Abstract
Background:
Acute promyelocytic leukemia (APL) is classically driven by the PML-RARA fusion oncogene and characterized by a maturation arrest of myeloid precursors. Variant APL (vAPL) with alternative RARA rearrangements presents diagnostic and therapeutic challenges.
Methods:
We report a novel case of TBL1XR1-RARB-positive vAPL and conducted a comprehensive literature review to synthesize clinical and molecular data from all previously reported cases of this rare entity.
Results:
Our patient presented with neutrophilic leukocytosis (15.44×10⁹/L) and an absence of peripheral promyelocytes, exhibiting fever as the sole symptom, an atypical CD45⁻/CD117⁻ immunophenotype, and a concurrent KRAS p.G12D mutation. Despite an initial response to ATRA/ATO therapy, relapse occurred during maintenance. Our literature review of all reported cases revealed key patterns: a predominant pediatric occurrence (median age 2.7 years), frequent ATRA/ATO resistance (55% response rate), and a high risk of relapse (44%).
Conclusion:
This study underscores the molecular heterogeneity and distinct clinical course of TBL1XR1-RARB-positive vAPL. It highlights significant therapeutic challenges, including a high rate of primary resistance and relapse, and provides critical guidance for the management of this rare but clinically significant disease.
Introduction
Acute promyelocytic leukemia is a distinct form of acute myeloid leukemia (AML) (1). It is typically precipitated by the emergence of the PML-RARA fusion which is caused by the t(15;17)(q24;q21) translocation and which results in the synthesis of the PML-RARA fusion protein and the obstruction of the typical differentiation process of myeloid cells. It frequently exhibits a proclivity for severe bleeding tendency and may induce disseminated intravascular coagulation (DIC), which may result in early mortality. With the standardized use of all-trans-retinoic acid (ATRA) and arsenic trioxide (ATO), the long-term survival rate of APL patients can reach more than 95% (2–4). However, approximately 5% of APL patients exhibit typical morphological features but are negative for the PML-RARA fusion, referred to as variant APL (5, 6). Variant APL is primarily caused by RARA rearrangements other than PML-RARA and RARB or RARG rearrangements. Additionally, certain MLL rearrangements, NPM1 rearrangements, or other gene rearrangements can also lead to the development of variant APL (6). Recent studies have found that X::RAR::X or X::RAR::Y three-part fusion forms can also cause the development of variant APL (7).
The molecular pathogenesis of variant APL remains poorly understood. Unlike classical APL, variant APL demonstrates heterogeneous responses to ATRA and ATO, posing significant therapeutic challenges. Due to the absence of standardized treatment protocols and frequent intrinsic drug resistance, variant APL is associated with an unfavorable prognosis. Current clinical guidelines recommend ATRA combined with chemotherapy as first-line therapy for most variant APL cases. However, in instances of confirmed drug resistance, alternative AML-type chemotherapy regimens are typically employed (6). Among the rare genetic drivers of variant APL, TBL1XR1-RARB represents an exceptionally uncommon fusion. To date, the clinical characteristics, optimal treatment strategies, and long-term outcomes of TBL1XR1-RARB-positive APL have not been systematically investigated.
TBL1XR1 is a WD40 repeat-containing protein and a core component of the NCoR (nuclear receptor corepressor)/SMRT (silencing mediator of retinoic acid and thyroid hormone receptors) corepressor complex (8). This complex regulates transcriptional repression by interacting with nuclear hormone receptors (NRs) and specific transcription factors (9). TBL1XR1 functions primarily as an “exchange factor,” recruiting ubiquitin-conjugating enzymes and the 19S proteasome complex to mediate the ubiquitination and degradation of corepressors such as NCoR/SMRT and CtBP, thereby relieving repression and activating target gene expression (9).
We report a rare case of variant APL characterized by fever as the primary clinical manifestation, in the absence of typical bleeding tendencies or coagulation abnormalities. The patient tested positive for the TBL1XR1-RARB fusion gene and exhibited an atypical flow cytometric immunophenotype due to the absence of CD117 (a typical APL marker) and localization of leukemic cells in the CD45- negative region, which is uncommon in classical APL. Despite receiving standard APL induction, consolidation, and re-consolidation therapy, the patient failed to achieve complete remission (CR). However, CR was successfully attained following the introduction of an AML-type consolidation regimen. Post-remission, the patient was maintained on a conventional high-risk APL therapy protocol. Unfortunately, relapse occurred during maintenance treatment, prompting consideration of hematopoietic stem cell transplantation (HSCT) as the next therapeutic step. Additionally, we conducted a comprehensive literature review to summarize the clinical features, genetic profiles, treatment responses, and prognostic outcomes of TBL1XR1-RARB-positive APL. This analysis aims to provide insights to guide future diagnosis and management of this rare and challenging disease variant.
Case presentation
Diagnostic evaluation
A previously healthy 1-year-and-11-month-old male presented to Mianyang Central Hospital on July 18, 2023, with a 15-day history of persistent high-grade fever (peaking at 39.5°C). The fever was unaccompanied by bleeding manifestations, significant anemia, or signs of extramedullary involvement. Despite out-of-hospital supportive treatment with antibiotics and intravenous human immunoglobulin (IVIG), the child continued to experience recurrent fever. Physical examination revealed an alert child with mild pharyngeal congestion but no cutaneous or mucosal hemorrhages. There was no hepatosplenomegaly or lymphadenopathy. The remainder of the physical examination was unremarkable, with normal cardiopulmonary and neurological findings. Cardiac evaluation showed right bundle branch block on ECG. Comprehensive imaging studies (cardiac/abdominal/testicular ultrasound, and CT of head/chest/abdomen) revealed no pathological findings. The child had no significant past medical history and his family history was negative for hematologic malignancies.
The patient’s pre-admission complete blood count (CBC) results from the referring hospital were as follows: Laboratory evaluation revealed leukocytosis (WBC 26.26×109/L) with neutrophilia (15.44×109/L), moderate anemia (Hb 85 g/L), and normal platelet count (240×109/L). Inflammatory markers showed mild elevation (CRP 17.47 mg/L). Coagulation studies demonstrated hypofibrinogenemia (FIB 1.43 g/L) with elevated fibrinolytic markers (FDP 37.40 µg/mL, D-dimer 10.82 mg/L), while PT (11.9 s) and APTT (27.5 s) remained within normal limits. Peripheral blood smear examination showed minimal abnormal cells (1%) without identifiable promyelocytes. The patient’s CBC performed at our hospital on July 19, 2023 revealed leukocytosis (WBC 19.82×109/L) with neutrophilia (10.98×109/L), moderate anemia (Hb 81 g/L), and a normal platelet count (192×109/L). Peripheral blood smear examination showed no evidence of abnormal cells or promyelocytes. The results of both tests are essentially consistent. Notably, this case presented with neutrophilic leukocytosis, which is atypical for classical APL characterized by maturation arrest.
Bone marrow evaluation demonstrated granulocytic hyperplasia (63.5% of ANC) with characteristic morphological features: abundant cytoplasm containing coarse purplish-red granules, showing strong MPO positivity (Figures 1A, B). Flow cytometry identified a predominant abnormal myeloid population (67.96% of nucleated cells) with a distinctive immunophenotype: positive for CD9, CD123, and MPO with heterogeneous CD33, CD13, and CD64 expression, while lacking CD45, CD117, CD34, HLA-DR, CD15, CD56, CD7, and CD2 expression (Figure 1C).
Figure 1
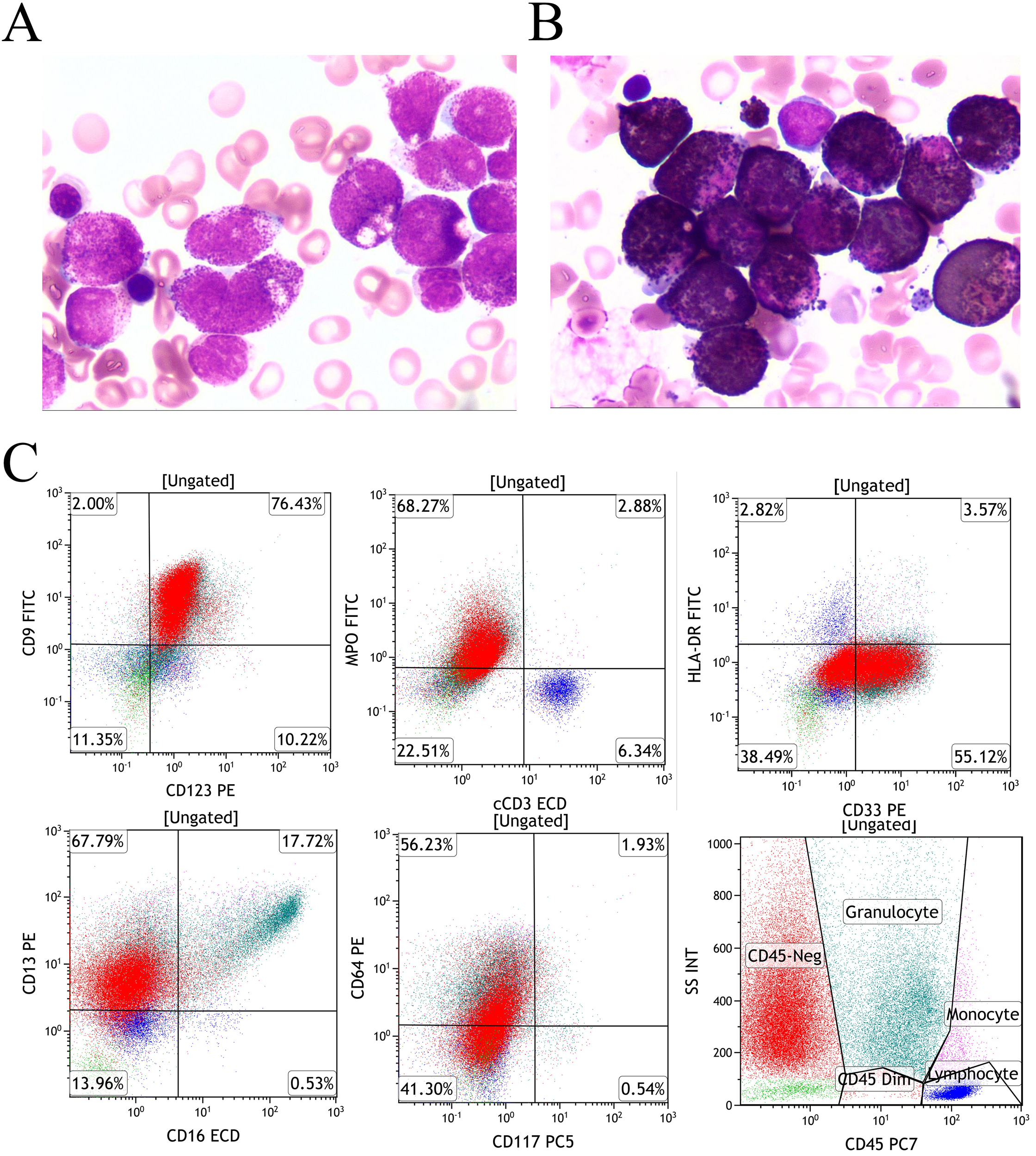
Morphology and immunophenotype (A) An increased number of promyelocytes, which contained large purplish-red granules (Wright-Giemsa staining of bone marrow, x1000); (B) Cytochemical demonstration of myeloperoxidase (MPO) activity using POX staining (Taiyang Biotechnology, China). Bone marrow smear showing intense POX positivity. (C) The leukemic population, residing in the C45-negative region, demonstrated positive expression of MPO, CD9, and CD123, with heterogeneous CD33, CD13, and CD64 expression, while remaining negative for HLA-DR and CD117.
At the time of initial diagnosis, the child’s bone marrow was evaluated through cytomorphometry and flow cytometric immunophenotyping, revealing an atypical immunophenotype consistent with acute promyelocytic leukemia (APL) (CD34-/HLA-DR-/CD45-CD117). Cytogenetic analysis via G-banded karyotyping, performed in accordance with the International System for Human Cytogenomic Nomenclature (2020) (10), showed no evidence of the classic t(15;17) translocation. Cytogenetic analysis revealed a karyotype of 47, XY, +6[20] (Figure 2A). Initial screening for PML-RARA fusion by RT-PCR was negative, prompting further molecular investigation.
Figure 2
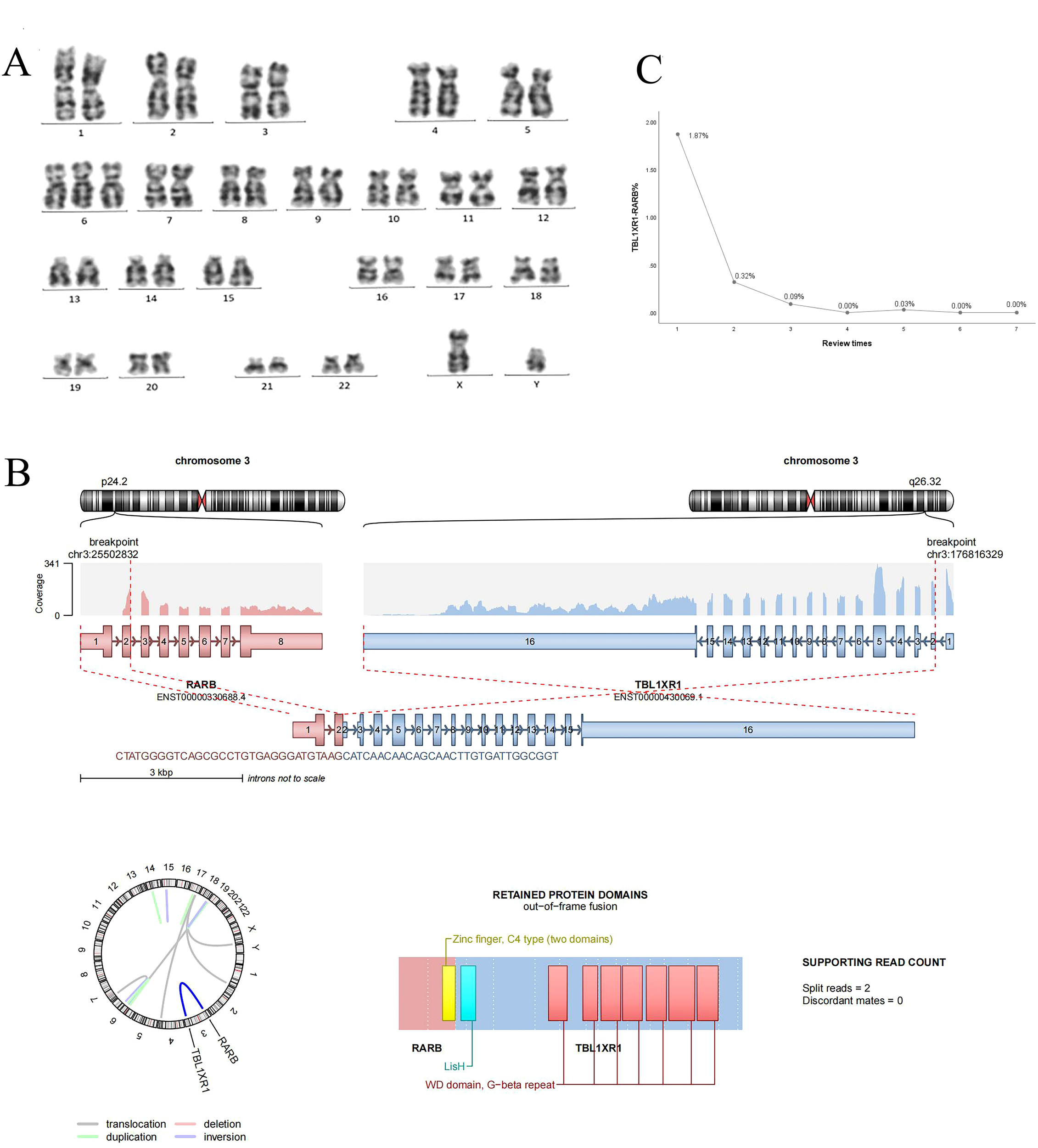
(A) Chromosome G band analysis revealing karyotype 46,XY,+6 [20]; (B) TBL1XR1-RARB fusion gene; (C) TBLIXRI-RARB fusion gene detected after induction, after consolidation l, after consolidation 2, after consolidation 3, before maintenance, after maintenance l and 2, respectively.
To identify the underlying genetic driver, whole transcriptome RNA sequencing (RNA-seq) was performed using the Illumina NovaSeq 6000 platform. Data analysis included: 1) Fusion detection using Arriba (GRCh37 reference); 2) Variant calling with VarScan (minimum VAF 1%); 3) Pathogenicity classification per AMP/ASCO/CAP tiers. All Tier-1 variants were confirmed by orthogonal methods. We identified a clinically significant (Tier-1) TBL1XR1-RARB fusion with 17 supporting reads (Figure 2B), classified as pathogenic based on AMP/ASCO/CAP guidelines. Concurrently, a KRAS p.G12D hotspot mutation was detected at 24.3% variant allele frequency, a known activating mutation in AML. Additionally, 12 variants of uncertain significance (VUS) were identified, including SPEN p.P2574L (56.6% VAF) and POT1 p.F565L (43.5% VAF), though their clinical relevance in APL remains unclear. No other pathogenic fusion genes were detected. Droplet digital PCR (ddPCR) was subsequently employed to confirmed the presence of TBL1XR1-RARB fusion.
Treatment course
The patient’s initial diagnosis of acute promyelocytic leukemia was established based on characteristic bone marrow morphological features and immunophenotypic profile. On July 18, 2023, treatment was initiated with a high-risk APL induction regimen consisting of ATRA, ATO, and daunorubicin, aiming to induce differentiation and apoptosis of leukemic cells. Subsequent molecular testing on July 21, 2023 revealed negative results for the PML-RARA fusion transcript, raising strong suspicion for variant APL. Confirmatory testing through comprehensive fusion analysis identified the rare TBL1XR1-RARB fusion transcript. Notably, despite existing literature suggesting resistance of TBL1XR1-RARB-positive APL to differentiating agents, our patient demonstrated a favorable clinical response to ATRA and ATO, manifested by gradual defervescence and the development of differentiation syndrome, which was successfully managed with dexamethasone. In light of this positive response, we continued treatment with the standard high-risk APL protocol. Supportive care included piperacillin-tazobactam for infection prophylaxis and hydroxyurea for leukocytosis management. Post-induction evaluation revealed complete morphological remission with negative minimal residual disease by flow cytometry. However, sensitive molecular monitoring detected persistent TBL1XR1-RARB fusion transcripts at 1.87% by digital PCR (Figure 2C), a level comparable to high-risk thresholds in classical APL (11). The patient subsequently underwent two cycles of consolidation therapy with ATO, ATRA, and daunorubicin, maintaining morphological and immunophenotypic remission with progressively decreasing but persistently detectable fusion transcript levels. Given the suboptimal molecular response and reported resistance patterns in variant APL, treatment was intensified with an AML-type consolidation regimen incorporating cytarabine and etoposide. This approach successfully achieved complete molecular remission with undetectable fusion transcripts. The observed response to initial ATRA/ATO therapy, while unexpected based on existing literature, suggested potential heterogeneity in treatment sensitivity among TBL1XR1-RARB-positive cases. Following achievement of complete remission, maintenance therapy was initiated using the standard high-risk APL protocol combining ATRA, realgar-indigo naturalis formula, 6-mercaptopurine, and methotrexate. Unfortunately, the patient experienced disease recurrence during maintenance therapy, prompting consideration of allogeneic hematopoietic stem cell transplantation as the next therapeutic approach. The complete treatment timeline is detailed in Figure 3.
Figure 3
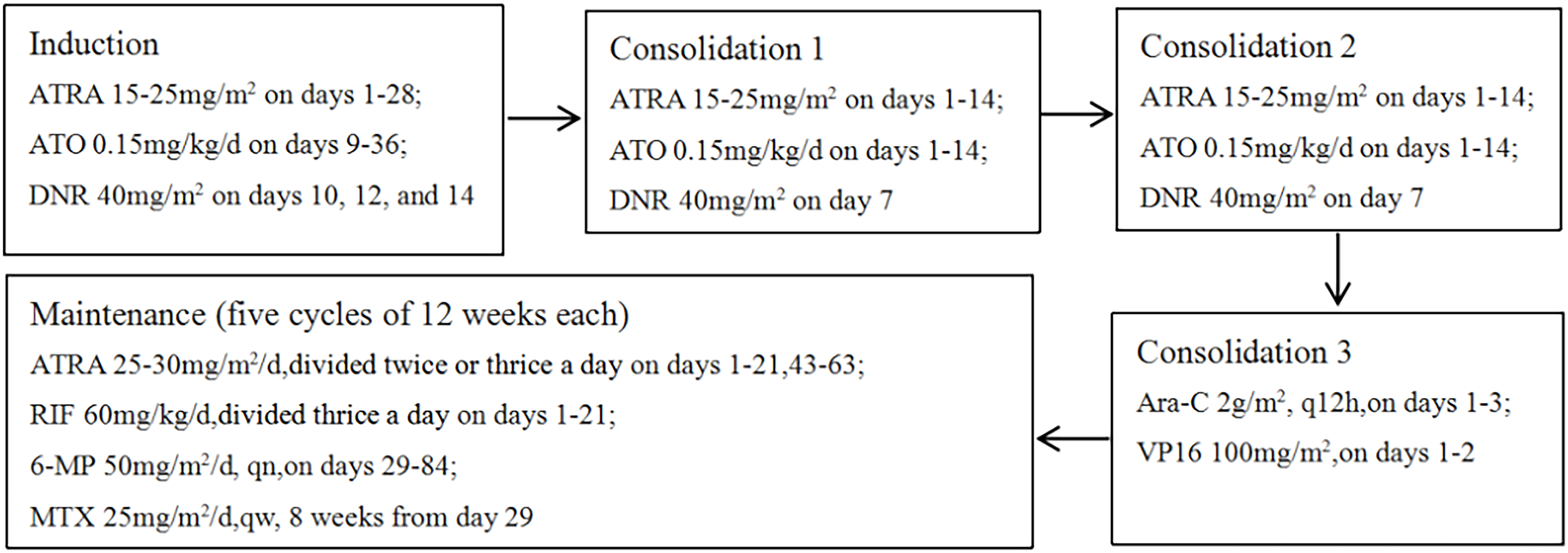
Treatment regimens. ATO, arsenic trioxide; ATRA, all-trans retinoic acid; DNR, daunorubicin; Ara-C, cytarabine; Vp16, etoposide; RlF, realgar-indigonaturalis; 6-MMP, 6-mercaptopurine; MTX, methotrexate.
Review of the literature
A comprehensive review of published literature through November 2024 identified six case reports documenting nine pediatric patients with TBL1XR1-RARB-positive variant APL. Borkovskaia et al. (2023) identified TBL1XR1-RARB and KMT2A-SEPT6 fusions in RARA-negative pediatric AML with APL-like morphology, noting resistance to ATRA/ATO and advocating for RNA-seq to improve diagnostic accuracy (12). Zhao et al. (2019) expanded this spectrum, revealing RARB/RARG fusions (e.g., TBL1XR1-RARB, CPSF6-RARG) and KMT2A rearrangements in morphologically similar cases, with RARA-negative patients showing inferior survival and higher relapse rates despite AML-style chemotherapy (13). Osumi et al. (2018) characterized TBL1XR1-RARB as a recurrent driver in RARA-negative APL, demonstrating its dominant-negative suppression of RAR signaling and clinical resistance to retinoids (14). These findings were corroborated by a 2021 case report of a 2-year-old boy with TBL1XR1-RARB APL, where initial ATRA/RIF therapy achieved transient remission, but subsequent AML-style chemotherapy (FLAG-IDA) and targeted maintenance ensured durable remission (15). Collectively, these studies underscore that RARB/RARG-rearranged and KMT2A-fused APL-like leukemias represent distinct entities with unique molecular drivers, necessitating early genetic profiling to guide therapy—typically AML regimens for RARA-negative cases—while highlighting TBL1XR1-RARB as a biomarker for ATRA resistance and potential HSCT candidacy. Standard immunophenotyping (CD13+/CD33+/HLA-DR+) cannot differentiate TBL1XR1-RARB from PML-RARA APL, underscoring the need for routine cytogenetic/molecular profiling in pediatric AML with APL-like features.
The cohort comprised five male and four female patients, with ages ranging from 11 months to 4.8 years. Clinical presentation data revealed that one-third of cases (3/9) presented with fever as a prominent symptom, while only one patient manifested concurrent bleeding tendencies and anemia at diagnosis. Hematologic parameters at presentation showed significant variability: leukocyte counts ranged from 6.1 to 128.3×109/L (median 32.4×109/L), with all patients demonstrating anemia. Thrombocytopenia was nearly universal, with only one case maintaining normal platelet levels. Morphologically, all cases fulfilled diagnostic criteria for acute promyelocytic leukemia (M3 morphology), confirmed by characteristic AML immunophenotypic profiles (Table 1). Therapeutic responses exhibited notable heterogeneity. Differentiation therapy outcomes showed that 55% (5/9) of patients responded to ATRA, while only 22% (2/9) demonstrated sensitivity to ATO, albeit with reduced efficacy compared to classical APL. All cases maintained chemosensitivity to conventional agents. Treatment outcomes revealed that 44% (4/9) of patients underwent hematopoietic stem cell transplantation (HSCT) at various disease stages. Clinical courses included one induction failure, four relapses (44%), and one mortality following multiple relapses. One patient was lost to follow-up, while the remaining seven patients (78%) achieved sustained remission (Table 2). These findings highlight both the clinical variability and therapeutic challenges associated with this rare APL variant.
Table 1
| Reference | No. | Age (years) | Gender | Clinical future | WBC (×109/L) | PLT (×109/L) | HB (g/L) | Morphology | Immunophenotype | Cytogenetics | Mutations |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Osumi (14) | 1 | 2.6 | M | NA | 53.11 | NA | NA | FAB-M3 | CD13+CD33+HLA-DR+CD34- | 46,XY,t(3;10;12)(q26.2;q22;q15)[20/20] | NA |
| 2 | 4.3 | F | NA | 6.1 | NA | NA | FAB-M3 | CD13+CD33+HLA-DR+CD34+ | 46,XX,del(2)(p)?,inv(4)(p16q12)[1/20],45,idem,-X[3/20],46,idem,del(3)(p25)[3/20],46,XX[13/2] | NA | |
| 3 | 4.1 | F | NA | 14.9 | NA | NA | FAB-M3 | CD13+CD33+HLA-DR+CD34+ | 47,XX,+3 [19/20] | NA | |
| Zhao (13) | 4 | 0.98 | M | NA | 128.3 | 34 | 98 | FAB-M3 | NA | 46,XY | ATXN3,MAP2K2 |
| 5 | 4.76 | M | NA | 24.3 | 54 | 84 | FAB-M3 | NA | 46,XY | STAG2 | |
| Shiba (16) | 6 | 0.9 | F | NA | 30.5 | NA | NA | FAB-M3 | NA | 47,XX,16[2]/46,XX | NA |
| Jiang (17) | 7 | 2 | F | Fever,ecchymosis,anemia | 41.0 | 29 | 74 | FAB-M3 | CD13+CD15+CD33+CD64+ MPO+ | 46,XX | EZH2, TBL1XR1 |
| Zhang (15) | 8 | 2.8 | M | Fever | 24.82 | 105 | 72 | FAB-M3 | NA | 46,XY | KRAS,NRAS, TBL1XR1 |
| Borkovskaia (12) | 9 | 1 | F | NA | 60 | NA | NA | FAB-M3 | CD33+ CD13+ CD15+ CD99+ MPO+ |
46,XX | NA |
| This study |
10 | 1.9 | M | Fever | 26.6 | 240 | 85 | FAB-M3 | CD9+CD123+CD33+CD64+CD13+MPO+ | 47,XY,+6[20] | KRAS |
Clinical features and laboratory tests.
NA, not applicable.
Table 2
| Reference | No. | Response to ATRA | Response To ATO | Chemotherapy | HSCT | Events | Time to relapse from diagnosis | Outcome |
|---|---|---|---|---|---|---|---|---|
| Osumi (14) | 1 | None | NA | Yes | Yes | relapse | 6 months | Alive |
| 2 | None | NA | Yes | Yes | Induction failure |
NA | Alive | |
| 3 | None | NA | Yes | Yes | relapse | 13 months | Alive | |
| Zhao (13) | 4 | Yes | NA | Yes | None | None | None | Alive |
| 5 | Yes | NA | Yes | None | relapse | 29.7 months | Death | |
| Shiba (16) | 6 | NA | NA | Yes | Yes | relapse | NA | Alive |
| Jiang (17) | 7 | Yes | Yes | Yes | None | None | NA | Alive |
| Zhang (15) | 8 | Yes | Yes | Yes | None | None | NA | Alive |
| Borkovskaia (12) | 9 | Yes | NA | Yes | None | NA | NA | NA |
| This study | 10 | Yes | Yes | Yes | None | relapse | 15 months | Alive |
Treatment response and prognosis.
ATRA, all-trans retinoic acid; ATO, arsenic trioxid; HSCT, Hematopoietic Stem Cell Transplantation, NA, not applicable.
Discussion
Consistency with previous studies
Classical PML-RARA-positive APL typically manifests with significant coagulopathy, characterized by severe bleeding diathesis and frequent development of DIC, which accounts for substantial early mortality in untreated cases (15, 17). In striking contrast, the TBL1XR1-RARB variant demonstrates a distinct clinical phenotype, typically presenting without coagulation abnormalities or overt hemorrhagic manifestations. Notably, approximately 11% of pediatric cases with this variant maintain normal platelet counts throughout their clinical course. The most common and frequently sole presenting symptom in TBL1XR1-RARB-positive APL is persistent fever, which serves as the primary clinical indicator in the majority of cases. In our case, fever was the primary symptom, while hemorrhagic manifestations—typical of classical APL—were absent. Current literature indicates that TBL1XR1-RARB-positive APL affects preschool-aged children, with reported cases ranging from 0.9 to 4.76 years (median 2.7 years) (13–17). Our patient’s age at onset (1.9 years) falls within this characteristic early-onset pattern. The suboptimal response to ATRA/ATO and eventual relapse mirror the resistance patterns observed in reported cases, underscoring the need for alternative strategies such as AML-type chemotherapy or HSCT (6, 13, 14).
Unique features of this case
Morphologically, our case demonstrated the classic M3 features typical of TBL1XR1-RARB-positive APL. The immunophenotypic profile showed some deviations from conventional APL patterns: while most APL cases exhibit strong CD117 expression (a well-established diagnostic marker), our patient surprisingly demonstrated CD117 negativity. This finding carries prognostic significance, as CD117-negative APL has been associated with poorer outcomes, including higher early mortality rates and reduced overall and progression-free survival (18, 19). The immunophenotype also showed absence of CD56, CD15, and CD2 - markers typically associated with adverse outcomes when expressed. CD56 positivity in particular has been correlated with increased risk of extramedullary involvement and treatment resistance (20–22). The CD45-/CD117-profile contrasts with classical APL (typically CD45+/CD117+) and may reflect origin from primitive progenitors, which not described in prior literature. This unique “CD45-CD117-” immunophenotype is exceptionally rare in clinical practice, suggesting the leukemic cells may have originated from more primitive hematopoietic progenitor cells than conventional APL.
KRAS mutations predominantly occur at critical amino acid residues (G12, G13, Q61, and A146) and are frequently implicated in various hematologic malignancies, including acute lymphoblastic leukemia (ALL), myelodysplastic syndromes (MDS), juvenile myelomonocytic leukemia (JMML), and AML (23). In this case, RNA sequencing of the patient’s bone marrow identified a KRAS p.G12D missense mutation—a well-characterized hotspot variant that constitutively activates RAS protein signaling. This gain-of-function mutation promotes sustained downstream pathway activation, contributing to dysregulated proliferation and survival of leukemic cells. The KRAS p.G12D variant, though not previously linked to TBL1XR1-RARB APL, aligns with its role in myeloid malignancies (13). Its functional impact warrants further study.
In contrast to classical APML, our patient presented with marked leukocytosis (26.26×109/L) and neutrophilia (ANC 15.44×109/L) with only rare abnormal cells identified on peripheral smear review. This atypical presentation may reflect either: 1) the presence of concurrent infection may have contributed to both the neutrophilic response and the obscuration of abnormal promyelocytes in peripheral blood smears, or 2) fundamental biological differences in the peripheral blood manifestations of TBL1XR1-RARB positive APML compared to classical PML-RARA positive disease, or 3) the typical APL cases are expected to exhibit maturation arrest; therefore, this case could potentially represent an extremely rare form of APL without maturation arrest. Multiple manual peripheral smear reviews confirmed the paucity of circulating abnormal promyelocytes (abnormal cell percentage 0.0%), supporting that this was not simply an artifact of automated analysis. This unusual hematologic presentation expands the known spectrum of variant APML manifestations and underscores the importance of molecular diagnostics when clinical and laboratory findings appear discordant with classical disease features.
Unlike most reported TBL1XR1-RARB cases with ATRA resistance (13, 14), our patient initially responded to ATRA/ATO, suggesting biologic heterogeneity. However, relapse during maintenance highlights the limitations of differentiation therapy alone.
Clinical implications
The RARB gene is located at chromosome 3p24, while TBL1XR1 resides at 3q26.32. The TBL1XR1-RARB fusion gene typically arises from t(3;3) or inv(3) rearrangements. However, unlike some other APL variants, TBL1XR1-RARB-positive cases have not been associated with loss of heterozygosity on chromosome 3, making this fusion particularly difficult to detect via conventional karyotyping. In our patient, cytogenetic analysis revealed a gain of chromosome 6 (+6) without structural abnormalities of chromosome 3, consistent with prior reports of this rare fusion. Structurally, RARB shares approximately 90% homology with RARA and RARG, all of which belong to the retinoic acid receptor (RAR) family and exhibit overlapping transcriptional regulatory functions. TBL1XR1 encodes an F-box/WD40 repeat-containing protein that serves as a critical component of the nuclear receptor corepressor complexes NCoR and SMRT. These complexes mediate transcriptional repression by unliganded NRs and other transcription factors (TFs) (24). Notably, TBL1XR1 can form homodimers that interfere with both RARA and RARB signaling. To date, TBL1XR1 is the only identified molecular chaperone for RARB. The TBL1XR1-RARB fusion oncoprotein mimics the leukemogenic mechanisms of PML-RARA by dysregulating the retinoic acid receptor pathway and blocking myeloid differentiation (11). Despite this mechanistic similarity, TBL1XR1-RARB-positive APL demonstrates intrinsic resistance to standard differentiating agents, including ATRA, ATO, and the synthetic retinoid tamibarotene (6, 11, 14, 25).
The therapeutic efficacy of ATRA in classical APL stems from its direct binding to the RARA moiety of PML-RARA, inducing conformational changes that facilitate co-repressor complex dissociation and co-activator recruitment, thereby reactivating the RARA transcriptional network. Concurrently, ATRA promotes PML-RARA degradation through the ubiquitin-proteasome system (26, 27). ATO exerts its effects by targeting specific cysteine residues in the zinc finger domains of PML, inducing sumoylation and subsequent proteasomal degradation of PML-RARA (28–30). The synergistic interaction between ATRA and ATO has been well-documented in clinical trials, contributing to the remarkable 95% long-term survival rate in APL, though approximately 5% of patients experience refractory or relapsed disease, particularly those with high-risk features or variant APL (3, 4). Patients harboring the TBL1XR1-RARB fusion typically demonstrate reduced sensitivity to ATRA/ATO therapy compared to classical APL (6). Our patient achieved morphological remission (negative bone marrow cytology and MRD) following induction therapy, yet persistent minimal residual disease was evident by quantitative detection of TBL1XR1-RARB (1.87%). While no established threshold exists for TBL1XR1-RARB in variant APL due to its rarity, insights can be drawn from PML-RARA monitoring in classical APL. Studies using ddPCR have demonstrated that MRD levels >0.1% post-consolidation correlate with increased relapse risk in PML-RARA-positive APL (11). Furthermore, pretreatment PML-RARA transcript levels >209.6 copies/ng by ddPCR independently predict relapse (31). In our case, persistent TBL1XR1-RARB at 1.87% post-induction prompted therapy escalation, supporting the hypothesis that low-level residual disease (even <1%) may be clinically significant. Given ddPCR’s superior sensitivity (LOD: 2.9 copies/μL vs. qPCR’s 98 copies/μL) (32), we propose: TBL1XR1-RARB levels >0.1% (ddPCR) should trigger closer surveillance. Transcripts >1% post-consolidation may warrant treatment intensification (e.g., HSCT). Further studies are needed to validate these thresholds in TBL1XR1-RARB-positive APL.
Subsequent consolidation therapy resulted in progressively declining fusion transcript levels, suggesting that while the TBL1XR1-RARB-positive leukemia retained some responsiveness to ATRA/ATO, this sensitivity was markedly diminished compared to classical APL. The cumulative daunorubicin dose of 200 mg/m² approached established cardiotoxicity thresholds, necessitating alternative therapeutic approaches. Current evidence supports the use of high-dose cytarabine consolidation for high-risk APL (33, 34) and AML-type regimens for treatment-resistant variant APL cases (34–38). All reported cases of TBL1XR1-RARB-positive APL have been managed with ATRA/ATO combined with chemotherapy, with three patients ultimately undergoing hematopoietic stem cell transplantation. Our patient achieved molecular remission following intensive consolidation incorporating high-dose cytarabine and etoposide. The observed gradual decline in fusion transcript levels during initial ATRA/ATO/daunorubicin therapy suggests some degree of preserved sensitivity to differentiating agents, albeit significantly reduced compared to classical APL. Given the substantial toxicity profile associated with intensive AML-type regimens, particularly the elevated risk of infectious complications, maintenance therapy with ATRA, RIF, 6-MP, and MTX was implemented. The emergence of disease relapse during maintenance therapy prompted consideration of hematopoietic stem cell transplantation as a definitive treatment approach. Notably, the treatment course was not complicated by severe infections or cardiotoxicity. While the combination of ATRA/ATO with chemotherapy may represent a viable initial approach for TBL1XR1-RARB-positive APL, the observed high relapse rate underscores the potential necessity of hematopoietic stem cell transplantation as a curative intervention for this variant form of APL.
Conclusion
We report a novel case of TBL1XR1-RARB-positive vAPL with fever as the sole symptom, atypical CD45-/CD117- immunophenotype, and KRAS p.G12D mutation. This case expands the phenotypic spectrum of TBL1XR1-RARB-positive vAPL by demonstrating neutrophilic leukocytosis —features suggestive of a maturation arrest-deficient subtype. Our case and literature review highlight the need for early molecular diagnosis, intensified chemotherapy, and consideration of HSCT due to high relapse risk. The absence of standardized MRD monitoring thresholds for this fusion underscores the urgent need for molecularly guided treatment protocols. Future efforts should focus on establishing international registries to facilitate multicenter collaborative studies, which will be critical for defining evidence-based diagnostic and therapeutic guidelines, elucidating the molecular mechanisms underlying treatment resistance and developing targeted approaches for this rare but clinically significant APL variant.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Mianyang Central Hospital and was conducted in accordance with the Declaration of Helsinki. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from primarily isolated as part of your previous study for which ethical approval was obtained. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Due to the anonymous and retrospective nature of the study, the Ethics Committee waived the requirement for written informed consent. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
CD: Data curation, Formal Analysis, Project administration, Writing – original draft, Writing – review & editing. FL: Data curation, Formal Analysis, Project administration, Writing – original draft, Writing – review & editing. LH: Writing – review & editing. RZ: Resources, Writing – review & editing. JF: Methodology, Supervision, Writing – review & editing. TH: Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We extend our sincere gratitude to Dr. Yakun Chen for her invaluable assistance and guidance throughout the submission process of this manuscript
Conflict of interest
Authors LH and JF were employed by Sichuan Kingmed Center for Clinical Laboratory CO., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Sanz MA Grimwade D Tallman MS Lowenberg B Fenaux P Estey EH et al . Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. (2009) 113:1875–91. doi: 10.1182/blood-2008-04-150250
2
Liquori A Ibañez M Sargas C Sanz MÁ Barragán E Cervera J . Acute promyelocytic leukemia: A constellation of molecular events around a single PML-RARA fusion gene. Cancers (Basel). (2020) 12:624. doi: 10.3390/cancers12030624
3
Zhu HH Wu DP Du X Zhang X Liu L Ma J et al . Oral arsenic plus retinoic acid versus intravenous arsenic plus retinoic acid for non-high-risk acute promyelocytic leukaemia: a non-inferiority, randomised phase 3 trial. Lancet Oncol. (2018) 19:871–79. doi: 10.1016/s1470-2045(18)30295-x
4
Zhu H Hu J Chen L Zhou W Li X Wang L et al . The 12-year follow-up of survival, chronic adverse effects, and retention of arsenic in patients with acute promyelocytic leukemia. Blood. (2016) 128:1525–8. doi: 10.1182/blood-2016-02-699439
5
Mannan A Muhsen IN Barragán E Sanz MA Mohty M Hashmi SK et al . Genotypic and phenotypic characteristics of acute promyelocytic leukemia translocation variants. Hematol Oncol Stem Cell Ther. (2020) 13:189–201. doi: 10.1016/j.hemonc.2020.05.007
6
Xiang Z Jiewen S Wenjuan Y Jie J . Current views on the genetic landscape and management of variant acute promyelocytic leukemia. biomark Res. (2021) 9:33–3. doi: 10.1186/s40364-021-00284-x
7
Zhou X Chen X Chen J Wen L Zhang Z Qin YZ et al . Critical role of tripartite fusion and LBD truncation in certain RARA- and all RARG-related atypical APL. Blood. (2024) 144:1471–85. doi: 10.1182/blood.2024023883
8
Hu Y Lauffer P Jongejan A Falize K Bruinstroop E van Trotsenburg P et al . Analysis of genes differentially expressed in the cortex of mice with the Tbl1xr1Y446C/Y446C variant. Gene. (2024) 927:148707. doi: 10.1016/j.gene.2024.148707
9
Perissi V Scafoglio C Zhang J Ohgi KA Rose DW Glass CK et al . TBL1 and TBLR1 phosphorylation on regulated gene promoters overcomes dual CtBP and NCoR/SMRT transcriptional repression checkpoints. Mol Cell. (2008) 29:755–66. doi: 10.1016/j.molcel.2008.01.020
10
McGowan-Jordan J Hastings R Moore S . Re: international system for human cytogenetic or cytogenomic nomenclature (ISCN): some thoughts, by T. Liehr. Cytogenet Genome Res. (2021) 161:225–6. doi: 10.1159/000516655
11
Brunetti C Anelli L Zagaria A Minervini A Minervini CF Casieri P et al . Droplet digital PCR is a reliable tool for monitoring minimal residual disease in acute promyelocytic leukemia. J Mol Diagn. (2017) 19:437–44. doi: 10.1016/j.jmoldx.2017.01.004
12
Borkovskaia A Bogacheva S Konyukhova T Dadakhanova E Gaskova M Soldatkina O et al . Molecular heterogeneity of pediatric AML with atypical promyelocytes accumulation in children-A single center experience. Genes (Basel). (2023) 14:675. doi: 10.3390/genes14030675
13
Zhao J Liang JW Xue HL Shen SH Chen J Tang YJ et al . The genetics and clinical characteristics of children morphologically diagnosed as acute promyelocytic leukemia. Leukemia. (2019) 33:1387–99. doi: 10.1038/s41375-018-0338-z
14
Osumi T Tsujimoto SI Tamura M Uchiyama M Nakabayashi K Okamura K et al . Recurrent RARB translocations in acute promyelocytic leukemia lacking RARA translocation. Cancer Res. (2018) 78:4452–58. doi: 10.1158/0008-5472.Can-18-0840
15
Zhang YJ Li QR Lin DN Wu L Cheng M Huang LL et al . Childhood acute promyelocytic leukemia (APL) with TBL1XR1-RARB fusion: a case report. Zhonghua Xue Ye Xue Za Zhi. (2022) 43:699. doi: 10.3760/cma.j.issn.0253-2727.2022.08.018
16
Shiba N Yoshida K Hara Y Yamato G Shiraishi Y Matsuo H et al . Transcriptome analysis offers a comprehensive illustration of the genetic background of pediatric acute myeloid leukemia. Blood Adv. (2019) 3:3157–69. doi: 10.1182/bloodadvances.2019000404
17
Mingyan J Jinrong L Jianrong W Yiping Z Ju G . Case report: A rare case of TBL1XR1-RARB positive acute promyelocytic leukemia in child and review of the literature. Front Oncol. (2022) 12:1028089. doi: 10.3389/fonc.2022.1028089
18
Zeng H He J Dong HB Zhou M Zhang Q Chen LX et al . Negative expression of CD117 predicted inferior OS and PFS in acute promyelocytic leukemia. Int J Lab Hematol. (2024) 46:1052–8. doi: 10.1111/ijlh.14326
19
Binoy Y Akanksha S Arthi S Frances C-C Adan R . POSTER: AML-103 clinical outcomes of patients with high-risk acute promyelocytic leukemia (APL) treated with all-trans retinoic acid (ATRA)/arsenic trioxide (ATO)–based induction and consolidation without maintenance therapy: A single-institution study. Clin Lymphoma Myeloma Leukemia. (2023) 23:S269–S69. doi: 10.1016/S2152-2650(23)00497-4
20
Fouzia S Pranay T Amitabh S Bhavika R Amar R Anita C et al . Analysing “tear-drop” prints of acute promyelocytic leukemia (APML): immunophenotypic prognostication of APML by FCM. Am J Blood Res. (2021) 11:446–57.
21
Breccia M Propris MSD Minotti C Stefanizzi C Raponi S Colafigli G et al . Aberrant phenotypic expression of CD15 and CD56 identifies poor prognostic acute promyelocytic leukemia patients. Leukemia Res. (2014) 38:194–97. doi: 10.1016/j.leukres.2013.11.008
22
Wojciech G . Acute promyelocytic leukemia: four distinct patterns by flow cytometry immunophenotyping. Polish J Pathol. (2012) 63:8–17.
23
Ma X Liu Y Liu Y Alexandrov LB Edmonson MN Gawad C et al . Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. (2018) 555:371–6. doi: 10.1038/nature25795
24
Li JY Daniels G Wang J Zhang X . TBL1XR1 in physiological and pathological states. Am J Clin Exp Urol. (2015) 3:13–23.
25
Chen Y Li S Zhou C Li C Ru K Rao Q et al . TBLR1 fuses to retinoid acid receptor α in a variant t(3;17)(q26;q21) translocation of acute promyelocytic leukemia. Blood. (2014) 124:936–45. doi: 10.1182/blood-2013-10-528596
26
Chen GQ Shen ZX Wu F Han JY Miao JM Zhong HJ et al . Pharmacokinetics and efficacy of low-dose all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Leukemia. (1996) 10:825–8.
27
Huang M Ye Y Chen S Chai J Lu J Zhoa L et al . Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. (1988) 72:567–72. doi: 10.1182/blood.V72.2.567.567
28
Chen SJ Zhou GB Zhang XW Mao JH de Thé H Chen Z . From an old remedy to a magic bullet: molecular mechanisms underlying the therapeutic effects of arsenic in fighting leukemia. Blood. (2011) 117:6425–37. doi: 10.1182/blood-2010-11-283598
29
Zhang XW Yan XJ Zhou ZR Yang FF Wu ZY Sun HB et al . Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. (2010) 328:240–3. doi: 10.1126/science.1183424
30
de Thé H Chen Z . Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer. (2010) 10:775–83. doi: 10.1038/nrc2943
31
Albano F Zagaria A Anelli L Coccaro N Tota G Brunetti C et al . Absolute quantification of the pretreatment PML-RARA transcript defines the relapse risk in acute promyelocytic leukemia. Oncotarget. (2015) 6:13269–77. doi: 10.18632/oncotarget.3773
32
Yuan D Cui M Yu S Wang H Jing R . Droplet digital PCR for quantification of PML-RARα in acute promyelocytic leukemia: a comprehensive comparison with real-time PCR. Anal Bioanal Chem. (2019) 411:895–903. doi: 10.1007/s00216-018-1508-6
33
Yang MH Wan WQ Luo JS Zheng MC Huang K Yang LH et al . Multicenter randomized trial of arsenic trioxide and Realgar-Indigo naturalis formula in pediatric patients with acute promyelocytic leukemia: Interim results of the SCCLG-APL clinical study. Am J Hematol. (2018) 93:1467–73. doi: 10.1002/ajh.25271
34
Zhang L Zou Y Chen Y Guo Y Yang W Chen X et al . Role of cytarabine in paediatric acute promyelocytic leukemia treated with the combination of all-trans retinoic acid and arsenic trioxide: a randomized controlled trial. BMC Cancer. (2018) 18:374. doi: 10.1186/s12885-018-4280-2
35
Sanz MA Fenaux P Tallman MS Estey EH Löwenberg B Naoe T et al . Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood. (2019) 133:1630–43. doi: 10.1182/blood-2019-01-894980
36
Issa GC Stein EM DiNardo CD . How I treat acute myeloid leukemia with differentiation therapy. Blood. (2025) 145:1251–9. doi: 10.1182/blood.2024024008
37
Kutny MA Alonzo TA Gerbing RB Wang YC Raimondi SC Hirsch BA et al . Arsenic trioxide consolidation allows anthracycline dose reduction for pediatric patients with acute promyelocytic leukemia: report from the children’s oncology group phase III historically controlled trial AAML0631. J Clin Oncol. (2017) 35:3021–9. doi: 10.1200/JCO.2016.71.6183
38
Lionel A Sylvie C Emmanuel R de Botton S Agnes G Arnaud P et al . Is cytarabine useful in the treatment of acute promyelocytic leukemia? Results of a randomized trial from the European Acute Promyelocytic Leukemia Group. J Clin Oncol. (2006) 24:5703–10. doi: 10.1200/JCO.2006.08.1596
Summary
Keywords
TBL1XR1-RARB fusion, atypical acute promyelocytic leukemia, clinical characteristics, immunophenotype, treatment
Citation
Du C, Liu F, Huang L, Zeng R, Fan J and Hu T (2025) A case report of TBL1XR1-RARB positive pediatric acute promyelocytic leukemia and literature review. Front. Oncol. 15:1566404. doi: 10.3389/fonc.2025.1566404
Received
24 January 2025
Accepted
08 August 2025
Published
29 August 2025
Volume
15 - 2025
Edited by
Alessandro Isidori, AORMN Hospital, Italy
Reviewed by
Alberto Olaya Vargas, National Institute of Pediatrics, Mexico
Nupur Das, Amrita School of Medicine, India
Updates
Copyright
© 2025 Du, Liu, Huang, Zeng, Fan and Hu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiamei Fan, 1126385817@qq.com; Tao Hu, 3087399@qq.com
†These authors share first authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.