar to those in their healthy counterparts (10). This raises the question whether targeting co-opted ECs and pericytes may be a desirable option, considering the possible effects on the healthy vasculature (10). On the other hand, the observed similarities in transcriptomic profiles of vascular cells of healthy and co-opting vessels may only apply to the investigated models, and further studies may unravel vascular differences of co-opted vessels compared to their healthy counterparts, when exploring different murine models, or in the clinical context. Interestingly, using a nanoparticle contrast agent in Computed Tomography Imaging in a murine model of breast cancer, co-opted vasculature was identified exhibit high vessel wall permeability (108). Thus, a leaky vasculature as a distinguishing characteristic of VCO-dependent tumor could hold great potential for future targeting strategies.
Immune cells in the tumor microenvironment might present another target to inhibit VCO. Tumor-associated macrophages, with both anti- and pro-tumor subsets, play pivotal roles in either promoting or inhibiting tumor surveillance and therefore tumor development (10, 109). An enrichment of immunosuppressive hypoxia-regulated macrophages was found in a preclinical VCO model, which likely resulted from the increased hypoxia level in tumors relying on VCO (10). Thus, inhibiting these cells may help regulate VCO (Figure 6C). Interestingly, M1-like macrophages, which present tumor-associated antigens and express pro-inflammatory cytokines, and thereby activate cytotoxic T lymphocytes, were highly detected in the same VCO-dependent tumor (10) (Figure 1). The enrichment of these pro-inflammatory macrophages may indicate that VCO can actually lead to anti-tumor immunity, which, however, was not strong enough to shrink the tumors, probably due to the existence of other immunosuppressive hypoxia-regulated and M2-like subsets. This raises the question whether further promoting M1-like macrophage polarization could potentially help to improve the regression of vessel co-opted tumors (Figure 6D). Indeed, a mathematical model of glioma suggested that enhanced oxygenation level could promote M1 abundance in tumors (110), while limiting the accumulation of the immunosuppressive hypoxia-related subset of macrophages. Similar to tumor-associated macrophages, tumor-associated neutrophils are also known for both their pro- and anti-tumor growth promoting roles (111). The accumulation of lysyl oxidase like 4 (LOXL-4)-expressing neutrophils in colorectal cancer lung metastases with replacement growth pattern, where VCO is the dominant pattern of vascularization, suggested their possible role in supporting the growth of vessel co-opted tumors, and indicated them as potential targets to control VCO (112) (Figure 1, Figure 6E). These studies suggest that targeting immune compartments of the tumor microenvironment might potentially contribute to VCO inhibition, thereby overcoming AAT resistance. The challenge will obviously be to selectively target VCO-promoting immune cells, while not eliminating anti-cancer immune cells.
Interestingly, cells in the brain stroma such as astrocytes and microglia express plasmin to suppress the adhesion-promoting activity of L1CAM produced by metastatic cancer cells (20). As explained in the above sections, plasmin is however suppressed by tumor cell-secreted serpins, such as neuroserpin and serpin B2. Thus, targeting serpins or enhancing plasmin production in the tumor microenvironment awaits further exploration to limit the adherence of cancer cells to the existing vessels, thereby preventing the formation of vessel co-opted tumors.
Healthy cells adjacent to the lesions also play a role in the tumor displacement process. When cancer cells approach the existing vasculature, their growth relies on the invasion of vessel-adjacent healthy tissue. Therefore, cells in close proximity to VCO lesions must undergo phenotypic alterations so that the displacement by cancer cells could occur. In a VCO-dependent CRCLM model, apoptosis, motility and EMT were induced in tumor-adjacent hepatocytes by the cancer cells via upregulation of proapoptotic cleaved caspase-3 and cleaved PARP-1, EMT-related vimentin and motility-mediated ARP2/3 (113). This resulted in hepatocyte displacement and formation of vessel co-opted metastases. Thus, the interactions between tumor and healthy cells should be further explored to identify potential strategies to interrupt this tumor promoting liaison.
Potential therapeutic approaches
As mentioned above, different cell types are involved in VCO-related tumorigenesis and tumor progression, thus it is essential to design an approach that specifically targets involved cells but shows no specificity to other cell types, to avoid/minimize off-target effects. Gene therapy using tumor-specific promoters has shown potential to suppress or to enhance the expression of genes in cancer cells with promising delivery and expression efficacies (114). Potential tumor-specific promoters such as human telomerase reverse transcriptase, α-fetoprotein promoter, thyroid transcription factor 1, or Mucin 1, are activated by a malignant process, but not in normal healthy cells (114–116). Gene delivery mediated by viral vectors has been exploited in clinical trials (117, 118). Recently, gene delivery systems using non-viral vectors have been developed for cancer therapy, in which gene carriers including lipids, polymers or peptides are investigated to transfer nucleic acids into target cells with low toxicity (119). By using non-viral gene carriers, limitations in delivery capacity and immunogenicity of classic engineered viruses can be overcome, as various types of nucleic acids could be packed in the novel carrier system (119, 120). The benefit of this system is the flexibility in carriers’ structure, which could be modified to increase their specificity and targeting (121). Thus, while partly still in development, opportunities exist that enable modification of molecular targets in individual cell types. Therefore, one may consider such strategies to seek blockade of motility- and adhesion-related molecules in cancer cells, or inducing a “hot” immune environment (e.g. containing immune cell populations with anti-tumor activities) in vessel co-opted tumors (4, 110, 122).
As tumors can switch between VCO- and angiogenesis-driven oxygenation (123), the combination of AAT with VCO inhibition may present a viably strategic approach to overcome AAT resistance and to improve cancer therapies. Interestingly, mathematical modeling of VCO indicated that sequential treatment of VCO inhibition followed by VEGFR blockade could reduce the tumor burden compared to simultaneous treatment (110). Notably, in this theoretical approach, drugs interfering with VCO were modeled computationally. It was also calculated that blocking both VEGF and VCO could enhance tumor oxygenation and increase M1 macrophage abundance, thus improving therapeutic outcomes (110). However, due to the heterogeneity between different tumors, more insight in the relationship between angiogenic and VCO tumor growth is needed to design specific treatments for each cancer type.
The immunosuppressive molecules programmed-cell death ligand 1 (PD-L1)/PD-L2 are upregulated in various cell types in the tumor microenvironment, including cancer cells, macrophages, lymphocytes and ECs among others (124–126). Moreover, in VCO-dependent tumors, PD-L1 and PD-L2 were respectively enriched in hypoxia-related and antigen-presenting/inflammatory macrophages (10). This observation underscores that targeting of non-vascular cell types in VCO tumors may present an attractive approach. Therefore, combining AAT with immunotherapies could result in a better antitumor effect, as the tumors’ nutrient and oxygen supply as well as their immune evasion mechanism are inhibited simultaneously. Indeed, there is increasing evidence of successful combination therapies of AAT and anti-PD-1/PD-L1 blockade when they were used in preclinical studies, partly proving their potential (127–130). Recently, this strategy was validated in several phase III clinical trials, where anti-angiogenic drugs combined with immune checkpoint inhibitors resulted in significant response rate and increased overall survival in cancer patients (131, 132).
It is important to note that such trials were originally designed on the premise that (i) VEGF signaling in angiogenic tumors can induce immunosuppression (133), and (ii) AAT causes vessel normalization, thereby enabling better intra-tumoral drug delivery. To what extent the success of these recent trials involves inhibition of VCO remains to be determined. Another interesting aspect to consider may be the emerging role of ECs as active participants in tumor immunity (134). For instance, several EC subsets, such as liver sinusoidal endothelial cells (135), or a fraction of ECs in lung cancer (136) were shown to express PD-L1, thereby exhibiting the potential to directly modulate immune cells. It will thus be intriguing to unravel how such types of ECs are impacted by immune checkpoint blockade, and what impact this targeting might have with respect to VCO-dependent tumors.
For most tumor types, chemotherapy has been considered the first-line of treatment for decades. Interestingly, some studies demonstrated a possibility to reduce vessel co-opted tumor growth when combining chemotherapy with AAT. Local invasion of metastatic breast tumors, which was worsened by AAT treatment alone, was blocked upon concurrent combination of AAT and Paclitaxel (137). The chemotherapy drug Topotecan improved the efficacy of AAT in a VCO-dependent triple negative breast cancer model (138). Therefore, combination of both therapies could be another option to overcome VCO and AAT resistance.
As it becomes obvious, while several possible (combinatorial) treatments might be considered and further investigated, an in-depth knowledge about all co-opted cell types and the mechanisms driving VCO is required to efficiently revise novel treatment strategies.
Approaches to study vessel co-option: old versus new
To date, approaches to study VCO have mainly been reduced to the identification of histological characteristics by light microscopy. The utilization of only one major methodology to investigate VCO, in addition to the scarcity of proposed molecular mechanisms, call for the exploitation of alternative novel experimental techniques, to shed more light on this thus far mysterious process.
Bulk and single-cell RNA sequencing technologies
Sequencing technologies have revolutionized our understanding of cancer and other diseases, by enabling insight into cells’ genomes, methylomes and transcriptomes in different contexts, leading to the discovery of novel molecular disease mechanisms, and vulnerabilities for potential targeting. However, sequencing studies focusing on VCO are rare. A few studies including RNA arrays, or bulk sequencing on cancer cells treated with AAT may hold some indications for changes in the transcriptome of co-opted cancer cells. In a 2009 study, glioma cells treated with AAT were subjected to a quantitative real-time reverse transcription PCR array (19), revealing a few transcriptional changes in cancer cells in co-opted tumors. The most upregulated genes in these glioma cells were metalloproteinase (MMP)-12, MMP-9, collagen type IV, α3 (COL4-A3), and CXC-chemokine ligand 9 (CXCL9), while laminin α1 (LAMA1), integrin β2 (ITGB2), MMP-1, and hyaluronan synthase 1 (HAS1) were downregulated after Bevacizumab treatment. Thus, in these cancer cells, genes related to angiogenesis seemed highly upregulated. Moreover, the deregulation of genes related to ECM remodeling may contribute to the increased invasive properties of the cancer cells. Importantly, when using the glioma cells in an in vivo model, alternative angiogenic mechanisms were found as resistance mechanisms, and VCO was not investigated, although increased invasiveness of the cancer cells was noted.
Recently, using animal models of VCO in lung metastasis, single-cell transcriptomic technology was used for the first time to identify the transcriptome of co-opted cell types (111). In both models of AAT-induced, as well as spontaneous VCO, co-opted vascular cell types (ECs and pericytes) displayed similar transcriptomic signatures to those of their healthy counterparts, distinct from tumor EC (TEC) signatures in angiogenic tumors. Previously reported EC subtypes (136), such as immature, capillary type 1 and 2, artery, vein, and lymphatic ECs, as well as capillary TECs, were found in lung metastases growing via angiogenesis, and via VCO, as well as in healthy lungs (10). However, capillary ECs expanded in both healthy and co-opted vessels, while angiogenic TECs (tip and proliferating ECs) largely disappeared in co-opted vessels, which may be (at least in part) a reason for the lack of effectiveness of AAT in patients with tumors growing via VCO (16, 139). Co-opted pericytes displayed gene signatures of quiescence and vasodilation, similar to those enriched in pericytes from healthy lungs, and distinct from the gene signatures of angiogenic pericytes (Figure 1), which involve genes related to activation, vessel sprouting, and matrix remodeling (10). Cancer cells were confirmed to upregulate invasive gene signatures, in line with previous observations (16, 52, 140, 141). Interestingly, in tumors relying on VCO, M1-like, antigen-presenting/inflammatory macrophages, as well as hypoxia-regulated and matrix-remodeling macrophages accumulated, in line with the characteristic hypoxic areas and invasive cancer cells in tumors relying on VCO (10) (Figure 1). On the other hand, M2-like, immunosuppressive macrophages were enriched in angiogenic tumors (10) (Figure 1). These intriguing observations may implicate potential novel treatment strategies based on macrophage targeting, as discussed above.
While providing important new insights, this study also generated many questions: is there a difference in EC transcriptomes between those in primary tumors or metastases relying on VCO? How do these findings translate into the clinical human setting? Based on the differences on ECs in different organs and vascular beds, are there differences in co-opted vessels, and other co-opted cell types based on location and type of the tumor? How do angiogenic ECs compare to those that are co-opted in (the same) metastatic lesions? Therefore, more single-cell transcriptomic studies, involving different cancer and metastasis models, as well as human samples, are required. Only with a more comprehensive understanding of the biology of ECs and other cell types during VCO, we can move forward to develop novel treatment strategies.
With this in mind, one could consider exploiting already existing single-cell transcriptomic studies, in particular those analyzing metastases, as VCO is thought to be present in a majority of metastatic lesions (16). Several studies have examined cancer cells as well as stromal cells (including vascular cell types) in metastatic lesions (142, 143), which could potentially be re-analyzed and compared to known transcriptome characteristics of co-opted cell types. Although such studies analyzed metastatic lesions at the single-cell level, sub-setting and an in-depth transcriptome analysis of ECs and other vascular cells have not yet been the focus of such studies. In order to gather a more complete picture of angiogenic and co-opted EC biology, one should compare vascular cell transcriptome profiles in primary angiogenic tumors, in angiogenic metastases, and in metastases relying on VCO. While this may be challenging with metastatic lesions relying on VCO, spatial transcriptomic options discussed below may be exploited to address this concern. Ultimately, original studies with confirmed VCO (via histology) are also required to gain full insight to confirm and expand interpretations from re-analyzed studies.
Where do we go from here?
While single-cell RNA sequencing provides large amounts of transcriptomic information on all cell types in tumors or metastases, spatial information is lost with this approach, preventing the investigation of cell-cell interactions. Nevertheless, several computational approaches, such as CellChat (144) and NicheNet (145), allow prediction of interactions between the different cell types analyzed in a single-cell study. CellChat relies on a database of known receptor-ligand complexes (CellChatDB). The program calculates a probability value of an interaction between two groups of cells, which differently over-express the ligand and the receptor, respectively. NicheNet additionally combines the predicted receptor-ligand interactions with expression data on interacting cells to project the impact of the interactions on the receiver cell’s gene expression, by integrating the anticipated receptor-ligand interactions with intracellular signaling pathways. In fact, CellPhoneDB was used to study interactions between the different cell types identified during VCO at single-cell level (10). Interestingly, different co-opted cell types seem to interact with each other. For instance, M1-like macrophages were predicted to interact with co-opted ECs via CXCL12-CXCR4, and oncostatin-M (OSM) receptor-OSM interactions; and with pericytes via TGF-β signaling (10). How such interactions may influence the functions and phenotypes of the involved cell types remains to be investigated.
The prediction of interactions of different cell types within a tissue may indeed also allow to speculate spatial relationships between cells. However, considering that the only recognized technique to identify VCO is histological analysis, one cannot omit this method when investigating co-opted vessels. In fact, some sophisticated approaches have been utilized to shed light on mechanisms of VCO. For instance, a 3-dimensional (3D) model of vessel growth in lung adenocarcinoma was established by combining hematoxylin and eosin, immunohistochemistry and multiplex immunofluorescence images of patient samples (146). The authors were able to analyze the spatial relationships of cancer cells with the vasculature and the lung parenchyma, identifying tumor nests in the alveolar spaces lacking CD31-positive vessels, as well as focal attachment of cancer cells (spread through air spaces (“STAS” cancer cells) to the alveolar walls, consistent with VCO. The model proposed that cancer cells can break loose from the tumor bulk, and journey through air spaces to attach to alveolar walls, which are surrounded by capillaries (146).
This 3D modeling strategy demonstrates the importance of spatial information, but on the other hand is limited to probing for a handful of proteins simultaneously. To gather the fine-grained single-cell transcriptomic information yet preserving the positional context of the cells within a tissue, “spatial transcriptomics” was developed. Spatial transcriptomics, which was selected as Nature Methods’ “Method of the Year” (147), comprises several approaches to perform RNA sequencing on tissue sections, in conjunction with imaging. Examples are fluorescent in situ RNA sequencing (FISSEQ) (148), RNA seqFISH+ (149, 150), Slide-seq (151), and the method termed “spatial transcriptomics” (152). The latter technology, which relies on barcoded reverse transcription primers allowing RNA sequencing data to be associated with a precise tissue location, was commercialized and is today known as the 10x Genomics product “Visium”. While spatial transcriptomics does not yet enable analysis at the single-cell level, the continuous progress in the field holds hope for single-cell spatiotemporal analysis in the coming years. In the meanwhile, different computational approaches exist to in situ map transcriptome information to spatial information (153–157).
Apart from investigating spatial relationships between cells with known transcriptomes, the VCO field may also benefit from additional novel technologies. For instance, cytometry by time of flight (“CYTOF” (158, 159), an application that uses antibodies conjugated to heavy metal isotopes that can be detected with a mass cytometer), allows simultaneous detection of 40-100 target proteins per cell. Although still not comparable to the vast gene expression information gained by single-cell RNA sequencing, one may argue that protein expression may translate better into cells’ phenotypes and their functions than RNA expression. In line with this, cite-seq is an approach that combines single-cell RNA sequencing with barcoded antibodies, thereby integrating protein and mRNA data (160). Lastly, to gather a comprehensive picture of the VCO process, one should seek to investigate the epigenome of co-opted cell types, to probe for DNA regulatory elements that may be changed in these cell types and provide an additional level of insights into the acquisition of co-opted phenotypes. Techniques, such as single-cell Assay for Transposase-Accessible Chromatin (ATAC) sequencing (161), or Methylome sequencing (162–164) are suitable for such endeavors.
In summary, the new and rapidly evolving era of sequencing and other novel technologies provide seemingly unlimited possibilities to explore VCO on many different levels. Ultimately, these techniques could enable us to understand the VCO process and develop successful therapeutic interventions.
Concluding remarks
VCO is a well-established, yet at the same time, a mysterious and barely understood process of tumor vascularization. For decades, it has been established that blood supply is vital for tumor growth and angiogenesis as a hallmark of cancer. While several additional vascularization mechanisms have long been recognized, most of them have not received much attention from the scientific community, one of which being VCO. The discovery of VEGF’s role in angiogenesis has led to groundbreaking achievements in cancer therapeutics, with approval of the first anti-angiogenic drug Bevacizumab for cancer in 2004. Unfortunately, although successful in some cancer patients, it became obvious that most cancer patients develop resistance mechanisms and become capable of ensuring their blood supply with alternative strategies. Almost two decades later, no vital tactics have been developed to efficiently counteract such resistance mechanisms. Given the high incidence of VCO in AAT-treated tumors, and in particular in metastases (which in most cancers are the cause of death), this mode of vascularization is of remarkable interest to overcome AAT resistance in tumors. It is thus surprising that VCO has received so little attention. However, with the advent of the single-cell transcriptomic era and the plethora of technologies, many opportunities exist to tackle the unraveling of the still cryptic process of VCO. There is great hope that future studies using novel technologies and analysis methods will illuminate the molecular mechanisms that initiate VCO, as well as the detailed characteristics of co-opted cell types. These insights will then facilitate target identification, and ultimately lead to development of unprecedented treatment strategies for tumors and metastases.
Author contributions
Conceptualization: PC. Writing manuscript and creating figures: AC, A-CKT, LMB, PS-G. All authors contributed to the article and approved the submitted version.
Funding
PC is supported by Grants from Methusalem funding (Flemish government), the Fund for Scientific Research-Flanders (FWO-Vlaanderen), the European Research Council ERC Advanced Research Grant EU- ERC74307 and the NNF Laureate Research Grant from Novo Nordisk Foundation (Denmark); LB is supported by a Marie-Sklodowska-Curie Individual Fellowship; AC, A-CKT, LMB are supported by ‘Fonds voor Wetenschappelijk Onderzoek’ (FWO) doctoral and postdoctoral fellowships.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviation
Ang-2, angiopoietin-2; AAT, anti-angiogenic therapy; ARP2/3, actin-related protein 2/3 complex; ATAC, assay for Transposase-Accessible Chromatin; B2R, B2 receptor; CA9, carbonic anhydrase 9 CAM chick chorioallantoic membrane; CDC42, cell division control protein 42 homolog; COL4-A3, collagen type IV, α3; CRCLM, colorectal cancer lung metastasis; CXCR4, CXC-chemokine receptor 4; CXCL9, CXC-chemokine ligand 9; CXCL12, CXC-chemokine ligand 12; CYTOF, cytometry by time of flight; ECM, extracellular matrix; ECs, endothelial cells; EGFR, epidermal growth factor receptor; EMT, epithelial-to-mesenchymal transition; ERK, extracellular signal-regulated kinase; FAK, focal adhesion kinase; FISSEQ, fluorescent in situ RNA sequencing; GBM, glioblastoma multiforme; GSK3β, glycogen synthase kinase 3β; HAS1, hyaluronan synthase 1; Hep3B-hCG, human hepatocellular carcinoma; HEVs, high endothelial venules; HGF, hepatocyte growth factor; HGP, histopathological growth pattern; HIF1α, hypoxia-inducible factor 1; IRE1, inositol-requiring enzyme 1; ITGB2, integrin β2; L1CAM, neural cell adhesion molecule L1; LAMA1, laminin α1; LOXL-4, lysyl oxidase like 4; MMP, metalloproteinase; OSM, oncostatin-M; P4HA1, hypoxia enhances prolyl-4-hydroxylase α1; PD-L1, programmed-cell death ligand 1; PD-L2, programmed-cell death ligand 2; PTP1B, protein tyrosine phosphatase 1B; RENCA, renal adenocarcinoma; RUNX1, runt-related transcription factor-1; STAS, spread through air spaces; TAZ, transcriptional co-activator with PDZ-binding motif; TEC, tumor EC; Tie-2, tyrosine kinase-receptor; VCO, tumor vessel co-option; VEGF-A, vascular endothelial growth factor-A; VEGFR2, vascular endothelial growth factor receptor; YAP, Yes-associated protein; ZEB2, Zinc finger E-box binding homeobox 2.
References
1. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature (2011) 473(7347):298–307. doi: 10.1038/nature10144
2. Pezzella F, Di Bacco A, Andreola S, Nicholson AG, Pastorino U, Harris AL. Angiogenesis in primary lung cancer and lung secondaries. Eur J Cancer (1996) 32A(14):2494–500. doi: 10.1016/S0959-8049(96)00377-2
3. Pezzella F, Pastorino U, Tagliabue E, Andreola S, Sozzi G, Gasparini G, et al. Non-small-cell lung carcinoma tumor growth without morphological evidence of neo-angiogenesis. Am J Pathol (1997) 151(5):1417–23.
4. Haas G, Fan S, Ghadimi M, De Oliveira T, Conradi LC. Different forms of tumor vascularization and their clinical implications focusing on vessel Co-option in colorectal cancer liver metastases. Front Cell Dev Biol (2021) 9:612774. doi: 10.3389/fcell.2021.612774
5. Qian CN, Tan MH, Yang JP, Cao Y. Revisiting tumor angiogenesis: vessel co-option, vessel remodeling, and cancer cell-derived vasculature formation. Chin J Cancer (2016) 35:10. doi: 10.1186/s40880-015-0070-2
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
7. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov (2022) 12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
8. Donnem T, Reynolds AR, Kuczynski EA, Gatter K, Vermeulen PB, Kerbel RS, et al. Non-angiogenic tumours and their influence on cancer biology. Nat Rev Cancer (2018) 18(5):323–36. doi: 10.1038/nrc.2018.14
9. Kuczynski EA, Vermeulen PB, Pezzella F, Kerbel RS, Reynolds AR. Vessel co-option in cancer. Nat Rev Clin Oncol (2019) 16(8):469–93. doi: 10.1038/s41571-019-0181-9
10. Teuwen LA, De Rooij L, Cuypers A, Rohlenova K, Dumas SJ, Garcia-Caballero M, et al. Tumor vessel co-option probed by single-cell analysis. Cell Rep (2021) 35(11):109253. doi: 10.1016/j.celrep.2021.109253
11. Montana V, Sontheimer H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J Neurosci (2011) 31(13):4858–67. doi: 10.1523/JNEUROSCI.3825-10.2011
12. Wang W, Zhou Y, Wei R, Jiang G, Li F, Chen X, et al. Bradykinin promotes proliferation, migration, and invasion of cervical cancer cells through STAT3 signaling pathways. Oncol Rep (2019) 42(6):2521–7. doi: 10.3892/or.2019.7380
13. Hardee ME, Zagzag D. Mechanisms of glioma-associated neovascularization. Am J Pathol (2012) 181(4):1126–41. doi: 10.1016/j.ajpath.2012.06.030
14. Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci (2007) 8(8):610–22. doi: 10.1038/nrn2175
15. Bridgeman VL, Vermeulen PB, Foo S, Bilecz A, Daley F, Kostaras E, et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J Pathol (2017) 241(3):362–74. doi: 10.1002/path.4845
16. Frentzas S, Simoneau E, Bridgeman VL, Vermeulen PB, Foo S, Kostaras E, et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat Med (2016) 22(11):1294–302. doi: 10.1038/nm.4197
17. Kuczynski EA, Kerbel RS. Implications of vessel co-option in sorafenib-resistant hepatocellular carcinoma. Chin J Cancer (2016) 35(1):97. doi: 10.1186/s40880-016-0162-7
18. Qian CN. Hijacking the vasculature in ccRCC–co-option, remodelling and angiogenesis. Nat Rev Urol (2013) 10(5):300–4. doi: 10.1038/nrurol.2013.26
19. Lucio-Eterovic AK, Piao Y, de Groot JF. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res (2009) 15(14):4589–99. doi: 10.1158/1078-0432.CCR-09-0575
20. Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell (2014) 156(5):1002–16. doi: 10.1016/j.cell.2014.01.040
21. Sardari Nia P, Hendriks J, Friedel G, Van Schil P, Van Marck E. Distinct angiogenic and non-angiogenic growth patterns of lung metastases from renal cell carcinoma. Histopathology (2007) 51(3):354–61.
22. Warth A, Muley T, Kossakowski CA, Goeppert B, Schirmacher P, Dienemann H, et al. Prognostic impact of intra-alveolar tumor spread in pulmonary adenocarcinoma. Am J Surg Pathol (2015) 39(6):793–801. doi: 10.1097/PAS.0000000000000409
23. Kadota K, Nitadori JI, Sima CS, Ujiie H, Rizk NP, Jones DR, et al. Tumor spread through air spaces is an important pattern of invasion and impacts the frequency and location of recurrences after limited resection for small stage I lung adenocarcinomas. J Thorac Oncol (2015) 10(5):806–14. doi: 10.1097/JTO.0000000000000486
24. Rini BI, Small EJ. Biology and clinical development of vascular endothelial growth factor-targeted therapy in renal cell carcinoma. J Clin Oncol (2005) 23(5):1028–43. doi: 10.1200/JCO.2005.01.186
25. Zhang Y, Wang S, Dudley AC. Models and molecular mechanisms of blood vessel co-option by cancer cells. Angiogenesis (2020) 23(1):17–25. doi: 10.1007/s10456-019-09684-y
26. Caspani EM, Crossley PH, Redondo-Garcia C, Martinez S. Glioblastoma: a pathogenic crosstalk between tumor cells and pericytes. PloS One (2014) 9(7):e101402. doi: 10.1371/journal.pone.0101402
27. Bernsen H, van der Laak J, Kusters B, van der Ven A, Wesseling P. Gliomatosis cerebri: quantitative proof of vessel recruitment by cooptation instead of angiogenesis. J Neurosurg (2005) 103(4):702–6. doi: 10.3171/jns.2005.103.4.0702
28. Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science (1999) 284(5422):1994–8. doi: 10.1126/science.284.5422.1994
29. Berghoff AS, Rajky O, Winkler F, Bartsch R, Furtner J, Hainfellner JA, et al. Invasion patterns in brain metastases of solid cancers. Neuro Oncol (2013) 15(12):1664–72. doi: 10.1093/neuonc/not112
30. Siam L, Bleckmann A, Chaung HN, Mohr A, Klemm F, Barrantes-Freer A, et al. The metastatic infiltration at the metastasis/brain parenchyma-interface is very heterogeneous and has a significant impact on survival in a prospective study. Oncotarget (2015) 6(30):29254–67. doi: 10.18632/oncotarget.4201
31. Hung T, Morin J, Munday WR, Mackenzie IR, Lugassy C, Barnhill RL. Angiotropism in primary cutaneous melanoma with brain metastasis: a study of 20 cases. Am J Dermatopathol (2013) 35(6):650–4. doi: 10.1097/DAD.0b013e31827e8315AD
32. Kusters B, Leenders WP, Wesseling P, Smits D, Verrijp K, Ruiter DJ, et al. Vascular endothelial growth factor-A(165) induces progression of melanoma brain metastases without induction of sprouting angiogenesis. Cancer Res (2002) 62(2):341–5.
33. Nakashima O, Sugihara S, Kage M, Kojiro M. Pathomorphologic characteristics of small hepatocellular carcinoma: a special reference to small hepatocellular carcinoma with indistinct margins. Hepatology (1995) 22(1):101–5.
34. Stessels F, Van den Eynden G, van der Auwera I, Salgado R, Van den Heuvel E, Harris AL, et al. Breast adenocarcinoma liver metastases, in contrast to colorectal cancer liver metastases, display a non-angiogenic growth pattern that preserves the stroma and lacks hypoxia. Br J Cancer (2004) 90(7):1429–36. doi: 10.1038/sj.bjc.6601727
35. Fernandez Moro C, Bozoky B, Gerling M. Growth patterns of colorectal cancer liver metastases and their impact on prognosis: a systematic review. BMJ Open Gastroenterol (2018) 5(1):e000217.
36. Jeong HS, Jones D, Liao S, Wattson DA, Cui CH, Duda DG, et al. Investigation of the lack of angiogenesis in the formation of lymph node metastases. J Natl Cancer Inst (2015) 107(9). doi: 10.1093/jnci/djv155
37. Naresh KN, Nerurkar AY, Borges AM. Angiogenesis is redundant for tumour growth in lymph node metastases. Histopathology (2001) 38(5):466–70. doi: 10.1046/j.1365-2559.2001.01061.x
38. Vermeulen PB, Sardari Nia P, Colpaert C, Dirix LY, Van Marck E. Lack of angiogenesis in lymph node metastases of carcinomas is growth pattern-dependent. Histopathology (2002) 40(1):105–7. doi: 10.1046/j.1365-2559.2002.1340c.x
39. Colpaert CG, Vermeulen PB, Van Beest P, Soubry A, Goovaerts G, Dirix LY, et al. Cutaneous breast cancer deposits show distinct growth patterns with different degrees of angiogenesis, hypoxia and fibrin deposition. Histopathology (2003) 42(6):530–40. doi: 10.1046/j.1365-2559.2003.01629.x
40. Lugassy C, Zadran S, Bentolila LA, Wadehra M, Prakash R, Carmichael ST, et al. Angiotropism, pericytic mimicry and extravascular migratory metastasis in melanoma: an alternative to intravascular cancer dissemination. Cancer Microenviron (2014) 7(3):139–52. doi: 10.1007/s12307-014-0156-4
41. Bentolila LA, Prakash R, Mihic-Probst D, Wadehra M, Kleinman HK, Carmichael TS, et al. Imaging of Angiotropism/Vascular Co-option in a murine model of brain melanoma: Implications for melanoma progression along extravascular pathways. Sci Rep (2016) 6:23834. doi: 10.1038/srep23834
42. Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med (2010) 16(1):116–22. doi: 10.1038/nm.2072
43. Carbonell WS, Ansorge O, Sibson N, Muschel R. The vascular basement membrane as "soil" in brain metastasis. PloS One (2009) 4(6):e5857. doi: 10.1371/journal.pone.0005857
44. Er EE, Valiente M, Ganesh K, Zou Y, Agrawal S, Hu J, et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat Cell Biol (2018) 20(8):966–78. doi: 10.1038/s41556-018-0138-8
45. Wang S, Liang K, Hu Q, Li P, Song J, Yang Y, et al. JAK2-binding long noncoding RNA promotes breast cancer brain metastasis. J Clin Invest (2017) 127(12):4498–515. doi: 10.1172/JCI91553
46. Leenders WP, Kusters B, Verrijp K, Maass C, Wesseling P, Heerschap A, et al. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin Cancer Res (2004) 10:6222–30. doi: 10.1158/1078-0432.CCR-04-0823
47. Szabo V, Bugyik E, Dezso K, Ecker N, Nagy P, Timar J, et al. Mechanism of tumour vascularization in experimental lung metastases. J Pathol (2015) 235(3):384–96. doi: 10.1002/path.4464
48. Werbeck JL, Thudi NK, Martin CK, Premanandan C, Yu L, Ostrowksi MC, et al. Tumor microenvironment regulates metastasis and metastasis genes of mouse MMTV-PymT mammary cancer cells in vivo. Vet Pathol (2014) 51(4):868–81. doi: 10.1177/0300985813505116
49. Kuczynski EA, Yin M, Bar-Zion A, Lee CR, Butz H, Man S, et al. Co-Option of liver vessels and not sprouting angiogenesis drives acquired sorafenib resistance in hepatocellular carcinoma. J Natl Cancer Inst (2016) 108(8). doi: 10.1093/jnci/djw030
50. Stoletov K, Strnadel J, Zardouzian E, Momiyama M, Park FD, Kelber JA, et al. Role of connexins in metastatic breast cancer and melanoma brain colonization. J Cell Sci (2013) 126:904–13. doi: 10.1242/jcs.112748
51. Chadwick EJ, Yang DP, Filbin MG, Mazzola E, Sun Y, Behar O, et al. A brain Tumor/Organotypic slice Co-culture system for studying tumor microenvironment and targeted drug therapies. J Vis Exp (2015) 105):e53304. doi: 10.3791/53304
52. Griveau A, Seano G, Shelton SJ, Kupp R, Jahangiri A, Obernier K, et al. A glial signature and Wnt7 signaling regulate glioma-vascular interactions and tumor microenvironment. Cancer Cell (2018) 33(5):874–89.e7. doi: 10.1016/j.ccell.2018.03.020
53. Jenkins MH, Steinberg SM, Alexander MP, Fisher JL, Ernstoff MS, Turk MJ, et al. Multiple murine BRaf(V600E) melanoma cell lines with sensitivity to PLX4032. Pigment Cell Melanoma Res (2014) 27(3):495–501. doi: 10.1111/pcmr.12220
54. Seifert S, Sontheimer H. Bradykinin enhances invasion of malignant glioma into the brain parenchyma by inducing cells to undergo amoeboid migration. J Physiol (2014) 592(22):5109–27. doi: 10.1113/jphysiol.2014.274498
55. Liu Y, G Y, Ng S, Deng Z, Lyon CJ, Koay EJ, et al. Circulating levels of hydroxylated bradykinin function as an indicator of tissue HIF-1α expression. Sci Bull (2020) 65(18):1570–9. doi: 10.1016/j.scib.2020.04.023
56. Kwaan HC, McMahon B. The role of plasminogen-plasmin system in cancer. Cancer Treat Res (2009) 148:43–66. doi: 10.1007/978-0-387-79962-9_4
57. Shao Y, Lu B. The emerging roles of circular RNAs in vessel co-option and vasculogenic mimicry: clinical insights for anti-angiogenic therapy in cancers. Cancer Metastasis Rev (2022) 41(1):173–91. doi: 10.1007/s10555-021-10000-8
58. Su CY, Li JQ, Zhang LL, Wang H, Wang FH, Tao YW, et al. The biological functions and clinical applications of integrins in cancers. Front Pharmacol (2020) 11:579068. doi: 10.3389/fphar.2020.579068
59. Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol (2015) 25(4):234–40. doi: 10.1016/j.tcb.2014.12.006
60. Garcia-Gomez P, Valiente M. Vascular co-option in brain metastasis. Angiogenesis (2020) 23(1):3–8. doi: 10.1007/s10456-019-09693-x
61. Fan J, Cai B, Zeng M, Hao Y, Giancotti FG, Fu BM. Integrin beta4 signaling promotes mammary tumor cell adhesion to brain microvascular endothelium by inducing ErbB2-mediated secretion of VEGF. Ann BioMed Eng (2011) 39(8):2223–41. doi: 10.1007/s10439-011-0321-6
62. Reymond N, Im JH, Garg R, Vega FM, Borda d'Agua B, Riou P, et al. Cdc42 promotes transendothelial migration of cancer cells through beta1 integrin. J Cell Biol (2012) 199(4):653–68. doi: 10.1083/jcb.201205169
63. Xiao XH, Lv LC, Duan J, Wu YM, He SJ, Hu ZZ, et al. Regulating Cdc42 and its signaling pathways in cancer: Small molecules and MicroRNA as new treatment candidates. Molecules (2018) 23(4):787–808. doi: 10.3390/molecules23040787
64. Morath I, Hartmann TN, Orian-Rousseau V. CD44: More than a mere stem cell marker. Int J Biochem Cell Biol (2016) 81:166–73. doi: 10.1016/j.biocel.2016.09.009
65. Rada M, Lazaris A, Kapelanski-Lamoureux A, Mayer TZ, Metrakos P. Tumor microenvironment conditions that favor vessel co-option in colorectal cancer liver metastases: A theoretical model. Semin Cancer Biol (2021) 71:52–64. doi: 10.1016/j.semcancer.2020.09.001
66. Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-tie system. Nat Rev Mol Cell Biol (2009) 10(3):165–77. doi: 10.1038/nrm2639
67. Coelho AL, Gomes MP, Catarino RJ, Rolfo C, Lopes AM, Medeiros RM, et al. Angiogenesis in NSCLC: is vessel co-option the trunk that sustains the branches? Oncotarget (2017) 8(24):39795–804. doi: 10.18632/oncotarget.7794
68. Dvorak HF. Tumor stroma, tumor blood vessels, and antiangiogenesis therapy. Cancer J (2015) 21(4):237–43. doi: 10.1097/PPO.0000000000000124
69. Yu Q, Stamenkovic I. Angiopoietin-2 is implicated in the regulation of tumor angiogenesis. Am J Pathol (2001) 158(2):563–70. doi: 10.1016/S0002-9440(10)63998-3
70. Nasarre P, Thomas M, Kruse K, Helfrich I, Wolter V, Deppermann C, et al. Host-derived angiopoietin-2 affects early stages of tumor development and vessel maturation but is dispensable for later stages of tumor growth. Cancer Res (2009) 69(4):1324–33. doi: 10.1158/0008-5472.CAN-08-3030
71. Auf G, Jabouille A, Guerit S, Pineau R, Delugin M, Bouchecareilh M, et al. Inositol-requiring enzyme 1alpha is a key regulator of angiogenesis and invasion in malignant glioma. Proc Natl Acad Sci USA (2010) 107(35):15553–8. doi: 10.1073/pnas.0914072107
72. Yeung KT, Yang J. Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol (2017) 11(1):28–39. doi: 10.1002/1878-0261.12017
73. Schlaepfer DD, Mitra SK. Multiple connections link FAK to cell motility and invasion. Curr Opin Genet Dev (2004) 14(1):92–101. doi: 10.1016/j.gde.2003.12.002
74. Yadav VN, Zamler D, Baker GJ, Kadiyala P, Erdreich-Epstein A, DeCarvalho AC, et al. CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget (2016) 7(50):83701–19. doi: 10.18632/oncotarget.13295
75. Yamaguchi H, Taouk GM. A potential role of YAP/TAZ in the interplay between metastasis and metabolic alterations. Front Oncol (2020) 10:928. doi: 10.3389/fonc.2020.00928
76. Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem (2009) 284(20):13355–62. doi: 10.1074/jbc.M900843200
77. Lehmann W, Mossmann D, Kleemann J, Mock K, Meisinger C, Brummer T, et al. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat Commun (2016) 7:10498. doi: 10.1038/ncomms10498
78. Depner C, Zum Buttel H, Bogurcu N, Cuesta AM, Aburto MR, Seidel S, et al. EphrinB2 repression through ZEB2 mediates tumour invasion and anti-angiogenic resistance. Nat Commun (2016) 7:12329. doi: 10.1038/ncomms12329
79. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi: 10.1038/nrm3758
80. Olsen JJ, Pohl SO, Deshmukh A, Visweswaran M, Ward NC, Arfuso F, et al. The role of wnt signalling in angiogenesis. Clin Biochem Rev (2017) 38(3):131–42.
81. Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, et al. A wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol (2006) 8(12):1398–406. doi: 10.1038/ncb1508
82. Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF, et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia (2000) 2(4):306–14. doi: 10.1038/sj.neo.7900102
83. di Tomaso E, Snuderl M, Kamoun WS, Duda DG, Auluck PK, Fazlollahi L, et al. Glioblastoma recurrence after cediranib therapy in patients: lack of "rebound" revascularization as mode of escape. Cancer Res (2011) 71(1):19–28. doi: 10.1158/0008-5472.CAN-10-2602
84. Fack F, Espedal H, Keunen O, Golebiewska A, Obad N, Harter PN, et al. Bevacizumab treatment induces metabolic adaptation toward anaerobic metabolism in glioblastomas. Acta Neuropathol (2015) 129(1):115–31. doi: 10.1007/s00401-014-1352-5
85. Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell (2012) 22(1):21–35. doi: 10.1016/j.ccr.2012.05.037
86. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell (2005) 8(4):299–309. doi: 10.1016/j.ccr.2005.09.005
87. Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell (2009) 15(3):232–9. doi: 10.1016/j.ccr.2009.01.021
88. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell (2009) 15(3):220–31. doi: 10.1016/j.ccr.2009.01.027
89. Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discovery (2012) 2(3):270–87. doi: 10.1158/2159-8290.CD-11-0240
90. Lengyel E, Prechtel D, Resau JH, Gauger K, Welk A, Lindemann K, et al. C-met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of Her2/neu. Int J Cancer (2005) 113(4):678–82. doi: 10.1002/ijc.20598
91. Sawada K, Radjabi AR, Shinomiya N, Kistner E, Kenny H, Becker AR, et al. C-met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res (2007) 67(4):1670–9. doi: 10.1158/0008-5472.CAN-06-1147
92. Xu L, Nilsson MB, Saintigny P, Cascone T, Herynk MH, Du Z, et al. Epidermal growth factor receptor regulates MET levels and invasiveness through hypoxia-inducible factor-1alpha in non-small cell lung cancer cells. Oncogene (2010) 29(18):2616–27. doi: 10.1038/onc.2010.16
93. Hu H, Chen Y, Tan S, Wu S, Huang Y, Fu S, et al. The research progress of antiangiogenic therapy, immune therapy and tumor microenvironment. Front Immunol (2022) 13:802846. doi: 10.3389/fimmu.2022.802846
94. Kunkel P, Ulbricht U, Bohlen P, Brockmann MA, Fillbrandt R, Stavrou D, et al. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res (2001) 61(18):6624–8.
95. Lamszus K, Brockmann MA, Eckerich C, Bohlen P, May C, Mangold U, et al. Inhibition of glioblastoma angiogenesis and invasion by combined treatments directed against vascular endothelial growth factor receptor-2, epidermal growth factor receptor, and vascular endothelial-cadherin. Clin Cancer Res (2005) 11(13):4934–40. doi: 10.1158/1078-0432.CCR-04-2270
96. Zhou J, Yi Q, Tang L. The roles of nuclear focal adhesion kinase (FAK) on cancer: a focused review. J Exp Clin Cancer Res (2019) 38(1):250. doi: 10.1186/s13046-019-1265-1
97. Vermeulen PB, Colpaert C, Salgado R, Royers R, Hellemans H, Van Den Heuvel E, et al. Liver metastases from colorectal adenocarcinomas grow in three patterns with different angiogenesis and desmoplasia. J Pathol (2001) 195(3):336–42. doi: 10.1002/path.966
98. Lie ALM, Marinopoulou E, Li Y, Patel R, Stefanska M, Bonifer C, et al. RUNX1 positively regulates a cell adhesion and migration program in murine hemogenic endothelium prior to blood emergence. Blood (2014) 124(11):e11–20. doi: 10.1182/blood-2014-04-572958
99. Rada M, Kapelanski-Lamoureux A, Petrillo S, Tabaries S, Siegel P, Reynolds AR, et al. Runt related transcription factor-1 plays a central role in vessel co-option of colorectal cancer liver metastases. Commun Biol (2021) 4(1):950. doi: 10.1038/s42003-021-02481-8
100. Sun S, Wu HJ, Guan JL. Nuclear FAK and its kinase activity regulate VEGFR2 transcription in angiogenesis of adult mice. Sci Rep (2018) 8(1):2550. doi: 10.1038/s41598-018-20930-z
101. Lu Y, Liu B, Liu Y, Yu X, Cheng G. Dual effects of active ERK in cancer: A potential target for enhancing radiosensitivity. Oncol Lett (2020) 20(2):993–1000. doi: 10.3892/ol.2020.11684
102. Byzova TV, Goldman CK, Pampori N, Thomas KA, Bett A, Shattil SJ, et al. A mechanism for modulation of cellular responses to VEGF: activation of the integrins. Mol Cell (2000) 6(4):851–60. doi: 10.1016/S1097-2765(00)00083-6
103. Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol (2018) 15(6):366–81. doi: 10.1038/s41571-018-0007-1
104. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene (2008) 27(45):5904–12. doi: 10.1038/onc.2008.271
105. Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell (2011) 19(4):512–26. doi: 10.1016/j.ccr.2011.02.005
106. Hillen F, Griffioen AW. Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev (2007) 26(3-4):489–502. doi: 10.1007/s10555-007-9094-7
107. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell (2014) 26(5):605–22. doi: 10.1016/j.ccell.2014.10.006
108. Ghaghada KB, Badea CT, Karumbaiah L, Fettig N, Bellamkonda RV, Johnson GA, et al. Evaluation of tumor microenvironment in an animal model using a nanoparticle contrast agent in computed tomography imaging. Acad Radiol (2011) 18(1):20–30. doi: 10.1016/j.acra.2010.09.003
109. Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-associated macrophages: Recent insights and therapies. Front Oncol (2020) 10:188. doi: 10.3389/fonc.2020.00188
110. Voutouri C, Kirkpatrick ND, Chung E, Mpekris F, Baish JW, Munn LL, et al. Experimental and computational analyses reveal dynamics of tumor vessel cooption and optimal treatment strategies. Proc Natl Acad Sci U S A (2019) 116(7):2662–71. doi: 10.1073/pnas.1818322116
111. Powell DR, Huttenlocher A. Neutrophils in the tumor microenvironment. Trends Immunol (2016) 37(1):41–52. doi: 10.1016/j.it.2015.11.008
112. Palmieri V, Lazaris A, Mayer TZ, Petrillo SK, Alamri H, Rada M, et al. Neutrophils expressing lysyl oxidase-like 4 protein are present in colorectal cancer liver metastases resistant to anti-angiogenic therapy. J Pathol (2020) 251(2):213–23. doi: 10.1002/path.5449
113. Rada M, Tsamchoe M, Kapelanski-Lamoureux A, Hassan N, Bloom J, Petrillo S, et al. Cancer cells promote phenotypic alterations in hepatocytes at the edge of cancer cell nests to facilitate vessel Co-option establishment in colorectal cancer liver metastases. Cancers (Basel) (2022) 14(5):1318–37. doi: 10.3390/cancers14051318
114. Montano-Samaniego M, Bravo-Estupinan DM, Mendez-Guerrero O, Alarcon-Hernandez E, Ibanez-Hernandez M. Strategies for targeting gene therapy in cancer cells with tumor-specific promoters. Front Oncol (2020) 10:605380. doi: 10.3389/fonc.2020.605380
115. Zhang P, Tan J, Yang DB, Luo ZC, Luo S, Chen P, et al. Gene therapy using the human telomerase catalytic subunit gene promoter enables targeting of the therapeutic effects of vesicular stomatitis virus matrix protein against human lung adenocarcinoma. Exp Ther Med (2012) 4(5):859–64. doi: 10.3892/etm.2012.679
116. Shepelev MV, Kopantzev EP, Vinogradova TV, Sverdlov ED, Korobko IV. hTERT and BIRC5 gene promoters for cancer gene therapy: A comparative study. Oncol Lett (2016) 12(2):1204–10. doi: 10.3892/ol.2016.4718
117. Giacca M, Zacchigna S. Virus-mediated gene delivery for human gene therapy. J Control Release (2012) 161(2):377–88. doi: 10.1016/j.jconrel.2012.04.008
118. Nayerossadat N, Maedeh T, Ali PA. Viral and nonviral delivery systems for gene delivery. Adv BioMed Res (2012) 1:27. doi: 10.4103/2277-9175.98152
119. Mohammadinejad R, Dehshahri A, Sagar Madamsetty V, Zahmatkeshan M, Tavakol S, Makvandi P, et al. In vivo gene delivery mediated by non-viral vectors for cancer therapy. J Control Release (2020) 325:249–75. doi: 10.1016/j.jconrel.2020.06.038
120. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet (2003) 4(5):346–58. doi: 10.1038/nrg1066
121. Ramamoorth M, Narvekar A. Non viral vectors in gene therapy- an overview. J Clin Diagn Res (2015) 9(1):GE01–6. doi: 10.7860/JCDR/2015/10443.5394
122. van Dam PJ, Daelemans S, Ross E, Waumans Y, Van Laere S, Latacz E, et al. Histopathological growth patterns as a candidate biomarker for immunomodulatory therapy. Semin Cancer Biol (2018) 52:86–93. doi: 10.1016/j.semcancer.2018.01.009
123. Kuczynski EA, Reynolds AR. Vessel co-option and resistance to anti-angiogenic therapy. Angiogenesis (2020) 23(1):55–74. doi: 10.1007/s10456-019-09698-6
124. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
125. Evrard D, Hourseau M, Couvelard A, Paradis V, Gauthier H, Raymond E, et al. PD-L1 expression in the microenvironment and the response to checkpoint inhibitors in head and neck squamous cell carcinoma. Oncoimmunology (2020) 9(1):1844403. doi: 10.1080/2162402X.2020.1844403
126. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res (2020) 10(3):727–42.
127. Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med (2017) 9(385). doi: 10.1126/scitranslmed.aak9679
128. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med (2017) 9(385). doi: 10.1126/scitranslmed.aak9670
129. Lee WS, Yang H, Chon HJ, Kim C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp Mol Med (2020) 52(9):1475–85. doi: 10.1038/s12276-020-00500-y
130. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun (2016) 7:12624. doi: 10.1038/ncomms12624
131. Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med (2019) 380(12):1116–27. doi: 10.1056/NEJMoa1816714
132. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med (2020) 382(20):1894–905. doi: 10.1056/NEJMoa1915745
133. Tamura R, Tanaka T, Akasaki Y, Murayama Y, Yoshida K, Sasaki H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: perspectives for therapeutic implications. Med Oncol (2019) 37(1):2. doi: 10.1007/s12032-019-1329-2
134. Amersfoort J, Eelen G, Carmeliet P. Immunomodulation by endothelial cells - partnering up with the immune system? Nat Rev Immunol (2022) 1–13. doi: 10.1038/s41577-022-00694-4
135. Yang M, Zhang C. The role of liver sinusoidal endothelial cells in cancer liver metastasis. Am J Cancer Res (2021) 11(5):1845–60.
136. Goveia J, Rohlenova K, Taverna F, Treps L, Conradi LC, Pircher A, et al. An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell (2020) 37(1):21–36.e13.
137. Paez-Ribes M, Man S, Xu P, Kerbel RS. Potential proinvasive or metastatic effects of preclinical antiangiogenic therapy are prevented by concurrent chemotherapy. Clin Cancer Res (2015) 21(24):5488–98. doi: 10.1158/1078-0432.CCR-15-0915
138. Di Desidero T, Xu P, Man S, Bocci G, Kerbel RS. Potent efficacy of metronomic topotecan and pazopanib combination therapy in preclinical models of primary or late stage metastatic triple-negative breast cancer. Oncotarget (2015) 6(40):42396–410. doi: 10.18632/oncotarget.6377
139. de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol (2010) 12(3):233–42. doi: 10.1093/neuonc/nop027
140. Dome B, Hendrix MJ, Paku S, Tovari J, Timar J. Alternative vascularization mechanisms in cancer: Pathology and therapeutic implications. Am J Pathol (2007) 170(1):1–15.
141. Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis (2014) 17(3):471–94. doi: 10.1007/s10456-014-9420-y
142. Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW, et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun (2020) 11(1):2285. doi: 10.1038/s41467-020-16164-1
143. Xu K, Wang R, Xie H, Hu L, Wang C, Xu J, et al. Single-cell RNA sequencing reveals cell heterogeneity and transcriptome profile of breast cancer lymph node metastasis. Oncogenesis (2021) 10(10):66. doi: 10.1038/s41389-021-00355-6
144. Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun (2021) 12(1):1088. doi: 10.1038/s41467-021-21246-9
145. Browaeys R, Saelens W, Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods (2020) 17(2):159–62. doi: 10.1038/s41592-019-0667-5
146. Yagi Y, Aly RG, Tabata K, Barlas A, Rekhtman N, Eguchi T, et al. Three-dimensional histologic, immunohistochemical, and multiplex immunofluorescence analyses of dynamic vessel Co-option of spread through air spaces in lung adenocarcinoma. J Thorac Oncol (2020) 15(4):589–600. doi: 10.1016/j.jtho.2019.12.112
147. Method of the year 2020: spatially resolved transcriptomics. Nat Methods (2021) 18(1):1. doi: 10.1038/s41592-020-01042-x
148. Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Ferrante TC, Terry R, et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat Protoc (2015) 10(3):442–58. doi: 10.1038/nprot.2014.191
149. Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L. Single-cell in situ RNA profiling by sequential hybridization. Nat Methods (2014) 11(4):360–1. doi: 10.1038/nmeth.2892
150. Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature (2019) 568(7751):235–9. doi: 10.1038/s41586-019-1049-y
151. Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science (2019) 363(6434):1463–7. doi: 10.1126/science.aaw1219
152. Stahl PL, Salmen F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science (2016) 353(6294):78–82. doi: 10.1126/science.aaf2403
153. Okochi Y, Sakaguchi S, Nakae K, Kondo T, Naoki H. Model-based prediction of spatial gene expression via generative linear mapping. Nat Commun (2021) 12(1):3731. doi: 10.1038/s41467-021-24014-x
154. Ren X, Zhong G, Zhang Q, Zhang L, Sun Y, Zhang Z. Reconstruction of cell spatial organization from single-cell RNA sequencing data based on ligand-receptor mediated self-assembly. Cell Res (2020) 30(9):763–78. doi: 10.1038/s41422-020-0353-2
155. Moriel N, Senel E, Friedman N, Rajewsky N, Karaiskos N, Nitzan M. NovoSpaRc: flexible spatial reconstruction of single-cell gene expression with optimal transport. Nat Protoc (2021) 16(9):4177–200. doi: 10.1038/s41596-021-00573-7
156. Abdelaal T, Mourragui S, Mahfouz A, Reinders MJT. SpaGE: Spatial gene enhancement using scRNA-seq. Nucleic Acids Res (2020) 48(18):e107. doi: 10.1093/nar/gkaa740
157. Cang Z, Nie Q. Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat Commun (2020) 11(1):2084. doi: 10.1038/s41467-020-15968-5
158. Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem (2009) 81(16):6813–22. doi: 10.1021/ac901049w
159. Tracey LJ, An Y, Justice MJ. CyTOF: An emerging technology for single-cell proteomics in the mouse. Curr Protoc (2021) 1(4):e118. doi: 10.1002/cpz1.118
160. Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods (2017) 14(9):865–8. doi: 10.1038/nmeth.4380
161. Stueve TR, Marconett CN, Zhou B, Borok Z, Laird-Offringa IA. The importance of detailed epigenomic profiling of different cell types within organs. Epigenomics (2016) 8(6):817–29. doi: 10.2217/epi-2016-0005
162. Cheung HH, Lee TL, Rennert OM, Chan WY. DNA Methylation of cancer genome. Birth Defects Res C Embryo Today (2009) 87(4):335–50. doi: 10.1002/bdrc.20163
163. Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol (2010) 28(10):1106–14. doi: 10.1038/nbt.1681
Keywords: vessel co-option, anti-angiogenic therapy resistance, tumor vascularization, mouse models, molecular mechanisms, state-of-the-art analysis
Citation: Cuypers A, Truong A-CK, Becker LM, Saavedra-García P and Carmeliet P (2022) Tumor vessel co-option: The past & the future. Front. Oncol. 12:965277. doi: 10.3389/fonc.2022.965277
Received: 09 June 2022; Accepted: 04 August 2022;
Published: 31 August 2022.
Edited by:
Francesco Pezzella, University of Oxford, United KingdomReviewed by:
Chao-Nan Qian, Sun Yat-sen University Cancer Center (SYSUCC), ChinaRobert Kerbel, Sunnybrook Research Institute (SRI), Canada
Copyright © 2022 Cuypers, Truong, Becker, Saavedra-García and Carmeliet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Carmeliet, cGV0ZXIuY2FybWVsaWV0QGt1bGV1dmVuLmJl
†These authors share first authorship