 Colin C. Young1*
Colin C. Young1* Ashley Lahr2Caroline Nestor2
Ashley Lahr2Caroline Nestor2 Ashley Kaminski1
Ashley Kaminski1 Marcy E. Richardson1*Georgianne L. Arnold2
Marcy E. Richardson1*Georgianne L. Arnold2- 1Ambry Genetics, 1 Enterprise, Aliso Viejo, CA, United States
- 2University of Pittsburgh Medical Center, Pittsburgh, PA, United States
Pathogenic alterations in BRCA1 are associated with autosomal dominant breast and ovarian cancer and autosomal recessive Fanconi Anemia Subtype S (FA-S). FA-S accounts for <1% of all reported cases of FA with only ten patients identified in the literature to-date. Here we describe an eleventh FA-S proband with severe microcephaly, growth failure, duodenal stenosis, hyperpigmented macules, dysmorphic features, and abnormal chromosomal breakage, consistent with other FA-S patients. Two pathogenic BRCA1 variants (c.191G>A, p.C64Y and c.3991C>T, p.Q1331*) were identified in trans. At four years old, this patient has not been diagnosed with cancer or bone marrow failure, which are hallmark features in other subtypes of FA. Like a majority of the literature-reported FA-S patients, this patient harbors a truncating variant in BRCA1 exon 11. This exon undergoes alternative splicing resulting in a protein with partial BRCA1 activity. The retained activity may be enough to rescue an otherwise lethal phenotype explaining the viability of FA-S patients. This retained functional activity may also modify clinical cancer risks and treatment implications for heterozygous carriers of exon 11 truncating variants. This work further characterizes the features of FA-S patients and discusses a molecular hypothesis for the rarity and viability of individuals with this condition.
Introduction
Pathogenic alterations in BRCA1 are linked to a high-penetrance, autosomal dominant cancer predisposition syndrome (MONDO: 0700268, OMIM: 604370) (1, 2). Women who are heterozygous carriers of pathogenic BRCA1 alterations have significantly increased lifetime risks of developing breast and ovarian cancer and both male and female heterozygous carriers are at increased risk for pancreatic cancer (3). The identification of patients who carry these alterations has significant implications on cancer screening recommendations, surgical management that includes prophylactic surgery, family planning, and familial testing to reduce cancer risk and decrease mortality.
More rarely discussed, however, is the risk of Fanconi Anemia Subtype S (FA-S; MONDO:0054748, OMIM:617883) which is an autosomal recessive disorder observed in patients with biallelic pathogenic alterations in BRCA1 (4). FA-S is an extremely rare condition and accounts for fewer than 1% of all FA cases (4). Thus far, only ten cases are described in the literature (5–11). The presentation of FA-S includes pre- and postnatal growth failure, microcephaly, developmental delay, hyperpigmented macules, and dysmorphic features. Other abnormalities include hip dysplasia, duodenal stenosis, and limb defects. None of the literature patients reported bone marrow failure, which is a common feature in other FA subtypes (12).
A chromosomal breakage test using either diepoxybutane (DEB) or mitomycin-C (MMC) can provide a molecular diagnosis of FA and eight of ten FA-S patients had abnormal chromosomal breakage (4). Eight of nine of these patients had at least one nonsense or frameshifting alteration in BRCA1 exon 11 (coding exon 9), and one had a frameshifting alteration in exon 10. BRCA1 knock-out mice are embryonic lethal (13), leading to questions about the mechanisms which lead to viability of FA-S patients in whom two loss-of-function alleles are identified (14).
Here we report an eleventh FA-S proband with biallelic pathogenic variants in BRCA1. The proband has physical and developmental characteristics consistent with the other FA-S patients reported in the literature to date. Molecular confirmation of FA-S was confirmed by chromosomal breakage analysis and phase confirmation was determined by parental testing. As with the majority of other FA-S patients, a nonsense alteration in exon 11 was identified as one of the alterations.
Patient and methods
Clinical testing and ethics
Peripheral blood samples and clinical histories were collected from the proband and both parents as part of diagnostic testing at Ambry Genetics and GeneDx. Chromosomal breakage analysis was performed on patient peripheral blood lymphocytes by the Comprehensive Center for Fanconi Anemia at Dana Farber Cancer Institute. An informed consent form was obtained by the patient’s parent/guardian for this publication.
Variant classification
Variant classification was performed using modified ACMG/AMP clinical classification guidelines as recommended by the ClinGen ENIGMA BRCA1 and BRCA2 Variant Curation Expert Panel (VCEP) (https://cspec.genome.network/cspec/ui/svi/doc/GN092).
Results
Patient phenotype
The female proband was born prematurely at 34 weeks to non-consanguineous parents. Prenatally, the proband was small for gestational age, had microcephaly (<3rd percentile), and intrauterine growth restriction. Prenatal karyotype, chromosome SNP microarray, and FISH were normal. The proband’s parents were of African American and Caucasian descent, were developmentally normal, and four maternal half-siblings were reported to be healthy.
After birth, the proband was identified to have two duodenal webs which required surgical repair. Ophthalmological exam showed unilateral retinal coloboma. A brain MRI noted simplified gyral pattern, colpocpehaly, and a partially dysplastic corpus callosum, which were expected to result in significant impacts on neurodevelopment. A cardiac exam showed a patent foramen ovale. The proband displayed developmental dysplasia of the left hip. Additional examinations noted the patient had bitemporal narrowing of the forehead, up-slanting palpebral fissures, a bulbous nose, bilateral clinodactyly, single palmar crease, and bilateral toe overlap.
Both whole exome sequencing and multi-gene panel testing were performed on the proband and identified a variant of uncertain significance in BMP4 (c.351G>A, p.V117V) and two pathogenic alterations in BRCA1: c.191G>A, p.C64Y (maternally inherited) and c.3991C>T, p.Q1331* (paternally inherited). No other variants were identified in FA-associated genes. The family cancer history, while is limited, includes a maternal grandmother with breast cancer at 38. Both are classified by multiple submitters as pathogenic in ClinVar.
Chromosomal breakage analysis determined the patient was positive for FA. Treatment of patient blood lymphocytes with MMC resulted in an abnormal result of 2.96 aberrations/cell (FA negative and FA positive controls were 0.2 and 4.84 aberrations/cell, respectively). 48% of patient cells had radial formations (negative and positive controls were 4% and 76%, respectively). Treatment with DEB showed similar results with 3.26 aberrations/cell (versus 0 and 4.40 aberrations/cell for positive and negative controls, respectively) and 30% of patient cells with radial formations (versus 0% and 64% for positive and negative controls, respectively. No spontaneous breakage was observed in the patient sample.
By age 2, the patient had 10 café-au-lait macules larger than 5 mm, delayed developmental milestones, and no other features of neurofibromatosis. The patient had persistent microcephaly (<2 SD) and was small in stature. The complete blood count showed mild macrocytosis, but normal leukocyte and platelet levels, supporting no evidence of bone marrow failure. As of writing, the patient is four years of age, has not developed cancer, and is living, but has been lost to follow up.
Discussion
The clinical characteristics of the proband detailed here closely match the clinical characteristics identified in the other FA-S patients, as recently reviewed (5, 15). This phenotype includes pre- and post-natal growth failure (10/11), microcephaly (11/11), developmental delay (9/11), and hyperpigmented spots (10/11) (5–11). Other common features include limb defects (8/11), microphthalmia (5/11), and structural and skeletal defects such as duodenal stenosis and hip dysplasia (8/11). In contrast to other FA subtypes, none of the 11 FA-S patients had bone marrow failure, including those who have reached adulthood. Due to the rarity of reported FA-S patients, cancer risk cannot be calculated, however six of the ten had been diagnosed with cancer by age 30 and included breast and ovarian in the older patients and leukemia, neuroblastoma, and a brain tumor with the earliest onset at age 13 months. While FA-S appears to result in early on-set of a spectrum of cancers, it does not appear to be as penetrant as FA-D1 (caused by biallelic loss of BRCA2), for example, in which malignancy has been reported to be observed in up to 97% of patients before age 6 (16).
It is expected that a human embryo with two fully penetrant, pathogenic BRCA1 variants would not be viable based on three observations: 1) the extreme dearth of FA-S cases; 2) the absence of reports of individuals homozygous or compound heterozygous for the two high-frequency founder pathogenic variants in BRCA1 [c.68_69delAG, p.E23Vfs*17: maximum allele frequency 0.4% in Ashkenazi Jewish-gnomAD v4.0) and c.5266dupC, p.Q1756Pfs*74: maximum allele frequency 0.1% in Ashkenazi Jewish-gnomAD v4.0] (17); and 3) the observation that brca1 knock-out mice are early embryonic lethal (13). One hypothesis for the viability of FA-S patients is the partial retained function of at least one of the pair of pathogenic alterations. The recurrent observation of FA-S patients with different pathogenic alterations in exon 11 supports this hypothesis as this exon is excluded in well-studied, alternative, in-frame splicing isoforms that whose protein products retain at least partial BRCA1 protein function (6, 7, 10, 15, 18, 19). The most predominant natural, in-frame events include 1) the splicing-out of exons 9 and 10 (r.548_670del); 2) the splicing-out of exons 9-11 (r.548_4096del); and 3) the splicing-out of part of exon 11 (r.788_4096del). The splicing-out of solely exon 11 (r.670_4096del) is observed but is less prevalent.
BRCA1 protein functional studies showed that, despite removing ~60% of the of the protein, loss of exon 11, which is not part of a known functional domain, retains partial homology-directed DNA repair activity (19, 20). In FA-S patients, the natural splicing-out of exon 11 removes the truncating alterations originating here in these patients, which in-turn could lead to the recovery of partial function of BRCA1 proteins originating from this allele. Thus, the recurrence of exon 11 loss of function alterations in FA-S patients is not by chance, but rather variant location is a mechanistically important factor for viability (Figure 1, Table 1).
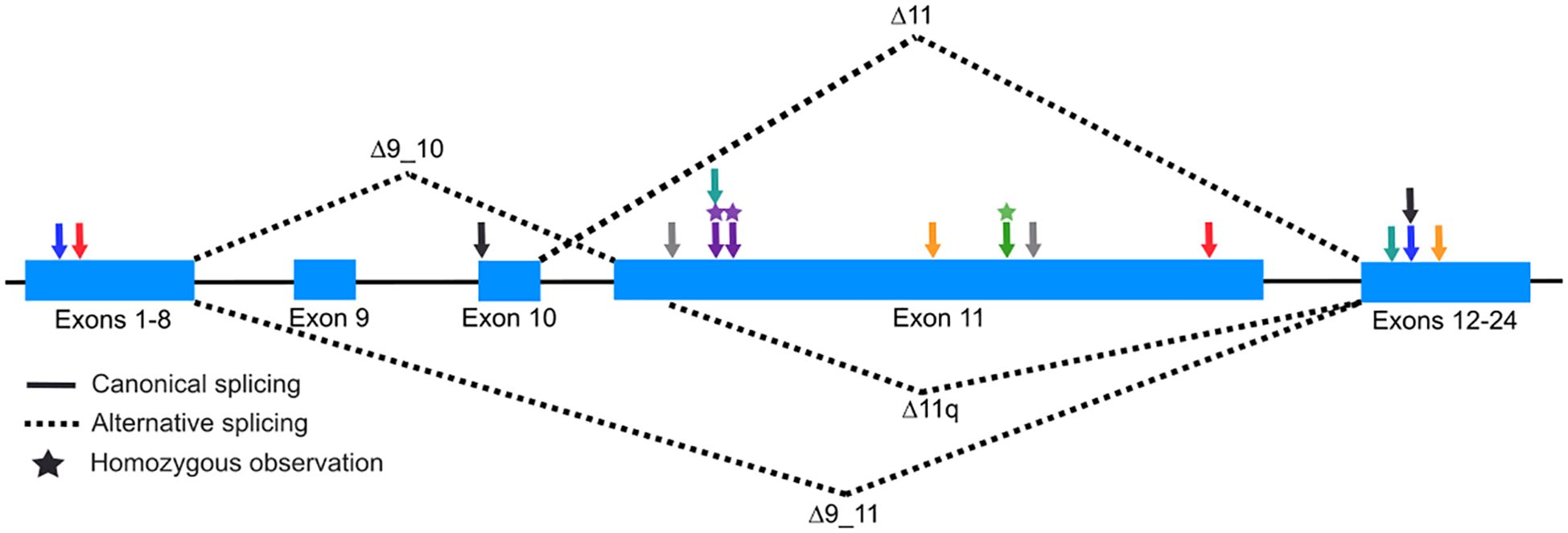
Figure 1. Illustration of BRCA1 alternative splicing isoforms. Schematic (not to scale) of BRCA1 highlighting exons 9 through 11 (coding exons 7 through 9). Solid lines between exons represent canonical splicing and dashed lines represent splicing isoforms which remove exons 9 and 10 (Δ9_10), exons 9-11 (Δ9_11), a majority of exon 11 (Δ11q), and exon 11 (Δ11). Arrows represent the approximate location of BRCA1 variants identified in FA-S patients (Black, Sawyer et al.; Blue, Kuepp et al.; Yellow, Domcheck et al.; Green, Freire et al; Purple, Seo et al.; Gray, Chirita-Emandi et al.; Teal, Borlin et al.; Red, this report).
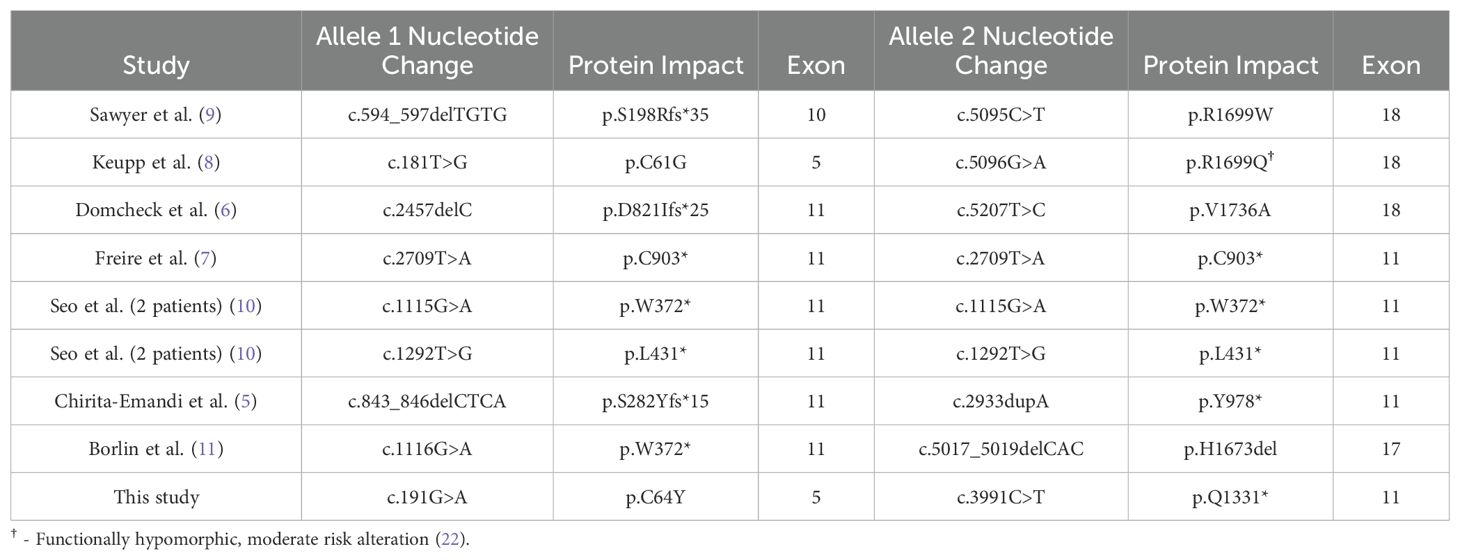
Table 1. BRCA1 germline alterations and protein impacts identified in FA-S patients.
The splicing, functional, and FA-S clinical and genetic data suggest that BRCA1 exon 11 nonsense and frameshifting alterations are definitionally hypomorphic and not complete loss-of-function alleles. The residual function of the alternative splicing isoforms could have an impact on the clinical risk associated with the development of cancers and the clinical management for carriers of these alterations in heterozygous individuals. Analyses of cancer risk conferred by pathogenic variants in BRCA1 exon 11 show a reduction in breast cancer risk, but a relative increase in ovarian cancer risk compared to the other regions of the protein (21). Additionally, both in vitro and in vivo murine functional studies have shown that alternative splicing of exon 11 leads to an increase in resistance to PARP inhibitors and cisplatin due to the expression and retained function of proteins arising from these isoforms (19). Interestingly, cells from the FA-S proband described in this work displayed intermediate sensitivity to DEB and MMC relative to the positive control, which may also be attributable to the retained function of proteins arising from alternative exon 11 splicing. This retained function may also be a partial explanation for why the phenotypic characteristics of FA-S patients are distinct from other FA subtypes, such as the absence of bone marrow failure (15).
Baseline ACMG/AMP variant interpretation guidelines suggest that loss-of-function variants can be assigned the PVS1 code when a variant results in a null allele, however, exon 11 frameshift and nonsense alterations do not strictly meet these criteria. While available evidence suggests that nonsense and frameshift alterations in BRCA1 exon 11 confer an increased risk for cancer, the risk profile and clinical management guidelines for heterozygous individuals may be attenuated relative to other complete loss-of-function alterations in BRCA1. Further study of the genotype-phenotype relationship for heterozygous carriers of BRCA1 exon 11 nonsense and frameshifting variants is warranted to help inform clinical care.
While this work has been mostly focused on alternative splicing as a mode for FA-S patient viability due to partial protein function, missense variants, such as those identified in the FA-S proband in Keupp et al. are another mode for partial protein function (8). The proband in that work had similar clinical features as other FA-S patients but was surprisingly negative by chromosomal breakage analysis. A proposed explanation for the normal chromosomal breakage results is the known reduced risk (relative to other truncating BRCA1 variants) of BRCA1 c.5096G>A, p.R1699Q which maintains partial BRCA1 function (22, 23).
Conclusions
Here we identify an eleventh FA-S patient with biallelic BRCA1 pathogenic alterations confirmed in trans through parental genetic testing. Phenotypic presentation in the patient was consistent with the other FA-S probands in the published literature including pre- and postnatal growth failure, developmental delay, severe microcephaly, dysmorphic features, and café-au-lait spots. Chromosomal breakage analysis was positive using both DEB and MMC. At four years of age, the patient is living and has not been diagnosed with cancer or bone marrow failure.
Most FA-S patients have a truncating variant in exon 11 and here we provide a hypothesis as-to how alternative splicing of this exon supports that this concentration of variants is not by chance, rather a mechanism for non-lethality of FA-S patients conferred by retained partial protein function. The partially retained function may also have implications for the cancer risk and clinical management of individuals heterozygous truncating variants originating in exon 11.
FA-S patients are at increased risk of developing cancers, including breast and ovarian, at an early age. The exact risk profile and spectrum of cancers associated with FA-S patients will require more time and identification of additional probands to fully characterize, but screening and management as with other FA patients is likely warranted.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, VCV000054400.69 https://www.ncbi.nlm.nih.gov/, VCV000037558.11.
Ethics statement
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
CY: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. AL: Conceptualization, Data curation, Investigation, Writing – review & editing. CN: Conceptualization, Investigation, Methodology, Writing – review & editing. AK: Conceptualization, Data curation, Investigation, Writing – review & editing. MR: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. GA: Conceptualization, Formal analysis, Investigation, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We would like to thank the patient’s family for participating in this study.
Conflict of interest
Authors CY, AK, and MR are full-time, salaried employees of Ambry Genetics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. (1994) 266:66–71. doi: 10.1126/science.7545954
2. Friedman LS, Ostermeyer EA, Szabo CI, Dowd P, Lynch ED, Rowell SE, et al. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat Genet. (1994) 8:399–404. doi: 10.1038/ng1294-399
3. Li S, Silvestri V, Leslie G, Rebbeck TR, Neuhausen SL, Hopper JL, et al. Cancer risks associated with BRCA1 and BRCA2 pathogenic variants. J Clin Oncol. (2022) 40:1529. doi: 10.1200/JCO.21.02112
4. Mehta PA and Ebens C. Fanconi Anemia. In: Adam MP, Feldman J, Mirzaa GM, et al, editors. GeneReviews®. University of Washington, Seattle, Seattle (WA (2002). p. 1993–2025. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1401/.
5. Chirita-Emandi A, Andreescu N, Popa C, Mihailescu A, Riza A-L, Plesea R, et al. Biallelic variants in BRCA1 gene cause a recognisable phenotype within chromosomal instability syndromes reframed as BRCA1 deficiency. J Med Genet. (2021) 58:648–52. doi: 10.1136/jmedgenet-2020-107198
6. Domchek SM, Tang J, Stopfer J, Lilli DR, Hamel N, Tischkowitz M, et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. (2013) 3:399–405. doi: 10.1158/2159-8290.CD-12-0421
7. Freire BL, Homma TK, Funari MF, Lerario AM, Leal AM, Velloso ED, et al. Homozygous loss of function BRCA1 variant causing a Fanconi-anemia-like phenotype, a clinical report and review of previous patients. Eur J Med Genet. (2018) 61:130–3. doi: 10.1016/j.ejmg.2017.11.003
8. Keupp K, Hampp S, Hübbel A, Maringa M, Kostezka S, Rhiem K, et al. Biallelic germline BRCA1 mutations in a patient with early onset breast cancer, mild Fanconi anemia-like phenotype, and no chromosome fragility. Mol Genet genomic Med. (2019) 7:e863. doi: 10.1002/mgg3.v7.9
9. Sawyer SL, Tian L, Kähkönen M, Schwartzentruber J, Kircher M, U.o.W.C.f.M. Genomics, et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. (2015) 5:135–42. doi: 10.1158/2159-8290.CD-14-1156
10. Seo A, Steinberg-Shemer O, Unal S, Casadei S, Walsh T, Gumruk F, et al. Mechanism for survival of homozygous nonsense mutations in the tumor suppressor gene BRCA1. Proc Natl Acad Sci. (2018) 115:5241–6. doi: 10.1073/pnas.1801796115
11. Borlin PR, Brazzola P, Frontzek K, Zanoni P, Morscher RJ, Hench J, et al. Cancer in children with biallelic BRCA1 variants and Fanconi anemia-like features: Report of a Malignant brain tumor in a young child. Pediatr Blood Cancer. (2022) 69:e29680. doi: 10.1002/pbc.v69.10
12. Dokal I. Fanconi’s anaemia and related bone marrow failure syndromes. Br Med Bull. (2006) 77:37–53. doi: 10.1093/bmb/ldl007
13. Gowen LC, Johnson BL, Latour AM, Sulik KK, and Koller BH. Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat Genet. (1996) 12:191–4. doi: 10.1038/ng0296-191
14. Woodward ER and Meyer S. Fanconi anaemia, childhood cancer and the BRCA genes. Genes. (2021) 12:1520. doi: 10.3390/genes12101520
15. Hughes T and Rose AM. The emergence of Fanconi anaemia type S: a phenotypic spectrum of biallelic BRCA1 mutations. Front Oncol. (2023) 13:1278004. doi: 10.3389/fonc.2023.1278004
16. Alter BP, Rosenberg PS, and Brody LC. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet. (2007) 44:1–9. doi: 10.1136/jmg.2006.043257
17. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
18. Jasiak A, Koczkowska M, Stukan M, Wydra D, Biernat W, Izycka-Swieszewska E, et al. Analysis of BRCA1 and BRCA2 alternative splicing in predisposition to ovarian cancer. Exp Mol Pathol. (2023) 130:104856. doi: 10.1016/j.yexmp.2023.104856
19. Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, et al. The BRCA1-Δ11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. (2016) 76:2778–90. doi: 10.1158/0008-5472.CAN-16-0186
20. Huber LJ, Yang TW, Sarkisian CJ, Master SR, Deng C-X, and Chodosh LA. Impaired DNA damage response in cells expressing an exon 11-deleted murine Brca1 variant that localizes to nuclear foci. Mol Cell Biol. (2001) 21:4005–15. doi: 10.1128/MCB.21.12.4005-4015.2001
21. Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L, et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. Jama. (2015) 313:1347–61. doi: 10.1001/jama.2014.5985
22. Spurdle AB, Whiley PJ, Thompson B, Feng B, Healey S, Brown MA, et al. BRCA1 R1699Q variant displaying ambiguous functional abrogation confers intermediate breast and ovarian cancer risk. J Med Genet. (2012) 49:525–32. doi: 10.1136/jmedgenet-2012-101037
Keywords: oncology, BRCA1, Fanconi anaemia, splicing, case report
Citation: Young CC, Lahr A, Nestor C, Kaminski A, Richardson ME and Arnold GL (2025) Case Report: Biallelic BRCA1 pathogenic alterations in a Fanconi Anemia patient and clinical implications of variant location. Front. Oncol. 15:1572310. doi: 10.3389/fonc.2025.1572310
Received: 06 February 2025; Accepted: 08 May 2025;
Published: 30 May 2025.
Edited by:
Hua Tan, National Human Genome Research Institute (NIH), United StatesReviewed by:
Tupa Basuroy, Cleveland Clinic Foundation Lerner Research Institute, United StatesYing Wai Chan, The University of Hong Kong, China
Copyright © 2025 Young, Lahr, Nestor, Kaminski, Richardson and Arnold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Colin C. Young, Y3lvdW5nQGFtYnJ5Z2VuLmNvbQ==; Marcy E. Richardson, bXJpY2hhcmRzb25AYW1icnlnZW4uY29t