 Corbin J. Eule
Corbin J. Eule Junxiao Hu
Junxiao Hu Kurtis D. Davies
Kurtis D. Davies Alkesh Jani4,5
Alkesh Jani4,5 Elaine T. Lam
Elaine T. Lam- 1Division of Medical Oncology, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 2Biostatistics Core, University of Colorado Cancer Center, Aurora, CO, United States
- 3Department of Pathology, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 4Division of Nephrology, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 5Rocky Mountain Regional Veterans Administration Medical Center, Aurora, CO, United States
- 6Division of Urology, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
Background: Patients with chronic kidney disease (CKD) or post-kidney transplant have an elevated risk of renal cell carcinoma (RCC). Despite increased understanding of genomic alterations in clear cell and papillary RCC, the most common subtypes, little is known about the effects of renal dysfunction on RCC pathogenesis.
Materials and methods: This retrospective study analyzed electronic medical records and pathology data from adult patients with renal cell carcinoma (RCC) who were evaluated for or received a kidney transplant at a single institution between 1995 and 2021. Molecular sequencing of RCC tissue samples was conducted to compare mutation rates in patients with renal dysfunction compared with those reported by The Cancer Genome Atlas (TCGA).
Results: This study identified 21 patients with RCC undergoing kidney transplant evaluation, mostly males with an increased occurrence of papillary RCC (38.1%) and early-stage disease (85.7%). Among clear cell RCC tumors, SDHA mutations were significantly more common compared to the TCGA cohort. For papillary RCC, genes such as BRD4 and those involved in DNA damage repair demonstrated increased mutation rates in patients with renal dysfunction
Conclusions: This study identifies key mutations in RCC among patients with CKD and post-kidney transplant, highlighting a complex relationship between renal dysfunction, inflammation, and tumorigenesis. Despite its limited sample size, the findings underscore the need for further research to understand the molecular drivers of RCC in high-risk populations, which could lead to more personalized treatment strategies.
Introduction
In 2023, 27,332 kidney transplants were performed in the United States, constituting 81% of solid organ transplants in that year (1). Following solid organ transplantation, the risk of malignancy increases despite an improvement in overall survival (2–4). In particular, renal cell carcinoma (RCC) was found to have a standard incidence ratio (SIR) of 4.7 as compared to the general population (p <0.001) in a cohort of over 175,000 solid organ transplant patients between 1987-2008. This is greater than the SIR of 2.10 (2.06-2.14 95% CI) of all malignancies in patients who have undergone a solid organ transplant (4). In a general population, the most prevalent histopathologic subtype of RCC is clear cell RCC (approximately 80 percent) (ccRCC) followed by papillary (15 percent) and chromophobe (5–10 percent) RCC (5–7). For kidney transplant recipients, the relative risk for RCC is highest for papillary RCC (SIR 13.3 vs. 3.98 for ccRCC) (8). While the increased frequency of abdominal imaging for patient undergoing a kidney transplant evaluation may facilitate RCC detection, this alone does not account for the increased RCC risk in the transplant patient population (9).
While the increased incidence of RCC following kidney transplant is an increasingly recognized phenomenon, the contribution of specific risk factors is not well elucidated. Chronic kidney disease (CKD) and acquired cystic kidney disease are associated with an increased risk of RCC (10, 11). End-stage renal disease (ESRD), kidney transplant, and impaired kidney graft function have also been shown to increase RCC risk (12, 13). However, RCC arising in patients with ESRD may have a more clinically indolent behavior with lower rates of metastasis and longer cancer-specific survival (14). Nonetheless, the genomic features of RCC arising in the context of CKD and ESRD and whether they differ from RCC in patients from the general population is not clearly understood (11, 15).
The Cancer Genome Atlas (TCGA) Research Network identified significantly mutated genes (SMGs) across over 500 ccRCC and papillary tumor specimens (16, 17). In ccRCC, the most common of these mutations occurred in the von Hippel Lindau (VHL) tumor suppressor gene implicated in cellular oxygen sensing and in the protein polybromo 1 (PBRM1) gene controlling the maintenance of chromatin (16). Clear cell RCC occurring in patients with ESRD may have lower rates of chromosome 3p loss, the location of VHL (18, 19). Studies of molecular alterations in RCC and ESRD have primarily been limited to chromosomal analysis (20). To our knowledge, no prior study has investigated the incidence of RCC gene mutations in the pre- and post-kidney transplant settings.
RCC is a significant contributor to the morbidity and mortality of patients with renal disease and following kidney transplant. Our study aims to characterize the clinical and genomic features of RCC as they relate to patients in the pre-kidney and post-kidney transplant settings at a kidney transplant center and identify potential differences in sporadic cases of RCC from TCGA cohort.
Materials and methods
Study design and patients
This is a retrospective study with collection of secondary use clinical and pathologic data. The electronic medical record (EMR) was queried for adult patients with a confirmed pathologic diagnosis of RCC who have (1) undergone formal evaluation for a kidney transplant and/or (2) received a kidney transplant at a single institution between 1995-2021. Patients who received multi-organ transplants were excluded from this study.
Demographic, clinical, pathologic, molecular, and outcomes data were extracted from the EMR. Standardized data collection templates were used to minimize inter-observer variation. Available RCC tissue specimens from the identified pre-kidney and post-kidney transplant were collected for molecular sequencing. The primary endpoint of this study was mutation rates of clear cell and papillary RCC in patients with CKD relative to mutation rates reported by TCGA (accessed September 27, 2023). This study was submitted to the Colorado Multiple Institutional Review Board (21-4627) and determined to be exempt from review due to the use of secondary data.
Molecular analysis
Archived tumor material was retrieved and then reviewed by a board-certified pathologist to determine adequacy for molecular testing. Tumor enrichment from samples was achieved by a pathologist-guided microdissection or macrodissection procedure prior to nucleic acid extraction. Mutational analysis of extracted total nucleic acid (TNA) was performed using a clinically validated next-generation sequencing (NGS)-based assay that assesses mutational status across all coding regions of 498 genes. Target enrichment for this assay was achieved through a hybrid-capture approach and sequencing was performed on the Illumina NextSeq platform. Raw NGS data was processed using a custom bioinformatic analysis pipeline and all variant calls were manually inspected by an expert (author KDD). Mutations in TCGA were identified using whole exome sequencing, copy-number analysis, messenger RNA and microRNA sequencing, DNA-methylation analysis, and proteomic analysis (16, 17).
Statistical methods
Summary statistics were reported for clinical and demographic characteristics. Continuous variables were presented with median and IQR. The categorical variables were presented with frequency and percentages. For each mutation and cohort, the frequencies and the percentages were calculated. Fisher’s exact test were conducted to compare the mutation rates between the two groups for clear cell tumors (all clear cell tumors biopsies were collected independently from patients). The permutation test based on log likelihood ratio were conducted compare the mutation rates between the two groups for papillary tumors (2 tumor biopsies were collected from one patient with bilateral papillary RCC at the same time, and the remaining biopsies were collected independently from patients), to address the clustered biopsies for the one patient with bilateral papillary RCC.
Statistical significance level was set to 0.05. All statistical analysis were conducted using R version 4.1.0, R Core Team (21).
Results
A total of 21 patients who underwent kidney transplant evaluation and had a diagnosis of RCC were identified (Table 1). The patients in the cohort include those with CKD, ESRD on dialysis, or post-kidney treansplant at the time of RCC diagnosis. The majority of patients were male (71.4%) and non-Hispanic white (61.9%). An increased proportion of papillary RCC histology was present (38.1%). RCC was most frequently diagnosed as early disease (stage I, 85.7%). Most RCC tumors were diagnosed in dialysis patients (76.2%) with a median dialysis duration of 64.7 months. The 24 RCC specimens, 12 ccRCC and 12 papillary RCC were collected from 21 patients. Two patients had metachronous clear cell and papillary RCCs, and one patient had bilateral papillary RCCs at the time of resection. Two patients had ccRCC of the allograft kidney and the remainder of RCCs were isolated from the native kidney. Both patients with ccRCC of the allograft kidney did not have BK virus detected on PCR. The majority (18 patients, 85.7%) were diagnosed with stage I tumors.
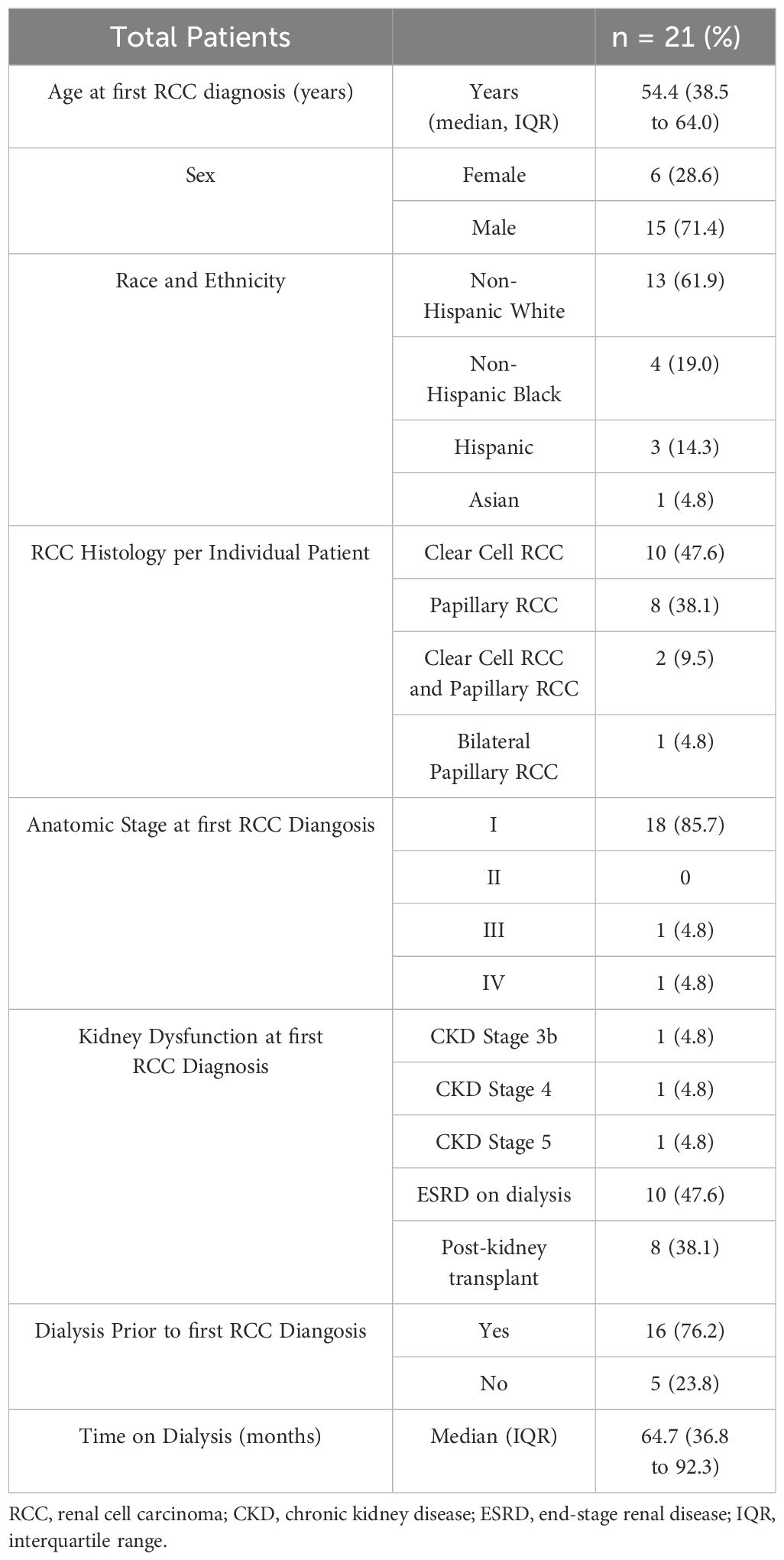
Table 1. Baseline clinical and demographic characteristics of patients evaluated for kidney transplant at time of RCC diagnosis.
Among the 12 ccRCC tumors collected from patients with renal dysfunction, there was no significant difference in the SMGs identified by TCGA (Table 2). The gene SDHA was mutated in 3 ccRCC tumors (25.0%) in patients with renal dysfunction but no patients from TCGA cohort, which was statistically significant (p <0.001). Five additional genes were mutated at greater frequency in patients with RCC and renal dysfunction compared to TCGA cohort.
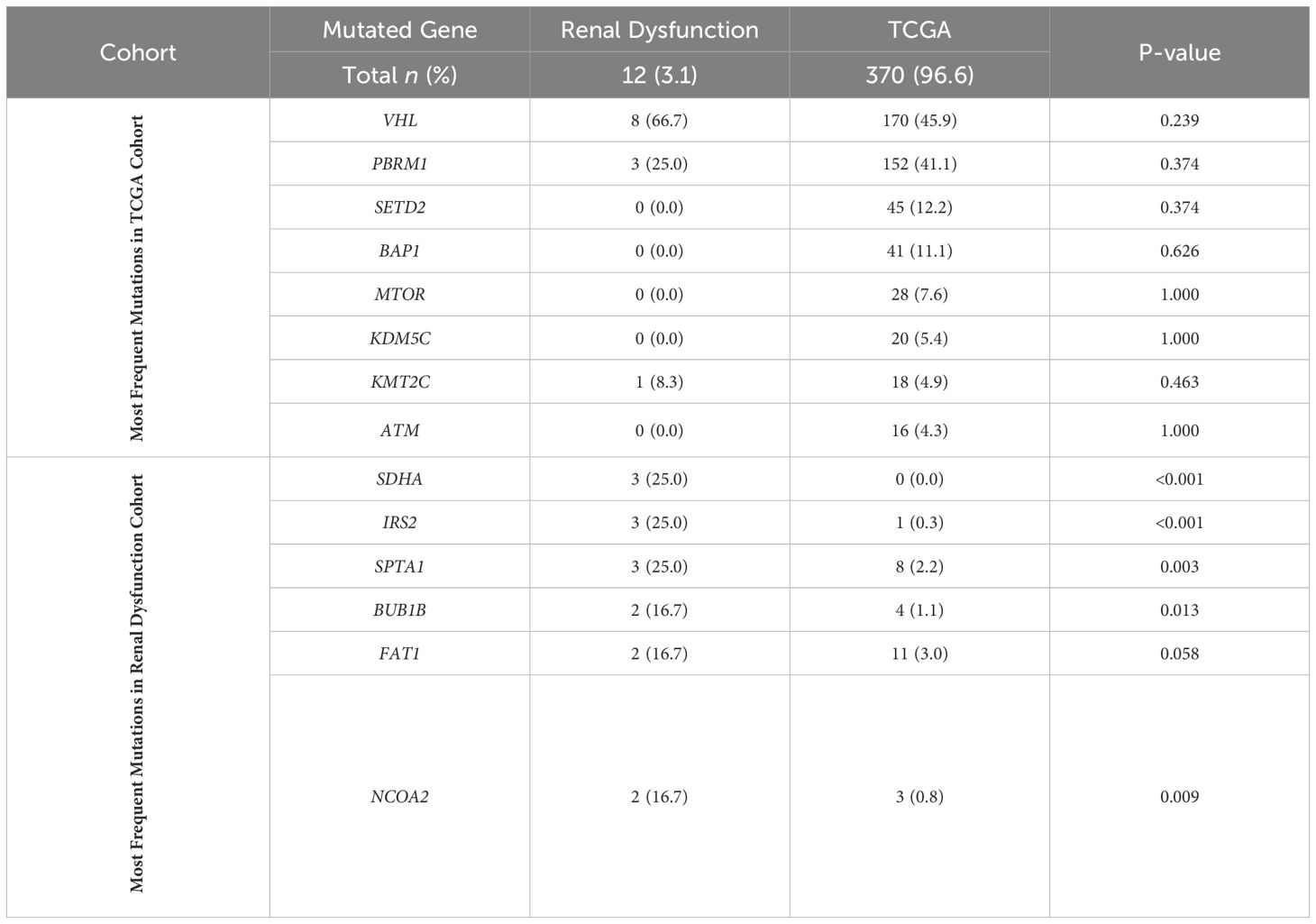
Table 2. Mutation rates in clear cell RCC of the most frequently mutated genes in TCGA and renal dysfunction cohorts.
Among patients with renal dysfunction and papillary RCC (12 tumors collected), again there was no significant difference from the most commonly mutated genes identified by TCGA (Table 3). In the renal dysfunction cohort, multiple genes had increased mutation rates relative to TCGA, with mutations in BRD4 and CHEK2 being the most frequent.
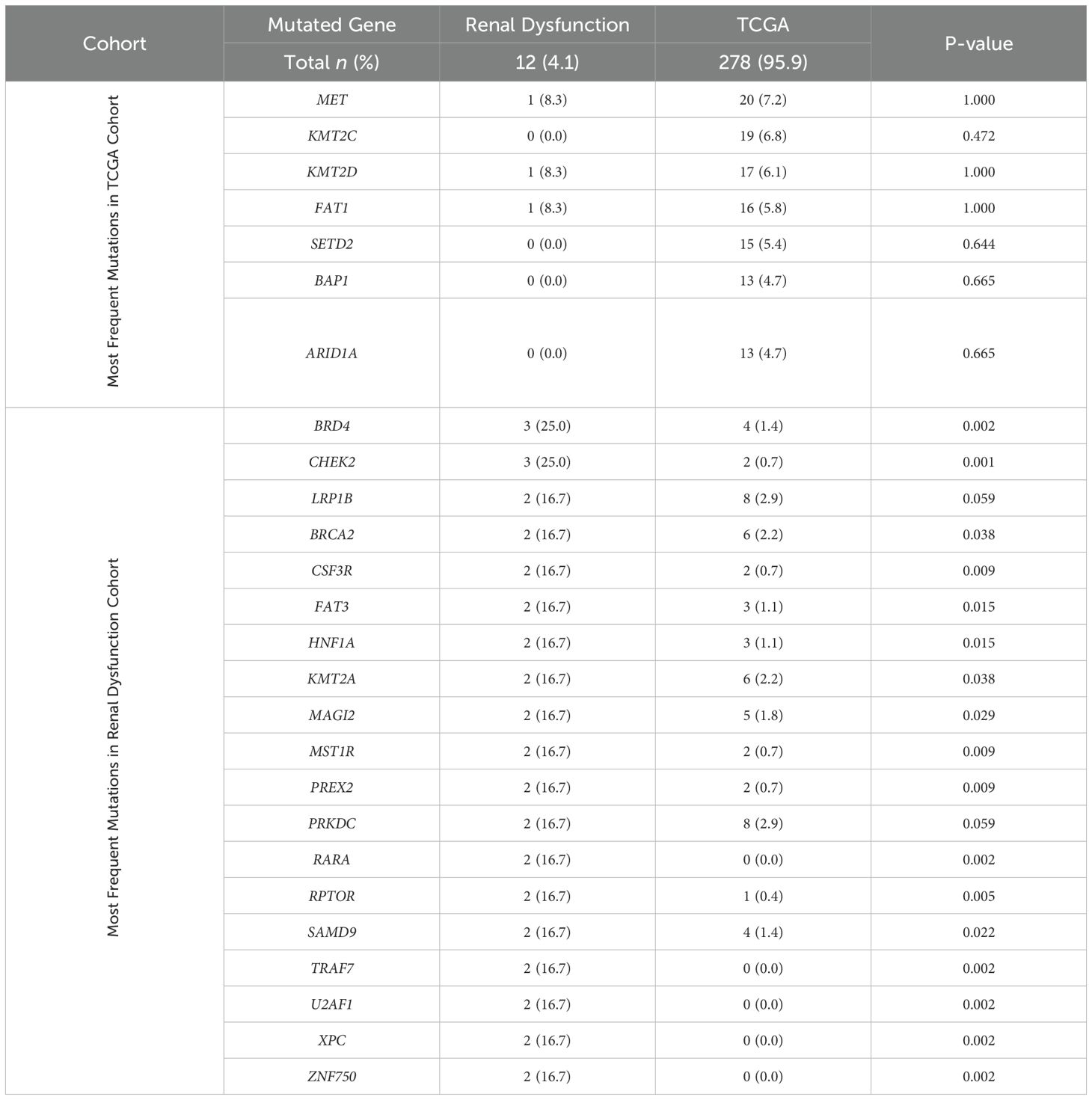
Table 3. Mutation rates in papillary RCC of the most frequently mutated genes in TCGA and renal dysfunction cohorts.
Discussion
This study investigated the molecular underpinnings of RCC in patients with CKD and post-kidney transplant, focusing on the genetic mutations that may influence tumorigenesis and treatment outcomes. Among 21 patients undergoing kidney transplant evaluation with RCC, the majority were male, non-Hispanic white, diagnosed with early-stage RCC prior to kidney transplantation and following long-term dialysis. TCGA cohort similarly showed a male predominance, although with a larger proportion of advanced RCC disease (22). The median time on hemodialysis was 64.7 months (IQR 36.8, 92.3). Notably, no BAP1 mutations were detected in the renal dysfunction cohort. BAP1 loss correlates with more clinically aggressive sporadic ccRCC given its association with high tumor grade and worse survival outcomes for patients (23). While not statistically significant, this finding corresponds with our previous work demonstrating lower rates of BAP1 mutations in RCC from patients with advanced CKD relative to patients without significant renal dysfunction (24).
SDH-deficient RCC is very rare, estimated to make up less than 0.2% of all RCCs (25). SDH-deficient RCC is most often due to a mutation in SHDB, while SDHA-deficient RCC is exquisitely rare (26). However, in ccRCC specimens from the renal dysfunction cohort, SDHA was mutated in 25.0% of patients. SDH, which encodes the succinate dehydrogenase complex, is a tumor suppressor gene involved in the mitochondrial respiratory chain and conversion of succinate to fumarate in the citric acid cycle (25, 26). Although SDH-deficient RCC in uncommon, functional SDH loss may play a broader role in RCC pathogenesis and was found to be a common adverse feature in approximately 80% of ccRCC cases (27). Succinate accumulation during renal ischemia is oxidized during reperfusion, yielding excessive reactive oxygen species and kidney damage (28). Our findings suggest that SDHA dysfunction may contribute to both CKD and ccRCC tumorigenesis through a similar underlying process.
BRD4 was one of the most frequently mutated genes in papillary RCC from patients with renal dysfunction. BRD4, which encodes bromodomain-containing protein 4, is a member of the bromodomain and extraterminal (BET) protein family and is essential to chromatin structure formation, transcription elongation, and epigenetic regulation (29). BRD4 binding leads to sustained activation of the transcription factor nuclear factor-κB (NF-κB), leading to aberrant cell proliferation (30). BRD4 and other BET proteins have been implicated in the upregulation of inflammatory genes in CKD, and BET inhibitors have been studies in preclinical setting for treatment of kidney injury and prevention of renal fibrosis (31). The impact of BRD4 in tumorigenesis and development of renal disease may illustrate an overlapping role in RCC formation in CKD.
In papillary RCC isolated from patients with renal dysfunctions, higher rates of CHEK2, BRCA2, and XPC mutations, all genes involved in DNA damage repair (DDR), were observed. Additionally, 3 other papillary RCC tumors were found to have a mutation in the DDR genes BRCA1, RAD51B, or MLH3. One papillary RCC had coexisting BRCA2 and NDN mutations. DDR response in renal tubular cells in response to acute injury has been shown to determine whether cellular recovery or irreversible damage occurs (32). In particular, loss of DDR is associated with irreversible kidney injury in cisplatin-exposed kidney organoid model (32). Considering the known association between CKD and papillary RCC, this raises the questions whether loss of DDR may be a mutually reinforcing process which facilitates progressive renal dysfunction and RCC tumorigenesis (8). The impact of DDR alterations on RCC is the premise of an ongoing clinical trial examining whether the poly ADP ribose polymerase inhibitor (PARPi) olarapib has antitumor activity in RCC harboring BAP1 or DDR mutations (NCT03786796).
Interestingly, in the renal dysfunction cohort, TRAF7 and RPTOR were mutations that were detected in the patient with bilateral papillary RCC diagnosed post-kidney transplant. TRAF7, TNF receptor associate factor 7, encodes an E3 ubiquitin ligase that potentiates MEKK3-mediated apoptosis (33). TRAF7 has previously been identified in NF2-independent meningiomas, malignant mesothelioma, and in small numbers of clear cell, papillary, and chromophobe RCCs (34, 35). TRAF7 has a role in the regulation of innate and adaptive immune responses, but it is indeterminate whether the post-transplant tumor environment or immunosuppression may enrich for this mutation (34). RPTOR, regulatory associated protein of mechanistic target of rapamycin (mTOR) complex 1, is a gene in the PI3K/AKT/mTOR pathway which is up regulated following loss of VHL (36). The mTOR inhibitor everolimus is an efficacious subsequent line option for the treatment of advanced RCC (37). Additionally, the mTOR inhibitors sirolimus and tacrolimus are commonly used immunosuppressive medication to prevent post-kidney transplant rejection (38). The degree to which immunosuppression impacts RCC risk, including potential molecular alterations, remains unclear (39). The most commonly used immunosuppressive regimen in this cohort was tacrolimus, mycophenolate, and prednisone.
Due to the small sample size, these results should be interpreted primarily as hypothesis generating, but collectively illustrate distinct molecular underpinnings for clear cell and papillary RCC pathogenesis in patients with renal dysfunction and following a kidney transplant. Mutational results for the renal dysfunction cohort were limited to those in the 498-gene panel, which by nature is less comphrehensive than bioinformatics studies utilizing a combination of whole genome, whole exome, transcriptome sequencing, or other molecular analysis. Germline genetic testing was not conducted for the patients in this study, limiting the ability to correlate it with the observed somatic mutations; however, this impact is expected to be minimal. At last follow-up, no patients had died as a result of their RCC, limiting the potential to detect a correlation between genes with high mutation rates and cancer-specific survival.
This study highlights distinct molecular characteristics of RCC in patients with CKD and post-kidney transplant, identifying key mutations such as SDHA, BRD4, and DDR-related genes CHEK2 and BRCA2. These mutations suggest a complex interplay between renal dysfunction, tumorigenesis, inflammation, and immune modulation. The findings illustrate that CKD and RCC may share overlapping mechanisms, potentially contributing to both renal injury and cancer development. Although the study is limited by its small sample size, it underscores the importance of continued research to better understand the molecular drivers of RCC in these high-risk populations. Future studies could ultimately inform more personalized and effective treatment strategies and clarify the influence of specific mutations on RCC-specific survival outcomes.
Data availability statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request and completion of a data sharing agreement.
Ethics statement
This study involved the secondary use of pathology specimens and clinical data obtained through retrospective chart review. The Colorado Multiple Institutional Review Board determined that the research was exempt from formal review. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by- product of routine care or industry. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
CE: Conceptualization, Funding acquisition, Methodology, Writing – original draft. JH: Formal analysis, Software, Visualization, Writing – review & editing. KD: Formal analysis, Software, Writing – review & editing. AJ: Methodology, Writing – review & editing, Conceptualization. TP: Methodology, Writing – review & editing, Conceptualization. EL: Methodology, Supervision, Writing – review & editing, Conceptualization, Funding acquisition.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by a pilot grant from the University of Colorado Cancer Center Support Grant P30CA046934.
Acknowledgments
Molecular testing was performed by the Molecular Pathology section (RRID: SCR_021995) of the Pathology Shared Resource (PSR) of the University of Colorado Cancer Center.
Conflict of interest
EL reports institutional research funding from Advaxis, Amgen, Argos Therapeutics, Arrowhead Pharmaceuticals, Astellas Pharma, Bristol-Myers Squibb, Calithera Biosciences, Constellation Pharmaceuticals, Decibel Therapeutics, Exelixis, FORMA, HiberCell, Pathos AI, Peloton Therapeutics, Harpoon Therapeutics, Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., OnQuality Pharmaceuticals, Pfizer, Phosplatin Therapeutics, Genentech, and F. Hoffmann-La Roche.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Organ procurement and transplantation network: national data [Online]. Available online at: https://optn.transplant.hrsa.gov/data/view-data-reports/national-data/ (Accessed August 16, 2021).
2. Wolfe RA, Ashby VB, Milford EL, Ojo AO, Ettenger RE, Agodoa LY, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med (1999) 341:1725–30. doi: 10.1056/NEJM199912023412303
3. Roberts MS, Angus DC, Bryce CL, Valenta Z, and Weissfeld L. Survival after liver transplantation in the United States: a disease-specific analysis of the UNOS database. Liver Transpl (2004) 10:886–97. doi: 10.1002/lt.20137
4. Engels EA, Pfeiffer RM, Fraumeni JF Jr., Kasiske BL, Israni AK, Snyder JJ, et al. Spectrum of cancer risk among US solid organ transplant recipients. JAMA (2011) 306:1891–901. doi: 10.1001/jama.2011.1592
5. Patard JJ, Leray E, Rioux-Leclercq N, Cindolo L, Ficarra V, Zisman A, et al. Prognostic value of histologic subtypes in renal cell carcinoma: a multicenter experience. J Clin Oncol (2005) 23:2763–71. doi: 10.1200/JCO.2005.07.055
6. Leibovich BC, Lohse CM, Crispen PL, Boorjian SA, Thompson RH, Blute ML, et al. Histological subtype is an independent predictor of outcome for patients with renal cell carcinoma. J Urol (2010) 183:1309–15. doi: 10.1016/j.juro.2009.12.035
7. Teloken PE, Thompson RH, Tickoo SK, Cronin A, Savage C, Reuter VE, et al. Prognostic impact of histological subtype on surgically treated localized renal cell carcinoma. J Urol (2009) 182:2132–6. doi: 10.1016/j.juro.2009.07.019
8. Karami S, Yanik EL, Moore LE, Pfeiffer RM, Copeland G, Gonsalves L, et al. Risk of renal cell carcinoma among kidney transplant recipients in the United States. Am J Transplant (2016) 16:3479–89. doi: 10.1111/ajt.13862
9. Al-Adra D, Al-Qaoud T, Fowler K, and Wong G. De novo Malignancies after kidney transplantation. Clin J Am Soc Nephrol. (2021) 17:434–43. doi: 10.2215/CJN.14570920
10. Truong LD, Krishnan B, Cao JT, Barrios R, and Suki WN. Renal neoplasm in acquired cystic kidney disease. Am J Kidney Dis (1995) 26:1–12. doi: 10.1016/0272-6386(95)90146-9
11. Lowrance WT, Ordonez J, Udaltsova N, Russo P, and Go AS. CKD and the risk of incident cancer. J Am Soc Nephrol (2014) 25:2327–34. doi: 10.1681/ASN.2013060604
12. Au EH, Chapman JR, Craig JC, Lim WH, Teixeira-Pinto A, Ullah S, et al. Overall and site-specific cancer mortality in patients on dialysis and after kidney transplant. J Am Soc Nephrol. (2019) 30:471–80. doi: 10.1681/ASN.2018090906
13. Yanik EL, Clarke CA, Snyder JJ, Pfeiffer RM, and Engels EA. Variation in Cancer Incidence among Patients with ESRD during Kidney Function and Nonfunction Intervals. J Am Soc Nephrol (2016) 27:1495–504. doi: 10.1681/ASN.2015040373
14. Neuzillet Y, Tillou X, Mathieu R, Long JA, Gigante M, Paparel P, et al. Renal cell carcinoma (RCC) in patients with end-stage renal disease exhibits many favourable clinical, pathologic, and outcome features compared with RCC in the general population. Eur Urol (2011) 60:366–73. doi: 10.1016/j.eururo.2011.02.035
15. Saly DL, Eswarappa MS, Street SE, and Deshpande P. Renal cell cancer and chronic kidney disease. Adv Chronic Kidney Dis (2021) 28460–468:e461. doi: 10.1053/j.ackd.2021.10.008
16. Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature (2013) 499:43–9. doi: 10.1038/nature12222
17. Cancer Genome Atlas Research, N, Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med (2016) 374:135–45. doi: 10.1056/NEJMoa1505917
18. Chudek J, Herbers J, Wilhelm M, Kenck C, Bugert P, Ritz E, et al. The genetics of renal tumors in end-stage renal failure differs from those occurring in the general population. J Am Soc Nephrol (1998) 9:1045–51. doi: 10.1681/ASN.V961045
19. Hughson MD, Schmidt L, Zbar B, Daugherty S, Meloni AM, Silva FG, et al. Renal cell carcinoma of end-stage renal disease: a histopathologic and molecular genetic study. J Am Soc Nephrol (1996) 7:2461–8. doi: 10.1681/ASN.V7112461
20. El-Zaatari ZM and Truong LD. Renal cell carcinoma in end-stage renal disease: A review and update. Biomedicines (2022) 10:657. doi: 10.3390/biomedicines10030657
21. R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing (2021) Available online at: https://www.R-project.org/.
22. Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell (2018) 173:400–416 e411. doi: 10.1016/j.cell.2018.02.052
23. Kapur P, Peña-Llopis S, Christie A, Zhrebker L, Pavía-Jiménez A, Rathmell WK, et al. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. Lancet Oncol (2013) 14:159–67. doi: 10.1016/S1470-2045(12)70584-3
24. Eule CJ, Hu J, Hedges D, Jani A, Pshak T, Manley BJ, et al. Clinical and genomic features of patients with renal cell carcinoma and advanced chronic kidney disease: analysis of a multi-institutional database. Cancers (Basel) (2024) 16:1920. doi: 10.3390/cancers16101920
25. Kumar R, Bonert M, Naqvi A, Zbuk K, and Kapoor A. SDH-deficient renal cell carcinoma - clinical, pathologic and genetic correlates: a case report. BMC Urol (2018) 18:109. doi: 10.1186/s12894-018-0422-8
26. Mcevoy CR, Koe L, Choong DY, Leong HS, Xu H, Karikios D, et al. SDH-deficient renal cell carcinoma associated with biallelic mutation in succinate dehydrogenase A: comprehensive genetic profiling and its relation to therapy response. NPJ Precis Oncol (2018) 2:9. doi: 10.1038/s41698-018-0053-2
27. Aggarwal RK, Luchtel RA, Machha V, Tischer A, Zou Y, Pradhan K, et al. Functional succinate dehydrogenase deficiency is a common adverse feature of clear cell renal cancer. Proc Natl Acad Sci U S A (2021) 118:e2106947118. doi: 10.1073/pnas.2106947118
28. Oh CJ, Kim MJ, Lee JM, Kim DH, Kim IY, Park S, et al. Inhibition of pyruvate dehydrogenase kinase 4 ameliorates kidney ischemia-reperfusion injury by reducing succinate accumulation during ischemia and preserving mitochondrial function during reperfusion. Kidney Int (2023) 104:724–39. doi: 10.1016/j.kint.2023.06.022
29. Wen Q, Liu H, Lou K, Zhang X, Chao W, Xin J, et al. Essential role of bromodomain proteins in renal cell carcinoma (Review). Mol Med Rep (2023) 28:139. doi: 10.3892/mmr.2023.13026
30. Zou Z, Huang B, Wu X, Zhang H, Qi J, Bradner J, et al. Brd4 maintains constitutively active NF-kappaB in cancer cells by binding to acetylated RelA. Oncogene (2014) 33:2395–404. doi: 10.1038/onc.2013.179
31. Morgado-Pascual JL, Rayego-Mateos S, Tejedor L, Suarez-Alvarez B, and Ruiz-Ortega M. Bromodomain and extraterminal proteins as novel epigenetic targets for renal diseases. Front Pharmacol (2019) 101315. doi: 10.3389/fphar.2019.01315
32. Gupta N, Matsumoto T, Hiratsuka K, Garcia Saiz E, Galichon P, Miyoshi T, et al. Modeling injury and repair in kidney organoids reveals that homologous recombination governs tubular intrinsic repair. Sci Transl Med (2022) 14:e:abj4772. doi: 10.1126/scitranslmed.abj4772
33. Xu LG, Li LY, and Shu HB. TRAF7 potentiates MEKK3-induced AP1 and CHOP activation and induces apoptosis. J Biol Chem (2004) 279:17278–82. doi: 10.1074/jbc.C400063200
34. Zotti T, Scudiero I, Vito P, and Stilo R. The emerging role of TRAF7 in tumor development. J Cell Physiol (2017) 232:1233–8. doi: 10.1002/jcp.v232.6
35. Yang Y, Vocke CD, Ricketts CJ, Wei D, Padilla-Nash HM, Lang M, et al. Genomic and metabolic characterization of a chromophobe renal cell carcinoma cell line model (UOK276). Genes Chromosomes Cancer (2017) 56:719–29. doi: 10.1002/gcc.v56.10
36. Ganner A, Gehrke C, Klein M, Thegtmeier L, Matulenski T, Wingendorf L, et al. VHL suppresses RAPTOR and inhibits mTORC1 signaling in clear cell renal cell carcinoma. Sci Rep (2021) 11:14827. doi: 10.1038/s41598-021-94132-5
37. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet (2008) 372:449–56. doi: 10.1016/S0140-6736(08)61039-9
38. Pilch NA, Bowman LJ, and Taber DJ. Immunosuppression trends in solid organ transplantation: The future of individualization, monitoring, and management. Pharmacotherapy (2021) 41:119–31. doi: 10.1002/phar.v41.1
Keywords: renal cell carcinoma, chronic kidney disease, end stage renal disease, kidney transplant, genomics
Citation: Eule CJ, Hu J, Davies KD, Jani A, Pshak T and Lam ET (2025) Clinical features and mutational frequency of renal cell carcinoma from patients undergoing kidney transplant evaluation. Front. Oncol. 15:1526545. doi: 10.3389/fonc.2025.1526545
Received: 11 November 2024; Accepted: 30 April 2025;
Published: 29 May 2025.
Edited by:
Umang Swami, The University of Utah, United StatesReviewed by:
Benjamin Maughan, The University of Utah, United StatesZheng Liu, Xiangtan Central Hospital, China
Copyright © 2025 Eule, Hu, Davies, Jani, Pshak and Lam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Corbin J. Eule, Q29yYmluLkV1bGVAQ1VBbnNjaHV0ei5lZHU=