 Rui Pan
Rui Pan Qiu Rao
Qiu Rao- Department of Pathology, Affiliated Jinling Hospital, Medical School of Nanjing University, Nanjing, China
Introduction: Epithelioid glioblastoma (eGBM) is one of the rare glioblastoma (GBM) variants in the current World Health Organization (WHO) categorization of central nervous system (CNS) tumours. However, the diagnostic basis and molecular features of eGBM have not been clearly defined to date. In this study, we aimed to molecularly characterize these tumours.
Methods: The clinicopathological, molecular, and immunohistochemical characteristics of 12 cases of eGBM were investigated.
Results: The tumours were found to be made up of epithelioid and rhabdoid cells when examined under a microscope. Six cases (50%) harboured the BRAF V600E mutation, and NF1 mutation was detected in 2 eGBM cases (16.7%). CDKN2A/B homozygous deletion was seen in 5 cases (41.7%). TP53 mutation was recognized in 2 instances (16.7%), and TERT promoter mutation was recognized in 5 cases (41.7%).
Discussion: eGBM is characterized by high molecular heterogeneity and has molecular overlaps between low-grade gliomas. Moreover, rather than being a variant or entity, the biological significance of the "epithelioid" appearance may be reduced to a simply morphological pattern. In order to target the proper treatment to suitable patients, molecular stratification via genome-wide molecular profiling will be crucial.
Introduction
GBM is the extremely frequent and aggressive tumour of the human brain. Epithelioid glioblastoma (eGBM) is the rare type of GBM variables in the 2021 WHO CNS tumours classification. This entity is mostly made up of epithelioid cells with abundant cytoplasm, eccentrically placed nuclei, and prominent nucleoli (1). Due to the lack of particular immunohistochemical or molecular markers for eGBM, diagnosis can be difficult. The BRAF V600E mutation has been identified in eGBMs at a relatively great frequency, despite being rare in conventional GBM (54%) (2–5). Moreover, low-grade glioma components in eGBM were reported in recent studies, and a few eGBM patients were previously diagnosed with pleomorphic xanthoastrocytoma (PXA) (6–9). Therefore, several studies have suggested that eGBM and PXA may be either the same entity or closely related (6, 10–13).
eGBM is commonly considered more devastating than classical GBM and has a higher molecular heterogeneity (12, 14). Nevertheless, the clinical features, pathological results and molecular characteristics of eGBM are still poorly understood. Moreover, the diagnostic basis and molecular features of eGBM have not been clearly defined to date. Wide panels of molecular and immunohistochemical markers are required to achieve the correct diagnosis. We described the clinicopathological and molecular characteristics of 12 eGBMs and discussed their molecular genetic features.
Methods
Data collection
The Institute Research Ethics Committee of Jinling Hospital approved this study. Slides from glioblastomas were retrieved from 2014 to 2022 surgical pathology files of the authors’ institution (Affiliated Jinling Hospital, Medical School of Nanjing University) and were involved in the study if they were diagnosed as eGBM on the basis of characteristic morphological and molecular features. Two pathologists performed a blinded review of the pathological materials according to the pathological and molecular definition of eGBM in the 2021 WHO categorization of CNS tumours. Thirteen GBM cases were consistent with epithelioid morphology. Case 13 was eliminated from the series because of the involvement of an IDH1 mutation. In total, 12 eGBMs were gathered in this study. The clinical, radiological and pathological data were obtained from the Department of Pathology, Affiliated Jingling Hospital, Medical School of Nanjing University. Reviewing electronic health records and attempting to contact referring pathologists and clinicians yielded clinical and demographic follow-up information.
Immunohistochemistry
Tumour tissues were embedded in paraffin after being fixed in 10% formalin. Sections were cut out at 3 μm thickness and immunohistochemically stained with conventional antibodies as well as several available commercially antibodies against gene expression targets identified throughout the gene expression analysis. The following proteins were chosen as targets: GFAP (MAB-0764, 1:150, Maixin Bio (MXB)), INI1 (ZA-0696, ready-to-use, Zhongshan (ZSGB)), IDH1 (ZM-0447, ready-to-use, ZSGB), BRAF V600E (790-5095, ready-to-use, Roche), CKpan (kit-0004, 1:200, MXB), ATRX (MAB-0855, ready-to-use, MXB), EMA(ZM-0095, ready-to-use, ZSGB) and TP53 (ZM-0408, 1:200, ZSGB).
TP53 immunostaining was identified as a missense mutation when higher than 10% nuclear positivity was exist (15). Immunostaining was defined as a frameshift when tumour cells demonstrated a full absence of nuclear staining, and intrinsic control cells showed focal nuclear staining (16, 17). Both missense and frameshift mutations were considered TP53 mutants (15, 16). Internal negative or positive controls, including endothelial cells and/or trapped cortical neurons, were identified in all immunostainings.
Targeted next-generation sequencing
Sequencing of a 425-gene panel was performed on the cases (Supplementary Table S1). Nucleic acid isolation for NGS was performed on formalin-fixed paraffin-embedded (FFPE) tumour tissue from a microdissected representative block. Following the generator’s instructions, five 10 μm tumour slices were utilized for DNA extraction utilizing the QIAamp DNA FFPE Kit (QIAGEN, Valencia, CA, USA). The quality of the DNA was determined using spectrophotometry with absorbance at 230, 260, and 280 nm, and the DNA was measured using Qubit 2.0. Sequencing libraries were created utilizing the KAPA Hyper Prep Kit (KAPA Biosystems) based on the manufacturer’s recommendations for various specimen types.
In summary, end repair, A-tailing, and ligation with indexed adapters were applied to 1 g of fragmented genomic DNA prior to size selection with Agencourt AMPure XP beads (Beckman Coulter). For hybridization-based target enrichment, the GeneseeqOneTM pan cancer gene panel (425 cancer-relevant genes, Geneseeq Technology Inc.) and the xGen Lockdown Hybridization and Wash Reagents Kit were utilized (Integrated DNA Technologies). Libraries captured by Dynabeads M-270 (Life Technologies) were amplified in KAPA HiFi HotStart ReadyMix (KAPA Biosystems), and their quantities were assessed by qPCR through KAPA Library Quantification Kit (KAPA Biosystems). On the Illumina HiSeq4000 platform, target-enriched libraries were sequenced with 2×150 bp paired-end reads. The Burrows-Wheeler Aligner was applied to match the sequencing dataset to the reference hg19 genome (Human Genome version 19). Sequencing data collected were demultiplexed by bcl2fastq (v2.19), analysed by Trimmomatic (18) to eliminate low-quality (quality <15) or N bases, and afterwards aligned to the reference hg19 genome (19). By using Picard (found at https://broadinstitute.github.io/picard/), PCR duplicates were eliminated. For base quality assurance and local realignments around indels, the Genome Analysis Toolkit (GATK) was used (20).SNPs and indels were identified by VarScan2 (21) and Haplotype Caller/Unified Genotyper in GATK, with a mutant allele frequency (MAF) cut-off of 0.5% for tissue cases and a least of three optimal mutant reads. Frequent variants were eliminated utilizing dbSNP and the 1000 Genome Project. An internal list of repeated sequencing errors generated from more than 10000 normal control cases sequenced on the same platform was used to further filter the resulting somatic variants. FACTERA identified gene fusions (22), and copy number variations (CNVs) were measured with ADTEx (23). For tissue samples, the log2 ratio cut-off for copy number gain was given as 2.0. All specimen types were used to detect copy number loss using a log2 ratio cut-off of 0.67. The thresholds were established from the absolute CNVs found by droplet digital PCR, which was used for earlier assay validation (ddPCR). FACETS (24) was used to estimate allele-specific CNVs with a 0.2 drift cut-off for unstable joint segments. By splitting the size of drifted segments by the overall segment size, the chromosomal instability’s percentage (CIN) was recorded.
Results
Clinical data
The clinical and histopathological data of eGBMs were tabulated and are presented in Table 1. There were 9 female and 3 male cases with ages varying from 28 to 70 years. The frontal lobe involving was 3, the temporal lobe involving was 5, the parietal lobe involving was 2, and the basal ganglia was 2. The most common symptoms were headaches and seizures. Radiological examination demonstrated gadolinium-enhancing, comparatively circumscribed lesions with significant perilesional oedema and central necrosis in all cases (Figure 1). In 1 case, there was a midline shift (8.33%). All patients had gross total resection. After surgery, 7 patients (58.3%) received radiation or chemotherapy. One patient received targeted therapy (case 12), and have not demonstrated tumour recurrence or metastatic disease to date. The follow-up period varied from 1 to 30 months. One patient was lost to followed-up. At the time of data cut-off, 4 cases developed local recurrences, and succumbed to complications (case 4, case 5, case 6 and case 7). One case developed a pulmonary metastasis (case 2). No radiological or histological evidence of cerebrospinal fluid dissemination was found.
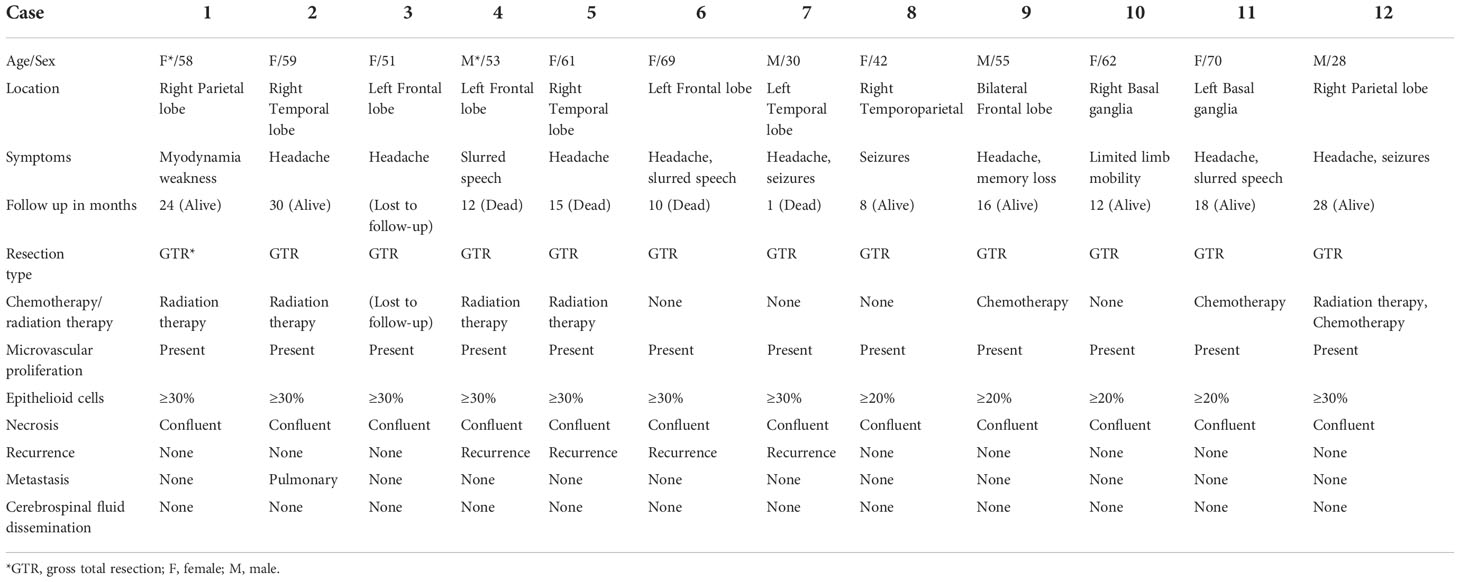
Table 1 Summary of the clinical parameters of 12 eGBM patients.
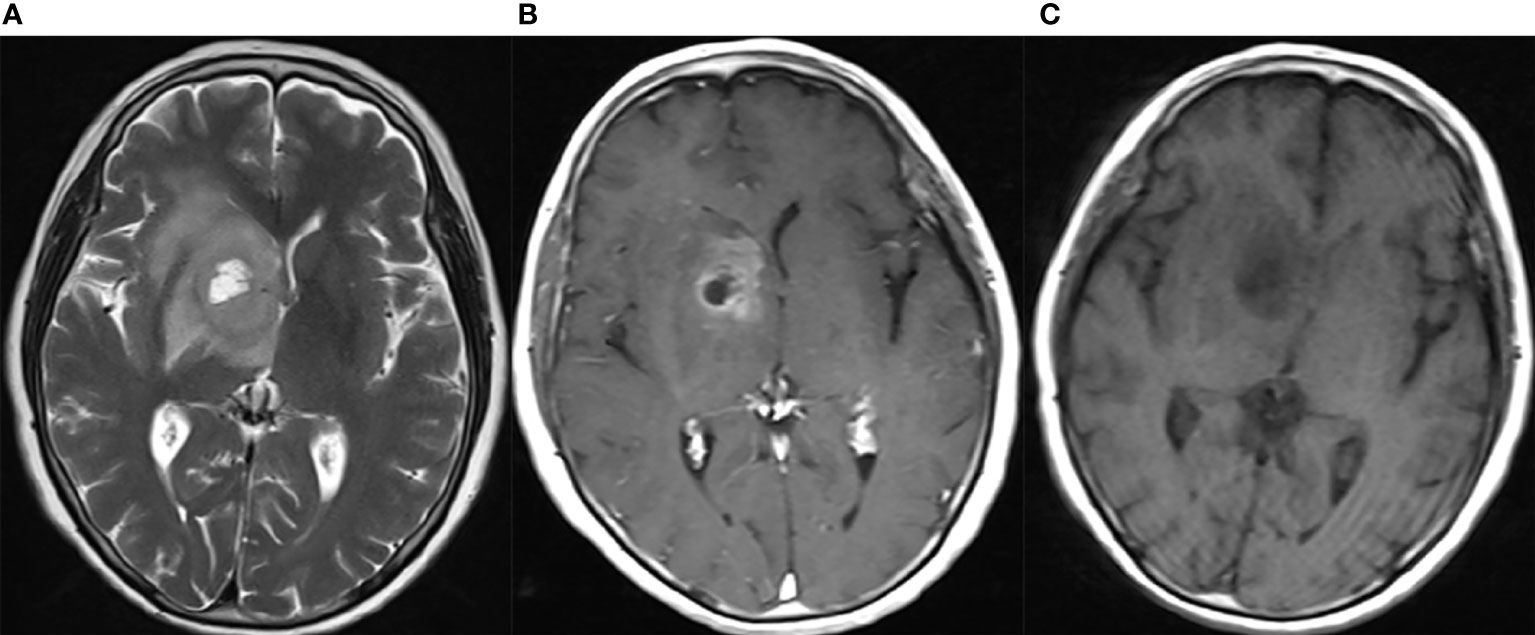
Figure 1 Neuroradiological findings for eGBM case 11. Neuroradiological results for eGBM case 11. (A) A heterogeneous lesion with necrosis and perilesional oedema on T1 in the right basal ganglia with significant midline shift, 23 mm × 28 mm × 17 mm in size. (B) A heterogeneous lesion with perilesional oedema (T2). (C) A rim-enhancing mass with perilesional oedema (T1-weighted enhanced).
Histopathological findings
The histological results are presented in Table 1 and Figure 2. The main notable features of most eGBMs were abundant epithelioid cells and extensive necrosis (Figure 2). In all 12 cases analyzed, microscopy revealed eGBM histopathological types (or melanoma or epithelioid-like cells’ sheets with abundant cytoplasm, eccentric nuclei, and loose cohesion). All tumours showed signs of microvascular proliferation, brisk mitotic activity, and necrosis. However, 4 cases had focal areas that resembled PXA (WHO grade 2) appearance (the set of spindled cells forming fascicles, single large bizarre cells, and vacuolated tumour cells with perivascular lymphocytic cuffing).
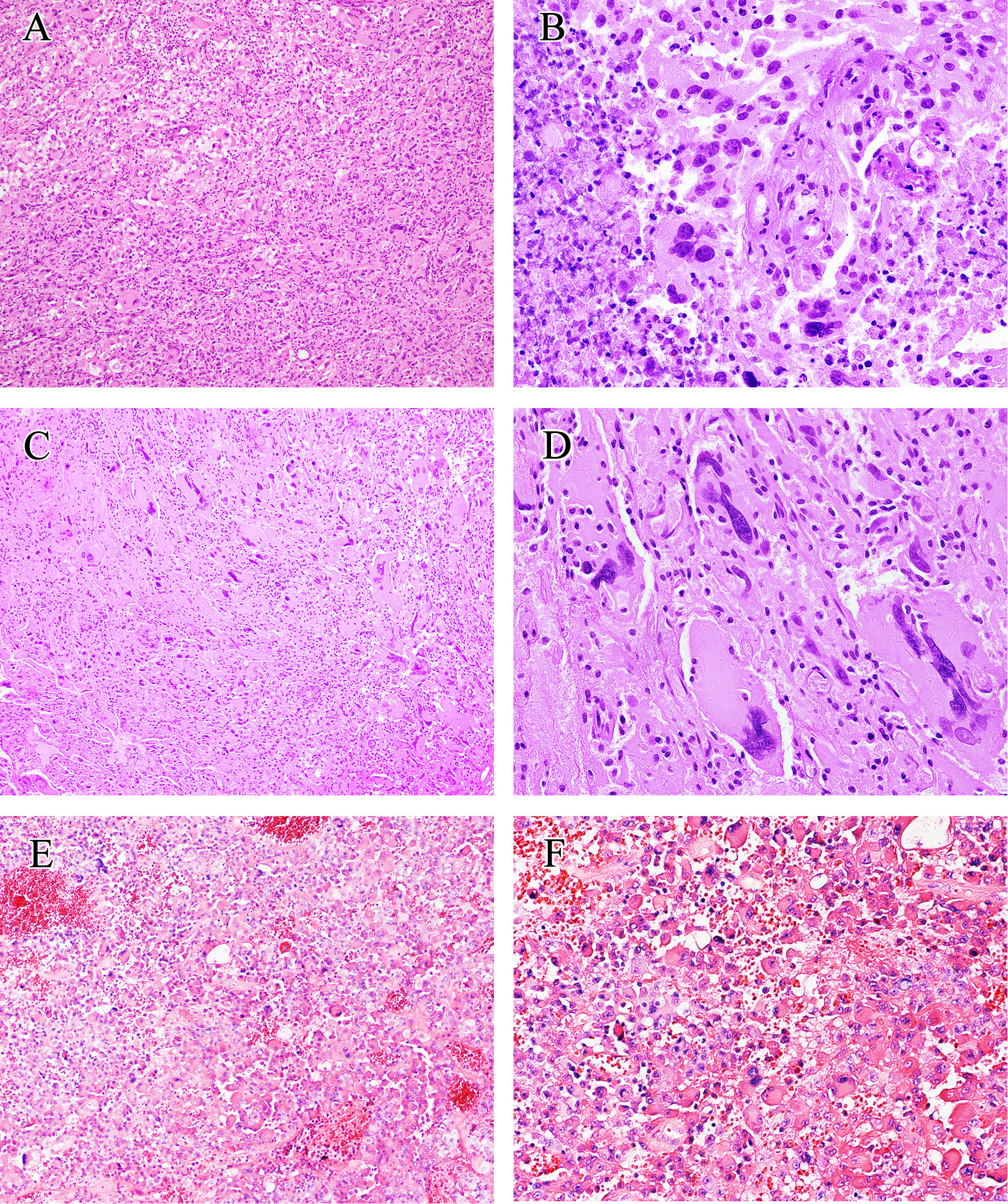
Figure 2 Histological findings of eGBM. (A) Patches of epithelioid and rhabdoid cells were presented (×200). (B) The tumour showed microvascular proliferation and zonal necrosis, and epithelioid and rhabdoid cells constituted abundant and uniformly eosinophilic cytoplasm and laterally located oval to pleomorphically shaped nuclei. Mitoses could easily be seen (×400). (C, D) PXA-like components in eGBM cases showed multinucleated pleomorphic cells, a fascicular arrangement of spindle-shaped cells and single large bizarre cells (×200 and ×400). (E, F) Histopathological findings of case 13 (IDH-mutant astrocytoma). The tumour presented epithelioid morphology (×200 and ×400).
Immunohistochemistry
The immunohistochemistry outcomes are presented in Table 2 and Figure 3. eGBM showed diffuse and strong staining with vimentin. GFAP (glial fibrillary acidic protein) immunoreactivity was diffusely observed in epithelioid cells and lower-grade glioma cells. eGBMs did not show cytokeratin (CK) or epithelial membrane antigen (EMA) staining. The SMARCB1 (INI1) staining was universally intact. Mutant TP53 was observed in 2 cases, and both cases were frameshift mutations. The ATRX loss expression was not observed in any case. IDH1 expression was also not observed in any case. BRAF V600E expression occurred in 50% (6/12) of cases.
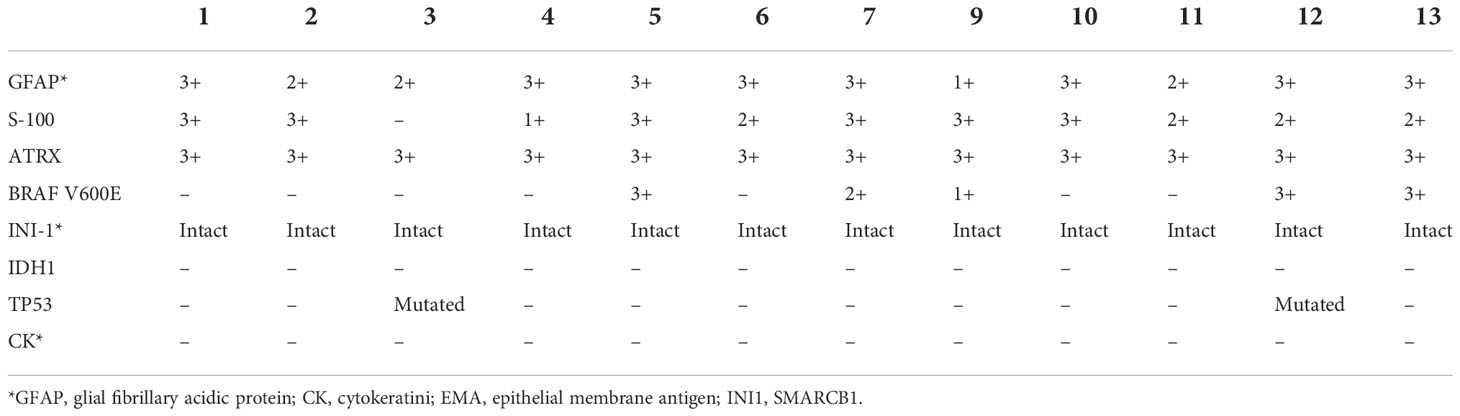
Table 2 Immunohistochemistry of 12 eGBM cases.
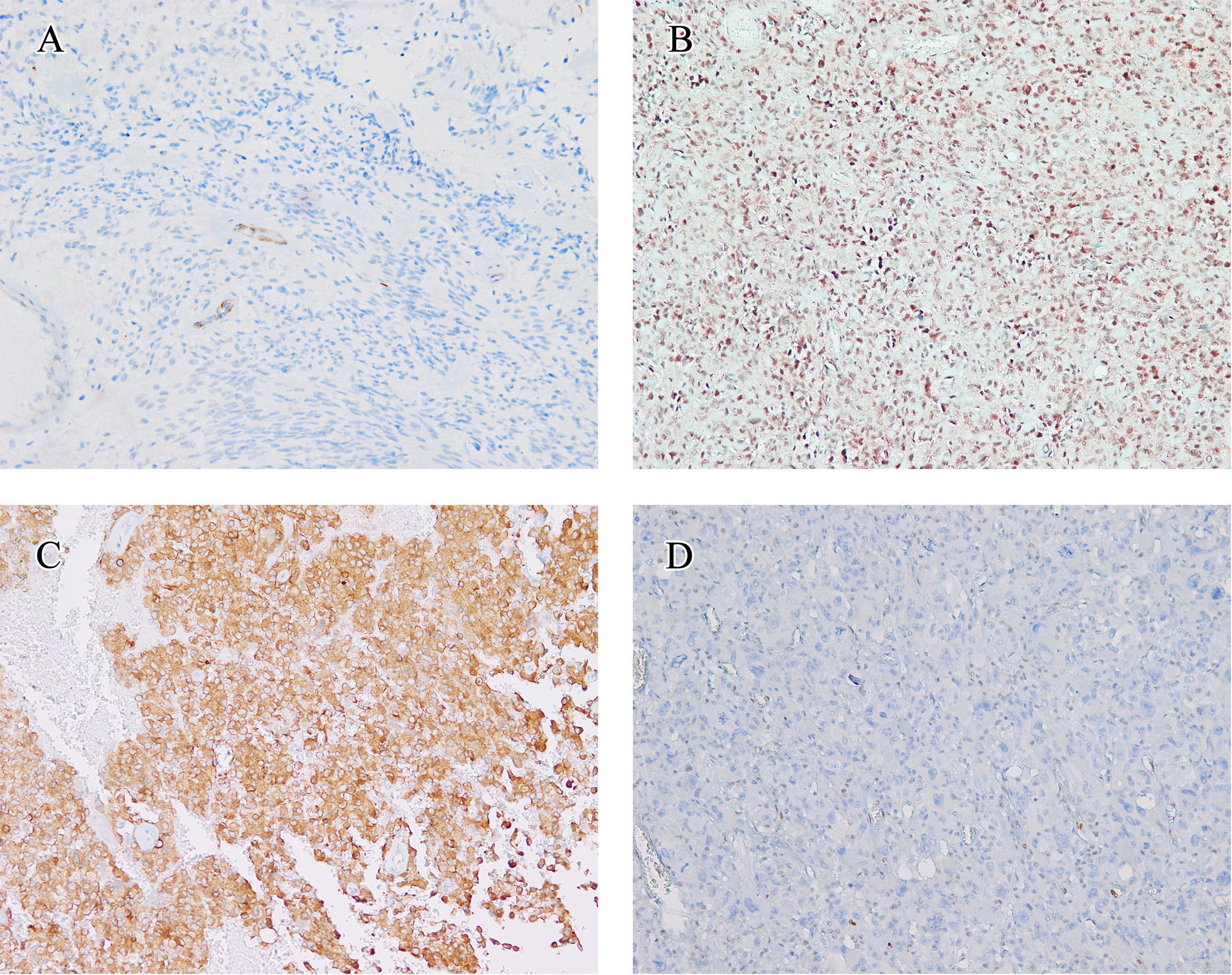
Figure 3 Immunohistochemical findings. The immunohistochemical findings of eGBMs. (A) Immunohistochemical studies showed negtive PTEN expression in 2 cases. Vascular endothelial cells provided an internal positive control (×200). (B) INI1 staining was universally intact (×200). (C) Positive expression of BRAF V600E in eGBM (×200). (D) The tumour cells demonstrated a complete absence of TP53 staining and lymphocytes showed TP53 nuclear staining focally (×200).
Genetic analysis
The findings of genetic analysis are outlined in Figure 4 and Supplementary Table S2. Six cases (50%) harboured the BRAF V600E mutation, and CDKN2A/B homozygous deletion was seen in 5 cases (41.7%). TP53 mutation was detected in 2 cases (16.7%), and TERT promoter mutation was detected in 5 cases (41.7%). PTEN deletion was detected in 2 cases (16.7%). Two of 6 cases without BRAF V600E mutation showed NF1 mutation. IDH and H3 K27M mutations were not found in any cases. In conclusion, eGBMs are complex and heterogeneous tumours, exhibiting multiple genetic mutations.
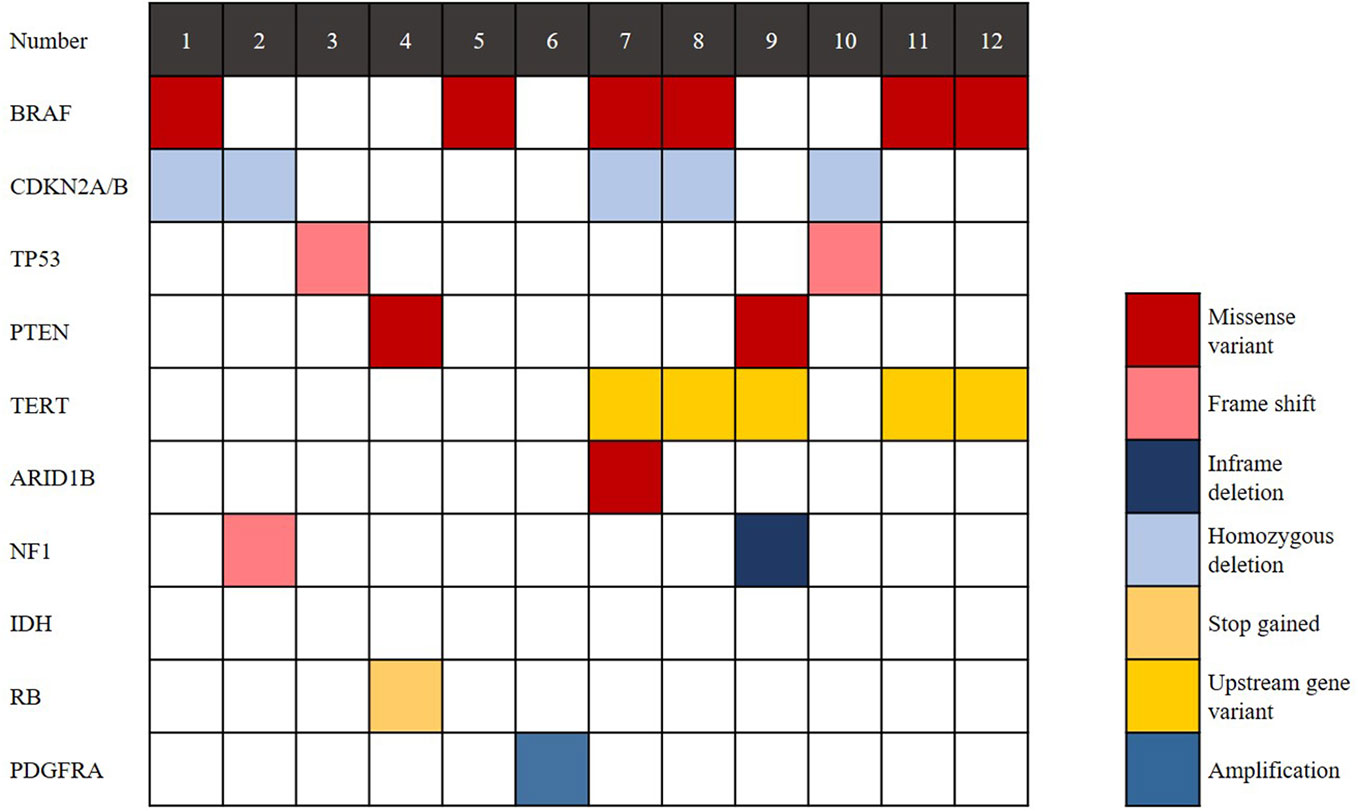
Figure 4 Genomic landscape of eGBM. Clinical and genomic features of 12 eGBM cases. In addition to BRAF V600E mutation, eGBM also showed TP53 mutation, CDKN2A/B homozygous deletions and NF1 mutation.
Discussion
Epithelioid glioblastoma is a rare and extremely aggressive variant of GBM. Kepes et al. first characterized it in 1982, and it was suggested as a histological subtype in the WHO classification of CNS tumours in 2021 (25, 26). However, the radiological, histological and molecular signature of eGBM have not been clearly defined (10, 27). In this study, we applied combined NGS, histology, radiology and immunohistochemistry to describe the clinicopathological and molecular characterization of eGBM.
Histologically, eGBMs are dominated by a population of epithelioid cells with focal discohension, eosinophilic cytoplasm, a differentiated cell membrane, and a nucleus placed laterally. The tumour is richly vascularized, involving thick- and thin-walled vessels with microvascular proliferation and hyaline degeneration, and also glomerulus-like vasculature. Extensive palisading necrosis has also been observed in eGBM. Although the exact aetiology and origin of epithelioid cells are unidentified, there have been numerous studies of eGBMs occurring concurrently with PXA, particularly tumours with anaplastic transformation and epithelioid characteristics, or occurring years after initial tumour resection (5, 10). Four eGBM cases in our series also presented PXA-like (WHO grade 2) morphological characteristics focally. PXA-like components (WHO grade 2) coexisting with eGBM demonstrated a spindle-shaped cells with some mono- or multinucleated pleomorphic cells (Figure 2). Intercellular reticlin meshwork and perivascular lymphocytic cuffing were noticed. Although eGBM is commonly considered to be a primary/de novo lesion, numerous cases of eGBM with a pre- or coexisting lower-grade component have been noted (2, 4, 6, 9, 13, 28). The majority of the lower-grade lesions identified thus far were PXA (WHO grade 2), and a few were low-grade diffuse glioma-like lesions (6–9). We speculate that these unique pathological features may be associated with the molecular heterogeneity.
Consistent with those reported in the literature, half of the eGBMs (50%) in our series were involved in the BRAF V600E mutation. NF1 mutation was detected in 2 eGBM cases (16.7%). The NF1 mutation was mutually exclusive to the BRAF V600E mutation. The codon 600 mutation (V600E) is the main mutation site for the BRAF gene, which is located on chromosome 7q34. BRAF is the gene that encodes cytoplasmic serine-threonine kinase. Subsequent activation of the mitogen-activated protein kinase (MAPK) signaling pathway occurs through the mutated BRAF protein, which in turn promotes tumourigenesis, cellular proliferation, as well as resistance to apoptosis (3, 14). The NF1 gene is located on 17q11.2 and encodes a tumour suppressor that works as a GTPase-activating protein to deactivate the RAS/MAPK signalling pathway, finally causing the occurrence of tumours (29, 30). Hence, both NF1 mutations and BRAF V600E mutations contribute to the constitutive stimulation of downstream RAS/MAPK signalling pathways (31–33), which may be associated with unique pathological features similar to eGBM and PXA (30, 34). Several studies have reported that part of wt-IDH glioblastomas with NF1 mutation also presented a xanthomatous histological appearance (34, 35). Consequently, in addition to BRAF V600E, NF1 mutation may be another meaningful biomarker for the diagnosis of eGBMs. However, the proportion of NF1 mutation in BRAF V600E negative eGBMs demands further investigation.
The work of Korshunov et al. has also illustrated the molecular heterogeneity of eGBM (11) (Table 3). They identified three distinct, previously described subtypes of tumours by combining data from methylation types, copy number alterations, as well as mutations analysis with outcomes from clinical trials. According to the authors, histopathologically defined eGBM is divided into at least 3 molecularly and biologically distinguishable classifications. Consequently, the outcome that eGBM molecularly shares overlaps with other subtypes of glioblastoma may reduce their epithelioid appearance to a morphological pattern, and decrease the biological significance of it.
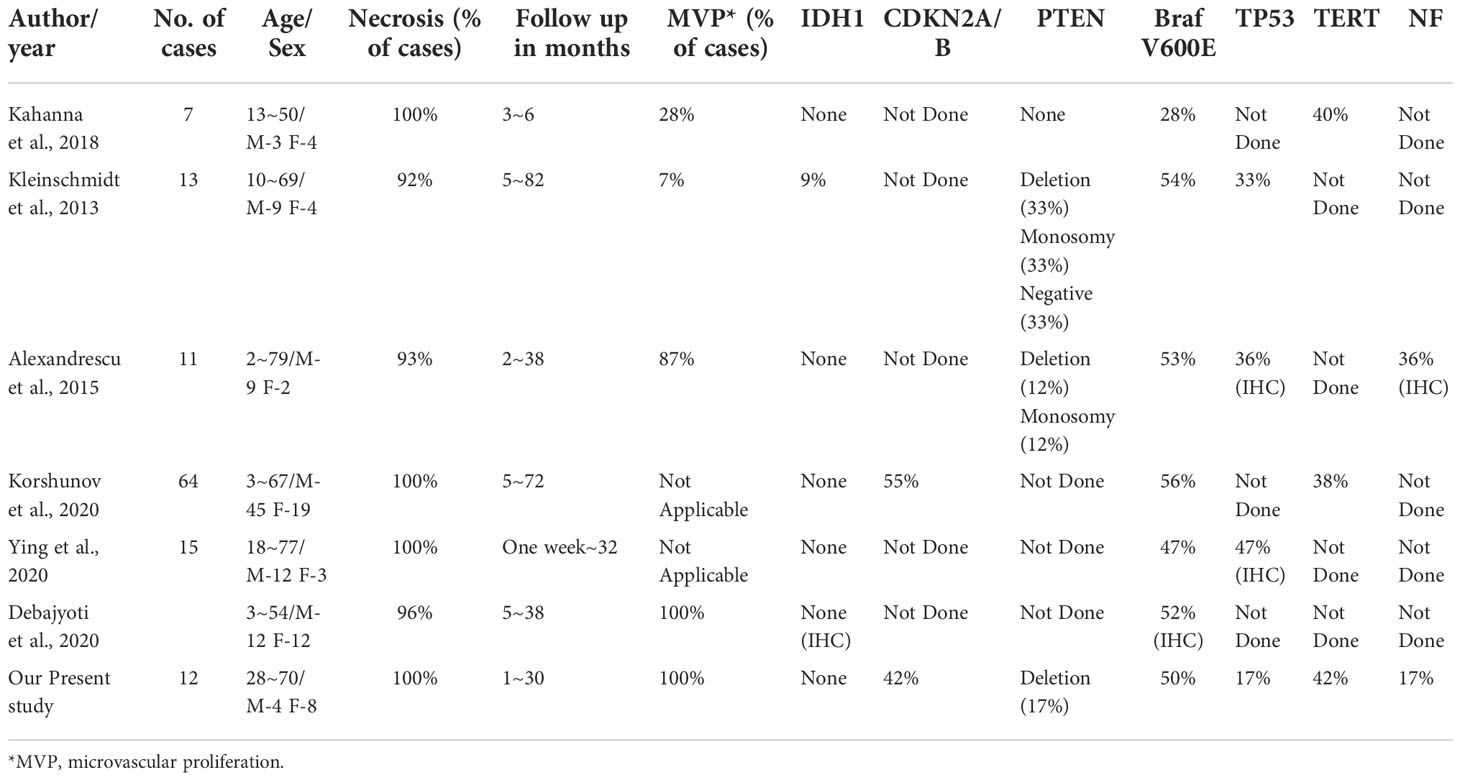
Table 3 Review of previous studies including mutational analysis.
Molecularly, in this series, TERT promoter mutation was detected in 41.7% (5/12) of cases. CDKN2A/B homozygous deletion was seen in 41.7% of cases and TP53 mutation was detected in 16.7% of cases. A total of 16.7% of cases were confirmed to have PTEN deletion (Figure 4). Some reports documented the TERT promoter mutation in GBMs, suggesting its role in the aggressive clinical course (4, 36). TERT promoter mutation is a poor prognostic indicator in wt-IDH gliomas. Moreover, the exitance of pTERT mutation partially clarifies the aggressive nature of GBMs, and its correlation with the tumour’s ability to overcome escape apoptosis and replicative senescence (the fundamental steps in tumourigenesis). CDKN2A is a tumour suppressor gene located on chromosome 9p21. It encodes the p16 protein, a negative regulator of cell cycle progression. The CDKN2B gene is located next to CDKN2A. The mutation to either CDKN2A or CDKN2B will lead to cellular proliferation and the disruption of proapoptotic pathways (37). In IDH-mutated gliomas, CDKN2A homozygous deletion is a strong adverse prognostic factor (38). PTEN is located on 10q23.3 and consists of 9 exons. PTEN deletion has been proven to correlate with poor survival in glioblastoma, suggesting that PTEN plays a role in patient outcomes (39). In this study, most cases (83.3%, 10/12) showed at least 1 mutation mentioned above, which has been detected frequently in gliomas and associated with poor prognosis. Even though, the prognosis of patients are quite different (Table 1), which further illustrates the clinical heterogeneity of eGBM.
Interestingly, case 13 in our study, which exhibited an epithelioid morphology (Figure 2), had both the BRAF V600E mutation and an IDH1 mutation. Consistent with the reports of IDH-mutated glioblastomas, this patient had a relatively long overall survival of up to 30 months. In consequence, this case should be diagnosed as IDH-mutant astrocytoma (WHO grade 4). Accordingly, when high-grade gliomas present epithelioid morphology, the diagnosis of eGBM may not be necessary. Another study also reported that K3 K27M-altered gliomas exhibited an epithelioid appearance (10).
In summary, we studied 12 eGBM cases and further described the clinicopathological and molecular features of the tumours. Our study indicates clinical and molecular heterogeneity among eGBMs. We propose that in addition to BRAF V600E, NF1 mutation may be another meaningful biomarker for the diagnosis of eGBMs. Instead of being a variant or entity, the “epithelioid” GBM phenotype might be a histologic subtype. In order to target the proper treatment to suitable patients, molecular stratification via genome-wide molecular profiling will be crucial in the upcoming years.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Dryad, doi: 10.5061/dryad.2280gb5w0.
Ethics statement
Approval for this study was granted by the Institute Research Ethics Committee of Jinling Hospital.
Author contributions
RP: Methodology, Formal analysis, Data curation, Writing-Original draft preparation. XW: Conceptualization, Formal analysis. RF: Data curation, Visualization. QX: Conceptualization. NW: Conceptualization, Project administration. QR: Conceptualization, Methodology, Project administration. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81802557 to QX and 81872095 to QR).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.980059/full#supplementary-material
References
1. Louis DN, Ohgaki H, Wiestler DO, Cavenee WK. WHO classification of tumours of the central nervous system. 4th ed. Revised Lyon: International Agency for Research on Cancer (2016) 33(6):710–1. doi: 10.1111/j.1365-2990.2007.00905.x
2. Kleinschmidt-Demasters BK, Aisner DL, Birks DK, Foreman NK. Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am J Surg Pathol (2013) 37:685–98. doi: 10.1097/PAS.0b013e31827f9c5e
3. Kleinschmidt-DeMasters BK, Aisner DL, Foreman NK. BRAF VE1 immunoreactivity patterns in epithelioid glioblastomas positive for BRAF V600E mutation. Am J Surg Pathol (2015) 39:528–40. doi: 10.1097/PAS.0000000000000363
4. Matsumura N, Nakajima N, Yamazaki T, Nagano T, Kagoshima K, Nobusawa S, et al. Concurrent TERT promoter and BRAF V600E mutation in epithelioid glioblastoma and concomitant low-grade astrocytoma. Neuropathology (2017) 37:58–63. doi: 10.1111/neup.12318
5. Tanaka S, Nakada M, Nobusawa S, Suzuki SO, Sabit H, Miyashita K, et al. Epithelioid glioblastoma arising from pleomorphic xanthoastrocytoma with the BRAF V600E mutation. Brain Tumour Pathol (2014) 31:172–6. doi: 10.1007/s10014-014-0192-2
6. Kuroda J, Nobusawa S, Nakamura H, Yokoo H, Ueda R, Makino K, et al. A case of an epithelioid glioblastoma with the BRAF V600E mutation colocalized with BRAF intact low-grade diffuse astrocytoma. Neuropathology (2016) 36:181–6. doi: 10.1111/neup.12258
7. Wang J, Liu Z, Cui Y, Liu Y, Fang J, Xu L, et al. Evaluation of EZH2 expression, BRAF V600E mutation, and CDKN2A/B deletions in epithelioid glioblastoma and anaplastic pleomorphic xanthoastrocytoma. J Neurooncol (2019) 144:137–46. doi: 10.1007/s11060-019-03212-0
8. Vaubel R, Zschernack V, Tran QT, Jenkins S, Caron A, Milosevic D, et al. Biology and grading of pleomorphic xanthoastrocytoma-what have we learned about it? Brain Pathol (2021) 31:20–32. doi: 10.1111/bpa.12874
9. Funata N, Nobusawa S, Yamada R, Shinoura N. A case of osteoclast-like giant cell-rich epithelioid glioblastoma with BRAF V600E mutation. Brain Tumour Pathol (2016) 33:57–62. doi: 10.1007/s10014-015-0239-z
10. Alexandrescu S, Korshunov A, Lai SH, Dabiri S, Patil S, Li R, et al. Epithelioid glioblastomas and anaplastic epithelioid pleomorphic xanthoastrocytomas–same entity or first cousins? Brain Pathol (2016) 26:215–23. doi: 10.1111/bpa.12295
11. Korshunov A, Chavez L, Sharma T, Ryzhova M, Schrimpf D, Stichel D, et al. Epithelioid glioblastomas stratify into established diagnostic subsets upon integrated molecular analysis. Brain Pathol (2017) 28(5):656–62. doi: 10.1111/bpa.12566
12. Nobusawa S, Hirato J, Kurihara H, Ogawa A, Okura N, Nagaishi M, et al. Intratumoural heterogeneity of genomic imbalance in a case of epithelioid glioblastoma with BRAF V600E mutation. Brain Pathol (2014) 24:239–46. doi: 10.1111/bpa.12114
13. Vaubel RA, Caron AA, Yamada S, Decker PA, Eckel Passow JE, Rodriguez FJ, et al. Recurrent copy number alterations in low-grade and anaplastic pleomorphic xanthoastrocytoma with and without BRAF V600E mutation. Brain Pathol (2018) 28:172–82. doi: 10.1111/bpa.12495
14. Nakajima N, Nobusawa S, Nakata S, Nakada M, Yamazaki T, Matsumura N, et al. BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions are frequent in epithelioid glioblastomas: a histological and molecular analysis focusing on intratumoural heterogeneity. Brain Pathol (2018) 28:663–73. doi: 10.1111/bpa.12572
15. Takami H, Yoshida A, Fukushima S, Arita H, Matsushita Y, Nakamura T, et al. Revisiting TP53 mutations and immunohistochemistry–a comparative study in 157 diffuse gliomas. Brain Pathol (2015) 25:256–65. doi: 10.1111/bpa.12173
16. Köbel M, Piskorz AM, Lee S, Lui S, LePage C, Marass F, et al. Optimized p53 immunohistochemistry is an accurate predictor of mutation in ovarian carcinoma. J Pathol Clin Res (2016) 2:247–58. doi: 10.1002/cjp2.53
17. Köbel M, Ronnett BM, Singh N, Soslow RA, Gilks CB, McCluggage WG. Interpretation of P53 immunohistochemistry in endometrial carcinomas: Toward increased reproducibility. Int J Gynecol Pathol (2019) 38 Suppl 1:S123–31. doi: 10.1097/PGP.0000000000000488
18. Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
19. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
20. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet (2011) 43:491–8. doi: 10.1038/ng.806
21. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res (2012) 22:568–76. doi: 10.1101/gr.129684.111
22. Newman AM, Bratman SV, Stehr H, Lee LJ, Liu CL, Diehn M, et al. FACTERA: A practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics (2014) 30:3390–3. doi: 10.1093/bioinformatics/btu549
23. Amarasinghe KC, Li J, Halgamuge SK. CoNVEX: copy number variation estimation in exome sequencing data using HMM. BMC Bioinformatics. (2013) 14(Suppl 2):S2. doi: 10.1186/1471-2105-14-S2-S2
24. Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res (2016) 44:e131–1. doi: 10.1093/nar/gkw520
25. Kepes JJ, Fulling KH, Garcia JH. The clinical significance of "adenoid" formations of neoplastic astrocytes, imitating metastatic carcinoma, in gliosarcomas. A Rev Five Cases Clin Neuropathol (1982) 1:139–50.
26. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumours of the central nervous system: A summary. Neuro Oncol (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
27. Broniscer A, Tatevossian RG, Sabin ND, Klimo P Jr, Dalton J, Lee R, et al. Clinical, radiological, histological and molecular characteristics of paediatric epithelioid glioblastoma. Neuropathol Appl Neurobiol (2014) 40:327–36. doi: 10.1111/nan.12093
28. Miyahara M, Nobusawa S, Inoue M, Okamoto K, Mochizuki M, Hara T. Glioblastoma with rhabdoid features: Report of two young adult cases and review of the literature. World Neurosurg (2016) 86:515.e511–519. doi: 10.1016/j.wneu.2015.10.065
29. Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med (2010) 12:1–11. doi: 10.1097/GIM.0b013e3181bf15e3
30. Bettegowda C, Agrawal N, Jiao Y, Wang Y, Wood LD, Rodriguez FJ, et al. Exomic sequencing of four rare central nervous system tumour types. Oncotarget (2013) 4:572–83. doi: 10.18632/oncotarget.964
31. Hsiao SJ, Karajannis MA, Diolaiti D, Mansukhani MM, Bender JG, Kung AL, et al. A novel, potentially targetable TMEM106B-BRAF fusion in pleomorphic xanthoastrocytoma. Cold Spring Harb Mol Case Stud (2017) 3:a001396. doi: 10.1101/mcs.a001396
32. Phillips JJ, Gong H, Chen K, Joseph NM, van Ziffle J, Jin LW, et al. Activating NRF1-BRAF and ATG7-RAF1 fusions in anaplastic pleomorphic xanthoastrocytoma without BRAF p.V600E mutation. Acta Neuropathol (2016) 132:757–60. doi: 10.1007/s00401-016-1616-3
33. Pratt D, Camelo-Piragua S, McFadden K, Leung D, Mody R, Chinnaiyan A, et al. BRAF activating mutations involving the beta3-alphaC loop in V600E-negative anaplastic pleomorphic xanthoastrocytoma. Acta Neuropathol Commun (2018) 6:24. doi: 10.1186/s40478-018-0525-1
34. Shibahara I, Sonoda Y, Suzuki H, Mayama A, Kanamori M, Saito R, et al. Glioblastoma in neurofibromatosis 1 patients without IDH1, BRAF V600E, and TERT promoter mutations. Brain Tumour Pathol (2018) 35:10–8. doi: 10.1007/s10014-017-0302-z
35. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17:98–110. doi: 10.1016/j.ccr.2009.12.020
36. Khanna G, Pathak P, Suri V, Sharma MC, Chaturvedi S, Ahuja A, et al. Immunohistochemical and molecular genetic study on epithelioid glioblastoma: Series of seven cases with review of literature. Pathol Res Pract (2018) 214:679–85. doi: 10.1016/j.prp.2018.03.019
37. Purkait S, Sharma V, Jha P, Sharma MC, Suri V, Suri A, et al. EZH2 expression in gliomas: Correlation with CDKN2A gene deletion/ p16 loss and MIB-1 proliferation index. Neuropathology (2015) 35:421–31. doi: 10.1111/neup.12201
38. Appay R, Dehais C, Maurage CA, Alentorn A, Carpentier C, Colin C, et al. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol (2019) 21:1519–28. doi: 10.1093/neuonc/noz126.000
Keywords: glioblastoma, epithelioid glioblastoma, BRAF V600E, molecular genetics, central nervous system tumour
Citation: Pan R, Wang X, Fang R, Xia Q, Wu N and Rao Q (2022) Epithelioid glioblastoma exhibits a heterogeneous molecular feature: A targeted next-generation sequencing study. Front. Oncol. 12:980059. doi: 10.3389/fonc.2022.980059
Received: 28 June 2022; Accepted: 04 November 2022;
Published: 24 November 2022.
Edited by:
Dengfeng Cao, Washington University in St. Louis, United StatesReviewed by:
Fuyou Guo, First Affiliated Hospital of Zhengzhou University, ChinaRenu Madan, Post Graduate Institute of Medical Education and Research (PGIMER), India
Copyright © 2022 Pan, Wang, Fang, Xia, Wu and Rao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiu Rao, cmFvcWl1MTEwM0AxMjYuY29t; Nan Wu, NTUxNTI1NzJAMTYzLmNvbQ==