 Griselda Metta Yvone1,2
Griselda Metta Yvone1,2 Joshua J. Breunig1,2,3,4,5*
Joshua J. Breunig1,2,3,4,5*- 1Board of Governors Regenerative Medicine Institute, Cedars-Sinai Medical Center, Los Angeles, CA, United States
- 2Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, United States
- 3Center for Neural Sciences in Medicine, Cedars-Sinai Medical Center, Los Angeles, CA, United States
- 4Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Medical Center, Los Angeles, CA, United States
- 5Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, United States
Pediatric low-grade gliomas represent the most common childhood brain tumor class. While often curable, some tumors fail to respond and even successful treatments can have life-long side effects. Many clinical trials are underway for pediatric low-grade gliomas. However, these trials are expensive and challenging to organize due to the heterogeneity of patients and subtypes. Advances in sequencing technologies are helping to mitigate this by revealing the molecular landscapes of mutations in pediatric low-grade glioma. Functionalizing these mutations in the form of preclinical models is the next step in both understanding the disease mechanisms as well as for testing therapeutics. However, such models are often more difficult to generate due to their less proliferative nature, and the heterogeneity of tumor microenvironments, cell(s)-of-origin, and genetic alterations. In this review, we discuss the molecular and genetic alterations and the various preclinical models generated for the different types of pediatric low-grade gliomas. We examined the different preclinical models for pediatric low-grade gliomas, summarizing the scientific advances made to the field and therapeutic implications. We also discuss the advantages and limitations of the various models. This review highlights the importance of preclinical models for pediatric low-grade gliomas while noting the challenges and future directions of these models to improve therapeutic outcomes of pediatric low-grade gliomas.
1 Introduction
Pediatric low-grade glioma (pLGG, WHO grade 1 or 2) is the most common brain tumor class in children (1–4). Although in general, prognosis, overall survival, and outcomes following treatment are better for pLGG than adult gliomas, the broad spectrum and heterogeneity of pLGG histopathology make these tumors challenging to treat (1, 3–6). For example, these gliomas may differ in their brain locations, histological spectrum, cell(s) of origin, genetic alterations, and the tumor microenvironment milieu (4). Additionally, since many pLGGs undergo senescence towards juvenile period or adulthood, a lot of pLGG patients end up not getting surgeries, making it harder to obtain tumor samples to be studied. For tumors located in brain areas accessible by surgery such as the cerebellum, total resection is a viable option and may be curative, but for less accessible tumors such as those in hypothalamus, midline areas, and optic pathway, additional treatments such as radiation and cytotoxic chemotherapies may be necessary (1, 7). Even with gross total resection, recurrence may still happen, and additional treatments may lead to allergies and long-term side effects (5, 7, 8). Furthermore, since pLGGs are chronic diseases, these children sustain significant life-long morbidity and general reduction in quality of life. Due to the complexity of the molecular landscape of pLGG, continuous efforts are directed towards characterizing the genomic and epigenomic alterations in these gliomas, and developing effective preclinical models are paramount in making informed decisions on therapeutic options.
2 Molecular landscape of pLGG
Advances in sequencing technologies have aided in classifying the diverse spectrum of pLGG histopathology. The various types of pLGG that are more often encountered are summarized in Table 1, including their locations and most common genetic alterations. pLGGs can be glial or mixed glial-neuronal tumors. Pilocytic astrocytomas (PAs) predominate in children younger than 15 years of age and is considered the most common type of pLGG (1, 7). However, it is still very challenging to accurately diagnose and stratify pLGG patients due to the broad spectrum of the histopathology and the complex molecular landscape (1, 4, 9, 14, 18). Moreover, in many cases, there are overlapping morphologies between the different groups of pLGGs, and even the more circumscribed tumors can contain infiltrative areas (4, 9, 14). The majority of pLGGs show convergence in MAPK (Mitogen-activated protein kinase)/ERK (Extracellular signal-regulated kinase) pathway alterations, as illustrated in Figure 1 and the next section will discuss in more details the genetic alterations found in various pLGGs based on advancement in molecular profiling studies (10, 11, 19).
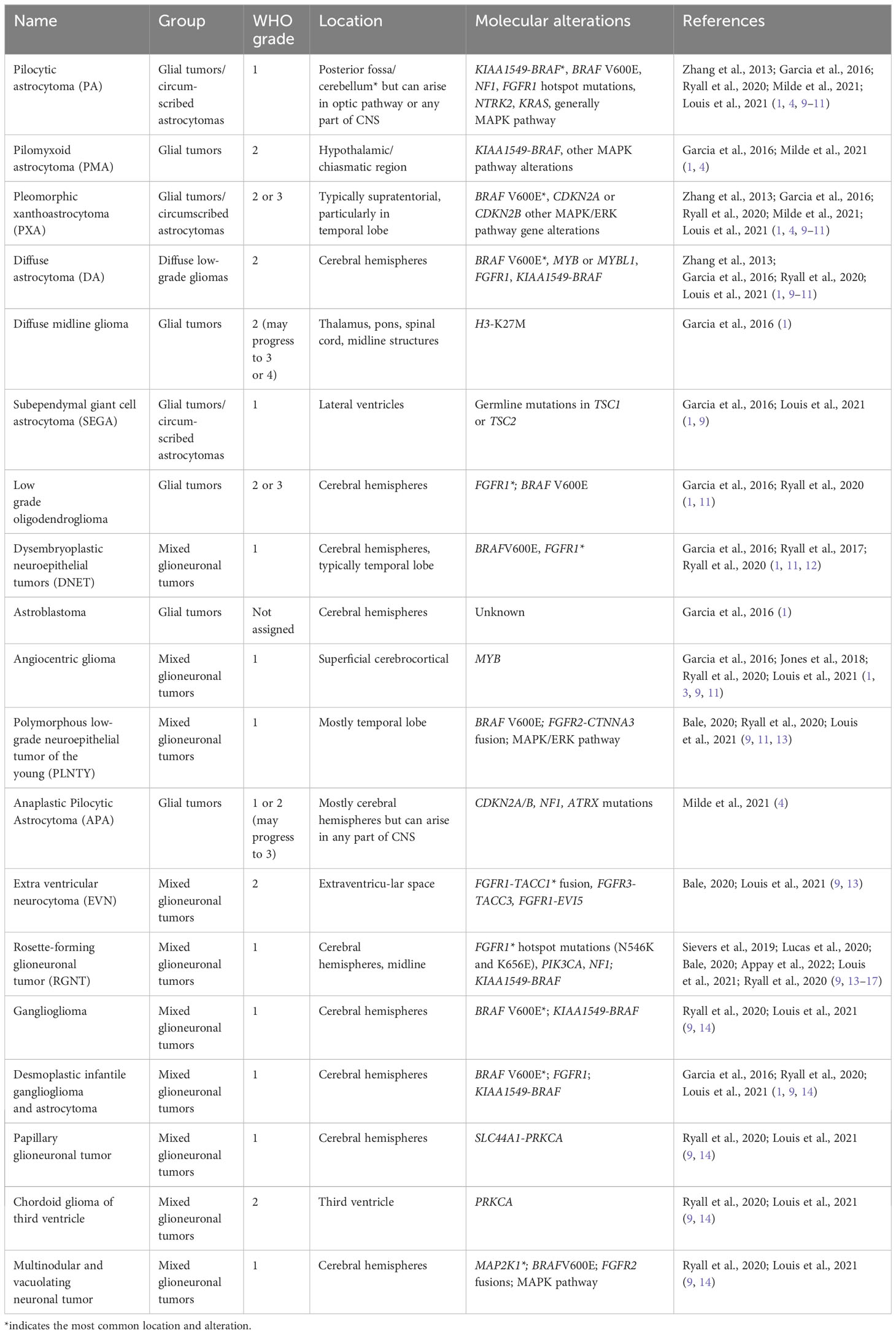
Table 1 pLGGs comprise heterogeneous histopathological and molecular alterations.
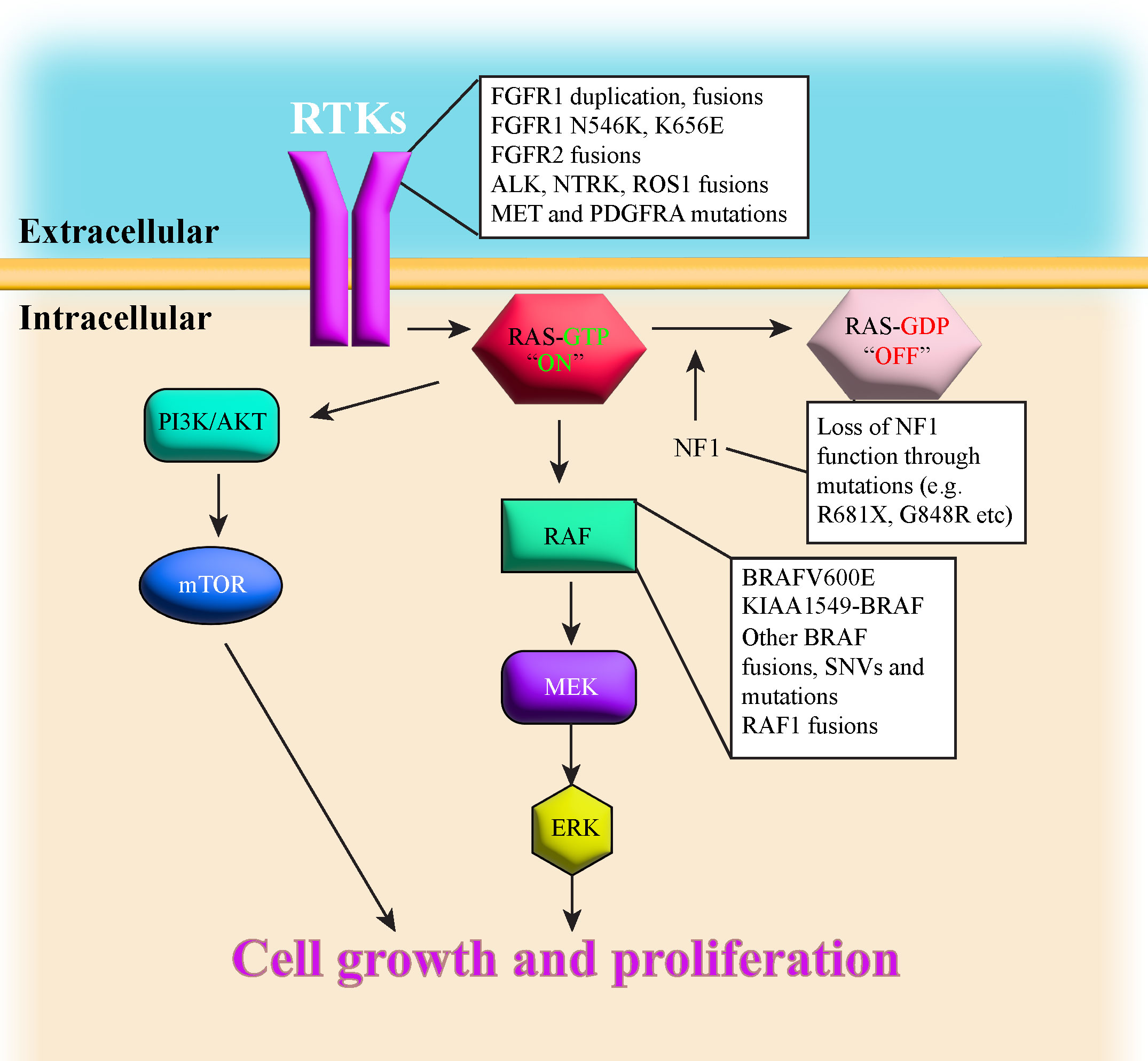
Figure 1 illustrates a simplified schematic of the MAPK signaling pathway. Many components of the MAPK pathway are altered in pLGGs.
2.1 BRAF
B-raf proto-oncogene (BRAF) encodes a protein belonging to the RAF family of serine/threonine protein kinases. This protein is part of the MAPK/ERK signaling pathway, which affects cell division, differentiation, transcription, and many other cellular processes (6).
Fusion with KIAA1549 gene (KIAA1549-BRAF) and BRAF V600E (Valine to Glutamic Acid) mutation are the most frequent BRAF alterations found in pLGGs, with KIAA1549-BRAF being almost exclusively a single-event driver (11). Other rarer BRAF alterations include fusions with partners other than KIAA1549 (FAM131B, RNF130, CLCN6, MKRN1, GNA11, QKI, FZR1, and MACF1) that mainly result in loss of the N-terminal regulatory region of the BRAF protein and retention of the kinase domain (19, 20), insertion at position 600 (V600ins), and single nucleotide variant (SNV) at position 594 (D594N (11). Jones et al. (2013) also found three amino acid insertion (Valine-Leucine-Arginine) that leads to stabilization of a dimeric form of BRAF and increased ERK phosphorylation, equivalent to the BRAF V600E mutant (19).
KIAA1549-BRAF fusion is the most common alteration found in PA and cerebellar tumors (10, 11, 19). This fusion is characteristic of sporadic juvenile PA (JPA) and does not occur in NF1-PA (PA with mutations in NF1 gene (21). The fusion occurs due to tandem duplication of BRAF gene that results in an in-frame fusion gene incorporating the BRAF kinase domain (22). The N-terminal end of the KIAA1549 protein replaces the N-terminal regulatory region of BRAF protein (5’ end of KIAA1549 gene and 3’ end of BRAF gene), leading to constitutive activation of the BRAF kinase domain (20, 22). The most common KIAA1549-BRAF fusion involved exons 1-16 of KIAA1549 gene and exons 9-18 of BRAF gene (16:9), although fusion between KIAA1549 exon 15 and BRAF exon 9 (15:9) was observed in hemispheric tumors and was associated with a worse progression-free survival compared with other fusions (11, 20, 22). A few infants with 15:11 fusion rapidly progressed and died (11).
BRAF V600E, another common mutation found in PAs, is often associated with additional alterations such as deletion in the tumor suppressor gene CDKN2A (Cyclin dependent kinase inhibitor 2A), SNVs in NF1, FGFR1 (Fibroblast growth factor receptor 1), KRAS (Kirsten rat sarcoma viral oncogene homolog), and H3F3A (H3 histone family member 3A), but never with fusion events. Interestingly, the combination of BRAF V600E and CDKN2A loss likely leads to tumor transformation and higher tumor grade (12). BRAF V600E mutation is found in multiple types of pLGGs such as ganglioglioma, diffuse astrocytoma (DA) and Pleomorphic Xanthoastrocytoma (PXA). BRAF V600E-driven pLGG most frequently occurs in the cerebral hemispheres and diencephalon (11).
Ryall et al. (2020) found that patients with KIAA1549-BRAF fusion have better outcome compared to those with BRAF V600E mutation in terms of progression-free survival. However, patients with BRAF V600E coupled with CDKN2A deletion tend to progress and succumb to their disease (11).
2.2 NF1
Patients with Neurofibromatosis type 1(NF1) suffer from a dominantly inherited genetic disease. These patients develop benign tumors, termed neurofibromas, along the nerves of body (23). Neurofibromatosis type 1 is considered as one of the most common genetic disorders in humans (7). Neurofibromin 1, the protein product of the NF1 gene, is a negative regulator of RAS (Rat sarcoma virus) in the MAPK/ERK pathway as it mediates the conversion of activated Ras-GTP to inactive Ras-GDP [ (24–27), Figure 1]. Loss of NF1 activity leads to hyperactivation of downstream RAS effectors (28), thus NF1 is considered a tumor suppressor gene. NF1-associated astrocytomas usually sustain germline mutation of the NF1 gene located on chromosome 17q and somatic loss of the remaining NF1 allele, resulting in bi-allelic inactivation and loss-of-function to the NF1 gene (7, 8, 29, 30). Children with NF1 are predisposed and may develop PAand optic pathway glioma (OPG (7, 10, 11, 23, 31), although they can also develop DA (8). NF1-associated PAs are most often found in the optic pathway or hypothalamus, whereas non NF1-PA are usually located in cerebellum (7). Sporadic OPGs tend to progress, are more aggressive and usually require treatment, but most NF1-associated OPGs grow slowly, are more indolent and thus don’t require treatment (8). NF1-associated OPGs are usually presented in early childhood and rarely continue to grow or cause symptoms after age 10 (32, 33). In general, the prognosis for children with NF1-associated LGGs is good, as most of these tumors are asymptomatic and require no therapeutic intervention. Additionally, children with NF1-LGGs who receive therapy tend to have better progression-free survival than their sporadic counterparts (8).
2.3 FGFR
The Fibroblast Growth Factor Receptor (FGFR) family consists of four transmembrane tyrosine kinase receptors (FGFR1-4) that dimerize in response to ligands, triggering downstream pathways including MAPK and phosphatidylinositol-3-kinase (PI3K)/AKT (Protein Kinase B) pathways implicated in tumorigenesis (13). FGFR signaling plays a role in angiogenesis, tumor cell migration, differentiation, proliferation and survival (13).
There are multiple types of FGFR1 alterations described in pLGGs including tyrosine kinase domain duplication (FGFR1 TKDD), fusions (FGFR1-TACC1), and FGFR1 hotspot mutations (N546K and K656E). FGFR1 TKDD appears to be largely absent in high-grade gliomas (HGG (10), but it’s been reported in rosette-forming glioneuronal tumor (RGNT), anaplastic PA, glioneuronal tumor with PA and PXA features (34, 35). FGFR1 TKDD includes exons 10-18, producing an in-frame fusion and duplicates the entire FGFR1 region encoding tyrosine kinase domain (10, 13, 36).
Meanwhile, FGFR1-TACC1 fusions have been reported in extraventricular neurocytoma (EVN) and in cerebral hemispheric PA (10, 13). Other fusion events involving FGFR1 (37) and FGFR2 (e.g. FGFR2-CTNNA3) have also been found in polymorphous low grade neuroepithelial tumor of the young (PLNTY (13). Other FGFR2 fusions reported include FGFR2-INA and FGFR2-ERC1 (11).
FGFR fusions or duplications usually lead to constitutive FGFR activity and activation of downstream pathways such as MAPK/PI3K/mechanistic target of rapamycin (mTOR (10, 12, 13, 19). FGFR1 TKD and FGFR2 fusions predominate in glioneuronal or oligodendroglial tumors and mostly occur in the cerebral hemispheres (11, 13).
Patients with FGFR1-TACC1 fusion or TKD duplications tend to have better progression-free survival compared to patients with FGFR1 SNVs (11).
FGFR1 hotspot mutations mainly consist of N546K (Asparagine to Lysine) and K656E (Lysine to Glutamic acid). These mutations are most commonly found in midline tumors such as dysembryoplastic neuroepithelial tumors (DNETs (10, 19, 38–40), but they have been reported in posterior fossa PA with widespread oligodendroglial features (15), extracerebellar PA (19), RGNTs (16, 17) and diffuse midline gliomas along with H3K27M mutations (41). These FGFR1 hotspot mutations can also co-occur with other genetic alterations including NF1, other FGFR1 point mutations or other RAS/MAPK pathway alterations (10, 13).
It appears that while pLGG patients with FGFR gene family alterations may progress and have worse outcome, the tumors rarely result in deaths (11, 42).
2.4 MYB
The MYB (Myeloblastosis) gene family consists of MYB, MYBL1 (MYB proto-oncogene like 1), and MYBL2, encoding the transcription factors MYB (c-MYB), MYBL1 (A-MYB), and MYBL2 (B-MYB (43). Since MYB proteins are essential for cellular growth, differentiation, and survival, they have been found to be aberrantly expressed in human cancers (43).
Tatevossian et al. (2010) reported alterations in MYB/MYBL1 in pediatric diffuse glioma (44). Rearrangements and copy number abnormality in MYB or MYBL1 resulting in upregulated MYB or MYBL1 are found in more infiltrative pLGGs including grade 2 DAs, angiocentric gliomas and oligodendroglioma (10, 44, 45). MYB alterations are associated with a deletion of a 3’ portion of MYB gene, involving either the negative regulatory region or 3’ UTR microRNA binding sites (10). Bandopadhayay et al. (2016) found a novel MYB-QKI (K-homology domain containing RNA binding) fusion event in angiocentric glioma (46). Interestingly, no MYB alterations are identified in pediatric HGGs or ependymomas (10). Although progressions are rare in MYB-altered tumors, they were more frequent in MYBL1-altered tumors (11).
2.5 Other mutations: NTRK2, TSC, H3K27M, KRAS
Fusions in other Receptor Tyrosine Kinases (RTKs) are rare in pLGGs and may include ALK (Anaplastic lymphoma kinase) gene fusions (CCDC88A-ALK, PPP1CB-ALK), ROS1 (ROS proto-oncogene) fusions (GOPC-ROS1), NTRK2 (Neurotrophic tyrosine receptor kinase)/TRKB (Tropomycin receptor kinase B) fusions (NTRK2-MID1, NTRK2-SF3B1) and PDGFB fusions (PDGFB-LRP1). These fusions mainly occur in cerebral hemispheric tumors, although ROS1 fusions are also seen in intraventricular space. Patients with these alterations rarely progress and/or succumb to their diseases (11).
Alterations in other members of the RAS/MAPK pathway have been documented in pLGG cases including RAF1 fusions, KRAS mutations, and MAP2K1 deletions (11).
Some pLGG patients also exhibit alterations in other RTKs including mutations in MET or PDGFRA, ALK, NTRK2 (TRKB). Jones et al. (2013) identified QKI-NTRK2 and NACC2-NTRK2 fusions in pLGGs, resulting in ligand-independent dimerization and indirectly activation of MAPK pathway (19).
Another small percentage of patients have H3F3A mutations (11), and it is most often K27M. The tumors are restricted to the midline and brainstem and tend to be DAs. H3K27M also often co-occurs with other alterations, most often with BRAF V600E. These patients tend to progress early (11).
IDH1 R132H mutations, while very common in adult lower-grade gliomas, are extremely rare in pLGGs. These tumors tend to occur in the cerebral hemispheres and are either oligodendroglioma or diffuse astrocytoma, and the patients may progress (11).
3 In vitro models of pLGG: the path to establishing patient-derived pLGG cell lines for preclinical testing
As sequencing technologies advance, more mutations are continuously discovered in various types of pLGGs, complicating their molecular landscape. Moreover, different mutations may cause the different tumors to respond variably to targeted therapeutic agents, making risk stratification and development of therapies for pLGG difficult. Efforts to develop preclinical models of pLGGs are mainly focused on the more common molecular alterations including but not limited to KIAA1549-BRAF, BRAF V600E, and NF1 mutations, but many have not been successful. Table 2 lists the in vitro models that have been developed for preclinical testing and their varying degrees of success. One obstacle in generating viable pLGG patient-derived cell lines is the lack of availability of patient tissues. Tumors located in areas difficult to surgically excise are often not biopsied, making it difficult to obtain sufficient samples from these patients to generate cell lines (4). Another major obstacle in generating viable cell lines for non-NF1-pLGG (e.g. PA) is the oncogene-induced-senescence (OIS) phenomenon [ (48, 49), Figure 2]. Expression of BRAF V600E and KIAA1549-BRAF fusion led to MAPK pathway activation and subsequent OIS in human cortical neurospheres, human immortalized astrocytes, fetal astrocytes, and low-passage primary PA cultures [ (48, 49), Table 2]. The OIS phenomenon, however, may be the rationale for the lack of progression of PA to higher-grade tumors, in the absence of additional cooperating mutations (48, 49). Factors like senescence-associated secretory phenotype (SASP) and microRNAs have been implicated in senescence of pLGG cell lines (58, 59). On the other hand, inactivation of CDKN2A gene encoding the tumor suppressor p16INK4a might facilitate escape from senescence in these cells, explaining the association between p16INK4a loss and worse outcomes in PA patients (48, 49).
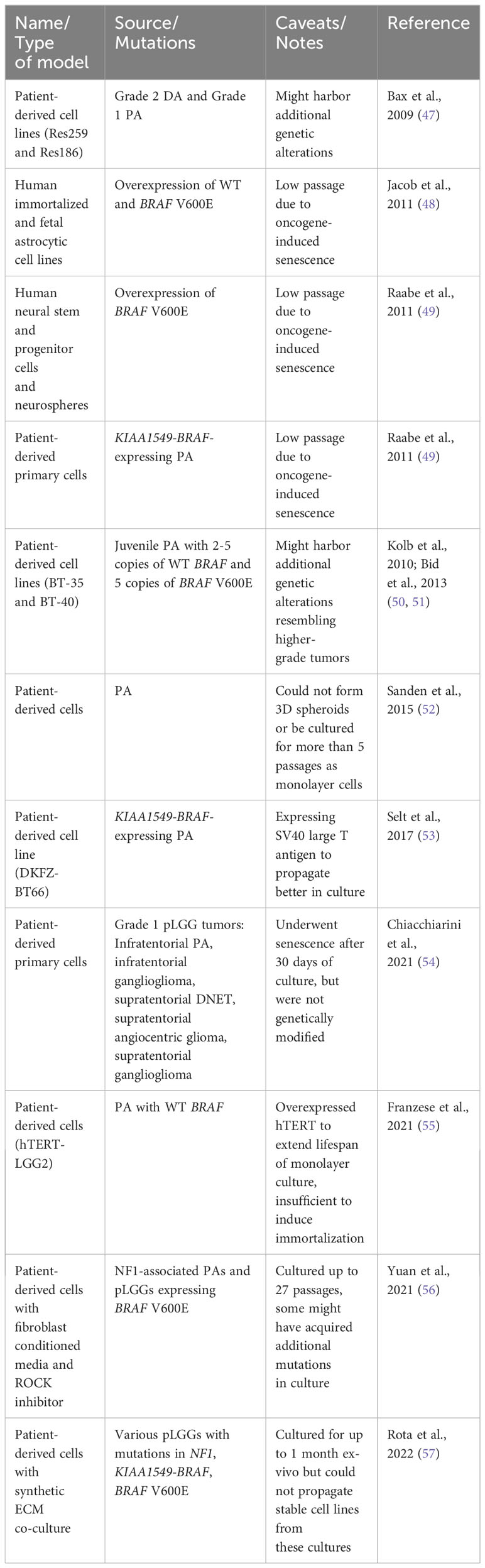
Table 2 List of in vitro models developed for preclinical testing.
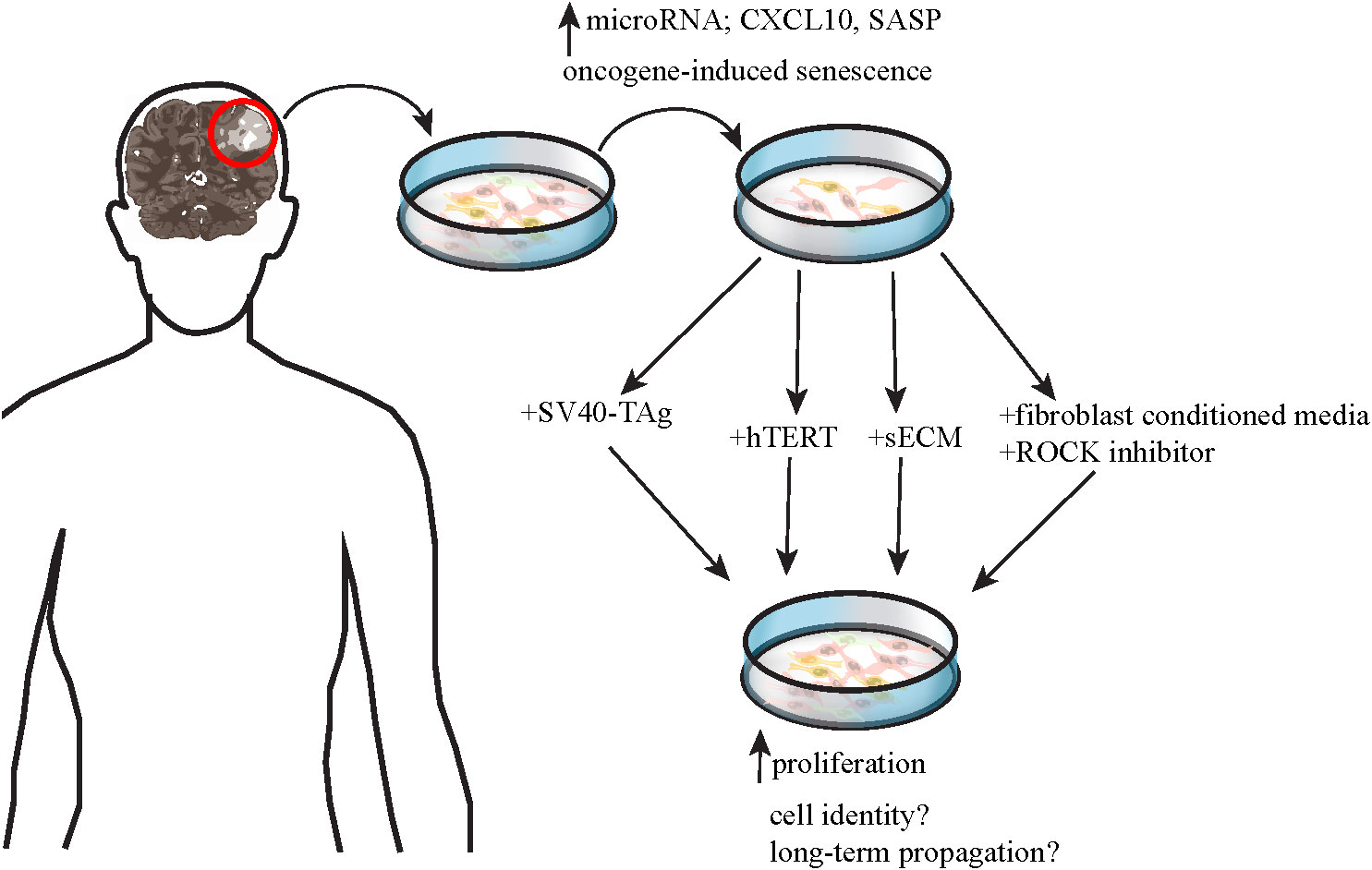
Figure 2 shows a schematic of the efforts made to establish patient-derived pLGG cell lines for preclinical testing, including OIS phenomenon which represents a major challenge to these endeavors. Factors like microRNAs, CXCL10, and SASP have been implicated in OIS. Several methods have been developed to extend lifespan of the pLGG cells in culture to varying degree of success and with remaining concerns on whether the cultured cells represent the original patient tumors and whether the cells can be stably propagated long-term for use in preclinical testing.
Attempts to generate pLGG patient-derived cell lines yielded varying success rates. Most of them could only be maintained as short-term cultures [ (52, 54, 60), Table 2] and/or they acquired additional alterations in culture that made them unrepresentative of the original tumors (47, 50). Bax et al. (2009) conducted molecular and phenotypic characterization of Res259 and Res186 cell lines derived from human pediatric astrocytomas to incorporate them in preclinical testing. Res259 and Res186 were derived from DA and PA patients, respectively. Although their immunophenotypes closely resembled low-grade lesions, both lines harbored genetic alterations reminiscent of higher-grade gliomas [ (47), Table 2].
As part of the Pediatric Preclinical Testing Program (PPTP), 2 astrocytoma cell lines were generated, BT-35 cells which had 2 to 5 copies of wild-type (WT) BRAF and BT-40 cells which had 5 copies of activated BRAF V600E [ (50, 51), Table 2)]. BT-40 was further characterized as grade 2-3 astrocytoma as these cells lost CDKN2A and wild-type P53 (50, 54, 61).
An important milestone in the development of patient-derived PA cell lines was when Selt et al. (2017) transduced cells derived from a 2-year-old PA patient with Dox inducible system coding for simian vacuolating virus 40 large T antigen (SV40-TAg, Figure 2). Expression of SV40-TAg inhibited 2 pathways critical for OIS induction and maintenance and enabled the generation of long-term PA cell line which could be especially useful for preclinical drug testing. This KIAA1549-BRAF-expressing cell line was named DKFZ-BT66 [ (53), Table 2)].
Chiacchiarini et al. (2021) reported additional pLGG cellular models which included primary cells derived from Grade I pLGG tumors. 3 of 9 tumors were infratentorial PAs, 1 of 9 tumors was infratentorial ganglioglioma, 3 of 9 tumors were supratentorial DNETs, and 1 of 9 was supratentorial angiocentric gliomas, and 1 of 9 was supratentorial ganglioglioma (54). Although these patient-derived cells also underwent senescence after about 30 days of culture, they were not genetically modified and thus were more representative of the tumors they were derived from [ (54), Table 2)]. Hence, these cells could be useful for short-term in vitro experiments.
The overexpression of hTERT (human telomerase reverse transcriptase, the catalytic subunit of telomerase) was proposed to counteract OIS in pLGG cell lines so they regain their proliferative potential for long-term cultures [ (55), Figure 2)]. Another method to extend lifespan of pLGG cell lines was proposed by Yuan et al. (2021). Instead of using feeder layer of 3T3 fibroblasts as first described (62), Yuan et al. (2021) co-cultured the pLGG cell lines in conditioned media from irradiated 3T3 cells and Rho kinase (ROCK) inhibitor Y-27632 [ (56), Figure 2)]. They found that these culture conditions led to reversible blockage of senescence, increased proliferation, and allowed these cell lines to propagate longer in culture while still maintaining signature genetic changes of the original tumors (56). This method was tried on multiple pLGG cell lines including 4 PAs, 2 gangliogliomas, 2 anaplastic gliomas, 1 anaplastic PXA and others. One cell line derived from NF1-PA (JHH-NF1-PA1) and another from BRAF V600E anaplastic PXA (JHH-PXA1) patients exhibited growth sufficient for preclinical testing in vitro. More recently, Rota et al. (2022) developed an ex vivo culture system that utilized synthetic extracellular matrices (sECMs) to partially mimic properties of extracellular matrix in vivo. This culture method promoted proliferation and enabled propagation of several patient-derived pLGG cells for up to 1 month ex vivo (57). Continuous efforts are made to generate more pLGG patient-derived cell lines that can be cultured long enough for preclinical testing and still resemble the original tumors, but these studies have made major advancements in the field.
4 In vivo models of pLGG
4.1 Generating non-NF1 pLGG xenograft models for preclinical drug testing
One of the ultimate goals for developing pLGG mouse models is for preclinical drug testing. The two patient/mouse heterografts of JPA generated by PPTP, BT-35 and BT-40, had WT BRAF and BRAF V600E mutation, respectively (50, 51). AZD6244 (Selumetinib), a MEK1/2 inhibitor showed efficacy in BT-40 xenografts, highlighting the MEK signaling pathway as a potential therapeutic target (51). However, although BT-40 xenografts were highly sensitive to Selumetinib and regressed completely during 6 weeks of treatment, the tumors regrew after treatment was stopped (50). Bid et al. (2013) selected and transplanted the resistant tumor clones into Severe Combined Immunodeficient (SCID) mice and resumed the Selumetinib treatment and showed that the resistance of BT-40 xenografts to Selumetinib was mediated by activation of STAT3 signaling, so STAT3 activation could be compensating for MEK inhibition to maintain proliferation and survival (50). Additionally, several studies have documented paradoxical activation of MAPK pathway during preclinical testing or clinical trials of BRAF inhibitors in pLGGs (63, 64). The studies by Kolb et al. (2010) and Bid et al. (2013) generated some of the few xenografts of pediatric low-grade astrocytomas available at that time. However, many of these models do not completely recapitulate patient tumors as they acquired additional genetic alterations, thus emphasizing the need to develop more pLGG in vivo models for more relevant preclinical testing. Table 3 lists the in vivo models that have been generated for non-NF1 pLGG and their contributions to elucidating the complexity of non-NF1 pLGG pathogenesis.
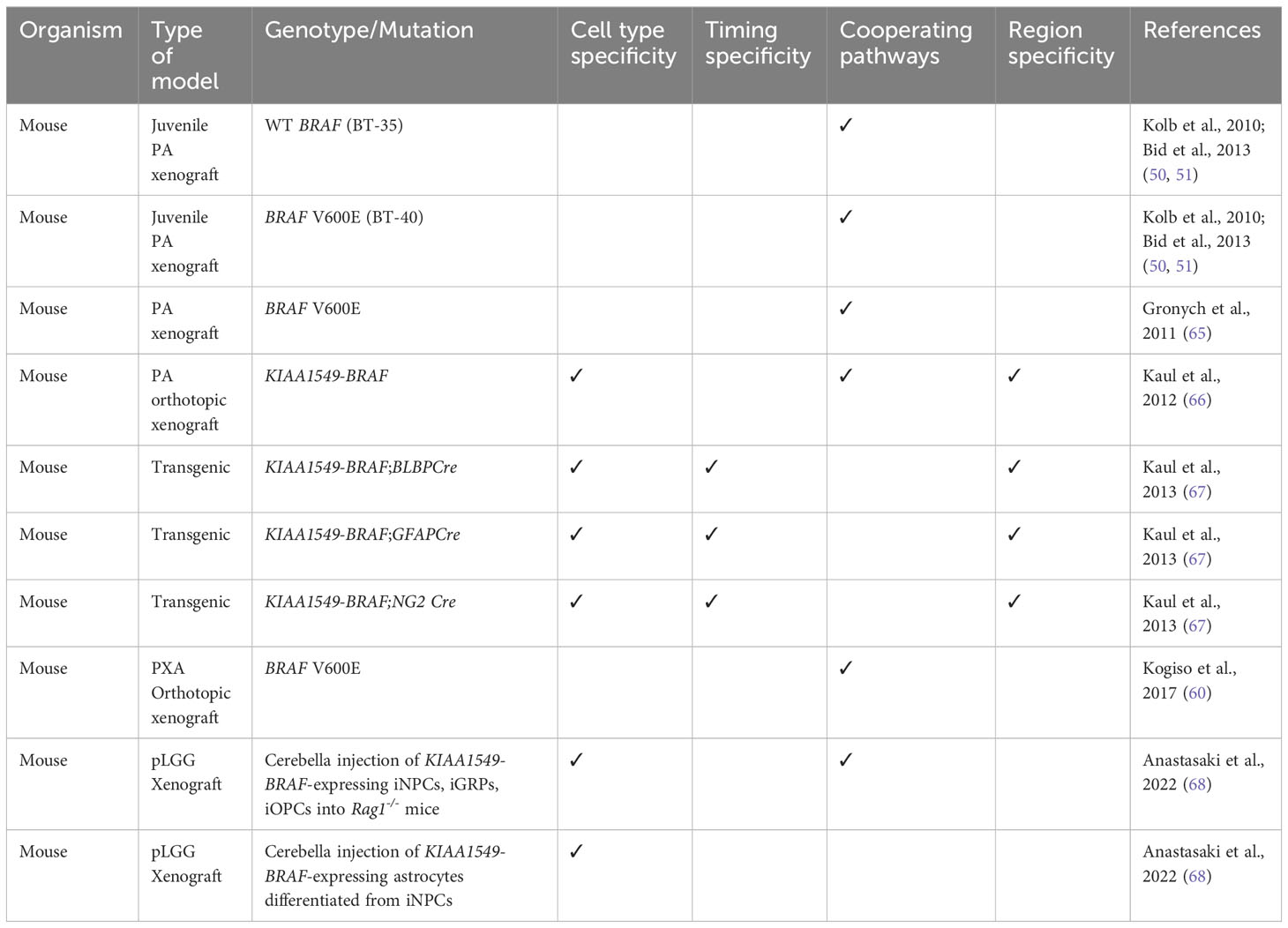
Table 3 List of in vivo models developed for non-NF1 pLGG.
Interestingly, intracranial overexpression of full-length BRAF V600E could not induce glioma formation alone, but a higher-grade glioma was formed with the additional loss of Ink4a/Arf or AKT activation (69). Consequently, Gronych et al. (2011) used the replication-competent avian leukosis virus with splice acceptor/Tv-a (RCAS/Tv-a) system to introduce different BRAF constructs into Nestin-expressing neural progenitor cells and found that while the full-length BRAF V600E did not induce glioma in vivo, the transgenic expression of only the BRAF V600E kinase domain in the cerebral hemispheres was sufficient to induce PA formation (65). Additionally, these tumors had MAPK activation and histopathological features reminiscent of those in PA patients (65).
A mouse model expressing KIAA1549-BRAF fusion was developed by Kaul et al. (2012) to investigate the role of fusion BRAF protein in sporadic PA (66). They first transduced KIAA1549-BRAF into cerebellar neural stem cells (NSCs) and injected these NSCs into the cerebella of 3-wk-old wild-type mice. They observed glioma-like lesions at 6 months post injection (66). Remarkably, KIAA1549-BRAF expression increased proliferation of third ventricle and brainstem NSCs, while not really affecting NSCs in the lateral ventricle and neocortex (66). They also found that the regulation of NSC proliferation by KIAA1549-BRAF was mediated by mTOR signaling (66).
Subsequently, Kaul et al. (2013) generated a conditional and regulatable KIAA1549-BRAF transgenic mouse strain (67). Mice expressing Lox-STOP-Lox-KIAA1549-BRAF with tetracycline-responsive (Tet-off) element were generated and intercrossed with Cre-transgenic mice in which Cre was expressed in Brain lipid binding protein (BLBP+ (70), Neuron glia-antigen 2 (NG2+ (71) and Glial fibrillary acidic protein (GFAP+ (72); cells. These led to the expression of KIAA1549-BRAF in NSCs starting at E9.5 (f-BRAFBLBP), neuroglial progenitor cells starting at E14.5 (f-BRAFNG2), and astroglial progenitor cells starting at E14.5 (f-BRAFGFAP). Corroborating their previous finding that ectopic KIAA1549-BRAF expression specifically increased NSC proliferation (66), KIAA1549-BRAF expression was highest in BLBP+ cells, and transgene expression was highest in the cerebellum of f-BRAFBLBP mice (67). Additionally, there was more KIAA1549-BRAF mRNA expression in astrocytes in the cerebellum compared to those in forebrain or brainstem (67). These conditional KIAA1549-BRAF mouse models will be useful tools in further interrogation of the spatial, temporal, and cell-type specificity of KIAA1549-BRAF expression. Interestingly, Chen et al. (2019) demonstrated that microglia recruitment was required for glioma-like lesion formation in vivo following injection of KIAA1549-BRAF-expressing cerebellar NSCs (73).
Although attempts to establish neurospheres and monolayer cultures from patient tumors failed due to low yields of tumor cells from patient samples, Kogiso et al. (2017) generated 1 pLGG patient-derived orthotopic xenograft (PDOX) out of 25 mice intracranially implanted with different cerebellar and cerebral pLGGs (60). This PDOX model, designated as IC-3635PXA, was confirmed to be a PXA as it still had low proliferation index without necrosis or aplasia, but it was moderately cellular and infiltrative (60). IC-3635PXA was then serially sub-transplanted in mouse brains four times, but by the third passage, the tumor began developing higher grade features, and by the fourth passage, it transformed into a high-grade tumor (60). Interestingly, BRAF V600E mutant allele frequency increased as IC-3635PXA was serially passaged, along with increasing trisomy 9, CDKN2A deletion, loss of GFAP and gain of Vimentin expression (60). Therefore, the PDOX model developed by Kogiso et al. (2017) had tumor progression resembling that of the original patient tumor and gave insights on the cellular drivers of tumor progression and molecular changes that occurred as a grade II PXA transformed into a higher-grade tumor.
4.2 NF1-LGG genetically engineered mouse models
Since NF1-associated LGG is also a very common group of pLGG, a lot of efforts were made to generate mouse models that recapitulate tumors seen in NF1 patients histopathologically and molecularly. Table 4 lists the various in vivo NF1 pLGG mouse models. Germline NF1 knockout mice (NF1-/-) were embryonic lethal, while NF1 heterozygous (NF1+/-) mice did not develop astrocytomas despite having increased astrocyte proliferation in vivo (74, 75). Conditional inactivation of NF1 in neurons (NF1SynIKO) led to astrogliosis, but the mice did not develop astrocytomas (76). Bajenaru et al. (2002) first developed astrocyte-specific NF1 conditional knockout mice using Cre/loxP technology in which Cre was expressed in astrocytes by E14.5, and the astrocytes in these mice were NF1 null (GFAPCre;NF1flox/flox or NF1GFAPCKO), but these mice also didn’t develop astrocytomas (72). Bajenaru et al. (2003) then generated another type of astrocyte-specific NF1 conditional knockout mice by first breeding NF1+/- mice with NF1flox/flox mice, and then breeding the progenies with GFAPCre mice to generate GFAPCre; NF1 flox/mut (NF1+/-GFAPCKO) mice. The development of optic nerve gliomas in these mice showed that astrocytoma formation requires NF1 heterozygosity and loss of NF1 in astrocytes and highlights this mouse model as one of the few preclinical models available for NF1-associated optic glioma (77). Zhu et al. (2005) described another mouse model for NF1-associated optic pathway glioma by crossing human hGFAPCre mice to NF1flox/- and NF1flox/flox mice to generate NF1hGFAPCKO mice. These mice exhibited hyperproliferation of glial progenitor cells, resulting in increased GFAP-expressing astrocytes in developing and adult brains (78). The optic nerve lesions in both mouse models lacked some common features of PA but displayed some morphological and pathological features reminiscent of the NF1-associated human tumors (78). The NF1hGFAPCKO mice exhibited fully penetrant glial cell hyperplasia and had more severe symptoms of optic pathway gliomas compared to NF1+/-GFAPCKO mice, possibly due to the timing of Cre activation, and hence loss of NF1 (E10.5 for NF1hGFAPCKO vs. E14.5 for NF1+/-GFAPCKO mice (77, 78). Dasgupta et al. (2005) also showed that activating KRAS specifically in astrocytes in NF1+/- mice similarly led to optic pathway glioma formation (79). Another NF1 mouse model was generated by inactivating NF1 in neuroglial progenitors starting at E9.5 (BLBPCre; NF1 flox/flox (70);. These mice exhibited increased neural stem cell proliferation and glial lineage differentiation and eventually developed optic glioma by 3 months (70, 82). Furthermore, Solga et al. (2014) generated genetically engineered mice with NF1 loss in NG2+ progenitor cells, which gave rise to oligodendrocytes and astrocytes in vivo, but these mice did not develop optic glioma (81). These mouse models emphasize the dependency of NF1-associated optic pathway glioma on timing of NF1 inactivation, cell-of-origin, and on the tumor microenvironment as NF1 heterozygosity was required for optic glioma formation.
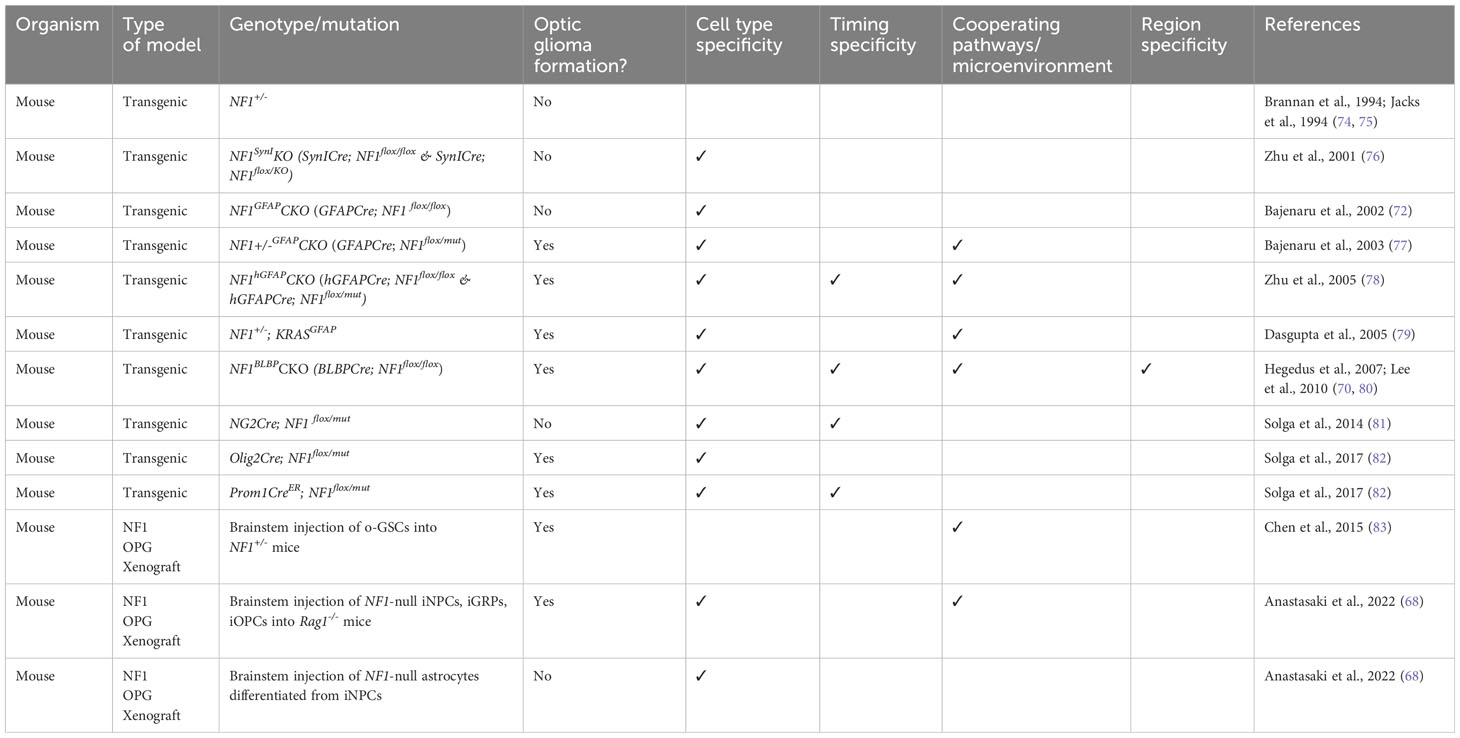
Table 4 List of in vivo NF1 LGG mouse models.
The generation of these NF1-pLGG mouse models paved the way for numerous investigations into multiple aspects of NF1-associated OPG including the pathways affected by loss of NF1 (79, 84, 85), the role of microenvironment in regulating these tumors (83, 86–90), the remarkable specificity of timing, region, and cell-of-origin of NF1-OPG (70, 80–82, 91–93). Importantly, the mouse models enable the preclinical testing of potential therapeutic compounds for this disease (94).
4.2.1 NF1-associated OPG displays temporal and spatial specificity and is highly dependent on cell-of-origin and MAPK pathway activation
The mTOR pathway had been well-implicated in NF1-associated disorders (85, 95, 96), and it was shown that MAPK pathway activation in NF1-deficient astrocytes resulted from RAS hyperactivation (78, 85). Dasgupta et al. (2005) showed that activating KRAS in astrocytes of NF1+/- mice was sufficient for the formation of NF1-OPG (79). They further demonstrated that NF1-deficient astrocytes exhibited high levels of mTOR pathway activation, and this was inhibited by blocking KRAS or PI3K.
Additionally, optic glioma formation was dependent on the type of germline NF1 mutation sustained as the nonsense mutation R681X resulted in greater reduction of Neurofibromin level and more proliferative optic glioma compared to the missense mutation G848R (97). Subsequently, the loss of Neurofibromin led to increased ERK, AKT, and in turn, mTOR activation, to drive NF1-deficient astrocyte proliferation in vitro and NF1 optic glioma growth in vivo (97). Furthermore, it became apparent that Neurofibromin regulation of mouse astrocyte and optic glioma growth was mediated by MEK and AKT signaling that all converged on the mTOR complex (96). These studies elucidated the role of MAPK and mTOR signaling in NF1-OPG.
Mouse OPG tumors were found to contain some neoplastic astrocytes that retained markers of astroglial progenitors such as nestin, BLBP, and contained GFAP- and Olig2-immunoreactive cells (86, 98). To investigate the possibility that these neural stem/progenitor cells gave rise to NF1-OPG, Dasgupta and Gutmann (2005) examined the relationship between Neurofibromin and NSCs. Inactivation or loss of NF1 led to hyperactivation of RAS, MAPK and AKT, and increased proliferation and survival of NSCs and facilitated engraftment and survival of NSCs in vivo (99). Remarkably, NSCs responded differently to NF1 inactivation depending on the brain regions they belonged to, such that NF1 loss led to increased NSC proliferation and gliogenesis in the brainstem but not in the cortex (80). This regional specificity in the response of NSC to NF1 loss was mediated by AKT and mTOR, as the expression of Rictor, an mTOR complex protein, was higher in brainstem compared to cortex (80). This differential Rictor expression in turn led to region-specific mTOR/Rictor-mediated AKT phosphorylation (80). Interestingly, in astrocytes, mTOR regulated cell growth by activating Rac1 instead of AKT (80, 84). Importantly, Lee et al. (2012) subsequently demonstrated that pediatric optic glioma in NF1+/-GFAPCKO mice (77) arose from third ventricle as NSCs from this region were the cells that hyperproliferated in response to mutations characteristic of pediatric glioma, and not NSCs from the lateral ventricle subventricular zone (92).
However, third ventricle NSCs were not the only cell population that can serve as initiating population of NF1-OPG. Subsequent research showed that NF1 loss in Olig2+ cells (Olig2Cre; NF1flox/mut), which also gave rise to astrocytes in murine optic nerve, also formed optic gliomas, albeit at 6 months (82) instead of 3 months as in NF1+/-GFAPCKO mice (77). Since in Olig2Cre mice, Cre recombinase was expressed by E12, this delay in optic glioma formation was likely due to cell-of-origin and not due to timing of NF1 loss. Since BLBP+ and GFAP+ neuroglial progenitor cells co-expressed CD133, a neural progenitor/stem cell marker, Chen et al. (2015) investigated whether CD133+ cells could serve as initiating cells for NF1-OPG. They first isolated CD133+ cells, which were characterized to be multipotent low grade optic glioma stem cells (o-GSCs) from tumor-bearing NF1+/-GFAPCKO mice (83). The transplantation of these o-GSCs into the brainstems of 3-week-old NF1+/- mice yielded optic gliomas within 6 months, but not transplantation of o-GSCs into brainstems of immunocompromised athymic mice, emphasizing the need for NF1+/- local microenvironment in glioma formation (83). Solga et al. (2017) generated inducible NF1 conditional knockout mice in which somatic NF1 was eliminated in CD133+ neural progenitor/stem cells at E15 (Prom1CreER; NF1flox/mut). The injection of tamoxifen and progesterone at E15 to control the timing of NF1 loss led to optic glioma formation at 3 months (82), like those in NF1+/-GFAPCKO mice (77) and BLBPCre; NF1flox/mut mice (70). These studies confirmed that cell of origin was a determinant of optic glioma formation and that neuroglial progenitor cells, including GFAP+, BLBP+, CD133+ cells, and pre-oligodendrocyte precursor cells (pre-OPCs) that are Olig2+ and negative for NG2 could serve as initiating cells for murine NF1-OPG (70, 77, 82, 83). The spatial and cell type specificity of optic glioma formation resembled that of KIAA1549-BRAF-expressing pLGG model (66).
4.2.2 NF1-LGG requires support of various cell types in the microenvironment for tumor growth and maintenance
In NF1+/-GFAPCKO mice, the formation of optic glioma only in NF1+/- mice with astroglial NF1 inactivation suggested that this tumor required microenvironment composed of cells heterozygous for NF1 mutation (77). Upon examination of the tumors in these mice, activated microglia in the tumor microenvironment was present (86). Daginakatte & Gutmann (2007) examined tumor specimens from human NF1-associated PAs and found microglia in all specimens (87). Microglia has been implicated in glioma as they were proposed to stimulate invasiveness of glioma (87, 100, 101). Daginakatte & Gutmann (2007) further found that NF1+/- brain microglia produced soluble factors, identified as hyaluronidase, that promoted NF1-/- astrocyte growth in vitro and in vivo (87). Since there were no low-grade glioma cell lines that simulated NF1-OPG, they utilized Adenovirus-Cre (Ad5-Cre) and NF1flox/flox astrocytes to generate NF1-/- astrocyte and microglia cultures (87). Microglia inactivation or genetic ablation consequently resulted in decreased optic glioma proliferation in NF1+/-GFAPCKO mice (87, 91). Furthermore, NF1+/- microglia exhibited increased c-Jun-NH2-kinase (JNK) pathway activation, without any significant changes in AKT, MAPK or p38-MAPK activity. Thus, JNK inhibition reduced proliferation, motility, and proinflammatory cytokine production of NF1+/- microglia, and inhibition of this pathway was sufficient to reduce optic glioma growth in vivo (88).
NF1 heterozygosity was sufficient to increase microglia proliferation and motility in vitro and in vivo (88), but remarkably, this effect had temporal and spatial specificity (91). Simmons et al. (2011) demonstrated that NF1 heterozygosity resulted in increase in microglia specifically within optic nerve, and not in brainstem or neocortex. Additionally, this increase in microglia numbers, which facilitated glial cell proliferation, occurred at a critical time during optic glioma development (91). Thus, they postulated that the increase in microglia in NF1+/- optic nerve likely resulted from defect in microglia homing and delay in dispersal of microglia from the optic nerve (91). Furthermore, the finding that CX3CR1-expressing stromal microglia were required for optic glioma formation in NF1-OPG mouse model established the role of microglia as essential drivers of optic gliomagenesis (93). Subsequent research demonstrated that NF1 mutation resulted in higher expression of the cytokine Midkine that activated CD8+ T-cells, which then produced Ccl4, a cytokine that induced microglia to express Ccl5 necessary for glioma growth and formation (89, 90). These studies established the mechanisms by which NF1 mutations affected the tumor microenvironment which contained various factors that regulated optic glioma growth.
5 Using hIPSCs to develop non-NF1 and NF1-pLGG xenograft models
A recent report by Anastasaki et al. (2022) utilized human induced pluripotent stem cells (hIPSCs) to generate LGG xenografts harboring NF1 loss and KIAA1549-BRAF fusion. First, they engineered different hIPSC lines with patient-derived germline NF1 mutations and with KIAA1549-BRAF fusion, then they differentiated these hIPSCs to multipotent human neural stem cells capable of generating both neuronal and glial lineage cells (hINPCs). These hINPC lines exhibited increased proliferation, and KIAA1549-BRAF-expressing hINPCs had increased MAPK pathway activation (68). Since injections into the mouse optic nerve, the most common site for NF1-pLGGs, caused a lot of tissue damage, NF1-null hINPCs were injected into the brainstem of immunocompromised Rag1-/- mice, which was the second most common site of NF1-pLGGs (68). Brainstem injections of NF1-null hINPCs and cerebellar injections of KIAA1549-BRAF-hINPCs formed LGGs at 1 month post injection. Moreover, these lesions exhibited many histopathologic features of human pLGGs. Mice with hIPSC-derived LGGs did not exhibit increased mortality, and the lesions had similar proliferative indices even though the sizes grew over time (68). These mirrored clinical observations in pLGG patients.
Orthotopic transplantation of NF1-null and KIAA1549-BRAF-expressing hIPSC-derived glial restricted progenitors (iGRPs) and oligodendrocyte progenitors (iOPCs), but not hIPSC-derived terminally differentiated astrocytes formed LGGs in Rag1-/- mice (68). This was reminiscent of the genetically engineered NF1-OPG mice in which neuroglial progenitor cells could serve as initiating cells of optic glioma formation (82, 92). Interestingly, iGRPs gave rise to tumors resembling optic pathway and brainstem gliomas, while iOPCs gave rise to tumors similar to many cerebellar human PAs (68).
Furthermore, Anastasaki et al. (2022) demonstrated that formation of LGGs required CD4+ T cell depletion and reduced astrocytic Cxcl10 expression. So LGGs could form in NOD/SCID, CD4-deficient, CD4/CD8-deficient mice, but not in CD8-deficient mice or other strains lacking expression of microglia or T cell chemokine receptors (68). They also showed that primary human PA cell lines including that with an NF1-PA (JHH-NF1-PA) and that with sporadic PA (Res186) could form LGGs in Rag1-/- and Cxcl10-/- mice (68). Although additional work is still required to create preclinical models that more closely recapitulate patient tumors, this model provides an important milestone in the development of a humanized pLGG orthotopic xenograft model.
6 Zebrafish as models for pLGG
Another in-vivo model that has been utilized to study pLGGs is the zebrafish. It is cost-effective, has short experimental timeframe, and enables rapid investigation into tumor growth, invasion, metastasis, and drug screening (56, 102–105). Fewer cells are needed to establish tumor xenografts, and the availability of transparent transgenic zebrafish or their translucent larvae made it possible to track tumor cells in real-time (102–104, 106). Several transgenic zebrafish models have also been generated to interrogate glioma pathogenesis. For example, using the Gal4-UAS system, Mayrhofer et al. (2017) generated zebrafish brain tumor model that expresses oncogenes that activate MAPK and PI3K signaling in neural progenitor cells and discovered that activation of YAP signaling pathway promotes development of aggressive brain tumors (107). Luo et al. (2021) used CRISPR/Cas9 to establish transgenic zebrafish lines that express mutated NF1, Rb1 or TP53 under GFAP promoter and showed that various combinations of NF1, TP53, and/or Rb1 mutations can induce gliomas of different grades and phenotypes (108). Using transgenic zebrafish, Lee et al. (2010) demonstrated that NF1 knockdown increased ERK signaling and increased OPC proliferation in the developing spinal cord (109). Orthotopic xenografts and immunodeficient zebrafish models have also been developed for brain tumors (110–113). Although many of the zebrafish xenograft models used glioblastoma cells, Yuan et al. (2021) injected JHH-NF1-PA1 cells into the midline in the optic tectum of zebrafish at 2 days post fertilization, and they found that the pLGG cells survived over 6 days and migrated in the brains of larval zebrafish before their adaptive immune system matured (56). Sigaud et al. (2023) also used zebrafish embryos to evaluate therapeutic options for pLGG xenografts generated by injecting DKFZ-BT66 and BT40 cells (105). While zebrafish models certainly have advantages over mouse models in terms of time and cost efficiency and are useful for rapid drug screening, development of more models and/or a combination of different types of models will be necessary to recapitulate the complexity of human pLGGs.
7 Development of NF1 genetically engineered minipigs
Isakson et al. (2018) used Transcription activator-like effector nucleases (TALENs) flanking a known NF1 nonsense mutation NF1R1947 to transfect fetal Ossabaw minipig fibroblasts. NF1 minipigs were generated through chromatin transfer and subsequent breeding (114). NF1 mutant allele exhibited germline transmission with Mendelian frequency and no evidence of reduced fitness (114). NF1 minipigs displayed features of NF1 patients such as skin abnormalities, neurofibromas, underwent biallelic inactivation of the NF1 gene, and they also developed OPG (114). Therefore, the generation of these NF1 minipigs paved a path for the field to interrogate NF1-related molecular pathogenesis, explore therapeutic options, and conduct preclinical testing in large animal models that hopefully present closer resemblance to human patients.
8 Leveraging the strengths and overcoming the challenges of generating pLGG in vitro and in vivo models
Although massive number of efforts have been directed at developing preclinical in vitro and in vivo models of pLGGs, there are many challenges to these endeavors. The generation of in vitro pLGG models has been hampered by many factors. First, the lack of availability of patient tissues as some pLGGs located in less accessible areas such as NF1-OPGs are rarely biopsied (4, 7), although a few NF1-PA cell lines are available (115). Second, pLGG samples obtained from young children as patients are usually small, hence only small number of cells can be obtained to culture. Third, the intrinsic slow growth and benign behavior of these tumors coupled with OIS made it difficult to grow these tumor cells in vitro (48, 49, 52, 54, 60). Several methods were developed to bypass OIS through genetic modifications of the pLGG cells so that they can be propagated long enough in culture (53, 55), but these methods might generate in vitro model systems that incompletely reflect the genetic/epigenetic background of the primary tumors. Nevertheless, as more research is being done into developing different ways to extend the lifespan of pLGG cells in vitro, certain methods such as conditional reprogramming culture conditions or using hIPSC-derived hINPCs might provide plausible solutions to generating viable in vitro models (56, 68).
Two main groups of pLGG in vivo models have been generated by several labs with varying degree of success: patient-derived xenograft models of mostly non-NF1-pLGGs (50, 51, 60, 65, 66, 116) and genetically engineered mouse models mostly for NF1-pLGGs especially NF1-OPGs (70, 72, 76–79, 82). However, some have also attempted to generate NF1-pLGG patient-derived xenograft models (68, 83) and transgenic non-NF1-pLGG model (67).
Genetically engineered NF1-OPG models have been extremely useful to investigate the pathogenesis of NF1-OPG, which could somewhat be extended to general NF1-pLGGs. In these models, tumors were readily detectable as they arose in predictable locations with near 100% penetrance, the histopathological features of these tumors resembled those in the patients, the initiating event (NF1 inactivation) was known, thus these models recapitulate many genetic and cellular abnormalities seen in NF1-OPG patients (94). Moreover, these tumors arose in immunocompetent mice allowing the interrogation into the tumor-immune axis that was necessary in NF1-OPG formation.
Nonetheless, there are many challenges presented to researchers in developing these pLGG in vivo models, and these reasons might vary depending on whether the pLGG is associated with NF1. For example, NF1-OPG formation depends on many factors including: temporal and spatial specificity (77, 78, 80, 91, 92), cell-of-origin (72, 76–78, 82, 83, 92), interaction with microenvironment (87–91, 93) and other signaling pathways (79, 84, 85, 117), nature of the NF1 mutation (97), among others. Meanwhile, the challenges in generating non-NF1 pLGG models include the low proliferation capacity and the specific permissive tumor microenvironment necessary for glioma formation (7). Interestingly, NF1-OPG requires NF1 heterozygous cells along with multiple different types of cells in the microenvironment to grow (77, 87–91, 93), whereas NF1 and non-NF1-pLGG xenografts needed loss of specific T-cell population to form (68). Consequently, the combination of patient-derived xenograft models and transgenic models are required to comprehensively interrogate the molecular pathogenesis of pLGGs and to assess potential immune response to therapeutic agents.
9 Future directions on pLGG modeling
Although most in vitro and in vivo models that were generated often expressed BRAF and NF1 mutations, there are other genetic alterations that have been highlighted as hallmarks of certain types of pLGGs. Some reports proposed that FGFR1 mutation may be a relevant prognostic marker in PAs as in some cases FGFR1 mutations were associated with more adverse outcomes in patients (42). FGFR1 hotspot mutations were also relatively frequent in pLGGs especially in mixed neuronal-glial tumors without known genetic drivers, providing an additional way to classify these tumors (118). Moreover, Egbivwie et al. (2019) generated 5 grade 1 PA patient-derived cell lines and found that FGFR1 overexpression alone was able to increase tumor cell migration and drive tumor progression. They also found that there was higher expression of membranous phosphorylated FGFR1 in grade 2 tumors, so the presence of pFGFR1 could be associated with malignancy and tumor grade (119). Future research could add more focus into generating pLGG models that interrogate the pathogenesis of rarer alterations such as FGFR, NTRK, and MYB mutations. Development of pLGG models could also consider epigenetic events that might promote tumor survival, maintenance and/or progression as much less is known about the role of epigenetics in pathogenesis of pLGGs (4).
Due to the difficulties in generating long-term cultures of pLGG patient-derived cell lines, more alternative methods could be explored. For example, since there is such strong dependence of pLGG formation in vivo on microenvironment, certain factors could be added into the cultures such as cytokines or tumor cells could be co-cultured with other cell types to provide more supportive microenvironment. The relatively successful sECM method to culture pLGG cells demonstrates that elements in the microenvironment will be necessary to generate viable in vitro pLGG models (57). There is also a need to develop more sophisticated technologies for genetic manipulation of tissues and cells. Indeed, emerging somatic transgenic in vivo mouse (120–123) and 3D cerebral organoid-based human glioma models (124) genetically manipulated using electroporation also hold promise for more flexible and renewable modeling. These models can employ plasmid or mRNA-delivered transgenes, and/or CRISPR/Cas elements to enable more rapid recapitulation of mutations in diverse spatiotemporal contexts but have been mostly employed to generate HGG models (120–123).
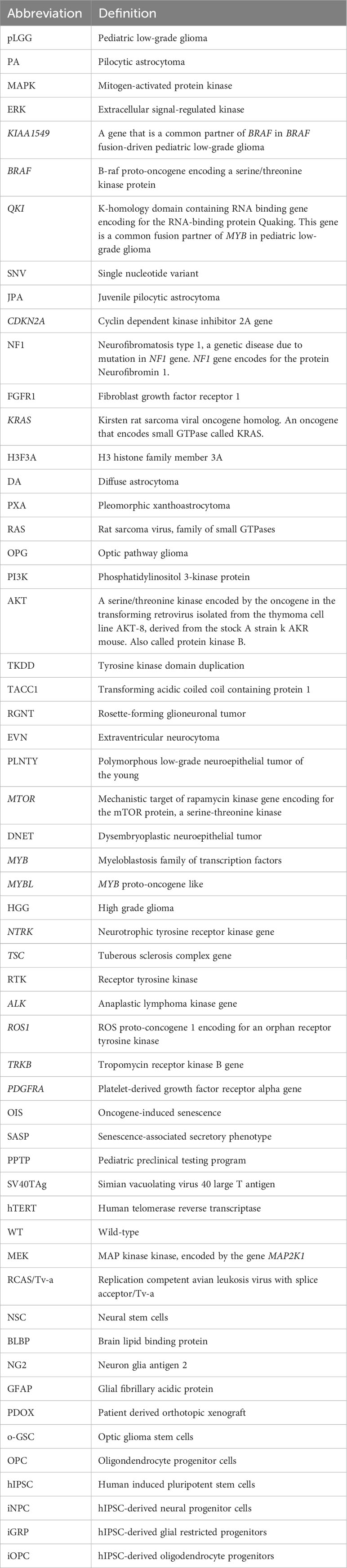
Table 5 List of abbreviations commonly used in the manuscript.
A truly useful pLGG model would ideally recapitulate the molecular pathogenesis of human pLGGs along with the dependency of these tumors on the microenvironment milieu and the inevitable activation of the MAPK pathway. However, despite the constant challenges, many important milestones were achieved in the development of in vitro and in vivo pLGG models. The combination of advanced sequencing technologies and prognostic methods and in vitro and in vivo models should be utilized to comprehensively investigate the pathogenesis of this highly heterogeneous group of pediatric brain tumors and identify viable therapeutic options.
Author’s note
After the completion of this review, a related work was published (125). Table 5 lists the abbreviations commonly used in the manuscript.
Author contributions
GY: Writing – original draft, Writing – review & editing. JB: Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We acknowledge support from the Board of Governors RMI of Cedars-Sinai (to JB). JB was supported by DoD NFRP grant HT9425-23-1-0269, NIH grant R33CA236687, American Cancer Society grant RSG-16-217-01-TBG. GY was supported by a CIRM Fellowship.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Garcia MA, Solomon DA, Haas-Kogan DA. Exploiting molecular biology for diagnosis and targeted management of pediatric low-grade gliomas. Future Oncol (2016) 12(12):1493–506. doi: 10.2217/fon-2016-0039
2. Packer RJ, Pfister S, Bouffet E, Avery R, Bandopadhayay P, Bornhorst M, et al. Pediatric low-grade gliomas: implications of the biologic era. Neuro Oncol (2017) 19(6):750–61. doi: 10.1093/neuonc/now209
3. Jones DTW, Kieran MW, Bouffet E, Alexandrescu S, Bandopadhayay P, Bornhorst M, et al. Pediatric low-grade gliomas: next biologically driven steps. Neuro Oncol (2018) 20(2):160–73. doi: 10.1093/neuonc/nox141
4. Milde T, Rodriguez FJ, Barnholtz-Sloan JS, Patil N, Eberhart CG, Gutmann DH. Reimagining pilocytic astrocytomas in the context of pediatric low-grade gliomas. Neuro Oncol (2021) 23(10):1634–46. doi: 10.1093/neuonc/noab138
5. Sturm D, Pfister SM, Jones DTW. Pediatric gliomas: current concepts on diagnosis, biology, and clinical management. J Clin Oncol (2017) 35(21):2370–7. doi: 10.1200/JCO.2017.73.0242
6. de Blank P, Fouladi M, Huse JT. Molecular markers and targeted therapy in pediatric low-grade glioma. J Neurooncol (2020) 150(1):5–15. doi: 10.1007/s11060-020-03529-1
7. Ricker CA, Pan Y, Gutmann DH, Keller C. Challenges in drug discovery for neurofibromatosis type 1-associated low-grade glioma. Front Oncol (2016) 6:259. doi: 10.3389/fonc.2016.00259
8. Khatua S, Gutmann DH, Packer RJ. Neurofibromatosis type 1 and optic pathway glioma: Molecular interplay and therapeutic insights. Pediatr Blood Cancer (2018) 65(3). doi: 10.1002/pbc.26838
9. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
10. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet (2013) 45(6):602–12. doi: 10.1038/ng.2611
11. Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J, et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell (2020) 37(4):569–583e5. doi: 10.1016/j.ccell.2020.03.011
12. Ryall S, Tabori U, Hawkins C. A comprehensive review of paediatric low-grade diffuse glioma: pathology, molecular genetics and treatment. Brain Tumor Pathol (2017) 34(2):51–61. doi: 10.1007/s10014-017-0282-z
13. Bale TA. FGFR- gene family alterations in low-grade neuroepithelial tumors. Acta Neuropathol Commun (2020) 8(1):21. doi: 10.1186/s40478-020-00898-6
14. Ryall S, Tabori U, Hawkins C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol Commun (2020) 8(1):30. doi: 10.1186/s40478-020-00902-z
15. Sievers P, Appay R, Schrimpf D, Stichel D, Reuss DE, Wefers AK, et al. Rosette-forming glioneuronal tumors share a distinct DNA methylation profile and mutations in FGFR1, with recurrent co-mutation of PIK3CA and NF1. Acta Neuropathol (2019) 138(3):497–504. doi: 10.1007/s00401-019-02038-4
16. Lucas CG, Gupta R, Doo P, Lee JC, Cadwell CR, Ramani B, et al. Comprehensive analysis of diverse low-grade neuroepithelial tumors with FGFR1 alterations reveals a distinct molecular signature of rosette-forming glioneuronal tumor. Acta Neuropathol Commun (2020) 8(1):151. doi: 10.1186/s40478-020-01027-z
17. Appay R, Bielle F, Sievers P, Barets D, Fina F, Boutonnat J, et al. Rosette-forming glioneuronal tumours are midline, FGFR1-mutated tumours. Neuropathol Appl Neurobiol (2022) 48(5):e12813. doi: 10.1111/nan.12813
18. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
19. Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet (2013) 45(8):927–32. doi: 10.1038/ng.2682
20. Collins VP, Jones DT, Giannini C. Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol (2015) 129(6):775–88. doi: 10.1007/s00401-015-1410-7
21. Jacob K, Albrecht S, Sollier C, Faury D, Sader E, Montpetit A, et al. Duplication of 7q34 is specific to juvenile pilocytic astrocytomas and a hallmark of cerebellar and optic pathway tumours. Br J Cancer (2009) 101(4):722–33. doi: 10.1038/sj.bjc.6605179
22. Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res (2008) 68(21):8673–7. doi: 10.1158/0008-5472.CAN-08-2097
23. Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell (2001) 104(4):593–604. doi: 10.1016/S0092-8674(01)00245-8
24. Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell (1990) 63(4):851–9. doi: 10.1016/0092-8674(90)90151-4
25. Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell (1990) 63(4):843–9. doi: 10.1016/0092-8674(90)90150-D
26. Xu GF, Lin B, Tanaka K, Dunn D, Wood D, Gesteland R, et al. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell (1990) 63(4):835–41. doi: 10.1016/0092-8674(90)90149-9
27. Xu GF, O'Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell (1990) 62(3):599–608. doi: 10.1016/0092-8674(90)90024-9
28. Lau N, Feldkamp MM, Roncari L, Loehr AH, Shannon P, Gutmann DH, et al. Loss of neurofibromin is associated with activation of RAS/MAPK and PI3-K/AKT signaling in a neurofibromatosis 1 astrocytoma. J Neuropathol Exp Neurol (2000) 59(9):759–67. doi: 10.1093/jnen/59.9.759
29. Gutmann DH, Donahoe J, Brown T, James CD, Perry A. Loss of neurofibromatosis 1 (NF1) gene expression in NF1-associated pilocytic astrocytomas. Neuropathol Appl Neurobiol (2000) 26(4):361–7. doi: 10.1046/j.1365-2990.2000.00258.x
30. Kluwe L, Hagel C, Tatagiba M, Thomas S, Stavrou D, Ostertag H, et al. Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocytomas. J Neuropathol Exp Neurol (2001) 60(9):917–20. doi: 10.1093/jnen/60.9.917
31. Listernick R, Charrow J, Gutmann DH. Intracranial gliomas in neurofibromatosis type 1. Am J Med Genet (1999) 89(1):38–44. doi: 10.1002/(SICI)1096-8628(19990326)89:1<38::AID-AJMG8>3.0.CO;2-M
32. Guillamo JS, Creange A, Kalifa C, Grill J, Rodriguez D, Doz F, et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain (2003) 126(Pt 1):152–60. doi: 10.1093/brain/awg016
33. Listernick R, Ferner RE, Piersall L, Sharif S, Gutmann DH, Charrow J. Late-onset optic pathway tumors in children with neurofibromatosis 1. Neurology (2004) 63(10):1944–6. doi: 10.1212/01.WNL.0000144341.16830.01
34. Ballester LY, Penas-Prado M, Leeds NE, Huse JT, Fuller GN. FGFR1 tyrosine kinase domain duplication in pilocytic astrocytoma with anaplasia. Cold Spring Harb Mol Case Stud (2018) 4(2):a002378. doi: 10.1101/mcs.a002378
35. Halfpenny A, Ferris SP, Grafe M, Woltjer R, Selden N, Nazemi K, et al. A case of recurrent epilepsy-associated rosette-forming glioneuronal tumor with anaplastic transformation in the absence of therapy. Neuropathology (2019) 39(5):389–93. doi: 10.1111/neup.12586
36. Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol (2016) 131(6):833–45. doi: 10.1007/s00401-016-1539-z
37. Galvin RT, Zheng C, Fitzpatrick G, Forster CL, Sandoval-Garcia C, Guillaume D, et al. MYO5A::FGFR1 represents a novel fusion event in pediatric low-grade glioma. Neurooncol Adv (2023) 5(1):vdad017. doi: 10.1093/noajnl/vdad017
38. Rivera B, Gayden T, Carrot-Zhang J, Nadaf J, Boshari T, Faury D, et al. Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol (2016) 131(6):847–63. doi: 10.1007/s00401-016-1549-x
39. Fina F, Barets D, Colin C, Bouvier C, Padovani L, Nanni-Metellus I, et al. Droplet digital PCR is a powerful technique to demonstrate frequent FGFR1 duplication in dysembryoplastic neuroepithelial tumors. Oncotarget (2017) 8(2):2104–13. doi: 10.18632/oncotarget.12881
40. Surrey LF, Jain P, Zhang B, Straka J, Zhao X, Harding BN, et al. Genomic analysis of dysembryoplastic neuroepithelial tumor spectrum reveals a diversity of molecular alterations dysregulating the MAPK and PI3K/mTOR pathways. J Neuropathol Exp Neurol (2019) 78(12):1100–11. doi: 10.1093/jnen/nlz101
41. Schuller U, Iglauer P, Dorostkar MM, Mawrin C, Herms J, Giese A, et al. Mutations within FGFR1 are associated with superior outcome in a series of 83 diffuse midline gliomas with H3F3A K27M mutations. Acta Neuropathol (2021) 141(2):323–5. doi: 10.1007/s00401-020-02259-y
42. Becker AP, Scapulatempo-Neto C, Carloni AC, Paulino A, Sheren J, Aisner DL, et al. KIAA1549: BRAF gene fusion and FGFR1 hotspot mutations are prognostic factors in pilocytic astrocytomas. J Neuropathol Exp Neurol (2015) 74(7):743–54. doi: 10.1097/NEN.0000000000000213
43. Ciciro Y, Sala A. MYB oncoproteins: emerging players and potential therapeutic targets in human cancer. Oncogenesis (2021) 10(2):19. doi: 10.1038/s41389-021-00309-y
44. Tatevossian RG, Tang B, Dalton J, Forshew T, Lawson AR, Ma J, et al. MYB upregulation and genetic aberrations in a subset of pediatric low-grade gliomas. Acta Neuropathol (2010) 120(6):731–43. doi: 10.1007/s00401-010-0763-1
45. Ramkissoon LA, Horowitz PM, Craig JM, Ramkissoon SH, Rich BE, Schumacher SE, et al. Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci U.S.A. (2013) 110(20):8188–93. doi: 10.1073/pnas.1300252110
46. Bandopadhayay P, Ramkissoon LA, Jain P, Bergthold G, Wala J, Zeid R, et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet (2016) 48(3):273–82. doi: 10.1038/ng.3500
47. Bax DA, Little SE, Gaspar N, Perryman L, Marshall L, Viana-Pereira M, et al. Molecular and phenotypic characterisation of paediatric glioma cell lines as models for preclinical drug development. PloS One (2009) 4(4):e5209. doi: 10.1371/journal.pone.0005209
48. Jacob K, Quang-Khuong DA, Jones DT, Witt H, Lambert S, Albrecht S, et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res (2011) 17(14):4650–60. doi: 10.1158/1078-0432.CCR-11-0127
49. Raabe EH, Lim KS, Kim JM, Meeker A, Mao XG, Nikkhah G, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res (2011) 17(11):3590–9. doi: 10.1158/1078-0432.CCR-10-3349
50. Bid HK, Kibler A, Phelps DA, Manap S, Xiao L, Lin J, et al. Development, characterization, and reversal of acquired resistance to the MEK1 inhibitor selumetinib (AZD6244) in an in vivo model of childhood astrocytoma. Clin Cancer Res (2013) 19(24):6716–29. doi: 10.1158/1078-0432.CCR-13-0842
51. Kolb EA, Gorlick R, Houghton PJ, Morton CL, Neale G, Keir ST, et al. Initial testing (stage 1) of AZD6244 (ARRY-142886) by the pediatric preclinical testing program. Pediatr Blood Cancer (2010) 55(4):668–77. doi: 10.1002/pbc.22576
52. Sanden E, Eberstal S, Visse E, Siesjo P, Darabi A. A standardized and reproducible protocol for serum-free monolayer culturing of primary paediatric brain tumours to be utilized for therapeutic assays. Sci Rep (2015) 5:12218. doi: 10.1038/srep12218
53. Selt F, Hohloch J, Hielscher T, Sahm F, Capper D, Korshunov A, et al. Establishment and application of a novel patient-derived KIAA1549:BRAF-driven pediatric pilocytic astrocytoma model for preclinical drug testing. Oncotarget (2017) 8(7):11460–79. doi: 10.18632/oncotarget.14004
54. Chiacchiarini M, Besharat ZM, Carai A, Miele E, Del Baldo G, Mastronuzzi A, et al. Pediatric low-grade gliomas: molecular characterization of patient-derived cellular models. Childs Nerv Syst (2021) 37(3):771–8. doi: 10.1007/s00381-020-04559-w
55. Franzese O, Di Francesco AM, Meco D, Graziani G, Cusano G, Levati L, et al. hTERT transduction extends the lifespan of primary pediatric low-grade glioma cells while preserving the biological response to NGF. Pathol Oncol Res (2021) 27:612375. doi: 10.3389/pore.2021.612375
56. Yuan M, White D, Resar L, Bar E, Groves M, Cohen A, et al. Conditional reprogramming culture conditions facilitate growth of lower-grade glioma models. Neuro Oncol (2021) 23(5):770–82. doi: 10.1093/neuonc/noaa263
57. Rota CM, Brown AT, Addleson E, Ives C, Trumper E, Pelton K, et al. Synthetic extracellular matrices and astrocytes provide a supportive microenvironment for the cultivation and investigation of primary pediatric gliomas. Neurooncol Adv (2022) 4(1):vdac049. doi: 10.1093/noajnl/vdac049
58. Buhl JL, Selt F, Hielscher T, Guiho R, Ecker J, Sahm F, et al. The senescence-associated secretory phenotype mediates oncogene-induced senescence in pediatric pilocytic astrocytoma. Clin Cancer Res (2019) 25(6):1851–66. doi: 10.1158/1078-0432.CCR-18-1965
59. Yuan M, Da Silva A, Arnold A, Okeke L, Ames H, Correa-Cerro LS, et al. MicroRNA (miR) 125b regulates cell growth and invasion in pediatric low grade glioma. Sci Rep (2018) 8(1):12506. doi: 10.1038/s41598-018-30942-4
60. Kogiso M, Qi L, Lindsay H, Huang Y, Zhao X, Liu Z, et al. Xenotransplantation of pediatric low grade gliomas confirms the enrichment of BRAF V600E mutation and preservation of CDKN2A deletion in a novel orthotopic xenograft mouse model of progressive pleomorphic xanthoastrocytoma. Oncotarget (2017) 8(50):87455–71. doi: 10.18632/oncotarget.20713
61. Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M, et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric Malignant astrocytomas. Cancer Res (2010) 70(2):512–9. doi: 10.1158/0008-5472.CAN-09-1851
62. Liu X, Krawczyk E, Suprynowicz FA, Palechor-Ceron N, Yuan H, Dakic A, et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc (2017) 12(2):439–51. doi: 10.1038/nprot.2016.174
63. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature (2010) 464(7287):427–30. doi: 10.1038/nature08902
64. Karajannis MA, Legault G, Fisher MJ, Milla SS, Cohen KJ, Wisoff JH, et al. Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro Oncol (2014) 16(10):1408–16. doi: 10.1093/neuonc/nou059
65. Gronych J, Korshunov A, Bageritz J, Milde T, Jugold M, Hambardzumyan D, et al. An activated mutant BRAF kinase domain is sufficient to induce pilocytic astrocytoma in mice. J Clin Invest (2011) 121(4):1344–8. doi: 10.1172/JCI44656
66. Kaul A, Chen YH, Emnett RJ, Dahiya S, Gutmann DH. Pediatric glioma-associated KIAA1549:BRAF expression regulates neuroglial cell growth in a cell type-specific and mTOR-dependent manner. Genes Dev (2012) 26(23):2561–6. doi: 10.1101/gad.200907.112
67. Kaul A, Chen YH, Emnett RJ, Gianino SM, Gutmann DH. Conditional KIAA1549:BRAF mice reveal brain region- and cell type-specific effects. Genesis (2013) 51(10):708–16. doi: 10.1002/dvg.22415
68. Anastasaki C, Chatterjee J, Cobb O, Sanapala S, Scheaffer SM, De Andrade Costa A, et al. Human induced pluripotent stem cell engineering establishes a humanized mouse platform for pediatric low-grade glioma modeling. Acta Neuropathol Commun (2022) 10(1):120. doi: 10.1186/s40478-022-01428-2
69. Robinson JP, VanBrocklin MW, Guilbeault AR, Signorelli DL, Brandner S, Holmen SL. Activated BRAF induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation. Oncogene (2010) 29(3):335–44. doi: 10.1038/onc.2009.333
70. Hegedus B, Dasgupta B, Shin JE, Emnett RJ, Hart-Mahon EK, Elghazi L, et al. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell (2007) 1(4):443–57. doi: 10.1016/j.stem.2007.07.008
71. Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development (2008) 135(1):145–57. doi: 10.1242/dev.004895
72. Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol (2002) 22(14):5100–13. doi: 10.1128/MCB.22.14.5100-5113.2002
73. Chen R, Keoni C, Waker CA, Lober RM, Chen YH, Gutmann DH. KIAA1549-BRAF expression establishes a permissive tumor microenvironment through NFkappaB-mediated CCL2 production. Neoplasia (2019) 21(1):52–60. doi: 10.1016/j.neo.2018.11.007
74. Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, et al. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev (1994) 8(9):1019–29. doi: 10.1101/gad.8.9.1019
75. Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet (1994) 7(3):353–61. doi: 10.1038/ng0794-353
76. Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev (2001) 15(7):859–76. doi: 10.1101/gad.862101
77. Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res (2003) 63(24):8573–7.
78. Zhu Y, Harada T, Liu L, Lush ME, Guignard F, Harada C, et al. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development (2005) 132(24):5577–88. doi: 10.1242/dev.02162
79. Dasgupta B, Li W, Perry A, Gutmann DH. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res (2005) 65(1):236–45. doi: 10.1158/0008-5472.236.65.1
80. Lee DY, Yeh TH, Emnett RJ, White CR, Gutmann DH. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev (2010) 24(20):2317–29. doi: 10.1101/gad.1957110
81. Solga AC, Gianino SM, Gutmann DH. NG2-cells are not the cell of origin for murine neurofibromatosis-1 (Nf1) optic glioma. Oncogene (2014) 33(3):289–99. doi: 10.1038/onc.2012.580
82. Solga AC, Toonen JA, Pan Y, Cimino PJ, Ma Y, Castillon GA, et al. The cell of origin dictates the temporal course of neurofibromatosis-1 (Nf1) low-grade glioma formation. Oncotarget (2017) 8(29):47206–15. doi: 10.18632/oncotarget.17589
83. Chen YH, McGowan LD, Cimino PJ, Dahiya S, Leonard JR, Lee DY, et al. Mouse low-grade gliomas contain cancer stem cells with unique molecular and functional properties. Cell Rep (2015) 10(11):1899–912. doi: 10.1016/j.celrep.2015.02.041
84. Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res (2005) 65(7):2755–60. doi: 10.1158/0008-5472.CAN-04-4058
85. Banerjee S, Crouse NR, Emnett RJ, Gianino SM, Gutmann DH. Neurofibromatosis-1 regulates mTOR-mediated astrocyte growth and glioma formation in a TSC/Rheb-independent manner. Proc Natl Acad Sci U.S.A. (2011) 108(38):15996–6001. doi: 10.1073/pnas.1019012108
86. Bajenaru ML, Garbow JR, Perry A, Hernandez MR, Gutmann DH. Natural history of neurofibromatosis 1-associated optic nerve glioma in mice. Ann Neurol (2005) 57(1):119–27. doi: 10.1002/ana.20337
87. Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet (2007) 16(9):1098–112. doi: 10.1093/hmg/ddm059
88. Daginakatte GC, Gianino SM, Zhao NW, Parsadanian AS, Gutmann DH. Increased c-Jun-NH2-kinase signaling in neurofibromatosis-1 heterozygous microglia drives microglia activation and promotes optic glioma proliferation. Cancer Res (2008) 68(24):10358–66. doi: 10.1158/0008-5472.CAN-08-2506
89. Pan Y, Xiong M, Chen R, Ma Y, Corman C, Maricos M, et al. Athymic mice reveal a requirement for T-cell-microglia interactions in establishing a microenvironment supportive of Nf1 low-grade glioma growth. Genes Dev (2018) 32(7-8):491–6. doi: 10.1101/gad.310797.117
90. Guo X, Pan Y, Xiong M, Sanapala S, Anastasaki C, Cobb O, et al. Midkine activation of CD8(+) T cells establishes a neuron-immune-cancer axis responsible for low-grade glioma growth. Nat Commun (2020) 11(1):2177. doi: 10.1038/s41467-020-15770-3
91. Simmons GW, Pong WW, Emnett RJ, White CR, Gianino SM, Rodriguez FJ, et al. Neurofibromatosis-1 heterozygosity increases microglia in a spatially and temporally restricted pattern relevant to mouse optic glioma formation and growth. J Neuropathol Exp Neurol (2011) 70(1):51–62. doi: 10.1097/NEN.0b013e3182032d37
92. Lee DY, Gianino SM, Gutmann DH. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell (2012) 22(1):131–8. doi: 10.1016/j.ccr.2012.05.036
93. Pong WW, Higer SB, Gianino SM, Emnett RJ, Gutmann DH. Reduced microglial CX3CR1 expression delays neurofibromatosis-1 glioma formation. Ann Neurol (2013) 73(2):303–8. doi: 10.1002/ana.23813
94. Hegedus B, Banerjee D, Yeh TH, Rothermich S, Perry A, Rubin JB, et al. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res (2008) 68(5):1520–8. doi: 10.1158/0008-5472.CAN-07-5916
95. Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U.S.A. (2005) 102(24):8573–8. doi: 10.1073/pnas.0503224102
96. Kaul A, Toonen JA, Cimino PJ, Gianino SM, Gutmann DH. Akt- or MEK-mediated mTOR inhibition suppresses Nf1 optic glioma growth. Neuro Oncol (2015) 17(6):843–53. doi: 10.1093/neuonc/nou329
97. Toonen JA, Anastasaki C, Smithson LJ, Gianino SM, Li K, Kesterson RA, et al. NF1 germline mutation differentially dictates optic glioma formation and growth in neurofibromatosis-1. Hum Mol Genet (2016) 25(9):1703–13. doi: 10.1093/hmg/ddw039
98. Otero JJ, Rowitch D, Vandenberg S. OLIG2 is differentially expressed in pediatric astrocytic and in ependymal neoplasms. J Neurooncol (2011) 104(2):423–38. doi: 10.1007/s11060-010-0509-x
99. Dasgupta B, Gutmann DH. Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J Neurosci (2005) 25(23):5584–94. doi: 10.1523/JNEUROSCI.4693-04.2005
100. Platten M, Kretz A, Naumann U, Aulwurm S, Egashira K, Isenmann S, et al. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann Neurol (2003) 54(3):388–92. doi: 10.1002/ana.10679
101. Markovic DS, Glass R, Synowitz M, Rooijen N, Kettenmann H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J Neuropathol Exp Neurol (2005) 64(9):754–62. doi: 10.1097/01.jnen.0000178445.33972.a9
102. Brown HK, Schiavone K, Tazzyman S, Heymann D, Chico TJ. Zebrafish xenograft models of cancer and metastasis for drug discovery. Expert Opin Drug Discovery (2017) 12(4):379–89. doi: 10.1080/17460441.2017.1297416
103. Wertman J, Veinotte CJ, Dellaire G, Berman JN. The zebrafish xenograft platform: evolution of a novel cancer model and preclinical screening tool. Adv Exp Med Biol (2016) 916:289–314. doi: 10.1007/978-3-319-30654-4_13
104. Sarmiento BE, Callegari S, Ghotme KA, Akle V. Patient-derived xenotransplant of CNS neoplasms in zebrafish: A systematic review. Cells (2022) 11(7):1204. doi: 10.3390/cells11071204
105. Sigaud R, Rosch L, Gatzweiler C, Benzel J, von Soosten L, Peterziel H, et al. The first-in-class ERK inhibitor ulixertinib shows promising activity in mitogen-activated protein kinase (MAPK)-driven pediatric low-grade glioma models. Neuro Oncol (2023) 25(3):566–79. doi: 10.1093/neuonc/noac183
106. Yang XJ, Cui W, Gu A, Xu C, Yu SC, Li TT, et al. A novel zebrafish xenotransplantation model for study of glioma stem cell invasion. PloS One (2013) 8(4):e61801. doi: 10.1371/journal.pone.0061801
107. Mayrhofer M, Gourain V, Reischl M, Affaticati P, Jenett A, Joly JS, et al. A novel brain tumour model in zebrafish reveals the role of YAP activation in MAPK- and PI3K-induced Malignant growth. Dis Model Mech (2017) 10(1):15–28. doi: 10.1242/dmm.026500
108. Luo J, Liu P, Lu C, Bian W, Su D, Zhu C, et al. Stepwise crosstalk between aberrant Nf1, Tp53 and Rb signalling pathways induces gliomagenesis in zebrafish. Brain (2021) 144(2):615–35. doi: 10.1093/brain/awaa404
109. Lee JS, Padmanabhan A, Shin J, Zhu S, Guo F, Kanki JP, et al. Oligodendrocyte progenitor cell numbers and migration are regulated by the zebrafish orthologs of the NF1 tumor suppressor gene. Hum Mol Genet (2010) 19(23):4643–53. doi: 10.1093/hmg/ddq395
110. Eden CJ, Ju B, Murugesan M, Phoenix TN, Nimmervoll B, Tong Y, et al. Orthotopic models of pediatric brain tumors in zebrafish. Oncogene (2015) 34(13):1736–42. doi: 10.1038/onc.2014.107
111. Pudelko L, Edwards S, Balan M, Nyqvist D, Al-Saadi J, Dittmer J, et al. An orthotopic glioblastoma animal model suitable for high-throughput screenings. Neuro Oncol (2018) 20(11):1475–84. doi: 10.1093/neuonc/noy071
112. Yan C, Brunson DC, Tang Q, Do D, Iftimia NA, Moore JC, et al. Visualizing engrafted human cancer and therapy responses in immunodeficient zebrafish. Cell (2019) 177(7):1903–1914e14. doi: 10.1016/j.cell.2019.04.004
113. Ai X, Ye Z, Xiao C, Zhong J, Lancman JJ, Chen X, et al. Clinically relevant orthotopic xenograft models of patient-derived glioblastoma in zebrafish. Dis Model Mech (2022) 15(4):dmm.049109. doi: 10.1242/dmm.049109
114. Isakson SH, Rizzardi AE, Coutts AW, Carlson DF, Kirstein MN, FIsher J, et al. Genetically engineered minipigs model the major clinical features of human neurofibromatosis type 1. Commun Biol (2018) 1:158. doi: 10.1038/s42003-018-0163-y
115. Poore B, Yuan M, Arnold A, Price A, Alt J, Rubens JA, et al. Inhibition of mTORC1 in pediatric low-grade glioma depletes glutathione and therapeutically synergizes with carboplatin. Neuro Oncol (2019) 21(2):252–63. doi: 10.1093/neuonc/noy150
116. Sun Y, Alberta JA, Pilarz C, Calligaris D, Chadwick EJ, Ramkissoon SH, et al. A brain-penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low-grade astrocytomas. Neuro Oncol (2017) 19(6):774–85. doi: 10.1093/neuonc/now261
117. Jentoft M, Giannini C, Cen L, Scheithauer W, Hoesley B, Sarkaria JN, et al. Phenotypic variations in NF1-associated low grade astrocytomas: possible role for increased mTOR activation in a subset. Int J Clin Exp Pathol (2010) 4(1):43–57.
118. Engelhardt S, Behling F, Beschorner R, Eckert F, Kohlhof P, Tatagiba M, et al. Frequent FGFR1 hotspot alterations in driver-unknown low-grade glioma and mixed neuronal-glial tumors. J Cancer Res Clin Oncol (2022) 148(4):857–66. doi: 10.1007/s00432-021-03906-x
119. Egbivwie N, Cockle JV, Humphries M, Ismail A, Esteves F, Taylor C, et al. FGFR1 expression and role in migration in low and high grade pediatric gliomas. Front Oncol (2019) 9:103. doi: 10.3389/fonc.2019.00103
120. Kim GB, Rincon Fernandez Pacheco D, Saxon D, Yang A, Sabet S, Dutra-Clarke M, et al. Rapid generation of somatic mouse mosaics with locus-specific, stably integrated transgenic elements. Cell (2019) 179(1):251–267.e24. doi: 10.1016/j.cell.2019.08.013
121. Li S, Mattar P, Dixit R, Lawn SO, Wilkinson G, Kinch C, et al. RAS/ERK signaling controls proneural genetic programs in cortical development and gliomagenesis. J Neurosci (2014) 34(6):2169–90. doi: 10.1523/JNEUROSCI.4077-13.2014
122. Chen F, Becker AJ, LoTurco JJ. Contribution of tumor heterogeneity in a new animal model of CNS tumors. Mol Cancer Res (2014) 12(5):742–53. doi: 10.1158/1541-7786.MCR-13-0531
123. Breunig JJ, Levy R, Antonuk CD, Molina J, Dutra-Clarke M, Park H, et al. Ets factors regulate neural stem cell depletion and gliogenesis in ras pathway glioma. Cell Rep (2015) 12(2):258–71. doi: 10.1016/j.celrep.2015.06.012
124. Bian S, Repic M, Guo Z, Kavirayani A, Burkard T, Bagley JA, et al. Genetically engineered cerebral organoids model brain tumor formation. Nat Methods (2018) 15(8):631–9. doi: 10.1038/s41592-018-0070-7
Keywords: pLGG, pediatric low grade glioma, NF1, BRAF, mouse models, preclinical model, KIAA-1549-fusion, optic glioma
Citation: Yvone GM and Breunig JJ (2024) Pediatric low-grade glioma models: advances and ongoing challenges. Front. Oncol. 13:1346949. doi: 10.3389/fonc.2023.1346949
Received: 30 November 2023; Accepted: 29 December 2023;
Published: 22 January 2024.
Edited by:
Ewa Krawczyk, Georgetown University, United StatesReviewed by:
Giuseppina Catanzaro, Sapienza University of Rome, ItalyJessica Tsai, Dana–Farber Cancer Institute, United States
Copyright © 2024 Yvone and Breunig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joshua J. Breunig, am9zaHVhLmJyZXVuaWdAY3Nocy5vcmc=