 Afsaneh Barzi1*
Afsaneh Barzi1* Caroline M. Weipert2
Caroline M. Weipert2 Carin R. Espenschied2
Carin R. Espenschied2 Victoria M. Raymond2
Victoria M. Raymond2 Andrea Wang-Gillam3Mohammad Amin Nezami4
Andrea Wang-Gillam3Mohammad Amin Nezami4 Eva J. Gordon5Daruka Mahadevan6Kabir Mody7
Eva J. Gordon5Daruka Mahadevan6Kabir Mody7- 1Department of Medical Oncology & Therapeutics Research, City of Hope National Medical Center, Duarte, CA, United States
- 2Guardant Health, Redwood City, CA, United States
- 3Division of Oncology, Siteman Cancer Center, St. Louis, MO, United States
- 4Orange Coast Medical Center of Hope, Newport Beach, CA, United States
- 5Private Health Management, Inc., Los Angeles, CA, United States
- 6Division of Hematology and Oncology, Department of Medicine, University of Texas Health, San Antonio, San Antonio, TX, United States
- 7Division of Hematology-Oncology, Department of Medicine, Mayo Clinic, Jacksonville, FL, United States
Purpose: Despite accumulating data regarding the genomic landscape of pancreatic ductal adenocarcinoma (PDAC), olaparib is the only biomarker-driven FDA-approved targeted therapy with a PDAC-specific approval. Treating ERBB2(HER2)-amplified PDAC with anti-HER2 therapy has been reported with mixed results. Most pancreatic adenocarcinomas have KRAS alterations, which have been shown to be a marker of resistance to HER2-targeted therapies in other malignancies, though the impact of these alterations in pancreatic cancer is unknown. We describe two cases of ERBB2-amplified pancreatic cancer patients treated with anti-HER2 therapy and provide data on the frequency of ERBB2 amplifications and KRAS alterations identified by clinical circulating cell-free DNA testing.
Methods: De-identified molecular test results for all patients with pancreatic cancer who received clinical cell-free circulating DNA analysis (Guardant360) between 06/2014 and 01/2018 were analyzed. Cell-free circulating DNA analysis included next-generation sequencing of up to 73 genes, including select small insertion/deletions, copy number amplifications, and fusions.
Results: Of 1,791 patients with pancreatic adenocarcinoma, 36 (2.0%) had an ERBB2 amplification, 26 (72.2%) of whom had a KRAS alteration. Treatment data were available for seven patients. Two were treated with anti-HER2 therapy after their cell-free circulating DNA result, with both benefiting from therapy, including one with a durable response to trastuzumab and no KRAS alteration detected until progression.
Conclusion: Our case series illustrates that certain patients with ERBB2-amplified pancreatic adenocarcinoma may respond to anti-HER2 therapy and gain several months of prolonged survival. Our data suggests KRAS mutations as a possible mechanism of primary and acquired resistance to anti-HER2 therapy in pancreatic cancer. Additional studies are needed to clarify the role of KRAS in resistance to anti-HER2 therapy.
1 Introduction
Pancreatic ductal adenocarcinoma (PDAC) is notoriously challenging to diagnose and treat, with over 80% of patients having regional or distant metastases at diagnosis, and over 87% surviving less than five years (1). There is currently only one United States Food and Drug Administration (FDA)-approved biomarker-driven targeted therapy approved specifically for PDAC (the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib), despite several PDAC sequencing studies identifying potential genomic targets, including homologous recombination deficiency (HRD) genes, BRAF, and receptor tyrosine kinases, such as ERBB2(HER2) (2–8).
Recent studies have identified ERBB2(HER2) amplifications in PDAC at rates of 1-7% (2–9). Targeted therapies are FDA-approved for ERBB2-amplified breast, colon, and gastric cancer (10–14). In the past, trastuzumab efficacy was examined in patients with ERBB2 positive PDAC, as assessed by IHC, with confirmed response rates of 6% to 23.5%, but numbers were small and tumors were not assessed for co-occurring mutations and potential mechanisms of resistance (15, 16). In the more recent Know Your Tumor study, four ERBB2-amplified patients with PDAC received trastuzumab in combination with various drugs and observed responses ranging from one month to over 12 months (5). The phase II MyPathway trial included nine ERBB2-amplified patients with PDAC who were treated with trastuzumab and pertuzumab, of whom two achieved a partial response (11). Additional case reports have shown mixed results, with a KRAS wild-type, ERBB2-amplified patient with PDAC progressing within one month of treatment with trastuzumab emtansine in the fifth-line setting, and another patient achieving stable disease with trastuzumab and pertuzumab, followed by complete response when treated with immunotherapy, radiation, and trastuzumab deruxtecan in the third-line setting (4, 17). KRAS mutations have been seen at the time of primary and acquired resistance to anti-HER2 therapy in other cancer types, including colorectal and gastroesophageal cancer; however, the impact of KRAS mutations co-occurring with potentially targetable alterations in PDAC has not been defined (11, 18–20).
While mutations in potentially targetable genes in PDAC are individually rare, recent studies suggest that 10-20% of PDAC tumors may harbor therapeutically actionable alterations (4, 21). Comprehensive genomic profiling with next-generation sequencing (NGS) provides the opportunity to identify patients who may benefit from a targeted therapy approach, and analysis of circulating cell-free DNA (cfDNA) could be particularly useful in PDAC where tissue specimens are limited and patients often have less time to wait for genomic test results given the urgency to initiate treatment (22, 23). Herein, we describe two cases of patients with ERBB2-amplified PDAC treated with anti-HER2 therapy following their cfDNA result, and assess the frequency of ERBB2 amplification and co-occurring KRAS mutations and/or amplification in clinical cfDNA NGS results in over 1,700 samples from patients with PDAC.
2 Materials and methods
2.1 Patients
We analyzed the Guardant360 deidentified database, containing results of patients who underwent clinical cfDNA testing between June 2014 and January 2018, and identified patients with a diagnosis of PDAC as reported by the ordering provider on the test requisition form. Patients with PDAC who had an ERBB2 amplification detected on at least one cfDNA test were included for further analysis. This research was approved by the Advarra Institutional Review Board (IRB) for the generation of deidentified data sets for research purposes. For select patients, additional details regarding treatments and outcomes were obtained from the treating physician as per local IRB guidelines.
2.2 cfDNA analysis
Blood draw, shipment, plasma isolation and cfDNA extraction procedures for the clinical cfDNA assay used in this study have been previously described (24). Guardant360 is a CLIA-certified, College of American Pathologists-accredited, New York State Department of Health-approved cfDNA NGS assay with analytic and clinical validation reported (24). Point mutations were analyzed in 68 to 73 genes, small insertions and/or deletions (indels) in up to 23 genes, copy number amplifications (CNA) in up to 18 genes, and fusions in up to six genes, depending on the panel version performed (Table 1) (24).
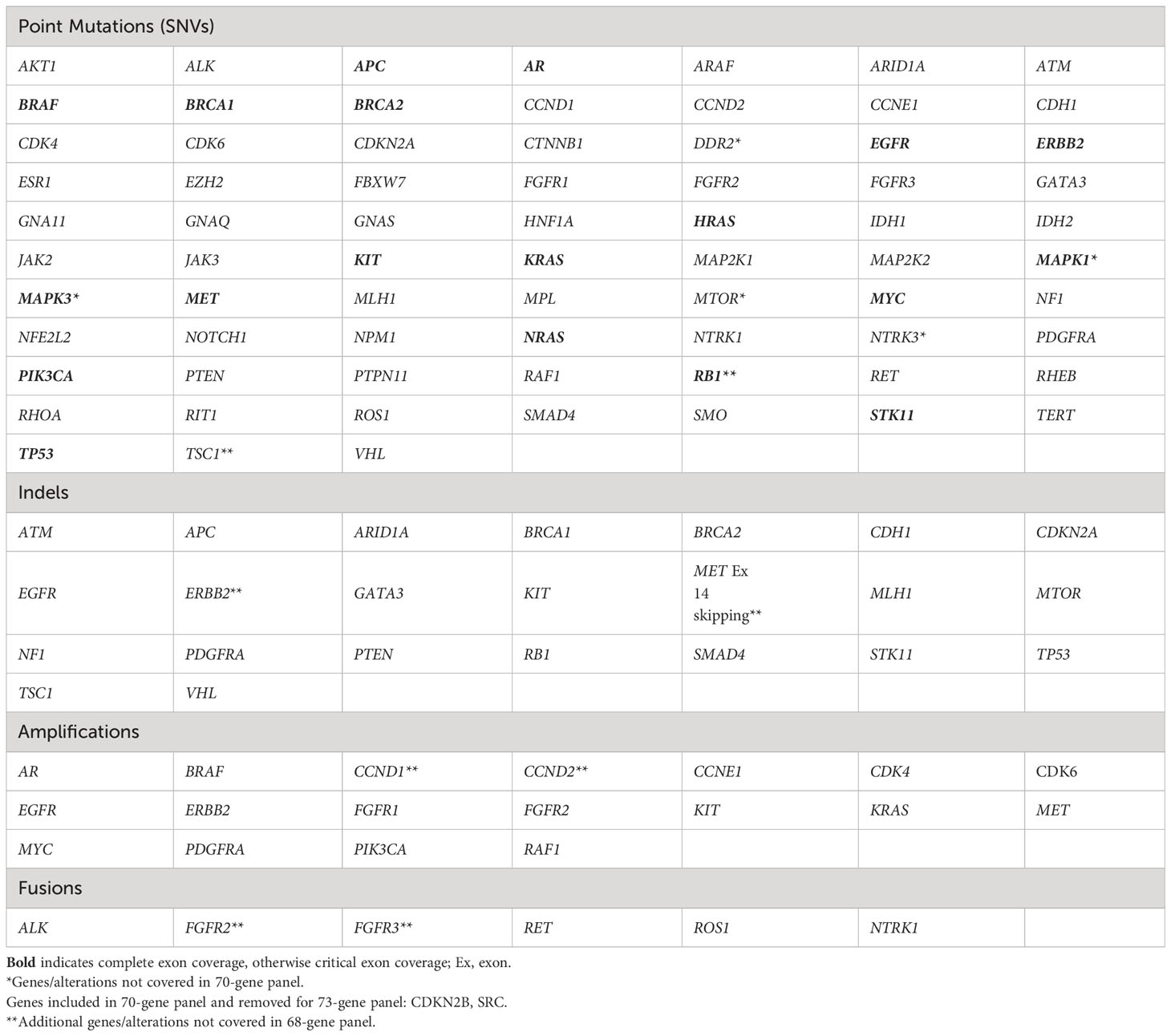
Table 1 Genes and mutation types analyzed in cfDNA analysis (Guardant360 73 gene panel).
2.3 Data analysis
cfDNA ERBB2 amplifications were classified by copy number and category (1+/2+/3+) and assessed in comparison with co-occurring cfDNA identified KRAS alterations. At the time of this study, amplifications were reported on a semi-quantitative scale given that the absolute number of copies in circulation is dependent on both tumor fraction and the magnitude of amplifications. The 1+ category applied to amplification magnitude in the lower 50th percentile of samples with amplifications, 2+ applied to amplification magnitude in the 50th to 90th percentile, and 3+ applied to amplification magnitude in the top 10th percentile. KRAS alterations were categorized as amplifications or single nucleotide variants (SNVs). All characterized pathogenic KRAS SNVs were included. For patients with cfDNA analysis at multiple time points, ERBB2 amplifications and KRAS alterations were assessed for changes over time.
3 Results
During the study period, 1,791 patients with PDAC had ≥1 alteration detected via cfDNA analysis, 36 (2.0%) of whom had an ERBB2 amplification (Figure 1). The cohort of patients with ERBB2-amplified PDAC was 44.4% female with a mean (median) age of 66.3 (65.5) years (range 32-91; Table 2). Among the 21 patients for whom date of initial diagnosis was available, the mean (median) time from diagnosis to blood draw for liquid biopsy was 582.4 (366.5) days (range 7-2,452). Most patients were receiving systemic therapy at the time of blood draw for liquid biopsy (Table 2).
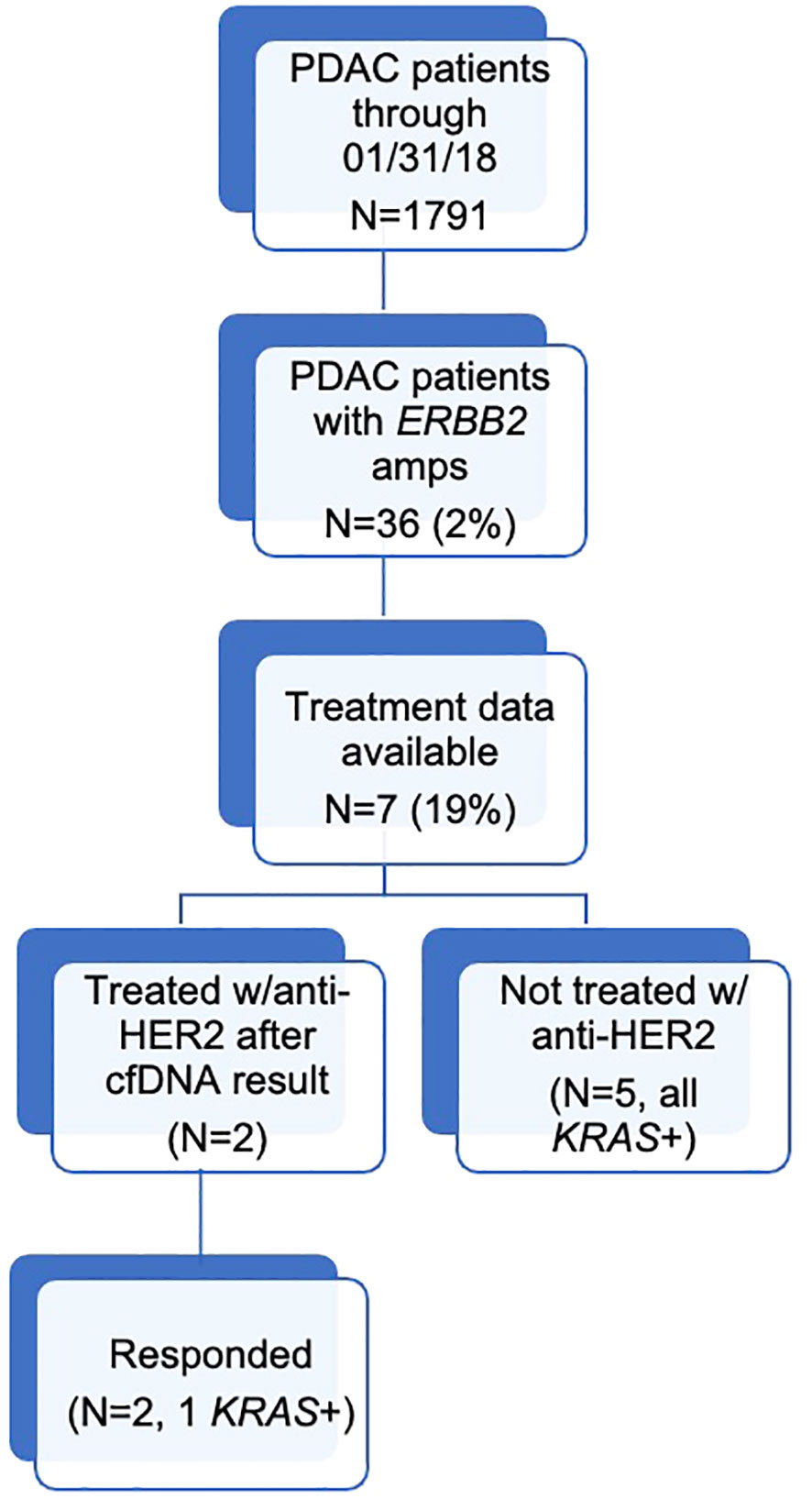
Figure 1 Diagram outlining the number of patients in each study category.
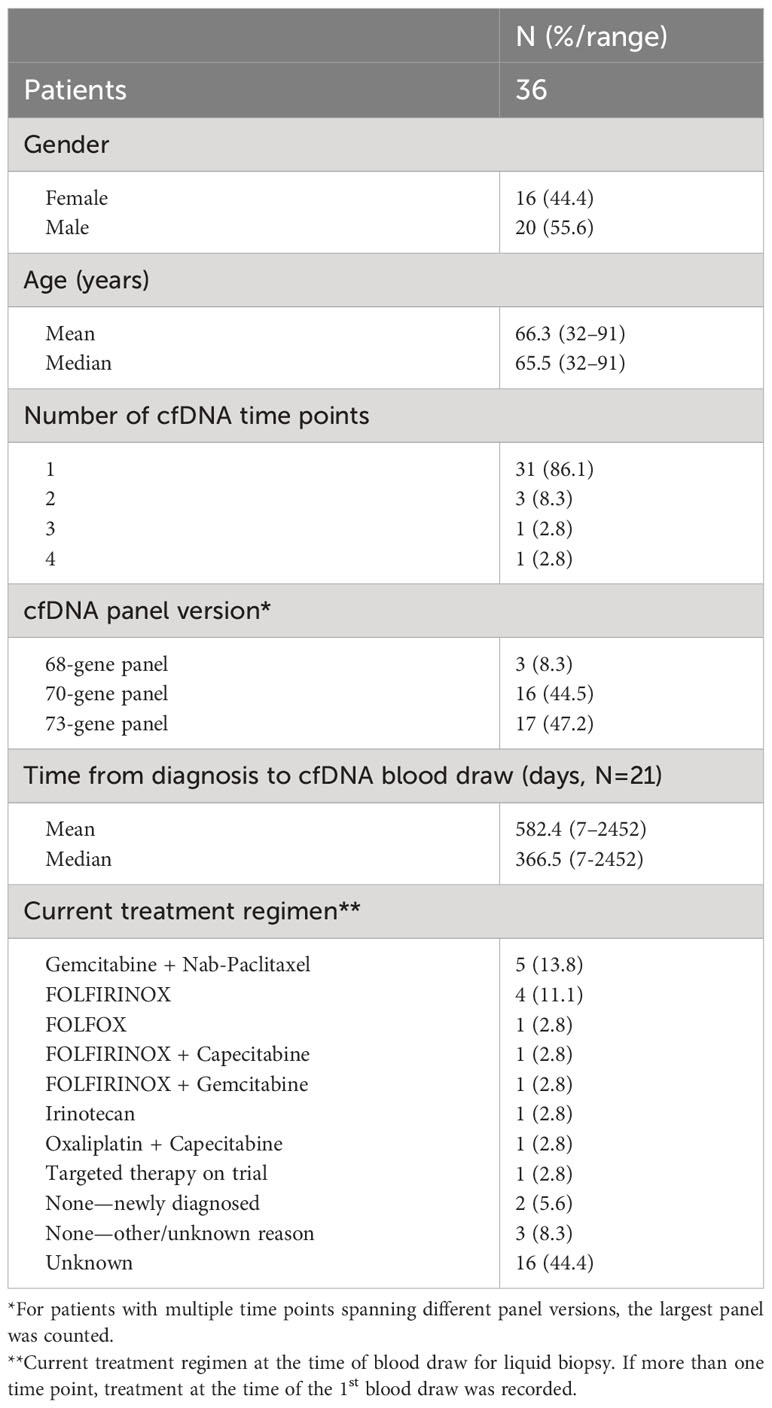
Table 2 Patient Demographics.
Among patients with ERBB2 amplifications (N=36), 19 (52.8%) were 1+, 15 (41.7%) were 2+, and 2 (5.6%) were 3+ (Table 2). Of these, 26 (72.2%) also had a KRAS mutation and/or amplification, with 14 of 26 patients having co-occurring KRAS SNV(s) without KRAS amplification (Table 3).
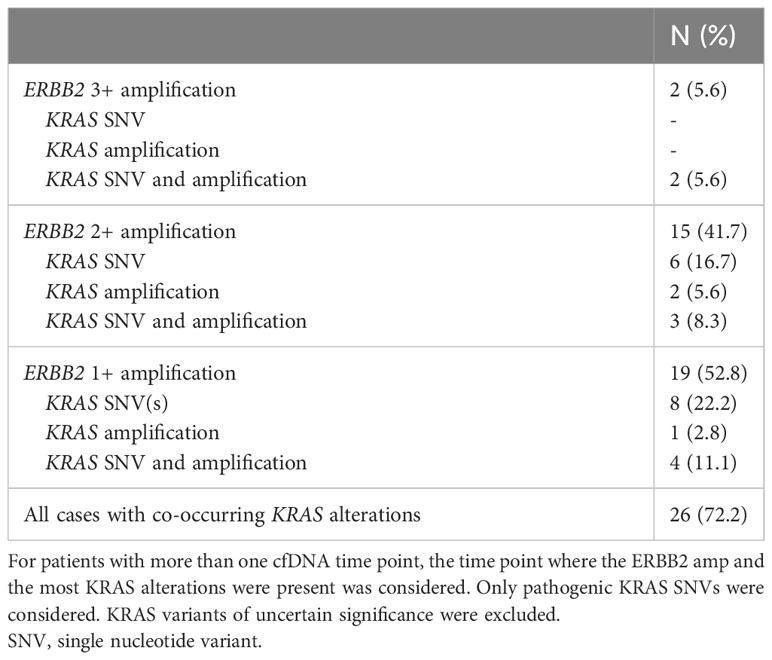
Table 3 ERBB2 amplification level (N=36) and co-occurring KRAS alterations.
Five patients with ERBB2-amplified PDAC had cfDNA analysis at multiple time points (Figure 2; Patient 17 shown in Figure 3). For all five patients, the ERBB2 amplification was detected at a single time point, and all had KRAS alterations at one or more time points. Three patients had at least one activating KRAS alteration and/or KRAS amplification detected in all samples, with the ERBB2 amplification occurring in the last sample, notably corresponding to an increase in tumor shed, as measured via the maximum variant allele frequency (maxVAF) in the sample. One patient had a KRAS alteration and an ERBB2 amplification detected in their first sample, with persistence of the KRAS alteration in their second sample but loss of the amplification. One patient (Patient 17 below) had ERBB2 amplification detected in their first sample, with neither ERBB2 amplification nor KRAS alterations detected in two subsequent blood draws, and a KRAS mutation detected in the fourth blood draw without identification of the ERBB2 amplification.
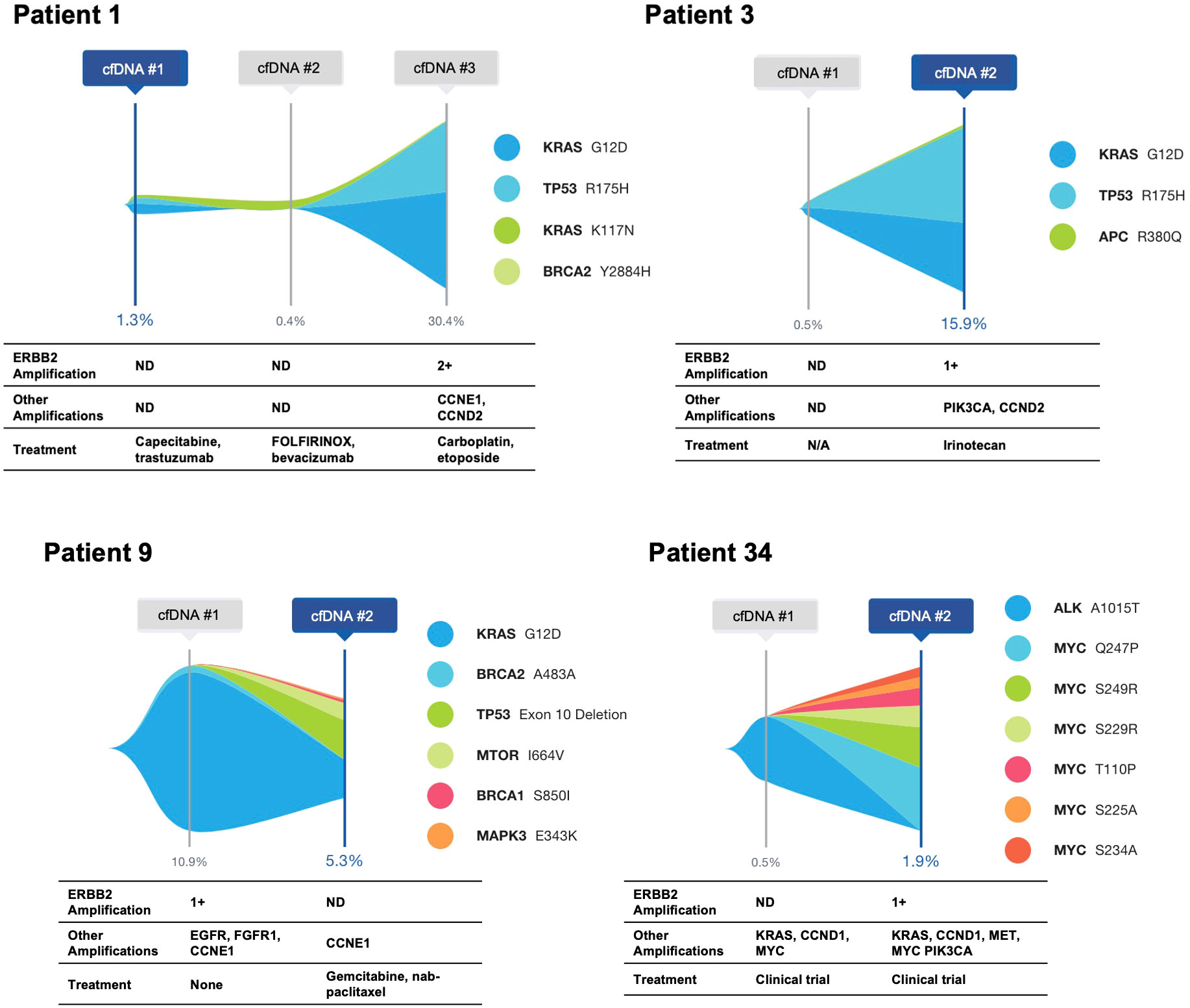
Figure 2 Serial cfDNA time points for patients with ERBB2 amplification identified in at least a single time point. Single nucleotide variants and insertion/deletion alterations are illustrated, with amplifications and treatment at the time of the cfDNA draw shown below.
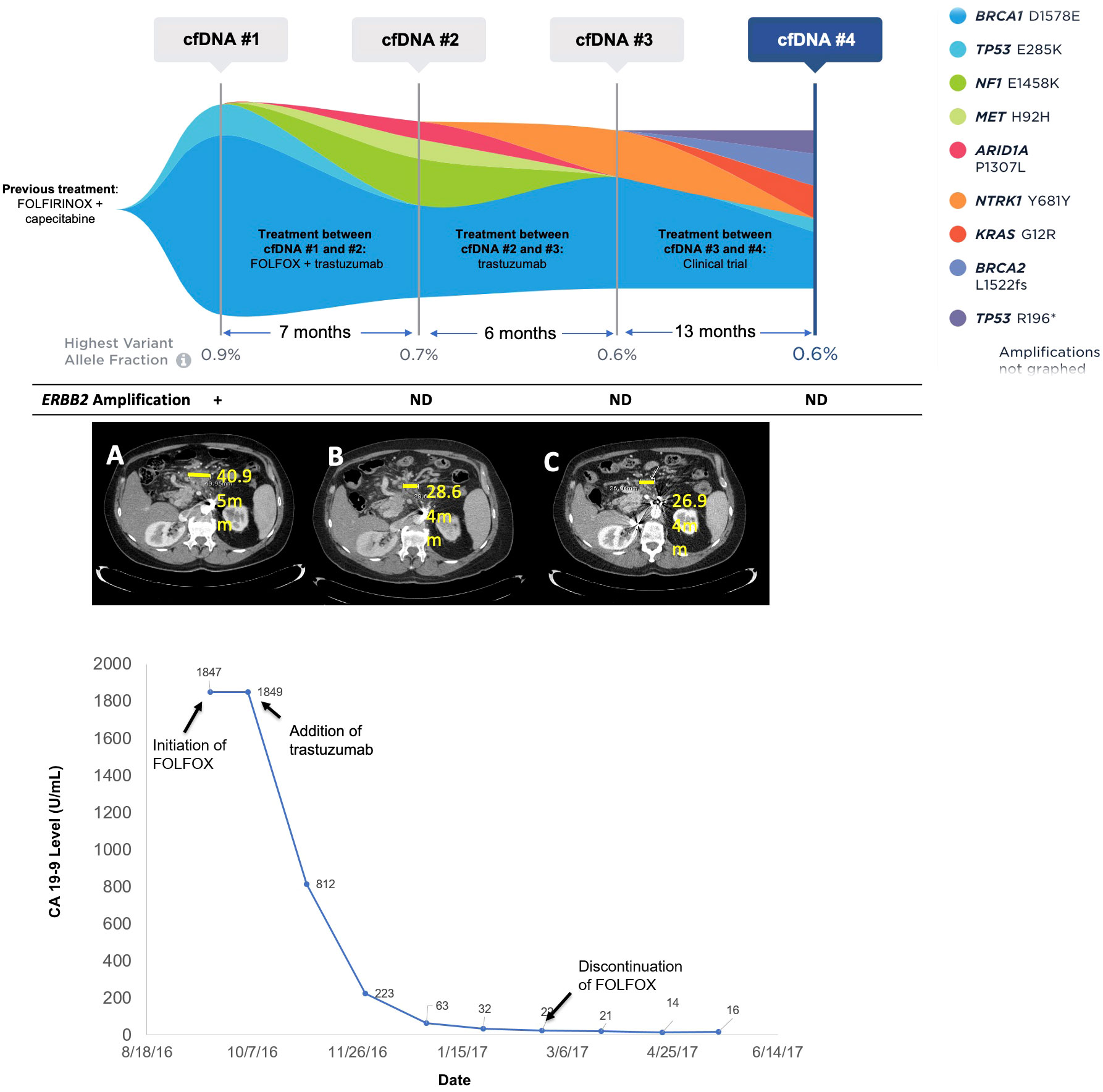
Figure 3 Molecular and clinical response to anti-HER2 therapy. Changes in mutations identified and variant allele fraction in Patient 17 as identified by cfDNA analysis along with corresponding systemic therapy and imaging results. The CA19-9 levels for Patient 17 over time corresponding to the systemic therapy and molecular results are also shown. Imaging shows CT scans at initation of FOLFOX (A), discontinuation of FOLOX (B), and three months after trastuzumab monotherapy (C).
Treatment data were available for seven patients, two of whom were treated with anti-HER2 therapy following their cfDNA result, with both benefiting from therapy. Patient 5 was a 56- year-old male with PDAC progressing after treatment with FOLFOX. cfDNA analysis identified an ERBB2 amplification at 2.3 copies (1+) as well as SMAD4 R361C at 35.1% VAF, KRAS G12D at 35.0% VAF, TP53 Q144* at 31.7% VAF, AR T878S at 0.2% VAF, and CDK6 amplification at 2.5 copies (1+). He was treated with the HER2/EGFR tyrosine kinase inhibitor afatinib in the second-line setting for one month and had stable disease with improved quality of life but was not a candidate for continued anti-HER2 or other subsequent therapy.
Patient 17 was a 64- year-old male, originally diagnosed with PDAC with metastases to the retroperitoneal lymph nodes. He was treated with first-line FOLFIRINOX and capecitabine maintenance for two years. At progression, liquid biopsy was ordered and was positive for an ERBB2 amplification at 2.2 copies (1+), TP53 E285K at 0.1% VAF, and a BRCA1 variant of uncertain significance (VUS) at 0.9% VAF (Figure 3). The patient was initiated on FOLFOX, and trastuzumab was added soon afterward. The patient experienced clinical improvement and significant decline in CA19-9 (1,847 to 22 U/mL) over a four-month period (Figure 3). Only VUS and synonymous variants were detected on a repeat liquid biopsy, with a maxVAF of 0.7% (Figure 3). FOLFOX was discontinued after about four months and the patient remained on trastuzumab alone with further clinical improvement and decrease in CA19-9 (16 U/mL). After 6 months on trastuzumab, the patient began to experience progression, and a third cfDNA test continued to only show VUS and synonymous variants with a maxVAF of 0.6%. FOLFOX was re-introduced with no biochemical or radiographic response. The patient then went on a clinical trial with an immunotherapy combination and did well on this trial. A final cfDNA test taken approximately one year after the previous test demonstrated the emergence of KRAS G12R at 0.3% VAF, BRCA2 L1522fs at 0.3% VAF, TP53 R196* at 0.2% VAF, and TP53 E285K at 0.1% VAF, with the ERBB2 amplification still not detected (Figure 3).
4 Discussion
Therapeutic options in patients with metastatic PDAC are limited. Patients with KRAS wildtype disease may have a different biology that may be more amenable to targeted therapies, but KRAS wildtype disease is present in a minority of patients with PDAC. Advancement of targeted therapies in PDAC has been challenging, with only one FDA-approved biomarker-directed targeted therapy (olaparib) currently available with a PDAC-specific approval. KRAS is the most frequently mutated gene in PDAC (70-95%), and until recently, was not generally considered a therapeutically targetable alteration (6, 25–28). Historic trials evaluating KRAS positive PDAC have primarily focused on a variety of MEK inhibitors and have shown limited benefit (29). More recently, the development of drugs specifically targeting KRAS G12C alterations, known to be present in a minority of PDAC cases, have shown promise (30, 31). Other KRAS alterations, such as G12D are much more common in PDAC, and drugs targeting other non-G12C alterations are in development (32).
Outside of KRAS, the efficacy of other targeted therapies continues to be explored in PDAC. Germline and/or somatic mutations in HRD genes may be present in up to 20% of advanced PDAC tumors (2). While a study of veliparib in patients with PDAC and germline pathogenic BRCA1/2 or PALB2 mutations had no confirmed responses, a study of rucaparib in patients with germline or somatic BRCA1/2 mutations had a 16% objective response rate (ORR) and 32% disease control rate (DCR) (33, 34). Additionally, a trial of olaparib in PDAC achieved a 22% ORR and 57% DCR, and the phase III POLO trial evaluating olaparib maintenance therapy versus placebo in patients with metastatic PDAC and germline BRCA1/2 mutations found significantly longer progression free survival (PFS) for olaparib versus placebo, resulting in its FDA approval (35–38). Additional targetable biomarkers in PDAC include NRG1 fusions, microsatellite instability (MSI), and BRAF V600E (39–47). The cumulative results of trials examining these biomarkers suggest identification of PDAC patients with targetable alterations, even if they are rare, may open up therapy options to patients who may have limited other options.
While studies have identified ERBB2(HER2) amplifications in PDAC at rates of 1-7%, most have shown mixed responses to HER2-targeted therapies in ERBB2 positive PDAC patients, with variable documentation of co-occurring RAS mutations (2–9, 15, 16). In the Know Your Tumor study, four ERBB2-amplified patients with PDAC received trastuzumab in combination with various drugs and observed responses ranging from one month to over 12 months (5). The phase II MyPathway trial included nine ERBB2-amplified patients with PDAC who were treated with trastuzumab and pertuzumab, of whom two achieved a partial response (11). Additional trials examining anti-HER2 therapy in PDAC, like the TAPUR trial, have yet to read out (48, 49). Among this clinical laboratory database of 1,791 patients with PDAC, ERBB2 amplifications were seen at rates consistent with previous, smaller, studies (2.0%), with the majority also harboring at least one KRAS activating alteration. Notably, we report a case in which a patient with ERBB2-amplified PDAC (in the absence of co-occurring KRAS mutations) responded to anti-HER2 therapy for several months with clinical improvement and decline of tumor markers, before demonstrating a KRAS alteration newly detected by cfDNA and loss of ERBB2 amplification via cfDNA after progression. Of note, this patient had relatively low tumor shed throughout their disease course (maxVAF at each time point did not exceed 1%), and thus it is difficult to say exactly when the patient acquired the KRAS G12R alteration as it was only seen in cfDNA approximately one year following trastuzumab discontinuation. Acquired KRAS alterations have been seen at the time of progression on trastuzumab in other cancer types (18). In contrast, a patient with co-occurring KRAS G12D and ERBB2 amplification prior to anti-HER2 therapy initiation achieved stable disease for one month.
The presence of KRAS alterations at the time of resistance to anti-HER2 therapy is consistent with recent data from a clinical trial of trastuzumab and lapatinib in metastatic colorectal cancer (19). In that study, RAS and RAF alterations were detected at baseline in the plasma of 6/7 (86%) patients refractory to anti-HER2 therapy, but in only 3/22 (14%) patients who benefited from anti-HER2 therapy. KRAS mutations were subsequently identified at progression in two patients who initially had SD and one with an initial PR (19). Notably, the MyPathway HER2 basket trial examining use of trastuzumab plus pertuzumab in ERBB2-amplified patients across solid tumors stratified patients by KRAS status, and showed that patients with a KRAS mutation had an ORR of 3.8% and DCR of 3.8%, compared to an ORR of 25.6% and DCR of 49.7% in patients who were KRAS wildtype, suggesting co-occurring KRAS mutations had a significant impact on likelihood of response (20). Taken together, these reports and our cases suggest that certain patients with ERBB2-amplified PDAC do benefit from anti-HER2 therapy. Though limited in size, our results are consistent with previous work suggesting that KRAS mutations may function as primary or acquired resistance to anti-HER2 therapy, similar to what has been observed in colorectal and gastroesophageal cancer.
Of note, two HER2-directed antibody-drug conjugates have now been FDA-approved in metastatic ERBB2-amplified breast cancer and are being explored in various other cancer types (50). These drugs have demonstrated significant improvements in patient outcomes in ERBB2-amplified breast cancer patients, and in the case of trastuzumab deruxtecan, are also approved for patients with “HER2-low” disease (50). Pan-cancer trials of these drugs have so far included a limited number of PDAC patients, with the phase II DESTINY-PanTumour02 trial including 25 PDAC patients, with three patients having response via independent central assessment, and the phase II KAMELEON trial including four PDAC patients, of whom one had a partial response (51, 52). Further exploration of the potential of these HER2-targeted antibody-drug conjugates in the PDAC population are needed.
Detection of copy number amplification via cfDNA is dependent on two factors: 1) the degree of tumor shed, and 2) the level of amplification. Thus, a low-level amplification may not be detected even in a patient with a high-degree of tumor shed (53). In each case where ERBB2 amplification was not originally detected, the sample with the amplification detected had a higher maxVAF, indicative of increased tumor shed. Thus, we cannot rule-out the possibility that the ERBB2 amplification was present in earlier samples but was occurring below the assay’s limit of detection. We also saw loss of ERBB2 expression in patients who were originally ERBB2-amplified, and in one of these instances the patient (Patient 17) was known to have been treated with anti-HER2 therapy. Loss of HER2 expression following anti-HER2 therapy has been reported in other cancer types, and thus in some instances may explain loss of HER2 expression over time (54–56). Attempts to account for the degree of tumor shed when examining plasma-based copy number calls are ongoing, but it remains a challenge to clearly account for tumor evolution/heterogeneity, treatment effects, and tumor shed when assessing changes in the amplification status of a particular tumor, especially when dealing with low and medium-level amplifications (53).
Overall, review of patients with serial cfDNA samples demonstrate tumor evolution in response to therapy, illustrating a well-known cause of tumor heterogeneity. Previous studies using rapid autopsy sampling of multiple metastatic sites have demonstrated that the molecular makeup of the primary tumor versus each metastatic site can vary (57). Thus, in the setting of disease progression, tissue biopsies may be limited in their ability to fully capture acquired resistance mutations unless multiple metastatic sites are biopsied at multiple time points, which is often not feasible. The difficulty of repeating tissue biopsies in patients progressing on therapy is especially acute in PDAC, where rapid clinical deterioration can create additional challenges to successful tissue biopsy (22). Liquid biopsy is much less invasive and has a demonstrated ability to capture a global picture of the alterations present throughout a patient’s disease burden, and thus may be optimally suited for tracking disease response and development of acquired resistance alterations leading to disease progression (58).
There are several limitations to our study, including the fact that it is a retrospective analysis, and our knowledge of patient clinical history is limited in most cases to what is provided by the ordering provider on the test acquisition form, which may not be wholly accurate and does not include orthogonal molecular testing information. Given this, we can only comment on the response to anti-HER2 therapy for a limited number of patients. For most of the patients with serial samples, we can only make educated guesses about why we see fluctuations in the appearance of ERBB2 amplification and/or KRAS activating mutations based on the degree of tumor shed, as detailed above. Additionally, it is possible that there are inherent biases in the cohort of patients selected to undergo testing via cfDNA by their treating physician (e.g., they could have more aggressive disease and/or have progressed through more lines of therapy). As such, the alterations seen here may not reflect the molecular landscape seen in a treatment naïve patient population and the cohort may not reflect the broader PDAC population.
In conclusion, this analysis of over 1,700 PDAC samples from patients undergoing clinical cfDNA testing demonstrates the utility of cfDNA in detecting targetable alterations in this patient population. Additionally, this case series of patients treated with anti-HER2 therapy based on ERBB2(HER2) amplification detected via cfDNA provides additional evidence to suggest that cfDNA may be an adequate tool to detect ERBB2(HER2) amplifications and identify patients who may benefit from anti-HER2 therapies, as demonstrated here by the patient who responded to anti-HER2 therapy for several months. Notably, there are a growing number of anti-HER2 therapies available, including multiple antibody-drug conjugates, meaning identification of these patients may become more relevant in the future (59). The importance of molecular testing in identifying patients with rare, but targetable, alterations across cancer types has been highlighted a number of times, and seems particularly relevant in PDAC given the lack of treatment options available and often rapid disease progression (22, 23). This case series supports the idea that co-occurring alterations may play a key role in determining which patients respond to targeted therapy or not, suggesting KRAS as a possible mechanism of innate and acquired resistance, and further demonstrating the need for patients to undergo comprehensive molecular profiling to identify optimal therapy. Given the well-known difficulties of obtaining tissue biopsy in PDAC, particularly following disease progression, cfDNA offers an attractive alternative (22). Moreover, cfDNA has a unique advantage in its ability to capture temporal and spatial tumor heterogeneity, both well-established causes of disease progression and mixed responses to treatment (58). This ability may be uniquely helpful in PDAC, as patients can progress quickly through lines of therapy and historic tissue biopsies may not accurately reflect the current molecular landscape, as shown here by the tumor evolution seen in serial cfDNA samples. Further studies reporting co-occurring mutations and clinical outcomes are needed to better clarify the role of KRAS and other potential mechanisms of resistance to anti-HER2 therapy to allow for the identification of PDAC patients most likely to benefit from this treatment, including further exploration of how molecular testing, applied more broadly in patients with PDAC, could aid in getting more patients with PDAC onto a growing number of targeted therapies.
Data availability statement
The datasets presented in this article are not readily available because they are derived from commercial testing. This data can be made available under a fully executed data use agreement. Requests to access the datasets should be directed to Y3dlaXBlcnRAZ3VhcmRhbnRoZWFsdGguY29t.
Ethics statement
The studies involving humans were approved by the Advarra Institutional Review Board (IRB) for the generation of deidentified data sets for research purposes. For select patients, additional details regarding treatments and outcomes were obtained from the treating physician as per local IRB guidelines. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
AB: Conceptualization, Methodology, Supervision, Writing – original draft, Writing – review & editing. CW: Formal analysis, Methodology, Writing – original draft, Writing – review & editing. CE: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. VR: Conceptualization, Methodology, Supervision, Writing – review & editing. AW-G: Data curation, Writing – review & editing. MN: Data curation, Writing – review & editing. EG: Data curation, Writing – review & editing. DM: Data curation, Writing – review & editing. KM: Data curation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
Authors CW, CE, and VR are employees and stockholders of Guardant Health, Inc. Author EG is employed by the company Private Health Management, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. National Cancer Institute Surveillance, Epidemiology, and End Result (SEER) Program. Cancer stat facts: pancreatic cancer. Available at: https://seer.cancer.gov/statfacts/html/pancreas.html. (Accessed January 25, 2024).
2. Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, et al. Real-time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discov (2018) 8(9):1096–111. doi: 10.1158/2159-8290.CD-18-0275
3. Chantrill LA, Nagrial AM, Watson C, Johns AL, Martyn-Smith M, Simpson S, et al. Precision medicine for advanced pancreas cancer: the individualized molecular pancreatic cancer therapy (IMPaCT) trial. Clin Cancer Res (2015) 21(9):2029–37. doi: 10.1158/1078-0432.CCR-15-0426
4. Lowery MA, Jordan EJ, Basturk O, Ptashkin RN, Zehir A, Berger MF, et al. Real-time genomic profiling of pancreatic ductal adenocarcinoma: potential actionability and correlation with clinical phenotype. Clin Cancer Res (2017) 23(20):6094–100. doi: 10.1158/1078-0432.CCR-17-0899
5. Pishvaian MJ, Bender RJ, Halverson D, Rahib L, Hendifar AE, Mikhail S, et al. Molecular profiling of pancreatic cancer patients: initial results from the know your tumor initiative. Clin Cancer Res (2018) 24(20):5018–27. doi: 10.1158/1078-0432.CCR-18-0531
6. Cancer Genome Atlas Research Network. Electronic address:YW5kcmV3X2FndWlycmVAZGZjaS5oYXJ2YXJkLmVkdSw= Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell (2017) 32(2):185–203.e13. doi: 10.1016/j.ccell.2017.07.007
7. Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun (2015) 6:7686. doi: 10.1038/ncomms8686
8. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature (2015) 518(7540):495–501. doi: 10.1038/nature14169
9. Chou A, Waddell N, Cowley MJ, Gill AJ, Chang DK, Patch AM, et al. Clinical and molecular characterization of HER2 amplified-pancreatic cancer. Genome Med (2013) 5(8):78. doi: 10.1186/gm482
10. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med (2001) 344(11):783–92. doi: 10.1056/NEJM200103153441101
11. Hainsworth JD, Meric-Bernstam F, Swanton C, Hurwitz H, Spigel DR, Sweeney C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from myPathway, an open-label, phase IIa multiple basket study. J Clin Oncol (2018) 36(6):536–42. doi: 10.1200/JCO.2017.75.3780
12. Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet (2010) 376(9742):687–97. doi: 10.1016/S0140-6736(10)61121-X
13. Strickler JH, Cercek A, Siena S, Andre T, Ng K, Van Cutsem E, et al. Additional analyses of MOUNTAINEER: a phase II study of tucatinib and trastuzumab for HER2-positive mCRC. Ann Oncol (2022) 33 (suppl_7), S808–69. doi: 10.1016/annonc/annonc108
14. Strickler JH, Zemla T, Ou FS, Cercek A, Wu C, Sanchez FA, et al. Trastuzumab and tucatinib for the treatment of HER2 amplified metastatic colorectal cancer (mCRC): Initial results from the MOUNTAINEER trial. Ann Oncol (2019) 30(Supp 5):v200. doi: 10.1093/annonc/mdz246.005
15. Safran H, Iannitti D, Ramanathan R, Schwartz JD, Steinhoff M, Nauman C, et al. Herceptin and gemcitabine for metastatic pancreatic cancers that overexpress HER-2/ neu. Cancer Invest (2004) 22(5):706–12. doi: 10.1081/CNV-200032974
16. Harder J, Ihorst G, Heinemann V, Hofheinz R, Moehler M, Buechler P, et al. Multicentre phase II trial of trastuzumab and capecitabine in patients with HER2 overexpressing metastatic pancreatic cancer. Br J Cancer (2012) 106(6):1033–8. doi: 10.1038/bjc.2012.18
17. King DA, Smith AR, Pineda G, Nakano M, Michelini F, Goedegebuure SP, et al. Complete remission of widely metastatic human epidermal growth factor receptor 2–amplified pancreatic adenocarcinoma after precision immune and targeted therapy with description of sequencing and organoid correlates. JCO Precis Oncol (2023) 7:e2100489. doi: 10.1200/PO.21.00489
18. Janjigian YY, Sanchez-Vega F, Jonsson P, Chatila WK, Hechtman JF, Ku GY, et al. Genetic predictors of response to systemic therapy in esophagogastric cancer. Cancer Discov (2018) 8(1):49–58. doi: 10.1158/2159-8290.CD-17-0787
19. Siravegna G, Lazzari L, Crisafulli G, Sartore-Bianchi A, Mussolin B, Cassingena A, et al. Radiologic and genomic evolution of individual metastases during HER2 blockade in colorectal cancer. Cancer Cell (2018) 34(1):148–162.e7. doi: 10.1016/j.ccell.2018.06.004
20. Meric-Bernstam F, Hainsworth J, Bose R, Burris HA III, Friedman CF, Kurzrock R, et al. MyPathway HER2 basket study: Pertuzumab (P) + trastuzumab (H) treatment of a large, tissue-agnostic cohort of patients with HER2-positive advanced solid tumors. JCO (2021) 39(15_suppl):3004–4. doi: 10.1200/JCO.2021.39.15_suppl.3004
21. Herbst B, Zheng L. Precision medicine in pancreatic cancer: treating every patient as an exception. Lancet Gastroenterol Hepatology (2019) 4(10):805–10. doi: 10.1016/S2468-1253(19)30175-X
22. Topham JT, Renouf DJ, Schaeffer DF. Circulating tumor DNA: toward evolving the clinical paradigm of pancreatic ductal adenocarcinoma. Ther Adv Med Oncol (2023) 15:175883592311576. doi: 10.1177/17588359231157651
23. Zhen DB, Safyan RA, Konick EQ, Nguyen R, Prichard CC, Chiorean EG. The role of molecular testing in pancreatic cancer. Therap Adv Gastroenterol (2023) 16:175628482311714. doi: 10.1177/17562848231171456
24. Odegaard JI, Vincent JJ, Mortimer S, Vowles JV, Ulrich BC, Banks KC, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin Cancer Res (2018) 24(15):3539–49. doi: 10.1158/1078-0432.CCR-17-3831
25. Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res (2017) 45(D1):D777–83. doi: 10.1093/nar/gkw1121
26. Hong SM, Vincent A, Kanda M, Leclerc J, Omura N, Borges M, et al. Genome-wide somatic copy number alterations in low-grade panINs and IPMNs from individuals with a family history of pancreatic cancer. Clin Cancer Res (2012) 18(16):4303–12. doi: 10.1158/1078-0432.CCR-12-1075
27. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6(269):pl1. doi: 10.1126/scisignal.2004088
28. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov (2012) 2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095
29. Heestand GM, Kurzrock R. Molecular landscape of pancreatic cancer: implications for current clinical trials. Oncotarget (2015) 6(7):4553–61. doi: 10.18632/oncotarget.2972
30. Hong DS, Kuo J, Sacher AG, Barlesi F, Besse B, Kuboki Y, et al. CodeBreak 100: Phase I study of AMG 510, a novel KRASG12C inhibitor, in patients (pts) with advanced solid tumors other than non-small cell lung cancer (NSCLC) and colorectal cancer (CRC). JCO (2020) 38(15_suppl):3511–1. doi: 10.1200/JCO.2020.38.15_suppl.3511
31. Waters AM, Der CJ. KRAS: the critical driver and therapeutic target for pancreatic cancer. Cold Spring Harbor Perspect Med (2018) 8(9):a031435. doi: 10.1101/cshperspect.a031435
32. Mullard A. Cracking KRAS. Nat Rev Drug Discov (2019) 18(12):887–91. doi: 10.1038/d41573-019-00195-5
33. Lowery MA, Kelsen DP, Capanu M, Smith SC, Lee JW, Stadler ZK, et al. Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur J Cancer (2018) 89:19–26. doi: 10.1016/j.ejca.2017.11.004
34. Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, et al. Rucaparib monotherapy in patients with pancreatic cancer and a known deleterious BRCA mutation. JCO Precis Oncol (2018) 2018:PO.17.00316. doi: 10.1200/PO.17.00316
35. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol (2015) 33(3):244–50. doi: 10.1200/JCO.2014.56.2728
36. Golan T, Oh DY, Reni M, Macarulla TM, Tortora G, Hall MJ, et al. POLO: A randomized phase III trial of olaparib maintenance monotherapy in patients (pts) with metastatic pancreatic cancer (mPC) who have a germline BRCA1/2 mutation (g BRCA m). J Clin Oncol (2016) 34(15_suppl):TPS4152–TPS4152. doi: 10.1200/JCO.2016.34.15_suppl.TPS4152
37. Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA -mutated metastatic pancreatic cancer. N Engl J Med (2019) 381(4):317–27. doi: 10.1056/NEJMoa1903387
38. Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Overall survival from the phase 3 POLO trial: Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. JCO (2021) 39(3_suppl):378–8. doi: 10.1200/JCO.2021.39.3_suppl.378
39. Jones MR, Williamson LM, Topham JT, Lee MKC, Goytain A, Ho J, et al. NRG1 gene fusions are recurrent, clinically actionable gene rearrangements in KRAS wild-type pancreatic ductal adenocarcinoma. Clin Cancer Res (2019) 25(15):4674–81. doi: 10.1158/1078-0432.CCR-19-0191
40. Aguirre AJ. Oncogenic NRG1 fusions: A new hope for targeted therapy in pancreatic cancer. Clin Cancer Res (2019) 25(15):4589–91. doi: 10.1158/1078-0432.CCR-19-1280
41. Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C, Mandelker D, et al. Microsatellite instability is associated with the presence of lynch syndrome pan-cancer. J Clin Oncol (2019) 37(4):286–95. doi: 10.1200/JCO.18.00283
42. Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, et al. Comprehensive analysis of hypermutation in human cancer. Cell (2017) 171(5):1042–1056.e10. doi: 10.1016/j.cell.2017.09.048
43. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi: 10.1056/NEJMoa1500596
44. Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med (2018) 7(3):746–56. doi: 10.1002/cam4.1372
45. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
46. Marabelle A, Le DT, Ascierto PA, Giacomo AMD, Jesus-Acosta AD, Delord JP, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair–deficient cancer: results from the phase II KEYNOTE-158 study. Clin Oncol (2019) 38(1):1–10. doi: 10.1200/JCO.19.02105
47. Salama AKS, Li S, Macrae ER, Park JI, Mitchell EP, Zwiebel JA, et al. Dabrafenib and trametinib in patients with tumors with BRAFV600E mutations: results of the NCI-MATCH trial subprotocol H. J Clin Oncol (2020) 38(33):3895–904. doi: 10.1200/JCO.20.00762
48. Mangat PK, Halabi S, Bruinooge SS, Garrett-Mayer E, Alva A, Janeway KA, et al. Rationale and design of the targeted agent and profiling utilization registry (TAPUR) study. JCO Precis Oncol (2018) 2018:10.1200/PO.18.00122. doi: 10.1200/PO.18.00122
49. American Society of Clinical Oncology. TAPUR study analysis plan and current status (2023). Available at: https://old-prod.asco.org/research-data/tapur-study/study-results (Accessed January 8, 2023).
50. Shastry M, Gupta A, Chandarlapaty S, Young M, Powles T, Hamilton E. Rise of antibody-drug conjugates: the present and future. Am Soc Clin Oncol Educ Book (2023) 43):e390094. doi: 10.1200/EDBK_390094
51. Meric-Bernstam F, Makker V, Oaknin A, Oh DY, Banerjee S, González-Martín A, et al. Efficacy and safety of trastuzumab deruxtecan in patients with HER2-Expressing solid tumors: primary results from the DESTINY-PanTumor02 phase II trial. JCO (2023) 42(1):47–58(2024). doi: 10.1200/JCO.23.02005
52. De Vries EGE, Rüschoff J, Lolkema M, Tabernero J, Gianni L, Voest E, et al. Phase II study ( KAMELEON ) of single-agent T-DM1 in patients with HER2 -positive advanced urothelial bladder cancer or pancreatic cancer/cholangiocarcinoma. Cancer Med (2023) 12(11):12071–83. doi: 10.1002/cam4.5893
53. Siravegna G, Sartore-Bianchi A, Nagy RJ, Raghav K, Odegaard JI, Lanman RB, et al. Plasma HER2 (ERBB2) copy number predicts response to HER2-targeted therapy in metastatic colorectal cancer. Clin Cancer Res (2019) 25(10):3046–53. doi: 10.1158/1078-0432.CCR-18-3389
54. Niikura N, Tomotaki A, Miyata H, Iwamoto T, Kawai M, Anan K, et al. Changes in tumor expression of HER2 and hormone receptors status after neoadjuvant chemotherapy in 21 755 patients from the Japanese breast cancer registry. Ann Oncol (2016) 27(3):480–7. doi: 10.1093/annonc/mdv611
55. Ignatov T, Gorbunow F, Eggemann H, Ortmann O, Ignatov A. Loss of HER2 after HER2-targeted treatment. Breast Cancer Res Treat (2019) 175(2):401–8. doi: 10.1007/s10549-019-05173-4
56. Zhao D, Klempner SJ, Chao J. Progress and challenges in HER2-positive gastroesophageal adenocarcinoma. J Hematol Oncol (2019) 12(1):50. doi: 10.1186/s13045-019-0737-2
57. Solomon BJ, Tan L, Lin JJ, Wong SQ, Hollizeck S, Ebata K, et al. RET solvent front mutations mediate acquired resistance to selective RET inhibition in RET-driven Malignancies. J Thorac Oncol (2020) 15(4):541–9. doi: 10.1016/j.jtho.2020.01.006
58. Corcoran RB, Chabner BA. Application of cell-free DNA analysis to cancer treatment. N Engl J Med (2018) 379(18):1754–65. doi: 10.1056/NEJMra1706174
Keywords: pancreatic cancer, HER2 amplification, ERBB2, ctDNA, liquid biopsy
Citation: Barzi A, Weipert CM, Espenschied CR, Raymond VM, Wang-Gillam A, Nezami MA, Gordon EJ, Mahadevan D and Mody K (2024) ERBB2 (HER2) amplifications and co-occurring KRAS alterations in the circulating cell-free DNA of pancreatic ductal adenocarcinoma patients and response to HER2 inhibition. Front. Oncol. 14:1339302. doi: 10.3389/fonc.2024.1339302
Received: 15 November 2023; Accepted: 16 January 2024;
Published: 08 February 2024.
Edited by:
William Matsui, The University of Texas at Austin, United StatesReviewed by:
Ming Yi, Zhejiang University, ChinaLaiba Arshad, Forman Christian College, Pakistan
Hongyan Xie, Harvard Medical School, United States
Weize Yuan, Massachusetts Institute of Technology, United States
Copyright © 2024 Barzi, Weipert, Espenschied, Raymond, Wang-Gillam, Nezami, Gordon, Mahadevan and Mody. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Afsaneh Barzi, YWJhcnppQGNvaC5vcmc=