 Li Shun Mak1,2
Li Shun Mak1,2 Xiuling Li3†Wilson Y. K. Chan2†
Xiuling Li3†Wilson Y. K. Chan2† Alex W. K. Leung2Daniel K. L. Cheuk2Liz Y. P. Yuen3Jason C. C. So3Shau Yin Ha2†
Alex W. K. Leung2Daniel K. L. Cheuk2Liz Y. P. Yuen3Jason C. C. So3Shau Yin Ha2† Anthony P. Y. Liu2,4*
Anthony P. Y. Liu2,4*- 1Department of Paediatrics and Adolescent Medicine, Princess Margaret Hospital, Hong Kong, Hong Kong SAR, China
- 2Department of Pediatrics and Adolescent Medicine, Hong Kong Children’s Hospital, Hong Kong, Hong Kong SAR, China
- 3Department of Pathology, Hong Kong Children’s Hospital, Hong Kong, Hong Kong SAR, China
- 4Department of Paediatrics and Adolescent Medicine, School of Clinical Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, Hong Kong SAR, China
Introduction: Medulloblastoma is the most common malignant brain tumor in children, often requiring intensive multimodal therapy, including chemotherapy with alkylating agents. However, therapy-related complications, such as therapy-related myeloid neoplasms (t-MNs), can arise, particularly in patients with genetic predisposition syndromes. This case report presents three pediatric cases of medulloblastoma with subsequent development of t-MNs, highlighting the potential role of genetic predisposition and the importance of surveillance for hematological abnormalities in long-term survivors.
Case presentation: We describe three cases of pediatric medulloblastoma who developed t-MNs after receiving chemotherapy, including alkylating agents. Two of the patients had underlying genetic predisposition syndromes (TP53 pathologic variants). The latency period between initial diagnosis of medulloblastoma and the development of secondary cancer varied among the cases, ranging from 17 to 65 months. The three cases eventually succumbed from secondary malignancy, therapy-related complications and progression of primary disease, respectively.
Conclusions: This report highlights the potential association between genetic predisposition syndromes and the development of therapy-related myeloid neoplasms in pediatric medulloblastoma survivors. It underscores the importance of surveillance for hematological abnormalities among such patients.
1 Introduction
Medulloblastoma is a common brain tumor among children (1). The standard treatment includes surgical resection and chemoradiotherapy. However, chemotherapy drugs used in its treatment, particularly alkylating agents and topoisomerase II inhibitors, have been linked to the development of secondary hematological malignancies (2, 3). Additionally, certain genetic predisposition syndromes, such as Li Fraumeni Syndrome (LFS), may increase the risk of multiple primary tumors (4, 5), and therapy-related malignancies. We present (Table 1) three children who first suffered from medulloblastoma and developed myeloid neoplasms shortly after treatment completion of the brain tumor.
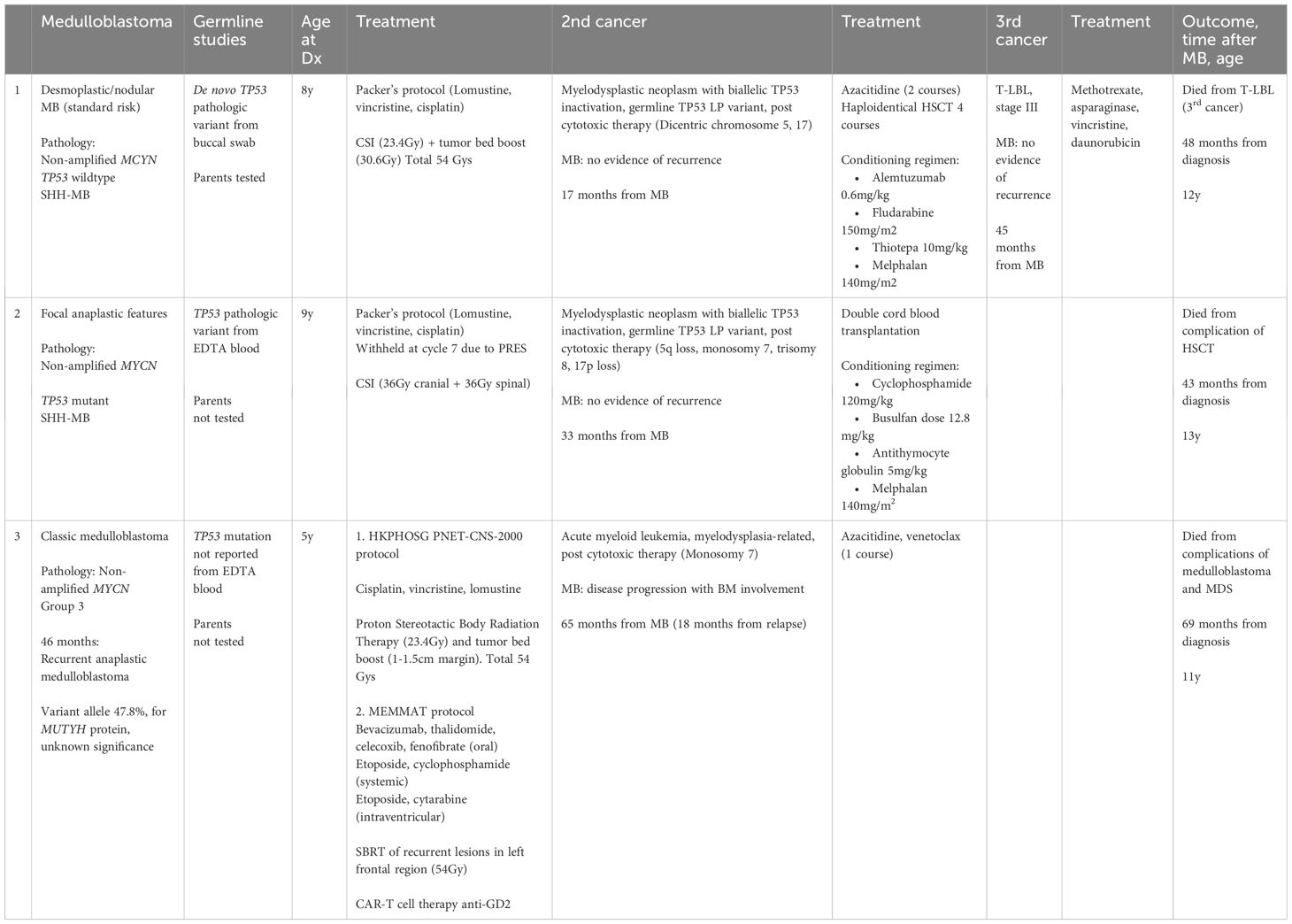
Table 1 Summary of clinical histories.
2 Case descriptions
2.1 Case 1
The first patient was an 8-year-old Chinese boy who underwent surgery for a localized tumor in the cerebellar vermis. Pathology confirmed desmoplastic/nodular medulloblastoma without MYCN amplification. The patient received craniospinal irradiation (CSI, 23.4Gy) with a tumor bed boost (30.6Gy) and concurrent vincristine (1.5mg/m2 8 doses), followed by chemotherapy according to Packer’s protocol, which comprised cisplatin (75mg/m2), lomustine (75mg/m2) and vincristine (1.5mg/m2 for 3 doses) for 8 cycles (6). Treatment was generally well-tolerated, with mild hearing loss and hypothyroidism as side effects.
At 13 months post-chemotherapy, the patient developed pancytopenia and circulating blasts at 11%. Marrow examination confirmed myelodysplastic neoplasm (with biallelic TP53 inactivation, germline TP53 LP variant, post cytotoxic therapy) (Figures 1A, B). Table 2 Cytogenetic analysis revealed a complex karyotype with dicentric chromosomes 5 and 17 (Figures 1C, D). Germline studies from the buccal swab confirmed a de novo heterozygous missense likely pathogenic variant in TP53 (NM_000546.5: c.326T>C, p.Phe109Ser). Genetic investigations for the parents were unremarkable confirming that the TP53 variant arose de novo in the patient. Methylation profiling of the medulloblastoma sample confirmed the Sonic-Hedgehog (SHH) group. The patient received 2 courses of azacytidine, lasting 7 days and 5 days respectively, followed by haploidentical hematopoietic stem cell transplantation with the father as the donor (Conditioning regimen outlined in Table 1). After the transplantation, there was no evidence of MDS on bone marrow examination.

Table 2 Pathological findings of myeloid neoplasms.
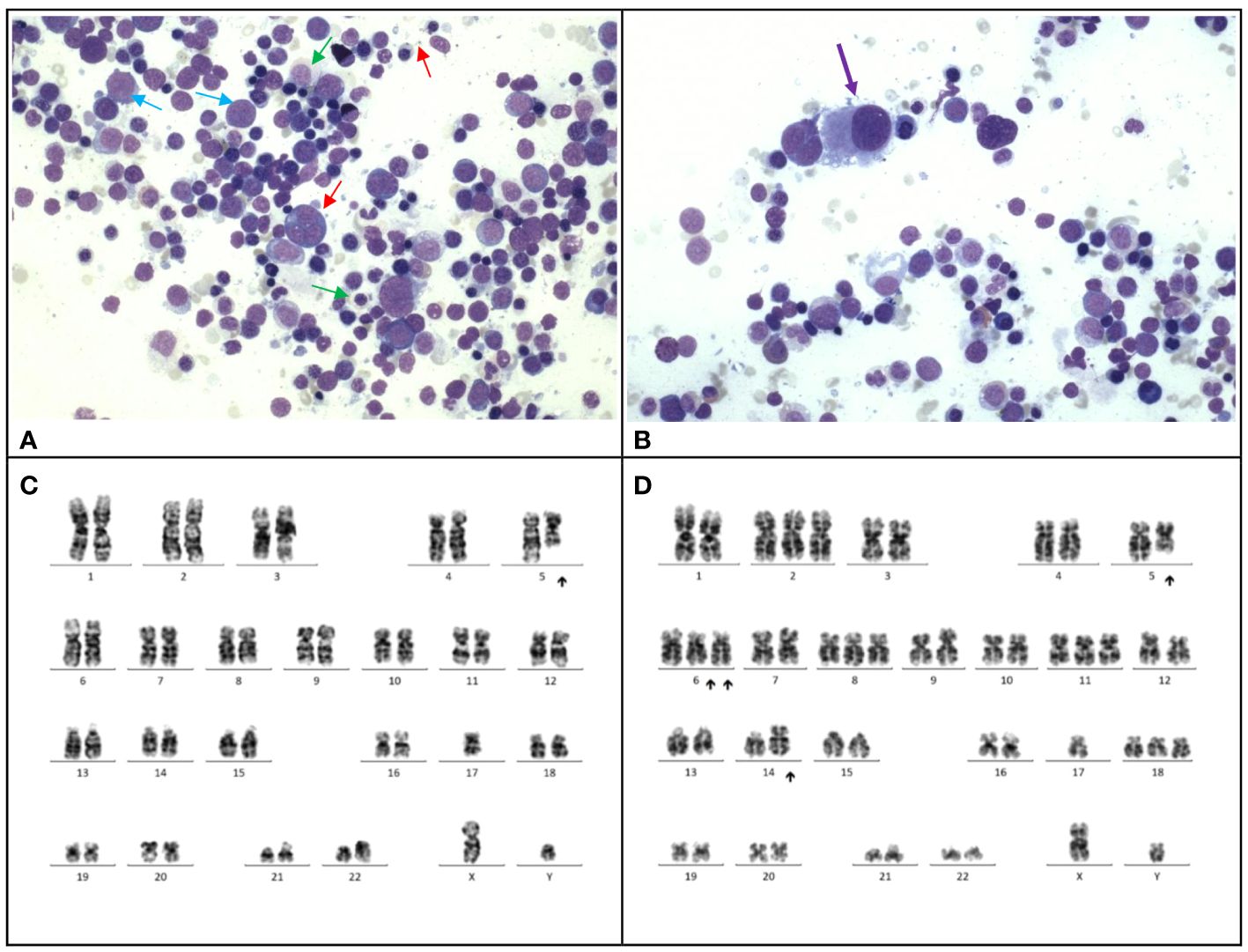
Figure 1 Case 1. (A), The bone marrow demonstrates dyserythropoiesis (red arrows), dysgranulopoiesis (green arrows) and increased blasts (blue arrow). Wright-Giemsa stain, 400 X magnification. (B), The bone marrow shows dysmegakaryopoiesis (purple arrow). Wright-Giemsa stain, 400 X magnification. (C, D), Karyotyping performed on the marrow reveals a complex karyotype with evidence of clonal evolution. (C), The stemline is 45,XY, dic(5;17)(q11.2;p11.2). (D), Clonal evolution to a complex karyotype 50,XY,+2,dic(5;17)(q11.2;p11.2);+6,add(6)(p21)x2,+8,+11,add(14)(p11.2),+18.
At 1 year 3 months post-hematopoietic stem cell transplantation (HSCT), the patient developed an anterior mediastinal mass, which was diagnosed as T-lymphoblastic lymphoma. The tumor sample showed a frameshift deletion in exon 34 encoding the PEST domain of the NOTCH gene (NM_017617.5:c.7386del p.(Ala2463Profs*14)), in addition to the known germline TP53 variant. He underwent chemotherapy with vincristine, daunorubicin, methotrexate, and asparaginase, which resulted in hepatorenal toxicities and acute pancreatitis, and only transient tumoral response. The patient passed away 2 months from the diagnosis of lymphoma and 4 years from the initial medulloblastoma diagnosis, due progression of mediastinal mass and subsequent respiratory failure with pleural effusion.
2.2 Case 2
The second patient was a 9-year-old Chinese girl diagnosed with a posterior fossa tumor in the superior vermis, extending into bilateral cerebellar hemispheres, with leptomeningeal enhancement and T4 drop metastasis. The patient underwent gross-total resection, and pathology confirmed medulloblastoma with focal anaplasia. TP53 immunohistochemistry indicated a mutant pattern, while MYCN amplification was not detected by fluorescence in situ hybridization (FISH). Methylation profiling of the medulloblastoma also revealed Sonic-Hedgehog (SHH) group. The patient received CSI (36Gy cranial + 36Gy spinal) along with concurrent vincristine, followed by chemotherapy according to the Packer’s protocol (6). However, she experienced posterior reversible encephalopathy syndrome after the seventh cycle of chemotherapy, leading to premature termination of treatment.
The patient experienced worsening thrombocytopenia and anemia 17 months after end of chemotherapy. The marrow examination revealed myelodysplastic neoplasm (with biallelic TP53 inactivation, germline TP53 LP variant, post cytotoxic therapy) (Figure 2A), (Table 2) which was evidenced by cytogenetic abnormalities including 5q loss, monosomy 7 and 17p loss (Figures 2B, C). Germline study obtained from the peripheral blood showed heterozygous missense likely pathogenic variant in TP53 (NM_000546.5: c.827C>A, p.Ala276Asp). The patient had a family history of leukemia in a maternal uncle, while parents opted not for germline testing. Methylation profiling confirmed the medulloblastoma molecular group as SHH activated. Double cord blood transplantation was performed (Conditioning regimen in Table 1), as near-full donor chimerism of 99-100% had been demonstrated in serial peripheral blood and marrow samples since post-transplant 1 month. The total nucleated cell (TNC) dose was 1.882 x 107/kg and stem cell dose 1.403 x 105 CD34+ cells/kg. Graft-versus-host disease (GVHD) prophylaxis was provided with cyclosporin and mycophenolate mofetil. However, the procedure was complicated by acute graft-versus-host disease (GVHD), adenovirus viremia, and thrombotic microangiopathy. The patient was treated with methylprednisolone, budesonide, infliximab, vedolizumab and extracorporeal photopheresis (ECP) for stage IV acute GVHD. The patient’s condition deteriorated and passed away 10 months after the MDS diagnosis due to fungal pneumonia, 3 years and 7 months since the initial diagnosis of medulloblastoma.
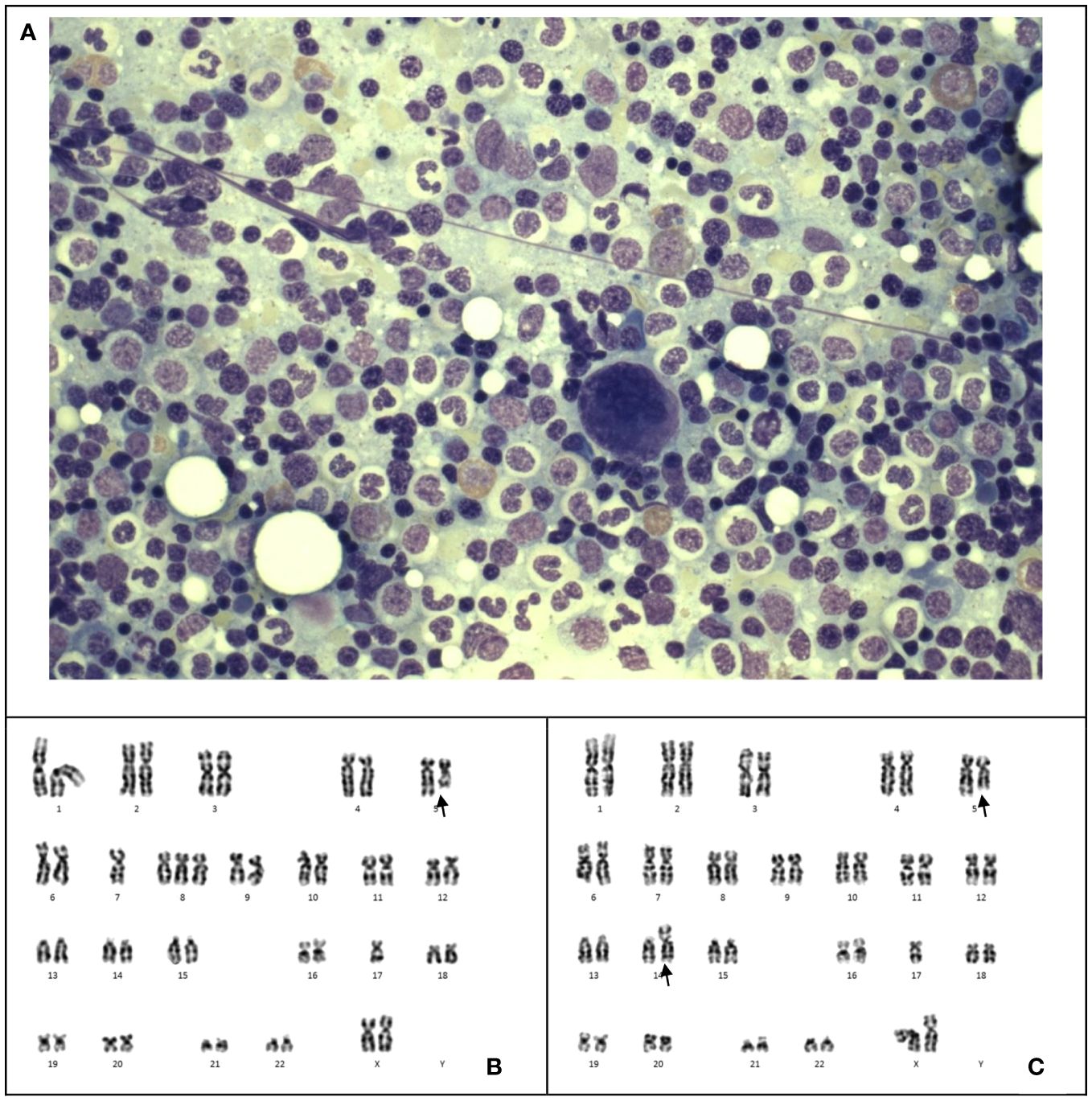
Figure 2 Case 2. (A) The bone marrow shows markedly hypercellular marrow with no dysplasia or increase in blasts. Wright-Giemsa stain, 1600 X magnification. (B, C), Karyotyping of the bone marrow reveals two unrelated clones 45,XX,del(5)(q31),der(14;17)(q10;q10)[3]/45,XX,der(5;17)(p10;q10),-7,+8[6]/46,XX[15]. The larger clone (B) shows monosomy 7, trisomy 8 and an unbalanced whole arm translocation of 5p and 17q. The latter results in loss of 5q and 17p. The smaller clone (C) shows deleted 5q and an unbalanced translocation of 14q and 17q with resultant loss of 17p.
2.3 Case 3
The third patient was a 5-year-old Chinese boy diagnosed with a localized cerebellar tumor. He underwent a gross total surgery resection and the pathology identified a group 3 medulloblastoma without MYC amplification. The patient underwent proton stereotactic body radiation therapy (23.4Gy) with tumor bed boost (Total 54Gy) and concurrent vincristine, followed by adjuvant cisplatin, lomustine and vincristine. Treatment lasted for 16 months and resulted in complications including hearing loss, growth hormone deficiency, and hypothyroidism.
46 months from initial diagnosis, metastatic relapse was detected with interval enlargement of a left caudate lesion, further confirmed on biopsy. Germline studies from the peripheral blood revealed a heterozygous variant in MUTYH (NM_001048171.1: c.892-2A>G). Genetic analysis of the relapsed tumor showed a variant allele frequency of 47.8% for the same mutation. DNA sequencing of the tumor sample showed no mutation in TP53. There was no significant family history of malignancies. The patient received chemotherapy according to the metronomic protocol MEMMAT (etoposide and cyclophosphamide containing) (7) and focal radiotherapy (54Gy).
However, 13 months after the relapse of medulloblastoma, chemotherapy was stopped due to pancytopenia. Marrow examination revealed acute myeloid leukemia (myelodysplasia-related, post cytotoxic therapy), with blasts constituting 23% of all cells through multiparametric flow cytometry, increased myeloblasts comprising >20% of nucleated cells in the trephine biopsy (Figures 3A, B), Table 2 and monosomy 7 by cytogenetic analysis (Figure 3C). A single course of azacitidine and venetoclax was administered, but chemotherapy was discontinued due to invasive fungal infection. A repeated marrow examination at 3 months from diagnosis of MDS showed marrow involvement by medulloblastoma (Figure 3D) positive for synaptophysin (Figure 3E) and chromogranin (Figure 3F), and negative for NeuN (Figure 3G). The patient was put on palliative care instead of curative approach. The patient’s condition deteriorated with progressive medulloblastoma, and passed away 4 months after the MDS diagnosis, marking 6 years and 9 months since the initial diagnosis of medulloblastoma.
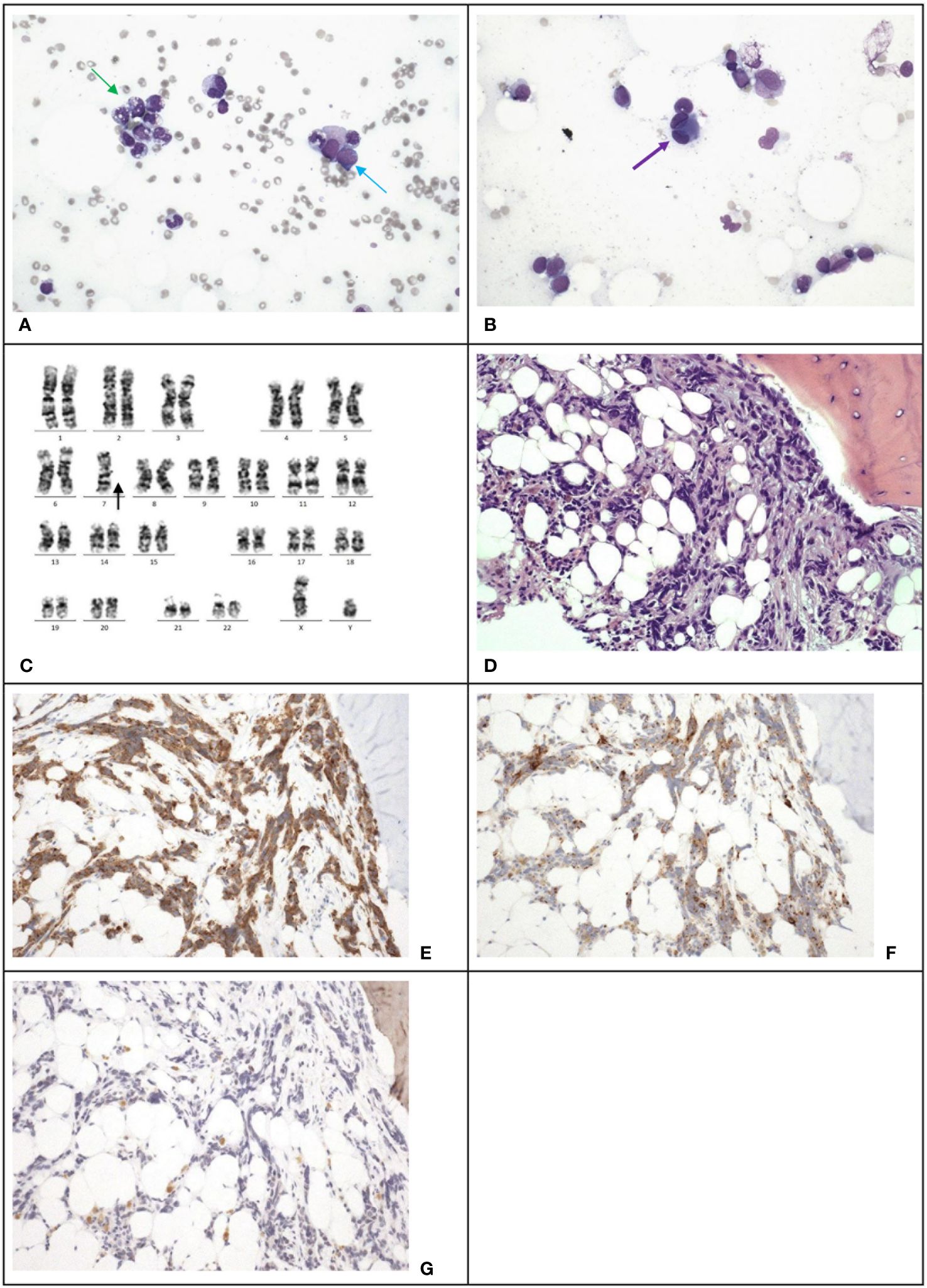
Figure 3 Case 3. (A), The bone marrow demonstrates dysgranulopoiesis (green arrow) and increase in blasts (blue arrow). Wright-Giemsa stain, 400 X magnification. (B), The bone marrow also demonstrates dysmegakaryopoiesis (purple arrow). Wright-Giemsa stain, 400 X magnification. (C) Karyotyping reveals a clone with monosomy 7. (D), The trephine biopsy shows non-hemic cellular infiltration. Hematoxylin & Eosin stain, 200 X magnification. Immunohistochemical staining shows the abnormal infiltration is positive for synaptophysin (E) and chromogranin (F), and negative for NeuN (G), which is compatible with marrow involvement by medulloblastoma. 200 X magnification.
3 Discussion
Treatment outcomes for medulloblastoma have improved with the systematic use of chemoradiation, with a 70-80% survival rate for localized disease (7). However, therapy-related myeloid neoplasms (t-MNs) can be a potential complication of treatment for medulloblastoma (8). MDS is a group of disorders characterized by abnormal blood cell production in the bone marrow, usually diagnosed in older adults. Therapy-related MDS (t-MDS) accounts for 10-15% of all MDS cases. The most common subtype of t-MDS is refractory anemia with excess blasts type (RAEB) (9), as was the case in our first patient.
Alkylating agents and topoisomerase II inhibitors are known to contribute to t-MNs. Previous studies have reported cases of t-MDS after treatment for primary brain tumors, and some cases progressed to secondary leukemia (2). In another long-term survival study of patients with recurrent medulloblastoma (10), with a MEMMAT-like approach, 5/29 patients developed secondary hematological malignancies. In general, the median time to development of t-MDS/AML is 3 to 5 years, with the risk decreasing after the first decade (11). Alkylating agents can affect marrow stem cells due to a relative deficiency in the DNA repair protein, O6-Alkylguanine-DNA alkyltransferase, resulting in chromosomal abnormalities with copy number changes. Our three cases received lomustine as standard upfront therapy and they all showed such karyotypic changes. Topoisomerase inhibitors, such as etoposide used in Case 3, generate DNA strand breaks and also chromosomal aberrations and increase the risk of secondary malignancies (12). Of note, topoisomerase inhibitors including etoposide are associated with increased risk of leukemia bearing balanced translocations and inversions which was not seen in case 3. Instead, monosomy 7 was detected in this case (Figure 3C) which is typically associated with alkylating agents used in the initial treatment. Radiotherapy may contribute to the development of t-MDS due to exposure of active bone marrow (13). The combination of chemotherapy and whole brain radiation was proposed to increase the risk of t-AML compared to chemotherapy alone (14). Few cases of t-MNs have been solely attributed to radiotherapy (15, 16).
Certain genetic predisposition syndromes also increase the risk of t-MNs (17). In the St. Jude series (18), germline abnormalities were found in 29% of t-MDS/t-AML patients, including LFS, mismatch repair deficiency, neurofibromatosis type 1, Fanconi anemia, and Down syndrome. In a case series, TP53 point mutations were present in 24% of t-MDS/t-AML cases (19). LFS is typically associated with hypodiploid ALL and AML, while it rarely involves t-MDS, with few reported cases (20). For the clinical characteristics of LFS associated t-MDS, the number of reported cases is small. Talwalkar et al. reported 3 cases of t-MDS with LFS, with latency period from primary cancer ranging from 1.5 years to 4 decades. In general, TP53 mutations in t-MDS are associated with complex cytogenetic findings, such as those seen in Case 2 (Figures 2B, C), and frequently involve chromosome 5 or 7, which was suggested to lead to a poorer prognosis (21). Moreover, TP53 mutations have been found to confer an inferior overall survival in patients with t-MDS. It was historically suggested that the cytotoxic effect of chemotherapy induced TP53 mutations, leading to t-MNs (22). However recent studies have proposed that pre-existing progenitors carrying prior TP53 mutation may be preferentially selected, leading to cytogenetic abnormalities and poor responses to chemotherapy, as seen in patients with t-MNs (23). The linkage between germline TP53 mutation and latency period to the development of t-MNs remains to be further elucidated. Nevertheless, it has been documented that TP53 mutations are enriched among medulloblastomas with SHH subgroup and are associated with poorer outcome (24), as shown in cases 1 and 2. Coupled with this, genetic predisposition syndromes such as TP53 carry a significant prognostic impact and account for treatment failure in these patients.
Case 3 showed a heterozygous germline MUTYH variant. The MUTYH gene, which encodes for the mutY DNA glycosylase protein, is involved in base excision repair in the DNA repair system. Biallelic inactivating mutations in MUTYH, either homozygous or compound heterozygous, are associated with gastrointestinal adenomas and carcinomas, known as the gastrointestinal polyposis syndrome (FAP2) (25). While brain tumors are rare in MUTYH mutations (26), Kline et al. (27) identified two pediatric patients with homozygous MUTYH mutations, one diagnosed with grade IV medulloblastoma, where loss of heterozygosity (LOH) as a result of chromosome 1p deletion was found. The MUTYH variant identified was NM 001048171.1 c.892-2 A>G, identical as that detected in our third patient. The oncogenicity this gene variant in the heterozygous setting warrants further evaluation (28).
In conclusion, we present three cases of early onset t-MN after treatment for medulloblastoma with confirmed or potential contribution by germline predisposition. This highlights the relevance of surveillance for hematological abnormalities in medulloblastoma survivors. While t-MNs are rare, it is important to recognize and monitor in particular for patients with underlying cancer genetic syndromes.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Hong Kong Children’s Hospital Research Ethics Committee (HKCH-REC-2020-068). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
LM: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis. VL: Writing – original draft, Writing – review & editing, Data curation, Formal analysis, Investigation, Visualization. WC: Writing – original draft, Writing – review & editing, Data curation. AWKL: Writing – original draft, Writing – review & editing, Data curation. DC: Writing – original draft, Writing – review & editing, Data curation. LY: Writing – original draft, Writing – review & editing, Data curation, Formal analysis, Investigation. JS: Writing – original draft, Writing – review & editing, Data curation, Formal analysis, Investigation. SH: Writing – original draft, Writing – review & editing, Conceptualization. APYL: Funding acquisition, Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Investigation, Supervision.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work is supported by the Health and Medical Research Fund (Commissioned Paediatric Research at Hong Kong Children’s Hospital, PR-HKU-6), Food and Health Bureau, Hong Kong SAR Government.
Acknowledgments
We would like to acknowledge Dr. Wong Wai Shan from the Department of Pathology, Queen Elizabeth Hospital, for providing the karyotyping figure for the second case.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fang FY, Rosenblum JS, Ho WS, Heiss JD. New developments in the pathogenesis, therapeutic targeting, and treatment of pediatric medulloblastoma. Cancers. (2022) 14:2285. doi: 10.3390/cancers14092285
2. Baehring JM, Marks PW. Treatment-related myelodysplasia in patients with primary brain tumors. Neuro-Oncology. (2012) 14:529–40. doi: 10.1093/neuonc/nos068
3. Kantarjian HM, Keating MJ, Walters RS, Smith TL, Cork A, McCredie KB, et al. Therapy-related leukemia and myelodysplastic syndrome: clinical, cytogenetic, and prognostic features. J Clin Oncol. (1986) 4:1748–57. doi: 10.1200/JCO.1986.4.12.1748
4. Kennedy AL, Shimamura A. Genetic predisposition to MDS: clinical features and clonal evolution. Blood. (2019) 133:1071–85. doi: 10.1182/blood-2018-10-844662
5. Quinn E, Nichols KE. Cancer predisposition syndromes associated with myeloid Malignancy. Semin Hematology. (2017) 54:115–22. doi: 10.1053/j.seminhematol.2017.04.003
6. Packer RJ, Sutton LN, Elterman R, Lange B, Goldwein J, Nicholson HS, et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurgery. (1994) 81:690–8. doi: 10.3171/jns.1994.81.5.0690
7. Winnicki C, Leblond P, Bourdeaut F, Pagnier A, Paluenzela G, Chastagner P, et al. Retrospective national “Real life” Experience of the SFCE with the metronomic MEMMAT and MEMMAT-like protocol. J Clin Med. (2023) 12:1415. doi: 10.3390/jcm12041415
8. Kollmannsberger C, Hartmann JT, Kanz L, Bokemeyer C. Risk of secondary myeloid leukemia and myelodysplastic syndrome following standard-dose chemotherapy or high-dose chemotherapy with stem cell support in patients with potentially curable Malignancies. J Cancer Res Clin Oncol. (1998) 124:207–14. doi: 10.1007/s004320050156
9. Sill H, Olipitz W, Zebisch A, Schulz E, Wölfler A. Therapy-related myeloid neoplasms: pathobiology and clinical characteristics. Br J Pharmacol. (2011) 162:792–805. doi: 10.1111/j.1476-5381.2010.01100.x
10. Slavc I, Mayr L, Stepien N, Gojo J, Maria Aliotti L, Azizi AA, et al. Improved long-term survival of patients with recurrent medulloblastoma treated with a “MEMMAT-like” Metronomic antiangiogenic approach. Cancers. (2022) 14:5128–8. doi: 10.3390/cancers14205128
11. Bhatia S. Therapy-related myelodysplasia and acute myeloid leukemia. Semin Oncol. (2013) 40:666–75. doi: 10.1053/j.seminoncol.2013.09.013
12. Le Deley M, Leblanc T, Shamsaldin A, Raquin M-A, Lacour B, Sommelet D, et al. Risk of secondary leukemia after a solid tumor in childhood according to the dose of epipodophyllotoxins and anthracyclines: A case-control study by the société Française d’Oncologie pédiatrique. J Clin Oncol. (2003) 21:1074–81. doi: 10.1200/jco.2003.04.100
13. Dracham CB, Shankar A, Madan R. Radiation induced secondary Malignancies: a review article. Radiat Oncol J. (2018) 36(2):85–94. doi: 10.3857/roj.2018.00290
14. Loöning L, Zimmermann M, Reiter A, Kaatsch P, Henze G, Riehm H, et al. Secondary neoplasms subsequent to Berlin-Frankfurt-Mönster therapy of acute lymphoblastic leukemia in childhood: significantly lower risk without cranial radiotherapy. Blood. (2000) 95(9):2770–5. doi: 10.1182/blood.V95.9.2770.009k16_2770_2775
15. Garcia-Manero G, Kantarjian H, Kornblau S, Estey E. Therapy-related myelodysplastic syndrome or acute myelogenous leukemia in patients with acute promyelocytic leukemia (APL). Leukemia. (2002) 16:1888–8. doi: 10.1038/sj.leu.2402616
16. Paulino AC, Ludmir EB, Grosshans DR, Su J, McGovern SL, Okcu MF, et al. Overall survival and secondary Malignant neoplasms in children receiving passively scattered proton or photon craniospinal irradiation for medulloblastoma. Cancer. (2021) 127:3865–71. doi: 10.1002/cncr.33783
17. Orazi A, Cattoretti G, Heerema NA, Sozzi G, John K, Neiman R. Frequent p53 overexpression in therapy related myelodysplastic syndromes and acute myeloid leukemias: an immunohistochemical study of bone marrow biopsies. PubMed. (1993) 6(5):521–5.
18. Broniscer A, Ke W, Fuller C, Wu J, Gajjar A, Kun LE. Second neoplasms in pediatric patients with primary central nervous system tumors. Cancer. (2004) 100(10):2246–52. doi: 10.1002/cncr.20253
19. Pedersen-Bjergaard J. Insights into leukemogenesis from therapy-related leukemia. New Engl J Med. (2005) 352:1591–4. doi: 10.1056/nejme048336
20. Talwalkar SS C, Yin C, Naeem R, John Hicks M, LC S, Abruzzo LV. Myelodysplastic syndromes arising in patients with germline TP53 mutation and li-fraumeni syndrome. Arch Pathol Lab Med. (2010) 134:1010–5. doi: 10.5858/2009-0015-oa.1
21. Kern W, Haferlach T, Schnittger S, Hiddemann W, Schoch C. Prognosis in therapy-related acute myeloid leukemia and impact of karyotype. J Clin Oncol. (2004) 22:2510–1. doi: 10.1200/JCO.2004.99.301
22. Chung J, Sallman DA, Padron E. TP53 and therapy-related myeloid neoplasms. Best Pract Res Clin Haematology. (2019) 32:98–103. doi: 10.1016/j.beha.2019.02.009
23. Chi Young Ok, Patel KP, Garcia-Manero G, Routbort MJ, Peng J, Tang G, et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. (2015) 8(1):1–8. doi: 10.1186/s13045-015-0139-z
24. Ray S, Chaturvedi NK, Bhakat KK, Rizzino A, Mahapatra S. Subgroup-specific diagnostic, prognostic, and predictive markers influencing pediatric medulloblastoma treatment. Diagnostics (Basel Switzerland). (2021) 12(1):61. doi: 10.3390/diagnostics12010061
25. Win AK, Cleary SP, Dowty JG, Baron JA, Young JP, Buchanan DD, et al. Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int J Cancer. (2011) 129:2256–62. doi: 10.1002/ijc.25870
26. Villy M-C, Warcoin M, Filser M, Bruno B, Golmard L, Suybeng V, et al. First report of medulloblastoma in a patient with MUTYH-associated polyposis. Neuropathology Appl Neurobiol. (2023) 49:1–7. doi: 10.1111/nan.12929
27. Kline CN, Joseph NM, Grenert JP, van Ziffle J, Yeh I, Bastian BC, et al. Inactivating MUTYH germline mutations in pediatric patients with high-grade midline gliomas. Neuro-Oncology. (2016) 18:752–3. doi: 10.1093/neuonc/now013
Keywords: case report, medulloblastoma (MB), therapy-related myelodysplastic syndrome/acute myeloid leukemia, chemotherapy, alkylating agent, genetic predisposition, Li-Fraumeni syndrome
Citation: Mak LS, Li X, Chan WYK, Leung AWK, Cheuk DKL, Yuen LYP, So JCC, Ha SY and Liu APY (2024) Case report: Therapy-related myeloid neoplasms in three pediatric cases with medulloblastoma. Front. Oncol. 14:1364199. doi: 10.3389/fonc.2024.1364199
Received: 01 January 2024; Accepted: 13 March 2024;
Published: 26 March 2024.
Edited by:
Angela Mastronuzzi, Bambino Gesù Children’s Hospital (IRCCS), ItalyReviewed by:
Martin Mynarek, University Medical Center Hamburg-Eppendorf, GermanyLucia Quaglietta, AORN Santobono-Pausilipon, Italy
Copyright © 2024 Mak, Li, Chan, Leung, Cheuk, Yuen, So, Ha and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony P. Y. Liu, YXB5bGl1QGhrdS5oaw==
†ORCID: Xiuling Li, orcid.org/0009-0008-9659-606X
Wilson Y. K. Chan, orcid.org/0000-0003-4178-2777
Shau Yin Ha, orcid.org/0000-0002-9191-8743