 Taniya Saha
Taniya Saha Kiven Erique Lukong
Kiven Erique Lukong- Department of Biochemistry, Microbiology, and Immunology, University of Saskatchewan, Saskatoon, SK, Canada
Classical estrogen receptors, ERα and ERβ, along with the membrane-bound G-protein-coupled estrogen receptor (GPER), play critical roles in driving ERα−positive breast cancer (BC). Clinical management of this subtype relies on endocrine therapy (ET), which targets ER signaling through selective estrogen receptors modulators (SERMs), degraders (SERDs), and aromatase inhibitors (AIs). While ET has significantly reduced recurrence and mortality rates, acquired resistance remains a major therapeutic challenge. Activating ESR1 mutations, which encode constitutively active ERα variants, are detected in 30-50% of therapy-resistant metastatic ERα−positive BC and serve as emerging biomarkers of poor prognosis. These hot-spot mutations stabilize ERα in its agonist conformation, thereby enabling ligand-independent transcriptional activation. Understanding the conformational constraints that keep wild-type ERα in an “off-state” in the absence of ligand—and how activating ESR1 mutations disrupt these regulatory mechanisms—is critical for developing effective targeted therapies. Concurrently, GPER-mediated non-genomic signaling, often inadvertently activated by SERMs and SERDs, contributes to tamoxifen resistance. This review explores the structural and functional intricacies of ERα, the impact of ESR1 mutations on its ligand-binding domain (ERα−LBD) and their contribution to ET resistance, and the role of GPER-mediated signaling in ERα−positive BC. We further highlight recent advances in next-generation therapeutics targeting both ERα mutants and GPER, which may offer a more effective, integrated strategy to overcome ET resistance.
1 Introduction
According to the American Cancer Society’s Breast Cancer Facts & Figures 2024–2025, an estimated 310,720 new cases of invasive breast cancer and 56,500 cases of ductal carcinoma in situ (DCIS) are expected to be diagnosed among U.S. women in 2024. The latest GLOBOCAN 2022 estimates from the International Agency for Research on Cancer identify breast cancer (BC) as the second most commonly diagnosed cancer worldwide—following lung cancer—and the most frequently diagnosed cancer in women, with approximately 2.3 million new cases, accounting for 11.5% of all cancer diagnoses (1–6). At the molecular level, genomic and transcriptomic profiling—based on the expression of estrogen receptors (ER), progesterone receptors (PR), and HER2—classifies breast tumors into four main subtypes: luminal A (ER+ and/or PR+, HER2−, Ki67 <14%), luminal B (ER+ and/or PR+, HER2+ or HER2−, Ki67 >14%), HER2-enriched (ER−, PR−, HER2+), and basal-like/triple-negative (ER−, PR−, HER2−) (7). Among these, luminal-A and luminal-B subtypes predominantly express ER, with approximately 70% of newly diagnosed breast cancers being ER-positive (ER+) (8, 9). In ER+ tumors, ERα serves as the principal oncogenic driver, typically requiring estrogen (E2) for activation. However, deregulated ER expression and aberrant E2-ER interactions contribute significantly to disease progression, making endocrine therapy (ET)—which works by blocking ERα activity—a mainstay treatment for this subtype. ERs are classified into two main families: (1) the classical ERs, ERα and ERβ, which are ligand-induced nuclear receptors with a high degree of amino acid homology, functioning through E2-mediated genomic signaling (10, 11); and (2) the G-protein-coupled receptor 30 (GPR30) or G protein-coupled estrogen receptor (GPER), a family of membrane receptors that mediate E2-induced rapid non-genomic signaling and function as transcription regulators via the second messenger system (12, 13). Conventionally, ET relies on six major therapeutic classes: selective estrogen receptor modulators (SERMs), selective estrogen receptor degraders (SERDs), aromatase inhibitors (AIs), CDK4/6 inhibitors, used in combination with SERDs/AIs, and mTORC1 inhibitors in combination with AIs, as discussed below (14–17).
Tamoxifen, the first SERM, is an ERα antagonist that competitively inhibits estrogen binding to ERα and was approved by the FDA in 1972 for both pre- and postmenopausal BC patients (18, 19). Orally administered tamoxifen is extensively metabolized into active forms—4-hydroxytamoxifen (4OHT) and 4-hydroxy-N-desmethyl-tamoxifen (endoxifen)—by cytochrome P450 (CYP) enzymes such as CYP3A4 and CYP2D6 (20). However, genetic polymorphisms in CYP2D6, observed in a significant number of BC patients, lead to variable tamoxifen metabolism, contributing to inconsistent therapeutic outcomes (21, 22). Notably, Z-endoxifen (ENDX), the most active isomer of endoxifen, has demonstrated promising antitumor activity and manageable toxicity compared to tamoxifen in ERα-positive metastatic breast cancer (MBC) patients harboring ESR1 mutations—the gene encoding ERα (23). Recognition of genetic variability in tamoxifen metabolism led to the development of toremifene, a first-generation SERM that differs from tamoxifen by a single chlorine atom (24, 25). SERMs are known for their tissue-specific dual activity—acting as ERα antagonists in breast tissue but agonists in the bone and uterus—which is associated with an increased risk of endometrial cancer and thromboembolism. To address these risks, tamoxifen was succeeded by second-generation SERMs such as raloxifene, arzoxifene, and idoxifene, and third-generation agents like lasofoxifene, which offer improved bioavailability, fewer side-effects, and a reduced risk of thromboembolism (26).
In contrast, fulvestrant (ICI 182,780)—the only FDA-approved SERD for hormone receptor-positive (HR+) MBC—competes with E2 for ER binding with 89% of E2’s binding affinity, significantly higher than tamoxifen, which has only 2.5% of E2’s binding affinity (27). The fulvestrant–ER interaction impairs receptor dimerization, disrupts both activating function domains (AF1 and AF2) of ERα, inhibits energy-dependent nucleo-cytoplasmic trafficking, and accelerates ERα degradation (28). Unlike SERMs, fulvestrant lacks agonist activity in non-breast tissues and uniquely downregulates cellular levels of both ER and PR. However, its clinical efficacy is limited by poor bioavailability, suboptimal systemic exposure and biodistribution, and extensive hepatic metabolism via CYP3A4, necessitating intramuscular administration for controlled release (29, 30).
AIs, in contrast, work by disrupting estrogen biosynthesis and are classified into steroidal (type I), such as exemestane, and non-steroidal (type II), such as anastrozole and letrozole. These agents are widely used as adjuvant therapies for both early-stage and metastatic ER-positive breast cancer in postmenopausal women (31, 32). However, acquired resistance to AIs—often due to a switch from ER-dependent signaling to growth factor-mediated pathways—has led to the emergence of combination therapies (33, 34). Notably, pairing fulvestrant or AIs with CDK4/6 inhibitors has proven to be a promising and well-tolerated strategy for treating MBC. Recent clinical trials—PALOMA-3, MONALEESA-3, and MONARCH-2 (fulvestrant combined with palbociclib, ribociclib, or abemaciclib) (35–38), as well as PALOMA-2, MONALEESA-2, and MONARCH-3 (AIs combined with the same CDK4/6 inhibitors)—have demonstrated significantly improved progression-free survival (PFS) and overall survival (OS) compared to fulvestrant or AI monotherapy (39–41) Additionally, targeting the PI3K/AKT/mTOR signaling cascade with mTOR inhibitors, such as everolimus, represents a significant advancement in BC therapy (42, 43). In 2012, the FDA approved everolimus in combination with exemestane for the treatment of HR+ but HER2− breast cancer, providing an effective option for improving patient outcomes (44, 45). Despite the success of ET, acquired resistance develops in approximately 30%-50% of patients undergoing prolonged treatment, ultimately compromising therapeutic response and contributing to disease progression, metastasis, and relapse (46–49). Among the various factors, point mutations in the ERα ligand-binding domain (ERα−LBD) significantly contribute to the emergence of acquired resistance.
Recent deep DNA sequencing studies have identified activating mutations in the ESR1 gene, which encodes ERα−LBD, in approximately 40% of recurrent, ET-resistant, ER-positive breast cancers (50–53). Most of these ESR1 mutations are ligand-independent activation mutations that stabilize the unliganded ER in an agonist-bound conformation, thereby conferring constitutive activity and driving resistance to current ERα−targeted therapies. Among these, Y537S and D538G are the two most prevalent mutations (53, 54). Metastatic, therapy-resistant ER-positive breast cancers driven by ESR1 mutations represent a significant clinical challenge and account for a substantial number of breast cancer-related deaths (55, 56). Deeper insights into the molecular mechanisms underlying mutant ERα activity is crucial for developing next-generation drugs targeting ESR1 mutations with improved pharmacokinetic properties. In this context, several clinical trials are evaluating the safety and efficacy of next-generation SERDs—including elacestrant (RAD1901) (57, 58), camizestrant (AZD9833) (59), imlunestrant (LY3484356) (29, 60, 61), and giredestrant (GDC-9545) (62)—either as monotherapy or in combination with other anti-cancer agents, for targeting both ESR1 wild-type and mutant ER+/HER2− locally advanced or MBC.
Other emerging therapeutic platforms, such as ER proteolysis-targeting chimeras (ER-PROTACs) like ARV-471 and ERD-3111 (63–66), complete estrogen receptor antagonists (CERANs) such as OP-1250 (Palazestrant) (67), and selective estrogen receptor covalent antagonists (SERCAs) like H3B-6545 (68), have demonstrated compelling preclinical anti-tumor efficacy and significant potency against clinically relevant ERα mutants, including Y537S and D538G. However, further studies are needed to evaluate long-term safety, side effect profiles, and recurrence prevention.
In parallel, GPER-mediated non-genomic signaling is emerging as a key contributor to ET resistance. Notably, the ability of both estrogen and anti-estrogens to activate GPER has led to findings that high GPER expression strongly correlates with tamoxifen resistance in BC patients (69–71). To address this, GPER-selective antagonists—such as G15 and G36—are being developed, offering further insights into the role of GPER in ER-positive breast cancer (72). This review emphasizes the structural features of ERs, particularly how the structure-function relationship of the ERα−LBD governs receptor activity, the role of activating ESR1 mutations in driving constitutive signaling, and the development of next-generation therapeutics—especially those targeting ERα mutants and GPER—to simultaneously antagonize both receptor classes implicated in ET resistance.
2 Structure of ERs
ERα, a 66 kDa protein composed of 595 amino acids, belongs to the nuclear hormone receptor (NHR) subfamily and is encoded by the ESR1 gene located on chromosome 6 (6q25.1). The ESR1 gene spans approximately 300 Kb and includes 8 exons that encode the full-length ERα protein (73). Structurally, ERα possesses conserved domains, including the N-terminal domain (NTD, ‘A/B’ domain), DNA-binding domain (DBD, ‘C’ domain), flexible hinge region (‘D’ domain) and ligand-binding domain (LBD, ‘E’ domain), followed by a short ‘F’ region (Figure 1A) (74–82). Two activation function domains, ligand-independent activation function (AF1) and ligand-dependent activation function (AF2), are located within the NTD and LBD, respectively, and mediate ER’s transcriptional activity. Alternative splicing of the ESR1 gene generates an exon-1-truncated ERα transcript, ERα46, which lacks the N-terminal 1–173 amino acids, including the AF1 domain, and acts as a dominant-negative inhibitor of full-length ERα (83–85). Additionally, another isoform, ERα36, lacks both the AF1 and AF2 transactivation domains but retains a unique 22-amino acid C-terminal sequence (86). Interestingly, ERα46, expressed in various normal and tumor cell types including BC, contributes to cancer cell growth arrest by interfering with the binding of ERα66 to DNA (84, 87, 88). However, its expression is diminished in tamoxifen-resistant breast cancer cells, and re-expression of ERα46 suppresses cell proliferation and ERα66-regulated signaling (88, 89). Although an earlier study reported that the ERα46/ERα66 expression ratio is negatively correlated with breast tumor grade, a recent investigation highlighted a cross-talk between ERα46 and insulin receptor (IR) signaling that promotes the growth and pulmonary metastasis of the naturally immortalized BCAHC-1 cell line. Notably, this cell line—derived from a patient with invasive ductal breast carcinoma—exhibits unique co-expression and bi-directional cooperation between ERα46 and IR. This receptor cross-talk activates interleukin-11 (IL-11) expression and function, promoting the expression of pro-tumorigenic genes such as ITGA5 and ICAM-1, and enhancing the migratory and invasive features of patient-derived breast cancer-associated fibroblasts (CAFs) (90). In contrast, tamoxifen acts as an agonist for ERα36 in breast cancer, enhancing stemness by upregulating ALDH1A1 and promoting ET resistance and metastasis (91).
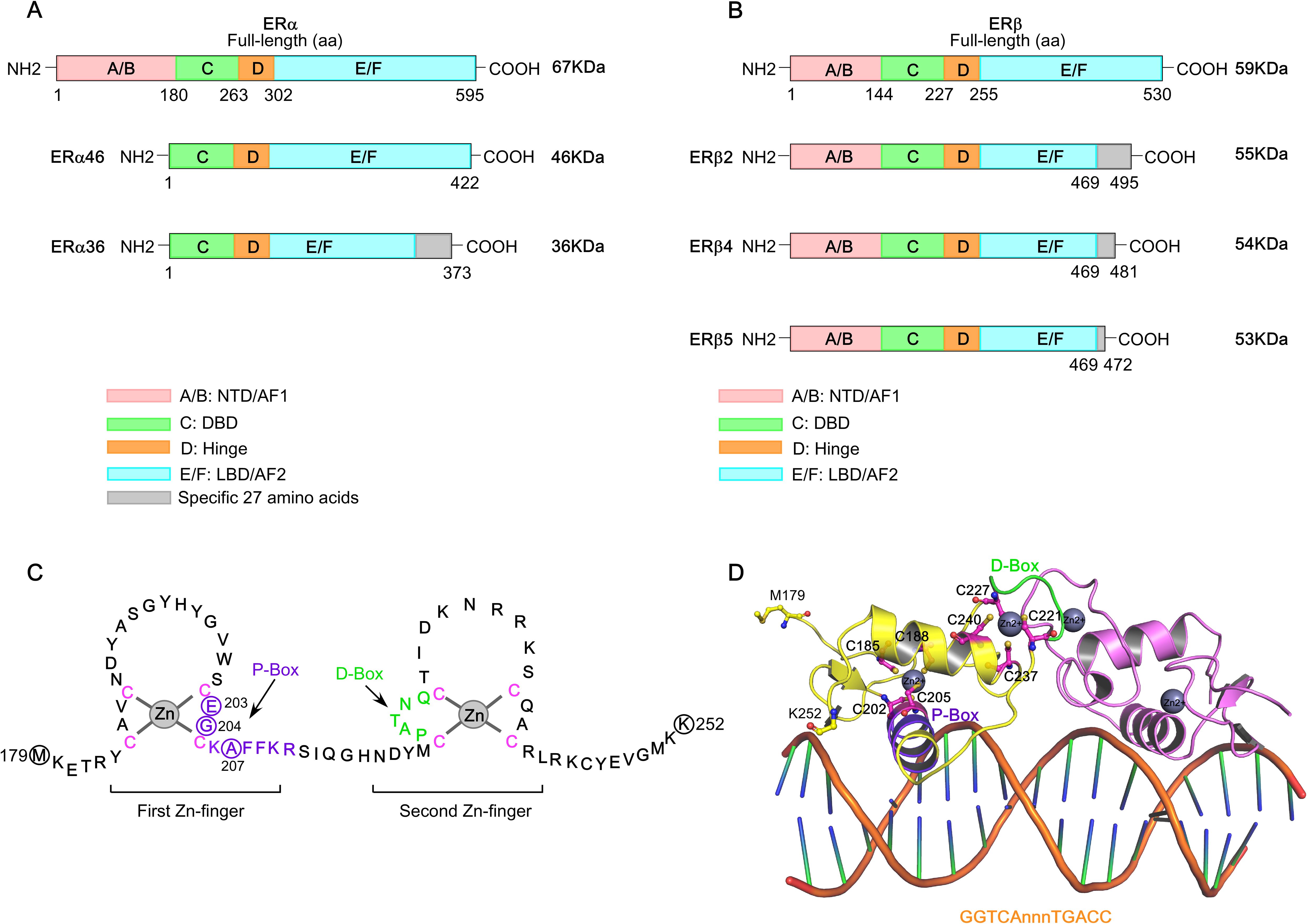
Figure 1. Structure of estrogen receptors (ERs). (A) Schematic figure of the structure of full-length ERα and its isoforms, ERα46 and ERα36. (B) Schematic figure of the structure of ERβ and its isoforms, ERβ2, ERβ4, and ERβ5. The domains of ERα and ERβ structures are color coded. (C) The DNA-binding domain (DBD) of ERα (Met179-Lys252). The ERα−DBD features two zinc-finger modules, each coordinated by a zinc ion (Zn2+) and four cysteine residues (indicated in pink). The amino acid residues forming the P-box and D-box are indicated in purple and green, respectively. (D) The cartoon structure of ERα−DBD dimer bound to consensus DNA sequence, GGTCAnnnTGACC (estrogen response element), with three non-specific (n) intervening bases (PDB: 1HCQ).
The ESR2 gene (spanning approximately 254 Kb), located on chromosome 14q23.2, encodes multiple ERβ isoforms due to alternative splicing or exon deletions of the last coding exon (exon 8), resulting in C-terminal truncations (92, 93). The full-length ERβ1 (60 kDa protein with 530 amino acids) is the only isoform capable of ligand-binding, whereas truncated isoforms such as ERβ2−β5 exhibit impaired ligand-binding activity due to the loss of AF2 function (Figure 1B) (94, 95). However, studies on ERβ isoform mRNA expression in breast cancer remain limited. Existing literature on the protein expression of different ERβ isoforms presents conflicting findings—some studies associate ERβ with favorable prognosis, while others report links to poor prognostic markers and reduced response to tamoxifen. Notably, ERβ2 mRNA expression is significantly correlated with better clinical outcomes in ERα−positive and node-negative tumors. A recent study further highlights that ERβ isoform mRNA and protein expressions are differentially associated with clinical characteristics and molecular subtypes of breast cancer (96). Simultaneous analysis of mRNA and protein expression levels of ERβ1, β2, and β5 across various BC subtypes revealed that ERβ isoform expression is significantly associated with Ki67 positivity (>15%), poor prognostic markers, and reduced OS. Specifically, high ERβ2 and β5 isoform expression is predictive of poor outcomes in ERα−negative breast cancer and TNBC.
The NTD, DBD, hinge, and LBD of ERα and ERβ share 17%, 97%, 36%, and 56% amino acid identity, respectively (97). Full-transcriptional activity of ERα is achieved through the synergism of AF1 and AF2, where AF1 is hormone-independent and mediates constitutive activation, while AF2 requires estrogen binding for activation. AF1 is activated by phosphorylation at Ser104, Ser106, Ser118, Ser167, and Ser305 via signaling pathways such as PI3K/AKT, PKA, MAPK, and CDK2/7. The ERα−DBD mediates interaction with the palindromic hexanucleotide sequence 5’-AGGTCAnnnTGACCT-3’ within estrogen response elements (EREs), with two ERα−DBD monomers binding to adjacent major grooves of the ERE. The ERα−DBD comprises two zinc ion (Zn)-binding motifs (98–102), each co-ordinated by four cysteine residues. The ‘P-box’ within the first Zn-finger module (Glu203, Gly204, and Ala207) defines DNA-binding specificity to the ERE, while the ‘D-box’ within the second Zn-finger module (Pro222, Ala223, Thr224, Asn225, Gln226) is essential for half-site spacing discrimination (Figures 1C, D) (97, 98, 100, 103–107). Following the DBD, the ‘D’ domain—also known as the hinge region—connects the DBD to the LBD and contains the nuclear localization signal (NLS), which becomes exposed upon estrogen binding. The ‘E’ domain encompasses the LBD, including the ligand-binding pocket (LBP), a dimerization interface, and sites for co-activator and suppressor interaction. The ERα−LBD (amino acids 304-554) has a globular structure with 12 α−helices (H1-H12). In the absence of a bound ligand (apo-receptor or unliganded state), the LBD is partially disordered or inactive, remains associated with heat shock proteins (HSPs, primarily HSP90), and is likely monomeric (Figure 2A) (108–110). Upon binding an agonist like estrogen (agonist-bound state), the LBD sheds the HSPs, dimerizes, and adopts a stabilized “agonistic conformation”. In this conformation, the terminal helix H12 folds over the LBP, creating a hydrophobic groove that facilitates co-activator binding (Figure 2B). By contrast, when an anti-estrogen like tamoxifen binds the LBD (antagonist-bound state), helix H12 repositions to block the co-activator binding groove, thereby inhibiting receptor activation (Figure 2C). Cartoon structures of ERα-LBD in the agonist (estrogen)-bound conformation (PDB: 1GWR) and the antagonist (4OHT)-bound conformation (PDB: 5W9C) are shown in Figures 2D, E, respectively. Notably, the nuclear export sequence (NES) is located within the DBD and LBD of ERα. Following ‘E’ domain, both ER isoforms contain an unstructured carboxyl-terminal ‘F’ region, which shares only 18% amino acid identity. Recent advancements in cryo-electron microscopy (cryo-EM) have significantly advanced our understanding of ERα transcriptional complexes, offering detailed architectural insights that overcome the limitations of traditional structural and biochemical approaches (111–113). Single-particle cryo-EM analyses have elucidated how the functional domain organization of ER supports its interaction with core-coactivators and how these co-activators collaborate to modify histones and initiate transcription. The structural organization of the ER/co-activator complex reveals that ERα recruits two SRC-3 molecules (SRC-3a and SRC-3b), each interacting with distinct regions of p300, thereby facilitating the recruitment of p300 to the ERα−binding site (111). Importantly, dimer formation is a pre-requisite for ERα function, and mutations that disrupt ERα dimerization render the receptor transcriptionally inactive.
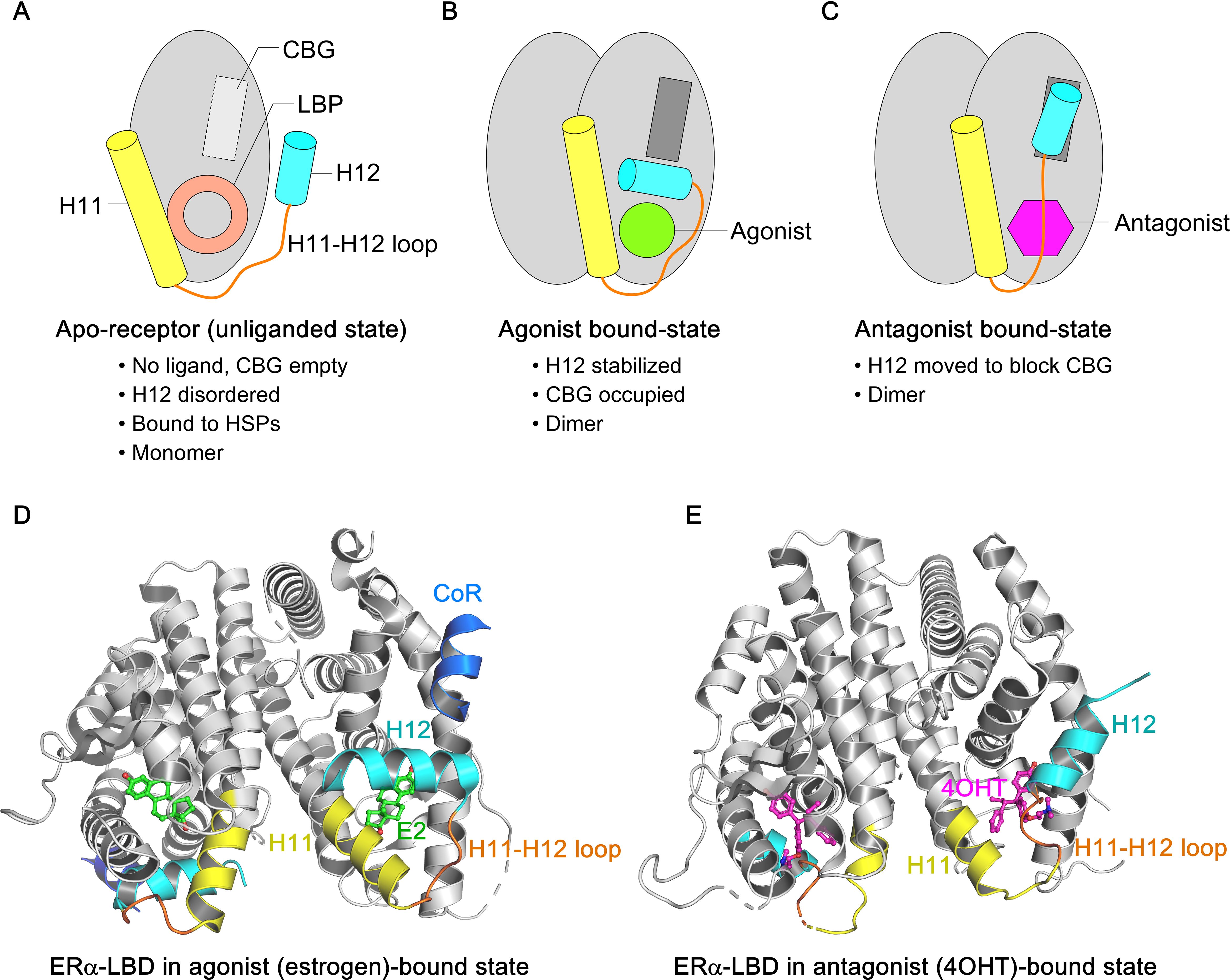
Figure 2. Overview of ligand-induced conformational states of ERα−LBD. (A-C) Schematic representation of three conformational states of ERα−LBD, highlighting the relative positions of H11 (yellow) and H12 (cyan) helices in the apo-state (no ligand), agonist-bound state (agonist in green), and antagonist-bound state (antagonist in purple), respectively. The H11–12 loop is shown in orange. (A) In the apo-state, both the ligand-binding pocket (LBP) and co-activator binding groove (CBG) are empty, preventing ER signaling. (B) In the agonist-bound state, H12 folds back to cover the LBP, enabling co-activator access to the CBG and initiating ER signaling. (C) In the antagonist-bound state, H12 shifts to block the CBG, inhibiting ER signaling. (D) The cartoon structure of wild-type ERα−LBD in complex with the agonist estrogen (in green sticks) and coregulator peptide (in blue) (PDB: 1GWR). (E) The cartoon structure of wild-type ERα−LBD in complex with the antagonist 4OHT (in purple sticks) (PDB: 5W9C). H11 and H12 helices are highlighted in yellow and cyan respectively, and the H11–12 loop in orange.
The topology of GPER is highly conserved and consists of an N-terminal extracellular domain, seven transmembrane α−helical regions connected by three extracellular loops and three intracellular loops, and a C-terminal intracellular domain (114). The N-terminal domain is essential for receptor maturation from the endoplasmic reticulum (ER) to the plasma membrane (PM). The GPER1 gene, located on chromosome 7 (7p22.3), encodes a 375-amino-acid protein with a molecular mass of 41 kDa. Upon binding ligands—including E2, SERMs, SERDs, or the GPER-selective agonist G-1—at either the extra-cellular surface or within the trans-membrane helices, GPER signals through a heterotrimeric G-protein. Estrogen or agonist binding activates the stimulatory Gαs subunit, thereby stimulating GPER, whereas antagonist binding activates the inhibitory Gαi subunit, leading to GPER inactivation (Figure 3A). Notably, both tamoxifen and fulvestrant exhibit significant binding affinity for GPER and can activate it in breast cancer. Interestingly, 43% of breast cancer biopsy samples co-express ER and GPER (Figure 3B). Moreover, physical interactions between GPER and both full-length ERα and ERα36 have been reported, suggesting a potential GPER-binding module in the ‘hinge’ region of both ERα (residues 295-311) and ERα36 (residues 123-139) (115, 116).
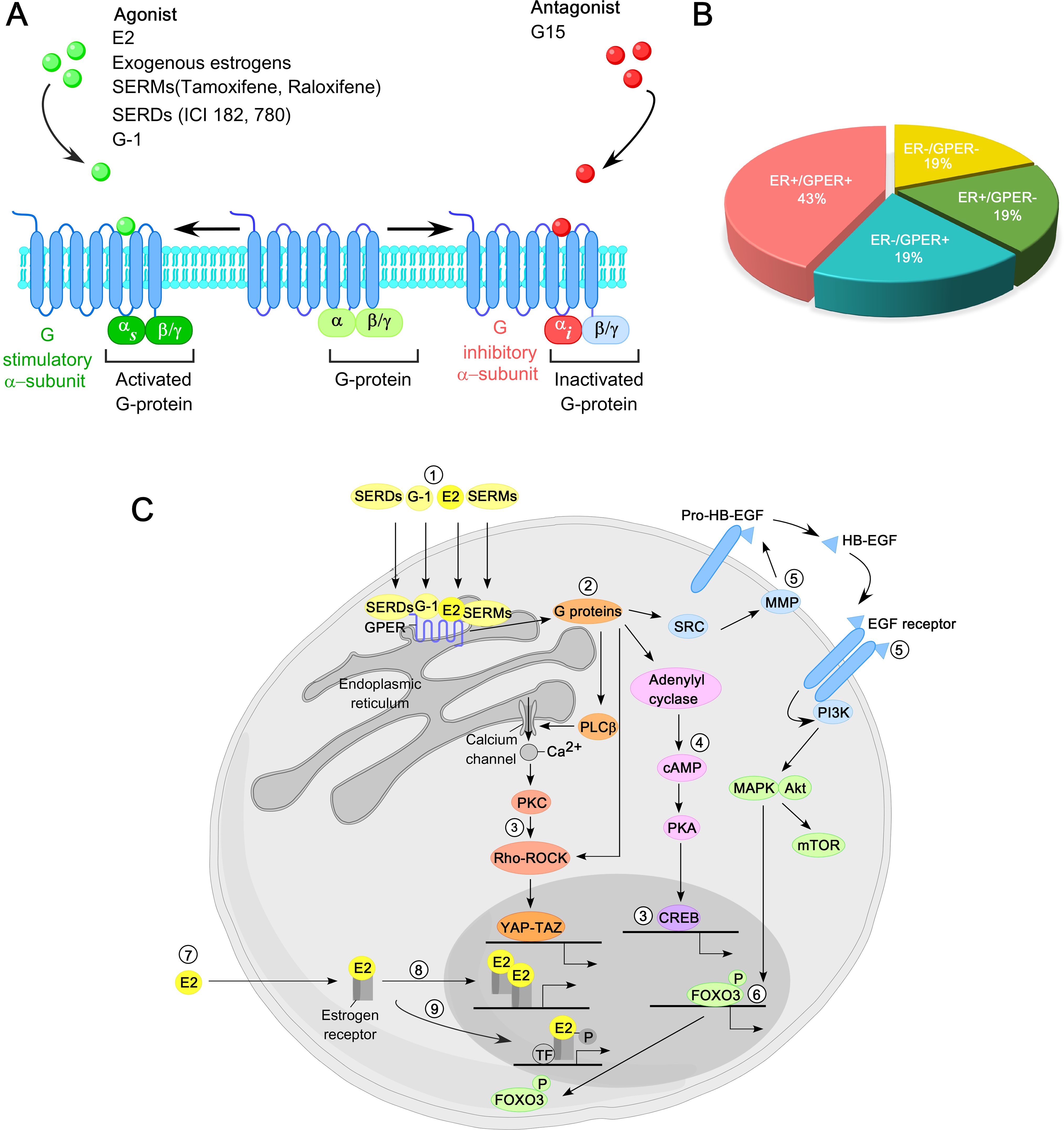
Figure 3. Overview of GPER function in breast cancer. (A) GPER is predominantly localized to the plasma membrane, featuring seven transmembrane helical domains, a ligand-binding pocket (LBP), and a G protein binding site. Upon interaction with estrogen or an agonist in its LBP, GPER activates a stimulatory G protein α−subunit (Gαs), leading to GPER activation. In contrast, interaction with antagonists triggers an inhibitory G protein α−subunit (Gαi), resulting in the inactivation of GPER. (B) Distribution of breast cancer based on the presence of ER and GPER in biopsy specimens. (C) Principal molecular pathways mediated by GPER in breast cancer. 17β−estradiol (E2), selective agonists such as G-1, SERMs, and SERDs activate GPER (1). GPER, in turn, activates heterotrimeric G proteins (2), triggering multiple downstream signaling cascades, including calcium mobilization from intra-cellular stores, activation of YAP-TAZ transcription factors via Rho/ROCK pathways (3), activation of Adenylyl cyclase-cAMP-PKA pathway (4), and activation of matrix metalloproteinases (MMPs) that cleave pro-heparin-binding epidermal growth factor (pro-HB-EGF) to release free HB-EGF, leading to EGFR trans-activation (5). This, in turn, activates MAPK (ERK1/2), Akt, and other signaling pathways. Activation of MAPK and Akt regulates gene transcription, including FOXO3 phosphorylation and degradation (6). In contrast, in the classical ER signaling, E2 binds to cytosolic or nuclear ERs (7), inducing receptor dimerization and binding to the promoter of ER-target genes (8). Additionally, activated ERs modulate the activity of other transcription factors (TFs) through protein-protein interactions (9).
3 ERα Post-translational modifications: defining stability and nucleo-cytoplasmic dynamics
Post-translational modifications (PTMs) of ERs, particularly ERα, play a crucial role in regulating its transcriptional activity in breast cancer and are fundamental to understanding ER biology (117). ERα undergoes PTMs under both ligand-dependent and ligand-independent conditions, often initiated by interactions with E2 or other ligands. The development of site-specific antibodies targeting post-translationally modified forms of ERα, along with advances in mass spectrometry, has greatly facilitated the identification of these PTM sites (118). To date, approximately 22 distinct PTM sites have been identified across the ERα structure, including phosphorylation, acetylation, sumoylation, and ubiquitination (Figure 4A). These modifications influence ERα’s stability (half-life), dimerization, transcriptional activity, subcellular localization, interactions with DNA and co-regulators, and degradation. In breast cancer cells, ERα is distributed across the cytoplasm and nucleus. Upon ligand binding (E2) to the ERα−LBD, ERα undergoes homo-dimerization and translocates to the nucleus, where the E2−ERα complex binds to EREs response elements (EREs) in the promoter regions of target genes. This binding facilitates co-regulator recruitment to the AF1/AF2 domains of ERα, driving gene expression. The schematic of ERα activation by estrogen is illustrated in Figure 4B.
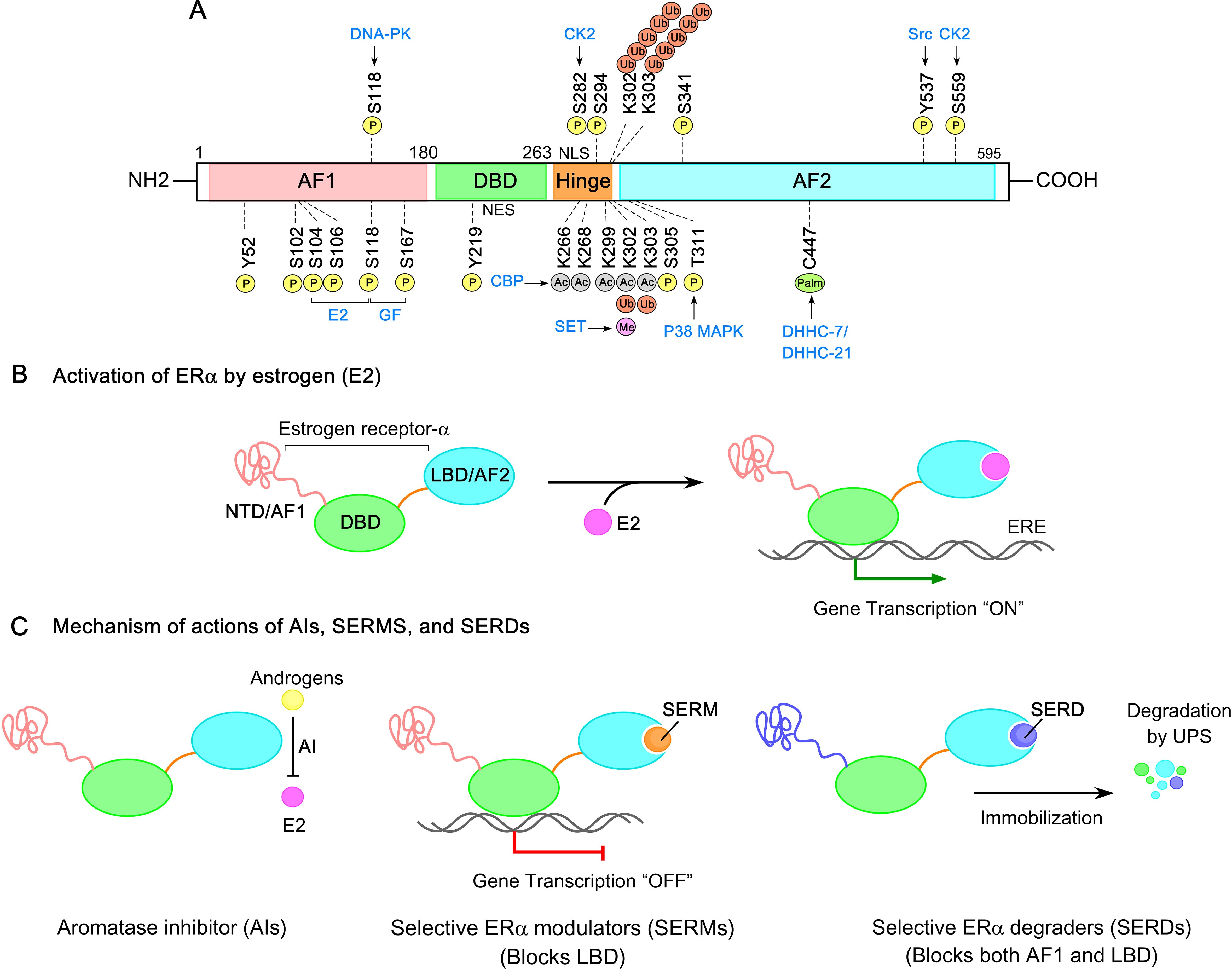
Figure 4. ERα Post-translational modifications (PTMs) and signaling pathways in breast cancer. (A) Amino acid residues in ERα subjected to phosphorylation, acetylation, palmitoylation, methylation, and ubiquitination are shown. Distinct post-translational modifications (PTMs) are color coded for clarity. (B) Estrogen-activated ERα initiates a transcriptional program that regulates target gene expression. (C) The mechanism of actions of aromatase inhibitors (AIs), selective ER modulators (SERMs), and selective ER degraders (SERDs). AIs block the conversion of androgens to estrogens, thereby reducing estrogen levels. SERMs inhibit the ERα−ligand binding domain (ERα−LBD) without affecting the DBD and AF1 domains. SERDs target both AF1 and LBD domains of ERα, leading to receptor immobilization, destabilization, and degradation.
Phosphorylation is a critical PTM of ERα, primarily targeting serine, threonine, and tyrosine residues. Among these, serine residues—particularly clustered within the N-terminal AF-1 region—are most frequently phosphorylated by MAPK, PI3K/AKT, and GSK-3, enabling ligand-independent transactivation of ERα. Key phosphorylation sites include Ser102, Ser104, Ser106, Ser118, Ser154, Ser167, Ser236, Ser294, Ser305, Ser559, Tyr52, Tyr219, Tyr537, and Thr311 (Figure 4A). Notably, Ser118, Ser167, and Ser305 are closely associated with ligand-independent ERα trans-activation and are often implicated in therapy-resistant ER-positive breast cancer.
Thomas et al. evaluated the relative significance of phosphorylation at Ser104, Ser106, and Ser118 for ERα activity, reporting the order of importance as Ser118>Ser104>Ser106 (119). Interestingly, substituting these serine residues with alanine had little effect, while replacement with glutamic acid (mimicking phosphorylation) markedly enhanced ERα activity, with the order of activity reversed—Ser106>Ser104>Ser118. Importantly, phosphorylation at Ser104/106 and Ser118 is essential for tamoxifen’s partial agonist activity, which has been linked to resistance in some breast cancers. Tamoxifen, exhibiting a dual role, inhibits the function of the LBD/AF-2 domain (antagonistic role) while simultaneously promoting ligand-independent activation of the AF-1 domain (agonistic role). The phosphorylation of Ser104/106 is estrogen-induced and mediated by kinases such as glycogen synthase kinase-3 (GSK3), cyclin-dependent kinase 2 (Cdk2), and MAPK (120). In contrast, Ser118 mediates both ligand-dependent and ligand-independent ERα activation, facilitating interactions with co-activators such as SRC-1 and CBP/p300, and is essential for ERα dimerization and RNA splicing (121–126). While estrogen induces Ser118 phosphorylation via kinases such as GSK3, IKKα, and CDK7, other stimuli—including epidermal growth factor (EGF) and insulin-like growth factor-1 (IGF-1)—can also trigger this modification through Ras-MAPK signaling. Recently, Du et al. showed that Ser118 phosphorylation triggers an unexpected conformational expansion of the intrinsically disordered ERα N-terminal domain (ERα−NTD), disrupting hydrophobic clustering between two aromatic-rich regions and promoting ligand-independent ERα activity (75, 127).
Phosphorylation of ERα at Ser305, mediated by protein kinase A (PKA) and p21 activated kinase 1 (PAK1), has been demonstrated to affect ER conformation, dimerization, interaction with coregulators, and DNA binding. Michalides et al. showed that this modification alters ERα conformation, contributing to tamoxifen resistance by preventing the receptor from adopting an inactive state despite tamoxifen binding (128). This conformational arrest shifts tamoxifen’s role from antagonist to agonist, promoting ERα−dependent transactivation. A phospho-mimetic ERα mutant, S305E, which mimics the constitutively phosphorylated state, exhibits increased binding to target gene promoters in the absence of ligand, suggesting that phosphorylation at Ser305 enables ligand-independent ERα activity (129). Thus, targeting PKA or blocking Ser305 phosphorylation offers a potential strategy to overcome endocrine resistance in breast cancer.
Conversely, phosphorylation at Ser167 is linked to favorable outcomes, including lower tumor grade, lymph node negativity, and longer relapse-free survival in BC patients (130–133). It also serves as a predictive marker for endocrine therapy response (134). In contrast, phosphorylation of ERβ remains less understood, with most identified sites located in the AF1 domain and the corresponding kinases yet to be identified.
Acetylation of ERα is a critical regulatory mechanism influencing its activity. ERα is acetylated by p300/CBP at five lysine residues—K266, K268, K299, K302, and K303 (Figure 4A) (135). Acetylation at K266 and K268 is estrogen-dependent and stimulatory, while modifications at K299, K302, and K303 are constitutive and suppress ERα transcriptional activity. Notably, the breast cancer susceptibility gene BRCA1 inhibits ERα acetylation by blocking p300 binding to ERα acetylation sites and/or by mono-ubiquitinating ERα at K302. Consequently, BRCA1 mutations increase the risk for BC development, while mutations at ERα acetylation sites—such as K266/268—confer resistance to BRCA1-mediated repression (136). Interestingly, K303 is a PTM hotspot, also subject to sumoylation and ubiquitination, and regulates methylation at adjacent K302. A recurrent K303R mutation, observed in ductal hyperplasia and invasive breast tumors, correlates with reduced relapse-free survival and confers resistance to tamoxifen and AIs by enhancing estrogen sensitivity (137–139). This mutation impairs K303 acetylation and promotes Ser305 phosphorylation. Barone et al. further showed that stable expression of a double K303R/S305A mutant receptor in MCF-7 cells induces AI resistance (137). Additionally, SET7-mediated methylation at K302 stabilizes ERα and enhances DNA binding, though acetylation at this site can hinder subsequent methylation (140). Notably, no acetylation sites have been identified for ERβ.
Palmitoylation — the reversible addition of palmitic acid to cysteine residues—regulates ERα stability, localization, activity, and membrane trafficking. ERα is palmitoylated at Cys447 by the acyltransferases DHHC-7 and DHHC-21 (Figure 4A), enhancing its hydrophobicity and anchoring it to membrane microdomains where it interacts with signaling molecules like Src (141, 142). This modification induces conformational changes that expose Src-binding sites, triggering rapid non-genomic estrogen signaling and promoting breast cancer cell proliferation. Upon E2 binding, ERα is depalmitoylated by acyl-protein thioesterases, leading to its dissociation from the membrane and translocation to the cytoplasm or nucleus. The dynamic palmitoylation-depalmitoylation cycle tightly regulates ERα function and represents a promising therapeutic target in ERα−positive BC.
Additionally, sumoylation of the ERα hinge region by SUMO-1 regulates its transcriptional activity (143). Notably, K266, K268, K299, K302, and K303 have been identified as key ERα sumoylation sites. Correspondingly, the double mutant (K266R/K268R) and the triple or five-lysine mutants (3K/R or 5K/R) exhibit significantly reduced levels of sumoylation compared to wild-type ERα, resulting in diminished transcriptional activity (144). However, sumoylation of ERβ has not yet been reported in the literature.
Furthermore, altered O-glycosylation of ERα is frequently observed in the majority of BC tissues, particularly in ERα−positive subtypes, where upregulated N-acetylgalactosaminyltransferase 6 (GALNT6 or GalNAc-T6) enzymatic activity is noted. Deng et al. demonstrated that GALNT6-mediated O-glycosylation at Ser573 is crucial for ERα stability and its nuclear trafficking in breast cancer cells (145). Consequently, targeting GALNT6 enzymatic activity or disrupting the GALNT6/ERα interaction with membrane-permeable peptides presents a promising therapeutic approach for ERα−positive breast cancer.
Ubiquitination adds another layer of complexity to ERα regulation. like other steroid receptors, ERα is subjected to ubiquitination via the 26S proteasome system, which governs both basal (ligand-independent) and ligand-induced degradation (146–148). In breast cancer cells, ERα degradation occurs through three distinct pathways: unliganded, ligand-bound (e.g., E2), and fulvestrant/other SERD-bound states. In its unliganded state, ERα is remarkably stable, with a half-life of up to five days. However, dynamic interactions with HSPs, co-chaperones, and E3 ubiquitin ligases (e.g. MDM2) target ERα for degradation (149), ensuring steady-state levels in the cytoplasm and maintaining homeostasis. Upon E2 binding, ERα’s half-life dramatically drops to 3–5 hours (150), as ligand-bound receptors are rapidly degraded to facilitate new protein synthesis. In contrast, fulvestrant and other SERDs induce ERα degradation independently of transcriptional activity or new protein synthesis (151, 152). Fulvestrant disrupts the HSP90-ERα complex and immobilizes ERα in the nuclear matrix, leading to its degradation (153). Berry et al. identified Lys302/303 as critical ubiquitination sites that protects against basal ERα degradation while promoting efficient E2- and fulvestrant-induced receptor turnover in BC cells (154). Key players in ERα ubiquitination include E3 ubiquitin ligases such as E6-AP, MDM2, EFP (estrogen-responsive finger protein), as well as the 26S proteasome and co-activators like SRC-1 and SRC-3. In the context of ERβ ubiquitination, the carboxy-terminus of HSP70-interacting protein interacts with N-terminus of ERβ receptor, facilitating its ubiquitination and eventual degradation.
Regarding GPER post-translational modifications, a recent study suggests that human GPER1 undergoes N-glycosylation, with asparagine 44 (Asn44) in the N-terminal domain being essential for receptor structure and activity (155). Mutating Asn44 to isoleucine inactivates the receptor, demonstrating that N-glycosylation at this site is critical for proper receptor maturation and trafficking to the plasma membrane. In contrast, residues 1–42 of the N-terminal domain do not appear to play a significant structural or functional role.
4 Regulatory factors governing ERα stability
Recent studies have identified key regulators that prolong ERα protein stability by inhibiting its polyubiquitination and degradation, thereby promoting ERα target gene expression and enhancing breast cancer cell proliferation. These ERα−polyubiquitination inhibitor proteins (EPIPs)—including kinases, transcriptional co-regulators, E3 ubiquitin ligases, and deubiquitinases—are often overexpressed in BC tissues, contributing to sustained ERα signaling and tamoxifen resistance. Notable EPIPs such as LMTK3, GSK3, cABL, TRIM family proteins, RNF8, RNF31, SHARPIN, and SMURF1 stabilize ERα by preventing its degradation (156). Collectively, these factors not only maintain elevated ERα levels and activity in breast tumors but also drive disease progression and therapeutic resistance.
4.1 Kinases and endonucleases
Several kinases—including LMTK3, DNA-PK, CK2, GSK3, and cABL—phosphorylate ERα, enhancing its stability and transcriptional activity while preventing degradation. LMTK3, a key ERα regulator in breast cancer, stabilizes ERα via direct phosphorylation and promotes its transcription by inhibiting PKC, reducing AKT phosphorylation, and facilitating FOXO3 binding to the ESR1 promoter (157–159). DNA-PK phosphorylates ERα at Ser-118, crucial for receptor stability and BC proliferation, with its inhibition leading to rapid ERα degradation (160). CK2 phosphorylates ERα at Ser167, Ser282 and Ser559, with Ser282 phosphorylation notably contributing to long-term receptor stabilization (161). Additionally, the endonuclease FEN1, often upregulated in tamoxifen-resistant breast cancer, enhances ERα transcription by supporting transcription complex assembly, and its inhibition leads to proteasome-mediated ERα degradation (162).
4.2 E3 ubiquitin ligases
Certain E3 ubiquitin ligases, especially members of the tripartite motif (TRIM) family, play critical roles in regulating ERα protein stability in breast cancer, by catalyzing the transfer of ubiquitin from E2 ubiquitin-conjugating enzymes to ERα lysine residues. While ubiquitination typically targets proteins for degradation, it can also modulate protein function and stability. Several TRIM proteins—including TRIM3, TRIM11, and TRIM56—enhance ERα stability (163, 164), whereas TRIM8 promotes its cytoplasmic degradation (165). For instance, TRIM56 interacts with the AF-1 domain of ERα and promotes K63-linked polyubiquitination, stabilizing ERα while inhibiting degradation-associated K48-linked ubiquitination (166). TRIM11, often overexpressed in BC, similarly stabilizes ERα, and its depletion impairs tumor cell proliferation and migration (163). Beyond TRIM proteins, atypical E3 ligases such as RNF31, RNF8, and SHARPIN mono-ubiquitinate ERα, shielding it from proteasomal degradation and enhancing ERα signaling (167–170). Additionally, SMURF1, HOIL-1, and RNF181 stabilize ERα by either inhibiting K48-linked ubiquitination or promoting K63-linked poly-ubiquitination (156, 171, 172). These findings highlight the crucial role of E3 ligases in modulating ERα turnover and activity, offering potential therapeutic targets for disrupting ERα−driven BC progression.
4.3 Ca2+ binding proteins
ERα transcriptional activity depends on its interaction with calmodulin (CaM), a ubiquitous Ca2+ sensor. Mutation of CaM (CaM1234), which disrupts Ca2+ binding, reduces E2-induced ERα transactivation in MCF7 cells. The interaction is mediated by ERα residues 298-303, particularly Lys-302 and Lys-303, which protect ERα from degradation and enhance its stability (173, 174). Additionally, calcineurin—a Ca2+ dependent phosphatase highly expressed in ERα−positive breast cancer with poor endocrine therapy response—stabilizes ERα by dephosphorylating Ser294, thereby preventing its degradation (175). Targeting the Ca2+/calmodulin complex or calcineurin, therefore, offers a potential therapeutic avenue for ERα−positive breast cancer.
4.4 Deubiquitinases
Deubiquitinases (DUBs) are proteases that regulate protein turnover by removing ubiquitin chains from substrate proteins, thereby influencing ERα stability in breast cancer. Several DUBs have been identified as key stabilizers of ERα, contributing to tumor progression and therapy resistance. USP7 shows a positive correlation with ERα levels in BC tissues and directly interacts with ERα to promote its deubiquitination and stabilization (176). Similarly, USP15 inhibits K48-linked ubiquitination of ERα, preventing its degradation, whereas USP15 depletion sensitizes ERα−positive breast cancer cells to tamoxifen (177). USP35 also stabilizes ERα, reducing the efficacy of tamoxifen and fulvestrant in ERα−positive breast cancer cells (178). Other DUBs, including OTUD7B and MINDY1, are over-expressed in breast cancer and support ERα stability by removing and K11- and K48-linked ubiquitin chains, with OTUD7B expression being associated with poor prognosis (179–181).
4.5 Concentration-inducible ERα function
The balance between ERα stability and degradation has significant implications for BC progression and therapeutic response. Fowler et al. demonstrated that elevated ERα concentrations can lead to its constitutive activation, driving aberrant promoter occupancy and gene expression even in the absence of estrogen (182). This phenomenon, termed “concentration-inducible ERα function”, involves serine 104/106/118-independent AF-1 transactivation and promotes breast tumor growth independently of estrogen, suggesting that ERα can drive transcription through mechanisms distinct from classical ligand-binding and phosphorylation-dependent pathways (182). High ERα concentration is often associated with poor prognosis and endocrine resistance in BC.
Besides, with the increasing use of AIs, breast cancer cells adapt to a low-estrogen environment, developing resistance through long-term estrogen deprivation (LTED). LTED induces estrogen hypersensitivity or super-sensitivity, enabling cells to respond to estrogen at concentrations 2–3 logs lower than those required for wild-type cells, or to grow in the absence of estrogen altogether (183–186). Both adaptations are characterized by elevated ERα expression, enhanced Ser118 phosphorylation, and activation of ERK1/2 and PI3K pathways, ultimately compensating for low estrogen levels. Paradoxically, ET resistance can also arise from reduced ERα levels due to enhanced degradation, as ERα is the primary target of SERMs and SERDs. For example, the ubiquitin-binding protein CUEDC2 promotes ERα degradation via the proteasome pathway; consequently, malignant mammary tumors with high CUEDC2 expression under tamoxifen-resistant conditions exhibit low ERα levels (187). These findings underscore that both prolonged ERα stability and accelerated degradation can disrupt the effectiveness of ET, highlighting the need for precise regulation of ERα homeostasis to optimize therapeutic outcomes.
In summary, ERα stability is not governed by a single linear pathway but by a dynamic and interconnected regulatory network of PTMs, protein-protein interactions, cellular signaling pathways, and subcellular trafficking mechanisms. PTMs—such as phosphorylation, mono-/poly-ubiquitination, deubiquitination mediated by kinases, E3 ubiquitin ligases, and deubiquitinases—play central roles in regulating ERα’s half-life, localization, transcriptional activity, and therapeutic resistance. These modifications often compete for the same sites on ERα, such as K303, underscoring the complexity of this tightly controlled system. Several cellular signaling pathways—including PI3K/AKT/mTOR and MAPK/ERK, Src, NF-κB and Wnt/β-catenin—are integral to maintain ERα stability and activity. Numerous studies have shown that ERα stability and nuclear export are critical for modulating both its nuclear and extra-nuclear functions, ultimately influencing BC progression and response to ET. Several proteins protect ERα from degradation while also impacting its subcellular distribution. For instance, elevated expression of dynein light chain 1 (DLC1) promotes E2-induced nuclear accumulation of ERα, enhancing its transcriptional activity (188). Conversely, the ERα mutant Y537F, which cannot bind the exportin protein CRM-1, accumulates in the nucleus and exhibits increased transcriptional activity. Normally, phosphorylation at Tyr537 by Src facilitates ERα interaction with CRM-1, promoting its nuclear export and subsequent degradation; The Y537F mutation disrupts this process, leading to ERα nuclear retention and heightened signaling (189). Collectively, these findings highlight the importance of both stability/degradation dynamics and subcellular trafficking in ERα regulation and endocrine resistance.
5 Structural insights into ERα hot-spot mutations & endocrine resistance:
Endocrine resistance—either de novo or acquired—is a major cause of relapse in ER-positive breast cancer. It reflects the tumor’s ability to evade or counteract therapies targeting the ERα signaling pathway, including tamoxifen, fulvestrant, and AIs (190). The mechanisms of action of these agents are illustrated in Figure 4C. Acquired resistance is frequently driven by emerging ESR1 mutations, noted in a significant proportion of patients with ER+ MBC (191, 192). Additionally, the increased proportion of therapy-resistant tumor-initiating breast cancer stem-like cells (BCSCs; CD44+CD24−/lowLineage−) contributes to treatment failure and poor survival, especially in tamoxifen-resistant tumors (193). Briefly, these resistant cells overexpress drug efflux transporters and display stem-like characteristics, including enhanced proliferation, increased mammospheres formation, upregulation of stemness-related proteins (OCT-4, SOX2, Nanog, CD133), and increased epithelial-mesenchymal transition (EMT) plasticity. Fulvestrant resistance is associated with activation of the MEK/ERK, NF-κB, EGFR, PI3K/AKT, and β-catenin pathways. In contrast, AI resistance—which affects over 20% of early-stage and most metastatic cases—is driven by both intrinsic (e.g., upregulation of FGFR, ERBB2, IGF1R, PI3K-AKT-mTOR, MAPK signaling) and extrinsic factors, including interactions with the tumor microenvironment (34).
Large-scale genomic studies, such as The Cancer Genome Atlas (TCGA) project, have provided critical insights into the genomic landscape and heterogeneity of breast cancer, revealing a higher frequency of ESR1 mutations in MBC. Constitutively active ERα mutants were first identified in the 1990s, through structure–function studies using random or site-directed mutagenesis of breast cancer cells in the absence of E2 or in the presence of anti-estrogens. Recent technological advancements, including next-generation sequencing (NGS) and droplet digital PCR (ddPCR), have enabled the detection of recurrent, missense, activating mutations clustered in ERα−LBD—particularly within the C-terminal H12 helix—in approximately 40% of BC patients previously treated with tamoxifen and AIs (51–53, 194–208). These activating ERα−LBD mutations are summarized in Table 1, including their proposed mechanisms of action, pharmacological phenotypes, and clinical implications. Since these mutations underscore the clinical need for more effective endocrine therapies, a detailed understanding of how the structure of ERα, particularly the ligand-induced conformation of its LBD, relates to its activity is essential (202). These mutations confer constitutive, ligand-independent activity at levels comparable to those induced by estrogen, implicating clonal selection as a key driver of endocrine resistance (48, 220, 221). Structural studies have shown that ERα−LBD mutations stabilize the receptor in an agonistic conformation, promoting ligand-independent ERα activation, altered gene expression, and changes in ERα−dependent cistrome (55, 222–224). The prevalence of common ESR1 mutations in tumor specimens from patients with endocrine-resistant, ER-positive breast cancer is depicted in Figure 5A.
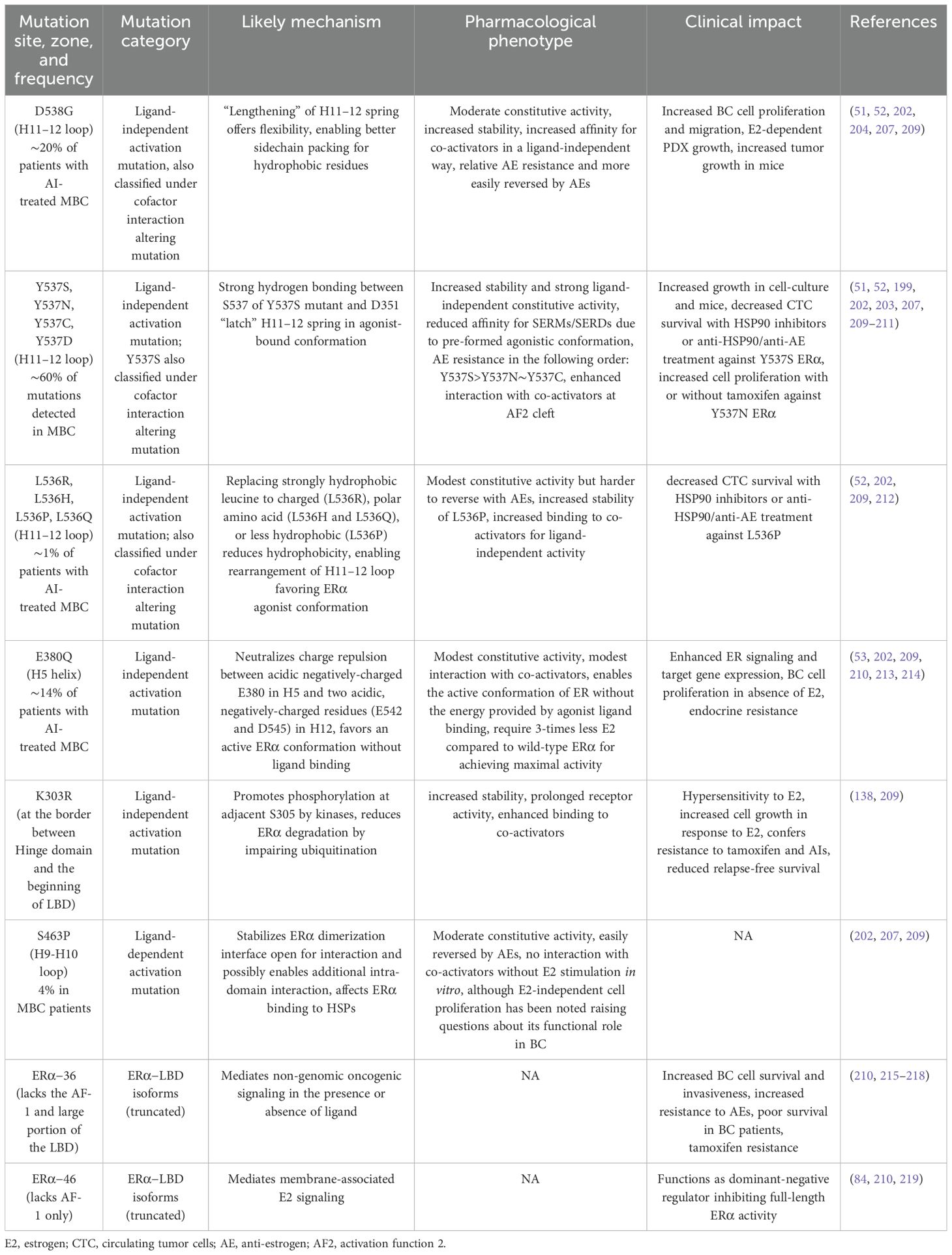
Table 1. Major ERα mutations, and their pharmacological phenotypes, mechanisms, and clinical impact.
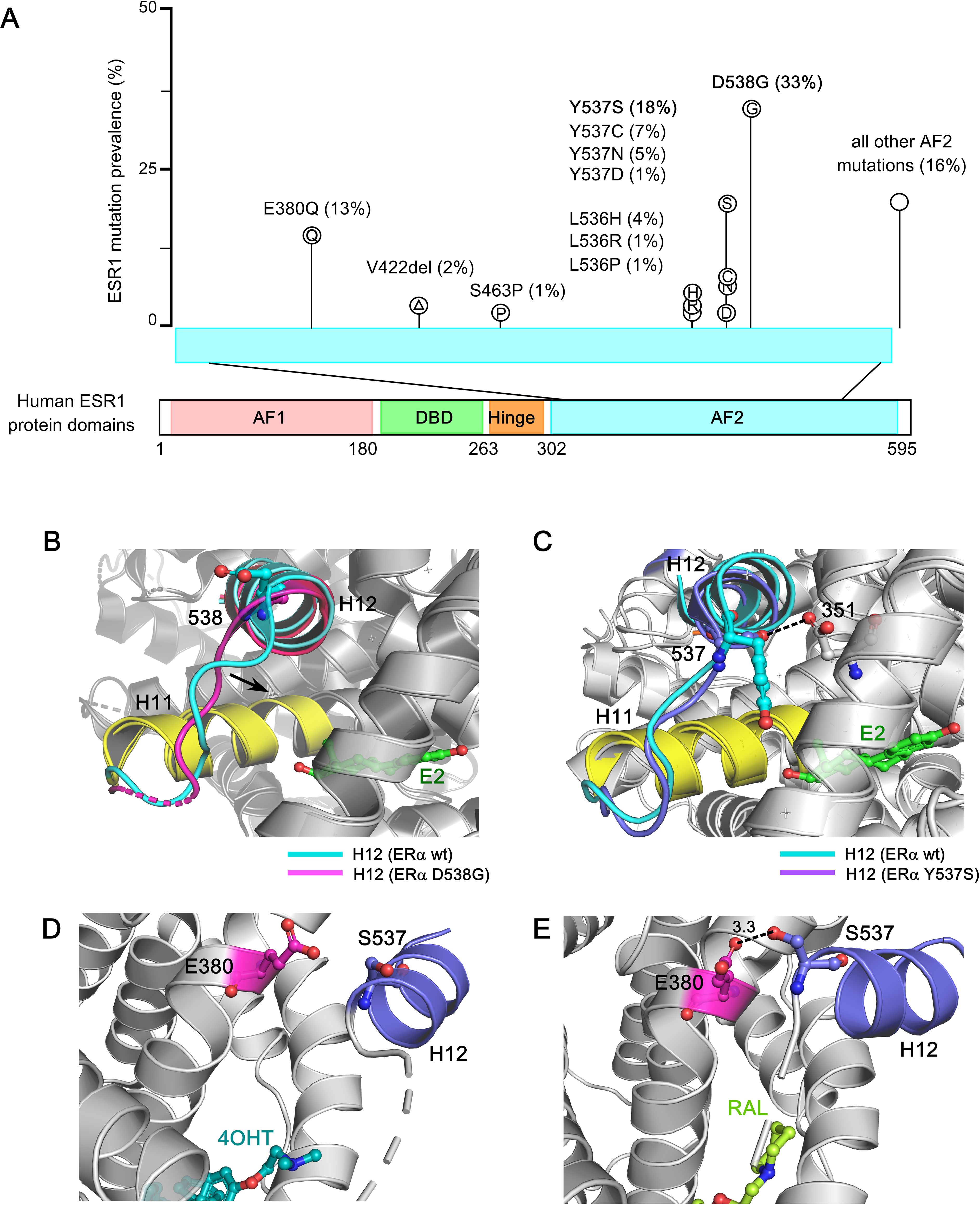
Figure 5. Structural basis of ESR1 activating mutations and resistance to inhibition by SERMs and SERDs. (A) Prevalence of common ESR1 mutations in breast tumor specimens from patients with endocrine-resistant ER+ breast cancer. The data is derived from two large retrospective studies, encompassing 2800 BC patients reflecting 283 ESR1 mutations. (B) Superposition based on alpha carbons of wild-type ERα−LBD in complex with E2 (PDB: 1GWR) and D538G ERα (PDB: 4Q13). The H12 helix in wild type ERα and D538G ERα are highlighted in cyan and purple respectively. The H11 helix is highlighted in yellow for both the wild-type and mutant structure, and the ligand (estrogen) is shown in green sticks. The arrow denotes the direction of new H11-H12 loop packing into the hydrophobic hormone-binding pocket in D538G ERα mutant. (C) Superposition of alpha carbons from the wild-type ERα−LBD in complex with E2 (PDB: 1GWR) and the Y537S ERα mutant (PDB: 2B23), highlighting the S537-D351 hydrogen bond with a dashed line. The H12 helix in Y537S ERα mutant is shown in violet, and the Y537S mutation is shown in violet sticks. In Y537S ERα, the strong hydrogen bond between S537 and D351 lock the H11-H12 region in an agonist conformation, turning on constitutive activity. (D) The ineffective SERM 4OHT (in cyan-blue sticks) in complex with Y537S ERα mutant (PDB: 6V87). The H12 helix is shown in violet, and the S537 amino acid is highlighted in violet sticks. In complex with 4OHT, H12 helix is displaced from the AF2 cleft, enhancing co-regulator binding at AF2 and leading to ERα activation. (E) The effective SERM/SERD Raloxifene (RAL) (in light green) in complex with the Y537S ERα (PDB: 7UJC), stabilizes the antagonist conformation by forming a new S537-E380 hydrogen bond (3.3 angstrom). The hydrogen bond is indicated with a dashed line.
Importantly, the dynamic nature of the H12 helix plays a critical role following the E2: ERα−LBD interaction. Among the most prevalent point mutations in ERα, Tyr537 is the most frequently mutated site, giving rise to four distinct variants: Y537S, Y537N, Y537C, and Y537D. These mutations interfere with receptor degradation, contributing to ET resistance and metastasis in breast cancer patients. Hot-spot mutations in the ERα structure—such as Y537S, Y537N, Y537C, D538G, and E380Q—differentially impact its structural integrity, promoting estrogen-independent activity. The ERα−LBD is an intrinsically disordered α−helical bundle that encapsulates a hydrophobic LBP, where estrogen binds, and the AF2 domain, which serves as the interaction site for ligand-dependent co-regulators. Access of co-regulators to the AF2 cleft depends on the structural dynamics of H12 helix within the ERα−LBD (225). In the apo or unliganded state, the H12 helix is highly dynamic, rendering the AF2 site inaccessible to coregulators (see Figure 2A). Estrogen binding provides favorable folding energies, allowing H12 helix to fold over the LBP, thereby opening the AF2 cleft for co-regulator interactions (PDB: 1GWR) (226) (see Figure 2B). Furthermore, this interaction exposes a hydrophobic patch in the loop between H11 and H12, resembling a “spring-like strained conformation” stabilized by estrogen. Mutations at leucine-536 (L536), tyrosine-537 (Y537), and aspartate-538 (D538) relieve this tension by reducing the hydrophobicity of this patch, stabilizing the unliganded ERα in an agonist-bound conformation (227). The D538G mutation, in the H11-H12 loop of ERα−LBD, is observed in ∼20% of BC patients with AI-treated metastatic disease and causes the “lengthening” of the H11-H12 spring in ERα, conferring constitutive activity (Figure 5B). (228). In contrast, high-resolution x-ray crystal structure reveals that in the Y537S mutation, S537 establishes a new hydrogen bond with D351, stabilizing the H12 helix in an agonist-bound conformation (PDB: 2B23) (229) (Figure 5C). This mutation confers greater therapeutic resistance to 4OHT by enhancing co-regulator binding at the AF2 cleft (PDB: 6V87), leading to ERα activation (Figure 5D). However, raloxifene (RAL) in complex with the Y537S ERα−LBD mutant favors the highly buried H12 antagonist conformation through the formation of a new S537-E380 hydrogen bond, effectively turning the receptor off (Figure 5E). Interestingly, the Y537S and D538G mutants exhibit a 3-10-fold reduced affinity for SERMs/SERDs due to their pre-formed agonistic conformation, contributing to ET resistance. These mutants also drive transcriptomic reprogramming, resulting in increased expression of metastasis-related genes. Notably, the E380Q mutant requires three times less estrogen than wild-type ER to achieve its maximal activity, while the S463P mutation leaves the ERα dimerization domain constitutively open for interaction. Additionally, mutations at leucine-536 (L536H/R/P/Q) compromise the structural integrity of the receptor, causing it to adopt a ligand-bound active conformational state (228).
As dynamic biomarkers of disease progression and endocrine resistance, ESR1 mutations present a valuable platform for improving clinical outcomes in ER-positive metastatic breast cancer. In this context, Goldberg et al. identified the most frequent ESR1 mutations−Y537S, D538G, and E380Q−as novel targets for developing breast cancer immunotherapies aimed at restoring endocrine sensitivity (230). Notably, mutations such as Y537N/C/S and D538G have been detected in circulating tumor DNA (ctDNA) in 39.1% of metastatic patients, showing a strong correlation with resistance to AIs (231). Furthermore, long-term estrogen deprivation (LTED), as previously discussed, promotes the selection of naturally occurring ESR1 mutations, including Y537C and Y537S, in ESR1-positive cell lines (221).
To further investigate the functional implications of these mutations, CRISPR-Cas9−engineered mutant breast cancer cell lines harboring L536R, Y537C, Y537N, Y537S, and D538G mutations demonstrated varying sensitivities to anti-estrogens such as tamoxifen and fulvestrant (227). Consistently, clinical data from the PALOMA-3 and MONARCH-2 trial control arms showed that fulvestrant was less effective in patients with ESR1 mutations compared to those with wild-type ER, highlighting increased resistance in this subset (232, 233). These acquired ESR1 mutations underscore the clinical need for developing next-generation ERα−targeted agents. Both the pharmaceutical industry and academia have been actively working to design novel ER inhibitors that block the ER signaling pathway, with each class operating through a distinct mechanism of action (see Table 2).
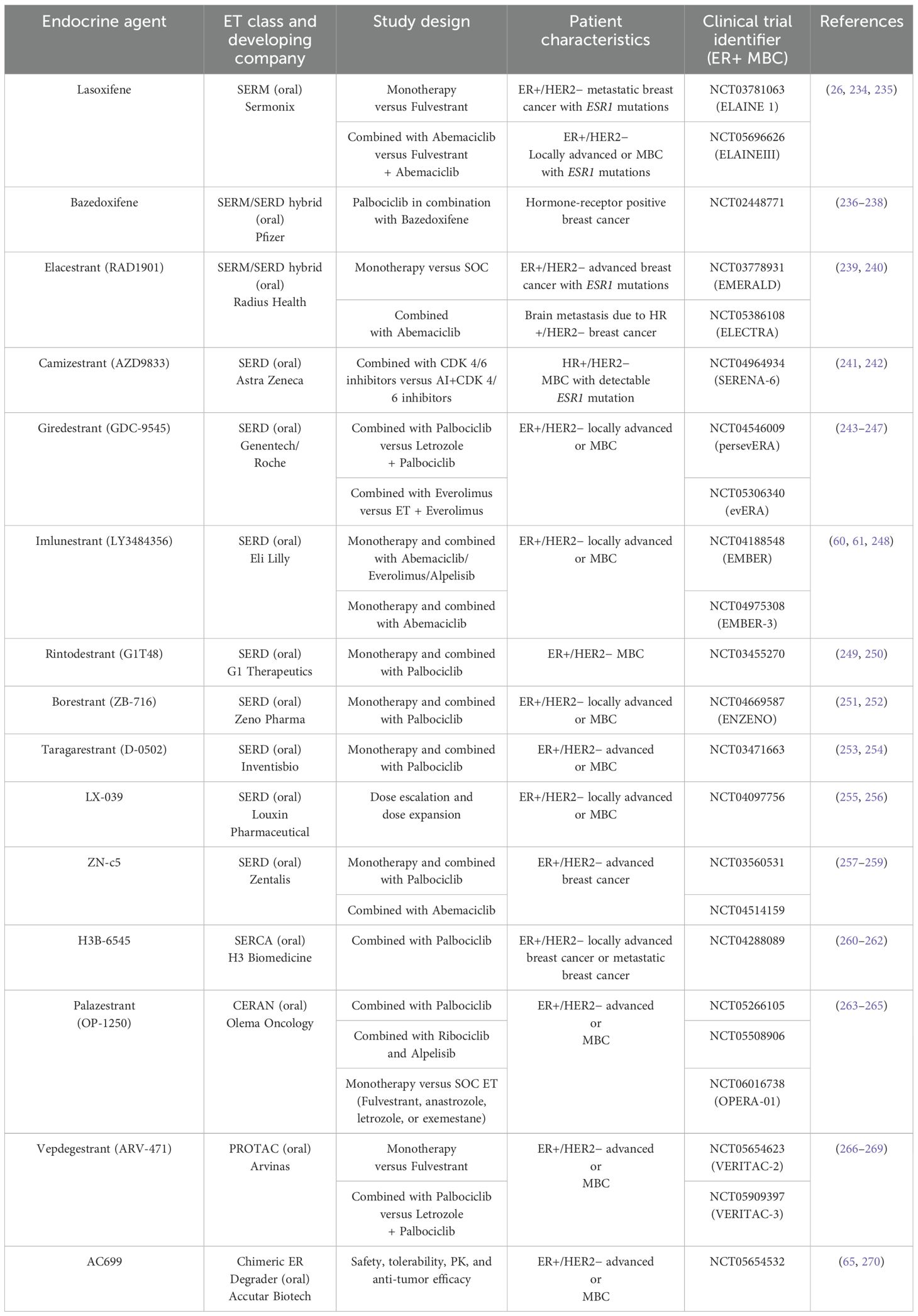
Table 2. Next-generation ER-targeting agents in clinical trials.
Importantly, the absence of detectable ESR1 mutations in primary breast tumors suggests that these mutations emerge through clonal selection during tumor evolution, enabling tumor cells to evade hormonal therapies. To monitor such adaptive genomic alterations, single-cell DNA sequencing of both tissues and serial plasma samples could enable real-time tracking of ESR1 mutation dynamics across disease stages. Early detection of ESR1 mutations in subclonal populations may help optimize adjuvant therapy decisions. Additionally, structural modeling of mutant ER could provide insight into conformational alterations and aid in designing peptide-based or alternative targeted therapies. Given the critical role of co-activators in the ligand-independent activity of mutant ERα, disrupting these interactions may represent a promising therapeutic strategy to reverse endocrine resistance.
6 Role of GPER in ERα–positive breast cancer
GPER is primarily localized to intra-cellular membranes, including the endoplasmic reticulum and Golgi apparatus, where it mediates non-genomic estrogen signaling (Figure 3C). In 2007, the International Union of Basic and Clinical Pharmacology officially designated GPR30 as GPER, recognizing it as a therapeutic target in breast cancer, including ERα−positive subtype (271–274). GPER is broadly expressed in breast cancer cell lines and primary tumors, with high expression levels correlating with increased tumor size, metastasis, tamoxifen resistance, and poor prognosis. Therefore, delineating ER−GPER crosstalk is crucial for understanding BC progression and ET resistance in ERα−positive tumors.
Notably, SERMs such as tamoxifen and raloxifene, and SERDs like fulvestrant, act as GPER agonists, inducing its expression and activating pro-survival signaling pathways (27, 69, 275–279). Due to GPER’s distinct pharmacological profile, the development of ERα-selective agents that do not cross-react with GPER is essential. Parallel efforts to develop GPER-selective ligands have deepened our understanding of its role in BC progression (see Table 3) (293). A notable example is G-1, a GPER-selective agonist identified through compound library screening in 2006 (280). Additional GPER- agonists include indole-thiazole derivatives such as GPER-L1 and GPER-L2 (282). The discovery of GPER-selective antagonists—G15 and G36 (290, 291)—has further illuminated GPER’s functions in breast cancer. Other antagonists include MIBE (Molecular Inhibitor for Breast Cancer Estrogen Receptor), pan-estrogen receptor antagonists, and CIMBA. MIBE targets both ERα and GPER, blocking their activation by estrogen and related agonists. Pan-estrogen receptor antagonists inhibit ERα, ERβ, and GPER, whereas G36 selectively targets GPER, blocking non-genomic signaling without significantly affecting ERα or ERβ. Its structural analogue, CIMBA, demonstrates even greater GPER-binding affinity and specificity (294). Two novel benzopyrroloxazine-based selective GPER antagonists, PBX1 and PBX2, inhibit GPER-dependent signaling in breast cancer cells and cancer-associated fibroblasts (CAFs), but require further validation in preclinical and clinical trials (295).
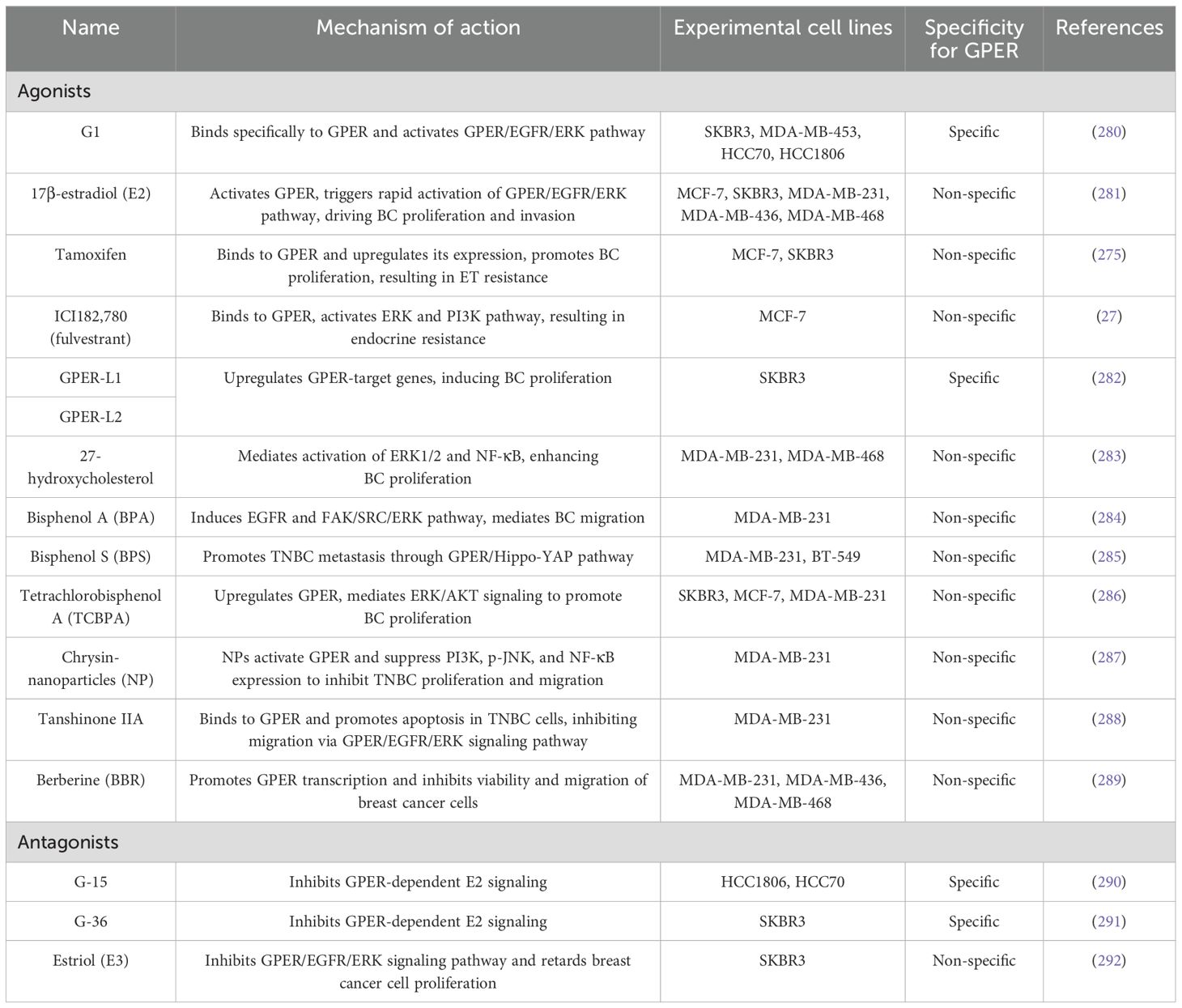
Table 3. GPER agonists and antagonists in breast cancer.
Recent studies emphasize the prognostic significance of GPER localization: plasma membrane-localized GPER correlates with poor outcomes, while its absence on the plasma membrane is associated with excellent long-term prognosis in tamoxifen-treated tumors (296). Cytoplasmic GPER is linked to non-ductal histologic subtypes, better differentiation, and lower tumor grades, while nuclear GPER is associated with poorly differentiated carcinomas and TNBC subtypes (297, 298). These findings underscore the need for precision therapies tailored to GPER expression levels and subcellular localization in BC patients.
6.1 GPER and phyto- and xeno-estrogens molecules
A wide range of phytoestrogens and xenoestrogens stimulate cAMP production, activate protein kinases, and drive GPER-dependent gene transcription in BC cells. Phytoestrogens—such as quercetin (299), genistein (300, 301), resveratrol (302), (-)-epicatechin, oleuropein, daidzein (303), equol, and icariin—are plant-derived compounds that mimic estrogen and target ERs. In contrast, xenoestrogens are synthetic, chemically stable endocrine-disrupting chemicals (EDCs) found in plastics, surfactants, pesticides, and pharmaceuticals. Examples include Bisphenol A (BPA), polychlorinated biphenyls (PCBs), diethylstilbestrol (DES), and Dichlorodiphenyltrichloroethane (DDT) and its metabolites. These compounds often act as GPER agonists and interact with both classical ERs and GPER, sometimes exerting opposing effects (285, 304, 305). For instance, 4OHT functions as an ERα antagonist but a GPER agonist, whereas estriol (E3) acts as an ERα agonist but a GPER antagonist.
6.2 GPER-mediated non-genomic signaling in breast cancer
GPER-mediated non-genomic signaling elicits rapid cellular responses independent of direct gene expression (306). Upon activation by E2 or ER antagonists, GPER initiates intracellular signaling cascades at the plasma membrane, leading to the production of second messengers such as cAMP, IP3, DAG, and Ca2+. These molecules activate downstream kinases including PKA, PKC, and MAPKs (Figure 3C), which drive cell proliferation, migration, and invasion. GPER also regulates the expression of genes such as c-FOS (299), CTGF, and EGR1, promoting tumor progression. It enhances motility via cyclins (A1, D, E), CTGF, CXCR1, and Notch signaling. For example, Chen et al. demonstrated that estrogen and fulvestrant enhance MCF-7 adhesion to the extracellular matrix via the GPER-calpain axis (307). GPER activation also promotes invasion of inflammatory BC cells by activating p-ERK1/2, suggesting its role in metastatic dissemination (308). Importantly, GPER expression is higher in metastatic lesions than in matched primary tumors, underscoring its role in disease progression. In TNBC, GPER has strong prognostic value, particularly in aggressive subtypes, including basal-like, immunomodulatory, mesenchymal-like, and luminal androgen receptor (LAR). Elevated GPER expression is strongly associated with reduced relapse-free survival (RFS) and distant metastasis-free survival (DMFS), especially in patients with additional risk factors such as lymph node metastasis (LNM), high tumor grade (G3), and advanced TNM stage (309). Zhu et al. further demonstrated that GPER activation enhances TNBC cell stemness, increasing the CD44+CD24−/low population and upregulating stemness-related genes in MDA-MB-468-derived mammospheres (310). These findings support the therapeutic potential of GPER-targeted inhibitors in managing aggressive BC subtypes, including TNBC (311).
6.3 GPER & tamoxifen resistance in ERα–positive breast cancer
Elevated GPER levels have been observed in BC patients primarily treated with tamoxifen, linking GPER signaling to tamoxifen resistance (69, 312). Early studies demonstrated that 4OHT exerts GPER agonistic activity, potentially inducing tamoxifen-resistant tumors instead of inhibiting them (301, 313). Through sustaining estrogen signaling in the presence of tamoxifen, GPER contributes to ET resistance, with AIs proving more effective than tamoxifen in ER+/GPER+ tumors. Ignatov et al. further reported that tamoxifen-treated patients with GPER-positive tumors exhibited increased GPER expression and decreased OS compared to those who did not receive tamoxifen (69). Mechanistically, tamoxifen cross-activates GPER, inducing proliferation of resistant breast cancer cells and promoting the nuclear expulsion of the pro-apoptotic transcription factor FOXO3a, thereby shifting cells toward a pro-survival state (314). Additionally, tamoxifen-mediated GPER cross-activation increases aromatase expression, further exacerbating resistance (275). Preclinical evidence supports targeting GPER as a strategy to overcome tamoxifen resistance: GPER knockdown or co-treatment with the GPER antagonist G15 attenuates breast cancer cell proliferation (70), and combining G15 with tamoxifen restores sensitivity in tamoxifen-resistant MCF-7 xenografts. Furthermore, G15 sensitizes epithelial breast cancer cells to doxorubicin by inhibiting EMT through GPER down-regulation (315). Collectively, these findings highlight the complex interplay between GPER and ERα signaling in driving gene expression changes that fuel ERα−positive BC progression. The non-genomic pathways mediated by GPER, along with critical intermediates and enzymes involved, are outlined below (refer to Figure 3C):
6.4 GPER, IP3-dependent calcium mobilization, and activation of the YAP-TAZ pathway
Upon activation by E2, G-1, SERMs, or SERDs, GPER interacts with hetero-trimeric G-proteins (Gα, Gβ, and Gγ) on the inner surface of the plasma membrane (316). G-protein activation leads to the dissociation of Gαq/11 from the Gβγ dimer. Activated Gαq/11 then stimulates phospholipase C (PLC), which catalyzes the hydrolysis of PIP2 (phosphatidylinositol 4,5-biphosphate) into IP3 (inositol triphosphate) and DAG (diacylglycerol). IP3 binds to its receptors on the endoplasmic reticulum, triggering Ca2+ release into the cytosol (Figure 3C), while DAG activates protein kinase C (PKC). The rise in cytosolic calcium concentration activates calcium-dependent kinases such as calcium/calmodulin-dependent protein kinase II (CaMKII) and promotes actin cytoskeleton reorganization. Simultaneously, GPER signaling activates Rho-GTPases, including RhoA, enhancing actin cytoskeleton assembly and increasing cellular tension. This mechanical tension inhibits the Hippo pathway, allowing unphosphorylated YAP (Yes-associated protein) and TAZ (transcriptional coactivator with PDZ-binding motif) to translocate into the nucleus (317). Nuclear YAP and TAZ drive the expression of genes involved in tumor cell proliferation, survival, angiogenesis, EMT, stemness, and drug resistance.
6.5 Activation of the Adenylyl Cyclase-cAMP-PKA pathway
GPER-mediated transcriptional regulation occurs indirectly through the cAMP and EGFR signaling pathways. Upon activation by E2, GPER signals via heterotrimeric G-protein, where the Gαs subunit undergoes activation and stimulates adenylyl cyclase to convert ATP into cAMP, thereby increasing intracellular cAMP levels (318). cAMP acts as a secondary messenger to activate PKA, which phosphorylates transcription factors such as CREB (cAMP response element-binding protein). Phosphorylated CREB then shuttles into the nucleus to induce the expression of genes involved in breast cancer cell proliferation, survival, metabolism, differentiation, metastasis, and therapeutic resistance (refer to Figure 3C) (316). In parallel, the Gβγ dimer activates SRC tyrosine kinase, which subsequently activates integrin α5β1 and matrix metalloproteinase (MMPs), leading to EGFR trans-activation (297, 316). These interconnected signaling events highlight the multifaceted role of GPER in driving BC progression.
6.6 GPER & EGFR trans-activation, activation of MAPK/ERK pathway
EGFR plays a pivotal role in GPER-mediated signaling in BC (319), particularly contributing to survival, proliferation, migration, and metastasis in ER-positive tamoxifen-resistant tumors. Upon GPER activation, MMPs cleave pro-heparin-binding epidermal growth factor (pro-HB-EGF), releasing HB-EGF, which binds to and activate EGFR (Figure 3C). This EGFR transactivation initiates downstream signaling pathways, including MAPK/ERK1/2 and PI3K/Akt, promoting breast cancer cell survival and proliferation (320). Moreover, EGFR ligands have been shown to upregulate GPER expression through the EGFR/ERK pathway, further reinforcing tamoxifen resistance in ER-positive BC. Hypoxic conditions within the tumor microenvironment also induce GPER upregulation via HIF-1α in an EGFR/ERK dependent manner (321). These findings highlight the interconnected nature of EGFR and GPER signaling in BC progression and therapy resistance. Consequently, dual-targeting strategies combining EGFR inhibitors (e.g., gefitinib or erlotinib) with GPER antagonists may offer a more effective approach for reducing tumor burden and overcoming tamoxifen resistance in ERα−positive BC.
6.7 GPER signaling in breast CAFs
Cancer-associated fibroblasts (CAFs), also referred to as myofibroblasts, constitute the most abundant stromal cell population within the breast tumor microenvironment (TME)—a dynamic and heterogeneous ecosystem comprising immune cells, blood vessels, extracellular matrix (ECM), and stromal elements that surround and interact with tumor cells. CAFs play a critical role in shaping the TME by orchestrating heterotypic cellular interactions and continuously secreting cytokines, chemokines, metabolites, and ECM-remodeling proteins. This contributes to an immunosuppressive or “immune-excluded” phenotype that facilitates tumor progression and promotes tumor immune escape.
CAFs secrete a diverse profile of cytokines (e.g., IL-6, TGF-β) and chemokines (CXCL1, CXCL12, CCL2, CCL5), which preferentially recruit immunosuppressive cell subsets such as myeloid-derived suppressor cells (MDSCs) and CD4+CD25+Foxp3+ regulatory T (Treg) cells, while inhibiting the cytotoxic activity of CD8+ T cells and natural killer (NK) cells. In addition, CAFs actively polarize tumor-associated macrophages (TAMs) and neutrophils (TANs) toward protumor phenotype (M2 and N2, respectively) via factors like IL-4, IL-6, IL-8, GM-CSF, CXCL8, and CXCL12 (322, 323).
GPER is highly expressed in CAFs and functions as a transcriptional regulator in response to estrogen or the GPER agonist G-1. Upon activation, GPER stimulates the paracrine secretion of chemotactic, angiogenic, and ECM-modulating factors, including IL-6, IL-8, VEGF, HGF, and matrix metalloproteinases (MMP-2, MMP-9) (324, 325), which collectively enhance processes such as F-actin reorganization, EMT, migration, and angiogenesis (326–328).
Under hypoxic conditions—commonly observed within tumors—CAFs upregulate HIF-1α, GPER, and α-SMA, leading to increased secretion of IL-6, VEGF, and connective tissue growth factor (CTGF). GPER activation promotes invasion through a CTGF-dependent mechanism, while silencing GPER in CAFs downregulates hypoxia-induced CTGF expression and suppresses BC invasion (329). Estrogen and G-1 have also been shown to elevate HIF-1α and VEGF levels, further promoting tumor angiogenesis (326, 330, 331).
Moreover, Pupo et al. demonstrated that estrogen induces nuclear translocation of GPER in CAFs, upregulating c-Fos and CTGF expression and enhancing fibroblast migration (332). Ligand-activated (E2 and G-1) GPER can also trigger a feedforward loop in both CAFs and MCF-7 cells through IL-1β/IL1R1 signaling, reinforcing invasive characteristics in breast cancer cells (333). Notably, GPER mediates tamoxifen-induced aromatase expression in both CAFs and tamoxifen-resistant BC cells, increasing local estrogen synthesis and driving resistance mechanisms (275, 324). Furthermore, CAF-derived CXCL12 facilitates tumor cell intravasation and metastasis by increasing vascular permeability and promoting leaky tumor vasculature (334). IL-6 from CAFs also promotes cancer stemness by inducing the formation of BCSCSs, which exhibit self-renewal capacity and therapy resistance.
Together, these findings highlight GPER’s central role in CAF biology, particularly in fostering a supportive TME that drives breast cancer progression. Targeting GPER in CAFs represents a promising therapeutic strategy to disrupt stromal support, attenuate immune evasion, and inhibit tumor advancement in ERα−positive BC. The use of GPER antagonists may be especially beneficial as an adjuvant therapy in ERα−positive breast cancer by enhancing immune infiltration and reducing tumor proliferation.
6.8 Controversies on GPER
Controversy remains regarding GPER’s role in pro-apoptotic signaling and its subcellular localization. While GPER is classified as a cell-surface transmembrane receptor, studies have reported its presence both at the plasma membrane and intra-cellularly, with distinct biological implications across BC subtypes. Thomas et al. and Filardo et al. observed that GPER primarily exhibits a cytoplasmic staining pattern in BC cells, with a minor fraction at the cell surface (277, 335). However, tumor specimens often show both nuclear and cytoplasmic GPER localization. Cheng et al. demonstrated that GPER accumulates in the perinuclear region and distributes in the cytoplasm via clathrin-coated vesicles (336), raising questions about its role as a membrane-localized estrogen receptor. Sjöström et al. reported that GPER over-expression and plasma membrane localization are key drivers of BC progression, with high membrane GPER correlating with poor histological grade, while its absence predicts excellent long-term prognosis in ER-positive tamoxifen-treated patients (296). In contrast, cytoplasmic GPER is linked to lower tumor stage and better differentiation, whereas nuclear GPER correlates with aggressive subtypes with poorly differentiated tumors (337). GPER’s role in pro-apoptotic signaling remains controversial, with its effects varying depending on the cellular context and signaling environment. Some studies suggest that GPER activation inhibits cancer cell growth (338), implying that high GPER expression may benefit the survival of BC patients, while others report that GPER induces the expression of genes involved in tumor cell migration and proliferation both in vitro and in vivo (339, 340). Moreover, high GPER expression correlates with increased tumor size and metastasis in breast malignancies (335). Additionally, GPER’s involvement in tamoxifen resistance adds further complexity; while some studies report that high GPER expression is negatively-associated with relapse-free survival in BC patients treated with tamoxifen, others suggest it may enhance treatment sensitivity. Collectively, these findings underscore the need for further investigation to clarify GPER’s dual role as both a pro- and anti-tumorigenic factor and to better understand its functions across diverse pathophysiological contexts, including ERα−positive BC.
7 Next-generation therapeutic strategies targeting ERα and GPER
Targeted protein degradation (TPD) has emerged as a promising front-line endocrine therapy, offering specific and irreversible silencing of ER by manipulating cellular proteostasis (341, 342). SERDs induce ERα degradation by binding to the ERα−LBP and recruiting the cellular degradation machinery. The first-generation SERD, fulvestrant (Faslodex™), features a core structure that fits into the ERα−LBP and a hydrophobic alkyl-side chain (degron) that binds to a hydrophobic pocket of ERα. This induces structural deformation of ERα, including the displacement or rearrangement of helix 12, which exposes hidden degradation signals. This facilitates the attachment of ubiquitin molecules to degron sequences, leading to ERα degradation. (343). In this section, we discuss recent advancements in fulvestrant and its analogues, highlighting novel innovations such as ER-targeting PROTACs, CERANs, SERCAs, and other emerging technologies (67, 344, 345).
7.1 Fulvestrant and its analogues
Presently, fulvestrant remains the only SERD approved for use in ET-resistant metastatic BC, both as a first-line and subsequent-line treatment (78). However, fulvestrant has several limitations, including low solubility, poor oral bioavailability requiring painful intramuscular administration, a bulky steroidal backbone that restricts chemical diversification, and the emergence of drug resistance due to mutations in ERα−LBP that impair binding and degradation (78, 346–349). These limitations have restricted the full clinical potential of fulvestrant, with ER blockade remaining below 75% even at a monthly dose of 500 mg, thereby spurring the development of second-generation oral SERDs with improved pharmacokinetics (PK) and efficacy (347, 350, 351). Consequently, pharmaceutical efforts have focused on utilizing non-steroidal scaffolds containing two types of chemical moieties—either an acid side chain or basic side chain—that perturb the ERα−LBD and interfere with co-activator binding (352). However, the clinical outcomes of these newly developed oral SERDs have varied so far.
Oral SERDs with acrylic acid side chains undergoing clinical trials include rintodestrant (G1T48), taragarestrant (D-0502), ZN-c5, and LX-039. The early SERD GW5638 was designed based on the tamoxifen core structure by substituting its piperidine side chain with acrylic acid side chain (64, 353). Rintodestrant, developed by G1 therapeutics, demonstrated excellent safety and tolerability in a Phase II clinical trial (NCT03455270) as a monotherapy and in combination with palbociclib in ER+/HER2− advanced BC patients with ESR1 mutations (354, 355). Similarly, the Phase Ib study of D-0502 (NCT03471663) showed promising anti-tumor activity and tolerable toxicity in patients with ER+/HER2− advanced or metastatic BC (254, 356). D-0502 is currently under evaluation in a Phase III study (CTR20190092). ZN-c5, developed by Zentalis, has demonstrated an excellent safety profile and is being evaluated in a Phase II trial as a monotherapy (NCT03560531) and in Phase I trials in combination with palbociclib (NCT03560531) and abemaciclib (NCT04514159) (257–259). LX-039, an indole-series compound from Luoxin Pharmaceuticals, demonstrated favorable pharmacokinetics and potent anti-tumor activity in wild-type and tamoxifen-resistant MCF-7 xenograft models (255, 357). It is currently in a Phase I trial (NCT04097756) for treating ER+/HER2− advanced or metastatic BC (256).
In contrast, oral SERDs with basic side chains include elacestrant (RAD-1901), imlunestrant (LY3484356), camizestrant (AZD9833), and giredestrant (GDC-9545). Elacestrant, a second-generation SERM-SERD hybrid developed by Stemline Therapeutics, received FDA approval under the brand name Orserdu® in 2023 (358, 359). The Phase III EMERALD trial (NCT03778931) demonstrated that elacestrant, as a single agent, significantly improved PFS compared to standard-of-care (AI or fulvestrant) in patients with ER+/HER2−, ESR1-mutated advanced or metastatic breast cancer previously treated with ET and a CDK4/6 inhibitor (57, 58, 360). The ongoing Phase Ib/II ELECTRA trial (NCT05386108) is evaluating elacestrant in combination with abemaciclib for treating brain metastases in ER+/HER2− breast cancer patients (361), suggesting that elacestrant could become a new standard-of-care in this setting.
Camizestrant (AZD9833), developed by AstraZeneca, demonstrated superior efficacy and tumor inhibition in patients with ER+/HER2− advanced breast cancer compared to fulvestrant in the Phase II SERENA-2 trial (NCT04214288) (241, 242, 362–365). The ongoing Phase-III SERENA-6 trial (NCT04964934) is evaluating its antitumor activity as a single agent or in combination with CDK4/6 or PI3K/AKT/mTOR inhibitors in fulvestrant-resistant, wild-type, and ESR1-mutated PDX models (59).
Imlunestrant (LY3484356), developed by Loxo Oncology at Eli Lilly Corp., is a next-generation brain-penetrant, oral selective ERα degrader that exhibits potent activity in both ESR1 wild-type and mutant breast cancers (29, 60). The ongoing Phase I/II EMBER trial (NCT04188548) is assessing the safety and efficacy of imlunestrant as monotherapy and in combination with other anticancer agents in patients with ER+ locally advanced or metastatic breast cancer (248). When combined with abemaciclib (a CDK4/6 inhibitor), alpelisib (a PI3K inhibitor), or everolimus (a mTOR inhibitor), imlunestrant demonstrates enhanced anti-tumor efficacy, including against brain metastases, irrespective of ESR1-mutation status (29). According to the ongoing Phase-III EMBER-3 trial (NCT04975308), the imlunestrant–abemaciclib combination significantly improves PFS compared to imlunestrant monotherapy in ER+/HER2− advanced breast cancer, regardless of ESR1 mutations (61).
Giredestrant (GDC-9545), developed by Genentech, is a highly potent, non-steroidal oral SERD and full ER antagonist. Phase I clinical data indicate that GDC-9545 is well tolerated and demonstrates promising efficacy both as a monotherapy and in combination with palbociclib (366, 367). Notably, at low doses, GDC-9545 induces tumor regression in both wild-type ERα tumor models and Y537S ERα mutant PDX models, either alone or in combination with a CDK4/6 inhibitor (62). Ongoing Phase III trials—persevERA (NCT04546009) and evERA (NCT05306340)—are evaluating its efficacy and safety in combination with palbociclib and everolimus, respectively, in ER+/HER2− locally advanced or metastatic breast cancer patients (245, 247).
However, clinical development of several new SERDs—such as AZD9496 (368–370), LSZ102 (371, 372), GDC-0810 (373–376), GDC-0927 (377, 378), SCO-120 (64, 379), SHR9549 (64) and SAR439859 (380–384)— has been suspended due to various concerns.
7.2 ER PROTACs
Proteolysis-targeting chimera (PROTAC) technology, first proposed by Sakamoto et al., is an emerging TPD strategy (66, 385–387). PROTACs are heterotrimeric bifunctional molecules consisting of three components: a ligand that binds to the protein of interest (POI), a ligand that binds to an E3 ubiquitin ligase, and a flexible linker connecting them. PROTACs induce the formation of a “POI-PROTAC-E3 ligase” ternary complex and, by “hijacking” the cellular ubiquitin-proteasome system (UPS), trigger POI ubiquitination and subsequent degradation via the proteasome pathway (388). In this context, orthosteric PROTACs target the active-site of the POI, whereas the allosteric PROTACs bind to a site distinct from the primary-ligand-binding pocket (Figure 6A). The rational design of small-molecule ER PROTACs—most notably the Von Hippel-Lindau (VHL)-based and Cereblon (CRBN)-based PROTACs—has driven the evolution of the ER PROTAC platform from conceptualization to clinical translation. In VHL-based PROTACs, HIF-1α or other small molecules serve as warheads (ligand-binding moieties) to recruit the VHL E3 ligase, whereas thalidomide and its derivatives act as warheads to engage the CRBN E3 ligase in CRBN-based PROTACs. The warhead for ERα generally includes E2, SERM/SERD, peptide, or DNA fragment. Notably, PROTACs are catalytic in nature, meaning they can be recycled after each degradation event to target additional POI molecules, distinguishing them from stoichiometric degraders. A key advantage of PROTACs over SERDs is that they do not require high-affinity binding to the ligand-binding pocket of the POI, allowing structural modifications to improve solubility without compromising efficacy.
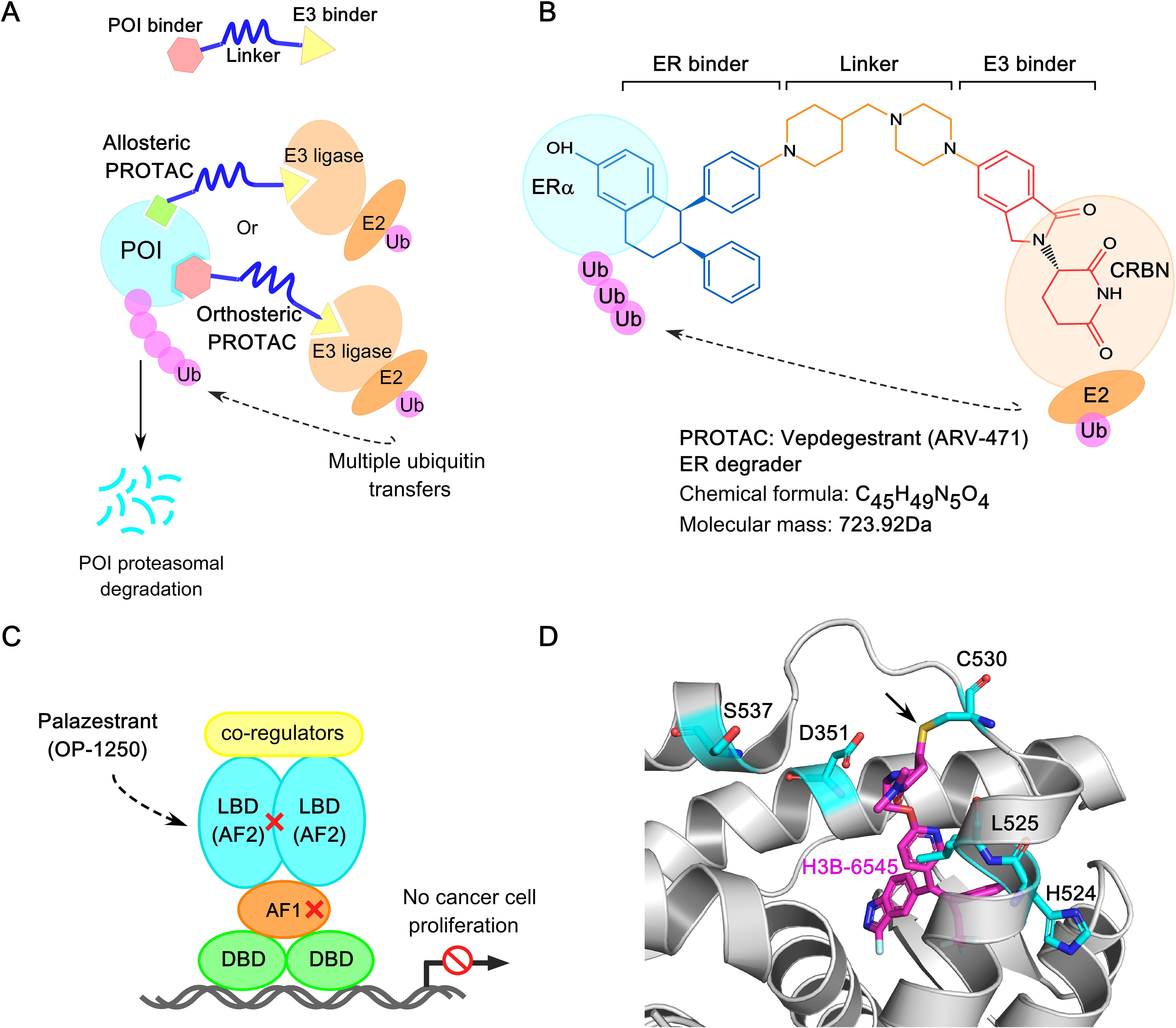
Figure 6. Next-generation protein degradation technologies for ER+ breast cancer therapy. (A) Mechanism of action of allosteric and orthosteric PROTACs, leading to proteasomal degradation of protein of interest (POI). (B) Chemical structure of oral ER-PROTAC ARV-471. (C) Mechanism of action of CERAN OP-1250. It completely turns off both AF1 and AF2 transcriptional activation function of ERα. (D) Crystal structure of H3B-6545 (in purple sticks) in complex with ER (PDB: 6OWC), highlighting the co-valent bonding between Cysteine 530 (C530) of ER and H3B-6545 (indicated with arrowhead).
7.3 CRBN-based ER PROTAC degraders
The pioneering ER PROTAC ARV-471 (Vepdegestrant), developed by Arvinas and Pfizer, entered clinical trials in 2019 and received FDA fast-track designation in February 2024 (64, 389, 390). ARV-471 is a CRBN-based PROTAC, incorporating a lasoxifene-derived ligand-binding moiety (Figure 6B) (65, 391). It simultaneously binds to the ER-LBD and the CRBN E3 ligase, facilitating the degradation of both wild-type and mutant ERα at nanomolar concentrations. Gough et al. reported that ARV-471 selectively and rapidly degraded ER, achieving >80% degradation within 4 hours across various ER+ cell lines, and demonstrating equal potency against clinically relevant ligand-independent ERα mutants (392). The phase III VERITAC-2 trial (NCT05654623) is currently evaluating the efficacy and safety of vepdegestrant versus fulvestrant, while VERITAC-3 (NCT05909397) is assessing vepdegestrant plus palbociclib versus letrozole plus palbociclib in patients with ER+/HER2− advanced breast cancer (266, 268, 269, 393). Additionally, ARV-471 is being explored in combination therapies with agents such as abemaciclib, ribociclib, everolimus, and Pfizer’s novel CDK-4 inhibitor (PF-07220060), expanding its potential applications for locally advanced or ER+/HER- metastatic BC (NCT06125522, NCT05573555, NCT0558127) (64).
ERD-3111 (compound 18) was reported as a novel CRBN-based ER PROTAC by the Wang group in 2023 (63, 64). This chimera utilized lasoxifene as the ER binder and incorporated a new CRBN ligand, TX-16. Notably, ERD-3111 demonstrated superior bioavailability and achieved significant tumor regression and complete growth inhibition in wild-type and two clinically relevant ESR1-mutated (Y537S and D538G) MCF-7 xenograft models, outperforming ARV-471. Based on these preclinical findings, ERD-3111 is being extensively evaluated as a highly potent ERα PROTAC for further development. Subsequently, the development of more potent and orally efficacious CRBN-based ER PROTACs led to ERD-1233 (compound 19) and ERD-12310A (compound 20), which utilize the lasoxifene scaffold as the ER-binding moiety and a novel CRBN ligand with high binding affinity (394, 395). Importantly, ERD-12310A exhibited significant inhibition of tumor growth in MCF-7 Y537S ERα mutant xenograft tumors without substantial weight loss or toxicity issues, making it more effective than ARV-471.
7.4 VHL-based ER PROTAC degraders
Besides CRBN ligands, the VHL ligand is also widely employed as an E3 ligase recruiter for designing ER PROTACs (396). A novel VHL-based ER PROTAC, AC0682, was developed by Accutar Biotech using an AI-empowered drug discovery platform with ACCU degron technology. Although AC0682 was reported to induce ERα degradation in wild-type and Y537S/D538G ERα−expressing MCF-7 cell lines with a sub-nanomolar DC50, its Phase I clinical trials (NCT05489679 and NCT05080842) were recently terminated. The next-generation AC699 is currently recruiting patients to evaluate its safety, tolerability, PK, and efficacy in ER+/HER2− advanced or metastatic BC, though its chemical structure remains undisclosed.
Other highly potent VHL-based ER PROTAC degraders, ERD-308 (compound 12) and ERD-148 (compound 11), were developed by the Wang group at the University of Michigan in 2019 (396–398). These compounds employed a raloxifene scaffold as the ER ligand and exhibited excellent ER-degrading potency. Notably, ERD-148 degrades both unphosphorylated and phosphorylated ERα, resulting in greater suppression of E2-dependent wild-type and E2-independent ESR1-mutated (Y537S and D538G) MCF-7 cells.
In 2022, another innovative class of ER PROTACs targeting the DBD of ERα—termed ERE-PROTACs (a nucleic acid conjugate)—was developed by the Tan group from Tsinghua university to overcome ET resistance (399). In a subsequent study, Feng et al. proposed an aptamer PROTAC strategy for targeting ERα−DBD to overcome drug resistance, using the aptamer as a ligand for ERα and the small-molecule VH032 for recruiting VHL E3 ligase (389). In 2023, another novel class of dual-targeting PROTAC degraders designed to simultaneously degrade ERα and aromatase (ARO) was introduced. Among these, 18c (compound 16) exhibited the most potent dual ERα/ARO degradation activity (400).
7.5 Complete estrogen receptor antagonists (CERANs)
OP-1250 (Palazestrant), developed by Olema, is the only orally bioavailable CERAN in clinical trials, effectively targeting both wild-type and mutant ERα (401). Unlike SERMs, CERANs are designed to completely suppress AF1 and AF2 activity, while also functioning as SERDs to promote ER degradation (Figure 6C). The Phase III OPERA-1 trial (NCT06016738) is currently evaluating the safety and efficacy of OP-1250 versus standard-of-care in patients with ER+/HER2− advanced breast cancer (265). Combination therapies of OP-1250 with palbociclib, ribociclib, and alpelisib are also being assessed in Phase I/II trials (NCT05266105, NCT05508906) (402).
7.6 Selective estrogen receptor covalent antagonists (SERCAs)
H3B-6545, a first-in-class oral SERCA, was discovered using a structure-based drug design strategy. It irreversibly inactivates both wild-type and mutant ERα through co-valent bond formation between the cysteine 530 (C530) in the ERα−LBD and the acrylamide warhead of H3B-6545 (PDB: 6OWC) (Figure 6D) (68). H3B-6545 demonstrates robust preclinical anti-tumor efficacy and superiority over fulvestrant across a wild-type and Y537S-mutant ERα−expressing models, including both palbociclib-sensitive and -resistant BC lines. Its clinical activity is being evaluated in ER+/HER2− metastatic BC, including patients harboring Y537S ERα, in trials NCT03250676, NCT04568902, and NCT04288089 (68, 260, 262, 403). While H3B-6545 enforces an antagonistic conformation without degrading ERα, compound 29c targets C530 covalently and engages in strong hydrophobic interactions with helix 11, demonstrating ERα degradation potency in both wild-type and ESR1-mutated BC cell lines (404).
7.7 Limitations of PROTACs
Despite the groundbreaking success of PROTAC technology, several technical challenges remain, including expanding the repertoire of E3 ligases, reducing off-target toxicity, and optimizing linker length—all of which hinder further development. The limited availability of E3 ligases further restricts its application. Similarly, other UPS-based modalities, such as autophagy-targeting chimeras (AUTACs), autophagosome-tethering compounds (ATTECs), molecular glues, dTAG, SNIPERs, and Trim-Away, face similar constraints (344, 405–409).
7.8 GPER-targeting strategies
Analysis of breast cancer biopsy samples based on ER and GPER expression reveals that 43% of cases are ER+/GPER+, 19% are either ER+/GPER- or ER-/GPER+, and 19% are ER-/GPER- (Figure 3B) (410). This suggests that standard ER-targeted therapies fully benefit only 19% of patients, partially benefit 43%, and overlook a substantial proportion of GPER-expressing tumors—highlighting a critical gap in current endocrine therapy.
Encouragingly, GPER-directed therapeutic strategies are emerging. For instance, the GPER agonist LNS8801 significantly inhibited tumor growth in uveal melanoma xenografts by inducing G2-M phase mitotic arrest and apoptosis (411). A Phase 1/1B clinical trial (NCT04130516) is currently evaluating LNS8801 as monotherapy and in combination with pembrolizumab in metastatic solid tumors, with early results demonstrating promising safety and efficacy.
In parallel, two dual ER/GPER-targeting PROTACs, UI-EP001 and UI-EP002, have shown nanomolar binding affinities and effectively degrade ERα, ERβ, and GPER (412). However, the broader application of such strategies remains limited by the scarcity of selective GPER modulators—both agonists and antagonists—constraining efforts to fully characterize GPER-mediated signaling in breast cancer.
While PROTACs have revolutionized intracellular protein degradation by harnessing UPS, they are generally ineffective against non-cytosolic and membrane-associated targets like GPER. To address this, novel degradation technologies such as antibody-based PROTACs (AbTACs) and lysosome-targeting chimeras (LYTACs) have gained traction. These approaches enable the selective degradation of transmembrane and extracellular proteins by directing them to the lysosomal pathway, potentially expanding the therapeutic options for previously “undruggable” targets. In the context of GPER, AbTAC and LYTAC strategies offer a promising avenue for overcoming the limitations of traditional degraders and hold significant clinical potential for ER+/GPER+ breast cancer (390, 413).
AbTAC are bispecific IgGs that simultaneously bind two distinct proteins (Figures 7A, B). Cotton et al. developed the first AbTAC, which targets RNF-43 (an E3 ligase) and programmed death-ligand 1 (PD-L1), promoting PD-L1 lysosomal degradation (414). Using Knobs-into-Holes (KIH) Fc engineering, one half-IgG incorporates the T366W ‘knob’ mutation—substituting threonine with the bulkier tryptophan—while the other half-IgG carries the T366S, L368A, and Y407V mutations to form the complementary ‘hole’. In addition, the N297G mutation is introduced to prevent Fc glycosylation, thereby silencing the Fc region and reducing antibody flexibility during AbTAC generation.
Figure 7. AbTAC- and LYTAC-based degradation strategies for targeting membrane-bound receptors (A) Generation of an AbTAC bispecific IgG that simultaneously binds to RNF43 and PD-L1, modified from Cotton et al. (414). The conditions for in vitro assembly of individually expressed half-IgGs to form a bispecific IgG with the desired point mutations are described. Using Knobs-into-holes (KIHs) Fc engineering, one half-IgG contains the T366W ‘knob’ mutation, substituting threonine with bulkier tryptophan, while the other half-IgG contains the T366S, L368A, Y407V mutations with a complementary ‘hole’. (B) Graphical representation of the AbTAC mode of action, recruiting RNF43 for lysosomal degradation of membrane-bound POI. (C) Structure of LYTAC utilizing glycopeptide ligand to target CI-M6PR/IGF2R. (D) Mechanism of action of first-generation and second-generation LYTACs for degrading extracellular POI or membrane-bound POI, recycling CI-M6PR and ASGPR receptors respectively.
In contrast, LYTACs, developed by the Bertozzi lab, consist of a small-molecule or antibody fused with a glycopeptide ligand recognized by cation-independent mannose-6-phosphate receptors (CI-M6PR), which shuttle M6P-tagged protein cargoes to lysosomes for degradation while recycling themselves (Figures 7C, D) (415, 416). Atezolizumab-derived LYTACs (anti-PD-L1-M6Pn) achieved ∼70% PD-L1 degradation via M6P recognition, while ASGPR-targeting LYTACs demonstrated liver-specific EGFR degradation (415, 417). Rational design of GPER-targeted warheads for LYTACs or AbTAC-drug conjugates (ATDCs) holds promise for degrading membrane-bound GPER, blocking downstream signaling, and enabling intracellular delivery of conjugated drugs in the treatment of ER+/HER2− advanced BC patients.
8 Conclusion
The reliance on ER signaling in ERα−positive breast cancer underscores the importance of ER-targeted therapies as the cornerstone of treatment for this tumor type. The high prevalence of ESR1 point mutations in ERα−positive metastatic tumors indicates that ER dependency persists throughout tumor progression, driving acquired resistance (418). Functional and structural studies have demonstrated that common mutations such as Y537S and D538G stabilize ERα in a conformation resembling the ligand-bound wild-type receptor, leading to constitutive, hormone-independent activity and resistance to conventional endocrine therapies (51, 52, 207, 210, 419). Crystallographic and modeling analyses reveal that helix 12 in the mutant receptor adopts an “on-state” conformation similar to the E2-bound wild-type ERα, emphasizing the need for novel therapeutics capable of overcoming this constitutively active state while preserving structural integrity to ensure inactivity in the absence of estrogen.
Substantial efforts thus have been directed toward the development of new-generation of ER-targeted agents, including oral SERDs and innovative strategies such as PROTACs, SERCAs, and CERANs. While next-generation SERDs and SERM/SERD hybrids have demonstrated efficacy in targeting ERs, their dependence on ligand binding and potential GPER agonism necessitate more comprehensive approaches. Rigorous evaluation of these agents is ongoing, with multiple preclinical and clinical trials underway in both primary and metastatic breast cancer. Currently, several candidates are in Phase III clinical trials, including camizestrant (AZD9833, AstraZeneca), Taragarestrant (D-0502, Inventis Bio), giredestrant (GDC-9545, Roche), Imlunestrant (LY3484356, Eli Lilly), and palazestrant (OP-1250, Olema Pharmaceuticals), either as monotherapy or in combination with CDK4/6 inhibitors, PI3K inhibitors, and mTOR inhibitors.
On contrary, targeted protein degradation (TPD) has emerged as a transformative strategy for addressing “undruggable” protein targets, with PROTAC technology revolutionizing traditional therapeutic paradigms. ARV-471 has demonstrated exceptional efficacy in Phase I/II trials and is currently in Phase III, positioning it as the first oral ER-targeting PROTAC with strong clinical potential (420). Nanoengineered-PROTACs (nano-PROTACs), such as ARV-loaded nanoparticles, have improved drug solubility, permeability, pharmacokinetics, and intracellular delivery—enhancing efficacy while minimizing systemic toxicity (421, 422). Additionally, surface modification of PLGA nanoparticles with PEG conferred high serum stability and extended half-life to c-Myc-targeting PROTACs in pancreatic cancer models (423, 424). Trastuzumab-conjugated PROTAC-loaded nanoparticles (MZ1-loaded polymeric antibody-conjugated nanoparticles) have also demonstrated enhanced specificity and cytotoxicity in HER-2 enriched BC (425).
Despite the advances, challenges remain in this evolving field, including optimization of linker length, ternary complex equilibria, pharmacokinetics, and the possibility of potential drug resistance of PROTACs. Emerging strategies such as AbTACs and LYTACs, supported by AI-driven platforms for high-throughput screening and rational designing, may represent the next frontier (426). Notably, the development of AbTAC-drug conjugates (ATDCs) targeting membrane proteins like GPER offers a dual benefit: receptor degradation and intracellular drug delivery. This approach addresses previously inaccessible targets and paves the way for more effective treatment options in ER+/HER2− breast cancer. Overall, continuous breakthroughs and refinements in PROTAC technology and related TPD strategies offer promise for developing safer, more precise, and controllable ER-targeting therapeutics—potentially transforming the treatment landscape for ERα−positive breast cancer patients.
Author contributions
TS: Conceptualization, Writing – original draft, Writing – review & editing. KL: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Research support funds (Fund number: 426143), College of Medicine, University of Saskatchewan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Arnold M, Morgan E, Rumgay H, Mafra A, Singh D, Laversanne M, et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast. (2022) 66:15–23. doi: 10.1016/j.breast.2022.08.010
2. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
3. Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Primers. (2019) 5:66. doi: 10.1038/s41572-019-0111-2
4. Saha T and Lukong KE. Breast cancer stem-like cells in drug resistance: A review of mechanisms and novel therapeutic strategies to overcome drug resistance. Front Oncol. (2022) 12:856974. doi: 10.3389/fonc.2022.856974
5. Siegel RL, Miller KD, Wagle NS, and Jemal A. Cancer statistics 2023. CA A Cancer J Clin. (2023) 73:17–48. doi: 10.3322/caac.21763
6. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
7. Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. (2001) 98:10869–74. doi: 10.1073/pnas.191367098
8. Ali S and Coombes RC. Estrogen receptor alpha in human breast cancer: occurrence and significance. J Mammary Gland Biol Neoplasia. (2000) 5:271–81. doi: 10.1023/a:1009594727358
9. Allred DC, Brown P, and Medina D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast Cancer Res. (2004) 6:240. doi: 10.1186/bcr938
10. Wittliff JL. Steroid-hormone receptors in breast cancer. Cancer. (1984) 53:630–43. doi: 10.1002/1097-0142(19840201)53:3+<630::aid-cncr2820531308>3.0.co;2-3
11. Zhao L, Zhou S, and Gustafsson J-Å. Nuclear receptors: recent drug discovery for cancer therapies. Endocr Rev. (2019) 40:1207–49. doi: 10.1210/er.2018-00222
12. Filardo EJ, Quinn JA, Bland KI, and Frackelton AR. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. (2000) 14:1649–60. doi: 10.1210/mend.14.10.0532
13. Molina Calistro L, Arancibia Y, Olivera MA, Domke S, and Torres RF. Interaction of GPER-1 with the endocrine signaling axis in breast cancer. Front Endocrinol. (2025) 16:1494411. doi: 10.3389/fendo.2025.1494411
14. Finn RS, Aleshin A, and Slamon DJ. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. (2016) 18:17. doi: 10.1186/s13058-015-0661-5
15. He T, Yang W, Zhang X, Li P, Yang D, Wu Y, et al. Comparative effectiveness of tamoxifen, toremifene, letrozole, anastrozole, and exemestane on lipid profiles in breast cancer patients: A network meta-analysis. Med (Baltimore). (2020) 99:e18550. doi: 10.1097/MD.0000000000018550
16. Patel HK and Bihani T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol Ther. (2018) 186:1–24. doi: 10.1016/j.pharmthera.2017.12.012
17. Patel R, Klein P, Tiersten A, and Sparano JA. An emerging generation of endocrine therapies in breast cancer: a clinical perspective. NPJ Breast Cancer. (2023) 9:20. doi: 10.1038/s41523-023-00523-4
18. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. (2005) 365:1687–717. doi: 10.1016/S0140-6736(05)66544-0
19. Johnston SRD. New strategies in estrogen receptor-positive breast cancer. Clin Cancer Res. (2010) 16:1979–87. doi: 10.1158/1078-0432.CCR-09-1823
20. Ahmad A, Ali SM, Ahmad MU, Sheikh S, and Ahmad I. Orally administered endoxifen is a new therapeutic agent for breast cancer. Breast Cancer Res Treat. (2010) 122:579–84. doi: 10.1007/s10549-009-0704-7
21. Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenom J. (2005) 5:6–13. doi: 10.1038/sj.tpj.6500285
22. Singh MS, Francis PA, and Michael M. Tamoxifen, cytochrome P450 genes and breast cancer clinical outcomes. Breast. (2011) 20:111–8. doi: 10.1016/j.breast.2010.11.003
23. Jayaraman S, Wu X, Kalari KR, Tang X, Kuffel MJ, Bruinsma ES, et al. Endoxifen downregulates AKT phosphorylation through protein kinase C beta 1 inhibition in ERα+ breast cancer. NPJ Breast Cancer. (2023) 9:101. doi: 10.1038/s41523-023-00606-2
24. Mustonen MV, Pyrhönen S, and Kellokumpu-Lehtinen P-L. Toremifene in the treatment of breast cancer. World J Clin Oncol. (2014) 5:393–405. doi: 10.5306/wjco.v5.i3.393
25. Vogel CL, Johnston MA, Capers C, and Braccia D. Toremifene for breast cancer: a review of 20 years of data. Clin Breast Cancer. (2014) 14:1–9. doi: 10.1016/j.clbc.2013.10.014
26. Lainé M, Fanning SW, Chang Y-F, Green B, Greene ME, Komm B, et al. Lasofoxifene as a potential treatment for therapy-resistant ER-positive metastatic breast cancer. Breast Cancer Res. (2021) 23:54. doi: 10.1186/s13058-021-01431-w
27. Osborne CK, Wakeling A, and Nicholson RI. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. Br J Cancer. (2004) 90 Suppl 1:S2–6. doi: 10.1038/sj.bjc.6601629
28. Robertson JF. ICI 182,780 (Fulvestrant)–the first oestrogen receptor down-regulator–current clinical data. Br J Cancer. (2001) 85 Suppl 2:11–4. doi: 10.1054/bjoc.2001.1982
29. Bhagwat SV, Mur C, Vandekopple M, Zhao B, Shen W, Marugán C, et al. Imlunestrant is an oral, brain-penetrant selective estrogen receptor degrader with potent antitumor activity in ESR1 wild-type and mutant breast cancer. Cancer Res. (2025) 85:777–90. doi: 10.1158/0008-5472.CAN-24-2608
30. Soleja M, Raj GV, and Unni N. An evaluation of fulvestrant for the treatment of metastatic breast cancer. Expert Opin Pharmacother. (2019) 20:1819–29. doi: 10.1080/14656566.2019.1651293
31. Milla-Santos A, Milla L, Portella J, Rallo L, Pons M, Rodes E, et al. Anastrozole versus tamoxifen as first-line therapy in postmenopausal patients with hormone-dependent advanced breast cancer: a prospective, randomized, phase III study. Am J Clin Oncol. (2003) 26:317–22. doi: 10.1097/01.COC.0000047126.10522.F9
32. Mukhopadhyay KD, Liu Z, Bandyopadhyay A, Kirma NB, Tekmal RR, Wang S, et al. Aromatase expression increases the survival and Malignancy of estrogen receptor positive breast cancer cells. PloS One. (2015) 10:e0121136. doi: 10.1371/journal.pone.0121136
33. Augusto TV, Correia-da-Silva G, Rodrigues CMP, Teixeira N, and Amaral C. Acquired resistance to aromatase inhibitors: where we stand! Endocr Relat Cancer. (2018) 25:R283–301. doi: 10.1530/ERC-17-0425
34. Ma CX, Reinert T, Chmielewska I, and Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer. (2015) 15:261–75. doi: 10.1038/nrc3920
35. Fasching PA, Beck JT, Chan A, De Laurentiis M, Esteva FJ, Jerusalem G, et al. Ribociclib plus fulvestrant for advanced breast cancer: Health-related quality-of-life analyses from the MONALEESA-3 study. Breast. (2020) 54:148–54. doi: 10.1016/j.breast.2020.09.008
36. Neven P, Fasching PA, Chia S, Jerusalem G, De Laurentiis M, Im S-A, et al. Updated overall survival from the MONALEESA-3 trial in postmenopausal women with HR+/HER2– advanced breast cancer receiving first-line ribociclib plus fulvestrant. Breast Cancer Res. (2023) 25:103. doi: 10.1186/s13058-023-01701-9
37. Sledge GW, Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. (2017) 35:2875–84. doi: 10.1200/JCO.2017.73.7585
38. Turner NC, Slamon DJ, Ro J, Bondarenko I, Im S-A, Masuda N, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. (2018) 379:1926–36. doi: 10.1056/NEJMoa1810527
39. Finn RS, Martin M, Rugo HS, Jones S, Im S-A, Gelmon K, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. (2016) 375:1925–36. doi: 10.1056/NEJMoa1607303
40. Rugo HS, Finn RS, Diéras V, Ettl J, Lipatov O, Joy AA, et al. Palbociclib plus letrozole as first-line therapy in estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer with extended follow-up. Breast Cancer Res Treat. (2019) 174:719–29. doi: 10.1007/s10549-018-05125-4
41. Wang X, Zhao S, Xin Q, Zhang Y, Wang K, and Li M. Recent progress of CDK4/6 inhibitors’ current practice in breast cancer. Cancer Gene Ther. (2024) 31:1283–91. doi: 10.1038/s41417-024-00747-x
42. Nunnery SE and Mayer IA. Targeting the PI3K/AKT/mTOR pathway in hormone-positive breast cancer. Drugs. (2020) 80:1685–97. doi: 10.1007/s40265-020-01394-w
43. Rodriguez MJ, Perrone MC, Riggio M, Palafox M, Salinas V, Elia A, et al. Targeting mTOR to overcome resistance to hormone and CDK4/6 inhibitors in ER-positive breast cancer models. Sci Rep. (2023) 13:2710. doi: 10.1038/s41598-023-29425-y
44. deGraffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, et al. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res. (2004) 10:8059–67. doi: 10.1158/1078-0432.CCR-04-0035
45. Steelman LS, Martelli AM, Cocco L, Libra M, Nicoletti F, Abrams SL, et al. The therapeutic potential of mTOR inhibitors in breast cancer. Br J Clin Pharmacol. (2016) 82:1189–212. doi: 10.1111/bcp.12958
46. Abad E, Graifer D, and Lyakhovich A. DNA damage response and resistance of cancer stem cells. Cancer Lett. (2020) 474:106–17. doi: 10.1016/j.canlet.2020.01.008
47. Fan P and Craig Jordan V. Acquired resistance to selective estrogen receptor modulators (SERMs) in clinical practice (tamoxifen & raloxifene) by selection pressure in breast cancer cell populations. Steroids. (2014) 90:44–52. doi: 10.1016/j.steroids.2014.06.002
48. Nardone A, De Angelis C, Trivedi MV, Osborne CK, and Schiff R. The changing role of ER in endocrine resistance. Breast. (2015) 24 Suppl 2:S60–66. doi: 10.1016/j.breast.2015.07.015
49. Radhi S. Molecular changes during breast cancer and mechanisms of endocrine therapy resistance. Prog Mol Biol Transl Sci. (2016) 144:539–62. doi: 10.1016/bs.pmbts.2016.09.009
50. Hosfield DJ, Weber S, Li N-S, Sauvage M, Joiner CF, Hancock GR, et al. Stereospecific lasofoxifene derivatives reveal the interplay between estrogen receptor alpha stability and antagonistic activity in ESR1 mutant breast cancer cells. Elife. (2022) 11:e72512. doi: 10.7554/eLife.72512
51. Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. (2014) 20:1757–67. doi: 10.1158/1078-0432.CCR-13-2332
52. Robinson DR, Wu Y-M, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. (2013) 45:1446–51. doi: 10.1038/ng.2823
53. Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. (2017) 7:277–87. doi: 10.1158/2159-8290.CD-15-1523
54. Pepermans RA and Prossnitz ER. ERα-targeted endocrine therapy, resistance and the role of GPER. Steroids. (2019) 152:108493. doi: 10.1016/j.steroids.2019.108493
55. Brett JO, Spring LM, Bardia A, and Wander SA. ESR1 mutation as an emerging clinical biomarker in metastatic hormone receptor-positive breast cancer. Breast Cancer Res. (2021) 23:85. doi: 10.1186/s13058-021-01462-3
56. Herzog SK and Fuqua SAW. ESR1 mutations and therapeutic resistance in metastatic breast cancer: progress and remaining challenges. Br J Cancer. (2022) 126:174–86. doi: 10.1038/s41416-021-01564-x
57. Bidard F-C, Kaklamani VG, Neven P, Streich G, Montero AJ, Forget F, et al. Elacestrant (oral selective estrogen receptor degrader) Versus Standard Endocrine Therapy for Estrogen Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: Results From the Randomized Phase III EMERALD Trial. JCO. (2022) 40:3246–56. doi: 10.1200/JCO.22.00338
58. Wardell SE, Nelson ER, Chao CA, Alley HM, and McDonnell DP. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr Relat Cancer. (2015) 22:713–24. doi: 10.1530/ERC-15-0287
59. Turner N, Huang-Bartlett C, Kalinsky K, Cristofanilli M, Bianchini G, Chia S, et al. Design of SERENA-6, a phase III switching trial of camizestrant in ESR1-mutant breast cancer during first-line treatment. Future Oncol. (2023) 19:559–73. doi: 10.2217/fon-2022-1196
60. Jhaveri KL, Lim E, Jeselsohn R, Ma CX, Hamilton EP, Osborne C, et al. Imlunestrant, an oral selective estrogen receptor degrader, as monotherapy and in combination with targeted therapy in estrogen receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: phase ia/ib EMBER study. J Clin Oncol. (2024) 42:4173–86. doi: 10.1200/JCO.23.02733
61. Jhaveri KL, Neven P, Casalnuovo ML, Kim S-B, Tokunaga E, Aftimos P, et al. Imlunestrant with or without abemaciclib in advanced breast cancer. N Engl J Med. (2024) 392(12):1189–202. doi: 10.1056/NEJMoa2410858
62. Liang J, Zbieg JR, Blake RA, Chang JH, Daly S, DiPasquale AG, et al. GDC-9545 (Giredestrant): A potent and orally bioavailable selective estrogen receptor antagonist and degrader with an exceptional preclinical profile for ER+ Breast cancer. J Med Chem. (2021) 64:11841–56. doi: 10.1021/acs.jmedchem.1c00847
63. Chen Z, Hu B, Rej RK, Wu D, Acharyya RK, Wang M, et al. Discovery of ERD-3111 as a potent and orally efficacious estrogen receptor PROTAC degrader with strong antitumor activity. J Med Chem. (2023) 66:12559–85. doi: 10.1021/acs.jmedchem.3c01186
64. Min J, Liu X, Peng R, Chen C-C, Wang W, and Guo R-T. New generation estrogen receptor-targeted agents in breast cancer: present situation and future prospectives. Acta Mater Med. (2024) 3:57–71. doi: 10.15212/amm-2024-0006
65. Peng R, Liu X, Chen C-C, Guo R-T, and Min J. Development of PROTACs targeting estrogen receptor: an emerging technique for combating endocrine resistance. RSC Med Chem. (2025) 16:1023–36. doi: 10.1039/D4MD00961D
66. Pettersson M and Crews CM. PROteolysis TArgeting Chimeras (PROTACs) — Past, present and future. Drug Discov Today: Technol. (2019) 31:15–27. doi: 10.1016/j.ddtec.2019.01.002
67. Parisian AD, Barratt SA, Hodges-Gallagher L, Ortega FE, Peña G, Sapugay J, et al. Palazestrant (OP-1250), A complete estrogen receptor antagonist, inhibits wild-type and mutant ER-positive breast cancer models as monotherapy and in combination. Mol Cancer Ther. (2024) 23:285–300. doi: 10.1158/1535-7163.MCT-23-0351
68. Furman C, Puyang X, Zhang Z, Wu ZJ, Banka D, Aithal KB, et al. Covalent ERα Antagonist H3B-6545 demonstrates encouraging preclinical activity in therapy-resistant breast cancer. Mol Cancer Ther. (2022) 21:890–902. doi: 10.1158/1535-7163.MCT-21-0378
69. Ignatov A, Ignatov T, Weissenborn C, Eggemann H, Bischoff J, Semczuk A, et al. G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res Treat. (2011) 128:457–66. doi: 10.1007/s10549-011-1584-1
70. Ignatov A, Ignatov T, Roessner A, Costa SD, and Kalinski T. Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res Treat. (2010) 123:87–96. doi: 10.1007/s10549-009-0624-6
71. Mo Z, Liu M, Yang F, Luo H, Li Z, Tu G, et al. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res. (2013) 15:R114. doi: 10.1186/bcr3581
72. D’Souza A, Spicer D, and Lu J. Overcoming endocrine resistance in metastatic hormone receptor-positive breast cancer. J Hematol Oncol. (2018) 11:80. doi: 10.1186/s13045-018-0620-6
73. Menasce LP, White GR, Harrison CJ, and Boyle JM. Localization of the estrogen receptor locus (ESR) to chromosome 6q25.1 by FISH and a simple post-FISH banding technique. Genomics. (1993) 17:263–5. doi: 10.1006/geno.1993.1320
74. Arao Y and Korach KS. The physiological role of estrogen receptor functional domains. Essays Biochem. (2021) 65:867–75. doi: 10.1042/EBC20200167
75. Du Z, Wang H, Luo S, Yun Z, Wu C, Yang W, et al. The sequence–structure–function relationship of intrinsic ERα disorder. Nature. (2025) 638:1130–8. doi: 10.1038/s41586-024-08400-1
76. Foo J, Gentile F, Massah S, Morin H, Singh K, Lee J, et al. Characterization of novel small molecule inhibitors of estrogen receptor-activation function 2 (ER-AF2). Breast Cancer Res. (2024) 26:168. doi: 10.1186/s13058-024-01926-2
77. Fuentes N and Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. (2019) 116:135–70. doi: 10.1016/bs.apcsb.2019.01.001
78. Guan J, Zhou W, Hafner M, Blake RA, Chalouni C, Chen IP, et al. Therapeutic ligands antagonize estrogen receptor function by impairing its mobility. Cell. (2019) 178:949–963.e18. doi: 10.1016/j.cell.2019.06.026
79. Kumar R, Zakharov MN, Khan SH, Miki R, Jang H, Toraldo G, et al. The dynamic structure of the estrogen receptor. J Amino Acids. (2011) 2011:812540. doi: 10.4061/2011/812540
80. Mader S, Chambon P, and White JH. Defining a minimal estrogen receptor DNA binding domain. Nucleic Acids Res. (1993) 21:1125–32. doi: 10.1093/nar/21.5.1125
81. Métivier R, Penot G, Flouriot G, and Pakdel F. Synergism between ERalpha transactivation function 1 (AF-1) and AF-2 mediated by steroid receptor coactivator protein-1: requirement for the AF-1 alpha-helical core and for a direct interaction between the N- and C-terminal domains. Mol Endocrinol. (2001) 15:1953–70. doi: 10.1210/mend.15.11.0727
82. Zwart W, de Leeuw R, Rondaij M, Neefjes J, Mancini MA, and Michalides R. The hinge region of the human estrogen receptor determines functional synergy between AF-1 and AF-2 in the quantitative response to estradiol and tamoxifen. J Cell Sci. (2010) 123:1253–61. doi: 10.1242/jcs.061135
83. Denger S, Reid G, Kos M, Flouriot G, Parsch D, Brand H, et al. ERalpha gene expression in human primary osteoblasts: evidence for the expression of two receptor proteins. Mol Endocrinol. (2001) 15:2064–77. doi: 10.1210/mend.15.12.0741
84. Flouriot G, Brand H, Denger S, Metivier R, Kos M, Reid G, et al. Identification of a new isoform of the human estrogen receptor-alpha (hER-alpha) that is encoded by distinct transcripts and that is able to repress hER-alpha activation function 1. EMBO J. (2000) 19:4688–700. doi: 10.1093/emboj/19.17.4688
85. Li L, Haynes MP, and Bender JR. Plasma membrane localization and function of the estrogen receptor alpha variant (ER46) in human endothelial cells. Proc Natl Acad Sci U.S.A. (2003) 100:4807–12. doi: 10.1073/pnas.0831079100
86. Shi L, Dong B, Li Z, Lu Y, Ouyang T, Li J, et al. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J Clin Oncol. (2009) 27:3423–9. doi: 10.1200/JCO.2008.17.2254
87. Chantalat E, Boudou F, Laurell H, Palierne G, Houtman R, Melchers D, et al. The AF-1-deficient estrogen receptor ERα46 isoform is frequently expressed in human breast tumors. Breast Cancer Res. (2016) 18:123. doi: 10.1186/s13058-016-0780-7
88. Penot G, Le Péron C, Mérot Y, Grimaud-Fanouillère E, Ferrière F, Boujrad N, et al. The human estrogen receptor-alpha isoform hERalpha46 antagonizes the proliferative influence of hERalpha66 in MCF7 breast cancer cells. Endocrinology. (2005) 146:5474–84. doi: 10.1210/en.2005-0866
89. Klinge CM, Riggs KA, Wickramasinghe NS, Emberts CG, McConda DB, Barry PN, et al. Estrogen receptor alpha 46 is reduced in tamoxifen resistant breast cancer cells and re-expression inhibits cell proliferation and estrogen receptor alpha 66-regulated target gene transcription. Mol Cell Endocrinol. (2010) 323:268–76. doi: 10.1016/j.mce.2010.03.013
90. Cirillo F, Pellegrino M, Talia M, Perrotta ID, Rigiracciolo DC, Spinelli A, et al. Estrogen receptor variant ERα46 and insulin receptor drive in primary breast cancer cells growth effects and interleukin 11 induction prompting the motility of cancer-associated fibroblasts. Clin Transl Med. (2021) 11:e516. doi: 10.1002/ctm2.516
91. Wang Q, Jiang J, Ying G, Xie X-Q, Zhang X, Xu W, et al. Tamoxifen enhances stemness and promotes metastasis of ERα36+ breast cancer by upregulating ALDH1A1 in cancer cells. Cell Res. (2018) 28:336–58. doi: 10.1038/cr.2018.15
92. Enmark E, Pelto-Huikko M, Grandien K, Lagercrantz S, Lagercrantz J, Fried G, et al. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab. (1997) 82:4258–65. doi: 10.1210/jcem.82.12.4470
93. Moore JT, McKee DD, Slentz-Kesler K, Moore LB, Jones SA, Horne EL, et al. Cloning and characterization of human estrogen receptor β Isoforms. Biochem Biophys Res Commun. (1998) 247:75–8. doi: 10.1006/bbrc.1998.8738
94. Swedenborg E, Power KA, Cai W, Pongratz I, and Rüegg J. Regulation of estrogen receptor beta activity and implications in health and disease. Cell Mol Life Sci. (2009) 66:3873–94. doi: 10.1007/s00018-009-0118-z
95. Warner M, Fan X, Strom A, Wu W, and Gustafsson J-Å. 25 years of ERβ: a personal journey. J Mol Endocrinol. (2022) 68:R1–9. doi: 10.1530/JME-21-0121
96. Choi Y, Kim H, and Pollack S. ERβ Isoforms have differential clinical significance in breast cancer subtypes and subgroups. Curr Issues Mol Biol. (2022) 44:1564–86. doi: 10.3390/cimb44040107
97. Yaşar P, Ayaz G, User SD, Güpür G, and Muyan M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod Med Biol. (2017) 16:4–20. doi: 10.1002/rmb2.12006
98. Helsen C, Kerkhofs S, Clinckemalie L, Spans L, Laurent M, Boonen S, et al. Structural basis for nuclear hormone receptor DNA binding. Mol Cell Endocrinol. (2012) 348:411–7. doi: 10.1016/j.mce.2011.07.025
99. Lutz PB, Coombs WR, and Bayse CA. Determination of structural factors contributing to protection of zinc fingers in estrogen receptor α through molecular dynamic simulations. J Phys Chem B. (2025) 129:2226–34. doi: 10.1021/acs.jpcb.4c05730
100. Mader S, Kumar V, de Verneuil H, and Chambon P. Three amino acids of the oestrogen receptor are essential to its ability to distinguish an oestrogen from a glucocorticoid-responsive element. Nature. (1989) 338:271–4. doi: 10.1038/338271a0
101. Muyan M. Dynamic transcriptional events mediated by estrogen receptor alpha. Front Biosci. (2019) 24:245–76. doi: 10.2741/4716
102. Schwabe JW, Chapman L, Finch JT, and Rhodes D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell. (1993) 75:567–78. doi: 10.1016/0092-8674(93)90390-c
103. Green S, Kumar V, Theulaz I, Wahli W, and Chambon P. The N-terminal DNA-binding “zinc finger” of the oestrogen and glucocorticoid receptors determines target gene specificity. EMBO J. (1988) 7:3037–44. doi: 10.1002/j.1460-2075.1988.tb03168.x
104. Green S, Walter P, Kumar V, Krust A, Bornert J-M, Argos P, et al. Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature. (1986) 320:134–9. doi: 10.1038/320134a0
105. Hewitt SC, Li L, Grimm SA, Winuthayanon W, Hamilton KJ, Pockette B, et al. Novel DNA motif binding activity observed in vivo with an estrogen receptor α Mutant mouse. Mol Endocrinol. (2014) 28:899–911. doi: 10.1210/me.2014-1051
106. Kumar V, Green S, Stack G, Berry M, Jin JR, and Chambon P. Functional domains of the human estrogen receptor. Cell. (1987) 51:941–51. doi: 10.1016/0092-8674(87)90581-2
107. Rawłuszko-Wieczorek AA, Romanowska K, and Nowicki M. Chromatin modifiers – Coordinators of estrogen action. Biomed Pharmacother. (2022) 153:113548. doi: 10.1016/j.biopha.2022.113548
108. Dhamad AE, Zhou Z, Zhou J, and Du Y. Systematic proteomic identification of the heat shock proteins (Hsp) that interact with estrogen receptor alpha (ERα) and biochemical characterization of the ERα-hsp70 interaction. PloS One. (2016) 11:e0160312. doi: 10.1371/journal.pone.0160312
109. Pratt WB and Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood). (2003) 228:111–33. doi: 10.1177/153537020322800201
110. Smith DF and Toft DO. Minireview: the intersection of steroid receptors with molecular chaperones: observations and questions. Mol Endocrinol. (2008) 22:2229–40. doi: 10.1210/me.2008-0089
111. Yi P, Yu X, Wang Z, and O’Malley BW. Steroid receptor-coregulator transcriptional complexes: new insights from CryoEM. Essays Biochem. (2021) 65:857–66. doi: 10.1042/EBC20210019
112. Yi P, Wang Z, Feng Q, Chou C-K, Pintilie GD, Shen H, et al. Structural and functional impacts of ER coactivator sequential recruitment. Mol Cell. (2017) 67:733–743.e4. doi: 10.1016/j.molcel.2017.07.026
113. Yi P, Wang Z, Feng Q, Pintilie GD, Foulds CE, Lanz RB, et al. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol Cell. (2015) 57:1047–58. doi: 10.1016/j.molcel.2015.01.025
114. Rosenbaum DM, Rasmussen SGF, and Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. (2009) 459:356–63. doi: 10.1038/nature08144
115. Abbas MA, Al-Kabariti AY, and Sutton C. Comprehensive understanding of the role of GPER in estrogen receptor-alpha negative breast cancer. J Steroid Biochem Mol Biol. (2024) 241:106523. doi: 10.1016/j.jsbmb.2024.106523
116. Acramel A and Jacquot Y. Deciphering of a putative GPER recognition domain in ERα and ERα36. Front Endocrinol (Lausanne). (2022) 13:943343. doi: 10.3389/fendo.2022.943343
117. Heo K-S. Regulation of post-translational modification in breast cancer treatment. BMB Rep. (2019) 52:113–8. doi: 10.5483/BMBRep.2019.52.2.017
118. Atsriku C, Britton DJ, Held JM, Schilling B, Scott GK, Gibson BW, et al. Systematic mapping of posttranslational modifications in human estrogen receptor-alpha with emphasis on novel phosphorylation sites. Mol Cell Proteomics. (2009) 8:467–80. doi: 10.1074/mcp.M800282-MCP200
119. Thomas RS, Sarwar N, Phoenix F, Coombes RC, and Ali S. Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-alpha activity. J Mol Endocrinol. (2008) 40:173–84. doi: 10.1677/JME-07-0165
120. Medunjanin S, Hermani A, De Servi B, Grisouard J, Rincke G, and Mayer D. Glycogen synthase kinase-3 interacts with and phosphorylates estrogen receptor alpha and is involved in the regulation of receptor activity. J Biol Chem. (2005) 280:33006–14. doi: 10.1074/jbc.M506758200
121. Chen D, Riedl T, Washbrook E, Pace PE, Coombes RC, Egly JM, et al. Activation of estrogen receptor alpha by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol Cell. (2000) 6:127–37. doi: 10.1016/S1097-2765(05)00004-3
122. Chen D, Pace PE, Coombes RC, and Ali S. Phosphorylation of human estrogen receptor alpha by protein kinase A regulates dimerization. Mol Cell Biol. (1999) 19:1002–15. doi: 10.1128/MCB.19.2.1002
123. Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. (1995) 270:1491–4. doi: 10.1126/science.270.5241.1491
124. Masuhiro Y, Mezaki Y, Sakari M, Takeyama K, Yoshida T, Inoue K, et al. Splicing potentiation by growth factor signals via estrogen receptor phosphorylation. Proc Natl Acad Sci U.S.A. (2005) 102:8126–31. doi: 10.1073/pnas.0503197102
125. Sheeler CQ, Singleton DW, and Khan SA. Mutation of serines 104, 106, and 118 inhibits dimerization of the human estrogen receptor in yeast. Endocr Res. (2003) 29:237–55. doi: 10.1081/erc-120022321
126. Weitsman GE, Li L, Skliris GP, Davie JR, Ung K, Niu Y, et al. Estrogen Receptor-α Phosphorylated at Ser118 Is Present at the Promoters of Estrogen-Regulated Genes and Is Not Altered Due to HER-2 Overexpression. Cancer Res. (2006) 66:10162–70. doi: 10.1158/0008-5472.CAN-05-4111
127. Peng Y, Cao S, Kiselar J, Xiao X, Du Z, Hsieh A, et al. A metastable contact and structural disorder in the estrogen receptor transactivation domain. Structure. (2019) 27:229–240.e4. doi: 10.1016/j.str.2018.10.026
128. Michalides R, Griekspoor A, Balkenende A, Verwoerd D, Janssen L, Jalink K, et al. Tamoxifen resistance by a conformational arrest of the estrogen receptor alpha after PKA activation in breast cancer. Cancer Cell. (2004) 5:597–605. doi: 10.1016/j.ccr.2004.05.016
129. Tharakan R, Lepont P, Singleton D, Kumar R, and Khan S. Phosphorylation of estrogen receptor alpha, serine residue 305 enhances activity. Mol Cell Endocrinol. (2008) 295:70–8. doi: 10.1016/j.mce.2008.07.018
130. Arnold SF, Obourn JD, Jaffe H, and Notides AC. Serine 167 is the major estradiol-induced phosphorylation site on the human estrogen receptor. Mol Endocrinol. (1994) 8:1208–14. doi: 10.1210/mend.8.9.7838153
131. Choi Y. Estrogen receptor β Expression and its clinical implication in breast cancers: favorable or unfavorable? J Breast Cancer. (2022) 25:75. doi: 10.4048/jbc.2022.25.e9
132. Jiang J, Sarwar N, Peston D, Kulinskaya E, Shousha S, Coombes RC, et al. Phosphorylation of estrogen receptor-alpha at Ser167 is indicative of longer disease-free and overall survival in breast cancer patients. Clin Cancer Res. (2007) 13:5769–76. doi: 10.1158/1078-0432.CCR-07-0822
133. Motomura K, Ishitobi M, Komoike Y, Koyama H, Nagase H, Inaji H, et al. Expression of estrogen receptor beta and phosphorylation of estrogen receptor alpha serine 167 correlate with progression-free survival in patients with metastatic breast cancer treated with aromatase inhibitors. Oncology. (2010) 79:55–61. doi: 10.1159/000319540
134. Yamashita H, Nishio M, Kobayashi S, Ando Y, Sugiura H, Zhang Z, et al. Phosphorylation of estrogen receptor alpha serine 167 is predictive of response to endocrine therapy and increases postrelapse survival in metastatic breast cancer. Breast Cancer Res. (2005) 7:R753–764. doi: 10.1186/bcr1285
135. Wilson BJ, Tremblay AM, Deblois G, Sylvain-Drolet G, and Giguère V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol Endocrinol. (2010) 24:1349–58. doi: 10.1210/me.2009-0441
136. Ma Y, Fan S, Hu C, Meng Q, Fuqua SA, Pestell RG, et al. BRCA1 regulates acetylation and ubiquitination of estrogen receptor-alpha. Mol Endocrinol. (2010) 24:76–90. doi: 10.1210/me.2009-0218
137. Barone I, Iacopetta D, Covington KR, Cui Y, Tsimelzon A, Beyer A, et al. Phosphorylation of the mutant K303R estrogen receptor α at serine 305 affects aromatase inhibitor sensitivity. Oncogene. (2010) 29:2404–14. doi: 10.1038/onc.2009.520
138. Fuqua SA, Wiltschke C, Zhang QX, Borg A, Castles CG, Friedrichs WE, et al. A hypersensitive estrogen receptor-alpha mutation in premalignant breast lesions. Cancer Res. (2000) 60:4026–9.
139. Herynk MH, Parra I, Cui Y, Beyer A, Wu M-F, Hilsenbeck SG, et al. Association between the estrogen receptor alpha A908G mutation and outcomes in invasive breast cancer. Clin Cancer Res. (2007) 13:3235–43. doi: 10.1158/1078-0432.CCR-06-2608
140. Subramanian K, Jia D, Kapoor-Vazirani P, Powell DR, Collins RE, Sharma D, et al. Regulation of estrogen receptor alpha by the SET7 lysine methyltransferase. Mol Cell. (2008) 30:336–47. doi: 10.1016/j.molcel.2008.03.022
141. Acconcia F, Ascenzi P, Fabozzi G, Visca P, and Marino M. S-palmitoylation modulates human estrogen receptor-alpha functions. Biochem Biophys Res Commun. (2004) 316:878–83. doi: 10.1016/j.bbrc.2004.02.129
142. Pedram A, Razandi M, Deschenes RJ, and Levin ER. DHHC-7 and -21 are palmitoylacyltransferases for sex steroid receptors. Mol Biol Cell. (2012) 23:188–99. doi: 10.1091/mbc.E11-07-0638
143. Geiss-Friedlander R and Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. (2007) 8:947–56. doi: 10.1038/nrm2293
144. Sentis S, Le Romancer M, Bianchin C, Rostan M-C, and Corbo L. Sumoylation of the estrogen receptor alpha hinge region regulates its transcriptional activity. Mol Endocrinol. (2005) 19:2671–84. doi: 10.1210/me.2005-0042
145. Deng B, Tarhan YE, Ueda K, Ren L, Katagiri T, Park J-H, et al. Critical role of estrogen receptor alpha O-glycosylation by N-acetylgalactosaminyltransferase 6 (GALNT6) in its nuclear localization in breast cancer cells. Neoplasia. (2018) 20:1038–44. doi: 10.1016/j.neo.2018.08.006
146. Han D, Wang L, Jiang S, and Yang Q. The ubiquitin-proteasome system in breast cancer. Trends Mol Med. (2023) 29:599–621. doi: 10.1016/j.molmed.2023.05.006
147. Helzer KT, Hooper C, Miyamoto S, and Alarid ET. Ubiquitylation of nuclear receptors: new linkages and therapeutic implications. J Mol Endocrinol. (2015) 54:R151–167. doi: 10.1530/JME-14-0308
148. Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. (2009) 37:937–53. doi: 10.1042/BST0370937
149. Duong V, Boulle N, Daujat S, Chauvet J, Bonnet S, Neel H, et al. Differential regulation of estrogen receptor alpha turnover and transactivation by Mdm2 and stress-inducing agents. Cancer Res. (2007) 67:5513–21. doi: 10.1158/0008-5472.CAN-07-0967
150. Alarid ET, Bakopoulos N, and Solodin N. Proteasome-mediated proteolysis of estrogen receptor: a novel component in autologous down-regulation. Mol Endocrinol. (1999) 13:1522–34. doi: 10.1210/mend.13.9.0337
151. Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir J-M, and Corbo L. Cracking the estrogen receptor’s posttranslational code in breast tumors. Endocr Rev. (2011) 32:597–622. doi: 10.1210/er.2010-0016
152. Wijayaratne AL and McDonnell DP. The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J Biol Chem. (2001) 276:35684–92. doi: 10.1074/jbc.M101097200
153. Long X and Nephew KP. Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-alpha. J Biol Chem. (2006) 281:9607–15. doi: 10.1074/jbc.M510809200
154. Berry NB, Fan M, and Nephew KP. Estrogen receptor-alpha hinge-region lysines 302 and 303 regulate receptor degradation by the proteasome. Mol Endocrinol. (2008) 22:1535–51. doi: 10.1210/me.2007-0449
155. Gonzalez de Valdivia E, Sandén C, Kahn R, Olde B, and Leeb-Lundberg LMF. Human G protein-coupled receptor 30 is N-glycosylated and N-terminal domain asparagine 44 is required for receptor structure and activity. Biosci Rep. (2019) 39:BSR20182436. doi: 10.1042/BSR20182436
156. Ding J and Kuang P. Regulation of ERα Stability and estrogen signaling in breast cancer by HOIL-1. Front Oncol. (2021) 11:664689. doi: 10.3389/fonc.2021.664689
157. Stebbing J, Shah K, Lit LC, Gagliano T, Ditsiou A, Wang T, et al. LMTK3 confers chemo-resistance in breast cancer. Oncogene. (2018) 37:3113–30. doi: 10.1038/s41388-018-0197-0
158. Stebbing J, Filipovic A, Ellis IO, Green AR, D’Silva TR, Lenz H-J, et al. LMTK3 expression in breast cancer: association with tumor phenotype and clinical outcome. Breast Cancer Res Treat. (2012) 132:537–44. doi: 10.1007/s10549-011-1622-z
159. Xu Y, Zhang H, Lit LC, Grothey A, Athanasiadou M, Kiritsi M, et al. The kinase LMTK3 promotes invasion in breast cancer through GRB2-mediated induction of integrin β1. Sci Signal. (2014) 7:ra58. doi: 10.1126/scisignal.2005170
160. Medunjanin S, Weinert S, Schmeisser A, Mayer D, and Braun-Dullaeus RC. Interaction of the double-strand break repair kinase DNA-PK and estrogen receptor-alpha. Mol Biol Cell. (2010) 21:1620–8. doi: 10.1091/mbc.e09-08-0724
161. Williams CC, Basu A, El-Gharbawy A, Carrier LM, Smith CL, and Rowan BG. Identification of four novel phosphorylation sites in estrogen receptor α: impact on receptor-dependent gene expression and phosphorylation by protein kinase CK2. BMC Biochem. (2009) 10:36. doi: 10.1186/1471-2091-10-36
162. Flach KD, Periyasamy M, Jadhav A, Dorjsuren D, Siefert JC, Hickey TE, et al. Endonuclease FEN1 coregulates ERα Activity and provides a novel drug interface in tamoxifen-resistant breast cancer. Cancer Res. (2020) 80:1914–26. doi: 10.1158/0008-5472.CAN-19-2207
163. Tang J, Luo Y, Tian Z, Liao X, Cui Q, Yang Q, et al. TRIM11 promotes breast cancer cell proliferation by stabilizing estrogen receptor α. Neoplasia. (2020) 22:343–51. doi: 10.1016/j.neo.2020.06.003
164. Zhuang T, Wang B, Tan X, Wu L, Li X, Li Z, et al. TRIM3 facilitates estrogen signaling and modulates breast cancer cell progression. Cell Commun Signal. (2022) 20:45. doi: 10.1186/s12964-022-00861-z
165. Tian Z, Tang J, Liao X, Gong Y, Yang Q, Wu Y, et al. TRIM8 inhibits breast cancer proliferation by regulating estrogen signaling. Am J Cancer Res. (2020) 10:3440–57. doi: 10.21203/rs.3.rs-42521/v1
166. Xue M, Zhang K, Mu K, Xu J, Yang H, Liu Y, et al. Regulation of estrogen signaling and breast cancer proliferation by an ubiquitin ligase TRIM56. Oncogenesis. (2019) 8:30. doi: 10.1038/s41389-019-0139-x
167. Wang S, Luo H, Wang C, Sun H, Sun G, Sun N, et al. RNF8 identified as a co-activator of estrogen receptor α promotes cell growth in breast cancer. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1615–28. doi: 10.1016/j.bbadis.2017.02.011
168. Zhu J, Zhuang T, Yang H, Li X, Liu H, and Wang H. Atypical ubiquitin ligase RNF31: the nuclear factor modulator in breast cancer progression. BMC Cancer. (2016) 16:538. doi: 10.1186/s12885-016-2575-8
169. Zhu J, Zhao C, Kharman-Biz A, Zhuang T, Jonsson P, Liang N, et al. The atypical ubiquitin ligase RNF31 stabilizes estrogen receptor α and modulates estrogen-stimulated breast cancer cell proliferation. Oncogene. (2014) 33:4340–51. doi: 10.1038/onc.2013.573
170. Zhuang T, Yu S, Zhang L, Yang H, Li X, Hou Y, et al. SHARPIN stabilizes estrogen receptor α and promotes breast cancer cell proliferation. Oncotarget. (2017) 8:77137–51. doi: 10.18632/oncotarget.20368
171. Yang H, Yu N, Xu J, Ding X, Deng W, Wu G, et al. SMURF1 facilitates estrogen receptor α signaling in breast cancer cells. J Exp Clin Cancer Res. (2018) 37:24. doi: 10.1186/s13046-018-0672-z
172. Zhu J, Li X, Su P, Xue M, Zang Y, and Ding Y. The ubiquitin ligase RNF181 stabilizes ERα and modulates breast cancer progression. Oncogene. (2020) 39:6776–88. doi: 10.1038/s41388-020-01464-z
173. Gallo D, Jacquemotte F, Cleeren A, Laïos I, Hadiy S, Rowlands MG, et al. Calmodulin-independent, agonistic properties of a peptide containing the calmodulin binding site of estrogen receptor alpha. Mol Cell Endocrinol. (2007) 268:37–49. doi: 10.1016/j.mce.2007.01.012
174. Li L, Li Z, and Sacks DB. The transcriptional activity of estrogen receptor-alpha is dependent on Ca2+/calmodulin. J Biol Chem. (2005) 280:13097–104. doi: 10.1074/jbc.M410642200
175. Masaki T, Habara M, Sato Y, Goshima T, Maeda K, Hanaki S, et al. Calcineurin regulates the stability and activity of estrogen receptor α. Proc Natl Acad Sci U.S.A. (2021) 118:e2114258118. doi: 10.1073/pnas.2114258118
176. Xia X, Liao Y, Huang C, Liu Y, He J, Shao Z, et al. Deubiquitination and stabilization of estrogen receptor α by ubiquitin-specific protease 7 promotes breast tumorigenesis. Cancer Lett. (2019) 465:118–28. doi: 10.1016/j.canlet.2019.09.003
177. Xia X, Huang C, Liao Y, Liu Y, He J, Shao Z, et al. The deubiquitinating enzyme USP15 stabilizes ERα and promotes breast cancer progression. Cell Death Dis. (2021) 12:329. doi: 10.1038/s41419-021-03607-w
178. Cao J, Wu D, Wu G, Wang Y, Ren T, Wang Y, et al. USP35, regulated by estrogen and AKT, promotes breast tumorigenesis by stabilizing and enhancing transcriptional activity of estrogen receptor α. Cell Death Dis. (2021) 12:619. doi: 10.1038/s41419-021-03904-4
179. Stanišić V, Malovannaya A, Qin J, Lonard DM, and O’Malley BW. OTU Domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) deubiquitinates estrogen receptor (ER) alpha and affects ERalpha transcriptional activity. J Biol Chem. (2009) 284:16135–45. doi: 10.1074/jbc.M109.007484
180. Tang J, Luo Y, Long G, and Zhou L. MINDY1 promotes breast cancer cell proliferation by stabilizing estrogen receptor α. Cell Death Dis. (2021) 12:937. doi: 10.1038/s41419-021-04244-z
181. Tang J, Wu Z, Tian Z, Chen W, and Wu G. OTUD7B stabilizes estrogen receptor α and promotes breast cancer cell proliferation. Cell Death Dis. (2021) 12:534. doi: 10.1038/s41419-021-03785-7
182. Fowler AM, Solodin N, Preisler-Mashek MT, Zhang P, Lee AV, and Alarid ET. Increases in estrogen receptor-alpha concentration in breast cancer cells promote serine 118/104/106-independent AF-1 transactivation and growth in the absence of estrogen. FASEB J. (2004) 18:81–93. doi: 10.1096/fj.03-0038com
183. Martin L-A, Farmer I, Johnston SRD, Ali S, and Dowsett M. Elevated ERK1/ERK2/estrogen receptor cross-talk enhances estrogen-mediated signaling during long-term estrogen deprivation. Endocr Relat Cancer. (2005) 12 Suppl 1:S75–84. doi: 10.1677/erc.1.01023
184. Martin L-A, Farmer I, Johnston SRD, Ali S, Marshall C, and Dowsett M. Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation. J Biol Chem. (2003) 278:30458–68. doi: 10.1074/jbc.M305226200
185. Santen RJ, Song RX, Zhang Z, Kumar R, Jeng M-H, Masamura A, et al. Long-term estradiol deprivation in breast cancer cells up-regulates growth factor signaling and enhances estrogen sensitivity. Endocr Relat Cancer. (2005) 12 Suppl 1:S61–73. doi: 10.1677/erc.1.01018
186. Traphagen NA, Hosford SR, Jiang A, Marotti JD, Brauer BL, Demidenko E, et al. High estrogen receptor alpha activation confers resistance to estrogen deprivation and is required for therapeutic response to estrogen in breast cancer. Oncogene. (2021) 40:3408–21. doi: 10.1038/s41388-021-01782-w
187. Pan X, Zhou T, Tai Y-H, Wang C, Zhao J, Cao Y, et al. Elevated expression of CUEDC2 protein confers endocrine resistance in breast cancer. Nat Med. (2011) 17:708–14. doi: 10.1038/nm.2369
188. Rayala SK, den Hollander P, Balasenthil S, Yang Z, Broaddus RR, and Kumar R. Functional regulation of oestrogen receptor pathway by the dynein light chain 1. EMBO Rep. (2005) 6:538–44. doi: 10.1038/sj.embor.7400417
189. Castoria G, Giovannelli P, Lombardi M, De Rosa C, Giraldi T, de Falco A, et al. Tyrosine phosphorylation of estradiol receptor by Src regulates its hormone-dependent nuclear export and cell cycle progression in breast cancer cells. Oncogene. (2012) 31:4868–77. doi: 10.1038/onc.2011.642
190. Ring A and Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer. (2004) 11:643–58. doi: 10.1677/erc.1.00776
191. Hermida-Prado F and Jeselsohn R. The ESR1 mutations: from bedside to bench to bedside. Cancer Res. (2021) 81:537–8. doi: 10.1158/0008-5472.CAN-20-4037
192. Jeselsohn R, Buchwalter G, De Angelis C, Brown M, and Schiff R. ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol. (2015) 12:573–83. doi: 10.1038/nrclinonc.2015.117
193. Ojo D, Wei F, Liu Y, Wang E, Zhang H, Lin X, et al. Factors promoting tamoxifen resistance in breast cancer via stimulating breast cancer stem cell expansion. Curr Med Chem. (2015) 22:2360–74. doi: 10.2174/0929867322666150416095744
194. Bahreini A, Li Z, Wang P, Levine KM, Tasdemir N, Cao L, et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. (2017) 19:60. doi: 10.1186/s13058-017-0851-4
195. Carlson KE, Choi I, Gee A, Katzenellenbogen BS, and Katzenellenbogen JA. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry. (1997) 36:14897–905. doi: 10.1021/bi971746l
196. Chandarlapaty S, Chen D, He W, Sung P, Samoila A, You D, et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: A secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. (2016) 2:1310–5. doi: 10.1001/jamaoncol.2016.1279
197. Chu D, Paoletti C, Gersch C, VanDenBerg DA, Zabransky DJ, Cochran RL, et al. ESR1 mutations in circulating plasma tumor DNA from metastatic breast cancer patients. Clin Cancer Res. (2016) 22:993–9. doi: 10.1158/1078-0432.CCR-15-0943
198. Clatot F, Perdrix A, Augusto L, Beaussire L, Delacour J, Calbrix C, et al. Kinetics, prognostic and predictive values of ESR1 circulating mutations in metastatic breast cancer patients progressing on aromatase inhibitor. Oncotarget. (2016) 7:74448–59. doi: 10.18632/oncotarget.12950
199. Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife. (2016) 5:e12792. doi: 10.7554/eLife.12792
200. Fribbens C, O’Leary B, Kilburn L, Hrebien S, Garcia-Murillas I, Beaney M, et al. Plasma ESR1 mutations and the treatment of estrogen receptor–positive advanced breast cancer. JCO. (2016) 34:2961–8. doi: 10.1200/JCO.2016.67.3061
201. Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. (2015) 7:302ra133. doi: 10.1126/scitranslmed.aab0021
202. Katzenellenbogen JA, Mayne CG, Katzenellenbogen BS, Greene GL, and Chandarlapaty S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat Rev Cancer. (2018) 18:377–88. doi: 10.1038/s41568-018-0001-z
203. Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. (2013) 4:1116–30. doi: 10.1016/j.celrep.2013.08.022
204. Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, et al. D538G mutation in estrogen receptor-α: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. (2013) 73:6856–64. doi: 10.1158/0008-5472.CAN-13-1197
205. Peng J, Sengupta S, and Jordan VC. Potential of selective estrogen receptor modulators as treatments and preventives of breast cancer. Anticancer Agents Med Chem. (2009) 9:481–99. doi: 10.2174/187152009788451833
206. Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, Savage H, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun. (2016) 7:11579. doi: 10.1038/ncomms11579
207. Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. (2013) 45:1439–45. doi: 10.1038/ng.2822
208. Wang P, Bahreini A, Gyanchandani R, Lucas PC, Hartmaier RJ, Watters RJ, et al. Sensitive detection of mono- and polyclonal ESR1 mutations in primary tumors, metastatic lesions, and cell-free DNA of breast cancer patients. Clin Cancer Res. (2016) 22:1130–7. doi: 10.1158/1078-0432.CCR-15-1534
209. Clusan L, Le Goff P, Flouriot G, and Pakdel F. A closer look at estrogen receptor mutations in breast cancer and their implications for estrogen and antiestrogen responses. Int J Mol Sci. (2021) 22:756. doi: 10.3390/ijms22020756
210. Thomas C and Gustafsson J-Å. Estrogen receptor mutations and functional consequences for breast cancer. Trends Endocrinol Metab. (2015) 26:467–76. doi: 10.1016/j.tem.2015.06.007
211. Weis KE, Ekena K, Thomas JA, Lazennec G, and Katzenellenbogen BS. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol Endocrinol. (1996) 10:1388–98. doi: 10.1210/mend.10.11.8923465
212. Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. (2014) 345:216–20. doi: 10.1126/science.1253533
213. Lazennec G, Ediger TR, Petz LN, Nardulli AM, and Katzenellenbogen BS. Mechanistic aspects of estrogen receptor activation probed with constitutively active estrogen receptors: correlations with DNA and coregulator interactions and receptor conformational changes. Mol Endocrinol. (1997) 11:1375–86. doi: 10.1210/mend.11.9.9983
214. Pakdel F, Reese JC, and Katzenellenbogen BS. Identification of charged residues in an N-terminal portion of the hormone-binding domain of the human estrogen receptor important in transcriptional activity of the receptor. Mol Endocrinol. (1993) 7:1408–17. doi: 10.1210/mend.7.11.8114756
215. Chaudhri RA, Olivares-Navarrete R, Cuenca N, Hadadi A, Boyan BD, and Schwartz Z. Membrane estrogen signaling enhances tumorigenesis and metastatic potential of breast cancer cells via estrogen receptor-α36 (ERα36). J Biol Chem. (2012) 287:7169–81. doi: 10.1074/jbc.M111.292946
216. Deng H, Yin L, Zhang X-T, Liu L-J, Wang M-L, and Wang Z-Y. ER-α variant ER-α36 mediates antiestrogen resistance in ER-positive breast cancer stem/progenitor cells. J Steroid Biochem Mol Biol. (2014) 144 Pt B:417–26. doi: 10.1016/j.jsbmb.2014.08.017
217. Deng H, Zhang X-T, Wang M-L, Zheng H-Y, Liu L-J, and Wang Z-Y. ER-α36-mediated rapid estrogen signaling positively regulates ER-positive breast cancer stem/progenitor cells. PloS One. (2014) 9:e88034. doi: 10.1371/journal.pone.0088034
218. Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, and Deuel TF. A variant of estrogen receptor-{alpha}, hER-{alpha}36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc Natl Acad Sci U.S.A. (2006) 103:9063–8. doi: 10.1073/pnas.0603339103
219. Barone I, Brusco L, and Fuqua SAW. Estrogen receptor mutations and changes in downstream gene expression and signaling. Clin Cancer Res. (2010) 16:2702–8. doi: 10.1158/1078-0432.CCR-09-1753
220. Alluri PG, Speers C, and Chinnaiyan AM. Estrogen receptor mutations and their role in breast cancer progression. Breast Cancer Res. (2014) 16:494. doi: 10.1186/s13058-014-0494-7
221. Martin L-A, Ribas R, Simigdala N, Schuster E, Pancholi S, Tenev T, et al. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat Commun. (2017) 8:1865. doi: 10.1038/s41467-017-01864-y
222. Arnesen S, Blanchard Z, Williams MM, Berrett KC, Li Z, Oesterreich S, et al. Estrogen receptor alpha mutations in breast cancer cells cause gene expression changes through constant activity and secondary effects. Cancer Res. (2021) 81:539–51. doi: 10.1158/0008-5472.CAN-20-1171
223. Liao H, Huang W, Pei W, and Li H. Detection of ESR1 mutations based on liquid biopsy in estrogen receptor-positive metastatic breast cancer: clinical impacts and prospects. Front Oncol. (2020) 10:587671. doi: 10.3389/fonc.2020.587671
224. Najim O, Seghers S, Sergoynne L, Van Gaver H, Papadimitriou K, Wouters K, et al. The association between type of endocrine therapy and development of estrogen receptor-1 mutation(s) in patients with hormone-sensitive advanced breast cancer: A systematic review and meta-analysis of randomized and non-randomized trials. Biochim Biophys Acta Rev Cancer. (2019) 1872:188315. doi: 10.1016/j.bbcan.2019.188315
225. Rastinejad F, Huang P, Chandra V, and Khorasanizadeh S. Understanding nuclear receptor form and function using structural biology. J Mol Endocrinol. (2013) 51:T1–T21. doi: 10.1530/JME-13-0173
226. Murakami S, Nagari A, and Kraus WL. Dynamic assembly and activation of estrogen receptor α enhancers through coregulator switching. Genes Dev. (2017) 31:1535–48. doi: 10.1101/gad.302182.117
227. Harrod A, Lai C-F, Goldsbrough I, Simmons GM, Oppermans N, Santos DB, et al. Genome engineering for estrogen receptor mutations reveals differential responses to anti-estrogens and new prognostic gene signatures for breast cancer. Oncogene. (2022) 41:4905–15. doi: 10.1038/s41388-022-02483-8
228. Grinshpun A, Chen V, Sandusky ZM, Fanning SW, and Jeselsohn R. ESR1 activating mutations: From structure to clinical application. Biochim Biophys Acta Rev Cancer. (2023) 1878:188830. doi: 10.1016/j.bbcan.2022.188830
229. Gu G, Tian L, Gao M, Rechoum Y, Gelsomino L, Dustin D, et al. Abstract 22: The Y537S ESR1 mutation is a dominant driver of distant ER-positive breast cancer metastasis. Cancer Res. (2018) 78:22–2. doi: 10.1158/1538-7445.AM2018-22
230. Goldberg J, Qiao N, Guerriero JL, Gross B, Meneksedag Y, Lu YF, et al. Estrogen receptor mutations as novel targets for immunotherapy in metastatic estrogen receptor-positive breast cancer. Cancer Res Commun. (2024) 4:496–504. doi: 10.1158/2767-9764.CRC-23-0244
231. Schiavon G, Hrebien S, Garcia-Murillas I, Cutts RJ, Pearson A, Tarazona N, et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med. (2015) 7:313ra182. doi: 10.1126/scitranslmed.aac7551
232. Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im S-A, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. (2016) 17:425–39. doi: 10.1016/S1470-2045(15)00613-0
233. Tolaney SM, Toi M, Neven P, Sohn J, Grischke E-M, Llombart-Cussac A, et al. Clinical significance of PIK3CA and ESR1 mutations in circulating tumor DNA: analysis from the MONARCH 2 study of abemaciclib plus fulvestrant. Clin Cancer Res. (2022) 28:1500–6. doi: 10.1158/1078-0432.CCR-21-3276
234. Goetz MP, Wander SA, Bachelot T, Batist G, Cortes J, Cristofanilli M, et al. Open-label, randomized, multicenter, phase 3, ELAINE 3 study of the efficacy and safety of lasofoxifene plus abemaciclib for treating ER+/HER2-, locally advanced or metastatic breast cancer with an ESR1 mutation. JCO. (2024) 42:TPS1127–TPS1127. doi: 10.1200/JCO.2024.42.16_suppl.TPS1127
235. Goetz MP, Bagegni NA, Batist G, Brufsky A, Cristofanilli MA, Damodaran S, et al. Lasofoxifene versus fulvestrant for ER+/HER2- metastatic breast cancer with an ESR1 mutation: results from the randomized, phase II ELAINE 1 trial. Ann Oncol. (2023) 34:1141–51. doi: 10.1016/j.annonc.2023.09.3104
236. Fanning SW, Jeselsohn R, Dharmarajan V, Mayne CG, Karimi M, Buchwalter G, et al. The SERM/SERD bazedoxifene disrupts ESR1 helix 12 to overcome acquired hormone resistance in breast cancer cells. Elife. (2018) 7:e37161. doi: 10.7554/eLife.37161
237. Komm BS, Kharode YP, Bodine PVN, Harris HA, Miller CP, and Lyttle CR. Bazedoxifene acetate: a selective estrogen receptor modulator with improved selectivity. Endocrinology. (2005) 146:3999–4008. doi: 10.1210/en.2005-0030
238. Tsuji J, Li T, Grinshpun A, Coorens T, Russo D, Anderson L, et al. Clinical efficacy and whole-exome sequencing of liquid biopsies in a phase IB/II study of bazedoxifene and palbociclib in advanced hormone receptor-positive breast cancer. Clin Cancer Res. (2022) 28:5066–78. doi: 10.1158/1078-0432.CCR-22-2305
239. Bihani T, Patel HK, Arlt H, Tao N, Jiang H, Brown JL, et al. Elacestrant (RAD1901), a selective estrogen receptor degrader (SERD), has antitumor activity in multiple ER+ Breast cancer patient-derived xenograft models. Clin Cancer Res. (2017) 23:4793–804. doi: 10.1158/1078-0432.CCR-16-2561
240. Kaklamani V, Bardia A, Wilks S, Weise A, Richards D, Harb W, et al. Abstract PD7-07: Final analysis of phase 1 study of elacestrant (RAD1901), a novel selective estrogen receptor degrader (SERD), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2-) advanced breast cancer. Cancer Res. (2020) 80:PD7–07-PD7-07. doi: 10.1158/1538-7445.SABCS19-PD7-07
241. Lawson M, Cureton N, Ros S, Cheraghchi-Bashi A, Urosevic J, D’Arcy S, et al. The next-generation oral selective estrogen receptor degrader camizestrant (AZD9833) suppresses ER+ Breast cancer growth and overcomes endocrine and CDK4/6 inhibitor resistance. Cancer Res. (2023) 83:3989–4004. doi: 10.1158/0008-5472.CAN-23-0694
242. Oliveira M, Pominchuk D, Nowecki Z, Hamilton E, Kulyaba Y, Andabekov T, et al. Camizestrant, a next-generation oral SERD, versus fulvestrant in post-menopausal women with oestrogen receptor-positive, HER2-negative advanced breast cancer (SERENA-2): a multi-dose, open-label, randomised, phase 2 trial. Lancet Oncol. (2024) 25:1424–39. doi: 10.1016/S1470-2045(24)00387-5
243. Jhaveri K, Winer EP, Lim E, Fidalgo JA, Bellet M, Mayer IA, et al. Abstract PD7-05: A first-in-human phase I study to evaluate the oral selective estrogen receptor degrader (SERD), GDC-9545, in postmenopausal women with estrogen receptor-positive (ER+) HER2-negative (HER2-) metastatic breast cancer. Cancer Res. (2020) 80:PD7–05-PD7-05. doi: 10.1158/1538-7445.SABCS19-PD7-05
244. Martín M, Lim E, Chavez-MacGregor M, Bardia A, Wu J, Zhang Q, et al. Giredestrant for estrogen receptor–positive, HER2-negative, previously treated advanced breast cancer: results from the randomized, phase II acelERA breast cancer study. JCO. (2024) 42:2149–60. doi: 10.1200/JCO.23.01500
245. Mayer EL, Tolaney S, Brufsky AM, Gradishar W, Jhaveri K, Martín M, et al. Abstract OT2-01-07: evERA Breast Cancer: A phase III study of giredestrant (GDC-9545) + everolimus vs exemestane + everolimus in patients with estrogen receptor+, HER2– locally advanced or metastatic breast cancer. Cancer Res. (2023) 83:OT2-01-07-OT2-01-07. doi: 10.1158/1538-7445.SABCS22-OT2-01-07
246. Schmid P, Geyer CE Jr., Harbeck N, Rimawi M, Hurvitz S, Martín M, et al. Abstract OT2-03-02: lidERA Breast Cancer: A phase III adjuvant study of giredestrant (GDC-9545) vs physician’s choice of endocrine therapy in patients with estrogen receptor+, HER2– early breast cancer. Cancer Res. (2023) 83:OT2-03-02-OT2-03-02. doi: 10.1158/1538-7445.SABCS22-OT2-03-02
247. Turner NC, Jhaveri KL, Bardia A, Niikura N, Dieras V, Barrios CH, et al. persevERA Breast Cancer (BC): Phase III study evaluating the efficacy and safety of giredestrant (GDC-9545) + palbociclib versus letrozole + palbociclib in patients (pts) with estrogen-receptor-positive, HER2-negative locally advanced or metastatic BC (ER+/HER2– LA/mBC). JCO. (2021) 39:TPS1103–TPS1103. doi: 10.1200/JCO.2021.39.15_suppl.TPS1103
248. Jhaveri KL, Lim E, Hamilton EP, Saura C, Meniawy T, Jeselsohn R, et al. A first-in-human phase 1a/b trial of LY3484356, an oral selective estrogen receptor (ER) degrader (SERD) in ER+ advanced breast cancer (aBC) and endometrial endometrioid cancer (EEC): Results from the EMBER study. JCO. (2021) 39:1050–0. doi: 10.1200/JCO.2021.39.15_suppl.1050
249. Andreano KJ, Wardell SE, Baker JG, Desautels TK, Baldi R, Chao CA, et al. G1T48, an oral selective estrogen receptor degrader, and the CDK4/6 inhibitor lerociclib inhibit tumor growth in animal models of endocrine-resistant breast cancer. Breast Cancer Res Treat. (2020) 180:635–46. doi: 10.1007/s10549-020-05575-9
250. Dees EC, Aftimos PG, Van Oordt H, De Vries EGE, Neven P, Pegram MD, et al. Dose-escalation study of G1T48, an oral selective estrogen receptor degrader (SERD), in postmenopausal women with ER+/HER2- locally advanced or metastatic breast cancer (ABC). Ann Oncol. (2019) 30:v121–2. doi: 10.1093/annonc/mdz242.035
251. Apostolidou K, Zografos E, Papatheodoridi MA, Fiste O, Dimopoulos MA, and Zagouri F. Oral SERDs alone or in combination with CDK 4/6 inhibitors in breast cancer: Current perspectives and clinical trials. Breast. (2024) 75:103729. doi: 10.1016/j.breast.2024.103729
252. Neupane N, Bawek S, Gurusinghe S, Ghaffary EM, Mirmosayyeb O, Thapa S, et al. Oral SERD, a novel endocrine therapy for estrogen receptor-positive breast cancer. Cancers (Basel). (2024) 16:619. doi: 10.3390/cancers16030619
253. Rej RK, Thomas JE, Acharyya RK, Rae JM, and Wang S. Targeting the estrogen receptor for the treatment of breast cancer: recent advances and challenges. J Med Chem. (2023) 66:8339–81. doi: 10.1021/acs.jmedchem.3c00136
254. Wang Y, Shi Z, Jiang Y, and Dai X. Abstract 5776: Pharmacologic and PK/PD study of D-0502: An orally bioavailable SERD with potent antitumor activity in ER-positive breast cancer cell lines and xenograft models. Cancer Res. (2018) 78:5776–6. doi: 10.1158/1538-7445.AM2018-5776
255. Lu J, Chan C, Sun D, Hu G, He H, Li J, et al. Discovery and preclinical profile of LX-039, a novel indole-based oral selective estrogen receptor degrader (SERD). Bioorg Med Chem Lett. (2022) 66:128734. doi: 10.1016/j.bmcl.2022.128734
256. Shen W, Hu X, Zhang J, Cao J, Yao H, Wang X, et al. 399P Results from a first-in-human phase Ia/b study of LX-039, an oral selective estrogen receptor (ER) degrader (SERD), in postmenopausal patients with ER+, HER2- advanced breast cancer (ABC). Ann Oncol. (2023) 34:S349. doi: 10.1016/j.annonc.2023.09.576
257. Kalinksy K, Abramson V, Chalasani P, Linden HM, Alidzanovic J, Layman RM, et al. Abstract P1-17-02: ZN-c5, an oral selective estrogen receptor degrader (SERD), in women with advanced estrogen receptor-positive (ER+)/human epidermal growth factor receptor 2 negative (HER2-) breast cancer. Cancer Res. (2022) 82:P1-17-02-P1-17-02. doi: 10.1158/1538-7445.SABCS21-P1-17-02
258. Keogh GP, Papish S, Piskorski W, Ulanska M, Jackson B, Suster M, et al. 564TiP A phase Ib dose-escalation study of ZN-c5, an oral selective estrogen receptor degrader (SERD), in combination with abemaciclib in patients with advanced estrogen receptor (ER)+/HER2- breast cancer. Ann Oncol. (2021) 32:S618–9. doi: 10.1016/j.annonc.2021.08.1086
259. Samatar AA, Li J, Hegde S, Huang P, Ma J, Bunker K, et al. Abstract 4373: Discovery of ZN-c5, a novel potent and oral selective estrogen receptor degrader. Cancer Res. (2020) 80:4373–3. doi: 10.1158/1538-7445.AM2020-4373
260. Hamilton EP, Pluard TJ, Wang JS, Morikawa A, Johnston SRD, Dees EC, et al. H3B-6545 in women with locally advanced/metastatic estrogen receptor-positive (ER+), HER2 negative (–) breast cancer (BC). JCO. (2024) 42:1015–5. doi: 10.1200/JCO.2024.42.16_suppl.1015
261. Hamilton EP, Wang JS, Pluard TJ, Johnston SRD, Morikawa A, Dees EC, et al. Phase I/II study of H3B-6545, a novel selective estrogen receptor covalent antagonist (SERCA), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2-) advanced breast cancer. JCO. (2021) 39:1018–8. doi: 10.1200/JCO.2021.39.15_suppl.1018
262. Johnston SRD, Pluard TJ, Wang JS, Hamilton EP, Song T, Rong Y, et al. H3B-6545 + palbociclib in patients (pts) with locally advanced/metastatic estrogen receptor-positive (ER+), HER2 negative (–) breast cancer (BC). JCO. (2024) 42:1051–1. doi: 10.1200/JCO.2024.42.16_suppl.1051
263. Borges VF, Chan A, Lin NU, Tonda ME, Shilkrut M, and Alemany CA. A phase 1b/2 dose escalation and expansion study of OP-1250 in combination with ribociclib or alpelisib in patients with advanced and/or metastatic estrogen receptor–positive (ER+)/HER2-negative (HER2-) breast cancer. JCO. (2023) 41:TPS1127–TPS1127. doi: 10.1200/JCO.2023.41.16_suppl.TPS1127
264. Chan A, Day D, Hui R, McCarthy N, Wilson R, Faltaos D, et al. Abstract P3-07-15: Preliminary data from a Phase 1b dose escalation study of OP-1250, an oral CERAN, in combination with palbociclib in patients with advanced and/or metastatic estrogen receptor (ER)-positive, HER2-negative breast cancer. Cancer Res. (2023) 83:P3-07-15-P3-07-15. doi: 10.1158/1538-7445.SABCS22-P3-07-15
265. Pistilli B, Bellet M, Del Mastro L, McArthur HL, Meisel JL, Schmid P, et al. OPERA-01: A randomized, open-label, phase 3 study of palazestrant (OP-1250) vs standard-of-care for ER+, HER2- advanced or metastatic breast cancer patients after endocrine therapy and CDK4/6 inhibitors. JCO. (2024) 42:TPS1135–TPS1135. doi: 10.1200/JCO.2024.42.16_suppl.TPS1135
266. Campone M, Ma CX, De Laurentiis M, Iwata H, Hurvitz SA, Wander SA, et al. VERITAC-2: A global, randomized phase 3 study of ARV-471, a proteolysis targeting chimera (PROTAC) estrogen receptor (ER) degrader, vs fulvestrant in ER+/human epidermal growth factor receptor 2 (HER2)- advanced breast cancer. JCO. (2023) 41:TPS1122–TPS1122. doi: 10.1200/JCO.2023.41.16_suppl.TPS1122
267. Flanagan J, Qian Y, Gough S, Andreoli M, Bookbinder M, Cadelina G, et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. (2019) 79:P5-04-18-P5-04-18. doi: 10.1158/1538-7445.SABCS18-P5-04-18
268. Hamilton EP, Ma C, De Laurentiis M, Iwata H, Hurvitz SA, Wander SA, et al. VERITAC-2: a Phase III study of vepdegestrant, a PROTAC ER degrader, versus fulvestrant in ER+/HER2- advanced breast cancer. Future Oncol. (2024) 20:2447–55. doi: 10.1080/14796694.2024.2377530
269. Hamilton EP, Han H, Schott A, Tan A, Nanda R, Juric D, et al. 390P Vepdegestrant, a proteolysis targeting chimera (PROTAC) estrogen receptor (ER) degrader, in ER+/human epidermal growth factor receptor 2 (HER2)- advanced breast cancer: Update of dose escalation results from a phase I/II trial. Ann Oncol. (2023) 34:S344. doi: 10.1016/j.annonc.2023.09.567
270. Patel MR, Layman RM, Danso MA, Cosgrove D, Zettler ME, Pehlivan KC, et al. Preliminary results from a phase 1 study of AC699, an orally bioavailable chimeric estrogen receptor degrader, in patients with advanced or metastatic breast cancer. JCO. (2024) 42:3074–4. doi: 10.1200/JCO.2024.42.16_suppl.3074
271. Carmeci C, Thompson DA, Ring HZ, Francke U, and Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. (1997) 45:607–17. doi: 10.1006/geno.1997.4972
272. Filardo EJ, Quinn JA, and Sabo E. Association of the membrane estrogen receptor, GPR30, with breast tumor metastasis and transactivation of the epidermal growth factor receptor. Steroids. (2008) 73:870–3. doi: 10.1016/j.steroids.2007.12.025
273. Filardo EJ and Thomas P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology. (2012) 153:2953–62. doi: 10.1210/en.2012-1061
274. Xu S, Yu S, Dong D, and Lee LTO. G protein-coupled estrogen receptor: A potential therapeutic target in cancer. Front Endocrinol (Lausanne). (2019) 10:725. doi: 10.3389/fendo.2019.00725
275. Catalano S, Giordano C, Panza S, Chemi F, Bonofiglio D, Lanzino M, et al. Tamoxifen through GPER upregulates aromatase expression: a novel mechanism sustaining tamoxifen-resistant breast cancer cell growth. Breast Cancer Res Treat. (2014) 146:273–85. doi: 10.1007/s10549-014-3017-4
276. Molina L, Bustamante F, Ortloff A, Ramos I, Ehrenfeld P, and Figueroa CD. Continuous exposure of breast cancer cells to tamoxifen upregulates GPER-1 and increases cell proliferation. Front Endocrinol (Lausanne). (2020) 11:563165. doi: 10.3389/fendo.2020.563165
277. Thomas P, Pang Y, Filardo EJ, and Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. (2005) 146:624–32. doi: 10.1210/en.2004-1064
278. Yin H, Zhu Q, Liu M, Tu G, Li Q, Yuan J, et al. GPER promotes tamoxifen-resistance in ER+ breast cancer cells by reduced Bim proteins through MAPK/Erk-TRIM2 signaling axis. Int J Oncol. (2017) 51:1191–8. doi: 10.3892/ijo.2017.4117
279. Yu T, Cheng H, Ding Z, Wang Z, Zhou L, Zhao P, et al. GPER mediates decreased chemosensitivity via regulation of ABCG2 expression and localization in tamoxifen-resistant breast cancer cells. Mol Cell Endocrinol. (2020) 506:110762. doi: 10.1016/j.mce.2020.110762
280. Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. (2006) 2:207–12. doi: 10.1038/nchembio775
281. Yu T, Liu M, Luo H, Wu C, Tang X, Tang S, et al. GPER mediates enhanced cell viability and motility via non-genomic signaling induced by 17β-estradiol in triple-negative breast cancer cells. J Steroid Biochem Mol Biol. (2014) 143:392–403. doi: 10.1016/j.jsbmb.2014.05.003
282. Lappano R, Rosano C, Santolla MF, Pupo M, De Francesco EM, De Marco P, et al. Two novel GPER agonists induce gene expression changes and growth effects in cancer cells. Curr Cancer Drug Targets. (2012) 12:531–42. doi: 10.2174/156800912800673284
283. Avena P, Casaburi I, Zavaglia L, Nocito MC, La Padula D, Rago V, et al. 27-hydroxycholesterol binds GPER and induces progression of estrogen receptor-negative breast cancer. Cancers. (2022) 14:1521. doi: 10.3390/cancers14061521
284. Castillo-Sanchez R, Ramirez-Ricardo J, Martinez-Baeza E, Cortes-Reynosa P, Candanedo-Gonzales F, Gomez R, et al. Bisphenol A induces focal adhesions assembly and activation of FAK, Src and ERK2 via GPER in MDA-MB-231 breast cancer cells. Toxicol Vitro. (2020) 66:104871. doi: 10.1016/j.tiv.2020.104871
285. Deng Q, Jiang G, Wu Y, Li J, Liang W, Chen L, et al. GPER/Hippo-YAP signal is involved in Bisphenol S induced migration of triple negative breast cancer (TNBC) cells. J Hazard Mater. (2018) 355:1–9. doi: 10.1016/j.jhazmat.2018.05.013
286. Lei B, Tang Q, Sun S, Zhang X, Huang Y, and Xu L. Insight into the mechanism of tetrachlorobisphenol A (TCBPA)-induced proliferation of breast cancer cells by GPER-mediated signaling pathways. Environ pollut. (2021) 275:116636. doi: 10.1016/j.envpol.2021.116636
287. Kim KM and Jung J. Upregulation of G protein-coupled estrogen receptor by chrysin-nanoparticles inhibits tumor proliferation and metastasis in triple negative breast cancer xenograft model. Front Endocrinol. (2020) 11:560605. doi: 10.3389/fendo.2020.560605
288. He Y, Yang K, Liu J, Shi D, Zhang Z, Yang J, et al. Tanshinone IIA inhibits triple-negative breast cancer cells MDA-MB-231 via G protein-coupled estrogen receptor- (GPER-) dependent signaling pathway. Dis Markers. (2023) 2023:1–14. doi: 10.1155/2023/8371623
289. Qi M, Liu X, Zhou Y, Wang H, Zhao Y, Ren J, et al. Berberine inhibits MDA-MB-231 cells as an agonist of G protein-coupled estrogen receptor 1. IJMS. (2021) 22:11466. doi: 10.3390/ijms222111466
290. Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, et al. In vivo effects of a GPR30 antagonist. Nat Chem Biol. (2009) 5:421–7. doi: 10.1038/nchembio.168
291. Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol. (2011) 127:358–66. doi: 10.1016/j.jsbmb.2011.07.002
292. Lappano R, Rosano C, De Marco P, De Francesco EM, Pezzi V, and Maggiolini M. Estriol acts as a GPR30 antagonist in estrogen receptor-negative breast cancer cells. Mol Cell Endocrinol. (2010) 320:162–70. doi: 10.1016/j.mce.2010.02.006
293. Rouhimoghadam M, Lu AS, Salem AK, and Filardo EJ. Therapeutic perspectives on the modulation of G-protein coupled estrogen receptor, GPER, function. Front Endocrinol (Lausanne). (2020) 11:591217. doi: 10.3389/fendo.2020.591217
294. DeLeon C, Wang HH, Gunn J, Wilhelm M, Cole A, Arnett S, et al. A novel GPER antagonist protects against the formation of estrogen-induced cholesterol gallstones in female mice. J Lipid Res. (2020) 61:767–77. doi: 10.1194/jlr.RA119000592
295. Maggiolini M, Santolla MF, Avino S, Aiello F, Rosano C, Garofalo A, et al. Identification of two benzopyrroloxazines acting as selective GPER antagonists in breast cancer cells and cancer-associated fibroblasts. Future Med Chem. (2015) 7:437–48. doi: 10.4155/fmc.15.3
296. Sjöström M, Hartman L, Grabau D, Fornander T, Malmström P, Nordenskjöld B, et al. Lack of G protein-coupled estrogen receptor (GPER) in the plasma membrane is associated with excellent long-term prognosis in breast cancer. Breast Cancer Res Treat. (2014) 145:61–71. doi: 10.1007/s10549-014-2936-4
297. Hsu L-H, Chu N-M, Lin Y-F, and Kao S-H. G-protein coupled estrogen receptor in breast cancer. Int J Mol Sci. (2019) 20:306. doi: 10.3390/ijms20020306
298. Zhang D, Wang J, Chen H, and Yan S. Cytoplasmic G protein-coupled estrogen receptor 1 as a prognostic indicator of breast cancer: A meta-analysis. Technol Cancer Res Treat. (2022) 21:15330338221131664. doi: 10.1177/15330338221131664
299. Maggiolini M, Vivacqua A, Fasanella G, Recchia AG, Sisci D, Pezzi V, et al. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J Biol Chem. (2004) 279:27008–16. doi: 10.1074/jbc.M403588200
300. Thomas P and Dong J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol. (2006) 102:175–9. doi: 10.1016/j.jsbmb.2006.09.017
301. Vivacqua A, Bonofiglio D, Recchia AG, Musti AM, Picard D, Andò S, et al. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17beta-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol. (2006) 20:631–46. doi: 10.1210/me.2005-0280
302. Dong W-H, Chen J-C, He Y-L, Xu J-J, and Mei Y-A. Resveratrol inhibits K(v)2.2 currents through the estrogen receptor GPR30-mediated PKC pathway. Am J Physiol Cell Physiol. (2013) 305:C547–557. doi: 10.1152/ajpcell.00146.2013
303. Kajta M, Rzemieniec J, Litwa E, Lason W, Lenartowicz M, Krzeptowski W, et al. The key involvement of estrogen receptor β and G-protein-coupled receptor 30 in the neuroprotective action of daidzein. Neuroscience. (2013) 238:345–60. doi: 10.1016/j.neuroscience.2013.02.005
304. Kajta M, Litwa E, Rzemieniec J, Wnuk A, Lason W, Zelek-Molik A, et al. Isomer-nonspecific action of dichlorodiphenyltrichloroethane on aryl hydrocarbon receptor and G-protein-coupled receptor 30 intracellular signaling in apoptotic neuronal cells. Mol Cell Endocrinol. (2014) 392:90–105. doi: 10.1016/j.mce.2014.05.008
305. Zhang X-L, Liu N, Weng S-F, and Wang H-S. Bisphenol A increases the migration and invasion of triple-negative breast cancer cells via oestrogen-related receptor gamma. Basic Clin Pharmacol Toxicol. (2016) 119:389–95. doi: 10.1111/bcpt.12591
306. Prossnitz ER and Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. (2011) 7:715–26. doi: 10.1038/nrendo.2011.122
307. Chen Y, Li Z, He Y, Shang D, Pan J, Wang H, et al. Estrogen and pure antiestrogen fulvestrant (ICI 182 780) augment cell–matrigel adhesion of MCF-7 breast cancer cells through a novel G protein coupled estrogen receptor (GPR30)-to-calpain signaling axis. Toxicol Appl Pharmacol. (2014) 275:176–81. doi: 10.1016/j.taap.2014.01.005
308. Ohshiro K, Schwartz AM, Levine PH, and Kumar R. Alternate estrogen receptors promote invasion of inflammatory breast cancer cells via non-genomic signaling. PloS One. (2012) 7:e30725. doi: 10.1371/journal.pone.0030725
309. Xu T, Ma D, Chen S, Tang R, Yang J, Meng C, et al. High GPER expression in triple-negative breast cancer is linked to pro-metastatic pathways and predicts poor patient outcomes. NPJ Breast Cancer. (2022) 8:100. doi: 10.1038/s41523-022-00472-4
310. Zhu D, Yang J, and Xu J. G-protein-coupled estrogen receptor enhances the stemness of triple-negative breast cancer cells and promotes Malignant characteristics. Oncologie. (2022) 24:471–82. doi: 10.32604/oncologie.2022.024062
311. Girgert R, Emons G, and Gründker C. Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: possible application in targeted therapy. Breast Cancer Res Treat. (2012) 134:199–205. doi: 10.1007/s10549-012-1968-x
312. Lappano R, Pisano A, and Maggiolini M. GPER function in breast cancer: an overview. Front Endocrinol (Lausanne). (2014) 5:66. doi: 10.3389/fendo.2014.00066
313. Vivacqua A, Bonofiglio D, Albanito L, Madeo A, Rago V, Carpino A, et al. 17beta-estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the g protein-coupled receptor GPR30. Mol Pharmacol. (2006) 70:1414–23. doi: 10.1124/mol.106.026344
314. Zekas E and Prossnitz ER. Estrogen-mediated inactivation of FOXO3a by the G protein-coupled estrogen receptor GPER. BMC Cancer. (2015) 15:702. doi: 10.1186/s12885-015-1699-6
315. Liu Y, Du F-Y, Chen W, Fu P-F, Yao M-Y, and Zheng S-S. G15 sensitizes epithelial breast cancer cells to doxorubicin by preventing epithelial-mesenchymal transition through inhibition of GPR30. Am J Transl Res. (2015) 7:967–75.
316. Prossnitz ER and Barton M. The G protein-coupled oestrogen receptor GPER in health and disease: an update. Nat Rev Endocrinol. (2023) 19:407–24. doi: 10.1038/s41574-023-00822-7
317. Zhou X, Wang S, Wang Z, Feng X, Liu P, Lv X-B, et al. Estrogen regulates Hippo signaling via GPER in breast cancer. J Clin Invest. (2015) 125:2123–35. doi: 10.1172/JCI79573
318. Filardo EJ, Quinn JA, Frackelton AR, and Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. (2002) 16:70–84. doi: 10.1210/mend.16.1.0758
319. Quinn JA, Graeber CT, Frackelton AR, Kim M, Schwarzbauer JE, and Filardo EJ. Coordinate regulation of estrogen-mediated fibronectin matrix assembly and epidermal growth factor receptor transactivation by the G protein-coupled receptor, GPR30. Mol Endocrinol. (2009) 23:1052–64. doi: 10.1210/me.2008-0262
320. Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, and Hathaway HJ. Estrogen signaling through the transmembrane G protein–coupled receptor GPR30. Annu Rev Physiol. (2008) 70:165–90. doi: 10.1146/annurev.physiol.70.113006.100518
321. Recchia AG, De Francesco EM, Vivacqua A, Sisci D, Panno ML, Andò S, et al. The G protein-coupled receptor 30 is up-regulated by hypoxia-inducible factor-1alpha (HIF-1alpha) in breast cancer cells and cardiomyocytes. J Biol Chem. (2011) 286:10773–82. doi: 10.1074/jbc.M110.172247
322. Cho H, Seo Y, Loke KM, Kim S-W, Oh S-M, Kim J-H, et al. Cancer-stimulated CAFs enhance monocyte differentiation and protumoral TAM activation via IL6 and GM-CSF secretion. Clin Cancer Res. (2018) 24:5407–21. doi: 10.1158/1078-0432.CCR-18-0125
323. Cohen N, Shani O, Raz Y, Sharon Y, Hoffman D, Abramovitz L, et al. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene. (2017) 36:4457–68. doi: 10.1038/onc.2017.65
324. Luo H, Yang G, Yu T, Luo S, Wu C, Sun Y, et al. GPER-mediated proliferation and estradiol production in breast cancer-associated fibroblasts. Endocr Relat Cancer. (2014) 21:355–69. doi: 10.1530/ERC-13-0237
325. Pupo M, Bodmer A, Berto M, Maggiolini M, Dietrich P-Y, and Picard D. A genetic polymorphism repurposes the G-protein coupled and membrane-associated estrogen receptor GPER to a transcription factor-like molecule promoting paracrine signaling between stroma and breast carcinoma cells. Oncotarget. (2017) 8:46728–44. doi: 10.18632/oncotarget.18156
326. De Francesco EM, Pellegrino M, Santolla MF, Lappano R, Ricchio E, Abonante S, et al. GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. (2014) 74:4053–64. doi: 10.1158/0008-5472.CAN-13-3590
327. Pupo M, Pisano A, Lappano R, Santolla MF, De Francesco EM, Abonante S, et al. Bisphenol A induces gene expression changes and proliferative effects through GPER in breast cancer cells and cancer-associated fibroblasts. Environ Health Perspect. (2012) 120:1177–82. doi: 10.1289/ehp.1104526
328. Yuan J, Liu M, Yang L, Tu G, Zhu Q, Chen M, et al. Acquisition of epithelial-mesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: a new role for G protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and β1-integrin signaling pathway in tumor cells. Breast Cancer Res. (2015) 17:69. doi: 10.1186/s13058-015-0579-y
329. Ren J, Guo H, Wu H, Tian T, Dong D, Zhang Y, et al. GPER in CAFs regulates hypoxia-driven breast cancer invasion in a CTGF-dependent manner. Oncol Rep. (2015) 33:1929–37. doi: 10.3892/or.2015.3779
330. Roskoski R. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncology/Hematol. (2007) 62:179–213. doi: 10.1016/j.critrevonc.2007.01.006
331. Zhang D, Chen H, Wang J, Ji J, Imam M, Zhang Z, et al. Current progress and prospects for G protein-coupled estrogen receptor in triple-negative breast cancer. Front Cell Dev Biol. (2024) 12:1338448. doi: 10.3389/fcell.2024.1338448
332. Pupo M, Vivacqua A, Perrotta I, Pisano A, Aquila S, Abonante S, et al. The nuclear localization signal is required for nuclear GPER translocation and function in breast Cancer-Associated Fibroblasts (CAFs). Mol Cell Endocrinol. (2013) 376:23–32. doi: 10.1016/j.mce.2013.05.023
333. De Marco P, Lappano R, De Francesco EM, Cirillo F, Pupo M, Avino S, et al. GPER signalling in both cancer-associated fibroblasts and breast cancer cells mediates a feedforward IL1β/IL1R1 response. Sci Rep. (2016) 6:24354. doi: 10.1038/srep24354
334. Ahirwar DK, Nasser MW, Ouseph MM, Elbaz M, Cuitiño MC, Kladney RD, et al. Fibroblast-derived CXCL12 promotes breast cancer metastasis by facilitating tumor cell intravasation. Oncogene. (2018) 37:4428–42. doi: 10.1038/s41388-018-0263-7
335. Filardo EJ, Graeber CT, Quinn JA, Resnick MB, Giri D, DeLellis RA, et al. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin Cancer Res. (2006) 12:6359–66. doi: 10.1158/1078-0432.CCR-06-0860
336. Cheng S-B, Quinn JA, Graeber CT, and Filardo EJ. Down-modulation of the G-protein-coupled estrogen receptor, GPER, from the cell surface occurs via a trans-Golgi-proteasome pathway. J Biol Chem. (2011) 286:22441–55. doi: 10.1074/jbc.M111.224071
337. Samartzis EP, Noske A, Meisel A, Varga Z, Fink D, and Imesch P. The G protein-coupled estrogen receptor (GPER) is expressed in two different subcellular localizations reflecting distinct tumor properties in breast cancer. PloS One. (2014) 9:e83296. doi: 10.1371/journal.pone.0083296
338. Weißenborn C, Ignatov T, Poehlmann A, Wege AK, Costa SD, Zenclussen AC, et al. GPER functions as a tumor suppressor in MCF-7 and SK-BR-3 breast cancer cells. J Cancer Res Clin Oncol. (2014) 140:663–71. doi: 10.1007/s00432-014-1598-2
339. Marjon NA, Hu C, Hathaway HJ, and Prossnitz ER. G protein-coupled estrogen receptor regulates mammary tumorigenesis and metastasis. Mol Cancer Res. (2014) 12:1644–54. doi: 10.1158/1541-7786.MCR-14-0128-T
340. Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, and Picard D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. (2009) 28:523–32. doi: 10.1038/emboj.2008.304
341. Fang Y, Wang S, Han S, Zhao Y, Yu C, Liu H, et al. Targeted protein degrader development for cancer: advances, challenges, and opportunities. Trends Pharmacol Sci. (2023) 44:303–17. doi: 10.1016/j.tips.2023.03.003
342. Grinshpun A. Clinician’s guide to targeted estrogen receptor degradation using PROTAC in patients with estrogen receptor-positive metastatic breast cancer. Curr Opin Oncol. (2023) 35:472–8. doi: 10.1097/CCO.0000000000000972
343. Wang L, Guillen VS, Sharma N, Flessa K, Min J, Carlson KE, et al. New class of selective estrogen receptor degraders (SERDs): expanding the toolbox of PROTAC degrons. ACS Med Chem Lett. (2018) 9:803–8. doi: 10.1021/acsmedchemlett.8b00106
344. Békés M, Langley DR, and Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. (2022) 21:181–200. doi: 10.1038/s41573-021-00371-6
345. Puyang X, Furman C, Zheng GZ, Wu ZJ, Banka D, Aithal K, et al. Discovery of selective estrogen receptor covalent antagonists for the treatment of ERαWT and ERαMUT breast cancer. Cancer Discov. (2018) 8:1176–93. doi: 10.1158/2159-8290.CD-17-1229
346. Robertson JFR. Fulvestrant (Faslodex) – how to make a good drug better. Oncologist. (2007) 12:774–84. doi: 10.1634/theoncologist.12-7-774
347. Robertson JFR, Lindemann J, Garnett S, Anderson E, Nicholson RI, Kuter I, et al. A good drug made better: the fulvestrant dose-response story. Clin Breast Cancer. (2014) 14:381–9. doi: 10.1016/j.clbc.2014.06.005
348. Robertson JFR and Harrison M. Fulvestrant: pharmacokinetics and pharmacology. Br J Cancer. (2004) 90 Suppl 1:S7–10. doi: 10.1038/sj.bjc.6601630
349. Van Kruchten M, De Vries EG, Glaudemans AW, Van Lanschot MC, Van Faassen M, Kema IP, et al. Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov. (2015) 5:72–81. doi: 10.1158/2159-8290.CD-14-0697
350. Blakely B, Shin S, and Jin K. Overview of the therapeutic strategies for ER positive breast cancer. Biochem Pharmacol. (2023) 212:115552. doi: 10.1016/j.bcp.2023.115552
351. Fanning SW and Greene GL. Next-generation ERα Inhibitors for endocrine-resistant ER+ Breast cancer. Endocrinology. (2019) 160:759–69. doi: 10.1210/en.2018-01095
352. Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engström O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. (1997) 389:753–8. doi: 10.1038/39645
353. Min J, Guillen VS, Sharma A, Zhao Y, Ziegler Y, Gong P, et al. Adamantyl antiestrogens with novel side chains reveal a spectrum of activities in suppressing estrogen receptor mediated activities in breast cancer cells. J Med Chem. (2017) 60:6321–36. doi: 10.1021/acs.jmedchem.7b00585
354. Aftimos P, Neven P, Pegram M, Van Oordt CWMDH, Dees EC, Schröder C, et al. Abstract PS12-04: Rintodestrant (G1T48), an oral selective estrogen receptor degrader in ER+/HER2- locally advanced or metastatic breast cancer: Updated phase 1 results and dose selection. Cancer Res. (2021) 81:PS12–04-PS12-04. doi: 10.1158/1538-7445.SABCS20-PS12-04
355. Beelen AP, Li C, Jarugula P, Gopalakrishnan M, Sorrentino JA, Wolfgang CD, et al. Abstract PS17-08: Population pharmacokinetic and exposure-response modeling of the oral selective estrogen receptor degrader, rintodestrant (G1T48), in patients with ER+/HER2- advanced breast cancer. Cancer Res. (2021) 81:PS17–08-PS17-08. doi: 10.1158/1538-7445.SABCS20-PS17-08
356. Wang J, Sun T, Zhang Q, Shi Y, Wang X, Chen Y, et al. Abstract PS15-02: A phase Ib study of D-0502 as monotherapy for advanced or metastatic ER-positive and HER2-negative breast cancer: results from the dose-expansion stage. Cancer Res. (2024) 84:PS15–02-PS15-02. doi: 10.1158/1538-7445.SABCS23-PS15-02
357. Dong J, Wang T-L, Lu J, Ding CZ, Hu L, Hu G, et al. Design, syntheses and evaluations of novel indole derivatives as orally selective estrogen receptor degraders (SERD). Bioorg Med Chem Lett. (2020) 30:127601. doi: 10.1016/j.bmcl.2020.127601
358. Hoy SM. Correction: elacestrant: first approval. Drugs. (2023) 83:1735. doi: 10.1007/s40265-023-01978-2
359. Shah M, Lingam H, Gao X, Gittleman H, Fiero MH, Krol D, et al. US food and drug administration approval summary: elacestrant for estrogen receptor–positive, human epidermal growth factor receptor 2–negative, ESR1 -mutated advanced or metastatic breast cancer. JCO. (2024) 42:1193–201. doi: 10.1200/JCO.23.02112
360. Bardia A, Cortés J, Bidard F-C, Neven P, Garcia-Sáenz J, Aftimos P, et al. Elacestrant in ER+, HER2– metastatic breast cancer with ESR1 -mutated tumors: subgroup analyses from the phase III EMERALD trial by prior duration of endocrine therapy plus CDK4/6 inhibitor and in clinical subgroups. Clin Cancer Res. (2024) 30:4299–309. doi: 10.1158/1078-0432.CCR-24-1073
361. Ibrahim N, Kim S-B, Lin N, Awada A, Gil EC, Tonini G, et al. Abstract PO2-05-05: ELECTRA: An open-label, multicenter, phase 1b/2 study of elacestrant in combination with abemaciclib in patients with brain metastasis (mets) from estrogen receptor-positive (ER+), HER2-negative (HER2-) breast cancer (BC). Cancer Res. (2024) 84:PO2-05-05-PO2-05–05. doi: 10.1158/1538-7445.SABCS23-PO2-05-05
362. Hamilton E, Oliveira M, Turner N, García-Corbacho J, Hernando C, Ciruelos EM, et al. A phase I dose escalation and expansion trial of the next-generation oral SERD camizestrant in women with ER-positive, HER2-negative advanced breast cancer: SERENA-1 monotherapy results. Ann Oncol. (2024) 35:707–17. doi: 10.1016/j.annonc.2024.04.012
363. Oliveira M, Hamilton EP, Incorvati J, Bermejo de la Heras B, Calvo E, García-Corbacho J, et al. Serena-1: Updated analyses from a phase 1 study (parts C/D) of the next-generation oral SERD camizestrant (AZD9833) in combination with palbociclib, in women with ER-positive, HER2-negative advanced breast cancer. JCO. (2022) 40:1032–2. doi: 10.1200/JCO.2022.40.16_suppl.1032
364. Scott JS, Moss TA, Balazs A, Barlaam B, Breed J, Carbajo RJ, et al. Discovery of AZD9833, a potent and orally bioavailable selective estrogen receptor degrader and antagonist. J Med Chem. (2020) 63:14530–59. doi: 10.1021/acs.jmedchem.0c01163
365. Scott JS, Moss TA, Barlaam B, Davey PRJ, Fairley G, Gangl ET, et al. Addition of fluorine and a late-stage functionalization (LSF) of the oral SERD AZD9833. ACS Med Chem Lett. (2020) 11:2519–25. doi: 10.1021/acsmedchemlett.0c00505
366. Lim E, Jhaveri KL, Perez-Fidalgo JA, Bellet M, Boni V, Perez Garcia JM, et al. A phase Ib study to evaluate the oral selective estrogen receptor degrader GDC-9545 alone or combined with palbociclib in metastatic ER-positive HER2-negative breast cancer. JCO. (2020) 38:1023–3. doi: 10.1200/JCO.2020.38.15_suppl.1023
367. Moore HM, Boni V, Bellet M, Bermejo De Las Heras B, Gión Cortés M, Oakman C, et al. Evaluation of pharmacodynamic (PD) and biologic activity in a preoperative window-of-opportunity (WOO) study of giredestrant (GDC-9545) in postmenopausal patients (pts) with estrogen receptor-positive, HER2-negative (ER+/HER2–) operable breast cancer (BC). JCO. (2021) 39:577–7. doi: 10.1200/JCO.2021.39.15_suppl.577
368. Hamilton EP, Patel MR, Armstrong AC, Baird RD, Jhaveri K, Hoch M, et al. A first-in-human study of the new oral selective estrogen receptor degrader AZD9496 for ER+/HER2– advanced breast cancer. Clin Cancer Res. (2018) 24:3510–8. doi: 10.1158/1078-0432.CCR-17-3102
369. Nardone A, Weir H, Delpuech O, Brown H, De Angelis C, Cataldo ML, et al. The oral selective oestrogen receptor degrader (SERD) AZD9496 is comparable to fulvestrant in antagonising ER and circumventing endocrine resistance. Br J Cancer. (2019) 120:331–9. doi: 10.1038/s41416-018-0354-9
370. Weir HM, Bradbury RH, Lawson M, Rabow AA, Buttar D, Callis RJ, et al. AZD9496: an oral estrogen receptor inhibitor that blocks the growth of ER-positive and ESR1-mutant breast tumors in preclinical models. Cancer Res. (2016) 76:3307–18. doi: 10.1158/0008-5472.CAN-15-2357
371. Jhaveri K, Juric D, Yap Y-S, Cresta S, Layman RM, Duhoux FP, et al. A phase I study of LSZ102, an oral selective estrogen receptor degrader, with or without ribociclib or alpelisib, in patients with estrogen receptor-positive breast cancer. Clin Cancer Res. (2021) 27:5760–70. doi: 10.1158/1078-0432.CCR-21-1095
372. Sharaf B, Hajahjeh A, Bani Hani H, and Abdel-Razeq H. Next generation selective estrogen receptor degraders in postmenopausal women with advanced-stage hormone receptors-positive, HER2-negative breast cancer. Front Oncol. (2024) 14:1385577. doi: 10.3389/fonc.2024.1385577
373. Bardia A, Mayer I, Winer E, Linden HM, Ma CX, Parker BA, et al. The oral selective estrogen receptor degrader GDC-0810 (ARN-810) in postmenopausal women with hormone receptor-positive HER2-negative (HR + /HER2 -) advanced/metastatic breast cancer. Breast Cancer Res Treat. (2023) 197:319–31. doi: 10.1007/s10549-022-06797-9
374. Dickler M, Bardia A, Mayer I, Winer E, Rix P, Hager J, et al. Abstract CT231: A first-in-human phase I study to evaluate the oral selective estrogen receptor degrader GDC-0810 (ARN-810) in postmenopausal women with estrogen receptor+ HER2-, advanced/metastatic breast cancer. Cancer Res. (2015) 75:CT231–1. doi: 10.1158/1538-7445.AM2015-CT231
375. Joseph JD, Darimont B, Zhou W, Arrazate A, Young A, Ingalla E, et al. The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER+ breast cancer. eLife. (2016) 5:e15828. doi: 10.7554/eLife.15828
376. Lai A, Kahraman M, Govek S, Nagasawa J, Bonnefous C, Julien J, et al. Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem. (2015) 58:4888–904. doi: 10.1021/acs.jmedchem.5b00054
377. Chandarlapaty S, Dickler MN, Perez Fidalgo JA, Villanueva-Vázquez R, Giltnane J, Gates M, et al. An open-label phase I study of GDC-0927 in postmenopausal women with locally advanced or metastatic estrogen receptor-positive breast cancer. Clin Cancer Res. (2023) 29:2781–90. doi: 10.1158/1078-0432.CCR-23-0011
378. Kahraman M, Govek SP, Nagasawa JY, Lai A, Bonnefous C, Douglas K, et al. Maximizing ER-α Degradation maximizes activity in a tamoxifen-resistant breast cancer model: identification of GDC-0927. ACS Med Chem Lett. (2019) 10:50–5. doi: 10.1021/acsmedchemlett.8b00414
379. Scott JS and Barlaam B. Selective estrogen receptor degraders (SERDs) and covalent antagonists (SERCAs): a patent review, (2015-present). Expert Opin Ther Pat. (2022) 32:131–51. doi: 10.1080/13543776.2022.2006185
380. Bardia A, Chandarlapaty S, Linden HM, Ulaner GA, Gosselin A, Cartot-Cotton S, et al. AMEERA-1 phase 1/2 study of amcenestrant, SAR439859, in postmenopausal women with ER-positive/HER2-negative advanced breast cancer. Nat Commun. (2022) 13:4116. doi: 10.1038/s41467-022-31668-8
381. Bardia A, Cortes J, Hurvitz SA, Delaloge S, Iwata H, Shao Z-M, et al. AMEERA-5: a randomized, double-blind phase 3 study of amcenestrant plus palbociclib versus letrozole plus palbociclib for previously untreated ER+/HER2- advanced breast cancer. Ther Adv Med Oncol. (2022) 14:17588359221083956. doi: 10.1177/17588359221083956
382. El-Ahmad Y, Tabart M, Halley F, Certal V, Thompson F, Filoche-Rommé B, et al. Discovery of 6-(2,4-dichlorophenyl)-5-[4-[(3S)-1-(3-fluoropropyl)pyrrolidin-3-yl]oxyphenyl]-8,9-dihydro-7H-benzo[7]annulene-2-carboxylic acid (SAR439859), a potent and selective estrogen receptor degrader (SERD) for the treatment of estrogen-receptor-positive breast cancer. J Med Chem. (2020) 63:512–28. doi: 10.1021/acs.jmedchem.9b01293
383. Meyskens T, Metzger O, Poncet C, Goulioti T, Xenophontos E, Carey LA, et al. Adjuvant study of amcenestrant (SAR439859) versus tamoxifen for patients with hormone receptor-positive (HR+) early breast cancer (EBC), who have discontinued adjuvant aromatase inhibitor therapy due to treatment-related toxicity (AMEERA-6). JCO. (2022) 40:TPS607–7. doi: 10.1200/JCO.2022.40.16_suppl.TPS607
384. Shomali M, Cheng J, Sun F, Koundinya M, Guo Z, Hebert AT, et al. SAR439859, a novel selective estrogen receptor degrader (SERD), demonstrates effective and broad antitumor activity in wild-type and mutant ER-positive breast cancer models. Mol Cancer Ther. (2021) 20:250–62. doi: 10.1158/1535-7163.MCT-20-0390
385. Jiang Y, Deng Q, Zhao H, Xie M, Chen L, Yin F, et al. Development of stabilized peptide-based PROTACs against estrogen receptor α. ACS Chem Biol. (2018) 13:628–35. doi: 10.1021/acschembio.7b00985
386. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, and Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U.S.A. (2001) 98:8554–9. doi: 10.1073/pnas.141230798
387. Tecalco-Cruz AC, Zepeda-Cervantes J, Ramírez-Jarquín JO, and Rojas-Ochoa A. Proteolysis-targeting chimeras and their implications in breast cancer. Explor Target Antitumor Ther. (2021) 2:496–510. doi: 10.37349/etat.2021.00060
388. Dale B, Cheng M, Park K-S, Kaniskan HÜ, Xiong Y, and Jin J. Advancing targeted protein degradation for cancer therapy. Nat Rev Cancer. (2021) 21:638–54. doi: 10.1038/s41568-021-00365-x
389. Feng Y, Zhang Z, Zhang H, Guo H, Tan C, Xu N, et al. Aptamer proteolysis-targeting chimeras (PROTACs): A novel strategy to combat drug resistance in estrogen receptor α-positive breast cancer. ACS Pharmacol Transl Sci. (2024) 7:3945–54. doi: 10.1021/acsptsci.4c00469
390. Lin J, Jin J, Shen Y, Zhang L, Gong G, Bian H, et al. Emerging protein degradation strategies: expanding the scope to extracellular and membrane proteins. Theranostics. (2021) 11:8337–49. doi: 10.7150/thno.62686
391. Han X and Sun Y. Strategies for the discovery of oral PROTAC degraders aimed at cancer therapy. Cell Rep Phys Sci. (2022) 3:101062. doi: 10.1016/j.xcrp.2022.101062
392. Gough SM, Flanagan JJ, Teh J, Andreoli M, Rousseau E, Pannone M, et al. Oral estrogen receptor PROTAC vepdegestrant (ARV-471) is highly efficacious as monotherapy and in combination with CDK4/6 or PI3K/mTOR pathway inhibitors in preclinical ER+ Breast cancer models. Clin Cancer Res. (2024) 30:3549–63. doi: 10.1158/1078-0432.CCR-23-3465
393. Iwata H, Hamilton EP, Ma CX, De Laurentiis M, Hurvitz SA, Wander SA, et al. 73TiP Global phase III studies evaluating vepdegestrant in estrogen receptor (ER)+/human epidermal growth factor receptor 2 (HER2)- advanced breast cancer: VERITAC-2 and VERITAC-3. Ann Oncol. (2023) 34:S1493. doi: 10.1016/j.annonc.2023.10.207
394. Acharyya RK, Rej RK, Hu B, Chen Z, Wu D, Lu J, et al. Discovery of ERD-1233 as a potent and orally efficacious estrogen receptor PROTAC degrader for the treatment of ER+ Human breast cancer. J Med Chem. (2024) 67:19010–37. doi: 10.1021/acs.jmedchem.4c01521
395. Rej RK, Hu B, Chen Z, Acharyya RK, Wu D, Metwally H, et al. Discovery of ERD-12310A as an exceptionally potent and orally efficacious PROTAC degrader of estrogen receptor α (ERα). J Med Chem. (2024) 67:20933–65. doi: 10.1021/acs.jmedchem.4c01401
396. Wang C, Zhang Y, Wang J, and Xing D. VHL-based PROTACs as potential therapeutic agents: Recent progress and perspectives. Eur J Med Chem. (2022) 227:113906. doi: 10.1016/j.ejmech.2021.113906
397. Hu B and Hu J. Complete elimination of estrogen receptor α by PROTAC estrogen receptor α degrader ERD-148 in breast cancer cells. Breast Cancer Res Treat. (2024) 203:383–96. doi: 10.1007/s10549-023-07136-2
398. Hu J, Hu B, Wang M, Xu F, Miao B, Yang C-Y, et al. Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER). J Med Chem. (2019) 62:1420–42. doi: 10.1021/acs.jmedchem.8b01572
399. Zhang X, Zhang Z, Xue X, Fan T, Tan C, Liu F, et al. PROTAC degrader of estrogen receptor α Targeting DNA-binding domain in breast cancer. ACS Pharmacol Transl Sci. (2022) 5:1109–18. doi: 10.1021/acsptsci.2c00109
400. Xin L, Wang C, Cheng Y, Wang H, Guo X, Deng X, et al. Discovery of novel ERα and aromatase dual-targeting PROTAC degraders to overcome endocrine-resistant breast cancer. J Med Chem. (2024) 67:8913–31. doi: 10.1021/acs.jmedchem.4c00196
401. Hodges-Gallagher L, Harmon CL, Sun R, Myles DC, and Kushner P. Abstract 4376: OP-1250, a complete estrogen receptor antagonist (CERAN) that shrinks estrogen receptor positive tumors and exhibits favorable pharmacokinetics. Cancer Res. (2020) 80:4376–6. doi: 10.1158/1538-7445.AM2020-4376
402. Parisian AD, Palanisamy GS, Ortega F, Sapugay JL, Bodell WJ, Kulp D, et al. Abstract P2-24-07: Combination of complete estrogen receptor antagonist, OP-1250, and CDK4/6 inhibitors enhances tumor suppression and inhibition of cell cycle-related gene expression. Cancer Res. (2023) 83:P2-24-07-P2-24–07. doi: 10.1158/1538-7445.SABCS22-P2-24-07
403. Johnston SRD, Pluard TJ, Wang JS, Hamilton EP, Juric D, Scholz CR, et al. Phase 1b study of H3B-6545 in combination with palbociclib in women with metastatic estrogen receptor–positive (ER+), human epidermal growth factor receptor 2 (HER2)-negative breast cancer. JCO. (2021) 39:e13025–5. doi: 10.1200/JCO.2021.39.15_suppl.e13025
404. Wang Y, Min J, Deng X, Feng T, Hu H, Guo X, et al. Discovery of novel covalent selective estrogen receptor degraders against endocrine-resistant breast cancer. Acta Pharm Sin B. (2023) 13:4963–82. doi: 10.1016/j.apsb.2023.05.005
405. Dai X-J, Ji S-K, Fu M-J, Liu G-Z, Liu H-M, Wang S-P, et al. Degraders in epigenetic therapy: PROTACs and beyond. Theranostics. (2024) 14:1464–99. doi: 10.7150/thno.92526
406. Li K and Crews CM. PROTACs: past, present and future. Chem Soc Rev. (2022) 51:5214–36. doi: 10.1039/D2CS00193D
407. Naito M, Ohoka N, Shibata N, and Tsukumo Y. Targeted protein degradation by chimeric small molecules, PROTACs and SNIPERs. Front Chem. (2019) 7:849. doi: 10.3389/fchem.2019.00849
408. Schreiber SL. The rise of molecular glues. Cell. (2021) 184:3–9. doi: 10.1016/j.cell.2020.12.020
409. Takahashi D, Moriyama J, Nakamura T, Miki E, Takahashi E, Sato A, et al. AUTACs: cargo-specific degraders using selective autophagy. Mol Cell. (2019) 76:797–810.e10. doi: 10.1016/j.molcel.2019.09.009
410. Hall KA and Filardo EJ. The G protein-coupled estrogen receptor (GPER): A critical therapeutic target for cancer. Cells. (2023) 12:2460. doi: 10.3390/cells12202460
411. Ambrosini G, Natale CA, Musi E, Garyantes T, and Schwartz GK. The GPER agonist LNS8801 induces mitotic arrest and apoptosis in uveal melanoma cells. Cancer Res Commun. (2023) 3:540–7. doi: 10.1158/2767-9764.CRC-22-0399
412. Lu AS, Rouhimoghadam M, Arnatt CK, Filardo EJ, and Salem AK. Proteolytic targeting chimeras with specificity for plasma membrane and intracellular estrogen receptors. Mol Pharm. (2021) 18:1455–69. doi: 10.1021/acs.molpharmaceut.1c00018
413. Zhao L, Zhao J, Zhong K, Tong A, and Jia D. Targeted protein degradation: mechanisms, strategies and application. Sig Transduct Target Ther. (2022) 7:113. doi: 10.1038/s41392-022-00966-4
414. Cotton AD, Nguyen DP, Gramespacher JA, Seiple IB, and Wells JA. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J Am Chem Soc. (2021) 143:593–8. doi: 10.1021/jacs.0c10008
415. Banik SM, Pedram K, Wisnovsky S, Ahn G, Riley NM, and Bertozzi CR. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature. (2020) 584:291–7. doi: 10.1038/s41586-020-2545-9
416. Ramadas B, Kumar Pain P, and Manna D. LYTACs: an emerging tool for the degradation of non-cytosolic proteins. ChemMedChem. (2021) 16:2951–3. doi: 10.1002/cmdc.202100393
417. Ahn G, Banik SM, Miller CL, Riley NM, Cochran JR, and Bertozzi CR. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat Chem Biol. (2021) 17:937–46. doi: 10.1038/s41589-021-00770-1
418. Jordan VC, Curpan R, and Maximov PY. Estrogen receptor mutations found in breast cancer metastases integrated with the molecular pharmacology of selective ER modulators. J Natl Cancer Inst. (2015) 107:djv075. doi: 10.1093/jnci/djv075
419. Nettles KW, Bruning JB, Gil G, Nowak J, Sharma SK, Hahm JB, et al. NFκB selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nat Chem Biol. (2008) 4:241–7. doi: 10.1038/nchembio.76
420. Schapira M, Calabrese MF, Bullock AN, and Crews CM. Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov. (2019) 18:949–63. doi: 10.1038/s41573-019-0047-y
421. Fam SY, Chee CF, Yong CY, Ho KL, Mariatulqabtiah AR, and Tan WS. Stealth coating of nanoparticles in drug-delivery systems. Nanomater (Basel). (2020) 10:787. doi: 10.3390/nano10040787
422. Liu H-J, Chen W, Wu G, Zhou J, Liu C, Tang Z, et al. Glutathione-scavenging nanoparticle-mediated PROTACs delivery for targeted protein degradation and amplified antitumor effects. Adv Sci (Weinh). (2023) 10:e2207439. doi: 10.1002/advs.202207439
423. Minko T. Nanoformulation of BRD4-degrading PROTAC: improving druggability to target the “Undruggable” MYC in pancreatic cancer. Trends Pharmacol Sci. (2020) 41:684–6. doi: 10.1016/j.tips.2020.08.008
424. Saraswat A, Patki M, Fu Y, Barot S, Dukhande VV, and Patel K. Nanoformulation of PROteolysis TArgeting Chimera targeting “undruggable” c-Myc for the treatment of pancreatic cancer. Nanomed (Lond). (2020) 15:1761–77. doi: 10.2217/nnm-2020-0156
425. Cimas FJ, Niza E, Juan A, Noblejas-López MDM, Bravo I, Lara-Sanchez A, et al. Controlled delivery of BET-PROTACs: in vitro evaluation of MZ1-loaded polymeric antibody conjugated nanoparticles in breast cancer. Pharmaceutics. (2020) 12:986. doi: 10.3390/pharmaceutics12100986
Keywords: breast cancer, estrogen signaling, ERα, GPER, endocrine resistance, SERM, SERD
Citation: Saha T and Lukong KE (2025) Decoding estrogen receptor and GPER biology: structural insights and therapeutic advances in ERα−positive breast cancer. Front. Oncol. 15:1513225. doi: 10.3389/fonc.2025.1513225
Received: 18 October 2024; Accepted: 26 May 2025;
Published: 26 June 2025.
Edited by:
Tamer Saad Kaoud, The University of Texas at Austin, Austin, United StatesReviewed by:
Anca Maria Cimpean, Victor Babes University of Medicine and Pharmacy, Timisoara, RomaniaAya Hamdy, Heliopolis University, Cairo, Egypt
Abhijit Debnath, Noida Institute of Engineering and Technology (NIET), Noida, India
Copyright © 2025 Saha and Lukong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Taniya Saha, dGFueXlhMTFAZ21haWwuY29t