 Nadja Ballin1*
Nadja Ballin1* Alexander Ott1Olga Seibel-Kelemen1
Alexander Ott1Olga Seibel-Kelemen1 Irina Bonzheim2Dominik Nann2Janina Beha3Stephan Spahn4Stephan Singer2
Irina Bonzheim2Dominik Nann2Janina Beha3Stephan Spahn4Stephan Singer2 Stephan Ossowski1Cristiana Roggia1Christopher Schroeder1Michael Bitzer3,4†Sorin Armeanu-Ebinger1†
Stephan Ossowski1Cristiana Roggia1Christopher Schroeder1Michael Bitzer3,4†Sorin Armeanu-Ebinger1†- 1Institute of Medical Genetics and Applied Genomics, University Hospital Tübingen, Tübingen, Germany
- 2Department of Pathology and Neuropathology, University Hospital Tübingen, Tübingen, Germany
- 3Center for Personalized Medicine, University Hospital Tübingen, Tübingen, Germany
- 4Department of Internal Medicine I, University Hospital Tübingen, Tübingen, Germany
FGFR2 fusions occur in up to 14% of patients with intrahepatic cholangiocarcinoma (iCCA) and have been considered as therapeutic target for FGFR inhibitors (FGFRi). However, response to targeted treatment may be limited due to the emergence of various resistance mechanisms. We report a case of recurrent iCCA in a 43-year-old patient with a FGFR2 fusion, who was treated with Lenvatinib. Next-generation sequencing (NGS) of tumor-normal DNA and tumor RNA under Lenvatinib treatment confirmed the FGFR2 fusion, however no further molecular resistance mutation was observed. After failure of FGFRi treatment (Lenvatinib and Infigratinib) ten months later, repeated NGS analysis revealed a new gain-of-function mutation in PIK3CA and a homozygous deletion of CDKN2A/B, potentially representing an acquired resistance mechanism. The emerging acquired resistance to FGFR inhibitor treatment has implications for subsequent treatment strategies.
Introduction
With the advent of precision medicine, the management of intrahepatic cholangiocellular carcinoma (iCCA) patients has been shaped in the last decade by molecular stratification of patients highlighting an enrichment of FGFR alterations in this tumor entity. Notably, oncogenic FGFR2 alterations constitute in up to 14% of iCCA cases the main tumor driver (1–6). Consequently, a variety of FGFR inhibitors (FGFRi) have been developed and achieved approval by the FDA as second line therapy in patients with unresectable locally advanced or metastatic iCCA with FGFR2 aberrations (7, 8). These are second generation FGFR inhibitors Pemigatinib and Infigratinib which are reversible competitive inhibitors of the ATP binding pocket of FGFR1-4 (9). Pemigatinib has also gained approval by the EMA. More recently, a third generation irreversible FGFR inhibitor, Futibatinib, has also been approved by the FDA and EMA for treatment of unresectable advanced or metastatic, chemotherapy refractory iCCA with FGFR2 rearrangements (10, 11).
Although 23 - 42% of FGFR2 fusion positive iCCA patients respond to FGFRi (7, 8, 11), secondary resistance and disease progression occur in most cases (9, 12). To date, different types of acquired resistance have been reported in iCCA patients after progression on FGFRi treatment. In up to 60% of cases resistance to FGFRi is mediated by secondary FGFR kinase domain mutations (13). Secondary resistance can also be mediated by acquired mutations with constitutive activation effect of FGFR2 itself and activation of alternate or downstream signaling pathways (MAPK or PIK3CA/MTOR). However, this type of acquired resistance has not been extensively studied in iCCA patients so far.
Here we present a patient with FGFR2 fusion positive iCCA that developed resistance to FGFRi treatment. Sequential NGS analysis of tumor and normal DNA and tumor RNA reveal potential causes of acquired resistance, which have not been described in patients with iCCA up to date. This case highlights the need for broad tumor characterization to identify potential resistance mechanisms and additional treatment options.
Case presentation
Clinical course
A 43-year-old patient presented at the department of internal medicine with hepatic, lymphogenic and pulmonary metastasized iCCA. He was first diagnosed with iCCA eleven months before and had already undergone treatment with gemcitabine/cisplatin (4 cycles, 3 months), followed by FOLFIRI (17 cycles, 8 months) (Figure 1). As part of his inclusion in the DKTK MASTER Trial (NCT05852522), the patient had received a first tumor DNA and RNA NGS analysis with identification of a FGFR2-SH3GLB1 fusion, likely presenting the main tumor driver. Consequently, he started treatment with Lenvatinib after progression on FOLFIRI. Clinical response of this patient to Lenvatinib treatment and in vitro characterization of the FGFR2-SH3GLB1 fusion have been reported in detail previously (14). After initial tumor regression, stable disease was observed after 5 months of treatment with Lenvatinib. At this point, a liver biopsy was taken. DNA and RNA were isolated and subjected to NGS analysis (panel sequencing of tumor/normal DNA and transcriptome sequencing of tumor RNA). Results were discussed in the local molecular tumor board with the recommendation of FGFR targeted therapy. Treatment with Lenvatinib was continued for 5 months until hepatic, lymphogenic and pulmonary progression was observed. Treatment was changed to FOLFOX for three months. Upon occurrence of new bone metastases, the patient was admitted to a phase II study with Infigratinib (BGJ398QED). After three months of treatment with Infigratinib stable disease was achieved with constant liver lesions, regression of pulmonary lesions and progression of bone metastases. Therefore, a bone biopsy was taken. NGS analysis of this specimen confirmed the presence of FGFR2-SH3GLB1 fusion and revealed new drivers including an activating PIK3CA mutation and a homozygous deletion of CDKN2A. Infigratinib treatment was continued for two months until progression of bone metastases. The patient died three months after termination of Infigratinib treatment.
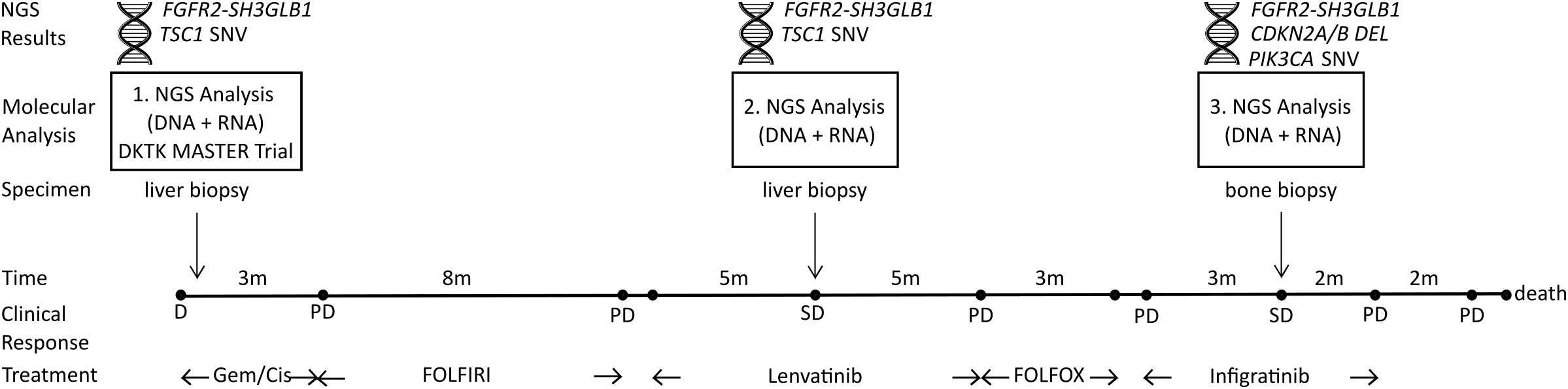
Figure 1. Clinical course and time points of three consecutive NGS analyses. First, DNA and RNA samples were obtained from a liver biopsy as part of DKTK MASTER Trial (NCT05852522) and NGS analysis identified a FGFR2-SH3GLB1 fusion and a TSC1 SNV as potential tumor drivers. The second NGS analysis was performed with DNA and RNA isolated from a liver biopsy during stable disease (SD) under Lenvatinib treatment. NGS results confirmed the FGFR2 fusion and TSC1 SNV. Eleven months later, a third NGS analysis was performed on DNA and RNA from a bone biopsy during SD under Infigratinib treatment. The NGS results confirmed the FGFR2 fusion and identified a new CDKN2A/B deletion together with an activating PIK3CA mutation. SNV, small/single nucleotide variant; DEL, homozygous deletion; PD, progressive disease; D, initial diagnosis; Gem/Cis, Gemcitabine and Cisplatin treatment.
Molecular analysis
At all-time points tested, the FGFR2-SH3GLB1 fusion was detected in tumor DNA and RNA (Supplementary Figure S1). The fusion protein results from a translocation event t(1;10)(p22.3;q26.13) with genomic breakpoints detected in tumor DNA in intron 17 of FGFR2 (ENST00000457416.7) and in intron 4 in SH3GLB1 (ENST00000616170.4). Transcriptome analysis further confirmed that the translocation leads to an in-frame fusion of both genes. The putative fusion protein contains the N-terminal part of FGFR2 with an intact protein kinase domain fused to the C-terminal part of SH3GLB1 with a Bar domain and variant SH3 domain. Both the genomic breakpoint in FGFR2 as well as the architecture of the FGFR2 fusion protein are typical of an oncogenic activation of FGFR2 via constitutive dimerization mediated by the BAR domain in the fusion partner SH3GLB1 (15). Therefore, FGFR2-SH3GLB1 fusion was determined as the main driver in this iCCA and FGFRi treatment was recommended.
In addition to the FGFR2-SH3GLB1 fusion, the first and second tumor-normal DNA analyses showed a likely oncogenic stop-gain variant in TSC1 together with a heterozygous deletion of the unaffected allele in the liver biopsy (Supplementary Figure S2). However, the variant was only present in a fraction of tumor cells and was therefore considered a less-important therapeutic target. This variant was absent in the third tumor DNA analysis from a bone biopsy.
The third NGS analysis of tumor-normal DNA from a progressive bone biopsy taken under Infigratinib treatment revealed a different somatic single nucleotide variant (SNV) profile with an oncogenic, activating PIK3CA c.3140A>T, p.(His1047Leu) variant. This PIK3CA variant represents a known somatic hotspot variant typically found in breast cancer patients (cancerhotspot.org, COSMIC). Although PIK3CA His1047Leu variant is less frequently observed than His1047Arg, it has been shown to constitutively activate PI3K which is downstream of FGFR signaling (16). Additionally, the non-mutated allele was lost by a copy-neutral loss of heterozygosity (LOH) event in the tumor cells (Supplementary Figure S2). Therefore, this variant likely represented an acquired oncogenic driver in this tumor acting downstream of the oncogenic FGFR2-SH3GLB1 fusion.
Comparison of the copy number variations (CNVs) between the two time points of the second and third NGS analysis performed at our institute showed an overall increase in number of CNVs (Supplementary Figure S3). The majority of these new CNVs were heterozygous deletions of complete or partial chromosome arms. On chromosome 9 a focal homozygous deletion of the CDKN2A/CDKN2B locus was detected in the bone biopsy, but not in the previous liver biopsies. This represented an additional acquired oncogenic event as the loss of CDKN2A leads to an activation of cell cycle (17).
Discussion
Knowledge of the type of therapy resistance is extremely important as it determines subsequent treatment options.
Regarding FGFR inhibitors secondary resistance is most frequently mediated by an acquisition of gatekeeper mutations that sterically block the binding of competitive inhibitors to the ATP binding pocket in the kinase domain (18, 19). Other kinase domain mutations that confer resistance to selective FGFR inhibitors affect the FGFR2 autoinhibitory domain. The residues N549, E565 and K641 form a molecular brake and, upon disruption via point mutations, FGFR2 becomes constitutively active (14, 20, 21). In this situation, switching to an irreversible FGFRi like Futibatinib or treatment with Lenvatinib as a multitargeted tyrosine kinase inhibitor may overcome therapy resistance (14, 22). In comparison to these direct mutations of FGFR, the activation of PIK3CA and loss of CDKN2A/B may induce resistance independent of FGFRi binding by activation of downstream signaling pathways.
Activation of a downstream or alternate signaling pathway may also confer resistance to targeted therapy/FGFR inhibition. Oncogenic FGFR2 fusions lead to a hyperactivation of FGFR2 signaling and consequently aberrant activation of RAS-RAF-ERK and PI3K-MTOR signaling, which promotes pro-tumorigenic processes such as proliferation, modulation of differentiation and survival (Figure 2). To escape the shutdown of these oncogenic signaling by FGFR inhibition tumors may gain activating alterations further downstream of the FGFR2 receptor. Yet, evidence for mutations activating downstream or alternate signaling pathways is still limited in iCCA patients.
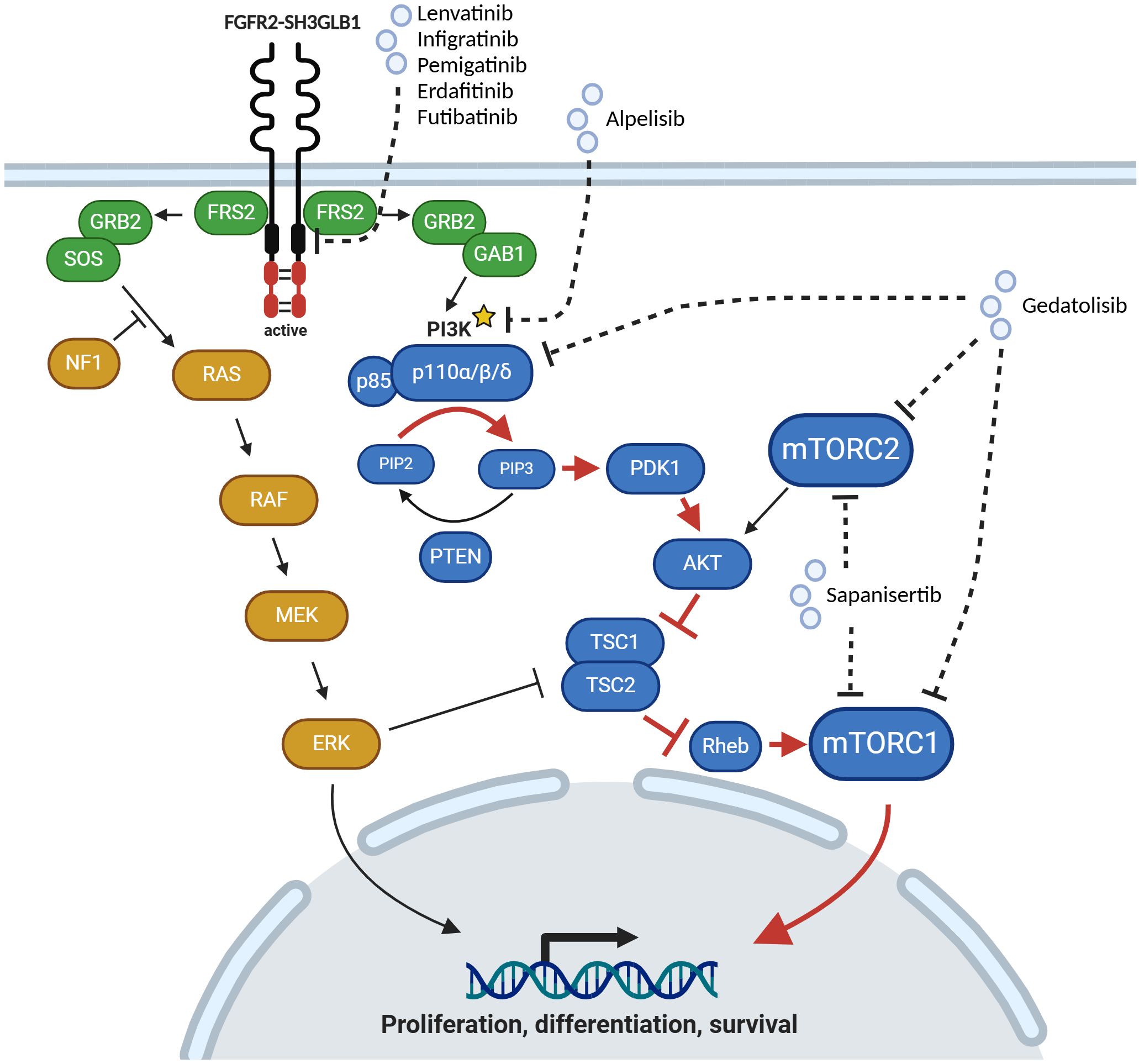
Figure 2. Activation of MAPK and PI3K/MTOR signaling pathways by oncogenic FGFR2 fusion protein. Upon inhibition of FGFR2 signaling by Lenvatinib and Infigratinib in this case of iCCA an activating PIK3CA (p110α) mutation (marked with an asterisk) was detected, which leads to an aberrant activation of downstream signaling pathway (red arrows). p110α can be targeted by Alpelisib. Alternatively, Gedatolisib, a pan-PI3K and dual mTOR inhibitor, or Sapanisertib, a dual mTOR inhibitor, could be used for targeted therapy. Created with BioRender.com.
So far, two independent studies of small cohorts of cholangiocarcinoma patients receiving FGFR targeted therapy reported the acquisition of MAPK or PI3K/MTOR pathway alterations. These alterations occurred in the majority of patients together with new FGFR mutations as secondary resistance (23, 24). Among these, three cases with FGFR2 fusions gained changes in PIK3CA unlikely to constitutively activate the kinase under FGFRi treatment together with alterations in other pathways or FGFR2 itself (23) and one patient harbored a PIK3CA H1047R mutation at baseline examination as an additional oncogenic driver (24). Targeting MAPK pathway co-alterations in KRAS and BRAF to enhance efficacy of FGFR inhibition revealed synergism with MEK inhibitors Trametinib and Binimetinib, and AKT inhibitors in vitro (23). Similarly, a case report of a FGFR2 altered iCCA patient showed secondary K/NRAS activation in addition to gatekeeper mutations in FGFR2 upon progression on treatment with Pemigatinib (25). However, a sole activation of PI3K/MTOR pathway after resistance to FGFRi treatment has not been shown.
Here, we present a case of FGFR2 fusion positive iCCA who acquired an oncogenic gain-of-function mutation in PIK3CA together with a homozygous deletion of CDKN2A/B upon treatment with Infigratinib and Lenvatinib. These alterations were detected by NGS analysis of a tumor biopsy taken during the treatment with Infigratinib. Consistent with the presence of a potential acquired resistance disease progression was observed two months later and targeting the resistance alterations as discussed in the molecular tumor board was not feasible.
The presence of new alterations in the bone metastasis could also be an expression of tumor heterogeneity. However, the bone metastasis progressed on FGFRi and was therefore accessible for analysis. In order to fully understand the causal relationship between the PIK3CA His1047Leu mutation and CDKN2A/B deletion in resistance to FGFRi treatment further functional studies in e.g. patient-derived organoids are needed.
Evidence for PIK3CA activation as a resistance mechanism to FGFR targeted therapy comes from FGFR driven urothelial cancer (26). Facchinetti et al. reported activation of MTOR signaling pathway in 58% (11/19) of FGFRi (Futibatinib or Erdafitinib) treated urothelial cancer patients with FGFR alterations. Two of these patients acquired an activating PIK3CA mutation at position E545K and E726K after progression on Erdafitinib treatment. Furthermore, they could show sensitivity of a patient derived xenograft with FGFR alteration and PIK3CA E545K to the combination treatment with Erdafitinib and Pictilisib (PI3K inhibitor). In addition, Arai et al. reported urothelial cell lines treated with Erdafitinib to gain resistance by PIK3CA mutation (27).
Moreover, activation of PI3K is an established mechanism of primary resistance to upstream HER2-targeted therapy in HER2+ breast cancer patients (28). In the SOLAR-1 study, HR+ HER2- advanced or metastatic breast cancer patients benefited from addition of PI3K inhibitor Alpelisib to Fulvestrant treatment if they harbored a PIK3CA mutation. Consequently, Alpelisib in combination with Fulvestrant gained FDA and EMA approval for this group of PIK3CA-mutated breast cancer patients. In FGFR2 rearranged iCCA, three of nine patients with PIK3CA mutation presented with concomitant inactivation of CDKN2A and had stable disease (one patient) as best response to Pemigatinib treatment (29).
Along with the gain of function variant in PIK3CA a homozygous deletion of CDKN2A and CDKN2B was detected by NGS analysis as an additional potentially acquired oncogenic event in our case of FGFR driven iCCA. FGFR signaling and CDKN2A/B both drive cell cycle progression via inactivation of p27 (30, 31), which supports our observation of emerging CDKN2A/B loss upon FGFRi treatment as potential resistance mechanism. Furthermore, co-occurrence of inactivation of CDKN2A/B with FGFR2 rearrangement significantly reduced median PFS in iCCA patients treated with Pemigatinib (29) and emphasizes the need for combination therapy. Interestingly, deletion of CDKN2A has been reported as concurrent resistance mechanism in two NSCLC patients treated with Abivertinib, a third generation tyrosine kinase/EGFR inhibitor (32). In these two cases CDKN2A deletion was detected together with acquired EGFR amplification. PI3K/mTOR pathway activation and cell cycle dysregulation jointly drive resistance (33). In breast cancer cells a combinatorial drug screening found that CDK4/6 inhibition exhibits the most significant synergism with PI3K inhibitors (34).
Dual oncogenic activation of signaling pathways argues for application of combination therapy. In this case, with three oncogenic drivers, in theory several combinations might be interesting.
Focusing on the FGFR fusion and activation of mTOR signaling by the PIK3CA alteration, combination of Infigratinib with an PIK3CA inhibitor (e.g. Alpelisib) would have been an option. This combination showed a manageable safety profile in a first in-human combination trial (35), however frequent dose interruptions or reductions were necessary and potentially limit their long-term application. A first assessment of synergistic effects in patients with solid tumors harboring PIK3CA and/or FGFR1-3 alterations failed to show any link to this molecular subtype. Yet, in respect to FGFR2 alterations this study was restricted to SNVs/Indels, which may already confer resistance to Infigratinib.
Interestingly, in vitro combination therapy of a FGFR2 fusion positive cell line with FGFR2 E565A molecular brake mutation with Infigratinib and dual mTOR inhibitor Sapanisertib (CB-228, formerly TAK-228/MLN0128) was able to reverse resistance to Infigratinib (21). Moreover, Facchinetti et al. reported one iCCA patient with a FGFR2 fusion together with PIK3CA H1047R activating mutation with progression on Pemigatinib treatment, who, however, benefited from second line treatment with Everolimus. Administration of Everolimus also showed anti-tumor activity in two phase II studies including iCCA patients (36, 37) and in a case report of PIK3CA E545G mutated iCCA patient (38).
Hence, it remains possible that mTOR inhibition or more specific PIK3CA inhibition would have been able to resensitize the tumor of the presented patient to Infigratinib treatment.
Considering the concomitant occurrence of an inactivation of CDKN2A, combination treatment with a CDK4/6 inhibitor (e.g. Palbociclib or Ribociclib) may also be relevant. From a mechanistic perspective, triple combination of a CDK4/6 inhibitor with an FGFR inhibitor and a PI3K/MTOR inhibitor might be ideal. However, clinical applicability of such a combination still has to be evaluated. So far, a phase Ib trial of Fulvestrant with Palbociclib and Erdafitinib in FGFR-amplified/ER+/HER2-negative metastatic breast cancer has been conducted and has reported on-target toxicities for Erdafitinib leading to treatment discontinuation (39). Combined inhibition of mTOR and CDK4/6 has been positively evaluated in patients with advanced breast cancer showing acceptable safety profiles. In this context, in a phase I/II trial of patients with ER+/HER2-negative metastatic breast cancer who had progressed on CDK4/6 targeted therapy the triplet combination of Ribociclib, Everolimus and Exemestane showed an acceptable safety profile (40). In addition, combination of PI3K/mTOR inhibitor Gedatolisib plus Palbociclib and endocrine therapy in women with HR+/HER2-negative advanced breast cancer was well tolerated with an acceptable safety profile (41). In the context of iCCA, pre-clinical studies have reported synergistic effects of combined CDK4/6 and mTOR inhibition (42).
To sum up, the presented case highlights the importance of sequential broad molecular tumor characterization in order to capture the full spectrum of potential resistance mechanisms to targeted therapy. Here, oncogenic changes in PIK3CA and CDKN2A/B were identified after progression on Infigratinib/Lenvatinib treatment. These are in principle targetable alterations that could be addressed by different combination therapies. Manageable safety profiles of drug combinations with maintenance of good quality of life in such complex molecularly altered tumors still require further assessment.
Methods
Patient
The patient provided written informed consent for blood collection, tissue biopsies, NGS analysis, treatment and publication of this report.
NGS analysis was performed in a diagnostic setting and already published in detail under (43–46).
DNA and RNA isolation
Genomic DNA was isolated from normal tissue (peripheral blood) using Qiagen FlexiGene (Qiagen, Hilden, Germany). Tumor DNA and RNA were extracted from macrodissected 5μm paraffin sections using the Maxwell RSC DNA FFPE Kit and Maxwell RSC RNA FFPE Kit and the Maxwell RSC Instrument (Promega, Madison, WI, USA) according to the manufacturer’s instructions. DNA concentrations were determined using the Qubit™ DNA BR kit (Thermo Fisher Scientific, Waltham, MA, USA) and DNA integrity was analyzed using the Agilent Genomic DNA Screen Tape System (Agilent, Santa Clara, CA, USA).
Next generation sequencing (NGS)
For each DNA sample 200 ng genomic DNA was sheared to 150-200 bp length using a Covaris E220 Ultrasonicator (Covaris, Woburn, MA, USA). Fragmented DNA was end repaired, A-tailed, adapter ligated and amplified with Agilent’s SureSelect XT Low Input Target Enrichment System for Illumina Paired-End Multiplexed Sequencing Library kit following the manufacturer’s instructions (Agilent Technologies, Santa Clara, CA, USA). Target enrichment was performed using a custom-designed hybrid capture panel covering 708 cancer-related genes, 7 promoter regions and selected fusions as well as microsatellite loci (Agilent Technologies, Santa Clara, CA, USA). The libraries were sequenced on a NovaSeq6000 sequencing platform (Illumina, San Diego, CA, USA) in paired-end mode as specified by the manufacturer.
Total tumor RNA was prepared for sequencing using New England Biolabs NEBNext Ultra II Directional RNA rRNA Depletion FFPE Kit according to the manufacturer’s instructions. Libraries were sequenced on a NovaSeq6000 sequencing platform (Illumina, San Diego, CA, USA) in paired-end mode.
Bioinformatic data analysis and variant interpretation
An in-house bioinformatics pipeline megSAP (https://github.com/imgag/megSAP) was used to analyze NGS data. Both tumor and normal samples were mapped with bwa mem2 (47), ABRA2 (48) for INDEL realignment and SeqPurge (49) for adapter trimming followed by variant calling with Freebayes (50) for the germline sample and Strelka2 (51) for the differential analysis, respectively. Variant annotation was performed using variant effect predictor (VEP) and various damage scores. Germline and somatic copy number variation (CNV) calling was performed with ClinCNV (52).
Our decision support application GSvar (https://github.com/imgag/ngs-bits) was used for variant interpretation and generation of final reports. Somatic variants were classified according to Variant Interpretation for Cancer Consortium (VICC) (53).
Transcriptome analysis
RNA data were demultiplexed using Illumina bcl2fastQ and then analyzed by our in-house bioinformatics pipeline megSAP. Normalized gene expression values FPKM (fragments per kilobase per million) and TPM (transcripts per kilobase per million) were calculated with Subreads Software (54). Fusions were identified with STAR-Fusion (55) and ARRIBA (56).
Data availability statement
Requests to access these datasets should be directed to NB, bmFkamEuYmFsbGluQG1lZC51bmktdHVlYmluZ2VuLmRl.
Ethics statement
The requirement of ethical approval was waived by BroadConsent V09, University Hospital Tuebingen, Germany for the studies involving humans. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
NB: Data curation, Formal Analysis, Visualization, Writing – original draft, Writing – review & editing. AO: Formal Analysis, Writing – review & editing. OS-K: Methodology, Writing – review & editing. IB: Resources, Writing – review & editing. DN: Investigation, Writing – review & editing. JB: Investigation, Writing – review & editing. SSp: Investigation, Writing – review & editing. SSi: Investigation, Writing – review & editing. SO: Formal Analysis, Writing – review & editing. CR: Investigation, Writing – review & editing. CS: Conceptualization, Formal Analysis, Writing – original draft, Writing – review & editing. MB: Conceptualization, Investigation, Writing – review & editing. SA-E: Conceptualization, Formal Analysis, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1527484/full#supplementary-material
References
1. Graham RP, Barr Fritcher EG, Pestova E, Schulz J, Sitailo LA, Vasmatzis G, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol. (2014) 45:1630–8. doi: 10.1016/j.humpath.2014.03.014
2. Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology. (2014) 59:1427–34. doi: 10.1002/hep.26890
3. Lowery MA, Ptashkin R, Jordan E, Berger MF, Zehir A, Capanu M, et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin Cancer Res. (2018) 24:4154–61. doi: 10.1158/1078-0432.CCR-18-0078
4. Ross JS, Wang K, Gay L, Al-Rohil R, Rand JV, Jones DM, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist. (2014) 19:235–42. doi: 10.1634/theoncologist.2013-0352
5. Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. (2017) 18:2780–94. doi: 10.1016/j.celrep.2017.02.033
6. Kendre G, Murugesan K, Brummer T, Segatto O, Saborowski A, Vogel A. Charting co-mutation patterns associated with actionable drivers in intrahepatic cholangiocarcinoma. J Hepatol. (2023) 78:614–26. doi: 10.1016/j.jhep.2022.11.030
7. Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. (2020) 21:671–84. doi: 10.1016/S1470-2045(20)30109-1
8. Javle M, Roychowdhury S, Kelley RK, Sadeghi S, Macarulla T, Weiss KH, et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol Hepatol. (2021) 6:803–15. doi: 10.1016/S2468-1253(21)00196-5
9. Lamarca A, Ostios L, McNamara MG, Garzon C, Gleeson JP, Edeline J, et al. Resistance mechanism to fibroblast growth factor receptor (FGFR) inhibitors in cholangiocarcinoma. Cancer Treat Rev. (2023) 121:102627. doi: 10.1016/j.ctrv.2023.102627
10. Bahleda R, Meric-Bernstam F, Goyal L, Tran B, He Y, Yamamiya I, et al. Phase I, first-in-human study of futibatinib, a highly selective, irreversible FGFR1–4 inhibitor in patients with advanced solid tumors. Ann Oncol. (2020) 31:1405–12. doi: 10.1016/j.annonc.2020.06.018
11. Goyal L, Meric-Bernstam F, Hollebecque A, Valle JW, Morizane C, Karasic TB, et al. Futibatinib for FGFR2-rearranged intrahepatic cholangiocarcinoma. N Engl J Med. (2023) 388:228–39. doi: 10.1056/NEJMoa2206834
12. Neureiter D, Ellinghaus P, Ocker M. FGFR inhibitor resistance in cholangiocarcinoma: current understanding and future directions. Expert Opin Pharmacother. (2023) 24:1833–7. doi: 10.1080/14656566.2023.2259802
13. Wu Q, Ellis H, Siravegna G, Michel AG, Norden BL, Fece de la Cruz F, et al. Landscape of clinical resistance mechanisms to FGFR inhibitors in FGFR2-altered cholangiocarcinoma. Clin Cancer Res. (2024) 30:198–208. doi: 10.1158/1078-0432.CCR-23-1317
14. Spahn S, Kleinhenz F, Shevchenko E, Stahl A, Rasen Y, Geisler C, et al. The molecular interaction pattern of lenvatinib enables inhibition of wild-type or kinase-mutated FGFR2-driven cholangiocarcinoma. Nat Commun. (2024) 15:1287. doi: 10.1038/s41467-024-45247-6
15. Neumann O, Burn TC, Allgäuer M, Ball M, Kirchner M, Albrecht T, et al. Genomic architecture of FGFR2 fusions in cholangiocarcinoma and its implication for molecular testing. Br J Cancer. (2022) 127:1540–9. doi: 10.1038/s41416-022-01908-1
16. Gymnopoulos M, Elsliger M-A, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci. (2007) 104:5569–74. doi: 10.1073/pnas.0701005104
17. Hélias-Rodzewicz Z, Lourenco N, Bakari M, Capron C, Emile JF. CDKN2A depletion causes aneuploidy and enhances cell proliferation in non-immortalized normal human cells. Cancer Invest. (2018) 36:338–48. doi: 10.1080/07357907.2018.1491588
18. Goyal L, DiToro D, Facchinetti F, Martin EE, Peng P, Baiev I, et al. A model for decoding resistance in precision oncology: acquired resistance to FGFR inhibitors in cholangiocarcinoma. Ann Oncol. (2024) 36(4):426–43. doi: 10.1016/j.annonc.2024.12.011
19. Katoh M, Loriot Y, Brandi G, Tavolari S, Wainberg ZA, Katoh M. FGFR-targeted therapeutics: clinical activity, mechanisms of resistance and new directions. Nat Rev Clin Oncol. (2024) 21:312–29. doi: 10.1038/s41571-024-00869-z
20. Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, et al. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell. (2007) 27:717–30. doi: 10.1016/j.molcel.2007.06.028
21. Krook MA, Lenyo A, Wilberding M, Barker H, Dantuono M, Bailey KM, et al. Efficacy of FGFR inhibitors and combination therapies for acquired resistance in FGFR2-fusion cholangiocarcinoma. Mol Cancer Ther. (2020) 19:847–57. doi: 10.1158/1535-7163.MCT-19-0631
22. Javle M, King G, Spencer K, Borad MJ. Futibatinib, an irreversible FGFR1-4 inhibitor for the treatment of FGFR-aberrant tumors. Oncologist. (2023) 28:928–43. doi: 10.1093/oncolo/oyad149
23. DiPeri TP, Zhao M, Evans KW, Varadarajan K, Moss T, Scott S, et al. Convergent MAPK pathway alterations mediate acquired resistance to FGFR inhibitors in FGFR2 fusion-positive cholangiocarcinoma. J Hepatol. (2024) 80:322–34. doi: 10.1016/j.jhep.2023.10.041
24. Facchinetti F, Loriot Y, Braye F, Vasseur D, Bahleda R, Bigot L, et al. Understanding and overcoming resistance to selective FGFR inhibitors across FGFR2-driven Malignancies. Clin Cancer Res. (2024) 30(21):4943–56. doi: 10.1158/1078-0432.Ccr-24-1834
25. Lim M, Lynch PT, Bai X, Hsiehchen D. Oncogenic RAS drives resistance to pemigatinib in cholangiocarcinoma harboring a FGFR2 delins disrupting ligand binding. JCO Precis Oncol. (2023) 7:e2200340. doi: 10.1200/PO.22.00340
26. Facchinetti F, Hollebecque A, Braye F, Vasseur D, Pradat Y, Bahleda R, et al. Resistance to selective FGFR inhibitors in FGFR-driven urothelial cancer. Cancer Discovery. (2023) 13:1998–2011. doi: 10.1158/2159-8290.CD-22-1441
27. Arai S, Sawada T, Ohtsu A, Onose M, Maeno Y, Sekine Y, et al. Abstract A014: PIK3CA mutation might confer resistance to an FGFR inhibitor, erdafitinib, in urothelial cancer cells. Clin Cancer Res. (2024) 30:A014–4. doi: 10.1158/1557-3265.BLADDER24-A014
28. Rasti AR, Guimaraes-Young A, Datko F, Borges VF, Aisner DL, Shagisultanova E. PIK3CA mutations drive therapeutic resistance in human epidermal growth factor receptor 2-positive breast cancer. JCO Precis Oncol. (2022) 6:e2100370. doi: 10.1200/po.21.00370
29. Silverman IM, Hollebecque A, Friboulet L, Owens S, Newton RC, Zhen H, et al. Clinicogenomic analysis of FGFR2-rearranged cholangiocarcinoma identifies correlates of response and mechanisms of resistance to pemigatinib. Cancer Discovery. (2021) 11:326–39. doi: 10.1158/2159-8290.CD-20-0766
30. Zhang Y, Lin Y, Bowles C, Wang F. Direct cell cycle regulation by the fibroblast growth factor receptor (FGFR) kinase through phosphorylation-dependent release of Cks1 from FGFR substrate 2. J Biol Chem. (2004) 279:55348–54. doi: 10.1074/jbc.M409230200
31. Matsuda Y, Ichida T, Genda T, Yamagiwa S, Aoyagi Y, Asakura H. Loss of p16 contributes to p27 sequestration by cyclin D(1)-cyclin-dependent kinase 4 complexes and poor prognosis in hepatocellular carcinoma. Clin Cancer Res. (2003) 9:3389–96. Available online at: https://aacrjournals.org/clincancerres/article/9/9/3389/204531/Loss-of-p16-Contributes-to-p27-Sequestration-by.
32. Zhang YC, Chen ZH, Zhang XC, Xu CR, Yan HH, Xie Z, et al. Analysis of resistance mechanisms to abivertinib, a third-generation EGFR tyrosine kinase inhibitor, in patients with EGFR T790M-positive non-small cell lung cancer from a phase I trial. EBioMedicine. (2019) 43:180–7. doi: 10.1016/j.ebiom.2019.04.030
33. Yip HYK, Shin SY, Chee A, Ang CS, Rossello FJ, Wong LH, et al. Integrative modeling uncovers p21-driven drug resistance and prioritizes therapies for PIK3CA-mutant breast cancer. NPJ Precis Oncol. (2024) 8:20. doi: 10.1038/s41698-024-00496-y
34. Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. (2014) 26:136–49. doi: 10.1016/j.ccr.2014.05.020
35. Hyman DM, Tran B, Paz-Ares L, Machiels JP, Schellens JH, Bedard PL, et al. Combined PIK3CA and FGFR inhibition with alpelisib and infigratinib in patients with PIK3CA-mutant solid tumors, with or without FGFR alterations. JCO Precis Oncol. (2019) 3:1–13. doi: 10.1200/PO.19.00221
36. Buzzoni R, Pusceddu S, Bajetta E, De Braud F, Platania M, Iannacone C, et al. Activity and safety of RAD001 (everolimus) in patients affected by biliary tract cancer progressing after prior chemotherapy: a phase II ITMO study. Ann Oncol. (2014) 25:1597–603. doi: 10.1093/annonc/mdu175
37. Kim ST, Lee J, Park SH, Park JO, Park YS, Kang WK, et al. Prospective phase II trial of everolimus in PIK3CA amplification/mutation and/or PTEN loss patients with advanced solid tumors refractory to standard therapy. BMC Cancer. (2017) 17:211. doi: 10.1186/s12885-017-3196-6
38. Bian JL, Wang MM, Tong EJ, Sun J, Li M, Miao ZB, et al. Benefit of everolimus in treatment of an intrahepatic cholangiocarcinoma patient with a PIK3CA mutation. World J Gastroenterol. (2017) 23:4311–6. doi: 10.3748/wjg.v23.i23.4311
39. Mayer IA, Haley BB, Abramson VG, Brufsky A, Rexer B, Stringer-Reasor E, et al. Abstract PD1-03: A phase Ib trial of fulvestrant + CDK4/6 inhibitor (CDK4/6i) palbociclib + pan-FGFR tyrosine kinase inhibitor (TKI) erdafitinib in FGFR-amplified/ER+/HER2-negative metastatic breast cancer (MBC). Cancer Res. (2021) 81:PD1–03-PD1-03. doi: 10.1158/1538-7445.SABCS20-PD1-03
40. Bardia A, Hurvitz SA, DeMichele A, Clark AS, Zelnak A, Yardley DA, et al. Phase I/II trial of exemestane, ribociclib, and everolimus in women with HR(+)/HER2(-) advanced breast cancer after progression on CDK4/6 inhibitors (TRINITI-1). Clin Cancer Res. (2021) 27:4177–85. doi: 10.1158/1078-0432.CCR-20-2114
41. Layman RM, Han HS, Rugo HS, Stringer-Reasor EM, Specht JM, Dees EC, et al. Gedatolisib in combination with palbociclib and endocrine therapy in women with hormone receptor-positive, HER2-negative advanced breast cancer: results from the dose expansion groups of an open-label, phase 1b study. Lancet Oncol. (2024) 25:474–87. doi: 10.1016/S1470-2045(24)00034-2
42. Song X, Liu X, Wang H, Wang J, Qiao Y, Cigliano A, et al. Combined CDK4/6 and pan-mTOR inhibition is synergistic against intrahepatic cholangiocarcinoma. Clin Cancer Res. (2019) 25:403–13. doi: 10.1158/1078-0432.CCR-18-0284
43. Renovanz M, Kurz SC, Rieger J, Walter B, Becker H, Hille H, et al. Clinical outcome of biomarker-guided therapies in adult patients with tumors of the nervous system. Neurooncol Adv. (2023) 5:vdad012. doi: 10.1093/noajnl/vdad012
44. Eckardt J, Schroeder C, Martus P, Armeanu-Ebinger S, Kelemen O, Gschwind A, et al. TMB and BRAF mutation status are independent predictive factors in high-risk melanoma patients with adjuvant anti-PD-1 therapy. J Cancer Res Clin Oncol. (2023) 149:833–40. doi: 10.1007/s00432-022-03939-w
45. Forschner A, Sinnberg T, Mroz G, Schroeder C, Reinert CP, Gatidis S, et al. Case report: combined CDK4/6 and MEK inhibition in refractory CDKN2A and NRAS mutant melanoma. Front Oncol. (2021) 11:643156. doi: 10.3389/fonc.2021.643156
46. Bitzer M, Ostermann L, Horger M, Biskup S, Schulze M, Ruhm K, et al. Next-generation sequencing of advanced GI tumors reveals individual treatment options. JCO Precis Oncol. (2020) 4:258–71. doi: 10.1200/PO.19.00359
47. Vasimuddin M, Misra S, Li H, Aluru S, editors. "Efficient architecture-aware acceleration of BWA-MEM for multicore systems", In: 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). Rio de Janeiro, Brazil: IEEE (2019).
48. Mose LE, Perou CM, Parker JS. Improved indel detection in DNA and RNA via realignment with ABRA2. Bioinformatics. (2019) 35:2966–73. doi: 10.1093/bioinformatics/btz033
49. Sturm M, Schroeder C, Bauer P. SeqPurge: highly-sensitive adapter trimming for paired-end NGS data. BMC Bioinf. (2016) 17:208. doi: 10.1186/s12859-016-1069-7
50. Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv:12073907v2. (2012). Available online at: https://galaxyproject.org/media/freebayes.pdf.
51. Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods. (2018) 15:591–4. doi: 10.1038/s41592-018-0051-x
52. Demidov G, Sturm M, Ossowski S. ClinCNV: multi-sample germline CNV detection in NGS data. bioRxiv. (2022) 2022.06.10.495642. doi: 10.1101/2022.06.10.495642
53. Horak P, Griffith M, Danos AM, Pitel BA, Madhavan S, Liu X, et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genet Med. (2022) 24:986–98. doi: 10.1016/j.gim.2022.01.001
54. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. (2014) 30:923–30. doi: 10.1093/bioinformatics/btt656
55. Haas BJ, Dobin A, Stransky N, Li B, Yang X, Tickle T, et al. STAR-fusion: fast and accurate fusion transcript detection from RNA-seq. bioRxiv. (2017) p:120295. doi: 10.1101/120295
Keywords: FGFR inhibition, secondary resistance, PIK3CA, intrahepatic cholangiocarcinoma (iCCA), molecular tumor board, CDKN2A/B
Citation: Ballin N, Ott A, Seibel-Kelemen O, Bonzheim I, Nann D, Beha J, Spahn S, Singer S, Ossowski S, Roggia C, Schroeder C, Bitzer M and Armeanu-Ebinger S (2025) Case Report: FGFR2 inhibitor resistance via PIK3CA and CDKN2A/B in an intrahepatic cholangiocarcinoma patient with FGFR2-SH3GLB1 fusion. Front. Oncol. 15:1527484. doi: 10.3389/fonc.2025.1527484
Received: 13 November 2024; Accepted: 20 March 2025;
Published: 07 April 2025.
Edited by:
Wenzhuo Zhuang, Soochow University, ChinaReviewed by:
Yun-Li Yu, Second Affiliated Hospital of Soochow University, ChinaXinjun Lu, Sun Yat-sen University, China
Copyright © 2025 Ballin, Ott, Seibel-Kelemen, Bonzheim, Nann, Beha, Spahn, Singer, Ossowski, Roggia, Schroeder, Bitzer and Armeanu-Ebinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nadja Ballin, bmFkamEuYmFsbGluQG1lZC51bmktdHVlYmluZ2VuLmRl
†These authors have contributed equally to this work