 Jwa Hoon Kim
Jwa Hoon Kim Boyeon Kim
Boyeon Kim Jiwon Lee
Jiwon Lee Jin Kim
Jin Kim Jung-Myun Kwak
Jung-Myun Kwak Soohyeon Lee
Soohyeon Lee- 1Division of Medical Oncology, Department of Internal Medicine, Korea University College of Medicine, Seoul, Republic of Korea
- 2Cancer Research Institute, Korea University College of Medicine, Seoul, Republic of Korea
- 3Department of Surgery, Korea University College of Medicine, Seoul, Republic of Korea
The gut microbiome plays a pivotal role in tumor–microenvironment interactions, inflammation modulation, and immune regulation, thereby affecting the response to anticancer therapy. This pilot study investigated gut composition according to clinical characteristics and its association with chemotherapy response in patients with metastatic colorectal cancer (mCRC). Seventeen patients were treated with first-line chemotherapy at Korea University Anam Hospital between 2021 and 2023. Stool samples were collected from 15 patients at baseline, during chemotherapy, or at the time of disease progression, and 16S rRNA sequencing was performed. As a result, among lifestyle factors affecting the development of CRC, smoking habits showed weak differences in beta diversity. Non-smokers predominantly harbored bacteria such as Butyricicoccaceae, Ruminococcaceae, Faecalibacterium, and Lachnospiraceae_NK4A136_group, whereas Smokers were associated with Actinomyces and Solobacterium. In terms of baseline characteristics and chemotherapy response, beta diversity exhibited notable differences according to RAS mutation status, and LEfSe analysis indicated that Holdemanella, Anaerostipes, and Collinsella were significantly enriched in patients with RAS mutations. Chemotherapy Responders harbored more beneficial bacteria, notably Lactobacillus, despite the lack of differences in diversity between the responder and Non-responder groups. According to disease control during follow-up, Bifidobacterium abundance significantly increased in the non-progressive disease group. This study suggests that gut microbiome composition is associated with smoking history, RAS mutation status, and chemotherapy response in patients with mCRC. These findings highlight the potential role of the gut microbiome as a biomarker to predict treatment response and prognosis, with its composition shaped by both host lifestyle factors and genetic mutations.
1 Introduction
Colorectal cancer (CRC) develops through the accumulation of mutations in colonic epithelial cells, which promote the transition from normal mucosa to adenocarcinoma (1). APC is a tumor suppressor gene that regulates cell adhesion and migration, maintenance of genome stability, and apoptosis (1, 2). It is known to be the gatekeeper gene during the adenoma–carcinoma sequence of CRC. In addition, several driver mutations, such as KRAS, BRAF, PIK3CA, SMAD4, and TP53, are associated with the development and progression of CRC (2). Several environmental factors contribute to the occurrence and accumulation of these somatic mutations. Numerous carcinogens, including fat, red and processed meat, alcohol, chemicals, and smoking, pass through the gastrointestinal (GI) tract and contribute to the development and progression of cancer, including CRC (3, 4). The increasing incidence of CRC in historically low-risk areas, such as Eastern Asia, is attributed to the so-called Western lifestyle (1, 5, 6).
The gut microbiome comprises a diverse group of microorganisms inhabiting the gastrointestinal (GI) tract that can be altered by daily lifestyle and dietary patterns (3, 7, 8). However, these habits cannot be determined using only one simple marker. Instead, the gut microbiome passing through the GI tract, which reflects the overall environment of the GI tract, can serve as a surrogate indicator. It plays a critical role in digestion, immunity, and overall health, and may influence cancer risk and progression (3, 7, 9). Certain bacterial species—including Fusobacterium nucleatum, Bacteroides fragilis, and Escherichia coli—are more abundant in CRC tissues than in normal tissues, indicating their potential role in cancer development (10). In contrast, beneficial bacteria, such as certain strains of Bifidobacterium and Lactobacillus, have shown anti-inflammatory and anti-carcinogenic effects in the colon (11).
Moreover, increasing evidence suggests that the gut microbiome is associated with cancer prognosis and response to chemotherapy. Specifically, Faecalibacterium and Ruminococcaceae were frequently observed in patients who responded well to cancer treatment (Responders), whereas Bacteroides was observed more frequently in Non-responders. Novel strategies for modulating the gut microbiome have been attempted in cancer treatment.
We aimed to investigate the characteristics of the gut microbiome in relation to clinical features and to explore its association with chemotherapy response in patients with locally advanced or metastatic CRC (mCRC).
2 Methods
We conducted a prospective pilot exploratory study to examine the diversity and composition of the gut microbiome according to clinicogenomic factors and chemotherapy responses in patients with mCRC. Written informed consent was obtained from all patients. This study was approved by our Institutional Review Board (approval number: 2021AN0403). The trial procedures were performed in accordance with the Declaration of Helsinki and the Guidelines for Good Clinical Practice.
2.1 Patients and data collection
This study enrolled patients with mCRC who were treated with first-line systemic chemotherapy between October 2021 and February 2023 at Korea University Anam Hospital. Eligible patients were those aged ≥19 years, with histologically or cytologically confirmed locally advanced or metastatic adenocarcinoma of the colon or rectum, Eastern Cooperative Oncology Group (ECOG) scores of 0–2, and no prior history of palliative systemic chemotherapy or antibiotic use.
The baseline clinical data included age; sex; smoking history; alcohol history; KRAS, NRAS, and BRAF mutation status; microsatellite instability (MSI) status; and prior radiotherapy. Data on chemotherapy regimens and responses were collected. RAS (KRAS and NRAS) and BRAF mutations were identified using polymerase chain reaction (PCR) or next-generation sequencing. 5-fluorouracil, leucovorin, and oxaliplatin (FOLFOX) or 5-fluorouracil, leucovorin, and irinotecan (FOLFIRI), with or without biological agents targeting vascular endothelial growth factor (VEGF) or epidermal growth factor receptor (EGFR), was administered every 2 weeks until disease progression or unacceptable toxicity. Tumor response was assessed according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 using computed tomography (CT) or magnetic resonance imaging (MRI) every 6 weeks during treatment. Short-course radiotherapy was administered to patients with rectal cancer.
2.2 Stool sample collection and 16S rRNA sequencing
Stool samples were self-collected by participants 1 day prior to their scheduled visit to Korea University Anam Hospital. Samples were obtained either before the initiation of systemic chemotherapy, during treatment, or at the time of disease progression. The OM-200 kit (DNA Genotek, Canada) was used according to the manufacturer’s instructions to collect stool samples. All stool samples were stored in the participant’s home freezer (−20°C), packed with ice packs, transported to the laboratory on ice, and stored in a deep freezer (−80°C), minimizing freeze–thaw cycles. After thawing, each sample was manually homogenized using a sterile tip, and small aliquots (0.2 g) were collected in a 2.0 mL microtube for microbiome and metabolome analyses. Fecal bacterial genomic DNA was extracted using the Mag-Bind® Universal Pathogen Kit (Omega Bio-tek, Norcross, GA, US). 16S rRNA sequencing was performed to analyze the composition and diversity of the gut microbiome.
2.3 Statistical analysis
Alpha diversity was quantified as the number of observed amplicon sequence variants (ASVs) and Chao1, Shannon, and Simpson indices. Wilcoxon signed-rank tests were used to evaluate differences in diversity among samples. Bray–Curtis and weighted UniFrac distance matrices for beta diversity were obtained, and q values were calculated using QIIME 2. These matrices were then imported into R to generate principal coordinate analysis plots. The linear discriminant analysis (LDA) effect size (LEfSe) algorithm was applied to identify taxonomic biomarkers and analyze them at the family and genus levels. Default parameters were used for significance (p < 0.05) and the LDA threshold (LDA score > 2.0).
Progression-free survival (PFS) was calculated from the date of initiation of first-line chemotherapy to the date of progression or death from any cause. Overall survival (OS) was calculated from the date of initiation of first-line chemotherapy to the date of death from any cause. Survival rates were estimated using the Kaplan–Meier method, and the log-rank test was used to compare differences between curves.
All analyses were performed using R packages (Qiime2R (version 0.99.6), Microbial (version 0.0.20), Microbiomeutilities (version 1.00.17)) and GraphPad Prism (version 9.0). Differences in the abundance of each microbial species were determined using the Mann–Whitney test, and p values < 0.05 were considered statistically significant.
3 Results
3.1 Patient characteristics
A total of 17 patients were included in this study. Table 1 summarizes the baseline patient characteristics. The median age was 61 years (range, 35–73 years), and 47.1% of the patients were men. The cohort included patients with RAS-mutated CRC (n = 8, 47.1%) and BRAF-mutated CRC (n = 1, 5.9%). All patients had microsatellite stable CRC. In terms of chemotherapy response, there were nine responders (complete response and partial response) and eight Non-responders (stable disease and progressive disease). At a median follow-up of 15.9 months (95% confidence interval [CI], 13.49–18.27), the median PFS was 10.9 months (95% CI, 7.12–14.64), and the median OS was not reached because none of the patients had died at the time of the analysis.
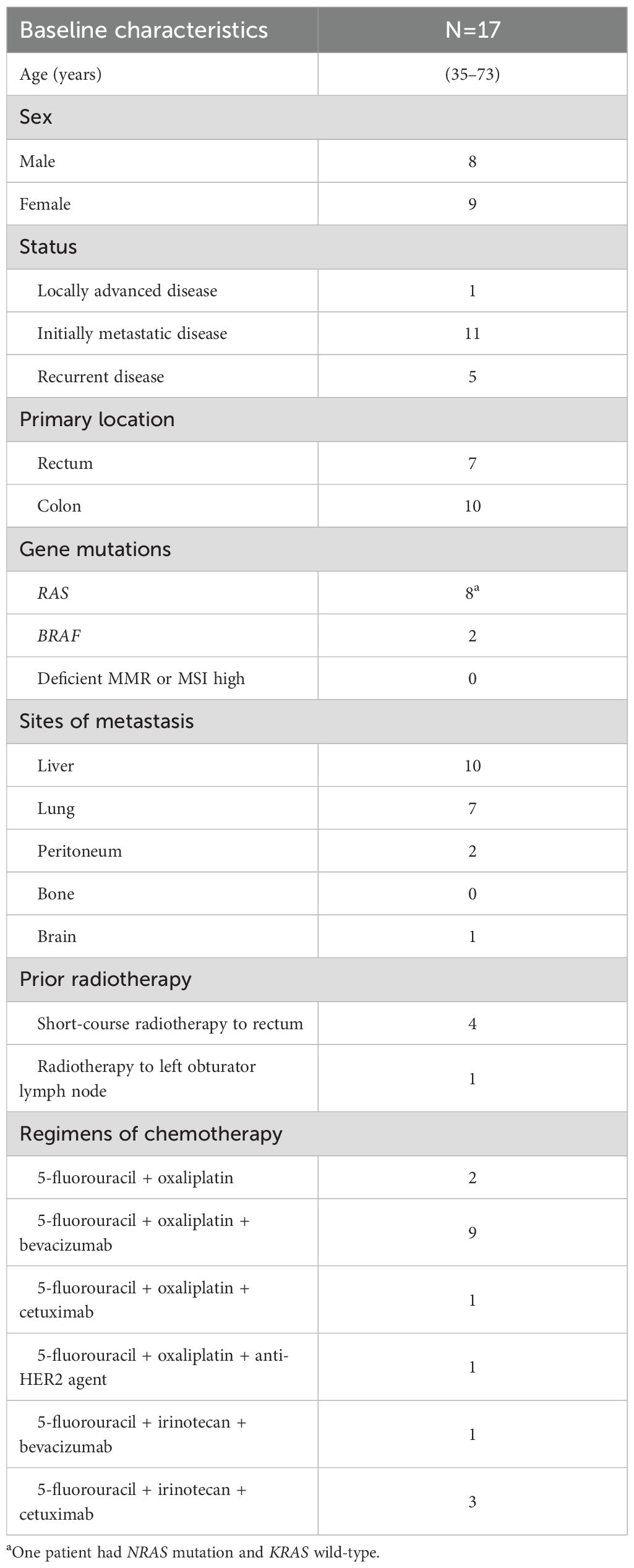
Table 1. Patients’ baseline characteristics.
Among the 17 patients, two failed to collect stool samples. Thirty stool samples were collected from 15 patients with mCRC: before chemotherapy (n = 15), during chemotherapy (n = 10), and at disease progression (n = 5). All samples met the QC requirements for 16S rRNA sequencing.
3.2 Comparing the gut microbiome by lifestyle factors: alcohol consumption and smoking habits
We compared the gut microbiome according to current alcohol intake and smoking status to assess its association with lifestyle habits known to potentially influence CRC development. No differences in alpha or beta diversity of the gut microbiome were found according to alcohol intake (Supplementary Figures 1A, B). Differences in alpha diversity according to smoking status were not significant; however, beta diversity indicated by Bray–Curtis and weighted UniFrac showed a weak difference (p = 0.058 and p = 0.056, respectively; Figures 1A, B). Furthermore, variations were observed in the microbial community based on smoking status. Non-smokers predominantly harbored bacteria such as Butyricicoccaceae, Ruminococcaceae, Faecalibacterium, and Lachnospiraceae_NK4A136_group, whereas Smokers mainly harbored Actinomyces and Solobacterium (Figure 1C, Supplementary Figure 2).
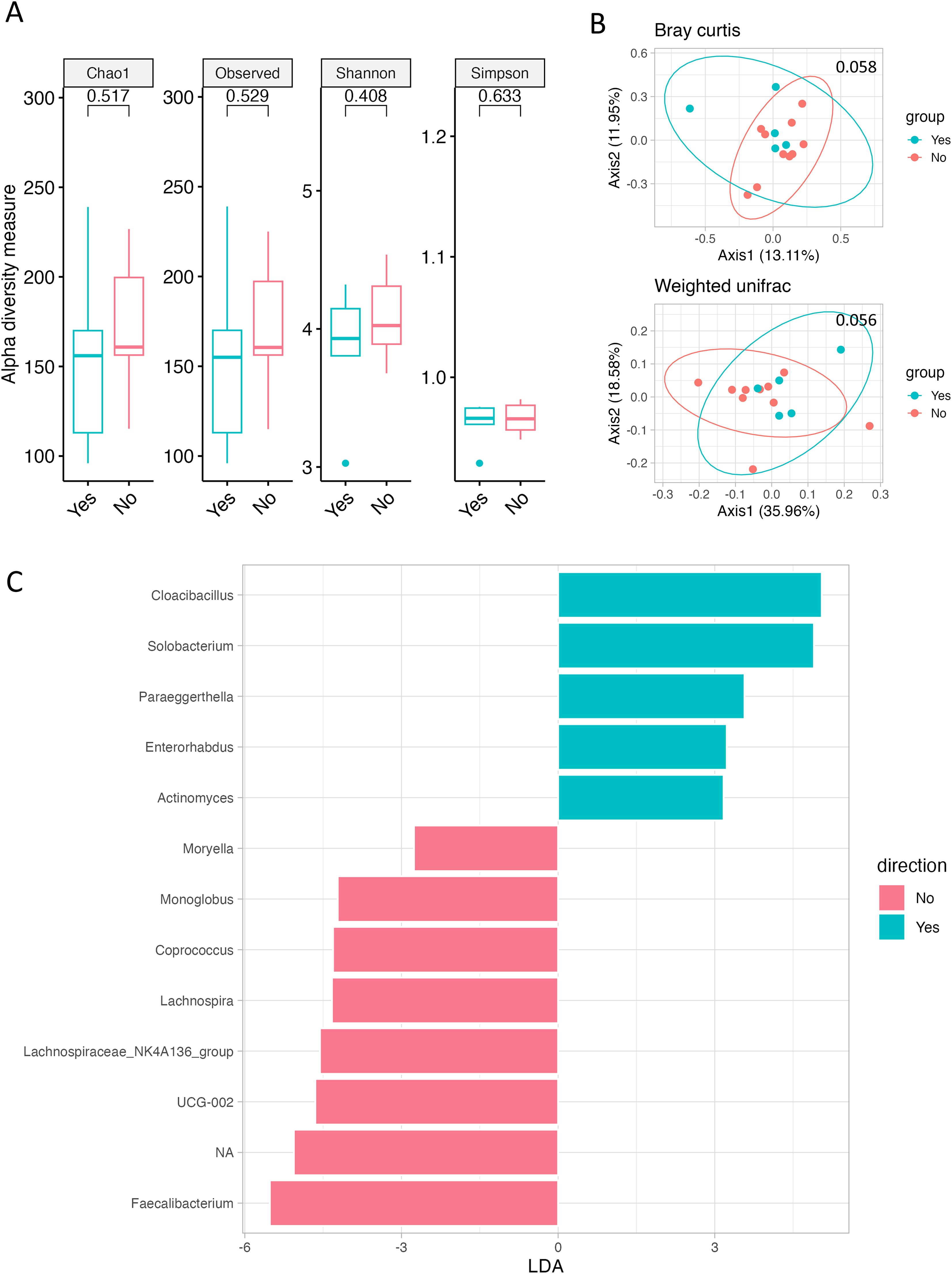
Figure 1. Gut microbiome according to smoking history. (A) Alpha diversity, as measured by the Chao1, Observed ASVs, Shannon, and Simpson indices, is depicted for both Smokers (green) and Never-smokers (red). (B) PCoA plots represent beta diversity, specifically Bray–Curtis (top) and weighted UniFrac (bottom). In these plots, each point represents a single sample, color-coded as Smokers (green) or Never-smokers (red). (C) Genus-level LEfSe analysis identified taxa with differential abundance in Smokers (green) and Never-smokers (red).
3.3 Analyzing gut microbial diversity and composition in relation to RAS mutation
We aimed to observe changes in the gut microbiome according to genetic mutations. However, due to the presence of BRAF mutations in only one patient and the absence of patients with deficient mismatch repair (MMR) or high MSI, statistical analysis for these factors was not feasible (data not shown). Regarding RAS mutations, although there were no significant changes in alpha diversity, beta diversity, as represented by Bray–Curtis and weighted UniFrac, exhibited notable differences (p = 0.042 and p = 0.047; Figures 2A, B). LEfSe analysis identified Holdemanella, Anaerostipes, and Collinsella as significantly enriched in patients with RAS mutations (Figure 2C, Supplementary Figure 3A). Additionally, at the family level, the frequency of Coriobacteriaceae increased in patients with RAS mutations (Supplementary Figures 3B, C).
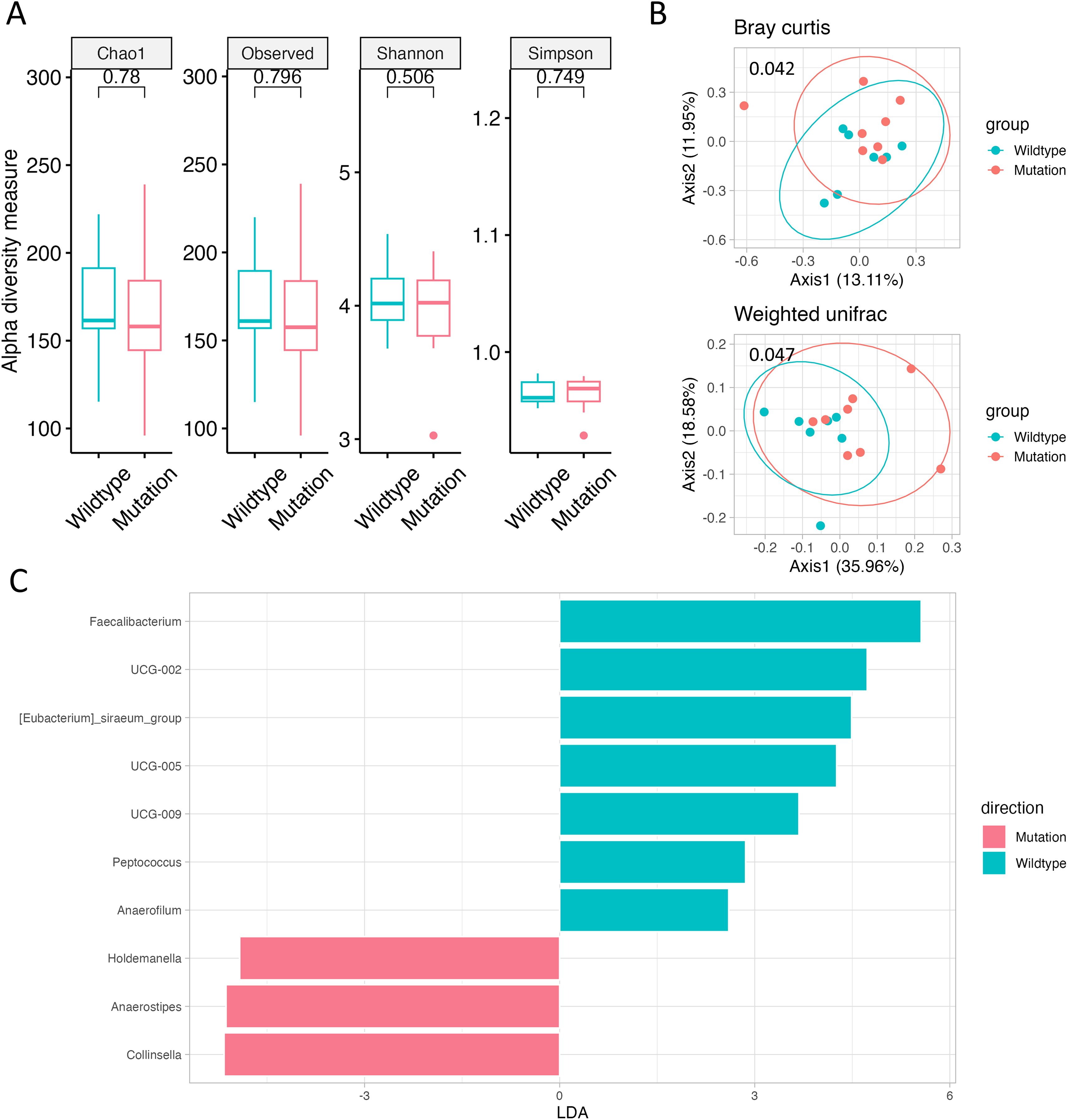
Figure 2. Gut microbiome according to RAS mutation. (A) Alpha diversity, as measured by the Chao1, Observed ASVs, Shannon, and Simpson indices, is depicted for both RAS Wild-type (green) and RAS-mutated (red) CRC. (B) Principal coordinate analysis (PCoA) plots represent beta diversity, specifically Bray–Curtis (top) and weighted UniFrac (bottom). In these plots, each point represents a single sample, color-coded as RAS Wild-type (green) or RAS-mutated (red) CRC. (C) Genus-level LEfSe analysis identified taxa with differential abundance in RAS Wild-type (green) and RAS-mutated (red) CRC.
3.4 Assessment of response to chemotherapy and baseline gut microbiome
After grouping the 15 patients into Responders (n = 9) and non-responders (n = 6) and analyzing the baseline gut microbiome before chemotherapy, no differences in alpha or beta diversity were found (Figures 3A, B). Nonetheless, the baseline samples of Responders contained numerous beneficial bacteria, including Lactobacillus spp. (Figure 3C, Supplementary Figure 4).
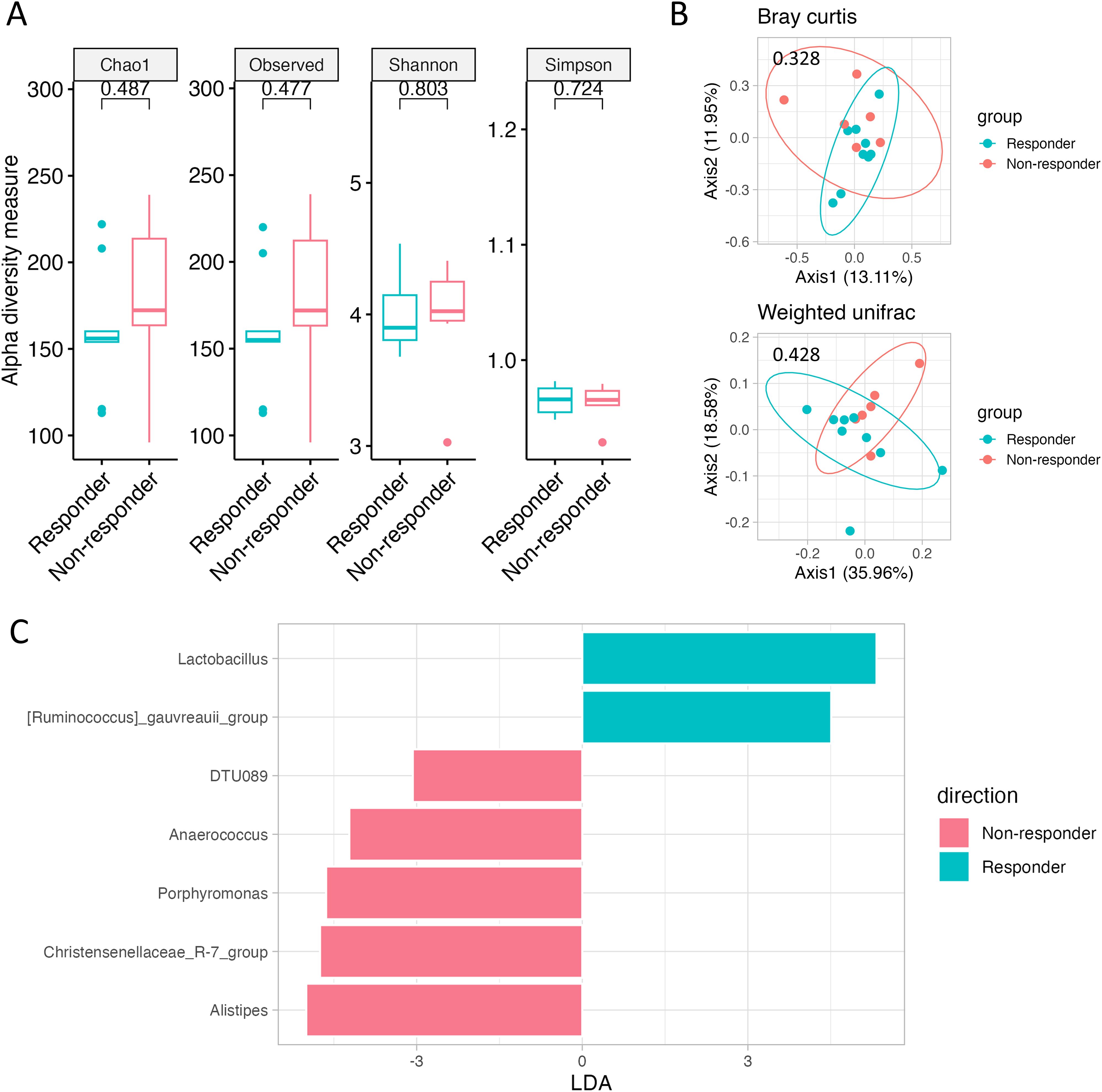
Figure 3. Gut microbiome according to chemotherapy response. (A) Alpha diversity, as measured by the Chao1, Observed ASVs, Shannon, and Simpson indices, is depicted for both Responders (green) and Non-responders (red). (B) PCoA plots represent beta diversity, specifically Bray–Curtis (top) and weighted UniFrac (bottom). In these plots, each point represents a single sample, color-coded as Responders (green) or Non-responders (red). (C) Genus-level LEfSe analysis identified taxa with differential abundance in Responders (green) and Non-responders (red).
3.5 Exploring changes of microbial abundance according to disease control with chemotherapy
Patients were grouped into progressive disease (PD) and non-PD groups based on whether their disease was controlled at the time of stool collection. We compared changes in the gut microbiome between baseline and follow-up. Among the investigated gut microbiota, there were no changes in the abundance of Enterococcus, Faecalibacterium, Peptostreptococcus, Streptococcus, or Lactobacillus between baseline and follow-up. Notably, Bacteroides tended to increase in the follow-up samples from the PD group (p = 0.2065), whereas Bifidobacterium significantly increased in the follow-up samples from the non-PD group (p = 0.0027; Figure 4 and Supplementary Figure 5).
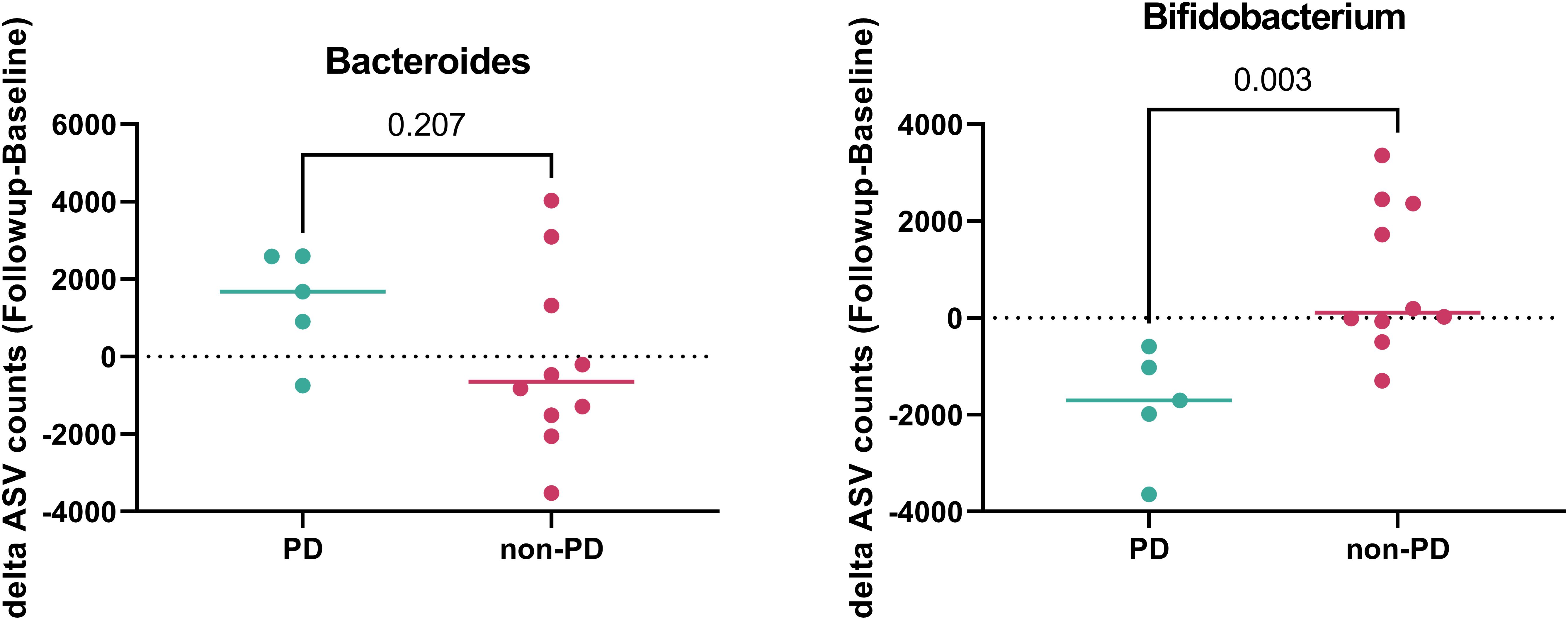
Figure 4. Gut microbiome according to disease control during follow-up. The abundance of Bacteroides and Bifidobacterium in patients with progressive disease (PD) (green) and non-PD (red).
4 Discussion
This exploratory pilot study investigated the gut microbiome in relation to lifestyle factors, clinicopathologic characteristics, and chemotherapy responses in patients with mCRC. The findings suggest that smoking habits alter the composition of the gut microbiome. Furthermore, this study highlights a potential association between RAS mutation status and the gut microbiome, which may ultimately influence chemotherapy response in patients with mCRC.
The gut microbiome comprises a diverse community of microorganisms residing in the gastrointestinal tract, and its composition can be influenced by various lifestyle factors. Our research team previously conducted a separate study involving colorectal cancer patients after surgical resection, in which a high-fiber diet was associated with an increased abundance of beneficial bacteria such as Prevotella. Furthermore, patients who experienced improvement in diarrhea symptoms showed elevated levels of Akkermansia and short-chain fatty acids (SCFAs) (12). In the present study, we also observed differences in gut microbiome composition according to smoking habits. Smokers had a higher prevalence of Actinomyces and Solobacterium—considered harmful microbiome components—than non-smokers among patients with mCRC. The diversity of the gut microbiome may also be more disrupted in Smokers than in non-smokers, given that this study showed a weak difference. Although the mechanisms by which smoking affects CRC development are not yet clear, the gut microbiome may play a mediating role (13). No relationship was found between alcohol consumption and the gut microbiome in this study. Although the composition of the gut microbiome cannot be explained by simple factors and requires comprehensive consideration of all environmental factors, these findings suggest that lifestyle factors, including diet and smoking, may contribute to alterations in the gut microbiome, which in turn could potentially influence clinical outcomes and prognosis in patients with CRC.
Notably, there were differences in the abundance and composition of the gut microbiome according to RAS mutation status in this study. Host genetics may influence the gut microbiome. A genome-wide association study (GWAS) of host genetic variation in microbiome taxa identified 31 loci affecting the gut microbiome, and the lactase gene locus was associated with Bifidobacterium abundance (14). In contrast, the gut microbiome has been shown to cause DNA damage, providing a driver for somatic mutations in preclinical studies (15). Colibactin produced by E. coli containing a pks island can cause double-strand breaks in mammalian DNA, promoting genome instability and increasing the mutation rate (16, 17). H. pylori and enteropathogenic E. coli can disrupt mismatch repair (MMR), leading to the deletion of MMR proteins (18, 19). A previous study reported a relationship between KRAS mutations and the gut microbiome in CRC (20). Roseburia, Parabacteroides, Metascardovia, Staphylococcus, and Bacillales are associated with KRAS mutation (20). As a proof of principle, tumor-associated species such as Fusobacterium and Bacteroides are positively correlated with cancer-related inflammatory pathways and negatively associated with cellular adhesion machinery (21). However, clinical data regarding the association between the gut microbiome and cancer-associated somatic mutations are lacking.
Our study provides clinical evidence for potential interactions between the gut microbiome and RAS mutations in mCRC. Anaerostipes, Collinsella, and Holdemanella were significantly more abundant in RAS-mutated CRC than in RAS Wild-type CRC, whereas Faecalibacterium and Eubacterium were significantly more abundant in RAS Wild-type CRC than in RAS-mutated CRC. Bacteroides, which is also associated with the development and progression of CRC, was more prevalent in RAS-mutated CRC than in RAS Wild-type CRC, although the difference was not statistically significant. Faecalibacterium, Eubacterium, as well as Anaerostipes, Collinsella, Holdemanella, and Bacteroides are representative gut microorganisms involved in CRC development and progression (1, 7, 11). In particular, Faecalibacterium is associated with a better treatment response, whereas Anaerostipes and Bacteroides are associated with poor treatment response (22). This may be one of the reasons for the poor prognosis of RAS-mutated CRC compared with RAS Wild-type CRC. Although preclinical functional studies are essential to elucidate the role of specific gut microbiota in prognosis, our study raises the possibility that differences in the gut microbiome between RAS-mutated and wild-type CRC may explain the different prognoses of CRC depending on RAS mutation status.
Chemotherapy Responders had a significantly higher prevalence of Ruminococcus and Lactobacillus, whereas Non-responders had a higher prevalence of Anaerococcus, Christensenellaceae, DTU089, and Porphyromonas. The association between chemotherapy response and Ruminococcus, Lactobacillus, and Anaerococcus has been reported previously (22). Porphyromonas can also promote colon cancer by activating MAPK/ERK, JNK kinase, and NF-kB signaling (23). Further investigations of Christensenellaceae and DTU089 are warranted to elucidate their clinical implications.
Notably, this study revealed individual changes in Bacteroides and Bifidobacterium—which are representative and well-known gut microorganisms in CRC (1, 7, 11), depending on whether the disease was controlled. Bifidobacterium, a beneficial microorganism, significantly decreased, and Bacteroides, a harmful microorganism, numerically increased during disease progression. Dynamic changes in the gut microbiome, as well as the baseline gut microbiome, may reflect cancer status.
Many studies investigating the interaction between the gut microbiome and cancer therapy have focused primarily on immune checkpoint inhibitors (24, 25). Several studies, including those reporting oxaliplatin-induced changes in gut microbiota and immune markers in the murine colon, as well as investigations of systemic immune responses following oxaliplatin-based neoadjuvant therapy, have suggested potential links between conventional chemotherapy, the gut microbiome, and host immunity (26, 27). However, the mechanistic interplay between conventional chemotherapeutic agents and the gut microbiota—particularly in modulating immune responses—has not been fully elucidated.
Our study offers preliminary observations that may contribute to a broader understanding of this area by exploring potential associations between gut microbial profiles and chemotherapy response in patients with CRC. Further research will be necessary to clarify whether chemotherapy-related immune modulation is influenced by the gut microbiota, which could potentially reveal novel aspects of treatment response or resistance.
This study had several limitations that should be acknowledged when interpreting the findings. First, the relatively small sample size constrained the statistical power of the analyses and limited our ability to conduct a comprehensive and robust assessment of changes in gut microbiome diversity across clinical subgroups. As a result, the observed associations should be considered exploratory and hypothesis-generating rather than conclusive. Further studies involving larger and independent cohorts are needed to validate these preliminary findings and better understand the potential clinical relevance of the microbiome signatures identified in this study.
Second, this study was conducted in a Korean population of East Asian descent, which may limit the generalizability of the findings to other ethnic groups. Moreover, data on key confounding variables—including socioeconomic status, marital status, use of medications or dietary supplements, and detailed lifestyle or dietary patterns—were not collected, which may have influenced gut microbiome composition.
Third, the timing of follow-up sample collection was not standardized across all patients due to practical and clinical considerations, potentially leading to inconsistencies in temporal microbiome data and limiting longitudinal interpretations.
Lastly, while our study observed potential associations between specific gut microbial species and clinical features, including RAS mutation status, we did not perform mechanistic experiments to investigate how these microbes might influence RAS-related pathways in gastrointestinal cancers. As a pilot exploratory study, our findings are preliminary and should be interpreted with caution. Further functional studies are warranted to elucidate the underlying biological mechanisms and to determine whether these microbes play a causal role in modulating RAS signaling or cancer progression. Future investigations should include shotgun metagenomics, metabolomics, and functional validation to clarify their relevance in CRC outcomes.
In conclusion, this pilot study sheds light on the intricate prognostic value of the gut microbiome and its association with RAS mutations in patients with mCRC treated with first-line chemotherapy. These findings provide a foundation for future studies of the gut microbiome.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA1214585.
Ethics statement
The studies involving humans were approved by Korea University Institutional Review Board (2021AN0403). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
JHK: Conceptualization, Data curation, Investigation, Resources, Writing – original draft. BK: Formal Analysis, Investigation, Methodology, Visualization, Writing – original draft. JL: Resources, Writing – review & editing. JK: Resources, Writing – review & editing. J-MK: Resources, Writing – review & editing. SL: Conceptualization, Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was supported by grants from Korea University, the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: RS-2022-KH130153) and Jeil Pharmaceutical Co., Ltd. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of interest
The authors declare that the research was conducted in theabsence of any commercial or financial relationships that could beconstrued as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1565661/full#supplementary-material
References
1. Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun. (2015) 6:6528. doi: 10.1038/ncomms7528
2. Huang D, Sun W, Zhou Y, Li P, Chen F, Chen H, et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. (2018) 37:173–87. doi: 10.1007/s10555-017-9726-5
3. Song M and Chan AT. Environmental factors, gut microbiota, and colorectal cancer prevention. Clin Gastroenterol Hepatol. (2019) 17:275–89. doi: 10.1016/j.cgh.2018.07.012
4. Davies NJ, Batehup L, and Thomas R. The role of diet and physical activity in breast, colorectal, and prostate cancer survivorship: a review of the literature. Br J Cancer. (2011) 105 Suppl 1:S52–73. doi: 10.1038/bjc.2011.423
5. Jemal A, Bray F, Center MM, Ferlay J, Ward E, and Forman D. Global cancer statistics. CA Cancer J Clin. (2011) 61:69–90. doi: 10.3322/caac.20107
6. Brenner H, Kloor M, and Pox CP. Colorectal cancer. Lancet. (2014) 383:1490–502. doi: 10.1016/s0140-6736(13)61649-9
7. Garrett WS. The gut microbiota and colon cancer. Science. (2019) 364:1133–5. doi: 10.1126/science.aaw2367
8. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. (2014) 505:559–63. doi: 10.1038/nature12820
9. Lopez LR, Bleich RM, and Arthur JC. Microbiota effects on carcinogenesis: initiation, promotion, and progression. Annu Rev Med. (2021) 72:243–61. doi: 10.1146/annurev-med-080719-091604
10. Whisner CM and Athena Aktipis C. The role of the microbiome in cancer initiation and progression: how microbes and cancer cells utilize excess energy and promote one another’s growth. Curr Nutr Rep. (2019) 8:42–51. doi: 10.1007/s13668-019-0257-2
11. Wong CC and Yu J. Gut microbiota in colorectal cancer development and therapy. Nat Rev Clin Oncol. (2023) 20:429–52. doi: 10.1038/s41571-023-00766-x
12. Kim B, Lee J, Jung ES, Lee S, Suh DH, Park YJ, et al. The impact of a modified microbiota-accessible carbohydrate diet on gut microbiome and clinical symptoms in colorectal cancer patients following surgical resection. Front Microbiol. (2024) 15:1282932. doi: 10.3389/fmicb.2024.1282932
13. Bai X, Wei H, Liu W, Coker OO, Gou H, Liu C, et al. Cigarette smoke promotes colorectal cancer through modulation of gut microbiota and related metabolites. Gut. (2022) 71:2439–50. doi: 10.1136/gutjnl-2021-325021
14. Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. (2021) 53:156–65. doi: 10.1038/s41588-020-00763-1
15. Barrett M, Hand CK, Shanahan F, Murphy T, and O’Toole PW. Mutagenesis by microbe: the role of the microbiota in shaping the cancer genome. Trends Cancer. (2020) 6:277–87. doi: 10.1016/j.trecan.2020.01.019
16. Chen B, Ramazzotti D, Heide T, Spiteri I, Fernandez-Mateos J, James C, et al. Contribution of pks(+) E. coli mutations to colorectal carcinogenesis. Nat Commun. (2023) 14:7827. doi: 10.1038/s41467-023-43329-5
17. Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. (2006) 313:848–51. doi: 10.1126/science.1127059
18. Maddocks OD, Scanlon KM, and Donnenberg MS. An Escherichia coli effector protein promotes host mutation via depletion of DNA mismatch repair proteins. mBio. (2013) 4:e00152–13. doi: 10.1128/mBio.00152-13
19. Jia K, Chen Y, Xie Y, Wang X, Hu Y, Sun Y, et al. Helicobacter pylori and immunotherapy for gastrointestinal cancer. Innovation (Camb). (2024) 5:100561. doi: 10.1016/j.xinn.2023.100561
20. Sui X, Chen Y, Liu B, Li L, Huang X, Wang M, et al. The relationship between KRAS gene mutation and intestinal flora in tumor tissues of colorectal cancer patients. Ann Transl Med. (2020) 8:1085. doi: 10.21037/atm-20-5622
21. Dohlman AB, Arguijo Mendoza D, Ding S, Gao M, Dressman H, Iliev ID, et al. The cancer microbiome atlas: a pan-cancer comparative analysis to distinguish tissue-resident microbiota from contaminants. Cell Host Microbe. (2021) 29:281–98.e5. doi: 10.1016/j.chom.2020.12.001
22. Yi Y, Shen L, Shi W, Xia F, Zhang H, Wang Y, et al. Gut microbiome components predict response to neoadjuvant chemoradiotherapy in patients with locally advanced rectal cancer: A prospective, longitudinal study. Clin Cancer Res. (2021) 27:1329–40. doi: 10.1158/1078-0432.Ccr-20-3445
23. Lu Y, Huang R, Zhang Y, Xiang W, Zhang X, Chen F, et al. Porphyromonas gingivalis induced UCHL3 to promote colon cancer progression. Am J Cancer Res. (2023) 13:5981–95.
24. Lee KA, Thomas AM, Bolte LA, Björk JR, de Ruijter LK, Armanini F, et al. Cross-cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma. Nat Med. (2022) 28:535–44. doi: 10.1038/s41591-022-01695-5
25. Li X, Zhang S, Guo G, Han J, and Yu J. Gut microbiome in modulating immune checkpoint inhibitors. EBioMedicine. (2022) 82:104163. doi: 10.1016/j.ebiom.2022.104163
26. Stojanovska V, McQuade RM, Fraser S, Prakash M, Gondalia S, Stavely R, et al. Oxaliplatin-induced changes in microbiota, TLR4+ cells and enhanced HMGB1 expression in the murine colon. PLoS One. (2018) 13:e0198359. doi: 10.1371/journal.pone.0198359
Keywords: colorectal cancer, gut microbiome, RAS mutation, chemotherapy response, prognosis
Citation: Kim JH, Kim B, Lee J, Kim J, Kwak J-M and Lee S (2025) Gut microbiome with RAS mutation and chemotherapy response in patients with advanced or metastatic colorectal cancer: a pilot, exploratory study. Front. Oncol. 15:1565661. doi: 10.3389/fonc.2025.1565661
Received: 23 January 2025; Accepted: 29 July 2025;
Published: 01 September 2025.
Edited by:
Sharon R. Pine, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Gratiela Gradisteanu Pircalabioru, University of Bucharest, RomaniaAlexander T. Sougiannis, The Ohio State University, United States
Copyright © 2025 Kim, Kim, Lee, Kim, Kwak and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Soohyeon Lee, c29vaHllb25fbGVlQGtvcmVhLmFjLmty
†These authors have contributed equally to this work and share first authorship