 Eman Elsabagh1†
Eman Elsabagh1† Rachel Gallant2†
Rachel Gallant2† Lior Goldberg3†
Lior Goldberg3† Aditya Sharma4†
Aditya Sharma4† Paul L. Martin1Timothy A. Driscoll1
Paul L. Martin1Timothy A. Driscoll1 Andrea Bauchat1Joanne Kurtzberg1LaTarsha Spencer1
Andrea Bauchat1Joanne Kurtzberg1LaTarsha Spencer1 Paibel I. Aguayo-Hiraldo5
Paibel I. Aguayo-Hiraldo5 Neena Kapoor5
Neena Kapoor5 Kris M. Mahadeo1‡
Kris M. Mahadeo1‡ Hisham Abdel-Azim4*‡
Hisham Abdel-Azim4*‡- 1Department of Pediatrics, Division of Transplantation and Cellular Therapy, Duke University, Durham, NC, United States
- 2Division of Pediatric Hematology-Oncology, University of Oklahoma Health Sciences Center, Oklahoma City, OK, United States
- 3Department of Pediatrics, T Cell Therapeutics Research Laboratories, City of Hope National Medical Center and Beckman Research Institute, Duarte, CA, United States
- 4Division of Transplant/Cell Therapy and Hematological Malignancies, Cancer Center, Departments of Pediatrics and Medicine, Loma Linda University School of Medicine, Children Hospital and Medical Center, Loma Linda, CA, United States
- 5Section of Blood & Marrow Transplantation, Cancer and Blood Disease Institute, Children’s Hospital Los Angeles, Los Angeles, CA, United States
Introduction: Allogeneic hematopoietic cell transplantation (HCT) is a potentially curative treatment for most children with juvenile myelomonocytic leukemia (JMML), but overall survival remains poor at 50%. Given its rarity and heterogeneity, there is no standard HCT conditioning regimen for JMML.
Methods: Retrospective study of consecutive patients with JMML who underwent HCT using a busulfan/ melphalan backbone conditioning regimen (n=17) at two academic centers.
Results: The median age at HCT was 1.9 (range 0.7-6.0) years. At a median follow up of 7.6 (range 2.9-21.5) years, 100% disease-free (DFS) and overall survival (OS), with prompt immune reconstitution were observed. This cyclophosphamide-sparing approach was associated with no transplant related mortality.
Discussion: Given excellent clinical outcomes at extended follow-up, prospective studies are needed to confirm our findings in this ultra-rare disease.
Introduction
There are approximately 25 cases/year of juvenile myelomonocytic leukemia (JMML) diagnosed in the United States, an aggressive myeloproliferative/myelodysplastic disorder characterized by infiltration of peripheral blood, bone marrow, and organs by abnormal myelomonocytic cells (1). Primarily a disease of infancy/childhood, the majority have somatic and/or germline mutations within the RAS/MAPK signaling pathway (NF1, PTPN11, KRAS, NRAS, or CBL), leading to pathologic activation and hypersensitivity of myeloid progenitor cells to the granulocyte-monocyte colony stimulating factor (GM-CSF) (1, 2). Prior to molecular characterization, in vitro hypersensitivity of monocyte/macrophage colonies to GM-CSF represented a diagnostic tool. Untreated, the median survival is less than 12 months (3). Neurofibromatosis type 1 (NF-1) mutations, found in up to 30% of JMML cases, significantly influence prognosis/treatment. This underscores the complexity of JMML and necessity for tailored therapeutic strategies (4). While affected children with germline CBL mutations may have spontaneous regression of myeloproliferation despite the persistence of LOH of CBL in hematopoietic cells, allogenic hematopoietic stem cell transplant (HCT) is recommended with disease progression (5).
HCT is the only potentially curative therapy for children with JMML. Yet, despite intensive therapy, 5-year overall survival (OS) following HCT is poor (50%); the primary cause of mortality following HCT, is relapse at a median of 4 months (6). Chemotherapy alone may provide temporary remission but is not curative (6, 7). Given the rarity and heterogeneity of JMML, no standard HCT approach has been established.
Total body irradiation (TBI) has been replaced by chemotherapy-based conditioning due to associated toxicity in younger children (6–8). Favorable outcomes with a TBI-sparing approach to JMML have been previously reported (8). This report characterizes long-term outcomes associated with that approach in an expanded cohort.
Material and methods
This retrospective study (approved by respective institutional review boards) includes all patients diagnosed with JMML undergoing first HCT between 2002-2022 at Children’s Hospital Los Angeles (CHLA) and Duke Children’s Hospital (Duke). Approach to diagnosis and indications for HCT are shown in Figure 1. Patients received myeloablative conditioning (MAC) with a busulfan/melphalan (Bu/Mel) backbone; Bu 1 mg/kg every 6 hours intravenously (IV) on days -8 to -5 (with therapeutic drug monitoring [TDM] targeting overall concentration steady state [CSS] of 800-1000 ng/mL) and Mel 45mg/m2/day IV on days -4 to -2. The backbone regimen was customized based on donor graft source and risk of graft-versus-host disease (GVHD) and/or graft rejection (Figure 1). Sinusoidal obstructive syndrome (SOS) was retrospectively graded (9). All patients either received intravenous immunoglobulin (IVIG) (1gm/kg) (CHLA) 48 hours prior to graft infusion to block the reticuloendothelial system (10) or underwent pre-HCT splenectomy (Duke) (6). Intended time for initiation of immunosuppression withdrawal was D+100, if no evidence GVHD.
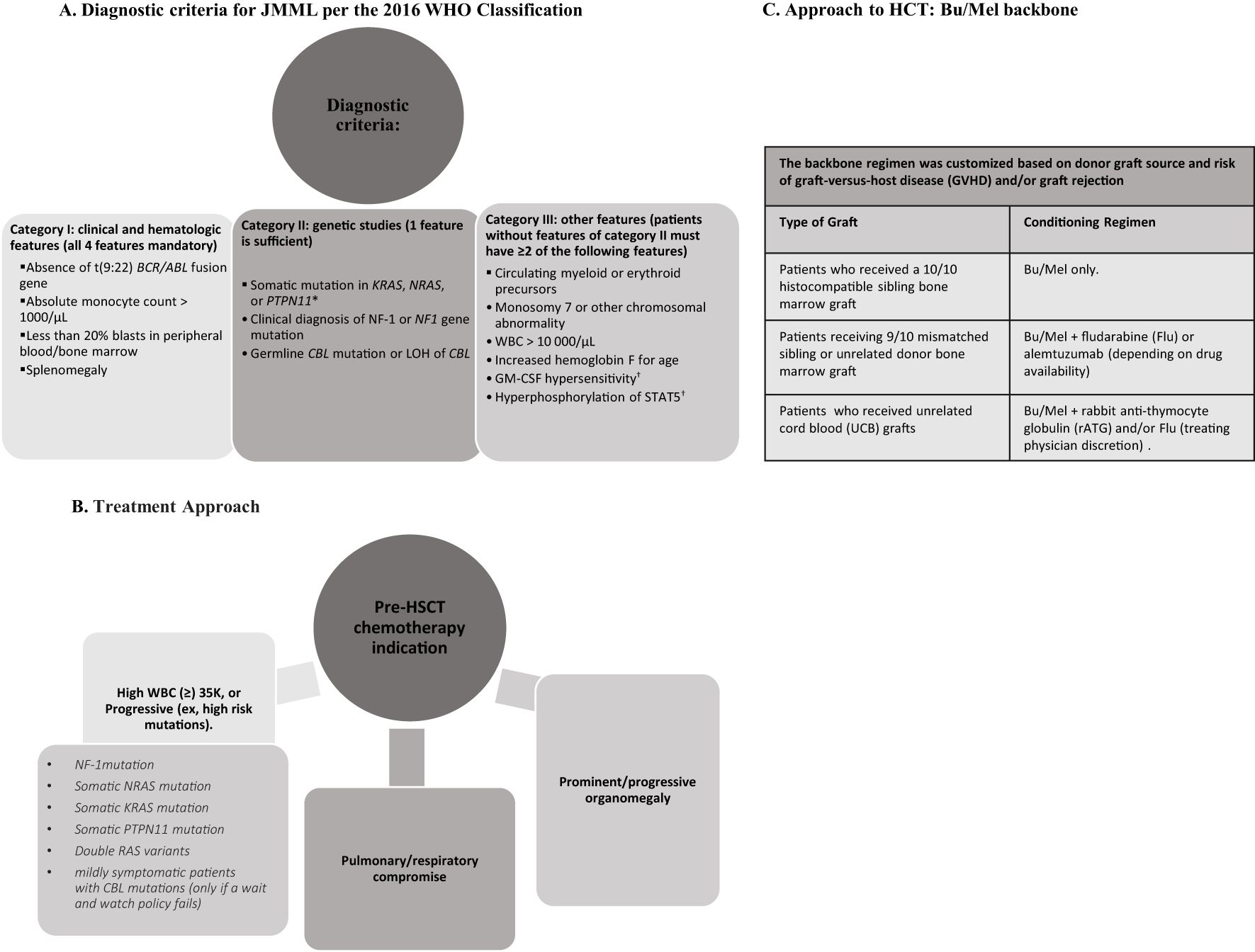
Figure 1. A comprehensive infographic summarizing diagnostic criteria, pre-treatment considerations, treatment approach for hematopoietic cell transplantation (HCT). (A) Diagnostic criteria for JMML based on clinical and hematologic features, genetic studies, and other supplementary features. (B) Pre-HCT chemotherapy indications in JMML. (C) Backbone regimen customized by donor graft source.
Results
Median age at diagnosis (n=17) was 0.9 (range 0.4-4.1) and age at HCT was 1.9 (range 0.7-6.0) years, respectively. Median time from diagnosis to HCT was 6 months with 76.5% patients receiving chemotherapy prior to HCT. Though underlying molecular lesions were not identified(n=8) in all patients due to the retrospective nature of the study and limitations of the assays available then, this cohort includes patients with high-risk molecular features such as NF1, KRAS, and PTPN11 mutations. All patients had clinical and morphological bone marrow progression of their disease, prior to HCT, irrespective of the molecular features (Table 1). Median (range) Bu CSS and area under the curve were 884 (560-1096) mg/L and 1293 (819-1601) mmol/L-minute, respectively. Median total nucleated cell counts and CD34 cell dose were 10 × 10^8 cells/kg (5-15 x10^8) and 9.5 × 10^6 cells/kg (2.2-15 x10^6), respectively. Median time to neutrophil and platelet engraftment were 17 (range: 12-43) and 52 (range: 15-133) days, respectively. No patients developed vasculitis pre- or post-HCT and no transplant related mortality was observed. Five (29%) patients developed SOS [Mild (n=4) and severe (n=1)] which resolved without specific therapy. Infectious complications [sepsis (n=4) and cytomegalovirus (CMV) reactivation (n=2)], resolved with therapy. Indications for PICU admission included severe SOS (n=1), respiratory distress (n=1), pericardial effusion (n=1), and sepsis (n=2). One patient developed grade IV acute GVHD which progressed to chronic GVHD requiring bowel resection and systemic therapy. At 13.9 years follow-up, this patient is alive with a performance score of 100%.
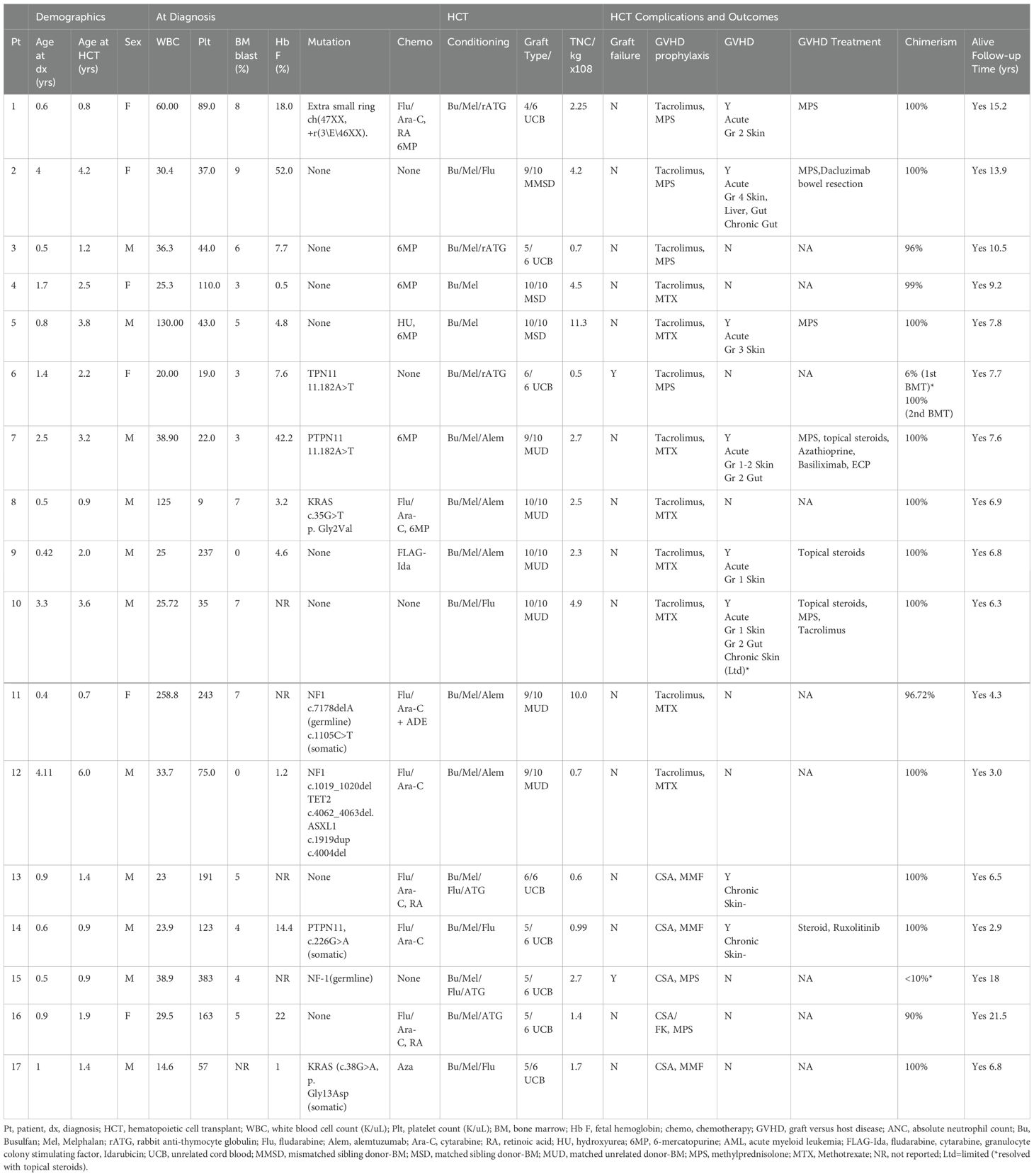
Table 1. Clinical characteristics and outcomes for pediatric patients with juvenile myelomonocytic leukemia (JMML) treated with hematopoietic cell transplant (HSCT) using busulfan/melphalan conditioning regimen.
Patient 6, experienced primary graft failure (PGF) at day +55 and received second (TCR- αβ/CD19-depleted related-haploidentical) transplant with TBI (1200 cGy), flu (160mg/m2), thiotepa (10mg/kg), rabbit antithymocyte globulin (3.75mg/kg) and rituximab (200mg/m2) and has maintained long-term remission. Patient 15 experienced PGF at day +41 with autologous recovery and remains in complete remission at 18 years follow-up.
At 1-year post-HCT, 92% and 69% of patients with immune-reconstitution data available (n=13) had normal B (CD19+) and T (CD3+) lymphocyte counts, respectively. Pre-HCT performance scores improved in almost all patients post-HCT. Patient 6 has a post-HCT performance score of 80% due to underlying congenital heart defect awaiting surgical repair. At a median follow-up 7.6 (range 2.9-21.5) years, 100% disease-free survival (DFS) and OS were observed. No patients have developed a secondary malignancy or HCT-related organ dysfunction.
Discussion
JMML is a predominantly aggressive and fatal disorder. In one analysis of patients diagnosed prior to molecular characterization, the probability of survival at 10 years in transplanted patients was 0.39 (standard error [SE] = 0.10) versus 0.06 (SE = 0.04) in non-transplanted patients (6). Prompt HCT is recommended for children with JMML and NF-1, somatic PTPN-11 and K-RAS mutations, and for most children with somatic N-RAS mutations (11).
Due to the rarity JMML and paucity of large clinical trials, there is no clear standard conditioning regimen for patients undergoing HCT. One of the largest reports (n=100) used MAC with oral Bu/Mel/cyclophosphamide (Cy), with the goal of avoiding TBI-associated toxicity (3). Transplant related mortality (TRM) was 13% and one-third of patients relapsed (3). Similar TRM/relapse rates were recently reported among patients who received MAC with Bu (oral/IV)/Mel/Flu (12). Therefore, further attempts to reduce transplant related morbidity/mortality without compromising survival have been employed (with conflicting results). The Children’s Oncology Group (COG) conducted a randomized study comparing 2 myeloablative regimens Bu/Flu (n=9) to Bu/Mel/Cy (n=6); however, the trial closed early due to high relapse rates in the Bu/Flu arm (13). In a Japanese registry study (n=129) with a variety of conditioning regimens, 5-year OS, cumulative relapse incidence and TRM were 64%, 34% and 21%, respectively, (with 73% 5-year OS and 26% cumulative relapse incidence in the subgroup (n=59) treated with myeloablative Bu/Flu/Mel) (8).
SOS disproportionately affects infants with a prevalence of 20–60% depending on age and primary disease indication for HCT, compared with 10% in adults (9, 14). In this study, 29% were infants at the time of HCT, and notably, these were the patients who developed SOS. In the COG study, both arms used a single alkylating agent, but SOS developed in 50% of patients on the Bu/Mel/Cy arm and in 22% on the Bu/Flu arm (13). Our results are comparable with Bu/Flu arm of the COG study, suggesting that patients with JMML have an inherently high risk for SOS (15). In 2016, defibrotide was approved for treatment of HCT patients diagnosed with severe SOS. In this study, most cases of SOS were mild, and all cases resolved without defibrotide therapy. Regardless, these collective findings may underscore the potential need for vigilant monitoring/aggressive mitigation strategies for SOS in JMML patients undergoing HCT in the current era. Approximately 35% of children undergoing HCT for various diseases require PICU support in the immediate post-transplant period with PICU mortality of 44% (15). The 100% long-term survival observed in this cohort, suggest that HCT patients with JMML requiring PICU support may have promising outcomes.
Mitigation efforts for PGF included either splenectomy or infusion of IVIG pre-HCT. Splenectomy was historically used for lack of other symptomatic control measures in patients with JMML as it was also associated with decreased transfusion requirements, albeit with increased risk of infection (6). The use of splenectomy to promote engraftment after HCT is no longer routine and typically reserved for the presence of hypersplenism/platelet refractoriness.6 In our study, patients received IVIG prior to graft infusion to block the reticuloendothelial system (10) aiming to enhance engraftment. However, the role of IVIG prior to HCT to promote engraftment may warrant prospective investigation. Our improved outcomes with Bu/Mel backbone conditioning regimen are consistent with findings of a recent Japanese study (16), although in this study 6/21 [28.6%] patients (compared to 2/17 [11.8%] patients in our study) had PGF. None of the patients in this Japanese study had splenectomy or IVIG pre-HCT.
The role of GVHD in relapse prevention in JMML is poorly defined. GVHD, particularly chronic GVHD, has been associated with improved survival and lower risk of relapse in JMML post-HCT presumably due to graft-versus-leukemia (GVL) effect (8, 11). However, one of the largest reports did not show benefit of either acute/chronic GVHD (3). The impact of GVHD could not be assessed in this study (100% OS).
While none of the patients in our study received maintenance therapy post-HCT, such therapy may significantly reduce relapse and improve survival in JMML patients. Although FLT3 mutations are not as common in JMML as in acute myeloid leukemia, they can occur and can be a target for maintenance therapy (17). Azacytidine or Decitabine maintenance post-HCT along with donor lymphocyte infusions to enhance GVL effect may salvage patient who have molecular evidence of disease Post-HCT (18, 19). Trametinib maintenance post-HCT for RAS mutated JMML is a promising approach (20).
Majority of patients in this cohort received chemotherapy for cytoreduction prior to HCT resulting in disease debulking, raising the possibility that the high DFS/OS could in part be due to a reduction of the pre-transplant disease burden. At the time of this report, all patients remain alive, disease free, GVHD free, and off all immunosuppression. Evidence of immune reconstitution was observed in most patients by 12 months post-transplant and there was no observed infection- related mortality. Targeted Bu CSS likely contributed to improved outcomes with no TRM, compared with prior reports with oral Bu and/or no TDM.
While conclusive inferences are limited by the small sample size and heterogeneity of the cohort (inherent with the rarity/heterogeneity of this disease), to our knowledge, with 100% OS and DFS, a Bu/Mel backbone conditioning regimen with or without serotherapy or Flu based on stem cell source and donor type, represents the most effective multi-center strategy published to date. This approach warrants prospective investigation.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The study involving humans was reviewed by CHLA IRB, Duke Medical Center IRB. The study was were conducted in accordance with the local legislation and institutional requirements. Because this is a retrospective study for existing data, written informed consent for participation in this specific study was not required.
Author contributions
EE: Data curation, Writing – review & editing. RG: Data curation, Formal analysis, Investigation, Writing – original draft, Writing – review & editing. LG: Data curation, Formal analysis, Investigation, Writing – original draft, Writing – review & editing. AS: Data curation, Formal analysis, Investigation, Writing – review & editing. PM: Data curation, Formal analysis, Investigation, Writing – review & editing. TD: Data curation, Formal analysis, Investigation, Writing – review & editing. AB: Data curation, Formal analysis, Investigation, Writing – review & editing. JK: Data curation, Formal analysis, Investigation, Writing – review & editing. LS: Data curation, Formal analysis, Investigation, Writing – review & editing. P-AH: Data curation, Formal analysis, Investigation, Writing – review & editing. NK: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. KM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. HA: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors acknowledge the patients, donors, and families and the nurses and inter-disciplinary staff involved in the care of patients.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. SEER hematopoietic and lymphoid neoplasm database. Bethesda, MD: SEER (2022). Available at: https://seer.cancer.gov/seertools/hemelymph/51f6cf57e3e27c3994bd5393/ (Accessed September 10, 2024).
2. Caye A, Strullu M, Guidez F, Cassinat B, Gazal S, Fenneteau O, et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet. (2015) 47:1334–40. doi: 10.1038/ng.3420
3. Locatelli F, Nöllke P, Zecca M, Korthof E, Lanino E, Working Group on Childhood MDS, et al. European Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG- MDS/EBMT trial. Blood. (2005) 105:410–9. doi: 10.1182/blood-2004-05-1944
4. Side LE, Emanuel PD, Taylor B, Franklin J, Thompson P, Castleberry RP, et al. Mutations of the NF1 gene in children with juvenile myelomonocytic leukemia without clinical evidence of neurofibromatosis, type 1. Blood. (1998) 92:267–72.
5. Oshrine B. Primary Graft Failure but Treatment Success: A Case of Reversion to Heterozygosity after Allogeneic Hematopoietic Cell Transplantation with Autologous Hematopoietic Recovery in a Child With CBL-related Juvenile Myelomonocytic Leukemia. J Pediatr Hematol Oncol. (2021) 43:e426. doi: 10.1097/MPH.0000000000001740
6. Gupta AK, Meena JP, Chopra A, Tanwar P, and Seth R. Juvenile myelomonocytic leukemia-A comprehensive review and recent advances in management. Am J Blood Res. (2021) 11:1–21.
7. Zhao Q, Mahadeo KM, Kapoor N, and Abdel-Azim H. Improved outcomes associated with hematopoietic stem cell transplantation for patients with juvenile myelomonocytic leukemia. Blood. (2015) 126:561–2.
8. Yoshida N, Sakaguchi H, Yabe M, Hasegawa D, Hama A, Hasegawa D, et al. Clinical outcomes after allogeneic hematopoietic stem cell transplantation in children with juvenile myelomonocytic leukemia: A report from the Japan society for hematopoietic cell transplantation. Biol Blood Marrow Transplant. (2020) 26:902–10. doi: 10.1016/j.bbmt.2019.11.029
9. Mahadeo KM, Bajwa R, Abdel-Azim H, Lehmann LE, Duncan C, Zantek N, et al. Diagnosis, grading, and treatment recommendations for children, adolescents, and young adults with sinusoidal obstructive syndrome: an international expert position statement. Lancet Haematol. (2020) 7:e61–72. doi: 10.1016/S2352-3026(19)30201-7
10. Crow AR, Song S, Semple JW, Freedman J, and Lazarus AH. IVIg inhibits reticuloendothelial system function and ameliorates murine passive-immune thrombocytopenia independent of anti-idiotype reactivity. Br J Haematol. (2001) 115:679–86. doi: 10.1046/j.1365-2141.2001.03136.x
11. Locatelli F and Niemeyer CM. How I treat juvenile myelomonocytic leukemia. Blood. (2015) 125:1083–90. doi: 10.1182/blood-2014-08-550483
12. Yabe M, Ohtsuka Y, Watanabe K, Inagaki J, Yoshida N, Sakashita K, and Japanese Pediatric Myelodysplastic Syndrome Study Group. Transplantation for juvenile myelomonocytic leukemia: a retrospective study of 30 children treated with a regimen of busulfan, fludarabine, and melphalan. Int J Hematol. (2015) 101:184–90. doi: 10.1007/s12185-014-1715-7
13. Dvorak CC, Satwani P, Stieglitz E, Cairo MS, Dang H, Pei Q, et al. Disease burden and conditioning regimens in ASCT1221, a randomized phase II trial in children with juvenile myelomonocytic leukemia: A Children’s Oncology Group study. Pediatr Blood Cancer. (2018) 65:e27034. doi: 10.1002/pbc.27034
14. Ragoonanan D, Abdel-Azim H, Sharma A, Bhar S, McArthur J, Madden R, et al. Retrospective analysis of veno-occlusive disease/sinusoidal obstruction syndrome in paediatric patients undergoing hematopoietic cell transplantation -a multicentre study. Lancet Reg Health Am. (2024) 33:100728. doi: 10.1016/j.lana.2024.100728
15. Zinter MS, Dvorak CC, Spicer A, Cowan MJ, and Sapru A. New insights into multicenter PICU mortality among pediatric hematopoietic stem cell transplant patients. Crit Care Med. (2015) 43:1986–94. doi: 10.1097/CCM.0000000000001085
16. Sakashita K, Yoshida N, Muramatsu H, Ohtsuka Y, Watanabe K, Yabe M, et al. Allogeneic hematopoietic cell transplantation for juvenile myelomonocytic leukemia with a busulfan, fludarabine, and melphalan regimen: JPLSG JMML-11. Transplant Cell Ther. (2024) 30:105.e1–105.e10. doi: 10.1016/j.jtct.2023.10.002
17. de Vries AC, Stam RW, Schneider P, Niemeyer CM, van Wering ER, Haas OA, et al. Role of mutation independent constitutive activation of FLT3 in juvenile myelomonocytic leukemia. Haematologica. (2007) 92:1557–60. doi: 10.3324/haematol.11201
18. Schroeder T, Rautenberg C, Haas R, Germing U, and Kobbe G. Hypomethylating agents for treatment and prevention of relapse after allogeneic blood stem cell transplantation. Int J Hematol. (2018) 107:138–50. doi: 10.1007/s12185-017-2364-4
19. Peng Z, Gao J, Huang L, He Y, Tang H, Zong S, et al. Decitabine-based treatment strategy improved the outcome of HSCT in JMML: a retrospective cohort study. Front Immunol. (2024) 15:1426640. doi: 10.3389/fimmu.2024.1426640
20. Li Z, Yang K, Xu T, Wang L, Wang X, Wen X, et al. Activity of trametinib as maintenance therapy after allogeneic hematopoietic stem cell transplantation in patients with relapsed or refractory RAS pathway-mutated hematologic Malignancies. Ann Hematol. (2025) 104:1295–301. doi: 10.1007/s00277-024-06046-7
Keywords: JMML, pediatric, conditioning regimen, hematopoietic stem cell transplant, busulfan, melphalan
Citation: Elsabagh E, Gallant R, Goldberg L, Sharma A, Martin PL, Driscoll TA, Bauchat A, Kurtzberg J, Spencer L, Aguayo-Hiraldo PI, Kapoor N, Mahadeo KM and Abdel-Azim H (2025) A reduced-toxicity myeloablative conditioning approach for hematopoietic cell transplant in juvenile myelomonocytic leukemia. Front. Oncol. 15:1541192. doi: 10.3389/fonc.2025.1541192
Received: 07 December 2024; Accepted: 12 May 2025;
Published: 03 June 2025.
Edited by:
Alaa Elhaddad, Children Cancer Hospital, EgyptReviewed by:
Mahmoud Hammad, Cairo University, EgyptYasser Elborai, Prince Sultan Military Medical City, Saudi Arabia
Copyright © 2025 Elsabagh, Gallant, Goldberg, Sharma, Martin, Driscoll, Bauchat, Kurtzberg, Spencer, Aguayo-Hiraldo, Kapoor, Mahadeo and Abdel-Azim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hisham Abdel-Azim, aGFiZGVsYXppbUBsbHUuZWR1
†These authors have contributed equally to this work
‡These authors have contributed equally to this work and share senior authorship