 Ella M. Dunderdale
Ella M. Dunderdale Evan R. Abt
Evan R. Abt- Department of Molecular and Medical Pharmacology, University of California Los Angeles, Los Angeles, CA, United States
Nucleoside metabolism regulates immune cell development and function, but the therapeutic implications of this link have yet to be fully realized. Evidence for the importance of nucleoside metabolism in immune system control was provided by observations of immunodeficiency and autoimmunity across patients with genetic errors that alter nucleoside synthesis or breakdown. Research over the past several decades has uncovered a multifaceted role for nucleosides in mediating immune responses that involves their function as metabolic precursors and as ligands for immune receptors. These findings prompted the development of treatments that block the production of the immunosuppressive nucleoside adenosine for cancer immunotherapy. Guanosine and pyrimidine nucleosides also mediate immune outcomes, and the key regulators of their metabolism are promising new targets to unleash anti-cancer immune responses or dampen autoimmune reactions. This review provides an overview of (i) recent research concerning the mechanisms underlying nucleoside-mediated immune regulation, (ii) the current landscape of therapeutic targets for immune modulation within nucleoside metabolism, and (iii) opportunities for developing improved preclinical models that recapitulate human nucleoside metabolism, which are needed to advance new metabolism-targeting therapies toward the clinic.
1 Introduction
Nucleosides critically regulate immune system function and targeting nucleoside metabolism has emerged as a promising approach to unleash anti-cancer immune responses or restrain autoimmune reactions. Nucleosides have a multifaceted role in immune system regulation that involves their function as metabolic precursors and signaling modifiers. Nucleosides are classical biosynthesis metabolites that fuel nucleotide production and nucleic acid synthesis. Nucleosides are also signaling molecules that regulate biological outcomes by engaging intracellular or cell surface-localized receptors. The immunosuppressive properties of the purine nucleoside adenosine are well-studied, and therapies that block adenosine production are currently under clinical investigation for cancer immunotherapy (1).
Recent research has revealed that nucleosides beyond adenosine also mediate immune-related outcomes. These nucleosides include (deoxy)guanosine and the pyrimidine nucleosides (deoxy)cytidine, uridine, and thymidine. The mechanisms underlying the immune-modifying properties of these metabolites are not as well-studied as adenosine, and therapeutic strategies to leverage their immune-modifying properties have not yet been systematically tested in the clinic. There has been a disproportionate focus on adenosine over pyrimidine or other purine nucleosides in the context of research related to immune system regulation. The striking manifestations of immune dysfunction in patients with diminished activity of the adenosine metabolizing enzyme adenosine deaminase (ADA), first described in the 1970s, may have contributed to this discrepancy. However, the proteins controlling pyrimidine or guanosine nucleoside metabolism may be equally crucial therapeutic targets to modify immune outcomes as those controlling adenosine-mediated immunosuppression.
New studies have highlighted the potential for targeting key regulators of guanosine and pyrimidine nucleoside synthesis, utilization, or breakdown to amplify immune responses against cancer or dampen autoimmune reactions. However, there is an incomplete understanding of the molecular mechanisms underlying the immune-regulatory properties of nucleosides beyond adenosine. This gap in knowledge may be addressed through future studies in improved preclinical models and the analysis of specimens from ongoing clinical trials testing inhibitors of adenosine metabolism for cancer treatment.
A challenge in the development of metabolism-targeting drugs for immune modification is the paucity of preclinical models that recapitulate human nucleoside metabolism. Significant differences exist in the nucleotide metabolism of humans and conventional laboratory models such as rodents. For example, the pyrimidine nucleosides deoxycytidine and thymidine are measured at a 100-fold higher concentration in murine sera compared to sera from humans or non-human primates (2). This discrepancy presents a major obstacle to implementing the findings from laboratory investigations in the design of clinical trials (3). Also contributing to the challenge of translating preclinical research findings are differences in the expression patterns of immune sensor proteins across humans and mouse models. Research using preclinical models that recapitulate both human nucleoside metabolism and immune responses may provide the insight needed to advance new therapies to alter immune-related outcomes in patients.
The goals of this review are to (i) highlight primary research articles that have demonstrated functions of nucleosides beyond adenosine in immune system regulation, (ii) provide an update on recent advances in targeting nucleoside metabolism for cancer immunotherapy, and (iii) summarize the challenges and opportunities related to the development of preclinical models for human nucleoside metabolism that are needed to advance new metabolism-targeting therapies toward the clinic.
2 Immune-regulatory functions of guanosine nucleosides
Evidence for the immune-regulatory roles of nucleosides was provided by the identification of immune dysfunction in patients with hereditary loss-of-function mutations in two genes responsible for the breakdown of purine nucleosides: ADA and purine nucleoside phosphorylase (PNP) (4, 5). A leader of these investigations was the physician-scientist Eloise Giblett (6), whose interest in purine metabolism began when she identified a complete lack of blood ADA activity in a patient with severe combined immunodeficiency (SCID). ADA is an enzyme within the purine salvage pathway that catalyzes the deamination of adenosine and deoxyadenosine nucleosides to inosine or deoxyinosine, respectively. Giblett and colleagues also identified that the mutational inactivation of PNP, an enzyme that is also involved in purine metabolism, produces a near-complete absence of T cells alongside altered phenotypes of other immune lineages (5). These foundational studies that associated defects in purine metabolism with the development of SCID provided compelling evidence for a role of nucleoside metabolism in regulating immune responses.
SCID is an established manifestation of PNP/ADA deficiency or defects in other genes that control immune responses. It is rare across the human population, occurring in 0.001-0.002% of births. ADA deficiency constitutes 10-15% of SCID cases (7). Enzyme replacement, hematopoietic stem cell transplantation (HSCT), and gene therapies enable the management of ADA-linked SCID (8). In contrast, only a very small fraction of SCID cases are due to defects in PNP; approximately 70 cases of PNP deficiency have been documented (9). The T cell deficiency associated with PNP inactivation is managed in the clinic with HSCT alongside other treatments.
PNP is a key regulator of the purine salvage pathway. PNP catalyzes the release of purine nucleobases that can be recycled by hypoxanthine phosphoribosyltransferase (HPRT), an enzyme which conjugates nucleobases with phosphoribosyl pyrophosphate (PRPP) to generate purine nucleotide monophosphate (10). In addition to enabling the intracellular purine salvage pathway, PNP controls the systemic levels of purine nucleosides. PNP deficiency results in the systemic accumulation of guanosine, adenosine, inosine, deoxyguanosine (dG), and deoxyadenosine (dA). PNP is also critical for the conversion of purine nucleosides to uric acid and their subsequent excretion (10).
The observations of T cell deficiency in PNP-deficient patients made by Giblett and colleagues have been confirmed by other groups who have expanded the catalog of altered immune phenotypes associated with PNP deficiency in humans (11). A subset of patients with PNP deficiency exhibit autoimmune phenotypes that include systemic lupus, autoimmune hemolytic anemia, and systemic juvenile idiopathic arthritis with macrophage activation syndrome (12, 13). Across patients with PNP deficiency, recurring PNP mutations have been identified that lead to immune dysfunction and susceptibility to infections (14). Partial PNP deficiency is associated with milder symptoms than complete inhibition, and patients with partial PNP activity can exhibit typical development and potentially near-normal immune activity (15).
The most profound phenotype observed in PNP-deficient patients, a near complete T cell immunodeficiency, is linked to the uncontrolled expansion of purine nucleotide pools in developing thymocytes following PNP inactivation. The accumulation of the PNP substrate deoxyguanosine and its subsequent metabolism in cells results in dNTP pool imbalance, DNA replication defects, and cell death. The stabilization of deoxyguanosine following PNP inhibition results in a massive expansion of the deoxyguanosine triphosphate (dGTP) pool in cells, which inhibits pyrimidine dNTP synthesis via ribonucleotide reductase (RNR) by an allosteric regulatory mechanism (Figure 1A) (16). The entry of deoxyguanosine nucleosides into cells and their subsequent phosphorylation to dGMP is mediated by the sequential activity of transmembrane nucleoside transporters and deoxycytidine kinase (dCK) (17). The preferred dCK substrate is dC. However, dCK also catalyzes the phosphorylation of the purine deoxyribonucleosides deoxyadenosine and deoxyguanosine to dAMP and dGMP, respectively (18). While dCK activity is suppressed via dCTP-mediated allosteric regulation, it is not susceptible to feedback regulation by purine deoxyribonucleotides. Therefore, additional mechanisms must function to counteract purine dNTP pool expansion in the context of PNP deficiency. Developing thymocytes are particularly vulnerable to PNP inactivation due to limited dNTP catabolism capacity, which exacerbates intracellular dGTP accumulation (10). The dNTP triphosphohydrolase SAM domain and HD domain-containing protein 1 (SAMHD1) is expressed at low levels across the early stages of thymocyte development, and this may explain the increased sensitivity of this lineage to PNP inhibition and the resulting uncontrolled expansion of dGTP pools (10). PNP inactivation is synthetically lethal with downregulation of SAMHD1, and this collateral dependency extends to SAMHD1-deficient cells from multiple lineages beyond lymphocytes (10). The transcriptional down-regulation of SAMHD1 during T cell development may be related to the increased dNTP demands of proliferating thymocytes for DNA replication, or the direct role of SAMHD1 in DNA repair by promoting homologous recombination (19). The lethal effects of purine dNTP imbalance are also apparent in T lymphoblastic leukemia cells with high levels of dCK expression alongside low levels of SAMHD1 expression (10).
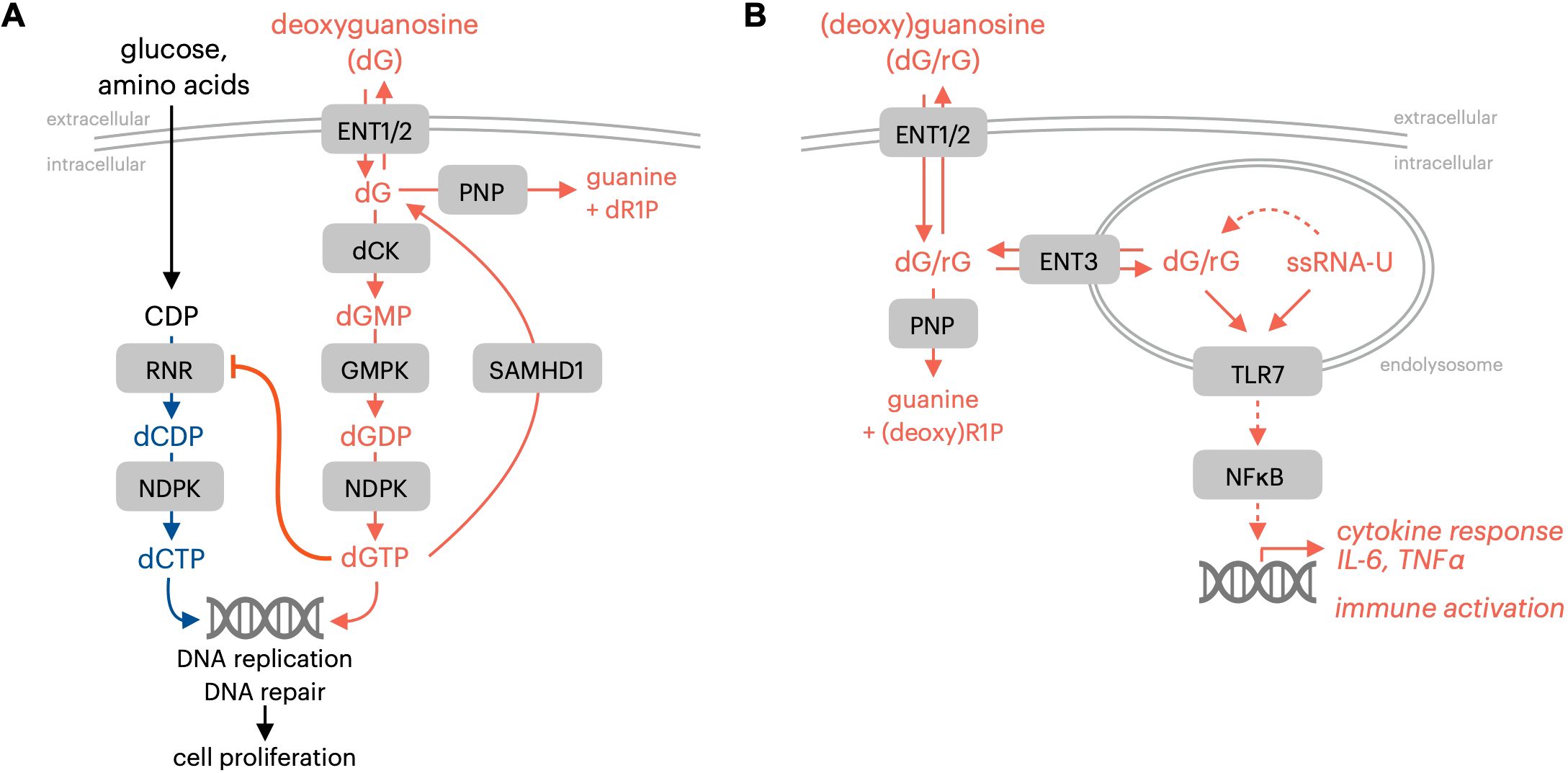
Figure 1. Immune-regulatory effects of guanosine nucleosides. (A) PNP and SAMHD1 prevent dGTP-mediated proliferation inhibition resulting from impaired dCTP synthesis. (B) PNP limits TLR7 activation by initiating guanosine nucleoside breakdown. RNR, ribonucleotide reductase; NDPK, nucleotide diphosphate kinase; ENT1/2, equilibrative nucleoside transporter 1/2 (SLC29A1/2); dCK, deoxycytidine kinase; GMPK, guanosine monophosphate kinase; SAMHD1, SAM and HD domain-containing protein 1; PNP, purine nucleoside phosphorylase; ENT3, equilibrative nucleoside transporter 3 (SLC29A3); TLR7, toll-like receptor 7; ssRNA-U, uridine-containing single-stranded RNA; dR1P, deoxyribose-1-phosphate; rG, guanosine; dG, deoxyguanosine.
The autoimmune manifestations related to PNP deficiency in humans are not explained solely by the cell-autonomous lethality that results from the intracellular expansion of purine dNTP pools and the resulting impairment of pyrimidine nucleotide synthesis. Recent research indicates that autoimmune consequences of PNP deficiency may be linked to the sensing of PNP substrates guanosine and deoxyguanosine by the endolysosomal pattern recognition receptor toll-like receptor 7 (TLR7; Figure 1B) (20). The toll-like receptor (TLR) family of proteins is a membrane-bound subset of pattern-recognition receptors that are responsible for sensing pathogens and initiating protective immune responses (21). TLRs are vital mediators of innate immune responses that detect pathogen-associated molecular patterns (PAMPs) and subsequently trigger a signaling cascade to activate cytokine production and stimulate immune responses (22). Endolysosomal TLR7 possesses two ligand binding sites that recognize either guanosine nucleosides or single-stranded uridine-containing ssRNA. Gain-of-function TLR7 mutations that result in enhanced guanosine sensing have been identified in patients with early onset systematic lupus erythematosus (23). This observation provided functional evidence for the role of guanosine nucleosides in regulating TLR7 activity in humans (23). Engineered mouse models with TLR7 mutations that increase its binding affinity for guanosine exhibit altered B and T cell function alongside autoimmune manifestations that recapitulate the clinical observations (23).
The effects of acute PNP inhibition on TLR7-mediated immune responses have been reported by multiple groups who have characterized immunological alterations in preclinical models triggered by PNP inhibitors (10, 24). The elevated systemic levels of guanosine nucleosides resulting from pharmacological PNP inactivation impact the function, proliferation, or survival of specific immune lineages as a function of their expression of SAMHD1, dCK, and TLR7. PNP inactivation promotes the expansion of germinal center B cells and populations of T follicular helper cells within secondary lymphoid tissues in the absence of exogenous antigen (10). PNP inhibitor-stabilized guanosine also triggers the production of inflammatory cytokines, such as IL-6 and TNFα, within TLR7-expressing macrophage populations when administered alongside single-stranded uridine-containing RNA (10).
A third manifestation of PNP deficiency is neurological alterations. Preclinical evidence highlights the critical role of PNP activity in neuron survival. The differentiation of induced pluripotent stem cells (iPSC) from PNP-deficient patients provided a platform to investigate the role of PNP in neurons (25). PNP deficiency is associated with enhanced p53-dependent intrinsic apoptosis in this setting, and RNR dysfunction was implicated as a mechanism underlying this effect. PNP also enables the utilization of inosine as a fuel for the pentose phosphate pathway, which has been implicated in the control of neuron function (26). In patients with partial PNP activity, neurological development was found to be normal (15).
Neurological manifestations are also produced by genetic defects in HPRT, a gene that functions downstream of PNP in the purine salvage pathway. Lesch-Nyhan syndrome, caused by the near-total impairment of HPRT, disrupts the synthesis of GMP and IMP nucleotides from guanine and hypoxanthine via the purine salvage pathway (27). HPRT deficiency blocks purine salvage, but spurs increased de novo pathway synthesis (27). Diminished guanosine salvage may alter the function of GTP-based secondary messenger systems operating in the central nervous system (27). The guanosine nucleotide GTP, which can be produced via the salvage of PNP products, functions in developmental neurology and controls cell migration, dendrite formation, and neurite outgrowth (28). Guanosine is also a regulator of glutamate re-uptake in glial cells. Therefore, altered guanosine metabolism could impact glutamatergic signaling (29). The phenotypes associated with PNP deficiency and HPRT deficiency reinforce the critical role of purine nucleoside salvage in a neurological context.
In summary, preclinical and clinical studies have pinpointed the role of the nucleoside PNP substrates as critical regulators of immune system function. These effects of guanosine (deoxy)ribonucleosides are related to their roles as (i) ligands for the intracellular pattern recognition receptor TLR7, (ii) substrates for dCK and subsequently fuel for dGTP synthesis, and (iii) substrates for the purine salvage pathway mediated by the sequential actions of PNP and HPRT.
3 Immune-regulatory functions of pyrimidine nucleosides
Recent preclinical studies have provided insights into the mechanisms underlying the immune-regulatory functions of pyrimidine nucleosides. Similar to guanosine, the function of pyrimidine nucleosides in immune system regulation involves their roles as metabolic precursors and as TLR ligands. Pyrimidine ribonucleotides, such as (deoxy)cytidine, uridine, and thymidine, can be produced by convergent de novo and salvage pathways in cells, and this redundancy allows for metabolic plasticity and adaption to alterations in the availability of environmental nutrients (30, 31). The de novo pathway utilizes glucose, glutamine, and aspartate precursors in a six-step biochemical process to produce pyrimidine nucleotides (32). An alternative salvage metabolic pathway for pyrimidine nucleotide synthesis utilizes preformed nucleosides and deoxyribonucleosides (dN) from the extracellular environment. Nucleotide synthesis via the salvage pathway requires the transport of pyrimidine nucleosides across the plasma membrane by nucleoside transporter proteins and their subsequent phosphorylation by intracellular nucleoside kinases (33, 34).
T cell activation is accompanied by the upregulation of multiple genes in the pyrimidine salvage pathway, including nucleoside transporters and the deoxyribonucleoside kinase dCK. This observation prompted the development of approaches that leverage enhanced nucleoside salvage pathway function in activated lymphocytes to non-invasively track immune responses. Radu and colleagues developed [18F]FAC, a pyrimidine deoxyribonucleoside-analog positron emission tomography (PET) probe, to visualize dCK activity as a surrogate marker for immune activation in preclinical mouse models and in humans (35). Following administration, deoxycytidine analog PET probes are transported into cells via nucleoside transporter proteins and are phosphorylated by the pyrimidine deoxyribonucleoside kinase dCK, which effectively traps the probe within cells with elevated dCK activity (35). The biodistribution of deoxycytidine-analog PET probes in preclinical models or in humans can be tracked using a PET scanner. Early studies testing deoxycytidine-analog PET probes in mouse models revealed a striking concentration of pyrimidine deoxyribonucleoside salvage in lymphoid tissues such as the spleen, lymph nodes, thymus, and bone marrow (35). This finding indicated enhanced pyrimidine salvage activity in immune cells in vivo. Prompted by this observation, a series of studies in engineered mouse models were performed to evaluate the functional role of dCK by evaluating immune phenotypes in mice where dCK was deleted. These studies provided evidence critically linking dCK function to hematopoiesis and lymphocyte proliferation. The analysis of dCK knockout mice highlighted a requirement for dCK in the development of multiple immune cell lineages, including CD4/CD8 T cells in the thymus, B cells, and erythrocytes (36). The requirement for dCK-mediated dC salvage in normal murine hematopoiesis was, in part, traced to a requirement for dCK to counteract the toxicity resulting from high levels of thymidine in specific tissues (Figure 2A) (37). Thymidine is phosphorylated and trapped in cells by thymidine kinase 1 (TK1), and high levels of environmental thymidine drive the expansion of intracellular thymidine triphosphate (dTTP) nucleotide pools. The unbalanced expansion of dTTP pools inhibits dCDP synthesis by RNR, which results in the depletion of dCTP and replication stress in the S-phase of the cell cycle. dCK-mediated dC salvage bypasses this metabolic block to enable proliferation under conditions of high environmental thymidine (37). Preventing thymidine salvage and dTTP pool expansion in dCK knockout mice by inhibiting TK1 prevents replication stress in thymocytes and restores T cell development (37). This data suggests that, in mouse models, dC and dT have major roles in immune cell development and proliferation by functioning as substrates of nucleoside salvage kinases. Similar to the consequences of elevated guanosine nucleoside abundance in PNP deficiency, elevated levels of dT nucleosides restrict hematopoiesis by cell-autonomous lethality. In contrast, dC itself does not appear to exert deleterious effects in hematopoiesis.
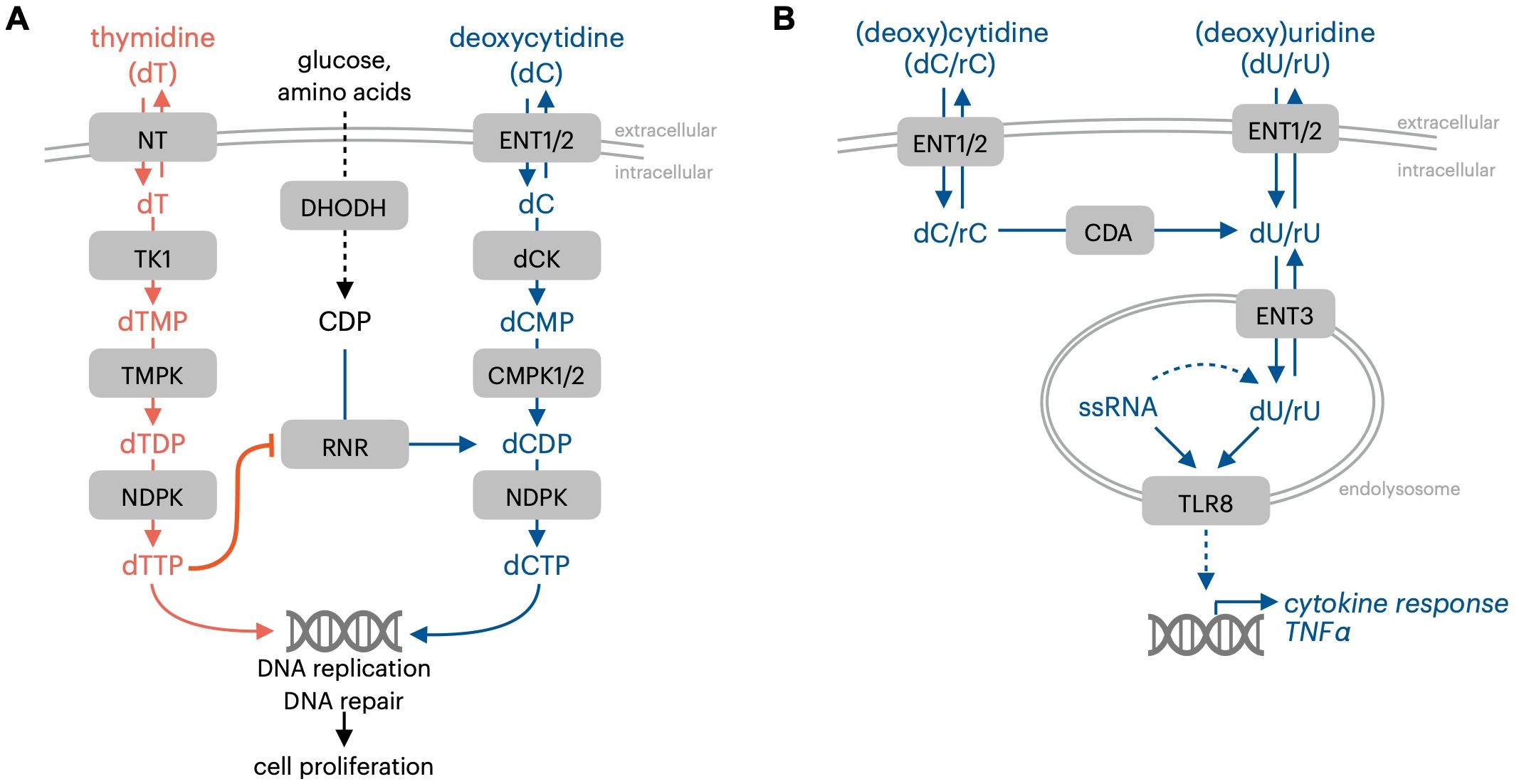
Figure 2. Immune-regulatory effects of pyrimidine nucleosides. (A) dCK and dC prevent thymidine-mediated proliferation inhibition resulting from impaired dCTP synthesis. (B) Pyrimidine nucleosides are TLR8 ligands. NT, nucleoside transporter TK1, thymidine kinase 1; RNR, ribonucleotide reductase; NDPK, nucleotide diphosphate kinase; ENT1/2, equilibrative nucleoside transporter 1/2 (SLC29A1/2); dCK, deoxycytidine kinase; TMPK, thymidine monophosphate kinase; CMPK1/2, cytidine monophosphate kinase 1/2; CDA,cytidine deaminase; ENT3, equilibrative nucleoside transporter 3 (SLC29A3); TLR8, toll-like receptor 8; ssRNA, single-stranded RNA; rC, cytidine; dC, deoxycytidine; rU, uridine; dU, deoxyuridine.
In addition to their role as substrates for the nucleoside kinases, nucleosides can be broken down and the resulting ribose can substitute for glucose under conditions of nutrient scarcity. Pancreatic ductal adenocarcinoma (PDAC) cells are resilient, resistant to treatment, and able to thrive in hostile environments by utilizing the pyrimidine nucleoside uridine (180). In low-glucose conditions, uridine phosphorylase 1 (UPP1) is over-expressed, driving the use of uridine as a carbon source that supports macromolecule synthesis and energy generation (180). In cancer cells, UPP1 is controlled by oncogenic KRAS-MAPK signaling and induced by nutrient restriction. Similarly, other nucleosides have been shown to serve as alternative carbon sources, including inosine in CD8 T cells (181) and dT in cancer cells (182).
Altered de novo pyrimidine synthesis in humans is associated with immune alterations in the context of the disorder Hereditary Orotic Aciduria (HOA) (38, 39). This rare condition results in defective pyrimidine nucleotide synthesis and is the only identified enzyme deficiency of the de novo pyrimidine biosynthetic pathway in humans. HOA is linked to mutational inactivation of uridine-5-monophosphate synthase (UMPS), which leads to decreased pyrimidine synthesis and increased excretion of orotic acid (39, 40). HOA was described as early as 1959 in an infant who passed away before a full investigation could be performed (41). Clinical evidence supports the notion that impaired pyrimidine synthesis results in immunodeficiency, suggesting that pyrimidine nucleotides have immune-strengthening effects. HOA is associated with weakened T cell responses, while humoral responses remain undamaged (38). Symptoms vary across HOA cases but have been reported to include megaloblastic anemia, a weakened immune system, delays in development, and failure to thrive (39). Some symptoms can be treated by supplementation with the pyrimidine nucleoside uridine administered as uridine triacetate.
Pyrimidine nucleosides have roles as metabolic precursors for nucleotide synthesis, and as direct signaling mediators. The endolysosomal pattern recognition receptor Toll-like receptor 8 (TLR8) mediates the signaling effects of pyrimidine nucleosides and possesses a binding pocket that accepts uridine (Figure 2B) (42). The nucleoside and oligonucleotide ligands for TLR8 are produced within the lysosomal compartment via RNA breakdown (43). Cytidine and deoxycytidine can be converted to uracil-containing nucleoside TLR8 ligands by cytidine deaminase (CDA) (44). The accumulation of nucleosides within endolysosomal compartments, where they are sensed by TLR7 or TLR8, is mediated by their transport across the endolysosomal membrane by equilibrative nucleoside transporter 3 (ENT3, encoded by the gene SLC29A3). Aberrant endolysosomal TLR signaling resulting from defective SLC29A3-mediated nucleoside transport is linked to H syndrome, an auto-inflammatory condition in humans (45).
Defective activity of the genes controlling the breakdown of pyrimidine nucleosides or nucleobases is linked to the development of various human pathologies. Dihydropyrimidine dehydrogenase (DPYD) catalyzes the first step of uracil and thymine degradation (46). DYPD deficiency (DPD) is a rare metabolic disorder resulting in seizures, developmental delay, microcephaly, and muscular hypotonia, although some patients who are carriers are asymptomatic (47). The activity of other genes and potential environmental factors likely dictate the severity of the manifestations of DPD, causing some individuals to be asymptomatic while others have life-altering manifestations (47). DPD is observed in approximately 3-5% of the population, but the prevalence and phenotypic manifestations vary across ethnic groups (48). DPYD is responsible for the catabolism of 80% of bodily 5-Fluorouracil, a chemotherapy widely used for cancer treatment (49). Therefore, 5-Fluorouracil cannot be used as a cancer treatment for those with DPD, as drug accumulation leads to toxic depletion of pyrimidine nucleotides in this patient population (46).
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is linked to heritable loss-of-function mutations in the gene encoding for thymidine phosphorylase (TYMP). TYMP is a pyrimidine nucleoside phosphorylase that regulates pyrimidine nucleoside salvage by controlling the breakdown of thymidine and deoxyuridine. MNGIE is linked to altered systemic pyrimidine nucleoside accumulation, large-scale disruption of nucleoside metabolism, alongside halted cholesterol, and fatty acid breakdown (50). TYMP deficiency in humans results in the accumulation of thymidine and deoxyuridine, which results in imbalanced nucleotide pools within mitochondria, disruption of mitochondrial DNA replication, and increased mutations. The resulting altered mitochondrial function underlies the manifestations of MNGIE, which include eye muscle weakness, muscle wasting, leukoencephalopathy, digestive dysmotility, microangiopathy, and occasional psychiatric symptoms (50–52). TYMP dysfunction is linked to nucleoside accumulation within lysosomes and the disruption of lysosomal transport proteins. However, it is not clear if any of the manifestations of TYMP deficiency are associated with altered endolysosomal nucleoside sensing by TLR7 or TLR8 (53). Mitochondrial pyrimidine nucleotide imbalance is also linked to the production of immuno-stimulatory type I interferon by triggering release of mitochondrial DNA to the cytosol and downstream cGAS/STING pathway activation (183). The mitochondrial protease YME1L preserves mitochondrial nucleotide pools by preventing pyrimidine nucleotide release to the cytosol via degradation of the mitochondrial nucleotide carrier SLC25A33.
4 Immune-regulatory functions of adenosine nucleosides
The nucleoside adenosine is a potent regulator of anti-tumor immune responses that weakens beneficial immune cell subsets and strengthens suppressive cell populations. The role of adenosine in immune system regulation is multifaceted and linked to its role as a signaling molecule and metabolic precursor. Tumor cells co-opt the immunosuppressive effects of adenosine to dampen immune responses by up-regulating the key metabolic enzymes responsible for its production. Therapies that block adenosine generation or sensing have emerged as promising therapeutic targets to reverse immunosuppression in the tumor microenvironment. The mechanisms underlying the effects of adenosine on immune system function and the landscape of therapies targeting the adenosine pathway have been reviewed (1).
Adenosine deaminase activity in humans is mediated by enzymes ADA1 and ADA2. Deficiency in ADA1 results in SCID, while patients with deficiency in ADA2 (DADA2) exhibit a variable clinical phenotype, including systemic inflammation, vasculopathy/vasculitis, and aplastic anemia, with dysregulation of immune, neural, and cardiovascular systems (54, 55). ADA1 does not compensate for dampened ADA2 activity in DADA2 patients. This difference in phenotype resulting from ADA1 or ADA2 deficiency is multifaceted and is linked to differential binding of soluble ADA1 or ADA2 proteins to immune cells (56).
One mechanism by which extracellular adenosine exerts immune-modifying effects is by activating specialized cell-surface receptors which control cell fate and function that are expressed across immune cell lineages. Multiple cell surface receptors for adenosine (A1, A2A, A2B, and A3) have been characterized (57). A2A is expressed across immune cell types and is well-studied for its role in mediating the immunosuppressive effects of adenosine in the context of anti-cancer immunity. Signaling downstream of A2A is known to exert pleiotropic immune-suppressive functions across immune lineages present in the tumor microenvironment (58, 59). The signaling effects of adenosine have been harnessed for therapy, and synthetic antagonists of adenosine receptors such as vipadenant (BIIB-014) and ST-1535 have shown signs of efficacy in clinical trials for Parkinson’s disease and other conditions (60, 61). Istradefyline, an A2A antagonist, is approved in Japan for Parkinson’s treatment (62). Adenosine signaling via A2A promotes the production of inflammatory cytokines and sustains inflammasome activation following initial activation (63).
The adenosine receptor A2B is also over-expressed in specific cancers (64). It is a low-affinity adenosine receptor compared to A2A and is activated in conditions of high environmental adenosine. It regulates the function of various cell types, including immune and stromal cells, and its inhibition suppresses the growth of tumors in mice (65). The small molecule A2B inhibitor PSB1115 blocks cytokine signaling in stromal cells to limit tumor growth in mouse models (64).
The production of extracellular adenosine in the tumor microenvironment is linked to poor patient outcomes and is driven by high expression of the ectonucleotidases CD39 (ENTPD1) and CD73 (NT5E) (57). CD39 generates AMP from ATP, and CD73 converts AMP into adenosine (66). The expression of adenosine-generating ectonucleotidases is positively regulated by hypoxia and inflammation in the tumor environment (57). The collective preclinical data suggests that therapies blocking CD39 and CD73 could help decrease adenosine production in tumors, thereby unleashing anti-cancer immune responses. This treatment strategy is supported by research testing the consequences of CD39 and CD73 inhibition in preclinical cancer models (57). Prostatic acid phosphatase (PAP) generates adenosine via the breakdown of AMP and may be responsible for immunosuppressive adenosine signaling in prostate cancer tumors via a metabolic pathway that bypasses CD73 (67).
Several approaches for CD73 inhibition are under clinical evaluation as strategies for immunotherapy in patients with solid tumors. Both monoclonal antibodies (68) and small molecule therapeutics (69) that block CD73 activity elicit anti-tumor immune responses and restrain metastasis in murine cancer models. Oleclumab is a CD73-targeting antibody currently under clinical investigation in multiple phase I and II trials and has exhibited promising signs of anti-tumor efficacy (70). Other CD73-targeting antibodies, CPI-006, SRF373/NZV930, and BMS-986179, as well as CD73-targeting small molecules, such as quemliclustat (AB680), are also under clinical evaluation (57).
The anti-cancer effects of preventing adenosine generation using CD39 blocking antibodies have been evaluated in murine models with success (71). POM-1 has been proven as an effective small-molecule CD39 inhibitor that increases cytotoxic T and NK cell activity (72). CD39 is emerging as a therapeutic target to induce anti-cancer immune responses, whereas CD73 and adenosine receptor inhibitors have a more substantial history of research focus (73). Nevertheless, multiple CD39 antagonistic antibodies are undergoing clinical investigation: AB598, TTX-030, IPH5201, and SRF-617, with more in development (74).
Ecto-nucleotide pyrophosphatase/phosphodiesterase (ENPP1) is an additional extracellular enzyme of interest in relationship to adenosine signaling as it degrades purine nucleotides, promoting adenosine production. ENPP1 is connected to the up-regulation of immunosuppressive adenosine signaling and is involved in the breakdown of ATP to AMP via a mechanism that parallels the activity of CD39 (75). ENPP1 also produces AMP via the hydrolysis of the cyclic dinucleotide immuno-transmitter 2’-3’-cGAMP, produced by the enzyme cGAS (76). Potent small-molecule ENPP1 inhibitors such as STF-1623/CM-3163 and AVA-NP-695 are being developed with the potential for cancer therapy (77, 78). Antibodies that target ENPP1 to prevent its enzymatic activity have been developed and tested in preclinical models of myocardial infarction to limit the cell death and fibrosis that is linked to increased ENPP1 activity following cardiac injury (79, 80).
Adenosine can be produced in the extracellular environment by the breakdown of nucleotides released by dying cells via CD39, ENPP1, and CD73. It is also released by live cells via equilibrative nucleoside transporters. Inhibition of nucleoside transport is currently under investigation as an alternative approach to ectonucleotidase inhibition to limit immunosuppressive adenosine signaling for cancer immunotherapy (81).
The purine nucleoside inosine, a product of adenosine deamination via ADA, has also been linked to cancer progression and metastasis, acting as a precursor for nucleotide synthesis in the tumor microenvironment during starvation (82, 83). In addition, inosine regulates the phenotype of T cells (84). One emerging tactic to leverage the immune-modifying properties of inosine is to improve CAR-T therapy by over-expressing ADA to promote the conversion of immunosuppressive adenosine to inosine. This approach increases the functionality and stem cell-like properties of CAR-T cells, which amplifies their anti-cancer effects (84).
The systemic inflammatory condition known as Still’s disease is spurred by genetic loss-of-function mutations in the gene FAMIN, which encodes an enzyme with a roles in adenosine, guanosine and inosine metabolism as well as the prevention of autoimmunity and pathogenic T cell activation. Still’s disease manifests in childhood, with recurrent fevers and arthritis being the most common phenotypes, although 20% of those with the condition develop macrophage activation syndrome. The enzymatic function of FAMIN overlaps with that of ADA, PNP and MTAP. Compromised FAMIN function in dendritic cells is linked to aberrant NAD/NADH metabolism antigen presentation, and inosine metabolism that together contribute to enhanced T cell priming (179).
5 Roles of nucleoside transporters in immune regulation
Systemic nucleoside abundance is tightly controlled by proteins that regulate nucleoside production, breakdown, and translocation across plasma membranes (85). Nucleoside uptake and release in live cells is mediated by specialized multi-pass transmembrane transporter proteins (85). In addition to their role in controlling the systemic levels of nucleosides, nucleoside transporters enable the nucleoside salvage pathway for nucleotide synthesis in cells. Equilibrative nucleoside transporters 1 and 2 (ENT1/2, encoded by the genes SLC29A1/2) mediate the passive transport of nucleosides across the plasma membrane along a concentration gradient, whereas concentrative nucleoside transporters CNT1 and CNT2 (encoded by the genes SLC28A1/2) mediate the sodium-coupled secondary active transport of nucleosides (86). The ENT family member SLC29A3 (ENT3) is located on the lysosomal membrane within cells and mediates the transfer of nucleosides across intracellular compartments (87). SLC29A4 (ENT4) functions as a plasma membrane polyamine transporter (88, 89).
Nucleoside transporters accept various substrates, including natural pyrimidine and purine nucleosides, synthetic anti-metabolite nucleoside analogs (such as gemcitabine, cytarabine, and clofarabine), and radionuclide-labeled nucleoside-analog PET imaging probes (such as [18F]FAC) (85). Cancer cells lacking nucleoside transporter activity are resistant to nucleoside-analog prodrugs (90). SLC29A1 (ENT1) is the predominantly expressed nucleoside transporter across normal and tumor cells and facilitates the utilization of environmental nucleosides for nucleotide synthesis. Beyond their ability to provide metabolic precursors to cells, nucleoside transporters also control the access of nucleosides to their sensors (such as adenosine receptors, TLR7, and TLR8). ENT1 mutations have been identified in human patients. The manifestations of impaired ENT1 activity in humans include ectopic mineralization, joint calcification, and dysregulated erythropoiesis (94, 95). The Augustine blood group system includes antigens encoded by various SLC29A1 alleles (96).
ENT1 mediates the immune-regulatory effects of adenosine in part by controlling its uptake in lymphocytes. Adenosine uptake is linked to pyrimidine synthesis inhibition via phosphoribosyl pyrophosphate synthetase (PRPS) and a resulting proliferation block in tumor-infiltrating T cells (91). Therefore, pharmacological ENT1 inhibition has been suggested as a strategy to enhance anti-cancer T cell responses and ENT inhibitors have emerged as a rational companion therapy for immune checkpoint blockade. Nucleoside transporters also have a critical role in dictating the immunological outcomes driven by guanosine nucleosides as their transport across the plasma membrane is mediated by ENTs (92, 93). The anti-proliferative effects of guanosine supplementation in culture are curbed by ENT1 inhibition.
Inactivation of lysosomal membrane nucleoside transport resulting from mutations in ENT3 is associated with hyperactive immune phenotypes in humans with genetically inherited disorders (97). ENT3 controls the abundance of guanosine and uridine nucleosides within lysosomes, which function as ligands for TLR7 and TLR8, respectively. Lymphocytes can use ENT3-mediated nucleoside recycling to support nucleic acid synthesis and sustain proliferation (97). ENT3 deficiency results in dysregulated nucleoside transport across lysosomal membranes, leading to nucleotide pool imbalance, metabolic stress, and aberrant TLR-driven cytokine responses (45). Germline loss-of-function SLC29A3 (encoding for ENT3) mutations in humans are notably associated with irregular histiocyte production and accumulation, causing autoimmune responses presenting as the genetic disorder H syndrome (98). Cases of H syndrome are rare, with patients presenting with pigmented hypertrichosis with insulin-dependent diabetes mellitus (PHID), Faisalabad histiocytosis, and sinus histiocytosis with massive lymphadenopathy (45, 97, 99, 100). These phenotypes are potentially linked to aberrant macrophage activation and accumulation in the spleen and other organs due to nucleoside accumulation in lysosomes and subsequent TLR7 or TLR8 activation (45, 101). Interestingly, while the manifestations of ENT3 deficiency in mice appear to be driven by aberrant TLR7 signaling, the consequences of ENT3 deficiency in human-derived cells are mediated by TLR8 (45).
6 Emerging therapeutic strategies to unleash the immune-stimulatory effects of nucleosides
6.1 PNP inhibition
Decades after the initial observations of T cell insufficiency in patients with ADA or PNP-linked SCID by Giblett and colleagues, their discovery was leveraged for therapy. Low PNP activity in humans is associated with decreased T cell counts, making T cell malignancies a natural place to examine the benefit of pharmacological PNP inhibition (102). PNP inhibition was hypothesized to selectively elevate deoxyguanosine levels in malignant T cells, leading to dGTP accumulation and cancer cell death (102, 103). In the 1990s, Schramm and colleagues applied their knowledge of the PNP transition state substrate-enzyme structure to design PNP inhibitors with exceptionally high potency (104, 105) (Table 1). Pharmacological PNP inhibition was found to selectively eradicate T cell leukemia cells in vitro, thus mirroring the observations of T cell deficiency in patients with PNP-linked SCID (105). These encouraging preclinical results prompted the testing of the PNP inhibitor forodesine (also known as BCX-1777 or Immucillin H) in clinical trials for relapsed/refractory T and B cell leukemias and lymphomas (106). Forodesine received approval for treating peripheral T cell lymphoma in Japan in 2017 (102, 107). Despite excellent tolerability and pharmacodynamic properties in humans, evidenced by plasma accumulation of PNP substrates and depletion of the downstream products of PNP (including uric acid), durable responses were observed only in a subset of patients. Additional PNP inhibitors, ulodesine and peldesine, with potency and bioavailability comparable to forodesine, have entered clinical trials for applications beyond cancer treatment, such as arthritis or limiting uric acid accumulation in gout (108). It has been noted that PNP inhibitors have lower efficacy against cancer cells in vivo compared to cell culture experimentation. This discrepancy may be due to the presence or absence of factors not accounted for in cell culture models or specific genetic differences of the cancer cells targeted in each study (106).
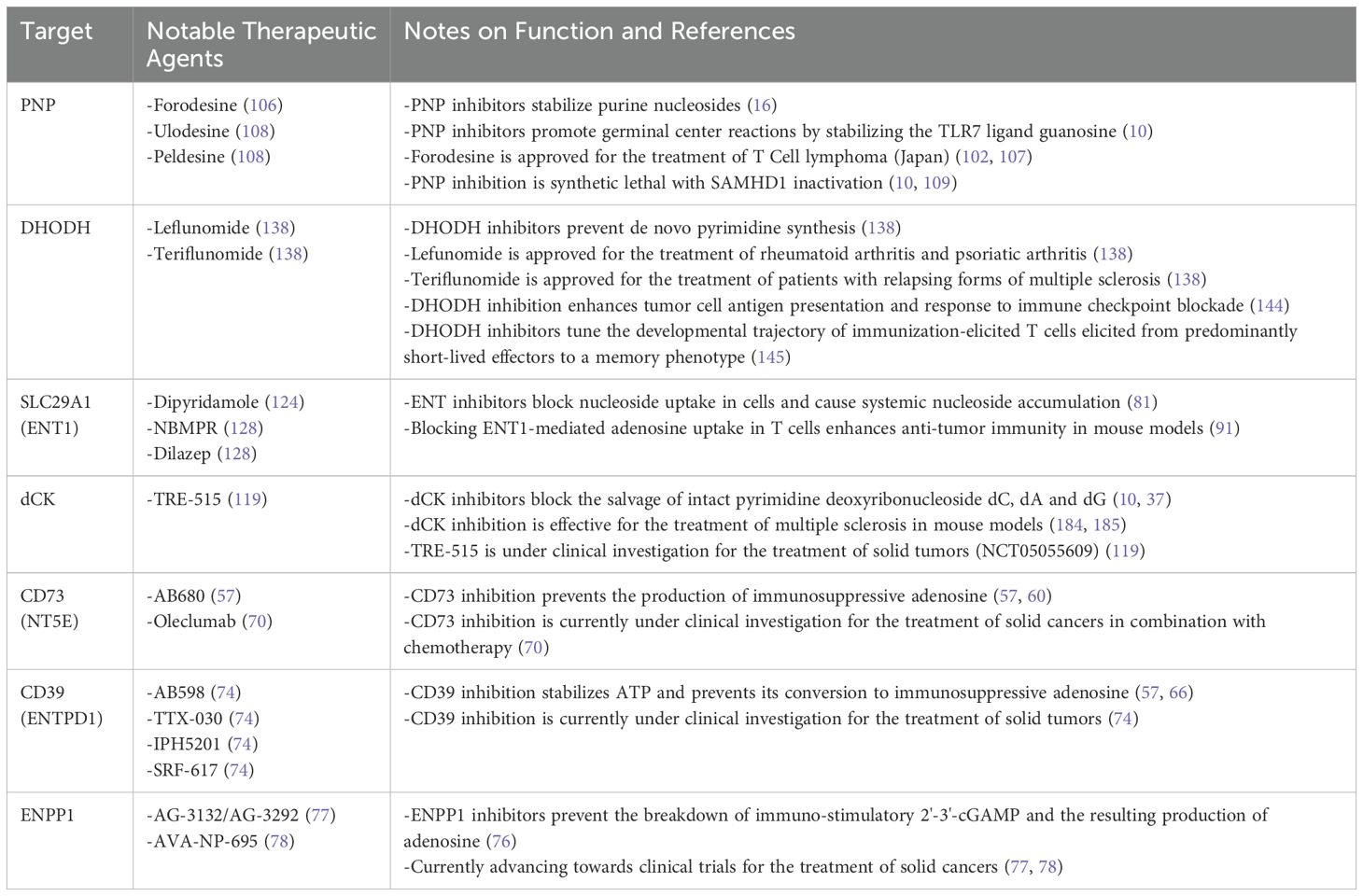
Table 1. Landscape of therapeutic targets for immune modulation within nucleoside metabolism.
PNP inhibitors appear to be most effective in inducing apoptosis in cancer cells deficient in the dNTP triphosphohydrolase SAMHD1 (10, 109). SAMHD1 degrades dNTPs to their corresponding nucleosides and prevents the expansion of intracellular dNTP pools (110). When challenged with PNP inhibitors, human and mouse cells without SAMHD1 are eradicated, while cells expressing SAMHD1 survive, indicating that SAMHD1 and PNP are a pair of synthetic lethal genes (109). SAMHD1 has, therefore, emerged as a crucial biomarker for the anti-cancer effects of PNP inhibitors. This insight has increased the potential clinical utility of PNP inhibitors by providing the rationale for treating solid tumors with low SAMHD1 expression (105). Loss-of-function SAMHD1 mutations occur in several cancer types, including lymphocytic leukemia, lung, and colon cancer (105, 111). SAMHD1 expression is controlled in part by transcriptional upregulation downstream of signaling driven by the cytokine interferon (112). PNP is up-regulated in certain cancers, such as pancreatic adenocarcinoma, where it may be a therapeutic target (113).
While PNP inhibitors were initially applied to eradicate malignant lymphocytes, immune-activating effects were noted in patients receiving this new type of treatment. These effects included enhanced responses to vaccines and beneficial effects in the context of post-HSCT relapse in patients with leukemia (114). Based on these observations, it was hypothesized that the immune stimulatory properties associated with PNP inhibitor treatment may result from the activation of immune sensor molecules such as TLRs. Consistent with this model, recent studies have indicated that the PNP substrates guanosine and deoxyguanosine activate TLR7.
Oral treatment with PNP inhibitors triggers transcriptional alterations in B cells, dendritic cells, and macrophages via TLR7 activation (10, 24). The transcriptional alterations in macrophages driven by PNP inhibitors, resulting from an accumulation of the endogenous TLR7 ligand guanosine, are distinct from those elicited by synthetic guanosine-analog TLR7 agonists, such as R848. An advantage for PNP inhibitors over synthetic agonists for therapeutic TLR7 agonism is the difference in the duration and magnitude of cytokine responses elicited by either therapy. While synthetic TLR7 agonists trigger an acute, transient high-level of TLR7 activation, PNP inhibitors may induce a lower level of activation but a long-lived response resulting from the sustained systemic accumulation of the purine nucleoside TLR7 ligands. Targeting the PNP-regulated immune checkpoint in patients may enhance anti-tumor immune responses or vaccine-driven humoral and T cell responses.
6.2 Nucleoside salvage kinase inhibition
The development of selective, potent, and orally bioavailable dCK inhibitors was guided by structural analysis and preclinical imaging studies that leveraged dCK-specific PET probes (115–119). The dCK inhibitor DI-87 (TRE-515) was developed to eradicate pathogenic cell types that rely on dCK activity (119). dCK inhibitors are well-tolerated in preclinical models and have minimal effects on normal cells. The first-in-class dCK inhibitor TRE-515 is currently under clinical investigation for the treatment of solid tumors (NCT05055609).
dCK inhibition is a promising anti-cancer treatment strategy, as tumor cells exhibit an increased demand for pyrimidine nucleotide synthesis to fuel DNA replication and repair. dCK inhibitors trigger replication stress alongside lethal DNA damage in tumor cells, and improve survival in mouse models of acute lymphoblastic leukemia (ALL) when administered alongside inhibitors of de novo dCTP synthesis, such as thymidine or triapine (117, 120). As a mono-therapy, dCK inhibitors may be most effective for treating specific tumors that exhibit a diminished capacity for de novo pyrimidine nucleotide synthesis resulting from transcriptional suppression, mutational inactivation, or nutrient scarcity (119).
Based on their strong safety profile and unique mechanism of action, dCK inhibitors are a promising companion for established treatments that induce DNA damage or restrict de novo pathway activity in tumor cells. dCK mediates radiation resistance by supplying the pyrimidine dNTP precursors needed for DNA repair (121). dCK inhibitors also have potent anti-cancer effects against cells deficient for the tumor suppressor gene BRCA2 (122). Therefore, dCK inhibitors are potentially a high-priority companion therapy for PARP inhibitors for this genetically-defined cancer type.
Preclinical observations of altered immune phenotypes in dCK knockout mice prompted the testing of a dCK inhibitor for treating autoimmune diseases. In this setting, dCK inhibitors may block a selective requirement of disease-driving lymphocyte populations on enhanced dCK activity while sparing normal cells that utilize the de novo pathway to satisfy their dNTP requirements. dCK inhibitors have demonstrated the potential to mitigate the manifestations of multiple sclerosis in mouse models, and dCK-specific PET probe accumulation has been proposed as a potential non-invasive biomarker for these inhibitors in patients (37, 123, 184, 185). Based on promising preclinical data, dCK inhibitors are progressing toward clinical development to curtail aberrant immune activation.
6.3 Nucleoside transport inhibition
Nucleoside transport across the plasma membrane is a critical step for nucleotide synthesis via salvage pathways and controlling the nucleoside levels in the extracellular environment. ENT inhibition is currently under evaluation as an approach to limit immunosuppressive adenosine signaling in tumors (81). Targeting ENT1 may enhance T cell-mediated tumor cell killing by (i) limiting the release of adenosine by tumor cells to prevent adenosine receptor signaling, and (ii) blocking the anti-proliferative effects resulting from adenosine uptake in immune cells. FDA-approved dipyridamole inhibits ENT1, effectively preventing adenosine uptake, particularly across inflammatory states with excessive adenosine production (124). The immune-stimulatory and anti-cancer effects of nucleoside transporter inhibition are also linked to the protection of tumor-infiltrating lymphocytes by preventing the pyrimidine de novo synthesis pathway defect triggered by excessive adenosine salvage (91).
Dipyridamole has cellular targets beyond ENT1. Therefore, its clinical utility in cancer immunotherapy is limited. NBMPR is a potent ENT1 inhibitor but has not been used directly as an anti-cancer therapeutic (127). The crystal structures of ENT1 in complex with two established inhibitors of adenosine re-uptake, NBMPR and Dilazep, have been solved (128), and this information may guide the development of new ENT inhibitors with improved target engagement and specificity that are suitable for clinical use. Nucleoside transport inhibitors may have anti-cancer effects when applied as a mono-therapy (125) and can potentially prevent resistance to DNA-damaging chemotherapeutics by limiting the synthesis of nucleotides via the salvage pathway that may support DNA repair (126).
6.4 TYMP inhibition
The link between MNGIE and altered TYMP activity prompted an investigation into the mechanisms linking thymidine metabolism to mitochondrial function in other contexts, such as cancer. TYMP up-regulation is associated with pro-tumor functions such as cancer cell proliferation, metabolic alterations, and increased angiogenesis (129). Many cancers utilize TYMP-mediated pathways to form 2-deoxyribose that can fuel biosynthetic processes (129). Therefore, blocking TYMP represents a potential anti-cancer treatment strategy. The TYMP inhibitor tipiracil hydrochloride (TPI) restrains basement membrane incursion to prevent metastasis and trigger apoptosis (130). In addition to their role in inhibiting pyrimidine salvage, TYMP inhibitors may have utility for restraining the production of the TLR8 ligand deoxyuridine from thymidine to limit uncontrolled immune responses.
6.5 MTAP inhibition
5’-methylthioadenosine (MTA) phosphorylase (MTAP) is an enzyme with a role in the metabolism of polyamine as well as the salvage pathway for the synthesis of adenine and methionine (131). MTAP degrades MTA into S-adenosyl-L-methionine (SAM) (132). The deleted form of the MTAP gene occurs in approximately 15% of cancers and has been linked to immune evasion (133, 134). MTDIA (Methylthio-DADMe-Immucillin-A) is an MTAP inhibitory molecule (133). In mouse models of lung and colorectal cancer, MTDIA therapy exhibited considerable anti-tumor effects, extending survival and reducing tumor growth (132, 135, 136). Unlike many other therapies, there is little high-dosage toxicity with MTDIA treatment, indicating that this therapy is suitable for extended use (132). When MTDIA is not administered, MTAP metabolizes MTA into adenosine and 5-methylthioribose-1-phosphate (MTR-1-P), allowing for cancer cell proliferation. When MTDIA is administered and MTAP is inhibited, PRMT5-mediated histone methylation and intron splicing are competitively decreased, resulting in the restraint of cancer growth (132).
6.6 DHODH inhibition
The increased requirement of activated lymphocytes on nucleotide synthesis has been leveraged therapeutically, as the inhibition of de novo pyrimidine nucleotide synthesis is an established treatment strategy for the management of autoimmune disorders (137). Inhibition of de novo pyrimidine synthesis using dihydroorotate dehydrogenase (DHODH) inhibitors is an FDA-approved approach to combat multiple sclerosis and rheumatoid arthritis (138). The DHODH inhibitor Leflunomide was approved in 1998 for treating rheumatoid arthritis. This was followed by the approval of Teriflunomide for multiple sclerosis in 2012. In these autoimmune disorders, pyrimidine synthesis-targeting drugs are administered to prevent the aberrant proliferation of immune cells that drive the autoimmune manifestations (139).
There is potential for DHODH inhibitors in cancer treatment, as DHODH has a central role in sustaining cancer cell proliferation and regulating anti-cancer immune activity. DHODH inhibition using small molecule drugs is effective for the treatment of preclinical cancer models such as small cell lung cancer (140), MYC-amplified medulloblastoma (141), and IDH1 mutant glioma (142). In addition to promoting nucleotide synthesis, DHODH activity strengthens cancer cells by providing defense against ferroptosis (143). DHODH inhibitors also reprogram myeloid differentiation, and this effect may be relevant for treating myeloid leukemias (137).
In mouse models, DHODH inhibition enhances the efficacy of immune checkpoint blockade using anti-CTLA-4 with anti-PD-1 antibodies by up-regulating the expression of antigen presentation pathway genes in cancer cells (144). The modulation of pyrimidine nucleotide synthesis using DHODH inhibitors also impacts T cells directly and has been shown to tune the developmental trajectory of immunization-elicited T cells elicited from predominantly short-lived effectors to a memory phenotype (145). The impact of nucleoside transport or salvage pathway inhibition on this process has yet to be defined.
6.7 Modified nucleoside therapies
While the structural basis for the sensing of guanosine by TLR7 has only recently been described, the immuno-stimulatory effect of small molecule guanosine analogs has been known for decades (146, 147). Guanosine analogs have immuno-stimulatory properties via the activation of TLR7, and guanosine-analog TLR7 agonists have been evaluated as a form of cancer immunotherapy (147). TLR7 activation by synthetic guanosine analogs bypasses the requirement for TLR7 binding to ssRNA. Guanosine derivatives, such as Loxoribine, have been developed as therapeutic agents to activate TLR7. This class of agonists initiate intracellular signaling cascade involving proteins such as p50 and p65, which drive the expression of pro-inflammatory cytokines (148, 149).
7 Lost in translation: differences between mouse and human metabolism is a significant obstacle in the preclinical study of the immune-regulatory functions of nucleosides
The disparities between mouse and human nucleoside metabolism limit the translational impact of the promising results obtained from experiments that use mouse models (150). These significant differences may produce confounding results and hinder the translation of new therapeutics. For example, pyrimidine deoxyribonucleoside concentrations are measured at levels that are orders of magnitude higher in rodent plasma than in humans (2, 151, 152). The variation in the systemic levels of nucleosides across species is related to differences in the expression and activity of enzymes involved in nucleoside breakdown. Distinct diet and behavioral patterns may also contribute to these differences.
The discrepancy in the expression and activity of nucleoside catabolism-related genes across species is a central contributor to the differences in the measured levels of systemic nucleosides. Mice are deficient for the enzyme ADA2 (encoded by the gene CECR1), which catalyzes the conversion of (deoxy)adenosine to (deoxy)inosine and has a ~100-fold lower affinity for free adenosine nucleosides than ADA1. ADA2 is broadly expressed in human cell types and is reported to function within endolysosomes to regulate TLR9 signaling with DNA as its primary substrate (153, 154). This cross-species metabolic incongruence complicates the extension of findings in mouse models regarding the links between adenosine deamination and immune activation in the human setting. The disconnect between human and mouse models is also highlighted by research involving the adenosine-generating enzyme CD73. CD73 deficiency in humans is associated with calcification of small joints, vascular calcification, and arteriomegaly; in contrast, CD73-deficient mice do not exhibit an apparent phenotype (155).
Studies of the bio-distribution of deoxyribonucleoside-analog PET probes across mice, dogs, non-human primates, and humans reinforce the differences in nucleoside metabolism across species. The thymidine analog PET probe [18F]FLT exhibits no specific tissue accumulation pattern in rodent models (156, 157). However, in humans, this probe accumulates in tumors and secondary lymphoid tissues characterized by high levels of cell proliferation and TK1 expression. One factor underlying this difference is differential systemic levels of plasma thymidine concentrations across mice and humans (2). Both thymidine and [18F]FLT require transport by plasma membrane transporters and phosphorylation by TK1 for their intracellular trapping. Thymidine competes with [18F]FLT for phosphorylation by TK1 as the fluorine substitution significantly decreases its affinity for TK1 (158, 159). One explanation for the difference in thymidine metabolism between mice and humans is the differential expression or activity of the enzyme responsible for thymidine breakdown, TYMP.
Differences in thymidine metabolism between mice and humans complicate the application of mice for MNGIE studies and result in diverging immune responses following TYMP inhibition (152). This discrepancy is due to several factors, including low TYMP levels in murine blood compared to humans, altered nucleoside levels in plasma, and the complementary role of uridine phosphorylase to TYMP in catabolizing dT and dU, providing a biochemical route to degrade these deoxyribonucleosides (152). In mice engineered to be deficient in uridine phosphorylase and TYMP, there is 1/10th the level of dU and dT increase compared to humans (160). This correlates with an incomplete pallet of symptoms in mice, which often lack the hallmark gastrointestinal and muscular manifestations (160). Furthermore, a heightened pyrimidine pool in mice could make specific cancer treatments appear more effective, as depletion of pyrimidines would result in a more drastic decrease in murine models than in humans. Similarly, while mice with PNP deficiency recapitulate the T cell deficiency observed in humans lacking PNP, mice experience a less severe phenotype, often lacking neurological symptoms (10, 161).
A similar challenge was encountered in translating the deoxycytidine-analog PET probes to monitor dCK activity non-invasively in vivo. While the first-generation dCK-specific PET probe [18F]FAC effectively visualized cell proliferation in lymphoid tissues in mice, it did not exhibit a specific uptake pattern in humans (2, 162). This species-specific tissue accumulation pattern of the dCK-specific PET probes was traced to the differential activity of CDA, the enzyme responsible for deoxycytidine catabolism, across mice and humans (163). Mice exhibit lower CDA activity, which may explain their higher plasma concentrations of pyrimidine nucleosides. This difference could account for variations in pyrimidine analog drug breakdown, as the slower breakdown in rodents is likely due to less active CDA (164). In addition to the natural pyrimidine deoxyribonucleosides, [18F]FAC is susceptible to CDA-mediated catabolism. This finding prompted the development of a next-generation dCK-specific PET probe resistant to CDA. [18F]CFA is a purine nucleoside analog that requires phosphorylation by dCK for its intracellular trapping but is not a substrate for CDA (2). [18F]CFA has shown promise for the noninvasive measurement of dCK activity in humans using PET imaging (2, 165).
Significant disparities also exist between mouse and human immune systems (150). While there are distinct patterns of expression or activity of genes within nucleoside metabolism between mice and humans, PRR-family nucleoside sensors also exhibit species-specific expression patterns. In particular, the uridine nucleoside sensor TLR8 is expressed at low levels in murine cells compared to human cells. This difference may underly the discrepancy in the manifestations of ENT3 deficiency across mice and humans, with humans presenting with auto-inflammation that is not fully recapitulated in SLC29A3 knockout mice (101). Transgenic mice have been developed to recapitulate the expression of TLR8 observed in humans (166). Nevertheless, this difference in PRR expression exemplifies the different biological environments of the two species that need to be considered when performing experiments in preclinical models.
8 Challenges and opportunities in the development of model systems to study the links between the human immune response and nucleoside metabolism
Improved preclinical tissue culture systems and mouse models that recapitulate both human nucleoside metabolism and immune responses are needed to facilitate the translation of new metabolism-targeting therapies. Promising advances have been made in engineering new mouse models for human immune responses. Interestingly, these models also recapitulate some aspects of human nucleoside metabolism, and may enable the evaluation of the effects of nucleoside metabolism-targeting therapies on immune system function. One approach for this is “humanized mouse models,” a system where mice are engineered with human tissues to recapitulate components of the human immune system, which is useful to study human tumor conditions and therapy responses (167). These models have been applied to evaluate antibodies, adoptive cell therapies, oncolytic viruses, and small molecule inhibitors (167). Immunodeficient mice are often the hosts for the immune engraftments, and there have been steady improvements to mouse strains and techniques over the last 50 years, allowing for decreased rejection of human cells upon transplantation (168). Multiple murine humanization techniques have been developed, including Hu-PBL, Hu-SRC, and Hu-BLT.
Hu-PBL is a relatively straightforward humanization method that involves the transplantation of human peripheral blood mononuclear cells (PBMC) into immunocompromised murine hosts (168). This engineering technique results in a human immune system mainly composed of T cells, albeit with diminished human cytokine levels and weak propagation of B and NK cells (168). This model is, therefore, best suited to test therapeutics and systems focusing on T cell behavior. A limitation of this model is that it often results in graft-versus-host disease (GVHD), limiting the scope and potential time frame for experiments (167).
The Hu-SRC technique more accurately captures the spectrum of human immune cell types (168). It involves the transfer of CD34+ hematopoietic stem cells (HSCs), which allows for the development of more complex innate and adaptive immune systems. Compared to Hu-PBL, it is a more stable model, with fewer instances of rejection (168). However, it may involve deficiencies of innate cell lineages and reduced B cell functionality (167).
Hu-BLT (bone marrow, liver, thymus) is a complex and more complete immune modeling system. It combines the Hu-SRC protocol of CD34+ hematopoietic stem and progenitor cell (HSPC) injection with particles of the human fetal thymus and fetal liver into immunodeficient mice (168, 169). This results in the growth of a human thymus analog within the mouse (167). However, there is still susceptibility to GVHD and rejection (168). Although certain strains of mice appear to resist rejection, obtaining sufficient human tissue for implantation complicates the engineering of this model (167). Notably, BLT mice recapitulate some aspects of human purine and pyrimidine metabolism, including lower systemic pyrimidine levels and enhanced pyrimidine catabolism (10, 169). BLT humanized mice, or next-generation humanized mice, may provide a powerful foundation for the investigation of new therapies that target nucleoside metabolism for immune modulation.
An alternative system for monitoring the interactions between human nucleoside metabolism and immune responses is the ex vivo culture of human tissue. These models involve the culture of primary human cells or explanted human donor material to recapitulate the heterotypic cellular composition of tissues, including tumors. These ex vivo models are an emerging platform to evaluate immune-based therapies and may be suitable for studying the immuno-modulatory effects of nucleoside metabolism-targeting therapies.
Patient-derived organoids are an ex vivo method for studying individual tumor responses to intervention. This method involves the collection of tissue from a patient, from which cancer cells are isolated and cultured to form 3D organoid structures (170). Organoids have been successfully formed from various tumor types (170). These models can potentially test whether a patient would respond to specific therapy (170).
Precision-cut tumor slices (PCTS) offer an experimental platform to model the intricate in vivo tumor environment in cell culture conditions (171). This system involves the culture of thinly sliced human or mouse tissue sections under specialized culture conditions. PCTS maintain integrity for 3–12 days, depending on culture methods and cancer type (171, 172). In contrast to organoid models, PCTS more completely encompass the heterotypic cellular composition of tissues. Several challenges with this model must be considered, including ischemia, hypoxia, loss of integrity during slicing, and the preservation of slices using cell culture methods (173). Multiple reports also suggest significant transcriptional changes in the hours after slices are prepared, and down-regulation cytokine production has been observed (173, 174). These models offer opportunities for developing personalized therapeutic approaches for cancer, as immunotherapies can be specified to the patient after tumor testing (173). The PCTS model has been applied to model immunosuppressive mechanisms operating in the tumor microenvironment and monitor the effects of immune-based anti-cancer therapies (175–178). PCTS models are a promising platform for future investigations of the immune-modifying properties of nucleoside metabolism-targeting therapies.
9 Conclusions
Over the past several years, substantial progress has been made in understanding the mechanisms underlying the immune-modifying effects of purine and pyrimidine nucleosides. This advancement was possible due to the commitment of scientists and physicians toward the development of new tools to measure and modify nucleoside metabolism in humans. However, the full therapeutic potential of nucleoside metabolism-targeting interventions for patient care has yet to be fully realized. Results from ongoing clinical trials evaluating the modification of adenosine signaling for cancer immunotherapy will undoubtedly provide new insight that may be applied in future clinical investigations. The development of new preclinical models that recapitulate human nucleoside metabolism is a central obstacle in translating new mechanistic insights from laboratory experiments into therapies. These models may provide insights into the therapeutic contexts and disease types where specific metabolism-targeting therapies will be most effective. New mouse models that possess a humanized nucleoside metabolism and immune system hold immense promise as a platform for these studies. Ex vivo cultures of primary human tissues may also serve as a valuable and relevant platform for future investigations of the intersections between nucleoside metabolism and immune system function.
Author contributions
ED: Writing – review & editing, Writing – original draft. EA: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. ED and EA were supported by a fellowship from the Hirshberg Foundation for Pancreatic Cancer Research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
PNP, purine nucleoside phosphorylase; ADA, adenosine deaminase; dCK, deoxcytidine kinase; TK1, thymidine kinase 1; HPRT, hypoxanthine-guanine phosphoribosyltransferase; TYMP, thymidine phosphorylase; PET, position emission tomography; dC, deoxycytidne; dA, deoxyadenosine; dG, deoxyguanosine; dT, thymidine; dU, deoxyuridine; U, uridine; A, adenosine; G, guanosine; C, cytidine; TLR7, toll-like receptor 7; TLR8, toll-like receptor 8; UPP1, uridine phosphorylase 1; CDA, cytidine deaminase; DHODH, dihydroorotate dehydrogenase; ENT1, equilibrate nucleoside transporter 1; ENT3, equilibrate nucleoside transporter 3; RNR, ribonucleotide reductase; DHODH, dihydroorotate dehydrogenase; SAMHD1, sterile alpha motif and histidine aspartate domain-containing protein 1; ADORA, adenosine receptor; dNTP, deoxyribonucleotide triphosphate; NBMPR, S-(4-nitrobenzyl)-6-thioinosine; ENTPD1, ectonucleoside triphosphate diphosphohydrolase 1; NT5E, `ecto-5′-nucleotidase.
References
1. Allard B, Allard D, Buisseret L, and Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol. (2020) 17:611–29. doi: 10.1038/s41571-020-0382-2
2. Kim W, Le TM, Wei L, Poddar S, Bazzy J, Wang X, et al. 18F]CFA as a clinically translatable probe for PET imaging of deoxycytidine kinase activity. Proc Natl Acad Sci U S A. (2016) 113:4027–32. doi: 10.1073/pnas.1524212113
3. Khalpey Z, Yuen AH, Lavitrano M, McGregor CG, Kalsi KK, Yacoub MH, et al. Mammalian mismatches in nucleotide metabolism: implications for xenotransplantation. Mol Cell Biochem. (2007) 304:109–17. doi: 10.1007/s11010-007-9491-9
4. Giblett ER, Anderson JE, Cohen F, Pollara B, and Meuwissen HJ. Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet. (1972) 2:1067–9. doi: 10.1016/s0140-6736(72)92345-8
5. Giblett ER, Ammann AJ, Wara DW, Sandman R, and Diamond LK. Nucleoside-phosphorylase deficiency in a child with severely defective T-cell immunity and normal B-cell immunity. Lancet. (1975) 1:1010–3. doi: 10.1016/s0140-6736(75)91950-9
6. Giblett ER. Back to the beginnings: an autobiography. Transfus Med Rev. (2006) 20:318–21. doi: 10.1016/j.tmrv.2006.05.005
7. Kuo CY, Garabedian E, Puck J, Cowan MJ, Sullivan KE, Buckley RH, et al. Adenosine deaminase (ADA)-deficient severe combined immune deficiency (SCID) in the US immunodeficiency network (USIDNet) registry. J Clin Immunol. (2020) 40:1124–31. doi: 10.1007/s10875-020-00857-9
8. Whitmore KV and Gaspar HB. Adenosine deaminase deficiency - more than just an immunodeficiency. Front Immunol. (2016) 7:314. doi: 10.3389/fimmu.2016.00314
9. Martín-Nalda A, Rivière JG, Català-Besa M, García-Prat M, Parra-Martínez A, Martínez-Gallo M, et al. Early diagnosis and treatment of purine nucleoside phosphorylase (PNP) deficiency through TREC-based newborn screening. Int J Neonatal Screen. (2021) 7:62. doi: 10.3390/ijns7040062
10. Abt ER, Rashid K, Le TM, Li S, Lee HR, Lok V, et al. Purine nucleoside phosphorylase enables dual metabolic checkpoints that prevent T cell immunodeficiency and TLR7-associated autoimmunity. J Clin Invest. (2022) 132:e160852. doi: 10.1172/JCI160852
11. Somech R, Lev A, Grisaru-Soen G, Shiran SI, Simon AJ, and Grunebaum E. Purine nucleoside phosphorylase deficiency presenting as severe combined immune deficiency. Immunol Res. (2013) 56:150–4. doi: 10.1007/s12026-012-8380-9
13. Al-Saud B, Al Alawi Z, Hussain FB, Hershfield M, Alkuraya FS, and Al-Mayouf SM. A case with purine nucleoside phosphorylase deficiency suffering from late-onset systemic lupus erythematosus and lymphoma. J Clin Immunol. (2020) 40:833–9. doi: 10.1007/s10875-020-00800-y
14. Grunebaum E, Zhang J, and Roifman CM. Novel mutations and hot-spots in patients with purine nucleoside phosphorylase deficiency. Nucleosides Nucleotides Nucleic Acids. (2004) 23:1411–5. doi: 10.1081/NCN-200027647
15. Grunebaum E, Campbell N, Leon-Ponte M, Xu X, and Chapdelaine H. Partial purine nucleoside phosphorylase deficiency helps determine minimal activity required for immune and neurological development. Front Immunol. (2020) 11:1257. doi: 10.3389/fimmu.2020.01257
16. Nassogne MC, Marie S, and Dewulf JP. Neurological presentations of inborn errors of purine and pyrimidine metabolism. Eur J Paediatr Neurol. (2024) 48:69–77. doi: 10.1016/j.ejpn.2023.11.013
17. Galmarini CM, Mackey JR, and Dumontet C. Nucleoside analogues: mechanisms of drug resistance and reversal strategies. Leukemia. (2001) 15:875–90. doi: 10.1038/sj.leu.2402114
18. Eriksson S, Munch-Petersen B, Johansson K, and Eklund H. Structure and function of cellular deoxyribonucleoside kinases. Cell Mol Life Sci. (2002) 59:1327–46. doi: 10.1007/s00018-002-8511-x
19. Daddacha W, Koyen AE, Bastien AJ, Head PE, Dhere VR, Nabeta GN, et al. SAMHD1 promotes DNA end resection to facilitate DNA repair by homologous recombination. Cell Rep. (2017) 20:1921–35. doi: 10.1016/j.celrep.2017.08.008
20. Shibata T, Ohto U, Nomura S, Kibata K, Motoi Y, Zhang Y, et al. Guanosine and its modified derivatives are endogenous ligands for TLR7. Int Immunol. (2016) 28:211–22. doi: 10.1093/intimm/dxv062
21. Sameer AS and Nissar S. Toll-like receptors (TLRs): structure, functions, signaling, and role of their polymorphisms in colorectal cancer susceptibility. BioMed Res Int. (2021) 2021:1157023. doi: 10.1155/2021/1157023
22. Takeda K and Akira S. Toll-like receptors. Curr Protoc Immunol. (2015) 109:14.12.1–14.12.10. doi: 10.1002/0471142735.im1412s109
23. Brown GJ, Cañete PF, Wang H, Medhavy A, Bones J, Roco JA, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. (2022) 605:349–56. doi: 10.1038/s41586-022-04642-z
24. Ikeda T, Sato K, Kawaguchi SI, Izawa J, Takayama N, Hayakawa H, et al. Forodesine Enhances Immune Responses through Guanosine-Mediated TLR7 Activation while Preventing Graft-versus-Host Disease. J Immunol. (2024) 212:143–53. doi: 10.4049/jimmunol.2300003
25. Tsui M, Biro J, Chan J, Min W, Dobbs K, Notarangelo LD, et al. Purine nucleoside phosphorylase deficiency induces p53-mediated intrinsic apoptosis in human induced pluripotent stem cell-derived neurons. Sci Rep. (2022) 12:9084. doi: 10.1038/s41598-022-10935-0
26. Miller A, York EM, Stopka SA, Martínez-François JR, Hossain MA, Baquer G, et al. Spatially resolved metabolomics and isotope tracing reveal dynamic metabolic responses of dentate granule neurons with acute stimulation. Nat Metab. (2023) 5:1820–35. doi: 10.1038/s42255-023-00890-z
27. Deutsch SI, Long KD, Rosse RB, Mastropaolo J, and Eller J. Hypothesized deficiency of guanine-based purines may contribute to abnormalities of neurodevelopment, neuromodulation, and neurotransmission in Lesch-Nyhan syndrome. Clin Neuropharmacol. (2005) 28:28–37. doi: 10.1097/01.wnf.0000152043.36198.25
28. Guibinga GH, Murray F, Barron N, Pandori W, and Hrustanovic G. Deficiency of the purine metabolic gene HPRT dysregulates microRNA-17 family cluster and guanine-based cellular functions: a role for EPAC in Lesch-Nyhan syndrome. Hum Mol Genet. (2013) 22:4502–15. doi: 10.1093/hmg/ddt298
29. Souza DG, Bellaver B, Bobermin LD, Souza DO, and Quincozes-Santos A. Anti-aging effects of guanosine in glial cells. Purinergic Signal. (2016) 12:697–706. doi: 10.1007/s11302-016-9533-4
30. Lane AN and Fan TW. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. (2015) 43:2466–85. doi: 10.1093/nar/gkv047
31. Evans DR and Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J Biol Chem. (2004) 279:33035–8. doi: 10.1074/jbc.R400007200
32. Abt ER, Rosser EW, Durst MA, Lok V, Poddar S, Le TM, et al. Metabolic modifier screen reveals secondary targets of protein kinase inhibitors within nucleotide metabolism. Cell Chem Biol. (2020) 27:197–205.e6. doi: 10.1016/j.chembiol.2019.10.012
33. Baldwin SA, Mackey JR, Cass CE, and Young JD. Nucleoside transporters: molecular biology and implications for therapeutic development. Mol Med Today. (1999) 5:216–24. doi: 10.1016/S1357-4310(99)01459-8
34. Van Rompay AR, Norda A, Lindén K, Johansson M, and Karlsson A. Phosphorylation of uridine and cytidine nucleoside analogs by two human uridine-cytidine kinases. Mol Pharmacol. (2001) 59:1181–6. doi: 10.1124/mol.59.5.1181
35. Radu CG, Shu CJ, Nair-Gill E, Shelly SM, Barrio JR, Satyamurthy N, et al. Molecular imaging of lymphoid organs and immune activation by positron emission tomography with a new [18F]-labeled 2’-deoxycytidine analog. Nat Med. (2008) 14:783–8. doi: 10.1038/nm1724
36. Toy G, Austin WR, Liao HI, Cheng D, Singh A, Campbell DO, et al. Requirement for deoxycytidine kinase in T and B lymphocyte development. Proc Natl Acad Sci U S A. (2010) 107:5551–6. doi: 10.1073/pnas.0913900107
37. Austin WR, Armijo AL, Campbell DO, Singh AS, Hsieh T, Nathanson D, et al. Nucleoside salvage pathway kinases regulate hematopoiesis by linking nucleotide metabolism with replication stress. J Exp Med. (2012) 209:2215–28. doi: 10.1084/jem.20121061
38. Girot R, Hamet M, Perignon JL, Guesnu M, Fox RM, Cartier P, et al. Cellular immune deficiency in two siblings with hereditary orotic aciduria. N Engl J Med. (1983) 308:700–4. doi: 10.1056/NEJM198303243081207
39. Al Absi HS, Sacharow S, Al Zein N, Al Shamsi A, and Al Teneiji A. Hereditary orotic aciduria (HOA): A novel uridine-5-monophosphate synthase (UMPS) mutation. Mol Genet Metab Rep. (2021) 26:100703. doi: 10.1016/j.ymgmr.2020.100703
40. Wortmann SB, Chen MA, Colombo R, Pontoglio A, Alhaddad B, Botto LD, et al. additional individual contributors. Mild orotic aciduria in UMPS heterozygotes: a metabolic finding without clinical consequences. J Inherit Metab Dis. (2017) 40:423–31. doi: 10.1007/s10545-017-0015-9
41. CM H, JA B, SL R, and Scoggins RB. Refractory megaloblastic anemia associated with excretion of orotic acid. Blood. (1959) 14:615–34. doi: 10.1182/blood.V14.6.615.615
42. Tanji H, Ohto U, Shibata T, Taoka M, Yamauchi Y, Isobe T, et al. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat Struct Mol Biol. (2015) 22:109–15. doi: 10.1038/nsmb.2943
43. Greulich W, Wagner M, Gaidt MM, Stafford C, Cheng Y, Linder A, et al. TLR8 is a sensor of RNase T2 degradation products. Cell. (2019) 179:1264–1275.e13. doi: 10.1016/j.cell.2019.11.001
44. Furusho K, Shibata T, Sato R, Fukui R, Motoi Y, Zhang Y, et al. Cytidine deaminase enables Toll-like receptor 8 activation by cytidine or its analogs. Int Immunol. (2019) 31:167–73. doi: 10.1093/intimm/dxy075
45. Shibata T, Sato R, Taoka M, Saitoh SI, Komine M, Yamaguchi K, et al. TLR7/8 stress response drives histiocytosis in SLC29A3 disorders. J Exp Med. (2023) 220:e20230054. doi: 10.1084/jem.20230054
46. Nyhan WL. Disorders of purine and pyrimidine metabolism. Mol Genet Metab. (2005) 86:25–33. doi: 10.1016/j.ymgme.2005.07.027
47. Fleger M, Willomitzer J, Meinsma R, Alders M, Meijer J, Hennekam RCM, et al. Dihydropyrimidine dehydrogenase deficiency: metabolic disease or biochemical phenotype? JIMD Rep. (2017) 37:49–54. doi: 10.1007/8904_2017_14
48. Saif MW, Syrigos K, Mehra R, Mattison LK, and Diasio RB. Dihydropyrimidine dehydrogenase deficiency (DPD) in gi malignancies: experience of 4-years. Pak J Med Sci. (2007) 23:832–9.
49. Kang J, Kim AH, Jeon I, Oh J, Jang IJ, Lee S, et al. Endogenous metabolic markers for predicting the activity of dihydropyrimidine dehydrogenase. Clin Transl Sci. (2022) 15:1104–11. doi: 10.1111/cts.13203
50. Du J, Zhang C, Liu F, Liu X, Wang D, Zhao D, et al. Distinctive metabolic remodeling in TYMP deficiency beyond mitochondrial dysfunction. J Mol Med (Berl). (2023) 101:1237–53. doi: 10.1007/s00109-023-02358-9
51. Martí R, Verschuuren JJ, Buchman A, Hirano I, Tadesse S, van Kuilenburg AB, et al. Late-onset MNGIE due to partial loss of thymidine phosphorylase activity. Ann Neurol. (2005) 58:649–52. doi: 10.1002/ana.20615
52. Garone C, Tadesse S, and Hirano M. Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain. (2011) 134:3326–32. doi: 10.1093/brain/awr245
53. Du J, Liu F, Liu X, Zhao D, Wang D, Sun H, et al. Lysosomal dysfunction and overload of nucleosides in thymidine phosphorylase deficiency of MNGIE. J Transl Med. (2024) 22:449. doi: 10.1186/s12967-024-05275-8
54. Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. (2014) 370:921–31. doi: 10.1056/NEJMoa1307362
55. Escherich C, Bötticher B, Harmsen S, Hömberg M, Schaper J, Lorenz MR, et al. The growing spectrum of DADA2 manifestations-diagnostic and therapeutic challenges revisited. Front Pediatr. (2022) 10:885893. doi: 10.3389/fped.2022.885893
56. Kaljas Y, Liu C, Skaldin M, Wu C, Zhou Q, Lu Y, et al. Human adenosine deaminases ADA1 and ADA2 bind to different subsets of immune cells. Cell Mol Life Sci. (2017) 74:555–70. doi: 10.1007/s00018-016-2357-0
57. Vigano S, Alatzoglou D, Irving M, Ménétrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol. (2019) 10:925. doi: 10.3389/fimmu.2019.00925
58. Visser SS, Theron AJ, Ramafi G, Ker JA, and Anderson R. Apparent involvement of the A(2A) subtype adenosine receptor in the anti-inflammatory interactions of CGS 21680, cyclopentyladenosine, and IB-MECA with human neutrophils. Biochem Pharmacol. (2000) 60:993–9. doi: 10.1016/s0006-2952(00)00414-7
59. Lappas CM, Rieger JM, and Linden J. A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J Immunol. (2005) 174:1073–80. doi: 10.4049/jimmunol.174.2.1073
60. Fredholm BB, IJzerman AP, Jacobson KA, Linden J, and Müller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors–an update. Pharmacol Rev. (2011) 63:1–34. doi: 10.1124/pr.110.003285
61. Pinna A. Novel investigational adenosine A2A receptor antagonists for Parkinson’s disease. Expert Opin Invest Drugs. (2009) 18:1619–31. doi: 10.1517/13543780903241615
62. Sitkovsky MV, Hatfield S, Abbott R, Belikoff B, Lukashev D, and Ohta A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol Res. (2014) 2:598–605. doi: 10.1158/2326-6066.CIR-14-0075
63. Ouyang X, Ghani A, Malik A, Wilder T, Colegio OR, Flavell RA, et al. Adenosine is required for sustained inflammasome activation via the A2A receptor and the HIF-1α pathway. Nat Commun. (2013) 4:2909. doi: 10.1038/ncomms3909
64. Sorrentino C, Miele L, Porta A, Pinto A, and Morello S. Activation of the A2B adenosine receptor in B16 melanomas induces CXCL12 expression in FAP-positive tumor stromal cells, enhancing tumor progression. Oncotarget. (2016) 7:64274–88. doi: 10.18632/oncotarget.11729
65. Iannone R, Miele L, Maiolino P, Pinto A, and Morello S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia. (2013) 15:1400–9. doi: 10.1593/neo.131748
66. Pacini ESA, Satori NA, Jackson EK, and Godinho RO. Extracellular cAMP-adenosine pathway signaling: A potential therapeutic target in chronic inflammatory airway diseases. Front Immunol. (2022) 13:866097. doi: 10.3389/fimmu.2022.866097
67. Zylka MJ, Sowa NA, Taylor-Blake B, Twomey MA, Herrala A, Voikar V, et al. Prostatic acid phosphatase is an ectonucleotidase and suppresses pain by generating adenosine. Neuron. (2008) 60:111–22. doi: 10.1016/j.neuron.2008.08.024
68. Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A. (2010) 107:1547–52. doi: 10.1073/pnas.0908801107
69. Forte G, Sorrentino R, Montinaro A, Luciano A, Adcock IM, Maiolino P, et al. Inhibition of CD73 improves B cell-mediated anti-tumor immunity in a mouse model of melanoma. J Immunol. (2012) 189:2226–33. doi: 10.4049/jimmunol.1200744
70. Bendell J, LoRusso P, Overman M, Noonan AM, Kim DW, Strickler JH, et al. First-in-human study of oleclumab, a potent, selective anti-CD73 monoclonal antibody, alone or in combination with durvalumab in patients with advanced solid tumors. Cancer Immunol Immunother. (2023) 72:2443–58. doi: 10.1007/s00262-023-03430-6
71. Hayes GM, Cairns B, Levashova Z, Chinn L, Perez M, Theunissen JW, et al. CD39 is a promising therapeutic antibody target for the treatment of soft tissue sarcoma. Am J Transl Res. (2015) 7:1181–8.
72. Bastid J, Regairaz A, Bonnefoy N, Déjou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res. (2015) 3:254–65. doi: 10.1158/2326-6066.CIR-14-0018
73. Liu Y, Li Z, Zhao X, Xiao J, Bi J, Li XY, et al. Review immune response of targeting CD39 in cancer. Biomark Res. (2023) 11:63. doi: 10.1186/s40364-023-00500-w
74. Moesta AK, Li XY, and Smyth MJ. Targeting CD39 in cancer. Nat Rev Immunol. (2020) 20:739–55. doi: 10.1038/s41577-020-0376-4
75. Ruiz-Fernández de Córdoba B, Martínez-Monge R, and Lecanda F. ENPP1 immunobiology as a therapeutic target. Clin Cancer Res. (2023) 29:2184–93. doi: 10.1158/1078-0432.CCR-22-1681
76. Li J, Duran MA, Dhanota N, Chatila WK, Bettigole SE, Kwon J, et al. Metastasis and immune evasion from extracellular cGAMP hydrolysis. Cancer Discov. (2021) 11:1212–27. doi: 10.1158/2159-8290.CD-20-0387
77. Carozza JA, Brown JA, Böhnert V, Fernandez D, AlSaif Y, Mardjuki RE, et al. Structure-aided development of small-molecule inhibitors of ENPP1, the extracellular phosphodiesterase of the immunotransmitter cGAMP. Cell Chem Biol. (2020) 27:1347–1358.e5. doi: 10.1016/j.chembiol.2020.07.007
78. Gangar M, Goyal S, Raykar D, Khurana P, Martis AM, Goswami A, et al. Design, synthesis and biological evaluation studies of novel small molecule ENPP1 inhibitors for cancer immunotherapy. Bioorg Chem. (2022) 119:105549. doi: 10.1016/j.bioorg.2021.105549
79. Li S, Tao B, Wan J, Montecino-Rodriguez E, Wang P, Ma F, et al. A humanized monoclonal antibody targeting an ectonucleotidase rescues cardiac metabolism and heart function after myocardial infarction. Cell Rep Med. (2024) 5:101795. doi: 10.1016/j.xcrm.2024.101795
80. Li S, Yokota T, Wang P, Ten Hoeve J, Ma F, Le TM, et al. Cardiomyocytes disrupt pyrimidine biosynthesis in nonmyocytes to regulate heart repair. J Clin Invest. (2022) 132:e149711. doi: 10.1172/JCI149711
81. Sanders TJ, Nabel CS, Brouwer M, and Hermant AL. Inhibition of equilibrative nucleoside transporter 1 relieves intracellular adenosine-mediated immune suppression. Cancer Res. (2024) 84. doi: 10.1158/1538-7445.AM2024-734
82. Lee H, Park S, Kim M, Park JY, Jin H, Oh K, et al. Integrative metabolomic and lipidomic profiling of lung squamous cell carcinoma for characterization of metabolites and intact lipid species related to the metastatic potential. Cancers (Basel). (2021) 13:4179. doi: 10.3390/cancers13164179
83. Li MX, Wu XT, Jing WQ, Hou WK, Hu S, and Yan W. Inosine enhances tumor mitochondrial respiration by inducing Rag GTPases and nascent protein synthesis under nutrient starvation. Cell Death Dis. (2023) 14:492. doi: 10.1038/s41419-023-06017-2
84. Klysz DD, Fowler C, Malipatlolla M, Stuani L, Freitas KA, Chen Y, et al. Inosine induces stemness features in CAR-T cells and enhances potency. Cancer Cell. (2024) 42:266–282.e8. doi: 10.1016/j.ccell.2024.01.002
85. Wright NJ and Lee SY. Toward a molecular basis of cellular nucleoside transport in humans. Chem Rev. (2021) 121:5336–58. doi: 10.1021/acs.chemrev.0c00644
86. Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, and Young JD. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. (2004) 447:735–43. doi: 10.1007/s00424-003-1103-2
87. Wei CW, Lee CY, Lee DJ, Chu CF, Wang JC, Wang TC, et al. Equilibrative nucleoside transporter 3 regulates T cell homeostasis by coordinating lysosomal function with nucleoside availability. Cell Rep. (2018) 23:2330–41. doi: 10.1016/j.celrep.2018.04.077
88. Engel K, Zhou M, and Wang J. Identification and characterization of a novel monoamine transporter in the human brain. J Biol Chem. (2004) 279:50042–9. doi: 10.1074/jbc.M407913200
89. Engel K and Wang J. Interaction of organic cations with a newly identified plasma membrane monoamine transporter. Mol Pharmacol. (2005) 68:1397–407. doi: 10.1124/mol.105.016832
90. Pastor-Anglada M, Molina-Arcas M, Casado FJ, Bellosillo B, Colomer D, and Gil J. Nucleoside transporters in chronic lymphocytic leukaemia. Leukemia. (2004) 18:385–93. doi: 10.1038/sj.leu.2403271
91. Allard D, Cormery J, Bricha S, Fuselier C, Abbas Aghababazadeh F, Giraud L, et al. Adenosine uptake through the nucleoside transporter ENT1 suppresses antitumor immunity and T cell pyrimidine synthesis. Cancer Res. (2024) 85: 692–703. doi: 10.1158/0008-5472.CAN-24-1875
92. Schneider EH, Hofmeister O, Kälble S, and Seifert R. Apoptotic and anti-proliferative effect of guanosine and guanosine derivatives in HuT-78 T lymphoma cells. Naunyn Schmiedebergs Arch Pharmacol. (2020) 393:1251–67. doi: 10.1007/s00210-020-01864-8
93. Hotani A, Kitabatake K, and Tsukimoto M. Extracellular guanosine and guanine nucleotides decrease viability of human breast cancer SKBR-3 cells. Biol Pharm Bull. (2024) 47:14–22. doi: 10.1248/bpb.b23-00402
94. Daniels G. Augustine blood group system and equilibrative nucleoside transporter 1. Transfus Med Hemother. (2022) 49:25–9. doi: 10.1159/000520596
95. Mikdar M, González-Menéndez P, Cai X, Zhang Y, Serra M, Dembele AK, et al. The equilibrative nucleoside transporter ENT1 is critical for nucleotide homeostasis and optimal erythropoiesis. Blood. (2021) 137:3548–62. doi: 10.1182/blood.2020007281
96. Zhong J, Mo C, Zhang Y, and Li L. A review of the Augustine blood group system. Int J Hematol. (2024) 120:44–9. doi: 10.1007/s12185-024-03791-3
97. Molho-Pessach V, Lerer I, Abeliovich D, Agha Z, Abu Libdeh A, Broshtilova V, et al. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet. (2008) 83:529–34. doi: 10.1016/j.ajhg.2008.09.013
98. Jacquot R, Jouret M, Valentin MG, Richard M, Jamilloux Y, Rousset F, et al. H syndrome treated with Tocilizumab: two case reports and literature review. Front Immunol. (2023) 14:1061182. doi: 10.3389/fimmu.2023.1061182
99. Melki I, Lambot K, Jonard L, Couloigner V, Quartier P, Neven B, et al. Mutation in the SLC29A3 gene: a new cause of a monogenic, autoinflammatory condition. Pediatrics. (2013) 131:e1308–13. doi: 10.1542/peds.2012-2255
100. Çağdaş D, Sürücü N, Tan Ç, Kayaoğlu B, Özgül RK, Akkaya-Ulum YZ, et al. Autoinflammation in addition to combined immunodeficiency: SLC29A3 gene defect. Mol Immunol. (2020) 121:28–37. doi: 10.1016/j.molimm.2020.02.014
101. Shiloh R, Lubin R, David O, Geron I, Okon E, Hazan I, et al. Loss of function of ENT3 drives histiocytosis and inflammation through TLR-MAPK signaling. Blood. (2023) 142:1740–51. doi: 10.1182/blood.2023020714
102. Maruyama D, Tsukasaki K, Uchida T, Maeda Y, Shibayama H, Nagai H, et al. Multicenter phase 1/2 study of forodesine in patients with relapsed peripheral T cell lymphoma. Ann Hematol. (2019) 98:131–42. doi: 10.1007/s00277-018-3418-2
103. Furman RR and Hoelzer D. Purine nucleoside phosphorylase inhibition as a novel therapeutic approach for B-cell lymphoid Malignancies. Semin Oncol. (2007) 34:S29–34. doi: 10.1053/j.seminoncol.2007.11.004
104. Miles RW, Tyler PC, Furneaux RH, Bagdassarian CK, and Schramm VL. One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry. (1998) 37:8615–21. doi: 10.1021/bi980658d
105. Kicska GA, Long L, Hörig H, Fairchild C, Tyler PC, Furneaux RH, et al. Immucillin H, a powerful transition-state analog inhibitor of purine nucleoside phosphorylase, selectively inhibits human T lymphocytes. Proc Natl Acad Sci U S A. (2001) 98:4593–8. doi: 10.1073/pnas.071050798
106. Balakrishnan K, Verma D, O’Brien S, Kilpatrick JM, Chen Y, Tyler BF, et al. Phase 2 and pharmacodynamic study of oral forodesine in patients with advanced, fludarabine-treated chronic lymphocytic leukemia. Blood. (2010) 116:886–92. doi: 10.1182/blood-2010-02-272039
107. Makita S, Maeshima AM, Maruyama D, Izutsu K, and Tobinai K. Forodesine in the treatment of relapsed/refractory peripheral T-cell lymphoma: an evidence-based review. Onco Targets Ther. (2018) 11:2287–93. doi: 10.2147/OTT.S140756
108. Chen Y, Li Y, Gao J, Yu Q, Zhang Y, and Zhang J. Perspectives and challenges in developing small molecules targeting purine nucleoside phosphorylase. Eur J Med Chem. (2024) 271:116437. doi: 10.1016/j.ejmech.2024.116437
109. Davenne T, Klintman J, Sharma S, Rigby RE, Blest HTW, Cursi C, et al. SAMHD1 Limits the Efficacy of Forodesine in Leukemia by Protecting Cells against the Cytotoxicity of dGTP. Cell Rep. (2020) 31:107640. doi: 10.1016/j.celrep.2020.107640
110. Davenne T and Rehwinkel J. PNP inhibitors selectively kill cancer cells lacking SAMHD1. Mol Cell Oncol. (2020) 7:1804308. doi: 10.1080/23723556.2020.1804308
111. Clifford R, Louis T, Robbe P, Ackroyd S, Burns A, Timbs AT, et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood. (2014) 123:1021–31. doi: 10.1182/blood-2013-04-490847
112. Abt ER, Le TM, Dann AM, Capri JR, Poddar S, Lok V, et al. Reprogramming of nucleotide metabolism by interferon confers dependence on the replication stress response pathway in pancreatic cancer cells. Cell Rep. (2022) 38:110236. doi: 10.1016/j.celrep.2021.110236
113. Vareed SK, Bhat VB, Thompson C, Vasu VT, Fermin D, Choi H, et al. Metabolites of purine nucleoside phosphorylase (NP) in serum have the potential to delineate pancreatic adenocarcinoma. PloS One. (2011) 6:e17177. doi: 10.1371/journal.pone.0017177
114. Bantia S. Purine nucleoside phosphorylase inhibitors as novel immuno-oncology agent and vaccine adjuvant. Int J Immunol Immunother. (2020) 7, 43. doi: 10.23937/2378-3672/1410043
115. Murphy JM, Armijo AL, Nomme J, Lee CH, Smith QA, Li Z, et al. Development of new deoxycytidine kinase inhibitors and noninvasive in vivo evaluation using positron emission tomography. J Med Chem. (2013) 56:6696–708. doi: 10.1021/jm400457y
116. Nomme J, Murphy JM, Su Y, Sansone ND, Armijo AL, Olson ST, et al. Structural characterization of new deoxycytidine kinase inhibitors rationalizes the affinity-determining moieties of the molecules. Acta Crystallogr D Biol Crystallogr. (2014) 70:68–78. doi: 10.1107/S1399004713025030
117. Nathanson DA, Armijo AL, Tom M, Li Z, Dimitrova E, Austin WR, et al. Co-targeting of convergent nucleotide biosynthetic pathways for leukemia eradication. J Exp Med. (2014) 211:473–86. doi: 10.1084/jem.20131738
118. Nomme J, Li Z, Gipson RM, Wang J, Armijo AL, Le T, et al. Structure-guided development of deoxycytidine kinase inhibitors with nanomolar affinity and improved metabolic stability. J Med Chem. (2014) 57:9480–94. doi: 10.1021/jm501124j
119. Poddar S, Capparelli EV, Rosser EW, Gipson RM, Wei L, Le T, et al. Development and preclinical pharmacology of a novel dCK inhibitor, DI-87. Biochem Pharmacol. (2020) 172:113742. doi: 10.1016/j.bcp.2019.113742
120. Le TM, Poddar S, Capri JR, Abt ER, Kim W, Wei L, et al. ATR inhibition facilitates targeting of leukemia dependence on convergent nucleotide biosynthetic pathways. Nat Commun. (2017) 8:241. doi: 10.1038/s41467-017-00221-3
121. Bunimovich YL, Nair-Gill E, Riedinger M, McCracken MN, Cheng D, McLaughlin J, et al. Deoxycytidine kinase augments ATM-Mediated DNA repair and contributes to radiation resistance. PloS One. (2014) 9:e104125. doi: 10.1371/journal.pone.0104125
122. Guantay L, Garro C, Siri S, Pansa MF, Ghidelli-Disse S, Paviolo N, et al. Deoxycytidine kinase (dCK) inhibition is synthetic lethal with BRCA2 deficiency. Drug Resist Updat. (2023) 67:100932. doi: 10.1016/j.drup.2023.100932
123. Chen BY, Ghezzi C, Villegas B, Quon A, Radu CG, Witte ON, et al. 18F-FAC PET visualizes brain-infiltrating leukocytes in a mouse model of multiple sclerosis. J Nucl Med. (2020) 61:757–63. doi: 10.2967/jnumed.119.229351
124. Ramakers BP, Riksen NP, Stal TH, Heemskerk S, van den Broek P, Peters WH, et al. Dipyridamole augments the antiinflammatory response during human endotoxemia. Crit Care. (2011) 15:R289. doi: 10.1186/cc10576
125. Spano D, Marshall JC, Marino N, De Martino D, Romano A, Scoppettuolo MN, et al. Dipyridamole prevents triple-negative breast-cancer progression. Clin Exp Metastasis. (2013) 30:47–68. doi: 10.1007/s10585-012-9506-0
126. Damaraju S, Damaraju VL, Mowles D, Sawyer MB, Damaraju S, and Cass CE. Cytotoxic activity of gemcitabine in cultured cell lines derived from histologically different types of bladder cancer: role of thymidine kinase 2. Biochem Pharmacol. (2010) 79:21–9. doi: 10.1016/j.bcp.2009.07.018
127. Zhang J, Visser F, King KM, Baldwin SA, Young JD, and Cass CE. The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev. (2007) 26:85–110. doi: 10.1007/s10555-007-9044-4
128. Wright NJ and Lee SY. Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat Struct Mol Biol. (2019) 26:599–606. doi: 10.1038/s41594-019-0245-7
129. Warfield BM and Reigan P. Multifunctional role of thymidine phosphorylase in cancer. Trends Cancer. (2022) 8:482–93. doi: 10.1016/j.trecan.2022.01.018
130. Fukushima M, Suzuki N, Emura T, Yano S, Kazuno H, Tada Y, et al. Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2’-deoxyribonucleosides. Biochem Pharmacol. (2000) 59:1227–36. doi: 10.1016/s0006-2952(00)00253-7
131. Fan N, Zhang Y, and Zou S. Methylthioadenosine phosphorylase deficiency in tumors: A compelling therapeutic target. Front Cell Dev Biol. (2023) 11:1173356. doi: 10.3389/fcell.2023.1173356
132. Firestone RS, Feng M, Basu I, Peregrina K, Augenlicht LH, and Schramm VL. Transition state analogue of MTAP extends lifespan of APCMin/+ mice. Sci Rep. (2021) 11:8844. doi: 10.1038/s41598-021-87734-6
133. Bedard GT, Gilaj N, Peregrina K, Brew I, Tosti E, Shaffer K, et al. Combined inhibition of MTAP and MAT2a mimics synthetic lethality in tumor models via PRMT5 inhibition. J Biol Chem. (2024) 300:105492. doi: 10.1016/j.jbc.2023.105492
134. Chang WH, Hsu SW, Zhang J, Li JM, Yang DD, Chu CW, et al. MTAP deficiency contributes to immune landscape remodelling and tumour evasion. Immunology. (2023) 168:331–45. doi: 10.1111/imm.13587
135. Basu I, Cordovano G, Das I, Belbin TJ, Guha C, and Schramm VL. A transition state analogue of 5’-methylthioadenosine phosphorylase induces apoptosis in head and neck cancers. J Biol Chem. (2007) 282:21477–86. doi: 10.1074/jbc.M702287200
136. Basu I, Locker J, Cassera MB, Belbin TJ, Merino EF, Dong X, et al. Growth and metastases of human lung cancer are inhibited in mouse xenografts by a transition state analogue of 5’-methylthioadenosine phosphorylase. J Biol Chem. (2011) 286:4902–11. doi: 10.1074/jbc.M110.198374
137. Lolli ML, Sainas S, Pippione AC, Giorgis M, Boschi D, and Dosio F. Use of human dihydroorotate dehydrogenase (hDHODH) inhibitors in autoimmune diseases and new perspectives in cancer therapy. Recent Pat Anticancer Drug Discov. (2018) 13:86–105. doi: 10.2174/1574892812666171108124218
138. Aly L, Hemmer B, and Korn T. From leflunomide to teriflunomide: drug development and immunosuppressive oral drugs in the treatment of multiple sclerosis. Curr Neuropharmacol. (2017) 15:874–91. doi: 10.2174/1570159X14666161208151525
139. Cherwinski HM, Cohn RG, Cheung P, Webster DJ, Xu YZ, Caulfield JP, et al. The immunosuppressant leflunomide inhibits lymphocyte proliferation by inhibiting pyrimidine biosynthesis. J Pharmacol Exp Ther. (1995) 275:1043–9. doi: 10.1016/S0022-3565(25)12093-4
140. Li L, Ng SR, Colón CI, Drapkin BJ, Hsu PP, Li Z, et al. Identification of DHODH as a therapeutic target in small cell lung cancer. Sci Transl Med. (2019) 11:eaaw7852. doi: 10.1126/scitranslmed.aaw7852
141. Gwynne WD, Suk Y, Custers S, Mikolajewicz N, Chan JK, Zador Z, et al. Cancer-selective metabolic vulnerabilities in MYC-amplified medulloblastoma. Cancer Cell. (2022) 40:1488–1502.e7. doi: 10.1016/j.ccell.2022.10.009
142. Shi DD, Savani MR, Levitt MM, Wang AC, Endress JE, Bird CE, et al. De novo pyrimidine synthesis is a targetable vulnerability in IDH mutant glioma. Cancer Cell. (2022) 40:939–956.e16. doi: 10.1016/j.ccell.2022.07.011
143. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. (2021) 593:586–90. doi: 10.1038/s41586-021-03539-7
144. Mullen NJ, Shukla SK, Thakur R, Kollala SS, Wang D, Chaika N, et al. DHODH inhibition enhances the efficacy of immune checkpoint blockade by increasing cancer cell antigen presentation. Elife. (2024) 12:RP87292. doi: 10.7554/eLife.87292
145. Scherer S, Oberle SG, Kanev K, Gerullis AK, Wu M, de Almeida GP, et al. Pyrimidine de novo synthesis inhibition selectively blocks effector but not memory T cell development. Nat Immunol. (2023) 24:501–15. doi: 10.1038/s41590-023-01436-x
146. Zhang Z, Ohto U, Shibata T, Krayukhina E, Taoka M, Yamauchi Y, et al. Structural analysis reveals that toll-like receptor 7 is a dual receptor for guanosine and single-stranded RNA. Immunity. (2016) 45:737–48. doi: 10.1016/j.immuni.2016.09.011
147. Lee J, Chuang TH, Redecke V, She L, Pitha PM, Carson DA, et al. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc Natl Acad Sci U S A. (2003) 100:6646–51. doi: 10.1073/pnas.0631696100
148. Heil F, Ahmad-Nejad P, Hemmi H, Hochrein H, Ampenberger F, Gellert T, et al. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur J Immunol. (2003) 33:2987–97. doi: 10.1002/eji.200324238
149. Rolfo C, Giovannetti E, Martinez P, McCue S, and Naing A. Applications and clinical trial landscape using Toll-like receptor agonists to reduce the toll of cancer. NPJ Precis Oncol. (2023) 7:26. doi: 10.1038/s41698-023-00364-1
150. Mestas J and Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
151. Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. (1994) 140:1–22. doi: 10.1007/BF00928361
152. Torres-Torronteras J, Gómez A, Eixarch H, Palenzuela L, Pizzorno G, Hirano M, et al. Hematopoietic gene therapy restores thymidine phosphorylase activity in a cell culture and a murine model of MNGIE. Gene Ther. (2011) 18:795–806. doi: 10.1038/gt.2011.24
153. Greiner-Tollersrud OK, Krausz M, Boehler V, Polyzou A, Seidl M, Spahiu A, et al. ADA2 is a lysosomal deoxyadenosine deaminase acting on DNA involved in regulating TLR9-mediated immune sensing of DNA. Cell Rep. (2024) 43:114899. doi: 10.1016/j.celrep.2024.114899
154. Signa S, Bertoni A, Penco F, Caorsi R, Cafaro A, Cangemi G, et al. Adenosine deaminase 2 deficiency (DADA2): A crosstalk between innate and adaptive immunity. Front Immunol. (2022) 13:935957. doi: 10.3389/fimmu.2022.935957
155. Joolharzadeh P and St Hilaire C. CD73 (Cluster of differentiation 73) and the differences between mice and humans. Arterioscler Thromb Vasc Biol. (2019) 39:339–48. doi: 10.1161/ATVBAHA.118.311579
156. Shields AF, Grierson JR, Dohmen BM, Machulla HJ, Stayanoff JC, Lawhorn-Crews JM, et al. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med. (1998) 4:1334–6. doi: 10.1038/3337
157. Shields AF. PET imaging with 18F-FLT and thymidine analogs: promise and pitfalls. J Nucl Med. (2003) 44:1432–4.
158. Munch-Petersen B, Cloos L, Tyrsted G, and Eriksson S. Diverging substrate specificity of pure human thymidine kinases 1 and 2 against antiviral dideoxynucleosides. J Biol Chem. (1991) 266:9032–8. doi: 10.1016/S0021-9258(18)31547-3
159. Liang K, Abt ER, Le TM, Cho A, Dann AM, Cui J, et al. STING-driven interferon signaling triggers metabolic alterations in pancreas cancer cells visualized by [18F]FLT PET imaging. Proc Natl Acad Sci U S A. (2021) 118:e2105390118. doi: 10.1073/pnas.2105390118
160. Pacitti D, Levene M, Garone C, Nirmalananthan N, and Bax BE. Mitochondrial neurogastrointestinal encephalomyopathy: into the fourth decade, what we have learned so far. Front Genet. (2018) 9:669. doi: 10.3389/fgene.2018.00669
161. Arpaia E, Benveniste P, Di Cristofano A, Gu Y, Dalal I, Kelly S, et al. Mitochondrial basis for immune deficiency. Evidence from purine nucleoside phosphorylase-deficient mice. J Exp Med. (2000) 191:2197–208. doi: 10.1084/jem.191.12.2197
162. Schwarzenberg J, Radu CG, Benz M, Fueger B, Tran AQ, Phelps ME, et al. Human biodistribution and radiation dosimetry of novel PET probes targeting the deoxyribonucleoside salvage pathway. Eur J Nucl Med Mol Imaging. (2011) 38:711–21. doi: 10.1007/s00259-010-1666-z
163. Shu CJ, Campbell DO, Lee JT, Tran AQ, Wengrod JC, Witte ON, et al. Novel PET probes specific for deoxycytidine kinase. J Nucl Med. (2010) 51:1092–8. doi: 10.2967/jnumed.109.073361
164. Mollick T and Laín S. Modulating pyrimidine ribonucleotide levels for the treatment of cancer. Cancer Metab. (2020) 8:12. doi: 10.1186/s40170-020-00218-5
165. Barrio MJ, Spick C, Radu CG, Lassmann M, Eberlein U, Allen-Auerbach M, et al. Human biodistribution and radiation dosimetry of 18F-clofarabine, a PET probe targeting the deoxyribonucleoside salvage pathway. J Nucl Med. (2017) 58:374–8. doi: 10.2967/jnumed.116.182394
166. Guiducci C, Gong M, Cepika AM, Xu Z, Tripodo C, Bennett L, et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J Exp Med. (2013) 210:2903–19. doi: 10.1084/jem.20131044
167. Chuprin J, Buettner H, Seedhom MO, Greiner DL, Keck JG, Ishikawa F, et al. Humanized mouse models for immuno-oncology research. Nat Rev Clin Oncol. (2023) 20:192–206. doi: 10.1038/s41571-022-00721-2
168. Yin L, Wang XJ, Chen DX, Liu XN, and Wang XJ. Humanized mouse model: a review on preclinical applications for cancer immunotherapy. Am J Cancer Res. (2020) 10:4568–84.
169. Carrillo MA, Zhen A, and Kitchen SG. The use of the humanized mouse model in gene therapy and immunotherapy for HIV and cancer. Front Immunol. (2018) 9:746. doi: 10.3389/fimmu.2018.00746
170. Yang H, Sun L, Liu M, and Mao Y. Patient-derived organoids: a promising model for personalized cancer treatment. Gastroenterol Rep (Oxf). (2018) 6:243–5. doi: 10.1093/gastro/goy040
171. Dimou P, Trivedi S, Liousia M, D’Souza RR, and Klampatsa A. Precision-cut tumor slices (PCTS) as an ex vivo model in immunotherapy research. Antibodies (Basel). (2022) 11:26. doi: 10.3390/antib11020026
172. Kenerson HL, Sullivan KM, Labadie KP, Pillarisetty VG, and Yeung RS. Protocol for tissue slice cultures from human solid tumors to study therapeutic response. STAR Protoc. (2021) 2:100574. doi: 10.1016/j.xpro.2021.100574
173. Trivedi S, Tilsed C, Liousia M, Brody RM, Rajasekaran K, Singhal S, et al. Transcriptomic analysis-guided assessment of precision-cut tumor slices (PCTS) as an ex-vivo tool in cancer research. Sci Rep. (2024) 14:11006. doi: 10.1038/s41598-024-61684-1
174. Bigaeva E, Gore E, Simon E, Zwick M, Oldenburger A, de Jong KP, et al. Transcriptomic characterization of culture-associated changes in murine and human precision-cut tissue slices. Arch Toxicol. (2019) 93:3549–83. doi: 10.1007/s00204-019-02611-6
175. Daniel SK, Sullivan KM, Dickerson LK, van den Bijgaart RJE, Utria AF, Labadie KP, et al. Reversing immunosuppression in the tumor microenvironment of fibrolamellar carcinoma via PD-1 and IL-10 blockade. Sci Rep. (2024) 14:5109. doi: 10.1038/s41598-024-55593-6
176. Labadie KP, Kreuser SA, Brempelis KJ, Daniel SK, Jiang X, Sullivan KM, et al. Production of an interleukin-10 blocking antibody by genetically engineered macrophages increases cancer cell death in human gastrointestinal tumor slice cultures. Cancer Gene Ther. (2023) 30:1227–33. doi: 10.1038/s41417-023-00632-z
177. Jabbari N, Kenerson HL, Lausted C, Yan X, Meng C, Sullivan KM, et al. Modulation of immune checkpoints by chemotherapy in human colorectal liver metastases. Cell Rep Med. (2020) 1:100160. doi: 10.1016/j.xcrm.2020.100160
178. Jiang X, Seo YD, Chang JH, Coveler A, Nigjeh EN, Pan S, et al. Long-lived pancreatic ductal adenocarcinoma slice cultures enable precise study of the immune microenvironment. Oncoimmunology. (2017) 6:e1333210. doi: 10.1080/2162402X.2017.1333210
179. Saveljeva S, Sewell GW, Ramshorn K, Cader MZ, West JA, Clare S, et al. A purine metabolic checkpoint that prevents autoimmunity and autoinflammation. Cell Metab. (2022) 34:106–124.e10. doi: 10.1016/j.cmet.2021.12.009
180. Nwosu ZC, Ward MH, Sajjakulnukit P, Poudel P, Ragulan C, Kasperek S, et al. Uridine-derived ribose fuels glucose-restricted pancreatic cancer. Nature. (2023) 618:151–8. doi: 10.1038/s41586-023-06073-w
181. Wang T, Gnanaprakasam JNR, Chen X, Kang S, Xu X, Sun H, et al. Inosine is an alternative carbon source for CD8+-T-cell function under glucose restriction. Nat Metab. (2020) 2:635–47. doi: 10.1038/s42255-020-0219-4
182. Yamamoto M, Goto H, Hirayama A, Ohishi M, Kuramoto T, Mitsuhashi A, et al. Thymidine catabolism as a metabolic strategy for cancer survival. Cell Rep. (2017) 19:1313–21. doi: 10.1016/j.celrep.2017.04.061
183. Sprenger HG, MacVicar T, Bahat A, Fiedler KU, Hermans S, Ehrentraut D, et al. Cellular pyrimidine imbalance triggers mitochondrial DNA-dependent innate immunity. Nat Metab. (2021) 3:636–50. doi: 10.1038/s42255-021-00385-9
184. Chen BY, Salas JR, Trias AO, Perez Rodriguez A, Tsang JE, Guemes M, et al. Targeting deoxycytidine kinase improves symptoms in mouse models of multiple sclerosis. Immunology. (2023) 168:152–69. doi: 10.1111/imm.13569
185. Salas JR, Ryan KM, Trias AO, Chen BY, Guemes M, Galic Z, et al. Blocking deoxycytidine kinase in activated lymphocytes depletes deoxycytidine triphosphate pools and alters cell cycle kinetics to yield less disease in a mouse multiple sclerosis model. Immunology. (2025) 174:247–63. doi: 10.1111/imm.13885
Keywords: metabolism, immune activation, cancer immunotherapy, nucleotide metabolism, autoimmune disease, immuno-metabolism
Citation: Dunderdale EM and Abt ER (2025) Landscape of targets within nucleoside metabolism for the modification of immune responses. Front. Oncol. 15:1483769. doi: 10.3389/fonc.2025.1483769
Received: 20 August 2024; Accepted: 25 April 2025;
Published: 30 May 2025.
Edited by:
Evanna Mills, Dana–Farber Cancer Institute, United StatesReviewed by:
Eunus S. Ali, University of Kentucky, United StatesRajeev Kumar Pandey, Johns Hopkins Medicine, United States
Copyright © 2025 Dunderdale and Abt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evan R. Abt, ZWFidEBtZWRuZXQudWNsYS5lZHU=