 Yixiao Liu1,2,3†
Yixiao Liu1,2,3† Bo Jin4†
Bo Jin4† Cheng Shen1,2,3†
Cheng Shen1,2,3† Xianshu Gao5Xin Qi5Mingwei Ma5
Xianshu Gao5Xin Qi5Mingwei Ma5 Hongzhen Li5Han Hao1,2,3
Hongzhen Li5Han Hao1,2,3 Qi Tang1,2,3Kaiwei Yang1,2,3Yue Mi1,2,3
Qi Tang1,2,3Kaiwei Yang1,2,3Yue Mi1,2,3 Jie Guan4Xuero Feng6Zhisong He1,2,3
Jie Guan4Xuero Feng6Zhisong He1,2,3 Haixia Li4*
Haixia Li4* Wei Yu1,2,3*
Wei Yu1,2,3*- 1Department of Urology, Peking University First Hospital, Peking University, Beijing, China
- 2Institute of Urology, Peking University, Beijing, China
- 3National Urological Cancer Center, Beijing, China
- 4Department of Clinical Laboratory, Peking University First Hospital, Beijing, China
- 5Department of Radiation Therapy, Peking University First Hospital, Beijing, China
- 6Department of Geriatrics, Peking University First Hospital, Beijing, China
Simple summary: Somatic and germline aberrations in homologous recombinant repair (HHR) genes are associated with increased incidence and poor prognosis for prostate cancer. Through next-generation sequencing of prostate cancer patients across all clinical states from north China, here the authors identified a somatic mutational rate of 3% and a germline mutational rate of 3.9% for HRR genes using 200 tumor tissues and 714 blood specimens. Thus, mutational rates in HRR genes were lower compared with previous studies.
Background: Homologous recombination repair deficiency is associated with higher risk and poorer prognosis for prostate cancer. However, the landscapes of somatic and germline mutations in these genes remain poorly defined in Chinese patients, especially for those with localized disease and those from north part of China. In this study, we explore the genomic profiles of these patients.
Methods: We performed next-generation sequencing with 200 tumor tissues and 714 blood samples from prostate cancer patients at Peking University First Hospital, using a 32 gene panel including 19 homologous recombination repair genes.
Results: TP53, PTEN, KRAS were the most common somatic aberrations; BRCA2, NBN, ATM were the most common germline aberrations. In terms of HRR genes, 3% (6/200) patients harbored somatic aberrations, and 3.8% (28/714) patients harbored germline aberrations. 98.0% (196/200) somatic-tested and 72.7% (519/714) germline tested patients underwent prostatectomy, of which 28.6% and 42.0% had Gleason scores ≥8 respectively. Gleason scores at either biopsy or prostatectomy were predictive for somatic aberrations in general and in TP53; while age of onset <60 years old, PSA at diagnosis, and Gleason scores at biopsy were clinical factors associated with positive germline aberrations in BRCA2/ATM.
Conclusions: Our results showed a distinct genomic profile in homologous recombination repair genes for patients with prostate cancer across all clinical states from north China. Clinicians may consider to expand the prostate cancer patients receiving genetic tests to include more individuals due to the weak guiding role by the clinical factors currently available.
1 Introduction
Prostate cancer (PCa) is the second most common cancer in men worldwide (1). Despite PCa incidence in China remains far lower than that in western countries, it has increased rapidly in recent years due to lifestyle changes and screenings (2). Genomic and molecular complexity in prostate cancer have been tremendously profiled (3–7). Although many correlations between different genetic variants and PCa clinical outcomes are yet to be revealed, patients with pathogenic variants in DNA damage repair genes, especially those from homologous recombinant repair (HRR) such as ATM, BRCA2, and BRCA1, are found to be closely associated with younger onset, increased risks, and poorer prognosis (8, 9). Furthermore, platin based regimens and poly (ADP-ribose) polymerase inhibitors have been proved to have additional benefits in this specific subgroup (10, 11).
However, most of these findings came from studies enriched with non-Asian population, and research data on domestic Chinese patients are still inadequate. Previous studies have found unique genomic and epigenomic features in Chinese PCa patients, implying the importance of taking population-specific contexts into consideration (12). There are a series of studies addressing the HRR genes in Chinese population (13–19). These studies have provided outlines of mutation landscape in HRR genes in Chinese PCa patients. However, most of these studies came from south part of China. Furthermore, these studies focused mostly on germline variants and metastatic cases. Integrative analysis of somatic variants somatic and germline alterations in Chinese PCa patients is still limited.
In this study, we sequenced 714 fresh blood samples and 200 formalin-fixed paraffin-embedded (FFPE) PCa tissues from a total of 720 PCa patients across all disease states at Peking University First Hospital (PUFH). The majority of patients in this PUFH cohort came from north China. We also analysed clinical characteristics and obtained the surgical results of these patients. Thus, PUFH cohort would serve as a unique supplementation to the current bank of HRR gene alterations in Chinese population.
2 Materials and methods
2.1 Patients
From September 2019 to February 2022, a total of 721 consecutive patients treated at Peking University First Hospital were enrolled in this study with histologically confirmed prostate adenocarcinoma. Among them, 194 patients underwent paired somatic and germline sequencing; 6 patients underwent somatic sequencing solely; 520 patients underwent germline sequencing solely. Thus, a total of 200 patients underwent somatic sequencing, and a total of 714 patients underwent germline sequencing. 196 out of the 200 somatic tested patients and 519 out of the 714 germline tested patients also underwent radical prostatectomy (RP). For the 196 patients underwent RP, somatic sequencing was carried out using RP specimens; for the 4 patients who did not undergo RP, somatic sequencing was done using biopsy specimens. For both types of specimens, pathologists checked under microscope to make sure the proportion of the tumor region accounted for >20% of total area. Peripheral blood drawn from patients was used for germline sequencing. Clinical data were collected from the institutional medical database. The study was approved by the Committee for Ethics at Peking University First Hospital.
2.2 Sample preparation, sequencing and variant classification
DNA samples were sequenced with a HRR 32-gene panel (including 19 HRR genes: ATM, ATR, BRCA1, BRCA2, BARD1, CHEK1, CHEK2, FANCA, FANCL, PALB2, RAD51B, RAD51C, RAD51D, RAD54L, CDK12, NBN, PPP2R2A, BRIP1, MRE11A, and 13 therapeutic related genes: AR, BRAF, CDH1, ESR1, HDAC2, KRAS, TP53, NRAS, PIK3CA, HOXB13, ERBB2, PTEN, STK11) provided by Amoy Diagnostics, Xiamen, China. DNA was extracted and sheared into 200 to 500 bp fragments and then used for library construction using the HRR gene combination detection library preparation kit (Amoy Diagnostics, Xiamen, China) according to the manufacturer’s protocol. DNA sequencing was performed on the NextSeq500 Illumina platform (Illumina, San Diego, CA, USA) at an average depth of 1000×.
The types of detected mutations included single nucleotide variants (SNVs) and small indels (<50 bp). Sequencing data was analyzed by Sequencing Data Analysis Software (Amoy Diagnostics, Xiamen, China). In terms of the quality control for calling, a sample would not pass the quality control if: the total depth of mutation and wild-type alleles was lower than 100× for somatic sequencing or 20× for germline sequencing; the depth of a mutation allele was lower than 5×; the allelic frequency was lower than 3% for somatic sequencing or 20% for germline sequencing; the base quality of a mutation was lower than 30; the base quality of mutation allele minus the average base quality of both mutation and wild-type alleles was smaller than -2; the read quality of mutation allele minus the average read quality of both mutation and wild-type alleles was smaller than -2; the mapping quality of mutation allele minus the average mapping quality of both mutation and wild-type alleles was smaller than -0.3. These parameters had been validated in previous studies (20, 21).
For annotation, we followed the evidence framework recommended by American College of Medical Genetics and Genomics (ACMG) guidelines (22). In terms of population data, we first searched for the presence and frequency of each variant in 1000G, ExAc, gnomAD (23–25). Then, for disease databases, we searched in ClinVar for each variant’s pathogenicity (26), and BRCA 1/2 variants were also searched in BRCA Exchange (27). Somatic variants were also searched in Cancer Hotspots, OncoKB, and JAX-CKB (28–30). The assessment also included searching the scientific and medical literature. Next, computational predictive programs were used to predict the function of each variant: Mutation Taster, PolyPhen-2, PROVEAN, and SIFT for non-synonymous mutations (31–34); HSF, NNsplice for synonymous mutations (35, 36). In this study, germline mutations included pathogenic (P) and likely pathogenic (LP) variants.
2.3 Statistical analysis
We used Mann-Whitney U test for clinical characteristics between different subgroups, including age at diagnosis, Gleason score (GS), et al. All reported P values were 2-tailed, and α <.05 was considered statistically significant. SPSS Statistics 25 was used for data analysis, and OriginPro 2020b and ComplexHeatmap package in R 4.2.1 were used for drawing figures.
We further compared the mutation frequency in this cohort with other public or published data. For somatic variants, we analysed PRAD patients from The Cancer Genome Atlas (TCGA) database, SU2C/PCF cohort, MSK-IMPACT cohort, and PCa patients from Chinese Prostate Cancer Genome and Epigenome Atlas (CPGEA) (4, 6, 12). For germline variants, we analysed results from studies by Nicolosi et al. which enriched with western PCa patients and Zhu et al. which enriched with south China PCa patients, as well as SU2C/PCF and MSK-IMPACT cohorts (4, 5, 7, 15). For SU2C/PCF and MSK-IMPACT cohorts, the data were accessed through cBioportal (37, 38).
3 Results
3.1 Patient characteristics
The characteristics of the 200 patients underwent somatic testing and 714 patients underwent germline testing were listed in Tables 1, 2 respectively. The majority of the patients came from north part of China. Patients mostly came from Beijing and Hebei, followed by Shandong, Liaoning, Inner Mongolia, and Shanxi. Most patients were Han Chinese, although patients who were not Han were also recruited. For somatic testing, 21% of the participants were below 60-year-old. 66.5% belonged to high to very high-risk groups, while 7.5% metastasised to regional lymph nodes and 2.5% to distant organs. For germline testing, 19.9% were younger than 60-year-old at time of diagnosis. 57.6% belonged to high to very high-risk groups according to NCCN clinical practice guideline, while 9.2% had regional lymph node involvement and 17.6% had remote metastasis.
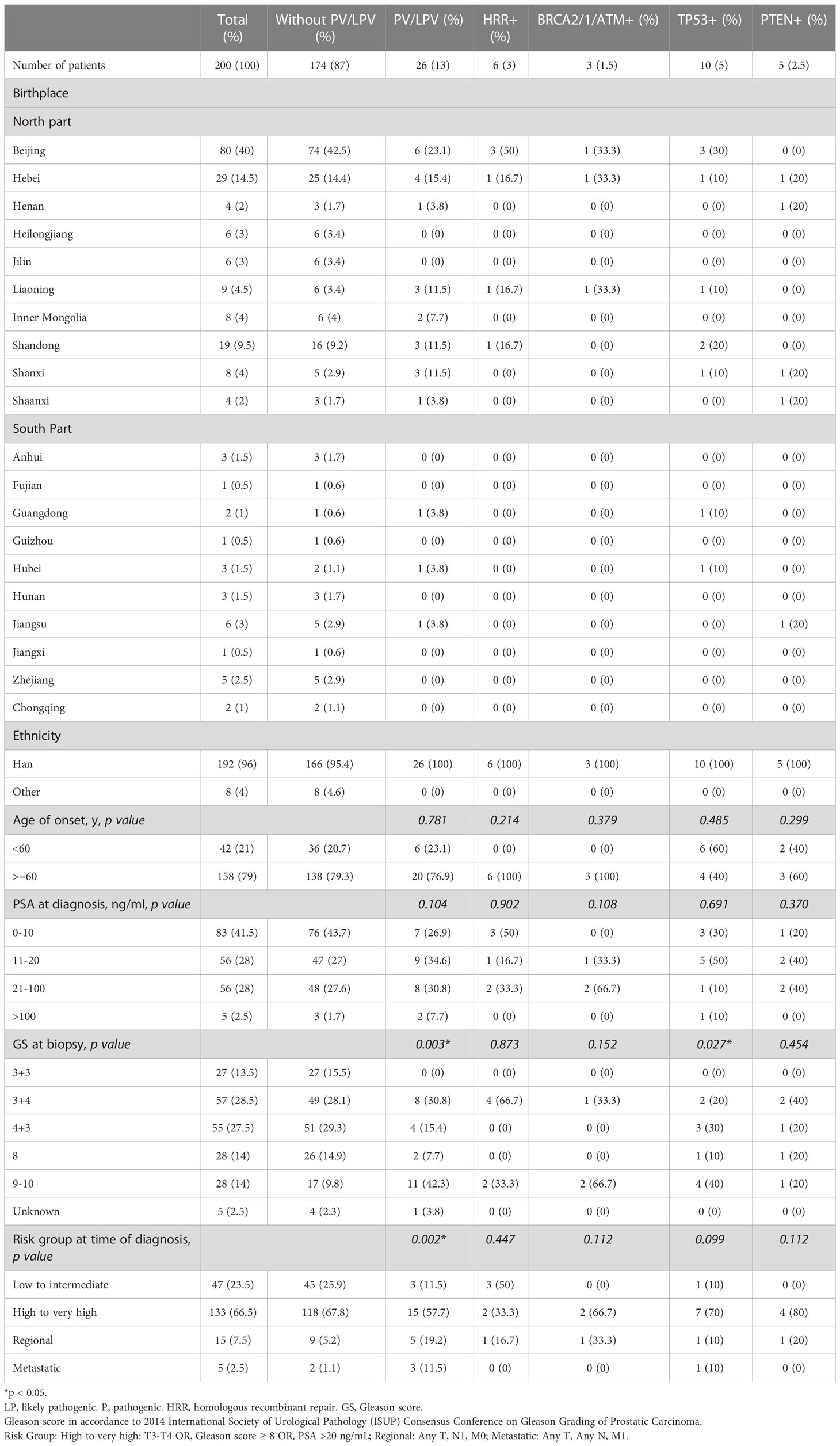
Table 1 Clinical characteristics of somatic testing at PKUFH.
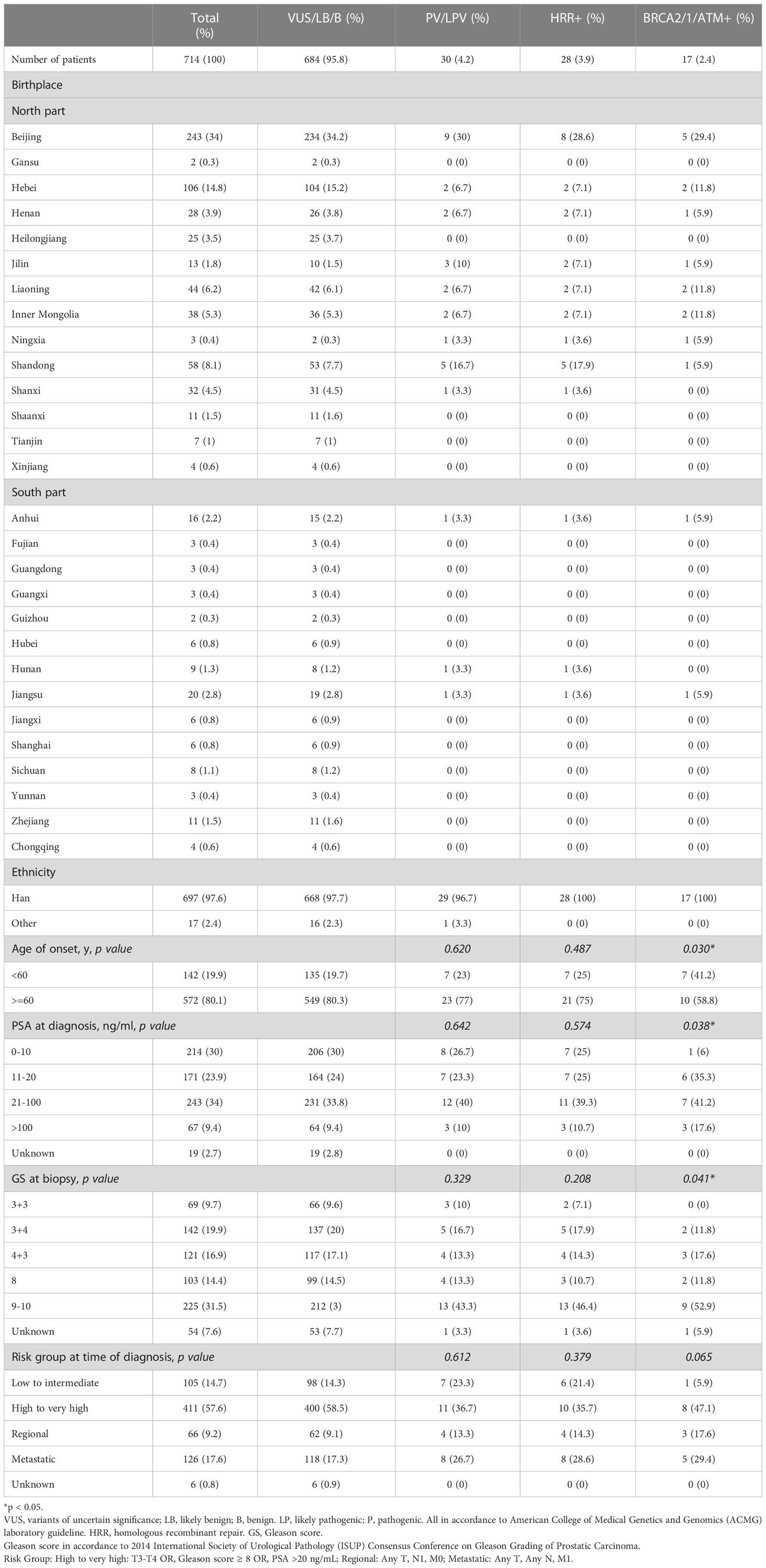
Table 2 Clinical characteristics of germline testing at PKUFH.
3.2 The landscape and comparative analysis of genomic aberrations
3% individuals harbored HHR somatic mutations. 1% demonstrated mutations in ATM and 0.5% in BRCA2 (Figure 1A). TP53 mutations (5%) were the most commonly identified alterations, followed by PTEN (2.5%), KRAS (1.5%), ATM (1%), NBN (1%), and CDK12 (1%). The mutational rates of these genes were lower than those previously reported (Figure 2A). For TP53, the most common type of mutations was missense; for PTEN, the most common type of mutations was frameshift (Figure 3A). GS at biopsy associated significantly with somatic aberrations in general (p=0.003) and in TP53 (p=0.027) (Table 1). VUS frequency was 53% for somatic sequencing (Supporting File).
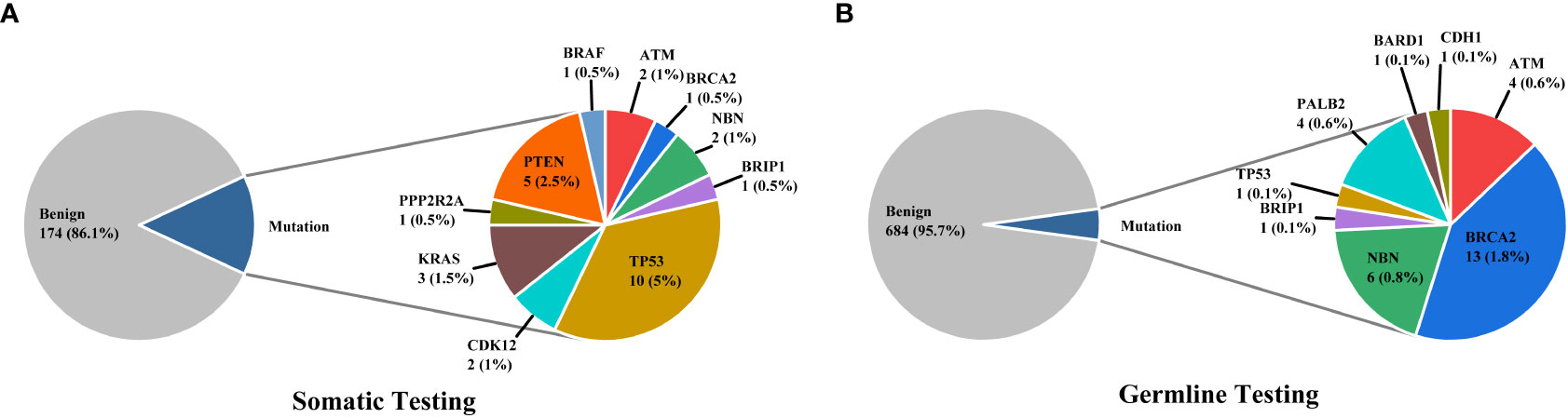
Figure 1 The distributions of somatic (A) and germline (B) mutations in our cohort.
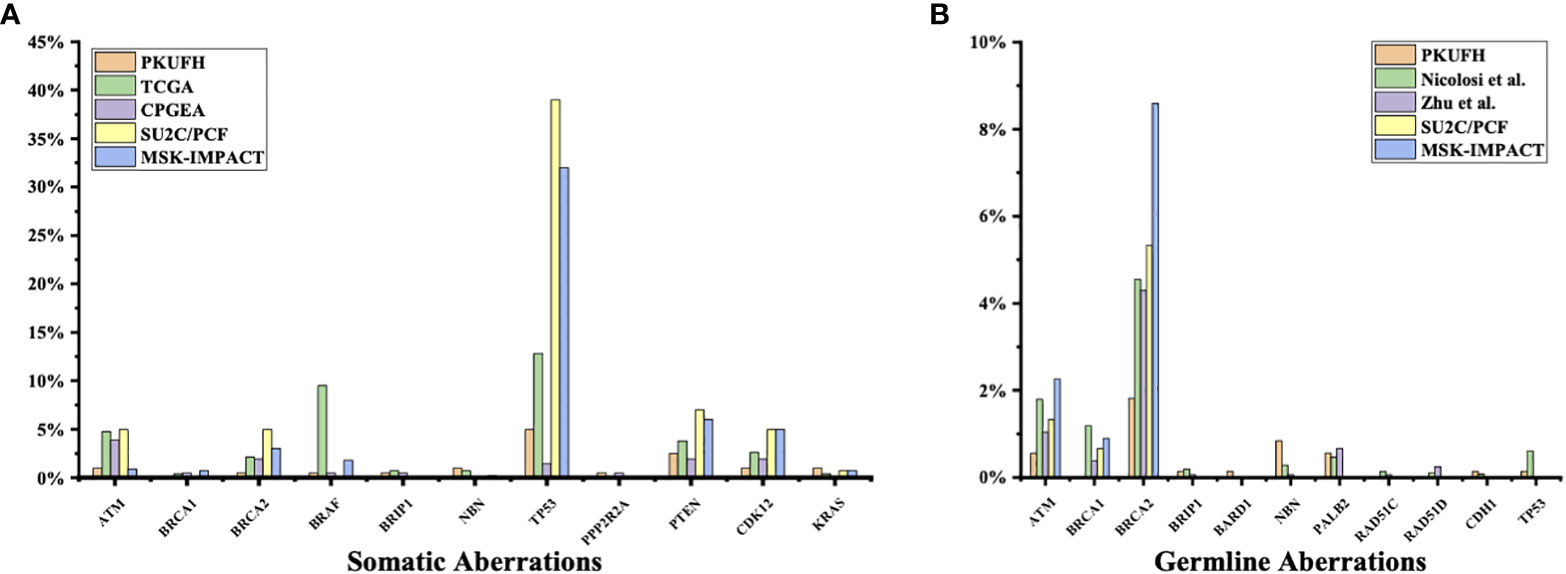
Figure 2 Comparison of somatic and germline variants between this study and previous ones (A) Somatic pathogenic variants in our study (Peking University First Hospital, PKUFH), PRAD from TCGA, Chinese Prostate Cancer Genome and Epigenome Atlas (CPGEA), SU2C/PCF, and MSK-IMPACT. CPGEA is a cohort with 208 pairs of tumor tissue samples and matched healthy control tissue from Chinese patients with primary prostate cancer. TCGA, SU2C/PCF, and MSK-IMPACT focused mostly on Caucasian populations. SU2C/PCF profiled only patients with metastatic castration resistant prostate cancer. (B) Germline Pathogenic variants (PV) and likely pathogenic variants (LPV) in PKUFH, studies by Nicolosi et al. and Zhu et al., SU2C/PCF, and MSK-IMPACT. The study by Nicolosi et al. recruited mostly patients with Caucasian descendance. The study by Zhu et al. recruited Chinese patients, mostly with metastatic disease.
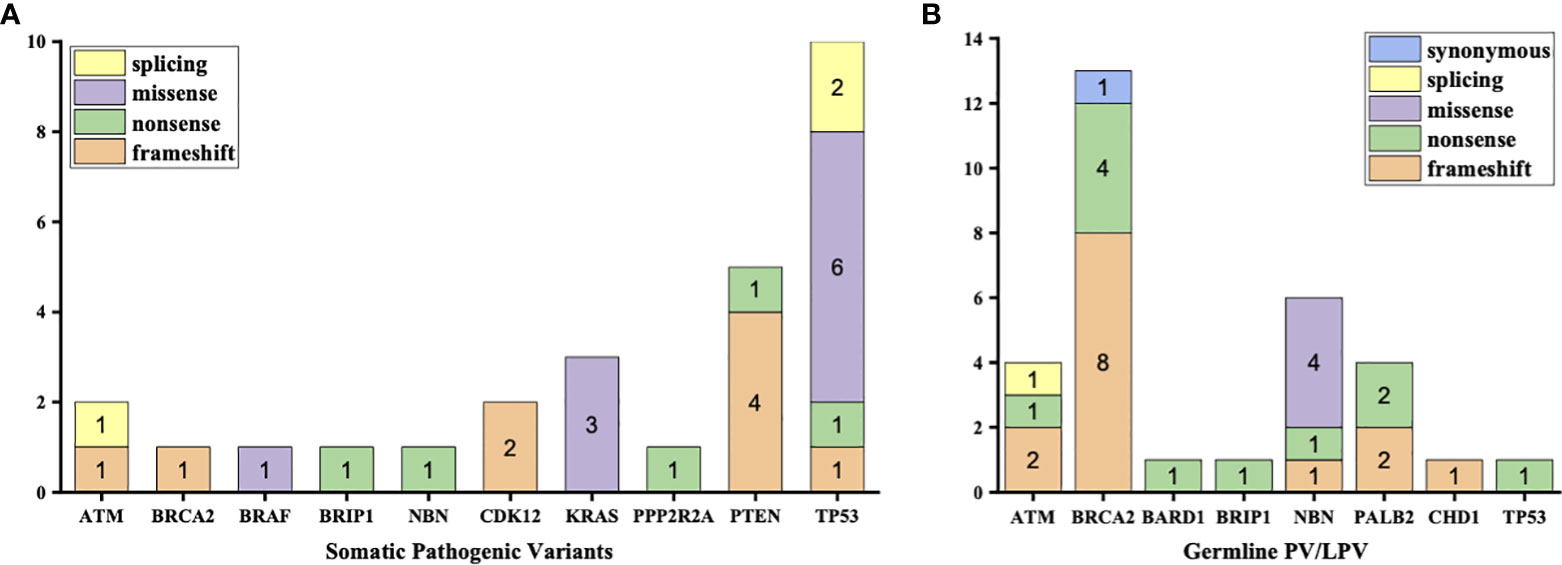
Figure 3 Types of mutations in each type of aberrations (A) Aberrations in somatic testing (B) Pathogenic variants (PV) and likely pathogenic variants (LPV) in germline testing.
3.9% individuals harbored HHR germline mutations, with 1.8% for BRCA2 and 0.6% for ATM (Figure 1B). These were lower compared with previous studies (Figure 2B). 2 mutations were not in HRR pathway: one for CDH1 and one for TP53. Most common types of mutations for ATM and BRCA2 were both frameshift (Figure 3B). For all clinical factors, age of onset (p=0.03), PSA at diagnosis (p=0.038), and GS at biopsy (p=0.041) were significantly associated with higher risk of germline mutations in BRCA2 and ATM in PCa (Table 2). VUS frequency was 50.3% for germline sequencing (Supporting File).
Particularly, no aberration was identified for BRCA1 in either germline or somatic tested patient.
3.3 Relation of needle biopsy to radical prostatectomy Gleason score
Tables 3, 4 detailed the corresponding RP GS for each of the five needle biopsy GS groups. For both, approximately 80% of cases were upgraded from a needle biopsy GS 6 to higher grade at RP. Approximately 50% of the cases had matching GS 3 + 4 and 4 + 3 at biopsy and RP. For the rest of the cases in these two groups, most cases upgraded after RP. When the biopsy was GS 8, there are almost 50% of the cases upgraded to GS 9-10 after RP. A biopsy of GS 9-10 led to nearly 70% equal grading at RP. Of note, somatic aberrations in general (p=0.006) and for TP53 (p=0.013) once again were significantly associated with higher RP grades, as for GS at biopsy mentioned above.
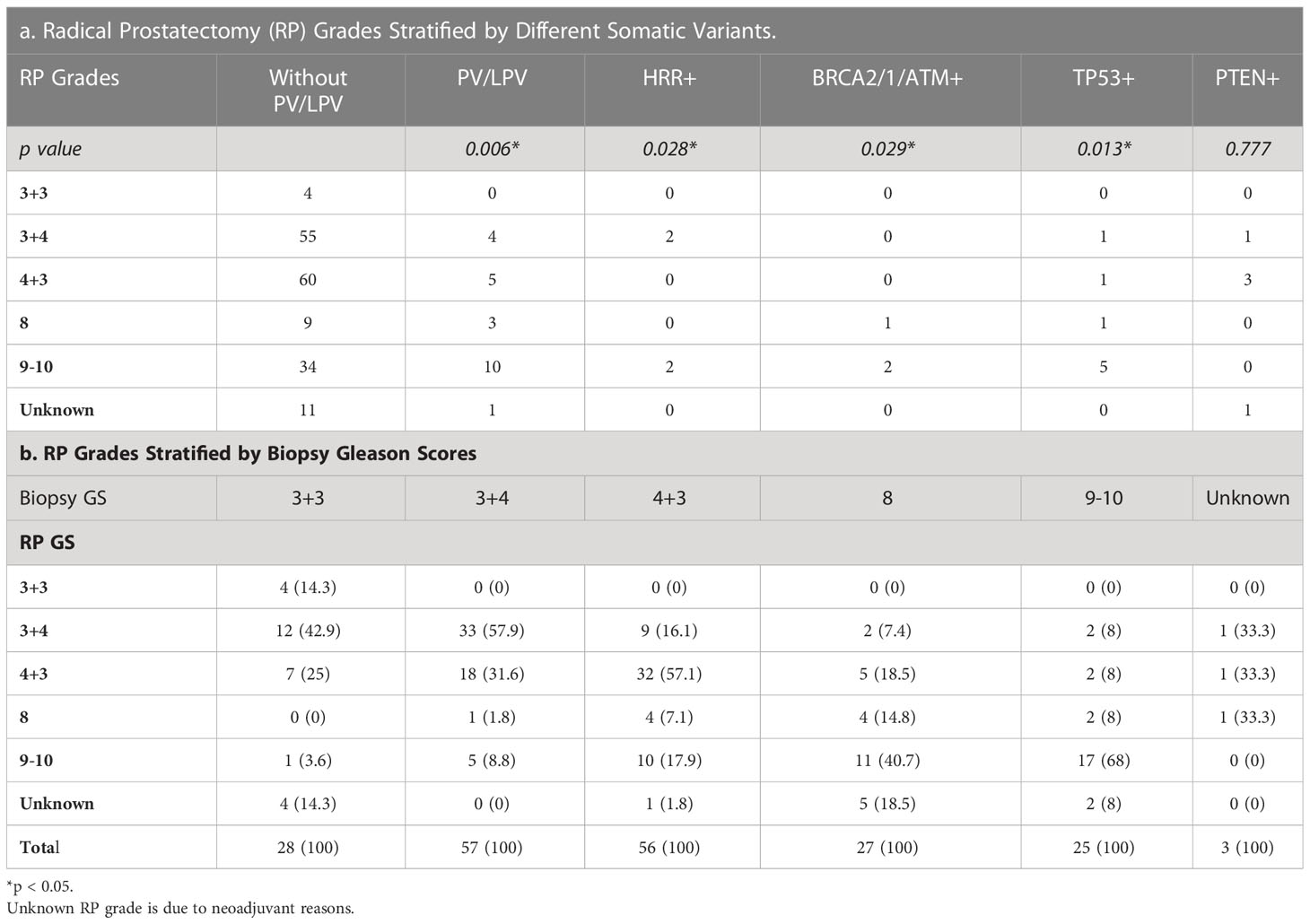
Table 3 Somatic tested patients underwent radical prostatectomy (n=196).
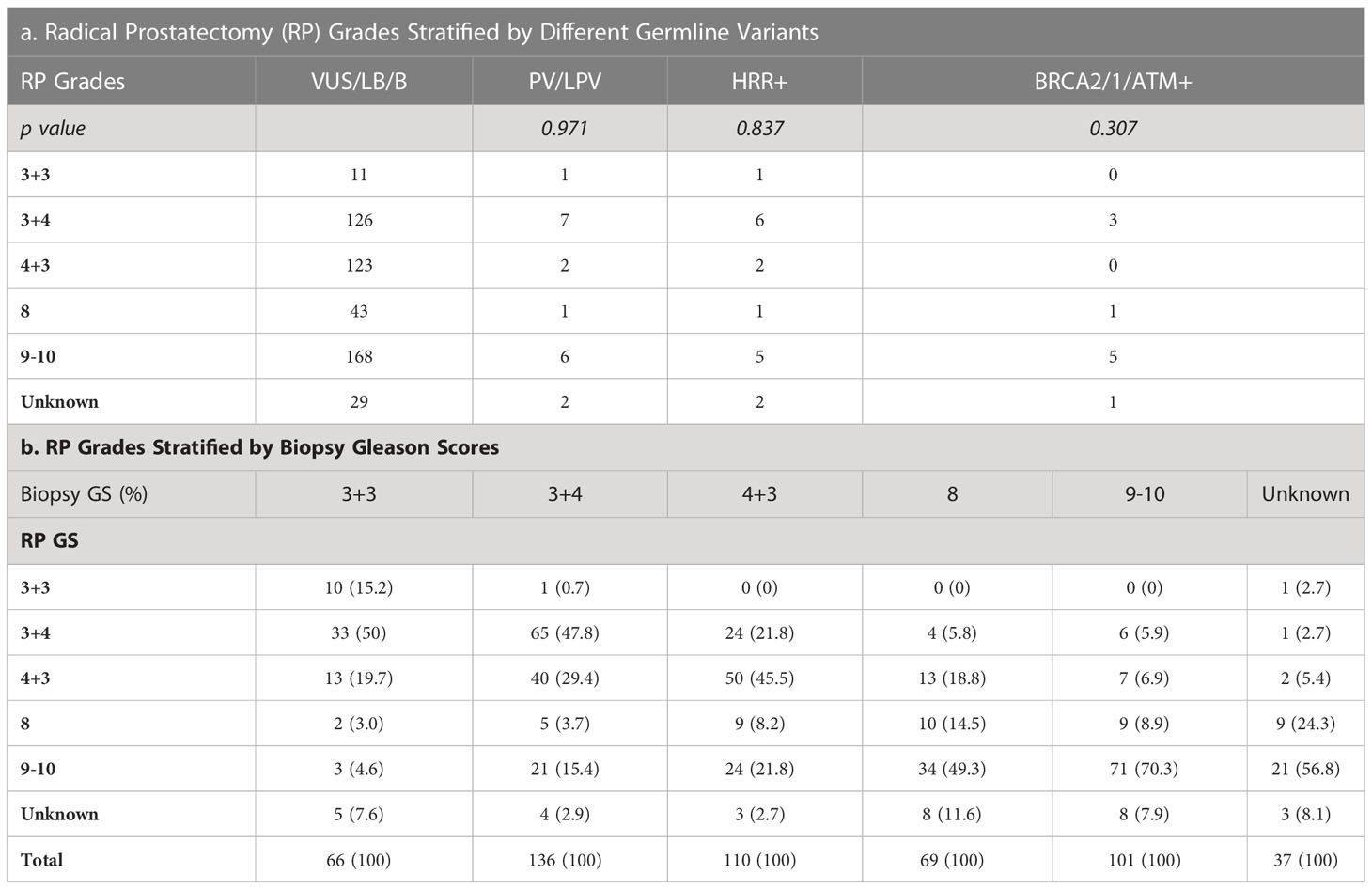
Table 4 Germline tested patients underwent radical prostatectomy (n=519).
4 Discussion
Through somatic sequencing of 200 cases and germline sequencing of 714 cases, we identified 53 patients harboring either somatic or germline mutations (Figure 4). Unlike previous studies in Chinese population, we profiled PCa in a range of clinical states—from locoregional to metastatic, and, thus, enabled comparison of genomic landscape across disease states using a single cohort. Furthermore, our study enriched PCa patients from north China, which shared a distinct genomic background but unequally represented before. Similar to previous studies, our results showed ATM and BRCA2 germline mutations were of great importance in Chinese population as were in western population. In addition, we echoed previous findings that TP53 accounted for the most common somatic mutation across all population, followed by PTEN and ATM.
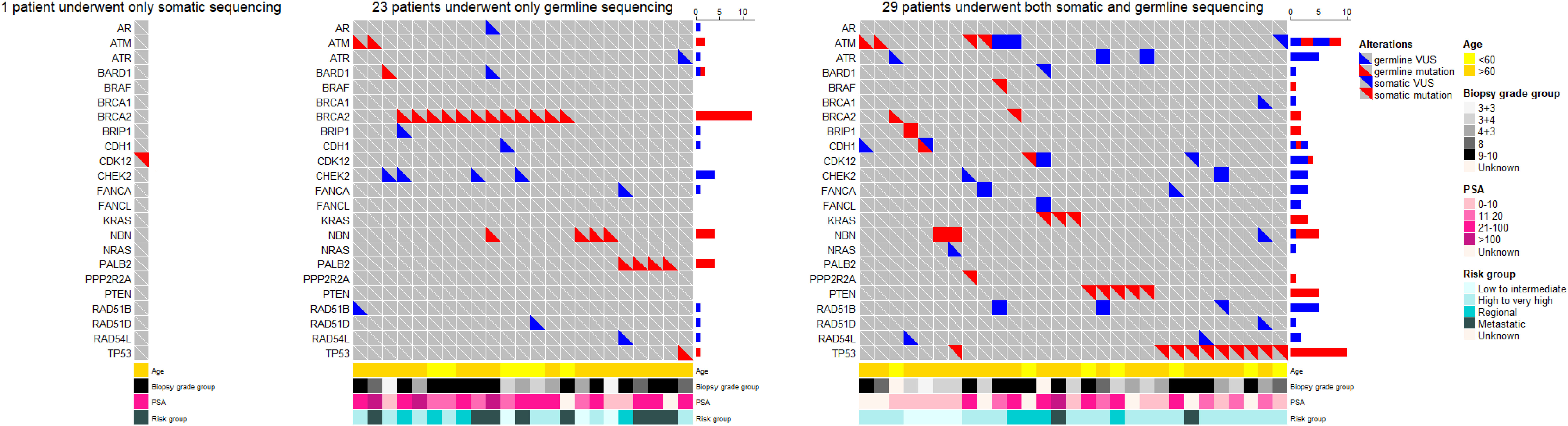
Figure 4 The oncoprints of somatic and germline aberrations tested using the 32-gene panel in our study. A total of 53 patients harbored either somatic or germline mutation. Among these patients, 1 patient underwent somatic sequencing only; 23 patients underwent germline sequencing only; and 29 patients underwent both somatic and germline sequencing. The VUS captured in these patients were also illustrated to provide a thorough view of genetic changes in these patients.
The somatic mutational rates of HRR genes in this study was 3% (6/200). By contrast, 8.68% of the cases in TCGA PARD and 9.4% in MSK-IMPACT harbored somatic aberrations in HRR genes (6). All the three studies included PCa from locoregional to metastatic states. The lower somatic mutational rates in our PUFH cohort confirmed the observation that mutational rates were generally lower in Asian patients compared to Caucasian patients. However, this 3% mutational rate was also lower than 7.89% in the CPGEA cohort which only included Chinese patients (15). This might be due to the fact that our study included a smaller proportion of metastatic (2.5% VS 8.65%) and GS 9-10 (14% VS 28.8%) patients. Furthermore, 17.8% patients in SU2C/PCF harbored somatic HRR gene mutations (4). As SU2C/PCF only included individuals at metastatic castration resistant state, the lower mutational rates in our cohort confirmed that aberrations in HRR genes were less common in early state diseases.
When it came to germline alterations, 3.9% (28/714) patients carried HRR gene mutations in PUFH cohort. The germline mutational rate was 1.8% for BRCA2 and 0.6% for ATM. This was lower compared to the results reported by Nicolosi et al., SU2C/PCF, and MSK-IMPACT in Caucasian population and by Zhu et al. (4.3% for BRCA2 and 1.04% for ATM) in south Chinese population (4, 5, 7, 15). Of note, in the study by Zhu et al, 6% PCa patients had reginal lymph node metastasis and 65% had remote metastasis. However, only 9.2% and 17.6% patients had reginal lymph node and remote metastasis in our cohort. Therefore, it was once again proved that the size of the dataset, stage of disease, and patient diversity—even for the same ethnicity from different regions—were of great importance when interpreting prostate cancer genomic data.
Due to the low mutational rates for HRR genes in patients from north China, a smarter protocol to select potential candidates for genetic testing is necessary. Broadly speaking, as HRR gene mutations were associated with poor outcomes, any clinical feature potentially linked to aggressive disease could serve as a candidate marker. Currently, most guidelines addressed the use of clinical characteristics including age of onset, family history, and personal cancer history to enhance the positive rate of genetic testing (39). Nicolosi et al. argued clinical factors frequently used to identify patients who qualified germline testing, including age, race, and family history, did not correlate with positive test results in their data (7). So was GS. However, their data revealed a higher likelihood of germline mutations in HRR genes with higher stages of diseases. These findings were not echoed by those of Wu et al. In their study, Wu et al. did not observe a relationship between the presence of germline HRR gene mutations and any clinical characteristics except age at diagnosis (14). In our study, Gleason grades at diagnostic biopsy were found to be correlated with somatic aberrations in general and in TP53, while age of onset, PSA at diagnosis, and GS at biopsy were factors correlated with germline ATM and BRCA2 mutations. Due to these conflicting evidences, clinicians should be cautious using these clinical factors when prescribing tailored genetic tests for PCa patients. Indeed, it would be a better idea to expand genetic tests to include wider range of men diagnosed with PCa as much as possible due to the fact that these mutation carriers demanded radical treatment as early as possible.
As a needle biopsy and corresponding RP might not always get the same GS, we also examined the results of RP GS. Our results showed a higher incidence for GS upgrading for most groups except GS 9-10 when compared to the benchmark from Epstein et al (40). Thus, it reflected a potential bias in using biopsy GS alone to pick patients with aggressive patients. However, some pathological features including Intraductal/ductal histology and lymphovascular invasion appeared to be associated with pathogenic germline DNA-repair gene mutations in men with prostate cancer (41). Therefore, it would be reasonable for future studies to investigate the underlying morphology in these specimens possessing HRR gene mutations, and, thus, could provide extra help to smarter screening.
There were puzzles waiting to be answered. We found no BRCA1 mutation in our cohort. Interestingly, in the only one previous study focused on north China population but with a smaller size, researchers found no BRCA1 mutation neither (19). This could possibly be a distinctive genomic feature for north China individuals. Future studies should validate this observation with multiple center studies and more powerful epidemiology tools.
Our study had some limitations. First, we treated VUS as negative alterations in this study. However, preliminary yet increasing data implies the pathogenicity of some of the current VUS. Second, our panel only tested variants including single-nucleotide polymorphisms and indels, but not copy number variants, which also could result in loss of function of proteins. Third, we interpreted the somatic variants with a variety of certified knowledge bases. Of note, there was currently no ‘gold standard’ somatic mutation database that was similar to what existed for germline. These concerns warranted future efforts and advancement in clinical genetics.
In summary, we investigated the genomic landscape of both somatic and germline HRR gene alterations in the PCa patients across all clinical states from north Chinese population. The lower HRR gene mutational rate in this PUFH cohort underlined the need for a more efficient pre-testing candidate-selection protocol in genetic testing.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The study was conducted in accordance with the Declaration of Helsinki. The studies involving human participants were reviewed and approved by Committee for Ethics at Peking University First Hospital (protocol code: 2022-199-001; date of approval: 2022-04-22). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
HXL and WY set up the experimental design and supervised the work. YL and BJ collected and selected the data. CS, XG, XQ, MM, HZL, HH, QT, KY, YM, and ZH organized the database. BJ and JG carried out the genetic testing. YL and XF performed the statistical analysis and wrote the manuscript. All authors reviewed the manuscript and agree with its contents. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by National Natural Science Foundation of China (81870518 to WY).
Acknowledgments
We are grateful to the patients and urological pathologists at the Peking University First Hospital. The results published here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. Yu Fan is acknowledged for sharing his insights on genomic testing in prostate cancer. Eliezer Van Allen is acknowledged for his kind advice on curation of somatic mutations. Likun Zhu is acknowledged for her advice and guidance on the illustration of figures. We express our sincere appreciation for the grant from the National Natural Science Foundation of China (81870518 to WY).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1086517/full#supplementary-material
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Qiu H, Cao S, Xu R. Cancer incidence, mortality, and burden in China: A time-trend analysis and comparison with the united states and united kingdom based on the global epidemiological data released in 2020. Cancer Commun (Lond) (2021) 41(10):1037–48. doi: 10.1002/cac2.12197
3. Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell (2015) 163(4):1011–25. doi: 10.1016/j.cell.2015.10.025
4. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer [published correction appears in cell. Cell. (2015) 161(5):1215–28. doi: 10.1016/j.cell.2015.05.001
5. Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol (2017) 17:00029. doi: 10.1200/PO.17.00029
6. Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A (2019) 116(23):11428–36. doi: 10.1073/pnas.1902651116
7. Nicolosi P, Ledet E, Yang S, Michalski S, Freschi B, O'Leary E, et al. Prevalence of germline variants in prostate cancer and implications for current genetic testing guidelines. JAMA Oncol (2019) 5(4):523–8. doi: 10.1001/jamaoncol.2018.6760
8. Cheng HH, Sokolova AO, Schaeffer EM, Small EJ, Higano CS. Germline and somatic mutations in prostate cancer for the clinician. J Natl Compr Canc Netw (2019) 17(5):515–21. doi: 10.6004/jnccn.2019.7307
9. Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol (2013) 31(14):1748–57. doi: 10.1200/JCO.2012.43.1882
10. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair defects and olaparib in metastatic prostate cancer. N Engl J Med (2015) 373(18):1697–708. doi: 10.1056/NEJMoa1506859
11. Cheng HH, Pritchard CC, Boyd T, Nelson PS, Montgomery B. Biallelic inactivation of BRCA2 in platinum-sensitive metastatic castration-resistant prostate cancer. Eur Urol (2016) 69(6):992–5. doi: 10.1016/j.eururo.2015.11.022
12. Li J, Xu C, Lee HJ, Ren S, Zi X, Zhang Z, et al. A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature. (2020) 580(7801):93–9. doi: 10.1038/s41586-020-2135-x
13. Wei Y, Wu J, Gu W, Qin X, Dai B, Lin G, et al. Germline DNA repair gene mutation landscape in Chinese prostate cancer patients. Eur Urol (2019) 76(3):280–3. doi: 10.1016/j.eururo.2019.06.004
14. Wu J, Wei Y, Pan J, Jin S, Gu W, Gan H, et al. Prevalence of comprehensive DNA damage repair gene germline mutations in Chinese prostate cancer patients. Int J Cancer (2021) 148(3):673–81. doi: 10.1002/ijc.33324
15. Zhu Y, Wei Y, Zeng H, Li Y, Ng CF, Zhou F, et al. Inherited mutations in Chinese men with prostate cancer. J Natl Compr Canc Netw (2021) 20(1):54–62. doi: 10.6004/jnccn.2021.7010
16. Chen W, Xia W, Xue S, Huang H, Lin Q, Liu Y, et al. Analysis of BRCA germline mutations in Chinese prostate cancer patients. Front Oncol (2022) 12:746102. doi: 10.3389/fonc.2022.746102
17. Fan L, Fei X, Zhu Y, Pan J, Sha J, Chi C, et al. Comparative analysis of genomic alterations across castration sensitive and castration resistant prostate cancer via circulating tumor DNA sequencing. J Urol (2021) 205(2):461–9. doi: 10.1097/JU.0000000000001363
18. Fei X, Fan L, Chen W, Chen W, Gong Y, Du X, et al. The prevalence and clinical implication of rare germline deleterious alterations in Chinese patients with prostate cancer: A real-world multicenter study. Clin Transl Med (2021) 11(10):e527. doi: 10.1002/ctm2.527
19. Jiang X, Hu X, Gu Y, Li Y, Jin M, Zhao H, et al. Homologous recombination repair gene mutations in Chinese localized and locally advanced prostate cancer patients. Pathol Res Pract (2021) 224:153507. doi: 10.1016/j.prp.2021.153507
20. Fumagalli C, Betella I, Ranghiero A, Guerini-Rocco E, Bonaldo G, Rappa A, et al. In-house testing for homologous recombination repair deficiency (HRD) testing in ovarian carcinoma: A feasibility study comparing AmoyDx HRD focus panel with myriad myChoiceCDx assay. Pathologica. (2022) 114(4):288–94. doi: 10.32074/1591-951X-791
21. Yuan W, Ni J, Wen H, Shi W, Chen X, Huang H, et al. Genomic scar score: A robust model predicting homologous recombination deficiency based on genomic instability. BJOG. (2022) 129 Suppl 2:14–22. doi: 10.1111/1471-0528.17324
22. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
23. The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature (2015) 526:68–74. doi: 10.1038/nature15393
24. Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, et al. The ExAC browser: Displaying reference data information from over 60 000 exomes. Nucleic Acids Res (2017) 45(D1):D840–5. doi: 10.1093/nar/gkw971
25. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
26. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res (2018) 46(D1):D1062–7. doi: 10.1093/nar/gkx1153
27. Cline MS, Liao RG, Parsons MT, Paten B, Alquaddoomi F, Antoniou A, et al. BRCA challenge: BRCA exchange as a global resource for variants in BRCA1 and BRCA2. PloS Genet (2018) 14(12):e1007752. doi: 10.1371/journal.pgen.1007752
28. Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discovery (2018) 8(2):174–83. doi: 10.1158/2159-8290.CD-17-0321
29. OncoKB: A precision oncology knowledge base. JCO Precis Oncol (2017) 17:00011. doi: 10.1200/PO.17.00011
30. Patterson SE, Liu R, Statz CM, Durkin D, Lakshminarayana A, Mockus SM. The clinical trial landscape in oncology and connectivity of somatic mutational profiles to targeted therapies. Hum Genomics (2016) 10:4. doi: 10.1186/s40246-016-0061-7
31. Schwarz J, Cooper D, Schuelke M, Schuelke M, Seelow D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat Methods (2014) 11:361–2. doi: 10.1038/nmeth.2890
32. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods (2010) 7(4):248–9. doi: 10.1038/nmeth0410-248
33. Choi Y, Chan AP. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. (2015) 31(16):2745–7. doi: 10.1093/bioinformatics/btv195
34. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res (2012) 40(Web Server issue):W452–7. doi: 10.1093/nar/gks539
35. Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in genie. J Comput Biol (1997) 4(3):311–23. doi: 10.1089/cmb.1997.4.311
36. Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res (2009) 37(9):e67. doi: 10.1093/nar/gkp215
37. cBioportal: cBio cancer genomics portal. Available at: https://www.cbioportal.org/study/summary?id=prad_mskcc_2017.
38. cBioportal: cBio cancer genomics portal. Available at: https://www.cbioportal.org/study/summary?id=prad_su2c_2019.
39. Mohler JL, Antonarakis ES, Armstrong AJ, D'Amico AV, Davis BJ, Dorff T, et al. Prostate cancer, version 2.2019, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw (2019) 17:479–505. doi: 10.6004/jnccn.2019.0023
40. Epstein JI, Feng Z, Trock BJ, Pierorazio PM. Upgrading and downgrading of prostate cancer from biopsy to radical prostatectomy: Incidence and predictive factors using the modified Gleason grading system and factoring in tertiary grades. Eur Urol (2012) 61(5):1019–24. doi: 10.1016/j.eururo.2012.01.050
Keywords: prostate cancer, DNA repair, mutation, China, genetic testing
Citation: Liu Y, Jin B, Shen C, Gao X, Qi X, Ma M, Li H, Hao H, Tang Q, Yang K, Mi Y, Guan J, Feng X, He Z, Li H and Yu W (2023) Somatic and germline aberrations in homologous recombination repair genes in Chinese prostate cancer patients. Front. Oncol. 13:1086517. doi: 10.3389/fonc.2023.1086517
Received: 01 November 2022; Accepted: 28 February 2023;
Published: 29 March 2023.
Edited by:
Jianzhong Ai, Sichuan University, ChinaReviewed by:
Sigve Nakken, Oslo University Hospital, NorwayRadka Kaneva, Medical University Sofia, Bulgaria
Copyright © 2023 Liu, Jin, Shen, Gao, Qi, Ma, Li, Hao, Tang, Yang, Mi, Guan, Feng, He, Li and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Yu, eXV3ZWlmQDEyNi5jb20=; Haixia Li, YmR5eWxoeEAxMjYuY29t
†These authors have contributed equally to this work and share first authorship