 Na Liang1,2†
Na Liang1,2† Changxian Chen1,2†
Changxian Chen1,2† Dan Yuan1,2Qiang Xu3
Dan Yuan1,2Qiang Xu3 Yali Zhan1,2Yi Zhao4Di Wu5Cheng Yang4
Yali Zhan1,2Yi Zhao4Di Wu5Cheng Yang4 Chunming Li1,2*
Chunming Li1,2*- 1Department of Pathology, School of Basic Medicine, Zunyi Medical University, Zunyi, China
- 2Department of Pathology, Affiliated Hospital of Zunyi Medical University, Zunyi, China
- 3Department of Urology, the Second Affiliated Hospital of Zunyi Medical University, Zunyi, China
- 4The First Clinical College of Zunyi Medical University, Zunyi, China
- 5School of Forensic Medicine, Zunyi Medical University, Zunyi, China
Histiocytic sarcoma (HS) is a rare malignant tumor that primarily affects the lymph nodes, intestines, skin, and soft tissues. Primary central nervous system histiocytic sarcoma (PCNSHS) is even rarer. We present the case of a 50-year-old Asian male with PCNSHS who was hospitalized after experiencing intermittent headaches and slurred speech for a week. During surgery, both tumor cells and purulent material were observed, and the diagnosis of PCNSHS was ultimately confirmed by immunohistochemistry. Furthermore, we conducted a literature review from 1952 to the present, screening and analyzing 49 related cases across 41 publications.
Introduction
Histiocytic sarcoma (HS) is a rare malignant tumor arising from the lymphohematopoietic system, commonly affecting the lymph nodes, intestines, skin and soft tissues (1). Primary central nervous system histiocytic sarcoma (PCNSHS) is extremely rare and can occur in the brain (2), cerebellum (3), leptomeninges (4) and spinal cord (5). According to the 2021 World Health Organization classification of central nervous system tumors, HS is classified as a histiocytic tumor (6). PCNSHS is often associated with a pronounced inflammatory response, such as lymphocyte infiltration, which may affect the diagnosis (7). Sometimes purulent material can also be found during surgery, which may be result from uncontrolled proliferation of monocytes or macrophages and can easily be misdiagnosed as an infectious abscess (3). The specific cause of PCNSHS has not yet been determined. However, studies suggest that PCNSHS may be associated with prior radiation therapy and complex cytogenetic abnormalities in the tumor (8). Currently, there is no standardized treatment plan for PCNSHS. Surgery is the main treatment, especially for patients with single lesions, and radiotherapy and chemotherapy are often used as adjuvant treatments (9). This study reports a 50-year-old Asian male patient with PCNSHS, focusing on the diagnosis and differential diagnosis of previously published cases. Through a comprehensive literature review and analysis, we aim to enhance clinicians’ diagnostic accuracy for PCNSHS, facilitating more effective identification and management of this condition.
Case presentation
The Medical Progress Timeline (Figure 1) summarizes three key phases:
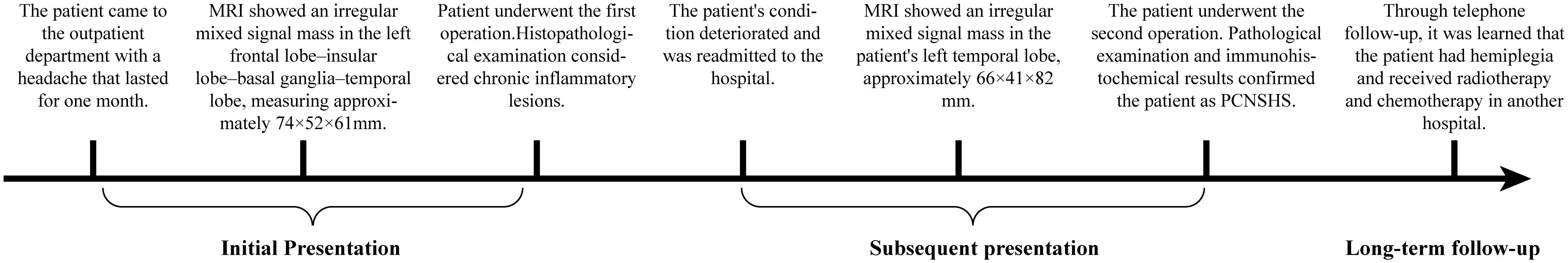
Figure 1. Timeline of the patient’s medical progress.
1. Initial Presentation (February 2024): A 50-year-old Asian male patient presented to our hospital’s outpatient department due to persistent headache for one month. Initial magnetic resonance imaging (MRI) showed an irregular mixed signal mass in the left frontal lobe–insular lobe–basal ganglia–temporal lobe, measuring approximately 74×52×61mm. T1WI showed an uneven high signal, and T2WI showed an uneven equal/slightly high signal. The enhanced scan showed obvious uneven enhancement of the tumor (Figures 2A–C). The patient underwent neuroendoscopic resection of ventricular lesions and neuronavigation resection of intracranial lesions. Histopathological examination showed that numerous lymphocytes and a small number of histiocytes with abundant cytoplasm, vacuolated nuclei, and pronounced nucleoli were seen in the excised tissue (Figures 3A, B). A chronic inflammatory lesion was initially considered.
2. Subsequent presentation (approximately 3 months after the initial presentation): After regular follow-up, the patient’s headache symptoms worsened again, accompanied by slurred speech for a week, and he was readmitted to the hospital. Preoperative MRI showed an irregular mixed-signal mass in the patient’s left temporal lobe, approximately 66×41×82 mm in size. It showed inhomogeneous high signal intensity on T1WI and inhomogeneous iso/slightly high signal intensity on T2WI. On enhanced scan, the solid part of the tumor showed obvious uneven enhancement, and DWI showed high signal intensity (Figures 2D–H). The patient underwent a second intracranial lesion resection. Intraoperatively, the patient’s left temporal lobe was found to be swollen with dura mater invasion, and about 30 mL of light yellow pus was drained from the tumor. The tumor measured approximately 5×5×6 cm. Intraoperative histopathology revealed that the tumor cells were arranged in sheets, had abundant cytoplasm, and exhibited eccentric nuclei, resembling gemistocytes (Figures 3C, D). The frozen section diagnosis was glioma. Postoperative computed tomography (CT) revealed a residual cavity, gas, and blood accumulation in the left temporal lobe, along with a large patchy hypodense area in the left cerebral hemisphere (Figure 2I). Finally, postoperative pathological examination was conducted (Figures 3E–P). Immunohistochemistry showed that the tumor cells were positive for molecules such as CD68 and CD163. After ruling out other negative markers and conducting an intra-departmental consultation, the patient was finally diagnosed with histiocytic sarcoma of the temporal lobe of the left lateral fissure cistern. The patient’s postoperative course was stable, with no complications, and he was discharged.
3. Long-term follow-up (approximately 12 months after the initial presentation): A follow-up call revealed that the patient’s tumor had recurred, and he had developed hemiplegia. The patient had received chemoradiotherapy at another hospital.
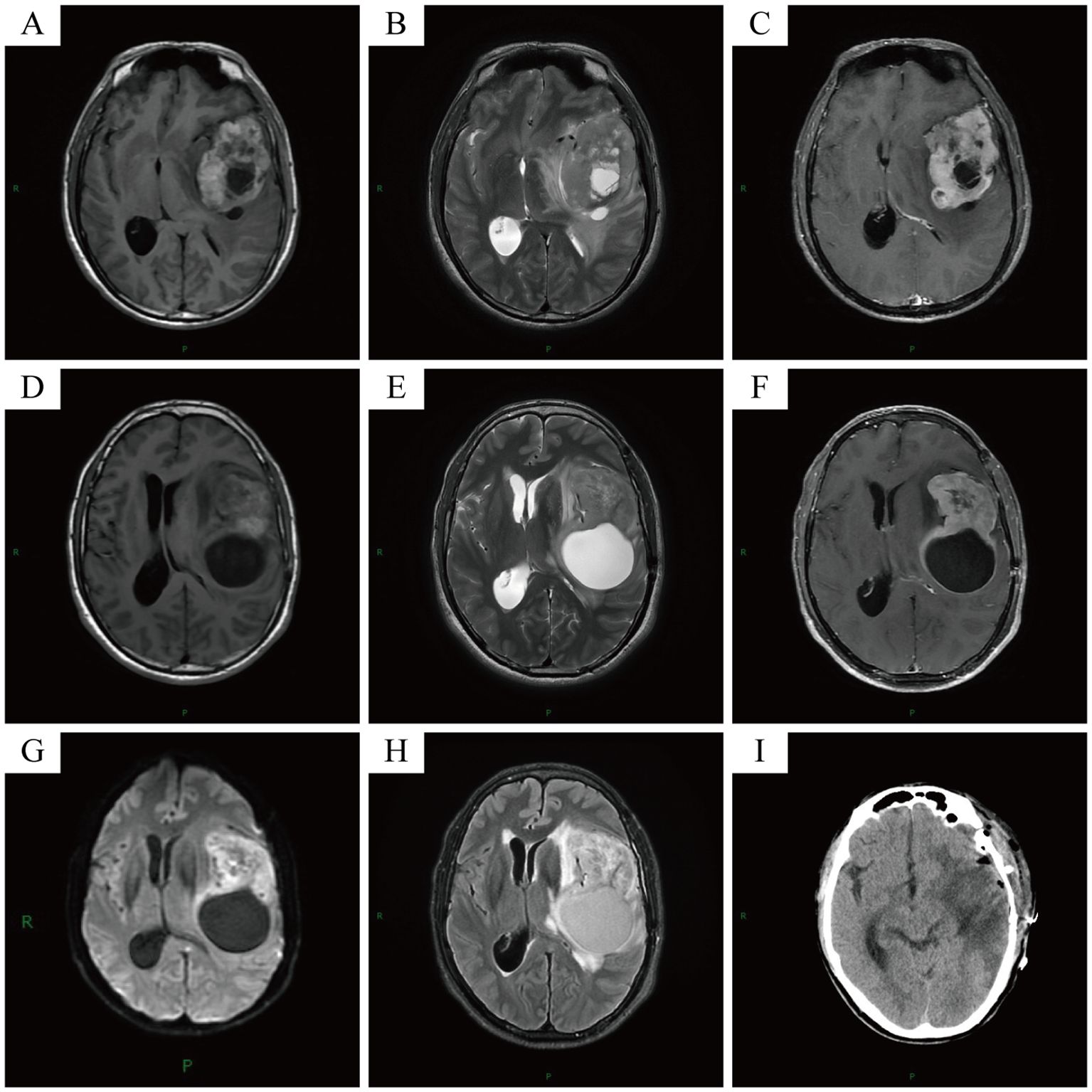
Figure 2. Initial MRI showed an irregular mixed signal mass in the patient’s left frontal lobe-insular lobe-basal ganglia region-temporal lobe, approximately 74×52×61mm in size (A-C). Pre-operative MRI showed an irregular mixed signal mass in the patient’s left temporal lobe, approximately 66×41×82mm in size (D-H). Post-operative CT showed post-operative changes in the patient’s left cerebral hemisphere (I).
Pathological findings
Histopathological examination of the second resection specimen showed diverse tumor cells with significant dysplasia, necrosis, mitotic figures, foam cells and inflammatory cell infiltration (Figures 3E–K).
Immunohistochemical staining of the second resection specimen showed that tumor cells were positive for CD68, CD163, CD4, INI-1 and ATRX (Figures 3L–N), Scattered focal positivity for lysozyme, and some positive for S-100 and LCA. The Ki-67 proliferation index was about 30%. The tumor cells were negative for CD34, MPO, CD117, CD43, CD21, GFAP, Oligo-2, IDH-1, EMA, PR, SSTR2, ALK, SMA, Desmin, H-caldesmon, MyoD1, TFE3, HMB45, Melan-A, and Langerin (Figures 3O, P). Based on histopathological and immunohistochemical results, PCNSHS was diagnosed.
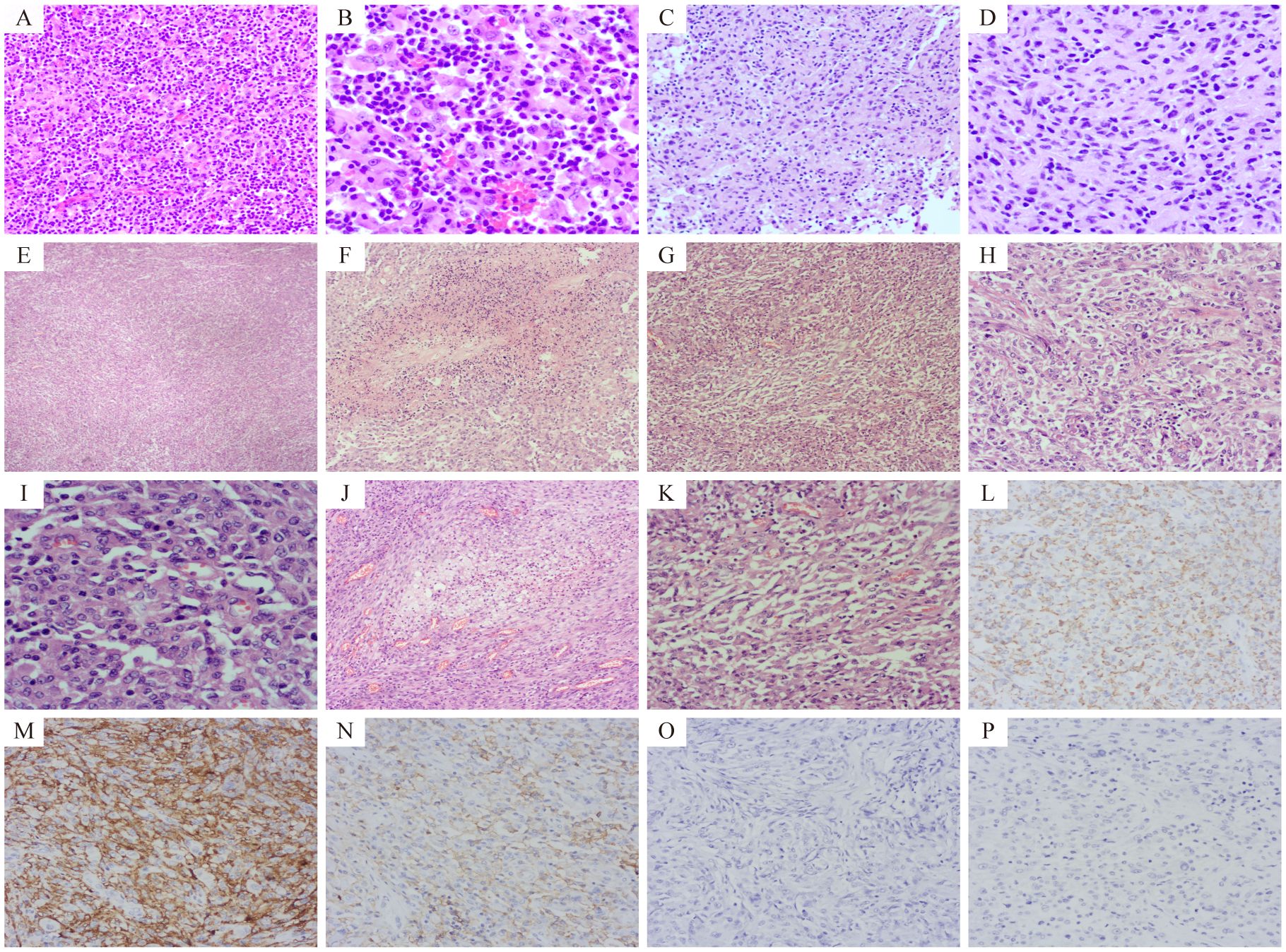
Figure 3. The results of hematoxylin-eosin (HE) staining and immunohistochemical (IHC) assays. Pathological sections after the first operation, showed a large number of lymphocytes and a small number of histiocytes (A, HE 200× and B, HE 400×). Frozen pathological sections during the second operation showed that the tumor cells were distributed in sheets and similar in shape to gemistocytes (C, HE 200× and D, HE 400×). Pathological examination after the second operation showed: Tumor cells were arranged in diffuse patches (E, HE 40×) with necrotic areas (F, HE 100×). Tumor cells appeared spindle-shaped (G, HE 100×) and epithelial-like (H, HE 200×). Tumor cells exhibited significant dysplasia and pleomorphism, and mitotic figures were common (I, HE 400×). Foam cells were seen in some areas (J, HE 100×), and a large number of inflammatory cell infiltrates were observed in some areas (K, HE 200×). Tumor cells were positive for CD68, CD163, and CD4 (L–N respectively, IHC 200×), and negative for GFAP and Oligo-2 (O, P, IHC 200×).
Systematic review of literature
By searching the Pubmed database for case reports on PCNSHS published between 1952 and 2024, a total of 191 results were obtained using the following keywords “((((nervous system) OR (neurological disorder)) OR (brain)) OR (neurological symptom)) AND (histiocytic sarcoma)”. After removing duplicates, excluding cases of metastatic central nervous system histiocytic sarcoma, and excluding animal-related studies, 49 cases reported in 41 articles were identified, excluding our own cases.
A summary of previously reported cases of PCNSHS (Table 1, Figure 4) showed that a total of 49 patients were included (male to female ratio of 0.88), with no significant gender differences. PCNSHS can develop at any age, ranging in age from 17 months to 71 years, with a median age of 43 years at diagnosis. The mortality rate was 61%, and the median survival was 10 months, indicating a poor prognosis. Tumors can affect all parts of the central nervous system, with the most common sites being the frontal lobe (12.3%), corpus callosum (8.2%), parietal lobe (6.8%), pia mater (6.8%), and cerebellum (5.5%). Patients can present a variety of clinical symptoms, with headache (21.6%) being the most common, followed by vomiting (12.9%), dizziness (8.6%), nausea (7.8%), and gait disturbance (6.9%). It should be noted that the symptoms of some patients gradually worsen over weeks to months, suggesting that PCNSHS may be difficult to detect in the early stages and require high vigilance for progressively worsening neurological symptoms.
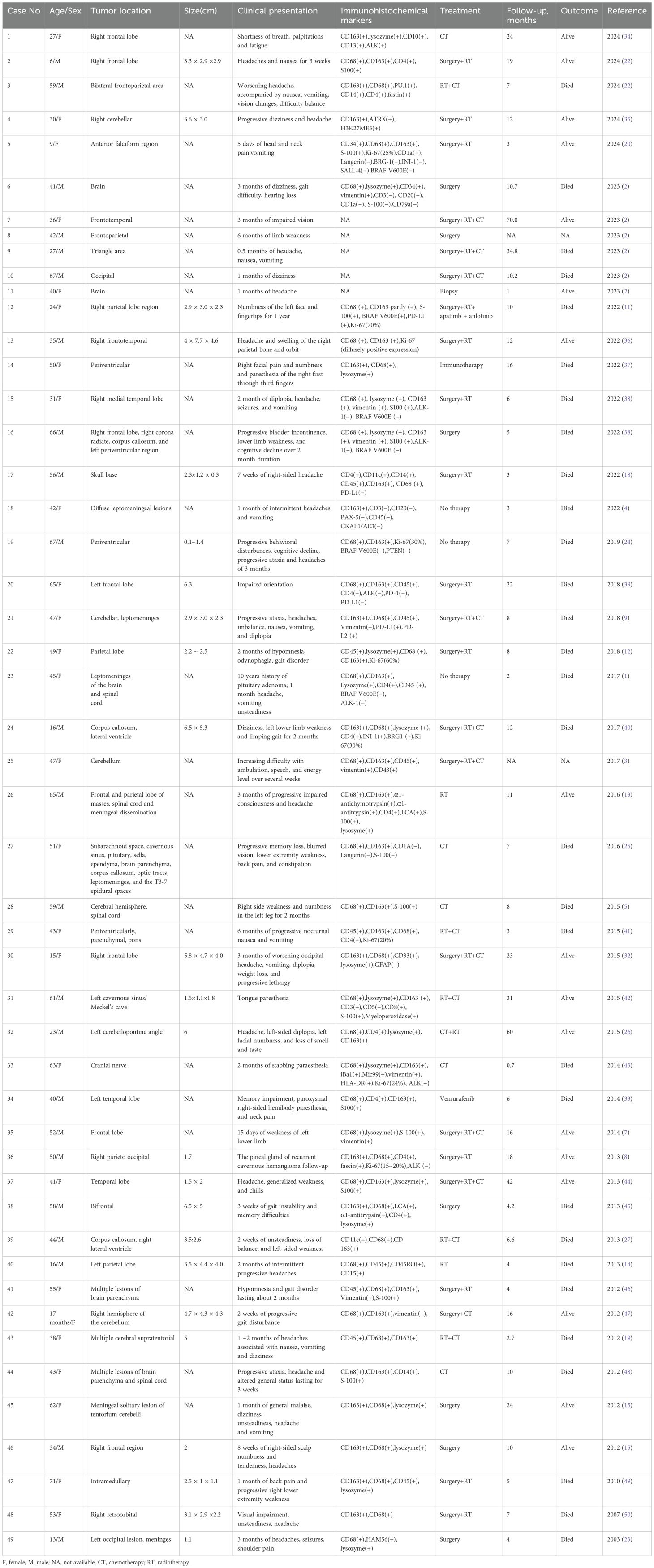
Table 1. Summary of previously published cases of primary central nervous system histiocytic sarcoma (PCNSHS).
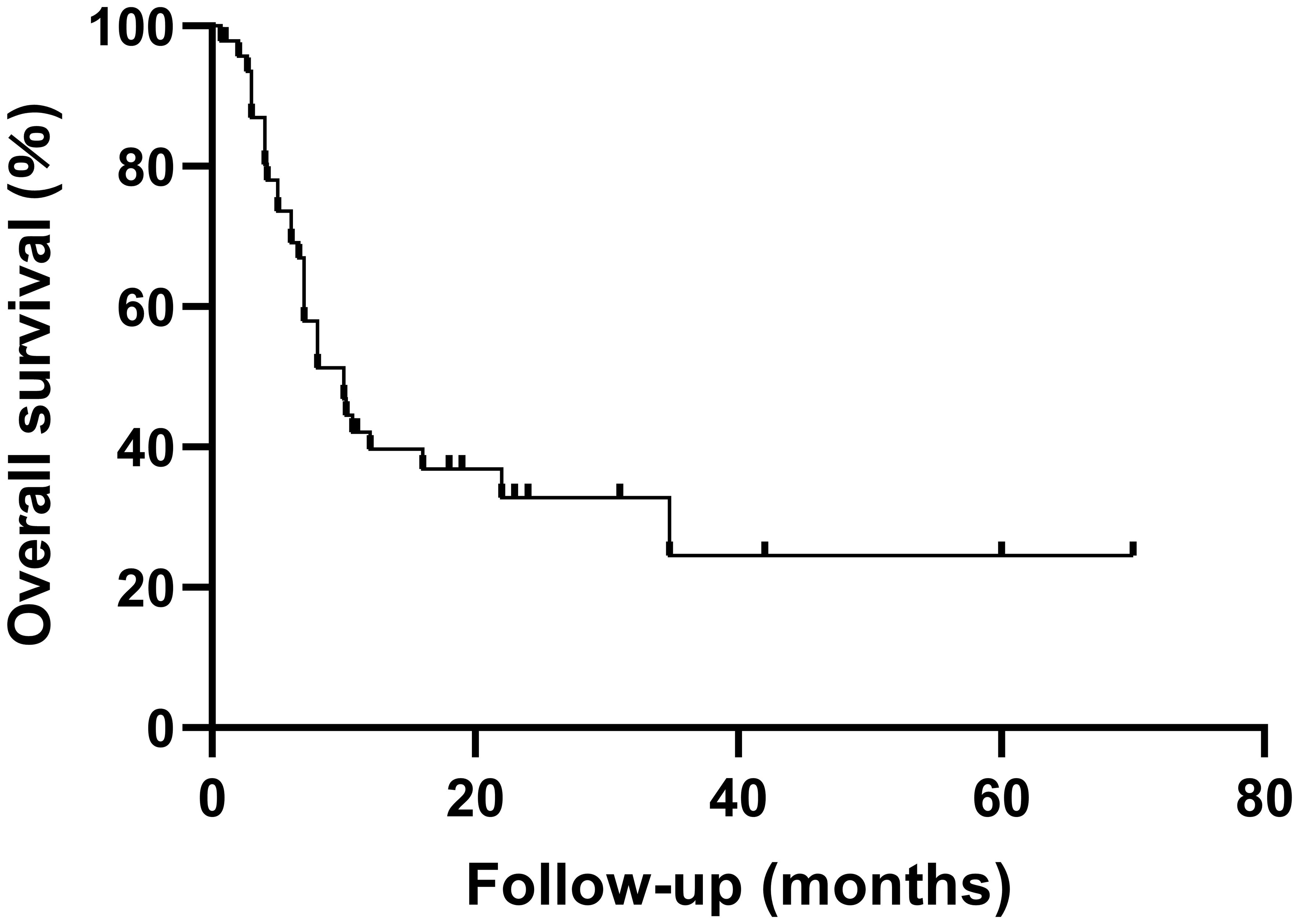
Figure 4. Kaplan-Meier survival curves for patients with PCNSHS.
Discussion
In 1970, Mathé et al. first reported HS, a malignant tumor originating from the lymphoid-hematopoietic system (10). HS is rare and typically occurs in lymph nodes, intestines, skin, and soft tissue. PCNSHS is even rarer and can occur in the brain, cerebellum, spinal cord, and leptomeninges (2–5). The histomorphology of PCNSHS is characterized by pleomorphic large cells, usually with abundant eosinophilic cytoplasm and irregular nuclei, accompanied by prominent nucleoli. Tumor cells proliferate actively, with common mitotic figures observed, and tumor necrosis may occur. In addition, tumor giant cells, red blood cell phagocytosis, or focal spindle cells can also be seen in some cases (1, 5, 11–13). PCNSHS is typically accompanied by significant inflammatory cell infiltration, including neutrophils, eosinophils, and plasma cells. This significant inflammatory response is an important feature that distinguishes PCNSHS from other forms of HS (14, 15). The histomorphology of this case was consistent with the histopathological results of previously published PCNSHS cases, exhibiting typical histological features and inflammatory reactions, further supporting the diagnosis of PCNSHS.
PCNSHS is a diagnosis of exclusion. Immunohistochemistry plays a crucial role in diagnosing PCNSHS by identifying the histiocytic lineage of tumor cells using specific markers. Diagnosis of HS (including PCNSHS) requires at least two positive markers, including CD68, CD163, CD4 and lysozyme, and negative markers are also needed to rule out other diseases (16). CD163 is a hemoglobin-clearance receptor that is expressed only on monocyte and histiocytic lineages. Compared to CD68, CD163 has stronger specificity in identifying histiocytic lineages and diagnosing HS (17). Interestingly, a study reported a case of PCNSHS with only weak and focal expression of CD163. In this regard, one hypothesis is that this case may show a similar phenotype to M1 macrophages, which typically have low CD163 expression; another hypothesis is that some HS cases may show partial loss of specific markers (18). In our case, CD4, CD68, and CD163 were all positive, and Lysozyme showed a small amount of sporadic positive expression, suggesting that the tumor cells originated from histiocytes.
The diagnosis of PCNSHS requires the systematic exclusion of tumors with overlapping morphological and molecular characteristics by integrating immunohistochemical results and molecular characteristics in multiple dimensions. Preliminary immunohistochemical assessment should focus on identifying histiocytes lineages, such as CD163, CD68 and other histiocytes markers. Secondly, tumors with overlapping morphological features must be excluded using ancillary techniques. For example: S-100 positivity is observed in melanoma, interdigitating dendritic cell sarcoma, and Rosai-Dorfman disease. However, S-100 is also expressed in approximately 33% of HS cases. Therefore, a positive S-100 does not rule out the diagnosis of HS (12). Negative EMA, CD34, SSTR2, and PR to exclude the possibility of most epithelial tumors and meningiomas; negative Langerin to exclude Langerhans cell-associated histiocytosis; negative CD21 to exclude out follicular dendritic cell sarcoma; negative GFAP and Oligo-2 to exclude glial tumors, such as glioblastoma and pleomorphic xanthoastrocytoma. For tumors with overlapping histological features, identifying molecular characteristics is crucial. For example: Both PCNSHS and high-grade gliomas are histologically characterized by pleomorphic tumor cells, abundant cytoplasm, significant mitotic activity, areas of necrosis, and spindle cells, which makes them difficult to distinguish (19). In addition, PCNSHS and glioma may also show overlapping molecular features, such as BRAF V600E mutation or BRAF gene fusion, CDKN2A/CDKN2B homozygous deletion, etc (20–22). However, gliomas typically do not express histiocytic markers and exhibit other molecular characteristics, including IDH mutations, ATRX alterations, 1p/19q co-deletions, EGFR amplification, TERT promoter mutations, H3 mutations, MYB/MYBL1 gene structural mutations, and FGFR1 mutations, etc (21). In conclusion, through a hierarchical strategy (first confirming the histiocytes origin, then systematically excluding tumors with overlapping morphological features, and finally verifying with molecular characteristics), the diagnostic specificity of PCNSHS can be improved.
It is well known that PCNSHS can manifest as suppurative inflammatory lesions and can therefore be easily confused with inflammatory lesions. This phenomenon can be repeatedly observed in numerous PCNSHS cases (3, 14, 15, 23). However, purulent material found during surgery is more likely a biological marker of active PCNSHS progression. Almefty et al. reported a case of PCNSHS in the left parietal lobe. The patient’s initial imaging and intraoperative findings showed purulent exudate, and pathology showed neutrophil infiltration and necrotizing inflammation. Recurrent purulent material post-surgery coexisted with heterotypic monocytic/multinucleated tumor cells in pathology. Despite repeated use of broad-spectrum antibiotics, purulent material and lesions reappeared (14). This suggests that the purulent material associated with PCNSHS is essentially secondary pathological changes accompanying tumor progression rather than an independent infection event. Clifton et al. also mentioned that this purulent substance may be a sign of disease progression (3). In our case, the initial post-operative pathology suggested inflammatory infiltration. The patient’s symptoms worsened only more than 2 months post-surgery, and pus along with an aggressive tumor was found during the second operation. It suggests that the persistence of inflammatory microenvironment may accelerate the recurrence process of PCNSHS. This may be due to chronic inflammation promoting tumor proliferation by releasing cytokines and angiogenesis factors, while the tumor itself induces secondary infection or inflammatory response, forming a vicious cycle.
Molecular diagnosis is often used as a supplement to tumor histology and pathological diagnosis. However, molecular diagnostic indicators for PCNSHS are limited and usually include routine tests such as T cell receptor gamma chain rearrangements, immunoglobulin heavy chain rearrangements, ALK alterations, and BRAF p.V600E mutations. The most common BRAF mutation in PCNSHS is BRAF p.V600E. However, Zhang et al. first reported a novel BRAF fusion variant called ARHGAP45::BRAF, found in pediatric PCNSHS (20). Marguet al. reported a case of multifocal PCNSHS. Molecular testing revealed extensive chromosomal abnormalities, particularly deletions of PTEN and CDKN2A genes, and emphasized that deletions of these genes may play a key role in the occurrence and progression of HS (24). May et al. reported a case of PCNSHS with a platelet-derived growth factor receptor (PDGFR) mutation and expression of PD-L1 and PD-L2, indicating the potential therapeutic effect of immune checkpoint inhibitors (9). Wang et al. reported a PCNSHS case with somatic NF2 mutations, enhancing our understanding of the molecular pathogenesis of this rare tumor (11). These findings suggest that the molecular characteristics and potential therapeutic targets of PCNSHS are complex and diverse, highlighting the need for further research to reveal its pathogenesis and explore more effective treatment strategies. Unfortunately, molecular testing was not performed in our case.
The etiology of PCNSHS is currently unclear and may be associated with a history of malignant hematological diseases and radiotherapy. One study reported a PCNSHS case with a prior history of chronic lymphoblastic leukemia (CLL). Due to limited sample materials, genetic rearrangement and further genetic testing could not be conducted, but clinical evidence strongly suggests that the PCNSHS may originate from previous CLL (25). Brown et al. reported on a 23-year-old male patient who developed PCNSHS seven years after remission of precursor B-cell acute lymphoblastic leukemia (B-ALL). Molecular testing revealed a clonal immunoglobulin heavy chain (IGH) gene rearrangement, suggesting that PCNSHS may have transformed from the patient’s prior B-ALL (26). Another study reported a 44-year-old male patient who developed PCNSHS 16 years after treatment for T-cell acute lymphoblastic leukemia (T-ALL) (27). These cases suggest that PCNSHS may be linked to prior hematological malignancies, with two potential theories proposed. One theory is that B-cell tumors and HS may originate from a common neoplastic progenitor cell, and B-cell tumors transform into HS through dedifferentiation and then redifferentiation. Another theory suggests that lymphoid tumors may transform into HS with different phenotypes but similar genotypes through direct transdifferentiation (28–31). In addition, a study reported a case of PCNSHS that occurred after radiotherapy. The authors established an etiological link between PCNSHS and prior radiotherapy, as well as its association with complex cytogenetic abnormalities in the tumors (8). Our patient had no previous history of lymphoma or other hematological malignancies and had not undergone tumor-related chemoradiotherapy.
PCNSHS is a highly aggressive malignant tumor with a poor prognosis. The median survival of patients is about 7 months, and the average survival is around 24 months (12). Although the optimal treatment plan for PCNSHS is unclear, surgery remains the primary treatment. For single lesions, surgery followed by postoperative radiotherapy is usually chosen; if the disease has turned multiple, more aggressive radiotherapy and combination chemotherapy are needed (9). Studies have shown that total resection is a key factor influencing overall survival and treatment outcomes in PCNSHS patients. Whether it is a single lesion or a solitary meningeal space-occupying lesion, complete resection of the tumor is closely associated with a better prognosis of the patient (2, 15, 32). Targeted therapy offers new hope for improving the prognosis of patients with PCNSHS. Vemurafenib is a targeted drug targeting the BRAF p.V600E mutation. Idbaih et al. reported a PCNSHS case with the BRAF p.V600E mutation. Within one month of receiving Vemurafenib treatment, the patient’s overall condition improved significantly. However, tumor recurrence and deterioration of neurological function ultimately led to the patient’s death (33). Another study reported a patient with PCNSHS with PDGFR mutation that was targeted by Dasatinib. However, the patient cannot tolerate Dasatinib due to side effects such as nausea, diarrhea and acute pancreatitis. In addition, the tumor also expressed PD-L1 and PD-L2, suggesting that immune checkpoint inhibitors may have potential therapeutic effects (9). In conclusion, although the treatment of PCNSHS still faces many challenges, the combination of surgery, chemoradiotherapy and targeted therapy offers a key treatment strategy to improve patient outcomes.
Conclusion
This article reports a rare case of PCNSHS, and provides an in-depth discussion of previously reported cases of PCNSHS, focusing on analyzing the key points of its diagnosis and differential diagnosis. By integrating the clinical manifestations and pathological characteristics of this case, the diagnostic difficulties of PCNSHS were further clarified, and the importance of immunohistochemical markers in differential diagnosis was emphasized. In addition, the presence of purulent material and inflammatory lesions during surgery further complicates the diagnosis. Therefore, a comprehensive pathological analysis and differential diagnosis are essential for the accurate diagnosis of PCNSHS.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
NL: Writing – review & editing. CC: Writing – original draft, Writing – review & editing. DY: Writing – review & editing. QX: Writing – original draft. YaZ: Writing – review & editing. YiZ: Writing – original draft. DW: Writing – original draft. CY: Writing – original draft. CL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the 2024 Guizhou Postgraduate Research Fund Project (2024YJSKYJJ310), the Guizhou Provincial College Students Innovation and Entrepreneurship Training Program (S202310661037), and the 2023 Zunyi Medical University Graduate Research Fund Project (ZYK240).
Acknowledgments
The authors are very grateful to the Zunyi Medical University and the related colleagues for their encouragement and support for this study, as well as to the patient and his family for their support and help for the study, and to the reviewers for their pertinent comments and careful review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
HS, Histiocytic sarcoma; PCNSHS, primary central nervous system histiocytic sarcoma; MRI, Magnetic resonance imaging; H&E, hematoxylin-eosin; IHC, immunohistochemical; PDGFR, platelet-derived growth factor receptor; CLL, chronic lymphoblastic leukemia; B-ALL, B-cell acute lymphoblastic leukemia; IGH, immunoglobulin heavy chain; T-ALL, T-cell acute lymphoblastic leukemia.
References
1. Zanelli M, Ragazzi M, Marchetti G, Bisagni A, Principi M, Fanni D, et al. Primary histiocytic sarcoma presenting as diffuse leptomeningeal disease: case description and review of the literature. Neuropathol: Off J Japanese Soc Neuropathol. (2017) 37:517–25. doi: 10.1111/neup.12390
2. Zuo P, Zhang M, Wu W, Wang Y, Li T, Sun T, et al. Primary intracranial histiocytic sarcomas: A report of six cases and a pooled analysis of individual patient data. J Cancer Res Clin Oncol. (2023) 149:12071–9. doi: 10.1007/s00432-023-05112-3
3. Clifton W, Akinduro OO, Lopez-Chiriboga S, Whitaker DA, Reimer R. Infection or glioma? The false dilemma of primary central nervous system histiocytic sarcoma. World Neurosurg. (2017) 106:1053.e1–.e5. doi: 10.1016/j.wneu.2017.07.001
4. Onyenekwu CP, Cunningham AM, Schilter K, Reddi HV, Cochran EJ. Diffuse leptomeningeal histiocytic sarcoma: histologic and molecular findings in an autopsy case. J Neuropathol Exp Neurol. (2022) 81:79–81. doi: 10.1093/jnen/nlab107
5. So H, Kim SA, Yoon DH, Khang SK, Hwang J, Suh CH, et al. Primary histiocytic sarcoma of the central nervous system. Cancer Res Treat. (2015) 47:322–8. doi: 10.4143/crt.2013.163
6. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 who classification of tumors of the central nervous system: A summary. Neuro-oncology. (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
7. Bai J, Li G, Shen M, Sui D, Lin S. Primary central nervous system histiocytic sarcoma mimicking glioma. Neurol India. (2014) 62:684–5. doi: 10.4103/0028-3886.149409
8. Wu W, Tanrivermis Sayit A, Vinters HV, Pope W, Mirsadraei L, Said J. Primary central nervous system histiocytic sarcoma presenting as a postradiation sarcoma: case report and literature review. Hum Pathol. (2013) 44:1177–83. doi: 10.1016/j.humpath.2012.11.002
9. May JM, Waddle MR, Miller DH, Stross WC, Kaleem TA, May BC, et al. Primary histiocytic sarcoma of the central nervous system: A case report with platelet derived growth factor receptor mutation and pd-L1/pd-L2 expression and literature review. Radiat Oncol (London England). (2018) 13:167. doi: 10.1186/s13014-018-1115-x
10. Mathé G, Gerard-Marchant R, Texier JL, Schlumberger JR, Berumen L, Paintrand M. The two varieties of lymphoid tissue “Reticulosarcomas”, histiocytic and histioblastic types. Br J Cancer. (1970) 24:687–95. doi: 10.1038/bjc.1970.82
11. Wang W, Zhang Y, Dong Y, Li S, Qin H. Primary central nervous system histiocytic sarcoma with somatic nf2 mutation: case report and review of literature. Clin Neuropathol. (2022) 41:253–62. doi: 10.5414/np301473
12. Ma S, Schild M, Tran D, Zhang X, Zhang WL, Shen S, et al. Primary central nervous system histiocytic sarcoma: A case report and review of literature. Medicine. (2018) 97:e11271. doi: 10.1097/md.0000000000011271
13. Ueno T, Nishijima H, Kurotaki H, Kurose A, Tomiyama M. An unusual case of chronic meningitis due to histiocytic sarcoma of the central nervous system with meningeal dissemination. Neurol Sci: Off J Ital Neurol Soc Ital Soc Clin Neurophysiol. (2016) 37:1875–7. doi: 10.1007/s10072-016-2636-4
14. Almefty RO, Tyree TL, Fusco DJ, Coons SW, Nakaji P. Primary histiocytic sarcoma of the brain mimicking cerebral abscess. J Neurosurg Pediatr. (2013) 12:251–7. doi: 10.3171/2013.6.Peds12533
15. Bell SL, Hanzely Z, Alakandy LM, Jackson R, Stewart W. Primary meningeal histiocytic sarcoma: A report of two unusual cases. Neuropathol Appl Neurobiol. (2012) 38:111–4. doi: 10.1111/j.1365-2990.2011.01205.x
16. Hung YP, Qian X. Histiocytic sarcoma. Arch Pathol Lab Med. (2020) 144:650–4. doi: 10.5858/arpa.2018-0349-RS
17. Nguyen TT, Schwartz EJ, West RB, Warnke RA, Arber DA, Natkunam Y. Expression of cd163 (Hemoglobin scavenger receptor) in normal tissues, lymphomas, carcinomas, and sarcomas is largely restricted to the monocyte/macrophage lineage. Am J Surg Pathol. (2005) 29:617–24. doi: 10.1097/01.pas.0000157940.80538.ec
18. Perez I, Gokden M, Day JD, Yaziji H, Pina-Oviedo S. Primary Histiocytic Sarcoma of the Clivus with Focal Extension into Central Nervous System and Neurologic Manifestations: First Description at an Unusual Site with an Overwhelming and Rapid Progression. Clin Neuropathol. (2022) 41:74–82. doi: 10.5414/np301400
19. Gill-Samra S, Ng T, Dexter M, Wong M, Nahar N, Allsopp K, et al. Histiocytic sarcoma of the brain. J Clin Neurosci: Off J Neurosurgical Soc Australasia. (2012) 19:1456–8. doi: 10.1016/j.jocn.2011.10.023
20. Zhang L, Zhang G, Zheng H, Jiang B, Ju Y, Duan Q, et al. A rare case of primary central nervous system histiocytic sarcoma harboring a novel arhgap45::Braf fusion: A case report and literature review. Brain Tumor Pathol. (2024) 41:18–29. doi: 10.1007/s10014-023-00471-8
21. Weller M, Wen PY, Chang SM, Dirven L, Lim M, Monje M, et al. Glioma. Nat Rev Dis Primers. (2024) 10:33. doi: 10.1038/s41572-024-00516-y
22. Cecchi R, Guptil D, Haslett N, Hristov A, Bledsoe JR, Tsai H, et al. Primary cns histiocytic sarcoma: two case reports highlighting a novel miga2::Braf gene fusion and genome-wide DNA methylation profiling results. J Neuropathol Exp Neurol. (2024) 83:882–6. doi: 10.1093/jnen/nlae061
23. Sun W, Nordberg ML, Fowler MR. Histiocytic sarcoma involving the central nervous system: clinical, immunohistochemical, and molecular genetic studies of a case with review of the literature. Am J Surg Pathol. (2003) 27:258–65. doi: 10.1097/00000478-200302000-00017
24. Marguet F, Piton N, Adle-Biassette H, Renaud F, Bohers E, Boyer T, et al. Molecular characteristics of multifocal brain histiocytic sarcoma. Neuropathol Appl Neurobiol. (2019) 45:309–13. doi: 10.1111/nan.12490
25. Curry RC, Faivre G, Akkari L, Joyce JA, Lin O, Rosenblum M, et al. High-dose methotrexate-based chemotherapy as treatment for histiocytic sarcoma of the central nervous system. Leukemia Lymphoma. (2016) 57:1961–4. doi: 10.3109/10428194.2015.1120867
26. Brown AF, Fan H, Floyd JR, Henry JM, Higgins RA. Primary central nervous system histiocytic sarcoma arising after precursor B-cell acute lymphoblastic leukemia. J Neuropathol Exp Neurol. (2015) 74:1120–6. doi: 10.1097/nen.0000000000000258
27. Chalasani S, Hennick MR, Hocking WG, Shaw GR, Lawler B. Unusual presentation of a rare cancer: histiocytic sarcoma in the brain 16 years after treatment for acute lymphoblastic leukemia. Clin Med Res. (2013) 11:31–5. doi: 10.3121/cmr.2012.1092
28. Skala SL, Lucas DR, Dewar R. Histiocytic sarcoma: review, discussion of transformation from B-cell lymphoma, and differential diagnosis. Arch Pathol Lab Med. (2018) 142:1322–9. doi: 10.5858/arpa.2018-0220-RA
29. Ansari J, Naqash AR, Munker R, El-Osta H, Master S, Cotelingam JD, et al. Histiocytic sarcoma as a secondary Malignancy: pathobiology, diagnosis, and treatment. Eur J Haematol. (2016) 97:9–16. doi: 10.1111/ejh.12755
30. Wang E, Papalas J, Hutchinson CB, Kulbacki E, Huang Q, Sebastian S, et al. Sequential development of histiocytic sarcoma and diffuse large B-cell lymphoma in a patient with a remote history of follicular lymphoma with genotypic evidence of a clonal relationship: A divergent (Bilineal) neoplastic transformation of an indolent B-cell lymphoma in a single individual. Am J Surg Pathol. (2011) 35:457–63. doi: 10.1097/PAS.0b013e3182098799
31. McClure R, Khoury J, Feldman A, Ketterling R. Clonal relationship between precursor B-cell acute lymphoblastic leukemia and histiocytic sarcoma: A case report and discussion in the context of similar cases. Leukemia Res. (2010) 34:e71–3. doi: 10.1016/j.leukres.2009.08.020
32. Foster M, Kamaly-Asl I, Stivaros S, Kelsey A, Gattamenini R, Kilday JP. Primary cerebral histiocytic sarcoma in childhood: A case report of protracted survival and review of the literature. Child’s Nervous System: ChNS: Off J Int Soc Pediatr Neurosurg. (2015) 31:2363–8. doi: 10.1007/s00381-015-2815-2
33. Idbaih A, Mokhtari K, Emile JF, Galanaud D, Belaid H, de Bernard S, et al. Dramatic response of a braf V600e-mutated primary cns histiocytic sarcoma to vemurafenib. Neurology. (2014) 83:1478–80. doi: 10.1212/wnl.0000000000000880
34. Bayram E, Pehlivan UA, Erdogan KE, Turker M, Yalniz H, Paydas S. Coexistence of acute severe leukocytosis and anaplastic lymphoma kinase−Positive histiocytic sarcoma, a rare entity with an unusual presentation: A case report. Oncol Lett. (2024) 28:516. doi: 10.3892/ol.2024.14649
35. Yanchu L, Li Z, Qiongwen Z, Jiayu D, Feng W. Case report: treatment of a rare primary cerebellum histiocytic sarcoma with surgery and radiotherapy. Front Oncol. (2024) 14:1398350. doi: 10.3389/fonc.2024.1398350
36. Shahrokh S, Rakhsha A, Shahin M, Javadzadegan A, Ahadi M, Azghandi S, et al. Successful treatment of central nervous system histiocytic sarcoma with craniectomy and adjuvant radiotherapy. Cureus. (2022) 14:e24690. doi: 10.7759/cureus.24690
37. Rogawski DS, Nirschl JJ, McDonald J, Nie E, Schwartz NU, Vogel H, et al. A rare neuromyelitis optica mimic: primary cns histiocytic sarcoma. Multiple Sclerosis (Houndmills Basingstoke England). (2022) 28:1651–4. doi: 10.1177/13524585221097564
38. Rajeshwari M, Suri V, Sarkar C, Garg A, Sharma MC. Primary histiocytic sarcoma of brain-illustration of two cases with varied histomorphological features. Neurol India. (2022) 70:1254–9. doi: 10.4103/0028-3886.349657
39. Takahashi E, Sakakibara A, Tsuzuki T, Nakamura S. Case of primary central nervous system histiocytic sarcoma with prominent proliferation of histiocytic cells between the trabeculae of reactive glial cells. Neuropathol: Off J Japanese Soc Neuropathol. (2018) 38:609–18. doi: 10.1111/neup.12510
40. Kim YH, Yie GT, Kim NR, Jeon IS, Cho HY, Seok JY, et al. Pediatric intracerebral histiocytic sarcoma with rhabdoid features: case report and literature review. Neuropathol: Off J Japanese Soc Neuropathol. (2017) 37:560–8. doi: 10.1111/neup.12396
41. Nieuwenhuis MB, van der Salm SM, Verhoeff JJ, van der Kooi AJ, Slavujecvic-Letic I, Pals ST, et al. A 43-year-old female with multifocal cerebral lesions. Histiocytic sarcoma. Brain Pathol (Zurich Switzerland). (2015) 25:371–2. doi: 10.1111/bpa.12260
42. Chen CJ, Williams EA, McAneney TE, Williams BJ, Mandell JW, Shaffrey ME. Histiocytic sarcoma of the cavernous sinus: case report and literature review. Brain Tumor Pathol. (2015) 32:66–71. doi: 10.1007/s10014-014-0191-3
43. Moulignier A, Mikol J, Heran F, Galicier L. Isolated iii cranial nerve palsies may point to primary histiocytic sarcoma. BMJ Case Rep. (2014). doi: 10.1136/bcr-2014-204663
44. Pérez-Ruiz E, Delgado M, Sanz A, Gil AM, Domínguez AR. Primary leptomeningeal histiocytic sarcoma in a patient with a good outcome: A case report and review of the literature. J Med Case Rep. (2013) 7:127. doi: 10.1186/1752-1947-7-127
45. Laviv Y, Zagzag D, Fichman-Horn S, Michowitz S. Primary central nervous system histiocytic sarcoma. Brain Tumor Pathol. (2013) 30:192–5. doi: 10.1007/s10014-012-0123-z
46. Wang J, Li T, Chen H, Liu Q. A case of primary central nervous system histiocytic sarcoma. Clin Neurol Neurosurg. (2012) 114:1074–6. doi: 10.1016/j.clineuro.2012.02.009
47. Gomi K, Tanaka M, Yoshida M, Ito S, Sonoda M, Iwasaki F, et al. Primary cerebellar histiocytic sarcoma in a 17-month-old girl. J Neurosurg Pediatr. (2012) 10:126–9. doi: 10.3171/2012.5.Peds11270
48. Devic P, Androdias-Condemine G, Streichenberger N, Berger F, Honnorat J, Broussolle E, et al. Histiocytic sarcoma of the central nervous system: A challenging diagnosis. QJM: Monthly J Assoc Physicians. (2012) 105:77–9. doi: 10.1093/qjmed/hcq244
49. Toshkezi G, Edalat F, O’Hara C, Delalle I, Chin LS. Primary intramedullary histiocytic sarcoma. World Neurosurg. (2010) 74:523–7. doi: 10.1016/j.wneu.2010.07.002
Keywords: histiocytic sarcoma, primary central nervous system, diagnosis and differential diagnosis, case report, immunohistochemistry
Citation: Liang N, Chen C, Yuan D, Xu Q, Zhan Y, Zhao Y, Wu D, Yang C and Li C (2025) Diagnostic challenges of primary central nervous system histiocytic sarcoma: case report and literature review. Front. Oncol. 15:1551157. doi: 10.3389/fonc.2025.1551157
Received: 24 December 2024; Accepted: 13 March 2025;
Published: 31 March 2025.
Edited by:
Brent T. Harris, Georgetown University, United StatesReviewed by:
Emily Sloan, MedStar Georgetown University Hospital, United StatesSakir Gultekin, University of Miami, United States
Copyright © 2025 Liang, Chen, Yuan, Xu, Zhan, Zhao, Wu, Yang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunming Li, bmFsaWFuZ0B6bXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship