 Yuanyang Sun
Yuanyang Sun Yun Wang2
Yun Wang2 Xiaofeng Wu
Xiaofeng Wu Suchun Yu
Suchun Yu- 1School of Health Science and Engineering, University of Shanghai for Science and Technology, Shanghai, China
- 2Department of Radiation Oncology, Shidong Hospital, Yangpu District, Shidong Hospital Affiliated to University of Shanghai for Science and Technology, Shanghai, China
- 3Department of Pharmacy, Shidong Hospital, Yangpu District, Shidong Hospital Affiliated to University of Shanghai for Science and Technology, Shanghai, China
Glioblastoma (GBM) is an extremely aggressive brain tumor. Its standard treatment currently involves surgery followed by radiotherapy and temozolomide. However, recurrence is frequently unavoidable, and the effect of various treatments is not ideal due to numerous inherent obstacles. Immunotherapy has demonstrated promising prospects in the management of various cancers. Despite several preclinical studies have shown that immunotherapy may improve the survival in GBM mouse model, the results from completed clinical trials reveal that it brings only limited benefit for GBM patients to date. Interestingly, several studies have demonstrated that radiotherapy not only eliminates tumor cells by inducing DNA damages but also improves the effect of immunotherapy by modulating immune response. Combining immunotherapy with radiotherapy for GBM has been evaluated extensively. Herein, we present the immunotherapy applied in GBM and highlight the importance of tumor microenvironment in immunotherapy for GBM. Moreover, we review the preclinical and clinical data for applying immunotherapy combined with radiotherapy for GBM. Finally, we also discuss the challenges facing combined treatment of immunotherapy and radiotherapy for GBM and further research aspects in the discussion section.
1 Introduction
Glioblastoma (GBM) generally has a poor prognosis with a median overall survival (mOS) of under 2 years and a 5-year survival rate of only 10% (1). The standard treatment for GBM includes surgery followed by concurrent radiotherapy combined with chemotherapy (2). However, the effect of this treatment is not satisfactory. Currently, immunotherapy has been investigated extensively in many malignant cancers (3–5). Immunotherapy has revolutionized the tumor treatment and brought new hope for the management of GBM. However, the BBB and immunosuppressive tumor microenvironment limit significantly the effect of immunotherapy for GBM (6). Radiotherapy is a common approach to treating various tumors. There is substantial evidence indicating that radiotherapy can exert systemic antitumor effect by enhancing the immune activation in many tumors (7–9). Several preclinical studies also reported that immunotherapy can exert radiosensitizing effect by normalizing tumor blood vascular system and alleviating hypoxia (10). A meta-analysis compared the effect and safety of radiochemotherapy alongside with immunotherapy against radiochemotherapy alone in newly diagnosed GBM (11). Nine clinical trials were included in this analysis. The results showed that immunotherapy was safely combined with radiochemotherapy. Nonetheless, combined treatment does not improve the survival greatly. Even so, combining immunotherapy with radiotherapy is still being actively researched in GBM (12, 13). Herein, we introduce the current situation of immunotherapy in GBM and biological rationale of combining immunotherapy with radiotherapy. Moreover, we summarize the recent progress of combined treatment of immunotherapy and radiotherapy for GBM and discuss certain current challenges that need to be addressed.
2 Immunotherapy
Immunotherapy for tumor treatment refers to utilizing immune system to identify and kill tumor cells. Currently, available immunotherapies for GBM treatment mainly consist of immune checkpoint inhibitors (ICIs), adoptive cell therapy (ACT), vaccine, oncolytic virus (Ov), and cytokine therapy (14, 15).
2.1 ICIs
Tumor cells generally employ immune checkpoints to evade the destructive impact of immune system (16). ICIs can inhibit this immunosuppressive effect and exert antitumor effect by blocking these immunosuppressive immune checkpoints. Pembrolizumab is a common ICI (anti-PD-1) for GBM. The outcomes of a clinical study revealed that giving neoadjuvant pembrolizumab, along with continued adjuvant treatment post-surgery, improved the mOS and mPFS in recurrent GBM, compared to those who only undergone adjuvant pembrolizumab (17). The mOS for neoadjuvant group and adjuvant group were 13.7 and 7.5 months, respectively. And the mPFS for neoadjuvant group and adjuvant group were 3.3 and 2.4 months, respectively. The neoadjuvant pembrolizumab enhanced the antitumor immune response both locally and systematically. Another phase II trial evaluated the effect of combining pembrolizumab with bevacizumab in contrast to administering pembrolizumab monotherapy in recurrent GBM (rGBM) (18). However, the outcomes showed that pembrolizumab, whether administered alone or alongside bevacizumab, had good tolerance but brought only modest benefits. Nivolumab is another anti-PD-1 antibody for treating GBM. A exploratory phase I trial assessed the tolerance of nivolumab, either alone or combined with ipilimumab, in rGBM (19). The outcomes revealed that the tolerance of nivolumab monotherapy was superior to that of dual therapy. The subsequent phase III trial compared nivolumab to bevacizumab in rGBM (20). The mOS for nivolumab group (9.8 months) was similar to that of bevacizumab group (10.0 months). The PFS and ORR for bevacizumab group were superior to those for nivolumab group. Another phase II trial evaluated the effect of neoadjuvant nivolumab in resectable GBM (21). The outcomes showed that no obvious clinical benefit was observed following surgery. Moreover, other immune checkpoints, such as CTLA-4 (22), CD47 (23), CD73 (24), and TIGIT (25), were also investigated in several preclinical studies or clinical trials.
2.2 ACT
ACT focuses on infusing immune cells to treat tumors, including genetically engineered T cells and tumor-infiltrating lymphocytes (TILs) (26). CAR T-cell therapy, which is a genetic engineering technique, adjusts T cells of patients to produce CARs aimed at tumor-associated proteins. There are preclinical evidence indicated that the CAR T-cells targeting IL-13Rα2 (27), EphA2 (28), HER2 (29), EGFR (30), and EGFRvIII (31) improved the survival in mouse GBM model. The results of clinical trials utilizing CAR T-cells for GBM also are promising. A phase I trial investigated the effect of the CAR- T cells targeting HER2 (VSTs) for progressive GBM (32). The outcomes showed that VSTs were well tolerated and brought clinical benefit for these patients. Among 16 patients, 1 patient exhibited a PR, 7 patients maintained SD and 8 patients progressed following VSTs infusion. Another trial evaluated the effect of CAR- T cells targeting IL13Ra2 for GBM (33). Reported results revealed that noticeable reduction of tumor lesions was observed, accompanied by increased cytokines and immune cells. Moreover, the CAR T-cells targeting EGFRvIII have also shown clinical benefit for GBM patients (34). Adoptive transfer of TILs is another common ACT, it has been demonstrated to lead to lasting regression in many tumors. A trial evaluated the safety of infusing autologous TILs and recombinant interleukin-2 locally for GBM. Six GBM patients received the infusion of TILs and recombinant interleukin-2 along with chemotherapy following surgery (35). The infusion of autologous TILs and recombinant interleukin-2 showed good therapeutic effect in treating GBM with acceptable toxicity. One patient showed a CR, two patients showed a PR, and three patients passed away due to progressive disease. No notable complications were observed in all patients. The clinical trial (NCT00331526) explored the effect of lymphokine-activated killer (LAK) cells for GBM (36). Thirty-three patients received adjuvant intralesional LAK cells therapy. LAK cells were well tolerated. The 1-year survival rate was 75%, with a mOS of 20.5 months. These outcomes demonstrated the potential of TILs transfer for GBM.
2.3 Vaccine
Several vaccines have been used to treat various tumor, with peptide and dendritic cell (DC) vaccines primarily used for treating GBM (37, 38). EGFRvIII, a deletion mutation of EGFR, can promote tumor development and enhance therapy resistance (39). A clinical study of rindopepimut (vaccine targeting EGFRvIII) plus bevacizumab for EGFRvIII-expressing rGBM indicated that rindopepimut plus bevacizumab brought potential PFS benefit (40). The 6-month PFS for rindopepimut cohort and control cohort were 28% and 16%, respectively. Another clinical study evaluated the effect of rindopepimut plus temozolomide in EGFRvIII-positive GBM (41). However, rindopepimut plus temozolomide did not improve the survival in contrast to temozolomide monotherapy. The mOS for rindopepimut plus temozolomide group and temozolomide group were 20.0months and 20.1months, respectively.
DC vaccine is another common vaccine in treating GBM. When activated by DCs, immune cells traverse BBB and reach tumor site to exert their antitumor functions (42). Moreover, DCs can promote the conversion of immunologically cold tumor into immunologically hot tumor (43). A research compared the OS for patients with nGBM and rGBM who underwent DCVax-L plus SOC vs external control patients who underwent SOC (44). Among 331 patients, 232 received DCVax-L, while 99 were given placebo. After experiencing recurrence, 64 patients from the placebo cohort switched to undergo DCVax-L. DCVax-L plus SOC improved the survival of GBM patients in contrast to external control patients. The mOS for 64 rGBM patients who underwent DCVax-L and control patients were 13.2 months and 7.8 months, respectively. The mOS for 232 nGBM patients who underwent DCVax-L and control patients were 19.3 months and 16.5 months, respectively.
2.4 OV therapy
OVs are genetically modified viruses that have a low level of pathogenicity and enhance the antitumor effect while sparing normal cells (45). The purposes for genetic modification of Ovs include (1): deleting virulence genes to enhance the safety (2), improving targeting to tumor cells (3), improving the effects against tumors. OVs can self-replicate inside tumor cells, directly destroying tumor cells. They also can activate immune responses by producing damage-related molecular patterns (DAMPs) and TAAs (46). Moreover, they can specifically suppress glioma stem cells (GSCs) that are crucial in promoting therapy resistance and tumor blood vessels formation (47). A clinical study of PVSRIPO (a nonpathogenic polio-rhinovirus chimera) for rGBM revealed that the 1- and 2- years survival rates of patients receiving PVSRIPO were superior to that of historical controls (48). Another clinical trial evaluated the effect of G47Δ (a oncolytic herpes simplex virus) for GBM following radiotherapy and temozolomide in Japan (49). The 1-year survival rate was 84.2%, with a mOS of 20.2 months. Due to the encouraging outcomes, G47Δ obtained temporary approval for GBM patients in Japan.
2.5 Cytokine therapy
Cytokines can improve immune response through adjusting the proliferation and differentiation immune cells (50). Several cytokines have been investigated as potential treatment to treat GBM, such as IFN-α, TGF-β, IL-2, IL-12, etc. A clinical trial investigated the efficacy of combining temozolomide with IFN-α compare to temozolomide monotherapy in nGBM (51). Temozolomide plus IFN-α greatly improved the mOS compared to temozolomide monotherapy. The mOS for patients receiving temozolomide in combination with IFN-α group was 26.7 months, significantly exceeding the standard group of 18.8 months. Another study investigated the effect of combining L19TNF (an antibody-conjugated cytokine) with CCNU (a chemotherapy drug) in mouse glioma model. L19TNF combined with CCNU exhibited strong anti-tumor effect. They discovered that L19TNF combined with CCNU led to tumor necrosis related to treatment, while also adjusting the immune microenvironment. Subsequently, they conducted a clinical trial for GBM patients who experienced first progression following chemotherapy and radiotherapy (52). The outcomes showed that L19TNF combined with CCNU was well tolerated. Common adverse events included symptomless rise in liver function text results and reduction in the counts of white blood cells and platelets. Among 6 patients, 1 showed a CR 9 months following treatment. And 1 showed a PR, with an 83% reduction in tumor lesions after 15 months. Other patients progressed ultimately at a distant site. The mPFS for all patients was 43.3 weeks, which is a significant improvement compared to the 4–12 weeks reported for CCNU alone. Increasing evidence demonstrated that cytokines may be a potential strategy for treating GBM.
3 Tumor microenvironment
Tumor microenvironment of GBM is intricate and heterogeneous, consisting of various components (Figure 1). The main components of tumor microenvironment of GBM include the glioma and glioma stem cells, nervous system, immune cells, signaling molecules, extracellular matrix, perivascular niche, and several chemical components (53). GBM is marked by a strongly immunosuppressive microenvironment, resulting in therapy resistance and tumor recurrence. The immunosuppressive microenvironment is caused by various factors, including the increase in immunosuppressive cells and immunosuppressive cytokines, low tumor-infiltrating lymphocyte density and high expression of inhibitory immune checkpoint molecules (54). Tregs can modulate immune homeostasis by exerting immunosuppressive effect. Tregs can be attracted to tumor microenvironment through various cytokines, increasing their viability and expansion and then contributing the immunosuppression (55). Glioma-associated macrophages and microglia (GAMs) also is essential for the microenvironment of GBM, influencing the tumor growth, spread and recurrence. GAMs can be categorized into M1 phenotype and M2 phenotype. M2 phenotype contributes to the immunosuppressive microenvironment by producing IL-10 and TGF-β. It also facilitates GSCs proliferation. GSCs can enhance resistance by regulating cell metabolism in tumor microenvironment in addition to facilitating the tissue remodeling by reprogramming related cells (56).
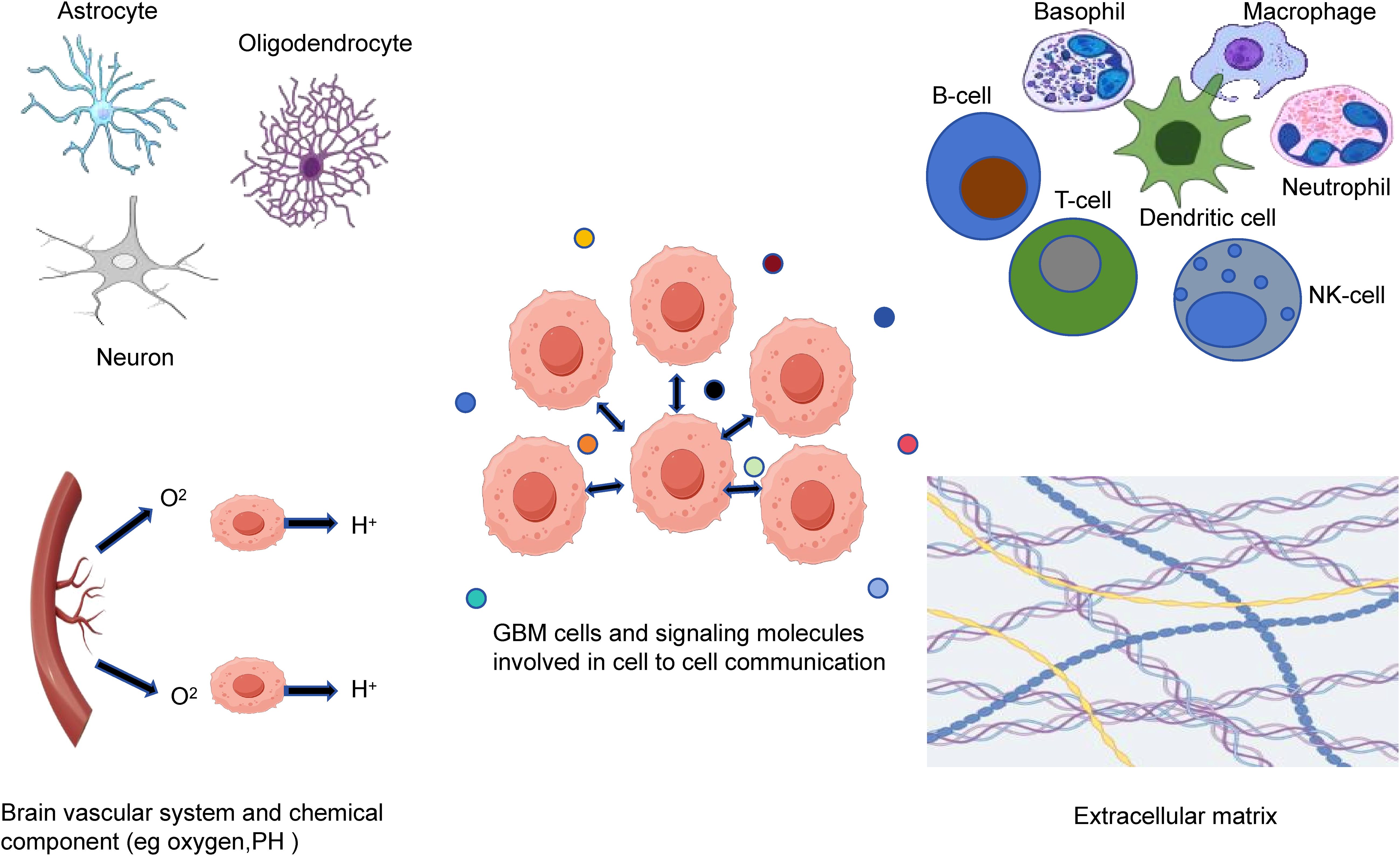
Figure 1. The tumor microenvironment components of GBM. Tumor microenvironment of GBM is a complex and heterogeneous system that consists of glioma and glioma stem cells, nervous system, immune cells, brain vascular system, extracellular matrix, signaling molecules and chemical components.
The tumor microenvironment of GBM appears to induce radioresistance (57). Several evidence has demonstrated that GSCs can obstruct chemotherapy and radiotherapy through various mechanisms, thereby developing resistance (58). The percentage of glioblastoma cells expressing CD133, which is a marker for GSCs, rises following radiotherapy. CD133-positive GSCs can activate DNA damage checkpoint proteins when exposed to radiation. This allows them to repair DNA damage caused by radiotherapy more efficiently, consequently promoting their radioresistance (59). GSCs also can contribute to radioresistance by overexpressing the PCNA-associated factor (PAF). This factor can help control how accessible DNA translesion synthesis enzymes are to PCNA. Following GSCs are irradiated, PAF interacts with PCNA to produce TLS Pol η, which restores error-free DNA synthesis, subsequently promoting the proliferation of GSCs and enhances their radioresistance (60).
4 The immunostimulatory effects of radiotherapy
Besides providing local control, radiotherapy also can exert systemic effect on distant tumor lesions that have not been irradiated, which known as the abscopal effect (61). Numerous works have demonstrated that abscopal effect is closely related to immune system (62). Moreover, immunotherapy may boost abscopal effect induced radiotherapy (63). Radiotherapy can induce systemic immune responses, providing the rationale for combining radiotherapy with immunotherapy. Radiotherapy can exert powerful antitumor immune responses by influencing nearly every stage of tumor-immunity cycle (64). These effects mainly involve promoting the release and presentation of tumor antigens, enhancing the activation of immune cells, increasing TILs density, aiding T cells in recognizing tumor cells and augmenting antitumor effect. Moreover, radiotherapy can modify tumor microenvironment by generating certain cytokines and causing stromal, immunological, and vascular changes (65).
Radiation is the primary trigger for immunogenic cell death (ICD) that can promote the elimination of target cells by effector T cells and activation of antigen-presenting cells (APCs). The production of TAAs and DAMPs is a significant feature of ICD (66). DCs are the most effective APCs due to their unique dendritic morphology, which is essential in connecting innate and adaptive immunity. When DCs are exposed to radiation, it promotes significantly the production of chemokines CCL19 and CCL21. These chemokines subsequently attach to CCR7, facilitating DCs movement. The release of TAAs induced by radiotherapy promotes the activation of DCs and T cells. DAMPs mainly consist of calreticulin, high-mobility group protein box 1, and ATP (67). Radiotherapy can promote the translocation of calreticulin to the cell membrane and then enhance the phagocytosis of tumor cell by DCs (68). High-mobility group protein box 1 can activate DCs to trigger targeted T-cell responses (69). ATP can bind to P2X7 receptors on DCs, activating DCs and IFNγ-producing CD8-positive T cells (70).
Radiotherapy can cause the release of cytosolic DNA, which will be detected by STING triggering immune responses by producing IFN-I (71). Cytosolic DNA induces structural alterations in the cGAS enzyme, resulting in the production of cGAMP and then activating STING. Upon activation, STING dimers move from endoplasmic reticulum to Golgi apparatus and microsomes. STING activates the kinases TBK1 and IKK, subsequently phosphorylating IRF3 and IκB family and activating NFκB. The simultaneous activation of IRF3 and NFκB can induce IFN-I generation and inflammasome activation. IFN-I can regulate DC function, activate T cells, enhance NK cell cytotoxicity, and generate long-lived memory cells (72).
Radiotherapy can transform “cold” into “hot” tumor through various mechanisms. The release of TAAs caused by radiotherapy makes them more detectable by immune systems (73). DNA damages induced by radiation also can increase the tumor mutational load, favoring immune recognition and elimination (74). In general, tumor cells can avoid the clearance by immune system by reducing MHC-I expression. Radiotherapy-induced DNA damage can upregulate MHC-I expression and boost TAAs presentation by activating mTOR. This process ultimately makes tumor cells more vulnerable to attacks from the immune system (75). ICD induced by radiotherapy can overcome immune evasion by promoting the expression of CXCL10 and CXCL16. It also can enhance the tumor blood vessels extravasation, which results in an increased tumor-infiltrating lymphocyte density in tumor tissue. Radiotherapy also is essential in expanding TCR repertoire and increasing CD8/CD4 ratio, primarily by increasing the CD80 costimulatory signal (76). NK cells can identify and destroy tumor cells. Radiotherapy can boost NK cells activity through FAS and FAS ligand, as well as ADCC (77).
The low-dose radiation promotes tumor vascular system normalization and the transformation of M2-type macrophage into M1-type iNOS+ phenotype (78). The iNOS+ macrophages can trigger Th1 chemokines generation, attracting T cells to tumor tissues and then enhancing anti-tumor effects. Radiotherapy also can induce oxidative stress and DNA damages in surrounding tumor tissues that have not been directly irradiated, a phenomenon referred to radiation-induced bystander effect. This bystander effect is mediated by microparticles derived from irradiated tumor cells and exhibits a unique and broad spectrum of antitumor effects. Tumor cells ingesting microparticles are easily sequestered by activated TAMs, which makes tumor cells susceptible to destruction by immune cells.
5 Combining immunotherapy with radiotherapy for GBM
5.1 Preclinical data
The preclinical studies of combination treatment of immunotherapy and radiotherapy for GBM mainly focused on the ICIs combined with radiotherapy, and a small number of other immunotherapies in combined with radiotherapy. Herein, we review the preclinical studies of combining various immunotherapy agents with radiotherapy for GBM. Zeng et al. (79) investigated the effect of anti-PD-1 combined with stereotactic radiosurgery (SRS) in a mouse GBM model. Combining anti-PD-1 with SRS improved greatly the survival by enhancing the tumor infiltration of cytotoxic T cells and reducing Tregs, compared with either treatment alone. 4-1BB (CD137), an antibody against co-stimulatory molecules, can facilitate CD8+ T cell proliferation and pro-inflammatory cytokines production when it interacts with specific ligand (80). A study of combining 4-1BB (CD137) activation with anti-CTLA-4 and focal radiotherapy in mice intracranial GBM models revealed that this combination treatment led to an increased TILs density, improving significantly the survival of intracranial GBM model (81). TIM-3, a negative immunomodulator, is upregulated in GBM similar to PD-1 (82). Kim et al. (83) investigated the effect of anti-TIM-3 monotherapy or combined with anti-PD-1 and SRS in mice GBM models. The results revealed that triple therapy improved greatly the survival compared to anti-TIM-3 monotherapy. Moreover, the GBM mice that received the combination of anti-CD47 and radiotherapy or temozolomide also improved greatly the survival in contrast to those that undergone monotherapy (84). In addition to ICIs, combining peripheral vaccination and radiotherapy also has been demonstrated to improve antitumor effect in the management of GBM by promoting MHC-I expression and increasing T-cells infiltration (85).
5.2 Clinical data
Many clinical studies focused on combining immunotherapy with radiotherapy for GBM, especially involving ICIs (Table 1). The common immunotherapy agents used in combination treatment for GBM clinical trials mainly include durvalumab (anti-PD-L1), atezolizumab (anti-PD-L1), avelumab (anti-PD-L1), pembrolizumab (anti-PD-1), nivolumab (anti-PD-1), and ipilimumab (anti-CTLA-4). Herein, we review the completed clinical trials of combining immunotherapy with radiotherapy for GBM according to different immunotherapy agents.

Table 1. Clinical trials involving the combination of immunotherapy and radiotherapy for GBM.
The phase II trial (NCT02336165) assessed the effect of durvalumab in 5 GBM groups. The reported results were from group A, which evaluated durvalumab plus radiotherapy followed by durvalumab in nGBM with unmethylated MGMT promoter after undergoing the maximum safe surgical resection (86). The outcomes showed that combining durvalumab with radiotherapy was well tolerated and appeared to be effective for nGBM. Among 40 enrolled patients, 35% experienced grade 3 or higher TRAEs. Twenty-four patients (60%) were alive at 12 months, outperforming the historical benchmarks of 50%. The mOS was 15.1 months, outperforming the historical benchmarks of 12.7 months. Another phase I trial investigated the safety of combining durvalumab with hypofractionated stereotactic radiotherapy (HFSRT) in 6 rGBM patients (87). The outcomes showed that combining durvalumab with HFSRT in rGBM was well tolerated. Only 1 patient experienced grade 3 vestibular neuritis associated with durvalumab. The mOS and mPFS were 16.7 and 2.3 months, respectively. The relevant phase II trial is ongoing.
A phase I/II trial investigated the effect of atezolizumab combined with radiotherapy and temozolomide followed by adjuvant atezolizumab and temozolomide in nGBM (88). Sixty patients were enrolled. The primary endpoints were safety and OS, with secondary endpoints of ORR and PFS. Atezolizumab exhibited good tolerance and modest efficacy in combination with radiochemotherapy. Thirty-three patients experienced grade 3 or higher TRAEs. The mOS was 17.1 months, with a mPFS of 9.7 months.
The phase II trial (NCT03047473) investigated the effect of avelumab plus standard treatment of radiotherapy and temozolomide in nGBM (89). The results revealed that avelumab combined with standard treatment was safe. However, this combination treatment failed to improve the OS. Among 30 patients, 3 patients experienced emergent adverse events related to avelumab. And 8 patients experienced one or more iRAEs. The mOS was 15.3 months, with a mPFS of 9.7 months. The tolerability results in GBM population of this trial failed to exhibit novel safety signals in contrast to previous avelumab research. The ORR was superior compared to that observed with other immunotherapies for GBM. In addition to the different mechanisms of action of avelumab, it may also be attributed to the early administration of avelumab as an addition to standard treatment in the disease course.
The phase I trial (NCT02313272) evaluated the effect of pembrolizumab plus bevacizumab plus HFSRT for rGBM (90). A total of 32 patients were enrolled. The combination of pembrolizumab with bevacizumab and HFSRT triple therapy exhibited encouraging modest efficacy with well tolerance. No neurological adverse events were detected. Only one patient discontinued the treatment because of the AST elevation. The tumor response rates (complete response + partial response) in bevacizumab-naïve group (20 of 24 patients) and bevacizumab-resistant group (5 of 8 patients) were 83% and 62%, respectively. The mOS in bevacizumab-naïve group and bevacizumab-resistant group were 13.45 and 9.3 months, respectively. The mPFS were 7.92 and 6.54 months, respectively.
The recently completed phase I trial (NCT02311920) evaluated the effect of adding ipilimumab and nivolumab to radiochemotherapy for nGBM (91). The outcomes showed that ipilimumab plus nivolumab exhibited was well tolerated in combination with radiochemotherapy. Among 32 patients, 16% experienced grade 4 events. The mOS for patients treated with combined treatment was 20.7 months, with a mPFS of 16.1 months. Another phase I trial investigated ipilimumab and nivolumab in combined with HFSRT and bevacizumab for bevacizumab-naïve rGBM (92). The primary endpoint was tolerability of this combined treatment. The secondary endpoints were 6- and 9-months survival. This combined treatment was well tolerated. Among 26 patients, 12 experienced grade 3 or 4 TRAEs. The 6- and 9-months survival were 92% and 75%, respectively. The phase III trial (NCT02617589) compared nivolumab plus radiotherapy to temozolomide plus radiotherapy in nGBM (93). A total of 560 patients were evenly assigned into nivolumab plus radiotherapy group or radiotherapy plus temozolomide group. The outcomes showed that nivolumab plus radiotherapy failed to improve the OS compared to temozolomide plus radiotherapy, suggesting that nivolumab should not be considered a substitute for temozolomide in this patient population. The mOS of temozolomide group and nivolumab group were 14.9 and 13.4 months, respectively. The mPFS were 6.2 and 6.0 months, respectively. The response rates were 7.8% and 7.2%, respectively. The grade 3 or higher TRAE rates of nivolumab group and temozolomide group were 21.9% and 25.1%, respectively. Another phase III trial (NCT02667587) investigated the effect of Stupp regime plus nivolumab or placebo in nGBM (94). A total of 716 patients were evenly assigned into nivolumab + radiotherapy + temozolomide or placebo + radiotherapy + temozolomide. Unfortunately, nivolumab plus Stupp regime did not improve the survival. The mOS for nivolumab group and placebo group were 28.9 and 32.1 months, respectively. The mPFS were 10.6 and 10.3 months, respectively. For patients without baseline corticosteroids, the mOS of nivolumab group and placebo group were 31.3 months and 33.0 months. Moreover, nivolumab plus Stupp regime resulted in increased rates of TRAEs. The grade 3 or higher TRAE rates of nivolumab group and placebo group were 52.4% vs 33.6%, respectively. The neurological TRAEs were observed in 23.1% of nivolumab group and 16.7% of placebo group. The most TRAEs in both groups were headache (9.3%/5.9%) and dysgeusia (5.6%/4.2%). Lymphopenia rates were 10.7% and 8.5% in nivolumab group and placebo group, respectively.
In addition to ICIs, other immunotherapies combined with radiotherapy for GBM also has been investigated. A clinical trial investigated the effect of DC vaccine targeting tumor stem cells for GBM (95). The outcomes showed that DC vaccine against glioblastoma stem cells was well tolerated and improved the PFS. The mPFS for vaccinated patients was 2.9 times greater than control groups. Of the 7 patients treated with DC vaccine immunotherapy, all patients experienced immune response induced by vaccination without negative side effects.
6 Discussion
Immunotherapy combined with radiotherapy appears to be a promising therapeutic strategy, backed by a solid biological foundation. However, the complexities underlying their synergetic modes still require further understanding. There are still several current challenges that require to be addressed or better elucidated regarding the combination of immunotherapy and radiotherapy for GBM.
For example, radiotherapy boosts anti-tumor immune responses while also leading to immunosuppression. Severe lymphopenia is observed frequently in GBM patients after radiotherapy. A prospective study revealed that the decrease in CD4 lymphocyte counts was observed in nGBM patients received radiochemotherapy (96). And the decrease in CD4 counts was linked to tumor progression. A large irradiation area and excessive fractionation may impair the immune function (97). Therefore, decreasing dose to healthy tissues, reducing radiotherapy target volume and applying the hypofractionated radiotherapy regimen may limit this effect. In many tumors, Tregs have immunosuppressive effects or even facilitate the progression of disease. It has been demonstrated radiotherapy can increase significantly the number of Tregs by various mechanisms. For example, radiotherapy can facilitate the production, expansion, differentiation, and development of Tregs by overexpressing TGF-β. Tumor-associated neutrophils (TANs) are typically categorized into two types: N1 neutrophils, which are antitumorigenic, and N2 neutrophils, which are protumorigenic (98). Radiotherapy also prompts TANs to show pro-tumor properties by TGF-β (99). N2 neutrophils cause tumor growth and immunosuppression by suppressing multiple immune cells while increasing Tregs. Tumor-associated macrophages (TAMs) also are categorized into two types, M1 (anti-tumor phenotype) and M2 (pro-tumor phenotype) (100). The activated p50–p50 dimer and elevated ROS levels induced by radiotherapy can promote the shift to M2 macrophage, subsequently releasing IL-10 and TGF-β that suppress DCs (101). To utilize the synergistic effect between radiotherapy and immunotherapy more effectively, additional research is needed to investigate how to balance stimulating and suppressing effects caused by radiotherapy.
Another potential challenge in combining immunotherapy with radiotherapy for GBM is the potentiating treatment related effects, commonly known as pseudoprogression. It is defined as radiographic changes associated with tumor progression that are related to treatment response and are transient (102). Several studies have suggested that radiotherapy and temozolomide may result in pseudoprogression in GBM treatment. It also is reported in GBM patients who underwent immunotherapy (103). Therefore, combining immunotherapy with radiotherapy for GBM may result in stronger pseudoprogression. In addition to the diagnostic challenge, pseudoprogression also may bring treatment challenges, since medications commonly applied to manage symptoms related to pseudoprogression may affect the effectiveness of immunotherapy. While existing imaging technologies appear to exhibit partial capacity in distinguishing between pseudoprogression and tumor progression, there remains a considerable demand for improving diagnostic accuracy. Radiomics is a technique that utilizes data characterization algorithms to extract numerous features from medical images, serving as a promising method for distinguishing between different conditions. Kim et al. (104) created a radiomics model that utilizes multiparametric MRI and compared its diagnostic effectiveness for pseudoprogression with that for single parameter and single imaging radiomics models. The results showed that this model outperformed single radiomics models. Moreover, it exhibited better accuracy in external and internal validation than other single models. In another research, the accuracy, specificity, and sensitivity of 72.78%, 61.33%, and 78.36% of radiomics model in distinguishing pseudoprogression from tumor progression still outperformed the performance of three radiologists (105). These preliminary data demonstrated the potential of radiomics models in differentiating between pseudoprogression from tumor progression.
Moreover, ascertaining optimal schedules, such as immunotherapy types, radiation dose, fractionation regimen and treatment sequence when combining immunotherapy with radiotherapy, is necessary to exert maximally synergistic effect. Combining ICIs with radiotherapy for GBM has been investigated extensively. However, the study of other immunotherapies combined with radiotherapy for GBM is still limited. The efficacy of several vaccines and OVs for GBM has been demonstrated in clinical studies. Therapeutic vaccine combined with radiotherapy also has been demonstrated to improve the antitumor effect in experimental high-grade gliomas model. Given the current situation of combining ICIs with radiotherapy for GBM, exploring other immunotherapies combined with radiotherapy for GBM may be a feasible direction. Moreover, we need to consider the effect of radiotherapy dose and fractionation on immune response. Low dose radiotherapy (LDRT) has been demonstrated to mobilize efficiently immune system even when an immune suppression pathway is present (106). Conventional radiotherapy may induce immunosuppressive factors generation; however, LDRT could be a viable approach to overcome these challenges. LDRT also can promote T cells recruitment by increasing the expression of certain chemokines, subsequently enhancing the potential of abscopal effect. The immunomodulatory effects of LDRT have been demonstrated in preclinical and clinical studies. Therefore, we have reason to believe that LDRT may further improve the effect of the existing combined treatment of immunotherapy and radiotherapy. Further study is needed to evaluate comprehensively its potential in GBM. Hypofractionated radiotherapy, such as stereotactic body radiotherapy (SBRT), can administer higher dose in fewer sessions. Several studies have demonstrated that hypofractionated radiotherapy may be more effective in inducing immune responses (107, 108). A phase I trial evaluated the effect of hypofractionated radiotherapy combined with bevacizumab and pembrolizumab for rGBM (109). Among 32 patients, 53% experienced CR or PR. The 12-months survival rate was 64%. To achieve a better immunomodulatory effect, further randomized clinical trials are necessary to ascertain the ideal radiotherapy dose and fractionation.
Moreover, it has been widely recognized that the treatment sequence will affect significantly the therapeutic efficacy of combined treatment. However, the optimal sequence for immunotherapy combined with radiotherapy is currently unclear. A preclinical research reported that the group that administrated anti-PD-L1 during fractional radiotherapy resulted in a better survival outcome compared to the group that underwent sequential administration after finishing radiotherapy (110). Another preclinical research revealed that the group receiving anti-CTLA-4 prior to radiotherapy experienced a better tumor response compared to the group receiving anti-CTLA-4 after radiotherapy (111). Another retrospective review indicated that immunotherapy may be more effective when administrated simultaneously with or after radiotherapy (112). A clinical study compared the effect of neoadjuvant and concurrent immunotherapy to adjuvant immunotherapy in nGBM (113). Twenty-two patients underwent the vaccine before starting chemoradiation and continued during treatment (group 1). Twenty-three patients (group 2) underwent the vaccine after finishing chemoradiation. Patients in group 1 exhibited a stronger initial response before chemoradiation, but this response diminished during treatment. In contrast, patients in group 2 exhibited a weaker initial response, but this response was more durable. Based on these outcomes, the optimal treatment sequence may differ according to the immunotherapy used and tumor type. On the one hand, the treatment sequence of radiotherapy followed by immunotherapy enables immunotherapy to partially overcome the tumor tolerance to radiotherapy and improve the effectiveness of radiotherapy. On the other hand, the treatment sequence of immunotherapy followed by radiotherapy may help stimulate the immune microenvironment, such as normalizing the tumor vasculature, alleviating hypoxia in tumor, increasing the radiotherapy sensitivity of tumor. Further research is required to elucidate more accurately the effect of different sequences on therapeutic efficacy.
Combining immunotherapy with radiotherapy for GBM has performed well in preclinical studies, but its efficacy remains to be limited in clinical trials, which may be due to the following reasons. Firstly, the tumor growth environment and physiological characteristics of animal models are different from those of humans. It is difficult for animal models to fully simulate the complex heterogeneity of human GBM, resulting in a reduced effect of effective treatment in animal models in human. Secondly, certain inherent obstacles in GBM patients, such as intricate tumor microenvironment and tumor heterogeneity, may influence the effect of combination treatment. Glioblastoma cells are highly heterogeneous in different patients and even within the same tumor, which is difficult to fully cover in preclinical studies. In clinical practice, certain tumor cells might not respond well to combined treatment, which may affect overall therapeutic effect. Human GBM will create a complex immunosuppressive microenvironment, which can weaken the immune response activated by combination treatment. Although radiotherapy can break immune tolerance to a certain extent, it is difficult to completely change this immunosuppressive state. Moreover, the observational indicators and endpoints of preclinical studies are different from clinical studies. Preclinical studies pay more attention to short-term indicators such as tumor growth inhibition, while clinical studies pay more attention to long-term indicators such as OS, PFS, and quality of life. The transformation from short-term effect to long-term benefit is affected by various factors, resulting in the results of clinical studies being worse than expected.
Although combined treatment of immunotherapy and radiotherapy has demonstrated advantages in many tumors, only limited patients benefit from the combined treatment. Therefore, exploring accurate biomarkers to predict and evaluate therapeutic responses is necessary. Considering the rationale behind the combined treatment of immunotherapy and radiotherapy, the DAMPs may be potential biomarkers for predicting the effectiveness of combination treatment. Radiotherapy can result in ICD by increasing calreticulin. Furthermore, calreticulin induced by radiotherapy may be crucial in tumor cells uptake and enhancing immune cell activity (114). Therefore, the amount of calreticulin following radiotherapy may be a potential biomarker for combination treatment. The circulating lymphocyte population plays a crucial role in anti-tumor immune responses. There is evidence that higher lymphocyte counts are linked to increased response rate in patients receiving ICIs (115). Moreover, there is some evidence that radiosensitivity may predict the effect of combined treatment of immunotherapy and radiotherapy. A research evaluated the correlation between radiosensitivity and immune responses in various tumors, including GBM (116). The outcomes showed that radiosensitivity index-low tumors exhibited a higher proportion of activated NK cells and M1 macrophages compared to radiosensitivity index-high tumors.
Finally, there is evidence that alterations in gut microbiota and its metabolites may affect the development of various diseases, including GBM. Gut microbiota establishes connections between gut and central nervous system via two-way signals along gut–brain axis. Gut microbiota dysregulation can promote ROS pro advancement duction by downregulating GM-CSF signaling, subsequently enhances the suppressive effects of MDSCs (117). Furthermore, gut microbiota dysregulation may result in the growth and apoptosis inhibition of tumor cells by downregulating Foxp3 (118). Short-chain fatty acids (SCFAs) are the main metabolites of gut microbiota. They have been demonstrated to improve disease activity by increasing Tregs and reducing Th1 and Th17 cells (119). Overall, the immunosuppressive microenvironment of GBM closely related to gut microbiota. A deeper understanding of gut microbiota may provide new opportunities for GBM treatment.
7 Conclusion
Currently, the performance of immunotherapy in treating GBM is still not ideal due to the immunosuppressive microenvironment. Combining immunotherapy with radiotherapy has consistently been an active research area, and GBM is no exception. Although preclinical studies have demonstrated the efficacy of this combination treatment for GBM, relative clinical evidence is still limited. Combining immunotherapy with radiotherapy for GBM showed modest efficacy only in several clinical studies of limited size, there are no successful phase III trials all over the world to date. Further studies that include the tumor heterogeneity and microenvironment are required to expound the mechanisms of success or failure.
Author contributions
YS: Writing – review & editing, Conceptualization, Investigation, Writing – original draft. YW: Investigation, Writing – original draft. SM: Investigation, Writing – original draft. XW: Investigation, Writing – original draft. SY: Investigation, Writing – original draft. ZW: Conceptualization, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Price M, Neff C, Nagarajan N, Kruchko C, Waite KA, Cioffi G, et al. CBTRUS statistical report: american brain tumor association & NCI neuro-oncology branch adolescent and young adult primary brain and other central nervous system tumors diagnosed in the United States in 2016-2020. Neuro Oncol. (2024) 26:iii1–iii53. doi: 10.1093/neuonc/noae047
2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
3. Ralli M, Botticelli A, Visconti IC, Angeletti D, Fiore M, Marchetti P, et al. Immunotherapy in the treatment of metastatic melanoma: current knowledge and future directions. J Immunol Res. (2020) 2020:9235638. doi: 10.1155/2020/9235638
4. Lahiri A, Maji A, Potdar PD, Singh N, Parikh P, Bisht B, et al. Lung cancer immunotherapy: progress, pitfalls, and promises. Mol Cancer. (2023) 22:40. doi: 10.1186/s12943-023-01740-y
5. Ferrall L, Lin KY, Roden RBS, Hung C-F, and Wu T-C. Cervical cancer immunotherapy: facts and hopes. Clin Cancer Res. (2021) 27:4953–73. doi: 10.1158/1078-0432.CCR-20-2833
6. Yasinjan F, Xing Y, Geng H, Guo R, Yang L, Liu Z, et al. Immunotherapy: a promising approach for glioma treatment. Front Immunol. (2023) 14:1255611. doi: 10.3389/fimmu.2023.1255611
7. Liu D, Wu L, Xu X, Yang S, Liu M, Hu M, et al. Regimens combining radiation and immunotherapy for cancer: latest updates from 2024 ASCO Annual Meeting. J Hematol Oncol. (2024) 17:84. doi: 10.1186/s13045-024-01587-w
8. Dagar G, Gupta A, Shankar A, Chauhan R, Macha MA, Bhat AA, et al. The future of cancer treatment: combining radiotherapy with immunotherapy. Front Mol Biosci. (2024) 11:1409300. doi: 10.3389/fmolb.2024.1409300
9. Lo Greco MC, Marano G, Milazzotto R, Liardo RLE, Finocchiaro I, La Rocca M, et al. The immunomodulatory potential of concurrent high-dose radiotherapy and immune checkpoint inhibitor cemiplimab in advanced cutaneous squamous cell carcinoma: initial results. J Pers Med. (2024) 14:581. doi: 10.3390/jpm14060581
10. Sharabi AB, Lim M, DeWeese TL, and Drake CG. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol. (2015) 16:e498–509. doi: 10.1016/S1470-2045(15)00007-8
11. Lara-Velazquez M, Shireman JM, Lehrer EJ, Bowman KM, Ruiz-Garcia H, Paukner MJ, et al. A comparison between chemo-radiotherapy combined with immunotherapy and chemo-radiotherapy alone for the treatment of newly diagnosed glioblastoma: A systematic review and meta-analysis. Front Oncol. (2021) 11:662302. doi: 10.3389/fonc.2021.662302
12. Chow KKH, Hara W, Lim M, and Li G. Combining immunotherapy with radiation for the treatment of glioblastoma. J Neurooncol. (2015) 123:459–64. doi: 10.1007/s11060-015-1762-9
13. Awada H, Paris F, and Pecqueur C. Exploiting radiation immunostimulatory effects to improve glioblastoma outcome. Neuro Oncol. (2023) 25:433–46. doi: 10.1093/neuonc/noac239
14. Lim M, Xia Y, Bettegowda C, and Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. (2018) 15:422–42. doi: 10.1038/s41571-018-0003-5
15. Yu MW and Quail DF. Immunotherapy for glioblastoma: current progress and challenges. Front Immunol. (2021) 12:676301. doi: 10.3389/fimmu.2021.676301
16. Rong L, Li N, and Zhang Z. Emerging therapies for glioblastoma: current state and future directions. J Exp Clin Cancer Res. (2022) 41:142. doi: 10.1186/s13046-022-02349-7
17. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. (2019) 25:477–86. doi: 10.1038/s41591-018-0337-7
18. Nayak L, Molinaro AM, Peters K, Clarke JL, Jordan JT, de Groot J, et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin Cancer Res. (2021) 27:1048–57. doi: 10.1158/1078-0432.CCR-20-2500
19. Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. (2018) 20:674–86. doi: 10.1093/neuonc/nox208
20. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkMate 143 phase 3 randomized clinical trial. JAMA Oncol. (2020) 6:1003–10. doi: 10.1001/jamaoncol.2020.1024
21. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. (2019) 25:470–6. doi: 10.1038/s41591-018-0339-5
22. Brown NF, Ng SM, Brooks C, Coutts T, Holmes J, Roberts C, et al. A phase II open label, randomised study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: the Ipi-Glio trial protocol. BMC Cancer. (2020) 20:198. doi: 10.1186/s12885-020-6624-y
23. Zhang M, Hutter G, Kahn SA, Azad TD, Gholamin S, Xu CY, et al. Anti-CD47 treatment stimulates phagocytosis of glioblastoma by M1 and M2 polarized macrophages and promotes M1 polarized macrophages. In Vivo. PloS One. (2016) 11:e0153550. doi: 10.1371/journal.pone.0153550
24. Azambuja JH, Schuh RS, Michels LR, Iser IC, Beckenkamp LR, Roliano GG, et al. Blockade of CD73 delays glioblastoma growth by modulating the immune environment. Cancer Immunol Immunother. (2020) 69:1801–12. doi: 10.1007/s00262-020-02569-w
25. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. (2018) 7:e1466769. doi: 10.1080/2162402X.2018.1466769
26. Wang J, Shen F, Yao Y, Wang L-L, Zhu Y, and Hu J. Adoptive cell therapy: A novel and potential immunotherapy for glioblastoma. Front Oncol. (2020) 10:59. doi: 10.3389/fonc.2020.00059
27. Krebs S, Chow KKH, Yi Z, Rodriguez-Cruz T, Hegde M, Gerken C, et al. T cells redirected to interleukin-13Rα2 with interleukin-13 mutein–chimeric antigen receptors have anti-glioma activity but also recognize interleukin-13Rα1. Cytotherapy. (2014) 16:1121–31. doi: 10.1016/j.jcyt.2014.02.012
28. Chow KKH, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. (2013) 21:629–37. doi: 10.1038/mt.2012.210
29. Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. (2016) 108. doi: 10.1093/jnci/djv375
30. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. (2015) 7:275ra22. doi: 10.1126/scitranslmed.aaa4963
31. Jiang H, Gao H, Kong J, Song B, Wang P, Shi B, et al. Selective targeting of glioblastoma with EGFRvIII/EGFR bitargeted chimeric antigen receptor T cell. Cancer Immunol Res. (2018) 6:1314–26. doi: 10.1158/2326-6066.CIR-18-0044
32. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
33. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
34. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
35. Quattrocchi KB, Miller CH, Cush S, Bernard SA, Dull ST, Smith M, et al. Pilot study of local autologous tumor infiltrating lymphocytes for the treatment of recurrent Malignant gliomas. J Neurooncol. (1999) 45:141–57. doi: 10.1023/a:1006293606710
36. Dillman RO, Duma CM, Ellis RA, Cornforth AN, Schiltz PM, Sharp SL, et al. Intralesional lymphokine-activated killer cells as adjuvant therapy for primary glioblastoma. J Immunother. (2009) 32:914–9. doi: 10.1097/CJI.0b013e3181b2910f
37. Swartz AM, Batich KA, Fecci PE, and Sampson JH. Peptide vaccines for the treatment of glioblastoma. J Neurooncol. (2015) 123:433–40. doi: 10.1007/s11060-014-1676-y
38. Li L, Zhou J, Dong X, Liao Q, Zhou D, and Zhou Y. Dendritic cell vaccines for glioblastoma fail to complete clinical translation: Bottlenecks and potential countermeasures. Int Immunopharmacol. (2022) 109:108929. doi: 10.1016/j.intimp.2022.108929
39. Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. (2013) 155:462–77. doi: 10.1016/j.cell.2013.09.034
40. Reardon DA, Desjardins A, Vredenburgh JJ, O’Rourke DM, Tran DD, Fink KL, et al. Rindopepimut with bevacizumab for patients with relapsed EGFRvIII-expressing glioblastoma (ReACT): results of a double-blind randomized phase II trial. Clin Cancer Res. (2020) 26:1586–94. doi: 10.1158/1078-0432.CCR-18-1140
41. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. (2017) 18:1373–85. doi: 10.1016/S1470-2045(17)30517-X
42. Wylie B, Macri C, Mintern JD, and Waithman J. Dendritic cells and cancer: from biology to therapeutic intervention. Cancers (Basel). (2019) 11:521. doi: 10.3390/cancers11040521
43. Frederico SC, Hancock JC, Brettschneider EES, Ratnam NM, Gilbert MR, and Terabe M. Making a cold tumor hot: the role of vaccines in the treatment of glioblastoma. Front Oncol. (2021) 11:672508. doi: 10.3389/fonc.2021.672508
44. Liau LM, Ashkan K, Brem S, Campian JL, Trusheim JE, Iwamoto FM, et al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: A phase 3 prospective externally controlled cohort trial. JAMA Oncol. (2023) 9:112–21. doi: 10.1001/jamaoncol.2022.5370
45. Liu P, Wang Y, Wang Y, Kong Z, Chen W, Li J, et al. Effects of oncolytic viruses and viral vectors on immunity in glioblastoma. Gene Ther. (2022) 29:115–26. doi: 10.1038/s41434-020-00207-9
46. Qi Z, Long X, Liu J, and Cheng P. Glioblastoma microenvironment and its reprogramming by oncolytic virotherapy. Front Cell Neurosci. (2022) 16:819363. doi: 10.3389/fncel.2022.819363
47. Zhu Z, Gorman MJ, McKenzie LD, Chai JN, Hubert CG, Prager BC, et al. Zika virus has oncolytic activity against glioblastoma stem cells. J Exp Med. (2017) 214:2843–57. doi: 10.1084/jem.20171093
48. Desjardins A, Gromeier M, Herndon JE, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. (2018) 379:150–61. doi: 10.1056/NEJMoa1716435
49. Todo T, Ito H, Ino Y, Ohtsu H, Ota Y, Shibahara J, et al. Intratumoral oncolytic herpes virus G47Δ for residual or recurrent glioblastoma: a phase 2 trial. Nat Med. (2022) 28:1630–9. doi: 10.1038/s41591-022-01897-x
50. Sooreshjani M, Tripathi S, Dussold C, Najem H, de Groot J, Lukas RV, et al. The use of targeted cytokines as cancer therapeutics in glioblastoma. Cancers (Basel). (2023) 15:3739. doi: 10.3390/cancers15143739
51. Guo C, Yang Q, Xu P, Deng M, Jiang T, Cai L, et al. Adjuvant temozolomide chemotherapy with or without interferon alfa among patients with newly diagnosed high-grade gliomas: A randomized clinical trial. JAMA Netw Open. (2023) 6:e2253285. doi: 10.1001/jamanetworkopen.2022.53285
52. Look T, Puca E, Bühler M, Kirschenbaum D, De Luca R, Stucchi R, et al. Targeted delivery of tumor necrosis factor in combination with CCNU induces a T cell-dependent regression of glioblastoma. Sci Transl Med. (2023) 15:eadf2281. doi: 10.1126/scitranslmed.adf2281
53. Dapash M, Hou D, Castro B, Lee-Chang C, and Lesniak MS. The interplay between glioblastoma and its microenvironment. Cells. (2021) 10:2257. doi: 10.3390/cells10092257
54. Jackson C, Ruzevick J, Phallen J, Belcaid Z, and Lim M. Challenges in immunotherapy presented by the glioblastoma multiforme microenvironment. Clin Dev Immunol. (2011) 2011:732413. doi: 10.1155/2011/732413
55. Vilgelm AE and Richmond A. Chemokines modulate immune surveillance in tumorigenesis, metastasis, and response to immunotherapy. Front Immunol. (2019) 10:333. doi: 10.3389/fimmu.2019.00333
56. Biserova K, Jakovlevs A, Uljanovs R, and Strumfa I. Cancer stem cells: significance in origin, pathogenesis and treatment of glioblastoma. Cells. (2021) 10:621. doi: 10.3390/cells10030621
57. Jamal M, Rath BH, Tsang PS, Camphausen K, and Tofilon PJ. The brain microenvironment preferentially enhances the radioresistance of CD133(+) glioblastoma stem-like cells. Neoplasia. (2012) 14:150–8. doi: 10.1593/neo.111794
58. Fidoamore A, Cristiano L, Antonosante A, d’Angelo M, Di Giacomo E, Astarita C, et al. Glioblastoma stem cells microenvironment: the paracrine roles of the niche in drug and radioresistance. Stem Cells Int. (2016) 2016:6809105. doi: 10.1155/2016/6809105
59. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. (2006) 444:756–60. doi: 10.1038/nature05236
60. Ong DST, Hu B, Ho YW, Sauvé C-EG, Bristow CA, Wang Q, et al. PAF promotes stemness and radioresistance of glioma stem cells. Proc Natl Acad Sci U.S.A. (2017) 114:E9086–95. doi: 10.1073/pnas.1708122114
61. Abuodeh Y, Venkat P, and Kim S. Systematic review of case reports on the abscopal effect. Curr Probl Cancer. (2016) 40:25–37. doi: 10.1016/j.currproblcancer.2015.10.001
62. Apetoh L, Tesniere A, Ghiringhelli F, Kroemer G, and Zitvogel L. Molecular interactions between dying tumor cells and the innate immune system determine the efficacy of conventional anticancer therapies. Cancer Res. (2008) 68:4026–30. doi: 10.1158/0008-5472.CAN-08-0427
63. Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. (2005) 11:728–34. doi: 10.1158/1078-0432.728.11.2
64. Park B, Yee C, and Lee K-M. The effect of radiation on the immune response to cancers. Int J Mol Sci. (2014) 15:927–43. doi: 10.3390/ijms15010927
65. McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer. (2020) 20:203–17. doi: 10.1038/s41568-020-0246-1
66. El-Saghire H, Michaux A, Thierens H, and Baatout S. Low doses of ionizing radiation induce immune-stimulatory responses in isolated human primary monocytes. Int J Mol Med. (2013) 32:1407–14. doi: 10.3892/ijmm.2013.1514
67. Rodriguez-Ruiz ME, Rodriguez I, Leaman O, López-Campos F, Montero A, Conde AJ, et al. Immune mechanisms mediating abscopal effects in radioimmunotherapy. Pharmacol Ther. (2019) 196:195–203. doi: 10.1016/j.pharmthera.2018.12.002
68. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. (2007) 13:54–61. doi: 10.1038/nm1523
69. Yamazaki T, Hannani D, Poirier-Colame V, Ladoire S, Locher C, Sistigu A, et al. Defective immunogenic cell death of HMGB1-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ. (2014) 21:69–78. doi: 10.1038/cdd.2013.72
70. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. (2009) 15:1170–8. doi: 10.1038/nm.2028
71. Cai X, Chiu Y-H, and Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell. (2014) 54:289–96. doi: 10.1016/j.molcel.2014.03.040
72. Fuertes MB, Domaica CI, and Zwirner NW. Leveraging NKG2D ligands in immuno-oncology. Front Immunol. (2021) 12:713158. doi: 10.3389/fimmu.2021.713158
73. Jiang W, Chan CK, Weissman IL, Kim BYS, and Hahn SM. Immune priming of the tumor microenvironment by radiation. Trends Cancer. (2016) 2:638–45. doi: 10.1016/j.trecan.2016.09.007
74. Schumacher TN and Schreiber RD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
75. Wan S, Pestka S, Jubin RG, Lyu YL, Tsai Y-C, and Liu LF. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PloS One. (2012) 7:e32542. doi: 10.1371/journal.pone.0032542
76. Rudqvist N-P, Pilones KA, Lhuillier C, Wennerberg E, Sidhom J-W, Emerson RO, et al. Radiotherapy and CTLA-4 blockade shape the TCR repertoire of tumor-infiltrating T cells. Cancer Immunol Res. (2018) 6:139–50. doi: 10.1158/2326-6066.CIR-17-0134
77. Pe F, Da M, Jf W, W X, Ah F, Ge A, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with Malignant glioma. Cancer Res. (2006) 66:3294–3302. doi: 10.1158/0008-5472.CAN-05-3773
78. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. (2013) 24:589–602. doi: 10.1016/j.ccr.2013.09.014
79. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. (2013) 86:343–9. doi: 10.1016/j.ijrobp.2012.12.025
80. Pollok KE, Kim YJ, Zhou Z, Hurtado J, Kim KK, Pickard RT, et al. Inducible T cell antigen 4-1BB. Analysis of expression and function. J Immunol. (1993) 150:771–81. doi: 10.4049/jimmunol.150.3.771
81. Belcaid Z, Phallen JA, Zeng J, See AP, Mathios D, Gottschalk C, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PloS One. (2014) 9:e101764. doi: 10.1371/journal.pone.0101764
82. Zhu C, Anderson AC, and Kuchroo VK. TIM-3 and its regulatory role in immune responses. Curr Top Microbiol Immunol. (2011) 350:1–15. doi: 10.1007/82_2010_84
83. Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. (2017) 23:124–36. doi: 10.1158/1078-0432.CCR-15-1535
84. Gholamin S, Youssef OA, Rafat M, Esparza R, Kahn S, Shahin M, et al. Irradiation or temozolomide chemotherapy enhances anti-CD47 treatment of glioblastoma. Innate Immun. (2020) 26:130–7. doi: 10.1177/1753425919876690
85. Newcomb EW, Demaria S, Lukyanov Y, Shao Y, Schnee T, Kawashima N, et al. The combination of ionizing radiation and peripheral vaccination produces long-term survival of mice bearing established invasive GL261 gliomas. Clin Cancer Res. (2006) 12:4730–7. doi: 10.1158/1078-0432.CCR-06-0593
86. Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn G, Lim M, et al. Phase II study to evaluate safety and efficacy of MEDI4736 (durvalumab) + radiotherapy in patients with newly diagnosed unmethylated MGMT glioblastoma (new unmeth GBM). JCO. (2019) 37:2032–2. doi: 10.1200/JCO.2019.37.15_suppl.2032
87. Pouessel D, Ken S, Gouaze-Andersson V, Piram L, Mervoyer A, Larrieu-Ciron D, et al. Hypofractionated stereotactic re-irradiation and anti-PDL1 durvalumab combination in recurrent glioblastoma: STERIMGLI phase I results. Oncologist. (2023) 28:825–e817. doi: 10.1093/oncolo/oyad095
88. Weathers S-PS, Kamiya-Matsuoka C, Harrison RA, Liu DD, Dervin S, Yun C, et al. Phase I/II study to evaluate the safety and clinical efficacy of atezolizumab (atezo; aPDL1) in combination with temozolomide (TMZ) and radiation in patients with newly diagnosed glioblastoma (GBM). JCO. (2020) 38:2511–1. doi: 10.1200/JCO.2020.38.15_suppl.2511
89. Jacques FH, Nicholas G, Lorimer IAJ, Sikati Foko V, Prevost J, Dumais N, et al. Avelumab in newly diagnosed glioblastoma. Neurooncol Adv. (2021) 3:vdab118. doi: 10.1093/noajnl/vdab118
90. Sahebjam S, Forsyth P, Arrington J, Jaglal M, Tran ND, Etame AB, et al. Atim-18. A phase i trial of hypofractionated stereotactic irradiation (hfsrt) with pembrolizumab and bevacizumab in patients with recurrent high grade glioma (nct02313272). Neuro-Oncology. (2017) 19:vi30. doi: 10.1093/neuonc/nox168.113
91. Sloan AE, Winter K, Gilbert MR, Aldape K, Choi S, Wen PY, et al. NRG-BN002: Phase I study of ipilimumab, nivolumab, and the combination in patients with newly diagnosed glioblastoma. Neuro Oncol. (2024) 26:1628–37. doi: 10.1093/neuonc/noae058
92. Sahebjam S, Raval RR, Forsyth PA, Enderling H, Tran ND, Arrington JA, et al. Phase 1 trial of hypofractionated stereotactic re-irradiation in combination with nivolumab, ipilimumab, and bevacizumab for recurrent high-grade gliomas. Neuro-Oncology Adv. (2025), 7:vdaf033. doi: 10.1093/noajnl/vdaf033
93. Omuro A, Brandes AA, Carpentier AF, Idbaih A, Reardon DA, Cloughesy T, et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol. (2023) 25:123–34. doi: 10.1093/neuonc/noac099
94. Lim M, Weller M, Idbaih A, Steinbach J, Finocchiaro G, Raval RR, et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. (2022) 24:1935–49. doi: 10.1093/neuonc/noac116
95. Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tønnesen P, Suso EMI, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother. (2013) 62:1499–509. doi: 10.1007/s00262-013-1453-3
96. Grossman SA, Ye X, Lesser G, Sloan A, Carraway H, Desideri S, et al. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin Cancer Res. (2011) 17:5473–80. doi: 10.1158/1078-0432.CCR-11-0774
97. Weeke E. Extracorporeal irradiation of the blood. Further investigations on the effect of varying transit dose, blood flow rate and frequency of treatment of the development of lymphopenia in uremic patients. Acta Med Scand. (1974) 195:149–54. doi: 10.1111/j.0954-6820.1974.tb08116.x
98. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
99. Schernberg A, Blanchard P, Chargari C, and Deutsch E. Neutrophils, a candidate biomarker and target for radiation therapy? Acta Oncol. (2017) 56:1522–30. doi: 10.1080/0284186X.2017.1348623
100. Funes SC, Rios M, Escobar-Vera J, and Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology. (2018) 154:186–95. doi: 10.1111/imm.12910
101. Wang N, Liang H, and Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. (2014) 5:614. doi: 10.3389/fimmu.2014.00614
102. Young JS, Al-Adli N, Scotford K, Cha S, and Berger MS. Pseudoprogression versus true progression in glioblastoma: what neurosurgeons need to know. J Neurosurg. (2023) 139:748–59. doi: 10.3171/2022.12.JNS222173
103. Li Y, Ma Y, Wu Z, Xie R, Zeng F, Cai H, et al. Advanced imaging techniques for differentiating pseudoprogression and tumor recurrence after immunotherapy for glioblastoma. Front Immunol. (2021) 12:790674. doi: 10.3389/fimmu.2021.790674
104. Kim JY, Park JE, Jo Y, Shim WH, Nam SJ, Kim JH, et al. Incorporating diffusion- and perfusion-weighted MRI into a radiomics model improves diagnostic performance for pseudoprogression in glioblastoma patients. Neuro Oncol. (2019) 21:404–14. doi: 10.1093/neuonc/noy133
105. Sun Y-Z, Yan L-F, Han Y, Nan H-Y, Xiao G, Tian Q, et al. Differentiation of pseudoprogression from true progressionin glioblastoma patients after standard treatment: A machine learning strategy combinedwith radiomics features from T1-weighted contrast-enhanced imaging. BMC Med Imaging. (2021) 21:17. doi: 10.1186/s12880-020-00545-5
106. Herrera FG, Ronet C, Ochoa de Olza M, Barras D, Crespo I, Andreatta M, et al. Low-dose radiotherapy reverses tumor immune desertification and resistance to immunotherapy. Cancer Discovery. (2022) 12:108–33. doi: 10.1158/2159-8290.CD-21-0003
107. Filatenkov A, Baker J, Mueller AMS, Kenkel J, Ahn G-O, Dutt S, et al. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin Cancer Res. (2015) 21:3727–39. doi: 10.1158/1078-0432.CCR-14-2824
108. Dovedi SJ, Cheadle EJ, Popple AL, Poon E, Morrow M, Stewart R, et al. Fractionated radiation therapy stimulates antitumor immunity mediated by both resident and infiltrating polyclonal T-cell populations when combined with PD-1 blockade. Clin Cancer Res. (2017) 23:5514–26. doi: 10.1158/1078-0432.CCR-16-1673
109. Sahebjam S, Forsyth PA, Tran ND, Arrington JA, Macaulay R, Etame AB, et al. Hypofractionated stereotactic re-irradiation with pembrolizumab and bevacizumab in patients with recurrent high-grade gliomas: results from a phase I study. Neuro Oncol. (2021) 23:677–86. doi: 10.1093/neuonc/noaa260
110. Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. (2014) 74:5458–68. doi: 10.1158/0008-5472.CAN-14-1258
111. Young KH, Baird JR, Savage T, Cottam B, Friedman D, Bambina S, et al. Optimizing timing of immunotherapy improves control of tumors by hypofractionated radiation therapy. PloS One. (2016) 11:e0157164. doi: 10.1371/journal.pone.0157164
112. Woody S, Hegde A, Arastu H, Peach MS, Sharma N, Walker P, et al. Survival is worse in patients completing immunotherapy prior to SBRT/SRS compared to those receiving it concurrently or after. Front Oncol. (2022) 12:785350. doi: 10.3389/fonc.2022.785350
113. Rampling R, Peoples S, Mulholland PJ, James A, Al-Salihi O, Twelves CJ, et al. A cancer research UK first time in human phase I trial of IMA950 (Novel multipeptide therapeutic vaccine) in patients with newly diagnosed glioblastoma. Clin Cancer Res. (2016) 22:4776–85. doi: 10.1158/1078-0432.CCR-16-0506
114. Liu Y, Dong Y, Kong L, Shi F, Zhu H, and Yu J. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J Hematol Oncol. (2018) 11:104. doi: 10.1186/s13045-018-0647-8
115. Diehl A, Yarchoan M, Hopkins A, Jaffee E, and Grossman SA. Relationships between lymphocyte counts and treatment-related toxicities and clinical responses in patients with solid tumors treated with PD-1 checkpoint inhibitors. Oncotarget. (2017) 8:114268–80. doi: 10.18632/oncotarget.23217
116. Dai Y-H, Wang Y-F, Shen P-C, Lo C-H, Yang J-F, Lin C-S, et al. Radiosensitivity index emerges as a potential biomarker for combined radiotherapy and immunotherapy. NPJ Genom Med. (2021) 6:40. doi: 10.1038/s41525-021-00200-0
117. Dehhaghi M, Kazemi Shariat Panahi H, Heng B, and Guillemin GJ. The gut microbiota, kynurenine pathway, and immune system interaction in the development of brain cancer. Front Cell Dev Biol. (2020) 8:562812. doi: 10.3389/fcell.2020.562812
118. Fan Y, Su Q, Chen J, Wang Y, and He S. Gut microbiome alterations affect glioma development and foxp3 expression in tumor microenvironment in mice. Front Oncol. (2022) 12:836953. doi: 10.3389/fonc.2022.836953
Keywords: glioblastoma, immunotherapy, radiotherapy, tumor microenvironment, clinical trial
Citation: Sun Y, Wang Y, Mu S, Wu X, Yu S and Wang Z (2025) Combining immunotherapy with radiotherapy for glioblastoma: recent advances and challenges. Front. Oncol. 15:1523675. doi: 10.3389/fonc.2025.1523675
Received: 06 November 2024; Accepted: 06 May 2025;
Published: 22 May 2025.
Edited by:
Cristiana Tanase, Victor Babes National Institute of Pathology (INCDVB), RomaniaReviewed by:
Corrado Spatola, University of Catania, ItalyVijay Kumar Ulaganathan, Université de Lorraine, France
Vinton Cheng, University of Birmingham, United Kingdom
Copyright © 2025 Sun, Wang, Mu, Wu, Yu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongming Wang, c2R3em0xMjkxQHVzc3QuZWR1LmNu