 Linnan Wu
Linnan Wu Weiji Xie1,2
Weiji Xie1,2 Juan Wang
Juan Wang Suying Lu
Suying Lu Yizhuo Zhang
Yizhuo Zhang Junting Huang
Junting Huang- 1Sun Yat-sen University Cancer Center; State Key Laboratory of Oncology in South China; Collaborative Innovation Center for Cancer Medicine, Guangzhou, China
- 2Department of Pediatric Oncology, Sun Yat-Sen University Cancer Center, Guangzhou, China
Neurotrophic tropomyosin receptor kinase (NTRK)-rearranged spindle cell tumors are often resistant to chemotherapy and radiotherapy. Fortunately, they are sensitive to targeted therapy of tropomyosin receptor kinase (TRK) inhibitors. However, the data on larotrectinib in Chinese children with NTRK-rearranged spindle cell tumor are still scarce. We reported 4 children with TRK fusion-positive solid tumors received larotrectinib in different clinical scenarios, including second-line treatment after progressive disease (patient #1), relapse after resection (patients #2 and #3), and metastatic disease (patient #4) and all of them benefited from the treatment. The patients harbored different TRK fusion genes (patient #1: TP53-NTRK1; #2: TPM3-NTRK1; #3: TPM3-NTRK1, DCST1-NTRK1, ZBTB7B-NTRK1, and NTRK1-DCST2; #4: LMNA-NTRK1). Our study provides new insights into the biology and management of NTRK-rearranged spindle cell tumors, contributing to the expanding evidence supporting the use of TRK inhibitors in these tumors. Further studies are needed to validate our findings and to explore the potential of second-generation TRK inhibitors in overcoming resistance.
1 Introduction
Neurotrophic tropomyosin receptor kinase (NTRK)-rearranged spindle cell tumors constitute a newly recognized group of soft tissue and bone tumors that are characterized by the fusion of NTRK1, NTRK2 or NTRK3 genes with other genes, resulting in the overexpression of neurotrophic receptor tyrosine kinases proteins (1). NTRK-rearranged spindle cell tumors are often resistant to conventional chemotherapy and radiotherapy, with a high risk of local recurrence and distant metastasis (2). However, these tumors are sensitive to targeted therapy of tropomyosin receptor kinase (TRK) inhibitors, which have shown high response rates and durable effects in solid tumors with NTRK fusions.
TRK signaling plays a crucial role in the nervous system under normal physiological conditions (3). However, translocation of the NTRK gene with fusion partners (e.g., ETV6, LMNA, TPM3) generates a novel fusion oncoprotein that results in aberrant activation of TRK kinase, triggers downstream pro-oncogenic pathways (e.g., RAS/MAPKs, MAPK, PI3K/AKT/mTOR), and ultimately leads to unregulated cell proliferation (4–6). This translocation is common in rare pediatric tumors such as infantile fibrosarcoma (IFS) (7), congenital mesoblastic nephroma (8), and papillary thyroid cancer (9).
Paediatric NTRK rearrangement-related tumours differ from adult counterparts in The epidemiology, histology, molecular, and biological characteristics of pediatric NTRK rearrangement-related tumors differ from those of their adult counterparts. The estimated incidence of NTRK fusion in pediatric tumors is approximately 2%. In general, these tumors have favorable outcomes, with 3-year overall survival (OS) rates > 90%. However, some tumors may recur or metastasize, especially those with high-grade histology or unfavorable sites (10).
Larotrectinib is a potent and orally available, highly selective small-molecule inhibitor of TRK (11). Its oral administration is more convenient compared to other similar therapies which require intravenous infusions at the hospital, thus contributing to a higher compliance, which is a better option for children. Recent phase 1/2 trials have demonstrated high overall response rate (ORR), sustained durable responses and favorable tolerability in different TRK fusion-positive solid tumors (12–14). The objective response rate (ORR) of these agents in pediatric patients ranges from 57.7% to 100% (12). However, data on larotrectinib in children with NTRK-rearranged spindle cell tumor are still scarce in China.
Herein, we report four cases of NTRK-rearranged spindle cell tumors in China, each harboring distinct TRK fusions achieved both durable response and good tolerability to larotrectinib.
2 Case description
Four cases of pediatric patients with TRK fusion-positive solid tumors treated with larotrectinib are presented (Figure 1, Table 1). NTRK fusions were identified via histopathology, immunohistochemistry, and whole-exome sequencing. Larotrectinib was administered as a second- or later-line therapy at 100 mg/m2 twice daily. The last follow-up was in April 2024. Patient #1 (facial mesenchymal tumor, TP53-NTRK1) received four cycles of alternating chemotherapy with vincristine-doxorubicin-cyclophosphamide plus ifosfamide-etoposide, but the disease progressed. Second-line larotrectinib induced partial response (PR) for 48 months, with a tumor regression rate of 44%. Patient #2 (spindle cell tumor in the thigh, TPM3-NTRK1), who relapsed after tumor resection, achieved PR with larotrectinib for 10 months with a tumor regression rate of 66%. Resistance developed due to a novel HDGF-TPM3 fusion gene, and subsequent treatment with ICP-723 (a second-generation TRK inhibitor) revealed tumor shrinkage. Patient #3 (retro-orbital spindle cell tumor, TPM3-NTRK1, DCST1-NTRK1, ZBTB7B-NTRK1, and NTRK1-DCST2) had local recurrence after surgery and achieved PR with larotrectinib for 36 months, with a tumor regression rate of 43%. Patient #4 (spindle cell tumor in the knee, LMNA-NTRK1) had multiple metastases 2 years after surgery and achieved PR with larotrectinib for 51 months. Treatment-related adverse events (TRAEs) in all four cases were grade 1, except one grade 2 aspartate aminotransferase elevation (Table 1). No serious TRAEs occurred. There was no treatment interruption or discontinuation due to TRAEs.
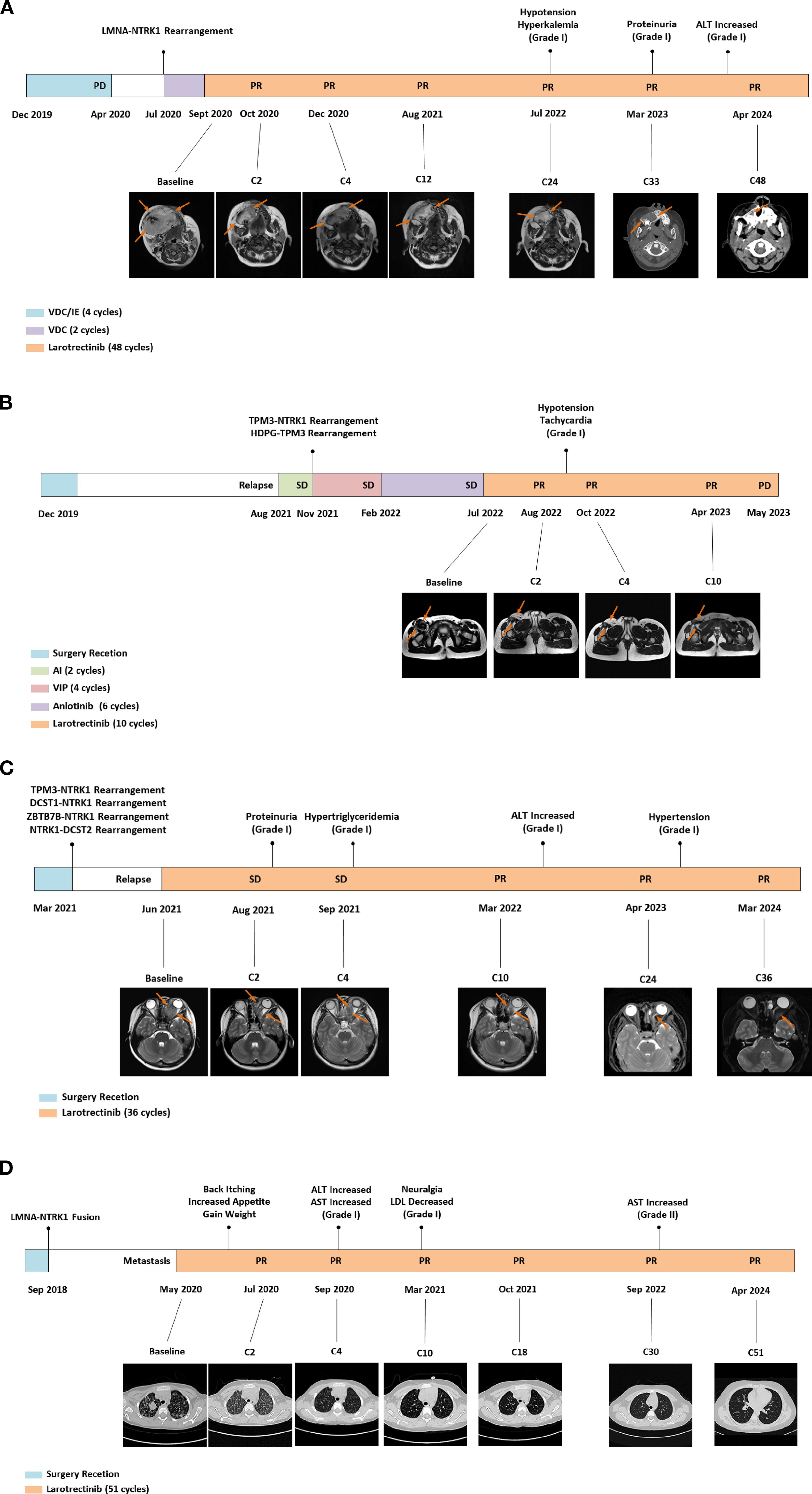
Figure 1. Diagnosis and therapeutic journey, imaging evaluations, genetic testing, and TRAEs of the four patients diagnosed with NTRK-rearranged spindle cell tumors. TRAEs, treatment-related adverse events; PD, progressive disease; PR, partial response; MR, magnetic resonance; VDC, vincristine-doxorubicin-cyclophosphamide; IE, isocyclophosphamide-etoposide; ALT, alanine aminotransferase; SD, stable disease; AI, pirarubicin-ifosfamide; VIP, etoposide-ifosfamide-cisplatin; CT, computed tomography; AST, aspartate aminotransferase; LDL, low-density lipoprotein.
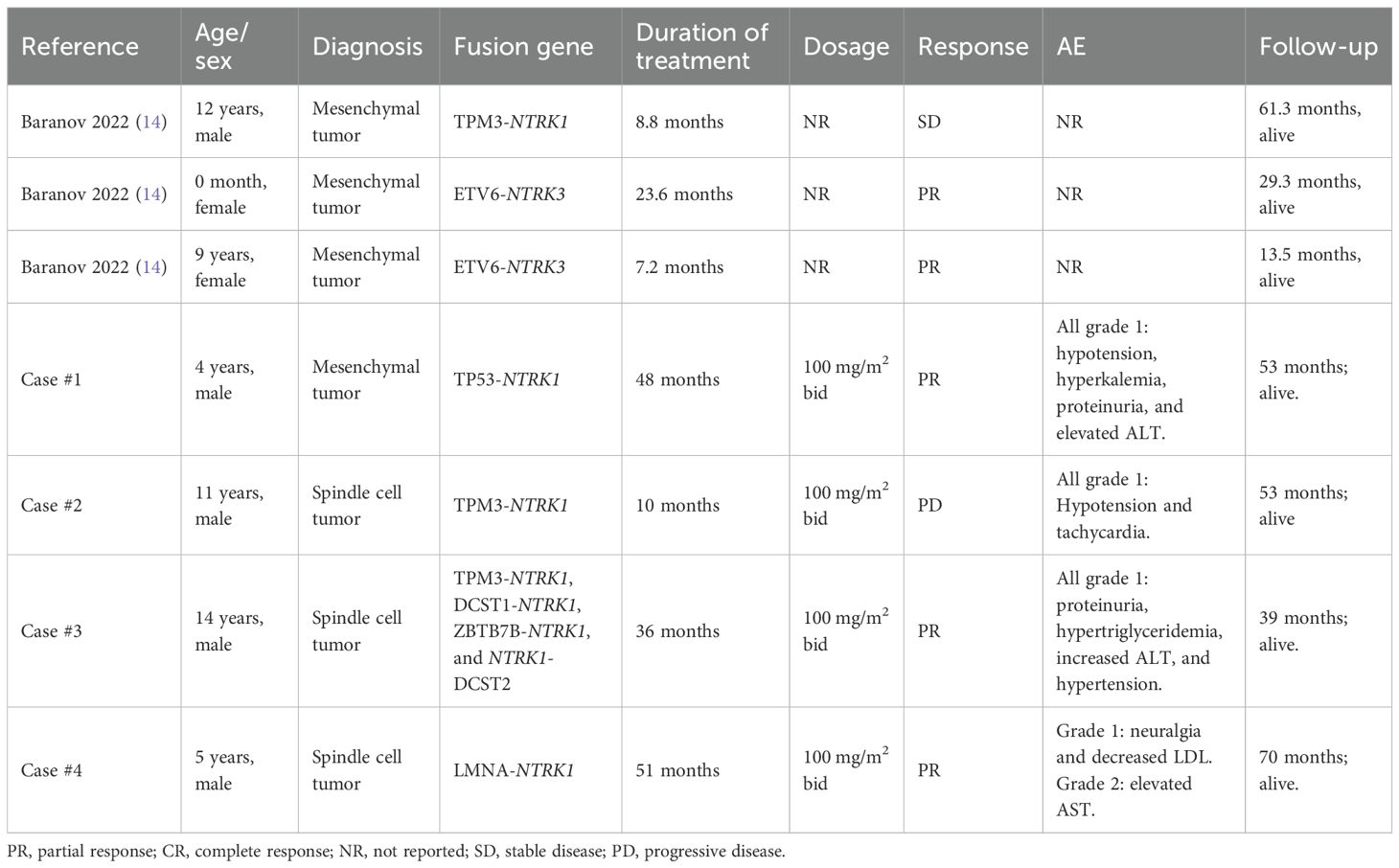
Table 1. Literature review of children treated with larotrectinib for solid tumors.
3 Discussion
Larotrectinib is a first-generation NTRK inhibitor indicated for patients with TRK fusion-positive solid tumors, regardless of age, tumor type, or fusion partner (12–14). The data on larotrectinib use in pediatric patients with NTRK-rearranged spindle cell tumors are still scarce in China. Here, we reported four cases under diverse clinical scenarios: second-line treatment after progressive disease (PD) (case #1), relapse after resection (cases #2 and #3), and metastatic disease (case #4). Larotrectinib treatment led to rapid and durable clinical responses. The longest treatment duration reached 51 cycles, and three patients maintained tumor control at the last follow-up. Notably, Case #2 developed disease progression after 10 cycles of larotrectinib, accompanied by the newly emergence of a novel HDGF-TPM3 fusion gene. Additionally, our cases exhibited distinct NTRK fusion profiles: DCST1-NTRK1, ZBTB7B-NTRK1, and NTRK1-DCST2 (Case #3)—all of which have not been previously reported in the literature. These findings expand the evidence base for larotrectinib’s clinical application in this patient population.
Larotrectinib has demonstrated robust efficacy in adult patients with TRK fusion-positive sarcomas, with a reported ORR of 58% and a median progression-free survival (PFS) of 28.3 months, the most common fusion partners named LMNA::NTRK1 and ETV6::NTRK3 (15). Recine et al. described a young adult with a rare TPM4-NTRK1 fusion-positive spindle cell neoplasm, who experienced rapid and durable regression of both visceral and bone metastases just 7 days after initiating larotrectinib, with no drug-related toxicities observed (16). Then, larotrectinib continues to durable responses with favorable safety in pediatric TRK fusion solid tumors (12, 17, 18). A global multicenter clinical trial further evaluated the safety and efficacy of larotrectinib in 91 pediatric sarcoma patients (including 21 with spindle cell tumors) with an ORR of 87%. Fifty percent of patients experienced treatment-related adverse events (TRAEs) of maximum grade 1 or 2, with no serious adverse reactions observed (17). Most TRAEs of larotrectinib are mild and tolerable, and common TRAEs include electrolyte disturbances, elevated liver enzymes, and blood pressure fluctuations. Among our four cases, all observed TRAEs were grade 1, except for one patient who developed grade 2 aspartate aminotransferase elevation. Notably, Case #4 experienced multiple neurological adverse eventsduring treatment, including back pruritus, increased appetite, weight gain, and persistent yet tolerable neuralgia. These neurological changes are likely attributable to disruption of TRK pathway signaling. When combined adults with the pediatric cases, these reports highlight the tumor-agnostic activity of TRK inhibition in diverse age groups and clinical disease settings.
Management of NTRK-rearranged spindle cell tumors requires a multidisciplinary approach, combining surgery with other modalities (e.g., radiotherapy, targeted therapy). However, the optimal timing and duration of TRK inhibitor therapy remain unclear. The risk of disease recurrence following TRK inhibitor discontinuation may depend on multiple factors, including NTRK fusion type, tumor histological grade, extent of surgical resection, and presence of residual disease. A Children’s Oncology Group (COG) study suggested that children with infantile fibrosarcoma (IFS) who achieved complete response (CR) with larotrectinib could discontinue treatment after 6 months without compromising prognosis (19). However, this recommendation may not extend to NTRK-rearranged spindle cell tumors. Notably, approximately one-third of patients who electively discontinued larotrectinib experienced disease relapse or progression; yet, 94% of these patients regained disease control upon reinitiating larotrectinib, with 69% achieving an objective response (17). An ongoing prospective COG trial (ADVL1823; NCT03834961) is evaluating response durability following larotrectinib discontinuation in pediatric solid tumors. These studies illustrated the complexity and diversity of clinical scenarios. Longer follow-up and individualized, closely monitored treatment decisions are therefore needed to determine the optimal larotrectinib treatment duration.
Mechanisms of acquired resistance to TRK inhibition are not completely understood. Point mutations in the kinase domain of TRK was the main resistance mechanisms to first-generation TRK inhibitors (6). Few literature reported details of the occurrence of NTRK fusion gene types in pediatric tumors. In our cohort, Case #2 developed larotrectinib resistance after 10 months of disease control, which was associated with the de novo emergence of the HDGF-TPM3 fusion gene. An adult patient with TPM4-NTRK1 fusion exhibited a durable response to larotrectinib (16). This finding suggests that different NTRK fusion partners may affect the efficacy of larotrectinib and the risk of developing resistance to them. Thus, it is essential to identify variations in fusion partners through comprehensive molecular testing and formulate individualized targeted therapy strategies to maximize the therapeutic benefits of TRK inhibitors. HDGF is a heparin-binding growth factor overexpressed in various cancers, where it promotes tumor growth, invasion, angiogenesis, and metastasis. TPM3—a tropomyosin family gene—encodes a cytoskeletal protein involved in muscle contraction and cell motility. Notably, TPM3 is a common fusion partner of NTRK1 in spindle cell tumors; fusion with HDGF may activate alternative signaling pathways or augment tumor aggressiveness. Further studies are necessary to delineate the potential mechanisms of HDGF-TPM3-mediated resistance.
The small sample size of this study limits the reliability of our findings. Additionally, our exploration of the underlying resistance mechanisms is insufficient, and future studies including larger cohorts and prospective designs are needed to validate these observations.
In summary, we reported the efficacy and safety of larotrectinib in four pediatric cases of NTRK-rearranged spindle cell tumors, and discussed the challenges in determining the optimal treatment duration. Notably, HDGF-TPM3 fusion gene may contribute to acquired resistance to larotrectinib. Further research is needed to explore the potential of second-generation TRK inhibitors for overcoming resistance.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethics Committee of Sun Yat-sen University Cancer Prevention Center. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
LW: Investigation, Methodology, Writing – original draft. WX: Conceptualization, Data curation, Formal Analysis, Writing – original draft. JW: Investigation, Writing – review & editing. SL: Investigation, Writing – review & editing. YZ: Funding acquisition, Project administration, Writing – review & editing. JH: Data curation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We would like to thank all the patients who participated in our study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Trubicka J, Grajkowska W, and Dembowska-Bagińska B. Molecular markers of pediatric solid tumors-diagnosis, optimizing treatments, and determining susceptibility: current state and future directions. Cells. (2022) 11:1238. doi: 10.3390/cells11071238
2. Dome JS, Rodriguez-Galindo C, Spunt SL, and Santana VM. Pediatric solid tumors. In: Niederhuber JE, Armitage JO, Kastan MB, Doroshow JH, Kastan MB, and Tepper JE, editors. Abeloff’s Clinical Oncology, 6th ed. Elsevier, Philadelphia, PA (2020). chap 92. doi: 10.1016/B978-0-323-47674-4.00092-X
3. Nakagawara A. Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett. (2001) 169:107–14. doi: 10.1016/s0304-3835(01)00530-4
4. Amatu A, Sartore-Bianchi A, and Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open. (2016) 1:e000023. doi: 10.1136/esmoopen-2015-000023
5. Cocco E, Scaltriti M, and Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. (2018) 15:731–47. doi: 10.1038/s41571-018-0113-0
6. Xiang S and Lu X. Selective type II TRK inhibitors overcome xDFG mutation mediated acquired resistance to the second-generation inhibitors selitrectinib and repotrectinib. Acta Pharm Sin B. (2024) 14:517–32. doi: 10.1016/j.apsb.2023.11.010
7. Bielack SS, Cox MC, Nathrath M, Apel K, Blattmann C, Holl T, et al. Rapid, complete and sustained tumour response to the TRK inhibitor larotrectinib in an infant with recurrent, chemotherapy-refractory infantile fibrosarcoma carrying the characteristic ETV6-NTRK3 gene fusion. Ann Oncol. (2019) 30:viii31–5. doi: 10.1093/annonc/mdz382
8. El Demellawy D, Cundiff CA, Nasr A, Ozolek JA, Elawabdeh N, Caltharp SA, et al. Congenital mesoblastic nephroma: a study of 19 cases using immunohistochemistry and ETV6-NTRK3 fusion gene rearrangement. Pathology. (2016) 48:47–50. doi: 10.1016/j.pathol.2015.11.007
9. Prasad ML, Vyas M, Horne MJ, Virk RK, Morotti R, Liu Z, et al. NTRK fusion oncogenes in pediatric papillary thyroid carcinoma in northeast United States. Cancer. (2016) 122:1097–107. doi: 10.1002/cncr.29887
10. Zhao X, Kotch C, Fox E, Surrey LF, Wertheim GB, Baloch ZW, et al. NTRK fusions identified in pediatric tumors: the frequency, fusion partners, and clinical outcome. JCO Precis Oncol. (2021) 1:PO.20.00250. doi: 10.1200/PO.20.00250
11. Doebele RC, Davis LE, Vaishnavi A, Le AT, Estrada-Bernal A, Keysar S, et al. An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov. (2015) 5:1049–57. doi: 10.1158/2159-8290.CD-15-0443
12. Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. (2018) 378:731–9. doi: 10.1056/NEJMoa1714448
13. Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. (2018) 19:705–14. doi: 10.1016/S1470-2045(18)30119-0
14. Hong DS, Bauer TM, Lee JJ, Dowlati A, Brose MS, Farago AF, et al. Larotrectinib in adult patients with solid tumours: a multi-centre, open-label, phase I dose-escalation study. Ann Oncol. (2019) 30:325–31. doi: 10.1093/annonc/mdy539
15. Kummar S, Shen L, Hong DS, McDermott R, Keedy VL, Casanova M, et al. Larotrectinib efficacy and safety in adult patients with tropomyosin receptor kinase fusion sarcomas. Cancer. (2023) 129:3772–82. doi: 10.1002/cncr.35036
16. Recine F, De Vita A, Fausti V, Pieri F, Bongiovanni A, Franchini E, et al. Case report: adult NTRK-rearranged spindle cell neoplasm: early tumor shrinkage in a case with bone and visceral metastases treated with targeted therapy. Front Oncol. (2021) 11:740676. doi: 10.3389/fonc.2021.740676
17. Mascarenhas L, DuBois SG, Albert CM, Bielack S, Orbach D, Federman N, et al. Elective discontinuation of larotrectinib in pediatric patients with TRK fusion sarcomas and related mesenchymal tumors. JCO. (2025) 43(10):1180–7. doi: 10.1200/JCO.24.00848
18. Baranov E, Winsnes K, O’Brien M, Voss SD, Church AJ, Janeway KA, et al. Histologic characterization of paediatric mesenchymal neoplasms treated with kinase-targeted therapy. Histopathology. (2022) 81:215–27. doi: 10.1111/his.14680
19. Laetsch TW, Ludwig K, Barkauskas DA, Dubois SG, Ronan J, Weigel B, et al. A phase II study of larotrectinib for children with newly diagnosed solid tumors and relapsed acute leukemias harboring TRK fusions: Children’s Oncology Group study ADVL1823. J Clin Oncol. (2020) 38:TPS10560–TPS10560. doi: 10.1200/JCO.2020.38.15_suppl.TPS10560
Keywords: larotrectinib, TRK inhibitor, NTRK-rearranged, children, spindle cell tumor
Citation: Wu L, Xie W, Wang J, Lu S, Zhang Y and Huang J (2025) Case Report: Novel findings of larotrectinib in children with NTRK-rearranged spindle cell tumor. Front. Oncol. 15:1561051. doi: 10.3389/fonc.2025.1561051
Received: 15 January 2025; Accepted: 15 September 2025;
Published: 29 September 2025.
Edited by:
Zheng Jin Tu, Cleveland Clinic, United StatesReviewed by:
Lorena Gurrieri, Scientific Institute of Romagna for the Study and Treatment of Tumors (IRCCS), ItalySilvia Vanni, IRST, Italy
Nem Kumar Jain, ITM University, India
Copyright © 2025 Wu, Xie, Wang, Lu, Zhang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junting Huang, aHVhbmdqdEBzeXN1Y2Mub3JnLmNu