 Soniya N. Pinto
Soniya N. Pinto Jason Chiang
Jason Chiang Ibrahim Qaddoumi
Ibrahim Qaddoumi David Livingston
David Livingston Asim Bag
Asim Bag- 1Department of Diagnostic Imaging, St. Jude Children’s Research Hospital, Memphis, TN, United States
- 2Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN, United States
- 3Departments of Global Pediatric Medicine, St. Jude Children’s Research Hospital, Memphis, TN, United States
- 4Department of Radiology, University of Tennessee Health Science Center, Memphis, TN, United States
The diencephalon is a complex midline structure consisting of the hypothalamus, neurohypophysis, subthalamus, thalamus, epithalamus, and pineal body. Tumors arising from each of these diencephalic components differ significantly in terms of biology and prognosis. The aim of this comprehensive review is to describe the epidemiology, clinical symptoms, imaging, histology, and molecular markers in the context of the 2021 WHO classification of central nervous system neoplasms. We will also discuss the current management of each of these tumors.
1 Introduction
The diencephalon, an area in the central part of the brain that encompasses crucial structures including the thalamus and hypothalamus, is a relatively uncommon site for primary pediatric brain tumors. Yet, when tumors do occur in this location, they present unique challenges in neuro-oncology due to their intricate location and diverse clinical manifestations (1).
This review article aims to provide a comprehensive overview of diencephalic tumors, shedding light on their epidemiology, clinical presentation, imaging, and histopathology and the current strategies employed in their management. We explore the unique neuroanatomical considerations and the wide-ranging symptomatology that these tumors can exhibit. Additionally, we delve into the molecular genetics underlying these neoplasms, recognizing the increasing role of molecular profiling in the classification and treatment of brain tumors. We further discuss the current standards and innovations in the diagnosis and management of these tumors, including neuroimaging techniques, surgical approaches, radiation therapy, and emerging targeted therapies. Acknowledging the multifaceted challenges posed by diencephalic tumors, this review also aims to highlight the existing gaps in our understanding and emphasize areas where further research is required. Through a comprehensive exploration of diencephalic tumors, this review hopes to provide insights into this complex domain, paving the way for improved patient outcomes.
The diencephalon is embryologically derived from the prosencephalon, which evolves to develop the epithalamus superiorly, the thalamus centrally, and the subthalamus and hypothalamus inferiorly (1). The epithalamus comprises the anterior and posterior paraventricular nuclei, the habenular nuclei and their interconnecting fibers, the habenular commissure, the stria medullaris, and the pineal body. The epithalamus connects the limbic system to the other parts of the brain and is responsible for melatonin secretion. The thalamus is the largest part of the diencephalon that surrounds the third ventricle and is subdivided into multiple nuclei that function as relay centers for sensory and motor information to and from the cerebral cortex. The subthalamus is a complex structure that comprises a group of subthalamic nuclei and tracts that mediate fine control of motor movement. The hypothalamus is located inferior and anterior to the thalamus, forming the inferolateral walls of the third ventricle. It consists of a group of nuclei that coordinate with the autonomic and endocrine system to control metabolism, energy balance, reproduction, thermoregulation, fluid and electrolyte balance, and immune and emotional responses. The hypothalamus is intimately connected with the neurohypophysis (posterior pituitary).
As diencephalic structures mediate important functions, tumors of this region often have widespread and lasting implications on neurological and other metabolic functions, particularly in the pediatric population (2). Surgical resection of tumors in this region is often technically challenging and fraught with morbidity (3). Therefore, clinical management requires a comprehensive approach with an astute recognition and knowledge of clinical symptoms, laboratory markers, cross-sectional imaging, histologic and molecular pathology, detailed surgical planning, radiation therapy, chemotherapy, and targeted molecular therapy.
Tumors arising from each diencephalic component differ histologically and genetically despite their common embryological origin. Based on tropism, diencephalic tumors can be broadly classified into four groups, hypothalamic tumors, tumors of the neurohypophysis, thalamic tumors, and tumors of the pineal region, and we will systematically approach tumors arising from these four groups.
2 Hypothalamic tumors
Hypothalamic tumors may arise from either the neuronal or glial components of the hypothalamus or may invade the hypothalamus from adjacent structures (Table 1) (5).
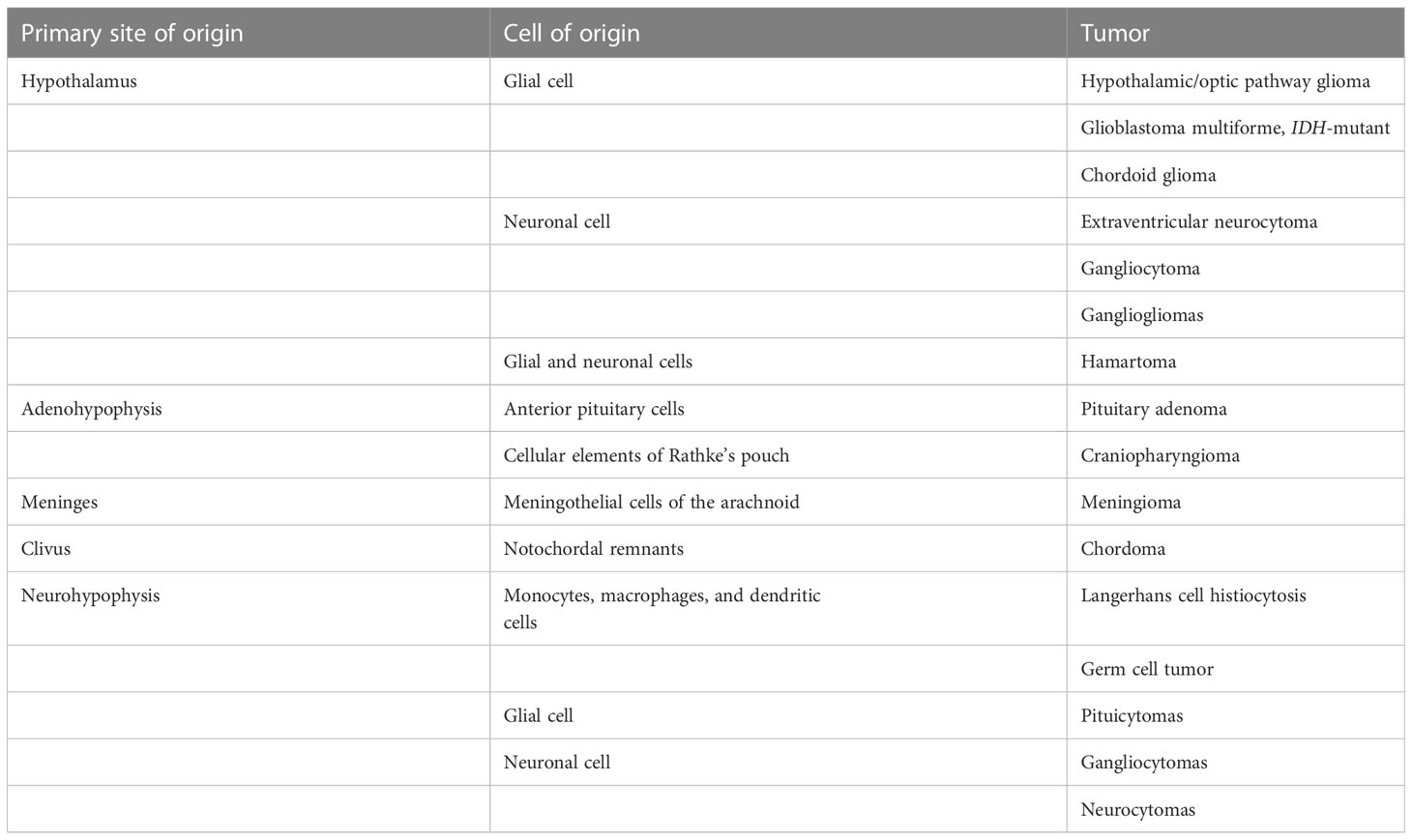
Table 1 Primary and infiltrating neoplasms of the hypothalamus in pediatric patients (4).
2.1 Primary hypothalamic tumors
2.1.1 Glial tumors
2.1.1.1 Hypothalamic/optic pathway gliomas
Gliomas are the most common primary tumors arising from the hypothalamus in the pediatric population and are usually low grade and slow growing. These tumors are related to either BRAF or NF1 aberrations. They can be limited to the optic pathway with secondary involvement of the hypothalamus or can involve both optic pathway and hypothalamus. Gliomas limited to the hypothalamus without any optic pathway involvement are possible but rare (5).
2.1.1.1.1 Clinical symptoms
Hypothalamic tumors clinically present with visual disturbances and endocrine dysfunctions. In children under the age of 3, this tumor may present with a unique diencephalic syndrome characterized by failure to thrive and emaciation despite normal caloric intake. Locomotor hyperactivity and increased alertness may also occur (6).
2.1.1.1.2 Imaging
These tumors typically demonstrate intrinsic T1 isointensity to hypointensity and T2/FLAIR hyperintensity. These tumors can be entirely solid or purely cystic or have mixed solid and cystic components. They can also be either well circumscribed or diffusely infiltrative, particularly if the tumors are associated with neurofibromatosis-1 (NF1). The enhancement pattern of these tumors is extremely variable. The solid component can demonstrate no enhancement, patchy mild enhancement, or intense enhancement. Similarly, the cyst wall can be enhancing or non-enhancing. Diffusion restriction is characteristically absent in these tumors (7). Extension along the cisternal, canalicular, and orbital segments of the optic nerves and the optic tracts is frequently seen (Figure 1) (8). The infiltrative tumors can involve bilateral medial temporal lobes and basal ganglia.
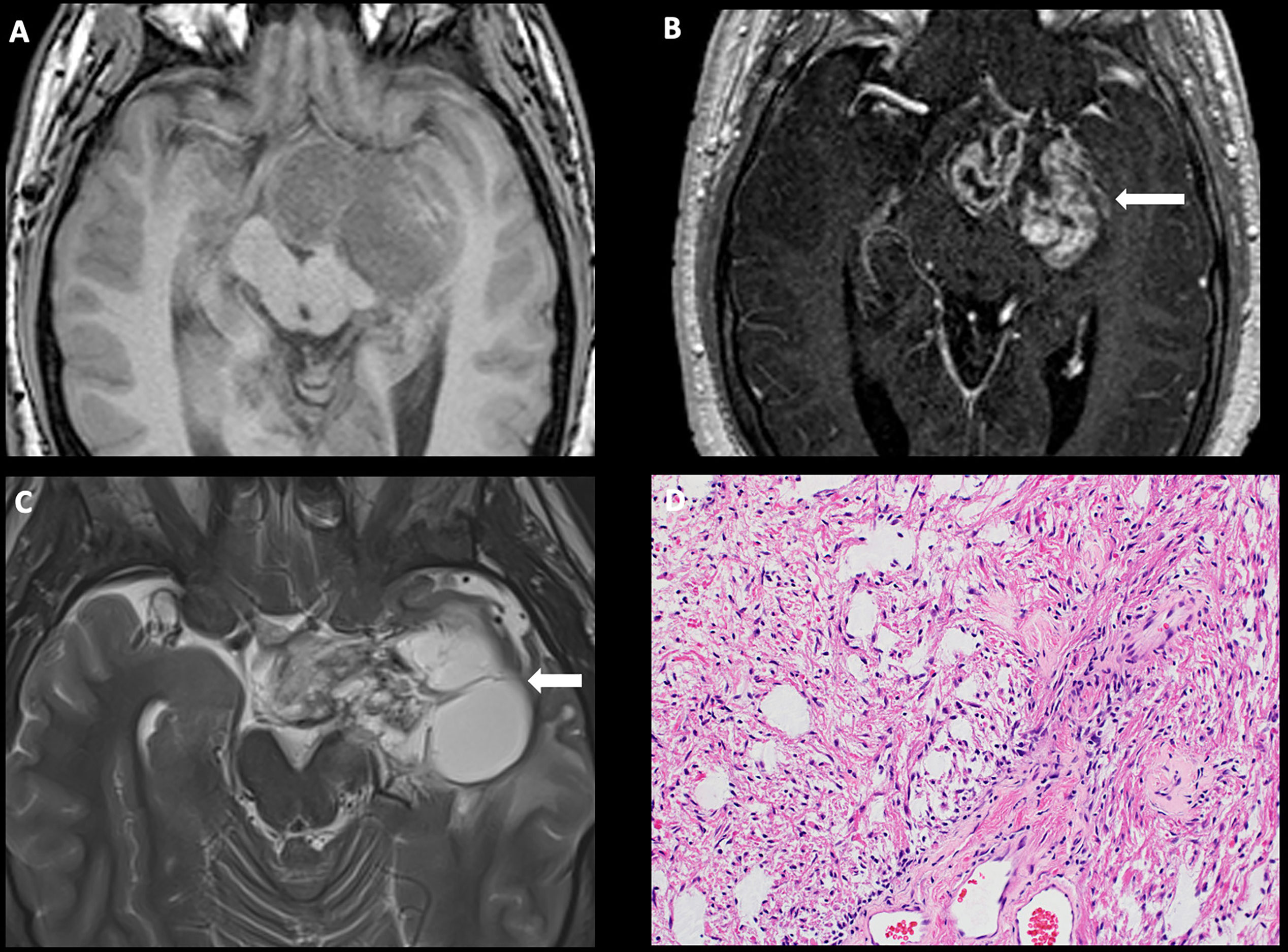
Figure 1 Obtained from a patient at St. Jude Children’s Research Hospital, Memphis, Tennessee. Axial pre-contrast T1-weighted (A) and post-contrast T1-weighted (B) images, demonstrating a complex mass centered in the left greater than right hypothalamus/optic chiasm, with extension into the left temporal lobe and enhancing solid components (arrow). (C) Axial T2-weighted sequence demonstrating a large cyst in the left temporal lobe (arrow). The histologic section (D) demonstrates a glial neoplasm with alternating loose and more compact architecture and low levels of mitotic activity. Many eosinophilic Rosenthal fibers are present in the background. Hyalinized vessels are typical findings.
2.1.1.1.3 Histology and molecular markers
It was found that 50%–70% of hypothalamic/optic pathway gliomas in the pediatric population are associated with NF1, an autosomal dominant disorder characterized by loss-of-function mutation in the NF1 gene, a negative regulator of the mitogen-activated protein kinase (MAPK) pathway (9). Most patients with NF1-associated hypothalamic/optic pathway gliomas arise in children younger than 7 years and may regress spontaneously without treatment (10). In contrast, sporadic hypothalamic/optic pathway gliomas (i.e., those not associated with NF1) arise throughout childhood, are more likely to progress, cause visual impairment, and require therapeutic intervention (11, 12). The majority of these tumors are WHO grade 1 pilocytic astrocytomas, with a few tumors demonstrating the more cellular pilomyxoid histology. The most common molecular aberration in these tumors is the activation of the BRAF oncogene, either via a KIAA1549::BRAF fusion or a BRAF p.V600E mutation, which in turn phosphorylates downstream targets MEK1 and MEK2 of the MAPK pathway (13). Compared to the posterior fossa pilocytic astrocytomas, the incidence of BRAF p.V600E point mutation is higher in hypothalamic/optic pathway tumors (14).
2.1.1.1.4 Management
MRI examination plays a central role in the diagnosis, assessment of extent, and monitoring of the response to therapy in patients with sporadic hypothalamic/optic pathway gliomas and surveillance in NF1-associated gliomas. Because the imaging appearance of these tumors is diagnostic, surgical biopsy is usually reserved for treatment-resistant cases in which a more detailed histopathologic and molecular analysis is warranted. These biopsies are usually performed via an open craniotomy with a transcallosal approach (15, 16). Complete surgical resection of these tumors without significant morbidity is not technically feasible, with chemotherapy remaining the mainstay of treatment (16). The most commonly used first-line chemotherapeutic regimens include carboplatin and vincristine (CV) combination therapy or thioguanine, procarbazine, lomustine, and vincristine (TPCV) combination therapy or vinblastine monotherapy (17). Depending on what first-line regimen was used (e.g., CV), patients can be switched to alternative regimens (e.g., vinblastine or TPCV) in cases of recurrent or progressive disease (18–20). Although radiotherapy is effective and has higher vision salvage rates, it is often avoided in young children due to a significant risk of intellectual disability, endocrinopathies, ototoxicity, and secondary malignant neoplasms (21). Molecular analysis of hypothalamic/optic pathway gliomas have helped identify valuable targets for therapy, specifically in patients with recurrent/progressive disease. Patients with fusions or duplications of BRAF often respond favorably to therapy with MEK inhibitors such as selumetinib and trametinib. On the other hand, point mutations in BRAFV600E represent a target for therapy with BRAF inhibitors such as vemurafenib and dabrafenib. In tumors with BRAFV600E mutations that progress on BRAF inhibitors or develop therapy-related toxicity, the addition of a MEK inhibitor often proves efficacious in disease control and enhancing tolerability (22, 23).
2.1.1.2 Chordoid glioma
Chordoid glioma is a rare, well-circumscribed, low-grade glioma characterized by PRKCA mutation that typically arises in the anterior third ventricle. Although this is typically an adult tumor (median age at presentation ~45 years), it is occasionally seen in the pediatric population. Pediatric chordoid glioma patients reported in the literature ranged from 7 to 13 years of age.
2.1.1.2.1 Clinical symptoms
Common presentations include obstructive hydrocephalus with symptoms of increased intracranial pressure. Endocrine abnormalities from mass effect on the hypothalamic nuclei and visual disturbances from mass effect to the optic pathway are other common presenting symptoms.
2.1.1.2.2 Imaging
These tumors are hyperdense on non-contrast computed tomography and usually well circumscribed, multilobulated, and enhance avidly with contrast administration. Connection to the hypothalamus can be seen in some tumors, suggesting the site of origin (24).
2.1.1.2.3 Histology and molecular markers
These tumors are WHO grade 2 neoplasms with well-defined borders without evidence of infiltration. Characteristic histology includes clusters and cords of epithelioid cells in a mucinous stroma (25–27). Mutation of the p.D463H in the PRKCA gene is now known to be a ubiquitous feature of chordoid gliomas.
2.1.1.2.4 Management
Gross-total resection is the treatment of choice, and a trans-lamina terminalis approach is associated with decreased overall postoperative morbidity as compared with the transcortical and transcallosal approaches. Radiation therapy is considered in patients with subtotal resection, on a case-by-case basis, but the benefit is not well established (27). Prognostic factors are not well elucidated due to rarity of this lesion.
2.1.1.3 High-grade glioma
High-grade gliomas arising from the hypothalamic regions are rare. To date, only one case of glioblastoma multiforme involving the hypothalamus and the optic chiasm has been reported in a pediatric patient who presented with progressive vision loss. On MRI, the lesion was T1 isointense, T2 hyperintense, with marked peripheral enhancement and central necrosis. The patient was treated with subtotal resection, and histopathology confirmed a WHO grade IV neoplasm with anaplastic hyperchromatic cells, atypical mitoses, and vascular proliferation (28).
2.1.2 Glioneuronal and neuronal tumors
2.1.2.1 Ganglioglioma
Gangliogliomas are common pediatric tumors that typically arise from the temporal lobe. However, there are many reports of gangliogliomas that primarily arise from the hypothalamus or secondarily involve the hypothalamus. The imaging characteristics and histopathology are similar to the gangliogliomas arising from elsewhere in the brain and are enumerated in detail in the thalamic tumor group.
2.1.2.2 Neuronal tumors
Neuronal tumors of the hypothalamus are relatively rare in the pediatric population. To date, there has been only one case report of a hypothalamic extraventricular neurocytoma in a child who presented with bitemporal hemianopsia. MRI revealed a circumscribed, multilobulated, avidly enhancing mass in the suprasellar region, and histopathology demonstrated homogeneous cells with a clear cytoplasm, a low mitotic index, and positive staining for synaptophysin and chromogranin A. The patient was treated with subtotal resection via a right frontotemporal approach and completed postoperative intensity-modulated radiation therapy (i.e., 30 Gy in 15 fractions) (29). Although molecular testing was not performed at the time, the FGFR1::TACC1 fusion is now known to be highly frequent in extraventricular neurocytomas; a few other FGFR alterations have also been identified (4).
2.2 Infiltrating tumors of the hypothalamus
2.2.1 Craniopharyngiomas
In the pediatric population, craniopharyngiomas are almost exclusively of adamantinomatous subtype (aCP), with papillary cases very rarely reported. Craniopharyngiomas arise from cellular elements related to the Rathke’s pouch (craniopharyngeal duct), which express oncogenic β-catenin during early embryonic development (30). The typical age of presentation is between 5 and 15 years without a definite sex predilection (31).
2.2.1.1 Clinical symptoms
Craniopharyngiomas usually present with symptoms of increased intracranial pressure such as headache, visual impairment, and endocrine deficits affecting the growth hormone (GH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), thyroid-stimulating hormone (TSH), adrenocorticotropic hormone (ACTH), or antidiuretic hormone (ADH). Almost half of the patients develop a hypothalamic syndrome characterized by morbid obesity, cognitive impairment, personality changes, and psychiatric symptoms either due to primary involvement of the hypothalamus by the mass or secondary to treatment-related complications (32).
2.2.1.2 Imaging
On imaging, craniopharyngiomas are coarsely calcified, usually cystic, heterogeneously enhancing neoplasms of the sellar, suprasellar, and parasellar regions. The cysts often demonstrate intrinsic T1 hyperintensity/T2 hypointensity due to proteinaceous debris (Figure 2) (33). aCPs follow a 90% rule: 90% are cystic, 90% demonstrate calcification, and 90% demonstrate contrast enhancement (34).
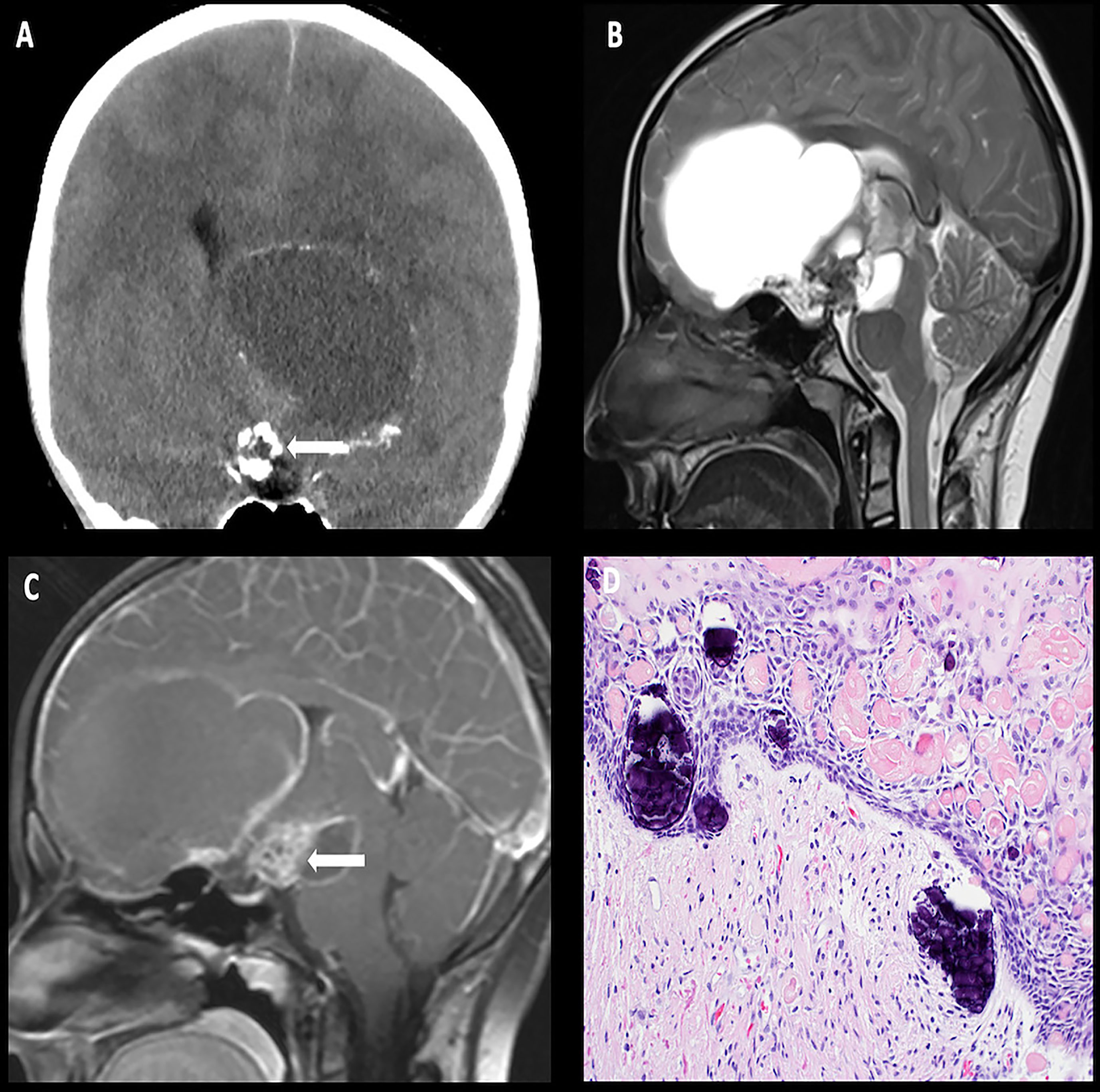
Figure 2 Obtained from a patient at St. Jude Children’s Research Hospital, Memphis, Tennessee. Coronal non-contrast head CT (A) demonstrating a calcified solid suprasellar mass (arrow), with a large cystic component extending into the third ventricle and the inferior frontal lobe on the left. Sagittal T2-weighted (B) and post-contrast T1-weighted (C) images demonstrating enhancing solid components (arrow). The histologic section (D) demonstrates an epithelial neoplasm with numerous fragments of “wet keratin” and microcalcifications in the background. The adjacent brain parenchyma shows reactive gliosis. Many Rosenthal fibers are present. Such reactive brain tissue may masquerade as pilocytic astrocytoma.
2.2.1.3 Histopathology and molecular markers
Histopathologic analysis has demonstrated well-differentiated tumor epithelium-forming cords, lobules, ribbons, nodular whorls, and irregular trabeculae, with degenerative features, such as fibrosis and calcification being commonly found. Occasionally, a xanthogranulomatous reaction to ruptured cyst material may be seen characterized by cholesterol clefts, hemosiderin deposits, xanthoma cells, and multinucleated giant cells. On molecular testing, demonstration of the CTNNB1 mutation and the absence of BRAF p.V600E may be helpful (4).
2.2.1.4 Management
The tumor’s imaging appearance is so characteristic that treatment usually proceeds without a surgical biopsy (34). The cystic components can often enlarge and cause symptoms secondary to mass effect; therefore, stereotactic cyst decompression with Ommaya catheter placement is often a first-line approach. The solid components of the tumor can usually be resected via an open transcranial technique or via newer minimally invasive techniques, such as the transsphenoidal, endoscopic endonasal, or supraorbital approaches (35). Although gross total resection is associated with a lower recurrence rate, it is also associated with significant hypothalamic and visual morbidity (30). Furthermore, many studies do not support a survival advantage of gross total resection over subtotal resection followed by adjuvant radiation therapy (36–38).
2.2.2 Pituitary adenoma/pituitary neuroendocrine tumor
Pituitary adenomas/pituitary neuroendocrine tumors are clonal neoplasms of the anterior pituitary hormone–producing cells. Only 5% of these tumors arise in the pediatric population.
2.2.2.1 Clinical symptoms
Clinical symptoms largely depend upon the size and functional activity of the tumor. Tumors less than 1 cm in size that are slow growing and do not secrete pituitary hormones are usually diagnosed incidentally. Larger non-secreting tumors present with symptoms related to mass effect, including headache and visual disturbances from compression of the optic chiasm. Hormone-secreting tumors are usually diagnosed early on as patients present with hormone excess syndromes such as hyperprolactinemia, Cushing’s disease, or hyperthyroidism. Occasionally, these tumors undergo acute hemorrhagic necrosis (apoplexy) and patients present with severe headache, lethargy, and signs of increased intracranial pressure (39).
2.2.2.2 Imaging
Most pituitary adenomas/pituitary neuroendocrine tumors are hypointense on T1-weighted imaging, with variable degrees of enhancement on post-contrast images, relative to native pituitary tissue (Figure 3). A dynamic sequence is frequently needed to diagnose pituitary microadenoma (<1 cm of size) as the rate of gadolinium-based contrast accumulation within the tumor lags behind that of the normal pituitary gland. The T2 signal is variable: densely granulated tumors demonstrate T2 hypointensity, and sparsely granulated tumors demonstrate T2 hyperintensity. Pituitary macroadenomas may locally invade the hypothalamus, the sphenoid, and cavernous sinuses (39).
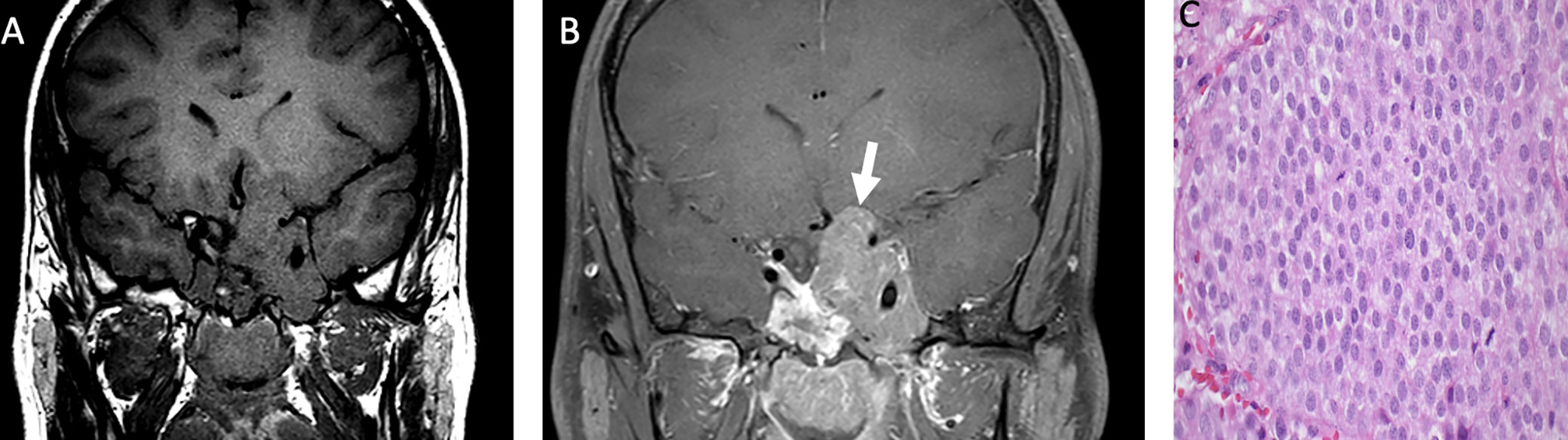
Figure 3 Obtained from a patient at St. Jude Children’s Research Hospital, Memphis, Tennessee. Coronal pre-contrast T1-weighted (A) and post-contrast T1-weighted (B) imaging demonstrating a T1 isointense, heterogeneously enhancing sellar/suprasellar mass invading the left greater than right cavernous sinus and the left hypothalamus (arrow). The histologic section (C) shows sheets of tumor cells with “salt-and-pepper” chromatin in the nuclei. There is a loss of the usual lobular architecture and mixed cell population of the normal adenohypophysis. Apparent mitotic activity can be found in biologically aggressive tumors.
2.2.2.3 Histopathology and molecular markers
On histopathology, pituitary adenomas demonstrate monomorphic cells arranged in diffuse, papillary, and trabecular arrangements. Cytologically, the cells may be acidophilic, basophilic, or chromophobic with densely or sparsely granulated cytoplasm and low mitotic activity. Molecular alterations in GNAS leading to upregulation of the cAMP/PKA pathway have been encountered in up to 40% of somatotroph adenomas, whereas mutations in USP8 and USP48 are identified in up to 50% of corticotroph adenomas (4). In addition, both functioning and non-functioning pituitary tumors have been encountered in a multitude of genetic syndromes, as described in Table 2.
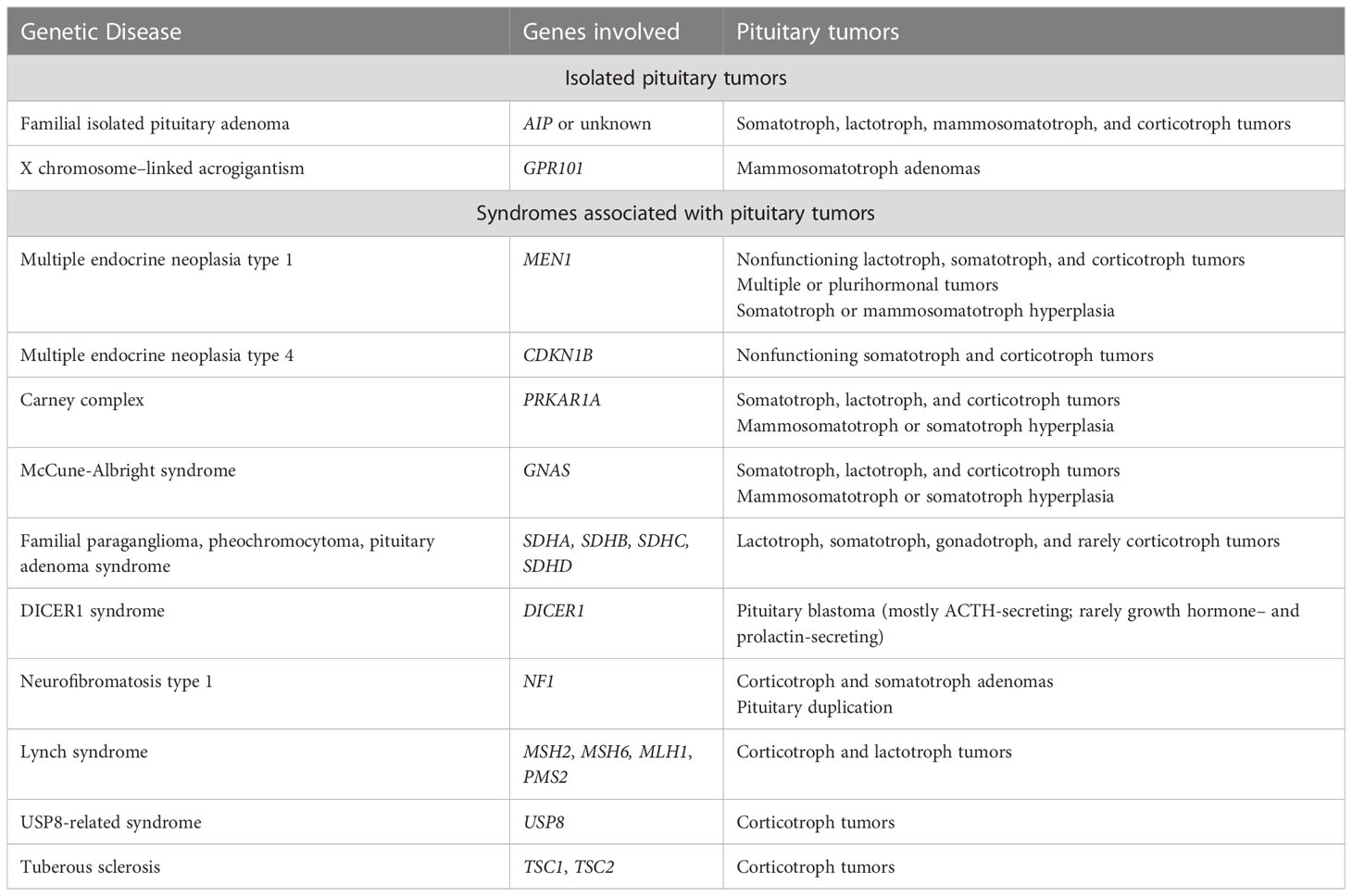
Table 2 Inherited genetic susceptibility to pituitary tumors (4).
2.2.2.4 Management
In tumors that are small (>1 cm) in diameter, slow growing, and/or incidentally diagnosed, watchful waiting with serial MRIs is a commonly employed strategy. Endoscopic transsphenoidal resection of the tumor is the preferred treatment (40). Open craniotomy is required for complete resection of the larger tumor. Invasion of the cavernous sinus, which is rare, precludes gross total resection and might warrant radiation therapy for tumor control (41).
2.2.3 Pituitary blastoma
Pituitary blastomas are exceptionally rare embryonal tumors composed of primitive blastemal cells, neuroendocrine cells, and Rathke’s pouch epithelium (42). They originate in the sellar region, with frequent invasion into the hypothalamus and cavernous sinuses. They usually occur in children younger than 2 years, with a slight female predilection (43).
2.2.3.1 Clinical symptoms
Patients usually present with Cushing syndrome, with an elevated non-suppressible ACTH level (43). Ophthalmoplegia may occur due to tumor extension into the parasellar region (44).
2.2.3.2 Imaging
Given the rarity of this entity, no hallmark imaging features have been described, with reported cases describing findings ranging from an enhancing solid mass limited to the sella to a mixed solid and cystic lesion with suprasellar/parasellar extension (45).
2.2.3.3 Histopathology and molecular markers
Histologically, pituitary blastomas are composed of three cellular components: large anterior pituitary neuroendocrine cells, cuboidal or columnar primitive Rathke’s pouch epithelial cells, and small undifferentiated blastemal cells (46). Pituitary blastomas are linked to a germline and somatic variation in DICER1, a key gene in microRNA processing, and may thereby be associated with other DICER1-related tumors, namely, ovarian Sertoli-Leydig cell tumor, renal tumors, soft tissue sarcomas, and several thyroid proliferations (47–49).
2.2.3.4 Management
Current treatment options include gross total resection when safe or subtotal resection with adjuvant chemotherapy or focal radiation therapy. Some of the commonly employed chemotherapy regimens include temozolomide alone or combination therapy with cyclophosphamide, vincristine, methotrexate, and carboplatin (49).
3 Tumors of the neurohypophysis
The neurohypophysis consists of the infundibulum and the pars nervosa (posterior lobe of the pituitary gland). The infundibulum consists of unmyelinated axons of neurosecretory cells, which originate in the hypothalamus and transport neuroendocrine hormones to the pars nervosa, where they are released into the systemic circulation via fenestrated sinusoidal capillaries. In the pediatric population, common neoplastic processes involving the infundibulum include Langerhans cell histiocytosis (LCH) and germ cell tumors. Tumors involving the pars nervosa, which are uncommon and include pituicytomas, gangliocytomas, and neurocytomas, will be discussed below.
3.1 Langerhans cell histiocytosis
LCH is characterized by an abnormal clonal proliferation of monocytes, macrophages, and dendritic cells.
3.1.1 Clinical symptoms
LCH presents as a solitary bone lesion or single-system or multi-system involvement. Children between the ages of 0 and 15 years are affected, with a peak incidence between 1 and 4 years and a slight male predilection. Infiltration of the infundibulum and hypothalamus is encountered in up to 20% of patients with the multisystem form of LCH. Diabetes insipidus is the most common presenting clinical symptom. Symptoms related to anterior pituitary hormone deficiencies are less common.
3.1.2 Imaging
Imaging often reveals nodular thickening and enhancement of the infundibulum, which measures more than 3.5 mm in transverse dimension and/or an enhancing suprasellar mass. Patients may have additional intracranial involvement with dural-based masses and lytic calvarial lesions preferentially involving the skull base and mastoid air cells (Figure 4) (50).
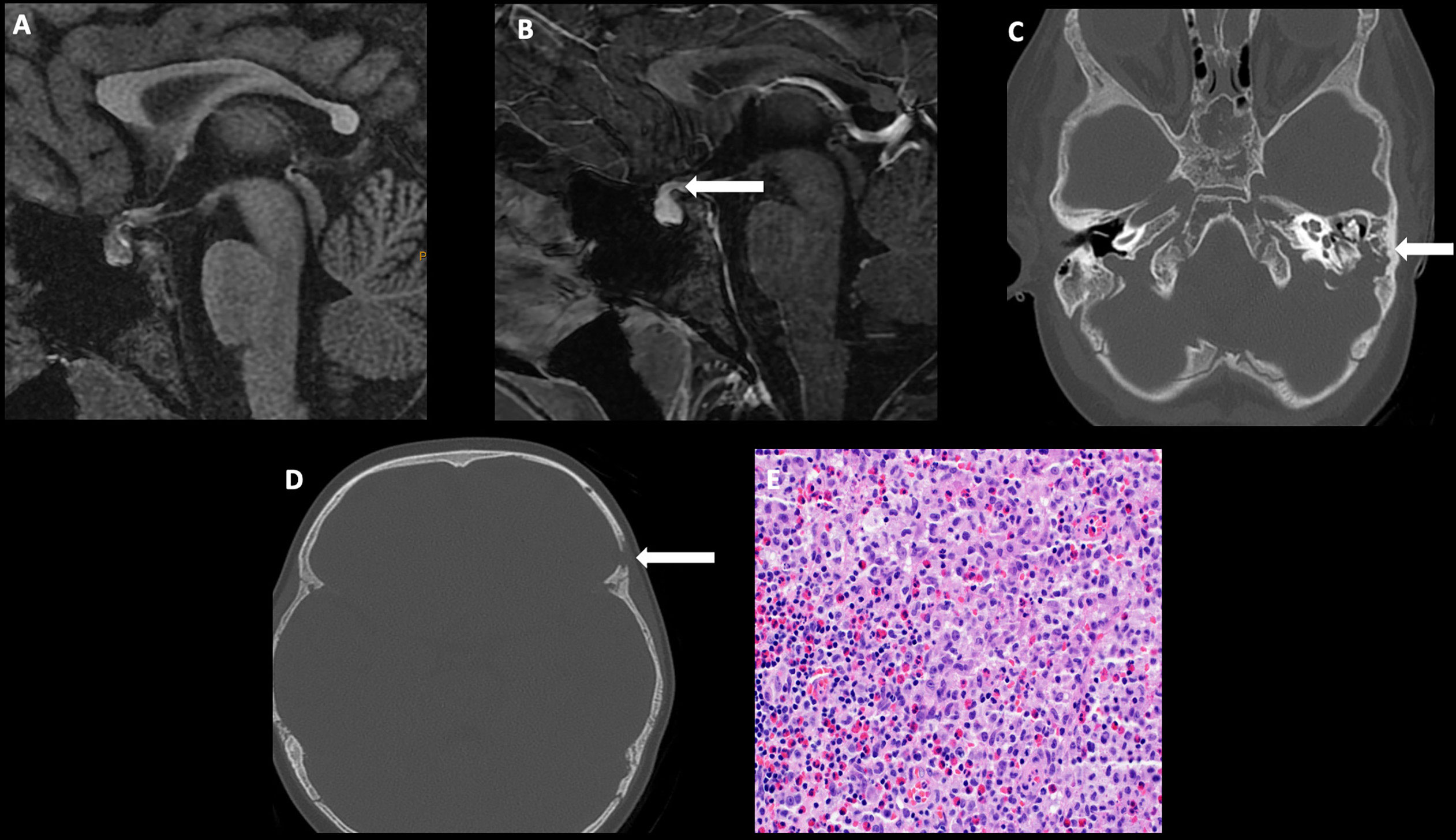
Figure 4 Obtained from a patient at St. Jude Children’s Research Hospital, Memphis, Tennessee. Sagittal T1 MRI images of pre- (A) and post- (B) contrast sequences demonstrating infundibular thickening >3 mm (arrow). Axial non-contrast CT of the temporal bone (C) demonstrating lytic-destructive change of the left mastoid air cells (arrow). (D) Axial CT image of the head, bone algorithm demonstrating a lytic lesion with a beveled edge in the left frontal calvarium (arrow). The histologic section (E) demonstrates numerous large and atypical tumor cells with abundant cytoplasm, prominent nucleoli, and irregular nuclear contour intermixed with many eosinophils and lymphocytes. The lesion can easily masquerade as an inflammatory or granulomatous process to inexperienced eyes.
3.1.3 Histopathology and molecular markers
Langerhans cells with variable reactive macrophages, lymphocytes, plasma cells, and eosinophils are characteristic histologic findings. The nuclei of Langerhans cells are slightly eccentric, ovoid, and reniform or convoluted, with linear grooves and inconspicuous nucleoli, with abundant pale to eosinophilic cytoplasm (51). The most frequent molecular alteration is BRAF p.V600E mutation encountered in approximately 50%–60% of cases (52). Other less frequent alterations include the BRAF p.V600D and ARAF mutations and BRAF fusions (53).
3.1.4 Management
Treatment of LCH in the central nervous system (CNS) depends on both the extent and severity of the disease. For example, solitary calvarial and extra-axial LCH masses are treated with surgical resection, and more extensive parenchymal and infundibular/suprasellar LCH involvement is treated with chemotherapy. The most common first-line chemotherapy regimen for LCH with CNS involvement includes a combination of vinblastine and prednisone, with purine analogs, such as cytarabine and cladribine reserved for patients whose disease does not respond to first-line treatment. The knowledge of BRAF and other MAPK pathway mutations has led to the use of BRAF and MEK inhibitors in prospective clinical trials of histiocytosis, including a small proportion of participants who have CNS LCH. Therefore, molecular-targeted therapies represent a valuable resource for patients with refractory LCH of the CNS (51–53).
3.2 Germ cell tumors of the infundibulum/suprasellar region
Germ cell tumors of the CNS comprise a group of immunophenotypic homologs of gonadal and other extra-CNS germ cell neoplasms that share certain molecular features. This group includes pure germinomas and nongerminomatous germ cell tumors, a group comprising mature and immature teratomas, yolk sac tumors, embryonal carcinomas, choriocarcinomas, and mixed germ cell tumors (4). Approximately 80%–90% of these tumors arise in the midline; the pineal region is most commonly affected, followed by the infundibulum/hypothalamus (54). Germ cell tumors of the infundibulum/hypothalamus are more prevalent in Asia than in Europe or the United States, with peak incidences reported at 10–14 years of age, with a slight female predilection (55).
3.2.1 Clinical symptoms
Clinically, these tumors present with symptoms secondary to mass effect, with visual disturbances due to impingement on the optic chiasm, diabetes insipidus and delayed growth, and sexual maturation due to disruption of the hypothalamic–pituitary axis, or headache and vomiting secondary to ventricular obstruction (55).
3.2.2 Imaging
On imaging, germinomas present as T1 isointense, T2 hyperintense, avidly enhancing masses involving the infundibular stalk and/or hypothalamus (Figure 5). On the other hand, nongerminomatous germ cell tumors are more heterogeneous in appearance, with areas of hemorrhage and intratumoral cysts identified in nearly all cases (Figure 6). Approximately 15% of germ cell tumors are metastatic at presentation, and imaging of the craniospinal axis along with CSF sampling is mandatory to assess for staging of disease. Although there is no dedicated staging system for intracranial germ cell tumors, most physicians utilize the TM/Chang system used for other brain tumors (56).
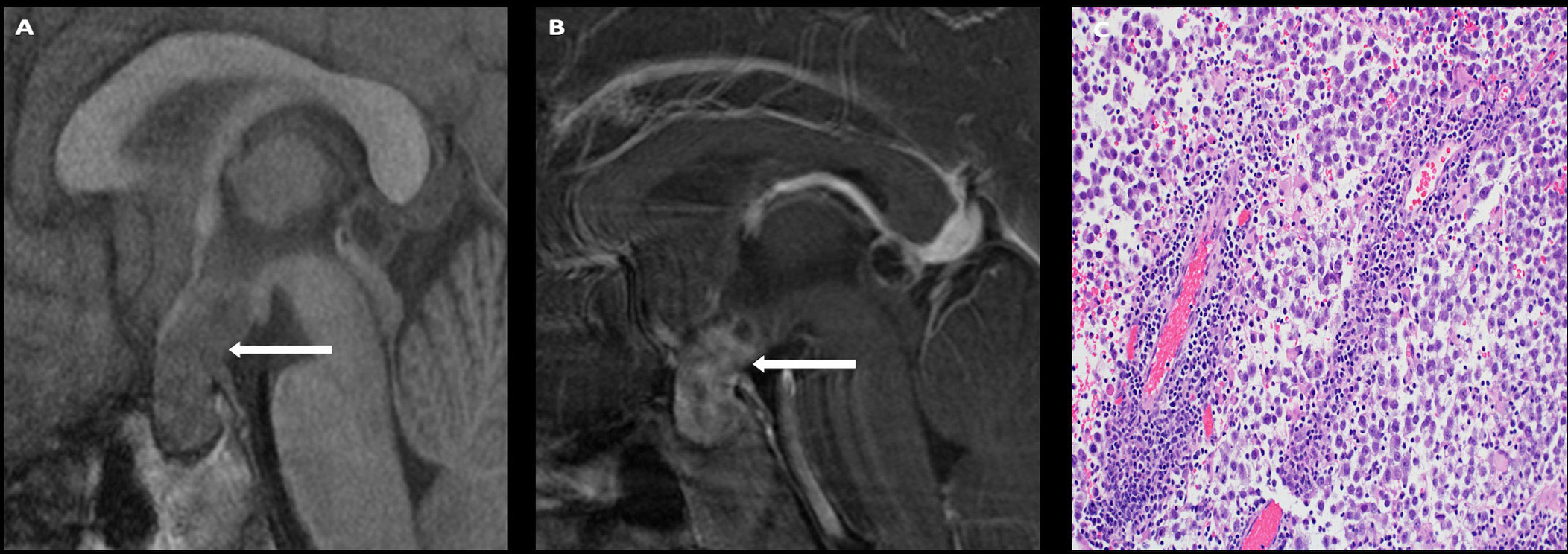
Figure 5 Obtained from a patient at St. Jude Children’s Research Hospital, Memphis, Tennessee. Sagittal T1 pre- (A) and post- (B) contrast sequences demonstrating a T1 isointense, avidly enhancing sellar/suprasellar mass (arrows). The native pituitary tissue and infundibulum are not distinctly identified. The histologic section (C) demonstrates numerous discohesive large tumor cells accompanied by many lymphocytes, a characteristic finding in germinoma.
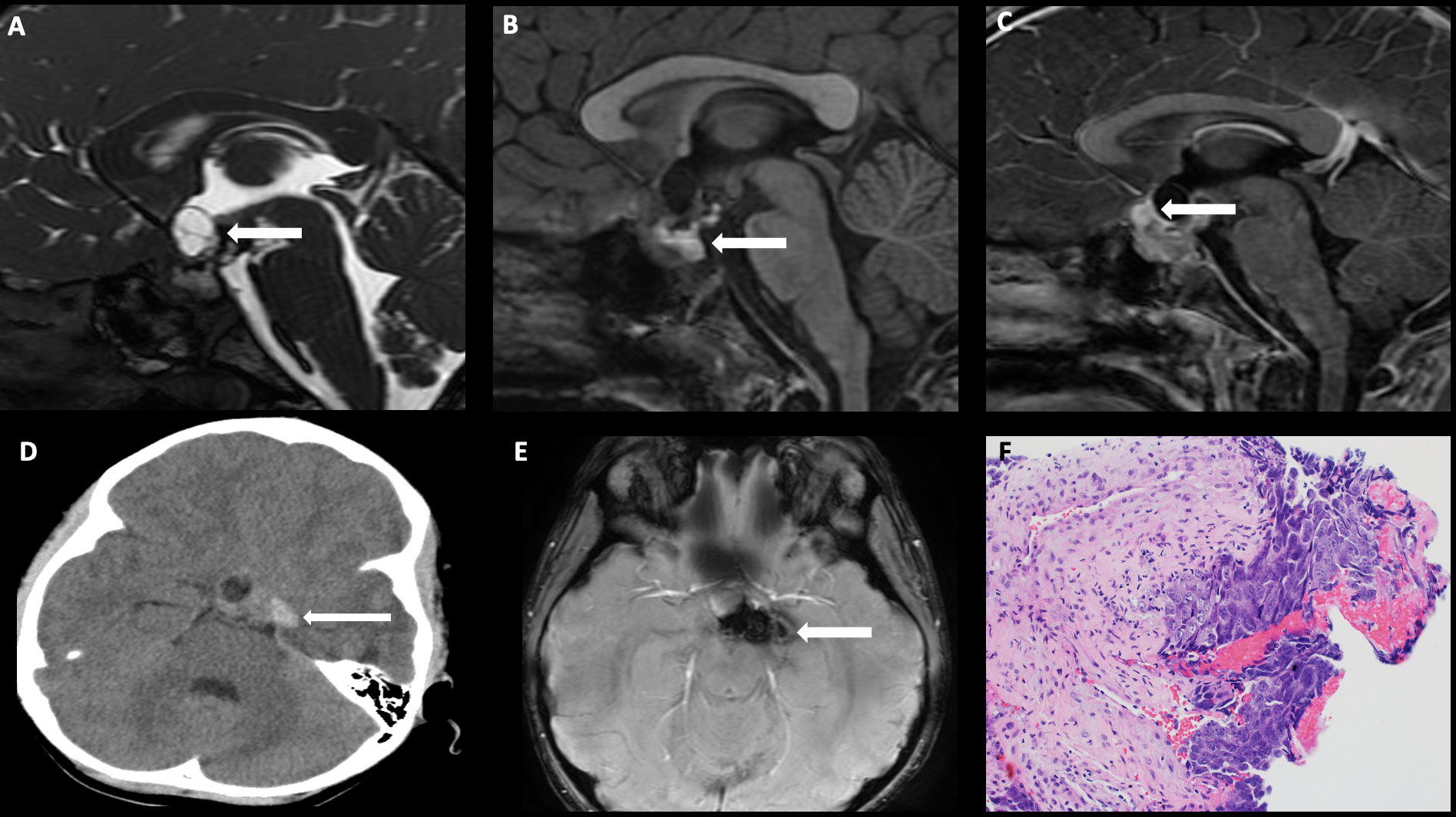
Figure 6 Obtained from a patient at St. Jude Children’s Research Hospital, Memphis, Tennessee. Sagittal FIESTA sequence demonstrating (A) a heterogeneous suprasellar mass with cystic components (arrow), (B) sagittal pre-contrast T1-weighted images demonstrating areas of intrinsic T1 hyperintensity compatible with blood products and/or proteinaceous debris (arrow), and (C) sagittal post-contrast T1-weighted images demonstrating an enhancing component in the anterior suprasellar cistern (arrow). (D) Axial non-contrast head CT demonstrating internal hemorrhage within the components to the left of midline (arrow), and (E) axial susceptibility-weighted imaging demonstrating a susceptibility artifact (arrow). The histologic section (F) shows embryonal carcinoma attached to the mesenchymal element of a teratoma. The large tumor cells in the embryonal carcinoma component have prominent nucleoli and basophilic cytoplasm.
3.2.3 Serum markers
The diagnosis involves careful interpretation of imaging findings in the context of laboratory markers, including beta-human chorionic gonadotropin (β-HCG), alpha fetoprotein (AFP), and placental alkaline phosphatase. The serum cutoff values for β-HCG vary by region, with more stringent European criteria suggesting serum levels as high as 50 mIU/ml for a diagnosis of pure germinoma and levels >50 mIU/ml for the diagnosis of nongerminomatous germ cell tumors. In contrast, the Children’s Oncology Group (COG) allows for serum levels as high as 100 mIU/ml for pure germinomas and >100 mIU/ml for nongerminomatous germ cell tumors. Serum levels of AFP >10 ng/ml and CSF levels of AFP >2 ng/ml, in the context of an intracranial mass, are considered diagnostic of nongerminomatous germ cell tumors. Levels of placental alkaline phosphatase are relatively sensitive and specific for a diagnosis of germinoma and are particularly helpful when serum and CSF levels of β-HCG and AFP are nondiagnostic. However, placental alkaline phosphatase alone is not considered a diagnostic criterion for germinoma, and a surgical biopsy may be needed particularly if the imaging is not diagnostic. In cases where the imaging and laboratory markers are diagnostic, a surgical biopsy is usually not warranted (57).
3.2.4 Histopathology and molecular markers
Histopathological features vary depending on the type of germ cell tumor and are summarized in Table 1. Despite this heterogeneity, most germ cell tumors demonstrate common molecular alterations of the MAPK and/or AKT/mTOR pathways, with various degrees of mutations in the KIT gene, which ultimately inhibit programmed cell death of primordial germ cells in the CNS (4).
3.2.5 Management
Treatment of pure germinomas has undergone significant reformation in recent years, with landmark clinical trials from COG and the International Society of Pediatric Oncology (SIOP) now advocating for the use of neoadjuvant chemotherapy, followed by maximal safe surgical resection and reduced-dose radiation for residual disease (58, 59). A response-based dose-reduction trial by COG (ACNS1123; Stratum 2) for patients with localized germinoma achieved a 3-year progression-free survival (PFS) of 94% ± 2.7% for patients treated with four courses of neoadjuvant carboplatin and etoposide, followed by gross total resection and 18 Gy whole-ventricular radiation, with a focal boost of 12 Gy to the tumor bed (58). In contrast, the SIOP CNS GCT II trial adopted 24 Gy whole-ventricular radiation in patients treated with neoadjuvant chemotherapy (alternating cycles of carboplatin + etoposide and ifosfamide + etoposide) followed by gross total resection (59). Treatment for nongerminomatous germ cell tumors is more intensive, given the higher rates of recurrence and poorer prognosis (60, 61). The SIOP CNS GCT-96 trial treated localized nongerminomatous germ cell tumors with four cycles of induction chemotherapy, followed by a focal boost of 54 Gy. The study achieved 5-year PFS and overall survival (OS) of 72% ± 4% and 82% ± 4%, respectively (60). On the other hand, the COG ACNS0122 study treated all patients with six cycles of induction chemotherapy, followed by 36 Gy craniospinal irradiation and a focal boost of 54 Gy to the tumor bed with excellent outcomes (61). Considering these data, the COG ACNS1123 Stratum 1 was developed, which used the same induction chemotherapy backbone as the ACNS0122 study but with a response-based radiation therapy plan. Per this plan, patients with a good response to chemotherapy were treated with 30.6 Gy whole-ventricular radiation with a focal boost of 23.4 Gy. Despite excellent results, this study was closed, as it met early stopping rules (62). For metastatic pure germinoma, the standard of care is 24 Gy craniospinal irradiation with a focal boost of 16 Gy to both primary and metastatic sites without chemotherapy, whereas metastatic nongerminomatous germ cell tumors are treated with four courses of chemotherapy, consisting of cisplatin, ifosfamide, and etoposide, followed by 30 Gy craniospinal irradiation and a focal boost of 16 Gy to the primary site (61, 63, 64).
3.3 Glial tumors of the pars nervosa
Pituicytoma, granular cell tumor of the sellar region, and spindle cell oncocytoma are a group of rare low-grade glial tumors that arise from the cells of the neurohypophysis or infundibulum. These group of tumors are likely representing variable manifestation of a single entity that overexpresses thyroid transcription factor-1 (TTF-1) (65, 66). These tumors typically present in adults with only a handful of case reports presenting in the first and second decades of life.
3.3.1 Clinical symptoms
Visual disturbances due to suprasellar extension and mass effect on the optic chiasm is common presentation (67–70). These tumors do not produce hypothalamic or pituitary hormones (68).
3.3.2 Imaging
On MRI, they demonstrate both solid and cystic components with early avid post-contrast enhancement, which persists into the venous phase (69).
3.3.3 Histopathology and molecular markers
The tumors uniformly demonstrated spindle cells with a low mitotic index, and immunohistochemical stains were uniformly positive for vimentin, glial fibrillary acid protein (GFAP), and S-100 (4).
3.3.4 Management
The hypervascular nature of these tumors and their proximity to the circle of Willis precluded gross total resection in these patients. These patients therefore underwent near total resection via an endoscopic transnasal, transsphenoidal, transcallosal, transseptal, or interforniceal approach with subsequent serial imaging to document stability (70).
3.4 Neuronal tumors of the pars nervosa
Other uncommon tumors of the pars nervosa include sellar neuronal tumors, such as gangliocytomas and neurocytomas (71). Gangliocytomas consist of larger, more mature neuronal elements and are more common in the adolescent and young adult population. Approximately 65% of gangliocytomas in the sellar region are associated with pituitary adenomas, and approximately 75% of them are associated with hypersecretion of a hypothalamic/pituitary hormone, most commonly GH-releasing hormone and ACTH-releasing hormone. These tumors most frequently present with endocrine abnormalities, such as acromegaly, Cushing disease, or syndrome of inappropriate antidiuretic hormone secretion confirmed by laboratory testing and immunohistochemical staining. On MRIs, these tumors are usually circumscribed, with homogeneous post-contrast enhancement (72, 73). Sellar neurocytomas, on the other hand, are extremely rare in the pediatric population, with only one case in an 8-year-old child reported in a case series of eight patients at a tertiary care institution (74). These tumors are WHO grade 2 lesions, with fibroblast growth factor receptor (FGFR) and isocitrate dehydrogenase wild type as their key molecular features (4). On imaging, these tumors are circumscribed, T1 isointense, T2 hyperintense, avidly enhancing lesions that can invade the adjacent suprasellar and parasellar structures. Surgical resection, either via a transcranial, transsphenoidal microscopic approach or an endoscopic endonasal approach, remains the mainstay of treatment, with adjuvant postoperative radiation reserved for patients with subtotal resection (74, 75).
4 Thalamic neoplasms
The thalamus consists of paired gray matter nuclei, which are located on either side of the third ventricle and are partitioned by a Y-shaped white matter structure, the internal medullary lamina (76). The subthalamus is an ovoid gray matter structure in the most caudal portion of the diencephalon, located lateral to the red nucleus and medial to the corticospinal tract. Tumors originating in the thalamus frequently extend into the subthalamic nuclei; therefore, these neoplasms will be discussed together (77). Thalamic tumors comprise 0.8%–5.2% of all pediatric brain tumors, with tumors of glial cell origin being the most common (78, 79). Less commonly, supratentorial ependymomas, embryonal tumors, and neuronal tumors may involve the thalamus and subthalamic nuclei. Surgical resection of tumors in this region is associated with significant postoperative morbidity and usually deferred for alternative treatments.
4.1 Pediatric-type diffuse astrocytoma
Pediatric-type diffuse astrocytomas are usually hemispheric tumors, which may occasionally involve the thalamus. They typically present in patients <19 years of age, with no definite sex predilection (80, 81).
4.1.1 Clinical symptoms
Sensory or motor or mixed sensorimotor symptoms may be present in patients with predominant thalamic involvement. Patients with larger tumors can present with symptoms of raised intracranial pressure due to mass effect on the adjacent third ventricle and obstructive hydrocephalus.
4.1.2 Imaging
These tumors demonstrate an infiltrative pattern, with intrinsic T1 isointensity to hypointensity, T2 heterogeneity or hyperintensity, with little to no contrast enhancement and no restricted diffusion. Differentiation of these tumors from its high-grade counterpart, diffuse midline glioma H3K27-altered, is not usually feasible on imaging. Surgical biopsy, which is usually performed under stereotactic guidance, is often needed for accurate histologic and molecular diagnosis (80).
4.1.3 Histopathology and molecular markers
Histopathology usually demonstrates tumor cells with bland nuclei and rare mitotic activity, barely elevating the cell density of the infiltrated brain parenchyma. On immunohistochemical staining, tumor cells express GFAP with variable immunoreactivity for MAP2 and were negative for SOX10. In the most recent WHO classification, these tumors are classified as either diffuse low-grade glioma MYB- or MYBL1-altered and diffuse low-grade glioma MAPK pathway-altered. The latter category can be further subdivided into diffuse low-grade glioma FGFR1-altered (fusion or mutation) or diffuse low-grade glioma BRAF p.V600E-mutant (4, 82).
4.1.4 Management
Treatment usually involves relief of obstructive hydrocephalus with a ventriculostomy or shunt, with surgical resection reserved for progressive disease. These tumors usually exhibit a benign clinical behavior, with 10-year PFS rates of 90% and 95% identified in one single-center study of 37 patients with MYB and MYBL1-altered tumors, respectively (81).
4.2 Pilocytic astrocytoma
Pilocytic astrocytomas are WHO grade 1 circumscribed astrocytic gliomas with variable proportions of bipolar hair-like pilocytic cells, compact and loose or myxoid regions, Rosenthal fibers, and eosinophilic granular bodies (4). A systematic review of 445 cases of thalamic gliomas in children revealed that pilocytic astrocytomas comprised approximately 33% of all of these tumors between the ages of 0 and 14 years (79).
4.2.1 Clinical symptoms
Clinically, pilocytic astrocytomas in the thalamus present with symptoms secondary to elevated intracranial pressure and/or motor weakness (79).
4.2.2 Imaging
Thalamic pilocytic astrocytomas are usually well-circumscribed T2 hyperintense tumors that can have both solid and cystic components (Figure 7) (83). Please refer to imaging of hypothalamic PAs for a detailed description.
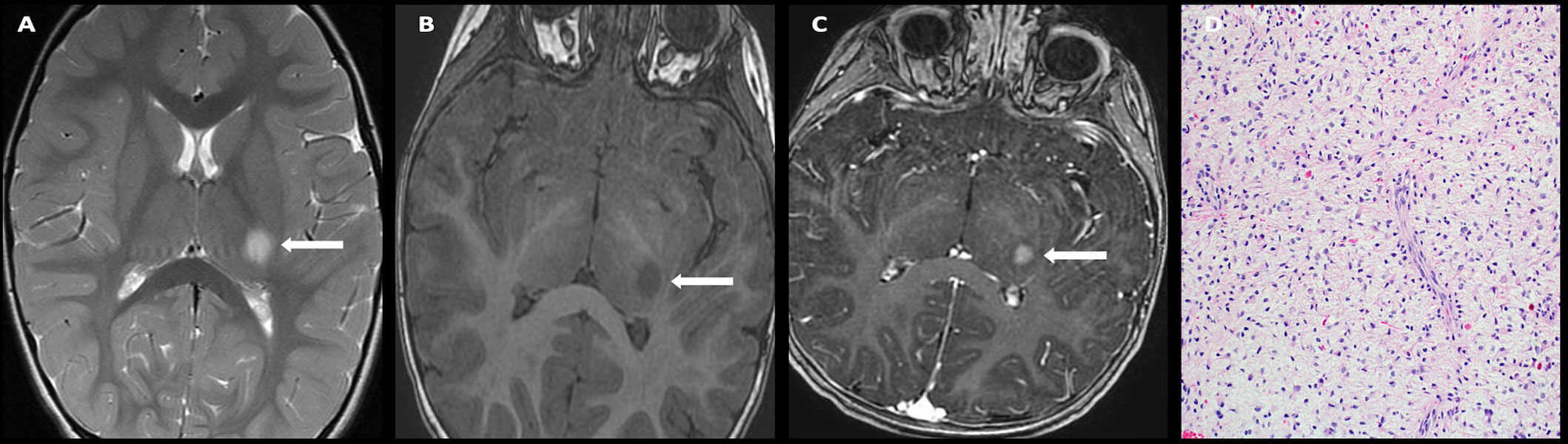
Figure 7 Obtained from a patient at St. Jude Children’s Research Hospital, Memphis, Tennessee. Axial T2 sequence (A) demonstrating a T2 hyperintense, circumscribed mass involving the left thalamus without significant mass effect. Axial pre-contrast T1-weighted (B) and post-contrast T1-weighted (C) images demonstrate a T1 hypointense, avidly enhancing mass. The histologic section (D) shows a piloid glial neoplasm with low levels of mild activity. Many eosinophilic Rosenthal fibers are present in the background. Prominent endothelial cells are a common finding in pilocytic astrocytomas.
4.2.3 Histopathology and molecular markers
Pilocytic astrocytomas have low to moderate cellularity and include varying portions of piloid and oligodendrocyte-type cells. Multinucleated cells with horseshoe-shaped nuclear clusters (pennies-on-a-plate pattern) are often seen. Rosenthal fibers and eosinophilic granular bodies are common but vary in prominence. The most frequent molecular aberration is a chromosome 7q34 rearrangement resulting in a KIAA1549:BRAF fusion. Other aberrations include alternative BRAF fusions, BRAF mutations (especially p.V600E), and NF1 and FGFR1 mutations (4). Compared to the cerebellar pilocytic astrocytomas, the KIAA1549:BRAF fusion is less common in thalamic pilocytic astrocytomas.
4.2.4 Management
Gross total resection is the treatment modality of choice and can often be achieved with preoperative diffusion tensor imaging to plan a corridor for approach, neuronavigation, tubular retractors to minimize adjacent white matter tract injury, and an exoscope for visualization (84). In cases where gross total resection cannot be achieved, targeted MEK inhibitor therapy is often employed, as most tumors harbor alterations in the MAPK genes (85).
4.3 Diffuse midline glioma, H3K27-altered
Diffuse midline H3K27-altered gliomas are WHO grade 4 pediatric high-grade gliomas that involve the brain stem, thalamus, and/or spinal cord. Thalamic involvement may be unilateral or bilateral; the unilateral pattern is more frequently seen in adolescents and young adults, and the bilateral pattern is more frequently seen in children. Thalamic diffuse midline gliomas represent 1%–5% of all pediatric brain tumors and as much as 25% of all pediatric thalamic tumors. The median age at presentation is 7–8 years with no clear sex predilection (86).
4.3.1 Clinical symptoms
Common presentations of thalamic DMGs include symptoms related to intracranial hypertension and motor or sensory or mixed motor and sensory deficits (87).
4.3.2 Imaging
Five distinct thalamic locations were observed in patients with thalamic DMGs enrolled in HERBY trial: thalamo-pulvinar, whole thalamus, uni-thalamic with diffuse spreads, anteromedial, and bithalamic in decreasing order of frequency (88). These tumors can be either well defined or infiltrative. They are usually T1 hypointense or isointense and T2 hyperintense with or without restricted diffusion (Figure 8). Peritumoral edema is characteristically absent in DMGs in contrast to other high-grade gliomas (89). Post-contrast enhancement is variable, with many tumors demonstrating little to no enhancement (88, 90). Radiographic necrosis can be present in up to 64% of tumors (88).
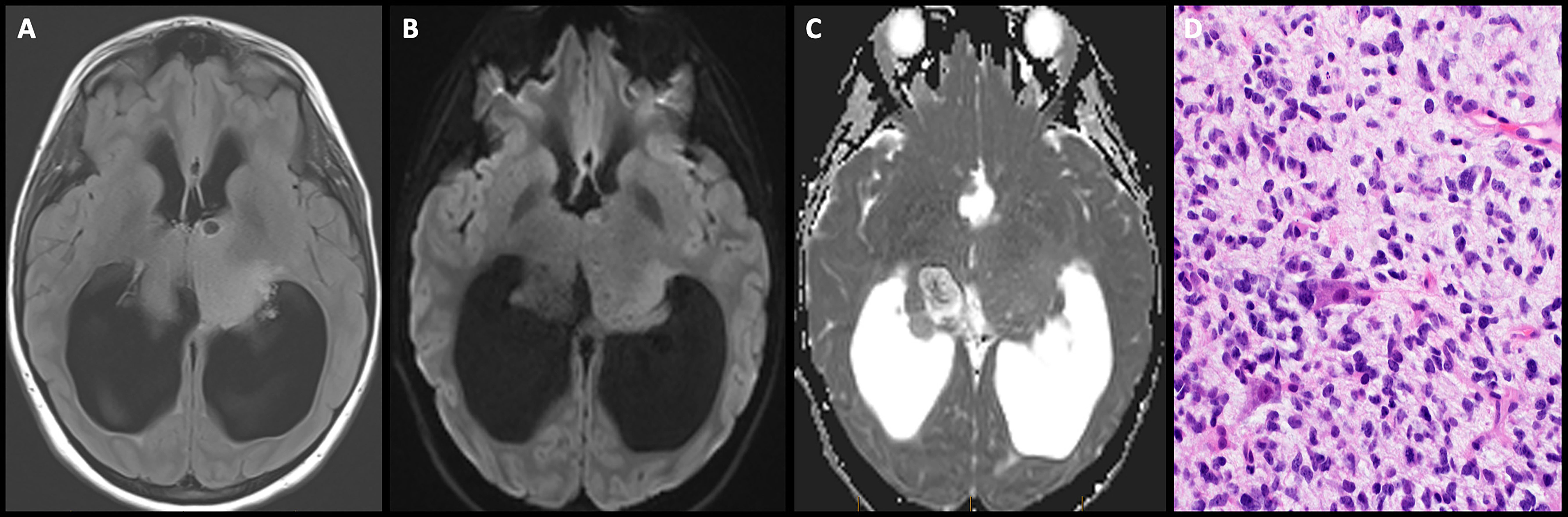
Figure 8 Obtained from a patient at St. Jude Children’s Research Hospital, Memphis, Tennessee. Axial FLAIR image (A) demonstrating a T2/FLAIR hyperintense infiltrative mass involving bilateral thalami with mass effect on the third ventricle and severe obstructive hydrocephalus. Axial diffusion-weighted (B) and apparent diffusion-coefficient maps (C) demonstrate no restricted diffusion. The histologic section (D) shows an infiltrative neoplasm with hyperchromatic nuclei and moderate nuclear pleomorphism. There are two large entrapped neurons.
4.3.3 Histopathology and molecular markers
Histopathology of these tumors demonstrates diffuse infiltration of the CNS parenchyma, without perivascular or perineuronal tropism. Most cells are small and monomorphic, but they can be polymorphous, showing astrocytic, piloid, oligodendroglial, giant cell, undifferentiated, or epithelioid cytology (4). The most common molecular abnormality of thalamic DMGs is an alteration in the 27th amino acid (lysine) on one of the Histone 3 (H3) isoforms, H3.1 or H3.2 or H3.3, and is present in up to 67% of thalamic DMGs (88). Other common molecular aberrations include H3-wild type with EZHIP overexpression or EGFR-mutant. The H3.1 or H3.2 K27-mutants are canonical sequence mutations, while H3.3 K-27 mutants are noncanonical in nature, both resulting in a widespread loss of H3 p. K28me3. In tumors with H3-wild-type molecular features, there is an overexpression of EZHIP, which acts as an endogenous H3 p.K28M mimic. EGFR-mutant diffuse midline glioma is characterized by abnormalities in the EGFR oncogene on chromosome band 7p11.2 (91).
4.3.4 Management
Due to the unfavorable location, surgical resection is often not an option. Surgical biopsy is usually performed under stereotactic guidance for accurate molecular characterization. After diagnosis is confirmed, these tumors are treated with radiation and adjuvant chemotherapy. The prognosis of DMGs is poor, with a 2-year survival rate less than 10% (89).
4.4 Gangliogliomas
Gangliogliomas of the thalamus are uncommon in children; only a few cases have been reported in the literature (92, 93). These tumors are usually slow growing and most commonly arise during the first two decades of life with a slight male predilection (92).
4.4.1 Clinical symptoms
Patients may present with sensory and/or motor deficits contralateral to the side of involvement (93).
4.4.2 Imaging
Gangliogliomas demonstrate a mixed solid and cystic appearance, with variable post-contrast enhancement of the solid component. Calcifications may be present. At present, there are no imaging features associated with morphological features or tumor genotype (94).
4.4.3 Histopathology and molecular markers
Histologically, glioneuronal tumors are biphasic, i.e., they consist of a variable mixture of neuronal and glial elements. The neuronal element consists of dysmorphic ganglion cells with abnormal clustering, a lack of cytoarchitectural organization, cytomegaly, perimembranous aggregation of Nissl substance, or binucleated forms. The glial element resembles a fibrillary astrocytoma, oligodendroglioma, or pilocytic astrocytoma. The most frequent molecular aberration is a BRAF p.V600E mutation, which is seen in 10%–60% of cases and activates the MAPK signaling pathway, which drives cellular proliferation. Gangliogliomas in the thalamus may exhibit concurrent BRAF p.V600E mutations and H3P.K28M mutations and are associated with an increased risk of local recurrence and lower overall survival (4).
4.4.4 Management
Gross total resection is the treatment of choice, with targeted therapy with either MEK inhibitors (in cases of BRAF duplications or fusions) or BRAF inhibitors (in cases of BRAFV600E mutations) reserved for patients with subtotal resection. Radiation is also a viable treatment option in patients with subtotal resection. Surgical resection may be performed via a transtemporal, subtemporal, anterior interhemispheric transcallosal or posterior interhemispheric transtentorial approach (95).
4.5 Supratentorial ependymomas
Supratentorial ependymomas are glial tumors that arise from the primitive ependymal cells that line the ventricles. These tumors are located within the brain parenchyma and may be due to trapping of embryonic ependymal cells during the development of the cerebral hemispheres (4). In the pediatric population, supratentorial ependymomas comprise approximately 3% of all thalamic neoplasms and harbor either the ZFTA (C11orf95) or the YAP1 fusion genes (4, 96). ZFTA fusion-positive ependymomas are more common, accounting for 66%–84% of all pediatric supratentorial ependymomas, with no specific gender predominance (96, 97). On the other hand, YAP1 fusion-positive ependymomas account for only 6%–7.4% of all pediatric supratentorial ependymomas and are restricted to young children, with a male:female ratio of 0.3:1 (98).
4.5.1 Clinical symptoms
Clinically, patients usually present with focal motor deficits, often accompanied by symptoms of elevated intracranial pressure such as headache and blurry vision. Altered sensorium may occur and is usually seen in the context of obstructive hydrocephalus. A few patients may present with an acute onset headache due to intratumoral and intraventricular hemorrhage (99).
Imaging: On imaging, both ZFTA and YAP1 fusion-positive ependymomas have circumscribed margins, with solid and cystic components (99). The solid components often restrict diffusion, particularly in the ZFTA fusion-positive subtype, and demonstrate heterogeneous post-contrast enhancement. Intratumoral hemorrhage and peritumoral edema are more common in the ZFTA fusion-positive subtype (100).
4.5.2 Histopathology and molecular markers
Histologically, supratentorial ependymomas may be WHO grade II or III tumors that demonstrate pseudorosettes and ependymal rosettes and comprise uniform small cells with round nuclei embedded in a fibrillary matrix. Both ZFTA and YAP1 fusion-positive ependymomas are demarcated from adjacent brain, with small- to medium-sized round nuclei. True ependymal rosettes may be encountered in the YAP1 fusion-positive subtype but are relatively rare in the ZFTA fusion-positive subtype. Molecularly, the ZFTA fusion-positive subtype is characterized by the fusion of the ZFTA gene with partner genes, mainly RELA, resulting in a pathological activation of NF-κB signaling. Homozygous deletions of CDKN2A indicate a disruption of cell cycle control in a subset of these tumors. In YAP1 fusion-positive ependymomas, fusions of YAP1 with MAMLD1 or other partner genes are the principal oncogenic drivers of the disease through the recruitment of nuclear factor I (NFI) and TEA domain (TEAD) family domains (4).
4.5.3 Management
Maximal safe surgical resection followed by focal radiation forms the mainstay of treatment for supratentorial ependymomas for patients over 12 months of age. The benefit of maintenance chemotherapy following radiation in this age group has been investigated by two phase III randomized clinical trials. The Children’s Oncology Group ACNS0831 trial evaluated the benefit of a 4-week maintenance chemotherapy regimen with vincristine, cisplatin, cyclophosphamide, and etoposide in newly diagnosed ependymoma patients over 12 months of age following maximal safe surgical resection and focal radiation. Patients were randomized to receive either focal radiation alone or radiation and maintenance chemotherapy post-resection. Their results at a median follow-up of 42.6 months suggested that patients who received any maintenance chemotherapy had a higher 3-year event-free survival as compared to those who did not receive any chemotherapy (p < 0.05). However, noncompliance rates were high in both the radiation and radiation + maintenance chemotherapy groups, and genomic testing is ongoing to determine which genetic subtypes may benefit the most from post-radiation maintenance chemotherapy (101). The International Society of Pediatric Oncology (SIOP) ependymoma II protocol is currently investigating the potential benefit of maintenance chemotherapy on stratum 1 in children over the age of 12 months treated with gross total resection and focal radiation by randomizing them to observation alone versus 16 cycles of maintenance chemotherapy with vincristine, etoposide, cyclophosphamide, and cisplatin. Stratum 2 of the protocol randomizes children over the age of 12 months of age with residual disease on imaging to induction chemotherapy with either vincristine, etoposide, and cyclophosphamide alone or with additional high-dose methotrexate. For patients with residual non-resectable disease following induction chemotherapy, patients will be further randomized to receive either focal radiation alone or with an additional 8 Gy boost to the tumor site (102).
In children below 12 months of age, chemotherapy is an integral component of adjuvant therapy, and radiation is typically delayed until the second year of life. Results from the SJYC07 trial demonstrated that four cycles of postoperative systemic chemotherapy followed by consolidative conformal focal radiation and 6 months of oral maintenance chemotherapy resulted in 4-year PFS rates of 72.6% ± 7.2% and overall survival rates of 92.6% ± 4.4%. Subtotal resection was associated with an inferior PFS rate at 4 years as compared to the gross total and near total resection groups (103).
5 Tumors of the pineal region
The epithalamus forms the posterior portion of the diencephalon and consists of the pineal gland, the habenular trigone, and the stria medullaris, a bundle of white matter axons connecting the hypothalamus to the habenular trigone. The pineal gland is a neuroendocrine organ involved in regulating the circadian rhythm. It is bounded superiorly by the splenium of the corpus callosum, inferiorly by the quadrigeminal plate cistern, and laterally by the thalamus. Tumors of the pineal gland account for approximately 3%–8% of all brain tumors in the pediatric population and are described in detail below (54).
5.1 Germ cell tumors of the pineal region
The most common pediatric pineal region tumors are germ cell tumors, which account for approximately 50%–75% of all pineal region tumors, with incidence peaks at 10–14 years of age and a clear male predominance for all histologic subtypes. These tumors are also more prevalent in Asian populations than in European or North American populations (54).
5.1.1 Clinical symptoms
Patients with germ cell pineal tumors present with headache due to obstructive hydrocephalus caused by compression of the cerebral aqueduct and/or upward gaze palsy with loss of convergence (Parinaud syndrome), resulting from tumor invasion of the tectal plate. Preserved pupillary accommodation with impaired constriction (Argyll Robertson pupil) is also frequent (54).
5.1.2 Imaging
these tumors are typically isointense to hypointense on T1-weighted imaging, isointense to hyperintense on T2-weighted imaging, with avid heterogeneous enhancement. These tumors usually restrict diffusion and may demonstrate susceptibility artifact secondary to calcification. Magnetic resonance spectroscopy of these tumors demonstrates an elevated choline/creatine ratio and a markedly elevated lipid/lactate peak (104).
5.1.3 Histopathology and molecular markers
The histopathological subtypes and molecular features of these tumors have been elaborated in the section on infundibular/hypothalamic germ cell tumors and are described in Table 3 (4).

Table 3 Histologic subtypes of CNS germ cell tumors (4).
5.1.4 Management
Treatment for pineal germ cell tumors parallels that of germ cell tumors of the infundibulum/suprasellar region. It involves a combination of neoadjuvant chemotherapy, surgical resection, and radiation therapy (58–63). In addition, because patients with pineal tumors often present with obstructive hydrocephalus, CSF-diversion procedures such as endoscopic third ventriculostomy (preferred method) or ventriculoperitoneal shunt placement (less-favored procedure due to the risk of peritoneal seeding) are performed. When CSF-diversion procedures are performed, surgical biopsies can be safely performed simultaneously, if indicated. Occasionally, these procedures can be avoided and a temporary ventriculostomy can be placed if chemotherapy is promptly instituted (104).
5.2 Pineocytoma
Pineocytoma is a well-differentiated WHO grade 1 pineal parenchymal tumor. This is a tumor of the adult but can present at any age group.
5.2.1 Clinical symptoms
Smaller lesions are usually incidentally diagnosed on imaging. Larger lesions can present with hydrocephalus due to obstruction of the cerebral aqueduct.
5.2.2 Imaging
The tumors are well circumscribed and may be uniformly solid or mixed solid and cystic. It is hypodense or isodense on CT and isointense to hypointense on T1-weighted imaging and hyperintense on T2-weighted imaging. The solid components enhance with intravenous contrast.
5.2.3 Histopathology and molecular markers
Pineocytoma is a well-differentiated moderately cellular neoplasm of the pinealocytes with occasional rosette formation. No chromosomal aberration has been associated with this tumor.
5.2.4 Management
Pineocytomas are treated with surgical resection with excellent prognosis if there is gross total resection (105).
5.3 Pineal parenchymal tumor of intermediate differentiation
Pineal parenchymal tumor of intermediate differentiation (PPTID) is a tumor of the pineal gland that demonstrates intermediate characteristics between pineocytoma and pineoblastoma. It is usually a tumor of adults but can present in pediatric patients (106, 107).
5.3.1 Clinical symptoms
Symptoms largely dependent upon size and are similar to the pineocytomas as described above.
5.3.2 Imaging
The imaging appearance is also similar to the pineocytomas. However, this tumor frequently demonstrates diffusion restriction due to hypercellularity. Invasion of the adjacent structure and leptomeningeal metastasis can be present.
5.3.3 Histopathology and molecular markers
This tumor is characterized by moderate to high cellularity and can be either diffuse or lobulated. Small in-frame insertion of KBTBD4 is recurrent and characteristic for PPTID.
5.3.4 Management
Gross total resection is the surgical goal. Usually, this tumor is treated with adjuvant radiation therapy following surgical removal with or without additional adjuvant chemotherapy. Tumor recurrence and metastasis are common (107).
5.4 Pineoblastoma
Pineoblastomas account for approximately 35% of all pineal parenchymal tumors with a median age of presentation of 6 years and a slight female preponderance (108).
5.4.1 Clinical symptoms
Clinically, they present with signs and symptoms due to increased intracranial pressure due to aqueductal obstruction, neuro-ophthalmological dysfunction (Parinaud syndrome), and brain stem or cerebellar dysfunction (109).
5.4.2 Imaging
On imaging, they present as large masses with invasion of the surrounding structures, are hyperdense on CT, isointense on T1-weighted imaging with heterogeneous post-contrast enhancement (Figure 9). Peripheral calcifications are often seen. These tumors restrict diffusion due to hypercellularity. Craniospinal dissemination is observed in 25%–33% of tumors at presentation (110).

Figure 9 Obtained from a patient at St. Jude Children's Research Hospital, Memphis, Tennessee. (A) Axial diffusion-weighted imaging and (B) apparent diffusion coefficient map demonstrating a hypercellular pineal region mass causing obstructive hydrocephalus. (C) Axial pre-contrast and (D) post-contrast T1 images demonstrating avid enhancement within the mass. (E) The histologic section demonstrates a hypercellular embryonal tumor with readily apparent mitotic activity. The tumor cells show nuclear hyperchromasia and an inconspicuous amount of cytoplasm.
5.4.3 Histopathology and molecular markers
On histopathology, pineoblastomas resemble other embryonal tumors of the CNS and are highly cellular with patternless sheets of densely packed small cells, irregular nuclei, and a high nuclear:cytoplasmic ratio. Pineoblastomas consist of distinct molecular subtypes, which impact overall prognosis and are summarized in Table 4 (4). Treatment strategies vary by age, with children younger than 3 years treated with maximal safe surgical resection followed by intensive chemotherapy regimens, including high-dose chemotherapy with autologous stem cell rescue. A younger age is associated with poor 5-year progression-free and overall survival rates, with the largest pooled outcome study of 178 pineoblastoma cases reporting rates of 13.5% ± 5.1% and 16.2% ± 5.3%, respectively. The poorer prognosis in younger children has been linked to unfavorable molecular subtypes, surgical resection challenges, and the omission of radiation therapy (111).
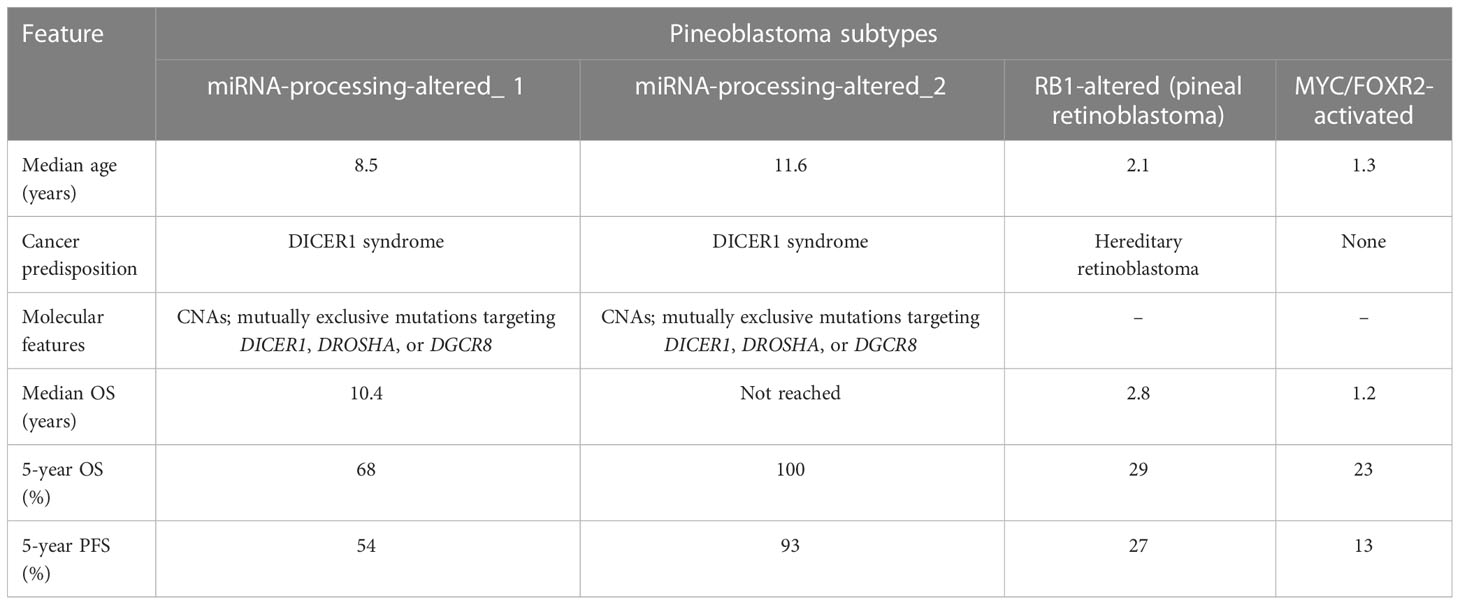
Table 4 Clinical and molecular features of pineoblastoma (4).
Management: Safe surgical resection of pineoblastomas in children <3 years of age is limited by several technical and tumor-related factors. Firstly, neurosurgeons often encounter a difficulty in ideal head positioning in this age group due to suboptimal bone thickness and open sutures. Secondly, the hemorrhagic nature of these tumors coupled with the low blood volume of a failure to thrive infant precludes an aggressive resection. Third, the proximity of the pineal gland to the deep cerebral veins and the midbrain can lead to catastrophic neurologic sequelae in case of injury (112). Recent studies employing focal proton beam radiation in children <3 years with pineoblastoma have demonstrated improved outcomes. On the other hand, children older than 3 years are treated with maximal safe surgical resection, craniospinal irradiation with a focal boost, and intensive alkylator- and platinum-based chemotherapy regimens, with more favorable outcomes (113). Less aggressive molecular subtypes, higher rates of gross total resection, and the addition of radiation therapy are largely responsible for superior progression-free and overall survival rates in this age category. The presence of disseminated disease at presentation is associated with lower 5-year and PFS rates in older children but not in infants (114).
5.5 Papillary tumors of the pineal region
Papillary tumors of the pineal region (PTPR) are neuroepithelial tumors with both solid and papillary components. The reported age range of this tumor is between 1 and 71 years. In the pediatric population, the median age of presentation is approximately 11.6 years, with a slight female predilection (115–118). This entity was first described in 2003 and subsequently enlisted in the WHO classification of CNS tumors in 2007 as Grade 2 or 3 lesions (115).
5.5.1 Clinical symptoms
Clinical presentations are similar to the other pineal parenchymal tumors. Imaging: On imaging, these tumors are heterogeneously enhancing, mixed solid and cystic masses that often obstruct the cerebral aqueduct, causing secondary hydrocephalus. Intrinsic T1 hyperintense proteinaceous secretions are also occasionally seen (115–118).
5.5.2 Histopathology and molecular markers
On histopathology, these tumors demonstrate an epithelium-like appearance with papillary features and more densely cellular areas, often exhibiting ependymal-like differentiation (i.e., true rosettes and tubes). They also often express high levels of genes expressed in the subcommisural organ, including SPDEF, ZFHX4, RFX3, TTR, and CALCA. Recurrent chromosomal imbalances include losses from chromosome 10 and gains on chromosomes 4 and 9. Genetic alterations of PTEN have also been reported that constitutively activate the PI3K/akt/mTOR pathway and apoptotic dysregulation (119).
5.5.3 Management
An effective treatment strategy has not been established for papillary tumors of the pineal region due to the scarcity of cases and the lack of randomized trials. However, gross total resection remains the mainstay of treatment in all age groups, with the largest multicenter study of 31 patients with papillary tumors reporting a 5-year overall survival of 73% in patients treated with gross total resection (120). In cases of subtotal resection, the propensity for tumor recurrence is high, and focal adjuvant radiation therapy with a boost to the tumor bed is recommended. Although currently, there is no definitive dose recommendation for adjuvant radiation therapy, 50 Gy is most frequently used (121, 122). The role of chemotherapy in papillary tumors remains controversial. Some studies have shown adjuvant chemotherapy regimens with carboplatin, vincristine, and temozolomide to be effective, whereas others have deemed them ineffective (123–125). Nevertheless, a systematic review in 2021 concluded that chemotherapy alone, both as a primary modality of treatment and as adjuvant therapy following resection, was not effective in preventing local recurrences in the pediatric population and should be used in conjunction with radiation (116).
6 Conclusions
Diencephalic tumors are complex midline tumors, and pediatric patients with these tumors usually present with symptoms secondary to mass effect on the hypothalamic–pituitary axis, optic nerves, or ventricles. Imaging plays a crucial role in localizing the primary tumor site and, occasionally, the cell of origin (e.g., glial, neuronal, primordial germ cell). Careful analysis of images often obviates the need for surgical biopsy and facilitates prompt institution of therapy. Surgical resection should be performed within maximal safe limits for most tumors, with neoadjuvant chemotherapy playing a key role in the preoperative management of germ cell tumors. Molecular analyses now enable the identification of potential targets in the treatment of certain entities, such as LCH and hypothalamic/optic pathway gliomas, and play a vital role in controlling disease. Although radiation therapy has several side effects, new lower-dose and conformal techniques have enabled higher treatment response rates with lower morbidity.
Author contributions
SP: writing, editing. JC: Image collection, editing. IQ: editing. DL: image collection. AB: Conceptualization. All authors contributed to the article and approved the submitted version.
Funding
JC receives support from the St. Jude Comprehensive Cancer Center (NCI grant P30CA021765), NCI Program Project P01CA096832. AB received supports from FDA grant (FD006368-01A1). This project was also supported, in part, by American Lebanese Syrian Associated Charities (ALSAC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Acknowledgments
We thank Angela J McArthur, PhD, for scientific editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lim Y, Golden JA. Patterning the developing diencephalon. Brain Res Rev (2007) 53(1):17–26. doi: 10.1016/j.brainresrev.2006.06.004
2. Yazici N, Varan A, Akalan N, Soylemezoglu F, Zorlu F, Kutluk T, et al. Diencephalic tumors in children: a 30-year experience of a single institution. Childs Nerv Syst (2011) 27(8):1251–6. doi: 10.1007/s00381-011-1395-z
3. Sharafeddine H, Hamideh D, Morsi RZ, Najjar MW. Surgical techniques in the management of supratentorial pediatric brain tumors: 10 years' experience at a tertiary care center in the middle East. Surg Neurol Int (2021) 12:269. doi: 10.25259/SNI_205_2021
4. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 who classification of tumors of the central nervous system: a summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
5. Schroeder JW, Vezina LG. Pediatric sellar and suprasellar lesions. Pediatr Radiol (2011) 41(3):287–98. doi: 10.1007/s00247-010-1968-0
6. de Blank P, Bandopadhayay P, Haas-Kogan D, Fouladi M, Fangusaro J. Management of pediatric low-grade glioma. Curr Opin Pediatr (2019) 31(1):21–7. doi: 10.1097/MOP.0000000000000717
7. Bag AK, Chiang J, Patay Z. Radiohistogenomics of pediatric low-grade neuroepithelial tumors. Neuroradiology (2021) 63(8):1185–213. doi: 10.1007/s00234-021-02691-1
8. Liu ZM, Liao CH, An X, Zhou WT, Ma ZY, Liu W, et al. The role of imaging features and resection status in the survival outcome of sporadic optic pathway glioma children receiving different adjuvant treatments. Neurosurg Rev (2022) 45(3):2277–87. doi: 10.1007/s10143-022-01743-1
9. Sturm D, Pfister SM, Jones DTW. Pediatric gliomas: current concepts on diagnosis, biology, and clinical management. J Clin Oncol (2017) 35(21):2370–7. doi: 10.1200/JCO.2017.73.0242
10. Packer RJ, Iavarone A, Jones DTW, Blakeley JO, Bouffet E, Fisher MJ, et al. Implications of new understandings of gliomas in children and adults with Nf1: report of a consensus conference. Neuro Oncol (2020) 22(6):773–84. doi: 10.1093/neuonc/noaa036
11. Parsa CF, Hoyt CS, Lesser RL, Weinstein JM, Strother CM, Muci-Mendoza R, et al. Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch Ophthalmol (2001) 119(4):516–29. doi: 10.1001/archopht.119.4.516
12. Wan MJ, Ullrich NJ, Manley PE, Kieran MW, Goumnerova LC, Heidary G. Long-term visual outcomes of optic pathway gliomas in pediatric patients without neurofibromatosis type 1. J Neurooncol (2016) 129(1):173–8. doi: 10.1007/s11060-016-2163-4
13. Rodriguez FJ, Ligon AH, Horkayne-Szakaly I, Rushing EJ, Ligon KL, Vena N, et al. Braf duplications and mapk pathway activation are frequent in gliomas of the optic nerve proper. J Neuropathol Exp Neurol (2012) 71(9):789–94. doi: 10.1097/NEN.0b013e3182656ef8
14. Collins VP, Jones DT, Giannini C. Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol (2015) 129(6):775–88. doi: 10.1007/s00401-015-1410-7
15. Fangusaro J, Witt O, Hernaiz Driever P, Bag AK, de Blank P, Kadom N, et al. Response assessment in paediatric low-grade glioma: recommendations from the response assessment in pediatric neuro-oncology (Rapno) working group. Lancet Oncol (2020) 21(6):e305–e16. doi: 10.1016/S1470-2045(20)30064-4
16. Goodden J, Pizer B, Pettorini B, Williams D, Blair J, Didi M, et al. The role of surgery in optic Pathway/Hypothalamic gliomas in children. J Neurosurg Pediatr (2014) 13(1):1–12. doi: 10.3171/2013.8.PEDS12546
17. Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the children's oncology group. J Clin Oncol (2012) 30(21):2641–7. doi: 10.1200/JCO.2011.36.6054
18. Lassaletta A, Scheinemann K, Zelcer SM, Hukin J, Wilson BA, Jabado N, et al. Phase ii weekly vinblastine for chemotherapy-naive children with progressive low-grade glioma: a Canadian pediatric brain tumor consortium study. J Clin Oncol (2016) 34(29):3537–43. doi: 10.1200/JCO.2016.68.1585
19. Gururangan S, Cavazos CM, Ashley D, Herndon JE 2nd, Bruggers CS, Moghrabi A, et al. Phase ii study of carboplatin in children with progressive low-grade gliomas. J Clin Oncol (2002) 20(13):2951–8. doi: 10.1200/JCO.2002.12.008
20. Gururangan S, Fangusaro J, Poussaint TY, McLendon RE, Onar-Thomas A, Wu S, et al. Efficacy of bevacizumab plus irinotecan in children with recurrent low-grade gliomas–a pediatric brain tumor consortium study. Neuro Oncol (2014) 16(2):310–7. doi: 10.1093/neuonc/not154
21. Roddy E, Mueller S. Late effects of treatment of pediatric central nervous system tumors. J Child Neurol (2016) 31(2):237–54. doi: 10.1177/0883073815587944
22. Bavle A, Jones J, Lin FY, Malphrus A, Adesina A, Su J. Dramatic clinical and radiographic response to braf inhibition in a patient with progressive disseminated optic pathway glioma refractory to mek inhibition. Pediatr Hematol Oncol (2017) 34(4):254–9. doi: 10.1080/08880018.2017.1360971
23. Mustansir F, Mushtaq N, Darbar A. Dabrafenib in Brafv600e mutant pilocytic astrocytoma in a pediatric patient. Childs Nerv Syst (2020) 36(1):203–7. doi: 10.1007/s00381-019-04346-2
24. Leeds NE, Lang FF, Ribalta T, Sawaya R, Fuller GN. Origin of chordoid glioma of the third ventricle. Arch Pathol Lab Med (2006) 130(4):460–4. doi: 10.5858/2006-130-460-OOCGOT
25. Castellano-Sanchez AA, Schemankewitz E, Mazewski C, Brat DJ. Pediatric chordoid glioma with chondroid metaplasia. Pediatr Dev Pathol (2001) 4(6):564–7. doi: 10.1007/s10024001-0087-1
26. Jain D, Sharma MC, Sarkar C, Suri V, Rishi A, Garg A, et al. Chordoid glioma: report of two rare examples with unusual features. Acta Neurochir (Wien) (2008) 150(3):295–300. doi: 10.1007/s00701-008-1420-x
27. Ampie L, Choy W, Lamano JB, Kesavabhotla K, Mao Q, Parsa AT, et al. Prognostic factors for recurrence and complications in the surgical management of primary chordoid gliomas: a systematic review of literature. Clin Neurol Neurosurg (2015) 138:129–36. doi: 10.1016/j.clineuro.2015.08.011
28. Cirak B, Unal O, Arslan H, Cinal A. Chiasmatic glioblastoma of childhood. a case report. Acta Radiol (2000) 41(4):375–6. doi: 10.1080/028418500127345505
29. Cho M, Joo JD, Kim BH, Choe G, Kim CY. Hypothalamic extraventricular neurocytoma (Evn) in a pediatric patient: a case of Evn treated with subtotal removal followed by adjuvant radiotherapy. Brain Tumor Res Treat (2016) 4(1):35–9. doi: 10.14791/btrt.2016.4.1.35
30. Martinez-Barbera JP. Molecular and cellular pathogenesis of adamantinomatous craniopharyngioma. Neuropathol Appl Neurobiol (2015) 41(6):721–32. doi: 10.1111/nan.12226
32. Müller HL. Disease and treatment-related hypothalamic alterations in craniopharyngioma: clinical presentation, prognostic impact, and implications for treatment strategies. In: Jouanneau E, Raverot G, editors. Adult craniopharyngiomas: differences and lessons from paediatrics. Cham: Springer International Publishing (2020). p. 157–86.
33. Muller HL. The diagnosis and treatment of craniopharyngioma. Neuroendocrinology (2020) 110(9-10):753–66. doi: 10.1159/000504512
34. Santagata S K-DB, Komori T, Müller HL, Pietsch T. Adamantinomatous craniopharyngiomas. In: Who classifcation of tumours: central nervous system tumours, 5th ed. Lyon (France: IARC (2021). p. 393–6. Board TWCoTR.
35. Hong CS, Omay SB. The role of surgical approaches in the multi-modal management of adult craniopharyngiomas. Curr Oncol (2022) 29(3):1408–21. doi: 10.3390/curroncol29030118
36. Soldozy S, Yeghyayan M, Yagmurlu K, Norat P, Taylor DG, Kalani MYS, et al. Endoscopic endonasal surgery outcomes for pediatric craniopharyngioma: a systematic review. Neurosurg Focus (2020) 48(1):E6. doi: 10.3171/2019.10.FOCUS19728
37. Akinduro OO, Izzo A, Lu VM, Ricciardi L, Trifiletti D, Peterson JL, et al. Endocrine and visual outcomes following gross total resection and subtotal resection of adult craniopharyngioma: systematic review and meta-analysis. World Neurosurg (2019) 127:e656–e68. doi: 10.1016/j.wneu.2019.03.239
38. Wang G, Zhang X, Feng M, Guo F. Comparing survival outcomes of gross total resection and subtotal resection with radiotherapy for craniopharyngioma: a meta-analysis. J Surg Res (2018) 226:131–9. doi: 10.1016/j.jss.2018.01.029
39. Molitch ME. Diagnosis and treatment of pituitary adenomas: a review. JAMA (2017) 317(5):516–24. doi: 10.1001/jama.2016.19699
40. Penn DL, Burke WT, Laws ER. Management of non-functioning pituitary adenomas: surgery. Pituitary (2018) 21(2):145–53. doi: 10.1007/s11102-017-0854-2
41. Bonneville JF, Bonneville F, Cattin F. Magnetic resonance imaging of pituitary adenomas. Eur Radiol (2005) 15(3):543–8. doi: 10.1007/s00330-004-2531-x
42. de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, et al. Pituitary blastoma: a pathognomonic feature of germ-line Dicer1 mutations. Acta Neuropathol (2014) 128(1):111–22. doi: 10.1007/s00401-014-1285-z
43. Scheithauer BW, Horvath E, Abel TW, Robital Y, Park SH, Osamura RY, et al. Pituitary blastoma: a unique embryonal tumor. Pituitary (2012) 15(3):365–73. doi: 10.1007/s11102-011-0328-x
44. Shih RY, Schroeder JW, Koeller KK. Primary tumors of the pituitary gland: radiologic-pathologic correlation. Radiographics (2021) 41(7):2029–46. doi: 10.1148/rg.2021200203
45. Guillerman RP, Foulkes WD, Priest JR. Imaging of Dicer1 syndrome. Pediatr Radiol (2019) 49(11):1488–505. doi: 10.1007/s00247-019-04429-x
46. Liu APY, Kelsey MM, Sabbaghian N, Park SH, Deal CL, Esbenshade AJ, et al. Clinical outcomes and complications of pituitary blastoma. J Clin Endocrinol Metab (2021) 106(2):351–63. doi: 10.1210/clinem/dgaa857
47. Kobalka PJ, Huntoon K, Becker AP. Neuropathology of pituitary adenomas and sellar lesions. Neurosurgery (2021) 88(5):900–18. doi: 10.1093/neuros/nyaa548
48. Chong AS, Han H, Albrecht S, Weon YC, Park SK, Foulkes WD. Dicer1 syndrome in a young adult with pituitary blastoma. Acta Neuropathol (2021) 142(6):1071–6. doi: 10.1007/s00401-021-02378-0
49. Nadaf J, de Kock L, Chong AS, Korbonits M, Thorner P, Benlimame N, et al. Molecular characterization of Dicer1-mutated pituitary blastoma. Acta Neuropathol (2021) 141(6):929–44. doi: 10.1007/s00401-021-02283-6
50. Demaerel P, Van Gool S. Paediatric neuroradiological aspects of langerhans cell histiocytosis. Neuroradiology (2008) 50(1):85–92. doi: 10.1007/s00234-007-0323-0
51. Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood (2020) 135(16):1319–31. doi: 10.1182/blood.2019000934
52. Jouenne F, Benattia A, Tazi A. Mitogen-activating protein kinase pathway alterations in langerhans cell histiocytosis. Curr Opin Oncol (2021) 33(2):101–9. doi: 10.1097/CCO.0000000000000707
53. Abla O, Weitzman S. Treatment of langerhans cell histiocytosis: role of Braf/Mapk inhibition. Hematol Am Soc Hematol Educ Program (2015) 2015:565–70. doi: 10.1182/asheducation-2015.1.565
54. Gittleman H, Cioffi G, Vecchione-Koval T, Ostrom QT, Kruchko C, Osorio DS, et al. Descriptive epidemiology of germ cell tumors of the central nervous system diagnosed in the United States from 2006 to 2015. J Neurooncol (2019) 143(2):251–60. doi: 10.1007/s11060-019-03173-4
55. Takami H, Graffeo CS, Perry A, Giannini C, Daniels DJ. Epidemiology, natural history, and optimal management of neurohypophyseal germ cell tumors. J Neurosurg (2020) 134(2):1–9. doi: 10.3171/2019.10.JNS191136
56. Juliano J, Melamed E, Christian E, Tamrazi B, Krieger MD. Imaging features predictive of recurrence in pediatric intracranial germ-cell tumors. Pediatr Neurosurg (2019) 54(3):173–80. doi: 10.1159/000493194
57. Allen J, Chacko J, Donahue B, Dhall G, Kretschmar C, Jakacki R, et al. Diagnostic sensitivity of serum and lumbar csf bhcg in newly diagnosed cns germinoma. Pediatr Blood Cancer (2012) 59(7):1180–2. doi: 10.1002/pbc.24097
58. Bartels U, Onar-Thomas A, Patel SK, Shaw D, Fangusaro J, Dhall G, et al. Phase ii trial of response-based radiation therapy for patients with localized germinoma: a children's oncology group study. Neuro Oncol (2022) 24(6):974–83. doi: 10.1093/neuonc/noab270
59. Calaminus G, Bison B, Faure-Conter C, Frappaz D, Peyrl A, Gerber N, et al. Gct-76. 24gy whole ventricular radiotherapy alone is sufficient for disease control in localised germinoma in cr after initial chemotherapy – early results of the siop cns gct ii study. Neuro-Oncology (2020) 22(Supplement_3):iii343–iii4. doi: 10.1093/neuonc/noaa222.292
60. Calaminus G, Frappaz D, Kortmann RD, Krefeld B, Saran F, Pietsch T, et al. Outcome of patients with intracranial non-germinomatous germ cell tumors-lessons from the siop-Cns-Gct-96 trial. Neuro Oncol (2017) 19(12):1661–72. doi: 10.1093/neuonc/nox122
61. Goldman S, Bouffet E, Fisher PG, Allen JC, Robertson PL, Chuba PJ, et al. Phase ii trial assessing the ability of neoadjuvant chemotherapy with or without second-look surgery to eliminate measurable disease for nongerminomatous germ cell tumors: a children's oncology group study. J Clin Oncol (2015) 33(22):2464–71. doi: 10.1200/JCO.2014.59.5132
62. Fangusaro J, Wu S, MacDonald S, Murphy E, Shaw D, Bartels U, et al. Phase ii trial of response-based radiation therapy for patients with localized cns nongerminomatous germ cell tumors: a children's oncology group study. J Clin Oncol (2019) 37(34):3283–90. doi: 10.1200/JCO.19.00701
63. Bajin IY, Foo JC, Bouffet E, Ertl-Wagner B, Tsang D, Laperriere N, et al. Gct-22. outcomes of children with localized and metastatic germinoma treated with chemotherapy followed by radiation therapy without primary tumor boost. Neuro-Oncology (2022) 24(Supplement_1):i59–i. doi: 10.1093/neuonc/noac079.216
64. Yeo KK, Nagabushan S, Dhall G, Abdelbaki MS. Primary central nervous system germ cell tumors in children and young adults: a review of controversies in diagnostic and treatment approach. Neoplasia (2023) 36:100860. doi: 10.1016/j.neo.2022.100860
65. Tian Y, Yue S, Jia G, Zhang Y. Childhood giant pituicytoma: a report and review of the literature. Clin Neurol Neurosurg (2013) 115(10):1943–50. doi: 10.1016/j.clineuro.2013.07.032
66. Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 who classification of pituitary tumors. Endocr Pathol (2022) 33(1):6–26. doi: 10.1007/s12022-022-09703-7
67. Cambiaso P, Amodio D, Procaccini E, Longo D, Galassi S, Camassei FD, et al. Pituicytoma and cushing's disease in a 7-Year-Old girl: a mere coincidence? Pediatrics (2015) 136(6):e1632–6. doi: 10.1542/peds.2015-0638
68. Yilmaz O, Turan A, Yigit H, Duymus M, Kosar U. Case of pituicytoma in childhood. Childs Nerv Syst (2012) 28(1):11–2. doi: 10.1007/s00381-011-1634-3
69. Chakraborti S, Mahadevan A, Govindan A, Sridhar K, Mohan NV, Satish IR, et al. Pituicytoma: report of three cases with review of literature. Pathol Res Pract (2013) 209(1):52–8. doi: 10.1016/j.prp.2012.10.006
70. Gibbs WN, Monuki ES, Linskey ME, Hasso AN. Pituicytoma: diagnostic features on selective carotid angiography and Mr imaging. AJNR Am J Neuroradiol (2006) 27(8):1639–42.
71. McCrea HJ, George E, Settler A, Schwartz TH, Greenfield JP. Pediatric suprasellar tumors. J Child Neurol (2016) 31(12):1367–76. doi: 10.1177/0883073815620671
72. Puchner MJ, Ludecke DK, Saeger W, Riedel M, Asa SL. Gangliocytomas of the sellar region–a review. Exp Clin Endocrinol Diabetes (1995) 103(3):129–49. doi: 10.1055/s-0029-1211342
73. Huang BY, Castillo M. Nonadenomatous tumors of the pituitary and sella turcica. Top Magn Reson Imaging (2005) 16(4):289–99. doi: 10.1097/01.rmr.0000224685.83629.18
74. Zhao X, Li M, Zhang G, Ren X, Yu S, Jiang H, et al. Extraventricular neurocytoma at the sellar region: report of 8 cases and literature review. J Clin Neurosci (2022) 99:379–86. doi: 10.1016/j.jocn.2022.03.038
75. Yang GF, Wu SY, Zhang LJ, Lu GM, Tian W, Shah K. Imaging findings of extraventricular neurocytoma: report of 3 cases and review of the literature. AJNR Am J Neuroradiol (2009) 30(3):581–5. doi: 10.3174/ajnr.A1279
76. Warsi NM, Yan H, Suresh H, Wong SM, Arski ON, Gorodetsky C, et al. The anterior and centromedian thalamus: anatomy, function, and dysfunction in epilepsy. Epilepsy Res (2022) 182:106913. doi: 10.1016/j.eplepsyres.2022.106913
77. Massey LA, Yousry TA. Anatomy of the substantia nigra and subthalamic nucleus on Mr imaging. Neuroimaging Clin N Am (2010) 20(1):7–27. doi: 10.1016/j.nic.2009.10.001
78. Puget S, Crimmins DW, Garnett MR, Grill J, Oliveira R, Boddaert N, et al. Thalamic tumors in children: a reappraisal. J Neurosurg (2007) 106(5 Suppl):354–62. doi: 10.3171/ped.2007.106.5.354
79. Gupta A, Shaller N, McFadden KA. Pediatric thalamic gliomas: an updated review. Arch Pathol Lab Med (2017) 141(10):1316–23. doi: 10.5858/arpa.2017-0249-RA
80. Kalelioglu T, Rama B, Cho BB, Lopes BM, Patel SH. Pediatric-type diffuse low-grade glioma with Myb/Mybl1 alteration: report of 2 cases. Neuroradiol J (2022) 19714009221126015:232–5. doi: 10.1177/19714009221126015
81. Chiang J, Harreld JH, Tinkle CL, Moreira DC, Li X, Acharya S, et al. A single-center study of the clinicopathologic correlates of gliomas with a myb or Mybl1 alteration. Acta Neuropathol (2019) 138(6):1091–2. doi: 10.1007/s00401-019-02081-1
82. Ryall S, Tabori U, Hawkins C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol Commun (2020) 8(1):30. doi: 10.1186/s40478-020-00902-z
83. Chourmouzi D, Papadopoulou E, Konstantinidis M, Syrris V, Kouskouras K, Haritanti A, et al. Manifestations of pilocytic astrocytoma: a pictorial review. Insights Imaging (2014) 5(3):387–402. doi: 10.1007/s13244-014-0328-2
84. Weiner HL, Placantonakis DG. Resection of a pediatric thalamic juvenile pilocytic astrocytoma with whole brain tractography. Cureus (2017) 9(10):e1768. doi: 10.7759/cureus.1768
85. Miller C, Moertel C, Guillaume D. Ptps-21mapk pathway inhibitor therapy for progressive pediatric astrocytomas. Neuro-Oncology (2015) 17(suppl_5):v183–v. doi: 10.1093/neuonc/nov228.21
86. Vuong HG, Le HT, Jea A, McNall-Knapp R, Dunn IF. Risk stratification of H3 K27m-mutant diffuse midline gliomas based on anatomical locations: an integrated systematic review of individual participant data. J Neurosurg Pediatr (2022) 30(1):1–8. doi: 10.3171/2022.3.PEDS2250
87. Wang Y, Feng LL, Ji PG, Liu JH, Guo SC, Zhai YL, et al. Clinical features and molecular markers on diffuse midline gliomas with H3k27m mutations: a 43 cases retrospective cohort study. Front Oncol (2020) 10:602553. doi: 10.3389/fonc.2020.602553
88. Rodriguez D, Calmon R, Aliaga ES, Warren D, Warmuth-Metz M, Jones C, et al. Mri and molecular characterization of pediatric high-grade midline thalamic gliomas: the Herby phase Ii trial. Radiology (2022) 304(1):174–82. doi: 10.1148/radiol.211464
89. Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell (2017) 32(4):520–37.e5. doi: 10.1016/j.ccell.2017.08.017
90. Aboian MS, Solomon DA, Felton E, Mabray MC, Villanueva-Meyer JE, Mueller S, et al. Imaging characteristics of pediatric diffuse midline gliomas with histone H3 K27m mutation. AJNR Am J Neuroradiol (2017) 38(4):795–800. doi: 10.3174/ajnr.A5076
91. Nishio S, Morioka T, Suzuki S, Takeshita I, Fukui M. Thalamic gliomas: a clinicopathologic analysis of 20 cases with reference to patient age. Acta Neurochir (Wien) (1997) 139(4):336–42. doi: 10.1007/BF01808830
92. Martinez-Lage JF, Perez-Espejo MA, Esteban JA, Poza M. Thalamic tumors: clinical presentation. Childs Nerv Syst (2002) 18(8):405–11. doi: 10.1007/s00381-002-0605-0
93. Johnson MD, Jennings MT, Toms ST. Oligodendroglial ganglioglioma with anaplastic features arising from the thalamus. Pediatr Neurosurg (2001) 34(6):301–5. doi: 10.1159/000056042
94. Steinbok P, Gopalakrishnan CV, Hengel AR, Vitali AM, Poskitt K, Hawkins C, et al. Pediatric thalamic tumors in the mri era: a Canadian perspective. Childs Nerv Syst (2016) 32(2):269–80. doi: 10.1007/s00381-015-2968-z
95. Cinalli G, Aguirre DT, Mirone G, Ruggiero C, Cascone D, Quaglietta L, et al. Surgical treatment of thalamic tumors in children. J Neurosurg Pediatr (2018) 21(3):247–57. doi: 10.3171/2017.7.PEDS16463
96. Zschernack V, Junger ST, Mynarek M, Rutkowski S, Garre ML, Ebinger M, et al. Supratentorial ependymoma in childhood: more than just rela or yap. Acta Neuropathol (2021) 141(3):455–66. doi: 10.1007/s00401-020-02260-5
97. Junger ST, Timmermann B, Pietsch T. Pediatric ependymoma: an overview of a complex disease. Childs Nerv Syst (2021) 37(8):2451–63. doi: 10.1007/s00381-021-05207-7
98. Andreiuolo F, Varlet P, Tauziede-Espariat A, Junger ST, Dorner E, Dreschmann V, et al. Childhood supratentorial ependymomas with Yap1-Mamld1 fusion: an entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol (2019) 29(2):205–16. doi: 10.1111/bpa.12659
99. Renedo D, Ferraro F, Johnson AR, Argañaraz R, Giovannini S, Zabala JP, et al. Thalamic tumors in children: case series from our institution and literature review. Childs Nerv Syst (2021) 37(2):457–63. doi: 10.1007/s00381-020-04830-0
100. Nowak J, Junger ST, Huflage H, Seidel C, Hohm A, Vandergrift LA, et al. Mri phenotype of rela-fused pediatric supratentorial ependymoma. Clin Neuroradiol (2019) 29(4):595–604. doi: 10.1007/s00062-018-0704-2
101. Smith A, Onar-Thomas A, Ellison D, Owens-Pickle E, Wu S, Leary SE, et al. EPEN-54. ACNS0831, phase III randomized trial of post-radiation chemotherapy in patients with newly diagnosed ependymoma ages 1 to 21 years. Neuro-oncology (2020) 22(3):318–9. doi: 10.1093/neuonc/noaa222.187
102. Leblond P, Massimino M, English M, Ritzmann TA, Gandola L, Calaminus G, et al. Toward improved diagnosis accuracy and treatment of children, adolescents, and young adults with ependymoma: the international SIOP ependymoma II protocol. Front Neurol (2022) 13:887544. doi: 10.3389/fneur.2022.887544
103. Upadhyaya SA, Robinson GW, Onar-Thomas A, Orr BA, Billups CA, Bowers DC, et al. Molecular grouping and outcomes of young children with newly diagnosed ependymoma treated on the multi-institutional SJYC07 trial. Neuro Oncol (2019) 21(10):1319–30. doi: 10.1093/neuonc/noz069
104. Attri G, Gosal JS, Khatri D, Das KK, Bhaisora KS, Mehrotra A, et al. Endoscopic third ventriculostomy and simultaneous tumor biopsy in pineal region tumors using the "Single burr hole" technique: an analysis of 34 cases. Asian J Neurosurg (2020) 15(4):976–82. doi: 10.4103/ajns.AJNS_194_20
105. Adams JW, Malicki D, Levy M, Crawford JR. Coincident pineocytoma and probable brainstem glioma in a child with 22q11.2 deletion syndrome. BMJ Case Rep (2022) 15(3). doi: 10.1136/bcr-2022-249232
106. Gupte A, Sood S, Kupsky WJ, Altinok D, Miller S, Roy S, et al. Pineal parenchymal tumor of intermediate differentiation and Dicer1 syndrome: a case report. J Pediatr Hematol Oncol (2022) 45(3):e406–9. doi: 10.1097/MPH.0000000000002518
107. Fomchenko EI, Erson-Omay EZ, Kundishora AJ, Hong CS, Daniel AA, Allocco A, et al. Genomic alterations underlying spinal metastases in pediatric H3k27m-mutant pineal parenchymal tumor of intermediate differentiation: case report. J Neurosurg Pediatr (2019) 25(2):1–10. doi: 10.3171/2019.8.PEDS18664
108. Greppin K, Cioffi G, Waite KA, Ostrom QT, Landi D, Takaoka K, et al. Epidemiology of pineoblastoma in the United States, 2000-2017. Neurooncol Pract (2022) 9(2):149–57. doi: 10.1093/nop/npac009
109. Gaillard F, Jones J. Masses of the pineal region: clinical presentation and radiographic features. Postgrad Med J (2010) 86(1020):597–607. doi: 10.1136/pgmj.2009.087460
110. Abdelbaki MS, Abu-Arja MH, Davidson TB, Fangusaro JR, Stanek JR, Dunkel IJ, et al. Pineoblastoma in children less than six years of age: the head start I, ii, and iii experience. Pediatr Blood Cancer (2020) 67(6):e28252. doi: 10.1002/pbc.28252
111. Hansford JR, Huang J, Endersby R, Dodgshun AJ, Li BK, Hwang E, et al. Pediatric pineoblastoma: a pooled outcome study of north American and Australian therapeutic data. Neurooncol Adv (2022) 4(1):vdac056. doi: 10.1093/noajnl/vdac056
112. Jin MC, Prolo LM, Wu A, Azad TD, Shi S, Rodrigues AJ, et al. Patterns of care and age-specific impact of extent of resection and adjuvant radiotherapy in pediatric pineoblastoma. Neurosurgery (2020) 86(5):E426–E35. doi: 10.1093/neuros/nyaa023
113. Mynarek M, Pizer B, Dufour C, van Vuurden D, Garami M, Massimino M, et al. Evaluation of age-dependent treatment strategies for children and young adults with pineoblastoma: analysis of pooled European society for paediatric oncology (Siop-E) and us head start data. Neuro Oncol (2017) 19(4):576–85. doi: 10.1093/neuonc/now234
114. Liu APY, Gudenas B, Lin T, Orr BA, Klimo P Jr., Kumar R, et al. Risk-adapted therapy and biological heterogeneity in pineoblastoma: integrated clinico-pathological analysis from the prospective, multi-center Sjmb03 and Sjyc07 trials. Acta Neuropathol (2020) 139(2):259–71. doi: 10.1007/s00401-019-02106-9
115. Nemir J, Maric LS, Trbojevic T, Zarkovic K, Jadrijevic-Cvrlje F. Papillary tumor of the pineal region in pediatric patient - a case report. Surg Neurol Int (2022) 13:488. doi: 10.25259/SNI_867_2022
116. Mathkour M, Hanna J, Ibrahim N, Scullen T, Kilgore MD, Werner C, et al. Papillary tumor of the pineal region in pediatric populations: an additional case and systematic review of a rare tumor entity. Clin Neurol Neurosurg (2021) 201:106404. doi: 10.1016/j.clineuro.2020.106404
117. Choque-Velasquez J, Colasanti R, Resendiz-Nieves J, Jahromi BR, Tynninen O, Collan J, et al. Papillary tumor of the pineal region in children: presentation of a case and comprehensive literature review. World Neurosurg (2018) 117:144–52. doi: 10.1016/j.wneu.2018.06.020
118. Gupta K, Khursheed S, Nayil K, Khursheed S, Makhdoomi R. Papillary tumor of pineal region in a 5-Year-Old Male child: a rare entity. Asian J Neurosurg (2021) 16(4):824–6. doi: 10.4103/ajns.AJNS_81_19
119. Heim S, Sill M, Jones DT, Vasiljevic A, Jouvet A, Fevre-Montange M, et al. Papillary tumor of the pineal region: a distinct molecular entity. Brain Pathol (2016) 26(2):199–205. doi: 10.1111/bpa.12282
120. Fevre-Montange M, Hasselblatt M, Figarella-Branger D, Chauveinc L, Champier J, Saint-Pierre G, et al. Prognosis and histopathologic features in papillary tumors of the pineal region: a retrospective multicenter study of 31 cases. J Neuropathol Exp Neurol (2006) 65(10):1004–11. doi: 10.1097/01.jnen.0000240462.80263.13
121. Abela L, Rushing EJ, Ares C, Scheer I, Bozinov O, Boltshauser E, et al. Pediatric papillary tumors of the pineal region: to observe or to treat following gross total resection? Childs Nerv Syst (2013) 29(2):307–10. doi: 10.1007/s00381-012-1935-1
122. Nakamura H, Makino K, Kochi M, Nakazato Y, Kuratsu J. Successful treatment of neoadjuvant therapy for papillary tumor of the pineal region. Brain Tumor Pathol (2009) 26(2):73–7. doi: 10.1007/s10014-009-0250-3
123. Lechapt-Zalcman E, Chapon F, Guillamo JS, Khouri S, Menegalli-Boggelli D, Loussouarn D, et al. Long-term clinicopathological observations on a papillary tumour of the pineal region. Neuropathol Appl Neurobiol (2011) 37(4):431–5. doi: 10.1111/j.1365-2990.2010.01133.x
124. Lorenzetti M, Motta F, Campanella R, Bauer D, Assi A, Arienta C, et al. Adjuvant temozolomide chemotherapy for treatment of papillary tumor of the pineal region. World Neurosurg (2011) 76(1-2):160–3. doi: 10.1016/j.wneu.2010.10.039
Keywords: adenoma, craniopharyngioma, diencephalon, germ cell tumor, langerhans cell histiocytosis, pineoblastoma
Citation: Pinto SN, Chiang J, Qaddoumi I, Livingston D and Bag A (2023) Pediatric diencephalic tumors: a constellation of entities and management modalities. Front. Oncol. 13:1180267. doi: 10.3389/fonc.2023.1180267
Received: 05 March 2023; Accepted: 27 June 2023;
Published: 13 July 2023.
Edited by:
Andrea Carai, Bambino Gesù Children’s Hospital (IRCCS), ItalyReviewed by:
Maura Massimino, Fondazione IRCCS Istituto Nazionale Tumori, ItalyAntonella Cacchione, Bambino Gesù Children’s Hospital (IRCCS), Italy
Copyright © 2023 Pinto, Chiang, Qaddoumi, Livingston and Bag. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Soniya N Pinto, c29uaXlhLnBpbnRvQHN0anVkZS5vcmc=