 Shahryar Khoshtinat Nikkhoi
Shahryar Khoshtinat Nikkhoi Geng Li
Geng Li Arash Hatefi
Arash Hatefi- 1Department of Pharmaceutics, Rutgers University, Piscataway, NJ, United States
- 2Cancer Pharmacology Program, Cancer Institute of New Jersey, New Brunswick, NJ, United States
This review article explores the rapidly evolving field of bi-, tri-, and multi-specific NK cell engagers (NKCEs), highlighting their potential as a cutting-edge approach in cancer immunotherapy. NKCEs offer a significant advancement over conventional monoclonal antibodies (mAbs) by enhancing Antibody-Dependent Cellular Cytotoxicity (ADCC). They achieve this by stably and selectively binding to both NK cell activating receptors and tumor-associated antigens (TAAs). Unlike traditional mAbs, which depend on the relatively transient interaction between their Fc region and CD16a, NKCEs establish more robust connections with a range of activating receptors (e.g., CD16a, NKG2D, NKp30, NKp46, NKG2C) and inhibitory receptors (e.g., Siglec-7) on NK cells, thereby increasing cancer cell killing efficacy and specificity. This review article critically examines the strategies for engineering bi-, tri-, and multi-specific NKCEs for cancer immunotherapy, providing an in-depth analysis of the latest advancements in NKCE platform technologies currently under development by pharmaceutical and biotech companies and discussing the preclinical and clinical progress of these products. While NKCEs show great promise, the review underscores the need for continued research to optimize their therapeutic efficacy and to overcome obstacles related to NK cell functionality in cancer patients. Ultimately, this article presents an overview of the current landscape and future prospects of NKCE-based cancer immunotherapy, emphasizing its potential to revolutionize cancer treatment.
1 Introduction
Antibody-mediated cancer immunotherapy is a type of treatment approach that harnesses the power of antibodies to bolster the immune system’s ability to fight cancer. These therapeutic antibodies are engineered to recognize and bind to specific antigens present on the surface of cancer cells. Upon binding to these antigens, initiate a series of immune responses, which directs the immune system to target and eliminate the cancer cells (1, 2). Structurally, antibodies are composed of two distinct regions that are integral to their function: the Fab and Fc regions (3). The Fab (fragment antigen-binding) region represents the variable portion of the antibody, which is responsible for recognizing and binding to specific antigens. This region comprises the two arms of the Y-shaped antibody molecule and contains the antigen-binding sites. In contrast, the Fc (fragment crystallizable) region is the constant portion of the antibody that mediates interactions with various components of the immune system (4). The Fc region determines the isotype of the antibody and its functional properties, including its capacity to bind to Fc gamma receptors (FcγRs) found on immune cells like natural killer (NK) cells, T cells, macrophages, neutrophils, and B cells (5, 6). In humans, FcγRs include several activating receptors, such as FcγRI (CD64), FcγRIIa (CD32a), FcγRIIc (CD32c), FcγRIIIa (CD16a), and FcγRIIIb (CD16b), along with the inhibitory receptor FcγRIIb (CD32b), which contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) (7). The expression and activity of these receptors are tightly regulated by factors such as cytokines, cell type, and activation state. For example, FcγRI expression is upregulated in response to interferon-gamma (IFN-γ), while FcγRIIb expression is modulated by signaling through the B cell receptor and other co-stimulatory pathways (8, 9). In tumors, FcγR expression patterns are often altered; tumor-associated macrophages frequently exhibit increased FcγRI and decreased FcγRIIb expression, shifting the balance toward an activating phenotype (10). Activating FcγRs possess immunoreceptor tyrosine-based activation motifs (ITAMs) that initiate downstream signaling cascades, leading to effector functions such as antibody-dependent cellular cytotoxicity (ADCC), phagocytosis, and pro-inflammatory cytokine production. Different IgG subclasses bind to FcγRs with varying affinities; for instance, FcγRI has high affinity for monomeric IgG, whereas FcγRIII preferentially binds IgG1 and IgG3 in immune complexes (11, 12).
NK cells are a critical subset of lymphocytes that play an essential role in the immune system’s defense against tumors and viral infections (13). They express several activating receptors, including CD16a, NKG2D, NKG2C, NKp46, and NKp30, as well as the inhibitory receptor Siglec-7. The CD16a receptor (FcγRIIIa) on NK cells is central to the ADCC mechanism, as it binds to the Fc region of IgG antibodies attached to the target cells. The clinical importance of CD16a interaction with antibody Fc region is highlighted by the success of several FDA-approved cancer therapies, including rituximab (14), trastuzumab (15), and cetuximab (16), all of which rely on CD16a receptor activation to achieve their anti-tumor effects (6). However, upon activation, CD16a can be shed from the cell surface through proteolytic cleavage by ADAM17, providing a mechanism to modulate NK cell activity and prevent excessive immune responses (17).
NKG2D is another activating receptor on NK cells that binds MICA/B and ULBP1-6 ligands, activating NK cells through DAP10 adaptor proteins and promoting cell cytotoxicity. Similarly, the NKG2C receptor pairs with CD94 and signals via DAP12, especially in response to cytomegalovirus (CMV) infection, enhancing NK cell activity and counteracting inhibitory signals from NKG2A. NKp46 is another interesting activating receptor on NK Cells which associates with CD3ζ or FcRγ to trigger cytolytic functions, while NKp30 induces NK cell activation through its isoforms when interacting with ligands such as B7-H6. Upon forming an immunological synapse with target cells, NK cells degranulate, releasing cytotoxic molecules like perforin and granzyme B, which induce lysis in target cells (18). Perforin creates pores in the target cell membrane, facilitating the entry of granzymes and granulysin, which activate apoptotic pathways leading to the destruction of the target cell (19). In contrast to these activating receptors, Siglec-7 is an inhibitory receptor that modulates NK cell activity by binding ligands and signaling through an ITIM motif, thus balancing NK cell function.
Despite the significant promise of ADCC-mediated therapies, several challenges must be addressed to fully harness their therapeutic potential. The efficacy of ADCC can be influenced by multiple factors, including the density of target antigen expression on cancer cells, the affinity and specificity of the antibodies employed, and the presence of inhibitory receptors on immune cells that can dampen the cytotoxic response. Furthermore, the expression and functionality of Fc receptors, such as CD16a, can be modulated by various factors, including cytokines and genetic polymorphisms, which may affect the overall therapeutic outcome (20, 21). Recent advancements have spurred a growing interest in developing strategies to enhance ADCC activity for the treatment of cancer and other diseases (22). One approach involves engineering the Fc region of therapeutic antibodies to improve their binding affinity for CD16a, thereby augmenting ADCC activity (23, 24). For instance, margetuximab, an Fc-engineered version of trastuzumab, has been modified to enhance its interaction with CD16a, leading to improved ADCC efficacy as demonstrated by both preclinical and clinical studies (25). However, a notable challenge with this strategy is the potential for increased interaction with other Fc receptors, which may lead to unintended effects. For example, engineering of the Fc domain in trastuzumab to create margetuximab resulted in enhanced CD16a (F176) binding affinity (~99 nM, a ten-fold improvement over trastuzumab’s ~1066 nM affinity), although this modification also increased affinity for the inhibitory receptor CD32b on B cells from ~33 nm to ~17 nM, potentially dampening B cell responses (26, 27).
An alternative strategy to improve ADCC efficiency involves using NK cell engagers (NKCEs) (28). NKCE therapies leverage the activating receptors to selectively direct NK cells towards tumor cells, amplifying the immune response by directing the NK cells to tumor-associated antigens (TAAs). By engaging multiple receptors simultaneously, NKCEs boost NK cell activation, helping to overcome immune evasion within the tumor microenvironment and representing a promising approach for cancer immunotherapy. Unlike the transient and relatively low-affinity interaction between the Fc domain of monoclonal antibodies (mAbs) and CD16a, NKCEs could form a stable connection with any activating receptor on NK cells, significantly enhancing ADCC efficiency (24, 29). Moreover, NKCEs are typically engineered to be highly selective and specific to activating receptors on NK cells, minimizing unwanted interactions with other receptors (30). There is considerable ongoing research focused on the development of novel bi-, tri-, and multi-specific NKCEs for NK cell-based cancer immunotherapy. While CD16a has traditionally been the receptor of choice for activating NK cells in antibody therapies, emerging studies suggest that NKCEs targeting other activating receptors can also effectively elicit NK cell responses to kill target cells. In this review, we explore various strategies for developing bi-, tri-, and multi-specific antibodies that engage NK cell surface receptors in cancer immunotherapy. We focus on NKCE platform technologies currently under development by pharmaceutical and biotech companies, discussing the preclinical and clinical progress of these products. The coverage of the literature is not encyclopedic; rather, select examples have been chosen to highlight certain important points. Finally, we explore potential strategies to further enhance the efficacy of NK cell engagers, offering insights into future directions in this rapidly evolving field.
2 NK cell engagers
In both peripheral and cord blood, NK cells are historically classified into two main subsets based on the expression of CD56 and CD16a: CD56bright CD16a¯ and CD56dim CD16a+. The CD56dim CD16a+ NK cells, which constitute approximately 90% of the NK cell population in the blood, are primarily responsible for executing innate anti-cancer effector functions (31, 32). In contrast, the CD56bright CD16a¯ NK cells, making up the remaining 10% of NK cells, are more specialized in cytokine release and exerting regulatory functions (32). The principal mechanism by which NK cells eliminate target cells via NK cell engagers is ADCC, predominantly mediated by theCD56dim CD16a+ NK cells (33). Various biotechnology companies have developed NKCEs that target activating receptors on NK cells other than CD16a receptors (Figure 1). For example, NKCEs can engage natural cytotoxicity receptors, including NKp30 and NKp46, to trigger NK cell effector functions. The subsequent sections will provide a detailed discussion of these platform technologies and their implications for cancer therapy.
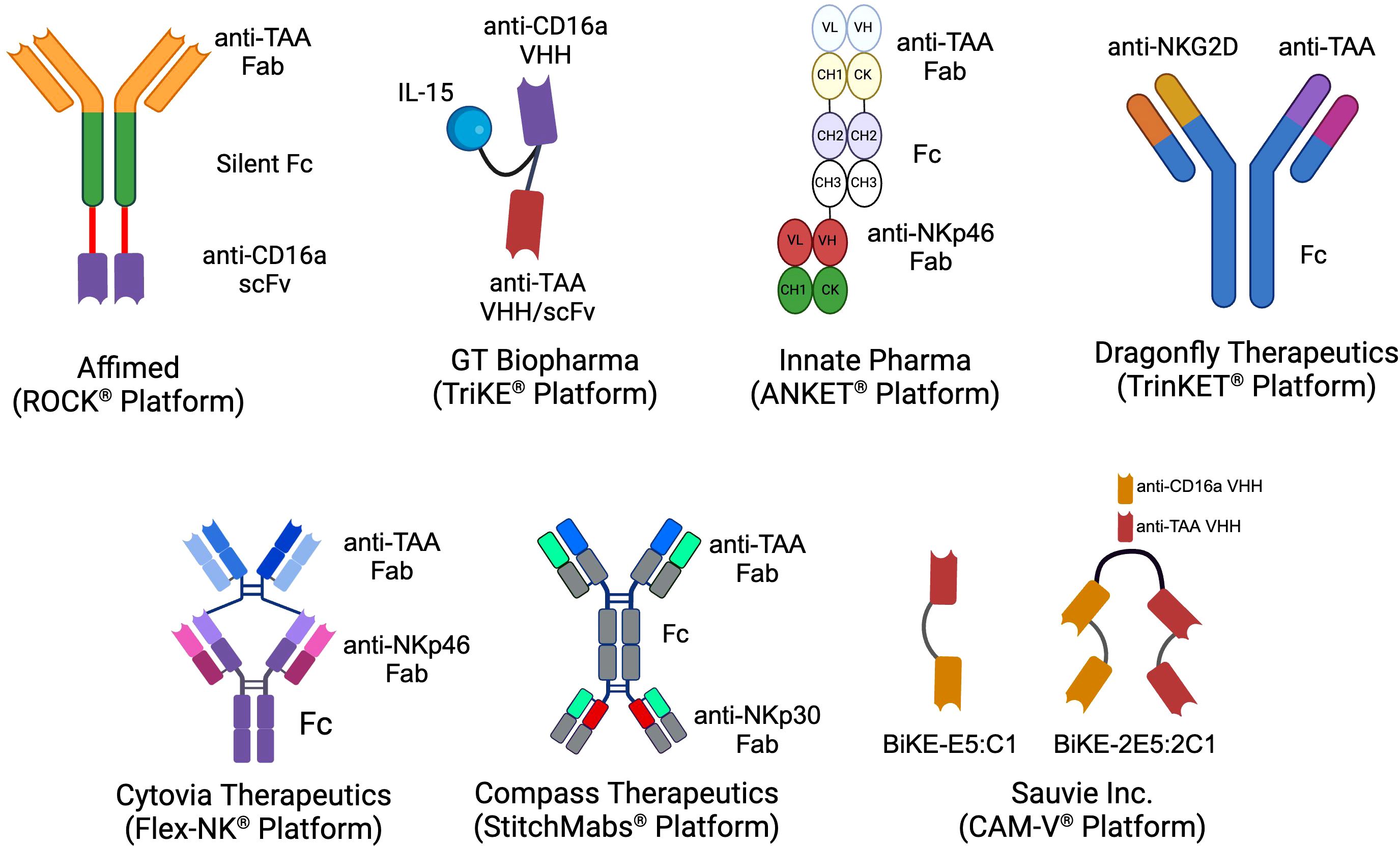
Figure 1. The schematics of the different NKCE platforms currently under development by biotech companies for cancer immunotherapy. (Created with BioRender.com).
2.1 scFv-based NKCEs targeting CD16a receptors
CD16a is an activating receptor comprising extracellular, transmembrane, and intracellular domains, each playing a critical role in its function. The extracellular portion contains two immunoglobulin-like domains, D1 and D2, which are responsible for binding to the Fc region of antibodies (34). The transmembrane domain facilitates connections with intracellular signaling molecules through polar and aromatic residues (35). The intracellular domain interacts with homodimers and heterodimers of CD3ζ (CD247) and/or FcϵR1γ, initiating and transducing activation signals within NK cells, ultimately leading to the full activation of NK cell effector functions (36). The high-affinity activation of CD16a receptors is crucial in cancer immunotherapy because it not only activates resting NK cells (37), but also helps them overcome inhibitory signals from HLA-E, which is often overexpressed on cancer cells (38). Consequently, many NKCEs under development are designed to target CD16a receptors.
For example, Uwe Reusch and colleagues (2014) successfully isolated a CD16a-specific single-chain variable fragment (scFv) through phage display and affinity maturation technologies. This engineered anti-CD16a scFv (code-named LSIV21) was later utilized by Affimed and incorporated into a tetravalent bispecific construct targeting CD30+ tumor cells. The resulting bispecific killer cell engager (BiKE), known as AFM13, demonstrated high affinity for CD16a without binding to CD16b-NA1 or CD16b-NA2 on neutrophils (39, 40). AFM13 exhibited superior potency in killing CD30+ cancer cells compared to both non-engineered and Fc-enhanced anti-CD30 mAbs (40). The anti-tumor efficacy of AFM13 was further enhanced when used in combination with peripheral or cord blood NK cells pre-conditioned with a cytokine cocktail of IL-12, IL-15, and IL-18 (41). Given its high efficacy in preclinical studies, AFM13 advanced to clinical trials. To date, seven clinical trials related to AFM13 (NCT03192202, NCT01221571, NCT02665650, NCT02321592, NCT04101331, NCT04074746, NCT05883449) have been registered, with three completed and results disclosed. AFM13 has been clinically evaluated as both a standalone treatment and in combination with other therapies in patients with CD30+ relapsed or refractory Hodgkin or non-Hodgkin lymphoma, including those who have previously undergone chemotherapy and/or autologous stem cell transplantation and have shown resistance to their most recent therapy (42–44). Although monotherapy with AFM13 yielded modest results (42, 43), its combination with the anti-PD-1 antibody pembrolizumab showed remarkable efficacy, with an overall response rate (ORR) of 83% at the highest evaluated dose (44). This was a significant improvement over monotherapy with pembrolizumab resulting in ORR of 69% as reported in Phase 2 clinical trial for relapsed or refractory Hodgkin lymphoma (45).
In another trial (NCT04074746), the effectiveness of AFM13 combined with cord blood NK cells was evaluated. In this study, AFM13 and cord blood NK cells were first mixed ex vivo and then freshly infused into patients (46). This approach resulted in an ORR of 92.8% and a complete response (CR) rate of 66.7% in patients resistant to CD30-directed brentuximab vedotin treatment (46). Later, Pharmacokinetic (PK) analysis revealed that AFM13 exhibited slightly greater than dose-proportional systemic exposure, with a half-life ranging from 9 to 19 hours (43). Anti-drug antibodies (ADA) and neutralizing antibodies (NAb) against AFM13 were detectable (43, 44), likely due to the murine-derived anti-CD30 sequence (scFv) within the AFM13 construct. Biomarker analysis of patients’ serum showed a reduction in soluble CD30 (sCD30) levels, indicative of AFM13-mediated CD30+ tumor cell lysis (43, 44). In addition to the changes in sCD30 serum levels, variations in the number and phenotype of NK cells in peripheral blood also served as pharmacodynamic biomarkers during therapy. Following AFM13 administration, a decrease in peripheral blood NK cell count was observed, likely due to AFM13-mediated NK cell redistribution (43). The authors describe the NK cell redistribution as the movement and redirection of NK cells from patient’s peripheral blood to the tumor sites following the administration of AFM13. Concurrently, an increase in the expression of CD69, a marker of NK cell activation, was observed on patients’ NK cells immediately after AFM13 infusion (43). In terms of safety, AFM13 was well tolerated, with the maximum tolerated dose not reached. Most AFM13-related adverse effects were mild and resolved with supportive treatment (43).
To date, Affimed has incorporated anti-CD16a scFv into various NK cell engager constructs (ROCK® platform) to create Immune Cell Engagers (ICE®) targeting tumor-associated antigens (TAAs) such as EGFR (47), BCMA (48, 49), and CD200 (48). The ROCK® platform has been shown to induce not only ADCC but also antibody-dependent cell-mediated phagocytosis (ADCP) through the activation of CD16a-expressing macrophages (Figure 2) (50).
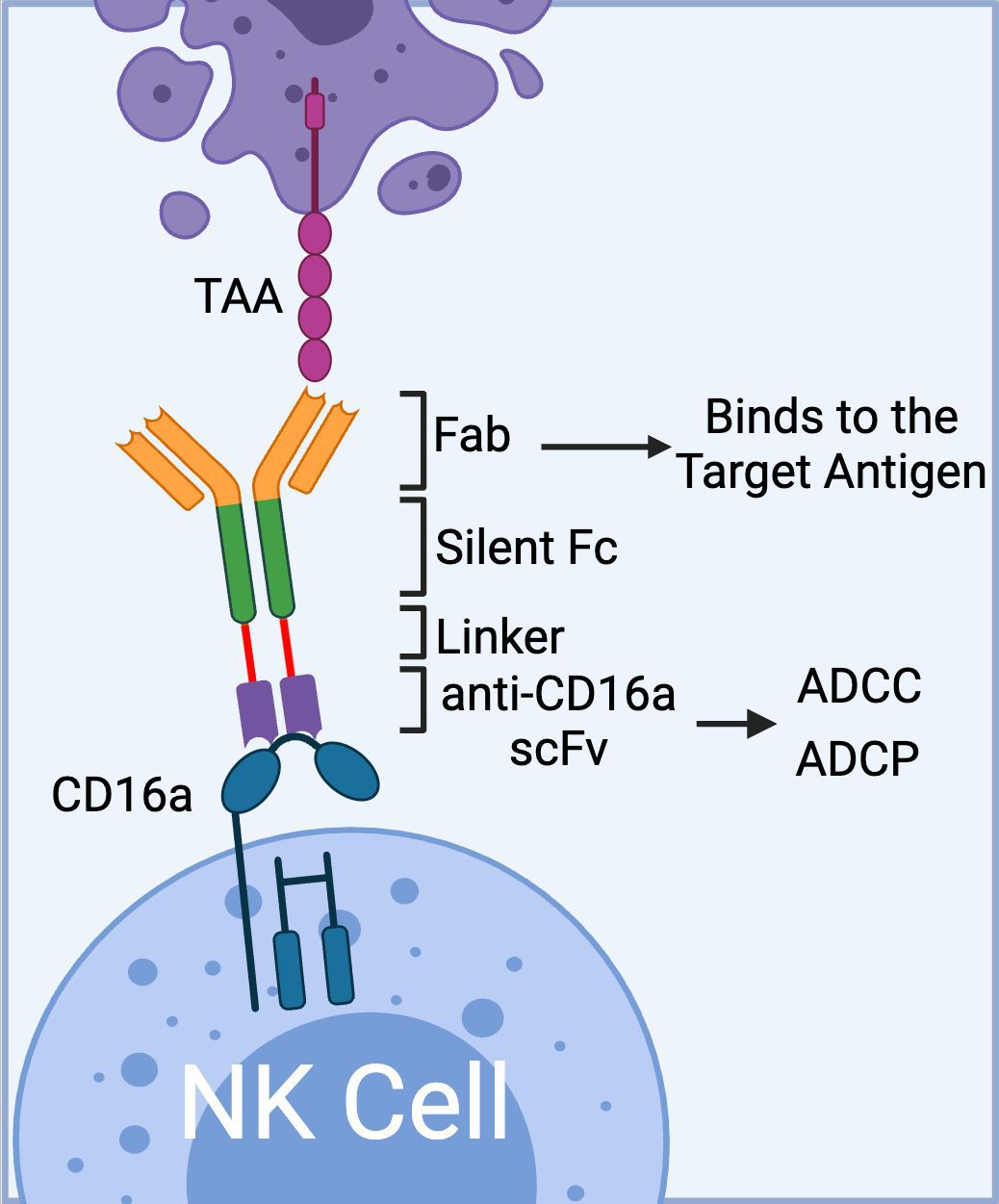
Figure 2. The schematics of Affimed’s ROCK® platform. The platform is composed of three domains: 1) Fab region that binds to tumor associated antigen, 2) Fc region (silent) that enhances blood circulation time; and 3) bivalent anti-CD16a scFv that binds to CD16a on NK cells with high affinity mediating ADCC. In addition to ADCC, the anti-CD16a scFv could mediate ADCP via binding to CD16a receptor on macrophages. (Created with BioRender.com).
In addition to AFM13, Affimed has successfully advanced another clinical trial with the AFM26 construct, targeting BCMA+ multiple myeloma cells. AFM26, which was recently licensed by Roche Inc. and rebranded as RO7297089, features an anti-CD16a scFv, P2C-47, engineered for enhanced binding affinity to both CD16a 158V and 158F allotypes (49, 51). CD16a receptor has three different allotypes based on the presence of phenylalanine (F) or valine (V) at amino acid residue 158. Each allotype (V/F, F/F, V/V, and V/F) allows the NK cells to interact with the Fc domain of mAbs with different affinities, which in turn can lead to a different clinical outcome. For more information on this subject, we would like to invite readers refer to the recent review articles by Barb et al. (2021), and Wemlinger et al. (2024) (5, 52). In vitro assays demonstrated that RO7297089 can effectively bind to NK cells even in the presence of high concentrations of human polyclonal IgG, mediating ADCC at a very low effector-to-target (E:T) ratio of 0.05. This led to the lysis of multiple myeloma cells with low BCMA expression, outperforming native and Fc-enhanced anti-BCMA IgGs (51).
RO7297089 has also undergone evaluation in a non-human primate (NHP) study to assess its PK profile and toxicity prior to its first-in-human clinical trial. Overall, RO7297089 exhibited a PK profile similar to that of human IgG1 antibodies (53). To correlate PK data with drug efficacy, the study quantified RO7297089’s binding to soluble BCMA or CD16a in serum and the formation of the BCMA-RO7297089-CD16a complex. These data, along with other parameters and considerations regarding interspecies differences between humans and monkeys, contributed to the initial establishment of the target-mediated drug disposition (TMDD) model for RO7297089. From this model, weekly administration of RO7297089 was recommended to ensure sustained BCMA engagement, a dosing interval that was ultimately applied in the phase 1 clinical study for multiple myeloma patients (54). In the phase 1 dose-escalation study, RO7297089 exhibited a favorable safety profile, with no maximum tolerated dose reached (54). Adverse effects during the clinical trial were manageable and did not lead to treatment discontinuation. Two cases of low-grade cytokine release syndrome induced by RO7297089 were observed, but both were resolved with supportive care. Notably, partial remission was achieved in patients receiving RO7297089 (54).
Other notable constructs from the ROCK® platform include AFM24, which targets EGFR, and AFM28, which targets CD123 (47, 55). The complete list of immune cell engagers currently under development by Affimed is presented in Table 1.
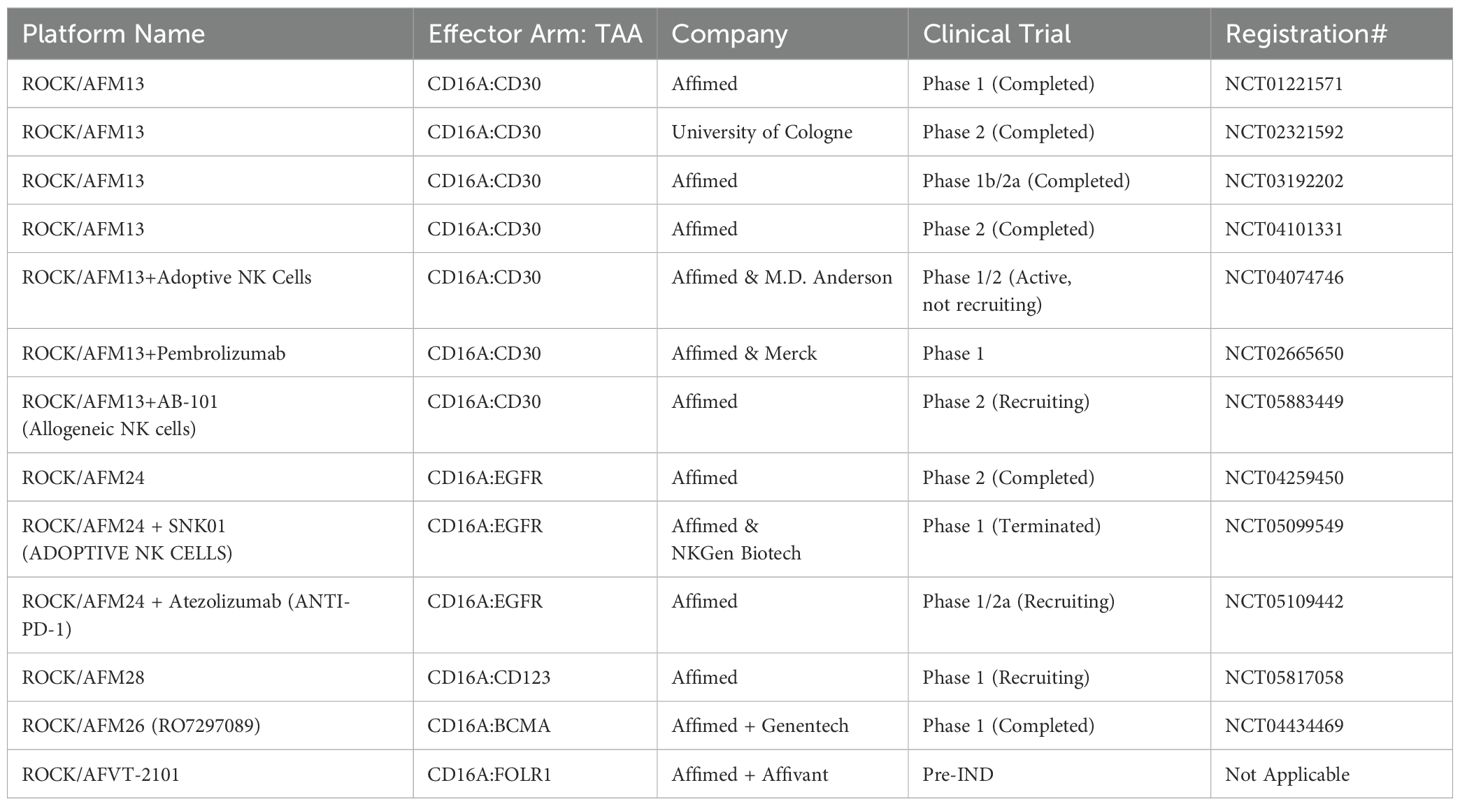
Table 1. List of Affimed’s immune cell engagers in preclinical/clinical trials.
2.2 VHH-based NKCEs targeting CD16a receptors
In addition to scFvs, single-domain antibodies derived from the variable domain of heavy chain-only antibodies in camelids, also known as VHHs or nanobodies (56), have been employed in the development of CD16a-targeting NK cell engagers. VHHs are the smallest intact antibody fragments, derived from camelid IgG2/IgG3 antibodies, and are notable for their lack of a light chain (57). For example, Nikkhoi et al. (2022) successfully isolated an anti-CD16a VHH (clone C1) that exhibited sub-nanomolar affinity for the CD16a receptor while avoiding interaction with the inhibitory CD32b receptor on B cells or CD16b-NA1 on neutrophils (30). Utilizing the C1 VHH, a bivalent bispecific killer cell engager (BiKE) was engineered by fusing C1 with a high-affinity anti-HER2 VHH (clone E5). In vitro ADCC assays revealed that the E5C1 BiKE could activate NK cells and was over 100-fold more potent than trastuzumab in killing HER2+ breast and ovarian cancer cells (30). Subsequent studies demonstrated that E5C1 BiKE could not only activate CD16-expressing NK cells and macrophages in vitro but also eradicate HER2+ metastatic ovarian tumors from the peritoneal cavity of NK-humanized hIL-15 and hIL-2 NOG mice (58). Additionally, the E5C1 BiKE was shown to activate CD16+ THP-1-derived M1 macrophages to eliminate HER2+ ovarian cancer cells through ADCP. Later, Yang et al. (2024), utilized the aforementioned bivalent BiKE as a template and engineered a tetravalent BiKE by recombinantly fusing two anti-CD16a and two anti-HER2 VHHs in tandem. It was demonstrated that the tetravalent BiKE achieved ultra-high affinity due to avidity and high anticancer activity without mediating NK cell fratricide. This NKCE construct developed by Nikkhoi et al. has been licensed to Sauvie Inc. (CAM-V® platform) for further development.
Similar to BiKEs, trispecific killer cell engagers (TriKEs) are being developed, capable of simultaneously targeting three antigens (28, 59). GT Biopharma currently has several candidates (TriKE® platform) in both clinical and preclinical pipelines. The first generation of TriKEs developed by GT Biopharma utilized anti-CD16a scFv, anti-tumor-associated antigen (TAA) scFv, and interleukin-15 (IL-15) (Figure 3).
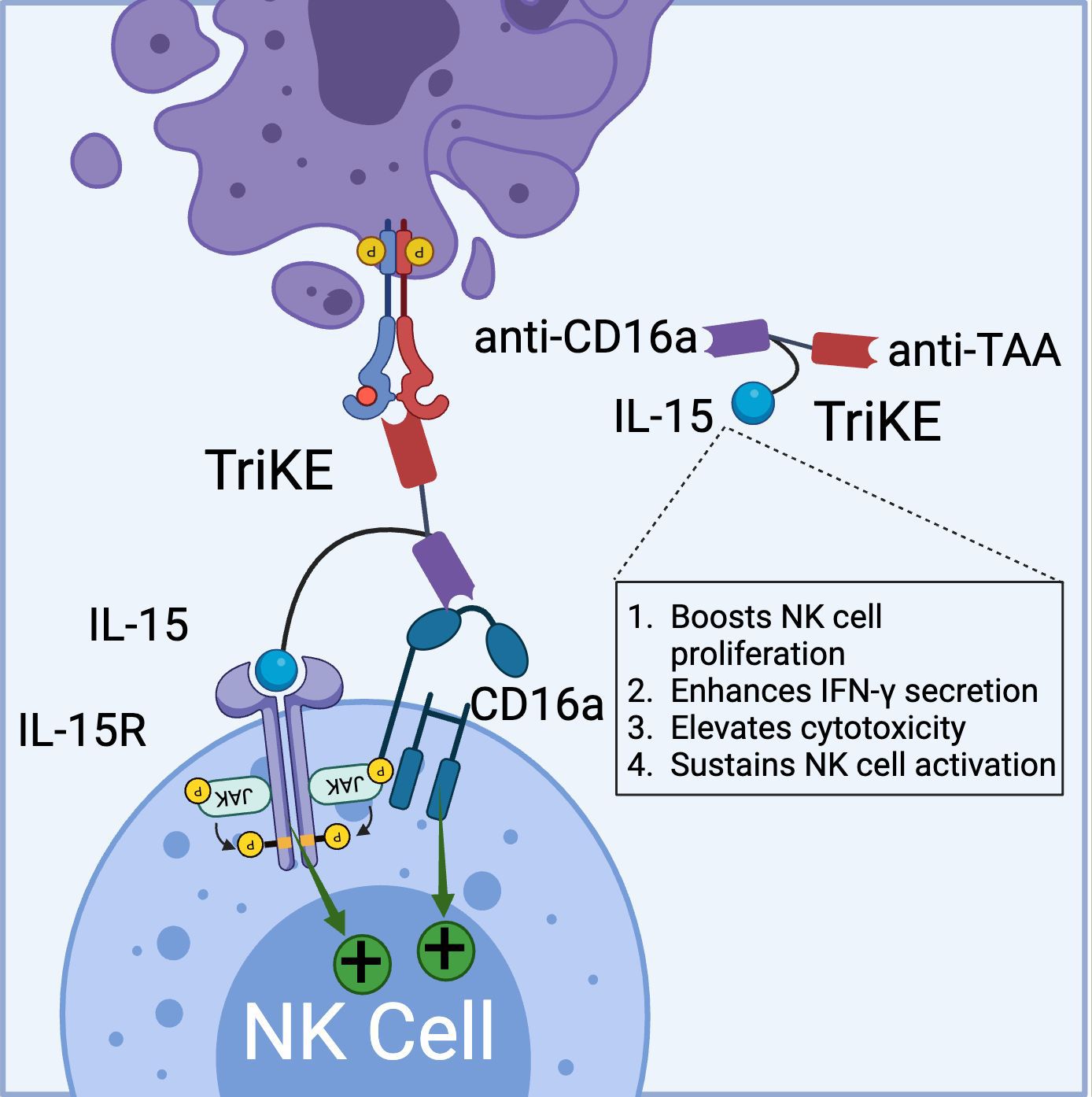
Figure 3. Schematic representation of TriKE composed of CD16a binding domain (VHH-based), tumor associated antigen binding domain (VHH-based), and IL-15 domain (binds to IL-15R). TriKE can bind and activate both CD16a and IL-15 signaling pathways to boost NK cell activation, proliferation and cytokine secretion. (Created with BioRender.com).
Targeting the IL-15 receptor is a strategic choice, as it can enhance NK cell proliferation, IFN-γ secretion, and cytotoxicity, positioning it as a promising candidate for NK-based cancer immunotherapy. Various studies have demonstrated the ability of these TriKEs to activate CD16a, induce NK cell proliferation, and increase the secretion of cytokines such as IFN-γ, GM-CSF, MIP1α, and TNF-α (60, 61). Building on this platform, GTB-3550, a first-generation TriKE candidate, entered a phase 2 clinical trial for the treatment of refractory/relapsed acute myeloid leukemia (AML) (62). Clinical trial data (NCT03214666) have shown minimal cytokine release syndrome in patients at doses ranging from 5-150 µg/kg/day. Due to its small molecular size (~60 kDa) and the absence of an Fc domain, GTB-3550 was administered daily, with a serum half-life calculated at 2.2-2.5 hours post-injection. Clinical benefits observed included a significant reduction of up to 63.7% in bone marrow plasmablast levels, proving effective in AML patients (62). Additionally, the treatment restored the patient’s endogenous NK cell function, proliferation, and immune surveillance. More recently, GT Biopharma has developed a second-generation TriKE by substituting the anti-CD16a scFv with an anti-CD16 VHH. This second-generation TriKE demonstrated superior functionality compared to the first generation, particularly in terms of binding and affinity, while maintaining a similar preclinical safety profile. Furthermore, NK cells activated by the second-generation TriKE expressed higher levels of CD107a, CD25, and CD69 compared to those activated by the first generation (63). Given the superior in vitro and in vivo results, GT Biopharma has terminated the GTB-3550 (first generation) clinical trial in favor of the GTB-3650 (second generation) trial. A comprehensive list of immune cell engagers currently under development by GT Biopharma is provided in Table 2.
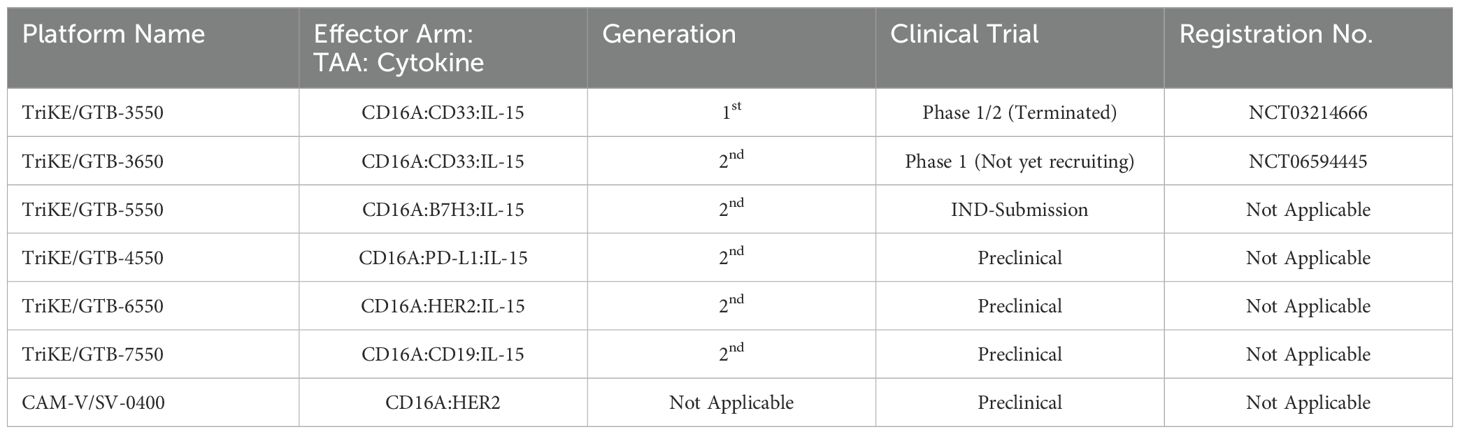
Table 2. List of GT Biopharma TriKE® and Sauvie’s CAM-V® platforms in preclinical/clinical trials.
2.3 NKCEs targeting NKG2D receptors
The natural killer group 2D (NKG2D) is an activating receptor that is abundantly present on all NK cells, NKT cells, and subsets of γδ T cells (64). For more detailed discussion on the role of NKG2D receptor as a regulator of cytotoxic immune cell responsiveness, we refer readers to an excellent review article by Wensveen et al. (2018) (65). To date, eight distinct ligands have been identified for NKG2D, which are classified into two groups: MHC class I-like proteins (MICA/B) and UL16 binding proteins (ULBP1-6) (66, 67). NKG2D is a type II transmembrane receptor characterized by a C-type lectin-like ectodomain that forms disulfide-bonded homodimers when binding to its ligands (66). In the cytoplasm, each NKG2D monomer associates with two DAP10 adaptor proteins, enabling the initiation of downstream signaling pathways (68). The activation of NK cells via the NKG2D receptor provides a compelling rationale for the development of NK cell engagers for cancer therapy.
For example, Chan et al. (2018) developed a BiKE that bridges CS1-expressing multiple myeloma cells and NKG2D receptors on NK cells. This BiKE successfully activated NK cells, leading to immune synapse formation, degranulation, and TNF-α secretion in IL-2-primed peripheral blood mononuclear cells (PBMCs) (69). Similarly, Raynaud et al. (2020) demonstrated that a VHH-based NKG2D-targeted BiKE could elicit effector functions in freshly isolated, resting NK cells by enhancing the tightness of the immune synapse. This increased tightness was attributed to the relatively small size of VHH-based immune cell engagers (70). Although the activation through NKG2D receptors is generally considered less robust than through CD16a receptors (37), NKG2D-targeted BiKEs offer promising alternative treatment options, particularly for patients with reduced CD16a expression (70).
Currently, Dragonfly Therapeutics is leveraging its TriNKETs® platform for cancer immunotherapy (Figure 4) (71–73). NKCEs generated from TriNKET® platform simultaneously engage two activating receptors, NKG2D and CD16a, on NK cells. This dual-engagement strategy enables NK cells to receive activation signals through distinct mediators. While CD16a transmits signals via CD3ζ and/or FcϵRIγ, NKG2D signals through DAP10. As a result, even if CD16 undergoes internalization or shedding, NK cells remain activated through the secondary receptor, NKG2D.
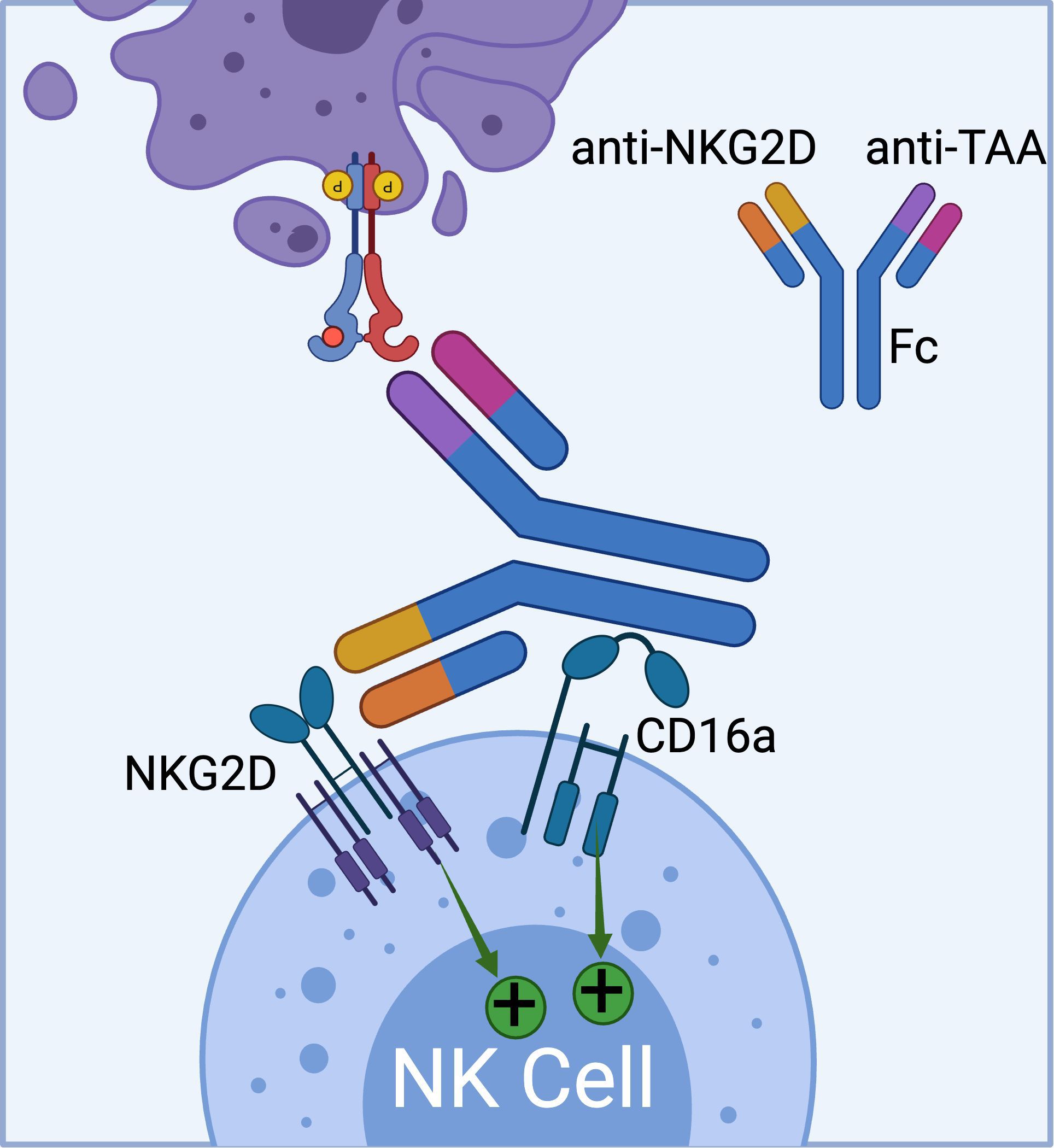
Figure 4. The schematics of TriNKET™ platform from Dragonfly (obtained from patent# AU2018220736A1), which can bind to both CD16a and NKG2D activating receptors on NK cells. (Created with BioRender.com).
TriNKET® NKCEs have been engineered to target various TAAs on cancer cells, including HER2, BCMA (72), and CD33 (73). CC-96191, also known as DF-2001, demonstrated maximum efficacy in eliminating CD33+ AML cells through the co-engagement of NKG2D and CD16a receptors. Notably, its maximal activity against AML cells was maintained even in the presence of high concentrations of the soluble NKG2D ligand, MICA (73). Dragonfly’s DF1001 candidate, which targets the HER2 receptor, has completed a Phase 1 clinical trial and reported positive outcomes in terms of safety and efficacy (NCT04143711). In terms of safety, no high-grade treatment-related adverse effects or dose-limiting toxicities were detected, and the maximum tolerated dose was not reached. A partial response was observed in 5 patients, with 22 patients achieving stable disease. In addition, the data demonstrated a reduction in tumor burden across various tumor types, particularly in heavily pre-treated patients with low HER2 expression. The overall clinical benefit rate was 39.7% (https://doi.org/10.1200/JCO.2023.41.16_suppl.2508). Dragonfly currently has several candidates in both clinical and preclinical pipelines, which are summarized in Table 3.
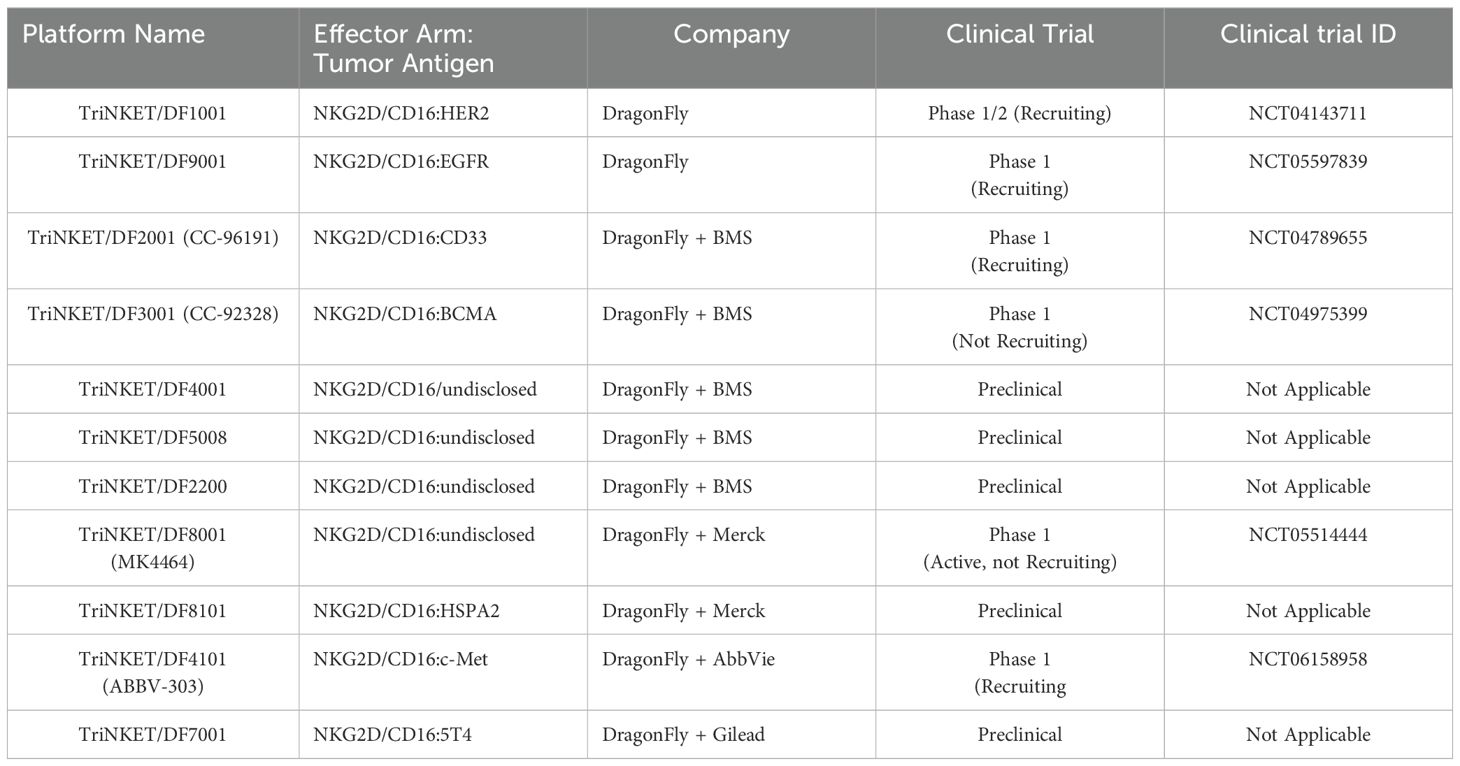
Table 3. List of Dragonfly’s TriNKET technology in preclinical/clinical trials.
Despite the promising efficacy demonstrated by NKG2D-targeted NKCEs, several challenges limit their effectiveness (74). One major obstacle is the ability of cancer cells to evade immune detection by shedding NKG2D ligands (e.g., MICA/B) through protease-mediated cleavage (75). The metalloproteinase ADAM10 plays a crucial role in this mechanism by cleaving these ligands under both normal and stress conditions in various cancer types (76). By shedding NKG2D ligands into the bloodstream, cancer cells prevent the immune cells from triggering an immune response, effectively bypassing NKG2D-mediated natural immune surveillance. It has been shown that soluble NKG2D ligands, primarily MICA, can bind to NKG2D receptors on NK cells resulting in their downregulation through endocytosis and degradation impairing their cytotoxic effects (74, 77, 78). For more information related to NKG2D ligand shedding and immune evasion of cancer cells, we refer the readers to the articles by Raffaghello et al. (2004) and Fantini et al. (2023) (74, 79). Additionally, tumor-derived cytokines, such as transforming growth factor beta (TGF-β), can downmodulate NKG2D expression on NK cells (80). Reduced NKG2D receptor expression has also been observed in tumor infiltrating NK cells isolated from renal cell carcinoma patients (81), which could further complicate the effectiveness of NKG2D-targeted NKCEs.
2.4 NKCEs targeting NKG2C receptors
NKG2C is another important activating receptor expressed on NK cells. Upon activation, the NKG2C receptor forms a heterodimer with CD94 and signals through the DAP12 adaptor protein (82). While NKG2C expression on NK cells is typically low, it can be significantly upregulated in response to cytomegalovirus (CMV) infection (83). The elevated surface density of NKG2C offers two key advantages: it enhances NK cell cytotoxicity and competes with the inhibitory receptor NKG2A for CD94 binding, thereby helping to sustain NK cell activation (84) (85). These findings have spurred efforts to develop NKCEs that trigger NK cell effector functions via NKG2C. For instance, Chiu et al. (2021) recently reported the development of an NKG2C-targeted NKCE that bridges NKG2C on NK cells with CD33 on target cells. This interaction successfully triggered NK cell degranulation, IFN-γ secretion, and lysis of the target cells (86). Remarkably, the engineered NKG2C-NKCE was as effective as a CD16-NKCE in controlling tumor formation in mice, even though NKG2C expression on NKG2C-positive adoptive NK cells was much lower than that of CD16 (86).
2.5 NKCEs targeting NKp46 receptors
NKp46 is a natural cytotoxicity receptor on NK cells, with its expression highly conserved across mammalian species, including humans, mice, and monkeys (87, 88). Like CD16a, NKp46 can associate with either CD3ζ or FcRγ to transduce activating signals via the ITAM motifs within NK cells (89). In humans, NKp46 is expressed on the family of innate immune cells and is considered a marker for mature NK cells (90). Activation through NKp46 can induce cytokine production and calcium ion influx, leading to NK cell degranulation and cytolytic activity (37, 90). Notably, unlike CD16a, NKp46 maintains sustained expression on NK cells that have infiltrated tumors, without significant downregulation (91). This unique characteristic has made NKp46 an attractive target for developing NKCEs. For example, Lipinski and colleagues isolated a variety of anti-NKp46 VHHs with differing affinities via yeast surface display (92). To develop a BiKE that targets both NKp46 and EGFR, the Fab region of cetuximab (an anti-EGFR monoclonal antibody) was fused to an anti-NKp46 VHH. This BiKE effectively mediated NK cell activation and later displayed enhanced EGFR-specific tumor cell lysis compared to FDA approved EGFR-targeted antibody cetuximab (92). Gauthier et al. (2019) also reported the structure of a NKCE targeting NKp46 (91). They also included an Fc domain in the NKCE structure to enhance the NK cell activation via the CD16a receptor. They observed a synergistic activity between CD16a and NKp46 receptor which was consistent with the reports from other groups (37). Based on this discovery, Innate Pharma developed a platform called Antibody-based NK cell Engager Therapeutics (ANKET™), while Cytovia Therapeutics developed FLEX-NK platform.
The ANKET™ platform is engineered through fusion of an anti-NKp46 Fab to an anti-TAA Fab through IgG Fc domain, which interacts with CD16a (Figure 5) (91). Preclinical data indicate that ANKET™ constructs can elicit robust NK cell effector functions, including specific target cell lysis and cytokine production. Furthermore, CD20-redirected NKCEs developed using this platform demonstrated superior efficacy compared to the FDA-approved anti-CD20 monoclonal antibody obinutuzumab, both in vitro and in vivo (93). Innate Pharma currently has several ANKET™-based candidates in its development pipeline. IPH6101/SAR’579 is currently undergoing a phase 1/2 clinical study in patients with relapsed or refractory AML, B-cell acute lymphoblastic leukemia (ALL), or high-risk myelodysplasia (HR-MDS). Results from the clinical study showed that IPH6101/SAR’579 has a favorable safety profile, with no dose-limiting toxicities that would necessitate treatment termination. Encouragingly, complete remission was observed in 5 out of 15 patients (https://doi.org/10.1182/blood-2023-173162). A summary of NKp46-targeted immune cell engagers can be found in Table 4.
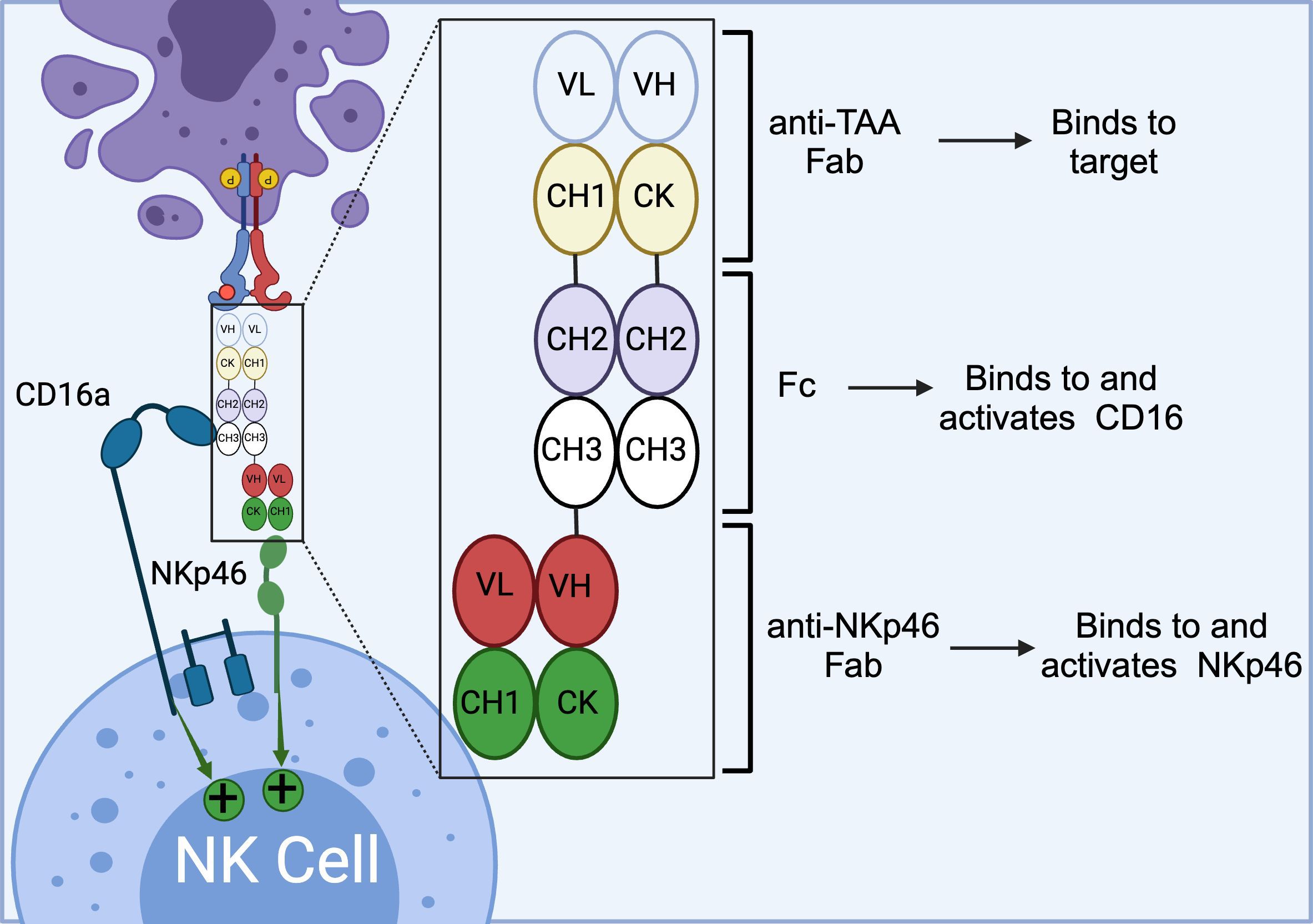
Figure 5. Schematics of ANKET™ technology. This NKCE, utilizes an anti-NKp46 Fab to activate NK cells via NKp46 receptor. It also boosts the ADCC via its Fc domain that engages CD16a receptor. (Created with BioRender.com).
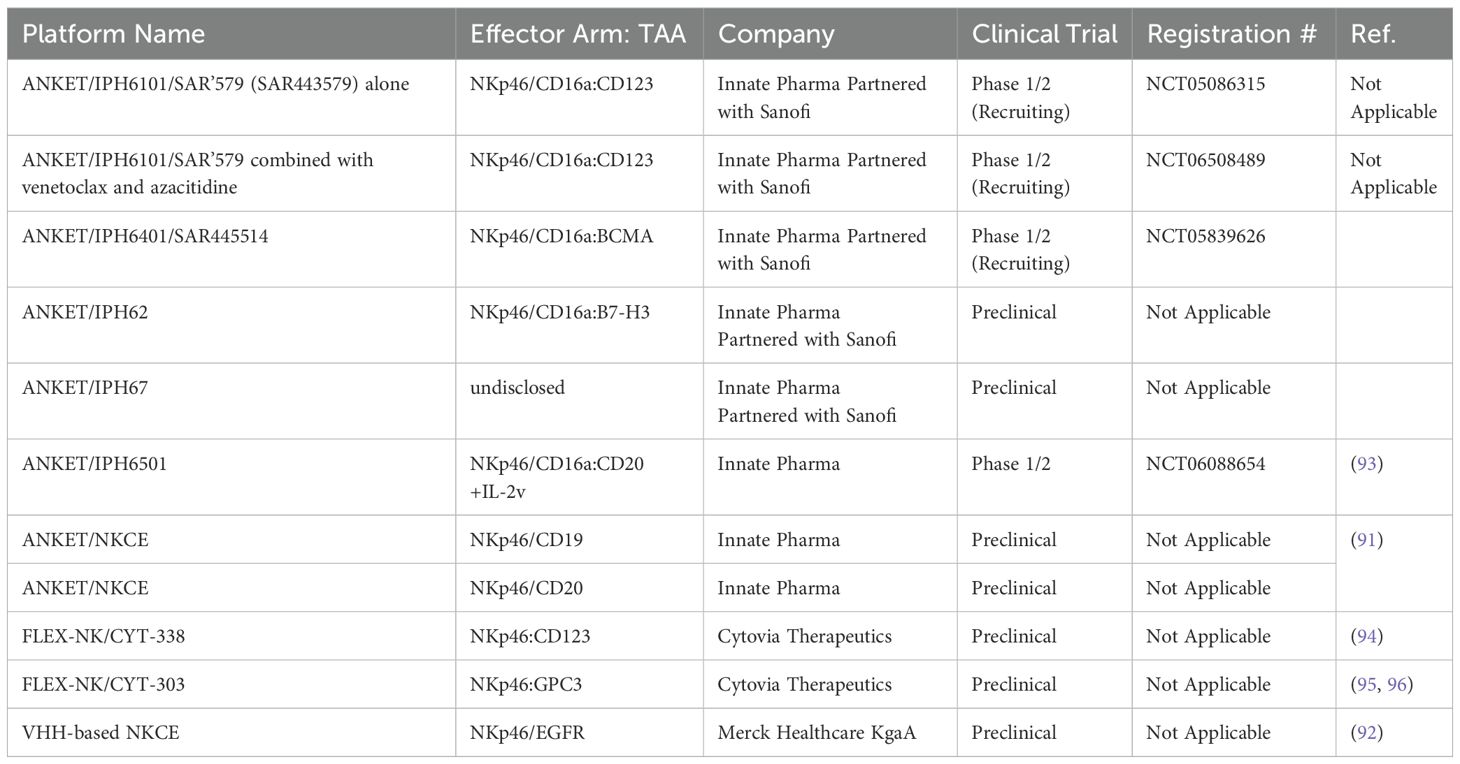
Table 4. The list of NKCEs that target NKp46 receptor on NK cells in preclinical/clinical studies.
Similar to Innate Pharma’s ANKET™ platform, Cytovia Therapeutics has leveraged its FLEX-NK platform to engineer CYT-303 and CYT-338 NKCEs, targeting the CD38 antigen for hematological malignancies and GPC-3 for hepatocellular carcinoma (95, 97). The CD38-targeted construct successfully restored the functionality of dysfunctional NK cells, resulting in the elimination of CD38+ multiple myeloma cells and significant tumor growth inhibition in mouse models (97). Additionally, this construct prevented NK cell fratricide, a common issue with anti-CD38 mAbs, such as daratumumab, by selectively activating NKp46 signaling (Table 4) (97).
2.6 NKCEs targeting NKp30 receptors
NKp30 is another pivotal natural cytotoxicity receptor that is constitutively expressed on NK cells. Upon binding to its ligands, B7-H6 or BAT3, NKp30 associates with the intracellular adaptor complex composed of CD3ζ and FcRγ, which transmits activating signals through ITAM domains (37). To date, six isoforms of the NKp30 receptor have been identified, with isoforms NKp30a and NKp30b being the most potent in inducing NK cell activation and cytolytic activities (89, 98). In patients with gastrointestinal stromal tumors, the expression levels of these NKp30 receptor isoforms on the NK cells have been shown to correlate with prognosis and survival outcomes (99, 100). In a study by Pekar et al. (2021), the potential of targeting the NKp30 receptor for the development of NK cell engagers (NKCEs) was demonstrated (101). As a first step, a panel of affinity-matured ΔB7-H6 ligands was isolated to efficiently target NKp30. These optimized ΔB7-H6 fragments exhibited up to a 45-fold higher binding affinity to NKp30 compared to the wild-type B7-H6. Subsequently, a bispecific antibody, termed an immunoligand, was engineered by fusing the affinity-matured ΔB7-H6 to the EGFR-binding Fab region derived from cetuximab. This engineered BiKE was shown to effectively activate NK cells and significantly enhance EGFR-specific tumor cell lysis compared to a BiKE constructed with wild-type B7-H6. The increased production of IFN-γ and TNF-α was also observed with the aforementioned immunoligands. In an interesting study by Klausz et al. (2022), a panel of anti-NKp30 VHHs with a wide range of binding affinities and epitope coverage for NKp30 receptors was isolated and used to create a series of EGFR-targeted NKCEs (102). All NKCEs generated in the study demonstrated the ability to eliminate EGFR-expressing cancer cells; however, the potency and efficacy of tumor cell killing varied significantly depending on the specific epitopes on NKp30 that each NKCE targeted. Importantly, the generated NKCEs were designed to bind to NKp30 receptor epitopes different from those recognized by natural ligands B7-H6. This approach allowed the NKCEs to circumvent the inhibitory effects of soluble B7-H6, which is commonly found in the serum of cancer patients (102). In a subsequent study by Boje et al. (2024), single-domain antibodies (sdAbs) were utilized to construct NKCEs targeting NKp30 to redirect NK cell cytotoxicity toward EGFR-expressing tumor cells (103). This study investigated the impact of several crucial parameters, including sdAb location, binding valencies, the targeted epitope on NKp30, and the overall antibody architecture, on the redirection capacity of NKCEs. Two NKp30-specific sdAbs were constructed: one targeting a similar epitope on NKp30 as its natural ligand B7-H6, and the other binding a non-competing epitope. For targeting EGFR-positive tumors, humanized antigen-binding domains of the therapeutic antibody cetuximab were employed. The results demonstrated that NKCEs that bivalently target both EGFR and NKp30 were superior to monovalent NKCEs in promoting NK cell-mediated tumor cell lysis. Importantly, the location and orientation of NKp30-targeting sdAbs within the NKCE sequence were found to be critical factors influencing the ultimate efficacy of NKCEs.
Given that both NKp46 and NKp30 serve as co-stimulatory activating receptors, a pertinent question arises: which receptor generates a more potent response when combined with an anti-CD16a engager? Colomar-Carando et al. conducted an intriguing study to address this question, comparing the efficacy of NKCEs that co-engage CD16a with either NKp30 or NKp46 in eliciting NK cell effector functions (104). The researchers generated TriKEs capable of targeting NK activating receptors CD16a-NKp30 or CD16a-NKp46, alongside CD19 or CD20 tumor-associated antigens (TAAs). These TriKEs were then evaluated for their ability to kill acute lymphoblastic leukemia cells expressing either CD19 or CD20. The study revealed that all NKCEs efficiently killed the NK cell-resistant MHH-CALL-4 cells, as evidenced by increased NK cell activity, including degranulation and IFN-γ production. Moreover, the CD19-targeting TriKEs were effective against primary BCP-ALL cells, even in patients who had undergone transplantation. Interestingly, the NKp30-CD16a and NKp46-CD16a NKCEs demonstrated equivalent capabilities in eliminating CD19 and CD20-expressing target cells, suggesting that NKp30 and NKp46 have comparable potency in activating NK cells (104). This observation could be attributed to the fact that the intracellular domains of both receptors are very similar, consisting of short intracellular fragments without intrinsic signaling domains, necessitating engagement with either CD3ζ or FcϵRIγ (89). A summary of immune cell engagers targeting NKp30 is provided in Table 5.
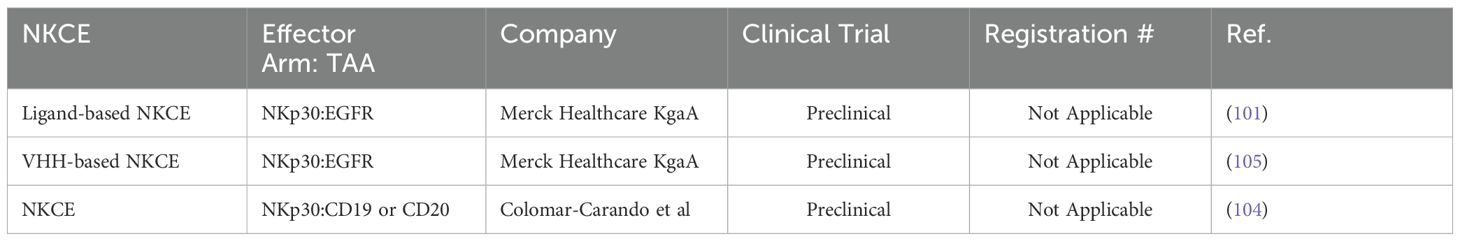
Table 5. List of NKCEs targeting NKp30 in preclinical studies.
3 Strategies to improve NKCE efficacy
NKCEs rely on the functionality of NK cells to achieve their intended therapeutic effects. However, it is common for NK cells in cancer patients to be dysfunctional or inept in recognizing tumor cells, presenting a significant challenge for effective treatment. To address this issue, various strategies have been employed to restore NK cell functionality and enhance the efficacy of NKCEs.
3.1 Targeting Siglec-7 (CD328) inhibitory receptors
Sialic acid-binding immunoglobulin-like lectin 7 (Siglec-7) has recently emerged as a promising target in cancer immunotherapy. Siglec-7 is primarily expressed on monocytes and NK cells, where it functions as an inhibitory receptor, playing a critical role in the regulation of NK cell activity (106, 107). Siglec-7 recognizes sialic acid residues present on sialylated glycoconjugates as its ligands and operates in a sialic acid-dependent manner (108). Upon binding to sialic acid, Siglec-7 transmits inhibitory signals through an immunoreceptor tyrosine-based inhibitory motif (ITIM) within NK cells (107).
Many tumors have been reported to exhibit significant levels of surface sialylation, which allows them to engage Siglec-7 and evade NK cell-mediated immunosurveillance (109, 110). Consequently, blocking the interaction between Siglec-7 and its ligands on cancer cells has been proposed as a viable strategy to enhance NK cell-mediated cytotoxicity (87). For instance, Bordoloi et al. (2023) explored the potential of this strategy by engineering a NKCE aimed at targeting ovarian cancer cells. The team constructed a BiKE that could simultaneously block Siglec-7 on NK cells and target the follicle-stimulating hormone receptor (FSHR) on ovarian cancer cells (111). This BiKE was demonstrated to effectively inhibit the negative signaling mediated by Siglec-7, thereby leading to NK cell activation and the subsequent elimination of ovarian cancer cells in both in vitro and in vivo models (112). These findings underscore the therapeutic potential of Siglec-7 blockade as a means to maintain NK cells in an activated state when they encounter target cells, ultimately enhancing their cytotoxic efficacy against cancer.
3.2 Inclusion of Fc domain in NCKE structure to exploit CDC and ADCP
An effective strategy to enhance the therapeutic activity of NK cell engagers (NKCEs) involves incorporating a functional Fc domain to leverage both complement-dependent cytotoxicity (CDC) and ADCP mechanisms. The CH2 domains within these functional Fc regions enable NKCEs to recruit complement proteins, initiating the classical CDC pathway. When an NKCE binds to a tumor cell, complement protein C1q first binds to the NKCE’s Fc domain, triggering a cascade that recruits additional complement proteins, including C4, C2, C3b, C5b, C6, C7, C8, and C9 (113). These complement complexes then assemble into membrane attack complexes, creating pores in the tumor cell membrane that ultimately lead to cell lysis (114). For instance, BL-01, a tetravalent Fc-bearing NKCE, has been demonstrated to efficiently induce CDC against CD20+ B-cell lymphoma cells (115). Similarly, cross-over dual-variable Ig-like proteins (CODV-Ig) with functional Fc domains have shown the ability to recruit complement proteins, effectively eliminating target cells via CDC mechanisms (116). NKCEs such as ISB1442 (117), CYT-338 (94), and CYT-303 (95) have also been reported to effectively induce CDC against CD38+ lymphoma cells and GPC-3+ hepatocellular carcinoma cells. Beyond the complement cascade, the CH2 domain within the Fc region can bind to Fcγ receptors on macrophages, stimulating them to engage in ADCP (118). In this process, activated macrophages engulf the opsonized cancer cells, degrading them within phagosomes through acidification (119). This ADCP-mediated cell elimination has been observed for CYT-338 and CYT-303 as previously discussed.
3.3 Inclusion of cytokines into NKCEs
In addition to complement recruitment, cytokines have been employed to further improve the efficacy of NKCEs. Beyond IL-15, which was previously discussed, other cytokines such as IL-2 have been explored for their potential to enhance NK cell activity. A variant of interleukin-2 (IL-2v) was incorporated into a tetrafunctional NKCE, known as IPH6501 (93). Notably, this IL-2 variant does not bind to the α-subunit of IL-2 receptor, thereby limiting the activation of regulatory T cells and reducing the risk of IL-2-mediated toxicity (93). Given the potent stimulatory effects of IL-2, IPH6501 has been shown to induce the proliferation and cytolytic activity of primary human NK cells, as well as the secretion of cytokines and chemokines in both in vitro and in vivo models of B cell malignancies (93).
3.4 Utilization of alternative NK cell sources
When NK cells from patients are insufficiently functional to support NKCE activity, alternative functional NK cell sources have been investigated. These sources include non-engineered allogeneic NK cells from healthy donors and iPSC-derived NK cells. For instance, AB-101, developed by Artiva Biotherapeutics, is an expandable, non-engineered allogeneic NK cell source isolated from the umbilical cord blood (UCB) of healthy donors. Most AB-101 NK cells express high-affinity CD16a (158 V/V) and other activating receptors at significant levels. In combination with monoclonal antibodies (mAbs), AB-101 has been shown to effectively induce ADCC and inhibit tumor growth in vivo (120). Moreover, AB-101 has demonstrated efficacy when combined with AFM-13 (Affimed), an NKCE targeting CD30+ T cell lymphoma. This combination therapy is currently being evaluated in a phase 2 clinical trial (NCT05883449) (121). However, a limitation of using NK cells from different donors is the potential for batch-to-batch variations and heterogeneity of the cell source, which may affect the reproducibility of therapeutic outcomes.
iPSC-derived NK cells offer an alternative with significant advantages, including homogeneity, engineerability, and an unlimited lifespan, minimizing batch-to-batch variations. Compared to NK cells isolated from UCB, iPSC-derived NK cells can be further engineered to enhance NKCE activity. For example, Chiu et al. (2021) engineered iPSC-derived NK cells to express NKG2C receptors and adaptor signaling molecules to improve the therapeutic efficacy of NKG2C-targeted NKCEs (86). Additionally, iPSC-derived NK cells can be modified to express non-cleavable high-affinity CD16a receptors, further promoting engagement with NKCEs and enhancing NK cell activity and proliferation (122). Fate Therapeutics is currently one of the leading companies in utilizing iPSC-derived NK cells for cancer therapy.
In a similar approach, Cytovia Therapeutics has utilized TALEN®-based gene-editing technology to modify iPSCs, inserting genes that enhance NK cell effector functions and deleting those that impair them. By leveraging the unlimited proliferative capacity of iPSCs, a selected single iPSC clone was expanded and differentiated into NK cells, referred to as iNK cells. As highlighted on their website, Cytovia Therapeutics is currently evaluating the efficacy and safety of iNK cells in combination with their FLEX-NK platform in preclinical studies for the treatment of multiple myeloma, hepatocellular carcinoma, and other cancers.
3.5 Targeting dual antigens to prevent tumor escape
While NKCEs have demonstrated their ability to effectively eliminate tumors, cancer cells may still evade immunosurveillance by introducing mutation to the NK cell binding site or downregulating TAAs. To counter these escape mechanisms, NKCEs have been designed to concurrently target two TAAs. These dual-targeting NKCEs have shown enhanced anti-tumor activities and the ability to block inhibitory signaling pathways (111, 123). For instance, Steinmetz et al. (2016) reported the development of a cross-over dual-variable Ig-like protein (CODV-Ig) platform, which features a circular, self-contained structure that maintains the parental antibody affinities for two antigens without positional effects (116). CODV-Ig was engineered to target both HER2 and HER3 antigens on the cell surface, effectively blocking their heterodimerization. This not only resulted in the blockade of mitogenic signaling but also in ADCC-mediated killing of cancer cells (116). Other examples of dual TAA-targeting NKCEs include those targeting EGFR/PD-L1 (59, 124, 125) and CD47/CD38 (117, 126), among others.
3.6 Optimizing the NKCE’s half-life
The half-life and systemic exposure of NKCEs are critical determinants of their therapeutic efficacy. Typically, the half-life of NKCEs is influenced by their molecular weight and sequence. Structurally, NKCEs can be categorized into two groups: those containing an Fc fragment and those without it. The PK profile of NKCEs that incorporate an Fc fragment is often influenced by the “sink effect.”. This phenomenon arises from the non-specific binding of the Fc fragment within the NKCE sequence to various activating and inhibitory Fcγ receptors on immune cells, such as CD16b on neutrophils or CD32b on B cells (50). In contrast, NKCEs engineered to selectively bind Fcγ receptors, particularly CD16a on NK cells, exhibit reduced non-specific binding and, consequently, a lower susceptibility to the sink effect. Similar to the Fc fragment, the antigen-binding domain of NKCEs can interact with soluble target antigens present in plasma, potentially altering the PK profile. These soluble antigens often result from the shedding of extracellular domains of membrane-bound receptors, including CD16b on neutrophils or tumor-associated antigens (TAAs) (127–129). Elevated levels of soluble target antigens are frequently observed in the serum of cancer patients. For instance, the concentration of soluble BCMA is significantly higher in the serum of multiple myeloma patients compared to healthy individuals, creating an “antigen sink” that impacts NKCE pharmacokinetics (128, 130). Studies have demonstrated an inverse relationship between the concentration of soluble antigens and both drug exposure and therapeutic efficacy in clinical settings (131, 132). Additionally, shedding of extracellular domains to generate soluble antigens is recognized as a major cancer drug resistance mechanism. To counteract the sink effect associated with soluble antigens, optimizing dosage and dosing intervals is essential (53, 54). Beyond tumor-derived soluble antigens, the expression of target antigens on healthy tissues can further complicate the sink effect, as their distribution across the body influences NKCE biodistribution (127). Although no studies have specifically examined NKCE disposition related to target antigens on healthy tissues, insights from T cell engager studies suggest potential strategies. For instance, NKCE tumor selectivity may be enhanced by targeting multiple co-expressed tumor antigens or increasing the binding valency to tumor-specific antigens. Overall, research into the sink effect on NKCEs remains limited, underscoring the need for further studies to address this challenge effectively.
In addition to the sink effect, NKCEs containing Fc fragments exhibit an extended half-life due to their relatively large molecular size and their capacity for neonatal Fc receptor (FcRn)-mediated recycling. FcRn plays a critical role in regulating the half-life and recycling of IgG and albumin, contributing to prolonged antibody persistence within the body (133, 134). Expressed across a variety of cells, including endothelial and epithelial cells, antigen-presenting cells, and even within tumor microenvironments, FcRn influences antibody distribution and therapeutic outcomes. At acidic pH, FcRn binds IgG with high affinity, facilitating recycling and transcytosis, while at physiological pH, it releases the antibody. Within tumors, FcRn expression can further impact therapeutic antibody delivery and efficacy, enhancing antigen presentation and immune activation through sustained exposure to IgG-bound antigens (135, 136). For instance, NKCEs with an IgG-like format, developed using Affimed’s ROCK® platform, demonstrate the longest serum half-life, followed by NKCEs with an Fc-fusion structure (50). Conversely, NKCEs derived from the ROCK® platform that consist solely of scFvs and lack the Fc domain display a markedly shorter half-life, sometimes as brief as 18 hours (50). This reduced half-life is attributed to the absence of FcRn-mediated recycling and the rapid renal clearance associated with their smaller molecular weight. To address this limitation, NKCEs with shorter half-lives can be engineered by fusing them with a high-affinity anti-human serum albumin (HSA) modality, thereby extending their half-life and improving systemic exposure (137). The NKCEs with longer half-life require less dosing frequency and are more likely to generate favorable therapeutic efficacy. However, it is believed that NKCEs with potent cytotoxic profiles may benefit from a shorter half-life to minimize the risk of acute adverse effects (50). Consequently, PK modulation of NKCEs through half-life adjustment must be carefully considered on a case-by-case basis to balance efficacy and safety.
Beyond molecular structure and weight, the immunogenicity of NKCEs also plays a pivotal role in determining their half-life and PK profile. NKCEs that incorporate moieties derived from non-human species, such as murine (scFvs) or camelid (VHHs), may exhibit immunogenicity, leading to the generation of anti-drug antibodies (ADAs). The presence of ADAs can result in rapid NKCE clearance and reduced systemic exposure. The mechanisms underlying ADA generation against monoclonal antibodies (mAbs) are explored in depth in a comprehensive review by Vaisman-Mentesh (138). Briefly, ADAs can be produced via T cell-dependent or T cell-independent B cell activation pathways. In the T cell-independent mechanism, therapeutic proteins cross-react with B cell receptors (BCRs) on B cells, stimulating them to differentiate into plasma cells that produce ADAs. ADAs generated through this mechanism are predominantly low-affinity IgM antibodies (139). In contrast, the T cell-dependent mechanism involves antigen-presenting cells, primarily dendritic cells, and T lymphocytes. Dendritic cells recognize therapeutic proteins as foreign antigens, process them, and present them in complex with MHC class II molecules to CD4+ helper T cells. These T cells then interact with B cells, inducing their transformation into plasma cells, which subsequently secrete high-affinity IgG ADAs (139).
To mitigate immunogenicity concerns, humanization of NKCE components is a viable strategy, particularly for scFv-based NKCEs derived from murine sources. Generally, VHH-based NKCEs exhibit low immunogenicity, owing to their small size and the high degree of homology (75-90%) between their encoding genes and those of the VH3 family of human antibodies (140–142). If an immunogenic response is observed, VHHs can be humanized using established strategies designed to reduce their immunogenic potential (143–145). However, it is important to recognize that humanization does not inherently guarantee reduced immunogenicity, as many humanized mAbs have still induced ADA formation in clinical settings. Therefore, efforts to enhance NKCEs’ systemic exposure by minimizing immunogenicity should be carefully planned and rigorously evaluated using various in vitro assays to predict the immunogenicity of biologics. For instance, assessing the activation and proliferation of CD4+ helper T cells, which play a crucial role in the T cell-dependent B cell activation pathway, after exposure to biotherapeutics, can provide valuable insights (146). Additionally, T-cell and B-cell epitope mapping can be employed to evaluate the immunogenic potential of therapeutic biologics. These approaches are integral to predicting and managing immunogenicity, ultimately ensuring the safe and effective use of NKCEs in clinical practice (139).
4 Conclusions and perspectives
In conclusion, NK cell engagers (NKCEs) have demonstrated promising efficacy and safety in both preclinical and clinical studies. Unlike conventional monoclonal antibodies (mAbs), NKCEs can be engineered to precisely target a broad range of NK cell-activating receptors and multiple tumor-associated antigens (TAAs) within a single construct, significantly enhancing their anticancer activity. By leveraging protein recombination technology, NKCEs can also be designed to incorporate various cytokines and chemokines that enhance NK cell functionality, thereby optimizing therapeutic efficacy. Moreover, compared to the complex manufacturing processes and high costs associated with chimeric antigen receptor NK (CAR-NK) cell therapy, NKCEs offer a more cost-effective alternative, making them accessible to a larger patient population. Importantly, NKCEs possess the unique ability to simultaneously target multiple TAAs using simple protein engineering techniques. This capability holds great promise for addressing challenges associated with tumor drug resistance, antigen heterogeneity, and tumor immune escape mechanisms. However, several challenges must be addressed to fully realize the therapeutic potential of NKCEs. The functionality of NK cells is paramount to the success of NKCE therapy, yet the reduced expression of activating receptors such as CD16 and NKG2D and enhanced expression of inhibitory checkpoint receptors such as LAG-3, NKG2A, and TIGIT are frequently observed in the NK cells of cancer patients (147, 148). Additionally, the immunosuppressive tumor microenvironment (TME) of solid tumors can further compromise the proliferation and functionality of NK cells. For instance, transforming growth factor-beta (TGF-β) present in the TME of solid tumors is known to downregulate the expression of granzyme B and perforin, significantly inhibiting the cytolytic activity of NK cells (149, 150). To overcome these barriers, the inclusion of functional NK cells as part of NKCE-based treatments is essential to ensure the anticipated therapeutic outcomes. The source of functional NK cells could be ex vivo expanded allogenic, autologous, or iPSC-derived. In the context of solid tumors, combining NKCEs with strategies that block immunosuppressive cytokines may help restore NK cell effector functions. Furthermore, combining NKCEs with other treatment modalities may lead to synergistic effects in cancer therapy. For example, the combination of chemotherapy with immunotherapy could be particularly beneficial in the treatment of solid tumors (151, 152). Chemotherapy not only eliminates a significant portion of cancer cells but also enhances tumor perfusion, facilitating the infiltration of immune cells into the tumor mass. Notably, mild chemotherapy has been shown to upregulate NKG2D ligands on cancer cells while preserving immune cells, thereby enhancing the efficiency of the immune response against cancer cells (53, 152). In addition to chemotherapy, combining NK cell engagers (NKCEs) with CAR-NK cell therapy presents a promising strategy with potential synergistic benefits. This dual approach harnesses CAR-NK cells as a source of highly functional NK cells, while NKCEs—particularly those with incorporated cytokine modules—promote CAR-NK cell proliferation and enhance their persistence in patients. This synergy can lead to improved anti-tumor efficacy. Furthermore, by allowing NKCEs and CAR-NK cells to target multiple TAAs, this combination minimizes the risk of tumor escape, potentially resulting in more durable therapeutic outcomes. In summary, NKCEs represent a versatile and potent class of therapeutic molecules that not only activate NK cells but also address the limitations of conventional monoclonal antibodies (mAbs) and CAR-based immunotherapies. NKCE, whether used alone or in combination with autologous NK cells, have shown limited effectiveness in some clinical trials, particularly when administered to heavily pre-treated cancer patients (e.g., NCT05099549, https://doi.org/10.1200/GO.2023.9.Supplement_1.26). These patients frequently undergo extensive chemotherapy, which can deplete NK cells and lead to lymphopenia, resulting in an immune system compromised by a reduced population of functional immune cells. In such immunocompromised conditions, NKCE therapies face challenges, as the weakened environment cannot fully support NK cells’ intended cytotoxic action against cancer cells. This underscores the importance of careful patient selection and timing of treatment to maximize the therapeutic potential of NKCE-based therapies. Published clinical data suggest a promising approach that combines NKCEs with allogeneic NK cells, enhancing immune responses against cancer cells and potentially improving outcomes for patients resistant to conventional treatments. This combination strategy leverages the advantages of NK cells, which naturally present a minimal risk of causing graft-versus-host disease (GvHD), a significant concern with T-cell therapies. Unlike T cells, NK cells do not require genetic modification to avoid GvHD, making them a safer and more feasible option for allogeneic applications. This characteristic allows NK cell therapies to be more broadly accessible, particularly benefiting patients who may not be suitable candidates for autologous immune cell treatments. Moreover, advancements in NKCE design have led to the development of CD16a variants resistant to shedding. Typically, activated NK cells upregulate the enzyme ADAM17, which mediates the shedding of CD16a to regulate immune responses. However, using CD16a-resistant variants in NKCEs helps maintain their efficacy, even in environments with high ADAM17 activity, thereby enhancing the persistence of NK cell engagement and cytotoxicity. Furthermore, Combining NKCEs with CAR-T cells offers a synergistic approach to cancer therapy by harnessing both innate and adaptive immune responses. This dual-cell strategy could provide a more robust and comprehensive immune assault on cancer cells, leveraging both the unique targeting abilities of CAR-T cells and the natural cytotoxicity of NK cells. One of the primary challenges in cancer immunotherapy is tumor heterogeneity, which can lead to recurrence, especially in solid tumors. CAR-T cells are highly target-driven, directly recognizing and eliminating cancer cells that express specific antigens. However, this specificity can be a limitation, as tumors may evade detection by downregulating target antigens or through antigen loss, resulting in relapse. In contrast, NK cells possess a multitude of innate activating receptors, enabling them to recognize and kill a wide range of cancer cells, including those that may escape CAR-T cell targeting. As the field continues to evolve and challenges are steadily addressed, NKCEs hold great promise as an alternative or complementary approach to existing cancer immunotherapies, potentially offering more effective and accessible treatment options for patients.
Author contributions
SN: Conceptualization, Investigation, Methodology, Validation, Visualization, Writing – original draft. GL: Conceptualization, Investigation, Methodology, Validation, Writing – original draft. AH: Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the NIH/NCI (R01CA251438), Department of Defense Ovarian Cancer Program (HT9425-23-1-0191), and NIH/NHLBI/Rutgers HealthAdvance joint program (U01HL150852).
Conflict of interest
AH and SN are named inventors on a patent WO2023129819 related to the E5C1 BiKE technology discussed in this review. The intellectual property IP rights for this patented technology are held by Rutgers University. Additionally, the BiKE technology for oncology applications has been licensed to Sauvie Inc.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. (2015) 6:368.
2. Lisi L, Lacal PM, Martire M, Navarra P, Graziani G. Clinical experience with CTLA-4 blockade for cancer immunotherapy: From the monospecific monoclonal antibody ipilimumab to probodies and bispecific molecules targeting the tumor microenvironment. Pharmacol Res. (2022) 175:105997.
3. Jin S, Sun Y, Liang X, Gu X, Ning J, Xu Y, et al. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduction Targeted Ther. (2022) 7:39.
5. Barb AW. Fc gamma receptor compositional heterogeneity: Considerations for immunotherapy development. J Biol Chem. (2021) 296:100057. doi: 10.1074/jbc.REV120.013168
6. Dixon KJ, Wu J, Walcheck B. Engineering anti-tumor monoclonal antibodies and fc receptors to enhance ADCC by human NK cells. Cancers (Basel). (2021) 13. doi: 10.3390/cancers13020312
7. Bournazos S, Gupta A, Ravetch JV. The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol. (2020) 20:633–43. doi: 10.1038/s41577-020-00410-0
8. Ravetch JV, Bolland S. IgG fc receptors. Annu Rev Immunol. (2001) 19:275–90. doi: 10.1146/annurev.immunol.19.1.275
9. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
10. Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. (2012) 119:5640–9. doi: 10.1182/blood-2012-01-380121
11. Coenon L, Villalba M. From CD16a biology to antibody-dependent cell-mediated cytotoxicity improvement. Front Immunol. (2022) 13:913215. doi: 10.3389/fimmu.2022.913215
12. Liu W. Evaluating human natural killer cells antibody-dependent cellular cytotoxicity (ADCC) using plate-bound anti-CD16 antibodies. Bio Protoc. (2022) 12:e4285. doi: 10.21769/BioProtoc.4285
13. Laskowski TJ, Biederstädt A, Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer. (2022) 22:557–75.
14. Hatjiharissi E, Xu L, Santos DD, Hunter ZR, Ciccarelli BT, Verselis S, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the FcγRIIIa-158 V/V and V/F polymorphism. Blood J Am Soc Hematol. (2007) 110:2561–4.
15. Kute TE, Savage L, Stehle JR, Kim-Shapiro JW, Blanks MJ, Wood J, et al. Breast tumor cells isolated from in vitro resistance to trastuzumab remain sensitive to trastuzumab anti-tumor effects in vivo and to ADCC killing. Cancer immunol Immunother. (2009) 58:1887–96.
16. Seo Y, Ishii Y, Ochiai H, Fukuda K, Akimoto S, Hayashida T, et al. Cetuximab-mediated ADCC activity is correlated with the cell surface expression level of EGFR but not with the KRAS/BRAF mutational status in colorectal cancer. Oncol Rep. (2014) 31:2115–22.
17. Srpan K, Ambrose A, Karampatzakis A, Saeed M, Cartwright ANR, Guldevall K, et al. Shedding of CD16 disassembles the NK cell immune synapse and boosts serial engagement of target cells. J Cell Biol. (2018) 217:3267–83. doi: 10.1083/jcb.201712085
18. Mandó P, Rivero SG, Rizzo MM, Pinkasz M, Levy EM. Targeting ADCC: A different approach to HER2 breast cancer in the immunotherapy era. Breast. (2021) 60:15–25.
19. Helmin-Basa A, Gackowska L, Balcerowska S, Ornawka M, Naruszewicz N, Wiese-Szadkowska M. The application of the natural killer cells, macrophages and dendritic cells in treating various types of cancer. Phys Sci Rev. (2020) 7(8):833–66.
20. Hsieh YT, Aggarwal P, Cirelli D, Gu L, Surowy T, Mozier NM. Characterization of FcgammaRIIIA effector cells used in in vitro ADCC bioassay: Comparison of primary NK cells with engineered NK-92 and Jurkat T cells. J Immunol Methods. (2017) 441:56–66. doi: 10.1016/j.jim.2016.12.002
21. Chung S, Lin YL, Reed C, Ng C, Cheng ZJ, Malavasi F, et al. Characterization of in vitro antibody-dependent cell-mediated cytotoxicity activity of therapeutic antibodies - impact of effector cells. J Immunol Methods. (2014) 407:63–75. doi: 10.1016/j.jim.2014.03.021
22. Heider K-H, Kiefer K, Zenz T, Volden M, Stilgenbauer S, Ostermann E, et al. A novel Fc-engineered monoclonal antibody to CD37 with enhanced ADCC and high proapoptotic activity for treatment of B-cell Malignancies. Blood J Am Soc Hematol. (2011) 118:4159–68.
23. Liu R, Oldham RJ, Teal E, Beers SA, Cragg MS. Fc-engineering for modulated effector functions—improving antibodies for cancer treatment. Antibodies. (2020) 9:64.
24. Kang TH, Jung ST. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp Mol Med. (2019) 51:1–9.
25. Rugo HS, Im S-A, Cardoso F, Cortés J, Curigliano G, Musolino A, et al. Efficacy of margetuximab vs trastuzumab in patients with pretreated ERBB2-positive advanced breast cancer: a phase 3 randomized clinical trial. JAMA Oncol. (2021) 7:573–84.
26. Stavenhagen JB, Gorlatov S, Tuaillon N, Rankin CT, Li H, Burke S, et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcγ receptors. Cancer Res. (2007) 67:8882–90.
27. Husna SMN, Wong KK. Margetuximab and trastuzumab deruxtecan: New generation of anti-HER2 immunotherapeutic agents for breast cancer. Mol Immunol. (2022) 152:45–54.
28. Gleason MK, Verneris MR, Todhunter DA, Zhang B, McCullar V, Zhou SX, et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine productionBiKEs and triKEs enhance NK cell effector function. Mol Cancer Ther. (2012) 11:2674–84.
29. Au KM, Park SI, Wang AZ. Trispecific natural killer cell nanoengagers for targeted chemoimmunotherapy. Sci Adv. (2020) 6:eaba8564.
30. Nikkhoi SK, Li G, Eleya S, Yang G, Vandavasi VG, Hatefi A. Bispecific killer cell engager with high affinity and specificity toward CD16a on NK cells for cancer immunotherapy. Front Immunol. (2022) 13:1039969. doi: 10.3389/fimmu.2022.1039969
31. Jacobs R, Hintzen G, Kemper A, Beul K, Kempf S, Behrens G, et al. CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur J Immunol. (2001) 31:3121–7. doi: 10.1002/1521-4141(2001010)31:10<3121::aid-immu3121>3.0.co;2-4
32. Poli A, Michel T, Theresine M, Andres E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology. (2009) 126:458–65. doi: 10.1111/j.1365-2567.2008.03027.x
33. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. (2017) 47:820–33. doi: 10.1016/j.immuni.2017.10.008
34. Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-Å crystal structure of the human IgG1 Fc fragment–FcγRIII complex. Nature. (2000) 406:267–73.
35. Blázquez-Moreno A, Park S, Im W, Call MJ, Call ME, Reyburn HT. Transmembrane features governing Fc receptor CD16A assembly with CD16A signaling adaptor molecules. Proc Natl Acad Sci. (2017) 114:E5645–E54.
36. Nagarajan S, Chesla S, Cobern L, Anderson P, Zhu C, Selvaraj P. Ligand binding and phagocytosis by CD16 (Fc γ Receptor III) isoforms: PHAGOCYTIC SIGNALING BY ASSOCIATED ζ AND γ SUBUNITS IN CHINESE HAMSTER OVARY CELLS (∗). J Biol Chem. (1995) 270:25762–70.
37. Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. (2006) 107:159–66. doi: 10.1182/blood-2005-04-1351
38. Bryceson YT, Ljunggren HG, Long EO. Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood. (2009) 114:2657–66. doi: 10.1182/blood-2009-01-201632
39. Tesar M, Ellwanger K, Fucek I, Reusch U, Ross T, Koch J, et al. . patent WO2019198051A2, US 11,001,633 B2 (2019).
40. Reusch U, Burkhardt C, Fucek I, Le Gall F, Le Gall M, Hoffmann K, et al. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs. (2014) 6(3):728–39.
41. Kerbauy LN, Marin ND, Kaplan M, Banerjee PP, Berrien-Elliott MM, Becker-Hapak M, et al. Combining AFM13, a bispecific CD30/CD16 antibody, with cytokine-activated blood and cord blood-derived NK cells facilitates CAR-like responses against CD30(+) Malignancies. Clin Cancer Res. (2021) 27:3744–56. doi: 10.1158/1078-0432.CCR-21-0164
42. Sasse S, Brockelmann PJ, Momotow J, Plutschow A, Huttmann A, Basara N, et al. AFM13 in patients with relapsed or refractory classical Hodgkin lymphoma: final results of an open-label, randomized, multicenter phase II trial. Leuk Lymphoma. (2022) 63:1871–8. doi: 10.1080/10428194.2022.2095623
43. Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood. (2015) 125:4024–31. doi: 10.1182/blood-2014-12-614636
44. Bartlett NL, Herrera AF, Domingo-Domenech E, Mehta A, Forero-Torres A, Garcia-Sanz R, et al. A phase 1b study of AFM13 in combination with pembrolizumab in patients with relapsed or refractory Hodgkin lymphoma. Blood. (2020) 136:2401–9. doi: 10.1182/blood.2019004701
45. Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic hodgkin lymphoma. J Clin Oncol. (2017) 35:2125–32. doi: 10.1200/JCO.2016.72.1316
46. Yago Nieto PB, Kaur I, Griffin L, Barnett M, Ganesh C, Borneo Z, et al. Innate cell engager (ICE®) AFM13 combined with preactivated and expanded (P+E) cord blood (CB)-derived natural killer (NK) cells for patients with refractory CD30-positive lymphomas: final results. Blood. (2023) 142.
47. Wingert S, Reusch U, Knackmuss S, Kluge M, Damrat M, Pahl J, et al. Preclinical evaluation of AFM24, a novel CD16A-specific innate immune cell engager targeting EGFR-positive tumors. MAbs. (2021) 13:1950264. doi: 10.1080/19420862.2021.1950264
48. Gantke T, Weichel M, Herbrecht C, Reusch U, Ellwanger K, Fucek I, et al. Trispecific antibodies for CD16A-directed NK cell engagement and dual-targeting of tumor cells. Protein Eng Des Sel. (2017) 30:673–84. doi: 10.1093/protein/gzx043
49. Giang KA, Boxaspen T, Diao Y, Nilvebrant J, Kosugi-Kanaya M, Kanaya M, et al. Affibody-based hBCMA x CD16 dual engagers for NK cell-mediated killing of multiple myeloma cells. N Biotechnol. (2023) 77:139–48. doi: 10.1016/j.nbt.2023.09.002
50. Ellwanger K, Reusch U, Fucek I, Wingert S, Ross T, Muller T, et al. Redirected optimized cell killing (ROCK(R)): A highly versatile multispecific fit-for-purpose antibody platform for engaging innate immunity. mAbs. (2019) 11:899–918. doi: 10.1080/19420862.2019.1616506
51. Kakiuchi-Kiyota S, Ross T, Wallweber HA, Kiefer JR, Schutten MM, Adedeji AO, et al. A BCMA/CD16A bispecific innate cell engager for the treatment of multiple myeloma. Leukemia. (2022) 36:1006–14. doi: 10.1038/s41375-021-01478-w
52. Wemlinger SM, Cambier JC. Therapeutic tactics for targeting B lymphocytes in autoimmunity and cancer. Eur J Immunol. (2024) 54:e2249947. doi: 10.1002/eji.202249947
53. Cai H, Kakiuchi-Kiyota S, Hendricks R, Zhong S, Liu L, Adedeji AO, et al. Nonclinical pharmacokinetics, pharmacodynamics, and translational model of RO7297089, A novel anti-BCMA/CD16A bispecific tetravalent antibody for the treatment of multiple myeloma. AAPS J. (2022) 24:100. doi: 10.1208/s12248-022-00744-8
54. Plesner T, Harrison SJ, Quach H, Lee C, Bryant A, Vangsted A, et al. Phase I study of safety and pharmacokinetics of RO7297089, an anti-BCMA/CD16a bispecific antibody, in patients with relapsed, refractory multiple myeloma. Clin Hematol Int. (2023) 5:43–51. doi: 10.1007/s44228-022-00023-5
55. Götz J-J, Pahl J, Schmitt N, Müller T, Haneke T, Kozlowska I, et al. AFM28, a novel bispecific innate cell engager (ICE®), designed to selectively re-direct NK cell lysis to CD123+ Leukemic cells in acute myeloid leukemia and myelodysplastic syndrome. Blood. (2021) 138:3344.
56. Steeland S, Vandenbroucke RE, Libert C. Nanobodies as therapeutics: big opportunities for small antibodies. Drug Discovery Today. (2016) 21:1076–113. doi: 10.1016/j.drudis.2016.04.003
57. Khaleghi S, Khoshtinat Nikkhoi S, Rahbarizadeh F. Camelid single-domain antibodies for targeting cancer nanotheranostics. Cancer Nanotheranostics. (2021) 1:93–123.
58. Khoshtinat Nikkhoi S, Yang G, Owji H, Grizotte-Lake M, Cohen RI, Gil Gonzalez L, et al. Bispecific immune cell engager enhances the anticancer activity of CD16+ NK cells and macrophages in vitro, and eliminates cancer metastasis in NK humanized NOG mice. J Immunother Cancer. (2024) 12. doi: 10.1136/jitc-2023-008295
59. Harwardt J, Bogen JP, Carrara SC, Ulitzka M, Grzeschik J, Hock B, et al. A generic strategy to generate bifunctional two-in-One antibodies by chicken immunization. Front Immunol. (2022) 13.
60. Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Natural Killer Cells. (2016) 1441:333–46.
61. Felices M, Kodal B, Hinderlie P, Kaminski MF, Cooley S, Weisdorf DJ, et al. Novel CD19-targeted TriKE restores NK cell function and proliferative capacity in CLL. Blood advances. (2019) 3:897–907.
62. Felices M, Warlick E, Juckett M, Weisdorf D, Vallera D, Miller S, et al. 444 GTB-3550 tri-specific killer engager TriKE™ drives NK cells expansion and cytotoxicity in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) patients. BMJ Specialist Journals. (2021) 9:A473–A.
63. Felices M, Lenvik TR, Kodal B, Lenvik AJ, Hinderlie P, Bendzick LE, et al. Potent cytolytic activity and specific IL15 delivery in a second-generation trispecific killer engagerSecond-generation triKE potentiates NK-cell activity. Cancer Immunol Res. (2020) 8:1139–49.
64. Fenis A, Demaria O, Gauthier L, Vivier E, Narni-Mancinelli E. New immune cell engagers for cancer immunotherapy. Nat Rev Immunol. (2024) 24:471–86. doi: 10.1038/s41577-023-00982-7
65. Wensveen FM, Jelencic V, Polic B. NKG2D: A master regulator of immune cell responsiveness. Front Immunol. (2018) 9:441. doi: 10.3389/fimmu.2018.00441
66. Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, et al. NKG2D and its ligands: “One for all, all for one. Front Immunol. (2018) 9:476. doi: 10.3389/fimmu.2018.00476
67. Carapito R, Aouadi I, Ilias W, Bahram S. Natural killer group 2, member D/NKG2D ligands in hematopoietic cell transplantation. Front Immunol. (2017) 8:368. doi: 10.3389/fimmu.2017.00368
68. Watzl C, Long EO. Signal transduction during activation and inhibition of natural killer cells. Curr Protoc Immunol. (2010) Chapter 11:Unit 11 9B. doi: 10.1002/0471142735.im1109bs90
69. Chan WK, Kang S, Youssef Y, Glankler EN, Barrett ER, Carter AM, et al. A CS1-NKG2D bispecific antibody collectively activates cytolytic immune cells against multiple myeloma. Cancer Immunol Res. (2018) 6:776–87. doi: 10.1158/2326-6066.CIR-17-0649
70. Raynaud A, Desrumeaux K, Vidard L, Termine E, Baty D, Chames P, et al. Anti-NKG2D single domain-based antibodies for the modulation of anti-tumor immune response. Oncoimmunology. (2020) 10:1854529. doi: 10.1080/2162402X.2020.1854529
71. Timmins P. Industry update: the latest developments in the field of therapeutic delivery, November 2021. Ther Deliv. (2022) 13:141–56. doi: 10.4155/tde-2022-0006
72. Tapia-Galisteo A, Alvarez-Vallina L, Sanz L. Bi- and trispecific immune cell engagers for immunotherapy of hematological Malignancies. J Hematol Oncol. (2023) 16:83. doi: 10.1186/s13045-023-01482-w
73. Lunn-Halbert MC, Laszlo GS, Erraiss S, Orr MT, Jessup HK, Thomas HJ, et al. Preclinical characterization of the anti-leukemia activity of the CD33/CD16a/NKG2D immune-modulating triNKET((R)) CC-96191. Cancers (Basel). (2024) 16. doi: 10.3390/cancers16050877
74. Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia. (2004) 6:558–68. doi: 10.1593/neo.04316
75. Schmiedel D, Mandelboim O. NKG2D ligands-critical targets for cancer immune escape and therapy. Front Immunol. (2018) 9:2040. doi: 10.3389/fimmu.2018.02040
76. Zingoni A, Vulpis E, Loconte L, Santoni A. NKG2D ligand shedding in response to stress: role of ADAM10. Front Immunol. (2020) 11:447. doi: 10.3389/fimmu.2020.00447
77. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. (2002) 419:734–8. doi: 10.1038/nature01112
78. Secchiari F, Nunez SY, Sierra JM, Ziblat A, Regge MV, Raffo Iraolagoitia XL, et al. The MICA-NKG2D axis in clear cell renal cell carcinoma bolsters MICA as target in immuno-oncology. Oncoimmunology. (2022) 11:2104991. doi: 10.1080/2162402X.2022.2104991
79. Fantini M, Arlen PM, Tsang KY. Potentiation of natural killer cells to overcome cancer resistance to NK cell-based therapy and to enhance antibody-based immunotherapy. Front Immunol. (2023) 14:1275904. doi: 10.3389/fimmu.2023.1275904
80. Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res. (2015) 3:575–82. doi: 10.1158/2326-6066.CIR-15-0098
81. Ziblat A, Iraolagoitia XLR, Nunez SY, Torres NI, Secchiari F, Sierra JM, et al. Circulating and tumor-infiltrating NK cells from clear cell renal cell carcinoma patients exhibit a predominantly inhibitory phenotype characterized by overexpression of CD85j, CD45, CD48 and PD-1. Front Immunol. (2021) 12:681615. doi: 10.3389/fimmu.2021.681615
82. Della Chiesa M, Sivori S, Carlomagno S, Moretta L, Moretta A. Activating KIRs and NKG2C in viral infections: toward NK cell memory? Front Immunol. (2015) 6:573.
83. Pupuleku A, Costa-García M, Farré D, Hengel H, Angulo A, Muntasell A, et al. Elusive role of the CD94/NKG2C NK cell receptor in the response to cytomegalovirus: novel experimental observations in a reporter cell system. Front Immunol. (2017) 8:1317.
84. Wada H, Matsumoto N, Maenaka K, Suzuki K, Yamamoto K. The inhibitory NK cell receptor CD94/NKG2A and the activating receptor CD94/NKG2C bind the top of HLA-E through mostly shared but partly distinct sets of HLA-E residues. Eur J Immunol. (2004) 34:81–90.
85. Lanier LL, Corliss B, Wu J, Phillips JH. Association of DAP12 with activating CD94/NKG2C NK cell receptors. Immunity. (1998) 8:693–701.
86. Chiu E, Felices M, Cichocki F, Davis Z, Wang H, Tuninga K, et al. Anti-NKG2C/IL-15/anti-CD33 killer engager directs primary and iPSC-derived NKG2C+ NK cells to target myeloid leukemia. Mol Ther. (2021) 29:3410–21.
87. Sivori S, Pende D, Bottino C, Marcenaro E, Pessino A, Biassoni R, et al. NKp46 is the major triggering receptor involved in the natural cytotoxicity of fresh or cultured human NK cells. Correlation between surface density of NKp46 and natural cytotoxicity against autologous, allogeneic or xenogeneic target cells. Eur J Immunol. (1999) 29:1656–66.
88. Walzer T, Bléry M, Chaix J, Fuseri N, Chasson L, Robbins SH, et al. Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc Natl Acad Sci. (2007) 104:3384–9.
89. Barrow AD, Martin CJ, Colonna M. The natural cytotoxicity receptors in health and disease. Front Immunol. (2019) 10:909.
90. Sivori S, Vitale M, Morelli L, Sanseverino L, Augugliaro R, Bottino C, et al. p46, a novel natural killer cell–specific surface molecule that mediates cell activation. J Exp Med. (1997) 186:1129–36.
91. Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell. (2019) 177:1701–13. e16. doi: 10.1016/j.cell.2019.04.041
92. Lipinski B, Arras P, Pekar L, Klewinghaus D, Boje AS, Krah S, et al. NKp46-specific single domain antibodies enable facile engineering of various potent NK cell engager formats. Protein Sci. (2023) 32(3):e4593.
93. Demaria O, Gauthier L, Vetizou M, Blanchard Alvarez A, Vagne C, Habif G, et al. Antitumor immunity induced by antibody-based natural killer cell engager therapeutics armed with not-alpha IL-2 variant. Cell Rep Med. (2022) 3:100783. doi: 10.1016/j.xcrm.2022.100783
94. Lin L, Chang H-M, Wu D, Titz B, Chen A, Chao Ma M, et al. Biological characterization and differential gene expression analysis of CYT-338 NK cell engager (NKE) against CD38 expressing tumors including multiple myeloma (MM). Blood. (2022) 140:7068–9.
95. Arulanandam A, Lin L, Chang H-M, Cerutti M, Choblet S, Gao P, et al. Derivation and preclinical characterization of CYT-303, a novel NKp46-NK cell engager targeting GPC3. Cells. (2023) 12:996.
96. Arulanandam A, Potu H, Khairnar V, Zou D, Triggiano M, Dilmac N, et al. 756P Glypican-3 (GPC3) and NKp46 directed FLEX-NK engager antibody (CYT-303) recruits natural killer (NK) cells to tumors in a preclinical hepatocellular carcinoma (HCC) mouse model. Ann Oncol. (2022) 33:S889.
97. Lin L, Chang H-M, Nakid C, Frankel S, Wu D, Kadouche J, et al. Abstract 3436: Novel multifunctional tetravalent CD38 NKp46 FLEX NK engagers actively target and kill multiple myeloma cells. Cancer Res. (2022) 82:3436. doi: 10.1158/1538-7445.Am2022-3436
98. Zhang S, Liu W, Hu B, Wang P, Lv X, Chen S, et al. Prognostic significance of tumor-infiltrating natural killer cells in solid tumors: A systematic review and meta-analysis. Front Immunol. (2020) 11:1242. doi: 10.3389/fimmu.2020.01242
99. Kim N, Yi E, Lee E, Park HJ, Kim HS. Interleukin-2 is required for NKp30-dependent NK cell cytotoxicity by preferentially regulating NKp30 expression. Front Immunol. (2024) 15:1388018. doi: 10.3389/fimmu.2024.1388018
100. Delahaye NF, Rusakiewicz S, Martins I, Menard C, Roux S, Lyonnet L, et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med. (2011) 17:700–7. doi: 10.1038/nm.2366
101. Pekar L, Klausz K, Busch M, Valldorf B, Kolmar H, Wesch D, et al. Affinity maturation of B7-H6 translates into enhanced NK Cell–mediated tumor cell lysis and improved proinflammatory cytokine release of bispecific immunoligands via NKp30 engagement. J Immunol. (2021) 206:225–36.
102. Klausz K, Pekar L, Boje AS, Gehlert CL, Krohn S, Gupta T, et al. Multifunctional NK cell-engaging antibodies targeting EGFR and NKp30 elicit efficient tumor cell killing and proinflammatory cytokine release. J Immunol. (2022) 209:1724–35. doi: 10.4049/jimmunol.2100970
103. Boje AS, Pekar L, Koep K, Lipinski B, Rabinovich B, Evers A, et al. Impact of antibody architecture and paratope valency on effector functions of bispecific NKp30 x EGFR natural killer cell engagers. Mabs. (2024). 16(1):2315640
104. Colomar-Carando N, Gauthier L, Merli P, Loiacono F, Canevali P, Falco M, et al. Exploiting Natural Killer cell engagers to control pediatric B-cell precursor acute lymphoblastic leukemia. Cancer Immunol Res. (2022) 10:291–302.
105. Klausz K, Pekar L, Boje AS, Gehlert CL, Krohn S, Gupta T, et al. Multifunctional NK cell–engaging antibodies targeting EGFR and NKp30 elicit efficient tumor cell killing and proinflammatory cytokine release. J Immunol. (2022) 209:1724–35.
106. Ikehara Y, Ikehara SK, Paulson JC. Negative regulation of T cell receptor signaling by Siglec-7 (p70/AIRM) and Siglec-9. J Biol Chem. (2004) 279:43117–25. doi: 10.1074/jbc.M403538200
107. Shao JY, Yin WW, Zhang QF, Liu Q, Peng ML, Hu HD, et al. Siglec-7 defines a highly functional natural killer cell subset and inhibits cell-mediated activities. Scand J Immunol. (2016) 84:182–90. doi: 10.1111/sji.12455
108. Yamakawa N, Yasuda Y, Yoshimura A, Goshima A, Crocker PR, Vergoten G, et al. Discovery of a new sialic acid binding region that regulates Siglec-7. Sci Rep. (2020) 10:8647. doi: 10.1038/s41598-020-64887-4
109. Bull C, den Brok MH, Adema GJ. Sweet escape: sialic acids in tumor immune evasion. Biochim Biophys Acta. (2014) 1846:238–46. doi: 10.1016/j.bbcan.2014.07.005
110. Bull C, Stoel MA, den Brok MH, Adema GJ. Sialic acids sweeten a tumor’s life. Cancer Res. (2014) 74:3199–204. doi: 10.1158/0008-5472.CAN-14-0728
111. Bordoloi D, Kulkarni AJ, Adeniji OS, Pampena MB, Bhojnagarwala PS, Zhao S, et al. Siglec-7 glyco-immune binding mAbs or NK cell engager biologics induce potent antitumor immunity against ovarian cancers. Sci Adv. (2023) 9:eadh4379.
112. Bordoloi D, Kulkarni AJ, Adeniji OS, Bhojnagarwala PS, Zhao S, Ionescu C, et al. Abstract PR-006: A novel bispecific NK cell engager targeting FSHR and Siglec-7 displays potent anti-tumor immunity against ovarian cancer. Cancer Res. (2024) 84:PR–006-PR.
113. Natsume A, In M, Takamura H, Nakagawa T, Shimizu Y, Kitajima K, et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res. (2008) 68:3863–72. doi: 10.1158/0008-5472.CAN-07-6297
114. Rispens T, Davies AM, Ooijevaar-de Heer P, Absalah S, Bende O, Sutton BJ, et al. Dynamics of inter-heavy chain interactions in human immunoglobulin G (IgG) subclasses studied by kinetic Fab arm exchange. J Biol Chem. (2014) 289:6098–109. doi: 10.1074/jbc.M113.541813
115. Interdonato A, Choblet S, Sana M, Valgardsdottir R, Cribioli S, Alzani R, et al. BL-01, an Fc-bearing, tetravalent CD20 x CD5 bispecific antibody, redirects multiple immune cells to kill tumors in vitro and in vivo. Cytotherapy. (2022) 24:161–71. doi: 10.1016/j.jcyt.2021.07.012
116. Steinmetz A, Vallée F, Beil C, Lange C, Baurin N, Beninga J, et al. CODV-Ig, a universal bispecific tetravalent and multifunctional immunoglobulin format for medical applications. MAbs. (2016) 8(5):867–78.
117. Grandclement C, Estoppey C, Dheilly E, Panagopoulou M, Monney T, Dreyfus C, et al. Development of ISB 1442, a CD38 and CD47 bispecific biparatopic antibody innate cell modulator for the treatment of multiple myeloma. Nat Commun. (2024) 15:2054. doi: 10.1038/s41467-024-46310-y
118. Chiu ML, Goulet DR, Teplyakov A, Gilliland GL. Antibody structure and function: the basis for engineering therapeutics. Antibodies (Basel). (2019) 8. doi: 10.3390/antib8040055
119. Kamen L, Myneni S, Langsdorf C, Kho E, Ordonia B, Thakurta T, et al. A novel method for determining antibody-dependent cellular phagocytosis. J Immunol Methods. (2019) 468:55–60. doi: 10.1016/j.jim.2019.03.001
120. Rogers P, Yang B, Kim H, Kim Y, Cho S, Morin B, et al. 306 Evaluation of AB-101, an allogeneic cord blood-derived natural killer (NK) cell therapy, as an ADCC enhancer in hematologic and solid tumors. BMJ Specialist J. (2022) 10:A321–A.
121. Moskowitz A, Harstrick A, Emig M, Overesch A, Pinto S, Ravenstijn P, et al. AFM13 in combination with allogeneic natural killer cells (AB-101) in relapsed or refractory hodgkin lymphoma and CD30+Peripheral T-Cell lymphoma: a phase 2 study (LuminICE). Blood. (2023) 142.
122. Ning J, Zachary D, Frank C, Hongbo W, Katie T, Ryan B, et al. IPSC-derived NK cells exhibit potent in vitro and in vivo tumorcidal activity against patient-derived glioblastoma stem cells (GSCs). Neurosurgery (2022) 69:87–8.
123. Huang S, van Duijnhoven SMJ, Sijts A, van Elsas A. Bispecific antibodies targeting dual tumor-associated antigens in cancer therapy. J Cancer Res Clin Oncol. (2020) 146:3111–22. doi: 10.1007/s00432-020-03404-6
124. Harwardt J, Carrara SC, Bogen JP, Schoenfeld K, Grzeschik J, Hock B, et al. Generation of a symmetrical trispecific NK cell engager based on a two-in-one antibody. Front Immunol. (2023) 14:1170042. doi: 10.3389/fimmu.2023.1170042
125. Bogen JP, Carrara SC, Fiebig D, Grzeschik J, Hock B, Kolmar H. Design of a trispecific checkpoint inhibitor and natural killer cell engager based on a 2 + 1 common light chain antibody architecture. Front Immunol. (2021) 12:669496. doi: 10.3389/fimmu.2021.669496
126. Piccione EC, Juarez S, Liu J, Tseng S, Ryan CE, Narayanan C, et al. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. MAbs. (2015) 7:946–56. doi: 10.1080/19420862.2015.1062192
127. Ball K, Dovedi SJ, Vajjah P, Phipps A. Strategies for clinical dose optimization of T cell-engaging therapies in oncology. mAbs. (2023) 15:2181016. doi: 10.1080/19420862.2023.2181016
128. Betts A, van der Graaf PH. Mechanistic quantitative pharmacology strategies for the early clinical development of bispecific antibodies in oncology. Clin Pharmacol Ther. (2020) 108:528–41. doi: 10.1002/cpt.1961
129. Wang Y, Wu J, Newton R, Bahaie NS, Long C, Walcheck B. ADAM17 cleaves CD16b (FcgammaRIIIb) in human neutrophils. Biochim Biophys Acta. (2013) 1833:680–5. doi: 10.1016/j.bbamcr.2012.11.027
130. Wiedemann A, Szita VR, Horvath R, Szederjesi A, Sebo A, Toth AD, et al. Soluble B-cell maturation antigen as a monitoring marker for multiple myeloma. Pathol Oncol Res. (2023) 29:1611171. doi: 10.3389/pore.2023.1611171
131. Zhang Y, Pastan I. High shed antigen levels within tumors: an additional barrier to immunoconjugate therapy. Clin Cancer Res. (2008) 14:7981–6. doi: 10.1158/1078-0432.CCR-08-0324
132. Yu B, Jiang T, Liu D. BCMA-targeted immunotherapy for multiple myeloma. J Hematol Oncol. (2020) 13:125. doi: 10.1186/s13045-020-00962-7
133. Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci U S A. (1996) 93:5512–6. doi: 10.1073/pnas.93.11.5512
134. Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, et al. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. (2006) 18:1759–69. doi: 10.1093/intimm/dxl110
135. Baker K, Rath T, Pyzik M, Blumberg RS. The role of fcRn in antigen presentation. Front Immunol. (2014) 5:408. doi: 10.3389/fimmu.2014.00408
136. Fabris C, Basso D, Piccoli A, Meggiato T, Del Favero G, Fogar P, et al. Role of local and systemic factors in increasing serum glycoprotein markers of pancreatic cancer. J Med. (1991) 22:145–56.
137. Hambach J, Fumey W, Stähler T, Gebhardt AJ, Adam G, Weisel K, et al. Half-life extended nanobody-based CD38-specific bispecific killercell engagers induce killing of multiple myeloma cells. Front Immunol. (2022) 13.
138. Vaisman-Mentesh A, Gutierrez-Gonzalez M, DeKosky BJ, Wine Y. The molecular mechanisms that underlie the immune biology of anti-drug antibody formation following treatment with monoclonal antibodies. Front Immunol. (2020) 11:1951. doi: 10.3389/fimmu.2020.01951
139. Sun R, Qian MG, Zhang X. T and B cell epitope analysis for the immunogenicity evaluation and mitigation of antibody-based therapeutics. MAbs. (2024) 16:2324836. doi: 10.1080/19420862.2024.2324836
140. Ackaert C, Smiejkowska N, Xavier C, Sterckx YGJ, Denies S, Stijlemans B, et al. Immunogenicity risk profile of nanobodies. Front Immunol. (2021) 12:632687. doi: 10.3389/fimmu.2021.632687
141. Su C, Nguyen VK, Nei M. Adaptive evolution of variable region genes encoding an unusual type of immunoglobulin in camelids. Mol Biol evolution. (2002) 19:205–15.
142. D’Huyvetter M, Vos J, Caveliers V, Vaneycken I, Heemskerk J, Duhoux FP, et al. Phase I trial of (131)I-GMIB-anti-HER2-VHH1, a new promising candidate for HER2-targeted radionuclide therapy in breast cancer patients. J Nucl Med. (2021) 62:1097–105. doi: 10.2967/jnumed.120.255679
143. Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. (2009) 284:3273–84. doi: 10.1074/jbc.M806889200
144. Sulea T. Humanization of camelid single-domain a ntibodies. Methods Mol Biol. (2022) 2446:299–312. doi: 10.1007/978-1-0716-2075-5_14
145. Rossotti MA, Belanger K, Henry KA, Tanha J. Immunogenicity and humanization of single-domain antibodies. FEBS J. (2022) 289:4304–27. doi: 10.1111/febs.15809
146. Walsh RE, Nix A, Ackaert C, Mazy A, Schockaert J, Pattyn S, et al. Preclinical immunogenicity risk assessment of biotherapeutics using CD4 T cell assays. Front Immunol. (2024) 15:1406040. doi: 10.3389/fimmu.2024.1406040
147. Wu SY, Fu T, Jiang YZ, Shao ZM. Natural killer cells in cancer biology and therapy. Mol Cancer. (2020) 19:120. doi: 10.1186/s12943-020-01238-x
148. Hasan MF, Croom-Perez TJ, Oyer JL, Dieffenthaller TA, Robles-Carrillo LD, Eloriaga JE, et al. TIGIT Expression on Activated NK Cells Correlates with Greater Anti-Tumor Activity but Promotes Functional Decline upon Lung Cancer Exposure: Implications for Adoptive Cell Therapy and TIGIT-Targeted Therapies. Cancers (Basel). (2023) 15. doi: 10.3390/cancers15102712
149. Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol. (2008) 181:3784–92. doi: 10.4049/jimmunol.181.6.3784
150. Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, et al. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PloS One. (2018) 13:e0191358. doi: 10.1371/journal.pone.0191358
151. Chao Y, Liang C, Tao H, Du Y, Wu D, Dong Z, et al. Localized cocktail chemoimmunotherapy after in situ gelation to trigger robust systemic antitumor immune responses. Sci Adv. (2020) 6:eaaz4204.
Keywords: NKCE platform technologies, NK activating receptors, bispecific trispecific multispecific antibodies, CD16a NKG2D NKG2C NKp30 NKp46, cancer immunotherapy, natural killer cells (NK cells)
Citation: Nikkhoi SK, Li G and Hatefi A (2025) Natural killer cell engagers for cancer immunotherapy. Front. Oncol. 14:1483884. doi: 10.3389/fonc.2024.1483884
Received: 20 August 2024; Accepted: 31 December 2024;
Published: 22 January 2025.
Edited by:
Sharon R. Pine, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Roberta Castriconi, University of Genoa, ItalyBruce Walcheck, University of Minnesota Twin Cities, United States
Olivier Demaria, Innate Pharma, France
Copyright © 2025 Nikkhoi, Li and Hatefi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arash Hatefi, YWhhdGVmaUBwaGFybWFjeS5ydXRnZXJzLmVkdQ==
†These authors have contributed equally to this work