 Yifan Wang1,2,3†
Yifan Wang1,2,3† Xinyu Dong2,3,4†
Xinyu Dong2,3,4† Shandong Tao1,2,3
Shandong Tao1,2,3 Qiuni Chen1,2,3
Qiuni Chen1,2,3 Yue Chen1,2,3
Yue Chen1,2,3 Lijuan Zhang1,2,3
Lijuan Zhang1,2,3 Yuye Shi1,2,3
Yuye Shi1,2,3 Zhengmei He1,2,3
Zhengmei He1,2,3 Liang Yu1,2,3,4*
Liang Yu1,2,3,4* Chunling Wang1,2,3,4*
Chunling Wang1,2,3,4*- 1Department of Hematology, The Affiliated Huaian No.1 People’s Hospital of Nanjing Medical University, Huai’an, China
- 2Northern Jiangsu Institute of Clinical Medicine, Nanjing Medical University, Huai’an, China
- 3Department of Hematology, Huaian Key Laboratory of Autoimmune Diseases, Huai’an, China
- 4Department of Hematology, The Huaian Clinical College of Xuzhou Medical University, Huai’an, China
The insulin-like growth factor (IGF) signaling system comprises functionally specific ligands (IGF-I and IGF-II), receptor (IR), and binding proteins (IGFBP). IGFs are activated by binding to their receptor, IGF-IR, which is a tyrosine kinase receptor. This activation initiates signaling cascades such as PI3K/Akt and MAPK/ErK pathways, which are essential for cell proliferation, differentiation, and survival. Growing evidence links the IGF system to various hematological disorders, yet comprehensive reviews on its role in Myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) are limited. To advance understanding in this area, we aim to summarize the emerging evidence on the involvement of IGF signaling in the pathogenesis of MDS and AML. Specifically, we highlight how dysregulation of IGF-I, IGF-IR, and IGFBPs contributes to disease progression, encompassing clonal hematopoietic abnormalities, ineffective hematopoiesis in MDS, and the development of AML. The potential therapeutic implications of targeting the IGF signaling pathway, including the role of NVP-AEW541 and NVP-ADW742 effectively suppressing AML cell proliferation and enhancing chemotherapy sensitivity, are also explored. By integrating current findings, this review provides novel insights into the mechanistic role of IGF signaling in MDS and AML and its therapeutic implications, thereby guiding future research and potential clinical applications. Given the challenges, such as pathway redundancy and therapy resistance, further investigations are necessary to validate IGF-targeted therapies and optimize their clinical utility in hematologic malignancies.
1 Introduction
IGF system is constituted by three ligands (IGF-I, IGF-II, and insulin), four receptors (IGF type I receptor [IGF-IR]), type II receptor [IGF-IIR], insulin receptor [IR], and a heterodimeric receptor between IGF-IR and IR), and six high-affinity IGF-binding proteins (1–4) (Figure 1). It regulates numerous physiological processes, including cell survival, proliferation, differentiation, migration, and short-term effects like glucose uptake and metabolism (5–7). Alterations in IGF axis expression or function can lead to various pathological conditions (8, 9).
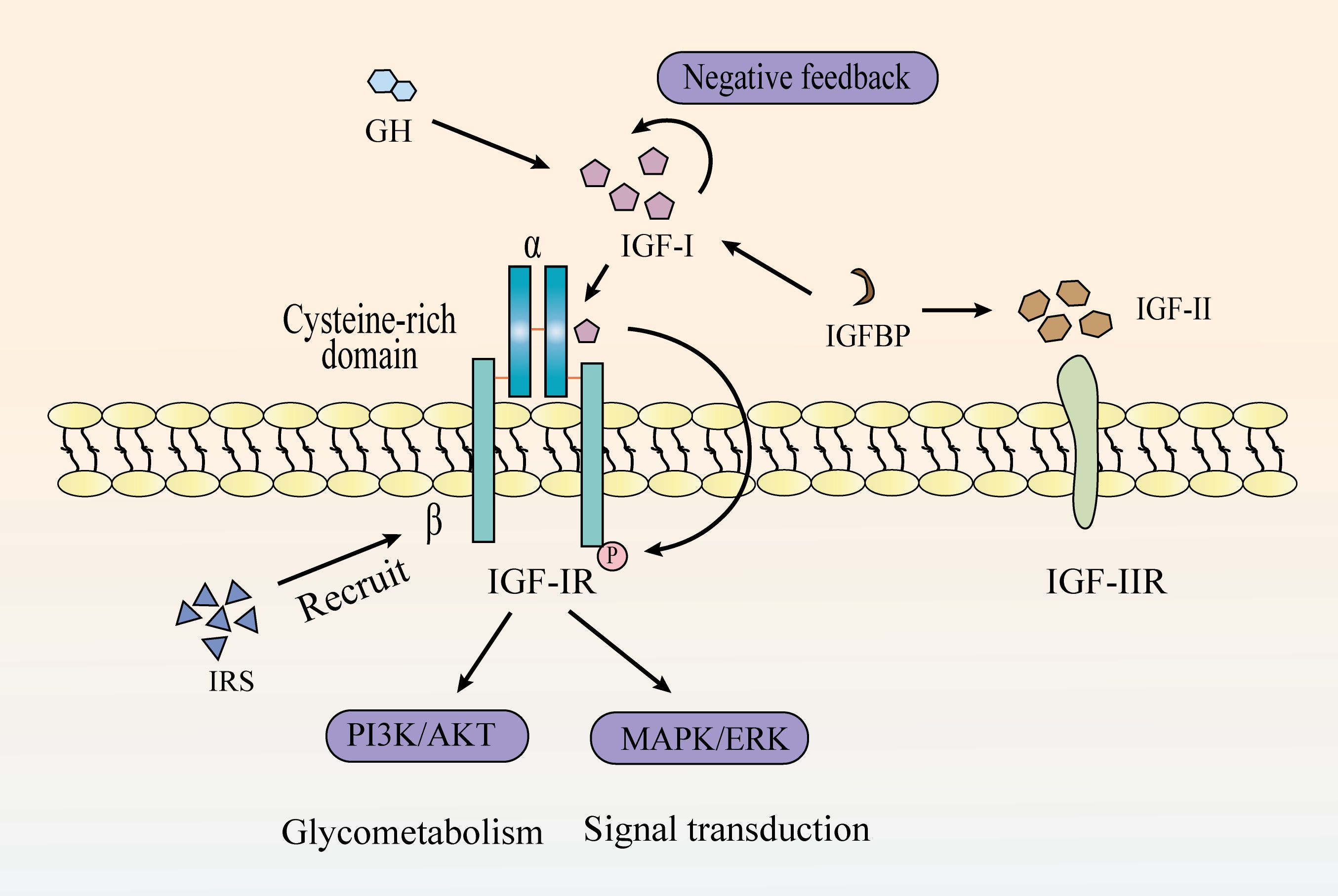
Figure 1. The mechanistic pathway of the IGF signaling system.
Dysregulation of the IGF axis is well documented in a variety of solid tumors, contributing to tumor progression, metastasis, and resistance to therapy (10, 11). However, growing evidence suggests that the IGF axis also plays a crucial role in hematologic malignancies, including myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). For instance, increased expression of IGF-I and IGF-IR has been observed in AML patient samples and cell lines, and correlates with enhanced PI3K/AKT pathway activation and poor clinical outcomes (12, 13). In MDS, elevated serum IGF-I levels and IGF-IR expression in CD34+ progenitor cells have been reported, suggesting a role in abnormal hematopoietic stem cell survival and clonal evolution (14). Moreover, bone marrow stromal cells in MDS and AML patients have been shown to secrete IGF ligands, further supporting a paracrine loop that promotes leukemic cell survival and chemoresistance (15).
MDS and AML are clonal disorders of the hematopoietic system with overlapping pathogenesis. MDS is characterized by ineffective hematopoiesis and multilineage cytopenias (16), while AML involves the rapid expansion of abnormal myeloid precursor cells that suppress normal hematopoiesis (17). MDS can progress to AML in 30%-40% of cases (18–20). The progression from MDS to AML is a multistep process characterized by the accumulation of genetic mutations and clonal selection (21, 22). Furthermore, alterations in the bone marrow microenvironment, such as elevated pro-inflammatory cytokines and dysfunctional stromal cells, also contribute to the transformation of MDS to AML (23, 24). Encouragingly, several IGF pathway–related inhibitors, such as NVP-AEW541, have shown promising preclinical activity and are being explored in early-phase clinical trials for AML (25).
Given the growing recognition of IGF signaling in hematologic malignancies, particularly in myeloid disorders such as MDS and AML, a focused review of its mechanistic relevance and therapeutic potential is timely. Although increasing attention has been paid to IGF-related pathways in these diseases, much of the current understanding is extrapolated from studies in solid tumors, and the specific roles of IGF signaling in the pathogenesis of myeloid neoplasms remain to be fully elucidated (26, 27). Therefore, this review aims to comprehensively examine the involvement of the IGF axis in MDS and AML, highlight its crosstalk with other oncogenic pathways, and summarize the current progress and challenges in targeting IGF signaling as a therapeutic strategy.
2 Regulation of IGF signaling system
2.1 IGF-I and IGF-IR
Insulin-like growth factor I (IGF-I) is a peptide hormone structurally similar to insulin but functionally distinct. Unlike insulin, which primarily regulates metabolic processes and maintains glucose homeostasis, IGF-I plays a central role in promoting cell growth and development, regulating proliferation, inhibiting apoptosis, and facilitating tissue repair (28). IGF-I mediates its biological effects mainly through autocrine and paracrine signaling. In the autocrine mode, IGF-I acts on the same cell that produces it; in the paracrine mode, it is secreted by neighboring stromal or niche cells and acts on adjacent target cells. These mechanisms are particularly critical in skeletal development and hematologic malignancies, where IGF-I contributes to cell proliferation, survival, and differentiation (29, 30). IGF-I signals through the IGF-IR receptor, activating intracellular pathways like PI3K/Akt and MAPK/Erk. These pathways are essential for cell proliferation, differentiation, and apoptosis regulation (31, 32). Upon IGF-I binding, IGF-IR undergoes conformational changes, leading to autophosphorylation of the β-subunits and recruitment of downstream signaling molecules, such as insulin receptor substrate (IRS) proteins. This activation triggers key oncogenic pathways, including the PI3K/Akt and MAPK/Erk pathways. Many researches showed that IGF-IR is highly expressed in a variety of tumor cells, and its expression level is closely related to the degree of malignancy and prognosis of a wide range of tumors (33). Given the oncogenic potential of IGF-IR, targeted therapeutic strategies have been explored, including Picropodophyllin (PPP) designed to block IGF-IR activation, has been tested in clinical trials for a range of tumors and has shown significant anti-tumor efficacy (34). The expression profiles and functional roles of IGF-IR, along with other IGF system components, are summarized in Table 1.
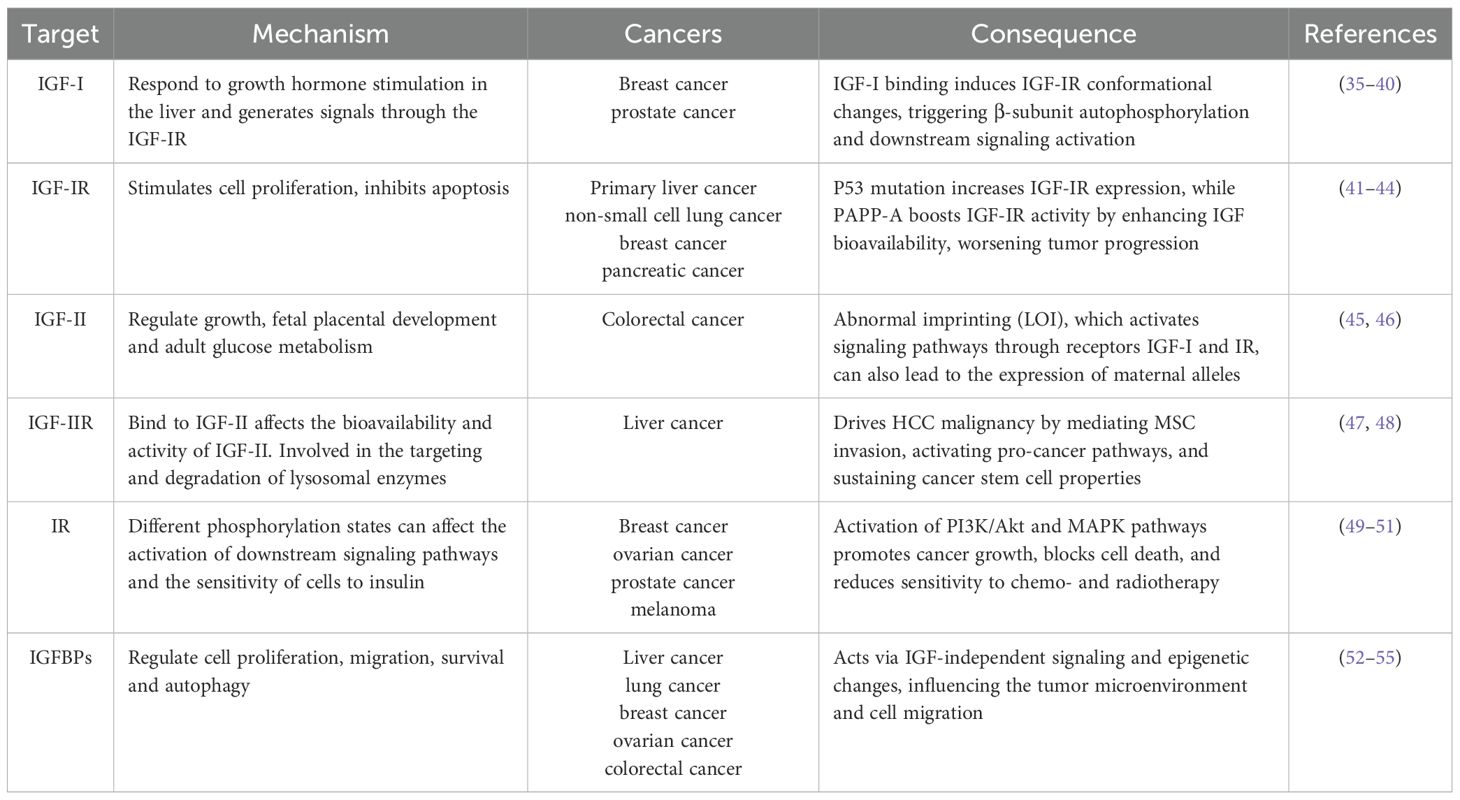
Table 1. Research progress of IGF signaling system related to cancer.
The overexpression or activation of IGF-I signaling axis is orchestrated by multiple mechanisms, including transcriptional regulation, microRNA-mediated modulation, receptor interactions, and influences from the tumor microenvironment. Transcriptionally, the c-Myb has been shown to upregulate the expression of IGF-I and its receptor IGF-IR, thereby enhancing cellular proliferation and survival (56). At the post-transcriptional level, several microRNAs, such as miR-7, miR-99a, and miR-145, directly target the 3’ untranslated region of IGF-IR mRNA, suppressing its expression (57). Additionally, IGF-IR can form heterodimers with other receptors, like the insulin receptor isoform A (IR-A), increasing sensitivity to IGF-I and activating downstream signaling pathways that promote cell proliferation (58). The tumor microenvironment also contributes to IGF-I axis activation (59). Cancer-associated fibroblasts secrete IGF-II, which acts in a paracrine manner to activate IGF-IR signaling in adjacent tumor cells, supporting cancer stem cell properties and facilitating tumor progression (60).
At present, a number of IGF-IR-targeted pharmaceutical agents have reached the stage of clinical trials, including small molecule inhibitors and monoclonal antibodies (61). Monoclonal antibodies that target either IGF-I or IGF-II have been developed, including dusigitumab and xentuzumab. These drugs inhibit the proliferation and survival of tumor cells by inhibiting the activation of IGF-IR and blocking downstream signaling.
2.2 IGF-II and IGF-IIR
Similar to IGF-I, IGF-II is a single-chain polypeptide molecule comprising a specific amino acid sequence (62). IGF-II has been demonstrated to exert a substantial influence on the proliferation of tumor cells, including those associated with hepatocellular colorectal cancer (63). Notably, IGF-II is an imprinted gene, and alterations in its imprinting state, such as loss of imprinting or imprinting relaxation, have been implicated in disease progression (64). In mammals, IGF-II also plays a pivotal role in the regulation of growth, foetal placental development and glucose metabolism in adults.
The principal function of Insulin-like growth factor II receptor (IGF-IIR) is to act as a cation-independent mannose 6-phosphate receptor (65), which is involved in the targeting of lysosomal enzymes. Unlike IGF-IR, IGF-IIR lacks tyrosine kinase activity and does not directly transduce signals (66). Although IGF-IIR does not transmit signals autonomously, it is capable of influencing the bioavailability and activity of IGF-II through its binding to IGF-II. Furthermore, it is involved in the targeting and degradation of lysosomal enzymes, which is essential for maintaining normal cellular physiological functions.
2.3 IR
The IR is a principal transmembrane tyrosine kinase receptor (67). When insulin binds to the α-subunit of the IR, it induces conformational changes that activate the kinase domain of the β-subunit (68). The activation triggers a cascade of signaling events through the phosphorylation of substrate proteins, including members of the IRS family. The diverse phosphorylation states of IRS protein family members can influence the activation of downstream signaling pathways and cellular sensitivity to insulin (69). Aberrant IR signaling has been implicated in promoting tumor cell proliferation and invasion in cancer, while IR dysfunction has also been associated with neuronal damage and apoptosis in neurodegenerative diseases (70). This suggests that dysregulation of IR signaling may play a crucial role in both cancer progression and neurodegeneration.
2.4 IGFBPs
Insulin-like growth factor-binding proteins (IGFBPs) are a family of soluble proteins that regulate IGF activity by modulating its bioavailability, transport, and half-life (71). Structurally, IGFBPs consist of three main regions: the N-terminus, C-terminus, and L-region (linker region). The N-terminus and the C-terminus exhibit high conservation and are hypothesized to be associated with the high affinity of IGF. In contrast, the L-region is specific and serves as a linker between the N-terminus and the C-terminus (72).
Among the IGFBP family, IGFBP-1 has emerged as a promising serum biomarker for disease burden in various conditions. For instance, in patients with chronic hepatitis and cirrhosis, serum IGFBP-1 levels decrease while growth hormone levels increase, suggesting a feedback regulatory mechanism. In this context, hepatic resistance to GH may lead to reduced IGFBP-1 levels, which in turn affects hepatocyte repair and metabolic functions (73). Furthermore, recent studies highlight the diagnostic and prognostic utility of IGFBP-1 in cardiovascular diseases. For example, serum IGF-I and IGFBP-1 levels have been shown to correlate with different types of heart failure (74), reflecting their roles in disease progression and potential value in assessing disease burden. These findings reinforce the notion that IGFBP-1 may serve as a useful indicator of disease severity or progression in a variety of clinical settings. Additionally, IGFBP-2 has been reported to promote the proliferation and survival of hematopoietic stem cells, thereby contributing to the maintenance of hematopoietic homeostasis (75).
IGFBP-3 has been extensively studied as a biomarker for a variety of diseases. These include type I diabetes mellitus and autoimmune diseases (76). The IGFBP-4, IGFBP-5, and IGFBP-6 proteins are also important members of the IGFBPs family, which binds to IGFs and regulates their activity and distribution (77). Overexpression of Skp2B can disrupt the prohibitin-p53 axis and upregulate the expression of Pregnancy-Associated Plasma Protein A (PAPP-A) (78). PAPP-A is a metalloproteinase that enhances IGF signaling by cleaving IGFBP-4, has been implicated in various cancers (79, 80). Furthermore, a class of proteins related to IGFBPs, including IGFBP-7 to IGFBP-10, has been identified (81–84).
3 Cross-talk between IGF signaling and other pathways in MDS and AML pathogenesis
IGF-I binds to IGF-IR, inducing a conformational change in the receptor. The activated IGF-IR subsequently recruits PI3K, positioning it proximal to the cell membrane (85). PI3K catalyzes the conversion of phosphatidylinositol-bisphosphate to phosphatidylinositol-trisphosphate (PIP3). Accumulation of PIP3 on the cell membrane facilitates the recruitment of Akt through interaction with the PH domain of Akt via its phosphate group (86). Akt is phosphorylated and activated by phosphatidylinositol-dependent kinase 1 and mechanistic target of rapamycin complex 2 (mTORC2) while localized at the membrane (87). Following activation, Akt translocates from the cell membrane into the cytoplasm and nucleus, where it phosphorylates various downstream target proteins, including Forkhead box O (FoxO) family transcription factors and mTOR (88). Notably, IGF-I activates the PI3K/Akt pathway to enhance Mouse Double Minute 2 (MDM2), which promotes p53 degradation and supports tumor growth (89). In turn, p53 suppresses IGF-IR and induces Phosphatase and Tensin Homolog (PTEN), forming a feedback loop that regulates tumor progression (90). Interestingly, MDMX, a homolog of MDM2, is overexpressed in preleukemic states and acts as a key driver of progression to AML. Independent of p53, MDMX binds to CK1α, leading to β-Catenin accumulation and activation of Wnt/β-Catenin signaling—a non-canonical pathway through which MDMX promotes leukemogenesis (91).
In AML, Ten-Eleven Translocation 2 (TET2) mutations have been shown to enhance mTORC1 signaling, linking epigenetic dysregulation to aberrant metabolic reprogramming (92, 93). Specifically, PIK3CA mutation, PTEN mutation or inactivation, and AKT hyperactivation have been shown to lead to sustained activation of the PI3K pathway, which has been demonstrated to promote AML and MDS cell proliferation, survival, and anti-apoptosis (94–96). The TET2 mutation has been observed to be prevalent among patients diagnosed with AML and MDS (97). The occurrence of a TET2 mutation may result in the abnormal methylation of PTEN and other genes (98), this, in turn, has the potential to inhibit their normal expression and to promote excessive activation of the PI3K/AKT pathway (99). Meanwhile, activation of the PI3K/Akt/mTOR pathway contributes to chemoresistance in AML and MDS by enhancing glycolysis and lipid synthesis while suppressing autophagy, ultimately reducing chemotherapy-induced cell death (100). Importantly, this pathway also converges with the Ras/MEK/ERK axis to reinforce oncogenic signaling and reduce treatment efficacy (101). The RAS/RAF/MEK/ERK cascade is activated when growth factors like Epidermal Growth Factor or Platelet-Derived Growth Factor bind to receptor tyrosine kinases on hematopoietic or stromal cells, leading to receptor dimerization, autophosphorylation, and recruitment of adaptor proteins that activate RAS via GDP-GTP exchange (102–104). Activated RAS, in turn, engages RAF kinases, initiating a phosphorylation cascade through MEK1/2 and ERK1/2. Once activated, ERK translocates to the nucleus to regulate transcription factors such as Elk-1, Myc, and AP-1, ultimately promoting genes involved in proliferation, metabolic remodeling, and inflammation (105).
Mutations in the RAS (KRAS/NRAS) gene, found in approximately 10–30% of AML and 5–15% of MDS cases, facilitate the progression of MDS to AML and are associated with poor prognosis (106, 107). These mutations drive sustained activation of signaling pathways that promote cell cycle progression and inhibit apoptosis, leading to clonal expansion in MDS and impaired differentiation of normal hematopoietic stem cells. Additionally, ERK activation promotes AML cell proliferation, metabolic reprogramming, and secretion of pro-inflammatory cytokines, worsening bone marrow inflammation and accelerating disease progression (108). However, sustained ERK activation also enhances DNA repair and inhibits drug-induced apoptosis, thereby increasing leukemia resistance and reducing MDS treatment efficacy (109). IGF-IR activation further amplifies disease progression by engaging both the Ras/Raf/MEK/ERK and PI3K/Akt pathways, forming a dual-pathway synergy that promotes tumor cell growth and exacerbates hematologic malignancies.
The canonical Wnt/β-catenin pathway is triggered when Wnt ligands bind to the Frizzled (FZD) receptor and its co-receptors LRP5/6 on the cell surface. This interaction recruits Dishevelled proteins, which disrupt the Axin–GSK3β complex and initiate downstream signaling (110, 111). The Wnt/β-catenin and IGF signaling pathways interact through multiple mechanisms, influencing cell behavior and contributing to various physiological and pathological processes. There is significant crosstalk between these pathways; for instance, β-catenin interacts with transcription factors such as TCF/LEF and FoxO, whose activity is regulated by IGF signaling (112). Additionally, Dishevelled, a key component of Wnt signaling, can influence IGF-induced Ras-Raf-MAPK signaling (113). IGF-I also plays a crucial role in modulating the location, stability, and transcriptional activity of β-catenin (114). In the context of AML, recent studies have uncovered distinct roles of Wnt signaling (115). Notably, cytoplasmic nuclear paraspeckle assembly transcript has been found to suppress AML progression by inhibiting Wnt signaling, in contrast to its oncogenic function in other cancers (115). Furthermore, TIM-3 signaling has been shown to hijack the canonical Wnt/β-catenin pathway, thereby promoting cancer stemness in AML. Given the critical involvement of Wnt signaling in AML pathogenesis and therapy resistance, targeting the mevalonate or Wnt pathways presents a promising strategy to overcome CAR T-cell resistance in TP53-mutant AML cells (116).
Additionally, activation of the PI3K/Akt pathway has been linked to non-canonical activation of the Hedgehog (Hh) pathway, which supports leukemia cell survival and self-renewal, thereby contributing to chemoresistance in myeloid leukemia (117). Cells that secrete Hh ligands process and release these ligands, which predominantly bind in a paracrine manner to the transmembrane receptors Patched 1 (PTCH1) and PTCH2 (118). This binding inhibits the suppressive activity of PTCH1 and PTCH2 on Smoothened (SMO) (119). Subsequently, the activation of Hh signaling cascades leads to the activation and nuclear localization of GLI transcription factors, driving the expression of Hh target genes (119). Targeting this pathway has been proposed as a strategy to reduce leukemia stem cell (LSC) populations, enhance treatment responses, and improve patient outcomes (120, 121). Cholesterol directly modifies the key Hh pathway ligand, Sonic Hedgehog (SHH), enhancing its release and activation of the downstream signal molecule, SMO. This leads to upregulation of Gli1 expression, a marker of Hh signaling, promoting tumor migration and metastasis (122). P53 regulates cholesterol metabolism, influencing this pathway (123). Specifically, it has been demonstrated that the upregulation of SHH and GLI1 expression in AML cells results in aberrant activation of the Hh pathway, particularly in CD34+ AML cells that are resistant to chemotherapeutic agents (124). Moreover, persistent activation of SMO in mouse models and patient samples has been shown to lead to the persistence of dormant BCR-ABL+ LSCs (125). Furthermore, the use of SMO inhibitors in mouse models and human leukemia has been demonstrated to inhibit Hh signalling (126). And the Gli3 transcription factor within the Hh pathway directly regulates IGF-I expression in its activator form, while its repressor function controls IGFBP-1 levels (127). This highlights a mechanistic link between the Hh and IGF pathways, underscoring the therapeutic potential of targeting this axis. Inhibiting the Hedgehog pathway, particularly in relapsed or refractory AML, has emerged as a promising strategy (128). Glasdegib, the first and only FDA-approved Hh pathway inhibitor for AML, is used in combination with low-dose cytarabine for patients ineligible for intensive chemotherapy (129). Moreover, targeting the Hh/IGF-IR/PI3K/Akt/MRP1 axis may offer an effective therapeutic approach for refractory AML (130). However, resistance mechanisms and the need for personalized treatment strategies remain critical challenges for future research.
In summary, IGF signaling interacts with multiple oncogenic pathways, including PI3K/Akt/mTOR, Ras/Raf/MEK/ERK, Wnt/β-catenin, and Hedgehog, collectively driving MDS and AML progression. These interactions contribute to enhanced proliferation, metabolic reprogramming, immune evasion, and chemoresistance. Figure 2 provides a schematic representation of these interconnected signaling pathways. Given these intricate connections, targeting IGF signaling in combination with inhibitors of these pathways may provide a more effective therapeutic approach for MDS and AML, warranting further investigation.
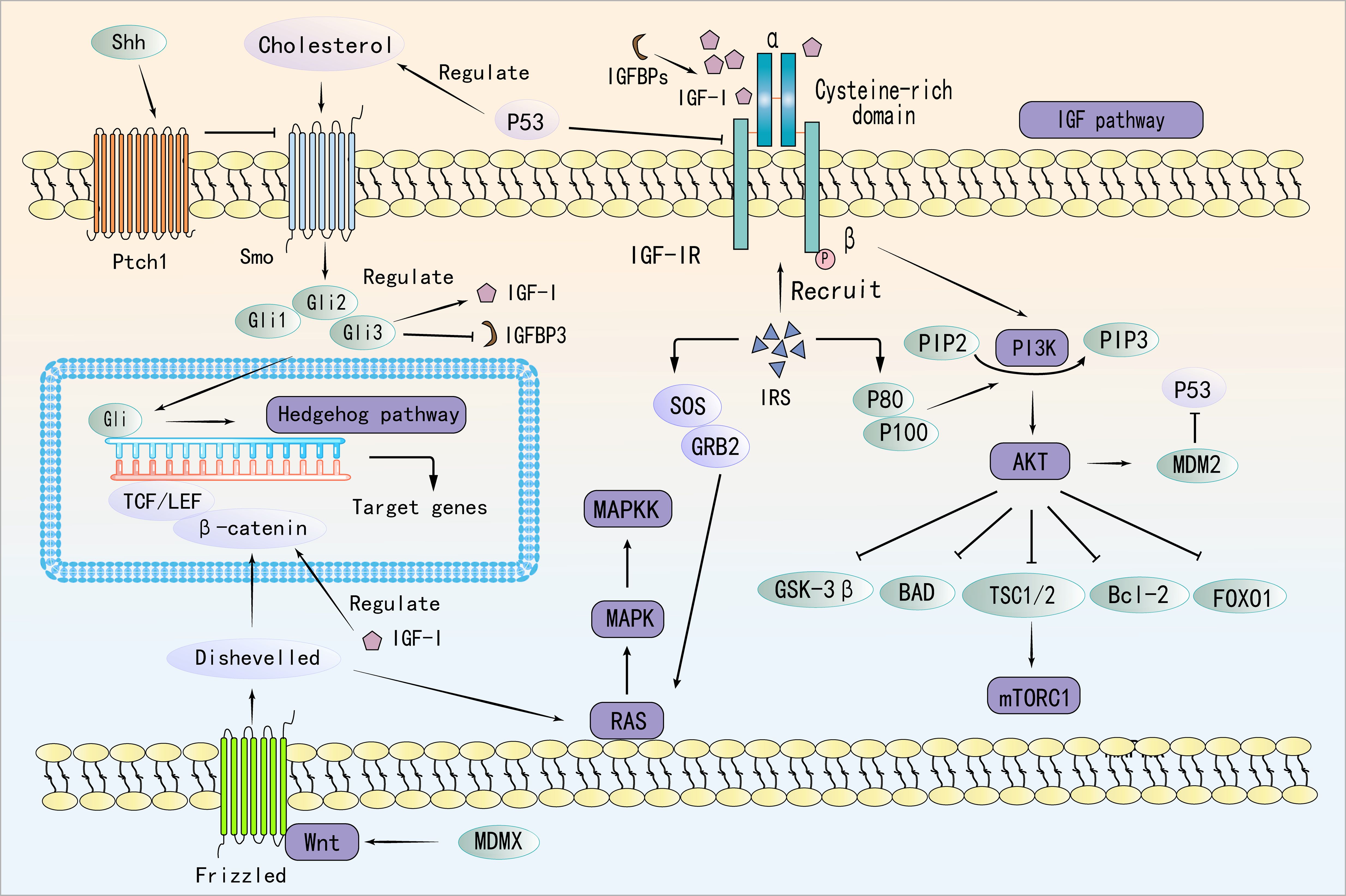
Figure 2. Molecular Interplay Between IGF, PI3K/Akt/mTOR, Ras/Raf/MEK/ERK, Wnt/β-Catenin, and Hedgehog Pathways.
4 IGF signaling system in the bone marrow microenvironment of MDS and AML
Unlike its role in promoting migration and invasion of MM cells (131–133), the IGF signaling pathway primarily exerts anti-apoptotic and pro-proliferative effects in MDS and AML. Comparative studies have shown significant alterations in the expression levels of certain genes within the IGF gene family in patients with AML compared to healthy bone marrow samples, with a notable increase in the expression of IGF-I, IGF-IR, and IGFBP-3 (134, 135).
Beyond its direct effects on leukemic cell proliferation and survival, IGF signaling also shapes the hematopoietic microenvironment. The bone marrow microenvironment (BMME), comprising mesenchymal stromal cells (MSCs), endothelial cells, and a spectrum of immune cells—plays a pivotal role in regulating hematopoiesis and supporting disease progression (136–138). A study found that IGF expression showed a downward trend in MDS-MSCs, suggesting that IGF dysregulation in the bone marrow niche may contribute to ineffective hematopoiesis and disease progression in MDS (139).
MSCs are central players in the BMME, and their interaction with leukemic cells is significantly influenced by IGF signaling. Bone marrow-derived MSCs have been shown to modulate the expression of ATP-binding cassette transporters in AML cells via the IGF-I pathway, thereby promoting chemotherapy resistance (15). Moreover, MSCs can secrete IGF-I to stimulate the proliferation of endothelial progenitor cells through PI3K/Akt activation, highlighting the pro-angiogenic and supportive roles of IGF signaling within the leukemia microenvironment (140).
Targeting the PI3K-Akt-mTOR axis in AML presents a particular challenge, as its dysregulation is not only driven by leukemic cell-intrinsic factors but also by cues from the microenvironment, including MSCs and immune cells (141). This complexity underscores the need to consider the BMME as an integral component of therapeutic strategies. Indeed, modulating IGF signaling has emerged as a promising approach to optimize MSC function and overcome microenvironment-induced resistance, potentially enhancing therapeutic efficacy (142).
Interestingly, studies from other tumor models support this concept. For example, human fetal MSCs inhibit liver cancer growth through secretion of IGFBPs, which suppress IGF-IR/PI3K/Akt signaling and reduce tumor proliferation (143). Similarly, M2-like tumor-associated macrophages in anaplastic thyroid carcinoma have been shown to promote tumor stemness and metastasis by secreting IGF-I/II and activating the IR-A/IGF-IR–mediated PI3K/AKT/mTOR pathway (144). These findings provide further evidence that IGF signaling within the tumor microenvironment can profoundly influence disease behavior.
Taken together, the IGF signaling axis contributes to both cell-intrinsic and microenvironmental mechanisms that drive MDS and AML progression. As research advances, targeting IGF signaling, either directly or via modulation of the BMME, may represent a cornerstone in the development of more effective treatment strategies for hematological malignancies (145).
5 Role of the IGF signaling system in MDS and AML
5.1 IGF-I in MDS and AML
IGF-I not only promotes the growth and development of the organism, but also regulates cell proliferation, differentiation and metabolism (146). In contrast to solid tumors, which have been extensively studied, relatively few studies have investigated the role of IGF-IR in leukemia, with the majority of these focusing on plasma cell myeloma (25, 147). Reduced IGF-I signaling is associated with ineffective hematopoietic features commonly observed in MDS (12). This dysregulation highlights the critical role of the IGF signaling system in maintaining hematopoietic homeostasis and its disruption in disease states (148). IGF-I exerts a significant influence on the clonal growth of hematopoietic cells in AML patients, particularly during active disease phases (13). Specifically, in vitro experiments confirm that IGF-I promotes the growth of AML cells primarily through the activation of the PI3K/Akt and ErK signaling pathways, a process that aligns with the fact that uncontrolled PI3K activation is present in 50% of AML cases (149, 150). AML cells can secrete IGF-I and express its receptor, IGF-IR, establishing an autocrine positive feedback loop that leads to constitutive activation of the PI3K/Akt signaling pathway. Studies have demonstrated that in approximately 70% of AML samples exhibiting PI3K activation, this persistent activation is attributable to autocrine IGF-I/IGF-IR signaling. Furthermore, treatment with neutralizing anti-IGF-IR antibodies significantly inhibits PI3K/Akt signaling and reduces the clonogenic capacity of leukemic progenitor cells (30).
IGF-I not only directly promotes cell growth but also influences the proliferation of AML cells through other mechanisms. For instance, ADAM28 degrades IGFBP-3, facilitating IGF-I-induced proliferation (151). Hematopoietic stem cell transplantation is a key therapeutic option for patients with AML (152). However, long-term survivors often face endocrine complications, which significantly impact their quality of life. Among these complications, approximately 10% involve dysfunction of the hypothalamic-pituitary-GH/IGF-I axis (153). This highlights the critical role of the IGF signaling system in post-transplant physiological regulation. Importantly, the IGF/IGF-IR axis is not only involved in endocrine homeostasis but also plays a fundamental role in regulating cancer stem cells by sustaining their stemness, survival, and proliferative capabilities (154), suggesting that targeting this axis could have dual benefits: mitigating post-transplant complications and suppressing leukemia recurrence.
5.2 IGF-IR in MDS and AML
IGF-IR is highly expressed and plays a key role in MDS clonal cells (155, 156). Compared with normal controls, the mean IGF-IR expression level was significantly increased in CD34+ cells of 100 MDS patients, suggesting its potential as a clonal cell marker for MDS (14). IGF-IR plays a dual role in MDS pathophysiology. Current studies have demonstrated that the expression rate of IGF-IR in nucleated cells from patients with MDS and AML is significantly higher than that in normal bone marrow (157). Furthermore, IGF-IR is more strongly expressed in advanced subtypes of MDS, such as refractory anemia with excess progenitor cells and its transformed form (158), which are associated with an increased proportion of blasts in the bone marrow—a critical marker indicating a heightened risk of progression from MDS to AML. IGF-IR facilitates the growth of MDS clonal cells, driving disease progression.
IGF-IR inhibits the MAPK signaling pathway, particularly p-p38 MAPK and p-p44/42 MAPK, which are critical regulators of cell proliferation and apoptosis (159). The activation of the IGF-IR pathway has been implicated in lenalidomide resistance among MDS patients. This resistance poses a significant challenge in therapeutic management and highlights the need for targeted interventions (160). A study found that IGF-IR inhibition reduces the proliferation and survival of del(5q) MDS cells both in vitro and in vivo. Furthermore, lenalidomide-resistant del(5q) MDS cells lacking TP53 or RUNX1 remain sensitive to IGF-IR inhibition. These findings suggest that targeting IGF-IR could be a promising strategy, particularly in MDS subtypes with genetic alterations that confer resistance to standard therapies (161). Knocking down IGF-IR in MDS cells results in increased phosphorylation of MAPK, reversing its inhibitory effects. Furthermore, the use of the IGF-IR inhibitor PPP effectively suppresses MDS cell proliferation and induces cell cycle arrest and apoptosis. These findings underscore the potential of IGF-IR inhibitors as therapeutic agents in MDS treatment (158).
In malignant bone marrow nucleated cells from AML patients, IGF-IR expression is as high as 92%, significantly exceeding levels found in other hematological disorders. Research indicates that IGF-IR-positive cells exhibit lower apoptosis rates compared to IGF-IR-negative cells, patients with high IGF-IR expression (>50%) demonstrate even lower rates of apoptosis, indicating a correlation between IGF-IR expression levels and resistance to apoptosis (162). In AML, miR-628 is downregulated, leading to the loss of its regulatory suppression on IGF-IR expression, thereby contributing to the upregulation of IGF-IR (163). In AML with RAS mutations, mutant RAS can upregulate the expression of IGF-IR, thereby enhancing the activity of the PI3K/Akt and MAPK signaling pathways (164). The study has demonstrated that HOXA9 directly induces IGF-IR expression, and that knockdown of HOXA9 leads to decreased IGF-IR levels, resulting in reduced leukemic cell growth and increased apoptosis (165). In certain AML cell lines, such as HL-60 and U937, the IGF-IR forms heterodimers with IR-A. These hybrid receptors enhance sensitivity to IGF-I and IGF-II, activating downstream signaling pathways like PI3K/Akt and MAPK/ERK, which promote leukemic cell proliferation (58).
IGF-IR not only plays a critical role in enhancing anti-apoptotic effects but also acts as a key regulator in inhibiting cell proliferation, particularly through its ability to activate natural killer cells, thereby suppressing the proliferation of leukemic cells (166). IGF-IR mediates growth through the PI3K/Akt/mTOR pathway and is influenced by SUMO-I modification; this pathway plays a pivotal role in the initiation and progression of AML (167). SUMOylation enhances autocrine signaling and activates downstream pathways such as PI3K/Akt, further promoting AML cell proliferation and survival. Dual inhibition of the mTOR C1 complex and the IGF-I/IGF-IR/PI3K/Akt pathway may enhance the efficacy of mTOR inhibitors in treating AML (168–170).
Collectively, these mechanisms underscore the multifaceted role of IGF-IR in AML pathogenesis and highlight potential therapeutic targets within the IGF-IR signaling axis.
5.3 IGF-II in AML
In leukemia cell samples from AML patients (n=32), a significant decrease in IGF-II expression was observed in both bone marrow biopsies and peripheral blood samples (171). The study demonstrates that IGF-II can induce Nanog expression through IGF-IR signaling, thereby enhancing the stem-like properties of LSCs in AML (172). The specific mechanisms of IGF-II in AML require further investigation. Studies have shown that IGF2BP-2 enhances the expression of genes associated with glutamine uptake and metabolism, thereby promoting the survival and growth of AML cells (27).
5.4 IGFBPs in MDS and AML
Bone marrow plasma from early-stage MDS patients exhibits significantly higher levels of IGFBP-3 compared to healthy controls. These elevated levels may contribute to the progression of the disease. IGFBP-3 promotes apoptosis in bone marrow cells, particularly under the influence of pro-apoptotic factors such as TNF-α. This apoptotic activity may exacerbate ineffective hematopoiesis and worsen cytopenias in MDS patients (173). Emerging studies suggest that adiponectin and resistin—two critical metabolic regulators—can influence the proliferation and survival of MDS cells. These effects are mediated by modulating the expression or activity of IGFBP-3. Understanding these interactions provides new insights into the metabolic regulation of MDS pathogenesis (174).
Members of the IGFBP family, particularly IGFBP-2 and IGFBP-7, play important roles in the progression of AML, treatment response, and prognostic evaluation (175). Research shows that high expression of IGFBP-2 in 99 adult AML patients is associated with the upregulation of leukemia-related genes and poor prognostic markers, as well as an increased rate of drug resistance (176). Patients with relapsed disease exhibit higher IGFBP-2 levels compared to those in complete clinical remission (177). Additionally, IGFBP-2 may indicate the risk of relapse after hematopoietic stem cell transplantation in pediatric AML patients (178). Experiments using mouse models of AML also demonstrate that the absence of IGFBP-2 can inhibit disease progression (179). In AML mouse models and human patients, elevated IGFBP-1 suppresses insulin and IGF-I activity, inducing systemic insulin resistance and supporting leukemic cell survival. Inflammatory mediators such as TNFSF13B and IL-8 may further enhance IGFBP-1 expression, aggravating metabolic dysfunction and promoting disease progression (180). In pre-treatment blood samples from non-M3 AML patients, low baseline levels of IGFBP-1 and IGFBP-6 correlate with better progression-free survival, while low baseline levels of IGFBP-2, IGFBP-2, IGFBP-6, and IGFBP-7 are closely associated with improved overall survival (181). IGFBP-7 is expressed at low levels in leukemic stem cells compared to normal hematopoietic stem cells (182). Notably, IGFBP-7 enhances the sensitivity of AML cells to chemotherapy, with higher expression levels correlating with better patient prognosis, indicating its positive role in AML treatment and outcomes (183, 184), while IGFBP-5 reduces cell proliferation and enhances chemotherapy efficacy by inhibiting the IGF-IR/AKT signaling pathway (185). The specific mechanisms of the IGF signaling pathway in MDS and AML are summarized in Figures 3, 4.
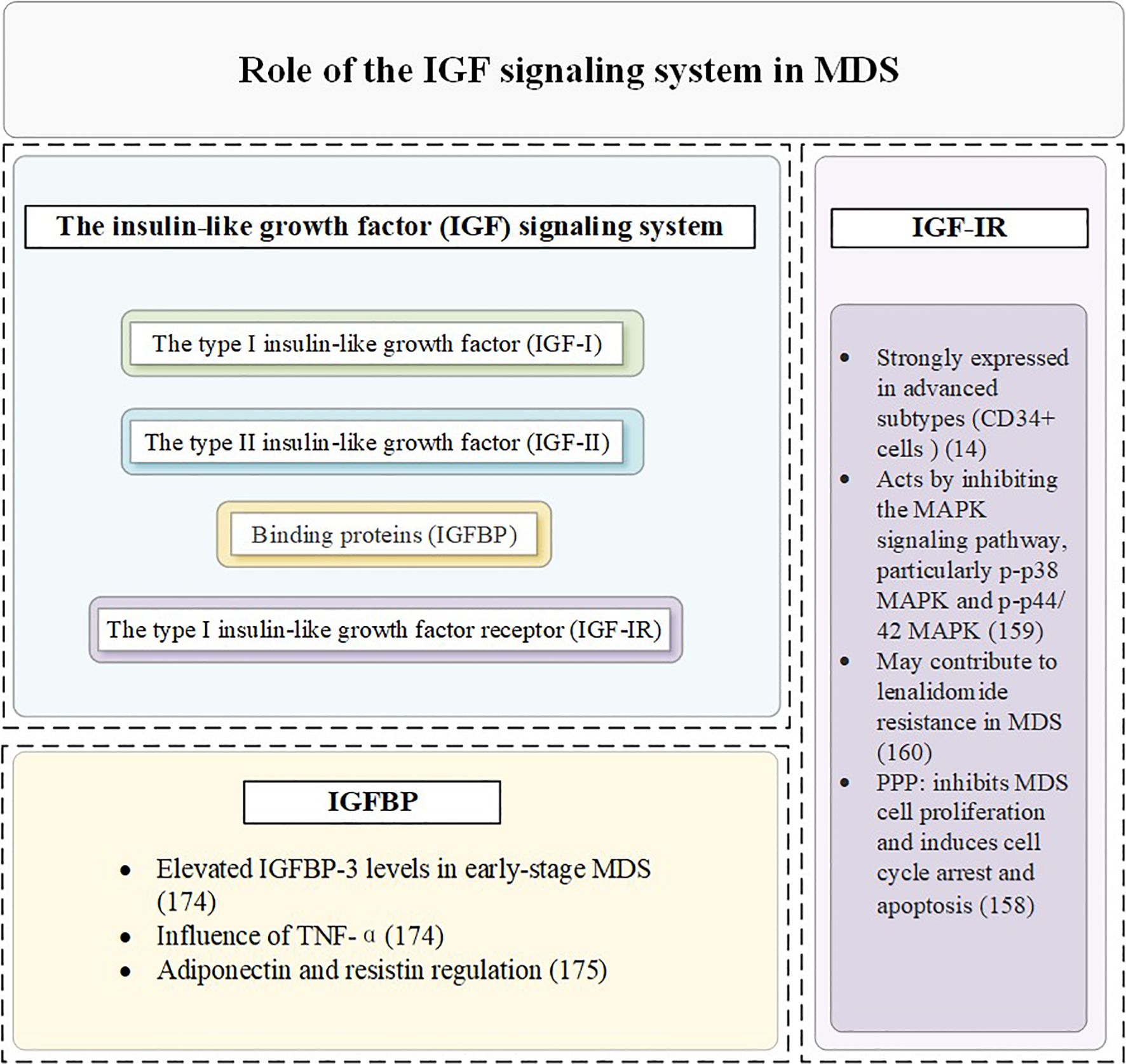
Figure 3. The role of the IGF signaling system in MDS.
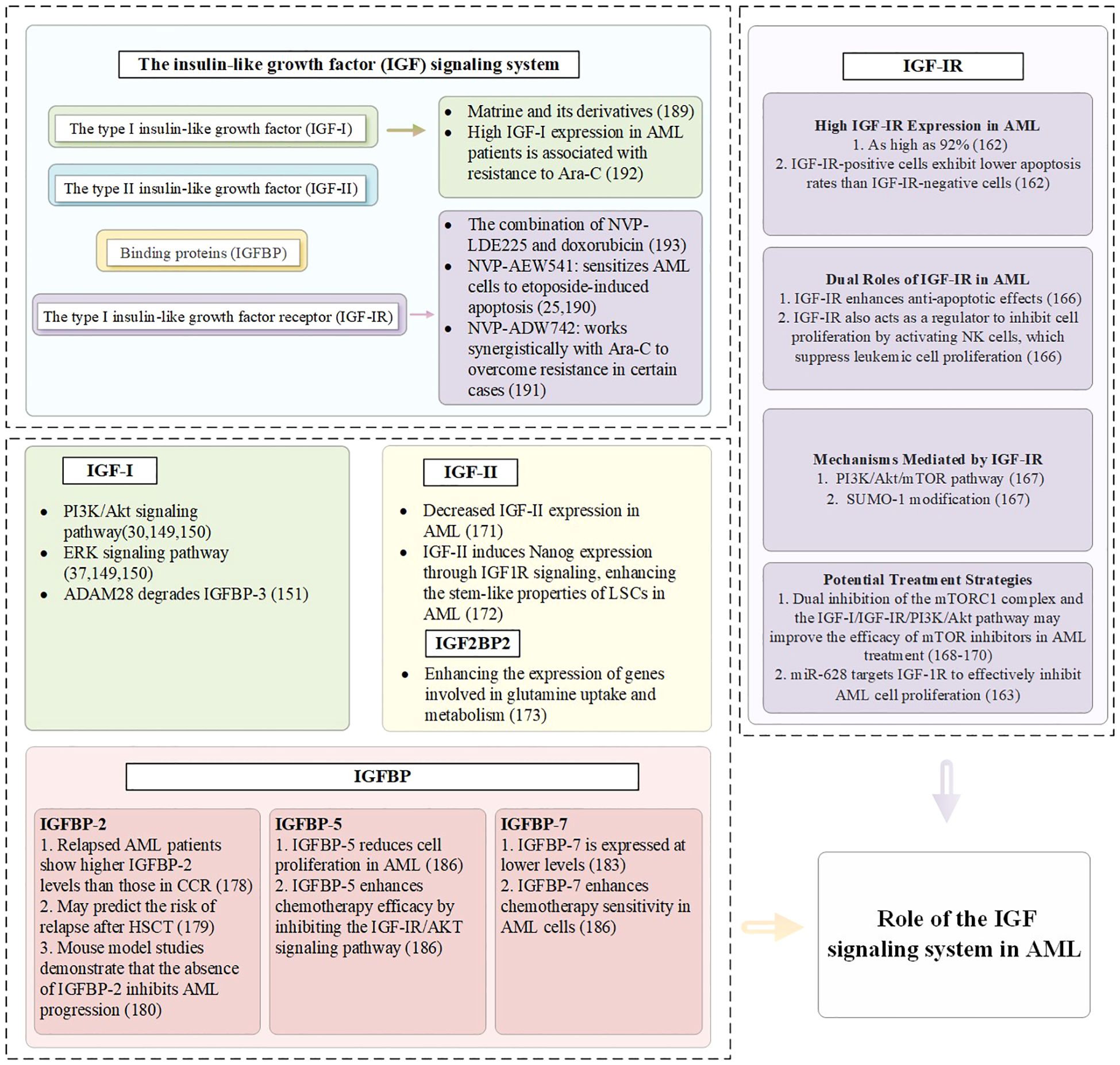
Figure 4. The role of the IGF signaling system in AML.
5.5 IGF signaling pathway as a therapeutic target in MDS and AML
IGF-I reverses the antiproliferative effects of chemotherapeutic agents, such as chidamide (CHI) combined with aspirin (ASA), on AML-MDS cells (186). The combined treatment with CHI and ASA was observed to significantly down-regulate the phosphorylation levels of PI3K and AKT, thereby inhibiting the activity of the PI3K/AKT pathway. This resulted in cell cycle arrest and the induction of apoptosis. However, IGF-I, acting as an agonist of PI3K/AKT, could reverse these effects, indicating potential resistance mechanisms via IGF signaling. Despite promising preclinical results, this combination therapy has not yet advanced to clinical trials.
Additionally, a phase 1b clinical study evaluated the safety and efficacy of IGF-methotrexate (IGF-MTX) in the treatment of high-grade MDS or oligoblastic AML. While two patients demonstrated prolonged survival and reduced clonal MDS burden, the trial was limited by its small sample size, lack of a control arm, and short follow-up duration. Furthermore, the study emphasized the need for further research to better define pharmacokinetics, optimal dosing, and long-term safety (187).
The primary reason for the extremely poor prognosis in AML is the treatment failure caused by chemotherapy resistance. Matrine and related compounds may help overcome resistance mechanisms mediated by high levels of IGF-I and p-Akt activation in AML cells by inhibiting the IGF signaling pathway (188). However, current evidence for matrine’s efficacy is primarily limited to in vitro and animal studies. Moreover, potential toxicities, pharmacokinetics, and long-term safety of matrine in humans remain unclear. Further translational and clinical investigations are warranted to determine its therapeutic potential in AML patients.
IGF-IR inhibitors provide new strategies for the treatment of AML. The small molecule inhibitors NVP-AEW541 and NVP-ADW742 exhibit significant anti-AML activity. NVP-AEW541 inhibits AML cell proliferation and sensitizes cells to etoposide-induced apoptosis (25, 189), while NVP-ADW742 induces apoptosis in AML cells and works synergistically with Ara-C in resistant specimens (190). High IGF-I expression in AML patients correlates with Ara-C resistance, confirming the role of IGF-I in resistance mechanisms (191). In mouse models of AML, the combination of NVP-LDE225 and doxorubicin demonstrates significant anti-tumor effects, potentially related to the inhibition of the Hh/IGF-IR/Akt/MRP1 pathway (192). NVP-LDE225, an inhibitor of the Hh signaling pathway, reduces the expression of p-IGF-IR and p-Akt, which inhibits the activity of this signaling pathway and consequently decreases cell survival signaling and promotes apoptosis. Furthermore, NVP-LDE225 diminished the expression of MRP1, a drug efflux pump, and augmented the sensitivity of cells to chemotherapeutic drugs such as paclitaxel, effectively reversing the drug resistance of tumor cells. Although these findings collectively highlight the critical roles of IGF-I and its receptor in the pathogenesis and resistance mechanisms of AML, most studies remain at the preclinical stage. The clinical translation of IGF-IR inhibitors faces several anticipated challenges, including potential off-target toxicity and limited efficacy in heterogeneous patient populations. Notably, due to the structural similarity between IGF-IR and the IR, some inhibitors may inadvertently interfere with metabolic regulation, a concern raised in previous drug development efforts (193, 194). Moreover, activation of compensatory pathways such as Ras/MAPK or mTOR has been suggested to attenuate therapeutic benefit (195).
To address this, combining IGF pathway inhibitors with other molecularly targeted therapies has been suggested as a potentially promising strategy. For instance, Fms-like tyrosine kinase 3 (FLT3) mutations, particularly FLT3-ITD, are prevalent in AML and contribute to disease progression (196). Combining FLT3 inhibitors with IGF-IR inhibitors may theoretically produce synergistic effects by concurrently targeting multiple proliferative and survival pathways (197, 198). Similarly, IDH1/2 mutations, which lead to epigenetic dysregulation and metabolic reprogramming in AML, may also represent actionable targets in combination regimens (199–201). While conclusive data in AML are lacking, preliminary evidence from other malignancies raises the possibility that IGF pathway inhibition, when combined with other targeted approaches, may enhance therapeutic efficacy. Future research is warranted to evaluate such strategies in AML. Such biomarker-driven combination therapies may enhance efficacy, overcome pathway redundancy, and improve patient outcomes.
Of particular note is the recent research has combined 188Re-antiCD20 radioimmunotherapy with stable silencing of IGF-IR for the treatment of Raji cells (a model for non-Hodgkin lymphoma), demonstrating potential efficacy (202). Blocking IGF-IR signaling reduces cell proliferation and sensitizes cancer cells to ionizing radiation (203). However, the efficacy of this therapeutic approach in MDS and AML requires further investigation. Collectively, these findings highlight the critical roles of IGF-I and its receptor in the pathogenesis and resistance mechanisms of AML, suggesting that their inhibitors offer promising new avenues for AML treatment. A summary of the key therapeutic agents targeting the IGF pathway in MDS and AML is provided in Table 2.
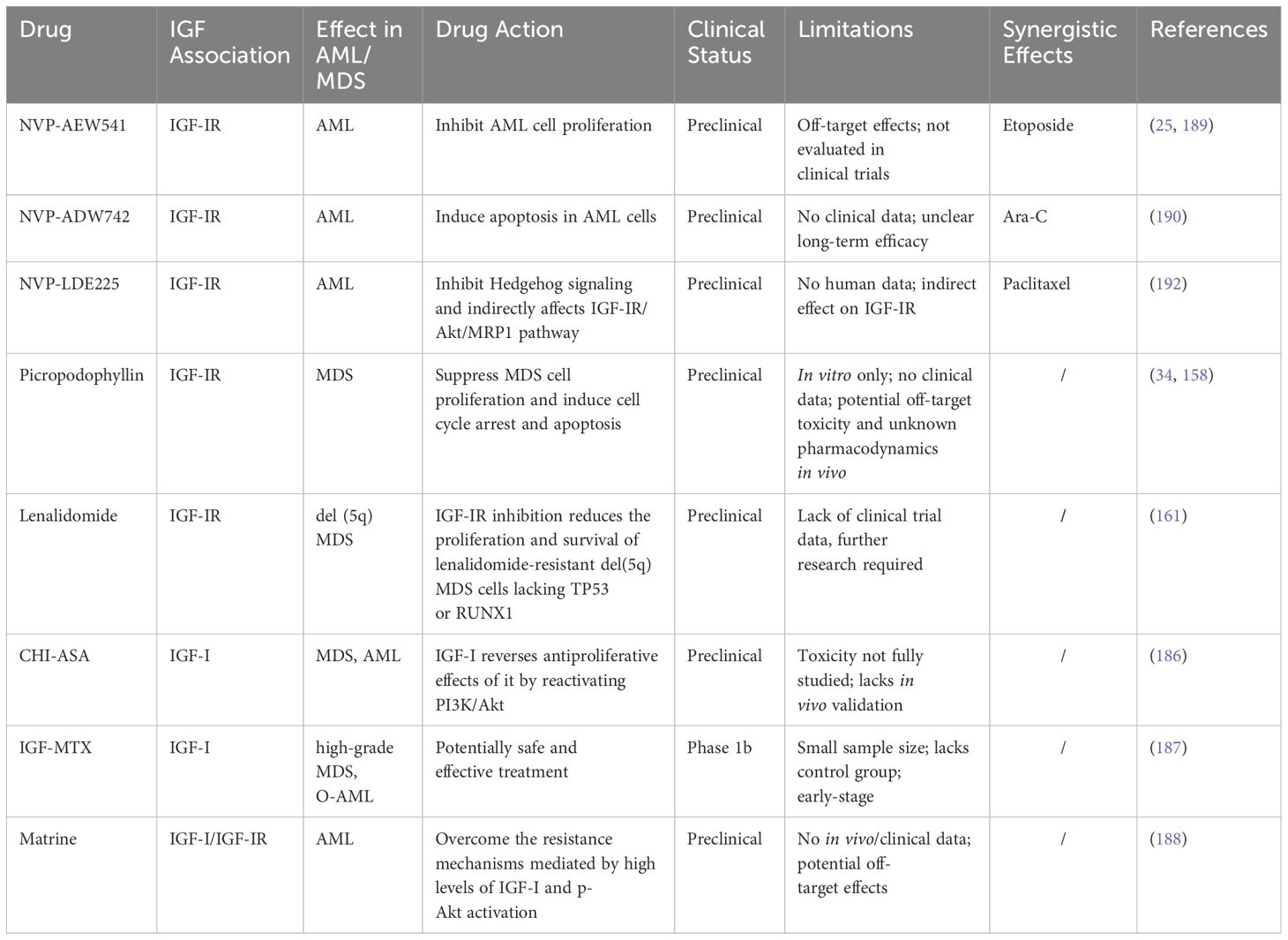
Table 2. IGF pathway-targeting drugs in MDS and AML: mechanisms and synergistic effects.
6 Conclusion
The IGF signaling pathway plays a pivotal role in the pathogenesis and progression of MDS and AML by promoting clonal proliferation, survival, and chemotherapy resistance. Dysregulation of IGF-I, IGF-IR, and IGFBPs, particularly IGFBP-2 and IGFBP-7, has profound implications for disease progression and patient prognosis. Targeting key components of this pathway, such as IGF-IR and IGFBPs, represents a promising therapeutic strategy.
However, clinical translation has been hampered by several challenges, including pathway redundancy with IR signaling, limited efficacy in genetically heterogeneous populations, and a lack of durable responses in clinical trials. Future strategies should prioritize biomarker-driven clinical trials to identify patient subsets most likely to benefit from IGF-targeted therapies. Combination regimens that integrate IGF pathway inhibitors with agents such as FLT3 or IDH1/2 inhibitors may enhance efficacy through synergistic effects. Furthermore, rational therapeutic design must address compensatory signaling and IR redundancy, potentially through selective dual inhibitors or pathway-specific degradation approaches. Ultimately, the successful clinical application of IGF-targeted therapies will depend on addressing these challenges and validating their efficacy in large-scale clinical trials.
Author contributions
YW: Writing – original draft. XD: Writing – original draft. ST: Writing – original draft. QC: Writing – original draft. YC: Writing – original draft. LZ: Writing – original draft. YS: Writing – original draft. ZH: Writing – original draft. LY: Writing – review & editing. CW: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Osher E and Macaulay VM. Therapeutic targeting of the IGF axis. Cells. (2019) 8(8):895. doi: 10.3390/cells8080895
2. Deyev IE, Mitrofanova AV, Zhevlenev ES, Radionov N, Berchatova AA, Popova NV, et al. Structural determinants of the insulin receptor-related receptor activation by alkali. J Biol Chem. (2013) 288:33884–93. doi: 10.1074/jbc.M113.483172
3. Takahashi SI. IGF research 2016-2018. Growth Horm IGF Res. (2019) 48-49:65–9. doi: 10.1016/j.ghir.2019.10.004
4. De-Freitas-Junior JC, Carvalho S, Dias AM, Oliveira P, Cabral J, Seruca R, et al. Insulin/IGF-I signaling pathways enhances tumor cell invasion through bisecting GlcNAc N-glycans modulation. an interplay with E-cadherin. PloS One. (2013) 8:e81579. doi: 10.1371/journal.pone.0081579
5. Obradovic M, Zafirovic S, Soskic S, Stanimirovic J, Trpkovic A, Jevremovic D, et al. Effects of IGF-1 on the cardiovascular system. Curr Pharm Des. (2019) 25:3715–25. doi: 10.2174/1381612825666191106091507
6. Belfiore A, Malaguarnera R, Vella V, Lawrence MC, Sciacca L, Frasca F, et al. Insulin receptor isoforms in physiology and disease: an updated view. Endocr Rev. (2017) 38:379–431. doi: 10.1210/er.2017-00073
8. Kasprzak A, Kwasniewski W, Adamek A, and Gozdzicka-Jozefiak A. Insulin-like growth factor (IGF) axis in cancerogenesis. Mutat Res Rev Mutat Res. (2017) 772:78–104. doi: 10.1016/j.mrrev.2016.08.007
9. Haywood NJ, Slater TA, Matthews CJ, and Wheatcroft SB. The insulin like growth factor and binding protein family: Novel therapeutic targets in obesity & diabetes. Mol Metab. (2019) 19:86–96. doi: 10.1016/j.molmet.2018.10.008
10. Hua J, Wang X, Ma L, Li J, Cao G, Zhang S, et al. CircVAPA promotes small cell lung cancer progression by modulating the miR-377-3p and miR-494-3p/IGF1R/AKT axis. Mol Cancer. (2022) 21:123. doi: 10.1186/s12943-022-01595-9
11. Song D, Wu Y, Li J, Liu J, Yi Z, Wang X, et al. Insulin-like growth factor 2 drives fibroblast-mediated tumor immunoevasion and confers resistance to immunotherapy. J Clin Invest. (2024) 134(22):e183366. doi: 10.1172/JCI183366
12. Zadik Z, Estrov Z, Karov Y, Hahn T, and Barak Y. The effect of growth hormone and IGF-I on clonogenic growth of hematopoietic cells in leukemic patients during active disease and during remission–a preliminary report. J Pediatr Endocrinol. (1993) 6:79–83. doi: 10.1515/JPEM.1993.6.1.79
13. Frostad S and Bruserud O. In vitro effects of insulin-like growth factor-1 (IGF-1) on proliferation and constitutive cytokine secretion by acute myelogenous leukemia blasts. Eur J Haematol. (1999) 62:191–8. doi: 10.1111/j.1600-0609.1999.tb01743.x
14. Zhang Q, He Q, Guo J, Xu F, Zhang Z, Yang L, et al. Expression of insulin-like growth factor recepter type I in CD34(+) cells of patients with myelodysplastic syndromes. Zhongguo Shi Yan Xue Ye Xue Za Zhi. (2018) 26:849–53. doi: 10.7534/j.issn.1009-2137.2018.03.036
15. Benabbou N, Mirshahi P, Bordu C, Faussat A, Tang R, Therwath A, et al. A subset of bone marrow stromal cells regulate ATP-binding cassette gene expression via insulin-like growth factor-I in a leukemia cell line. Int J Oncol. (2014) 45:1372–80. doi: 10.3892/ijo.2014.2569
16. Hasserjian RP, Germing U, and Malcovati L. Diagnosis and classification of myelodysplastic syndromes. Blood. (2023) 142:2247–57. doi: 10.1182/blood.2023020078
17. Shimony S, Stahl M, and Stone RM. Acute myeloid leukemia: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol. (2023) 98:502–26. doi: 10.1002/ajh.26822
18. Menssen AJ and Walter MJ. Genetics of progression from MDS to secondary leukemia. Blood. (2020) 136:50–60. doi: 10.1182/blood.2019000942
19. Granfeldt ØL, Medeiros BC, Sengeløv H, Nørgaard M, Andersen MK, Dufva IH, et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: A national population-based cohort study. J Clin Oncol. (2015) 33:3641–9. doi: 10.1200/JCO.2014.60.0890
20. Hulegårdh E, Nilsson C, Lazarevic V, Garelius H, Antunovic P, Rangert DÅ, et al. Characterization and prognostic features of secondary acute myeloid leukemia in a population-based setting: a report from the Swedish Acute Leukemia Registry. Am J Hematol. (2015) 90:208–14. doi: 10.1002/ajh.23908
21. Bănescu C, Tripon F, and Muntean C. The genetic landscape of myelodysplastic neoplasm progression to acute myeloid leukemia. Int J Mol Sci. (2023) 24(6):5734. doi: 10.3390/ijms24065734
22. Chen J, Kao YR, Sun D, Todorova TI, Reynolds D, Narayanagari SR, et al. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat Med. (2019) 25:103–10. doi: 10.1038/s41591-018-0267-4
23. Chakraborty S, Shapiro LC, de Oliveira S, Rivera-Pena B, Verma A, and Shastri A. Therapeutic targeting of the inflammasome in myeloid Malignancies. Blood Cancer J. (2021) 11:152. doi: 10.1038/s41408-021-00547-8
24. Porwit A and Saft L. The AML–MDS interface—leukemic transformation in myelodysplastic syndromes. J Hematop. (2011) 4:69–79. doi: 10.1007/s12308-011-0088-6
25. Tazzari PL, Tabellini G, Bortul R, Papa V, Evangelisti C, Grafone T, et al. The insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 induces apoptosis in acute myeloid leukemia cells exhibiting autocrine insulin-like growth factor-I secretion. Leukemia. (2007) 21:886–96. doi: 10.1038/sj.leu.2404643
26. Ghobrial IM, Detappe A, Anderson KC, and Steensma DP. The bone-marrow niche in MDS and MGUS: implications for AML and MM. Nat Rev Clin Oncol. (2018) 15:219–33. doi: 10.1038/nrclinonc.2017.197
27. Weng H, Huang F, Yu Z, Chen Z, Prince E, Kang Y, et al. The m(6)A reader IGF2BP2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell. (2022) 40:1566–1582.e10. doi: 10.1016/j.ccell.2022.10.004
28. Basu R and Kopchick JJ. GH and IGF1 in cancer therapy resistance. Endocr Relat Cancer. (2023) 30(9):e220414. doi: 10.1530/ERC-22-0414
29. Wang Y, Bikle DD, and Chang W. Autocrine and paracrine actions of IGF-I signaling in skeletal development. Bone Res. (2013) 1:249–59. doi: 10.4248/BR201303003
30. Chapuis N, Tamburini J, Cornillet-Lefebvre P, Gillot L, Bardet V, Willems L, et al. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica. (2010) 95:415–23. doi: 10.3324/haematol.2009.010785
31. Yoshida T and Delafontaine P. Mechanisms of IGF-1-mediated regulation of skeletal muscle hypertrophy and atrophy. Cells. (2020) 9(9):1970. doi: 10.3390/cells9091970
32. Zhang W and Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. (2002) 12:9–18. doi: 10.1038/sj.cr.7290105
33. Yeo CD, Kim YA, Lee HY, Kim JW, Lee SH, Kim SJ, et al. Inhibiting IGF-1R attenuates cell proliferation and VEGF production in IGF-1R over-expressing EGFR mutant non-small cell lung cancer cells. Exp Lung Res. (2017) 43:29–37. doi: 10.1080/01902148.2017.1282994
34. Wu Q, Tian AL, Li B, Leduc M, Forveille S, Hamley P, et al. IGF1 receptor inhibition amplifies the effects of cancer drugs by autophagy and immune-dependent mechanisms. J Immunother Cancer. (2021) 9(6):e002722. doi: 10.1136/jitc-2021-002722
35. Mazziotti G, Lania AG, and Canalis E. Skeletal disorders associated with the growth hormone-insulin-like growth factor 1 axis. Nat Rev Endocrinol. (2022) 18:353–65. doi: 10.1038/s41574-022-00649-8
36. Wang B, Xu Y, Liu X, Liu Q, Liu Y, Zhang Y, et al. Molecular characterization and expression profiles of insulin-like growth factors in yellowtail kingfish (Seriola lalandi) during embryonic development. Fish Physiol Biochem. (2019) 45:375–90. doi: 10.1007/s10695-018-0570-5
37. Simpson A, Petnga W, Macaulay VM, Weyer-Czernilofsky U, and Bogenrieder T. Insulin-like growth factor (IGF) pathway targeting in cancer: role of the IGF axis and opportunities for future combination studies. Target Oncol. (2017) 12:571–97. doi: 10.1007/s11523-017-0514-5
38. Guo B, Lv Z, Cui C, and Wang W. IGF-1R transported to the cell nuclei to regulate the proliferation of breast cancer cells. Cell Biochem Biophys. (2021) 79:801–13. doi: 10.1007/s12013-021-00989-8
39. Cao Y, Nimptsch K, Shui IM, Platz EA, Wu K, Pollak MN, et al. Prediagnostic plasma IGFBP-1, IGF-1 and risk of prostate cancer. Int J Cancer. (2015) 136:2418–26. doi: 10.1002/ijc.v136.10
40. Nieto-Estévez V, Defterali Ç, and Vicario-Abejón C. IGF-I: A key growth factor that regulates neurogenesis and synaptogenesis from embryonic to adult stages of the brain. Front Neurosci. (2016) 10:52. doi: 10.3389/fnins.2016.00052
41. Ungewitter E and Scrable H. Delta40p53 controls the switch from pluripotency to differentiation by regulating IGF signaling in ESCs. Genes Dev. (2010) 24:2408–19. doi: 10.1101/gad.1987810
42. Wang Z, Wang Z, Liang Z, Liu J, Shi W, Bai P, et al. Expression and clinical significance of IGF-1, IGFBP-3, and IGFBP-7 in serum and lung cancer tissues from patients with non-small cell lung cancer. Onco Targets Ther. (2013) 6:1437–44. doi: 10.2147/OTT.S51997
43. Enriori PJ, Fischer CR, Gori JR, Etkin AE, Calandra RS, and Lüthy IA. Augmented serum levels of the IGF-I/IGF-binding protein-3 ratio in pre-menopausal patients with type I breast cysts. Eur J Endocrinol. (2003) 148:177–84. doi: 10.1530/eje.0.1480177
44. Samani AA and Brodt P. The receptor for the type I insulin-like growth factor and its ligands regulate multiple cellular functions that impact on metastasis. Surg Oncol Clin N Am. (2001) 10:289–312, viii. doi: 10.1016/S1055-3207(18)30066-8
45. Lopez-Tello J, Yong H, Sandovici I, et al. Fetal manipulation of maternal metabolism is a critical function of the imprinted Igf2 gene. Cell Metab. (2023) 35:1195–1208.e6. doi: 10.1016/j.cmet.2023.06.007
46. Kasprzak A and Adamek A. Insulin-like growth factor 2 (IGF2) signaling in colorectal cancer-from basic research to potential clinical applications. Int J Mol Sci. (2019) 20(19):4915. doi: 10.3390/ijms20194915
47. Zhang B, Brahma RK, Zhu L, Feng J, Hu S, Qian L, et al. Insulin-like growth factor 2 (IGF2)-fused lysosomal targeting chimeras for degradation of extracellular and membrane proteins. J Am Chem Soc. (2023) 145:24272–83. doi: 10.1021/jacs.3c08886
48. Zhu XY, Liu WT, Hou XJ, Zong C, Yu W, Shen ZM, et al. CD34(+)CLDN5(+) tumor associated senescent endothelial cells through IGF2-IGF2R signaling increased cholangiocellular phenotype in hepatocellular carcinoma. J Adv Res. (2024) 12:S2090-1232(24)00564-2. doi: 10.1016/j.jare.2024.12.008
49. White MF. Insulin signaling in health and disease. Science. (2003) 302:1710–1. doi: 10.1126/science.1092952
50. Conover CA. The IGF-p53 connection in cancer. Growth Horm IGF Res. (2018) 39:25–8. doi: 10.1016/j.ghir.2017.11.007
51. Yang Y, Chen X, and Ma C. Insulin receptor is implicated in triple-negative breast cancer by decreasing cell mobility. J BioMed Res. (2020) 35:189–96. doi: 10.7555/JBR.34.20200082
52. Pohlman AW, Moudgalya H, Jordano L, Lobato GC, Gerard D, Liptay MJ, et al. The role of IGF-pathway biomarkers in determining risks, screening, and prognosis in lung cancer. Oncotarget. (2022) 13:393–407. doi: 10.18632/oncotarget.28202
53. Yang C, Tao H, Zhang H, Xia Y, Bai J, Ge G, et al. TET2 regulates osteoclastogenesis by modulating autophagy in OVX-induced bone loss. Autophagy. (2022) 18:2817–29. doi: 10.1080/15548627.2022.2048432
54. Thomas D and Radhakrishnan P. Role of tumor and stroma-derived IGF/IGFBPs in pancreatic cancer. Cancers (Basel). (2020) 12(5):1228. doi: 10.3390/cancers12051228
55. Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer. (2014) 14:329–41. doi: 10.1038/nrc3720
56. Reiss K, Ferber A, Travali S, Porcu P, Phillips PD, and Baserga R. The protooncogene c-myb increases the expression of insulin-like growth factor 1 and insulin-like growth factor 1 receptor messenger RNAs by a transcriptional mechanism. Cancer Res. (1991) 51:5997–6000.
57. Jung HJ and Suh Y. Regulation of IGF -1 signaling by microRNAs. Front Genet. (2014) 5:472. doi: 10.3389/fgene.2014.00472
58. Wahner HA, Haluska P, Schneider PA, Loegering DA, Peterson KL, Attar R, et al. Expression of insulin receptor isoform A and insulin-like growth factor-1 receptor in human acute myelogenous leukemia: effect of the dual-receptor inhibitor BMS-536924 in vitro. Cancer Res. (2009) 69:7635–43. doi: 10.1158/0008-5472.CAN-09-0511
59. Chen PC, Kuo YC, Chuong CM, and Huang YH. Niche modulation of IGF-1R signaling: its role in stem cell pluripotency, cancer reprogramming, and therapeutic applications. Front Cell Dev Biol. (2020) 8:625943. doi: 10.3389/fcell.2020.625943
60. Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA, Ling TY, et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat Commun. (2014) 5:3472. doi: 10.1038/ncomms4472
61. Kuse Y, Matsumoto S, Tsuzuki S, Carolina E, Okumura T, Kasai T, et al. Placenta-derived factors contribute to human iPSC-liver organoid growth. Nat Commun. (2025) 16:2493. doi: 10.1038/s41467-025-57551-w
62. Liu GL, Jiang SW, Xiong YZ, and Qu YC. The character of heredity and biological function of insulin-like growth factor 2. Yi Chuan. (2002) 24:211–3.
63. Chen W, Wu C, Li Y, Wang T, Huang M, Wang M, et al. Mir-483-5p-mediated activating of IGF2/H19 enhancer up-regulates IGF2/H19 expression via chromatin loops to promote the Malignant progression of hepatocellular carcinoma. Mol Cancer. (2025) 24:10. doi: 10.1186/s12943-024-02204-7
64. Pan Y, He B, Lirong Z, Nie Z, Chen L, Gu L, et al. Gene therapy for cancer through adenovirus vector−mediated expression of the Ad5 early region gene 1A based on loss of IGF2 imprinting. Oncol Rep. (2013) 30:1814–22. doi: 10.3892/or.2013.2646
65. Sélénou C, Brioude F, Giabicani E, Sobrier ML, and Netchine I. IGF2: development, genetic and epigenetic abnormalities. Cells. (2022) 11(12):1886. doi: 10.3390/cells11121886
66. Alberini CM. IGF2 in memory, neurodevelopmental disorders, and neurodegenerative diseases. Trends Neurosci. (2023) 46:488–502. doi: 10.1016/j.tins.2023.03.007
67. Lee J and Pilch PF. The insulin receptor: structure, function, and signaling. Am J Physiol. (1994) 266:C319–34. doi: 10.1152/ajpcell.1994.266.2.C319
68. Lizcano JM and Alessi DR. The insulin signalling pathway. Curr Biol. (2002) 12:R236–8. doi: 10.1016/S0960-9822(02)00777-7
69. Gorgisen G, Gulacar IM, and Ozes ON. The role of insulin receptor substrate (IRS) proteins in oncogenic transformation. Cell Mol Biol (Noisy-le-grand). (2017) 63:1–5. doi: 10.14715/cmb/2017.63.1.1
70. Pomytkin I, Costa-Nunes JP, Kasatkin V, Veniaminova E, Demchenko A, Lyundup A, et al. Insulin receptor in the brain: Mechanisms of activation and the role in the CNS pathology and treatment. CNS Neurosci Ther. (2018) 24:763–74. doi: 10.1111/cns.2018.24.issue-9
71. Hu Q, Zhou Y, Ying K, and Ruan W. IGFBP, a novel target of lung cancer? Clin Chim Acta. (2017) 466:172–7. doi: 10.1016/j.cca.2017.01.017
72. Ding H and Wu T. Insulin-like growth factor binding proteins in autoimmune diseases. Front Endocrinol (Lausanne). (2018) 9:499. doi: 10.3389/fendo.2018.00499
73. Walenkamp MJE and Wit JM. Genetic disorders in the GH IGF-I axis in mouse and man. Eur J Endocrinol. (2007) 157 Suppl 1:S15–26. doi: 10.1530/EJE-07-0148
74. Adamson C, Welsh P, Docherty KF, de Boer RA, Diez M, Drożdż J, et al. IGFBP-7 and outcomes in heart failure with reduced ejection fraction: findings from DAPA-HF. JACC heart fail. (2023) 11:291–304. doi: 10.1016/j.jchf.2022.09.004
75. Huynh H, Kaba M, Rudra S, Zheng J, Wu CJ, Lodish HF, et al. IGFBP2 Supports ex vivo Expansion of Hematopoietic Stem Cells. IGFs: Local Repair Survival Fact Throughout Life Span. (2010) 2010:21–41. doi: 10.1007/978-3-642-04302-4_3
76. Monzavi R and Cohen P. IGFs and IGFBPs: role in health and disease. Best Pract Res Clin Endocrinol Metab. (2002) 16:433–47. doi: 10.1053/beem.2002.0212
77. Daza DO, Sundström G, Bergqvist CA, Duan C, and Larhammar D. Evolution of the insulin-like growth factor binding protein (IGFBP) family. Endocrinology. (2011) 152:2278–89. doi: 10.1210/en.2011-0047
78. Chander H, Halpern M, Resnick-Silverman L, Manfredi JJ, and Germain D. Skp2B overexpression alters a prohibitin-p53 axis and the transcription of PAPP-A, the protease of insulin-like growth factor binding protein 4. PloS One. (2011) 6:e22456. doi: 10.1371/journal.pone.0022456
79. Conover CA. Key questions and answers about pregnancy-associated plasma protein-A. Trends Endocrinol Metab. (2012) 23:242–9. doi: 10.1016/j.tem.2012.02.008
80. Laursen LS, Kjaer-Sorensen K, Andersen MH, and Oxvig C. Regulation of insulin-like growth factor (IGF) bioactivity by sequential proteolytic cleavage of IGF binding protein-4 and -5. Mol Endocrinol. (2007) 21:1246–57. doi: 10.1210/me.2006-0522
81. Sun S, Li Y, Li Y, Niu Y, Hu Z, Deng C, et al. Delayed administration of IGFBP7 improved bone defect healing via ZO-1 dependent vessel stabilization. Adv Sci (Weinh). (2025) 12:e2406965. doi: 10.1002/advs.202406965
82. Yang DH, Kim HS, Wilson EM, Rosenfeld RG, and Oh Y. Identification of glycosylated 38-kDa connective tissue growth factor (IGFBP-related protein 2) and proteolytic fragments in human biological fluids, and up-regulation of IGFBP-rP2 expression by TGF-beta in Hs578T human breast cancer cells. J Clin Endocrinol Metab. (1998) 83:2593–6. doi: 10.1210/jcem.83.7.5097
83. Tao L, Chen J, Zhou H, Qin C, Li P, Cao Q, et al. A functional polymorphism in the CYR61 (IGFBP10) gene is associated with prostate cancer risk. Prost Cancer Prost Dis. (2013) 16:95–100. doi: 10.1038/pcan.2012.41
84. Li D, Xia L, Huang P, Wang Z, Guo Q, Huang C, et al. Cancer-associated fibroblast-secreted IGFBP7 promotes gastric cancer by enhancing tumor associated macrophage infiltration via FGF2/FGFR1/PI3K/AKT axis. Cell Death Discov. (2023) 9:17. doi: 10.1038/s41420-023-01336-x
85. Khezri MR, Jafari R, Yousefi K, and Zolbanin NM. The PI3K/AKT signaling pathway in cancer: Molecular mechanisms and possible therapeutic interventions. Exp Mol Pathol. (2022) 127:104787. doi: 10.1016/j.yexmp.2022.104787
86. He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW, et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther. (2021) 6:425. doi: 10.1038/s41392-021-00828-5
87. Hermida MA, Dinesh Kumar J, and Leslie NR. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv Biol Regul. (2017) 65:5–15. doi: 10.1016/j.jbior.2017.06.003
88. Lin A, Piao H, Zhuang L, Sarbassov DD, Ma L, and Gan B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacologic inhibition of the PI3K-AKT pathway. Cancer Res. (2014) 74:1682–93. doi: 10.1158/0008-5472.CAN-13-1729
89. Fulcher LJ, Sobajima T, Batley C, Gibbs-Seymour I, and Barr FA. MDM2 functions as a timer reporting the length of mitosis. Nat Cell Biol. (2025) 27:262–72. doi: 10.1038/s41556-024-01592-8
90. Bergholz JS, Wang Q, Wang Q, Ramseier M, Prakadan S, Wang W, et al. PI3Kβ controls immune evasion in PTEN-deficient breast tumours. Nature. (2023) 617:139–46. doi: 10.1038/s41586-023-05940-w
91. Ueda K, Kumari R, Schwenger E, Wheat JC, Bohorquez O, Narayanagari SR, et al. MDMX acts as a pervasive preleukemic-to-acute myeloid leukemia transition mechanism. Cancer Cell. (2021) 39:529–547.e7. doi: 10.1016/j.ccell.2021.02.006
92. Glaviano A, Foo A, Lam HY, Fan Z, Zhang J, He G, et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. (2023) 22:138. doi: 10.1186/s12943-023-01827-6
93. Li H, Wen X, Ren Y, Fan Z, Zhang J, He G, et al. Targeting PI3K family with small-molecule inhibitors in cancer therapy: current clinical status and future directions. Mol Cancer. (2024) 23:164. doi: 10.1186/s12943-024-02072-1
94. Li Y, Guo Y, Feng Z, Bergan R, Li B, Qin Y, et al. Involvement of the PI3K/akt/nrf2 signaling pathway in resveratrol-mediated reversal of drug resistance in HL-60/ADR cells. Nutr Cancer. (2019) 71:1007–18. doi: 10.1080/01635581.2019.1578387
95. Tlili H, Macovei A, Buonocore D, Lanzafame M, Najjaa H, Lombardi A, et al. The polyphenol/saponin-rich Rhus tripartita extract has an apoptotic effect on THP-1 cells through the PI3K/AKT/mTOR signaling pathway. BMC Complement Med Ther. (2021) 21:153. doi: 10.1186/s12906-021-03328-9
96. Nyåkern M, Tazzari PL, Finelli C, Bosi C, Follo MY, Grafone T, et al. Frequent elevation of Akt kinase phosphorylation in blood marrow and peripheral blood mononuclear cells from high-risk myelodysplastic syndrome patients. Leukemia. (2006) 20:230–8. doi: 10.1038/sj.leu.2404057
97. Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell. (2017) 170:1079–1095.e20. doi: 10.1016/j.cell.2017.07.032
98. Abdel-Wahab O, Patel J, and Levine RL. Clinical implications of novel mutations in epigenetic modifiers in AML. Hematol Oncol Clin North Am. (2011) 25:1119–33. doi: 10.1016/j.hoc.2011.09.013
99. Shen Q, Zhang Q, Shi Y, Shi Q, Jiang Y, Gu Y, et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature. (2018) 554:123–7. doi: 10.1038/nature25434
100. Bertacchini J, Heidari N, Mediani L, Capitani S, Shahjahani M, Ahmadzadeh A, et al. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol Life Sci. (2015) 72:2337–47. doi: 10.1007/s00018-015-1867-5
101. Asati V, Mahapatra DK, and Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur J Med Chem. (2016) 109:314–41. doi: 10.1016/j.ejmech.2016.01.012
102. Bahar ME, Kim HJ, and Kim DR. Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies. Signal Transduct Target Ther. (2023) 8:455. doi: 10.1038/s41392-023-01705-z
103. Ritt DA, Abreu-Blanco MT, Bindu L, Durrant DE, Zhou M, Specht SI, et al. Inhibition of ras/raf/MEK/ERK pathway signaling by a stress-induced phospho-regulatory circuit. Mol Cell. (2016) 64:875–87. doi: 10.1016/j.molcel.2016.10.029
104. Geest CR and Coffer PJ. MAPK signaling pathways in the regulation of hematopoiesis. J Leukoc Biol. (2009) 86:237–50. doi: 10.1189/jlb.0209097
105. Huang Y, Zhen Y, Chen Y, Sui S, and Zhang L. Unraveling the interplay between RAS/RAF/MEK/ERK signaling pathway and autophagy in cancer: From molecular mechanisms to targeted therapy. Biochem Pharmacol. (2023) 217:115842. doi: 10.1016/j.bcp.2023.115842
106. Patnaik MM and Tefferi A. Chronic myelomonocytic leukemia: 2024 update on diagnosis, risk stratification and management. Am J Hematol. (2024) 99:1142–65. doi: 10.1002/ajh.27271
107. Zhang Y, Wu J, Qin T, Xu Z, Qu S, Pan L, et al. Comparison of the revised 4th (2016) and 5th (2022) editions of the World Health Organization classification of myelodysplastic neoplasms. Leukemia. (2022) 36:2875–82. doi: 10.1038/s41375-022-01718-7
108. Samatar AA and Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. (2014) 13:928–42. doi: 10.1038/nrd4281
109. Sun J, Zhao W, Zhang L, Wu S, Xue S, Cao H, et al. Centromere protein U mediates the ubiquitination and degradation of RPS3 to facilitate temozolomide resistance in glioblastoma. Drug Resist Updat. (2025) 80:101214. doi: 10.1016/j.drup.2025.101214
110. Akoumianakis I, Polkinghorne M, and Antoniades C. Non-canonical WNT signalling in cardiovascular disease: mechanisms and therapeutic implications. Nat Rev Cardiol. (2022) 19:783–97. doi: 10.1038/s41569-022-00718-5
111. Yu F, Yu C, Li F, Zuo Y, Wang Y, Yao L, et al. Wnt/β-catenin signaling in cancers and targeted therapies. Signal Transduct Target Ther. (2021) 6:307. doi: 10.1038/s41392-021-00701-5
112. Lapointe E, Boyer A, Rico C, Paquet M, Franco HL, Gossen J, et al. FZD1 regulates cumulus expansion genes and is required for normal female fertility in mice. Biol Reprod. (2012) 87:104. doi: 10.1095/biolreprod.112.102608
113. Siegle L, Schwab JD, Kühlwein SD, Lausser L, Tümpel S, Pfister AS, et al. A Boolean network of the crosstalk between IGF and Wnt signaling in aging satellite cells. PloS One. (2018) 13:e0195126. doi: 10.1371/journal.pone.0195126
114. Playford MP, Bicknell D, Bodmer WF, and Macaulay VM. Insulin-like growth factor 1 regulates the location, stability, and transcriptional activity of beta-catenin. Proc Natl Acad Sci U S A. (2000) 97:12103–8. doi: 10.1073/pnas.210394297
115. Yan H, Wang Z, Sun Y, Hu L, and Bu P. Cytoplasmic NEAT1 suppresses AML stem cell self-renewal and leukemogenesis through inactivation of wnt signaling. Adv Sci (Weinh). (2021) 8:e2100914. doi: 10.1002/advs.202100914
116. Mueller J, Schimmer RR, Koch C, Schneiter F, Fullin J, Lysenko V, et al. Targeting the mevalonate or Wnt pathways to overcome CAR T-cell resistance in TP53-mutant AML cells. EMBO Mol Med. (2024) 16:445–74. doi: 10.1038/s44321-024-00024-2
117. Sicurella M, De Chiara M, and Neri LM. Hedgehog and PI3K/akt/mTOR signaling pathways involvement in leukemic Malignancies: crosstalk and role in cell death. Cells. (2025) 14(4):269. doi: 10.3390/cells14040269
118. Clara JA, Monge C, Yang Y, and Takebe N. Targeting signalling pathways and the immune microenvironment of cancer stem cells - a clinical update. Nat Rev Clin Oncol. (2020) 17:204–32. doi: 10.1038/s41571-019-0293-2
119. Han Y, Zhou M, Wang B, and Jiang J. Morphogen-induced kinase condensates transduce Hh signal by allosterically activating Gli. Sci Adv. (2025) 11:eadq1790. doi: 10.1126/sciadv.adq1790
120. Queiroz KC, Ruela-de-Sousa RR, Fuhler GM, Aberson HL, Ferreira CV, Peppelenbosch MP, et al. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene. (2010) 29:6314–22. doi: 10.1038/onc.2010.375
121. Lainez-González D, Serrano-López J, and Alonso-Domínguez JM. Understanding the hedgehog signaling pathway in acute myeloid leukemia stem cells: A necessary step toward a cure. Biol (Basel). (2021) 10(4):255. doi: 10.3390/biology10040255
122. Qiu ZP, Lin ZC, Hu A, Liu YB, Zeng WE, Zhao X, et al. GRAMD1/ASTER-mediated cholesterol transport promotes Smoothened cholesterylation at the endoplasmic reticulum. EMBO J. (2023) 42:e111513. doi: 10.15252/embj.2022111513
123. Sun H, Li L, Li W, Yang F, Zhang Z, Liu Z, et al. p53 transcriptionally regulates SQLE to repress cholesterol synthesis and tumor growth. EMBO Rep. (2021) 22:e52537. doi: 10.15252/embr.202152537
124. Long B, Wang L, Zheng F, Lai S, Xu D, Hu Y, et al. Targeting GLI1 suppresses cell growth and enhances chemosensitivity in CD34+ Enriched acute myeloid leukemia progenitor cells. Cell Physiol Biochem. (2016) 38:1288–302. doi: 10.1159/000443075
125. Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. (2009) 458:776–9. doi: 10.1038/nature07737
126. Katagiri S, Tauchi T, Okabe S, Minami Y, Kimura S, Maekawa T, et al. Combination of ponatinib with Hedgehog antagonist vismodegib for therapy-resistant BCR-ABL1-positive leukemia. Clin Cancer Res. (2013) 19:1422–32. doi: 10.1158/1078-0432.CCR-12-1777
127. Matz-Soja M, Aleithe S, Marbach E, Böttger J, Arnold K, Schmidt-Heck W, et al. Hepatic Hedgehog signaling contributes to the regulation of IGF1 and IGFBP1 serum levels. Cell Commun Signal. (2014) 12:11. doi: 10.1186/1478-811X-12-11
128. Shallis RM, Bewersdorf JP, Boddu PC, and Zeidan AM. Hedgehog pathway inhibition as a therapeutic target in acute myeloid leukemia. Expert Rev Anticancer Ther. (2019) 19:717–29. doi: 10.1080/14737140.2019.1652095
129. Thompson DL and Moore DC. Glasdegib: A novel hedgehog pathway inhibitor for acute myeloid leukemia. J Adv Pract Oncol. (2020) 11:196–200. doi: 10.6004/jadpro.2020.11.2.8
130. Huang K, Ding B, Zhong Q, Jiang X, Li X, Wang Z, et al. Hh/IGF-1R/PI3K/Akt/MRP1 pathway induce refractory acute myeloid leukemia and its targeting therpy. Molecular Pharmacology and Drug Resistance in Myeloid Diseases: Poster III. (2014). doi: 10.1182/blood.V124.21.3612.3612
131. Peng Y, Li F, Zhang P, Wang X, Shen Y, Feng Y, et al. IGF-1 promotes multiple myeloma progression through PI3K/Akt-mediated epithelial-mesenchymal transition. Life Sci. (2020) 249:117503. doi: 10.1016/j.lfs.2020.117503
132. Qiang YW, Yao L, Tosato G, and Rudikoff S. Insulin-like growth factor I induces migration and invasion of human multiple myeloma cells. Blood. (2004) 103:301–8. doi: 10.1182/blood-2003-06-2066
133. Rø TB, Holien T, Fagerli UM, Hov H, Misund K, Waage A, et al. HGF and IGF-1 synergize with SDF-1α in promoting migration of myeloma cells by cooperative activation of p21-activated kinase. Exp Hematol. (2013) 41:646–55. doi: 10.1016/j.exphem.2013.03.002
134. Moonesi M, Mehdizade H, Zakakhosravi S, Molaei Ramshe S, Allahbakhshian M, and Solali S. IGF gene family expression: A comparative study in acute myeloid leukemia (AML) and healthy bone marrow. Iran J Blood Cancer. (2024) 16:32–9. doi: 10.61186/ijbc.16.3.32
135. Moonesi M, Omran HM, Khosravi SZ, Ramshe SM, Allahbakhshian Farsani M, Solali S, et al. Comparison of the relative expression of the IGF gene family in the bone marrow of patients with AML compared with normal individuals. (2023). doi: 10.21203/rs.3.rs-2989442/v1
136. Giallongo S, Duminuco A, Dulcamare I, Zuppelli T, La Spina E, Scandura G, et al. Engagement of mesenchymal stromal cells in the remodeling of the bone marrow microenvironment in hematological cancers. Biomolecules. (2023) 13. doi: 10.3390/biom13121701
137. Wang A and Zhong H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology. (2018) 23:729–39. doi: 10.1080/10245332.2018.1486064
138. Azizidoost S, Babashah S, Rahim F, Shahjahani M, and Saki N. Bone marrow neoplastic niche in leukemia. Hematology. (2014) 19:232–8. doi: 10.1179/1607845413Y.0000000111
139. Yin C, Yan X, Ren J, Zhang C, Liu J, Wang Z, et al. MSCs with upregulated lipid metabolism block hematopoietic stem cell differentiation via exosomal CTP-1A in MDS. Stem Cell Res Ther. (2025) 16:53. doi: 10.1186/s13287-025-04154-3
140. Hou J, Peng X, Wang J, Zhang H, Xia J, Ge Q, et al. Mesenchymal stem cells promote endothelial progenitor cell proliferation by secreting insulin−like growth factor−1. Mol Med Rep. (2017) 16:1502–8. doi: 10.3892/mmr.2017.6741
141. Brenner AK, Andersson TT, and Bruserud Ø. The complexity of targeting PI3K-akt-mTOR signalling in human acute myeloid leukaemia: the importance of leukemic cell heterogeneity, neighbouring mesenchymal stem cells and immunocompetent cells. Molecules. (2016) 21(11):1512. doi: 10.3390/molecules21111512
142. Youssef A, Aboalola D, and Han VK. The roles of insulin-like growth factors in mesenchymal stem cell niche. Stem Cells Int. (2017) 2017:9453108. doi: 10.1155/2017/9453108
143. Yulyana Y, Ho IA, Sia KC, Newman JP, Toh XY, Endaya BB, et al. Paracrine factors of human fetal MSCs inhibit liver cancer growth through reduced activation of IGF-1R/PI3K/Akt signaling. Mol Ther. (2015) 23:746–56. doi: 10.1038/mt.2015.13
144. Lv J, Liu C, Chen FK, Feng ZP, Jia L, Liu PJ, et al. M2−like tumour−associated macrophage−secreted IGF promotes thyroid cancer stemness and metastasis by activating the PI3K/AKT/mTOR pathway. Mol Med Rep. (2021) 24(2):604. doi: 10.3892/mmr.2021.12249
145. Moonesi M, Zaka Khosravi S, Molaei Ramshe S, Allahbakhshian Farsani M, Solali S, Mohammadi MH, et al. IGF family effects on development, stability, and treatment of hematological Malignancies. J Cell Physiol. (2021) 236:4097–105. doi: 10.1002/jcp.v236.6
146. Kuang HY, Zou W, Zhang GB, Gao XY, and Yin HQ. The relationship of blood glucose and the expression of insulin-like growth factor-1 receptor mRNA in diabetic cardiomyopathy: a preliminary study with rats. Zhonghua Yi Xue Za Zhi. (2007) 87:1249–51.
147. Zhou H, Rao J, Lin J, Yin B, Sheng H, Lin F, et al. The insulin-like growth factor-I receptor kinase inhibitor NVP-ADW742 sensitizes medulloblastoma to the effects of chemotherapy. Oncol Rep. (2011) 25:1565–71. doi: 10.3892/or.2011.1233
148. Bouronikou E, Georgoulias P, Giannakoulas N, Valotassiou V, Palassopoulou M, Vassilopoulos G, et al. Metabolism-related cytokine and hormone levels in the serum of patients with myelodysplastic syndromes. Acta Haematol. (2013) 130:27–33. doi: 10.1159/000345427
149. Wiese W, Barczuk J, Racinska O, Siwecka N, Rozpedek-Kaminska W, Slupianek A, et al. PI3K/akt/mTOR signaling pathway in blood Malignancies-new therapeutic possibilities. Cancers (Basel). (2023) 15(21):5297. doi: 10.3390/cancers15215297
150. Darici S, Alkhaldi H, Horne G, Jørgensen HG, Marmiroli S, and Huang X. Targeting PI3K/akt/mTOR in AML: rationale and clinical evidence. J Clin Med. (2020) 9(9):2934. doi: 10.3390/jcm9092934
151. Zhang JM, Wang CC, Zhang GC, Jiang Q, Yang SM, Fu HX, et al. ADAM28 promotes tumor growth and dissemination of acute myeloid leukemia through IGFBP-3 degradation and IGF-I-induced cell proliferation. Cancer Lett. (2019) 442:193–201. doi: 10.1016/j.canlet.2018.10.028
152. Sumbly V, Landry I, Sneed C, Iqbal Q, Verma A, Dhokhar T, et al. Leukemic stem cells and advances in hematopoietic stem cell transplantation for acute myeloid leukemia: a narrative review of clinical trials. Stem Cell Investig. (2022) 9:10. doi: 10.21037/sci-2022-044
153. Niedzielska E, Wójcik D, Barg E, Pietras W, Sega-Pondel D, Doroszko A, et al. Evaluation of selected endocrine complications in patients treated with auto- and allo-haematopoietic stem cell transplantation. Med Wieku Rozwoj. (2008) 12:761–6.
154. Liu F, Ye S, Zhao L, and Niu Q. The role of IGF/IGF-1R signaling in the regulation of cancer stem cells. Clin Transl Oncol. (2024) 26:2924–34. doi: 10.1007/s12094-024-03561-x
155. He Q, Li X, Zhang Z, Zhang QX, and Yang LP. The type I insulin-like growth factor receptor highly expressed in clonal cells of myelodysplastic syndromes. Zhonghua Xue Ye Xue Za Zhi. (2011) 32:744–7.
156. He Q, Li X, Zhang Z, Zhang Q, Xu F, Yang L, et al. Overexpression of IGF-IR in Malignant clonal cells in bone marrow of myelodysplastic syndromes. Cancer Invest. (2010) 28:983–8. doi: 10.3109/07357907.2010.489537
157. He Q, Li X, Tao Y, Liu YZ, Yang LP, and Ying SX. Expression of insulin-like growth factor receptor type I in marrow nucleated cells from hematologic Malignancies and its anti-apoptotic effect. Zhongguo Shi Yan Xue Ye Xue Za Zhi. (2005) 13:483–7.
158. He Q, Chang CK, Xu F, Zhang QX, Shi WH, and Li X. Purification of bone marrow clonal cells from patients with myelodysplastic syndrome via IGF-IR. PloS One. (2015) 10:e0140372. doi: 10.1371/journal.pone.0140372
159. He Q, Zheng Q, Xu F, Shi W, Guo J, Zhang Z, et al. IGF−IR promotes clonal cell proliferation in myelodysplastic syndromes via inhibition of the MAPK pathway. Oncol Rep. (2020) 44:1094–104. doi: 10.3892/or.2020.7652
160. Gharaee N. Overcoming lenalidomide resistance in del (5q) myelodysplastic syndrome. The University of British Columbia (2022). doi: 10.14288/1.0415838
161. Gharaee N, Wegrzyn-Woltosz J, Jiang J, Akhade VS, Bridgers J, Stubbins RJ, et al. Haploinsufficiency of miR-143 and miR-145 reveal targetable dependencies in resistant del(5q) myelodysplastic neoplasm. Leukemia. (2025) 39(4):917–28. doi: 10.1038/s41375-025-02537-2
162. Qi H, Xiao L, Lingyun W, Ying T, Yi-Zhi L, Shao-Xu Y, et al. Expression of type 1 insulin-like growth factor receptor in marrow nucleated cells in Malignant hematological disorders: correlation with apoptosis. Ann Hematol. (2006) 85:95–101. doi: 10.1007/s00277-005-0031-y
163. Chen L, Jiang X, Chen H, Han Q, Liu C, and Sun M. RETRACTED ARTICLE: microRNA-628 inhibits the proliferation of acute myeloid leukemia cells by directly targeting IGF-1R. Onco Targets Ther. (2019) 2019:907–19.doi: 10.2147/OTT.S192137
164. Weisberg E, Nonami A, Chen Z, Nelson E, Chen Y, Liu F, et al. Upregulation of IGF1R by mutant RAS in leukemia and potentiation of RAS signaling inhibitors by small-molecule inhibition of IGF1R. Clin Cancer Res. (2014) 20:5483–95. doi: 10.1158/1078-0432.CCR-14-0902
165. Whelan JT, Ludwig DL, and Bertrand FE. HoxA9 induces insulin-like growth factor-1 receptor expression in B-lineage acute lymphoblastic leukemia. Leukemia. (2008) 22:1161–9. doi: 10.1038/leu.2008.57
166. Wang S, Wang X, Shen K, Wei C, and Li J. Insulin-like growth factor 1 receptor inhibits the proliferation of acute myeloid leukaemia cells via NK cell activation. Ann Hematol. (2023) 102:2353–64. doi: 10.1007/s00277-023-05378-0
167. Nepstad I, Hatfield KJ, Grønningsæter IS, and Reikvam H. The PI3K-akt-mTOR signaling pathway in human acute myeloid leukemia (AML) cells. Int J Mol Sci. (2020) 21(8):2907. doi: 10.3390/ijms21082907
168. Doepfner KT, Boller D, and Arcaro A. Targeting receptor tyrosine kinase signaling in acute myeloid leukemia. Crit Rev Oncol Hematol. (2007) 63:215–30. doi: 10.1016/j.critrevonc.2007.05.005
169. Zhang J, Huang FF, Wu DS, Li WJ, Zhan HE, Peng MY, et al. SUMOylation of insulin-like growth factor 1 receptor, promotes proliferation in acute myeloid leukemia. Cancer Lett. (2015) 357:297–306. doi: 10.1016/j.canlet.2014.11.052
170. Tamburini J, Chapuis N, Bardet V, Park S, Sujobert P, Willems L, et al. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood. (2008) 111:379–82. doi: 10.1182/blood-2007-03-080796
171. Tessema M, Länger F, Bock O, Seltsam A, Metzig K, Hasemeier B, et al. Down-regulation of the IGF-2/H19 locus during normal and Malignant hematopoiesis is independent of the imprinting pattern. Int J Oncol. (2005) 26:499–507. doi: 10.3892/ijo.26.2.499
172. Xu D, Wang Y, Zhou P, Qin S, Zhang R, Zhang Y, et al. The IGF2/IGF1R/Nanog signaling pathway regulates the proliferation of acute myeloid leukemia stem cells. Front Pharmacol. (2018) 9:687. doi: 10.3389/fphar.2018.00687
173. Wilson H, Lesnikov V, Plymate SR, Ward J, and Deeg HJ. High IGFBP-3 levels in marrow plasma in early-stage MDS: effects on apoptosis and hemopoiesis. Leukemia. (2005) 19:580–5. doi: 10.1038/sj.leu.2403672
174. Dalamaga M, Karmaniolas K, Nikolaidou A, Chamberland J, Hsi A, Dionyssiou-Asteriou A, et al. Adiponectin and resistin are associated with risk for myelodysplastic syndrome, independently from the insulin-like growth factor-I (IGF-I) system. Eur J Cancer. (2008) 44:1744–53. doi: 10.1016/j.ejca.2008.04.015
175. Nabawy S, El-taweel M, Darwish AD, and Mokhtar D. Expression of insulin-like growth factor–binding protein-2 and 7 (IGFBP2 and IGFBP7) genes in acute myeloid leukemia. Journal of the Egyptian Society of Haematology & Research. (2018) 14:17–24.
176. Kühnl A, Kaiser M, Neumann M, Fransecky L, Heesch S, Radmacher M, et al. High expression of IGFBP2 is associated with chemoresistance in adult acute myeloid leukemia. Leuk Res. (2011) 35:1585–90. doi: 10.1016/j.leukres.2011.08.006
177. Dawczynski K, Steinbach D, Wittig S, Pfaffendorf N, Kauf E, and Zintl F. Expression of components of the IGF axis in childhood acute myelogenous leukemia. Pediatr Blood Cancer. (2008) 50:24–8. doi: 10.1002/pbc.21294
178. Dawczynski K, Kauf E, Schlenvoigt D, Gruhn B, Fuchs D, and Zintl F. Elevated serum insulin-like growth factor binding protein-2 is associated with a high relapse risk after hematopoietic stem cell transplantation in childhood AML. Bone Marrow Transplant. (2006) 37:589–94. doi: 10.1038/sj.bmt.1705281
179. Chen X, Zheng J, Zou Y, Song C, Hu X, and Zhang CC. IGF binding protein 2 is a cell-autonomous factor supporting survival and migration of acute leukemia cells. J Hematol Oncol. (2013) 6:72. doi: 10.1186/1756-8722-6-72
180. Ye H, Adane B, Khan N, Alexeev E, Nusbacher N, Minhajuddin M, et al. Subversion of systemic glucose metabolism as a mechanism to support the growth of leukemia cells. Cancer Cell. (2018) 34:659–673.e6. doi: 10.1016/j.ccell.2018.08.016
181. Karmali R, Larson ML, Shammo JM, Basu S, Christopherson K, Borgia JA, et al. Impact of insulin-like growth factor 1 and insulin-like growth factor binding proteins on outcomes in acute myeloid leukemia. Leuk Lymph. (2015) 56:3135–42. doi: 10.3109/10428194.2015.1022767
182. Verhagen H, van Gils N, Martiañez T, van Rhenen A, Rutten A, Denkers F, et al. IGFBP7 induces differentiation and loss of survival of human acute myeloid leukemia stem cells without affecting normal hematopoiesis. Cell Rep. (2018) 25:3021–3035.e5. doi: 10.1016/j.celrep.2018.11.062
183. Verhagen HJ, de Leeuw DC, Roemer MG, Denkers F, Pouwels W, Rutten A, et al. IGFBP7 induces apoptosis of acute myeloid leukemia cells and synergizes with chemotherapy in suppression of leukemia cell survival. Cell Death Dis. (2014) 5:e1300. doi: 10.1038/cddis.2014.268
184. Niu Y, Wu S, and Hu S. IGFBP7 as a potential therapeutic target for AML. Blood. (2018) 132:5134. doi: 10.1182/blood-2018-99-119528
185. Zhang B, Deng X, You R, Liu J, Hou D, Wang X, et al. Secreted insulin-like growth factor binding protein 5 functions as a tumor suppressor and chemosensitizer through inhibiting insulin-like growth factor 1 receptor/protein kinase B pathway in acute myeloid leukemia. Neoplasia. (2024) 47:100952. doi: 10.1016/j.neo.2023.100952
186. Liang S, Zhou X, Cai D, Rodrigues-Lima F, Chi J, and Wang L. Network pharmacology and experimental validation reveal the effects of chidamide combined with aspirin on acute myeloid leukemia-myelodysplastic syndrome cells through PI3K/AKT pathway. Front Cell Dev Biol. (2021) 9:685954. doi: 10.3389/fcell.2021.685954
187. Alkhateeb HB, Patnaik MM, Al-Kali A, Zblewski DL, Wallerich S, McTavish H, et al. Phase 1b study of IGF-methotrexate conjugate in the treatment of high-grade myelodysplastic syndromes. Anticancer Res. (2020) 40:3883–8. doi: 10.21873/anticanres.14378
188. Zhang S, Zhang Y, Zhuang Y, Zblewski DL, Wallerich S, McTavish H, et al. Matrine induces apoptosis in human acute myeloid leukemia cells via the mitochondrial pathway and Akt inactivation. PloS One. (2012) 7:e46853. doi: 10.1371/journal.pone.0046853
189. Doepfner KT, Spertini O, and Arcaro A. Autocrine insulin-like growth factor-I signaling promotes growth and survival of human acute myeloid leukemia cells via the phosphoinositide 3-kinase/Akt pathway. Leukemia. (2007) 21:1921–30. doi: 10.1038/sj.leu.2404813
190. He Y, Zhang J, Zheng J, Du W, Xiao H, Liu W, et al. The insulin-like growth factor-1 receptor kinase inhibitor, NVP-ADW742, suppresses survival and resistance to chemotherapy in acute myeloid leukemia cells. Oncol Res. (2010) 19:35–43. doi: 10.3727/096504010X12828372551821
191. Abe S, Funato T, Takahashi S, Yokoyama H, Yamamoto J, Tomiya Y, et al. Increased expression of insulin-like growth factor i is associated with Ara-C resistance in leukemia. Tohoku J Exp Med. (2006) 209:217–28. doi: 10.1620/tjem.209.217
192. Huang K, Sun Z, Ding B, Jiang X, Wang Z, Zhu Y, et al. Suppressing Hedgehog signaling reverses drug resistance of refractory acute myeloid leukemia. Onco Targets Ther. (2019) 12:7477–88. doi: 10.2147/OTT.S216628
193. Buck E, Gokhale PC, Koujak S, Brown E, Eyzaguirre A, Tao N, et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther. (2010) 9:2652–64. doi: 10.1158/1535-7163.MCT-10-0318
194. Gualberto A and Pollak M. Emerging role of insulin-like growth factor receptor inhibitors in oncology: early clinical trial results and future directions. Oncogene. (2009) 28:3009–21. doi: 10.1038/onc.2009.172
195. Bertrand FE, Steelman LS, Chappell WH, Abrams SL, Shelton JG, White ER, et al. Synergy between an IGF-1R antibody and Raf/MEK/ERK and PI3K/Akt/mTOR pathway inhibitors in suppressing IGF-1R-mediated growth in hematopoietic cells. Leukemia. (2006) 20:1254–60. doi: 10.1038/sj.leu.2404217
196. Jalte M, Abbassi M, El MH, Daha BH, Ahakoud M, and Bekkari H. FLT3 mutations in acute myeloid leukemia: unraveling the molecular mechanisms and implications for targeted therapies. Cureus. (2023) 15:e45765. doi: 10.7759/cureus.45765
197. Short NJ, Daver N, Dinardo CD, Kadia T, Nasr LF, Macaron W, et al. Azacitidine, venetoclax, and gilteritinib in newly diagnosed and relapsed or refractory FLT3-mutated AML. J Clin Oncol. (2024) 42:1499–508. doi: 10.1200/JCO.23.01911
198. Zhang R, Xu J, Shi Z, Zhang H, Zhang A, and Zhang Y. Identification of ANGPT2, FLT3, IGF1 and SPP1 related to glycolysis and PI3K/Akt signaling pathway in hepatocellular carcinoma. (2023). doi: 10.21203/rs.3.rs-3522986/v1
199. Nassereddine S, Lap CJ, Haroun F, and Tabbara I. The role of mutant IDH1 and IDH2 inhibitors in the treatment of acute myeloid leukemia. Ann Hematol. (2017) 96:1983–91. doi: 10.1007/s00277-017-3161-0
200. Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. (2017) 130:722–31. doi: 10.1182/blood-2017-04-779405
201. Chen CH, Chen PY, Lin YY, Feng LY, Chen SH, Chen CY, et al. Suppression of tumor growth via IGFBP3 depletion as a potential treatment in glioma. J Neurosurg. (2020) 132:168–79. doi: 10.3171/2018.8.JNS181217
202. Nasehi L, Abdolhossein ZB, Rahimi H, and Hossein GM. Radio-immunotherapy by (188)Re-antiCD20 and stable silencing of IGF-IR in Raji cells, new insight in treatment of lymphoma. Gene. (2023) 882:147638. doi: 10.1016/j.gene.2023.147638
Keywords: IGF-I, IGF-II, IGF receptor, IGF binding proteins, MDS, AML
Citation: Wang Y, Dong X, Tao S, Chen Q, Chen Y, Zhang L, Shi Y, He Z, Yu L and Wang C (2025) Advances in the role of the IGF signaling system in myelodysplastic syndromes and acute myeloid leukemia. Front. Oncol. 15:1540426. doi: 10.3389/fonc.2025.1540426
Received: 05 December 2024; Accepted: 27 May 2025;
Published: 24 June 2025.
Edited by:
Alessandro Isidori, AORMN Hospital, ItalyReviewed by:
Dangdang Li, Northeast Forestry University, ChinaAthanasios Tasis, Democritus University of Thrace, Greece
Ioanna Mosialou, Columbia University, United States
Copyright © 2025 Wang, Dong, Tao, Chen, Chen, Zhang, Shi, He, Yu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang Yu, eXVsaWFuZ2hhQDE2My5jb20=; Chunling Wang, d2NsNjUwNkAxNjMuY29t
†These authors have contributed equally to this work and share first authorship