 Magdalena Mroczek1*††‡
Magdalena Mroczek1*††‡ Jakub Liu2†‡Mateusz Sypniewski1‡Tadeusz Pieńkowski1,3‡Bartosz Itrych1‡Joanna Stojak1,4‡
Jakub Liu2†‡Mateusz Sypniewski1‡Tadeusz Pieńkowski1,3‡Bartosz Itrych1‡Joanna Stojak1,4‡ Bartosz Pronobis-Szczylik1‡Maria Stępień5‡
Bartosz Pronobis-Szczylik1‡Maria Stępień5‡ Elżbieta Kaja6‡
Elżbieta Kaja6‡ Maciej Dąbrowski7‡
Maciej Dąbrowski7‡ Tomasz Suchocki2,8‡Marzena Wojtaszewska9,10‡Paweł Zawadzki7‡
Tomasz Suchocki2,8‡Marzena Wojtaszewska9,10‡Paweł Zawadzki7‡ Anna Mach11‡
Anna Mach11‡ Paweł Sztromwasser7‡Zbigniew J. Król1‡
Paweł Sztromwasser7‡Zbigniew J. Król1‡ Joanna Szyda2,8‡
Joanna Szyda2,8‡ Paula Dobosz1‡
Paula Dobosz1‡- 1Central Clinical Hospital of Ministry of the Interior and Administration in Warsaw, Warsaw, Poland
- 2Biostatistics Group, Wrocław University of Environmental and Life Sciences, Wrocław, Poland
- 3Postgraduate Medical Education Center, Warsaw, Poland
- 4Department of Experimental Embryology, Institute of Genetics and Animal Biotechnology, Polish Academy of Science, Jastrzębiec, Poland
- 5Department of Sports Medicine, Doctoral School, Medical University of Lublin, Lublin, Poland
- 6Department of Medical Chemistry and Laboratory Medicine, Poznan University of Medical Sciences, Poznan, Poland
- 7MNM Bioscience Inc., Cambridge, MA, United States
- 8National Research Institute of Animal Production, Balice, Poland
- 9Department of Haematology, Institute of Medical Sciences, College of Medical Sciences, University of Rzeszów, Rzeszów, Poland
- 10Department of Haematology, Frederic Chopin Provincial Specialist Hospital, Rzeszów, Poland
- 11Department of Psychiatry, Medical University of Warsaw, Warsaw, Poland
Introduction: Population-based cancer screening has raised many controversies in recent years, not only regarding the costs but also regarding the ethical nature and issues related to variant interpretation. Nowadays, genetic cancer screening standards are different in every country and usually encompass only individuals with a personal or family history of relevant cancer.
Methods: Here we performed a broad genetic screening for cancer-related rare germline variants on population data from the Thousand Polish Genomes database based on 1076 Polish unrelated individuals that underwent whole genome sequencing (WGS).
Results: We identified 19 551 rare variants in 806 genes related to oncological diseases, among them 89% have been located in non-coding regions. The combined BRCA1/BRCA2 pathogenic/likely pathogenic according to ClinVar allele frequency in the unselected population of 1076 Poles was 0.42%, corresponding to nine carriers.
Discussion: Altogether, on the population level, we found especially problematic the assessment of the pathogenicity of variants and the relation of ACMG guidelines to the population frequency. Some of the variants may be overinterpreted as disease-causing due to their rarity or lack of annotation in the databases. On the other hand, some relevant variants may have been overseen given that there is little pooled population whole genome data on oncology. Before population WGS screening will become a standard, further studies are needed to assess the frequency of the variants suspected to be pathogenic on the population level and with reporting of likely benign variants.
Introduction
It has been over 20 years since the very first version of the entire human genome was released. Not a long time afterward, the scientific community agreed that cancer is a disease of the genome. Today, although advanced sequencing methods are available at a reasonable price and the role of significant genetic variants localized along the whole genome is quite well defined, the clinical implementation of whole-genome sequencing (WGS) in diagnosis and treatment remains in its infancy. Tumor sequencing (somatic variants) can provide valuable information regarding treatment response and outcomes, however, cannot explain all cancer cases. Around 5-14% of all cancers are caused by germline (inherited) variants (1). Usually, screening for germline variants related to oncological diseases encompasses only individuals with a personal or family history of relevant cancer. Here, we present the first whole-genome sequencing-based cancer population screening in the 1076 individuals from the Polish population.
Cancer remains one of the major epidemiological challenges in Poland as the number of cancer deaths is increasing. In the last 50 years, it has risen by 2.5 times, and the survival rate is one of the lowest in Europe (2). In Poland, women are affected most often by breast (22.9%), lung (9.9%), corpus uteri (7.0%), colon (5.9%), and ovary (4.3%) cancers (2). Among Polish men, the most common are prostate (20.6%), lung (16.1%), colon (6.8%), and urinary bladder (6.4%) cancers (2). The Polish population has an especially low cancer survival rate with over 10 percentage points lower than the 5-year survival rate of other European countries (3). The most significant reasons for this are a too-late diagnosis caused by the lack of screening programs and poor access to the healthcare system (4). Other characteristics of the Polish cancer-related genetic landscape are relative homogeneity and a strong influence of the founder effects (5).
The benefits of screening and preventive treatments for individuals with pathogenic cancer-related variants in different populations depend on the prevalence and penetrance of the mutation, the mortality associated with the disease, the age of the person screened, the potential effects of preventive measures on the risk of developing the disease, quality of life, and costs (6).
Some rare genetic variants have already been included in the populational screening, especially in isolated populations. Breast cancer prevention is one of the most broadly applied examples of population screening. The current screening strategy encompasses individuals with a personal or family history of relevant cancer and the criteria and screening for founder mutations in isolated populations, such as Ashkenazi Jews. In the Ashkenazi population, 3% of women carry one of the three ancient mutations: BRCA1 185delAG (c.68_69delAG), BRCA1 5382insC (c.5266dupC), or BRCA2 6174delT (c.5946delT) (7, 8). In Poland breast cancer mutations BRCA1 and BRCA2 show a strong founder effect. The incidence of the founder breast cancer mutations in the unselected early-onset breast Polish population is around 6% (8). Around 80%-90% of the BRCA1 mutations in Polish cancer families can be explained by one of three founder mutations (5382insC, C61G and 4153delA) (6). Although the identification of carriers of hereditary breast and ovarian cancer gene variants through family cancer history alone is suboptimal, population screening is still controversial. In one of the few population screening studies, it was shown that as many as 38 of 5908 women (0.64%) carried a clinically actionable pathogenic variant (9). 42% of pathogenic variant carriers did not have a first-degree relative with breast or ovarian cancer and the family history was very often not informative (9) Polish population has been examined mainly in the context of selected genes playing a role as a control for cancer patient studies. The population prevalence for combined founder BRCA1 5382insC and C61G variants in Poland was 0.25% (1/400) (10). The following prevalence for BRCA1 variants has been found: 0.17% for c.5266dupC, 0.1% for c.3819del5, and 0.08% for C61G variant. The testing strategy is suboptimal and detects only a fraction of BRCA pathogenic/likely pathogenic (P/LP) variants carriers (11). In the Greater London Area, only 2.6% of total estimated carriers and 5.1% of detectable carriers have been identified in the general population with current NHS strategies (12). Given the rising number of risk management options and the availability of new sequencing technologies, a future screening strategy requires an urgent discussion.
The aim of this study is to investigate germline cancer-related gene variants on the population level in Poland, to assess the type of variation (i.e., point or structural), to compare the frequencies with the frequency of corresponding variants estimated for the non-Finnish European population (NFE), and to interpret their relevance in the context of the family history-based testing. Our study is an extension of the study presented in Kaja et al. (13), however here we focus exclusively on the variants related to oncology, the frequency of these germline variants in a healthy genetically homogenous population and their potential significance on the clinical level. Further, we discuss the application of local genomic databases in oncology and the feasibility of populational cancer testing with WGS.
Materials and methods
The cohort
The cohort was described in detail in Kaja et al., 2022 (13). Briefly, the studied population was recruited through the project “Search for genomic markers predicting the severity of the response to COVID- 19” to the genetic predisposition to COVID-19 severity. Samples were collected from 1222 individuals of Polish origin (extended cohort), between April 2020 and April 2021. Among them 1076 were unrelated and did not suffer from any significant disease (core cohort), for a detailed cohort description see Kaja et al., 2022 (13). Samples encompassed the whole territory of Poland. All participants, guardians, or parents of the participants under 18 years of age, provided informed consent before the collection of blood samples and filling in the clinical data form, which included a questionnaire about the country of origin and chronic diseases. All participants signed informed consent. The ethical approval for the study was obtained from the Ethics Committee of the Central Clinical Hospital of the Ministry of Interior and Administration in Warsaw (decision nr: 41/2020 from 3 April 2020 and 125/2020 from 1 July 2020). The database containing anonymized genetic variants is available and can be downloaded as a VCF file from https://naszegenomy.pl/ after accepting the policy and filling in a short form.
The core cohort consisted of 1076 unrelated individuals of Polish origin. The median age of participants was 45.4 (2–99) years and there was a slight predominance of males (697 vs. 525). This is comparable to the median age in Poland. The core cohort characteristics are presented in Table 1. The patients have initially been recruited for the COVID studies, independent of their chronic disease status. The analysis of clinical data demonstrated that the most common chronic self-reported diseases in the survey were: hypertension (13.0%), cancer (4.6%), diabetes (4.0%), and hypothyroidism or Hashimoto’s disease (3.0%). For hypertension (13 vs. 60.28% (14)) and diabetes (4.0% vs. 6.97% (15)) the self-reported prevalence was lower than those reported in the studies, whereas for cancer mostly incidence data are available. The types of tumours identified in individuals from the extended cohort of 1222 people are reported in Supplementary Table 1. Because access to the phenotypic information is restricted, we were not able to extract the cancer type and genetic variant information for the core cohort of 1076 only, due to technical reasons. The information on whether cancer has been confirmed pathologically was not collected. Also, the patient reported both benign and malignant tumours under the name “cancer”. No chronic disorders were reported by 86% of participants from the core cohort.
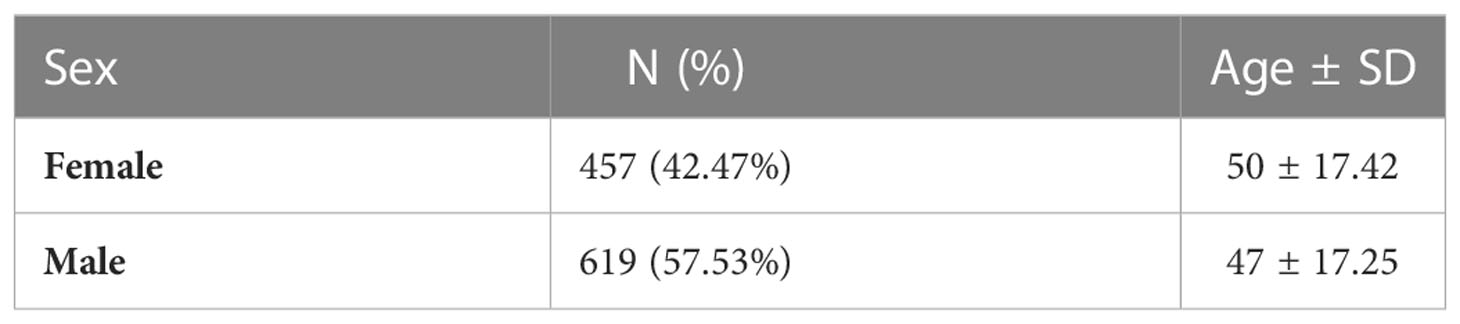
Table 1 Sex and age characteristics of analyzed core cohort.
Whole-genome sequencing and data processing
DNA was isolated from the patient’s peripheral blood. The library has been prepared using a TruSeq DNA PCR-free kit (Illumina Inc., San Diego, CA, USA) and 550 bp inserts. Sequencing was performed on the Illumina NovaSeq 6000 platform with the following parameters: 150 bp paired-end reads, an average read depth of 35.26×. The quality has been assessed with FastQC v0.11.7 and mapped to the GRCh38 human reference genome. Single-nucleotide variants and short indels have been identified with DeepVariant v0.8.0 and genotyped all together with GL nexus v1.2.6-0-g4d057dc. Multiallelic variant calls were decomposed into monoallelic and normalized groups using BCFtools v1.9 (16). The variants have been annotated using the following resources: Ensembl Variant Effect Predictor v.107], including references to databases of genomic variants from ClinVar v. 20220624 and dbSNP build 151, variant population frequencies from the 1000 Genomes Project, and GnomAD v2.0.1 and v3.0, as well as pathogenicity scores, such as Polyphen-2, SIFT, DANN and CADD (13). All gene coordinates were padded with variants in the 10 kb range at both ends of the genes.
Cancer variant extraction and analysis
The variants associated with cancer were chosen based on the COSMIC database (Catalogue of Somatic Mutations in Cancer (17)) and literature review based on the PubMed search (Supplementary Table 2). These variants were classified as having a high, moderate, or low impact based on Sequence Ontology terms. The annotation has been performed with a SNPEff software (for details see In-depth-NGS-Data-Analysis-Course). Furthermore, the variants were filtered to identify those with known pathogenicity in cancer, following the classification available in the ClinVar (version from 2022-06-24) database. The frequency of the variants in the Polish population (this study) was compared to the frequency estimated for the non-Finnish European (NFE EUR) population obtained from the gnomAD (accessed on 2022-07-02) database. NFE population is the most similar to the Polish population. The list of variants was filtered out for moderate and high impact as a first step, and then only variants annotated in gnomAD for the NFE were further considered. For better comparison, the differences in alternative allele frequencies (AF) between the Polish cohort and gnomAD NFE were expressed in terms of odds ratio defined as where AFPL is alternative allele frequency in polish cohort and AFNFE is alternative allele frequency in gnomAD NFE. Statistical significance of odds ratios for each variant was estimated with Fisher’s exact test and corrected for Bonferroni adjustments. The prevalence of carriers was counted as a ratio of positive samples to the total number of samples tested.
To analyze genetic diversity AF from our database have been compared to exomes from gnomAD (gnomADe), genomes from gnomAD (gnomADg) and 1000 Genomes Project (1KGP) database (https://www.internationalgenome.org/, accessed on 2023-01-13). The African (AFR), Admixed American (AMR), East Asian (EAS), South Asian (SAS) and European, gnomADe and gnomADg NFE and EUR for 1KGP, ancestries were considered. Variants were filtered according to Combined Annotation-Dependent Depletion CADD (https://cadd.gs.washington.edu/,CADD>=20), which is a deleteriousness measure.
In the manuscript the conventional variant naming has been used. The HGVS nomenclature and genomic locations are available in the Supplementary Table 3.
Results
The landscape of identified variants in the European context
Overall, 19551 variants related to oncological diseases have been identified in 806 different genes in the core cohort (Supplementary Table 2). Among them, 316 variants were of high impact and 9835 variants were moderate impact variants. Importantly, in our study 9739 (75.87%) of the 12836 rare variants defined by MAF< 0.01 were classified as singletons (Figure 1). Among the variants, 1552 were located in introns and 17353 in exons. The MAF of variants of high, moderate and low impact (as assigned by VEP) was compared between NFE (gnomAD) and Polish populations (this study). The variant enrichment divided into medium and high frequency is presented in Figure 2. High-impact variant for FANCC had a significant OR (p<0.05) in the Polish population. Moderate impact variants with the highest OR (p<0.05) in the Polish population were: NUTM2B, MUC4, NOTCH1, NUTM2B, USP6 (Table 2). Their functions and pathways they are involved in are presented in Table 3.
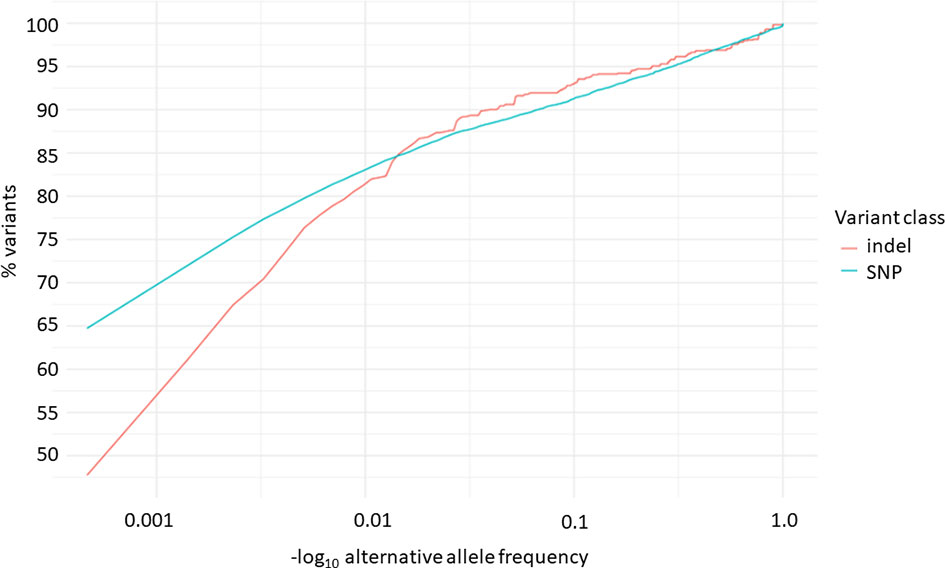
Figure 1 Distribution of alternative allele frequencies of indels and SNPs related to oncological diseases.
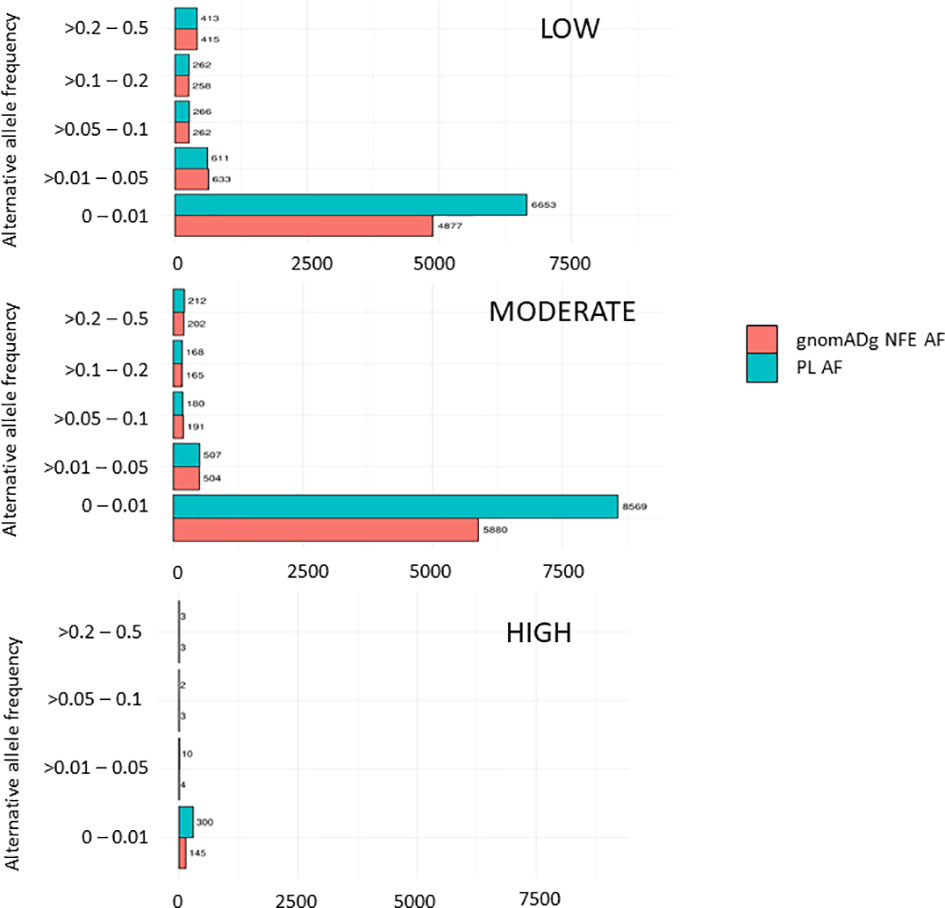
Figure 2 Distribution variants related to cancers in the PL population based on their impact.
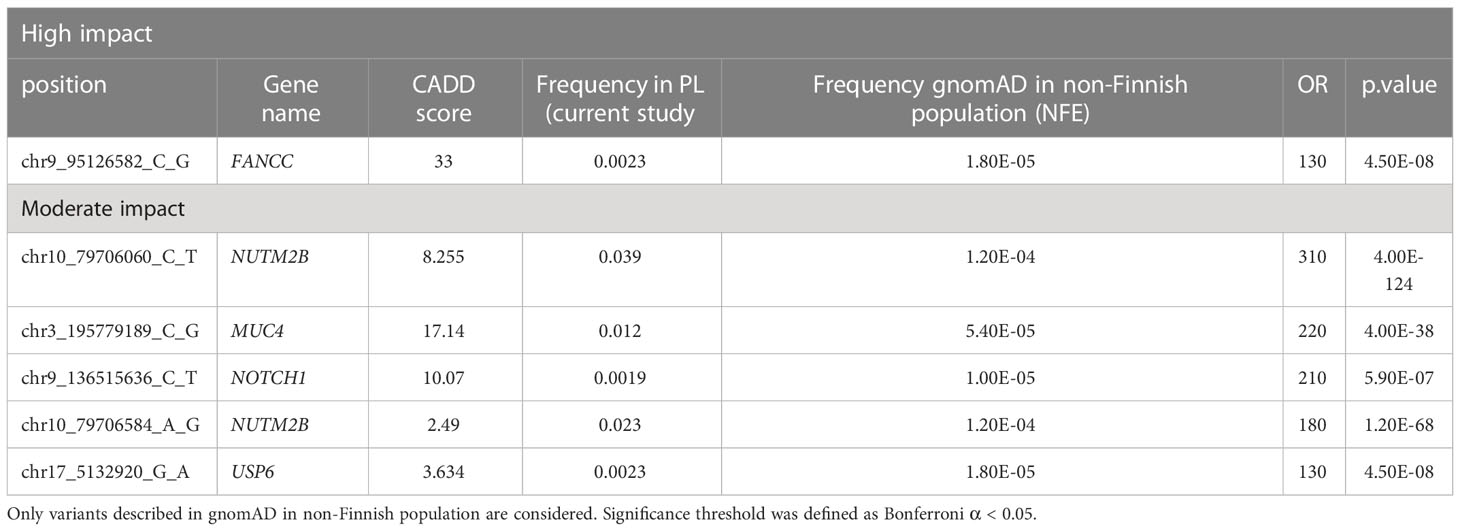
Table 2 High and moderate impact variants with a significantly higher frequency in the PL population as compared to the gnomAD in NFE population.
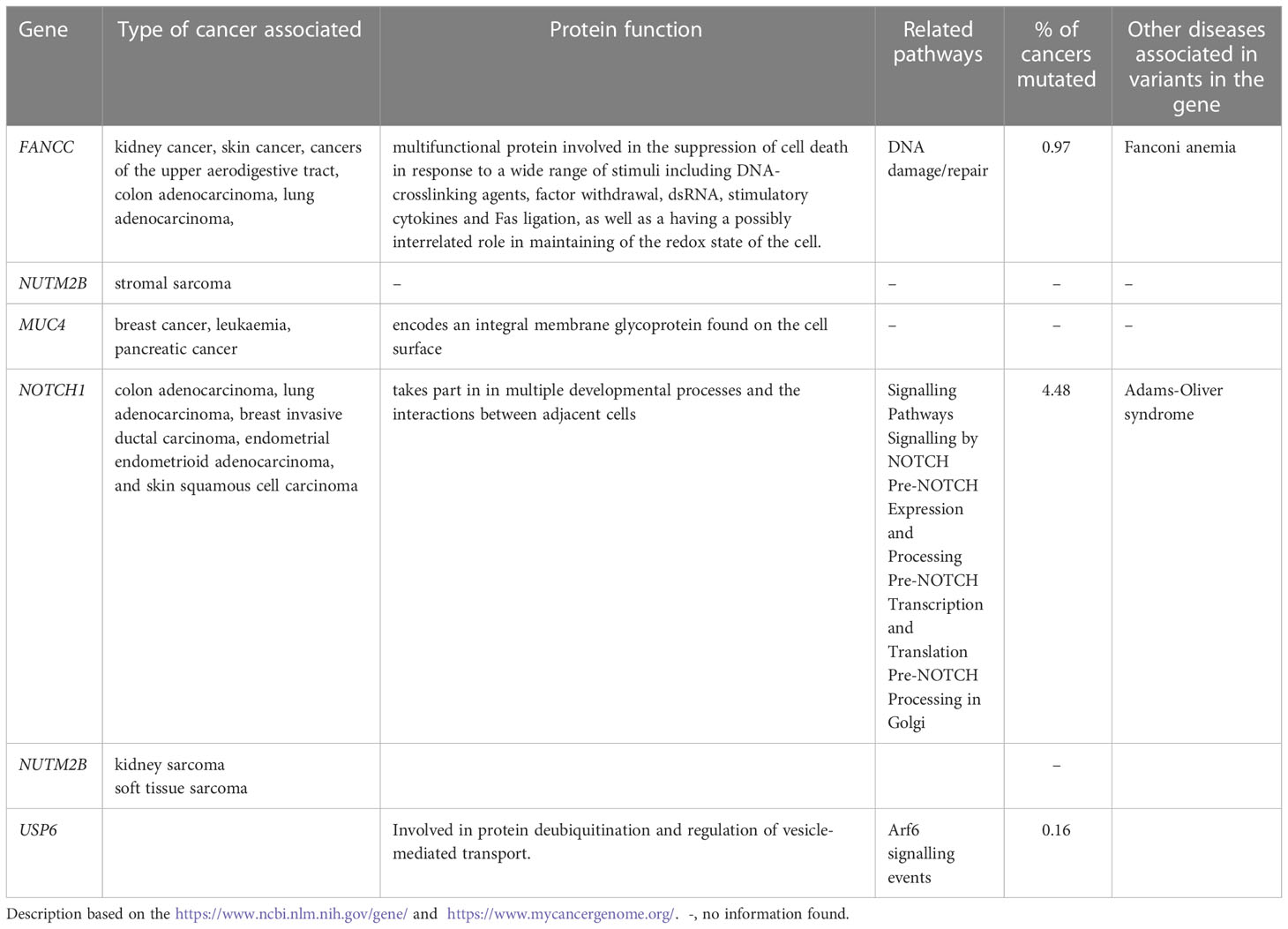
Table 3 Protein's function, pathways and types of cancers related to the high and moderate impact variants with the highest OR.
Polish population in the global context
To present variants related to oncology identified in our database in regard to global genetic diversity and other genetic and genomic databases, we compared the number of variants identified in the Polish population to different ancestries and from three databases. In the first step the analysis was performed for the BRCA1/BRCA2 P/LP variants identified in the our database in the Polish population. In the context of 1KGP database, all these variants were unique across ancestries. The number of P/LP BRCA1/BRCA2 variants shared according to gnomADg between Polish population (this study) and other ancestries presented as follows: NFE (n=5/7), AMR (n=3/7), AFR (n=2/7), EAS (n=1/7), SAS (n=1/7).
In the second step, we analyzed the variants related to oncology in Poland in the broader perspective. Not to exclude common variants, but at the same time to use computational resources effectively, we analyzed the variants with CADD > = 20 from the variants related o oncological diseases list. These included 8491 variants, among them 840 rare (MAF<0.01) and 7651 common (MAF>0.01). Among 8491 variants, 5647 were unique for Polish population (monomorphic) according to 1KGP database, 4392 according to gnomADe and 2315 according to gnomADg. The number of variants related to oncology shared between Polish cohort and particular ancestries is shown in Figure 3.
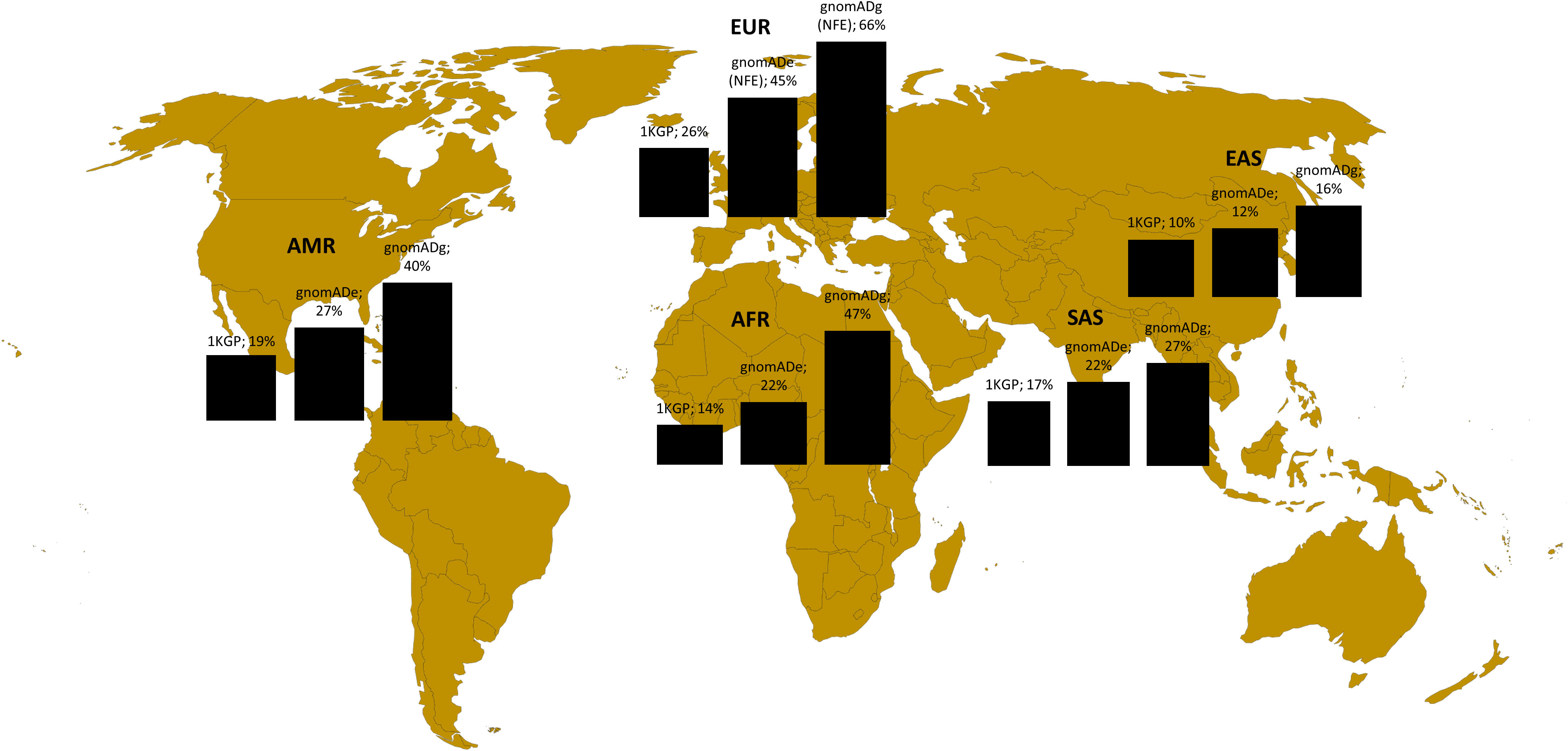
Figure 3 Variants shared between Polish population (this study) and different ancestries according to 1KGP, gnomADe and gnomADg. Legend: AFR- African, AMR- Admixed American, EAS-East Asian, SAS-South Asian and EUR (European-EUR for 1KGP and NFE-European non-Finnish for gnomADe and gnomADg).
Variants in BRCA1/BRCA2
In the Polish population, 45 different rare variants were identified within the BRCA1 gene and 72 in the BRCA2. Among them in BRCA1 two were of a high impact, 30 of a moderate impact and 13 of a low impact. Among BRCA2 five were of high impact, 46 of moderate impact and 21 of low impact. According to ClinVar three variants in BRCA1 were classified as P/LP (c.5266dupC, 3819del5, and c.181T>G (p.Cys61Gly)) and four variants in BRCA2 (c.658_659delGT, c.1813dupA, c.5238dup and c.9371A>T) were classified as P/LP. The BRCA1/BRCA2 variants and their AF are shown in Supplementary Table 4. Altogether, the AF for the BRCA1/BRCA2 P/LP variants according to ClinVar (18) in the unselected populations of 1076 patients in a genetically homogenous Polish population was 0.42%. That means that we identified 9 P/LP BRCA1/BRCA2 variants in the unselected Polish population. AF for the three pathogenic BRCA1 founder variants was 0.23%. None of the BRCA1/BRCA2 variants reached a statistically relevant higher allele frequency in the Polish population comparing to other European populations, suggesting that this kind of studies require a much larger cohort.
Variants in non-BRCA1/BRCA2
Several genes in which the variants were identified, are related to hereditary cancer syndromes, such as PALB2, related to ovarian and breast cancer or JAK2 (c.3323A>G, p.Asn1108Ser) associated with lung adenocarcinoma, myeloproliferative neoplasm, breast invasive ductal carcinoma, polycythemia vera, and colon adenocarcinoma (Supplementary Table 5). RAD51 protein product is involved in DNA repair and is altered in 0.65% of all cancers with lung adenocarcinoma, glioblastoma and glioblastoma multiforme (19). Germline mutations in the CDKN2A strongly predispose to cutaneous melanoma (16, 20). Although we identified a variant (c.194-3652G>C) in the CDKN2A gene located only one bp distant from the pathogenic CDKN2A variant c.-34G>T (21, 22), after a thorough analysis the variant proved to be benign. Other variants reported as pathogenic/likely pathogenic/with conflicting interpretations of pathogenicity according to the ClinVar were located in the following genes: MSH6 associated with Lynch Syndrome i.e. hereditary non-polyposis colorectal cancer (23), PIK3CB, which is altered in 1.62% of all cancers such as colon adenocarcinoma, prostate adenocarcinoma, lung adenocarcinoma, breast invasive ductal carcinoma; RPN1 altered in 0.19% of all cancers with high grade ovarian serous adenocarcinoma, colon adenocarcinoma, oesophageal adenocarcinoma, bladder urothelial carcinoma (24). Although the variants in these genes have been described as enriched in cancer populations or considered pathogenic by expert panels, the evidence of variant pathogenicity is often reported in very small sample sizes comprising a few families in control and a diseased group and thus not being representative of the whole population.
Phenotype correlation
Variants in 5 genes (RPN1, CDKN2A, HRAS, PALB2, CBFA2T3) have been identified in 32 individuals with tumor from the extended cohort and classified as pathogenic, likely pathogenic, and VUS (Supplementary Table 1). Given the limited information on cancer in our study cohort, e.g., in 4 cases there is no information about the cancer type, it is difficult to correlate the cancer type with genetic variants. Variants in three genes (RPN1, HRAS and PALB2) were carried by several individuals with cancer in extended cohort. Ribophorin 1 (RPN1) is a major part of oligosaccharyltransferase complex, which takes part in the glycosylation process. RPN1 is upregulated in breast cancer and has also been associated with several other cancers, such as high-grade ovarian serous adenocarcinoma, colon adenocarcinoma, oesophageal adenocarcinoma, and bladder urothelial carcinoma. As much as 7 out of 12 individuals from our cohort with cancer had a likely pathogenic variant on both alleles. Another recurrent variant was localized in HRAS gene. HRAS is a known oncogene related to several different cancers, among them Costello syndrome, a rare condition predisposing to tumours in different parts of the body, epidermal tumours, head and neck cancer, bladder carcinoma, and with ovarian carcinoma patients, but was also identified in the healthy controls The variants frequency is not annotated in ClinVar so that it also requires further characterization on the populational level. The variant p.Ile1035Val in PALB2, associated with breast and ovarian cancers, was identified in two out of eight individuals with breast cancers and in one ovarian cancer person from the extended cohort. All these variants will be further followed up in the affected cohorts in Poland in further studies.
Discussion
Oncology remains the major field that can benefit most from whole genome sequencing (25). The first collection of over 350 cancer-related genes has been created, protein-coding genes to be more specific, with new genes being added over time (26). More and more WGS is being performed in a variety of different populations, supporting the notion that WGS of different cohorts enhances the power to identify genetic associations (24, 27–30). Recently, in light of rising population consciousness in terms of cancer population testing and with sinking costs of genetic testing it has been suggested that genetic testing should be offered on the population level. Cancer-susceptibility-gene (CSG) testing might shift from bespoke tests towards whole genome or whole exome analysis as a part of comprehensive population-wide programmes; incorporating such strategy into healthcare systems, with equitable access for the entire population, will be challenging. These issues should especially be discussed, together with variant-of-unknown-significance (VUS) interpretation, penetrance, and genetic counseling. The Thousand Polish Genomes database can find several applications in oncology and show benefits in comparison to the other genomic databases in the following areas: analysis of the intron variants, calculating a polygenic risk score (PRS) and being a reference database for the Polish studies on cancer patients.
Traditionally, most of the studies were focused on the identification of cancer mutations solely in protein-coding genes, ignoring the remaining 99% of the genome. Gene panels can detect only mutations in 0.01 – 0.10% of the genome, at best. The main reason for it was that a large proportion of early NGS-based studies was constrained to whole exome sequencing - a more cost-effective way of ascertaining large samples, as well as the fact that the functional annotation of the non-protein coding part of the genome has constantly been updated and thus made more informative (31). In our study, 89% of the variants in cancer-related genes were identified in non-coding regions, although most of the database reported variants derived from the exons. Whole Genome Sequence (WGS) based analyses of thousands of genomes representing various cancer tissues revealed multiple cancer-driver events localised in non-coding regions of DNA such as promoters and introns, to name just a few. Such events include not only single nucleotide variations (SNV) but also small indels and larger structural changes (32). There are already some attempts to better understand the function of the non-coding regions. For example, on the large UK Biobank sample a depletion rank has been applied to characterize the function and importance of the non-coding variants. Surprisingly, among the 1% of regions with the lowest DR, 13.0% were coding and 87.0% were non-coding (33). This suggests the still not understood importance of non-coding regions.
Rare variants in high/medium penetrance cancer-related genes, although significant, cannot explain the cancer genetic risk sufficiently. Some common variants (polymorphisms, SNPs with a frequency >1%) may contribute to the individual high or moderate risks of developing cancer or alternatively may imply a lower risk of developing the malignant disease. Polygenic Risk Score (PRS) aggregates the effects of many genetic variants across the human genome into a single score, being an estimate of an individual’s genetic liability to a trait or disease (34). PRS can be useful in the prediction of the progression and recurrence of cancer, which allows improving the efficiency of population-level screening. Recent studies showed that common variants may sufficiently assess personal risk, at least for some types of cancers, such as breast, colon, lungs, thyroid and prostate (35). For prostate cancer the combination of rare and common genetic variants has been reported to be the most efficient in the disease risk prediction (36). Black et al. (37) reported that a set of 72 SNPs PRS was predictive of prostate cancer and could be used to identify unaffected individuals at high risk of developing prostate cancer. In the case of breast cancer, Kapoor et al. (38) highlighted that the lifetime risk of breast cancer associated with classical risk factors was greater for women with higher PRS. The estimation of genome wide PRS for breast and prostate cancers showed that having a high PRS led from 21% to 38% higher lifetime risk, and 4 to 9 years earlier disease onset. PRSs improved model calculations over age, sex and clinical risk and substantiated the application of PRS in population screening (39). PRS may additionally play the role of biomarker or response markers for chemotherapy and be taken into consideration to reduce the costs of cancer screening (34). There are also critical voices on the use of PRS, especially in a clinical setting. Before being introduced to the clinics, PRS must provide significant discrimination and be applicable in terms of early detection and prevention. One of the concerns regarding PRS is the fact that most analyses performed, are based on European populations and may not be generalizable to other (e.g., Asian) populations (40). That is why understanding the cancer-related variants in the context of the country’s population is crucial for the interpretation at the population level and later on in an individual context. Although the data gathered as part of this study do not include at the moment strictly cancer patients, they can, later on, serve as a population control for assessing PRS.
To check the representation of the variants in genes related to oncology in the Polish cohorts, we compared them to the data of multiple ancestries from the global databases. For the BRCA1/BRCA2 analysis, the pathogenic variants identified in this study for the Polish population were mostly shared with the NFE ancestry and just one common variant within the EAS and SAS ancestry was identified. As Polish BRCA1/BRCA2 P/LP variants have a strong founder effect that is why as expected there are shared mainly with the European NFE population, which is the closest genetically. Most of these variants are not represented in the other ancestries. In the context of variants with a highest CADD (mostly common variants) located in the genes associated with cancer, when compared to the gomADg database, encompassing most of the variants, as many as 27% were monomorphic for Poles, but this was strongly dependent on the database. The result highlights the importance of creating a local database due to the fact that a large part of the variants related to oncological diseases is population specific and not shared not only between the various ancestries, but also between the same continent (PL vs. NFE). Moreover, the size of the database has a considerable meaning, while a low number of individuals can lead to bias. For example, 1KGP database encompasses around 3202 WGS individuals from different ancestries (41), while gnomAD has aggregated 15.708 genomes and 125.748 exomes (42). Although 1KGP database has several advantages, such as a broad representation of the human genetic variation among ancestries and inclusion of only healthy individuals, however considering only a small number of individuals may not properly illustrate the similarities and diversity between different ancestries in terms of germline variants related to cancer.
The Thousand Polish Genomes database can be used as a reference database for germline cancer variants. To the best of our knowledge, this is the first study on the Polish population using a whole-genome sequencing approach for cancer predisposition screening. Previous studies focussed on selected genes and known founder mutation, although they have encompassed large cohorts, such as 16 229 healthy persons. The Thousand Polish Genomes database encompass also genetic modifiers and genetic biomarkers related to treatment outcomes. In Poland several projects targeted the discovery of new genes related to cancer and cancer susceptibility genes (43, 44). The Thousand Polish Genomes database can provide population references for such studies. Also, recently it has been shown that it may be possible to construct germline variants from the discarded tumor sequencing reads (45). The local population can help to validate such a result. One of the most relevant issues is in assessing variant pathogenicity. This is a challenge also for the variants already reported in ClinVar. For example, the variant in PALB2 (c.3103A>G (p.Ile1035Val)) gene reported as having conflicting interpretations of pathogenicity in ClinVar and suggested of being pathogenic in the literature (46) was present in healthy individuals as well as in breast cancer individuals from the extended cohort Our study confirmed the presence of this PALB2 (c.3103A>G (p.Ile1035Val)) variant in the Polish core cohort in a very high AF (12.3%).
Are we ready for population cancer screening using WGS? Given the sinking costs of sequencing technologies and the rising availability of prevention management alternatives, we are heading towards it. We identified nine P/LP variants in the BRCA1/BRCA2 variants only in the unselected Polish population. However, especially on the individual level we are not able to interpret data reliably, for example in terms of PRS, and that is why we cannot make any prediction based on them. As a scientific and clinical community, we should further try to provide results of the negative association in databases like ClinVar, not only the positive ones. We should be aware of differences between populations, even within the same ethnicity group, and to be able to identify risk alleles we advocate for large cohorts. For now, cancer screening with WGS on a research basis in larger populations will show us how feasible the method could be in clinics.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository can be found below: https://1000polishgenomes.com.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Central Clinical Hospital of the Ministry of Interior and Administration in Warsaw (decision nr: 41/2020 from 3 April 2020 and 125/2020 from 1 July 2020). The patients/participants provided their written informed consent to participate in this study.
Author contributions
Conceptualization, MM, PD, BI, JL. Formal analysis, MM, TS, JL. Data curation, TP, PD, PT, TS, JSt. Data generation, PD, PT, PZ, ZK, EK, MD, MW. Methodology, MM, MatS, TS, JSt, BP-S. Visualisation and writing—original draft preparation, MM, JL, BI, JSz, BP-S, MarS. Writing—reviewing and editing: JSz, PD, JSt, MD, BP-S, TP. Funding acquisition, ZK. Supervision: PD, JSz. All authors contributed to the article and approved the submitted version.
Funding
This research was partially funded by the The National Centre for Research and Development project SZPITALE-JEDNOIMIENNE/2/2020 “Development of an innovative diagnostic test to assess the course of COVID-19 and post-death complications with the aid of whole genome analysis".
Acknowledgments
We thank the patients for consenting to take part in this study.
Conflict of interest
Author PS and MD are employed by MNM Bioscience. Author MatS, EK and PD were employed by MNM Bioscience.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1045817/full#supplementary-material
References
1. Yap TA, Ashok A, Stoll J, Mauer E, Nepomuceno VM, Blackwell KL, et al. Prevalence of germline findings among tumors from cancer types lacking hereditary testing guidelines. JAMA netw Open (2022) 5(5):e2213070. doi: 10.1001/jamanetworkopen.2022.13070
2. Didkowska J, Wojciechowska U, Michalek IM, Caetano dos Santos FL. Cancer incidence and mortality in Poland in 2019. Sci Rep (2022) 12(1):10875.3. doi: 10.1038/s41598-022-14779-6
3. Burzyńska M, Maniecka-Bryła I, Pikala M. Trends of mortality due to breast cancer in Poland, 2000–2016. BMC Public Health (2020) 20(1):120. doi: 10.1186/s12889-020-8256-1
4. de Angelis R, Sant M, Coleman MP, Francisci S, Baili P, Pierannunzio D, et al. Cancer survival in Europe 1999–2007 by country and age: results of EUROCARE-5–a population-based study. Lancet Oncol (2014) 15(1):23–34. doi: 10.1016/S1470-2045(13)70546-1
5. Górski B, Byrski T, Huzarski T, Jakubowska A, Menkiszak J, Gronwald J, et al. Founder mutations in the BRCA1 gene in polish families with breast-ovarian cancer. Am J Hum Genet (2000) 66(6):1963–8. doi: 10.1086/302922
6. Górski B, Jakubowska A, Huzarski T, Byrski T, Gronwald J, Grzybowska E, et al. A high proportion of founder BRCA1 mutations in polish breast cancer families. Int J Cancer (2004) 110(5):683–6. doi: 10.1002/ijc.20162
7. Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among ashkenazi jews. New Engl J Med (1997) 336(20):1401–8. doi: 10.1056/NEJM199705153362001
8. Lubiński J, Górski B, Huzarski T, Byrski T, Gronwald J, Serrano-Fernández P, et al. BRCA1-positive breast cancers in young women from Poland. Breast Cancer Res Treat (2006) 99(1):71–6. doi: 10.1007/s10549-006-9182-3
9. Rowley SM, Mascarenhas L, Devereux L, Li N, Amarasinghe KC, Zethoven M, et al. Population-based genetic testing of asymptomatic women for breast and ovarian cancer susceptibility. Genet Med (2019) 21(4):913–22. doi: 10.1038/s41436-018-0277-0
10. Brozek I, Cybulska C, Ratajska M, Piatkowska M, Kluska A, Balabas A, et al. Prevalence of the most frequent BRCA1 mutations in polish population. J Appl Genet (2011) 52(3):325–30. doi: 10.1007/s13353-011-0040-6
11. Foulkes WD, Knoppers BM, Turnbull C. Population genetic testing for cancer susceptibility: founder mutations to genomes. Nat Rev Clin Oncol (2016) 13(1):41–54. doi: 10.1038/nrclinonc.2015.173
12. Manchanda R, Blyuss O, Gaba F, Gordeev VS, Jacobs C, Burnell M, et al. Current detection rates and time-to-detection of all identifiable BRCA carriers in the greater London population. J Med Genet (2018) 55(8):538–45. doi: 10.1136/jmedgenet-2017-105195
13. Kaja E, Lejman A, Sielski D, Sypniewski M, Gambin T, Dawidziuk M, et al. The thousand polish genomes–a database of polish variant allele frequencies. Int J Mol Sci (2022) 23(9):4532. doi: 10.3390/ijms23094532
14. Szuba A, Martynowicz H, Zatońska K, Ilow R, Regulska-Ilow B, Różańska D, et al. Prevalence of hypertension in polish population of PURE Poland study. J Health Inequal (2016) 2:157–62. doi: 10.5114/jhi.2016.65356
15. Topor-Madry R, Wojtyniak B, Strojek K, Rutkowski D, Bogusławski S, Ignaszewska-Wyrzykowska A, et al. Prevalence of diabetes in Poland: a combined analysis of national databases. Diabetic Med (2019) 36(10):1209–16. doi: 10.1111/dme.13949
16. Liu L, Dilworth D, Gao L, Monzon J, Summers A, Lassam N, et al. Mutation of the CDKN2A 5’ UTR creates an aberrant initiation codon and predisposes to melanoma. Nat Genet (1999) 21(1):128–32. doi: 10.1038/5082
17. COSMIC. The Catalogue Of Somatic Mutations In Cancer. https://cancer.sanger.ac.uk/cosmic (accessed on 01.03.2022).
18. Szwiec M, Jakubowska A, Górski B, Huzarski T, Tomiczek-Szwiec J, Gronwald J, et al. Recurrent mutations of BRCA1 and BRCA2 in Poland: an update. Clin Genet (2015) 87(3):288–92. doi: 10.1111/cge.12360
19. André F, Arnedos M, Baras AS, Baselga J, Bedard PL, Berger MF, et al. AACR project GENIE: Powering precision medicine through an international consortium. Cancer Discovery (2017) 7(8):818–31. doi: 10.1158/2159-8290.CD-17-0151
20. Harland M, Cust AE, Badenas C, Chang Y-M, Holland EA, Aguilera P, et al. Prevalence and predictors of germline CDKN2A mutations for melanoma cases from Australia, Spain and the united kingdom. Hered Cancer Clin Pract (2014) 12(1):20. doi: 10.1186/1897-4287-12-20
21. Eliason MJ, Larson AA, Florell SR, Zone JJ, Cannon-Albright LA, Samlowski WE, et al. Population-based prevalence of CDKN2A mutations in Utah melanoma families. J Invest Dermatol (2006) 126(3):660–6. doi: 10.1038/sj.jid.5700094
22. Puig S, Potrony M, Cuellar F, Puig-Butille JA, Carrera C, Aguilera P, et al. Characterization of individuals at high risk of developing melanoma in Latin America: bases for genetic counseling in melanoma. Genet Med (2016) 18(7):727–36. doi: 10.1038/gim.2015.160
23. Lee S-C, Guo J-Y, Lim R, Soo R, Koay E, Salto-Tellez M, et al. Clinical and molecular characteristics of hereditary non-polyposis colorectal cancer families in southeast Asia. Clin Genet (2005) 68(2):137–45. doi: 10.1111/j.1399-0004.2005.00469.x
24. Besenbacher S, Liu S, Izarzugaza JMG, Grove J, Belling K, Bork-Jensen J, et al. Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios. Nat Commun (2015) 6(1):5969. doi: 10.1038/ncomms6969
25. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature (2009) 458(7239):719–24. doi: 10.1038/nature07943
26. Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev Cancer (2004) 4(3):177–83. doi: 10.1038/nrc1299
27. Gilly A, Suveges D, Kuchenbaecker K, Pollard M, Southam L, Hatzikotoulas K, et al. Cohort-wide deep whole genome sequencing and the allelic architecture of complex traits. Nat Commun (2018) 9(1):4674. doi: 10.1038/s41467-018-07070-8
28. Boomsma DI, Wijmenga C, Slagboom EP, Swertz MA, Karssen LC, Abdellaoui A, et al. The genome of the Netherlands: design, and project goals. Eur J Hum Genet (2014) 22(2):221–7. doi: 10.1038/ejhg.2013.118
29. Telenti A, Pierce LCT, Biggs WH, di Iulio J, Wong EHM, Fabani MM, et al. Deep sequencing of 10,000 human genomes. Proc Natl Acad Sci (2016) 113(42):11901–6. doi: 10.1073/pnas.1613365113
30. Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, Oddson A, Gylfason A, et al. Large-Scale whole-genome sequencing of the icelandic population. Nat Genet (2015) 47(5):435–44. doi: 10.1038/ng.3247
31. Sun Y, Ruivenkamp CAL, Hoffer MJV, Vrijenhoek T, Kriek M, van Asperen CJ, et al. Next-generation diagnostics: Gene panel, exome, or whole genome? Hum Mutat (2015) 36(6):648–55. doi: 10.1002/humu.22783
32. Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer (2018) 18(1):5–18. doi: 10.1038/nrc.2017.99
33. Halldorsson B, Eggertsson HP, Moore KHS, Hauswedell H, Eiriksson O, Ulfarsson MO, et al. The sequences of 150,119 genomes in the UK biobank. Nature (2022) 607(7920):732–40. doi: 10.1038/s41586-022-04965-x
34. Sud A, Turnbull C, Houlston R. Will polygenic risk scores for cancer ever be clinically useful? NPJ Precis Onc (2021) 5:40. doi: 10.1038/s41698-021-00176-1
35. Lambert SA, Abraham G, Inouye M. Towards clinical utility of polygenic risk scores. Hum Mol Genet (2019) 28(R2):R133–42. doi: 10.1093/hmg/ddz187
36. Darst BF, Sheng X, Eeles RA, Kote-Jarai Z, Conti DV, Haiman CA. Combined effect of a polygenic risk score and rare genetic variants on prostate cancer risk. Eur Urol (2021) 80(2):134–8. doi: 10.1016/j.eururo.2021.04.013
37. Black MH, Li S, LaDuca H, Lo MT, Chen J, Hoiness R, et al. Validation of a prostate cancer polygenic risk score. Prostate (2020) 80(15):1314–21. doi: 10.1002/pros.24058
38. Kapoor PM, Mavaddat N, Choudhury PP, Wilcox AN, Lindström S, Behrens S, et al. Combined associations of a polygenic risk score and classical risk factors with breast cancer risk. J Natl Cancer Inst (2021) 113(3):329–37. doi: 10.1093/jnci/djaa056
39. Mars N, Koskela JT, Ripatti P, Kiiskinen T, Havulinna AS, Lindbohm JV, et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat Med (2020) 26(4):549–57. doi: 10.1038/s41591-020-0800-0
40. Cavazos TB, Witte JS. Inclusion of variants discovered from diverse populations improves polygenic risk score transferability. HGG Adv (2021) 2(1):100017. doi: 10.1016/j.xhgg.2020.100017
41. Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, et al. High-coverage whole-genome sequencing of the expanded 1000 genomes project cohort including 602 trios. Cell (2022) 185(18):3426–3440.e19. doi: 10.1016/j.cell.2022.08.004
42. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature (2020) 581(7809):434–43. doi: 10.1038/s41586-020-2308-7
43. Cybulski C, Carrot-Zhang J, Rivera B, Kashyap A, Wokołorczyk D, Giroux S, et al. Germline RECQL mutations are associated with breast cancer susceptibility. Nat Genet (2015) 47(6):643–6. doi: 10.1038/ng.3284
44. Cybulski C, Górski B, Debniak T, Gliniewicz B, Mierzejewski M, Masojć B, et al. NBS1 is a prostate cancer susceptibility gene. Cancer Res (2004) 64(4):1215–9. doi: 10.1158/0008-5472.CAN-03-2502
45. Gusev A, Groha S, Taraszka K, Semenov YR, Zaitlen N. Constructing germline research cohorts from the discarded reads of clinical tumor sequences. Genome Med (2021) 13(1):179. doi: 10.1186/s13073-021-00999-4
Keywords: genetics, cancer risk, Poland, population cancer screening, cancer
Citation: Mroczek M, Liu J, Sypniewski M, Pieńkowski T, Itrych B, Stojak J, Pronobis-Szczylik B, Stępień M, Kaja E, Dąbrowski M, Suchocki T, Wojtaszewska M, Zawadzki P, Mach A, Sztromwasser P, Król ZJ, Szyda J and Dobosz P (2023) The cancer-risk variant frequency among Polish population reported by the first national whole-genome sequencing study. Front. Oncol. 13:1045817. doi: 10.3389/fonc.2023.1045817
Received: 16 September 2022; Accepted: 20 January 2023;
Published: 10 February 2023.
Edited by:
Elzbieta Pluciennik, Medical University of Lodz, PolandReviewed by:
Radka Kaneva, Medical University Sofia, BulgariaAndrea Degasperi, University of Cambridge, United Kingdom
Copyright © 2023 Mroczek, Liu, Sypniewski, Pieńkowski, Itrych, Stojak, Pronobis-Szczylik, Stępień, Kaja, Dąbrowski, Suchocki, Wojtaszewska, Zawadzki, Mach, Sztromwasser, Król, Szyda and Dobosz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magdalena Mroczek, bS5tcm9jemVrODg4QGdtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship
‡ORCID: Magdalena Mroczek, orcid.org/0000-0002-8731-0540
Jakub Liu, orcid.org/0000-0003-4788-8417
Mateusz Sypniewski, orcid.org/0000-0002-0108-0255
Tadeusz Pieńkowski, orcid.org/0000-0002-0110-5950
Bartosz Itrych, orcid.org/0000-0002-2608-1289
Joanna Stojak, orcid.org/0000-0003-2423-5026
Bartosz Pronobis-Szczylik, orcid.org/0000-0001-5220-9859
Maria Stępień, orcid.org/0000-0002-3377-4665
Elżbieta Kaja, orcid.org/0000-0003-1277-6140
Maciej Dąbrowski, orcid.org/0000-0003-4150-3985
Tomasz Suchocki, orcid.org/0000-0002-8530-3486
Marzena Wojtaszewska, orcid.org/0000-0002-8979-2723
Paweł Zawadzki, orcid.org/0000-0002-9032-2315
Anna Mach, orcid.org/0000-0002-2453-5320
Paweł Sztromwasser, orcid.org/0000-0002-0661-8800
Zbigniew J. Król, orcid.org/0000-0003-3162-8813
Joanna Szyda, orcid.org/0000-0001-9688-0193
Paula Dobosz, orcid.org/0000-0002-5134-0975