 Daniel Antunes Moreno1
Daniel Antunes Moreno1 Murilo Bonatelli2
Murilo Bonatelli2 Augusto Perazzolo Antoniazzi3Flávia Escremim de Paula2
Augusto Perazzolo Antoniazzi3Flávia Escremim de Paula2 Leticia Ferro Leal1,5
Leticia Ferro Leal1,5 Felipe Antônio de Oliveira Garcia1
Felipe Antônio de Oliveira Garcia1 André Escremim de Paula2Gustavo Ramos Teixeira4,5Iara Viana Vidigal Santana4Fabiano Saggioro6Luciano Neder6
André Escremim de Paula2Gustavo Ramos Teixeira4,5Iara Viana Vidigal Santana4Fabiano Saggioro6Luciano Neder6 Elvis Terci Valera7
Elvis Terci Valera7 Carlos Alberto Scrideli7João Stavale8
Carlos Alberto Scrideli7João Stavale8 Suzana Maria Fleury Malheiros8Matheus Lima9
Suzana Maria Fleury Malheiros8Matheus Lima9 Glaucia Noeli Maroso Hajj9
Glaucia Noeli Maroso Hajj9 Hernan Garcia-Rivello10
Hernan Garcia-Rivello10 Silvia Christiansen10Susana Nunes11Maria João Gil-da-Costa11Jorge Pinheiro12
Silvia Christiansen10Susana Nunes11Maria João Gil-da-Costa11Jorge Pinheiro12 Flavia Delgado Martins13
Flavia Delgado Martins13 Carlos Almeida Junior14Bruna Minniti Mançano15
Carlos Almeida Junior14Bruna Minniti Mançano15 Rui Manuel Reis1,2,16,17*
Rui Manuel Reis1,2,16,17*- 1Molecular Oncology Research Center, Barretos Cancer Hospital, Barretos, Brazil
- 2Molecular Diagnosis Laboratory, Barretos Cancer Hospital, Barretos, Brazil
- 3Cancer Genetics Department, Barretos Cancer Hospital, Barretos, Brazil
- 4Barretos School of Health Sciences Dr. Paulo Prata, Barretos Cancer Hospital, Barretos, Brazil
- 5Pathology Department, Barretos Cancer Hospital, Barretos, Brazil
- 6Department of Pathology and Forensic Medicine, University of São Paulo, Ribeirão Preto, Brazil
- 7Department of Pediatrics of Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto, Brazil
- 8Department of Neurology and Neurosurgery, Federal University of São Paulo (UNIFESP), São Paulo, Brazil
- 9Oncology Department, AC Camargo Hospital, São Paulo, Brazil
- 10Pathology Department, Italian Hospital of Buenos Aires, Buenos Aires, Argentina
- 11Pediatric Oncology Department, Centro Hospitalar Universitário São João, Porto, Portugal
- 12Department of Pathology, Centro Hospitalar Universitário São João, Porto, Portugal
- 13Brasília Children’s Hospital, Brasília, Brazil
- 14Pediatric Neurosurgery Department, Barretos Cancer Hospital, Barretos, Brazil
- 15Pediatric Oncology Department, Barretos Cancer Hospital, Barretos, Brazil
- 16ICVS/3B’s – PT Government Associate Laboratory, Braga/Guimarães, Portugal
- 17Life and Health Sciences Research Institute (ICVS), School of Medicine, University of Minho, Braga, Portugal
Purpose: Medulloblastomas are the most common primary malignant brain tumors in children. They are divided into molecular subgroups: WNT-activated, SHH-Activated, TP53 mutant or wild type, and non-WNT/non-SHH (Groups 3 and 4). WNT-activated medulloblastomas are usually caused by mutations in the CTNNB1 gene (85%–90%), and most remaining cases of CTNNB1 wild type are thought to be caused by germline mutations in APC. So far, the frequencies of CTNNB1 have been reported mainly in North American and European populations. The aim of this study was to report the frequency of CTNNB1 mutations in WNT-activated medulloblastomas in a Latin-Iberian population and correlate with their clinicopathological characteristics.
Methods: A total of 266 medulloblastomas from seven different institutions from Brazil (n=211), Portugal (n=38), and Argentina (n=17) were evaluated. Following RNA and DNA isolation from formalin-fixed, paraffin-embedded (FFPE) tumor tissues, the molecular classification and CTNNB1 mutation analysis were performed by nCounter and Sanger sequencing, respectively.
Results: WNT-activated medulloblastomas accounted for 15% (40/266) of the series. We observed that 73% of WNT-activated medulloblastomas harbored CTNNB1 mutations. CTNNB1 wild-type cases (27%) were more prevalent in female individuals and suggested to be associated with a worse outcome. Among the CTNNB1 wild-type cases, the available analysis of family history revealed two cases with familiar adenomatous polyposis, harboring APC germline variants.
Conclusion: We observed a lower incidence of CTNNB1 mutations in WNT-activated medulloblastomas in our Latin-Iberian cohort compared to frequencies previously described in other populations. Considering that CTNNB1 wild-type cases may exhibit APC germline mutations, our study suggests a higher incidence (~30%) of hereditary WNT-activated medulloblastomas in the Latin-Iberian population.
Introduction
Medulloblastomas are a group of heterogeneous embryonal tumors considered as the most common primary malignant brain tumor in children, with an annual incidence of 0.4 in 100,000 population in children and young adults aged 0–19 years (1). Histologically, medulloblastomas are divided into classic (40%–45%), desmoplastic nodular (30%–35%), anaplastic/large cell (15%–20%), and with extensive nodularity (10%) (2). These tumors are commonly divided into four molecular subgroups: WNT activated, SHH activated, Group 3, and Group 4 (2, 3). In the 2021 WHO classification, the SHH-activated group is subdivided into TP53 wild type and TP53 mutant, and Groups 3 and 4 are merged in a non-WNT/non-SHH subgroup (4). The standard therapy based on surgery, craniospinal irradiation, and chemotherapy may vary according to the molecular subgroup, the patient age, leptomeningeal dissemination status, and the extension of surgical resection (2).
The wingless (WNT)-activated group accounts for 10% of all medulloblastomas, is commonly observed in children older than 4 years, in an equal proportion of boys and girls, and usually shows no metastasis at diagnosis (3). This molecular subgroup is a particular entity with a distinctly better outcome in children, with more than 95% of 5-year overall survival when these children are submitted to standard therapy and displays a distinct molecular pattern of gene expression (5) and methylation profile (6).
The WNT is a conserved pathway that may induce cell proliferation and growth during development via regulation by beta-catenin, which is translocated to the nucleus for binding to transcriptional factors inducing the expression of cyclins and proto-oncogenes (7). In differentiated cells, the WNT pathway is mostly in a non-activated state, in which a disruptive complex comprised APC, AXIN, GSK3, and CK1, enabling beta-catenin phosphorylation, triggering its ubiquitination and degradation (7).
It has been reported that 85%–90% of WNT-activated medulloblastomas harbor hotspot mutations in the CTNNB1 gene, which encodes beta catenin (8). Hotspot mutations are located at the exon 3, which corresponds to the phosphorylation site of the beta-catenin. These mutations in the exon 3 inhibits the phosphorylation of beta-catenin, triggering escape from degradation, resulting in cytoplasmatic accumulation of beta-catenin, which translocates to the nucleus inducing activation of genes involved in cell proliferation (7, 9). Most of the remaining 10%–15% of WNT-activated (CTNNB1 wild type) tumors carry germline APC variants (8, 10). This later condition is observed in Turcot syndrome, when primary brain tumors such as medulloblastomas may co-occur with multiple colorectal adenomas, observed in families with familial adenomatosis polyposis (FAP) (11).
For patients with WNT-activated medulloblastomas CTNNB1 wild type, referral for genetic risk cancer assessment and germline APC sequencing is recommended. It is reported that 70% of these patients will have the FAP diagnosis, triggering prevention measures such as total colectomy for the patient and germline tests to the relatives (8). Identifying these cases, patients with WNT-activated medulloblastomas and CTNNB1 wild type have a high chance of harboring germline mutations in APC and should improve the patient’s treatment by differentiated surveillance and early cancer detection in patients and relatives resulting in more effective therapy (8).
The incidence of the WNT-activated CTNNB1 wild type is currently well established for the North American and European population (8), and little is known about the frequency of these mutations in Brazilian and other Latin-Iberian countries. In the present study, we aimed to analyze the frequency of CTNNB1 mutations in WNT-activated medulloblastomas in a large Latin Iberian medulloblastoma cohort.
Materials and methods
Patient cohorts
In the present retrospective study, we evaluated 266 FFPE medulloblastoma specimens collected between 2001 and 2022 from naive-treated patients from seven different institutions in Brazil, Argentina, and Portugal: Barretos Cancer Hospital, Brazil (n=119); Federal University of São Paulo UNIFESP (n=20), Brazil; AC Camargo Hospital, Brazil (n=15), Ribeirão Preto Medical School, Brazil (n=37); Child Hospital of Brasília, Brazil (n=20); Italian Hospital of Buenos Aires, Argentina (n=17); and Centro Hospitalar Universitário São João, Portugal (n=38). The frequency of molecular subgroups was previously reported in a subset of cohort5,19. The patient’s clinical and molecular features were collected on medical reports and stored in the Research Electronic Data Capture (RedCap). This study was approved by the institutional review board from Barretos Cancer Hospital (CAAE: 59979816.6.1001.5437). Informed consent was obtained from the patients or familiars before APC germline evaluation.
RNA and DNA isolation
The tumor area was previously marked by an experienced pathologist, ensuring the presence of >80% of tumor cells and the absence of microvascular proliferation and necrosis. For CTNNB1 gene analysis, DNA isolation from macrodissected formalin-fixed, paraffin-embedded (FFPE) was performed using the QIAamp DNA Mini Kit (Qiagen, Venlo, The Netherlands), following the manufacturer’s recommendations. For APC gene analysis, DNA was isolated from peripheral blood samples using the QIAamp DNA Blood Mini Kit (Qiagen, Venlo, The Netherlands), following the manufacturer’s instructions. The DNA quantification was performed using the NanoDropVR 2000 (Thermo Fisher Scientific, Waltham, USA) (12). The RNA isolation was performed using the deparaffinization solution (Qiagen, Venlo, The Netherlands) and the RNeasy Mini Kit (Qiagen, Venlo, The Netherlands), and the Qubit 2.0 Fluorometer (RNA HS Assay kit, Life Technologies, Thermo Fisher Scientific, Waltham, USA) was applied for RNA quantification following the manufacturer’s recommendations (13, 14).
Molecular classification by gene expression
Gene expression assays were performed in the nCounter® FLEX Analysis System available in the Molecular Oncology Research Center of Barretos Cancer Hospital (BCH) using the nCounter® Elements custom panel (NanoString Technologies, Seattle, USA). The panel comprises three reference genes (ACTB, GAPDH, and LDHA) and 22 targets for WNT (WIF1, TNC, GAD1, DKK2, and EMX2), SHH (PDLIM3, EYA1, HHIP, ATOH1, and SFRP1), Groups 3 (IMPG2, GABRAS, EGFL11, NRL, MAB21L2, and NPR3), and Group 4 classification (KCNA1, EOMES, KHDRBS2, RBM24, UNC5D, and OAS1) as previously described (15, 16).
CTNNB1 and APC sequencing
The CTNNB1 mutation was evaluated by Sanger sequencing using the following CTNNB1 primers: forward, GCTGATTTGATGGAGTTGGA; reverse, GCTACTTGTTCTTGAGTGAA as reported (17). The PCR reactions were optimized using 7.2 µL of HotStarTaq Master Mix (Qiagen, Hilden, Germany), 5.6 µL of sterile nuclease-free water, 0.6 µL of magnesium chloride (5 mM), 0.3 µL of each forward and reverse primers (10 µM), and 1 µL of DNA. The PCR was performed in the Veriti 96-well thermocycler (Applied Biosystems, model 9902, Singapore) in the following conditions: 40 cycles of denaturation at 96°C for 45 s, annealing at 53°C for 45 s, and extension at 72°C for 45 s.
APC gene (NM_00038.5) mutations were evaluated by NGS (next-generation sequencing) in patients with a family history of colon cancer and/or polyps who have provided consent for germline molecular analysis. Library construction was carried out according to the Barretos Cancer Hospital Hereditary Rare Cancer Solution kit (Sophia Genetics, Switzerland), which includes the genes APC, BRCA2, CEBPA, DICER1, GATA2, SMARCB1, MEN1, NF1, NF2, PALB2, PTCH1, PTEN, RB1, RET, RUNX1, SUFU, TP53, TSC1, TSC2, and VHL, according to the manufacturer’s protocol. Briefly, DNA fragments were generated using an enzymatic fragmentation step. The three subsequent enzymatic steps, end-repair, A-tailing, and ligation to Illumina adapters, were performed in order to produce NGS libraries. A capture-based target enrichment was carried out on the pooled libraries. The quantitation of the final pool of libraries was performed using Qubit dsDNA HS fluorimetric assays (Life Technologies, USA). Quality control of fragment size was assessed using DNA ScreenTape analysis (4150 TapeStation System, Agilent). Sequencing was achieved with the final library concentration of 10 pM onto a 600-cycle format V3 flow cell, via Illumina MiSeq platform (Illumina, San Diego, CA, USA).
Data analysis was performed in order to detect single nucleotide variants (SNVs), insertions/deletions (indels), and copy number alterations (CNAs). Sequencing FASTQ data were analyzed by the Sophia DDM® platform (Sophia Genetics, Switzerland).
The classification of each genomic variant into five different categories, namely, benign (B), likely benign (LB), variant of uncertain significance (VUS), likely pathogenic (LP), and pathogenic (P), were performed according to the American College of Medical Genetics and Genomics (ACMG) guidelines.
In silico analysis of medulloblastomas molecular subgroups and CTNNB1 mutation
To access the literature frequency of molecular subgroups and the CTNNB1 mutation, we downloaded the clinical and molecular information on medulloblastomas from The Cancer Genome Atlas (TCGA) consortium at cBioPortal (https://www.cbioportal.org/). It included five different whole exome and genome datasets from the Pediatric Cancer Genome Project (PCGP) (whole genome, Nature 2012, n=37) (18), the Sickkids (whole genome, Nature 2016, n=46) (19), International Cancer Genome Consortium (ICGC) (whole exome, Nature 2012, n=125) (20), The German Cancer Research Center (Deutsches Krebsforschungszentrum, DKFZ) (whole exome, Nature 2017, n=491) (21), and from the Broad Institute (whole exome, Nature, 2012, n=92). We excluded duplicate registers, alterations (mutations, structural variants, and copy number) of unknown significance, and samples not classified in the molecular subgroups, totalizing 617 patients with both information regarding molecular subgroups and CTNNB1 mutation in the WNT-activated subgroup.
Statistical analysis
The statistical analysis was performed using the software IBM SPSS statistics version 25. The chi-square or ANOVA tests were applied for qualitative variables, and the Mann–Whitney test was applied for quantitative variables, rejecting the null hypothesis with p<0.05. For survival analyses, the Kaplan–Meier method and the log-rank test were applied.
Results
Higher frequency of WNT-activated medulloblastomas with CTNNB1 wild type in Latin-Iberians population
We evaluated 266 Latin-Iberian medulloblastomas from Brazil (n=212), Portugal (n=38), and Argentina (n=16). All cases were molecularly classified into the four main medulloblastomas subgroups, namely, WNT activated (n=40, 15%), SHH activated (n=122, 46%), Group 3 (n=42, 16%), and Group 4 (n=62, 23%) (Figure 1A). The clinicopathological features of WNT-activated medulloblastomas from the Latin-Iberian population (n=40) are outlined in detail in Supplementary Tables 1, 2. Among the 40 WNT-activated, seven cases were inconclusive for CTNNB1 mutations due to the low quantity and quality of DNA for Sanger sequencing. From the remaining 33 WNT-activated cases (24 from Brazil, seven from Portugal, and two from Argentina), we detected CTNNB1 mutation in 24 (73%) cases, and nine (27%) medulloblastomas were CTNNB1 wild type (Figure 1A).
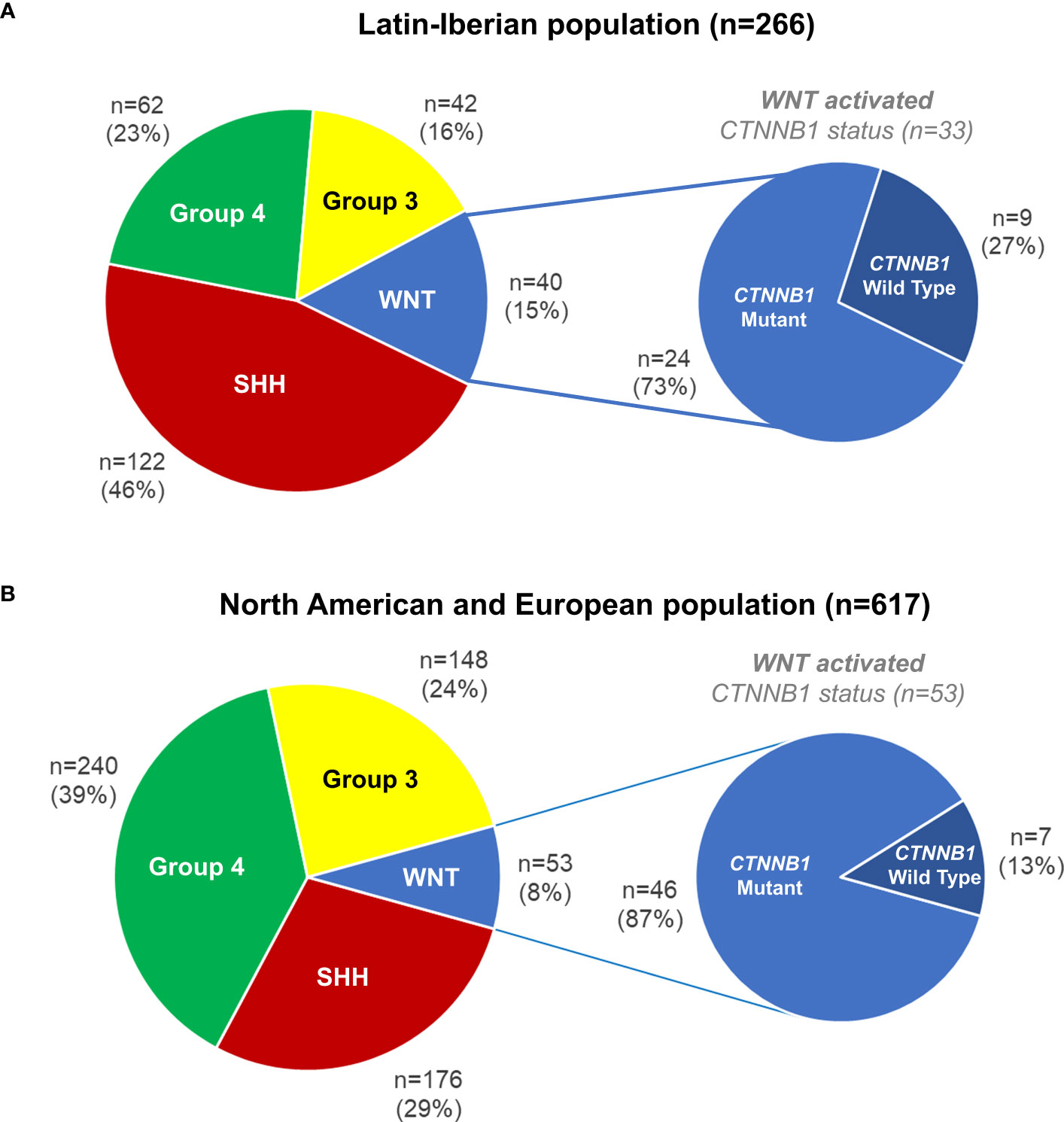
Figure 1 Medulloblastomas molecular subgroups and CTNNB1 mutations in WNT-activated medulloblastomas. (A) Latin-Iberian population (n=266, including patients from Brazil, Portugal, and Argentina). (B) In silico analysis in medulloblastomas from the North American and European populations (n=617).
The in silico analysis of medulloblastomas from the North American and European (NAM/EU) populations showed that 29% (n=176) was SHH activated, 39% (n=240) Group 4, 24% (n=148) Group 3, and 8% (n=53) WNT activated. In the WNT-activated subgroup, 87% (n=46) showed hotspot mutation in the CTNNB1, and 13% (n=7) were CTNNB1 wild type (Figure 1B).
The frequency of WNT-activated medulloblastomas in Latin-Iberian population (Figure 1A) was significantly higher (15%) compared to the frequency observed in NAM/EU populations of 8% (Figure 1B), (p=0.000023).
CTNNB1 variants in the WNT-activated medulloblastomas from Latin-Iberian population
In our Latin-Iberian series of 24 CTNNB1 mutant medulloblastomas, we found a total of 25 pathogenic variants in the hotspot region of the exon 3, being 24 missense mutations, and one in-frame deletion (Figures 2A, B). More detailed information about the mutational status of CTNNB1 in WNT-activated medulloblastomas in Latin-Iberian patients is described in Supplementary Table S2. The most frequent CTNNB1 variant observed was the p.(Ser33Tyr) found in five cases, followed by the p.(Gly34Val) found in three cases, and variants p.(Ser37Tyr), p.(Asp32Tyr), p.(Ser33Cys), and p.(Ser33Phe) were found in two cases each one. The remaining variants were observed only in one case (Figure 2B, Supplementary Table S2). In one case (ID88), we observed two CTNNB1 variants, p.(Ser33Tyr) and p.(Ala43Thr) (Supplementary Table S2).
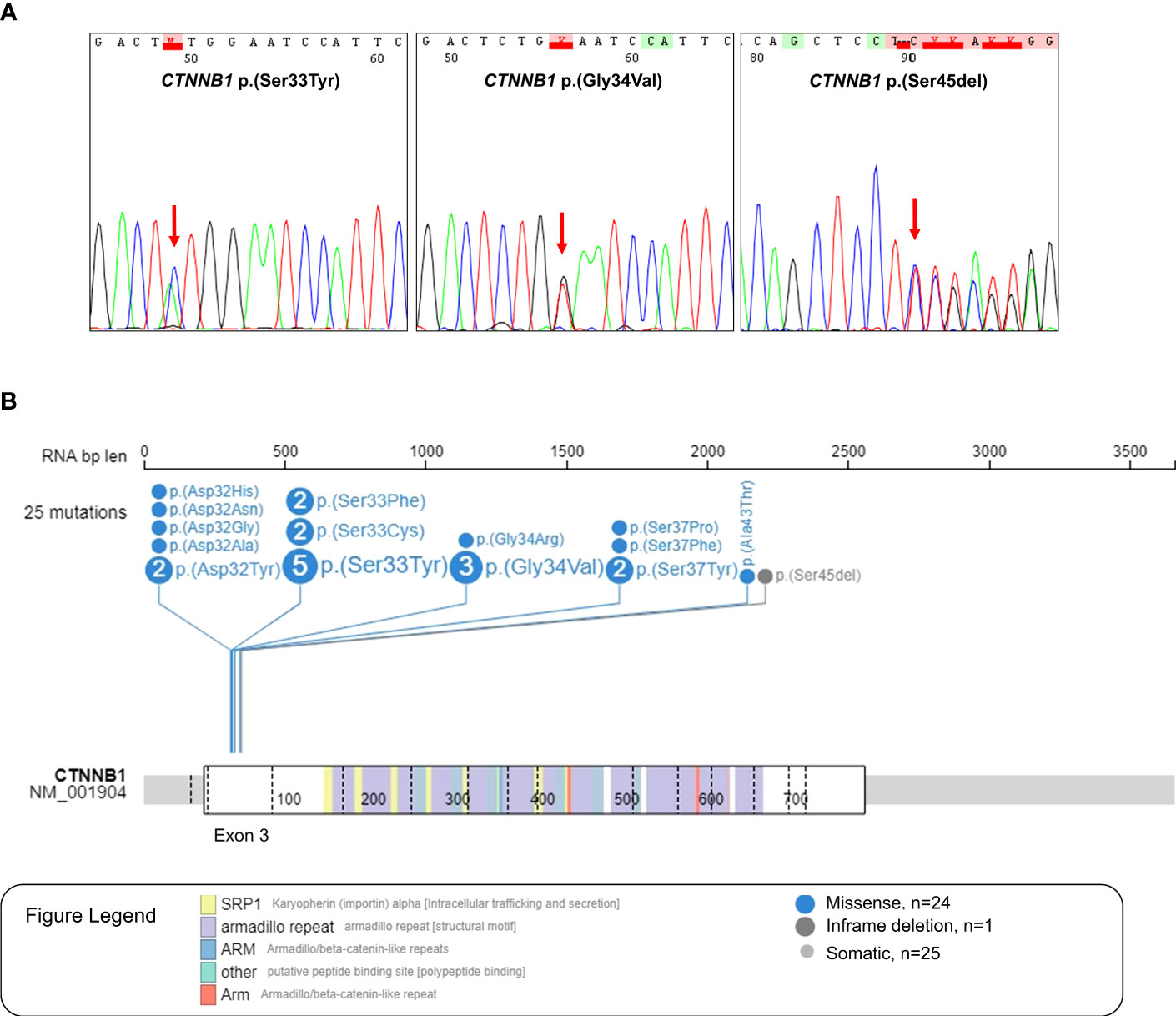
Figure 2 CTNNB1 variants found in the Latin-Iberian population. (A) Electropherogram of Sanger sequencing of the hotspot region in the exon 3 of CTNNB1 (chr3:41224525 + 41224751), which codes the phosphorylation site of the protein, showing three different CTNNB1 variants: p.(Ser33Tyr), p.(Gly34Val) and p.(Ser45del). (B) Lollipop showing the 25 CTNNB1 variants observed in the WNT-activated medulloblastomas from the Latin-Iberian population.
The in silico analysis of medulloblastomas from the North American and European (NAM/EU) populations reveals 47 CTNNB1 variants in 46 WNT-activated medulloblastomas, with all variants being located in the exon 3 of the CTNNB1 gene (Supplementary Figure S1). One case showed two mutations ICGC_MB113, p.(Thr41Ala) and p.(Ser33Cys). Similarly to what was observed in our series, the p.(Ser33Cys) variant was one of the most frequent detected variants (Supplementary Figure S1).
The frequency of WNT-activated medulloblastomas with CTNNB1 wild type was significantly higher in Latin-Iberian population (27%) (Figure 1A) compared to those observed in NAM/EU populations (13%) (Figure 1B), (p=0.014769).
WNT-activated medulloblastomas with CTNNB1 wild type were prevalent in females and showed worse outcome in the Latin-Iberian population
We further associated the CTNNB1 mutational status with our patients’ clinical–pathological features (Table 1). WNT-activated medulloblastoma patients with CTNNB1 mutant showed an older median age at diagnosis of 11.3 years, compared with 10.0 years of CTNNB1 wild type, yet not statistically significant. The CTNNB1 wild-type cases were prevalent in female individuals (p=0.04), and no significant associations were observed regarding histology, surgery extension, and metastasis (Table 1).
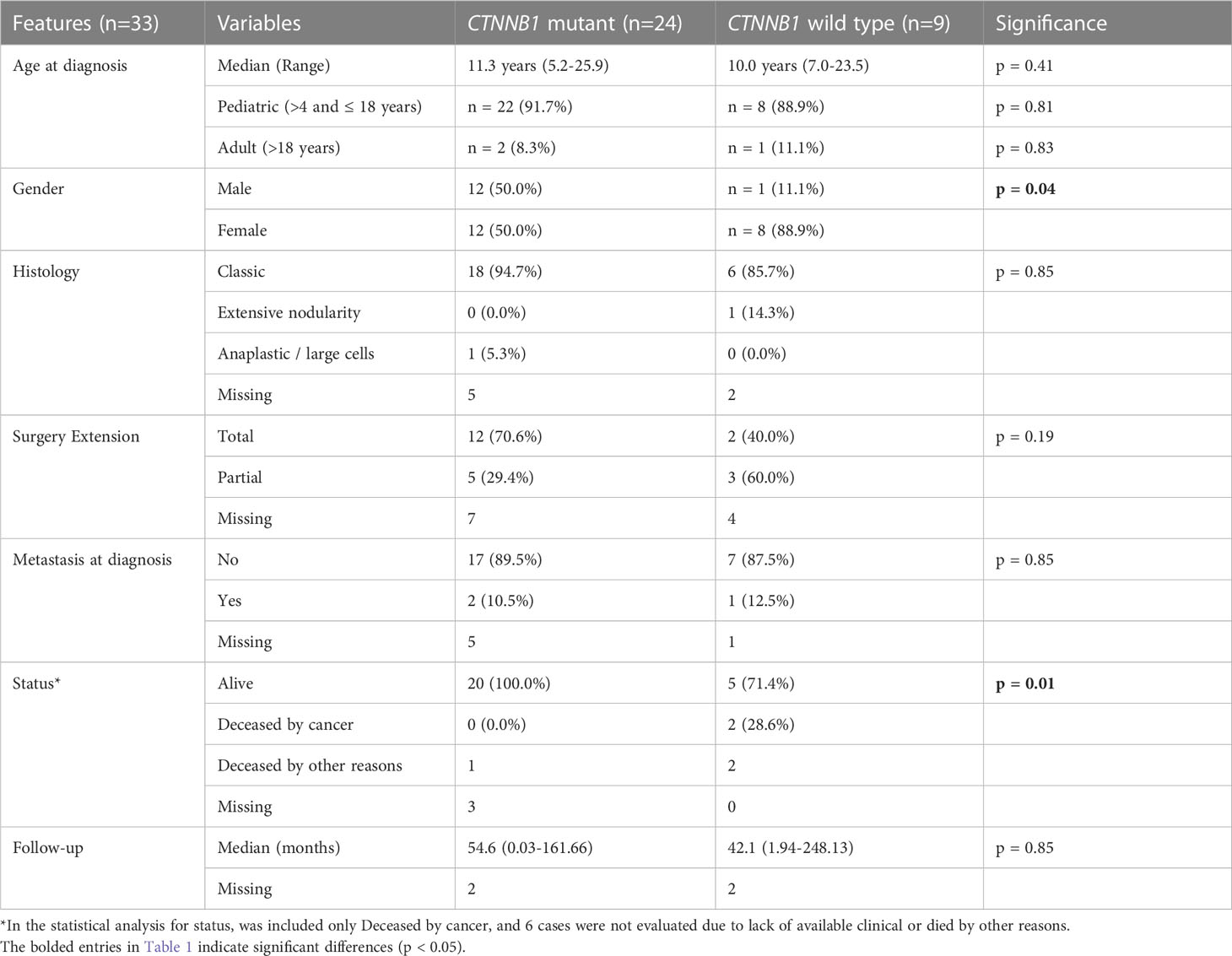
Table 1 Association of CTNNB1 status with clinicopathological features of 33 WNT-activated medulloblastomas from a Latin-Iberian population.
Despite not being statistically significant, we observed that the CTNNB1 mutant had a better outcome, with a 54.6-month median follow-up, compared with 42.1 months in wild-type cases (Table 1). We observed that 28.6% (2/7) of CTNNB1 wild-type patients died of cancer, contrasting with CTNNB1 mutant cases, where any patient died of cancer (p=0.01) (Table 1). Three CTNNB1 mutants were lost to follow-up (missing: ID97, 197, and 260), and additionally, three patients died due to other reasons (ID94, ID95, and ID96), thus were not included in the survival analysis (Supplementary Table S2).
The Kaplan–Meier analysis showed that patients with WNT-activated medulloblastomas CTNNB1 wild type had worse outcome, with 71.4% of overall survival compared to 100% of CTNNB1 mutant cases (log rank: p=0.031) (Figure 3).
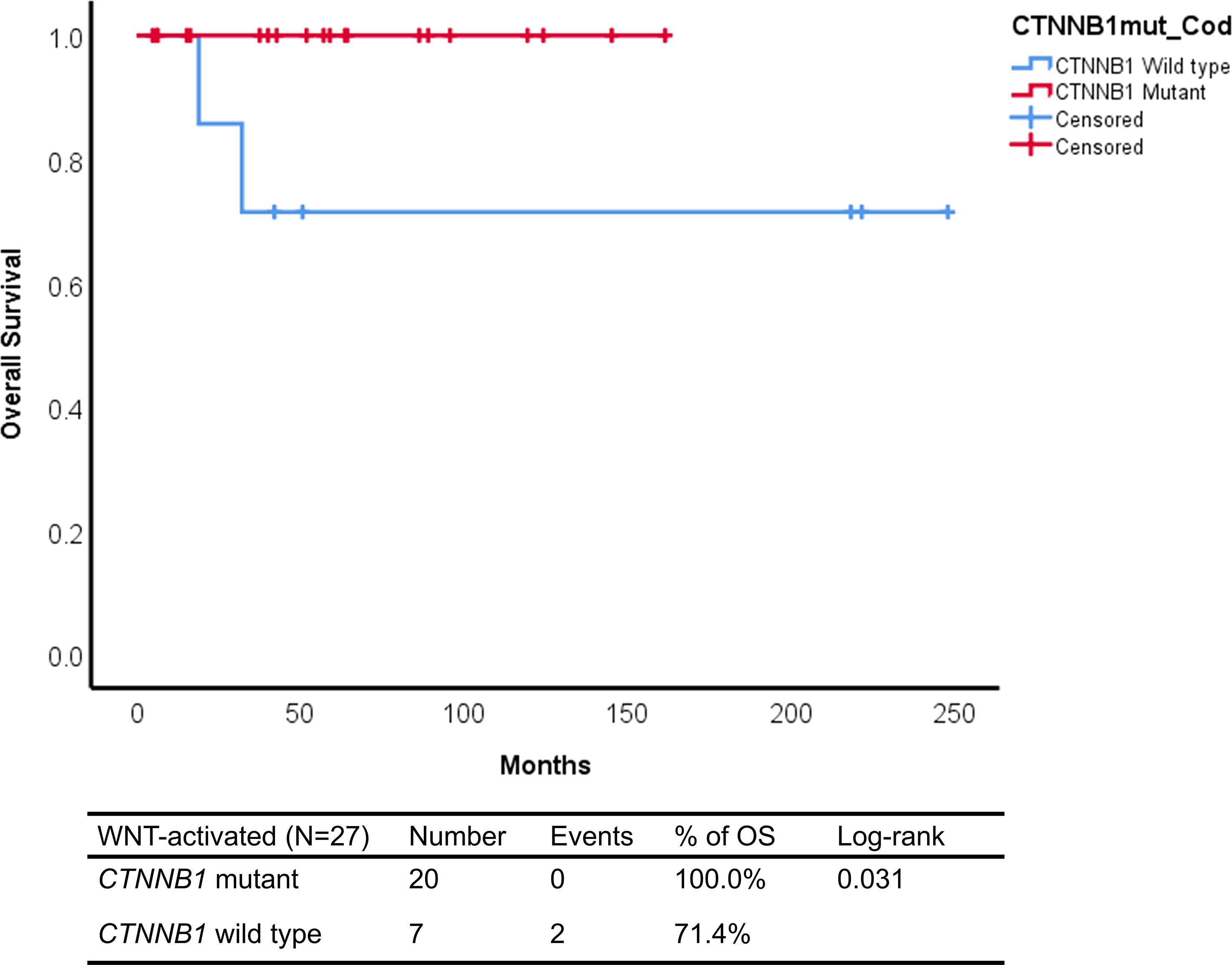
Figure 3 Kaplan–Meier curves of CTNNB1 mutant and CTNNB1 wild-type WNT-activated medulloblastomas from Latin-Iberian population.
APC germline mutation associated with WNT-activated medulloblastomas
We found nine WNT-activated medulloblastomas CTNNB1 wild type in our Latin-Iberian population. The analysis of the clinical records available showed the existence of two reports of familial adenomatous polyposis, one from Barretos Cancer Hospital (Brazil) (Figure 4) and one from Centro Hospitalar Universitário São João (Portugal).
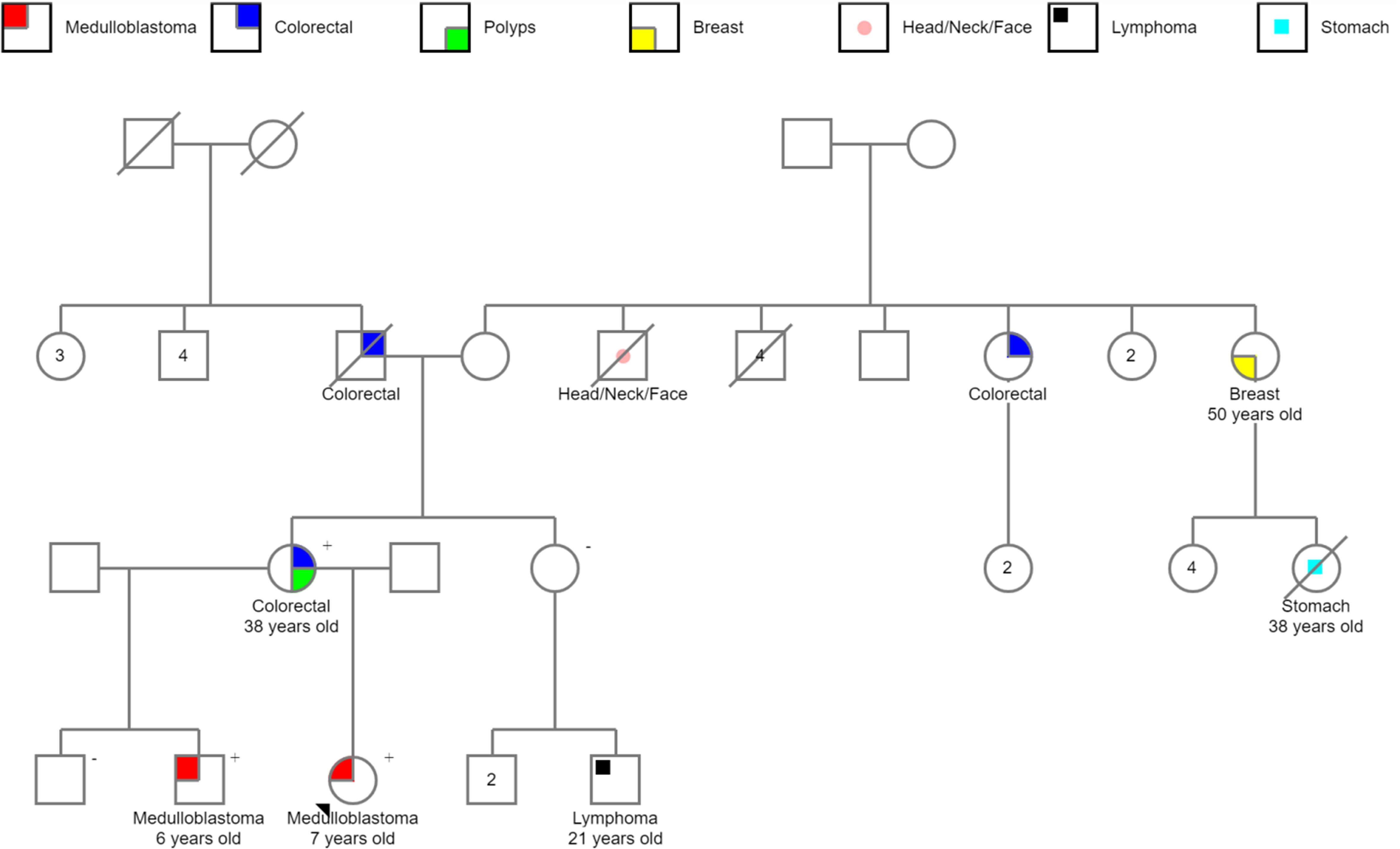
Figure 4 Pedigree of familial adenomatous polyposis showing the occurrence of APC-associated WNT-activated medulloblastomas in siblings. The APC germline mutation was tested, and the “+” indicates APC mutation and “−,” APC wild type.
Of note, the Brazilian case, a germline APC c.3183_3147delACAAA-p.(Gln1062Ter) pathogenic variant was detected. The proband of this family is a 7-year-old girl diagnosed with medulloblastoma; her brother was also diagnosed with medulloblastoma when he was 6 years old. Their mother had approximately 100 polyps and developed colorectal cancer at the age of 38, fulfilling the criteria of FAP (also known as Turcot syndrome type 2) (Figure 4).
The Portuguese patient harbored a c.3183_3187delACAAA-p.(Gln1062Ter) APC germline variant, similarly to the variant detected in the Brazilian family. The patient was a 9.7-year-old girl diagnosed with WNT-activated, CTNNB1-wild type medulloblastoma. At 24 years old, she presented gastrointestinal tumors with hepatic metastasis. Currently, she is 30 years old, and in total remission of the brain tumor.
Discussion
The origin of the WNT-activated medulloblastomas is attributed to molecular alterations that promote nuclear accumulation of beta-catenin products, inducing cell proliferation and tumor growth (22). Landmark genomic studies have shown that 97% of WNT-driven medulloblastomas can be explained by somatic mutations in the CTNNB1 gene (~90%) and germline mutations in APC (~10%), which are mutually exclusive (8, 10).
In the present study, we report for the first time, the frequency of CTNNB1 mutations in WNT-activated medulloblastomas in a large cohort of Latin-Iberian patients. The WNT-activated medulloblastoma subgroup in our Latin-Iberian population was of 15% (n=40), which is higher than those reported in North American and European populations (7%–10%) (2). A higher proportion of WNT-activated medulloblastomas were also described in previous Brazilian cohorts, 16.1% (24/149) (5) and 27% (24/92) (23). Another notable distinction observed in our cohort was the higher frequency of 46% for SHH-activated medulloblastomas. A potential reason for these distinct frequencies observed may lay in the methodologies used (24). In our study, we used a robust 22-gene panel assay by nCounter (25). However, DNA methylation assays have been the most recent approach recommended for medulloblastoma classification (26). A comparison study among methodologies showed that up to 10% of WNT medulloblastomas previously determined by nCounter were further classified as high-grade neuroepithelial tumor with BCOR alteration and anaplastic pilocytic astrocytoma by the DNA methylation assay (24). Further studies are needed to understand whether there is methodological issue, a hospital-based bias selection, or true epidemiological differences of medulloblastoma molecular subgroups among populations.
Among the 40 WNT-activated medulloblastomas included in the present study, we successfully evaluated CTNNB1 mutations in 33 cases and found 73% (24/33) of CTNNB1-mutated cases and 27% (9/33) CTNNB1 wild type. Of note, our frequency of CTNNB1 wild-type WNT-activated medulloblastomas is significantly higher than that reported in North American and European populations, varying from 6.8% (8) to 13% (in silico analysis). Waszak and colleagues performed whole exome sequencing in 66 WNT-activated medulloblastomas and found somatic CTNNB1 mutations in 89.4% and CTNNB1 wild type in 10.6% of cases8. A recent German study evaluated a large cohort of 191 WNT-activated medulloblastomas and reported 92.2% (176/191) CTNNB1 mutants and 7.8% (15/191) wild-type cases (10). Our in-silico analysis of CTNNB1 mutations at cBioPortal showed that 13% (7/53) of WNT-activated medulloblastomas are CTNNB1 wild type.
The reason for this discrepancy in the frequency of CTNNB1 mutations in different populations needs to be clarified. In the present study, we performed Sanger sequencing of the exon 3 of the CTNNB1 gene, and the studies mentioned above used whole genome or whole exome sequencing (10, 27). Nevertheless, all mutations reported by NGS were located in the exon 3, covered by our Sanger sequencing assay. Moreover, the CTNNB1 variants identified in our Latin-Iberian study is very similar to the variants reported in the North American and European populations.
We found that patients’ CTNNB1 wild type was more frequent in female individuals and was associated with a worse outcome. Nevertheless, caution should be taken, since these findings are based on a few patients and in a very heterogeneously treated population. Our findings contrast with other North American and European studies that did not observe any association of CTNNB1 status with WNT-activated medulloblastoma clinicopathological features (8, 10). Therefore, further studies with more extensive series from non-European populations are warranted to explore the clinical impact of CTNNB1 mutations in WNT-activated medulloblastomas. Moreover, other molecular features, namely, somatic alterations in TP53, OTX2, and monosomy for chromosome 6, have been associated with prognosis in WNT-activated medulloblastomas (10, 28). Additionally, addressing these alterations is needed to fully characterize the present Latin-Iberian cohort.
Considering that in WNT-activated medulloblastomas, CTNNB1 wild-type cases can harbor APC germline mutations, our study suggests that up to 27% of Latin-Iberian WNT-activated medulloblastomas can occur in the context of FAP, contrasting with the reported approximately 10% in North American and European populations. The rates of germline mutations can vary between different populations (29). A cross-sectional study evaluated the frequencies of germline mutations in APC in more than six thousand individuals with a history of colorectal cancer in their families. It showed that the APC mutation rate was higher in Asians than in Caucasians (Western/Northern European, Central/Eastern European, and Ashkenazi ancestry), African American, and others (Latin American/Caribbean, Near/Middle Eastern, and Native American) (30).
Disparities in genomic studies due to the under-representation of some populations, such as from South America, were previously demonstrated (31). Consequently, genomic data from North America and the European population may only partially capture the genetic variability range in low- and middle-income countries (32). In this context, the data from our current study may contribute to the characterization of WNT-activated medulloblastomas in a poorly explored population.
It is estimated that germline APC mutations are associated with a 92 times higher risk for developing medulloblastomas than in the general population (11). Medulloblastoma was reported to be the most common brain tumor (79%, 11/14) observed in families with FAP10. Waszak and coworkers reported that all APC mutation carriers with available medical records (n=4) had a family history of FAP and associated cancers. Additional malignancies were observed in three patients with APC germline mutations (8).
Based on the available medical records, the present study identified two families fulfilling the FAP criteria. Due to the study’s retrospective nature, several clinical records are very omissive in the familiar history description, not allowing for an accurate assessment of the putative hereditary nature of the CTNNB1 wild-type cases. Nevertheless, an active search of the CTNNB1 wild-type patients will be done, and genetic counseling and potential confirmation of its germline nature will be offered. Importantly, the possibility of a new or founder APC mutation cannot be entirely ruled out (33). These data demonstrate the importance of CTNNB1 genetic testing and should indicate that patients with WNT-activated medulloblastomas CTNNB1 wild type must be monitored by a multidisciplinary team, due to possible hereditary nature of the disease and propensity to develop other tumor types.
Interestingly, one of our FAP exhibited a rare co-occurrence of medulloblastomas in siblings. The APC variant identified in this family, p.(Gln1062Ter), has been previously detected in families with classic FAP (34–36) and reported founder in Spanish and Greek populations (27, 37). This variant is located in a region of the APC gene associated with a higher risk of developing extracolonic tumors (38), which may explain the development of the two reported medulloblastomas. To our knowledge, only one case of siblings with APC-associated WNT-activated medulloblastomas was reported, involving an 11-year-old girl and her 19-year-old brother exhibiting both APC germline mutation p.(R213*) (10).
In conclusion, the reported higher incidence of CTNNB1 wild type in our Latin-Iberian patients may be associated with a worse outcome and suggests a higher prevalence of hereditary WNT-activated medulloblastomas in this poorly characterized population. We also reported a rare case of siblings with WNT-activated medulloblastomas associated with APC germline mutation in a South American patient.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
This study was approved by the institutional review board from Barretos Cancer Hospital (CAAE: 59979816.6.1001.5437). Informed consent was obtained from the patients or familiars before APC germline evaluation.
Author contributions
DM: data collection, experimental procedures, data analysis, and manuscript writing; MB: experimental procedures, results discussion, and manuscript review. AA: oncogenetic analysis, results discussion, manuscript writing, and review. FP: experimental procedures, results discussion, and manuscript review. LL: experimental procedures, manuscript writing, and review. FG: experimental analysis, medical reports review, manuscript writing, and review. AP: experimental procedures, manuscript writing, and review. GT: pathological review of tumor samples from Barreto’s cancer hospital, Brazil, results discussion, manuscript review. IS: pathological review of tumor samples from Barreto’s cancer hospital, Brazil, results discussion, manuscript review. FS: pathological review of tumor, results discussion, and manuscript review. LN: pathological review of tumor, results discussion, and manuscript review. EV: patient’s clinical data collection, and manuscript review. CS: patient’s clinical data collection, and manuscript review. JS: patient’s clinical data collection and manuscript review. SM: patient’s clinical data collection, and manuscript review; ML: Patient’s clinical data collection and manuscript review; GH: patient’s clinical data collection and manuscript review; HG-R: Pathological review of tumor and patient’s clinical data collection, manuscript review. SC: pathological review of the tumor and patient’s clinical data collection and manuscript review. SN: patient’s clinical data collection and manuscript review. MG-d-C: patient’s clinical data colection and manuscript review. JP: pathological review of tumor and manuscript review. FM: patient’s clinical data colection and manuscript review. CJ: medical reports analysis, patients’ treatment protocol review, and manuscript review. BM: medical reports analysis, patients’ treatment protocol review, and manuscript review. RR: supervisor and project coordinator, results discussion, and manuscript writing. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Barretos Cancer Hospital and the Public Ministry of Labor Campinas (Research, Prevention, and Education of Occupational Cancer, Brazil). LL was supported by the Public Ministry of Labor Campinas (Research, Prevention, and Education of Occupational Cancer) in Campinas, Brazil. RMR is a recipient of CNPq Productivity (Brazil). DM is supported by a scholarship from National Oncology Care Support Program (PRONON), Brazil.
Acknowledgments
The authors want to thank the teams of Molecular Oncology Research Center, Neurooncology Pediatric Department of Barretos Cancer Hospital, Ribeião Preto Medical School, Federal University of São Paulo, AC Camargo Hospital, Brasília Children’s Hospital, Italian Hospital of Buenos Aires, and Centro Hospitalar Universitário São João, for contributions and for discussing the results.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1237170/full#supplementary-material
Supplementary Figure 1 | Lollipop showing the 47 CTNNB1 variants observed in the 46 WNT-activated medulloblastomas from the North American and European populations. Data collected at cBioPortal.
References
1. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2014-2018. Neuro Oncol (2021) 23:III1–III105. doi: 10.1093/NEUONC/NOAB200
2. Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC, et al. Medulloblastoma. Nat Rev Dis Primers (2019) 5(1):11. doi: 10.1038/s41572-019-0063-6
3. Hovestadt V, Ayrault O, Swartling FJ, Robinson GW, Pfister SM, Northcott PA. Medulloblastomics revisited: biological and clinical insights from thousands of patients. Nat Rev Cancer (2020) 20:42. doi: 10.1038/S41568-019-0223-8
4. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol (2021) 23:1231–51. doi: 10.1093/NEUONC/NOAB106
5. da Silva LS, Mançano BM, de Paula FE, dos Reis MB, de Almeida GC, Matsushita M, et al. Expression of GNAS, TP53, and PTEN improves the patient prognostication in sonic hedgehog (SHH) medulloblastoma subgroup. J Mol Diagnostics (2020) 22:957–66. doi: 10.1016/j.jmoldx.2020.04.207
6. Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell (2017) 31:737–754.e6. doi: 10.1016/j.ccell.2017.05.005
7. Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis (2008) 4:68. doi: 10.4161/ORG.4.2.5851
8. Waszak SM, Northcott PA, Buchhalter I, Robinson GW, Sutter C, Groebner S, et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol (2018) 19:785–98. doi: 10.1016/S1470-2045(18)30242-0
9. Gao C, Wang Y, Broaddus R, Sun L, Xue F, Zhang W. Exon 3 mutations of CTNNB1 drive tumorigenesis: a review. Oncotarget (2018) 9:5492. doi: 10.18632/ONCOTARGET.23695
10. Goschzik T, Mynarek M, Doerner E, Schenk A, Spier I, Warmuth-Metz M, et al. Genetic alterations of TP53 and OTX2 indicate increased risk of relapse in WNT medulloblastomas. Acta Neuropathol (2022) 144:1143–56. doi: 10.1007/S00401-022-02505-5
11. Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, et al. The molecular basis of Turcot’s syndrome. N Engl J Med (1995) 332:839–47. doi: 10.1056/NEJM199503303321302
12. Gomes I, Moreno DA, dos Reis MB, da Silva LS, Leal LF, Gonçalves GM, et al. Low MGMT digital expression is associated with a better outcome of IDH1 wildtype glioblastomas treated with temozolomide. J Neurooncol (2021) 151:135–44. doi: 10.1007/s11060-020-03675-6
13. Moreno DA, da Silva LS, Zanon MF, Bonatelli M, de Paula FE, de Medeiros Matsushita M, et al. Single nCounter assay for prediction of MYCN amplification and molecular classification of medulloblastomas: a multicentric study. J Neurooncol (2022) 157:27–35. doi: 10.1007/S11060-022-03965-1
14. Moreno DA, da Silva LS, Gomes I, Leal LF, Berardinelli GN, Gonçalves GM, et al. Cancer immune profiling unveils biomarkers, immunological pathways, and cell type score associated with glioblastoma patients’ survival. Ther Adv Med Oncol (2022) 14:175883592211276. doi: 10.1177/17588359221127678
15. Northcott PA, Shih DJH, Remke M, Cho YJ, Kool M, Hawkins C, et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol (2012) 123:615–26. doi: 10.1007/S00401-011-0899-7
16. Leal LF, Evangelista AF, de Paula FE, Caravina Almeida G, Carloni AC, Saggioro F, et al. Reproducibility of the NanoString 22-gene molecular subgroup assay for improved prognostic prediction of medulloblastoma. Neuropathology (2018) 38:475–83. doi: 10.1111/neup.12508
17. Leal LF, Mermejo LM, Ramalho LZ, Martinelli CE, Yunes JA, Seidinger AL, et al. Wnt/beta-catenin pathway deregulation in childhood adrenocortical tumors. J Clin Endocrinol Metab (2011) 96:3106–14. doi: 10.1210/JC.2011-0363
18. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature (2012) 488:43–8. doi: 10.1038/NATURE11213
19. Morrissy AS, Garzia L, Shih DJH, Zuyderduyn S, Huang X, Skowron P, et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature (2016) 529:351–7. doi: 10.1038/NATURE16478
20. Jones DTW, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature (2012) 488:100–5. doi: 10.1038/NATURE11284
21. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature (2017) 547:311–7. doi: 10.1038/NATURE22973
22. Eberhart CG, Tihan T, Burger PC. Nuclear localization and mutation of beta-catenin in medulloblastomas. J Neuropathol Exp Neurol (2000) 59:333–7. doi: 10.1093/JNEN/59.4.333
23. Cruzeiro GAV, Salomão KB, de Biagi CAO, Baumgartner M, Sturm D, Lira RCP, et al. A simplified approach using Taqman low-density array for medulloblastoma subgrouping. Acta Neuropathol Commun (2019) 7:33. doi: 10.1186/S40478-019-0681-Y
24. Korshunov A, Chavez L, Northcott PA, Sharma T, Ryzhova M, Jones DTW, et al. DNA-methylation profiling discloses significant advantages over NanoString method for molecular classification of medulloblastoma. Acta Neuropathol (2017) 134:965–7. doi: 10.1007/S00401-017-1776-9
25. Korshunov A, Sahm F, Zheludkova O, Golanov A, Stichel D, Schrimpf D, et al. DNA methylation profiling is a method of choice for molecular verification of pediatric WNT-activated medulloblastomas. Neuro Oncol (2019) 21:214–21. doi: 10.1093/NEUONC/NOY155
26. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature (2018) 555:469–74. doi: 10.1038/NATURE26000
27. Fostira F, Thodi G, Sandaltzopoulos R, Fountzilas G, Yannoukakos D. Mutational spectrum of APC and genotype-phenotype correlations in Greek FAP patients. BMC Cancer (2010) 10:389. doi: 10.1186/1471-2407-10-389
28. Richardson S, Hill RM, Kui C, Lindsey JC, Grabovksa Y, Keeling C, et al. Emergence and maintenance of actionable genetic drivers at medulloblastoma relapse. Neuro Oncol (2022) 24:153. doi: 10.1093/NEUONC/NOAB178
29. Campbell CD, Eichler EE. Properties and rates of germline mutations in humans. Trends Genet (2013) 29:575. doi: 10.1016/J.TIG.2013.04.005
30. Inra JA, Steyerberg EW, Grover S, McFarland A, Syngal S, Kastrinos F. Racial variations in frequency and phenotypes of APC and MUTYH mutations in 6,169 individuals undergoing genetic testing. Genet Med (2015) 17:815. doi: 10.1038/GIM.2014.199
31. Fatumo S, Chikowore T, Choudhury A, Ayub M, Martin AR, Kuchenbaecker K. A roadmap to increase diversity in genomic studies. Nat Med (2022) 28:243–50. doi: 10.1038/S41591-021-01672-4
32. Sirugo G, Williams SM, Tishkoff SA. The missing diversity in human genetic studies. Cell (2019) 177:26. doi: 10.1016/J.CELL.2019.02.048
33. Ripa R, Bisgaard ML, Bülow S, Nielsen FC. De novo mutations in familial adenomatous polyposis (FAP). Eur J Hum Genet (2002) 10:631–7. doi: 10.1038/sj.ejhg.5200853
34. Zhang S, Qin H, Lv W, Luo S, Wang J, Fu C, et al. Novel and reported APC germline mutations in Chinese patients with familial adenomatous polyposis. Gene (2016) 577:187–92. doi: 10.1016/J.GENE.2015.11.034
35. Kerr SE, Thomas CB, Thibodeau SN, Ferber MJ, Halling KC. APC germline mutations in individuals being evaluated for familial adenomatous polyposis: a review of the Mayo Clinic experience with 1591 consecutive tests. J Mol Diagn (2013) 15:31–43. doi: 10.1016/J.JMOLDX.2012.07.005
36. Rivera B, González S, Sánchez-Tomé E, Blanco I, Mercadillo F, Letón R, et al. Clinical and genetic characterization of classical forms of familial adenomatous polyposis: a Spanish population study. Ann Oncol (2011) 22:903–9. doi: 10.1093/ANNONC/MDQ465
37. González S, Blanco I, Campos O, Julià M, Reyes J, Llompart A, et al. Founder mutation in familial adenomatous polyposis (FAP) in the Balearic Islands. Cancer Genet Cytogenet (2005) 158:70–4. doi: 10.1016/J.CANCERGENCYTO.2004.07.003
Keywords: medulloblastomas, WNT activated, CTNNB1, APC, Latin-Iberian
Citation: Moreno DA, Bonatelli M, Antoniazzi AP, de Paula FE, Leal LF, Garcia FAO, de Paula AE, Teixeira GR, Santana IVV, Saggioro F, Neder L, Valera ET, Scrideli CA, Stavale J, Malheiros SMF, Lima M, Hajj GNM, Garcia-Rivello H, Christiansen S, Nunes S, Gil-da-Costa MJ, Pinheiro J, Martins FD, Junior CA, Mançano BM and Reis RM (2023) High frequency of WNT-activated medulloblastomas with CTNNB1 wild type suggests a higher proportion of hereditary cases in a Latin-Iberian population. Front. Oncol. 13:1237170. doi: 10.3389/fonc.2023.1237170
Received: 13 June 2023; Accepted: 31 July 2023;
Published: 04 September 2023.
Edited by:
Daniel Moreira, St. Jude Children’s Research Hospital, United StatesReviewed by:
Andres Morales La Madrid, Sant Joan de Déu Hospital, SpainEric Bouffet, University of Toronto, Canada
Copyright © 2023 Moreno, Bonatelli, Antoniazzi, de Paula, Leal, Garcia, de Paula, Teixeira, Santana, Saggioro, Neder, Valera, Scrideli, Stavale, Malheiros, Lima, Hajj, Garcia-Rivello, Christiansen, Nunes, Gil-da-Costa, Pinheiro, Martins, Junior, Mançano and Reis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui Manuel Reis, cnVpcmVpcy5oY2JAZ21haWwuY29t; cnJlaXNAbWVkLnVtaW5oby5wdA==