 Benigno C. Valdez1*
Benigno C. Valdez1* Bin Yuan1David Murray2
Bin Yuan1David Murray2 Jeremy Leon Ramdial1
Jeremy Leon Ramdial1 Yago Nieto1Uday Popat1
Yago Nieto1Uday Popat1 Xiaowen Tang3
Xiaowen Tang3 Borje S. Andersson1
Borje S. Andersson1- 1Department of Stem Cell Transplantation and Cellular Therapy, University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Department of Oncology, University of Alberta, Edmonton, AB, Canada
- 3Department of Hematology, The First Affiliated Hospital of Soochow University, Jiangsu Institute of Hematology, Suzhou, China
Combinations of nucleoside analog(s) and DNA alkylating agent(s) are used for cancer treatment as components of pre-transplant regimens used in hematopoietic stem cell transplantation. Their efficacies are enhanced by combining drugs with different mechanisms of action, which also allows a reduction in the individual drug dosages and thus potentially in toxicity to the patient. We hypothesized that addition of SAHA and olaparib, an HDAC- and a PARP-inhibitor, respectively, to the established combination of fludarabine, clofarabine and busulfan would enhance AML cell cytotoxicity. Exposure of the AML cell lines KBM3/Bu2506, MV4-11, MOLM14 and OCI-AML3 to the 5-drug combination resulted in synergistic cytotoxicity with combination indexes < 1. Increased protein acetylation and decreased poly(ADP-ribosyl)ation were observed, as expected. Activation of apoptosis was suggested by cleavage of Caspase 3 and PARP1, DNA fragmentation, increased reactive oxygen species, and decreased mitochondrial membrane potential. The reduction in poly(ADP-ribosyl)ation was independent of caspase activation. Several proteins involved in DNA damage response and repair were downregulated, which may be contributing factors for the observed synergism. The increased phosphorylation of DNAPKcs suggests inhibition of its kinase activity and diminution of its role in DNA repair. A similar synergism was observed in patient-derived cell samples. These findings will be important in designing clinical trials using these drug combinations as pre-transplant conditioning regimens for AML patients.
1 Introduction
Preclinical and clinical studies have demonstrated the synergistic cytotoxicity of fludarabine (Flu), clofarabine (Clo), and busulfan (Bu) towards acute myeloid leukemia (AML) cells (1–5). The antineoplastic activities of Bu, a DNA alkylating agent, and the two nucleoside analogs Flu and Clo involve different mechanisms; Bu forms DNA adducts/crosslinks while Flu and Clo become incorporated into newly replicating DNA strands during DNA synthesis, and they transiently arrest the cell cycle at G2- and S-phase, respectively (6, 7). Both processes trigger a chain of events including formation of DNA strand breaks and subsequent activation of pro-apoptotic pathways (7–9). Combinations of DNA alkylator(s) and nucleoside analog(s) are effective pre-transplant regimens for hematopoietic stem cell transplantation (HSCT) for AML patients, provided that the drugs are sequenced properly, with the nucleoside analog(s) being administered before the alkylating agent (1, 7, 10).
The cytotoxicity of [Flu+Clo+Bu] is enhanced with histone deacetylase (HDAC) inhibitors (11–14), hypomethylating agents (13), BCL-2 inhibitors (15), and FLT3 inhibitors (16). HDAC inhibitors (HDACi) such as SAHA/vorinostat restore appropriate gene expression, resulting in induction of cell differentiation, cell cycle arrest and apoptosis (17). Despite their preclinical efficacy, HDACi do not seem to have high clinical effectiveness as monotherapy, and potentially more effective anti-tumor activity is observed when they are combined with other anti-cancer drugs (18–20). In this context, the differential effects of HDACi on the expression of cellular drug transporters must also be considered before applying them in combination chemotherapy, e.g., they are known to decrease MRP1 protein and increase MDR1 protein levels in human hematologic cancer cell lines (21). Such mechanisms may partly explain the lack of clinical efficacy when the HDACi romidepsin was combined with MDR1 ligands such as doxorubicin or vincristine (22, 23).
The efficacy of HDACi in combination chemotherapy may be attributed in part to its ability to induce DNA double-strand breaks (DSBs); in fact, HDACi-mediated changes in chromatin structure directly activate the DNA damage response (24, 25). HDACi affect the acetylation status of proteins involved in different DNA repair pathways and may have an impact on the genomic instability of cancer cells (26).
Another class of drugs which affect genomic instability are the poly(ADP-ribose) polymerase (PARP) inhibitors. PARP enzymes catalyze protein poly(ADP-ribosyl)ation (PARylation); they bind to DNA strand breaks, self-ribosylate, and recruit and PARylate DNA repair proteins (27). Inhibition of PARP enzymes compromises the localization, stability and activity of chromatin factors including histones, topoisomerases and DNA repair proteins, thereby affecting the DNA damage response (28, 29).
HDACi interact significantly with PARP inhibitors (PARPi) (30, 31). We recently showed that HDACi inhibit protein PARylation and exhibit synergistic cytotoxicity with chemotherapeutic agents such as gemcitabine (Gem), Bu and melphalan (Mel) in lymphoma cell lines (31). Clinically, the combination [Gem+Bu+Mel+SAHA+Olaparib (Ola)] provided ~90% event-free and overall survival rates for lymphoma patients undergoing autologous HSCT (32). These results prompted us to expand this rationale into myeloid malignancies and determine the synergistic cytotoxicity of an alkylating agent, nucleoside analog(s), HDACi, and PARPi in AML cells. Using the proven clinical efficacy of [Flu+Clo+Bu] in AML as a basis, this three-drug combination was combined with the HDACi SAHA/vorinostat and the PARPi, Ola. We now report the synergistic cytotoxicity of these five drugs in AML cell lines and cell samples derived from patients with acute leukemia. The results provide another level of mechanistic insight into the previously reported observations on the HDACi- and PARPi-mediated inhibition of DNA repair in hematologic cancers and its potential exploitation for therapeutic purposes in allogeneic stem cell transplantation.
2 Methods
2.1 Cell culture
The AML cell lines MV4-11, MOLM14 and OCI-AML3 were obtained from Dr. Michael Andreeff at the University of Texas MD Anderson Cancer Center (UTMDACC). The busulfan-resistant KBM3/Bu2506 (KBu) cell line was established in our laboratory by serial exposure of KBM3 cells (33) to increasing concentrations of Bu. All cells were cultured in RPMI 1640 (Mediatech, Inc., Manassas, VA) with 10% heat-inactivated fetal bovine serum (FBS: Gemini Bio Products, West Sacramento, CA), 100 U/ml penicillin and 100 μg/ml streptomycin (Mediatech) at 37°C in a fully humidified atmosphere of 5% CO2 in air.
2.2 Patient samples
Leukemia cell samples were isolated from patients’ peripheral blood using lymphocyte separation medium (Mediatech) and incubated in suspension in the RPMI 1640 medium described above. Patient 1 had T-cell prolymphocytic leukemia (T-PLL), patient 2 had mixed phenotype acute leukemia and patient 3 had AML, as shown in Figure 1. Patient samples were collected after obtaining written informed consent, and all studies using these samples were performed under a protocol approved by the Institutional Review Board of the UTMDACC, in accordance with the Declaration of Helsinki.
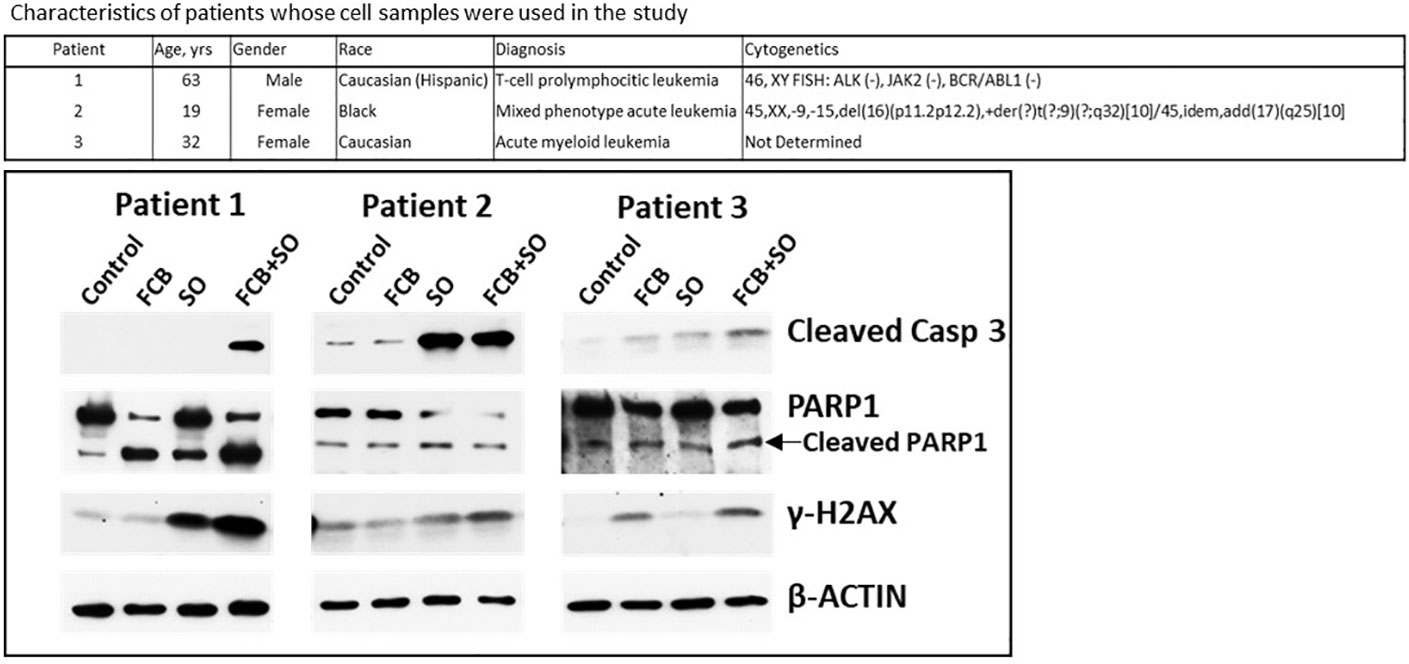
Figure 1 Effects of drugs on molecular markers of apoptosis in patient-derived cell samples. Mononuclear cells were isolated from peripheral blood of patients with leukemia (upper panel) and exposed to the indicated drugs for 48 h prior to analysis by Western blotting (lower panel). F: 0.15 µM fludarabine; C: 15 nM clofarabine; B: 15 µg/ml busulfan; S: 0.6 µM SAHA/vorinostat; O: 7 µM olaparib. Casp, caspase.
2.3 Drugs
Fludarabine (Flu), clofarabine (Clo), SAHA/vorinostat, olaparib (Ola), NU7741, LTURM34 and AZD7648 were purchased from Selleck Chemicals LLC (Houston, TX), and busulfan (Bu) was obtained from Sigma-Aldrich Chemical Sciences Corporation (St. Louis, MO). Z-VAD-FMK was purchased from Cayman Chemical Co. (Ann Arbor, MI). Drug stock solutions were dissolved in dimethyl sulfoxide (DMSO) and the final concentration of DMSO was <0.08% by volume, a level that does not induce differentiation of these cell lines.
2.4 Cell proliferation and cell death assays
Cell proliferation assays were done in triplicate using 96-well plates. Cell aliquots (100 µl of 0.5 X 106 cells/ml) were analyzed for proliferation using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay as previously described (34). The inhibition of cell proliferation after a 48-h drug exposure was determined relative to the control cells exposed to solvent alone. Graphical analyses including calculations of IC10–IC20 values (the concentration of drug required for 10–20% growth inhibition) were done using Prism 5 (GraphPad Software, San Diego, CA). Programmed cell death was determined by flow cytometric measurements of phosphatidylserine externalization with Annexin-V-FLUOS (Roche Diagnostics, Indianapolis, IN) and 7-aminoactinomycin D (BD Biosciences, San Jose, CA) using a Muse Cell Analyzer (MilliporeSigma, St. Louis, MO).
Drug combination effects were estimated based on the combination index (CI) values calculated using the CompuSyn software (Combo Syn, Inc., Paramus, NJ). This program was developed based on the median-effect method: CI < 1 indicates synergy, CI ≈ 1 is additive, and CI > 1 suggests antagonism.
2.5 Western blot analysis
Cells were exposed continuously to drug(s) for 48 h, harvested and washed with cold phosphate-buffered saline (PBS). Cells were lysed with lysis buffer (Cell Signaling Technology, Danvers, MA). Total protein concentrations in the cell lysates were determined using a BCA Protein Assay kit (Thermo Fisher Scientific, Rockford, IL). Western blot analysis was done by separating protein extracts on polyacrylamide-SDS gels and blotting onto nitrocellulose membranes (Bio-Rad, Hercules, CA). Immunoblot analyses were done using the Immobilon Western Chemiluminescent HRP Substrate (MilliporeSigma). The sources of the antibodies and their optimum dilutions are available upon request. The β-actin protein was used as an internal control.
2.6 Determination of the level of PARylation
The levels of total PARylated proteins were determined by Western blot analysis (as described above) and enzyme-linked immunosorbent assay (ELISA) using the poly(ADP-ribose) ELISA kit from Cell Biolabs, Inc. (San Diego, CA). The monoclonal anti-PAR antibody used for Western blotting was obtained from R&D Systems, Inc. (Minneapolis, MN). The antibody is specific for PAR polymers 2 to 50 units long, but does not recognize structurally related RNA, DNA, ADP-ribose monomers, NAD, or other nucleic acid monomers.
2.7 Analysis of reactive oxygen species
Cells were analyzed for production of ROS using CM-H2DCFDA (5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester), an ROS indicator that diffuses into cells where it is oxidized to a fluorescent product (Thermo Fisher Scientific). Briefly, cells were aliquoted (0.4 ml) into tubes and CM-H2DCFDA (3 μl of 0.12 mM solution in DMSO) was added. Cells were incubated at 37°C for 1 h and immediately analyzed with a Gallios Flow Cytometer (Beckman Coulter, Inc., Indianapolis, IN) using excitation/emission wavelengths of 492/520 nm. Arithmetic means of the fluorescence intensities were compared and the relative fold increase in ROS production was calculated.
2.8 Analysis of mitochondrial membrane potential
An MMP kit (Cayman Chemical Co.) was used to determine changes in the MMP using the JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) reagent. Cells to be analyzed were aliquoted (0.4 ml) into flow cytometry tubes. Diluted (1:10 with cell growth medium, 3 μl) MMP-sensitive fluorescent dye JC-1 was added to each tube, incubated at 37°C for 20 min, and analyzed by flow cytometry (λex = 488 nm) using the 530 nm (FL-1 channel, green) and 585 nm (FL-2 channel, red) band-pass filters. Healthy cells with functional mitochondria and high MMP exhibit red fluorescence (aggregated JC-1), whereas cells with disrupted mitochondria and low MMP show green fluorescence (monomeric JC-1).
2.9 Statistical analysis
Results are presented as the mean ± standard deviation of at least three independent experiments and statistical analysis was performed using Student’s paired t-test with a two-tailed distribution (Microsoft Excel, Redmond, WA, USA). A P value of <0.05 was considered statistically significant.
3 Results
3.1 [Flu+Clo+Bu+SAHA+Ola] combination shows synergistic cytotoxicity in AML cells
Cells were exposed to individual drugs to calculate their IC10-20 values, which were used in the drug combination experiments to determine their effects on cell proliferation and apoptosis. Exposure of KBu, MV4-11, MOLM14, and OCI-AML3 cells to Flu, Clo, Bu, SAHA or Ola resulted in 9-28% growth inhibition. [Flu+Clo+Bu] inhibited growth by 42%, 45%, 52%, and 56% in KBu, MV4-11, MOLM14, and OCI-AML3 cells, respectively (Figure 2A). Addition of SAHA and Ola to the three-drug combinations inhibited cell proliferation by 66%-71% versus control (Figure 2A). The [Flu+Clo+Bu+Ola] combination decreased cell proliferation more than [Flu+Clo+Bu+SAHA] in AML cells (data not shown). All of these results correlate with the corresponding Annexin V data. The [Flu+Clo+Bu] combination caused ~28%-56% cell death as indicated by Annexin V positivity. Exposure to the [Flu+Clo+Bu+SAHA+Ola] combination increased Annexin V positivity to 38%-72% of control (Figure 2A). To test for a synergistic interaction, cells were exposed to different concentrations of the individual drugs or to the five-drug combination at a constant concentration ratio, and the MTT assay was performed. The calculated CI at increasing drug effects was graphically analyzed. Figure 2B shows combination index (CI) values <1 at fraction affected (Fa) > 0.5 in all cell lines tested, suggesting synergism for drug concentrations where potentially clinically relevant cytotoxicity was apparent. The graphs also suggest that drug antagonism (CI > 1) may exist at lower drug concentrations where lower Fa values were observed, and which resulted in relatively little cytotoxicity (Figure 2B).
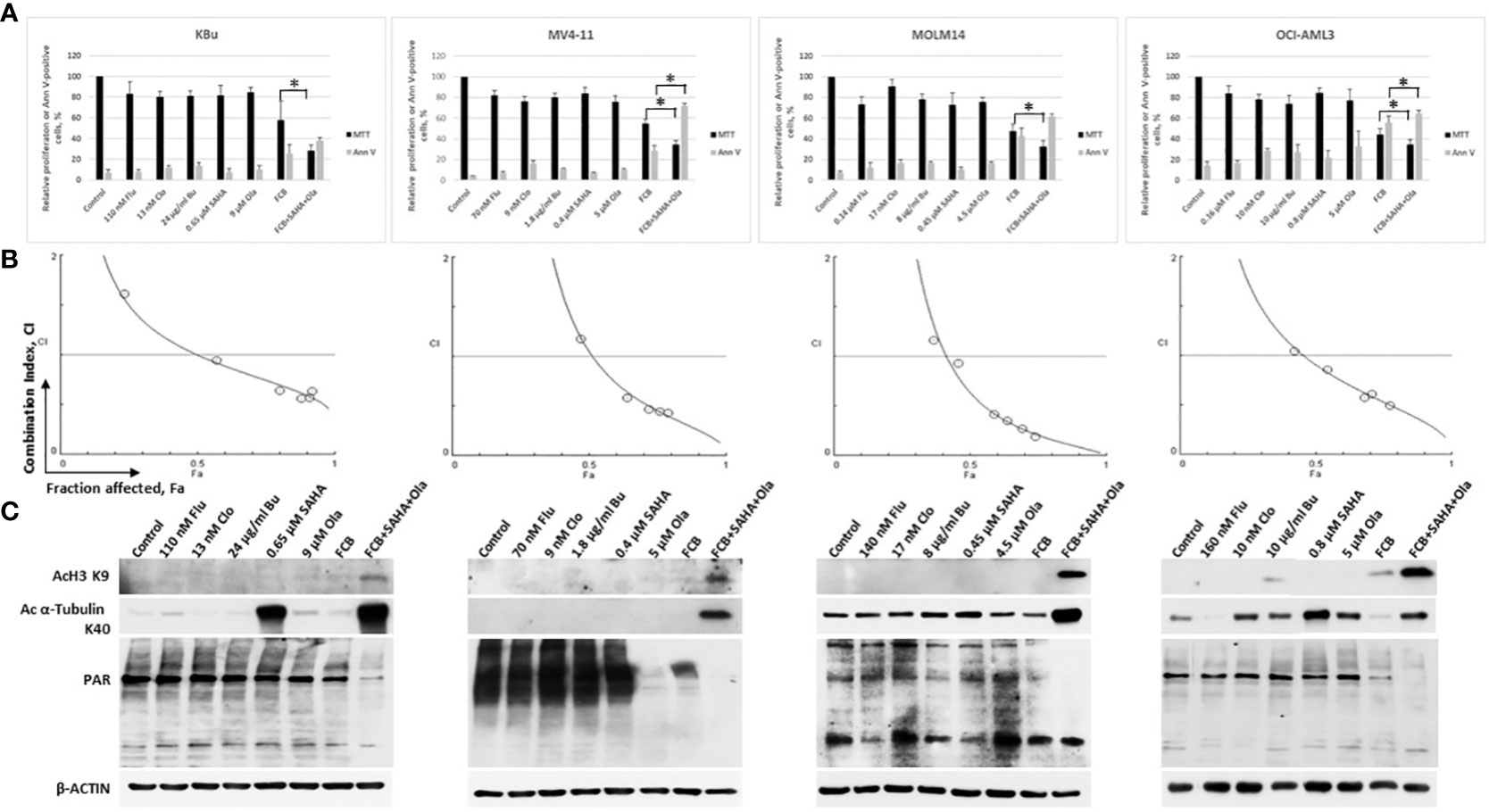
Figure 2 Cytotoxicity of fludarabine (Flu/F), clofarabine (Clo/C), busulfan (Bu/B), vorinostat (SAHA/S), and olaparib (Ola/O) in AML cell lines. Cells were exposed to the indicated concentrations of the drugs, alone or in combination, for 48 h prior to determination of relative cell proliferation by the MTT assay and apoptosis by the Annexin V (Ann V) assay (A). The results are averages of three independent experiments and each experiment was done in duplicate. P values less than 0.05 are indicated by an asterisk (*). (B) To determine drug synergism, cells were exposed to various drug combinations at constant ratio concentrations for 48 h prior to the MTT assay. The relationships between the calculated combination indexes (CI) and fraction affected (Fa) are shown; CI <1.0 indicates synergism. The graphs are representatives of two independent experiments. (C) After 48-h drug exposure, cells were lysed, boiled and analyzed for the levels of protein acetylation and poly(ADP-ribosyl)ation by Western blotting.
SAHA is known to inhibit histone deacetylases, and Ola is a potent inhibitor of PARP. We, therefore, sought to determine their effects on the acetylation of histone 3 and α-tubulin as well as on the PARylation of proteins. Exposure of AML cells to low levels of SAHA (0.4 µM – 0.8 µM) did not cause any significant increase in the level of AcH3K9 but resulted in a modest increase in the level of Ac α-tubulin K40 (Figure 2C). The [Flu+Clo+Bu] combination had minimal effect on the acetylation of histone 3 and α-tubulin; addition of [SAHA+Ola] to this combination increased the acetylation of both proteins (Figure 2C). Olaparib alone at the concentrations used here (4.5 µM – 9 µM) did not decrease the PARylation levels except in MV4-11 cells; its addition to [Flu+Clo+Bu+SAHA] combination, however, markedly decreased the levels of PAR in all four AML cell lines tested (Figure 2C). These observations are consistent with the previously reported synergistic interactions between HDAC and PARP inhibitors (27, 30, 31); addition of [Flu+Clo+Bu] apparently enhances the inhibition of PARylation.
3.2 [Flu+Clo+Bu+SAHA+Ola] induces apoptosis
Whether the observed increases in Annexin V positivity (Figure 2A) might be associated with apoptotic cell death was assessed by analyzing the cleavage of Caspase 3 and PARP1, which are indicators of apoptosis. While [Flu+Clo+Bu] caused cleavage of Caspase 3 and PARP1, addition of SAHA and Ola to this triple-drug combination markedly enhanced their cleavage (Figure 3A). Phosphorylation of histone 2AX also increased in cells exposed to [Flu+Clo+Bu] and even more so after [Flu+Clo+Bu+SAHA+Ola], suggesting DSB formation and/or activation of the DNA-damage response, consistent with the observed cleavage of genomic DNA as shown by agarose gel analysis (Figure 3B), which was probably due to caspase-mediated activation of nuclear DNases (35).
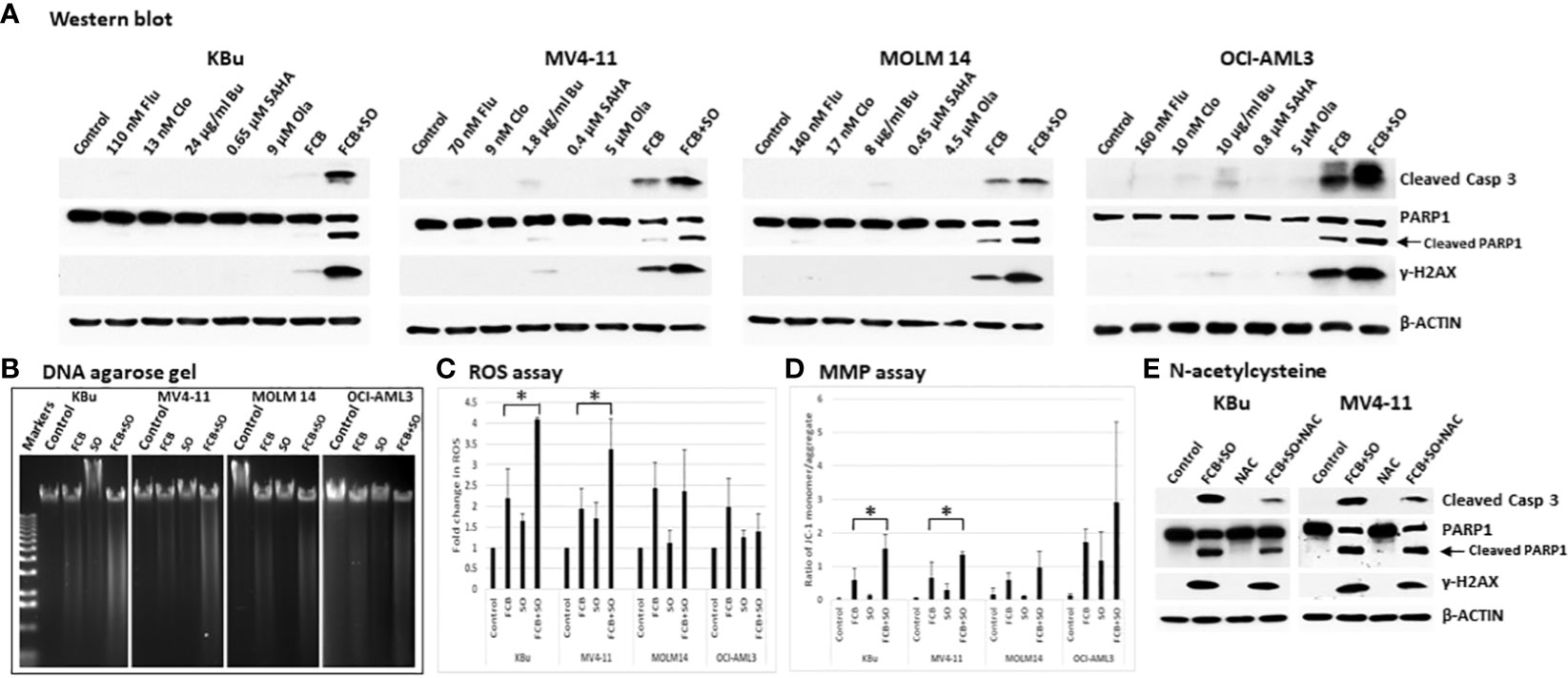
Figure 3 The five-drug combinations strongly activate apoptosis. Cells were exposed to the indicated drugs for 48 h, harvested, and analyzed for (A) molecular markers of apoptosis by Western blotting, (B) cleavage of DNA by agarose gel electrophoresis, (C) reactive oxygen species (ROS) production, and (D) changes in mitochondrial membrane potential (MMP). The results in panels (C, D) are averages of three independent experiments. P values less than 0.05 are indicated by an asterisk (*). (E) Cells were pre-exposed to 5 mM N-acetylcysteine (NAC) for 1 h prior to addition of the 5-drug combination and cells were analyzed by Western blotting after 48 h. Casp: Caspase; other abbreviations are the same as in Figure 2.
3.3 [Flu+Clo+Bu+SAHA+Ola] induces production of ROS
To further identify possible mechanisms of apoptosis activation, we examined the production of ROS in AML cells exposed to [Flu+Clo+Bu+SAHA+Ola]. Flow cytometric analysis showed an ~2-fold increase in ROS levels in KBu and MV4-11 cells exposed to [Flu+Clo+Bu] versus control cells, which further increased to ~3.5- to 4-fold versus control cells after exposure to the [Flu+Clo+Bu+SAHA+Ola] 5-drug combination (P < 0.05 in both cell lines); in contrast, no significant change (P > 0.05 in both cell lines) was observed when SAHA and Ola were added to the [Flu+Clo+Bu] combination in MOLM14 and OCI-AML3 cells (Figure 3C). Mitochondrial membrane potential (MMP) decreased in all four cell lines after exposure to [Flu+Clo+Bu+SAHA+Ola] whereas more modest effects were seen with the [Flu+Clo+Bu] combination, as suggested by the relative increases in monomer JC-1/aggregate JC-1 ratio (Figure 3D); the increase in the JC-1 ratio between [Flu+Clo+Bu] and [Flu+Clo+Bu+SAHA+Ola] treatments was statistically significant (P < 0.05) in KBu and MV4-11 cell lines. The decrease in MMP may cause pro-apoptotic factors to leak from the mitochondria and activate caspases. Indeed, the level of active Caspase 3 (based on its cleavage) also increased more in cells exposed to [Flu+Clo+Bu+SAHA+Ola] than to [Flu+Clo+Bu] (Figure 3A). This Caspase 3 activation and decreased MMP closely paralleled the triggering of apoptosis as shown by the Annexin V assay (Figure 2A). Overall, cell exposure to [Flu+Clo+Bu+SAHA+Ola] induced ROS production and decreased MMP, potentially contributing to the leakage of pro-apoptotic factors and activation of caspases, and committing cells to apoptosis as shown by the cleavage of Caspase 3 and PARP1, as well as by genomic DNA fragmentation (Figures 3A, B). The conclusion that ROS production and mitochondrial dysfunction are in part responsible for apoptosis induction was supported by the observation that treating KBu or MV4-11 cells with 5 mM N-acetylcysteine, an ROS scavenger, for 1 h prior to the 5-drug combination treatment did not completely inhibit apoptosis as shown by partial decrease in the levels of cleaved caspase 3 and γ-H2AX (Figure 3E).
3.4 The inhibition of PARylation mediated by [Flu+Clo+Bu+SAHA+Ola] does not depend on caspase activation
Since Caspase 3 was activated by the [Flu+Clo+Bu+SAHA+Ola] combination, we wished to determine if the observed inhibition of PARylation was caspase-dependent. To this effect, cells were exposed to an irreversible pan-caspase inhibitor, Z-VAD-FMK, 30 min prior to addition of the five-drug combination and analyzed by Western blotting and ELISA. Figure 4A shows PARylation of multiple proteins in both the control and [Flu+Clo+Bu] treatment groups. The [SAHA+Ola] and [Flu+Clo+Bu+SAHA+Ola] combinations significantly inhibited PARylation; however, addition of Z-VAD-FMK did not affect this drug-mediated inhibition (Figure 4A). Again, significant cleavage of Caspase 3 and PARP1 was observed in cells exposed to [Flu+Clo+Bu+SAHA+Ola] and addition of Z-VAD-FMK inhibited Caspase 3 cleavage in MV4-11 and MOLM14 cells but had little effect on PARP1 cleavage (Figure 4A). Interestingly, the phosphorylation of histone 2AX seen after [Flu+Clo+Bu+SAHA+Ola] treatment was markedly inhibited by Z-VAD-FMK in MV4-11 cells but not in MOLM14 cells (Figure 4A). Quantitative determination of the levels of PARylation by ELISA provided similar results (Figure 4B), suggesting that the observed inhibition of PARylation is caspase independent.
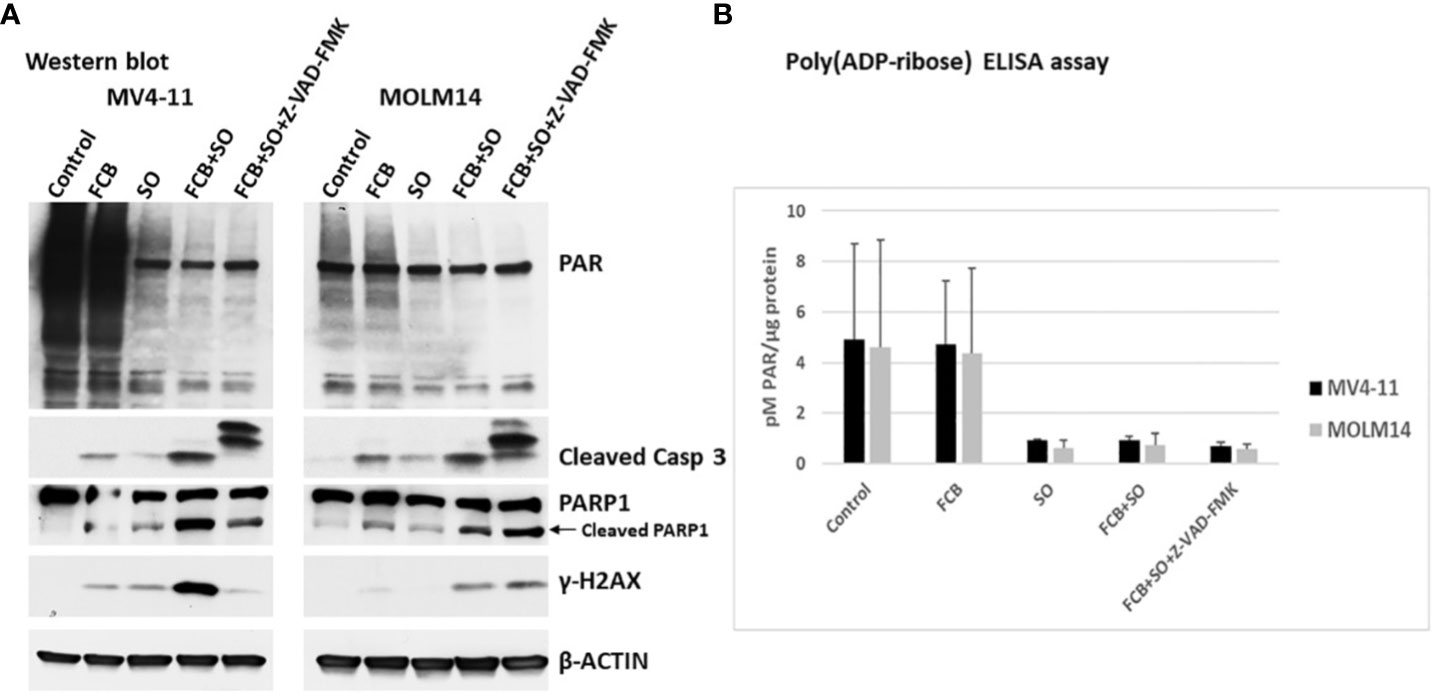
Figure 4 The inhibition of poly(ADP-ribosyl)ation by [Flu+Clo+Bu+SAHA+Ola] is caspase independent. Cells were exposed to the indicated drug combinations with or without 40 µM caspase inhibitor Z-VAD-FMK (added 30 min before the other drugs) for 48 h prior to (A) Western blot analysis and (B) Poly(ADP-ribose) ELISA assay. The results in panel B are averages of two independent experiments. Abbreviations and drugs concentrations are the same as in Figure 2. Casp, Caspase.
3.5 [Flu+Clo+Bu+SAHA+Ola] has similar synergistic effects in patient-derived leukemia cells
To assess the potential clinical extension of our results, cells from patients with leukemia were exposed to [Flu+Clo+Bu], [SAHA+Ola], or [Flu+Clo+Bu+SAHA+Ola] and analyzed by Western blotting. Increased cleavage of Caspase 3 was apparent in patient samples 1 and 3 exposed to the five-drug combination (patient 2 had protein loading problems as indicated by unequal levels of β-ACTIN), suggesting robust activation of the apoptosis pathway; a modest cleavage of PARP1 was observed in cells exposed to the 5-drug combination (Figure 1). Phosphorylation of histone 2AX was also observed in cells exposed to the 5-drug combination, suggesting DSB formation and/or activation of the DNA-damage response. These findings suggest a synergism of [Flu+Clo+Bu+SAHA+Ola] in cells derived from patients with leukemia involving mechanisms similar to those seen in the cultured cell lines. Additional patient samples may need to be analyzed to confirm the clinical relevance of these results.
3.6 [Flu+Clo+Bu+SAHA+Ola] combination affects the levels and phosphorylation of proteins involved in DNA damage response and repair
Acetylation and PARylation are known to occur in some proteins involved in DNA repair (27, 36). These post-translational modifications affect the stability of the proteins, as previously shown for UHFR1 and BRCA1 (37, 38). We, therefore, examined the effects of Flu, Clo, Bu, SAHA and Ola, individually or in combinations, on DNA damage response protein levels (total and phosphorylated) in AML cell lines. In general, the levels of phosphorylated and pan ATM (which functions in DNA DSB repair and cell cycle checkpoint activation) decreased in cells exposed to the five-drug combination; the level of BRCA1 (which functions in homologous recombination (HR) repair) greatly decreased (Figure 5). ATRX is a chromatin remodeling protein involved in HR (39) while DAXX has multiple functions in human cells, including regulation of DNA repair (40). The levels of both proteins decreased in AML cells exposed to the five-drug combination (Figure 5). While the level of the non-homologous end joining (NHEJ) repair protein DNAPKcs decreased in cells exposed to [Flu+Clo+Bu+SAHA+Ola], its phosphorylation at serine 2056 dramatically increased; the level of another NHEJ protein, Artemis, also decreased (Figure 5).
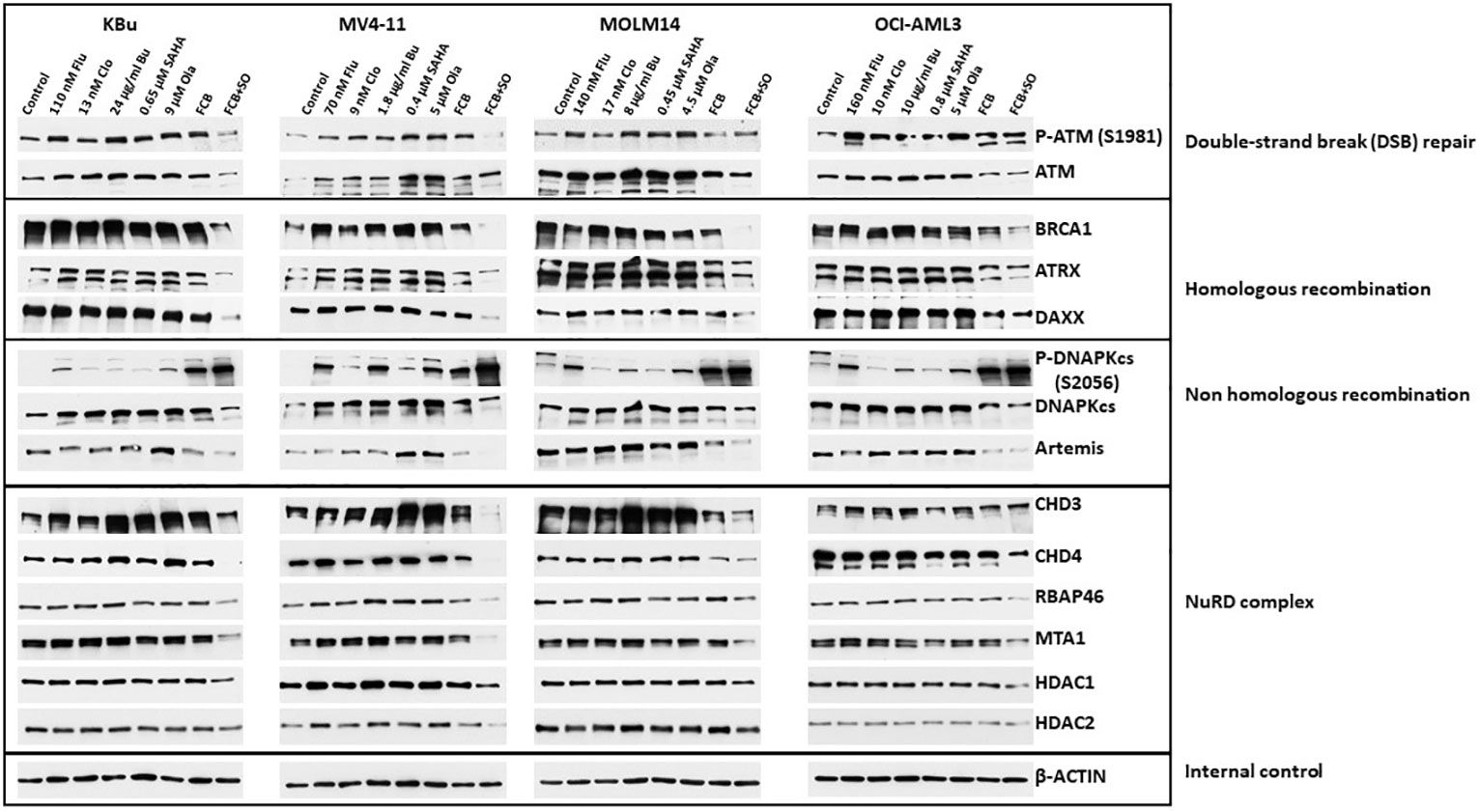
Figure 5 Effects of drugs on the levels and phosphorylation status of various proteins involved in DNA repair/DNA damage response. Cells were exposed to the indicated drug concentrations for 48 h prior to analysis by Western blotting. NuRD, Nucleosome Remodeling and Deacetylase.
The NuRD complex is involved in chromatin remodeling and deacetylation processes (41) and plays a key role in the cellular DNA damage response by regulating DNA damage signaling and repair events (42). The levels of the CHD3, CHD4, RBAP46, and MTA1 subunits of NuRD decreased in all cell lines exposed to [Flu+Clo+Bu+SAHA+Ola]; the HDAC1 subunit also modestly decreased, but HDAC2 was only minimally affected (Figure 5).
Of all the DNA damage response/repair protein events analyzed, the increased phosphorylation of DNAPKcs at serine 2056 was most intriguing. We, therefore, analyzed the kinetics of its phosphorylation. Figure 6A shows a marked increase in the level of P-DNAPKcs (S2056) within 6 h of exposure to [Flu+Clo+Bu+SAHA+Ola], similar to the kinetics of P53 phosphorylation at serine 15 and phosphorylation of histone 2AX in the MV4-11 and MOLM14 cell lines. These results are consistent with the reported function of DNAPKcs, which is to control the repair of the broken DNA ends and transmit the damage signal through P53 protein to induce cell cycle arrest and apoptosis (43). Whereas the level of P-DNAPKcs increased with drug treatment, the level of unphosphorylated DNAPKcs decreased within 24 h, which coincides with the cleavage of Caspase 3. DNAPKcs autophosphorylation at Ser2056 results in inactivation of its kinase activity and DNA repair ability (44–46). These results suggest significant effects of [Flu+Clo+Bu+SAHA+Ola] on the complex interactions among proteins involved in the DNA damage response, DNA repair, cell cycle, and programmed cell death.
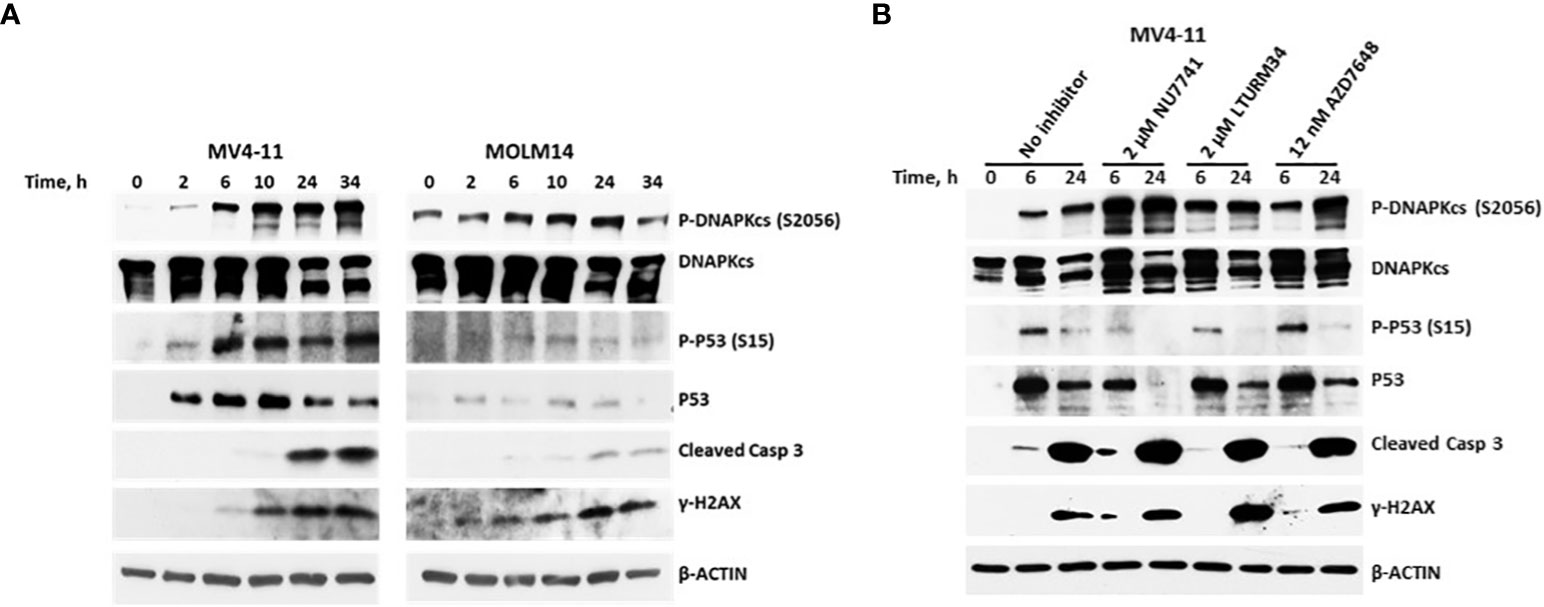
Figure 6 Kinetics of the phosphorylation of the DNA-dependent protein kinase catalytic subunit (DNAPKcs). (A) Cells were exposed to five-drug combinations for various times and analyzed by Western blotting. MV4-11 cells: 70 nM fludarabine, 9 nM clofarabine, 1.8 µg/ml busulfan, 0.4 µM SAHA and 5 µM olaparib. MOLM14 cells: 140 nM fludarabine, 17 nM clofarabine, 8 µg/ml busulfan, 0.45 µM SAHA and 4.5 µM olaparib. (B) MV4-11 cells were pre-exposed to the indicated DNAPK inhibitors for 1 h prior to addition of [Flu+Clo+Bu+SAHA+Ola] and analyzed by Western blotting after 6 h and 24 h. Casp, caspase.
To further analyze the possible interaction between DNAPK and P53, MV4-11 cells were exposed to DNAPK inhibitors prior to addition of the chemotherapy drugs. The inhibitors Nu7741, LTURM34 and AZD7648 increased the level of P-DNAPKcs (S2056) but decreased the level of pan P53 relative to the control (i.e., no inhibitor) after the 24 h five-drug exposure (Figure 6B), suggesting that the stability of P53 was compromised in the presence of DNAPK inhibitors. These findings are consistent with DNA-PK phosphorylating and stabilizing P53 in this setting as previously reported (47, 48).
4 Discussion
We recently reported the in vitro synergistic cytotoxicity of Bu, gemcitabine (Gem), melphalan (Mel), SAHA and Ola in lymphoma cells (31). A clinical trial of [Gem+Bu+Mel+SAHA+Ola] as a pre-transplant (salvage) regimen for autologous HSCT for 35 patients with refractory Hodgkin’s and Non-Hodgkin’s lymphomas was very promising, yielding ~90% event-free and overall survival rates with a median follow-up of 16 months, without adding any discernible risk to patient safety by the addition of SAHA+Ola (32). The significant efficacy of such a combination of chemotherapeutic agents, HDACi, and PARPi in lymphoma prompted us to determine their activity in leukemia cells and the potential benefit of this combination as part of pre-transplant regimens in the context of patients with acute leukemia who typically receive allogeneic HSCT. Using the combination [Flu+Clo+Bu] as a backbone, which has proven to be an efficacious pre-transplant regimen for AML patients receiving allogeneic HSCT (2–5, 49), the HDACi SAHA and the PARPi Ola were added with the expectation of further enhancing drug cytotoxicity. Indeed, the present study shows a synergistic cytotoxicity of [Flu+Clo+Bu+SAHA+Ola] in AML cells, which may be attributed to a combination of drug-induced histone modifications, decreased protein PARylation, DNA damage, increased ROS production, decreased MMP, and down-regulation of the levels of DNA repair proteins.
The low individual concentrations of Flu, Clo and SAHA used in these combination treatments with the expectation of ultimately minimizing patient toxicity in clinical trials may have provided the initial histone acetylation needed for chromatin remodeling, which in turn may have resulted in increased exposure of the DNA to Bu-mediated alkylation, as we described previously in the context of the so-called “Loop of Death” model for synergy between alkylating agents and nucleoside analogs in hematological malignancies (7, 50). The resulting DNA adducts may provide signals for additional histone acetylation to further open up the chromatin and increase the accessibility of the DNA to further alkylation, as per the “Loop of Death” model (Figure 7). These effects are presumably enhanced when Flu and Clo are incorporated into growing nascent DNA strands during replication via the same mechanism(s). This futile cycle of histone acetylation, DNA alkylation and nucleoside analog incorporation into DNA exacerbates DNA damage and potentially increases its complexity, making it more challenging for the DNA damage response to process correctly. This model does suggest the importance of drug sequencing for optimal anticancer efficacy, with the nucleoside analogs ideally being added prior to the alkylating agents. However, in the present study the cells were exposed concurrently to the various drugs, which thus represents a limitation in terms of its mechanistic implications.
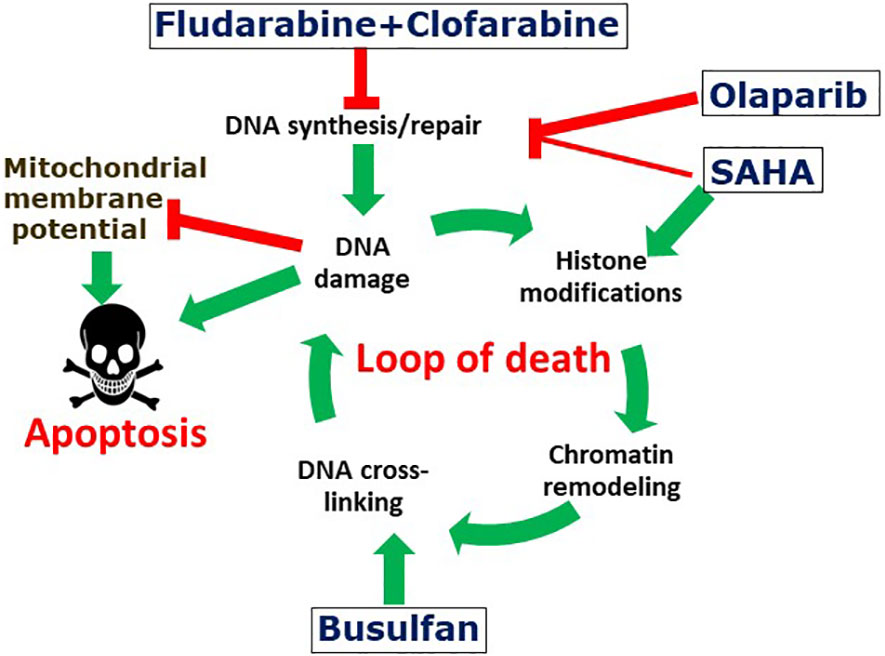
Figure 7 Suggested mechanism of the synergistic cytotoxicity of fludarabine, clofarabine, busulfan, SAHA and olaparib. The activated nucleoside analogs inhibit DNA synthesis and DNA repair, which results in complex DNA lesions. DNA strand breaks may cause histone modifications and chromatin remodeling, which may expose genomic DNA and make it potentially more accessible to DNA alkylation with busulfan. Attempts to repair DNA cross-links results in additional DNA strand breaks and the cycle is further amplified by additional DNA alkylations. The increased levels and complexity of DNA damage results in enhanced cell death. Olaparib and SAHA inhibit protein PARylation and histone deacetylation, respectively, resulting in inhibition of DNA repair and further enhancement of the cycle of DNA damage-repair and cell death. DNA damage is associated with decreased mitochondrial potential and consequently with increased apoptosis.
Consistent with this model, the complex DNA damage invoked by [Flu+Clo+Bu] strongly activates the DNA damage response as indicated by increased phosphorylation of histone 2AX, a response that is even more strongly activated in combination with the HDACi and PARPi (Figure 4). Repair of the damaged DNA is known to require the initial binding of PARP1 at the damage site(s) (51), after which PARP1 PARylates itself, histones and certain chromatin-associated proteins to provide a scaffold for recruitment of the DNA repair machinery (52). This process could be abrogated by the combined effects of SAHA and Ola through inhibition of PARylation via PARP1 trapping to chromatin, a mechanism that explains the cytotoxicity of the HDACi trichostatin A and the PARPi talazoparib in leukemia cells (53). The DNA damage inflicted by [Flu+Clo+Bu] combined with the high levels of oxidative and replication stress characteristic of many human cancers may render these AML cells highly dependent on protein acetylation and PARylation, and therefore extremely sensitive to the added [HDACi+PARPi] exposure (Figure 7).
One disadvantage of using high dosages of chemotherapeutic agents is their known tissue toxicities. Our data show that combining low dosages of these drugs and agents with different mechanisms of action enhanced synergistic cytotoxicity against leukemia cells. Addition of SAHA and Ola to [Flu+Clo+Bu] significantly induced apoptosis as indicated by increased Annexin V positivity and activation of Caspase 3 (Figures 2, 3). Since our clinical trial on [Gem+Bu+Mel+SAHA+Ola] in lymphoma resulted in minimal normal tissue toxicities, and notably with no cases of serious veno-occlusive disease (32), we anticipate that the [Flu+Clo+Bu+SAHA+Ola] combination will potentially result in low toxicities in AML patients undergoing HSCT. Further investigations, notably clinical trials, will be required to establish the therapeutic relevance of this drug combination. It should be noted however, that for such clinical trials, high-risk patients with relapsed/refractory AML and a low comorbidity index should be favored as the most optimal group in which to test the novel combination (4).
In summary, our data with [Flu+Clo+Bu+SAHA+Ola] provide evidence of interrelated mechanisms that commit AML cells to apoptosis following complex genotoxic insult. Since similar synergistic cytotoxicity of this 5-drug combination was observed in established AML cell lines and patient-derived cell samples, our results may be used as a hypothetical basis to justify a clinical trial to evaluate these drugs as part of pre-conditioning regimens for high-risk AML patients undergoing allogeneic HSCT.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Institutional Review Board of the UTMDACC. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
BV: Conceptualization, Formal Analysis, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing. BY: Data curation, Investigation, Methodology, Writing – original draft. DM: Data curation, Writing – review & editing. JR: Conceptualization, Formal Analysis, Writing – review & editing. YN: Conceptualization, Formal Analysis, Writing – review & editing. UP: Conceptualization, Formal Analysis, Writing – review & editing. XT: Conceptualization, Formal Analysis, Writing – review & editing. BA: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Project administration, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Part of this research was performed in the Flow Cytometry & Cellular Imaging Facility, which is supported in part by the National Institutes of Health through M.D. Anderson’s Cancer Center Support Grant CA016672. This work was also supported by the Stephen L. and Lavinia Boyd Fund for Leukemia Research and funds donated by grateful patients.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Valdez BC, Li Y, Murray D, Champlin RE, Andersson BS. The synergistic cytotoxicity of clofarabine, fludarabine and busulfan in AML cells involves ATM pathway activation and chromatin remodeling. Biochem Pharm (2011) 81:222–32. doi: 10.1016/j.bcp.2010.09.027
2. Andersson BS, Valdez BC, de Lima M, Wang X, Thall P, Worth LL, et al. Clofarabine ± Fludarabine with once daily IV busulfan as pretransplant conditioning therapy for advanced myeloid leukemia and MDS. Biol Blood Marrow Transplant. (2011) 17:893–900. doi: 10.1016/j.bbmt.2010.09.022
3. Rakszawski K, Miki K, Claxton D, Wagner H, Shike H, Mineishi S, et al. Clofarabine followed by haploidentical stem cell transplant using fludarabine, busulfan, and total-body irradiation with post-transplant cyclophosphamide in non-remission AML. Int J Hematol (2018) 108:348–50. doi: 10.1007/s12185-018-2431-5
4. Andersson BS, Thall PF, Ma J, Valdez BC, Bassett C Jr., Chen J, et al. A randomized phase III study of pretransplant conditioning for AML/MDS with fludarabine and once daily IV busulfan ± clofarabine in allogeneic stem cell transplantation. Bone Marrow Transplant. (2022) 57:1295–303. doi: 10.1038/s41409-022-01705-7
5. Versluijs AB, de Koning CCH, Lankester AC, Nierkens S, Kollen WJ, Bresters D, et al. Clofarabine-fludarabine-busulfan in HCT for pediatric leukemia: an effective, low toxicity, TBI-free conditioning regimen. Blood Adv (2022) 6:1719–30. doi: 10.1182/bloodadvances.2021005224
6. Shi Z, Azuma A, Sampath D, Li YX, Huang P, Plunkett W. S-Phase arrest by nucleoside analogues and abrogation of survival without cell cycle progression by 7-hydroxystaurosporine. Cancer Res (2001) 61:1065–72.
7. Valdez BC, Andersson BS. Interstrand crosslink inducing agents in pretransplant conditioning therapy for hematologic Malignancies. Environ Mol Mutagen (2010) 51:659–68. doi: 10.1002/em.20603
8. Huang P, Chubb S, Plunkett W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. A mechanism for cytotoxicity. J Biol Chem (1990) 265:16617–25. doi: 10.1016/S0021-9258(17)46267-3
9. Galaup A, Paci A. Pharmacology of dimethanesulfonate alkylating agents: busulfan and treosulfan. Expert Opin Drug Metab Toxicol (2013) 9:333–47. doi: 10.1517/17425255.2013.737319
10. Rambaldi A, Grassi A, Masciulli A, Boschini C, Micò MC, Busca A, et al. Busulfan plus cyclophosphamide versus busulfan plus fludarabine as a preparative regimen for allogeneic haemopoietic stem-cell transplantation in patients with acute myeloid leukaemia: an open-label, multicentre, randomised, phase 3 trial. Lancet Oncol (2015) 16:1525–36. doi: 10.1016/S1470-2045(15)00200-4
11. Valdez BC, Murray D, Nieto Y, Li Y, Wang G, Champlin RE, et al. Synergistic cytotoxicity of the DNA alkylating agent busulfan, nucleoside analogs and suberoylanilide hydroxamic acid in lymphoma cell lines. Leuk Lymphoma. (2012) 53:973–81. doi: 10.3109/10428194.2011.634043.1.e2
12. Song G, Valdez BC, Li Y, Dominguez JR, Corn P, Champlin RE, et al. The histone deacetylase inhibitor SAHA sensitizes acute myeloid leukemia cells to a combination of nucleoside analogs and the DNA-alkylating agent busulfan. Leuk Lymphoma. (2014) 55:1625–34. doi: 10.3109/10428194.2013.856007
13. Valdez BC, Li Y, Murray D, Ji J, Liu Y, Popat U, et al. Comparison of the cytotoxicity of cladribine and clofarabine when combined with fludarabine and busulfan in AML cells: Enhancement of cytotoxicity with epigenetic modulators. Exp Hematol (2015) 43:448–56. doi: 10.1016/j.exphem.2015.02.001
14. Valdez BC, Brammer JE, Li Y, Murray D, Teo EC, Liu Y, et al. Romidepsin enhances the cytotoxicity of fludarabine, clofarabine and busulfan combination in Malignant T-cells. Leuk Res (2016) 47:100–8. doi: 10.1016/j.leukres.2016.05.019
15. Valdez BC, Murray D, Yuan B, Nieto Y, Popat U, Andersson BS. ABT199/venetoclax potentiates the cytotoxicity of alkylating agents and fludarabine in acute myeloid leukemia cells. Oncotarget (2022) 13:319–30. doi: 10.18632/oncotarget.28193
16. Song G, Valdez BC, Li Y, Liu Y, Champlin RE, Andersson BS. Synergistic cytotoxicity of sorafenib with busulfan and nucleoside analogs in human FMS-like tyrosine kinase 3 internal tandem duplications-positive acute myeloid leukemia cells. Biol Blood Marrow Transplant. (2014) 20:1687–95. doi: 10.1016/j.bbmt.2014.08.003
17. Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res (2011) 3:166–79.
18. Montalban-Bravo G, Garcia-Manero G. Novel drugs for older patients with acute myeloid leukemia. Leukemia (2015) 29:760–9. doi: 10.1038/leu.2014.244
19. Nieto Y, Valdez BC, Thall PF, Jones RB, Wei W, Myers A, et al. Double epigenetic modulation of high-dose chemotherapy with azacitidine and vorinostat for patients with refractory or poor-risk relapsed lymphoma. Cancer (2016) 122:2680–8. doi: 10.1002/cncr.30100
20. San José-Enériz E, Gimenez-Camino N, Agirre X, Prosper F. HDAC nhibitors in acute myeloid leukemia. Cancers (Basel). (2019) 11:1794. doi: 10.3390/cancers11111794
21. Valdez BC, Li Y, Murray D, Brammer J, Liu Y, Hosing C, et al. Differential effects of histone deacetylase inhibitors on cellular drug transporters and their implications for using epigenetic modifiers in combination chemotherapy. Oncotarget (2016) 7:63829–38. doi: 10.18632/oncotarget.11561
22. Chiappella A, Dodero A, Evangelista A, Re A, Orsucci L, Usai SV, et al. Romidepsin-CHOEP followed by high-dose chemotherapy and stem-cell transplantation in untreated Peripheral T-Cell Lymphoma: results of the PTCL13 phase Ib/II study. Leukemia (2023) 37:433–40. doi: 10.1038/s41375-022-01780-1
23. Ramdial JL, Valdez BC, Andersson BS, Nieto Y. Romidepsin antagonism with CHOEP. Leukemia (2023) 37:1160–1. doi: 10.1038/s41375-023-01873-5
24. Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature (2003) 421:499–506. doi: 10.1038/nature01368
25. Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol (2013) 20:317–25. doi: 10.1038/nsmb.2499
26. Gkotzamanidou M, Terpou E, Kentepozidis N, Terpos E. Targeting the interplay between HDACs and DNA damage repair for myeloma therapy. Int J Mol Sci (2021) 22:10406. doi: 10.3390/ijms221910406
27. Liszczak G, Diehl KL, Dann GP, Muir TW. Acetylation blocks DNA damage-induced chromatin ADP-ribosylation. Nat Chem Biol (2018) 14:837–40. doi: 10.1038/s41589-018-0097-1
28. Caldecott KW. Protein ADP-ribosylation and the cellular response to DNA strand breaks. DNA Repair (Amst). (2014) 19:108–13. doi: 10.1016/j.dnarep.2014.03.021
29. Tallis M, Morra R, Barkauskaite E, Ahel I. Poly(ADP-ribosyl)ation in regulation of chromatin structure and the DNA damage response. Chromosoma (2014) 123:79–90. doi: 10.1007/s00412-013-0442-9
30. Min A, Im SA, Kim DK, Song SH, Kim HJ, Lee KH, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res (2015) 17:33. doi: 10.1186/s13058-015-0534-y
31. Valdez BC, Nieto Y, Yuan B, Murray D, Andersson BS. HDAC inhibitors suppress protein poly(ADP-ribosyl)ation and DNA repair protein levels and phosphorylation status in hematologic cancer cells: implications for their use in combination with PARP inhibitors and chemotherapeutic drugs. Oncotarget (2022) 13:1122–35. doi: 10.18632/oncotarget.28278
32. Nieto Y, Valdez BC, Thall PF, Ramdial JL, Srour SA, Hosing C, et al. High efficacy of the PARP inhibitor olaparib combined with high-dose chemotherapy and autologous stem-cell transplant for refractory lymphomas. Blood (2022) 140(Supplement 1):1183–4. doi: 10.1182/blood-2022-156121
33. Andersson BS, Bergerheim US, Collins VP, Childs C, Beran M, Sen S, et al. KBM-3, an in vitro model of human acute myelomonocytic leukemia. Exp Hematol (1992) 20:361–7.
34. Valdez BC, Li Y, Murray D, Liu Y, Nieto Y, Bashir Q, et al. Panobinostat and venetoclax enhance the cytotoxicity of gemcitabine, busulfan, and melphalan in multiple myeloma cells. Exp Hematol (2020) 81:32–41. doi: 10.1016/j.exphem.2020.01.003
35. Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell (1997) 89:175–84. doi: 10.1016/s0092-8674(00)80197-x
36. Curtin NJ, Szabo C. Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat Rev Drug Discovery (2020) 19:711–36. doi: 10.1038/s41573-020-0076-6
37. Ha K, Fiskus W, Choi DS, Bhaskara S, Cerchietti L, Devaraj SG, et al. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget (2014) 5:5637–50. doi: 10.18632/oncotarget.2154
38. Yin L, Liu Y, Peng Y, Peng Y, Yu X, Gao Y, et al. Correction to: PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/. BRCA1 DNA damage repair complex in prostate cancer cells. J Exp Clin Cancer Res (2022) 41:72. doi: 10.1186/s13046-022-02290-9
39. Elbakry A, Löbrich M. Homologous recombination subpathways: A tangle to resolve. Front Genet (2021) 12:723847. doi: 10.3389/fgene.2021.723847
40. Mahmud I, Liao D. DAXX in cancer: phenomena, processes, mechanisms and regulation. Nuc Acids Res (2019) 47:7734–52. doi: 10.1093/nar/gkz634
41. Gursoy-Yuzugullu O, House N, Price BD. Patching broken DNA: Nucleosome dynamics and the repair of DNA breaks. J Mol Biol (2016) 428:1846–60. doi: 10.1016/j.jmb.2015.11.021
42. Smeenk G, Wiegant WW, Vrolijk H, Solari AP, Pastink A, van Attikum H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J Cell Biol (2010) 190:741–9. doi: 10.1083/jcb.201001048
43. Yue X, Bai C, Xie D, Ma T, Zhou P-K. DNA-PKcs: A multi-faceted player in DNA damage response. Front Genet (2020) 11:607428. doi: 10.3389/fgene.2020.607428
44. Chan DW, Lees-Miller SP. The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J Biol Chem (1996) 271:8936–41. doi: 10.1074/jbc.271.15.8936
45. Douglas P, Sapkota GP, Morrice N, Yu Y, Goodarzi AA, Merkle D, et al. Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA-dependent protein kinase. Biochem J (2002) 368(Pt 1):243–51. doi: 10.1042/BJ20020973
46. Jiang W, Crowe JL, Liu X, Nakajima S, Wang Y, Li C, et al. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol Cell (2015) 58:172–85. doi: 10.1016/j.molcel.2015.02.024
47. Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell (1997) 91:325–34. doi: 10.1016/s0092-8674(00)80416-x
48. Boehme KA, Kulikov R, Blattner C. p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc Natl Acad Sci U S A. (2008) 105:7785–90. doi: 10.1073/pnas.0703423105
49. Alatrash G, Saberian C, Bassett R, Thall PF, Ledesma C, Lu Y, et al. Vorinostat combined with busulfan, fludarabine, and clofarabine conditioning regimen for allogeneic hematopoietic stem cell transplantation in patients with acute leukemia: Long-term study outcomes. Transplant Cell Ther (2022) 28:501. doi: 10.1016/j.jtct.2022.05.021
50. Andersson BS, Valdez BC, Jones RB. Pharmacologic basis for high-dose chemotherapy. In: Forman SJ, Negrin RS, Antin JH, Appelbaum FR, editors. Thomas’ Hematopoietic Cell Transplantation, 5th Ed. West Sussex, UK: John Wiley & Sons, Ltd (2016). p. 211–22.
51. Schützenhofer K, Rack JGM, Ahel I. The making and breaking of serine-ADP-ribosylation in the DNA damage response. Front Cell Dev Biol (2021) 9:745922. doi: 10.3389/fcell.2021.745922
52. De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol (2012) 84:137–46. doi: 10.1016/j.bcp.2012.03.018
Keywords: alkylating agent, nucleoside analog, HDAC inhibitor, PARP inhibitior, stem cell transplant (SCT), pre-transplant regimen
Citation: Valdez BC, Yuan B, Murray D, Ramdial JL, Nieto Y, Popat U, Tang X and Andersson BS (2023) Synergistic cytotoxicity of fludarabine, clofarabine, busulfan, vorinostat and olaparib in AML cells. Front. Oncol. 13:1287444. doi: 10.3389/fonc.2023.1287444
Received: 01 September 2023; Accepted: 08 November 2023;
Published: 23 November 2023.
Edited by:
Jean El Cheikh, American University of Beirut Medical Center, LebanonReviewed by:
Dali Zong, National Cancer Institute (NIH), United StatesTamim Alsuliman, Hôpital Saint-Antoine, France
Copyright © 2023 Valdez, Yuan, Murray, Ramdial, Nieto, Popat, Tang and Andersson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benigno C. Valdez, YnZhbGRlekBtZGFuZGVyc29uLm9yZw==