 Chinmay S. Sankhe
Chinmay S. Sankhe Lisa Hall
Lisa Hall Genevieve C. Kendall
Genevieve C. Kendall- 1Center for Childhood Cancer Research, The Abigail Wexner Research Institute, Nationwide Children’s Hospital, Columbus, OH, United States
- 2Division of Hematology and Oncology, Nationwide Children’s Hospital, Columbus, OH, United States
- 3Department of Pediatrics, The Ohio State University College of Medicine, Columbus, OH, United States
Rhabdomyosarcoma (RMS) contributes to 3% of all childhood cancers with roughly 400-500 cases diagnosed each year in the United States. The World Health Organization classifies rhabdomyosarcoma into four histological subtypes which include alveolar, embryonal, spindle-cell and pleomorphic. The primary genetic drivers in a subset of alveolar and spindle-cell histological subtypes are gene fusions. This review explores the fusion oncogenes identified in RMS such as PAX-and NCOA2-based fusions, along with discussing studies defining fusion oncogene biology and tumorigenic mechanisms. Focus areas include data around transformation events and progression along with dysregulated biological processes. Furthermore, we summarize model systems, ranging from cell to animal models, that have been implemented to study fusion oncogenes identified in RMS. With the constant identification of novel fusion oncogenes, this review also emphasizes the need for genetically characterizing RMS tumors and rapidly developing new model systems. These models are critical to study fusion oncogene activity and to delineate key regulatory players and potential therapeutic targets that suppress tumorigenesis. The identification of RMS fusion oncogenes and integration with animal and cell culture models will help identify conserved molecular targets, optimize therapeutic approaches, and ultimately improve clinical outcomes for children with RMS.
1 Introduction
Rhabdomyosarcoma (RMS) is a rare soft-tissue sarcoma that affects predominantly children but can also present in adulthood [reviewed in (1)]. RMS is associated with significant diagnostic and therapeutic challenges due to the diverse genetic drivers and varying disease aggressiveness. Understanding the biological mechanisms driving RMS subtypes will help identify therapeutic opportunities with the goal of improving clinical outcomes. The most aggressive forms of RMS are genetically driven by fusion oncogenes that engage different transcriptional signatures, which ultimately converges on tumors with molecular features of arrested skeletal muscle differentiation. Traditionally, fusion-positive RMS has focused on PAX-based fusion oncogenes of PAX3::FOXO1 and PAX7::FOXO1. However, new RMS fusion oncogenes are being rapidly identified from clinical sequencing efforts with little to no knowledge of their tumorigenic mechanisms. Here, fusion-positive refers to the PAX3/7::FOXO1 fusions and fusion driven refers to the collection of fusions found in RMS. In this review, we will discuss RMS fusion oncogenes and summarize studies of disease-driving mechanisms and functional targets. First, for context, we briefly describe clinical presentations of RMS. Next, we detail RMS fusion oncogenes and segregate the discussion based on if the fusion partners are PAX3/7 or NCOA2. We summarize key studies describing fusion oncogene function and how it contributes to RMS initiation and progression. Finally, we focus on approaches to functionally validate and model fusion oncogenes by describing experimental strategies and available genetic animal tumor models. Such integrated bench to bedside approaches are critically needed for fusion driven pediatric sarcoma patients. The goal of this review is to discuss progress and identify remaining challenges in our understanding of fusion oncogene-driven RMS, providing a basis for future studies to support identifying novel therapeutic targets.
1.1 Clinical presentation
RMS accounts for 50% of all pediatric soft-tissue sarcoma cases and the tumors express proteins such as myogenin (MyoG), desmin and MyoD that are indicative of immature skeletal muscle [reviewed in (1–3)]. RMS presents in soft tissues in body regions such as the head, neck, chest, bladder, prostate, arms, legs, and trunk, with specific subtypes having more common presentation sites. RMS typically metastasizes to the lung, bone marrow and lymph nodes (4). Roughly 400-500 new cases of RMS are diagnosed in the United States annually. Of these cases, 59% occur in children (<19 years) and 41% occur in adults (>19 years) (1, 2, 5). In children, more than 50% of cases are seen in the first decade of life (6). A subset of these patients will have a germline cancer predisposition syndrome such as Li-Fraumeni or DICER1 (7, 8). Five-year survival rates vary based on risk stratification, a combination of site, nodal involvement, metastases, age, surgical resection, and FOXO1 fusion presence (9). These risk stratification factors for RMS patients, including the presence of the PAX3/7::FOXO1 fusion, were also the basis of a clinical trial developed by the European pediatric soft tissue sarcoma study group (10). These rates decrease from 70-90% in low-risk pediatric groups to 20-30% in high risk. Adults have poorer outcomes compared to the pediatric groups, largely based on the presence of metastases at diagnosis. Adult 5 year overall survival is 20% (5, 11). Overall survival also varies with histological subtype (embryonal, alveolar, spindle-cell, and pleomorphic) (12). Current treatments for RMS include chemotherapy, radiation therapy, surgery, or multimodal therapy; the specific treatment plans are often guided by risk stratification and recently have included molecular diagnostics once available [reviewed in (1)]. There are no therapies that directly target the primary oncogenic drivers of the disease.
RMS has four histological subtypes, some of which are driven by gene fusions. Embryonal RMS is the most common subtype contributing to 60% of all RMS cases and also has the most favorable prognosis [reviewed in (13)]. Most embryonal RMS cases occur in children under 10 years of age and originate in the head and neck, prostate, urinary bladder, and abdomen; this includes tissues of the ear, tongue, and nasopharynx [reviewed in (2, 14)]. Embryonal RMS is not genetically fusion driven, but rare fusions have been described, including PAX3::NCOA1/2. Alveolar RMS is a more aggressive subtype of RMS that affects primarily teens and young adults; it contributes to roughly 20% of all RMS cases. This subtype is associated with unfavorable prognosis and commonly originates in regions such as limbs, trunks, head, and neck [reviewed in (2, 15)]. The most common genetic drivers are PAX3/7::FOXO1. Spindle-cell or sclerosing RMS is a rare RMS subtype that accounts for 10% of all RMS cases and has a broader presentation spectrum. It affects both children (and infants) and adults, with a more favorable prognosis in children. This subtype typically presents in the head, neck, and brain region (16, 17). In infants, spindle cell RMS is typically driven by NCOA2 fusions. Pediatric and adult cases of spindle cell RMS are also driven by a collection of fusion oncogenes, a subset which is detailed in Figure 1. Pleomorphic RMS is an aggressive RMS subtype that often metastasizes to distant tissues. It is associated with 10% of all RMS cases and occurs in adults with an age group spanning from the early twenties to 80. This subtype is associated with unfavorable prognosis and is found in the chest, abdomen, shoulder, and lower extremities (18–21), and is not known to be driven by gene fusions. Understanding the genetics of the disease, especially the presence or absence of gene fusions, guides clinical treatment plans (among other factors) and has prognostic importance. Here, we will focus on the fusion-positive RMS and fusion driven spindle cell/sclerosing RMS.
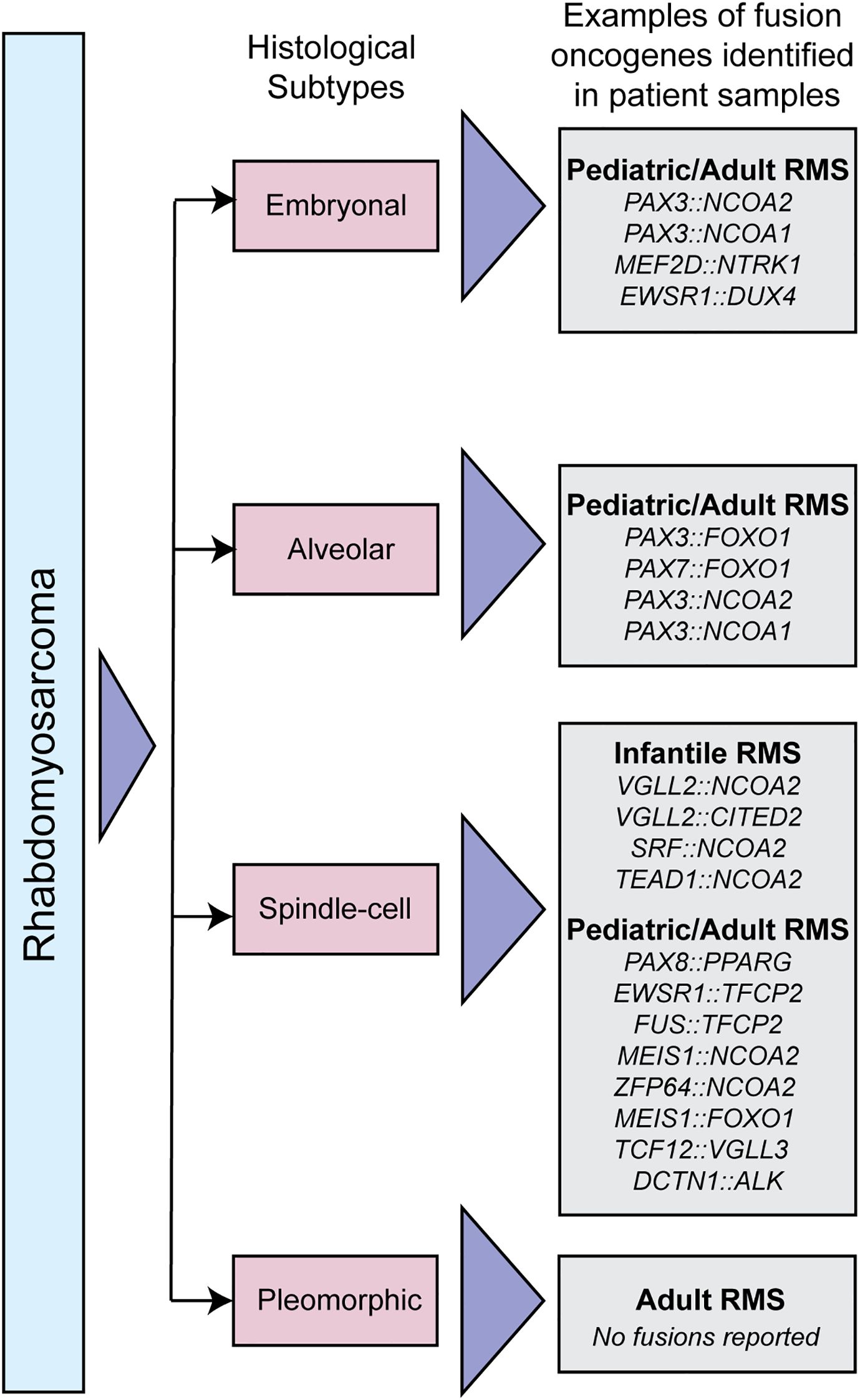
Figure 1. Classification of RMS subtypes. Schematic depicting different histological RMS subtypes, and examples of fusion oncogenes in each subtype that have been identified in patient samples.
2 PAX-based fusion oncogenes
Approximately 80% of alveolar RMS are genetically driven by a paired box (PAX) gene-associated chromosomal translocations (22–24). In 60% of alveolar RMS cases, the PAX3 DNA-binding domain on chromosome 2 is fused to the forkhead box O1 (FOXO1) transactivation domain on chromosome 13 (referred to as PAX3::FOXO1). In 20% of alveolar RMS cases, the PAX7 gene on chromosome 1 is fused to the FOXO1 gene on chromosome 13 (referred to as PAX7::FOXO1) (2, 24–26). In 20% of alveolar RMS cases, there is no fusion oncogene that has been detected to date. This subset is indistinguishable from embryonal RMS with no fusion oncogenes in terms of gene expression signatures and clinical outcomes (27).
Normally, PAX3/PAX7 and FOXO1 both act as transcription factors with critical roles in neural crest specification and differentiation, skeletal muscle development, and in cell proliferation processes. PAX3 directly regulates the transcriptional activity of MYF5, MYOD1, and FGFR4, which play a role in skeletal muscle development. It also regulates MITF, TYRP1, RET, TBX2, NGN2, HES1, and NRCAM, which contribute to neural development. Additionally, PAX3 plays a role in chromatin structure regulation (28). PAX7 is a myogenic transcription factor important for renewal and maintenance of satellite cells seen in postnatal skeletal muscles. Pax7 deficient mice were significantly smaller than wildtype and had reduced muscle mass indicating that knockout of Pax7 inhibited satellite cell-mediated growth of skeletal muscle (29, 30). Importantly, PAX3 and PAX7 are in-part functionally redundant but not completely. In a study investigating the functional roles of Pax3 and Pax7 in limb muscle development, the Pax3 gene was replaced by Pax7 in mice. Pax7 was able to compensate for the loss of Pax3 for neural tube closure, neural crest cell development and migration; however, these mice had a loss of forelimb muscle development (31). FOXO1 is a member of the forkhead box protein O family of transcription factors and is expressed in most muscle types. It regulates muscle regulatory roles of growth, glucose metabolism, and differentiation (32). Subcellular localization of FOXO1 is crucial for regulating myogenic differentiation; nuclear export of FOXO1 is especially important for early skeletal muscle differentiation (33). One hypothesis is that the fusion oncogenes retain the activity of their normal fusion partners.
The PAX3/7::FOXO1 fusions juxtapose the PAX3 or PAX7 5’ DNA binding paired box and paired-type homeodomains to the FOXO1 3’ transactivation domain (Figure 2). PAX3::FOXO1 and PAX7::FOXO1 fusions likely function differently. This is supported from both clinical and experimental data. Clinically, PAX3::FOXO1 RMS patients have reduced overall survival compared to PAX7::FOXO1 patients (22, 24, 41–43). Further, genetic animal model studies show PAX3 and PAX7 are not functionally redundant or fully compensatory (31), and a direct comparison of PAX3::FOXO1 and PAX7::FOXO1 activity highlights key differences, suggesting shared and divergent functions (44). In patient tumors, PAX7::FOXO1 often has copy number amplification which is not true for PAX3::FOXO1 (45). One interesting note is that molecularly, PAX3::FOXO1 patient tumors have enhanced staining for proliferation marker MIB1, along with higher apoptosis, as compared to PAX7::FOXO1 tumors (46). In the subsequent sections, we summarize the studies examining the neomorphic functions of PAX3::FOXO1, which is more prevalent, aggressive, and more studied than PAX7::FOXO1.
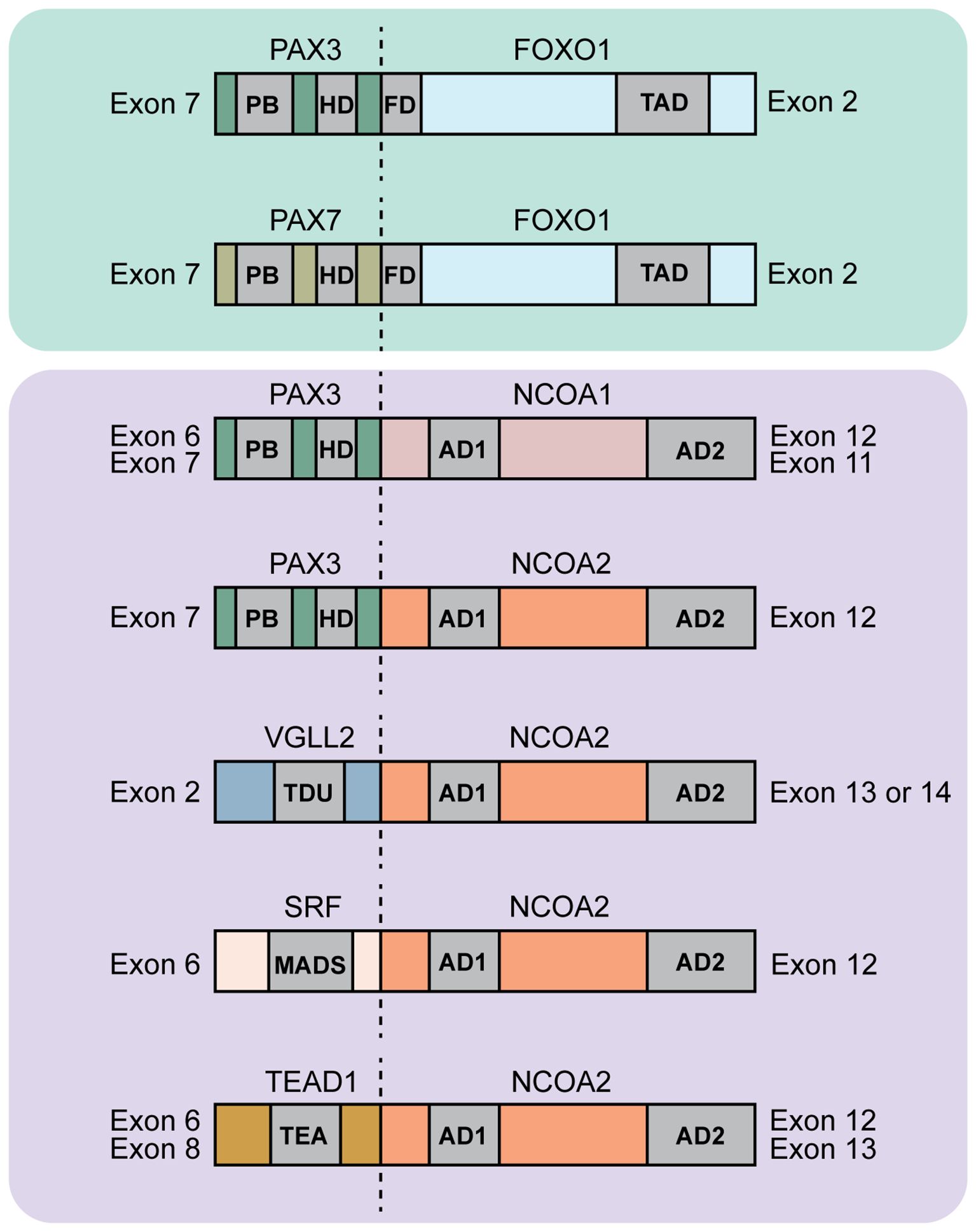
Figure 2. Diagram of protein domains found in rhabdomyosarcoma fusions. Shown are a subset of FOXO1 and NCOA2 fusions. The dotted lines indicate the fusion junction. For the PAX-based fusion oncogenes the abbreviations indicate the following: Paired box gene 3/7 (PAX3/7), forkhead box O1 (FOXO1), PB: paired box domain, HD: homeodomain, FD: forkhead domain, TAD: Transactivation domain. PAX3/PAX7::FOXO1 fuses exon 7 of PAX3/7 to exons 2-3 of FOXO1 [ref (34, 35)]. For the NCOA-based fusion oncogenes the abbreviations indicate the following: Paired box gene 3 (PAX3), nuclear receptor coactivator 1/2 (NCOA1/2), AD1, AD2: activating domains 1 and 2 of NCOA genes. PAX3::NCOA1 fuses either exons 1-6 (type 1) or exons 1-7 (type 2) of PAX3 to exons 12-20 (type 1) or exons 11-20 (type 2) respectively of NCOA1. Type 1 has 894 amino acids with 319 amino acids of PAX3 and 575 amino acids of NCOA1. Type 2 has 1026 amino acids with 391 amino acids of PAX3 and 635 amino acids of NCOA1. PAX3::NCOA2 fuses exons 1-7 of PAX3 to exons 12-23 of NCOA2. This fusion protein is 1057 amino acid long with 391 amino acids of PAX3 and 666 amino acids of NCOA2 [ref (36)]. For the VGLL2::NCOA2 fusion oncogene the abbreviations indicate the following: Vestigial-like family member 2 (VGLL2), TDU: Tondu domain. VGLL2::NCOA2 fuses exons 1-2 of VGLL2 to exon 13 or 14-23 of NCOA2 [ref (37, 38)]. For the SRF::NCOA2 fusion oncogene, abbreviations indicate the following: Serum response factor (SRF), MADS: MCM1, AGAMOUS, DEFICIENS, and SRF domain. SRF::NCOA2 fuses exon 6 of SRF to exon 12 of NCOA2 [ref (39)]. For the TEAD1::NCOA2 fusion oncogene, abbreviations indicate the following: TEA domain transcription factor 1 (TEAD1) also called transcriptional enhancer factor (TEF1). TEAD1::NCOA2 fuses either exon 8 (type 1) or exon 6 (type 2) of TEAD1 to exon 13 (type 1) or exon 12 (type 2) respectively of NCOA2 [ref (39, 40)].
2.1 PAX3::FOXO1 activity in cellular contexts
PAX3::FOXO1 has been studied in diverse contexts with functions ranging from transcriptional to epigenetic. In NIH3T3 cells, transient transfection of PAX3::FOXO1 and PAX3 identified that PAX3::FOXO1 is a more potent transcriptional activator than PAX3 (47). This suggests PAX3::FOXO1 can lead to enhanced or persistent activation of normal PAX3 target genes. In human SaOS-2 and U2-OS osteosarcoma cells, ectopic PAX3 expression initiated mesenchymal-epithelial transition, yet PAX3::FOXO1 expression had an even more pronounced effect (48). This study also identified distinct PAX3::FOXO1 gene signatures compared to PAX3, suggesting that not all targets overlap with PAX3 and that PAX3::FOXO1 has neomorphic activity. A study using alveolar RMS cells (RH30 and RH4) found that PAX3::FOXO1 can inhibit the activity of the endogenous FOXO1 transcription factor, as cells transfected with siRNA inhibiting PAX3::FOXO1 had an increase in FOXO1 protein levels. Knocking down PAX3::FOXO1 in human RMS cells resulted in decreased cell proliferation, lower motility, increased protein levels of Desmin and Myosin Heavy Chain (MHC), down-regulation of mRNA levels of MYOD1, MYF5, MRF4, and increased myogenin protein levels, highlighting the importance of PAX3::FOXO1 in tumor maintenance and in suppressing myogenic differentiation (49).
In fibroblast and epithelial-like cells, studies have supported varying roles of PAX3::FOXO1 in regulating proliferation and provided a platform for comparing PAX3/7::FOXO1 activity. Transduction of retroviral PAX3::FOXO1 into chicken embryo fibroblasts increased growth and colony formation in soft agar (50). In a separate study, PAX3::FOXO1 expression in mouse fibroblasts supported a transformed phenotype with increased contractility and anchorage-independent growth (51). The level of PAX3::FOXO1 protein can also induce discrete phenotypes, with low PAX3::FOXO1 expression in mouse fibroblasts resulting in transformation, while high expression of PAX3::FOXO1 supported growth inhibition (52). In human foreskin fibroblasts, PAX3::FOXO1 and PAX7::FOXO1 expression had discrete transcriptomic landscapes with higher deposition of H3K27 acetylation in the case of PAX7::FOXO1 binding compared to PAX3::FOXO1 (44). A recent publication reports a dual inducible model system to study PAX3::FOXO1 and HES3 genetic cooperation using HEK293T cells. Cells expressing only PAX3::FOXO1 formed fewer spheres compared to dual-induced cells with both PAX3::FOXO1 and HES3 expression. This supports that HES3 promotes PAX3::FOXO1 transformation in vitro (53).
PAX3::FOXO1’s oncogenic activity have also been investigated in mesenchymal stem cells and skeletal muscle myoblast cells, which have been proposed as a potential RMS cell(s) of origin. In mouse mesenchymal stem cells (MSCs), expression of PAX3::FOXO1 and PAX7::FOXO1 resulted in enhanced growth and alveolar RMS tumors in mouse allografts with cooperating mutations. Required cooperating mutations include activated RAS, tp53 mutation and expression of SV40 early region (54). C2C12 mouse myoblast cells can tolerate PAX3::FOXO1 expression, and exhibit increased proliferation and inhibition of myogenic differentiation (55). In primary human myoblasts, PAX3::FOXO1 expression enhanced proliferation and drove cell growth past the senescence stage. This increase in cell proliferation was accompanied by a reduction in protein levels of tumor suppressor CDKN2A (p16INK4A) (56). PAX3::FOXO1 can also act as a pioneer factor as shown biochemically in RMS cell lines and genomically in a human inducible myoblast model. PAX3::FOXO1 binds to regions of closed chromatin in addition to accessible chromatin, allowing it to control distinct cell-fate decisions (57). PAX3::FOXO1 pioneering activity and functional consequences were also shown with an in vivo embryonic zebrafish model. These studies identified that PAX3::FOXO1 utilizes partial homeobox motif recognition to bind to closed chromatin. Whereas, binding to its composite paired-box/homeobox motif resulted in increased chromatin accessibility and activation of neural gene signatures (58).
In immortalized human myoblast cells, PAX3::FOXO1 expression promoted proliferation, inhibited myogenic differentiation, and formed xenografts in SCID mice, albeit slow growing. PAX3::FOXO1 activity was augmented by MYCN overexpression, and xenografting these cells resulted in a faster tumor growth rate than PAX3::FOXO1 alone (59). In an inducible PAX3::FOXO1 human myoblast system, the fusion-oncogene cooperated with MYCN to inhibit differentiation and promote proliferation during transformation; however, in this context, PAX3::FOXO1 is not required for tumor recurrence (60). In a follow-up study, PAX3::FOXO1 genetically cooperates with transcriptional target, FGF8 (with constitutive MYCN expression), to enhance cell proliferation and tumor growth (61).
Taken together, these studies suggest that PAX3::FOXO1 has diverse regulatory functions depending on the cell type, thereby highlighting the need to understand the potential RMS cell(s) of origin and how PAX3::FOXO1 might function in those contexts.
2.2 PAX3::FOXO1 targets
PAX3::FOXO1 binds and activates target genes that make up its core regulatory circuitry (62). Furthermore, studies support that PAX3::FOXO1 utilizes super enhancers such as FOXO1 cis-regulatory domains in association with other transcription factors of MYCN, MYOD and myogenin to inhibit skeletal muscle differentiation (63, 64). This results in locking RMS cells in an undifferentiated myogenic-like stage that is highly proliferative. PAX3::FOXO1-target genes likely have diverse pro-tumorigenic functions such as stimulating proliferation and invasion, inhibiting differentiation, and promoting cancer cell survival by repressing tumor suppressors. Understanding PAX3::FOXO1 targets and their functional requirements for the disease could represent potential therapeutic opportunities. Table 1 outlines key genes that are upregulated by ectopic PAX3::FOXO1 expression.
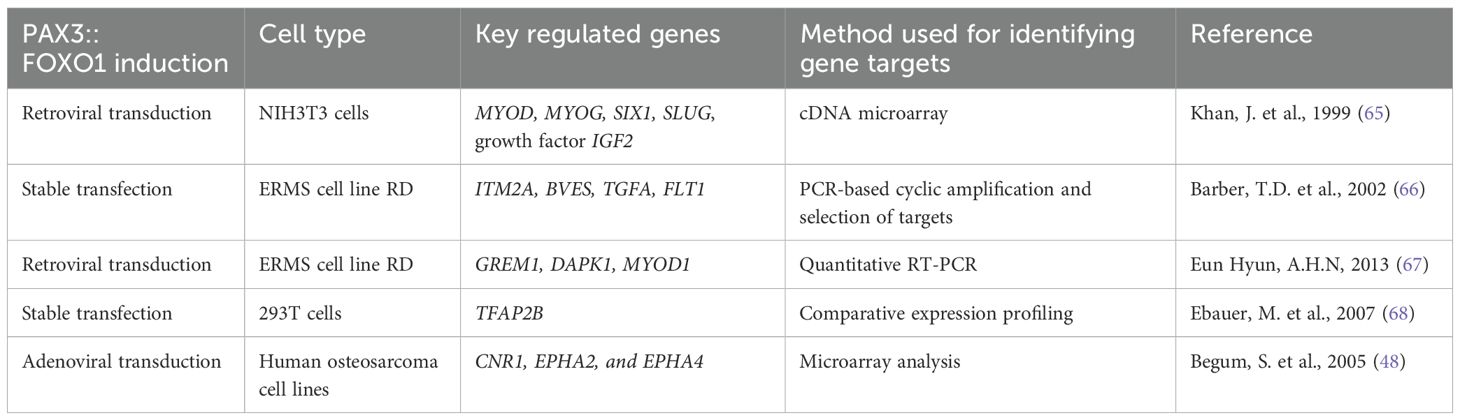
Table 1. Key differentially regulated genes following ectopic PAX3::FOXO1 expression in cells.
Different cellular contexts modify PAX3::FOXO1 activity. For example, in NIH3T3 cells, transduction of PAX3::FOXO1 upregulated gene expression of MYOD, MYOG, SIX1, SLUG, and growth factor IGF2 (65). Given the toxicity of the fusion, another strategy is introducing PAX3::FOXO1 into an embryonal or fusion-negative RMS cell line. This approach identified activation of genes such as ITM2A, BVES, TGFA, and FLT1, shared gene targets of the PAX3 gene (66). Viral transduction of PAX3::FOXO1 into embryonal RMS cells identified direct targets including tumor suppressor gremlin 1 (GREM1), death-associated protein kinase-1 (DAPK1), and MYOD1 (67). A parallel study using inducible PAX3::FOXO1 expressed in human fusion-negative embryonal cell line RD confirmed this finding along with identifying target genes involved in apoptosis, development, and signal transduction (69). Comparative expression profiling was utilized to determine PAX3::FOXO1 specific genes, and TFAP2B was identified as the direct target regulating PAX3::FOXO1’s anti-apoptotic roles (68). Examining the global gene signatures of alveolar and embryonal RMS resulted in the identification of genes that are uniquely overexpressed in alveolar RMS: cannabinoid receptor 1 (CNR1), PIPOX, DCX, ABAT, JAKMIP2, NRCAM, DKFZp762M127, and FOXF1 (70). Using chromatin immunoprecipitation (ChIP), this study found that PAX3::FOXO1 directly binds to the promoter of CNR1, EPHA2, and EPHA4, thereby, regulating their expression (48). Another study implemented ChIP sequencing on RMS PAX3::FOXO1-positive cell lines and fusion-negative cells transfected with PAX3::FOXO1 and found that the binding sites of PAX3::FOXO1 are majorly located distal to the transcription start sites and highly correlated with genes upregulated in PAX3::FOXO1-positive cells. This study also identified genes FGFR4 and IGF1R to be direct targets of PAX3::FOXO1 (71). In addition, ChIP sequencing on RH4 RMS cells revealed PAX3::FOXO1 genome-wide binding patterns were different than core regulatory transcription factors of MYCN, MYOG, MYOD1, and that PAX3::FOXO1 binding sites were adjacent to repressive histone marks H3K9me3 and H3K27me3 (57). These data suggest that not all the PAX3::FOXO1 ChIP binding sites are conserved across cell lines, indicating different RMS tumors might have unique biology.
PAX3::FOXO1 plays a dual role of regulating induction and inhibition of myogenesis. NIH3T3 cells transduced with PAX3::FOXO1 had elevated MyoD and myogenin protein expression; however, the cells did not differentiate or form myotubes, suggesting that PAX3::FOXO1 inhibited complete myogenic differentiation. This inhibition of myogenic differentiation was mediated by fibroblast growth factor receptors (FGFRs) (72). In another study examining PAX3/7::FOXO1’s role in myogenesis, expression of PAX3/7::FOXO1 inhibited terminal differentiation and suppressed the MyoD-target gene, MyoG. However, PAX3/7::FOXO1 did not affect MyoD transcriptional activity or binding at the MyoG promoter. Instead, PAX3/7::FOXO1 decreased histone H4 acetylation and reduced RNA polymerase II binding at the MyoG promoter, indirectly impacting MyoD’s normal function (73). In a study investigating genetic cooperation of PAX3::FOXO1 and the cannabinoid receptor 1 (CNR1) in a panel of RMS cell lines, CNR1 mRNA expression levels were only upregulated in alveolar RMS cells. CNR1 did not contribute to the PAX3::FOXO1 cell proliferation and differentiation, but it was essential for regulating PAX3::FOXO1-induced invasion and metastasis, suggesting that CNR1 could be considered a therapeutic opportunity (74).
PAX3::FOXO1 also has epigenetic targets that support its oncogenic activity. For example, jumonji and AT-rich interaction domain-containing 2 (JARID2) gene levels are increased in PAX3::FOXO1 fusion-positive RMS patient tumors, with JARID2 expression being dependent on PAX3::FOXO1. JARID2 knockdown decreased cell proliferation, increased cell elongation, and activated protein expression of myogenin and myosin light chain, both differentiation markers. Mechanistically, JARID2 bound the promoter region of myogenin and myosin light chain, and together with Polycomb Repressive Complex 2 (PRC2) complex, increased H3K27Me3 deposition leading to their repression and maintenance of an undifferentiated cell state (75). KDM4B, a histone lysine demethylase, also plays a role in regulating PAX3::FOXO1 activity in cell culture models. Induction of PAX3::FOXO1 in two systems (an immortalized human myoblast and human fusion-negative embryonal RD cells) led to elevated levels of KDM4B, elongated morphology, and transformation. Depletion of KDM4B in RH30 and Rh41 resulted in reduced cell growth (76), decreased colony formation, and delayed tumor formation in xenografts (77). PAX3::FOXO1 and KDM4B form a complex and regulate the expression of MYOD1 (77). In a recently published study, another histone lysine demethylase, KDM3B, was shown to be crucial for the tumorigenic activity of PAX3::FOXO1. KDM3B suppression inhibits PAX3::FOXO1 activity, with the readouts being cell growth in vitro and in vivo (78).
These data suggest that PAX3::FOXO1 acts as a transcriptional activator regulating genes involved in inhibiting terminal skeletal muscle differentiation, promoting proliferation and invasion, and driving RMS tumorigenesis. PAX3::FOXO1 targets such as IGF1R, CNR1, JARID2, KDM4B and KDM3B could be further explored as therapeutic approaches for PAX3::FOXO1-driven RMS.
3 NCOA2-based fusion oncogenes
3.1 PAX3::NCOA1/NCOA2 fusion oncogene structure
Nuclear receptor coactivator 2 (NCOA2) is a nuclear hormone receptor and a member of the p160 steroid nuclear receptor coactivator family (39, 79). The nuclear hormone receptor plays essential roles in various cellular processes such as cell growth, differentiation, inflammatory and metabolic pathways (80, 81). NCOA2 consists of basic helix-loop-helix and Per-Arnt-Sim domain (bHLH/PAS) receptor nuclear translocator domain along with transcriptional activation domains (AD) CID/AD1 and AD2 (82). NCOA2 and its close family member, NCOA1, are common fusion partners in cancer encompassing soft tissue sarcoma, prostate and breast cancer and acute myeloid leukemia as examples (83–88). PAX3::NCOA1 and PAX3::NCOA2 fusions are found in rhabdomyosarcoma. The resulting fusion protein consisted of the paired-box and DNA binding domain of PAX3 along with the CID/AD1 domain, the Q-rich region, and the AD2 domain of NCOA1 or NCOA2 (Figure 2). Normally, the NCOA2 CID/AD1 domains bind CBP/p300, whereas the AD2 domain binds with CARM1/PRTM1, among other interactions (82, 89, 90). Reverse transcriptase polymerase chain reaction (RT-PCR) analysis of patient tumor samples revealed two different PAX3::NCOA1 translocations with fusion of the PAX3 exon 7 to NCOA1 exon 11 and fusion of the PAX3 exon 6 to NCOA1 exon 12 (36). The PAX3 exon 6 fusion to NCOA1 exon 12 was also identified by an earlier study by Wachtel M. et al. (91). In the case of PAX3::NCOA2, the fusion incorporates the PAX3 exons 1-7 and NCOA2 exons 12-23, both of which contain the protein domains previously highlighted.
Functional studies of these PAX3::NCOA1/2 fusion oncogenes have been performed in fibroblast and myoblast cell culture systems. Transfection of NIH3T3 mouse fibroblast cells with the fusion oncogenes resulted in colony formation, whereas deletion of CID/AD1 and the AD2 domain of the NCOA proteins resulted in a significant decrease in colony formation capacity (36). Another study identified PAX3::NCOA2 in a patient tumor diagnosed with embryonal RMS. Transfection of PAX3::NCOA2 in C2C12 cells resulted in increased proliferation and higher motility as compared to the control-transfected cells; however, the increase in proliferation and motility was still less compared to cells transfected with PAX3::FOXO1. PAX3::NCOA2-transfected C2C12 cells also had reduced capacity for myogenic differentiation in culture. This study also showed that PAX3::NCOA2 expressing C2C12 cells were able to form allograft tumors in nude mice at a slower rate than PAX3::FOXO1 allograft tumors (92). These studies indicate that fusion oncogenes with one shared fusion partner can both be transforming with different capacity; suggesting, the 3’ fusion partner supports unique tumorigenic mechanisms.
3.2 Infantile/pediatric rhabdomyosarcoma NCOA2 fusion oncogene structure
Characterizing pediatric and adult spindle-cell RMS specimens via next-generation RNA sequencing led to identification of two NCOA2-based RMS fusion oncogenes (39). The first NCOA2 fusion was with the serum response factor (SRF) gene with exon 6 of SRF fused to the exon 12 of NCOA2 (Figure 2). SRF is a transcription factor that is highly expressed in skeletal muscle and regulates genes involved in muscle development and differentiation (93). The second NCOA2 fusion was with the TEA domain transcription factor 1 (TEAD1) with exon 8 of TEAD1 fused to the exon 13 of NCOA2 (Figure 2). An Archer Anchored Multiplex PCR analysis on a 16-month-old spindle-cell RMS patient identified exon 6 of TEAD1 fused with exon 12 of NCOA2 (40). TEAD1 is expressed in skeletal muscle and regulates genes involved in metabolism and developmental processes (94). In a follow-up study characterizing spindle-cell infantile/pediatric RMS tumor specimens, additional fusions were identified including VGLL2::NCOA2, VGLL2::CITED2, TEAD1::NCOA2, and SRF::NCOA2. Further analysis revealed that intron 3 of VGLL2 fused with exon 2 of CITED2 in four infantile/pediatric spindle-cell RMS cases, while exon 2 of VGLL2 fused with exon 14 of NCOA2 in two RMS cases (37, 95) (Figure 2). Normally, vestigial-like family member-2 (VGLL2) promotes skeletal muscle differentiation via translocating to the nucleus and interacting with the TEAD1 transcription factor (96). VGLL2 is also co-expressed with myogenin in differentiating muscle cells (97). Thus, the VGLL2::NCOA2 fusion might disrupt and co-opt the muscle differentiation cascade for tumorigenesis.
NCOA2 is a common fusion partner, especially in sarcoma. Table 2 summarizes a subset of the different NCOA2-based fusions that have been identified to date. Some fusions, like MEIS1::NCOA2, were identified in patients with spindle-cell RMS, intraosseous spindle-cell RMS, and spindle-cell sarcoma of the kidney in adults, highlighting that the same fusion can have different presentations (98–100). This is likely a collaborative process between fusion acquisition and cell of origin.
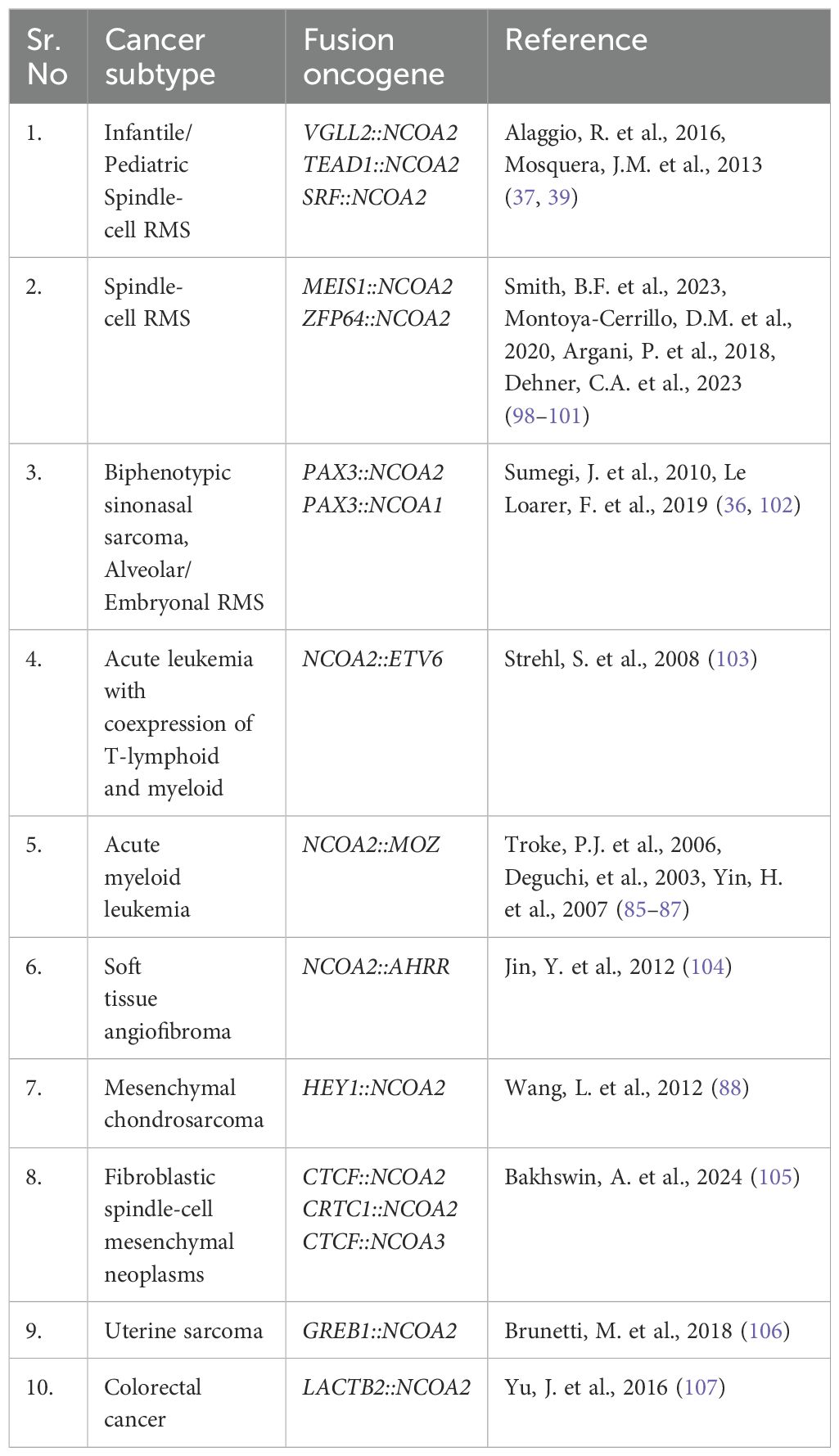
Table 2. NCOA2-based fusion oncogenes identified in different cancer subtypes.
4 Modeling fusion oncogene driven rhabdomyosarcoma
To understand fusion oncogene activity and guide targeted treatment, it is essential to functionally validate their activity using model systems including cell and animal models. Clinical sequencing efforts are rapidly identifying new fusion oncogenes in RMS with limited models to mechanistically study them. We have summarized some examples of the different fusion oncogenes found in RMS and some sarcoma subtypes in Table 3, many of which have no cell or animal models of the disease. This section will discuss the different types of model systems (cell and animal based) that are used for functional genomics of RMS-specific fusion oncogenes. Every model has its benefits and challenges, and the choice of model depends on the research question being asked. Additional factors to consider when deciding on a model include generation time, efficiency, relevance to human disease, cost, experience, and infrastructure and ease of experimental setup.
Table 3. Examples of fusion oncogenes in RMS and other sarcoma subtypes.
4.1 Cell culture and PDX models
4.1.1 PAX3::FOXO1 cell models
There are a variety of cell culture strategies and patient-derived cell lines used to study fusion oncogene-driven RMS [reviewed in (121)]. Patient-derived CW9019 harbors the PAX7::FOXO1 translocation (122) while cell lines such as KFR, RH10, RH30, and TC212 harbor the PAX3::FOXO1 fusion (121). Another RMS cell line, RUCH-2, was derived from a primary botryoid RMS patient with positive expression for protein markers of PAX3, MYF3 and MYF5, but lost expression of MYF3 and MYF5 after 3.5 months in culture. However, the cells became metastatic with increased invasiveness and had the ability to form faster tumors in nude mice. Thus, this cell line facilitated comparison between the primary and metastatic phase in RMS (123).
To complement patient cell lines, studies have also induced the expression of PAX3::FOXO1 in human immortalized myoblast cells to understand transformation and changes in proliferative state. Human skeletal muscle myoblast cells were used that had been immortalized with overexpression of hTERT and absence of Ink4a/ARF. In these myoblasts, PAX3::FOXO1 expression in combination with MYCN generated RMS-like tumors in SCID/beige mice, suggesting the role of these components in driving myoblast cells to a tumorigenic state (124). In several other studies, dual expression of PAX3::FOXO1 and MYCN in immortalized human myoblast cells has been shown to develop rapid tumors in SCID mice by maintaining the cells in a proliferative state and preventing cellular differentiation compared to PAX3::FOXO1 expression alone (59–61).
RMS 3D cell culture models have also been explored to better recapitulate an in vivo environment (125–127). In a RH28 and RH30 3D rhabdosphere model of PAX3::FOXO1-driven RMS, non-adhered spheres had increased expression of stemness markers such as SOX2, NANOG, and OCT4. The spheres could also generate tumors at a faster rate than adherent monolayer RH30 cells when injected into immunodeficient mice (125). RH30 cells have also been cultured on a collagen sponge inside a bioreactor system with perfusion flow leading to a 3D organotypic cell culture RMS model. The cells under perfusion flow exhibited higher proliferation with elevated levels of matrix metalloprotease 2 and invasive RMS protein marker of LAMA1/2 as compared to the static cultured cells (125, 127). These systems could represent cost-efficient strategies to complement in vivo animal modeling.
4.1.2 PAX3/7::FOXO1 single cell RNA sequencing in cell and PDX models
In the past few years, there have been multiple single cell RNA sequencing transcriptomic analyses that have aided in identifying muscle-specific lineages and cell subpopulations represented in RMS. A single-cell atlas of patient tumor samples, patient-derived cell lines and xenograft models revealed that RMS tumors consist of proliferative, apoptotic and differentiating cell subpopulations along with quiescent progenitor cells. This study also found that PAX3::FOXO1 and PAX7::FOXO1 tumors have an unique neuronal cell subpopulation expressing genes such as DCX, L1CAM, SYP and CHGA. Importantly, these same neural genes were activated in response to chemotherapy suggesting they have a role in therapeutic resistance (128). Another study on RMS patient-derived xenograft cultures and alveolar RMS cell lines found three different cell states. The first subpopulation resembled early myogenic muscle stem cell-like state with high expression of cell adhesion genes. The seconds subpopulation was proliferating cells with enrichment in cell division genes, and the third subpopulation was differentiated cells with expression of myotube and terminal differentiation genes (129).
Another single cell RNA sequencing study using RMS patient-derived xenografts found similar heterogeneity in RMS tumors with a differentiated cell subpopulation expressing muscle genes of MYLPF, ACTC1, LRNN1, TNNT3 and TSPAN33; progenitor cells expressing extracellular matrix genes of MMP2, and the majority of RMS cell states expressing MYOD and DES (130). On investigating the tumor microenvironment in RMS, this study found that the presence of differentiated macrophages in the M2 polarization state are linked with angiogenesis and suppression of inflammation. There was also a presence of dendritic and undifferentiated macrophages subpopulations in this tumor microenvironment, indicative of immune dysfunction (131).
Investigating primary embryonal and alveolar RMS patient tumor tissues using single cell RNA sequencing revealed that tumors exhibited subpopulations of paraxial mesoderm expressing MEOX2 and PAX3, myoblasts expressing MYF5 and MSC, and myocytes expressing MYOG and MEF2C. In addition, there were differences between alveolar RMS and embryonal RMS, where a greater percentage of tumor cells in alveolar RMS exhibited myocyte-like state expressing MYOG, and a lesser percentage exhibited paraxial mesoderm with MEOX2 expression (132). Thus, RMS-based tumors are heterogenous with multiple cell subpopulations contributing to tumor-related activities that can be aligned with developmental timepoints. Further, fusion-positive RMS is less adhered to the myogenic hierarchy and tumors and PDX samples express markers of neural subpopulations.
4.1.3 Rhabdomyosarcoma patient-derived xenograft and organoid models
Researchers have also developed models to better reflect the immune environment or use primary patient tumor samples. For example, a mouse xenograft system was established in an immunocompetent mouse to model immunotherapy-based approaches in RMS. This system integrated a humanized immune system into mice with a severe lack of innate immunity. To do this, human hematopoietic stem cells were transplanted into the mice followed by subcutaneously xenografting human RMS cell lines, RD or RH30. Histological analysis of the tumors revealed integration of human RMS cells with the human immune system containing few monocytes and B and T cells located in peripheral blood. This study serves as an exciting approach to evaluate immunotherapeutic strategies in RMS-based tumorigenesis (133). In another study, a pleomorphic patient-derived RMS tumor was used to develop an orthotopic xenograft model (PDOX) and subcutaneous transplant model. Tumors that formed in the PDOX model were significantly faster growing and more invasive, suggesting a malignant state, compared to the subcutaneous transplant tumors, indicative of a benign state (134).
Xenograft models have also been used to evaluate efficacy of chimeric antigen receptor (CAR)-T based approaches. For example, FGFR4 has been identified as overexpressed in RMS and this overexpression is predictive of reduced overall survival (135, 136). FGFR4 is a cell surface signaling molecule and amenable for CAR-T based targeting. As such, using in vitro and in vivo CAR-T based approaches, FGFR4 has been targeted causing specific cytotoxicity in vitro and decreased tumor burden in vivo in an intramuscular PAX3::FOXO1 xenograft model (137, 138). Further, there were few observed off-target effects such as weight loss and fur loss, and the mice tolerated the therapy. This strategy is now being explored as an antibody drug conjugate (139), and has promising translational potential for treating children with RMS.
Another approach for modeling RMS is using tumor organoid models which offers benefits of faster processing and supporting high-throughput drug screening. These organoids are formed by plating the minced patient tumors in basement membrane extract or extracellular matrix component. This study developed 19 pediatric alveolar and embryonal RMS tumor organoids that genetically and histologically resembled the original tumors (140). A limitation is being at a large medical center to obtain primary tissue for these rare diseases. Efforts have been undertaken to develop patient derived tumor organoid screening platform to test drug resistance and generate personalized therapeutic approaches in various sarcomas (141, 142). This approach can be further explored to answer biological and clinical research questions pertaining to RMS.
4.1.4 Infantile/pediatric spindle cell rhabdomyosarcoma models
S-RMS1 was developed from resected fresh tumor extracted after surgery from a 4-month-old boy with infantile/pediatric spindle-cell RMS. This patient derived cell line harbored the SRF::NCOA2 fusion and expresses RMS markers of MyoD, desmin and myogenin. The cell line extensively overlapped with the tumor at diagnosis using whole genome sequencing. The study also compared this cell line with other human RMS cell lines of RH30 and RD18 and found that S-RMS1 had a lower doubling time than RH30 and RD18 along with higher mRNA transcript levels of neovascularization marker endoglin and GATA-6. Other markers such as skeletal muscle differentiation markers of MEF2A, MEF2B, MEF2C, and MEF2D in S-RMS1 were similar to the mRNA levels seen in RH30 and RD18 (143). A recent study developed a VGLL2::NCOA2 cell culture model using C2C12 mouse myoblast cells, where the cells were transfected to stably express human VGLL2::NCOA2. C2C12-VGLL2::NCOA2 expressing cells were allografted into immunodeficient mice and generated aggressive and rapid tumors compared to C2C12 pcDNA3.1 controls (38). This study found that C2C12 expressing VGLL2:NCOA2 could transform and induced a developmental gene program that highlighted potential therapeutic opportunities. This strategy was expanded upon by another group that showed that expressing the TEAD1::NCOA2 fusion in C2C12 mouse myoblast cells also generated colonies in vitro and tumors in a mouse allograft model. The study also demonstrated that inhibiting p300 activity via a small molecule inhibitor led to reduction in TEAD1::NCOA2 colony formation along with suppressing tumor growth in the allograft model (144).
4.2 Animal models
4.2.1 PAX7::FOXO1 drosophila model
A drosophila model is the only animal model for PAX7::FOXO1. In this strategy, PAX7::FOXO1 is expressed in myosin heavy chain positive cells. This is lineage restricted by using a UAS/GAL4 genetic system. After crossing UAS/GAL4 flies, PAX7::FOXO1 is conditionally expressed in Drosophila syncytial myofibers. Further monitoring revealed that PAX7::FOXO1 expression resulted in the formation of nucleated cells that separated from the Drosophila syncytial myofibers and spread to non-muscular regions and the central nervous system (145). In a follow-up study utilizing the PAX7::FOXO1 Drosophila fly model as the basis for a genetic screen, the authors found that Mef2 is a target gene of the PAX7::FOXO1 fusion and that inhibiting Mef2 suppressed PAX7::FOXO1-dependent phenotypes (146).
4.2.2 PAX3::FOXO1 chick neural tube model
PAX3::FOXO1 expression in neural crest cells derived from chick embryos is a strategy to understand the tumorigenic early response and transformation potential. In this system, PAX3::FOXO1 decreased expression of neurogenic markers of SOX2 and PAX6 and caused reorganization of the ventricular and mantle regions of the neural tube as compared to PAX3 overexpression and control embryos. In addition, after extracting chick neural cells from the neural tube at 48 hours post electroporation, PAX3::FOXO1 expression elevated the levels of fusion-positive RMS genes ALK, ARHGAP25 and FGFR4, and transcription factors such as EYA2, FOXF1, LMO4, MEOX1, MYOD1, PITX2, PAX2, PRDM12 and TFAP2β. In contrast, PAX3 overexpression did not regulate these genes. Thus, induction of PAX3::FOXO1 resulted in reprogramming chick neural cells to exhibit fusion-positive RMS-like features (147).
4.2.3 Pax3::Foxo1 mouse model
The Pax3::Foxo1 mouse model was developed in 2004. This model has exons 2-3 of Foxo1 conditionally knocked in to the endogenous Pax3 allele after exon 7. After crossing with a Myf6-Cre driver, Pax3::Foxo1 is expressed in Myf6+ cells and the normal Pax3 allele is disrupted. This led to the formation of 1 tumor out of 228 animals at 12 months without cooperating mutations. The addition of Ink4a/ARF (or CDKN2A) or Trp53 mutations increased Pax3::Foxo1 tumor latency and frequency (148). In a separate study credentialing the Pax3::Foxo1 mouse model, induction of Pax3::Foxo1 expression in mice along with mutations in Ink4a/ARF (or CDKN2A) or Trp53 resulted in increased tumor penetrance. This tumor model recapitulates the aggressive nature of human alveolar RMS pediatric cancer and demonstrates rapid growth, invasion, involvement of regional lymph nodes along with distant hematogenous metastasis (149). The Pax3::Foxo1 genetically engineered mouse model has been used to understand genetic cooperation in the disease including the role of Hippo signaling in regulating Pax3::Foxo1-driven RMS tumorigenesis. Hippo signaling is suppressed in alveolar RMS cell lines and tumors (150), and genetically inhibiting the Hippo/MST signaling by knocking out kinases MST1 and MST2 (Stk4, Stk3), led to more rapid Pax3::Foxo1 tumor onset and higher penetrance. This indicates a dual regulatory mechanism where loss of MST kinases along with expression of Pax3::Foxo1 and mutational loss of Cdkn2a promotes more aggressive disease (151).
The Pax3::Foxo1 mouse model with inactivation of the p53 pathway has been leveraged to understand transformation capacity of a subset of myogenic and endothelial lineages. Evaluated lineages include Pax3 (MCre-Pax3), embryonic and fetal lineage (Myf6Cre), and postnatal satellite cell lineage (Pax7CreER). MCre-Pax3 and Myf6Cre resulted in RMS tumors resembling alveolar RMS histology, while Pax7CreER led to pleomorphic or spindle cell-like tumors. Notably, Myf6Cre showed the highest tumor incidence, indicating that Pax3::Foxo1 expression in specific lineages can drive more aggressive tumor behavior (152). The expression of Myf6 in this Cre driver is postulated to have both an embryonic expression with unclear timing and postnatal expression. In a recent study, expression of Pax3::Foxo1 in a Tek-Cre-driven mice endothelial lineage generated tumors similar to RMS. Pax3::Foxo1 tumors expressed desmin, MyoD1 and myogenin at the mRNA and protein level. In another lineage of Fabp4-Cre, induction of Pax3::Foxo1 drove high penetrance tumors that were consistent with RMS (153). These data suggest that there could be multiple cell(s) of origin outside of a myogenic lineage, and that Pax3::Foxo1 has the capacity to transdifferentiate an endothelial cell to a tumor cell expressing skeletal muscle proteins.
4.2.4 PAX3::FOXO1 zebrafish model
Zebrafish are vertebrates, have high genetic conservation to humans, and are a rapid model to perform functional validation and study of novel RMS fusion oncogenes (154, 155). Previous work has utilized transgenic zebrafish models for PAX3::FOXO1 RMS and infantile/pediatric VGLL2::NCOA2 RMS. Generated tumors recapitulate the human RMS disease transcriptionally and histologically. In the zebrafish PAX3::FOXO1 tumor model, the effect of many promoters/restricted cell lineages was investigated to understand transformation capacity on a broader scale. The PAX3::FOXO1 sequence and a viral 2A fluor of GFP was integrated with the promoter sequences using the Gateway cloning approach to the Tol2 transposon backbone (156) (Figure 3A). This allows for expression on the same mRNA but translation of GFP and PAX3::FOXO1 as independent proteins. These constructs were injected at the single-cell stage of zebrafish embryos and are mosaically but stably integrated into the zebrafish genome. Injected zebrafish were then monitored for tumor formation using phenotypic analysis and GFP expression as a proxy for fusion oncogene expression.
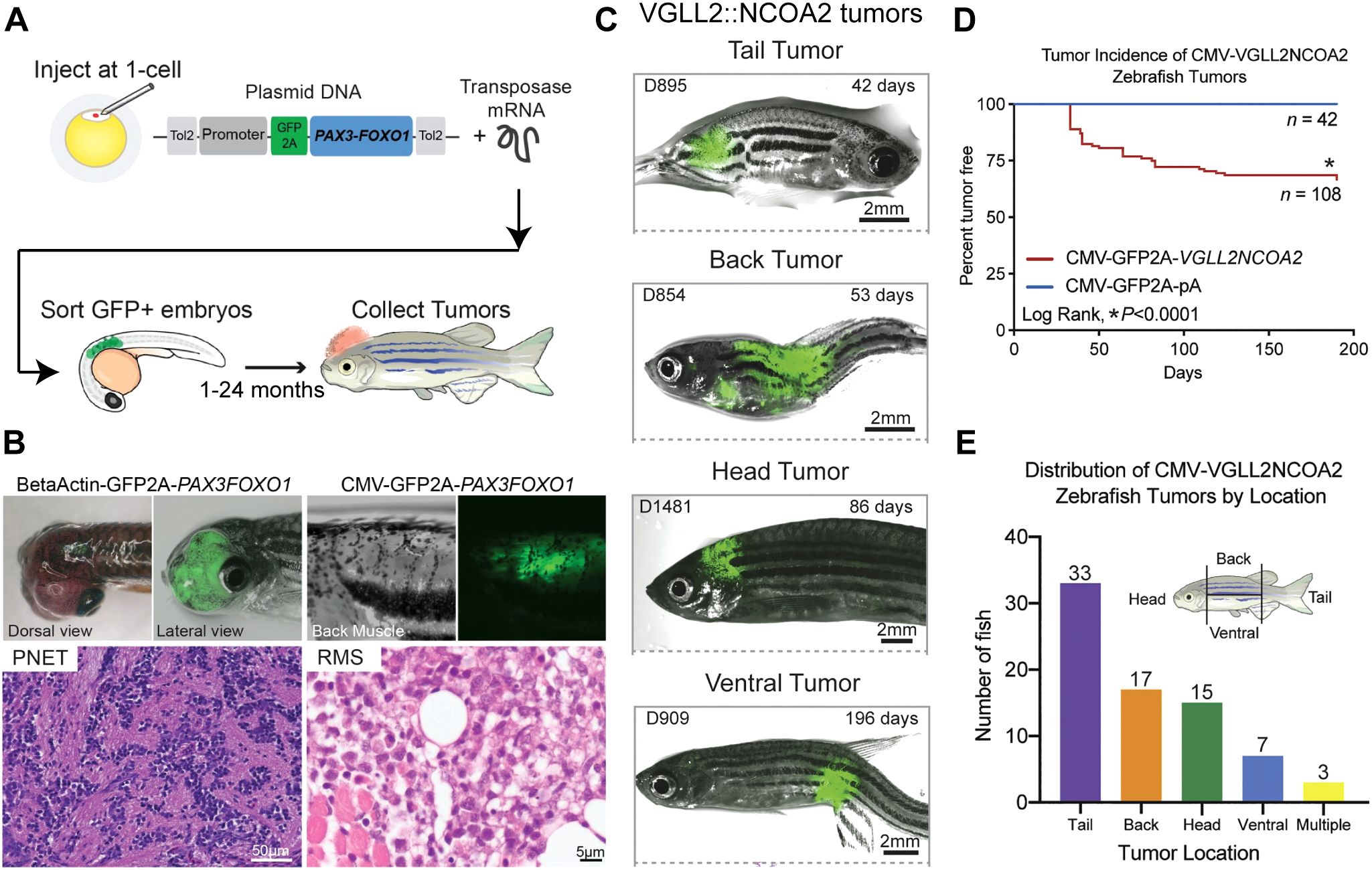
Figure 3. Functional genomics of rhabdomyosarcoma fusion oncogenes using zebrafish. (A) Schematic depicting the integration of the fusion oncogene and the viral 2A linked fluorescent protein into the Tol2 transposon backbone. Zebrafish embryos are injected at the single-cell stage and then GFP+ embryos are sorted after 24 hours and monitored for tumor formation. (B) The β-actin promoter driving PAX3::FOXO1 led to formation of tumors in zebrafish consistent with primitive neuroectodermal tumors. CMV promoter driven PAX3::FOXO1 formed RMS tumors, but required a cooperating tp53 missense mutation. (C) Representative zebrafish tumors formed using the CMV promoter driven VGLL2::NCOA2 fusion and classified by location of the tumor. (D) Tumor incidence curve comparing the tumor formation in VGLL2::NCOA2 injected zebrafish with the control-injected zebrafish. (E) Distribution of zebrafish VGLL2::NCOA2-injected tumors segregated as per the tumor location on the fish. Panels (A, B) are originally from Kendall, G.C. et al., 2018 (157). Panels (C-E) are originally from Watson, S., LaVigne, CA et al., 2023 (38).
Two promoters that supported transformation included the β-actin promoter and CMV promoter. β-actin driven PAX3::FOXO1 tumors were consistent with primitive neuroectodermal tumors and did not require a tp53 mutation to form tumors. On the other hand, CMV-driven PAX3::FOXO1 tumors required a tp53 missense mutation (158) to generate rhabdomyosarcoma that recapitulate the human disease (Figure 3B). Using this PAX3::FOXO1 zebrafish tumor model, this group also identified a novel PAX3::FOXO1 target gene her3, the human ortholog HES3, that is upregulated in fusion positive RMS patients (157). They investigated the functional role of her3/HES3 in neural development utilizing the zebrafish model and generated a stable her3 zebrafish knockout. RNA-seq analysis of the transcriptome suggested that her3 loss resulted in impairment in organ development and in matrix metallopeptidase function, along with regulating genes involved in apoptosis of the nervous system during development (159). Thus, zebrafish as a model system offers complementary advantages to study transformation capacity and genetic cooperation of PAX3::FOXO1 in RMS.
4.2.5 VGLL2::NCOA2 zebrafish model
VGLL2::NCOA2 is also transforming in zebrafish (38). In this transgenic zebrafish system, the CMV promoter drives expression of the VGLL2::NCOA2 human coding sequence. This fusion oncogene is linked to a GFP viral 2A sequence, allowing translation as independent proteins. The entire construct is then integrated into the zebrafish genome with the Tol2 mRNA and injection at the single-cell stage (156), and GFP expression (a proxy for fusion oncogene expression) is detected by 24 hours post fertilization. In a wildtype genetic background, 20% of the injected zebrafish developed tumors after 50 days and this percentage increased to 30% after 6 months. This highlights that the VGLL2::NCOA2 fusion oncogene does not require secondary cooperating genes and is sufficient for transformation in vivo (Figures 3C-E). Generated tumors histologically and transcriptionally recapitulate the human disease. VGLL2::NCOA2 expression led to repression of skeletal muscle differentiation, reactivation or inappropriate persistence of developmental genes, and the model identified potential druggable targets, such as the small GTPase ARF6. Further, ARF6 is overexpressed in VGLL2::NCOA2 driven zebrafish, mouse allograft, and patient tumors compared to normal mature skeletal muscle. Mechanistically, Arf6 knockout in C2C12 mouse myoblast cells alleviated the VGLL2::NCOA2 differentiation block and suppressed VGLL2::NCOA2 driven colony formation. This supports the rationale to further explore ARF6 as a therapeutic target for VGLL2::NCOA2-driven rhabdomyosarcoma and highlights the zebrafish system as a model to understand new biology.
5 Concluding remarks
In this review, we highlighted fusion oncogene-driven RMS as a major clinical and scientific challenge. Since they are often the most aggressive RMS subtype, it is critical to understand fusion oncogene biology to identify new molecular targets. We outlined different fusion oncogenes identified in RMS tumors and summarized the research studies that have been performed. The PAX3::FOXO1 fusion is the most common fusion found in alveolar RMS patients, and there have been more studies than in other RMS fusions. In comparison, NCOA2-based fusion oncogenes are newly identified and more effort is needed to understand their biological basis for tumorigenesis. We highlight with the increase in genomic characterization of tumors, there will be an increase in newly identified fusion oncogenes. Therefore, there is a need to functionally validate novel fusion oncogenes using animal and cell culture models, with the anticipation that these models will highlight key biology and therapeutic opportunities. Fusion oncogene expression can also be induced in cells, and these genetically modified cells can then be used in conjunction with animal models to generate tumors that recapitulate the human disease. Tumors can be analyzed for skeletal muscle and RMS protein markers and sequenced using approaches such as RNA sequencing, ChIP sequencing and assay for transposase-accessible chromatin (ATAC) sequencing. Such studies allow for more in-depth understanding of the tumor’s transcriptomic and epigenomic landscape. A challenge is understanding the correct cell type(s) to express the fusion in, or if there is convergent biology across multiple cell types that highlights core mechanisms of fusion oncogene activity. The experimental studies described in this review have increased our understanding of RMS biology along with establishing a platform for future studies. Our proposed workflow for studying fusion oncogene RMS, outlined in Figure 4, will determine key molecular targets that can be explored to inhibit tumorigenesis. The overall goal of these systems is to improve therapeutic approaches and clinical outcomes for children with RMS.
Figure 4. Workflow depicting strategies to model fusion oncogene driven RMS. The approach is to rapidly translate identified fusion oncogenes into tractable model systems and leverage the underlying biology to identify new therapeutic opportunities.
Author contributions
CSS: Conceptualization, Writing – original draft, Writing – review & editing. LH: Writing – review & editing. GCK: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. GK. is grateful for support from an NIH/NCI R01 CA272872 grant, an Alex’s Lemonade Stand Foundation “A” Award, a CancerFree Kids New Idea Award, and Startup Funds from The Abigail Wexner Research Institute at Nationwide Children’s Hospital. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
1. Skapek SX, Ferrari A, Gupta AA, Lupo PJ, Butler E, Shipley J, et al. Rhabdomyosarcoma. Nat Rev Dis Prime. (2019) 5:1. doi: 10.1038/s41572-018-0051-2
2. Kashi VP, Hatley ME, and Galindo RL. Probing for a deeper understanding of rhabdomyosarcoma: insights from complementary model systems. Nat Rev Cancer. (2015) 15:426–39. doi: 10.1038/nrc3961
3. Pomella S, Danielli SG, Alaggio R, Breunis WB, Hamed E, Selfe J, et al. Genomic and epigenetic changes drive aberrant skeletal muscle differentiation in rhabdomyosarcoma. Cancers. (2023) 15:2823. doi: 10.3390/cancers15102823
4. Kaseb H KJ and Babiker HM. Rhabdomyosarcoma. StatPearls Publishing LLC: National Library of Medicine (2022). Available at: https://www.ncbi.nlm.nih.gov/books/NBK507721/.
5. Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo C, and Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. (2009) 27:3391–7. doi: 10.1200/JCO.2008.19.7483
6. Ognjanovic S, Linabery AM, Charbonneau B, and Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer. (2009) 115:4218–26. doi: 10.1002/cncr.24465
7. Doros L, Yang J, Dehner L, Rossi CT, Skiver K, Jarzembowski JA, et al. DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer. (2012) 59:558–60. doi: 10.1002/pbc.24020
8. Li H, Sisoudiya SD, Martin-Giacalone BA, Khayat MM, Dugan-Perez S, Marquez-Do DA, et al. Germline cancer predisposition variants in pediatric rhabdomyosarcoma: A report from the children's oncology group. J Natl Cancer Inst. (2021) 113:875–83. doi: 10.1093/jnci/djaa204
9. Haduong JH, Heske CM, Allen-Rhoades W, Xue W, Teot LA, Rodeberg DA, et al. An update on rhabdomyosarcoma risk stratification and the rationale for current and future Children's Oncology Group clinical trials. Pediatr Blood Cancer. (2022) 69:e29511. doi: 10.1002/pbc.29511
10. Chisholm J, Mandeville H, Adams M, Minard-Collin V, Rogers T, Kelsey A, et al. Frontline and relapsed rhabdomyosarcoma (FaR-RMS) clinical trial: A report from the European paediatric soft tissue sarcoma study group (EpSSG). Cancers (Basel). (2024) 16. doi: 10.3390/cancers16050998
11. ASCO CN. Rhabdomyosarcoma. Childhood: Statistics American Society of Clinical Oncology: Cancer.Net (2020). Available at: https://www.cancer.net/cancer-types/rhabdomyosarcoma-childhood/statistics (Accessed March, 2024).
12. Parham DM, Barr FG, Montgomery E, and Nascimento AF. Skeletal muscle tumors. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, and Mertens F, editors. WHO classification of tumours of soft tissue and bone. Lyon: IARC Press (2013) p. 123–135.
13. Lindberg MR, Lucas D, Cassarino DS, Gardner JM, and Stallings-Archer K eds. Embryonal Rhabdomyosarcoma. In: Diagnostic Pathology: Soft Tissue Tumors, 2nd ed. Elsevier, Philadelphia. p. 372–7.
14. Folpe AL. Chapter 8 - Soft-Tissue Tumors of the Head and Neck. In: Gnepp DR, editor. Diagnostic Surgical Pathology of the Head and Neck, 2nd ed. W.B. Saunders, Philadelphia (2009). p. 647–727.
15. Qian X. Chapter 16 - Soft Tissue. In: Cibas ES and Ducatman BS, editors. Cytology, 3rd ed. W.B. Saunders, Philadelphia (2009). p. 451–94.
16. Jakkampudi A, Kaliyath S, Hegde P, Mathias M, and Shetty V. Spindle cell rhabdomyosarcoma in the adult: A rare case report. J Oral Maxillofac Pathol. (2022) 26:S103–s6. doi: 10.4103/jomfp.jomfp_251_21
17. Whittle S, Venkatramani R, Schönstein A, Pack SD, Alaggio R, Vokuhl C, et al. Congenital spindle cell rhabdomyosarcoma: An international cooperative analysis. Eur J Cancer. (2022) 168:56–64. doi: 10.1016/j.ejca.2022.03.022
18. Furlong MA, Mentzel T, and Fanburg-Smith JC. Pleomorphic rhabdomyosarcoma in adults: A clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Modern Pathol. (2001) 14:595–603. doi: 10.1038/modpathol.3880357
19. Siregar KB and Azrah A. Pleomorphic rhabdomyosarcoma on the chest wall which infiltrated intercostal muscles: A case report. Int J Surg Case Rep. (2020) 75:380–4. doi: 10.1016/j.ijscr.2020.09.040
20. Tomida A, Chiyonobu T, Tokuda S, Miyachi M, Murashima K, Hirata M, et al. Pleomorphic rhabdomyosarcoma in a young adult harboring a novel germline MSH2 variant. Hum Genome Var. (2022) 9:8. doi: 10.1038/s41439-022-00185-x
21. Xi S and Tong W. Pleomorphic rhabdomyosarcoma metastasis to small intestine causing intussusception: A case report. Med (Baltimore). (2018) 97:e13648. doi: 10.1097/MD.0000000000013648
22. Heske CM, Chi Y-Y, Venkatramani R, Li M, Arnold MA, Dasgupta R, et al. Survival outcomes of patients with localized FOXO1 fusion-positive rhabdomyosarcoma treated on recent clinical trials: A report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. Cancer. (2021) 127:946–56. doi: 10.1002/cncr.33334
23. Rudzinski ER, Teot LA, Anderson JR, Moore J, Bridge JA, Barr FG, et al. Dense pattern of embryonal rhabdomyosarcoma, a lesion easily confused with alveolar rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. Am J Clin Pathol. (2013) 140:82–90. doi: 10.1309/AJCPA1WN7ARPCMKQ
24. Raze T, Lapouble E, Lacour B, Guissou S, Defachelles AS, Gaspar N, et al. PAX-FOXO1 fusion status in children and adolescents with alveolar rhabdomyosarcoma: Impact on clinical, pathological, and survival features. Pediatr Blood Cancer. (2023) 70:e30228. doi: 10.1002/pbc.30228
25. Doyle LA. Surgical Pathology of Sarcomas. In: McManus LM and Mitchell RN, editors. Pathobiology of Human Disease. Academic Press, San Diego (2014). p. 3546–62.
26. Barr FG, Galili N, Holick J, Biegel JA, Rovera G, and Emanuel BS. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet. (1993) 3:113–7. doi: 10.1038/ng0293-113
27. Williamson D, Missiaglia E, de Reyniès A, Pierron G, Thuille B, Palenzuela G, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol. (2010) 28:2151–8. doi: 10.1200/JCO.2009.26.3814
28. Boudjadi S, Chatterjee B, Sun W, Vemu P, and Barr FG. The expression and function of PAX3 in development and disease. Gene. (2018) 666:145–57. doi: 10.1016/j.gene.2018.04.087
29. Oustanina S, Hause G, and Braun T. Pax7 directs postnatal renewal and propagation of myogenic satellite cells but not their specification. EMBO J. (2004) 23:3430–9–9. doi: 10.1038/sj.emboj.7600346
30. Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, and Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell. (2000) 102:777–86. doi: 10.1016/S0092-8674(00)00066-0
31. Relaix F, Rocancourt D, Mansouri A, and Buckingham M. Divergent functions of murine Pax3 and Pax7 in limb muscle development. Genes Dev. (2004) 18:1088–105. doi: 10.1101/gad.301004
32. Xu M, Chen X, Chen D, Yu B, and Huang Z. FoxO1: a novel insight into its molecular mechanisms in the regulation of skeletal muscle differentiation and fiber type specification. Oncotarget. (2017) 8:10662–74. doi: 10.18632/oncotarget.12891
33. De Alvaro C, Nieto-Vazquez I, Rojas JM, and Lorenzo M. Nuclear exclusion of forkhead box O and Elk1 and activation of nuclear factor-kappaB are required for C2C12-RasV12C40 myoblast differentiation. Endocrinology. (2008) 149:793–801. doi: 10.1210/en.2007-0657
34. Barr FG, Nauta LE, and Hollows JC. Structural analysis of PAX3 genomic rearrangements in alveolar rhabdomyosarcoma. Cancer Genet Cytogen. (1998) 102:32–9. doi: 10.1016/S0165-4608(97)00287-2
35. Fitzgerald JC, Scherr AM, and Barr FG. Structural analysis of PAX7 rearrangements in alveolar rhabdomyosarcoma. Cancer Genet Cytogen. (2000) 117:37–40. doi: 10.1016/S0165-4608(99)00130-2
36. Sumegi J, Streblow R, Frayer RW, Dal Cin P, Rosenberg A, Meloni-Ehrig A, et al. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes Chromosomes Cancer. (2010) 49:224–36. doi: 10.1002/gcc.20731
37. Alaggio R, Zhang L, Sung YS, Huang SC, Chen CL, Bisogno G, et al. A molecular study of pediatric spindle and sclerosing rhabdomyosarcoma: identification of novel and recurrent VGLL2-related fusions in infantile cases. Am J Surg Pathol. (2016) 40:224–35. doi: 10.1097/PAS.0000000000000538
38. Watson S, LaVigne CA, Xu L, Surdez D, Cyrta J, Calderon D, et al. VGLL2-NCOA2 leverages developmental programs for pediatric sarcomagenesis. Cell Rep. (2023) 42:112013. doi: 10.1016/j.celrep.2023.112013
39. Mosquera JM, Sboner A, Zhang L, Kitabayashi N, Chen CL, Sung YS, et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer. (2013) 52:538–50. doi: 10.1002/gcc.22050
40. Tan GZL, Saminathan SN, Chang KTE, Odoño EG, Kuick CH, Chen H, et al. A rare case of congenital spindle cell rhabdomyosarcoma with TEAD1-NCOA2 fusion: A subset of spindle cell rhabdomyosarcoma with indolent behavior. Pathol Int. (2020) 70:234–6. doi: 10.1111/pin.12908
41. Selfe J, Olmos D, Al-Saadi R, Thway K, Chisholm J, Kelsey A, et al. Impact of fusion gene status versus histology on risk-stratification for rhabdomyosarcoma: Retrospective analyses of patients on UK trials. Pediatr Blood Cancer. (2017) 64:e26386. doi: 10.1002/pbc.26386
42. Sorensen PHB, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, Maurer HM, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: A report from the children’s oncology group. J Clin Oncol. (2002) 20:2672–9. doi: 10.1200/JCO.2002.03.137
43. Koscielniak E, Stegmaier S, Ljungman G, Kazanowska B, Niggli F, Ladenstein R, et al. Prognostic factors in patients with localized and metastatic alveolar rhabdomyosarcoma. A report from two studies and two registries of the Cooperative Weichteilsarkom Studiengruppe CWS. Cancer Med. (2025) 14:e70215. doi: 10.1002/cam4.70215
44. Manceau L, Richard Albert J, Lollini P-L, Greenberg MVC, Gilardi-Hebenstreit P, and Ribes V. Divergent transcriptional and transforming properties of PAX3-FOXO1 and PAX7-FOXO1 paralogs. PloS Genet. (2022) 18:e1009782. doi: 10.1371/journal.pgen.1009782
45. Barr FG, Nauta LE, Davis RJ, Schafer BW, Nycum LM, and Biegel JA. In vivo amplification of the PAX3-FKHR and PAX7-FKHR fusion genes in alveolar rhabdomyosarcoma. Hum Mol Genet. (1996) 5:15–21. doi: 10.1093/hmg/5.1.15
46. Collins MH, Zhao H, Womer RB, and Barr FG. Proliferative and apoptotic differences between alveolar rhabdomyosarcoma subtypes: A comparative study of tumors containing PAX3-FKHR or PAX7-FKHR gene fusions. Med Pediatr Oncol. (2001) 37:83–9. doi: 10.1002/mpo.1174
47. Fredericks WJ, Galili N, Mukhopadhyay S, Rovera G, Bennicelli J, Barr FG, et al. The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol. (1995) 15:1522–35. doi: 10.1128/MCB.15.3.1522
48. Begum S, Emani N, Cheung A, Wilkins O, Der S, and Hamel PA. Cell-type-specific regulation of distinct sets of gene targets by Pax3 and Pax3/FKHR. Oncogene. (2005) 24:1860–72. doi: 10.1038/sj.onc.1208315
49. Kikuchi K, Tsuchiya K, Otabe O, Gotoh T, Tamura S, Katsumi Y, et al. Effects of PAX3-FKHR on Malignant phenotypes in alveolar rhabdomyosarcoma. Biochem Biophys Res Commun. (2008) 365:568–74. doi: 10.1016/j.bbrc.2007.11.017
50. Scheidler S, Fredericks WJ, Rauscher FJ 3rd, Barr FG, and Vogt PK. The hybrid PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma transforms fibroblasts in culture. Proc Natl Acad Sci U S A. (1996) 93:9805–9. doi: 10.1073/pnas.93.18.9805
51. Scuoppo C, Riess I, Schmitt-Ney M, Allegra P, Forni PE, Bersani F, et al. The oncogenic transcription factor PAX3-FKHR can convert fibroblasts into contractile myotubes. Exp Cell Res. (2007) 313:2308–17. doi: 10.1016/j.yexcr.2007.02.037
52. Xia SJ and Barr FG. Analysis of the transforming and growth suppressive activities of the PAX3-FKHR oncoprotein. Oncogene. (2004) 23:6864–71. doi: 10.1038/sj.onc.1207850
53. Kent MR, Jay AN, and Kendall GC. New dual inducible cellular model to investigate temporal control of oncogenic cooperating genes. Sci Rep. (2024) 14:20773. doi: 10.1038/s41598-024-71227-3
54. Ren Y-X, Finckenstein FG, Abdueva DA, Shahbazian V, Chung B, Weinberg KI, et al. Mouse mesenchymal stem cells expressing PAX-FKHR form alveolar rhabdomyosarcomas by cooperating with secondary mutations. Cancer Res. (2008) 68:6587–97. doi: 10.1158/0008-5472.CAN-08-0859
55. Wang W, Slevin M, Kumar S, and Kumar P. The cooperative transforming effects of PAX3-FKHR and IGF-II on mouse myoblasts. Int J Oncol. (2005) 27:1087–96. doi: 10.3892/ijo.27.4.1087
56. Linardic CM, Naini S, Herndon JE II, Kesserwan C, Qualman SJ, and Counter CM. The PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescence. Cancer Res. (2007) 67:6691–9. doi: 10.1158/0008-5472.CAN-06-3210
57. Sunkel BD, Wang M, LaHaye S, Kelly BJ, Fitch JR, Barr FG, et al. Evidence of pioneer factor activity of an oncogenic fusion transcription factor. iScience. (2021) 24:102867. doi: 10.1016/j.isci.2021.102867
58. Kucinski J, Tallan A, Taslim C, Wang M, Cannon MV, Silvius KM, et al. Rhabdomyosarcoma fusion oncoprotein initially pioneers a neural signature in vivo. bioRxiv. (2024), 603270. doi: 10.1101/2024.07.12.603270
59. Xia SJ, Holder DD, Pawel BR, Zhang C, and Barr FG. High expression of the PAX3-FKHR oncoprotein is required to promote tumorigenesis of human myoblasts. Am J Pathol. (2009) 175:2600–8. doi: 10.2353/ajpath.2009.090192
60. Pandey PR, Chatterjee B, Olanich ME, Khan J, Miettinen MM, Hewitt SM, et al. PAX3-FOXO1 is essential for tumour initiation and maintenance but not recurrence in a human myoblast model of rhabdomyosarcoma. J Pathol. (2017) 241:626–37. doi: 10.1002/path.2017.241.issue-5
61. Boudjadi S, Pandey PR, Chatterjee B, Nguyen TH, Sun W, and Barr FG. A fusion transcription factor–driven cancer progresses to a fusion-independent relapse via constitutive activation of a downstream transcriptional target. Cancer Res. (2021) 81:2930–42. doi: 10.1158/0008-5472.CAN-20-1613
62. Linardic CM. PAX3-FOXO1 fusion gene in rhabdomyosarcoma. Cancer Lett. (2008) 270:10–8. doi: 10.1016/j.canlet.2008.03.035
63. Gryder BE, Wachtel M, Chang K, El Demerdash O, Aboreden NG, Mohammed W, et al. Miswired enhancer logic drives a cancer of the muscle lineage. iScience. (2020) 23:101103. doi: 10.1016/j.isci.2020.101103
64. Gryder BE, Yohe ME, Chou H-C, Zhang X, Marques J, Wachtel M, et al. PAX3–FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Disc. (2017) 7:884–99. doi: 10.1158/2159-8290.CD-16-1297
65. Khan J, Bittner ML, Saal LH, Teichmann U, Azorsa DO, Gooden GC, et al. cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc Natl Acad Sci U S A. (1999) 96:13264–9. doi: 10.1073/pnas.96.23.13264
66. Barber TD, Barber MC, Tomescu O, Barr FG, Ruben S, and Friedman TB. Identification of target genes regulated by PAX3 and PAX3–FKHR in embryogenesis and alveolar rhabdomyosarcoma. Genomics. (2002) 79:278–84. doi: 10.1006/geno.2002.6703
67. Eun Hyun AHN. Regulation of target genes of PAX3–FOXO1 in alveolar rhabdomyosarcoma. Anticancer Res. (2013) 33:2029.
68. Ebauer M, Wachtel M, Niggli FK, and Schäfer BW. Comparative expression profiling identifies an in vivo target gene signature with TFAP2B as a mediator of the survival function of PAX3/FKHR. Oncogene. (2007) 26:7267–81. doi: 10.1038/sj.onc.1210525
69. Ahn EH, Mercado GE, Laé M, and Ladanyi M. Identification of target genes of PAX3-FOXO1 in alveolar rhabdomyosarcoma. Oncol Rep. (2013) 30:968–78. doi: 10.3892/or.2013.2513
70. Laé M, Ahn EH, Mercado GE, Chuai S, Edgar M, Pawel BR, et al. Global gene expression profiling of PAX-FKHR fusion-positive alveolar and PAX-FKHR fusion-negative embryonal rhabdomyosarcomas. J Pathol. (2007) 212:143–51. doi: 10.1002/path.2170
71. Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, et al. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. (2010) 70:6497–508. doi: 10.1158/0008-5472.CAN-10-0582
72. Graf Finckenstein F, Shahbazian V, Davicioni E, Ren YX, and Anderson MJ. PAX-FKHR function as pangenes by simultaneously inducing and inhibiting myogenesis. Oncogene. (2008) 27:2004–14. doi: 10.1038/sj.onc.1210835
73. Calhabeu F, Hayashi S, Morgan JE, Relaix F, and Zammit PS. Alveolar rhabdomyosarcoma-associated proteins PAX3/FOXO1A and PAX7/FOXO1A suppress the transcriptional activity of MyoD-target genes in muscle stem cells. Oncogene. (2013) 32:651–62. doi: 10.1038/onc.2012.73
74. Marshall AD, Lagutina I, and Grosveld GC. PAX3-FOXO1 induces cannabinoid receptor 1 to enhance cell invasion and metastasis. Cancer Res. (2011) 71:7471–80. doi: 10.1158/0008-5472.CAN-11-0924
75. Walters ZS, Villarejo-Balcells B, Olmos D, Buist TW, Missiaglia E, Allen R, et al. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene. (2014) 33:1148–57. doi: 10.1038/onc.2013.46
76. Walters ZS, Aladowicz E, Villarejo-Balcells B, Nugent G, Selfe JL, Eve P, et al. Role for the histone demethylase KDM4B in rhabdomyosarcoma via CDK6 and CCNA2: compensation by KDM4A and apoptotic response of targeting both KDM4B and KDM4A. Cancers (Basel). (2021) 13. doi: 10.3390/cancers13071734
77. Singh S, Abu-Zaid A, Jin H, Fang J, Wu Q, Wang T, et al. Targeting KDM4 for treating PAX3-FOXO1–driven alveolar rhabdomyosarcoma. Sci Trans Med. (2022) 14:eabq2096. doi: 10.1126/scitranslmed.abq2096
78. Kim YY, Gryder BE, Sinniah R, Peach ML, Shern JF, Abdelmaksoud A, et al. KDM3B inhibitors disrupt the oncogenic activity of PAX3-FOXO1 in fusion-positive rhabdomyosarcoma. Nat Commun. (2024) 15:1703. doi: 10.1038/s41467-024-45902-y
79. Xu J and O'Malley BW. Molecular mechanisms and cellular biology of the steroid receptor coactivator (SRC) family in steroid receptor function. Rev Endocr Metab Disord. (2002) 3:185–92. doi: 10.1023/A:1020016208071
80. Rollins DA, Coppo M, and Rogatsky I. Minireview: Nuclear Receptor Coregulators of the p160 Family: Insights into Inflammation and Metabolism. Mol Endocrinol. (2015) 29:502–17. doi: 10.1210/me.2015-1005
81. Hong H, Kohli K, Garabedian MJ, and Stallcup MR. GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol Cell Biol. (1997) 17:2735–44. doi: 10.1128/MCB.17.5.2735
82. Onate SA, Boonyaratanakornkit V, Spencer TE, Tsai SY, Tsai M-J, Edwards DP, et al. The steroid receptor coactivator-1 contains multiple receptor interacting and activation domains that cooperatively enhance the activation function 1 (AF1) and AF2 domains of steroid receptors*. J Biol Chem. (1998) 273:12101–8. doi: 10.1074/jbc.273.20.12101
83. Qin J, Lee H-J, Wu S-P, Lin S-C, Lanz RB, Creighton CJ, et al. Androgen deprivation–induced NCoA2 promotes metastatic and castration-resistant prostate cancer. J Clin Invest. (2014) 124:5013–26. doi: 10.1172/JCI76412
84. Cai M, Liang X, Sun X, Chen H, Dong Y, Wu L, et al. Nuclear receptor coactivator 2 promotes human breast cancer cell growth by positively regulating the MAPK/ERK pathway. Front Oncol. (2019) 9. doi: 10.3389/fonc.2019.00164
85. Troke PJ, Kindle KB, Collins HM, and Heery DM. MOZ fusion proteins in acute myeloid leukaemia. Biochem Soc Symp. (2006) 73:23–39. doi: 10.1042/bss0730023
86. Deguchi K, Ayton PM, Carapeti M, Kutok JL, Snyder CS, Williams IR, et al. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell. (2003) 3:259–71. doi: 10.1016/S1535-6108(03)00051-5
87. Yin H and Glass J. The MOZ portion of the leukemia oncogenic protein MOZ-TIF2 inhibits nuclear receptor-mediated transcription. Cancer Res. (2007) 67(9_Supplement):2113.
88. Wang L, Motoi T, Khanin R, Olshen A, Mertens F, Bridge J, et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer. (2012) 51:127–39. doi: 10.1002/gcc.20937
89. Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, et al. Regulation of transcription by a protein methyltransferase. Science. (1999) 284:2174–7. doi: 10.1126/science.284.5423.2174
90. Chen SL, Dowhan DH, Hosking BM, and Muscat GE. The steroid receptor coactivator, GRIP-1, is necessary for MEF-2C-dependent gene expression and skeletal muscle differentiation. Genes Dev. (2000) 14:1209–28. doi: 10.1101/gad.14.10.1209
91. Wachtel M, Dettling M, Koscielniak E, Stegmaier S, Treuner J, Simon-Klingenstein K, et al. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res. (2004) 64:5539–45. doi: 10.1158/0008-5472.CAN-04-0844
92. Yoshida H, Miyachi M, Sakamoto K, Ouchi K, Yagyu S, Kikuchi K, et al. PAX3-NCOA2 fusion gene has a dual role in promoting the proliferation and inhibiting the myogenic differentiation of rhabdomyosarcoma cells. Oncogene. (2014) 33:5601–8. doi: 10.1038/onc.2013.491
93. Pipes GC, Creemers EE, and Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. (2006) 20:1545–56. doi: 10.1101/gad.1428006
94. Qiu H, Wang F, Liu C, Xu X, and Liu B. TEAD1-dependent expression of the FoxO3a gene in mouse skeletal muscle. BMC Mol Biol. (2011) 12:1. doi: 10.1186/1471-2199-12-1
95. Watson S, Perrin V, Guillemot D, Reynaud S, Coindre J-M, Karanian M, et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol. (2018) 245:29–40. doi: 10.1002/path.5053
96. Honda M, Hidaka K, Fukada SI, Sugawa R, Shirai M, Ikawa M, et al. Vestigial-like 2 contributes to normal muscle fiber type distribution in mice. Sci Rep. (2017) 7:7168. doi: 10.1038/s41598-017-07149-0
97. Maeda T, Chapman DL, and Stewart AF. Mammalian vestigial-like 2, a cofactor of TEF-1 and MEF2 transcription factors that promotes skeletal muscle differentiation. J Biol Chem. (2002) 277:48889–98. doi: 10.1074/jbc.M206858200
98. Smith BF, Doung Y-C, Beckett B, Corless CL, Davis LE, and Davis JL. Intraosseous spindle cell rhabdomyosarcoma with MEIS1::NCOA2 fusion – case report with substantial clinical follow-up and review of the literature. Cancer Invest. (2023) 41:704–12. doi: 10.1080/07357907.2023.2255668
99. Montoya-Cerrillo DM, Diaz-Perez JA, and Rosenberg AE. Novel gene fusions in rhabdomyosarcoma. Am J Clin Pathol. (2020) 154:S156–S. doi: 10.1093/ajcp/aqaa161.341
100. Argani P, Reuter VE, Kapur P, Brown JE, Sung YS, Zhang L, et al. Novel MEIS1-NCOA2 gene fusions define a distinct primitive spindle cell sarcoma of the kidney. Am J Surg Pathol. (2018) 42:1562–70. doi: 10.1097/PAS.0000000000001140
101. Dehner CA, Broski SM, Meis JM, Murugan P, Chrisinger JSA, Sosa C, et al. Fusion-driven spindle cell rhabdomyosarcomas of bone and soft tissue: A clinicopathologic and molecular genetic study of 25 cases. Modern Pathol. (2023) 36. doi: 10.1016/j.modpat.2023.100271
102. Le Loarer F, Laffont S, Lesluyes T, Tirode F, Antonescu C, Baglin AC, et al. Clinicopathologic and molecular features of a series of 41 biphenotypic sinonasal sarcomas expanding their molecular spectrum. Am J Surg Pathol. (2019) 43:747–54. doi: 10.1097/PAS.0000000000001238
103. Strehl S, Nebral K, König M, Harbott J, Strobl H, Ratei R, et al. ETV6-NCOA2: a novel fusion gene in acute leukemia associated with coexpression of T-lymphoid and myeloid markers and frequent NOTCH1 mutations. Clin Cancer Res. (2008) 14:977–83. doi: 10.1158/1078-0432.CCR-07-4022
104. Jin Y, Möller E, Nord KH, Mandahl N, Von Steyern FV, Domanski HA, et al. Fusion of the AHRR and NCOA2 genes through a recurrent translocation t(5;8)(p15;q13) in soft tissue angiofibroma results in upregulation of aryl hydrocarbon receptor target genes. Genes Chromosomes Cancer. (2012) 51:510–20. doi: 10.1002/gcc.21939
105. Bakhshwin A, Armstrong SM, Duckworth LA, Stoehr R, Konishi E, Rubin BP, et al. Novel NCOA2/3-rearranged low-grade fibroblastic spindle cell tumors: A report of five cases. Genes Chromosomes Cancer. (2024) 63:e23203. doi: 10.1002/gcc.23203
106. Brunetti M, Panagopoulos I, Gorunova L, Davidson B, Heim S, and Micci F. RNA-sequencing identifies novel GREB1-NCOA2 fusion gene in a uterine sarcoma with the chromosomal translocation t(2;8)(p25;q13). Genes Chromosomes Cancer. (2018) 57:176–81. doi: 10.1002/gcc.22518
107. Yu J, Wu WKK, Liang Q, Zhang N, He J, Li X, et al. Disruption of NCOA2 by recurrent fusion with LACTB2 in colorectal cancer. Oncogene. (2016) 35:187–95. doi: 10.1038/onc.2015.72
108. Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ 3rd, Emanuel BS, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. (1993) 5:230–5. doi: 10.1038/ng1193-230
109. Shapiro DN, Sublett JE, Li B, Downing JR, and Naeve CW. Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res. (1993) 53:5108–12.
110. Davis RJ, D'Cruz CM, Lovell MA, Biegel JA, and Barr FG. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. (1994) 54:2869–72.
111. Barr FG, Qualman SJ, Macris MH, Melnyk N, Lawlor ER, Strzelecki DM, et al. Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res. (2002) 62:4704–10.
112. Dermawan JK, Malik F, Gross JM, Baraban E, Pratilas C, Mneimneh W, et al. Novel PAX3::MAML3 fusion identified in alveolar rhabdomyosarcoma, using DNA methylation profiling to expand the genetic spectrum of “Fusion-positive” Cases. Modern Pathol. (2024) 37:100594. doi: 10.1016/j.modpat.2024.100594
113. Wang X, Bledsoe KL, Graham RP, Asmann YW, Viswanatha DS, Lewis JE, et al. Recurrent PAX3-MAML3 fusion in biphenotypic sinonasal sarcoma. Nat Genet. (2014) 46:666–8. doi: 10.1038/ng.2989
114. Li N-M, Jiang S-H, Zhou P, and Li X-H. Case Report: An NTRK1 fusion-positive embryonal rhabdomyosarcoma: clinical presentations, pathological characteristics and genotypic analyses. Front Oncol. (2023) 13. doi: 10.3389/fonc.2023.1178945
115. Sirvent N, Trassard M, Ebran N, Attias R, and Pedeutour F. Fusion of EWSR1 with the DUX4 facioscapulohumeral muscular dystrophy region resulting from t(4;22)(q35;q12) in a case of embryonal rhabdomyosarcoma. Cancer Genet Cytogen. (2009) 195:12–8. doi: 10.1016/j.cancergencyto.2009.06.011
116. Bourgeois JM, Knezevich SR, Mathers JA, and Sorensen PHB. Molecular detection of the ETV6-NTRK3 gene fusion differentiates congenital fibrosarcoma from other childhood spindle cell tumors. Am J Surg Pathol. (2000) 24:937–46. doi: 10.1097/00000478-200007000-00005
117. Kawamura-Saito M, Yamazaki Y, Kaneko K, Kawaguchi N, Kanda H, Mukai H, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet. (2006) 15:2125–37. doi: 10.1093/hmg/ddl136
118. Szuhai K, Ijszenga M, de Jong D, Karseladze A, Tanke HJ, and Hogendoorn PC. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res. (2009) 15:2259–68. doi: 10.1158/1078-0432.CCR-08-2184
119. Pierron G, Tirode F, Lucchesi C, Reynaud S, Ballet S, Cohen-Gogo S, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. (2012) 44:461–6. doi: 10.1038/ng.1107
120. Rakheja D, Park JY, Alhasan M, and Uddin N. Spindle cell/sclerosing rhabdomyosarcoma with PAX8::PPARG fusion. Int J Surg Pathol. (2022) 30:950–5. doi: 10.1177/10668969221095170
121. Hinson AR, Jones R, Crose LE, Belyea BC, Barr FG, and Linardic CM. Human rhabdomyosarcoma cell lines for rhabdomyosarcoma research: utility and pitfalls. Front Oncol. (2013) 3:183. doi: 10.3389/fonc.2013.00183
122. Missiaglia E, Selfe J, Hamdi M, Williamson D, Schaaf G, Fang C, et al. Genomic imbalances in rhabdomyosarcoma cell lines affect expression of genes frequently altered in primary tumors: An approach to identify candidate genes involved in tumor development. Genes Chromosomes Cancer. (2009) 48:455–67. doi: 10.1002/gcc.20655
123. Scholl FA, Betts D, Niggli FK, and Schafer BW. Molecular features of a human rhabdomyosarcoma cell line with spontaneous metastatic progression. Br J Cancer. (2000) 82:1239–45. doi: 10.1054/bjoc.1999.1069
124. Naini S, Etheridge KT, Adam SJ, Qualman SJ, Bentley RC, Counter CM, et al. Defining the cooperative genetic changes that temporally drive alveolar rhabdomyosarcoma. Cancer Res. (2008) 68:9583–8. doi: 10.1158/0008-5472.CAN-07-6178
125. Slemmons KK, Deel MD, Lin YT, Oristian KM, Kuprasertkul N, Genadry KC, et al. A method to culture human alveolar rhabdomyosarcoma cell lines as rhabdospheres demonstrates an enrichment in stemness and Notch signaling. Biol Open. (2021) 10. doi: 10.1242/bio.050211
126. Stefanek E, Samiei E, Kavoosi M, Esmaeillou M, Roustai Geraylow K, Emami A, et al. A bioengineering method for modeling alveolar Rhabdomyosarcoma and assessing chemotherapy responses. MethodsX. (2021) 8:101473. doi: 10.1016/j.mex.2021.101473
127. Saggioro M, D’Agostino S, Veltri G, Bacchiega M, Tombolan L, Zanon C, et al. A perfusion-based three-dimensional cell culture system to model alveolar rhabdomyosarcoma pathological features. Sci Rep. (2023) 13:9444. doi: 10.1038/s41598-023-36210-4
128. Danielli SG, Wei Y, Dyer MA, Stewart E, Sheppard H, Wachtel M, et al. Single cell transcriptomic profiling identifies tumor-acquired and therapy-resistant cell states in pediatric rhabdomyosarcoma. Nat Commun. (2024) 15:6307. doi: 10.1038/s41467-024-50527-2
129. Danielli SG, Porpiglia E, De Micheli AJ, Navarro N, Zellinger MJ, Bechtold I, et al. Single-cell profiling of alveolar rhabdomyosarcoma reveals RAS pathway inhibitors as cell-fate hijackers with therapeutic relevance. Sci Adv. (2023) 9:eade9238. doi: 10.1126/sciadv.ade9238
130. Wei Y, Qin Q, Yan C, Hayes MN, Garcia SP, Xi H, et al. Single-cell analysis and functional characterization uncover the stem cell hierarchies and developmental origins of rhabdomyosarcoma. Nat Cancer. (2022) 3:961–75. doi: 10.1038/s43018-022-00414-w
131. DeMartino J, Meister MT, Visser LL, Brok M, Groot Koerkamp MJA, Wezenaar AKL, et al. Single-cell transcriptomics reveals immune suppression and cell states predictive of patient outcomes in rhabdomyosarcoma. Nat Commun. (2023) 14:3074. doi: 10.1038/s41467-023-38886-8
132. Patel AG, Chen X, Huang X, Clay MR, Komorova N, Krasin MJ, et al. The myogenesis program drives clonal selection and drug resistance in rhabdomyosarcoma. Dev Cell. (2022) 57:1226–40.e8. doi: 10.1016/j.devcel.2022.04.003
133. Seitz G, Pfeiffer M, Fuchs J, Warmann SW, Leuschner I, Vokuhl C, et al. Establishment of a rhabdomyosarcoma xenograft model in human-adapted mice. Oncol Rep. (2010) 24:1067–72. doi: 10.3892/or.2010.1067
134. Igarashi K, Kawaguchi K, Kiyuna T, Murakami T, Miwa S, Nelson SD, et al. Patient-derived orthotopic xenograft (PDOX) mouse model of adult rhabdomyosarcoma invades and recurs after resection in contrast to the subcutaneous ectopic model. Cell Cycle. (2017) 16:91–4. doi: 10.1080/15384101.2016.1252885
135. Khan J, Wei JS, Ringnér M, Saal LH, Ladanyi M, Westermann F, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. (2001) 7:673–9. doi: 10.1038/89044
136. Taylor J, Cheuk AT, Tsang PS, Chung JY, Song YK, Desai K, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest. (2009) 119:3395–407. doi: 10.1172/JCI39703
137. Tian M, Wei JS, Cheuk AT-C, Milewski D, Zhang Z, Kim YY, et al. CAR T-cells targeting FGFR4 and CD276 simultaneously show potent antitumor effect against childhood rhabdomyosarcoma. Nat Commun. (2024) 15:6222. doi: 10.1038/s41467-024-50251-x
138. Tian M, Wei JS, Shivaprasad N, Highfill SL, Gryder BE, Milewski D, et al. Preclinical development of a chimeric antigen receptor T cell therapy targeting FGFR4 in rhabdomyosarcoma. Cell Rep Med. (2023) 4:101212. doi: 10.1016/j.xcrm.2023.101212
139. Tian M, Wei JS, Cheuk A, Milewski D, Zhang Z, Kim YY, et al. Abstract 1784: FGFR4 and CD276 dual targeting CAR T cells demonstrate synergistic antitumor activity in childhood rhabdomyosarcoma. Cancer Res. (2023) 83:1784. doi: 10.1158/1538-7445.AM2023-1784
140. Meister MT, Groot Koerkamp MJA, de Souza T, Breunis WB, Frazer-Mendelewska E, Brok M, et al. Mesenchymal tumor organoid models recapitulate rhabdomyosarcoma subtypes. EMBO Mol Med. (2022) 14:e16001. doi: 10.15252/emmm.202216001
141. Al Shihabi A, Tebon PJ, Nguyen HTL, Chantharasamee J, Sartini S, Davarifar A, et al. The landscape of drug sensitivity and resistance in sarcoma. Cell Stem Cell. (2024) 31:1524–42.e4. doi: 10.1016/j.stem.2024.08.010
142. Nguyen HTL and Soragni A. Patient-derived tumor organoid rings for histologic characterization and high-throughput screening. STAR Protoc. (2020) 1:100056. doi: 10.1016/j.xpro.2020.100056
143. Colletti M, Galardi A, Miele E, Di Paolo V, Russo I, De Stefanis C, et al. Establishment and characterization of a cell line (S-RMS1) derived from an infantile spindle cell rhabdomyosarcoma with SRF-NCOA2 fusion transcript. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22115484
144. Guo S, Hu X, Cotton JL, Ma L, Li Q, Cui J, et al. VGLL2 and TEAD1 fusion proteins drive YAP/TAZ-independent transcription and tumorigenesis by engaging p300. eLife Sci Public Ltd. (2024) 13:RP98386. doi: 10.7554/eLife.98386.3
145. Galindo RL, Allport JA, and Olson EN. A Drosophila model of the rhabdomyosarcoma initiator PAX7-FKHR. Proc Natl Acad Sci U S A. (2006) 103:13439–44. doi: 10.1073/pnas.0605926103
146. Galindo KA, Endicott TR, Avirneni-Vadlamudi U, and Galindo RL. A rapid one-generation genetic screen in a Drosophila model to capture rhabdomyosarcoma effectors and therapeutic targets. G3 (Bethesda). (2014) 5:205–17. doi: 10.1534/g3.114.015818
147. Gonzalez Curto G, Der Vartanian A, Frarma YE-M, Manceau L, Baldi L, Prisco S, et al. The PAX-FOXO1s trigger fast trans-differentiation of chick embryonic neural cells into alveolar rhabdomyosarcoma with tissue invasive properties limited by S phase entry inhibition. PloS Genet. (2020) 16:e1009164. doi: 10.1371/journal.pgen.1009164
148. Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, and Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. (2004) 18:2614–26. doi: 10.1101/gad.1244004
149. Nishijo K, Chen QR, Zhang L, McCleish AT, Rodriguez A, Cho MJ, et al. Credentialing a preclinical mouse model of alveolar rhabdomyosarcoma. Cancer Res. (2009) 69:2902–11. doi: 10.1158/0008-5472.CAN-08-3723
150. Crose LE, Galindo KA, Kephart JG, Chen C, Fitamant J, Bardeesy N, et al. Alveolar rhabdomyosarcoma-associated PAX3-FOXO1 promotes tumorigenesis via Hippo pathway suppression. J Clin Invest. (2014) 124:285–96. doi: 10.1172/JCI67087
151. Oristian KM, Crose LES, Kuprasertkul N, Bentley RC, Lin YT, Williams N, et al. Loss of MST/hippo signaling in a genetically engineered mouse model of fusion-positive rhabdomyosarcoma accelerates tumorigenesis. Cancer Res. (2018) 78:5513–20. doi: 10.1158/0008-5472.CAN-17-3912
152. Abraham J, Nuñez-Álvarez Y, Hettmer S, Carrió E, Chen HI, Nishijo K, et al. Lineage of origin in rhabdomyosarcoma informs pharmacological response. Genes Dev. (2014) 28:1578–91. doi: 10.1101/gad.238733.114
153. Searcy MB, Larsen RK, Stevens BT, Zhang Y, Jin H, Drummond CJ, et al. PAX3-FOXO1 dictates myogenic reprogramming and rhabdomyosarcoma identity in endothelial progenitors. Nat Commun. (2023) 14:7291. doi: 10.1038/s41467-023-43044-1
154. Kent MR, Silvius K, Kucinski J, Calderon D, and Kendall GC. Functional genomics of novel rhabdomyosarcoma fusion-oncogenes using zebrafish. Methods Mol Biol. (2024) 2707:23–41. doi: 10.1007/978-1-0716-3401-1_2
155. Kendall GC and Amatruda JF. Zebrafish as a model for the study of solid Malignancies. Methods Mol Biol. (2016) 1451:121–42. doi: 10.1007/978-1-4939-3771-4_9
156. Urasaki A, Morvan G, and Kawakami K. Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics. (2006) 174:639–49. doi: 10.1534/genetics.106.060244
157. Kendall GC, Watson S, Xu L, LaVigne CA, Murchison W, Rakheja D, et al. PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. eLife. (2018) 7:e33800. doi: 10.7554/eLife.33800
158. Berghmans S, Murphey RD, Wienholds E, Neuberg D, Kutok JL, Fletcher CDM, et al. tp53 mutant zebrafish develop Malignant peripheral nerve sheath tumors. Proc Natl Acad Sci. (2005) 102:407–12. doi: 10.1073/pnas.0406252102
Keywords: pediatric sarcomas, PAX3/7::FOXO1, VGLL2::NCOA2, skeletal muscle, oncogenic drivers, animal models
Citation: Sankhe CS, Hall L and Kendall GC (2025) Fusion oncogenes in rhabdomyosarcoma: model systems, mechanisms of tumorigenesis, and therapeutic implications. Front. Oncol. 15:1570070. doi: 10.3389/fonc.2025.1570070
Received: 02 February 2025; Accepted: 30 May 2025;
Published: 17 June 2025.
Edited by:
Ewa Krawczyk, Georgetown University, United StatesReviewed by:
Zoë Walters, University of Southampton, United KingdomKyle L MacQuarrie, Ann & Robert H. Lurie Children's Hospital of Chicago, United States
Copyright © 2025 Sankhe, Hall and Kendall. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Genevieve C. Kendall, Z2VuZXZpZXZlLmtlbmRhbGxAbmF0aW9ud2lkZWNoaWxkcmVucy5vcmc=