 Kento Kawata
Kento Kawata Owen S. Chapman2
Owen S. Chapman2 Daisuke Kawauchi
Daisuke Kawauchi- 1Department of Pediatrics, Keio University School of Medicine, Tokyo, Japan
- 2Department of Neuro-oncology, Institute of Brain Science, Graduate School of Medical Sciences, Nagoya City University, Aichi, Nagoya, Japan
Pediatric brain tumors, the most devastating cancers affecting children, are believed to originate from neural stem/progenitor cells in developing brain. In precise timing and specific regions during the brain development, chromatin deregulation plays crucial roles in redirecting normal neuronal differentiation pathways toward tumorigenesis. Indeed, epigenomic abnormalities are thought to be more important for brain tumor formation especially in children than adults, as pediatric brain tumors generally exhibit fewer genetic mutations compared to adult brain tumors. Given the small number of mutations, targeting such limited alterations involved in cancer epigenomes is expected to be more effective in pediatric brain tumors. The mechanisms of cancer epigenomes include mutation or dysregulation of chromatin remodelers, histone modifiers, histones themselves, and DNA methylation enzymes. Furthermore, genomic rearrangements and/or higher-order chromatin topology also contribute to these epigenomic mechanisms. These mechanisms are commonly observed in various types of pediatric brain tumors. However, alterations in chromatin regulatory factors differ across tumor types, reflecting the unique epigenetic landscapes shaped by their tumor origins. Accordingly, clarifying their functional similarities and differences across tumor types could offer valuable insights for finding new therapeutic strategies. Thus, this review article focuses on elucidating how pediatric brain tumors arise from epigenomic deregulation and what epigenetic molecules or mechanisms could serve as therapeutic targets.
1 Introduction
Pediatric cancers develop when cells gain abnormal proliferative capacities due to the disruption of genetic programs for cellular differentiation during development (1, 2). While both familial and sporadic genetic mutations are recognized as primary causes, cancers are not solely driven by mutations in protein-coding genes directly relevant to cell proliferation. Recent cancer genome sequencing efforts have uncovered recurrent mutations in genes responsible for chromatin regulation (3, 4). These findings suggest that cancer progression may require not only aberrant upregulation of the genetic programs responsible for cellular growth signaling but also specific genomic alterations known as the cancer epigenome, which plays a pivotal role in tumorigenesis (5). In fact, pediatric solid tumors generally have fewer genetic mutations compared to adult tumors; therefore, epigenomic abnormalities are believed to be more important in tumor formation in children than adults (1). The same holds true for the differences between pediatric and adult brain tumors (6).
The epigenome consists of reversible molecular modifications to genomic chromatin. Chromatin, comprising the ~3 billion base pairs of human genomes bound to histones and other proteins, forms a highly organized structure. Within chromatin, the human genome is efficiently organized and compacted as 146 base pairs of DNA strands wrapped around histone protein octamers, creating repeating units called nucleosomes (7). Such chromatin structures are modified by epigenetic mechanisms including DNA methylation, histone occupancy and modifications, and higher-order chromatin topology (Figure 1a). The epigenome plays an essential role in regulating normal cell division and differentiation (8, 9). Disruption of proper epigenomic functions can lead to cellular senescence and/or apoptosis, resulting in developmental abnormalities such as Coffin-Siris syndrome, Nicolaides-Baraitser syndrome, and CHARGE syndrome, which arise from germline mutations in essential chromatin regulatory factors (10, 11). These mutations cause impaired neuronal differentiation, increased cell death, and disrupted neural circuitry, underscoring the critical role of cell-specific epigenomes in maintaining neuronal viability and defining cellular characteristics. Conversely, accumulating evidence indicates that epigenomic changes may also promote abnormal survival and proliferation of brain tumors. Therefore, strategies focused on elucidating mechanisms underlying the cancer-specific epigenome, and subsequent targeting of these alterations, offer promising potential for inhibiting cancer cell proliferation and inducing cancer-specific senescence and apoptosis. Although the path to fully realizing this approach remains challenging, it represents a critical area of research with the potential to lead to innovative therapeutic interventions.
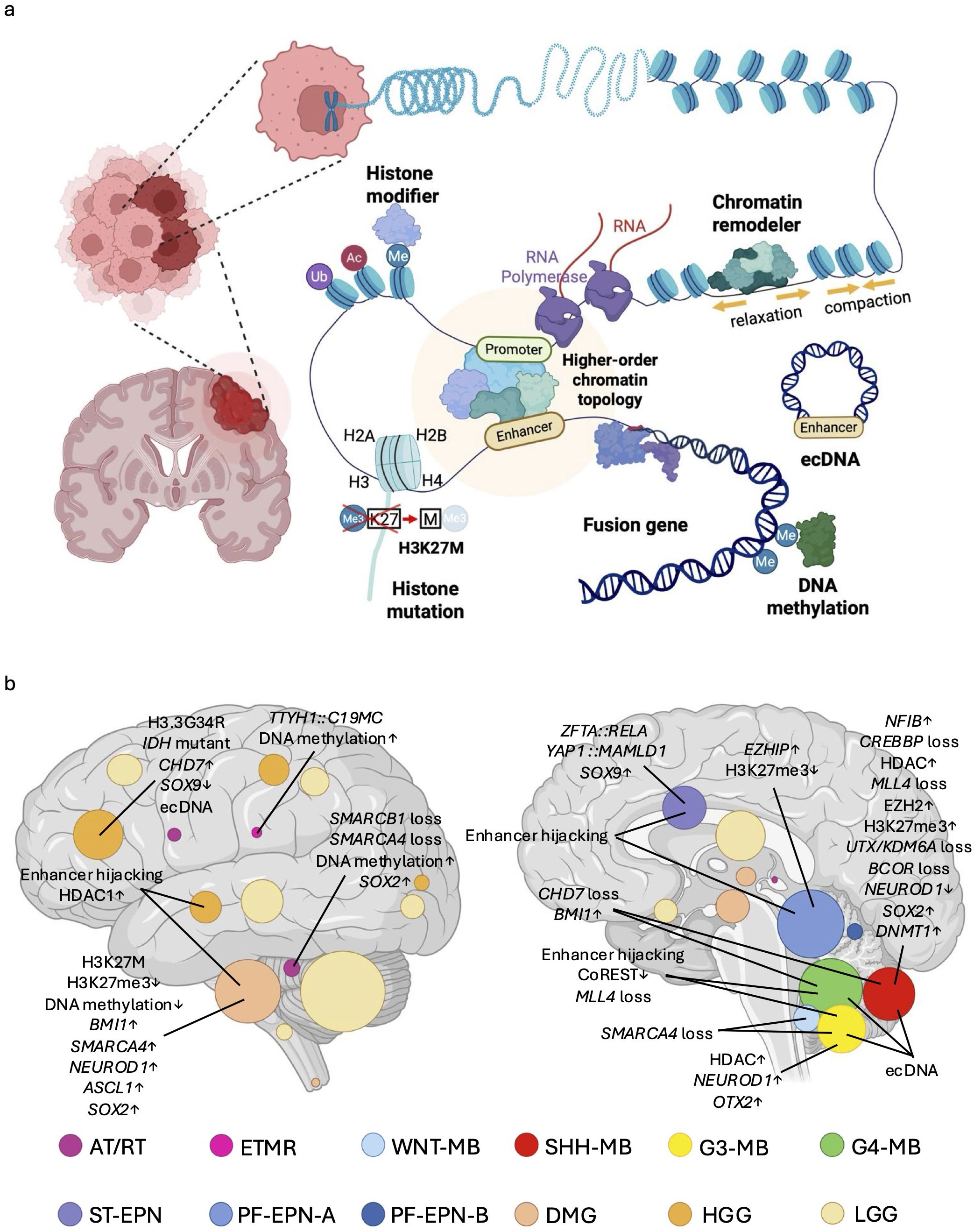
Figure 1. Overview of epigenetic regulatory mechanisms in pediatric brain tumors and their tumor-type-specific distribution. (a) Pediatric brain tumors develop through various kinds of epigenetic mechanisms, including dysregulation of chromatin remodelers, histone modifiers, histone mutations, and DNA methylation. Genomic rearrangements may generate gene fusions, extrachromosomal DNA (ecDNA) or alterations in higher-order chromatin topology which also often contribute to the cancer epigenome. (b) Schematic illustration graphically depicts the anatomical origins and distribution of each tumor type (26), including Atypical teratoid/rhabdoid tumor (AT/RT) (19), Embryonal tumors with multilayered rosette (ETMR) (25), Wingless medulloblastoma (WNT-MB) (20), Sonic hedgehog medulloblastoma (SHH-MB) (20), Group 3 medulloblastoma (G3-MB) (20), Group 4 medulloblastoma (G4-MB) (20), Supratentorial ependymoma (ST-EPN) (23), Posterior fossa A ependymoma (PF-EPN-A) (23), Circle area is proportional to the number of new diagnoses at each anatomical location. Posterior fossa B ependymoma (PF-EPN-B) (23), Diffuse midline glioma (DMG) (21, 22), High grade glioma (HGG) (21, 22), Low grade glioma (LGG) (24). This image also shows major epigenomic alterations associated with pediatric brain tumors discussed in this study.
Pediatric brain tumors are the most lethal form of pediatric cancer, arising from both genetic and epigenetic defects during critical stages of brain development (12). Similar to other cancers, global cancer genome sequencing initiatives have identified cancer-specific loss-of-function (LOF) mutations in chromatin-modifying genes, as well as altered genomic rearrangements leading to aberrant epigenetic regulation of cancer-related genes (13, 14). Given that different brain cells at distinct developmental stages give rise to distinct tumor types (15, 16), it is hypothesized that life stage-dependent and region-specific epigenomes underpin the gene expression programs necessary for tumor initiation (17, 18). Thus, it is unsurprising that genetic alterations in chromatin regulatory factors vary across cancer types, reflecting the unique epigenetic landscapes from which these tumors originate (19–26) (Figure 1b, Table 1). Collectively, it is likely that pediatric brain tumors acquire unique epigenetic regulation that drives tumorigenesis.
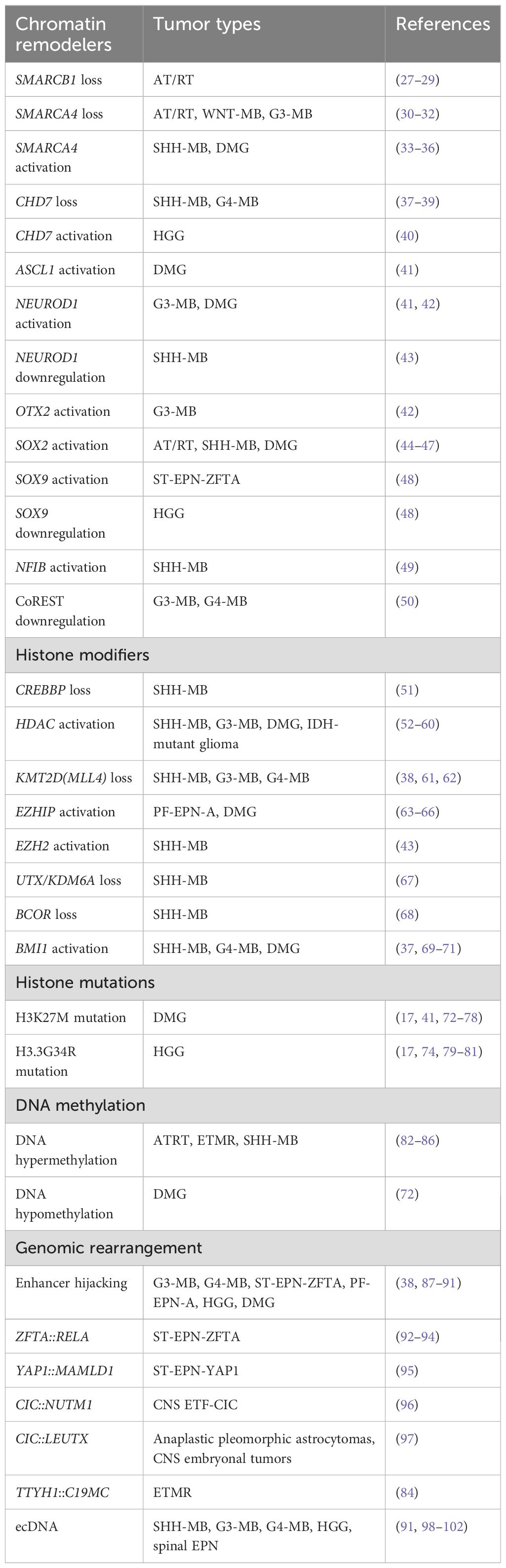
Table 1. Tumor-type-specific roles of epigenetic deregulation reviewed in this study.
One of the fundamental biological questions that emerges here is whether dysregulation of distinct chromatin regulatory factors across various types of pediatric brain tumors exert similar effects on the epigenome, and what the shared mechanisms and key differences might be. Investigating these aspects could yield valuable insights into the molecular pathways driving tumor formation. However, our current understanding remains limited, and efforts must begin by elucidating the specific epigenetic modifications involved in the formation of individual pediatric brain tumors and their subsequent consequences. Accordingly, this review highlights the molecular mechanisms that influence the epigenome during pediatric brain tumor development.
2 Chromatin modifications
Chromatin modifications are essential epigenetic mechanisms that alter chromatin architecture and regulate gene expression. Dysregulation of these processes plays a pivotal role in the pathogenesis of pediatric brain tumors. For instance, chromatin remodelers modify chromatin structure to either open it, forming transcriptionally active euchromatin, or close it, forming transcriptionally inactive heterochromatin, by depositing, removing, or shifting nucleosomes bound to genomic DNA (103, 104). They serve as gatekeepers by modulating access of DNA-binding transcription factors to the genome to regulate cell type-specific gene expression programs.
Additionally, certain processes directly modify chromatin components including histones and DNA. Molecular modifications to histones including methylation, acetylation, and ubiquitination serve as binding sites for cellular transcriptional machinery to up- or downregulate transcription of nearby genes (105–107). Mutations to histone modifiers or the histone proteins themselves can disrupt proper histone modification, leading to abnormal chromatin configurations (17, 108). Similarly, DNA methyltransferases (DNMTs) repress gene expression by direct methylation of cytosine residues of genomic DNA (109). The subsequent sections will explore how these chromatin modification mechanisms contribute to the development of pediatric brain tumors.
2.1 Chromatin remodelers
Chromatin remodelers are multiprotein complexes that utilize the energy of ATP hydrolysis to mobilize and restructure nucleosomes for gene regulation (103, 104). So far, four subfamilies of the ATP-dependent chromatin remodeling complexes have been identified: SWItch/Sucrose Non-Fermentable (SWI/SNF), Imitation SWItch (ISWI), Chromodomain Helicase DNA binding protein (CHD) and INOsitol 80 (INO80) subfamilies (11, 104). In pediatric brain tumors, abnormal alterations in chromatin structure suppress neuronal differentiation programs, enhancing susceptibility of transformation into malignant cells by maintaining cell proliferation signals (10, 27, 37, 110).
The SWI/SNF complex is one of the frequently mutated chromatin remodelers in pediatric brain tumors (7, 111, 112). In normal cells, the SWI/SNF complex functions to modify chromatin structure by recruiting other proteins that add epigenetic marks, including histone acetylation and methylation, to achieve proper chromatin compaction (Figure 2a). Such chromatin modification affects gene expression patterns essential for normal neurodevelopmental processes including cell cycle regulation (11), which is reflected in cancer genome sequencing data that reveal various genetic alterations in individual components of the SWI/SNF complex (111–113). For example, somatic mutation-based biallelic inactivation of SMARCB1 (Figure 2a), a core component of the SWI/SNF complex, is frequently observed in Atypical Teratoid/Rhabdoid Tumor (AT/RT) (approximately 98%) (28). In line with this, loss of SMARCB1 expression between embryonic day 6 and 10, but not during any other developmental periods, induced AT/RT formation in genetically engineered mouse models (27). Besides, SMARCB1-deficient human induced pluripotent stem cells (iPSCs) gave rise to AT/RT-like tumors (29), implying that embryonic stem cell (ESC)-like signature plays a crucial role in driving the malignant characteristics of AT/RT. LOF mutations in SMARCA4 (Figure 2a), another key component of the SWI/SNF complex, have also been identified in AT/RT. SMARCB1 and, rarely, SMARCA4 are mutated in a mutually exclusive manner (30). This implies that dysfunction of the SWI/SNF complex is a main cause of AT/RT formation, and even single mutations in one of the main components in the SWI/SNF complex are enough to affect the division and differentiation of brain cells.
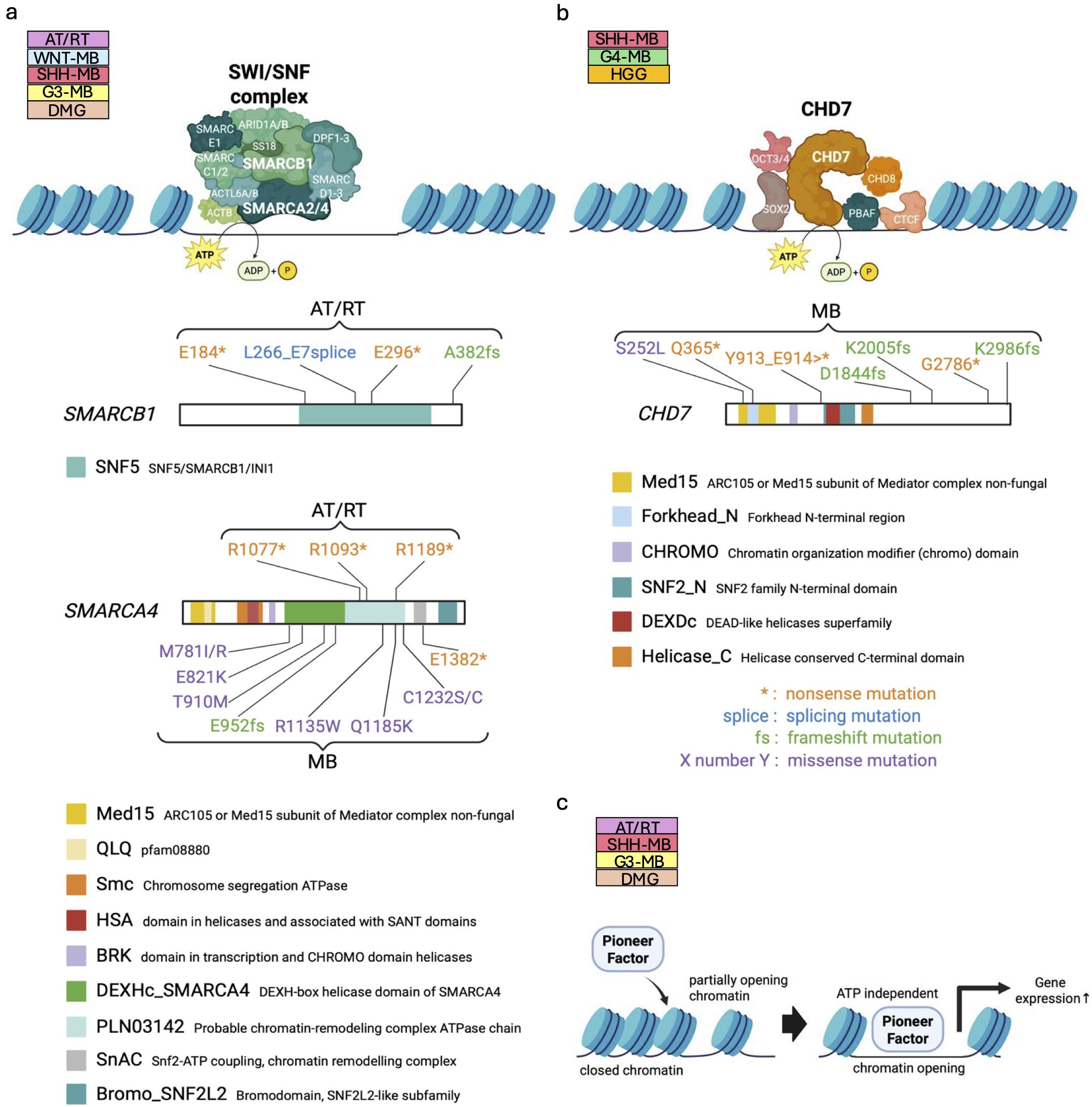
Figure 2. Dysregulation of chromatin remodelers in pediatric brain tumors. (a, b) Structure of ATP-dependent chromatin remodelers, the SWI/SNF complex (upper panel in a) and CHD7 (upper panel in (b)). The core subunits of the SWI/SNF complex, SMARCB1 and SMARCA4 are mutated in human patients bearing AT/RTs and MBs (111) (lower panels in (a)), while mutations of CHD7 are found in MBs (38) (lower panel in (b)). The known functional domains of the respective proteins are highlighted and labeled with their names. The patterns of genetic mutations are shown in the figure. Data sourced from previous studies (38, 111) and St. Jude Cloud Pediatric Brain Tumor Portal (https://pbtp.stjude.cloud). (c) Schematic diagram of regulation of chromatin compaction by ATP-independent pioneer factors.
LOF of the SWI/SNF complex often collaborates with oncogenic signaling for tumorigenesis. In the initial phase of medulloblastoma (MB), cooperative interaction between SMARCA4 loss and CTNNB1 mutation promotes proliferation of embryonic cerebellar ventricular zone cells in mice, resulting in Wingless (WNT)-MB formation (31). Similarly, combinatorial loss of SMARCA4 with overexpression of MYC increases proliferation of cerebellar granule neuron precursors (GNPs), leading to Group 3 (G3)-MB formation in mice (32). In addition, SMARCA4 interacts with DNA topoisomerase II α to facilitate DNA decatenation and its loss is associated with anaphase bridges that often result in partial chromosome gain or loss as well as polyploidy, which may predispose aneuploidy in MB (114). Meanwhile, no MB formation is observed with the loss of either SMARCA4 or SMARCB1 alone in mice (33). Rather, SMARCA4 deletion in the murine cerebellum inhibited Sonic hedgehog (SHH)-MB formation, as SMARCA4 is required for activation of SHH signaling (33, 34). Similarly, as found in adult glioblastomas (35), CRISPR-based LOF and pharmacologic inhibition of SMARCA4 revealed that SMARCA4 is required to maintain characteristics of H3K27M-driven diffuse midline glioma (DMG) (36). Thus, the SWI/SNF complex does not always function independently in tumor formation; instead, it sometimes contributes to establishing an epigenetic landscape that facilitates tumorigenesis driven by other cancer signals, with its role varying depending on the tumor type.
Alongside the SWI/SNF complex, dysfunction of CHD subfamily chromatin remodelers is also associated with failure of neural cell differentiation. CHD7 plays a key role in maintaining euchromatin (Figure 2b) via recruiting DNA topoisomerase II β to target genes required for differentiation of cerebellar GNPs (10). In addition to the hypothesis that imbalance between GNP differentiation and proliferation could result in SHH-MB formation in cerebellum (115), the observation of LOF mutations in CHD7 (38) (Figure 2b) in MB warrants further investigation of the oncogenic mechanisms of CHD7 mutations. To date, evidence that LOF mutations in CHD7 promote pediatric brain tumors been limited to SHH-MB (39) and Group 4 (G4)-MBs (37). However, further elucidation of differentiation mechanisms in normal cells, together with a deeper understanding of CHD7 mutations in tumorigenesis, is expected to lead to the identification of new molecular targets.
Conversely, functional CHD7 is suspected to trigger tumorigenesis in gliomas. CHD7 is highly expressed in gliomas, and CHD7 overexpression increased proliferation and maintained the stemness of neural stem cells (NSCs) and neural precursor cells (NPCs) (40). This study also revealed that silencing CHD7 diminished the proliferation of glioma initiating cells, suggesting that CHD7 could be a potential therapeutic target of gliomas.
As shown above, the SWI/SNF and CHD chromatin remodeler complexes are involved in formation of some pediatric brain tumors. In contrast, little is known about the possible roles of the ISWI and INO80 complexes in pediatric brain tumors, and reports of their mutation are exceedingly rare. This could be explained by the possibility that the functions of these chromatin regulators are compensated by other molecules in their cells of origin, or that these regulators are crucial for the survival of these cells. These intriguing possibilities warrant further investigation, and we look forward to future research shedding light on them.
Pioneer factors (PFs) are known not only to function as transcription factors but also to modify chromatin structure independently of ATP. Mechanistically, PFs directly recognize nucleosome motifs, bind to closed chromatin, and alter DNA accessibility, thus enabling the reprogramming of cell fate decisions (44) (Figure 2c). Among the many known pioneer factors, ASCL1 (116), NEUROD1 (117, 118), OTX2 (42), SOX2 (45, 119, 120), and SOX9 (48) play crucial roles in pediatric brain tumor formation as well as tissue-specific chromatin regulation and are also involved in a range of neural developmental processes. These PFs are also closely associated with pediatric brain tumor formation. In cellular models, chromatin modifications mediated by ASCL1 and NEUROD1 are implicated in transcriptional circuitry of H3.3K27M-driven DMG (41) (see section 1.3). In G3-MB, genome-wide chromatin and expression profiling have shown that NEUROD1 acts as a key transcriptional mediator for tumor growth. NEUROD1 cooperates with another pioneer factor, OTX2, to shape the regulatory landscape of G3-MB through cooperative activity at enhancer elements and promotes the expression of target genes (42). Conversely, in SHH-MB, mouse models have demonstrated that NEUROD1 overexpression enhances differentiation of tumor cells and inhibits tumor growth (43). These findings suggest that NEUROD1 has different functions depending on the tumor type as also seen in other chromatin remodelers. Similarly, SOX9 also exhibits distinct epigenomic regulation across different tumor types. SOX9 suppresses high grade glioma (HGG) growth and expands acetylation of histone H3 at lysine 27 (H3K27ac), but facilitates zinc finger translocation-associated (ZFTA) fusion-positive supratentorial ependymoma (ST-EPN-ZFTA) development by altering H3K27ac occupancy (48). These tumor-type-specific function of pioneer factors may reflect differences in epigenomic landscapes of cellular origins of each tumor. In addition to these factors, SOX2 is also a crucial component of the transcriptional circuitry in some kinds of brain tumor such as DMG (44, 46), AT/RT (44, 47), and SHH-MB (45), and maintains neural stemness and developmental potency in tumor cells. However, it remains unknown what specific chromatin changes SOX2 induces during tumorigenesis.
Nuclear Factor I (NFI) family proteins not only regulate gene expression as a transcription factor but also maintain open chromatin architecture by binding to open chromatin regions, albeit little evidence as PFs (121). NFIB, a member of the NFI family, plays an important role in brain development, including cerebellar formation and neuronal migration (122–124). Along with its role in normal brain development, we have recently discovered that SHH-MB-specific NFI-binding open chromatin regions emerge in precancerous GNPs, then NFIB binds to these open chromatin regions and maintains the chromatin structure. These chromatin alterations mainly occur at the transition from GNPs to hyperplasia during SHH-MB progression, then strengthening oncogenic pathways including the SHH signaling pathway (49). Thus, ATP-independent chromatin modulators are also emerging as an essential factor for pediatric brain tumor formation, although there is still much unclear about functions in detail.
Aside from transcriptional activators as discussed above, repressive chromatin regulators also play a role in neurodevelopment and cancer. For example, repressor element 1 silencing transcription factor (REST) serves as a key regulatory factor by repressing the transcription of genes involved in neuronal differentiation and maturation. REST forms a complex with the REST corepressor (CoREST), and it recruits chromatin-modifying enzymes to induce a condensed chromatin state. CoREST functions not only as a corepressor but also plays distinct roles in neuronal differentiation and maturation independently of REST (125). During normal development, the CoREST complexes comprising SMARCA4, lysine-specific histone demethylase 1A (LSD1) and histone deacetylase 1/2 (HDAC1/2) methyl and acetyl groups from histone H3 at lysine 4 (H3K4) and lysine 9 (H3K9), respectively. These modifications facilitate chromatin condensation through the recruitment of heterochromatin protein 1 (HP1) and methyl CpG-binding protein 2 (MeCP2), thus leading to transcriptional silencing of target genes (125–127). In the context of pediatric brain tumor formation, the CoREST complex is degraded in G3/G4 MBs due to KBTBD4 mutations, which impair its E3 ubiquitin ligase function. Consequently, CoREST target genes, including those involved in stemness, become aberrantly activated, ultimately promoting G3/G4 MB tumor growth in vitro (50). In addition, CoREST and the NuRD chromatin remodeling complex have been shown to contribute to EGFR silencing and may function as tumor suppressors in the breast cancer cells (128–130). Thus, dysregulation of repressive chromatin remodelers may not only disrupt normal neurodevelopmental trajectories but also contribute to oncogenic transformation in pediatric brain tumors.
2.2 Histone modifiers
Another well-studied epigenetic gene regulatory mechanism for chromatin remodeling is direct modification of the histones. Histones are octamer complexes composed of two subunits each of H2A, H2B, H3, and H4. Histone modifiers regulate gene expression through various modifications of histones. Methylation and acetylation primarily occur on H3 and H4, while ubiquitination mainly takes place on H2A and H2B. Acetylation of H3K27 is associated with open chromatin, as weakening the binding of DNA to histones by adding a negatively charged carboxyl group to the lysine residue. Due to this, the genomic regions marked by H3K27ac are accessible for gene transcription and are defined as active enhancers of transcription. Histone acetyltransferases (HATs) catalyze such histone acetylation, while HDACs negatively regulate this modification, often resulting in gene silencing. Unlike histone acetylation, histone methylation occurs at lysine or arginine residues in the histone tail and is regulated by histone methyltransferases (HMTs), with gene transcription being either activated or repressed depending on the type of amino acid residue modified and the site of methylation. HMT-based modification comprises mono-, di-, and tri-methylation of histones, whereas histone demethylase facilitates the removal of methyl groups. Specifically, trimethylation of H3K4 (H3K4me3) are strongly associated with active promoters. On the other hand, H3K27me3 is enriched in heterochromatin and is linked to gene silencing. Mono-ubiquitination of histone H2A is also known to be responsible for gene silencing (105–107). Therefore, mutation or dysregulation of proteins responsible for histone modifications can disrupt cellular gene expression patterns, often contributing to cancer progression. So far, various studies have employed brain tumor models to explore which alterations or dysregulation of histone-modifying enzymes contribute to tumor development. This section introduces several notable histone modifiers and their roles in pediatric brain tumor development.
Tumor initiation is sometimes induced by inactivation of differentiation-associated genes due to HAT dysfunction. For example, in SHH-MB, mutations in the HAT domain of CREB-binding protein (CREBBP) downregulate brain-derived neurotrophic factor (BDNF) (Figure 3a), in turn inhibiting proper migration of GNPs and retaining them in the germinal zone on the cerebellar surface known as a proliferative niche (131). Indeed, postnatal loss of CREBBP synergizes with activation of SHH signaling to accelerate SHH-MB growth (51). Of interest, HATs acetylate not only histones to regulate chromatin compaction, but also some proteins to directly modulate their functions. Many HATs such as CREBBP, E1A-associated protein p300 (p300), and p300/CREBBP associated factor (PCAF) activate p53 tumor suppressor functions by acetylation (132, 133). Since SHH-MBs display enhanced aggressiveness in Trp53-deficient background (134) and CREBBP or p300 loss (51, 133, 135, 136), epigenetic regulation-independent regulatory mechanisms may also need to be considered for this type of entity. Thus, HAT mutation promotes tumor formation by epigenetic and non-epigenetic mechanisms.
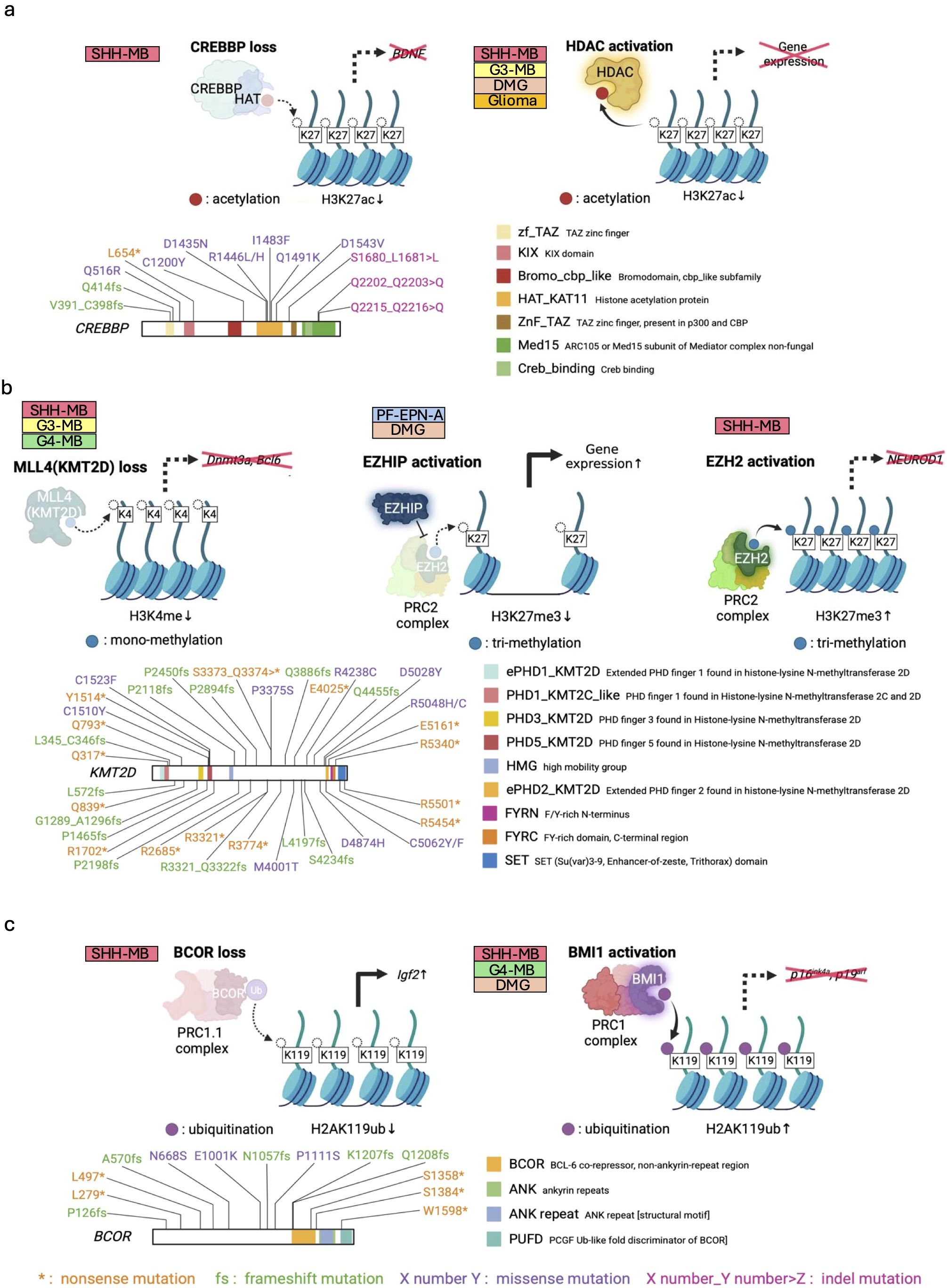
Figure 3. Failure of appropriate histone modification in pediatric brain tumors. (a) Inhibition of chromatin opening by loss of HAT function (e.g., CREBBP) (upper left panel) and aberrant activation of HDAC (upper right panel). Pathogenic loss-of-function (LOF) mutations of CREBBP in MB (38, 51) (lower panel). (b) Histone H3 demethylation by loss of KMT function (upper left panel) and EZHIP activation (upper middle panel) for tumorigenesis. Conversely, in some tumors, EZH2 activation often functions to prevent proper differentiation and contributes to oncogenesis (upper right panel). Mutations of KMT2D found in MBs (lower panel) (38). (c) Histone H2A ubiquitination regulating cancer-related genes. BCOR loss activates Igf2 oncogene (upper left panel), while BMI activation inhibits tumor suppressor genes (upper right panel). LOF mutations of BCOR in MBs (68) (lower panel). The known functional domains of the respective proteins are highlighted and labeled with their names. The patterns of genetic mutations are shown in the figure. Data sourced from previous studies (38, 51, 68) and St. Jude Cloud Pediatric Brain Tumor Portal (https://pbtp.stjude.cloud).
Similar to LOF mutations of HATs, HDAC activation also attenuates histone acetylation, resulting in gene silencing and tumorigenesis (137), although no reports of HDAC amplification have been reported in pediatric brain tumors. (Figure 3a). In MYC-amplified G3-MBs, HDAC2 and MYC are co-bound in H3K27ac open chromatin regions, adjusting expression of MYC-dependent genes via histone deacetylation (52, 53). Moreover, as predicted from the functions of HAT described above, HDACs also directly deacetylate and activate proliferation-related molecules, such as GLI1 and GLI2 in SHH-MB formation (54, 55). Besides MB, isocitrate dehydrogenase (IDH)-mutant gliomas, occasionally observed in adolescent and young adult (138), exhibit upregulation of genes associated with HDAC activity (56) and show significant anti-tumor responses upon knock-down of HDAC1 and HDAC6 (57). Furthermore, HDAC is also associated with the H3K27M DMG in several preclinical models (58–60). Thus, HDACs contribute to the maintenance of cancer cells through epigenetic mechanisms, across various cancer types.
In addition to dysregulation of histone acetylation, mutations in some HMTs also promote brain tumor formation. Specifically, H3K4 methyltransferase Mixed-lineage leukemia 4 (Mll4) in mice, also known as Lysine Methyltransferase 2D (KMT2D) in human, regulates neuronal differentiation and tumor suppression, and its loss triggers G3-like MB development with upregulating oncogenic Ras and Notch pathways, and downregulating tumor suppressor genes (e.g., Dnmt3a, Bcl6) (61) (Figure 3b). Another investigation of SHH-MB mouse models has shown that heterozygous Kmt2d loss, combined with abnormal SHH pathway activation, increases hindbrain invasion and spinal cord metastasis through downregulation of differentiation-associated and tumor suppressor genes, and upregulation of progression- and metastasis-related pathways/genes (e.g., TGFβ-signaling, NOTCH-signaling, Atoh1, Sox2, and Myc) (62). Notably, KDM6A, KMT2C, and KMT2D mutations also frequently occur in human SHH- and G4-MBs (38). Given that these molecules form the core nuclear regulatory structure, so-called the COMPASS complex (139), how deficiency of the COMPASS complex function regulates MB formation remains to be investigated.
Another HMT pivotal in brain tumor formation is Enhancer of Zeste Homolog 2 (EZH2), a key component of Polycomb Repressive Complex 2 (PRC2). EZH2 drives chromatin silencing by catalyzing the trimethylation of H3K27 (H3K27me3). Conversely, EZH Inhibitory Protein (EZHIP) disrupts this process by inhibiting EZH2 (Figure 3b). Alterations in H3K27me3 are linked to posterior fossa A ependymoma (PF-EPN-A), the most aggressive EPN subgroup, and DMG. PF-EPN-A exhibits global reduction in H3K27me3 alongside elevated EZHIP expression (63, 64). Previous studies demonstrated that EZHIP knockout inhibits PF-EPN-A cell growth in vitro (65). Although further in vivo validation using animal models is still required, the high expression of EZHIP and the resulting H3K27me3 reduction might be potential drivers of PF-EPN-A tumorigenesis. In DMGs, the hallmark mutation of H3K27M inhibits H3K27 histone trimethylation by PRC2 (see section 1.3). This mutation mimics the function of EZHIP, and in some cases of DMGs, elevated EZHIP levels have been reported as an alternative to the H3K27M mutation (66). Of interest, recent study has demonstrated that such common molecular features between PF-EPN-A and DMG are linked closely to expression patterns of genes (e.g., CRABP1) in human hindbrain development (140). Whether the failure of EZH2/EZHIP-mediated epigenetic histone regulation mimics an epigenetic state normally restricted to the developing hindbrain for their tumorigenesis remains to be further studied.
Aside from H3K27me3 loss, H3K27me3-based gene repression is inversely implicated in other types of brain tumors. In IDH-mutant gliomas, IDH mutations promote histone methylation marks including H3K27me3 by epigenetic reprogramming (141) Meanwhile, in SHH-MB, EZH2-mediated H3K27me3 marks accumulate at NEUROD1 regulatory elements, suppressing NEUROD1 expression and maintaining tumor cells in an undifferentiated state (Figure 3b). Consistently, pharmacological inhibition of EZH2 reduces H3K27me3, upregulates NEUROD1, and drives tumor cell differentiation, thereby inhibiting tumor growth (43). Remarkably, once SHH-MB cells are differentiated through such mechanisms, they permanently lose their proliferative ability and tumorigenic functions (43). Another study showed that complete deletion of PRC2 reduced occurrence of SHH-MB because PRC2 is required for maintenance of GNPs, but partial deletion of PRC2 led to SHH-MB growth through increased expression of oncogenes such as Igf2 and non-cell autonomous mechanism with paracrine IGF2 signaling (142). Additionally, H3K27me3 demethylase UTX/KDM6 plays crucial roles in GNP differentiation through NEUROD2 expression and recruits immune cells to the tumor microenvironment, thereby UTX/KDM6A deletion contributes to SHH-MB development by maintaining undifferentiated and immunologically cold states (67). Overall, epigenetic modification changes of H3K27me3 by dysregulation of EZH2/EZHIP and/or histone demethylases are intimately involved in pediatric brain tumors.
The gene silencing mechanism by histone H2A mono-ubiquitination is also closely associated with cancer. Both canonical PRC1 and non-canonical PRC1.1 ubiquitinate histone H2AK119 via RING1A/B, an E3 ligase within the complex (Figure 3c). Canonical PRC1 represses expression of tumor suppressor genes (e.g., p16Ink4a, p19Arf) through histone ubiquitination (143) and BMI1, a core component of PRC1, is upregulated in various types of cancers (144). Hyper-physiological expression of BMI1 downregulates tumor suppressor genes and disrupts normal developmental signaling in the cerebellum (Figure 3c), thereby contributing to SHH-MB tumorigenesis (69). In xenograft models of G4-MB, BMI1 knockdown suppress their tumor growth and invasion (37, 70). Besides MBs, H3K27M DMGs also upregulate BMI1 and are susceptible to its inhibition (71). In contrast, LOF mutations in non-canonical PRC1.1 components occasionally activate oncogenes for pediatric brain tumor formation. For example, our previous study demonstrated that LOF mutations in BCL6-Co-Repressor (BCOR), a component of PRC1.1, enhance the aggressiveness of SHH-MBs via upregulation of Insulin-like growth factor 2 (Igf2), a strong mitogen for GNPs, due to failure in gene silencing mediated by histone ubiquitination (68). (Figure 3c). As with other histone modifications, understanding of which genes are regulated by histone ubiquitination is crucial for the phenotype of tumor cells.
Notably, a recent study has further identified that neurotransmitters such as serotonin also modify histones. Mouse models of ST-EPN-ZFTA have demonstrated that serotonin secreted from serotonergic neurons is transported into the nucleus of tumor cells, in turn modifying histones. This histone serotonylation promotes the expression of Etv5 by opening chromatin. The elevated ETV5 then transcriptionally represses the tumor suppressor neuropeptide Y (NPY), facilitating EPN tumor formation (145). Such an entirely new mechanism of histone modification by neurotransmitters is now gradually becoming clear from the cancer neuroscience field and could pave the way for novel therapeutic avenues.
2.3 Histone mutations
Histone modifications play a vital role in the precise regulation of gene expression. Mutations at these modification sites can disrupt cellular gene expression networks, sometimes driving tumorigenesis. Pediatric brain tumors are no exception, with some histone mutations reported. Indeed, pathogenic histone mutations are largely restricted to H3K27 and H3G34, which are linked to DMGs and pediatric HGGs (pHGGs). In recent years, multifaceted research approaches are advancing our understanding of the oncogenic signaling pathways triggered by these mutations. The following section describes, based on the latest knowledge, how histone mutations influence the formation and growth of pediatric brain tumors.
H3K27M, missense mutation in histone H3 at amino acid 27, lysine (K) to methionine (M), is one of the best-studied histone mutations in pediatric brain tumors (Figure 4a). H3K27M mutations are seen in about 80% of DMG. Approximately 75% of H3K27M mutations are found in H3-3A (H3F3A) encoding H3.3, while the remaining 25% are seen in H3C2 (HIST1H3B) encoding H3.1 (17, 22). Although H3K27M mutations occur only in 5-17% of the total H3 expressed (17), they potentially have a dominant negative effect to inhibit PRC2, resulting in global loss of H3K27me3 and enhanced gene expression in those loci. This can be explained by the mechanism through which H3K27M exhibits higher binding affinity to PRC2 than wild-type H3K27 and reduces EZH2 auto-methylation, further limiting the methyltransferase activity of PRC2 (17, 72). However, H3K27me3 are still retained in several genes by residual PRC2, and DMGs also require PRC2 for proliferation. Thus, inhibition of PRC2 could be a potential therapeutic strategy for DMG patients (146). The H3K27M mutation-induced H3K27me3 reduction causes chromatin structure opening, leading to increased expression of PFs, such as NEUROD1 and ASCL1. This processes subsequently enhance chromatin accessibility and upregulate the expression of neurogenesis- and oncogenesis-related genes (e.g., COBL, OLIG2), thereby triggering tumorigenesis (41) (Figure 4a). In line with this, the H3K27M mutation disrupts normal differentiation, while promoting proliferation and stemness phenotypes in human ESCs (73) as well as human fetal NSC models (74). Even so, H3K27M alone is insufficient for tumorigenesis. In human fetal NSC models, H3.3K27M enhanced clonogenicity and reduced senescence only in the brainstem but not in the forebrain, implying that regionally specific developmental cellular characteristics and their microenvironment could be required (74). It has also been demonstrated that H3K27M in combination with PDGF signaling activation and TP53 knockdown causes tumorigenesis from hES-derived NSCs, NPCs (75–77), and OPCs (76). In these genetic backgrounds, the oncogenic effects of H3K27M depend on the cell of origin, with H3K27M being more tumorigenic in NSCs or NPCs than in OPCs (75–77). Our previous research using the models derived from human iPSCs demonstrated that induction of H3.3K27M and TP53 inactivation gave rise to DMG only from NSCs but not from OPCs (78). Collectively, the H3.3K27M-driven epigenetic state seems to collaborate with region-specific and cell-type specific epigenetic programs for transformation into DMGs.
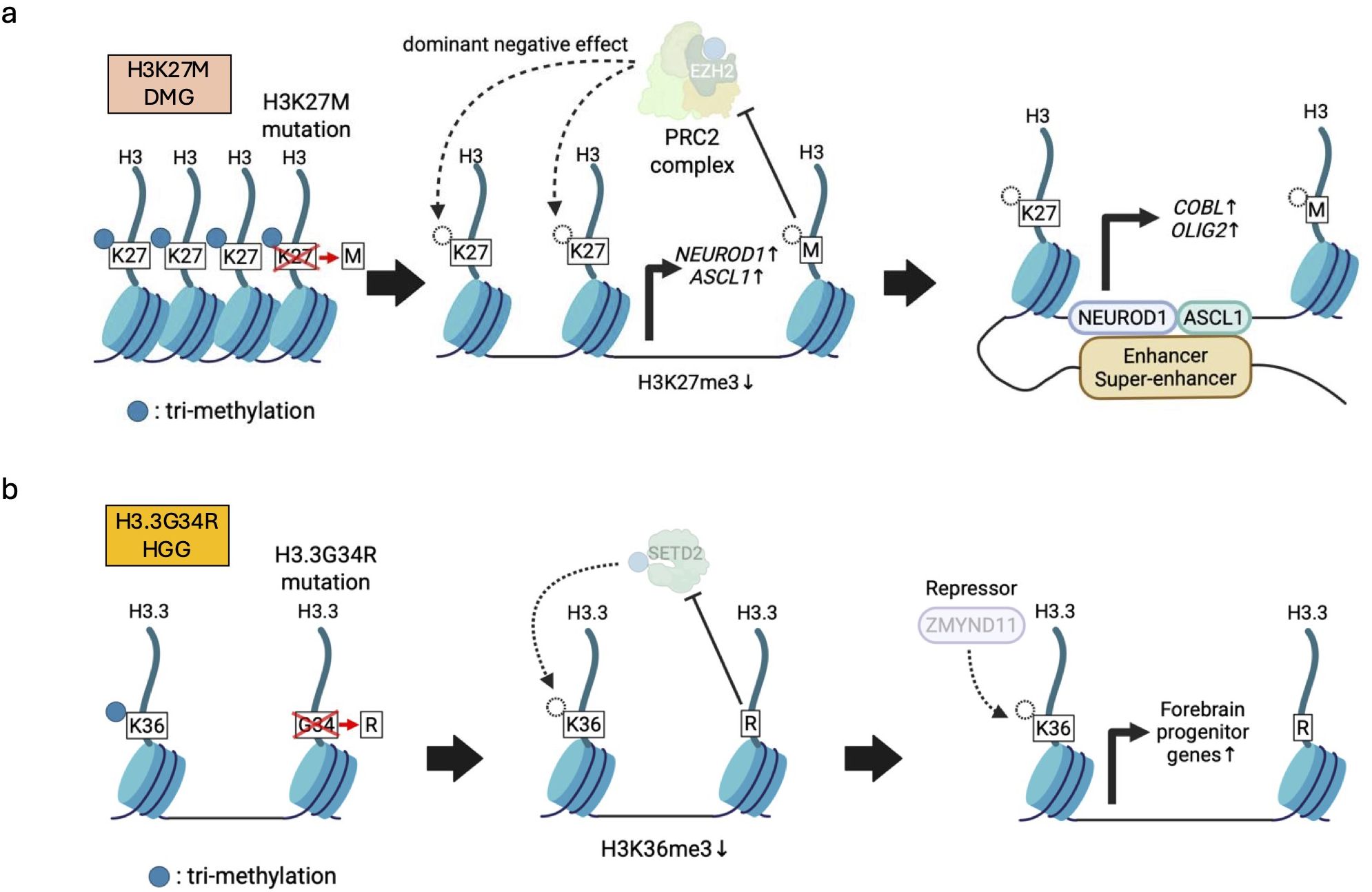
Figure 4. Histone mutations and the resulting abnormalities in histone modifications. (a) H3K27M-mutant H3 exhibits higher binding affinity than wild-type histone H3 to PRC2, leading to global reduction of PRC2-mediated H3K27me3 (left). Decreased H3K27me3 induces expression of pioneer factors (e.g., NEUROD1, ASCL1) (middle) that subsequently cooperate with enhancer/super-enhancers to further enhance abnormal chromatin accessibility (right). (b) H3.3G34R represses a lysine 36 methyltransferase SETD2, leading to low H3.3K36me3 levels, which disrupts interaction between a repressor ZMYND11 and H3.3. Reduced ZMYND11 function, in turn, activates forebrain progenitor genes involved in hemispheric pHGG formation.
H3G34R/V is another remarkable histone mutation found in pediatric glioma. H3G34R/V indicates missense mutations in histone H3 at amino acid 34, glycine (G) to arginine (R) or valine (V). H3G34R/V mutations are identified exclusively in H3-3A (H3F3A) encoding H3.3 (17) (Figure 4b). More than 30% of pHGGs arising in the cerebral hemispheres contain H3.3G34R/V mutations (79, 80). Although the function of H3.3G34R/V mutations are not fully understood, hESC models have shown that H3.3G34R along with knockout of ATRX and TP53 blocked differentiation and enhanced proliferation of tumor cells resembling interneuron progenitor cells in the ventral forebrain, but had no effect in ventral hindbrain spheroids (81). This suggests that H3.3G34R also confers a selective oncogenic advantage in specific regions. Consistent with this idea, H3.3G34R/V pHGGs arise in hemispheric or cortical regions, while H3K27M DMGs are found in pontine or supratentorial midline regions (17). At the molecular level, H3.3G34R, ATRX, and TP53 mutations cooperate to affect RNA splicing through suppression of intron retention, leading to increased expression of NOTCH2NL and eventually promoting tumor growth and survival (81). Furthermore, H3.3G34R mutations cause the loss of adjacent H3.3K36me3 by inhibiting SETD2, a lysine 36 methyltransferase, at the promoters and genetic regions of forebrain-associated genes, and disrupt interactions between H3.3 and ZMYND11, a transcriptional repressor that specifically reads H3.3K36me3. As a result, the failure of ZMYND11 possibly enhances the expression of forebrain progenitor genes (74) (Figure 4b). Thus, H3.3G34R/V is involved in the pathogenesis of hemispheric pHGGs in a different manner from H3.3K27M.
2.4 DNA methylation
Histone modifications, as mentioned above, regulate transcription from genomic DNA either positively or negatively. Direct modifications of genomic DNA itself also affect gene transcription. DNA methylation is a well-known molecular machinery that negatively regulates transcription. Distinct DNA methylation regions are characteristic of each cancer type, likely reflecting the identity of the tumors themselves or the cells of origin, and diagnostic methods utilizing these patterns (147) have become increasingly popular.
DNMTs add methyl groups to cytosine bases in genomic DNA, altering transcription factor binding kinetics and recruiting transcriptional repressor complexes such as PRC2 to silence transcription (109). Recently, genome-wide CRISPR-Cas9 knockout screens in murine MB models illustrated that DNMTs are vital for normal murine cerebellar development and required for SHH-MB tumorigenesis (82) (Figure 5a). Pharmacological inhibition of DNMT1 reduced tumor growth in a cell line and in vivo mouse models of SHH-MB (82). Furthermore, TTYH1::C19MC-driven abnormal activation of DNMT3B is a hallmark of embryonal tumors with multilayered rosettes (ETMRs) and accounts for 90% of ETMR tumors (83, 84). In preclinical settings, DNMT inhibitors have been shown to suppress the growth of ETMR cell lines by inducing cell death and differentiation (85). As such, the role of DNMTs in tumorigenesis and their potential as therapeutic targets are increasingly gaining attention.
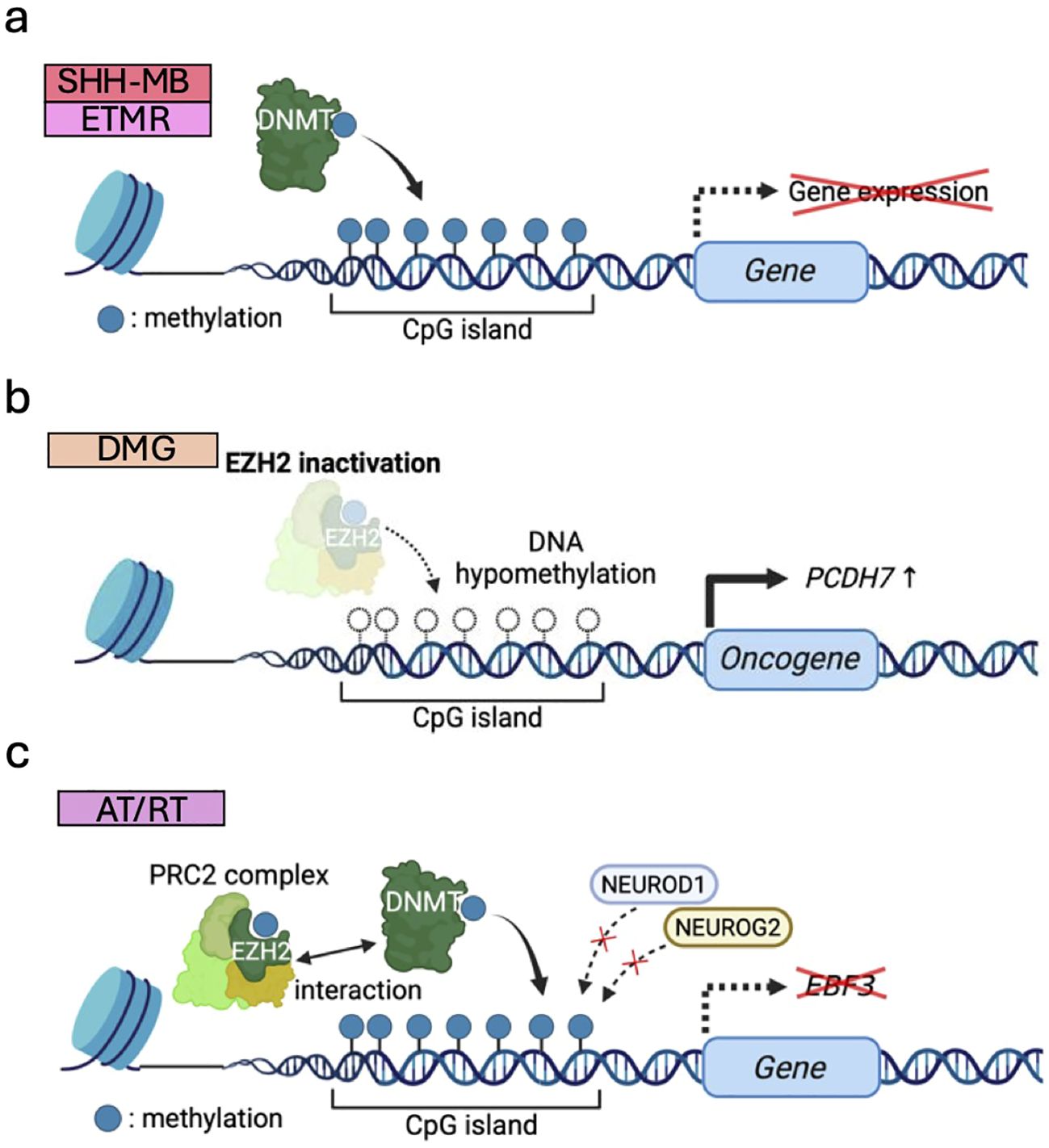
Figure 5. DNA methylation dysregulation associated with pediatric brain tumor formation. (a) Upregulated DNMTs methylate CpG islands of genomic DNA and silence transcription in SHH-MB and ETMR. (b) Global DNA hypomethylation due to EZH2 inactivation induces oncogene expression (e.g., PCDH7) to drive DMG tumorigenesis. (c) DNA hypermethylation caused by EZH2 and DNMTs blocks the binding of transcription factors (e.g., NEUROG2/NEUROD1), repressing differentiation-associated genes (e.g., EBF3) and maintaining stemness of AT/RTs.
DNMT-mediated DNA methylation has been reported to be modified by EZH2 through their direct interaction with each other (148). In DMG, reduced H3K27me3 and DNA hypomethylation, possibly due to failure of proper EZH2 recruitment, cooperate to activate ectopic gene expression (e.g., PCDH7) and drive tumorigenesis (72) (Figure 5b). It is suggested that decreased levels of H3K27me3 by H3.3K27M mutation may predispose to global DNA hypomethylation (72). This may be explained by the mechanisms in which H3K27M exhibits higher binding affinity to PRC2 than wild-type H3K27, thereby limiting the methyltransferase activity of PRC2 and resulting in global DNA hypomethylation (17). In AT/RT, DNA hypermethylation is frequently observed partially due to DNMT1 and DNMT3A upregulation, and DNMT inhibitors impaired tumor growth in vitro and in vivo (149). EZH2 also interacts with DNMTs and promotes DNA hypermethylation, leading to suppression of neural differentiation factors like NEUROG2/NEUROD1. This downregulates neural differentiation-associated genes (e.g., EBF3) and maintains PSC-like DNA methylation and gene expression patterns specific to this type of tumor (86) (Figure 5c). Accordingly, EZH2-mediated DNA methylation at specific sites is closely related with tumorigenesis.
3 Genomic rearrangement
Genomic rearrangement is one of the characteristic phenomena often caused by erroneous double strand break repair, chromosomal segregation failures, and chromothripsis (150–152), and it can sometimes contribute to tumor formation by inducing abnormal gene expression. Recent advancements in understanding epigenome-regulated gene expression have increasingly elucidated novel cancer epigenomic mechanisms arising from genomic rearrangements (152, 153). Genomic rearrangement can modify chromatin topology, including chromatin looping and distal chromatin interactions, links oncogenes to distal transcriptional regulatory elements called enhancers. Structural rearrangement may allow oncogenes to utilize enhancers of other genes, a phenomenon called enhancer hijacking. In addition, oncoproteins or microRNA (miRNA) from fusion genes occasionally caused by genomic rearrangement have been revealed to epigenetically regulate tumor development. As another finding, genomic rearrangement can generate extra chromosomal DNA (ecDNA), circular chromatin that exists outside chromosome structures within the cell nucleus and plays a crucial role in shaping cancer epigenome. This chapter focuses on cancer signaling pathways linked to epigenomic changes that stem from such genomic rearrangement.
3.1 Enhancer hijacking
Enhancer hijacking events are caused by abnormal genomic rearrangements and chromatin configurations, where cancer-related genes are aberrantly activated by enhancers that are originally designated for other genes. For instance, enhancers of BARHL1/DDX31 activate GFI1/GFI1B transcription following genomic rearrangements for G3/G4-MB growth (87) (Figure 6a). A follow-up study has revealed physical interaction between GFI1 and LSD1 for tumorigenesis, identifying an LSD1 inhibitor as a new potential therapeutic drug for GFI1/MYC-driven MB (88) Thus, understanding of the mechanisms underlying abnormal enhancer activities within cancer genomes leads to the discovery of novel therapeutic targets.
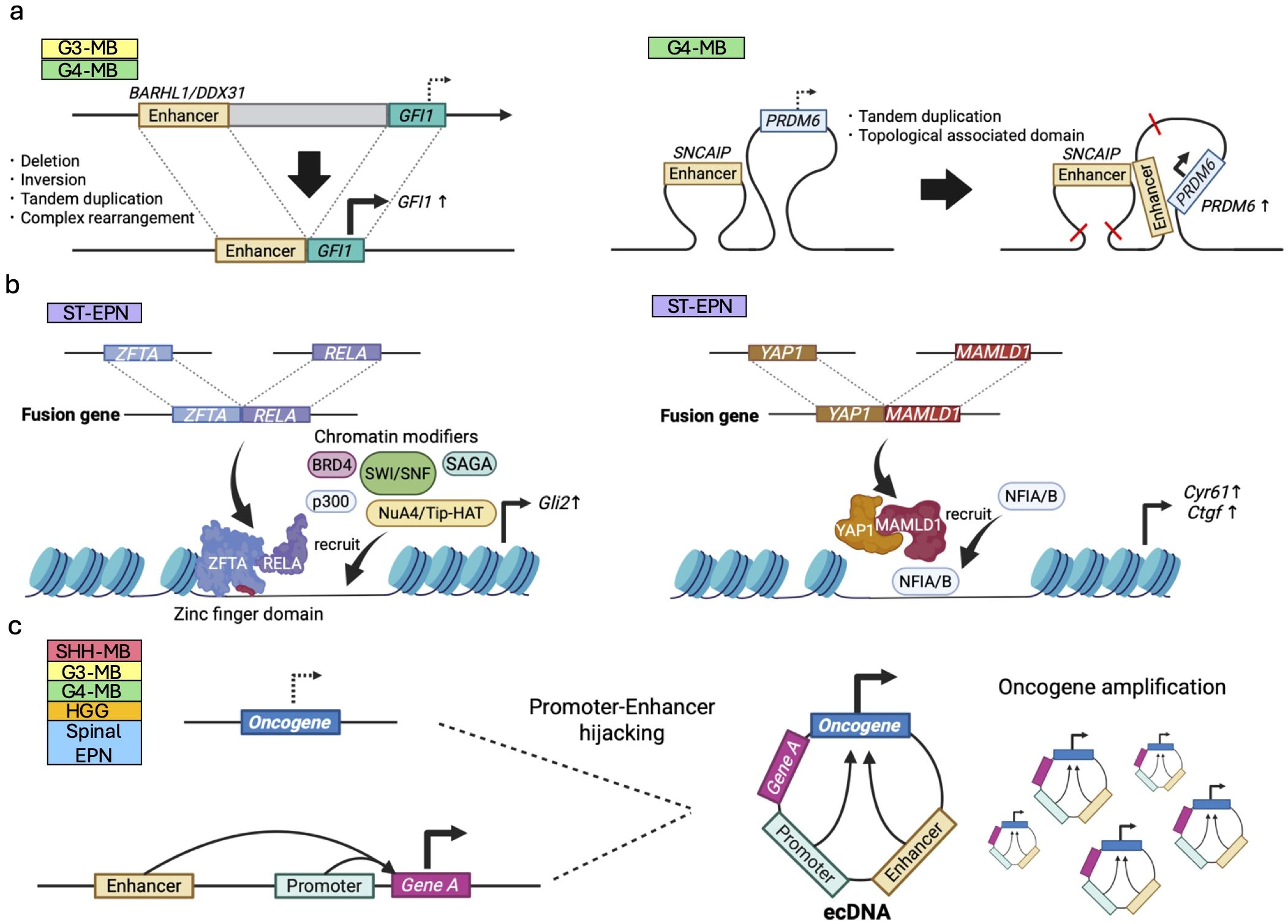
Figure 6. Epigenetic dysregulation induced by genomic rearrangement in pediatric brain tumors. (a) Enhancer hijacking; oncogenes (e.g., GFI1) are aberrantly activated by enhancers of other genes (e.g., BARHL1/DDX31) via deletion, inversion, tandem duplication due to genomic rearrangement in G3/G4-MBs (left panel). Topological associated domains and chromatin looping can also lead to the enhancer hijacking (right panel). (b) Fusion oncoproteins; ZFTA::RELA (left panel) and YAP1::MAMLD1 (right panel) observed in ST-EPNs expressed from these fusion genes modulate chromatin states in combination with other chromatin regulators. (c) Structural rearrangements leading to ecDNA formation may juxtapose oncogenes with ectopic enhancer elements, leading to transcriptional dysregulation of the ecDNA-amplified oncogene.
Following this line of research, extensive efforts have been made to elucidate enhancer regions in various pediatric brain tumors. For instance, some G4-MBs upregulate PRDM6 expression by SNCAIP-mediated G4-specific enhancer hijacking (38) (Figure 6a). Consistent with the fact that PRDM6 is known to be a histone methyltransferase, the latest study has demonstrated that PRDM6 binds to H3K27me3 and exerts widespread repression of chromatin accessibility. Overexpression of PRDM6 alone in iPSC-derived neuroepithelial stem cells led to tumor formation albeit with molecular characteristics resembling those of G3-MBs (89), highlighting the oncogenic potential of PRDM6 in a human genetic background.
In addition, recent high-throughput chromosome conformation capture (Hi-C) analyses on supratentorial ependymoma with ZFTA fusion (ST-EPN-ZFTA) and PF-EPN-A have found that 3D genome conformation activates the genes essential for their survival through enhancer hijacking, thus often causing cancer type-specific vulnerabilities (90). In pHGGs and DMGs, structural variants (SVs) drive MYC activation primarily through enhancer hijacking rather than gene amplification. Regulatory elements near PVT1 and CCDC26 are frequently co-opted, leading to aberrant MYC overexpression and highlighting enhancer reorganization as a key mechanism of tumorigenesis (91). Another recent study showed that somatic SVs enriched for enhancer hijacking also play a major role in shaping the cancer DNA methylome and regulating the expression of nearby genes, such as MYC, MYCN, TERT, ZFTA, KIAA1549, ATRX, and CDKN2A, in pediatric brain tumors (154).
Of note, enrichment of the cells carrying such enhancer hijacking events within cancer may provide some clues about the cellular origins of cancer. If this event is important for tumor initiation, the abnormally used enhancers could be active in their cellular origins (155). An in vivo reporter assay in mice (156, 157) would be a powerful tool to identify the cell of origin by analyzing the activity of the identified enhancers in the developing brain.
3.2 Fusion genes
Another epigenomic regulatory mechanism resulting from genomic rearrangements is the formation of cancer-specific fusion genes by combining two previously separate genes. The resulting abnormal fusion proteins or miRNA can function as oncogenic molecules for the development and progression of brain tumors. While a wide range of fusion genes have been identified in pediatric brain tumors (97), we highlight here those that are particularly involved in epigenetic regulation.
Unlike enhancer hijacking events, this type of mechanism regulates cancer epigenomes indirectly. ZFTA fusion genes, recently identified in ST-EPNs, are composed of a segment of the ZFTA gene fused with various transcription activators including RELA. ZFTA fusions have been reported to have an oncogenic capacity in vivo using animal models and to drive epigenetic changes and activate downstream oncogenic transcription programs, including Gli2 activation (92, 93). The portion of ZFTA plays a crucial role in chromatin binding and remodeling via its zinc finger DNA-binding domains, as well as its translocation into the nucleus (94). This fusion interacts other chromatin modifiers such as SWI/SNF, Spt-Ada-Grn5 acetyltransferase (SAGA) and NuA4/Tip60 histone acetyltransferase (NuA4/Tip-HAT) (94) (Figure 6b), hypothesizing its contribution to profound chromatin landscape modification, in turn converting various genes to a transcriptionally active state.
Another commonly detected fusion gene in ST-EPNs involves the activity of Yes-associated protein 1 (YAP1), a component of the Hippo signaling pathway that promotes cell growth and prevents cell death. Our previous study has shown that the segment of its primary fusion partner, MAMLD1, directs YAP1::MAMLD1 to the cell nucleus and attracts chromatin modifiers NFIA/B to specific loci on YAP1 target genes, such as Cyr61 and Ctgf (Figure 6b). This process amplifies the cancer-promoting YAP1 signaling pathway (95). Among the various fusion genes, CIC fusions are observed in certain types of pediatric brain tumors, such as CIC::NUTM1 in CNS Ewing sarcoma family tumors with CIC alterations (CNS EFT-CIC) (96) and CIC::LEUTX in anaplastic pleomorphic astrocytomas and CNS embryonal tumors (97). Of note, CIC::DUX4 fusions, which are detected in Ewing sarcomas but not in CNS tumors, have recently been revealed to function as transcriptional activators and to modify chromatin states through direct interaction with the acetyltransferase p300 (158). Such chromatin regulatory mechanisms driven by CIC fusions may also be involved in CNS tumors.
As another example, tumorigenesis of ETMRs also seems to be triggered by specific fusion genes. ETMRs are characterized by the miRNA cluster amplification caused by recurrent gene fusions of chromosome 19 miRNA cluster (C19MC) and tweety family member 1 chloride ion channel (TTYH1). The TTYH1::C19MC fusion structure enhances the expression of C19MC miRNAs, which downregulate the transcriptional repressor RBL2 and in turn, upregulate DNMT3B, leading to ETMR development as mentioned above (84). Collectively, cancer-specific fusion genes not only regulate molecules involved in cell proliferation and survival but also possess functions that create a cancer-supportive epigenome conducive to cellular growth. They achieve this by interacting with various epigenomic factors to establish an environment favorable for tumor progression.
In addition to the fusion genes presented in this study, some other fusion genes, such as those involving BCOR, p300 and BEND2, may also contribute to the cancer epigenome, given that their original functions are related to chromatin modifications (97, 159, 160). However, further studies are needed to elucidate the mechanisms by which these fusion genes promote tumor progression.
3.3 Extrachromosomal DNA
Genomic rearrangements in cancer occasionally produce circular chromatin, lacking centromeric sequences and usually 50kbp to 10Mbp in length, called extrachromosomal DNA (ecDNA) or double minutes (dm). Replication and segregation of ecDNA is decoupled from that of chromosomal DNA, enabling a tumor to accumulate high-copy amplification of ecDNA under positive selection (161). In human cancers, ecDNA is believed to drive malignant tumors by various mechanisms including amplification and overexpression of oncogenic sequences (162) including fusion oncogenes (163, 164), tumor evolution (161), chromatin remodeling (165), enhancer hijacking (166–168), and promoter hijacking (169, 170) (Figure 6c). Here we briefly review the role of ecDNA in epigenetic dysregulation of pediatric brain tumors and refer the reader to a recent review of transcriptional regulation by ecDNA across human cancers (171).
ecDNA in pediatric brain tumors has been reported in MBs (98, 99), pHGGs (91, 100, 101), and spinal EPNs (102). MYCN was most frequently amplified on ecDNA in MBs and spinal EPNs, although in MB ecDNA amplifications may alternately target epigenetic regulators including SETBP1 and KMT2E (98). To our knowledge, no ecDNA amplifications of ZFTA nor YAP fusion genes in EPNs have been reported to date.
In addition to oncogene amplification, the genomic rearrangements which produce ecDNA may also result in enhancer hijacking. In a small cohort of eight MB patient tumors profiled by Hi-C, half showed evidence of enhancer hijacking events between genomic loci from distal locations on the reference genome but juxtaposed on the ecDNA sequence due to genomic rearrangement (98). The assays required to detect regulatory interactions and ecDNA are not yet part of the standard of care for pediatric brain tumors. Thus, in our view, these observations probably represent an incomplete sample of the oncogenic amplifications and enhancer hijacking events which occur on MB and other pediatric brain tumors. We anticipate that future work will further illuminate the frequency and diversity of epigenetic dysregulation in rare pediatric brain tumors.
4 Toward therapies targeting epigenetic regulation
It has been recognized that pediatric brain tumors are often caused by epigenetic dysregulation. This growing understanding has opened new avenues for therapeutic intervention, and recent preclinical studies have demonstrated some promising strategies targeting epigenetic regulation (Table 2). These approaches aim to reverse or mitigate the epigenetic changes driving tumor growth, offering potential for more precise and effective treatments.
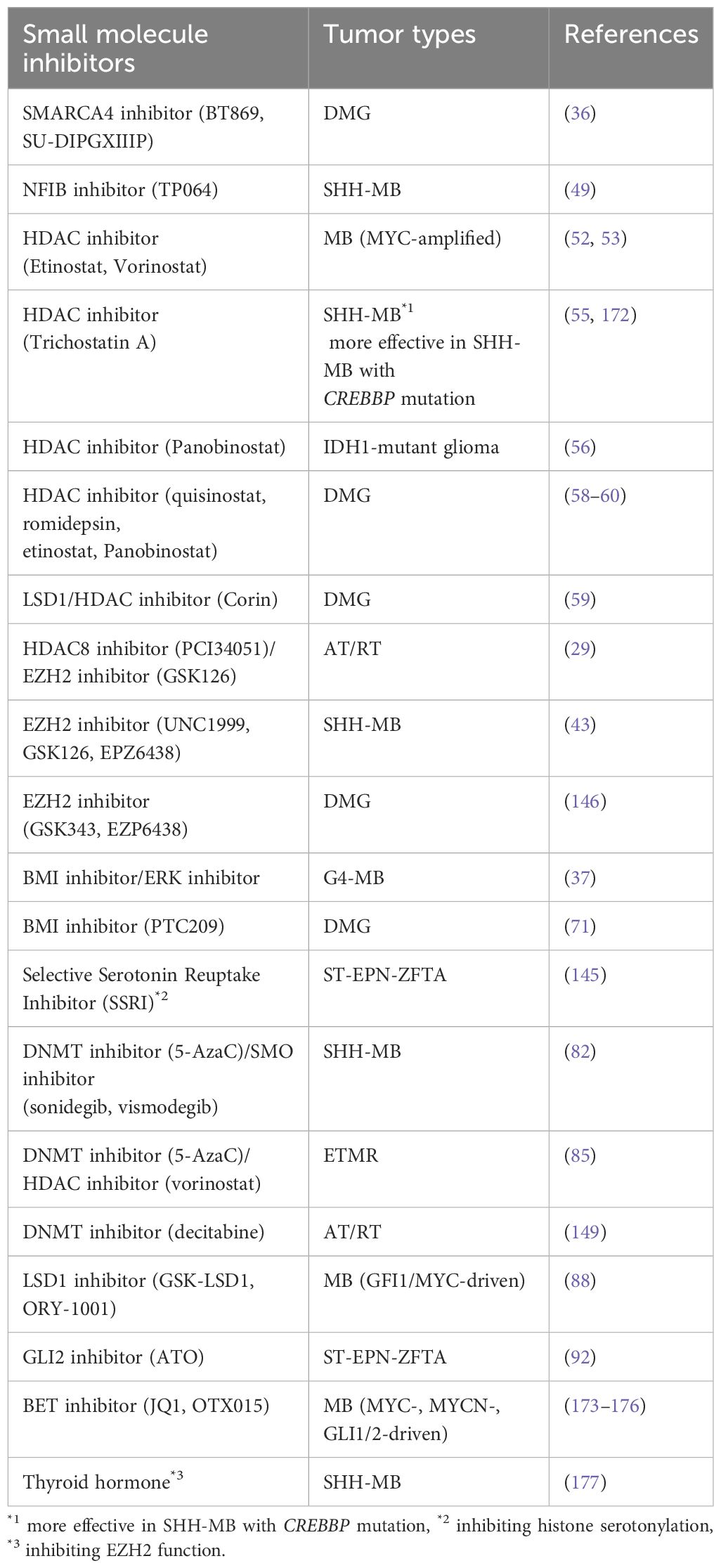
Table 2. Pre-clinical studies on treatments targeting epigenetic mechanisms.
One approach is the identification of molecules responsible for the epigenetic regulation of target genes followed by administering drugs that specifically act on these molecules. Recent successful examples are bromodomain-containing protein 4 (BRD4) inhibitor and thyroid hormone treatments. BRD4 is an epigenetic reader that recognizes histone acetylation motifs on ϵ-N-terminal lysine residues (178). At histone acetylated regions, BRD4 often promotes oncogene expression by interacting with various transcriptional factors and chromatin remodeling proteins, forming a bridge between super enhancer and promoter, and recruiting RNA polymerase II (179). In preclinical models of MB, JQ1 and OTX015, bromodomain and extraterminal (BET) inhibitors suppress oncogenic pathways in MBs driven by MYC (173, 174), GLI1 and GLI2 (175) and MYCN (176) via functional inhibition of BRD4. As the second example, thyroid hormone can inhibit EZH2 function by blocking the interaction between EZH2 and thyroid hormone receptor (TRα1). Thyroid hormone-mediated EZH2 inhibition reduces H3K27me3 histone marks at NEUROD1 regulatory regions and enhances NEUROD1 expression, eventually driving tumor cell differentiation into postmitotic cells and suppressing MB growth irreversibly (43, 177). These strategies hold promise as they target key epigenetic regulators directly, potentially enhancing therapeutic specificity and efficacy in cancer treatment.
The development of epigenetic drugs has consequently presented an attractive therapeutic approach in clinical setting. For example, HDAC inhibitors (e.g., Vorinostat, Panobinostat, Fimepinostat, and MTX110), EZH2 inhibitors (e.g., Tazemetostat), BMI inhibitors (PTC028, PTC596), and BET inhibitors (e.g., BMS-986158 and BMS-986378) are being utilized, and assessed their efficacy and safety in clinical trials (Table 3) (180–186). Although these therapies face issues such as poor blood-brain barrier (BBB) penetration, resistance due to tumor heterogeneity, and systemic toxicity, recent studies have been addressing these issues (185, 187, 188). However, challenges persist due to the lack of mechanistic clarity for individual cancer types, as well as off-target effects, which refer to the unintentional and broad impacts these drugs may have on non-target gene expression. These factors complicate efforts to mitigate side effects and address the lack of efficacy observed in some cases.
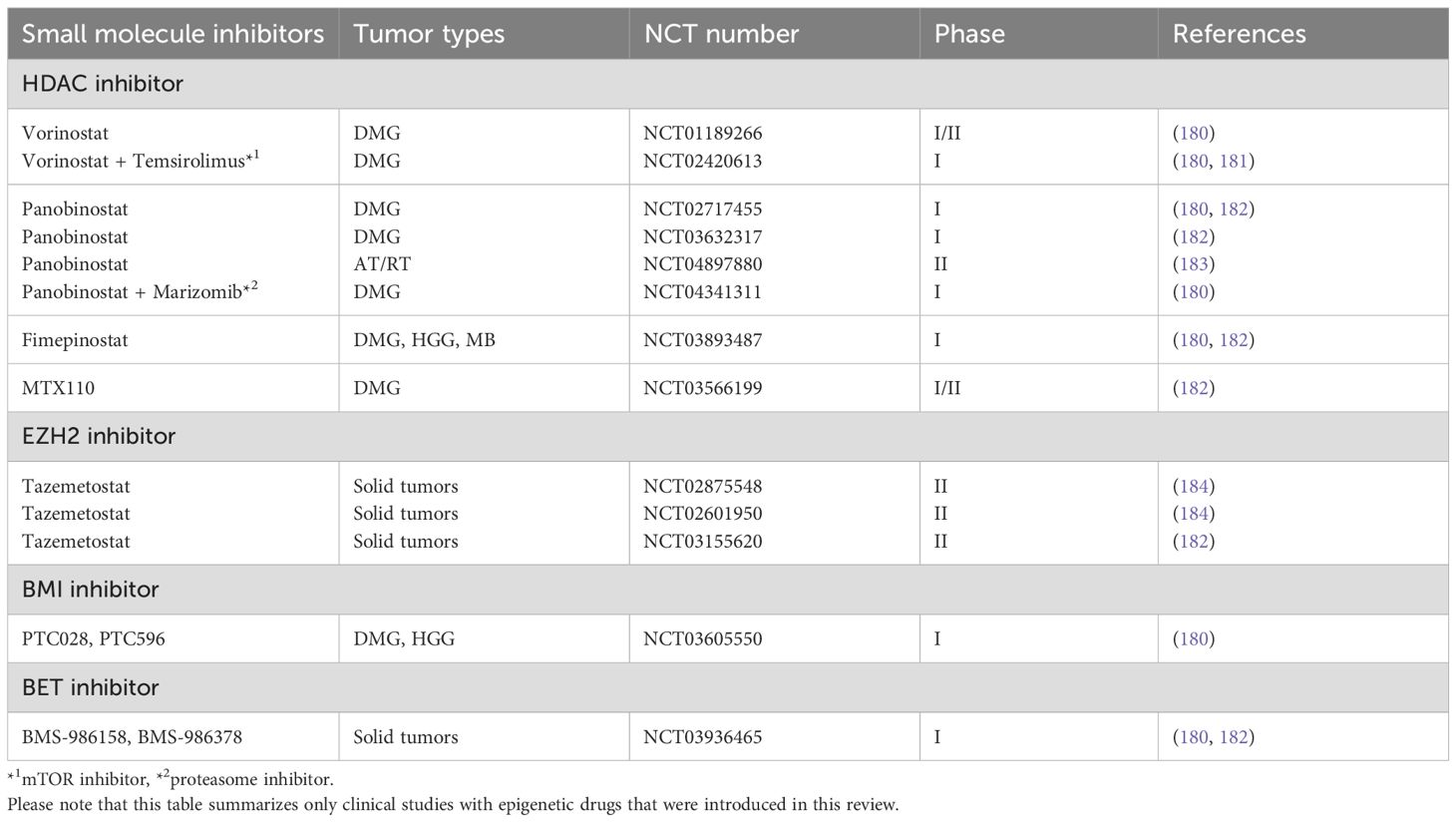
Table 3. Inhibitors used for clinical trials of epigenome-targeted therapy on pediatric brain tumors.
Given that several genes are regulated by multiple chromatin regulatory factors, synthetic lethality approaches could be an alternative option to eliminate cancer by dysregulation of cancer epigenomes (189). Synthetic lethality occurs when the simultaneous impairment of two or more genes or pathways results in cell death, whereas the impairment of either one alone does not. This strategy leverages the unique vulnerabilities of cancer cells by targeting specific chromatin regulators in combination, aiming to induce cancer cell death while minimizing effects on normal cells. Furthermore, the synthetic lethality approach enables treatment of LOF mutations that were once considered undruggable (189). For example, loss of SMARCB1 and inhibition of EZH2 cause synthetic lethality in malignant rhabdoid tumor (MRT) cells. EZH2 inhibition induces apoptosis, differentiation, and the expression of the tumor suppressor gene p16Ink4a specifically in SMARCB1-deficient MRT cells, due to epigenetic antagonism between SWI/SNF and PRC2 (190). In addition, HDAC inhibitors also cause synthetic lethality for SMARCB1 loss. These inhibitors partially complement the histone acetylation function of the SWI/SNF complex in SMARCB1-deficient MRTs, enhancing the expression of differentiation markers and inhibiting the proliferation signaling of tumor cells (191, 192). Accordingly, EZH2 inhibitors and/or HDAC inhibitors are also applicable to SMARCB1-deficient AT/RT (29).
Synthetic lethality has also been explored in relation to the balance of histone acetylation. HDACs and HAT domain of CREBBP have opposing functions; therefore, HDAC inhibitors complement HAT activity, which makes them more effective in SHH-MB with CREBBP mutation than CREBBP wild-type (172). Alternatively, interaction between CREBBP loss and p300 inhibition also induces synthetic lethality through distinct mechanisms. Both CREBBP and p300 have HAT activity; hence, p300 inhibitors can induce synthetic lethality for CREBBP-deficient tumor cells by inhibiting compensatory p300 activation in lung and hematopoietic cancer cells through the regulation of MYC promoter activity (193). A similar effect of p300 inhibitors could also be expected in CREBBP-deficient MBs. Thus, synthetic lethality-based functional screening would be one of the powerful approaches for identification of druggable targets.
A recent novel sophisticated approach for seeking a potential cancer therapy is the use of CRISPRi to precisely target epigenetic loci. This method involves expressing histone regulatory proteins, such as p300 or Krüppel-Associated Box (KRAB), fused with dead CRISPR-associated nuclease 9 (dCas9) and recruiting these complexes to the regulatory regions of target genes, specifically promoter regions, via single-guide RNA (sgRNA), leading to either activation by p300 or repression by KRAB of gene expression (49, 194, 195). Unlike the synthetic lethality approach, this method specifically targets the expression of genes critical to cancer proliferation or cell death, potentially reducing side effects. However, there are still several challenges associated with CRISPR/dCas9 such as off-target effects, delivery efficiency, lack of persistence due to instability, and cancer resistance. Recent studies have been addressing these issues through improved sgRNA design with high-fidelity dCas9 variants, efficient and stable delivery systems (e.g., viral vectors, lipid nanoparticles), and combination therapy with immunotherapy and molecular targeted drugs (196). Further research to overcome these challenges will be essential for advancing CRISPR/dCas9-based therapies toward clinical applications.
5 Conclusion
Over the past few decades, epigenetic dysregulation has been increasingly recognized as a critical factor in the tumorigenesis of pediatric brain tumors. Chromatin regulatory dysfunction, with proper timing and regional specificity, plays a pivotal role in diverting normal neuronal differentiation signaling toward oncogenesis. Insights into epigenetically driven tumorigenesis could pave the way for novel treatments that shift oncogenic pathways back toward normal differentiation signaling. The chromatin modifications underlying tumor development consist of various mechanisms, including mutation or dysregulation of chromatin remodelers, histone modifiers, histones themselves, and DNA methyltransferases. Genomic rearrangement events disrupt epigenetic regulation by generating new fusion genes or placing existing genes in new regulatory contexts. These mechanisms are commonly seen in multiple brain tumor types, and elucidating functional similarities and differences across tumor types may provide clues to finding new common therapeutic approaches. Indeed, certain targeted therapies could even be applied to multiple tumor types as seen in HDAC inhibitors for SHH-MB, MYC-amplified G3-MB, and DMG (52–55, 58–60, 137, 172). Furthermore, the impact of tumor microenvironment on epigenetics has been gaining attention as an emerging field. As seen in the histone modification by neurotransmitters (e.g., histone serotonylation), other elements of tumor microenvironment may also regulate chromatin states and could serve as druggable targets. When considering potential treatments targeting epigenomes, incorporating the latest knowledge (e.g., synthetic lethality) and cutting-edge technologies (e.g., CRISPR/dCas9) into therapeutic strategies may also be crucial for maximizing the efficacy while minimizing the side effects. In conclusion, further multifaceted studies will be required to elucidate the epigenetic mechanisms and advance innovative therapies for pediatric brain tumors.
Author contributions
KK: Conceptualization, Writing – original draft, Writing – review & editing, Visualization. OSC: Writing – original draft, Writing – review & editing. SN: Supervision, Writing – review & editing. DK: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work is supported by the Kawano Masanori Memorial Public Interest Incorporated Foundation for Promotion of Pediatrics (DK) and JSPS postdoctoral fellowship grant number #P24404 (OSC).
Acknowledgments
We thank IBS-NCU lab members for fruitful discussion. The figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lawlor ER and Thiele CJ. Epigenetic changes in pediatric solid tumors: promising new targets. Clin Cancer Res. (2012) 18:2768–79. doi: 10.1158/1078-0432.Ccr-11-1921
2. Maris JM and Denny CT. Focus on embryonal Malignancies. Cancer Cell. (2002) 2:447–50. doi: 10.1016/s1535-6108(02)00206-4
3. Kumar R, Li DQ, Müller S, and Knapp S. Epigenomic regulation of oncogenesis by chromatin remodeling. Oncogene. (2016) 35:4423–36. doi: 10.1038/onc.2015.513
4. Wilson BG and Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. (2011) 11:481–92. doi: 10.1038/nrc3068
5. Baylin SB and Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. (2016) 8. doi: 10.1101/cshperspect.a019505
6. Abedalthagafi M, Mobark N, Al-Rashed M, and AlHarbi M. Epigenomics and immunotherapeutic advances in pediatric brain tumors. NPJ Precis Oncol. (2021) 5:34. doi: 10.1038/s41698-021-00173-4
7. Mittal P and Roberts CWM. The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat Rev Clin Oncol. (2020) 17:435–48. doi: 10.1038/s41571-020-0357-3
8. Ronan JL, Wu W, and Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet. (2013) 14:347–59. doi: 10.1038/nrg3413
9. Juliandi B, Abematsu M, and Nakashima K. Chromatin remodeling in neural stem cell differentiation. Curr Opin Neurobiol. (2010) 20:408–15. doi: 10.1016/j.conb.2010.04.001
10. Feng W, Kawauchi D, Körkel-Qu H, Deng H, Serger E, Sieber L, et al. Chd7 is indispensable for mammalian brain development through activation of a neuronal differentiation programme. Nat Commun. (2017) 8:14758. doi: 10.1038/ncomms14758
11. Sokpor G, Xie Y, Rosenbusch J, Tuoc T, and Chromatin Remodeling BAF. (SWI/SNF) complexes in neural development and disorders. Front Mol Neurosci. (2017) 10:243. doi: 10.3389/fnmol.2017.00243
12. McNeill KA. Epidemiology of brain tumors. Neurol Clin. (2016) 34:981–98. doi: 10.1016/j.ncl.2016.06.014
13. McLeod C, Gout AM, Zhou X, Thrasher A, Rahbarinia D, Brady SW, et al. St. Jude cloud: A pediatric cancer genomic data-sharing ecosystem. Cancer Discov. (2021) 11:1082–99. doi: 10.1158/2159-8290.Cd-20-1230
14. Shapiro JA, Gaonkar KS, Spielman SJ, Savonen CL, Bethell CJ, Jin R, et al. OpenPBTA: the open pediatric brain tumor atlas. Cell Genom. (2023) 3:100340. doi: 10.1016/j.xgen.2023.100340
15. Swartling FJ, Savov V, Persson AI, Chen J, Hackett CS, Northcott PA, et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell. (2012) 21:601–13. doi: 10.1016/j.ccr.2012.04.012
16. Kawauchi D, Ogg RJ, Liu L, Shih DJH, Finkelstein D, Murphy BL, et al. Novel MYC-driven medulloblastoma models from multiple embryonic cerebellar cells. Oncogene. (2017) 36:5231–42. doi: 10.1038/onc.2017.110
17. Ocasio JK, Budd KM, Roach JT, Andrews JM, and Baker SJ. Oncohistones and disrupted development in pediatric-type diffuse high-grade glioma. Cancer Metastasis Rev. (2023) 42:367–88. doi: 10.1007/s10555-023-10105-2
18. Okonechnikov K, Joshi P, Sepp M, Leiss K, Sarropoulos I, Murat F, et al. Mapping pediatric brain tumors to their origins in the developing cerebellum. Neuro Oncol. (2023) 25:1895–909. doi: 10.1093/neuonc/noad124
19. Guo G, Zhuang J, Zhang K, Zhou Z, Wang Y, and Zhang Z. Atypical teratoid/rhabdoid tumor of the central nervous system in children: case reports and literature review. Front Surg. (2022) 9:864518. doi: 10.3389/fsurg.2022.864518
20. Klonou A, Spiliotakopoulou D, Themistocleous MS, Piperi C, and Papavassiliou AG. Chromatin remodeling defects in pediatric brain tumors. Ann Transl Med. (2018) 6:248. doi: 10.21037/atm.2018.04.08
21. Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. (2012) 22:425–37. doi: 10.1016/j.ccr.2012.08.024
22. Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell. (2017) 32:520–537.e5. doi: 10.1016/j.ccell.2017.08.017
23. Andreiuolo F, Puget S, Peyre M, Dantas-Barbosa C, Boddaert N, Philippe C, et al. Neuronal differentiation distinguishes supratentorial and infratentorial childhood ependymomas. Neuro Oncol. (2010) 12:1126–34. doi: 10.1093/neuonc/noq074
24. Sievert AJ and Fisher MJ. Pediatric low-grade gliomas. J Child Neurol. (2009) 24:1397–408. doi: 10.1177/0883073809342005
25. Korshunov A, Ryzhova M, Jones DT, Northcott PA, van Sluis P, Volckmann R, et al. LIN28A immunoreactivity is a potent diagnostic marker of embryonal tumor with multilayered rosettes (ETMR). Acta Neuropathol. (2012) 124(6):875–81. doi: 10.1007/s00401-012-1068-3
26. Basheer F, Dhar, and Samarasinghe RM. Zebrafish models of paediatric brain tumours. Int J Mol Sci. (2022) 23:875–81. doi: 10.3390/ijms23179920
27. Han ZY, Richer W, Fréneaux, Chauvin C, Lucchesi C, Guillemot D, et al. The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat Commun. (2016) 7:10421. doi: 10.1038/ncomms10421
28. Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. (2016) 29:379–93. doi: 10.1016/j.ccell.2016.02.001
29. Terada Y, Jo N, Arakawa Y, Sakakura M, Yamada Y, Ukai T, et al. Human pluripotent stem cell-derived tumor model uncovers the embryonic stem cell signature as a key driver in atypical teratoid/rhabdoid tumor. Cell Rep. (2019) 26:2608–2621.e6. doi: 10.1016/j.celrep.2019.02.009
30. Hasselblatt M, Gesk S, Oyen F, Rossi S, Viscardi E, Giangaspero F, et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol. (2011) 35:933–5. doi: 10.1097/PAS.0b013e3182196a39
31. Göbel C, Schoof M, Holdhof D, Spohn M, and Schüller U. SMARCA4 loss and mutated β-catenin induce proliferative lesions in the murine embryonic cerebellum. J Neurosci. (2024) 44. doi: 10.1523/jneurosci.1605-23.2024
32. Göbel C, Godbole S, Schoof M, Holdhof D, Kresbach C, Loose C, et al. MYC overexpression and SMARCA4 loss cooperate to drive medulloblastoma formation in mice. Acta Neuropathol Commun. (2023) 11:174. doi: 10.1186/s40478-023-01654-2
33. Moreno N, Schmidt C, Ahlfeld J, Pöschl J, Dittmar S, Pfister SM, et al. Loss of Smarc proteins impairs cerebellar development. J Neurosci. (2014) 34:13486–91. doi: 10.1523/jneurosci.2560-14.2014
34. Shi X, Wang Q, Gu J, Xuan Z, and Wu JI. SMARCA4/Brg1 coordinates genetic and epigenetic networks underlying Shh-type medulloblastoma development. Oncogene. (2016) 35:5746–58. doi: 10.1038/onc.2016.108
35. Wang Y, Yang CH, A.Schultz MM, Miller DD, and Pfeffer LM. Brahma-Related Gene-1 (BRG1) promotes the Malignant phenotype of glioblastoma cells. J Cell Mol Med. (2021) 25:2956–66. doi: 10.1111/jcmm.16330
36. Panditharatna E, Marques JG, Wang T, Trissal MC, Liu I, Jiang L, et al. BAF complex maintains glioma stem cells in pediatric H3K27M glioma. Cancer Discov. (2022) 12:2880–905. doi: 10.1158/2159-8290.Cd-21-1491
37. Badodi S, Dubuc A, Zhang X, Rosser G, Da Cunha Jaeger M, Kameda-Smith MM, et al. Convergence of BMI1 and CHD7 on ERK signaling in medulloblastoma. Cell Rep. (2017) 21:2772–84. doi: 10.1016/j.celrep.2017.11.021
38. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. (2017) 547:311–7. doi: 10.1038/nature22973
39. Wang W, Kumegawa K, Chapman OS, Shiraishi R, Xiao Z, Okonechnikov K, et al. Chromatin modification abnormalities by CHD7 and KMT2C loss promote medulloblastoma progression. Cell Rep. (2025) 44(5):115673. doi: 10.1016/j.celrep.2025.115673
40. Ohta S, Yaguchi T, Okuno H, Chneiweiss H, Kawakami Y, and Okano H. CHD7 promotes proliferation of neural stem cells mediated by MIF. Mol Brain. (2016) 9:96. doi: 10.1186/s13041-016-0275-6
41. Lewis NA, Klein RH, Kelly C, Yee J, and Knoepfler PS. Histone H3.3 K27M chromatin functions implicate a network of neurodevelopmental factors including ASCL1 and NEUROD1 in DIPG. Epigenet Chromatin. (2022) 15:18. doi: 10.1186/s13072-022-00447-6
42. Boulay G, Awad ME, Riggi N, Archer TC, Iyer S, Boonseng WE, et al. OTX2 activity at distal regulatory elements shapes the chromatin landscape of group 3 medulloblastoma. Cancer Discov. (2017) 7:288–301. doi: 10.1158/2159-8290.Cd-16-0844
43. Cheng Y, Liao S, Xu G, Hu J, Guo D, Du F, et al. NeuroD1 dictates tumor cell differentiation in medulloblastoma. Cell Rep. (2020) 31:107782. doi: 10.1016/j.celrep.2020.107782
44. Sunkel BD and Stanton BZ. Pioneer factors in development and cancer. iScience. (2021) 42(5):103132. doi: 10.1016/j.isci.2021.103132
45. Selvadurai HJ, Luis E, Desai K, Lan X, Vladoiu MC, Whitley O, et al. Medulloblastoma arises from the persistence of a rare and transient sox2(+) granule neuron precursor. Cell Rep. (2020) 31:107511. doi: 10.1016/j.celrep.2020.03.075
46. Anderson JL, Muraleedharan R, Oatman N, Klotter A, Sengupta S, Waclaw RR, et al. The transcription factor Olig2 is important for the biology of diffuse intrinsic pontine gliomas. Neuro Oncol. (2017) 19:1068–78. doi: 10.1093/neuonc/now299
47. Torchia J, Golbourn B, Feng S, Ho KC, Sin-Chan, Vasiljevic A, et al. Integrated (epi)-genomic analyses identify subgroup-specific therapeutic targets in CNS rhabdoid tumors. Cancer Cell. (2016) 30:891–908. doi: 10.1016/j.ccell.2016.11.003
48. Sardar D, Chen HC, Reyes A, Varadharajan S, Jain A, Mohila C, et al. Sox9 directs divergent epigenomic states in brain tumor subtypes. Proc Natl Acad Sci U.S.A. (2022) 119:e2202015119. doi: 10.1073/pnas.2202015119
49. Shiraishi R, Cancila G, Kumegawa K, Torrejon J, Basili I, Bernardi F, et al. Cancer-specific epigenome identifies oncogenic hijacking by nuclear factor I family proteins for medulloblastoma progression. Dev Cell. (2024) 59:2302–2319.e12. doi: 10.1016/j.devcel.2024.05.013
50. Chen Z, Ioris RM, Richardson S, Van Ess AN, Vendrell I, Kessler BM, et al. Disease-associated KBTBD4 mutations in medulloblastoma elicit neomorphic ubiquitylation activity to promote CoREST degradation. Cell Death Differ. (2022) 29:1955–69. doi: 10.1038/s41418-022-00983-4
51. Merk DJ, Ohli J, Merk ND, Thatikonda V, Morrissy S, Schoof M, et al. Opposing effects of CREBBP mutations govern the phenotype of rubinstein-taybi syndrome and adult SHH medulloblastoma. Dev Cell. (2018) 44:709–724.e6. doi: 10.1016/j.devcel.2018.02.012
52. Ecker J, Thatikonda V, Sigismondo G, Selt F, Valinciute G, Oehme I, et al. Reduced chromatin binding of MYC is a key effect of HDAC inhibition in MYC amplified medulloblastoma. Neuro Oncol. (2021) 23:226–39. doi: 10.1093/neuonc/noaa191
53. Shofuda T and Kanemura Y. HDACs and MYC in medulloblastoma: how do HDAC inhibitors control MYC-amplified tumors? Neuro Oncol. (2021) 23:173–4. doi: 10.1093/neuonc/noaa292
54. Canettieri G, Di Marcotullio L, Greco A, Coni S, Antonucci L, Infante, et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. (2010) 12:132–42. doi: 10.1038/ncb2013
55. Lee SJ, Lindsey S, Graves B, Yoo S, Olson JM, and Langhans SA. Sonic hedgehog-induced histone deacetylase activation is required for cerebellar granule precursor hyperplasia in medulloblastoma. PloS One. (2013) 8:e71455. doi: 10.1371/journal.pone.0071455
56. Sears TK, Horbinski CM, and Woolard KD. IDH1 mutant glioma is preferentially sensitive to the HDAC inhibitor panobinostat. J Neurooncol. (2021) 154:159–70. doi: 10.1007/s11060-021-03829-0
57. Garrett MC, Albano R, Carnwath T, Elahi L, Behrmann CA, Pemberton M, et al. HDAC1 and HDAC6 are essential for driving growth in IDH1 mutant glioma. Sci Rep. (2023) 13:12433. doi: 10.1038/s41598-023-33889-3
58. Noll A, Myers C, Biery MC, Meechan M, Tahiri S, Rajendran A, et al. Therapeutic HDAC inhibition in hypermutant diffuse intrinsic pontine glioma. Neoplasia. (2023) 43:100921. doi: 10.1016/j.neo.2023.100921
59. Anastas JN, Zee BM, Kalin JH, Kim M, Guo R, Alexandrescu S, et al. Re-programing chromatin with a bifunctional LSD1/HDAC inhibitor induces therapeutic differentiation in DIPG. Cancer Cell. (2019) 36:528–544.e10. doi: 10.1016/j.ccell.2019.09.005
60. Vitanza NA, Biery MC, Myers C, Ferguson E, Zheng Y, Girard EJ, et al. Optimal therapeutic targeting by HDAC inhibition in biopsy-derived treatment-naïve diffuse midline glioma models. Neuro Oncol. (2021) 23:376–86. doi: 10.1093/neuonc/noaa249
61. Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, et al. MLL4 is required to maintain broad H3K4me3 peaks and super-enhancers at tumor suppressor genes. Mol Cell. (2018) 70:825–841.e6. doi: 10.1016/j.molcel.2018.04.028
62. Sanghrajka RM, Koche R, Medrano H, El Nagar S, Stephen DN, Lao Z, et al. KMT2D suppresses Sonic hedgehog-driven medulloblastoma progression and metastasis. iScience. (2023) 26:107831. doi: 10.1016/j.isci.2023.107831
63. Hübner JM, Müller T, Papageorgiou DN, Mauermann M, Krijgsveld J, Russell RB, et al. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol. (2019) 21:878–89. doi: 10.1093/neuonc/noz058
64. Kardian AS and Mack S. The intersection of epigenetic alterations and developmental state in pediatric ependymomas. Dev Neurosci. (2024) 46:365–72. doi: 10.1159/000537694
65. Pajtler KW, Wen J, Sill M, Lin T, Orisme W, Tang B, et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. (2018) 136:211–26. doi: 10.1007/s00401-018-1877-0
66. Castel D, Kergrohen T, Tauziède-Espariat A, Mackay A, Ghermaoui S, Lechapt E, et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol. (2020) 139:1109–13. doi: 10.1007/s00401-020-02142-w
67. Yi J, Shi X, Xuan Z, and Wu J. Histone demethylase UTX/KDM6A enhances tumor immune cell recruitment, promotes differentiation and suppresses medulloblastoma. Cancer Lett. (2021) 499:188–200. doi: 10.1016/j.canlet.2020.11.031
68. Kutscher LM, Okonechnikov K, Batora NV, Clark J, Silva PBG, Vouri M, et al. Functional loss of a noncanonical BCOR-PRC1.1 complex accelerates SHH-driven medulloblastoma formation. Genes Dev. (2020) 34:1161–76. doi: 10.1101/gad.337584.120
69. Leung C, Lingbeek M, Shakhova O, Liu J, Tanger E, Saremaslani, et al. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. (2004) 428:337–41. doi: 10.1038/nature02385
70. Merve A, Dubuc AM, Zhang X, Remke M, Baxter PA, Li XN, et al. Polycomb group gene BMI1 controls invasion of medulloblastoma cells and inhibits BMP-regulated cell adhesion. Acta Neuropathol Commun. (2014) 2:10. doi: 10.1186/2051-5960-2-10
71. Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, Mathewson ND, et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science. (2018) 360:331–5. doi: 10.1126/science.aao4750
72. Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. (2013) 24:660–72. doi: 10.1016/j.ccr.2013.10.006
73. Kfoury-Beaumont N, Prakasam R, Pondugula S, Lagas JS, Matkovich S, Gontarz, et al. The H3K27M mutation alters stem cell growth, epigenetic regulation, and differentiation potential. BMC Biol. (2022) 20:124. doi: 10.1186/s12915-022-01324-0
74. Bressan RB, Southgate B, Ferguson KM, Blin C, Grant V, Alfazema N, et al. Regional identity of human neural stem cells determines oncogenic responses to histone H3. 3 mutants Cell Stem Cell. (2021) 28:877–893.e9. doi: 10.1016/j.stem.2021.01.016
75. Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, Pahlavan, et al. H3.3(K27M) cooperates with trp53 loss and PDGFRA gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas. Cancer Cell. (2017) 32:684–700.e9. doi: 10.1016/j.ccell.2017.09.014
76. Tomita Y, Shimazu Y, Somasundaram A, Tanaka Y, Takata N, Ishi Y, et al. A novel mouse model of diffuse midline glioma initiated in neonatal oligodendrocyte progenitor cells highlights cell-of-origin dependent effects of H3K27M. Glia. (2022) 70:1681–98. doi: 10.1002/glia.24189
77. Funato K, Major T, Lewis PW, Allis CD, and Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3. 3K27M histone mutation Sci. (2014) 346:1529–33. doi: 10.1126/science.1253799
78. Haag D, Mack N, Benites Goncalves da Silva, Statz B, Clark J, Tanabe K, et al. H3.3-K27M drives neural stem cell-specific gliomagenesis in a human iPSC-derived model. Cancer Cell. (2021) 39:407–422.e13. doi: 10.1016/j.ccell.2021.01.005
79. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. (2012) 482:226–31. doi: 10.1038/nature10833
80. Chen CCL, Deshmukh S, Jessa S, Hadjadj D, Lisi V, Andrade AF, et al. Histone H3.3G34-mutant interneuron progenitors co-opt PDGFRA for gliomagenesis. Cell. (2020) 183:1617–1633.e22. doi: 10.1016/j.cell.2020.11.012
81. Funato K, Smith RC, Saito Y, and Tabar V. Dissecting the impact of regional identity and the oncogenic role of human-specific NOTCH2NL in an hESC model of H3. 3G34R-mutant glioma Cell Stem Cell. (2021) 28:894–905.e7. doi: 10.1016/j.stem.2021.02.003
82. Tsiami F, Lago C, Pozza N, Piccioni F, Zhao X, Lülsberg F, et al. Genome-wide CRISPR-Cas9 knockout screens identify DNMT1 as a druggable dependency in sonic hedgehog medulloblastoma. Acta Neuropathol Commun. (2024) 12:125. doi: 10.1186/s40478-024-01831-x
83. Lambo S, von Hoff K, Korshunov A, Pfister SM, and Kool M. ETMR: a tumor entity in its infancy. Acta Neuropathol. (2020) 140:249–66. doi: 10.1007/s00401-020-02182-2
84. Kleinman CL, Gerges N, Papillon-Cavanagh S, Sin-Chan, Pramatarova A, Quang DA, et al. Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet. (2014) 46:39–44. doi: 10.1038/ng.2849
85. Sin-Chan and Huang A. DNMTs as potential therapeutic targets in high-risk pediatric embryonal brain tumors. Expert Opin Ther Targets. (2014) 18:1103–7. doi: 10.1517/14728222.2014.938052
86. Pekkarinen M, Nordfors K, Uusi-Mäkelä J, Kytölä V, Hartewig A, Huhtala L, et al. Aberrant DNA methylation distorts developmental trajectories in atypical teratoid/rhabdoid tumors. Life Sci Alliance. (2024) 7. doi: 10.26508/lsa.202302088
87. Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. (2014) 511:428–34. doi: 10.1038/nature13379
88. Lee C, Rudneva VA, Erkek S, Zapatka M, Chau LQ, Tacheva-Grigorova SK, et al. Lsd1 as a therapeutic target in Gfi1-activated medulloblastoma. Nat Commun. (2019) 10:332. doi: 10.1038/s41467-018-08269-5
89. Schmidt C, Cohen S, Gudenas BL, Husain S, Carlson A, Westelman S, et al. PRDM6 promotes medulloblastoma by repressing chromatin accessibility and altering gene expression. Sci Rep. (2024) 14:16074. doi: 10.1038/s41598-024-66811-6
90. Okonechnikov K, Camgöz A, Chapman O, Wani S, Park DE, Hübner JM, et al. 3D genome mapping identifies subgroup-specific chromosome conformations and tumor-dependency genes in ependymoma. Nat Commun. (2023) 14:2300. doi: 10.1038/s41467-023-38044-0
91. Dubois FPB, Shapira O, Greenwald NF, Zack T, Wala J, Tsai JW, et al. Structural variants shape driver combinations and outcomes in pediatric high-grade glioma. Nat Cancer. (2022) 3:994–1011. doi: 10.1038/s43018-022-00403-z
92. Zheng T, Ghasemi DR, Okonechnikov K, Korshunov A, Sill M, Maass KK, et al. Cross-species genomics reveals oncogenic dependencies in ZFTA/C11orf95 fusion-positive supratentorial ependymomas. Cancer Discov. (2021) 11:2230–47. doi: 10.1158/2159-8290.Cd-20-0963
93. Arabzade A, Zhao Y, Varadharajan S, Chen HC, Jessa S, Rivas B, et al. ZFTA-RELA dictates oncogenic transcriptional programs to drive aggressive supratentorial ependymoma. Cancer Discov. (2021) 11:2200–15. doi: 10.1158/2159-8290.Cd-20-1066
94. Kupp R, Ruff L, Terranova S, Nathan E, Ballereau S, Stark R, et al. ZFTA translocations constitute ependymoma chromatin remodeling and transcription factors. Cancer Discov. (2021) 11:2216–29. doi: 10.1158/2159-8290.Cd-20-1052
95. Pajtler KW, Wei Y, Okonechnikov K, Silva PBG, Vouri M, Zhang L, et al. YAP1 subgroup supratentorial ependymoma requires TEAD and nuclear factor I-mediated transcriptional programmes for tumorigenesis. Nat Commun. (2019) 10:3914. doi: 10.1038/s41467-019-11884-5
96. Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell. (2016) 164:1060–72. doi: 10.1016/j.cell.2016.01.015
97. Roosen M, Odé Z, Bunt J, and Kool M. The oncogenic fusion landscape in pediatric CNS neoplasms. Acta Neuropathol. (2022) 143:427–51. doi: 10.1007/s00401-022-02405-8
98. Chapman OS, Luebeck J, Sridhar S, Wong IT, Dixit D, Wang S, et al. Circular extrachromosomal DNA promotes tumor heterogeneity in high-risk medulloblastoma. Nat Genet. (2023) 55:2189–99. doi: 10.1038/s41588-023-01551-3
99. Bigner SH, Mark J, Friedman HS, Biegel JA, and Bigner DD. Structural chromosomal abnormalities in human medulloblastoma. Cancer Genet Cytogenet. (1988) 30:91–101. doi: 10.1016/0165-4608(88)90096-9
100. Bigner SH, McLendon RE, Fuchs H, McKeever PE, and Friedman HS. Chromosomal characteristics of childhood brain tumors. Cancer Genet Cytogenet. (1997) 97:125–34. doi: 10.1016/s0165-4608(96)00404-9
101. Xu K, Ding L, Chang TC, Shao Y, Chiang J, Mulder H, et al. Structure and evolution of double minutes in diagnosis and relapse brain tumors. Acta Neuropathol. (2019) 137:123–37. doi: 10.1007/s00401-018-1912-1
102. Raffeld M, Abdullaev Z, Pack SD, Xi L, Nagaraj S, Briceno N, et al. High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol Commun. (2020) 8:101. doi: 10.1186/s40478-020-00973-y
103. Clapier CR, Iwasa J, Cairns BR, and Peterson CL. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol. (2017) 18:407–22. doi: 10.1038/nrm.2017.26
104. Zhang FL and Li DQ. Targeting chromatin-remodeling factors in cancer cells: promising molecules in cancer therapy. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232112815
105. Bannister AJ and Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. (2011) 21:381–95. doi: 10.1038/cr.2011.22
106. Zhang Y, Sun Z, Jia J, Du T, Zhang N, Tang Y, et al. Overview of histone modification. Adv Exp Med Biol. (2021) 1283:1–16. doi: 10.1007/978-981-15-8104-5_1
107. Kimura H. Histone modifications for human epigenome analysis. J Hum Genet. (2013) 58:439–45. doi: 10.1038/jhg.2013.66
108. Espinoza Pereira KN, Shan J, Licht JD, and Bennett RL. Histone mutations in cancer. Biochem Soc Trans. (2023) 51:1749–63. doi: 10.1042/bst20210567
109. Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. (2018) 19:81–92. doi: 10.1038/nrg.2017.80
110. Erkek S, Johann PD, Finetti MA, Drosos Y, Chou HC, Zapatka M, et al. Comprehensive analysis of chromatin states in atypical teratoid/rhabdoid tumor identifies diverging roles for SWI/SNF and polycomb in gene regulation. Cancer Cell. (2019) 35:95–110.e8. doi: 10.1016/j.ccell.2018.11.014
111. Navickas SM, Giles KA, Brettingham-Moore KH, and Taberlay PC. The role of chromatin remodeler SMARCA4/BRG1 in brain cancers: a potential therapeutic target. Oncogene. (2023) 42:2363–73. doi: 10.1038/s41388-023-02773-9
112. Cooper GW and Hong AL. SMARCB1-deficient cancers: novel molecular insights and therapeutic vulnerabilities. Cancers (Basel). (2022) 14. doi: 10.3390/cancers14153645
113. Tolstorukov MY, Sansam CG, Lu, Koellhoffer EC, Helming KC, Alver BH, et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc Natl Acad Sci U.S.A. (2013) 110:10165–70. doi: 10.1073/pnas.1302209110
114. Dykhuizen EC, Hargreaves DC, Miller EL, Cui K, Korshunov A, Kool M, et al. BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature. (2013) 497:624–7. doi: 10.1038/nature12146
115. Behesti H and Marino S. Cerebellar granule cells: insights into proliferation, differentiation, and role in medulloblastoma pathogenesis. Int J Biochem Cell Biol. (2009) 41:435–45. doi: 10.1016/j.biocel.2008.06.017
116. Raposo A, Vasconcelos FF, Drechsel D, Marie C, Johnston C, Dolle D, et al. Ascl1 coordinately regulates gene expression and the chromatin landscape during neurogenesis. Cell Rep. (2015) 10:1544–56. doi: 10.1016/j.celrep.2015.02.025
117. Pataskar A, Jung J, Smialowski, Noack F, Calegari F, Straub T, et al. NeuroD1 reprograms chromatin and transcription factor landscapes to induce the neuronal program. EMBO J. (2016) 35:24–45. doi: 10.15252/embj.201591206
118. Smith DK, Yang J, Liu ML, and Zhang CL. Small molecules modulate chromatin accessibility to promote NEUROG2-mediated fibroblast-to-neuron reprogramming. Stem Cell Rep. (2016) 7:955–69. doi: 10.1016/j.stemcr.2016.09.013
119. Hagey DW, Klum S, Kurtsdotter I, Zaouter C, Topcic D, Andersson O, et al. SOX2 regulates common and specific stem cell features in the CNS and endoderm derived organs. PloS Genet. (2018) 14:e1007224. doi: 10.1371/journal.pgen.1007224
120. Hagey DW, Bergsland M, and Muhr J. SOX2 transcription factor binding and function. Development. (2022) 149. doi: 10.1242/dev.200547
121. Denny SK, Yang D, Chuang CH, Brady JJ, Lim JS, Grüner BM, et al. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell. (2016) 166:328–42. doi: 10.1016/j.cell.2016.05.052
122. Fraser J, Essebier A, Brown AS, Davila RA, Harkins D, Zalucki O, et al. Common regulatory targets of NFIA, NFIX and NFIB during postnatal cerebellar development. Cerebellum. (2020) 19:89–101. doi: 10.1007/s12311-019-01089-3
123. Betancourt J, Katzman S, and Chen B. Nuclear factor one B regulates neural stem cell differentiation and axonal projection of corticofugal neurons. J Comp Neurol. (2014) 522:6–35. doi: 10.1002/cne.23373
124. Steele-Perkins G, Plachez C, Butz KG, Yang G, Bachurski CJ, Kinsman SL, et al. The transcription factor gene Nfib is essential for both lung maturation and brain development. Mol Cell Biol. (2005) 25:685–98. doi: 10.1128/mcb.25.2.685-698.2005
125. Maksour S, Ooi L, and Dottori M. More than a corepressor: the role of coREST proteins in neurodevelopment. eNeuro. (2020) 7. doi: 10.1523/eneuro.0337-19.2020
126. Lunyak VV, Burgess R, Prefontaine GG, Nelson C, Sze SH, Chenoweth J, et al. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science. (2002) 298:1747–52. doi: 10.1126/science.1076469
127. Fuks F, Hurd PJ, Wolf D, Nan X, Bird A, and Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. (2003) 278:4035–40. doi: 10.1074/jbc.M210256200
128. Denslow SA and Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene. (2007) 26:5433–8. doi: 10.1038/sj.onc.1210611
129. Li R, Zhang H, Yu W, Chen Y, Gui B, Liang J, et al. ZIP: a novel transcription repressor, represses EGFR oncogene and suppresses breast carcinogenesis. EMBO J. (2009) 28:2763–76. doi: 10.1038/emboj.2009.211
130. Li L, Liu X, He L, Yang J, Pei F, Li W, et al. ZNF516 suppresses EGFR by targeting the CtBP/LSD1/CoREST complex to chromatin. Nat Commun. (2017) 8:691. doi: 10.1038/s41467-017-00702-5
131. Choi Y, Borghesani PR, Chan JA, and Segal RA. Migration from a mitogenic niche promotes cell-cycle exit. J Neurosci. (2005) 25:10437–45. doi: 10.1523/jneurosci.1559-05.2005
132. Jin Y, Zeng SX, Dai MS, Yang XJ, and Lu H. MDM2 inhibits PCAF (p300/CREB-binding protein-associated factor)-mediated p53 acetylation. J Biol Chem. (2002) 277:30838–43. doi: 10.1074/jbc.M204078200
133. Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, et al. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. (2001) 20:1331–40. doi: 10.1093/emboj/20.6.1331
134. Tamayo-Orrego L, Wu CL, Bouchard N, Khedher A, Swikert SM, Remke M, et al. Evasion of cell senescence leads to medulloblastoma progression. Cell Rep. (2016) 14:2925–37. doi: 10.1016/j.celrep.2016.02.061
135. Shiraishi R and Kawauchi D. Epigenetic regulation in medulloblastoma pathogenesis revealed by genetically engineered mouse models. Cancer Sci. (2021) 112:2948–57. doi: 10.1111/cas.14990
136. Kool M, Jones DT, Jäger N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. (2014) 25:393–405. doi: 10.1016/j.ccr.2014.02.004
137. Perla A, Fratini L, Cardoso PS, Nör C, Brunetto AT, Brunetto AL, et al. Histone deacetylase inhibitors in pediatric brain cancers: biological activities and therapeutic potential. Front Cell Dev Biol. (2020) 8:546. doi: 10.3389/fcell.2020.00546
138. Roux A, Pallud J, Saffroy R, Edjlali-Goujon M, Debily MA, Boddaert N, et al. High-grade gliomas in adolescents and young adults highlight histomolecular differences from their adult and pediatric counterparts. Neuro Oncol. (2020) 22:1190–202. doi: 10.1093/neuonc/noaa024
139. Lavery WJ, Barski A, Wiley S, Schorry EK, and Lindsley AW. KMT2C/D COMPASS complex-associated diseases [K(CD)COM-ADs]: an emerging class of congenital regulopathies. Clin Epigenet. (2020) 12:10. doi: 10.1186/s13148-019-0802-2
140. Pun M, Pratt D, Nano PR, Joshi PK, Jiang L, Englinger B, et al. Common molecular features of H3K27M DMGs and PFA ependymomas map to hindbrain developmental pathways. Acta Neuropathol Commun. (2023) 11:25. doi: 10.1186/s40478-023-01514-z
141. Han S, Liu Y, Cai SJ, Qian M, Ding J, Larion M, et al. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br J Cancer. (2020) 122:1580–9. doi: 10.1038/s41416-020-0814-x
142. Yi J, Kim B, Shi X, Zhan X, Lu QR, Xuan Z, et al. PRC2 heterogeneity drives tumor growth in medulloblastoma. Cancer Res. (2022) 82:2874–86. doi: 10.1158/0008-5472.Can-21-4313
143. Jacobs JJ, Kieboom K, Marino S, DePinho RA, and van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. (1999) 397:164–8. doi: 10.1038/16476
144. Glinsky GV, Berezovska O, and Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. (2005) 115:1503–21. doi: 10.1172/jci23412
145. Chen HC, He, McDonald M, Williamson MR, Varadharajan S, Lozzi B, et al. Histone serotonylation regulates ependymoma tumorigenesis. Nature. (2024) 632:903–10. doi: 10.1038/s41586-024-07751-z
146. Mohammad F, Weissmann S, Leblanc B, Pandey D, Højfeldt JW, Comet I, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. (2017) 23:483–92. doi: 10.1038/nm.4293
147. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature. (2018) 555:469–74. doi: 10.1038/nature26000
148. Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. (2006) 439:871–4. doi: 10.1038/nature04431
149. Graf M, Interlandi M, Moreno N, Holdhof D, Göbel C, Melcher V, et al. Single-cell transcriptomics identifies potential cells of origin of MYC rhabdoid tumors. Nat Commun. (2022) 13:1544. doi: 10.1038/s41467-022-29152-4
150. Ly P, Brunner SF, Shoshani O, Kim DH, Lan W, Pyntikova T, et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat Genet. (2019) 51:705–15. doi: 10.1038/s41588-019-0360-8
151. Gu W, Zhang F, and Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. (2008) 1:4. doi: 10.1186/1755-8417-1-4
152. Krupina K, Goginashvili A, and Cleveland DW. Scrambling the genome in cancer: causes and consequences of complex chromosome rearrangements. Nat Rev Genet. (2024) 25:196–210. doi: 10.1038/s41576-023-00663-0
153. Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature. (2020) 578:112–21. doi: 10.1038/s41586-019-1913-9
154. Chen F, Zhang Y, Shen L, and Creighton CJ. The DNA methylome of pediatric brain tumors appears shaped by structural variation and predicts survival. Nat Commun. (2024) 15:6775. doi: 10.1038/s41467-024-51276-y
155. Luo Z, Xia M, Shi W, Zhao C, Wang J, Xin D, et al. Human fetal cerebellar cell atlas informs medulloblastoma origin and oncogenesis. Nature. (2022) 612:787–94. doi: 10.1038/s41586-022-05487-2
156. Saba R, Johnson JE, and Saito T. Commissural neuron identity is specified by a homeodomain protein, Mbh1, that is directly downstream of Math1. Development. (2005) 132:2147–55. doi: 10.1242/dev.01781
157. Kvon EZ. Using transgenic reporter assays to functionally characterize enhancers in animals. Genomics. (2015) 106:185–92. doi: 10.1016/j.ygeno.2015.06.007
158. Bakaric A, Cironi L, Praz V, Sanalkumar R, Broye LC, Favre-Bulle K, et al. CIC-DUX4 chromatin profiling reveals new epigenetic dependencies and actionable therapeutic targets in CIC-rearranged sarcomas. Cancers (Basel). (2024) 16. doi: 10.3390/cancers16020457
159. Dai Q, Ren A, Westholm JO, Serganov AA, Patel DJ, and Lai EC. The BEN domain is a novel sequence-specific DNA-binding domain conserved in neural transcriptional repressors. Genes Dev. (2013) 27:602–14. doi: 10.1101/gad.213314.113
160. Torre M, Meredith DM, Dubuc A, Solomon DA, Perry A, Vasudevaraja V, et al. Recurrent EP300-BCOR fusions in pediatric gliomas with distinct clinicopathologic features. J Neuropathol Exp Neurol. (2019) 78:305–14. doi: 10.1093/jnen/nlz011
161. Lange JT, Rose JC, Chen CY, Pichugin Y, Xie L, Tang J, et al. The evolutionary dynamics of extrachromosomal DNA in human cancers. Nat Genet. (2022) 54:1527–33. doi: 10.1038/s41588-022-01177-x
162. Bailey C, Pich O, Thol K, Watkins TBK, Luebeck J, Rowan A, et al. Origins and impact of extrachromosomal DNA. Nature. (2024) 635:193–200. doi: 10.1038/s41586-024-08107-3
163. Morel F, Bris MJ, Herry A, Calvez GL, Marion V, Abgrall JF, et al. Double minutes containing amplified bcr-abl fusion gene in a case of chronic myeloid leukemia treated by imatinib. Eur J Haematol. (2003) 70:235–9. doi: 10.1034/j.1600-0609.2003.00046.x
164. Barr FG, Nauta LE, Davis RJ, Schäfer BW, Nycum LM, and Biegel JA. In vivo amplification of the PAX3-FKHR and PAX7-FKHR fusion genes in alveolar rhabdomyosarcoma. Hum Mol Genet. (1996) 5:15–21. doi: 10.1093/hmg/5.1.15
165. Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature. (2019) 575:699–703. doi: 10.1038/s41586-019-1763-5
166. Helmsauer K, Valieva ME, Ali S, Chamorro González R, Schöpflin R, Röefzaad C, et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat Commun. (2020) 11:5823. doi: 10.1038/s41467-020-19452-y
167. Morton AR, Dogan-Artun N, Faber ZJ, MacLeod G, Bartels CF, Piazza MS, et al. Functional enhancers shape extrachromosomal oncogene amplifications. Cell. (2019) 179:1330–1341.e13. doi: 10.1016/j.cell.2019.10.039
168. Zhu Y, Gujar AD, Wong CH, Tjong H, Ngan CY, Gong L, et al. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell. (2021) 39:694–707.e7. doi: 10.1016/j.ccell.2021.03.006
169. Pang J, Nguyen N, Luebeck J, Ball L, Finegersh A, Ren S, et al. Extrachromosomal DNA in HPV-mediated oropharyngeal cancer drives diverse oncogene transcription. Clin Cancer Res. (2021) 27:6772–86. doi: 10.1158/1078-0432.Ccr-21-2484
170. Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. (2012) 488:49–56. doi: 10.1038/nature11327
171. Hung KL, Mischel PS, and Chang HY. Gene regulation on extrachromosomal DNA. Nat Struct Mol Biol. (2022) 29:736–44. doi: 10.1038/s41594-022-00806-7
172. Hellwig M, Merk DJ, Lutz B, and Schüller U. Preferential sensitivity to HDAC inhibitors in tumors with CREBBP mutation. Cancer Gene Ther. (2020) 27:294–300. doi: 10.1038/s41417-019-0099-5
173. Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res. (2014) 20:912–25. doi: 10.1158/1078-0432.Ccr-13-2281
174. Kling MJ, Kesherwani V, Mishra NK, Alexander G, McIntyre EM, Ray S, et al. A novel dual epigenetic approach targeting BET proteins and HDACs in Group 3 (MYC-driven) Medulloblastoma. J Exp Clin Cancer Res. (2022) 41:321. doi: 10.1186/s13046-022-02530-y
175. Tang Y, Gholamin S, Schubert S, Willardson MI, Lee A, Bandopadhayay, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. (2014) 20:732–40. doi: 10.1038/nm.3613
176. Henssen A, Althoff K, Odersky A, Beckers A, Koche R, Speleman F, et al. Targeting MYCN-driven transcription by BET-bromodomain inhibition. Clin Cancer Res. (2016) 22:2470–81. doi: 10.1158/1078-0432.Ccr-15-1449
177. Yang Y, Valdés-Rives SA, Liu Q, Gao T, Burudpakdee C, Li Y, et al. Thyroid hormone suppresses medulloblastoma progression through promoting terminal differentiation of tumor cells. Cancer Cell. (2024) 42:1434–1449.e5. doi: 10.1016/j.ccell.2024.07.008
178. Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. (2012) 149:214–31. doi: 10.1016/j.cell.2012.02.013
179. Donati B, Lorenzini E, and Ciarrocchi A. BRD4 and Cancer: going beyond transcriptional regulation. Mol Cancer. (2018) 17:164. doi: 10.1186/s12943-018-0915-9
180. Groves A and Cooney TM. Epigenetic programming of pediatric high-grade glioma: Pushing beyond proof of concept to clinical benefit. Front Cell Dev Biol. (2022) 10:1089898. doi: 10.3389/fcell.2022.1089898
181. Stitzlein LM, Baig MU, Chandra J, McGovern S, Paulino A, Ketonen LM, et al. Phase I study of vorinostat and temsirolimus in newly diagnosed or progressive diffuse intrinsic pontine glioma. Pediatr Blood Cancer. (2025) 72:e31619. doi: 10.1002/pbc.31619
182. Mueller T, Stucklin ASG, Postlmayr A, Metzger S, Gerber N, Kline C, et al. Advances in targeted therapies for pediatric brain tumors. Curr Treat Options Neurol. (2020) 22. doi: 10.1007/s11940-020-00651-3
183. Geethadevi A and Raabe EH. Approaches for prevention of tumors in patients with rhabdoid tumor predisposition syndrome. Neurooncol Adv. (2024) 6:vdae158. doi: 10.1093/noajnl/vdae158
184. Duan R, Du W, and Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol. (2020) 13:104. doi: 10.1186/s13045-020-00937-8
185. Eckschlager T, Plch J, Stiborova M, and Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. (2017) 18. doi: 10.3390/ijms18071414
186. Mueller S, Kline C, Stoller S, Lundy S, Christopher L, Reddy AT, et al. PNOC015: Repeated convection-enhanced delivery of MTX110 (aqueous panobinostat) in children with newly diagnosed diffuse intrinsic pontine glioma. Neuro Oncol. (2023) 25:2074–86. doi: 10.1093/neuonc/noad105
187. Zhou Z, Singh R, and Souweidane MM. Convection-enhanced delivery for diffuse intrinsic pontine glioma treatment. Curr Neuropharmacol. (2017) 15:116–28. doi: 10.2174/1570159x14666160614093615
188. Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, Zhang L, et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell. (2017) 31:635–652.e6. doi: 10.1016/j.ccell.2017.03.011
189. Yang H, Cui W, and Wang L. Epigenetic synthetic lethality approaches in cancer therapy. Clin Epigenet. (2019) 11:136. doi: 10.1186/s13148-019-0734-x
190. Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered Malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U.S.A. (2013) 110:7922–7. doi: 10.1073/pnas.1303800110
191. Algar EM, Muscat A, Dagar V, Rickert C, Chow CW, Biegel JA, et al. Imprinted CDKN1C is a tumor suppressor in rhabdoid tumor and activated by restoration of SMARCB1 and histone deacetylase inhibitors. PloS One. (2009) 4:e4482. doi: 10.1371/journal.pone.0004482
192. Muscat A, Popovski D, Jayasekara WS, Rossello FJ, Ferguson M, Marini KD, et al. Low-dose histone deacetylase inhibitor treatment leads to tumor growth arrest and multi-lineage differentiation of Malignant rhabdoid tumors. Clin Cancer Res. (2016) 22:3560–70. doi: 10.1158/1078-0432.Ccr-15-2260
193. Ogiwara H, Sasaki M, Mitachi T, Oike T, Higuchi S, Tominaga Y, et al. Targeting p300 Addiction in CBP-Deficient Cancers Causes Synthetic Lethality by Apoptotic Cell Death due to Abrogation of MYC Expression. Cancer Discov. (2016) 6:430–45. doi: 10.1158/2159-8290.Cd-15-0754
194. Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, and Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. (2013) 10:977–9. doi: 10.1038/nmeth.2598
195. Laufer BI and Singh SM. Strategies for precision modulation of gene expression by epigenome editing: an overview. Epigenet Chromatin. (2015) 8:34. doi: 10.1186/s13072-015-0023-7
Keywords: pediatric brain tumor, epigenetics, chromatin remodeler, histone modification, genomic rearrangement, extrachromosomal DNA (ecDNA), fusion oncogene, DNA methylation
Citation: Kawata K, Chapman OS, Narumi S and Kawauchi D (2025) Epigenetic modifications and their roles in pediatric brain tumor formation: emerging insights from chromatin dysregulation. Front. Oncol. 15:1569548. doi: 10.3389/fonc.2025.1569548
Received: 01 February 2025; Accepted: 28 May 2025;
Published: 17 June 2025.
Edited by:
Vidya Gopalakrishnan, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Donghang Cheng, University of Texas MD Anderson Cancer Center, United StatesJiang Wu, University of Texas Southwestern Medical Center, United States
Copyright © 2025 Kawata, Chapman, Narumi and Kawauchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daisuke Kawauchi, a2F3YXVjaGlAbWVkLm5hZ295YS1jdS5hYy5qcA==