 Michael D. Diamantidis
Michael D. Diamantidis- Thalassemia and Sickle Cell Disease Unit, Department of Hematology, General Hospital of Larissa, Larissa, Greece
The use of the BCL2 inhibitor venetoclax in combination with hypomethylating agents (HMA) is a revolution for the treatment of frail and elderly acute myeloid leukemia (AML) patients. This effective treatment strategy is increasingly more and more applicable for other subsets of AML patients and is currently being tested in numerous clinical trials in combination with other drugs in all treatment lines. In particular, venetoclax combinations can also serve as a definitive therapy or as an effective bridge to allogeneic hematopoietic stem cell transplantation (HSCT). However, the factors affecting response to venetoclax in the abovementioned AML patients are not completely clear and understood until today. The aim of this review is to describe the molecular and clinical patterns of response and durable remission of venetoclax-based combinations in AML patients. Hence, mutations in IDH1, IDH2, ASXL1, NPM1, DDX41, chromatin-cohesin complex and splicing-factor genes predict superior response to venetoclax, while inferior response to the drug has been observed for FLT3-ITD, KRAS, NRAS and TP53 gene mutations. Intriguingly, the achievement of measurable residual disease (MRD) negativity in the first four cycles of venetoclax administration characterizes a subgroup of NPM1-mutated AML patients with a more favorable outcome. Even though focus will be given on factors influencing response to the drug in this review, the main mechanisms of resistance to venetoclax in AML patients will also be discussed.
Introduction
The combination therapy of venetoclax and azacitidine (VEN-AZA) has demonstrated substantial benefits for patients with acute myeloid leukemia (AML) who are ineligible for intensive chemotherapy, particularly for those with specific molecular characteristics, such as NPM1-mutated AML or IDH-mutated AML (1). This regimen [Venetoclax 400 mg per os; days 1-28) and azacitidine (75 mg/m2; subcutaneously; days 1-7/28-day cycle] became the cornerstone and standard of care for the elderly and frail patients with AML, irrespective of the mutational status, based on the results of the VIALE-A randomized clinical trial (2). The VEN-AZA combination was associated with a statistically significant longer median overall survival (OS) of 14.7 months versus only 9.6 months for those who had received AZA alone (2). The latest 3-year follow-up of the study was also in favor of the combination, showing a three-year survival rate of 25% for VEN-AZA versus 10% for AZA alone (3).
However, the combination VEN-AZA often exhibits limited durability in response for numerous patients. In fact, response patterns to VEN remain poorly understood. Certain patients with AML demonstrate a remarkable response to the initial cycles of the drug, whereas others display various levels of response or no response at all. Consequently, research has increasingly concentrated on how more complex regimens may enhance outcomes, particularly through the incorporation of targeted drugs, such as FLT-3 or IDH inhibitors to VEN-AZA.
Moreover, despite the introduction of innovative techniques such as BH3 profiling to stratify AML patients undergoing VEN treatment in clinical trial settings (4), there is an absence of clinically relevant biomarkers, that predict the response to the combination of VEN-AZA. Conversely, resistance to the treatment is observed with the emergence of novel mutations compared to the initial ones. Despite the association of anti-apoptotic BCL2 family proteins with AML pathogenesis, the specific functional significance of individual proteins, including BCL2, BCL-xL, MCL1, BAX, and BFL1, remains inadequately clarified. The expression of these proteins is altered during the progression of AML by the administration of the selective BCL2 inhibitor VEN (5–8). The latter offers insights into the mechanisms of resistance to VEN in AML, suggesting potential treatment targets.
This review aims to elucidate the complex mechanisms of response and resistance to VEN in AML patients unfit for intensive therapy.
Response to venetoclax
The use of BCL2 inhibitors, like VEN, in combination with hypomethylating agents (HMAs) has been associated with increased overall survival (OS) in AML patients over 65 years, who have BCL2 overexpression (9). The observed synergy between AZA and VEN can be attributed to various mechanisms. AZA induces pro-apoptotic modifications, including a reduction in MCL1 protein level, thereby facilitating the apoptotic properties of VEN (10, 11). Furthermore, through an increase in NOXA levels, AZA primes leukemic cells to VEN-induced apoptosis (12, 13). In addition, HMA and VEN combinations induce reactive oxygen species (ROS) accumulation and mitochondrial ROS production in leukemic cells, triggering the apoptotic mechanisms and facilitating the oxidative death of AML cells (14–16).
An intriguing observation was that AML patients with IDH mutations (either IDH1 or IDH2) showed high remission rates with an OS of 24.5 months and a composite complete remission CRc (CR + CRi) rate of 79% when treated with VEN-AZA (17). These percentages were much higher compared to the respective data of the VIALE-A study, which initiated VEN plus HMAs in frail or elderly AML patients ineligible for intensive chemotherapy (median OS: 14.7 months, CR/CRi: 66.4%) (2). Despite the small sample size in the molecular subgroups, the limited number of patients in the AZA group and the lack of thorough analysis between the IDH-mutant isoforms and concomitant mutations, the combination of VEN-AZA emerged as a very good competitive candidate for the IDH-mutated AML, apart from treatment option with the IDH inhibitors [enasidenib, ivosidenib (IVO), olutasidenib] (17, 18). Hence, one of the major factors predicting a good response to VEN-AZA in AML patients is the presence of IDH mutations (3, 18–20).
Intriguingly, among the IDH2-mutated subgroup a CRc rate of 86%, a CR rate of 56% and a not reached median OS have been observed, demonstrating a unique benefit for these AML patients with VEN-AZA. In addition, patients harboring the IDH1 mutation and receiving the combination of VEN-AZA also show a favorable response (CRc: 67%, CR: 27%, median OS: 15 months) (17). The latter is superior to the IDH1 inhibitor IVO in the first line treatment of AML, as the IVO data indicate a CRc rate of 42% and an OS of 12.6 months (21). Nevertheless, the approved combination of IVO plus AZA in IDH1-mutated AML demonstrates a median OS of 24 months, an event-free survival (EFS) of 37% at 12 months, a CRc rate of 53%, along with a median duration of response of 22.1 months (22). AML patients have nowadays many treatment options with the approval of novel targeted therapies, except for the combination of VEN-AZA. The most important clinical trials involving VEN, AZA and their combinations in AML patients are shown in Table 1. Regarding toxicity, grade ≥3 is provided, unless otherwise stated for any grade side effects.
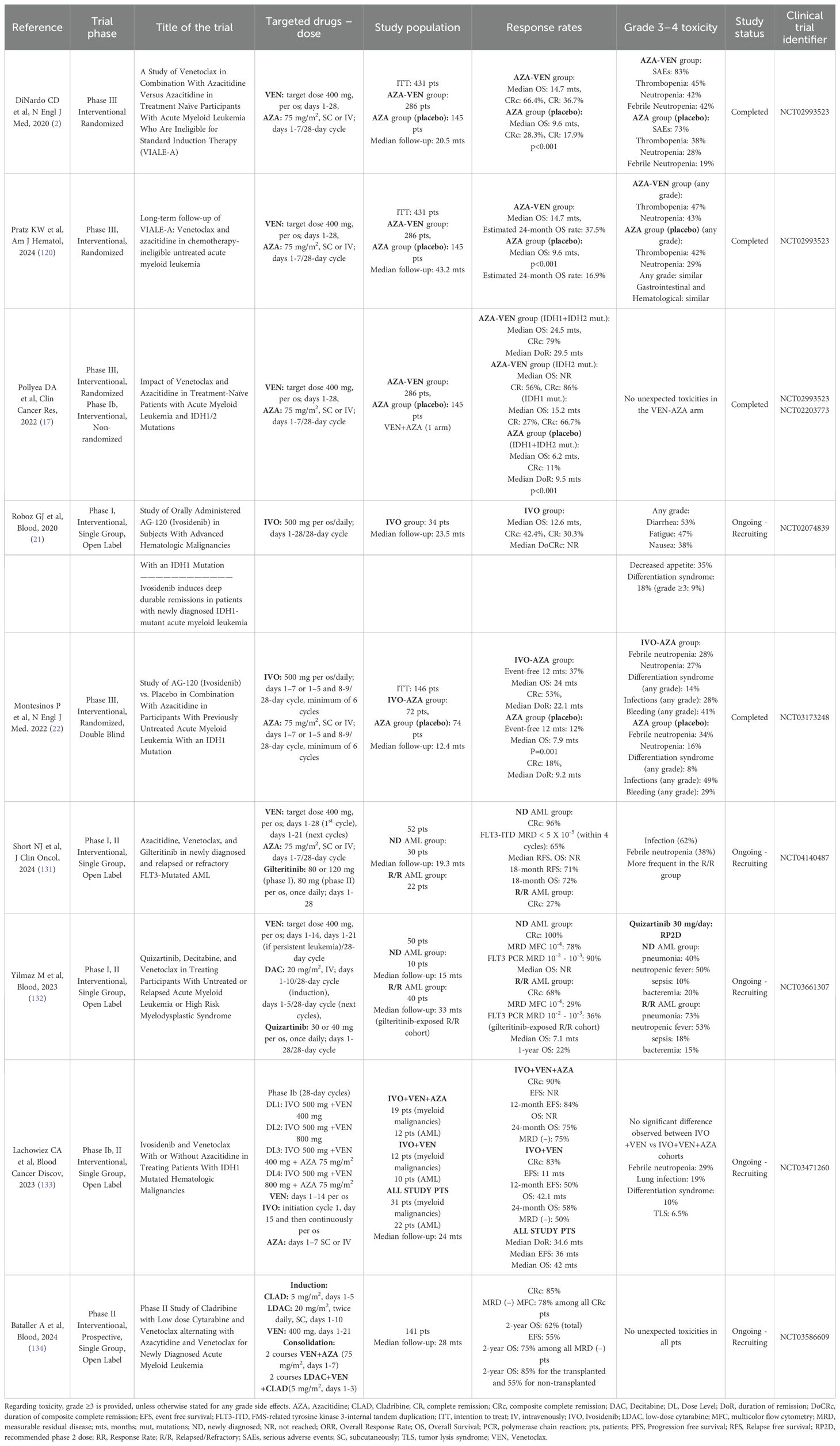
Table 1. Clinical trials involving venetoclax, azacitidine, decitabine, ivosidenib, gilteritinib, quizartinib, cladribine and their combinations in patients with acute myeloid leukemia (AML) ineligible for intensive chemotherapy.
The biological basis for the favorable response of the IDH-mutated AML (especially IDH2-mutated AML) to VEN-AZA is largely unknown. It has been documented that the oncometabolite (R)-2-hydroxyglutarate (2-HG) disrupts mitochondrial function via inhibition of cytochrome c oxidase, thereby increasing the efficacy of VEN in IDH-mutated AML (23, 24). The latter is a possible theory explaining the remarkable response of IDH-mutated AML to VEN.
It has also been reported that R140 IDH2 mutations have a more favorable prognosis, compared to R172 IDH2 mutations and R132 IDH1 mutations in AML patients (25–27). However, such an association has not been observed in a recent large cohort evaluating the type and incidence of IDH mutations in AML (28). Intriguingly, R140 IDH2 mutations are highly associated with nucleophosmin-1 (NPM1) mutations (27). NPM1 is mutated in almost one third of AML patients and has been linked with a favorable prognosis in de novo AML, but with a dismal prognosis in relapsed or refractory (R/R) AML (28–30). The association of NPM1 with R140 IDH2 might justify the favorable prognosis of R140 IDH2 mutated AML, especially in patients receiving VEN-AZA (18, 19). Moreover, triplet regimens targeting IDH1/IDH2 mutations in combination with VEN/AZA, either at the presence of IDH mutations at diagnosis or at AML relapse, are analyzed in the future directions part of this work.
Furthermore, an NPM1-mutated AML is an excellent target for VEN-based therapies (18, 30). Patients with this leukemic subset receiving low-dose cytarabine (LDAC) plus VEN exhibit a response rate of 78% and a median OS of more than 2 years (31, 32). Furthermore, bone marrow blast reductions >50% might be the case only after 7 days of treatment with VEN in the NPM1-mutated AML (33, 34). Interestingly, a higher benefit group has been observed with a median OS expectation of 39 months for a subset of patients with NPM1-mutated AML lacking either TP53, FLT3-ITD, KRAS or NRAS variants (18, 32). The combinations of VEN plus NPM1 downstream targets, such as menin and exportin 1 (XPO1) inhibitors are described in the future directions part of the present article.
A possible molecular mechanism linking VEN with NPM1-mutated AML derives from studies in the human monocytic leukemia cell line THP-1 harboring the NPM1 mutation. VEN inhibits the anti-apoptotic activity of the transcription factor NF-κB and suppresses BCL2, which selectively sequesters the apoptotic protein BAX, thereby targeting the anti-apoptotic pathways observed in NPM1-mutated AML (35, 36). VEN blocks the ability of the anti-apoptotic protein BCL2 to bind to BAX/BAK, acting as a BH3-mimetic, because of its unique structural similarity to BH3-containing proteins, resulting in competitive inhibition (36, 37).
It has been proposed that the evaluation of BCL2 levels at AML diagnosis by mRNA quantification or immunohistochemistry, could serve as a biomarker predicting the response to VEN or anti-BCL2 treatment, because elevated BCL2 protein levels correlate with higher sensitivities to VEN (38). However, the amount of BCL2 capable of binding and sequestering proapoptotic proteins (BAX, BIM) provides a better explanation for this sensitivity, rather than high BCL2 levels alone (39, 40). Intriguingly, biomarkers predicting response to VEN or anti-apoptotic therapy in AML patients might involve BCL2 mutations of the VEN binding site or other BCL2 family gene mutations (8). Moreover, patients with core binding factor (CBF) AMLs exhibit CR/CRi response rates of 80% (41) or 70% (42) when treated with VEN/HMAs, with a significant better response for patients with inv (16) compared to t (8, 21) patients (100% vs 30% achieving CR/CRi after the first cycle) (39, 42). Clinical trials are testing the latter finding (NCT04628026). Finally, CR rates over 90% with measurable residual disease (MRD) clearance have been reported for the favorable risk subgroup with the combination of VEN plus CLIA (cladribine, idarubicin, cytarabine) or FLAG-Ida (fludarabine, cytarabine, granulocyte colony-stimulating factor, idarubicin) (43).
The ELN risk classifications of 2017 (44) and 2022 (45) failed to stratify prognostic outcomes for frail AML patients ineligible for allogeneic hematopoietic stem cell transplantation (HSCT) receiving non-intensive chemotherapy, such as VEN-AZA (46). These classifications were formed incorporating the intensive treatment protocols applied to AML patients and excluding less-intensive treatments, like VEN-AZA, because the latter therapeutic approaches are more novel comparatively (46).
The molecular signature of the four genes TP53, KRAS, NRAS and FLT3-ITD guides prognosis and classifies AML patients who received VEN-AZA to three different prognostic groups. A favorable risk group with all the aforementioned four genes negative (median OS: 26.5 months), an intermediate risk group by positive FLT3-ITD and/or mutated KRAS and/or mutated NRAS (median OS: 12.1 months) and an adverse risk group with mutated TP53 (median OS: 5.5 months) (46). Moreover, this four-gene prognostic combination was further confirmed by Bataller et al. (47). Novel therapeutic avenues are necessary for AML patients harboring KRAS, NRAS and FLT3-ITD mutations (also named activating signaling gene mutations), because they exhibit a relatively unfavorable outcome with VEN-AZA (47). Intriguingly, this evaluation of response to VEN with the contribution of the four abovementioned genes does not include AML patients with a prior myelodysplastic syndrome (MDS) or a prior myeloproliferative neoplasm (MPN) or prior exposure to HMAs before the initiation of VEN and HMAs, due to the exclusion of such patients from the relevant clinical trials of VEN in AML.
Intriguingly, assessment of NPM1 MRD evaluated by quantitative real-time polymerase chain reaction (qRTPCR) provides valuable prognostic intuition in AML patients who received VEN combinations. The achievement of bone marrow (BM) MRD negativity after 4 cycles of combination treatment with VEN and HMAs demonstrated a 2-year OS of 84%, compared with 46% for MRD positive by the end of cycle 4 for AML patients (48). Thus, a patient group with a better outcome has been described for NPM1-mutated AML patients who received VEN. VEN-based combination treatment is a very promising targeted therapy for NPM1-mutated AML, because it has been associated with durable molecular remission and increased OS (18, 48). Even in patients with NPM1 molecular failure, VEN combinations serve either as a bridge to transplant or as definitive therapy, as 71% of these patients become MRD negative (49). MRD monitoring is emerging and plays a complementary role in defining prognosis for AML patients receiving VEN-AZA (50). Thus, AML patients with CRc and MRD < 10-3 after treatment with VEN-AZA had longer duration of response, EFS and OS, compared to responding patients with an MRD ≥ 10-3 (50).
Another key element for the prognosis and the response to VEN-AZA in NPM1-mutated AML is the co-existence of activating signaling mutations (FLT3-ITD, KRAS, NRAS). NPM1-mutated AML without these mutations has a median OS of 39 months, in contrast to only 9.9 months when these kinase signaling mutations are present in parallel with the NPM1 mutation (51). The results are similar for IDH2 mutated AML, RUNX1 mutated AML and AML with myelodysplasia related (MR) gene mutations, depending on the absence or presence of an activated kinase pathogenic variant (51).
Additionally, patients with DDX41 mutated AML exhibit a particularly favorable outcome with 91.1% of OS probability at 2 years (median OS not reached) after being treated with VEN-AZA (52, 53). Moreover, Weng G et al. demonstrated that ASXL1, NPM1 and chromatin-cohesin complex genes predict superior response to VEN (54). Improved responses of AML patients with ASXL1 mutations (55) or with splicing factor gene mutations (56), along with exceptional positive outcomes for those harboring MR gene mutations with the combination of VEN plus HMAs have also been reported (57).
Interestingly, the elimination of a population of leukemic stem cells (LSCs) determines the response to the combination of VEN-AZA. The proteinic expression of BCL2, BCL-xL, and MCL1 in these LSCs, not only significantly predicts the response to the combination with high sensitivity and specificity, but correlates the combinatorial levels of BCL2 family members with an increased EFS in AML patients as well (58). The flow cytometry-based “Mediators of apoptosis combinatorial score” (MAC-Score) incorporates a combination of the expression of the abovementioned BCL2 family members, reliably predicting the response of AML patients to VEN-AZA. LSCs of refractory or relapsed patients to the combination exhibit perturbed apoptotic dependencies (58).
The relevant factors contributing to a favorable response to VEN-AZA in AML patients are shown in the upper part of Table 2.
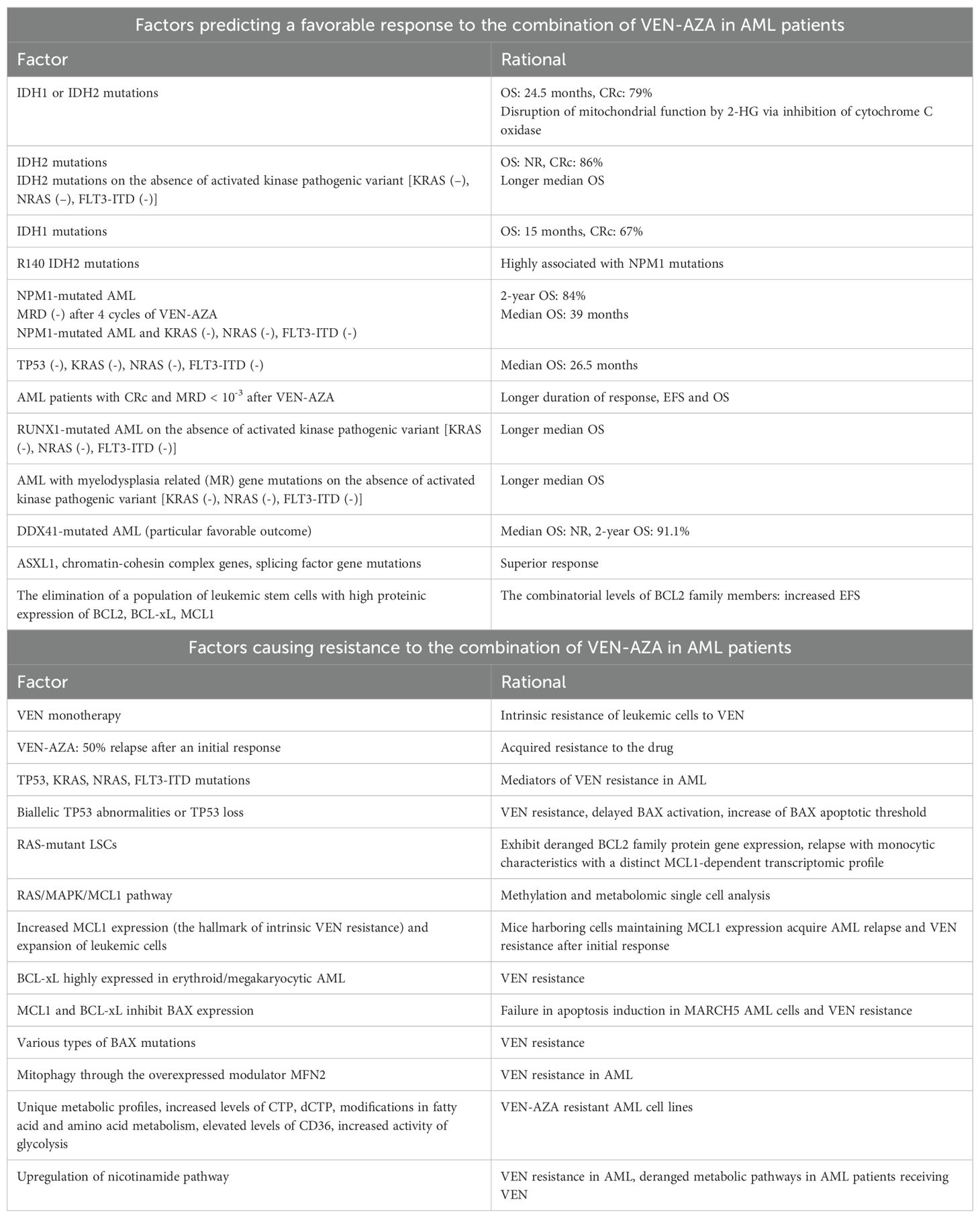
Table 2. Factors predicting a favorable response (upper part of the table) and factors causing resistance (lower part of the table) to the combination of VEN-AZA in AML patients.
Resistance to venetoclax
Even though VEN targets BCL2 overexpression in AML patients, the monotherapy of the drug has minimal therapeutic properties, hinting a mechanism of intrinsic resistance of the leukemic cells to the drug (59). Moreover, almost 27% of AML patients show primary resistance and more than 50% relapse after an initial response to VEN-AZA, suggesting the existence of acquired resistance, in parallel with the intrinsic one (2). Nevertheless, the precise molecular mechanisms driving resistance to VEN in AML patients have yet to be identified.
The molecular signature of the four genes TP53, KRAS, NRAS and FLT3-ITD not only defines response to VEN-AZA, but also determines resistance to VEN (60, 61). In other words, TP53, KRAS, NRAS and FLT3-ITD are mediators of VEN resistance in AML (5, 60, 62, 63). Adaptive resistance is often associated with biallelic TP53 abnormalities or TP53 loss or kinase activation, in particular FLT3 ITD or RAS mutations (18, 60, 62). Interestingly, RAS mutations are late events in leukemogenesis and can originate from a different clone, compared to the ancestral ones. RAS-mutant leukemic stem cells (LSCs) are resistant to VEN, exhibit deranged BCL2 family proteins gene expression and relapse with monocytic characteristics (64, 65). Resistant monocytic AML exhibits a distinct transcriptomic profile and is MCL1-dependent to maintain survival (65, 66). DNA sequencing experiments also provided evidence of selection of RAS-mutated clones in AML patients treated with VEN, conferring resistance to the drug (65, 67). This resistance and AML relapse is mainly driven by the RAS-mutant leukemic stem cells (LSCs) alone, rather than the monocytic differentiation state of the LSCs, which is the result of the initially subclonal, oncogenic RAS driver variants after selective clonal pressure, caused by VEN treatment (64). Finally, the activation of RAS/MAPK/MCL1 pathway has been recognized as a main mechanism of VEN resistance in AML through gene and protein expression, along with methylation and metabolomic single cell analysis (67).
More precisely, AML patients receiving VEN-AZA, might harbor FLT3-ITD mutation at diagnosis, or may develop the same mutation at relapse, demonstrating secondary or acquired VEN resistance during the course of the disease (68). Moreover, VEN resistance can be observed in AML patients, through activation of intracellular signaling pathways by mutations in genes involving key kinases, such as RAS or PTPN11 (68, 69). FLT3-ITD triggers PI3K-protein kinase B (Akt), RAS-MAPK and STAT5 pathways. STAT5 controls BCL-xL and Akt controlling MCL1 stabilization in FLT3-ITD leukemic cells (70). The presence of FLT3-ITD mutations either at diagnosis or driving AML relapse after VEN-AZA treatment, exhibits apparent therapeutic applications with triplet regimens targeting FLT3-ITD, highlighted in the future directions part of this work.
Defective TP53 has a major role in determining VEN resistance in AML, because TP53 knockout impairs apoptotic cell death caused by VEN (71). An initial activity of small degree with a rapid evolution and growth of subsequent TP53-mutant clones causing a quick relapse has been observed in TP53-mutated AML (18). Suboptimal concentrations of VEN cause delayed BAX activation and the latter increases BAX apoptotic threshold (72). In this regard, it is more difficult for VEN to induce apoptosis of leukemic cells, due to the elevated apoptotic threshold. TP53 knockout delays activation of the apoptotic proteins BAX and BAK and thus apoptosis, because the target genes of TP53, such as NOXA, BIM and PUMA are implicated in BAX and BAK activation (62, 72). Overall, VEN is not effective in TP53-mutated AML, as defective TP53 confers resistance to the drug (73).
Because VEN is a selective inhibitor of BCL2 protein expressed in AML, other anti-apoptotic BCL2 proteins, such as BCL-xL (74), MCL1 (66, 67, 75), BCL2A1 (76, 77) and BFL1 serve as mediators for primary or acquired secondary resistance to VEN by leukemic cells, via alterations of the apoptotic pathways (5–7, 69). These anti-apoptotic proteins through specific interactions cause sequestration of BH3-only proteins, preventing them from synergizing with BAK or BAX proteins to initiate apoptosis (78). BAX inactivation, along with TP53, leads to VEN resistance in AML (71). It has been observed that AML relapse arises in mice harboring cells that maintain MCL1 expression, after an initial successful inhibition of MCL1 and impermanent eradication of leukemic cells (79). Additionally, MCL1 is the highest expressed BCL2 family protein in AML, thereby proving the relation between MCL1 and the progression of the disease, along with the expansion of leukemic cells (60, 80–84). Selective BCL2 inhibition by VEN leads to the death of leukemic cells. Nevertheless, this BCL2 inhibition is antagonized via an increase of MCL1 expression by AML, which is the hallmark of intrinsic VEN resistance (38, 67, 85). It has been observed that genetic silencing of MCL1 reverses the resistance to VEN and restores the anti-leukemic properties of the drug (38, 67). In addition, BCL-xL is highly expressed in erythroid/megakaryocytic AML and it is responsible for VEN resistance in this leukemic subset (74).
As VEN is directly attached to the BCL2 BH3-binding grooves of the anti-apoptotic proteins (38, 86), the protein BAX is released from BCL2 and upon its self-assembly, novel pores are constructed, permeabilizing the outer membrane of the mitochondria, which becomes vulnerable to external and internal stress factors (87, 88). The subsequent release of cytochrome c from the mitochondria, induces cellular apoptosis, via activation of the caspase programmed cell death pathway (87, 88).
Intriguingly, mutations in the BCL2 family members after initial response to VEN treatment is another major mechanism driving VEN resistance in AML. Resistance-associated point mutations in the BCL2 protein, such as BCL2 Asp103Glu, Val148Leu and Phe104Leu have recently been identified in AML (89). These BCL2 variants are polyclonal, arise during VEN therapy and reduce the affinity of BCL2 with the drug, establishing secondary resistance to VEN in AML, despite VEN dose escalation (81, 89). Moreover, secondary inactivating frameshift/nonsense or missense mutations involving the BAX apoptotic gene have also been described in relapsed AML patients who received VEN (90).
It has been documented that the MARCH5 mitochondrial E3 ligase complex, which consists of MARCH5, UBE2K and UBE2J2 controls VEN resistance and sensitivity, through modulation of BAX saturation of the anti-apoptotic proteins (91). When genome-wide CRISPR/Cas9 techniques were applied in a resistant to VEN AML mouse model, the levels of MARCH5, UBE2K and UBE2J2 were significantly decreased after exposure to VEN, suggesting a combined lethal effect. However, after VEN treatment, MCL1 and BCL-xL entrapped BAX, which leads to failure in the induction of apoptosis in MARCH5 AML cells (91). Furthermore, BAX knockdown resulted in VEN resistance (75). Nevertheless, in MARCH5 knockout cells, the released BAX from BCL2, failed to attach to MCL1, because another molecule (NOXA) occupied the respective MCL1 BH3-binding domains. In this way, BAX effectively caused mitochondrial apoptosis and overcame the MCL1-induced resistance to VEN in AML (91). Intriguingly, the leading role of MCL1 in the development of resistance to VEN treatment in AML has also been confirmed in the aforementioned experiments. Finally, various types of BAX mutations, leading to different alterations in the structure and the function of the BAX protein, have been established in relapsed AML patients who received VEN (90).
Another interesting mechanism involving the mitochondrial axis and promoting resistance to VEN in AML is mitophagy, which is a selective type of autophagy in which mitochondria are destructed, through autophagic degradation (92, 93). MFN2, a mitophagy modulator, is responsible for inducing resistance in BH3 mimetics, like VEN in AML (93). Thus, MFN2 overexpression leads to increased interactions between mitochondria and endoplasmic reticulum, causing enhanced mitochondrial clearance and resistance to VEN, which in the abovementioned cell background of mitophagy, cannot function as an effective drug in AML (93). Furthermore, a gene implicated in mitochondrial metabolism is CLPB. This gene interacts with the OPA1 protein, maintains mitochondrial cristae structure and it is overexpressed upon VEN resistance in AML (94).
Unique VEN-AZA resistant AML cell lines have been developed showing more than 300-fold persistent resistance compared to the parental lines. These cells have unique metabolic profiles, increased levels of cytidine triphosphate (CTP) and deoxycytidine triphosphate (dCTP), modifications in fatty acid and amino acid metabolism, elevated levels of the fatty caid transporter CD36, increased levels of MCL1, decreased levels of BAX and increased utilization and reliance on glycolysis (75). Intriguingly, pharmaceutical inhibition of glycolysis re-sensitized the resistant cells to VEN-AZA (75). AML cells generate the energy needed for their metabolic functions from the citric acid cycle and the oxidative phosphorylation (OXPHOS). VEN inhibits the electron transport chain complexes I, II and IV, thereby decreasing OXPHOS and destroying leukemic cells (84, 94). However, AML cells develop resistance to VEN, because they upregulate the nicotinamide pathway (95) or the fatty acid metabolism (96).
Another biological mechanism driving VEN resistance in AML involves ATP-binding cassette (ABC) transporters, which are major regulators of drug efflux. They are implicated in detoxification, cell signaling and metabolism by transporting substrates, like metabolites, nucleosides, ions, hormones, lipids and cytotoxic drugs across the cell membrane (97). ABC transporters, such as ABCC1, have the potential to efflux anticancer agents, as their overexpression has been associated with multidrug resistance and poor response to chemotherapy (98). It has been demonstrated that ABCC1 overexpression induces resistance to VEN in AML by reducing the intracellular drug levels, thereby predicting poor response to VEN (99). Conversely, ABCC1-specific export of glutathionylated substrates leading to the inhibition of glutathione metabolism, increases the response to VEN in AML patients. In other words, glutathione metabolism and ABCC1 limit VEN efficacy in AML (99). The molecular mechanisms describing the deranged metabolic dependency in AML patients receiving VEN have been extensively reported and are beyond the scope of this review (81, 82, 84, 100, 101).
Finally, the immune microenvironment of AML causes increased production of pro-inflammatory cytokines, driving leukemic progression (102). In particular, aberrant myeloid cell proliferation of monocytic origin, observed in del7/7q AML patients, leads to enhanced IFNγ signaling, which is a key feature of VEN resistance in AML (103).
The corresponding mechanisms causing resistance to VEN-AZA in AML patients are described in the lower part of Table 2.
Discussion - concluding remarks – future directions
Therapeutic ways of overcoming VEN resistance in AML target MCL1 and BCL-xL (5, 60–62). There are direct MCL1 inhibitors, like S63845/S64315 (MIK665) (104), AZD5991 (105), AMG-176 (tapotoclax) (106) and AMG-397 (murizatoclax) (107) or indirect MCL1 inhibitors, such as CDK9 inhibitors [alvosidib (108, 109), dinaciclib (110), AZD4573 (111, 112), PIK-75 (113)], deubiquitinase inhibitors (114) and ceramide (SPHK1 inhibitor) (115, 116).
However, the results of direct MCL1 inhibitors in clinical trials were not satisfactory, due to side effects, like myelosuppression (104, 106, 117) or cardiotoxicity (107). Importantly, MCL1 knockout mice show fatal cardiac failure (118, 119). Conversely, heterozygous mice for MCL1 exhibit no cardiac manifestations, thereby rendering decreased dosing a possible effective future therapeutic strategy for direct MCL1 inhibitors in AML. Alternatively, indirect MCL1 inhibitors have a better tolerability with less toxicities and are under extensive research as potent drugs overcoming VEN resistance in AML (112). Moreover, targeting BCL-xL is a potential treatment option in erythroid/megakaryoblastic leukemias resistant to VEN (74).
An updated safety analysis regarding long-term use of VEN-AZA in elderly AML patients or those with comorbidities identified no novel safety concerns. Treatment emergent adverse events (TEAEs) were similar between the VEN-AZA arm versus the placebo-AZA arm. Slightly higher rates of hematologic TEAEs were reported in the VEN-AZA arm, compared to the placebo-AZA arm. These were grade ≥3 thrombocytopenia (46% vs 40%), neutropenia (43% vs 29%) and febrile neutropenia (43% vs 19%) (120). With a median follow-up of 43.2 months, 86% of the VEN-AZA arm and 77% of the control arm had serious TEAEs. Fatal TEAEs reached 25% in the VEN-AZA arm, compared to 22% in the placebo-AZA arm (120). Since VEN-AZA is applied for a prolonged use or in combination with other drugs/therapies in elderly AML patients, a retrospective study evaluated the possible cardiac complications. Cardiomyopathy, pericarditis/effusions, along with non-ST elevation myocardial infarction were the major cardiac complications observed in the 7.6% of a cohort of newly diagnosed (ND) AML patients. Males were more likely and DNMT3A-mutated patients less likely to be affected. These cardiac complications were associated with a trend towards shorter OS and CRc (121). Finally, the general guidelines for preventing tumor lysis syndrome (TLS), along with the dose modifications for VEN with concomitant use of CYP3A (protection from fungal infections) and P-gp inhibitors are well known and applied from the clinicians (122).
Potential strategies to overcome resistance to VEN-AZA in AML include combination with other agents like FLT3, IDH, RAS, menin, XPO1 and immune checkpoint inhibitors. Current triplet regimens under clinical trials involving VEN/HMAs with FLT3 inhibitors are either with quizartinib (NCT03661307, NCT04687761) or gilteritinib [NCT04140487 (Table 1), NCT05010122, NCT05520567, NCT06317649, NCT06696183] in the frontline setting or in the R/R disease. Gilteritinib is tested in most trials with VEN/AZA and in one trial with VEN plus ASTX727 [oral decitabine (DAC) and the cytidine deaminase inhibitor cedazuridine], whereas quizartinib with DAC plus VEN (NCT03661307, Table 1) or either with VEN/AZA or with low-dose cytarabine (LDAC)/VEN (NCT04687761, VEN-A-QUI trial).
Moreover, triplet regimens with IDH inhibitors under clinical trials are the combinations of IVO plus VEN/AZA or VEN in the R/R setting (NCT03471260, Table 1) and the combination of ASTX727 plus VEN plus any type of IDH inhibitor (NCT04774393). Another trial involves the combinations of IVO plus VEN (NCT06611839) both in the first line and in R/R AML. Furthermore, the addition of VEN to enasidenib and AZA (NCT03683433) has been associated with 100% CRc in elderly patients with IDH2-mutant AML in the frontline setting (123). Finally, olutasidenib with VEN/AZA (NCT06782542) is under evaluation in IDH1-mutated ND AML patients eligible for intensive induction chemotherapy (phase II OLUVENAZA trial). Another phase Ib/II clinical trial is under conduction testing olutasidenib plus VEN/DAC (NCT06445959) in ND and R/R AML patients.
Menin inhibitors are also under clinical trials to treat ND or R/R KMT2Ar, NUP98r or NPM1c AML. The therapeutic combinations applied in clinical trials involve the triplets of Revumenib plus VEN plus ASTX727 (NCT05360160), Revumenib plus VEN/AZA (NCT06652438), Ziftomenib plus VEN/AZA (NCT05735184, KOMET-007 study) (124) or Bleximenib plus VEN/AZA (NCT05453903). The promising results of these combinations in specific leukemic subsets are being evaluated. Based on the results of the AUGMENT-101 clinical trial (NCT04065399), Revumenib was approved in the United States for R/R acute leukemia with a KMT2A translocation in adult and pediatric patients of 1 year and older (125).
Clinical trials with XPO1 inhibitors, like selinexor or eltanexor in combination with VEN or with VEN/AZA have been completed or are under conduction, either in frontline AML or in the R/R setting. The most important are the combinations of selinexor plus VEN (NCT03955783), selinexor plus VEN/AZA (NCT05736965) or eltanexor plus VEN (NCT06399640). The initial encouraging results are important, and a thorough analysis is underway to observe the efficacy and safety of the abovementioned therapeutic combinations in AML.
Because RAS mutations emerge as a major mechanism driving VEN resistance in AML (64, 81, 101, 126), RAS inhibitors have also been tested. RAS mutations exert their leukemogenic actions through activation of the PI3K, Akt, mTOR, RAF, MEK and ERK downstream signaling pathways (126). Trametinib, a MEK inhibitor, in combination with VEN/AZA showed modest activity and substantial toxicity in R/R AML (NCT04487106), with similar responses to trametinib monotherapy (127). Another MEK inhibitor, cobimetinib, acting through indirect RAS inhibition, along with VEN, showed limited preliminary efficacy similar to VEN monotherapy with significant toxicity in the R/R setting (NCT02670044) (128). However, the combination of VEN with the murine double minute 2 (MDM2) inhibitor idasanutlin exhibited manageable safety and encouraging efficacy in unfit patients with R/R AML (NCT02670044) (129). Future clinical trials targeting RAS and MEK pathways in R/R AML with improved efficacy and acceptable toxicities are essential.
Immune checkpoint (PD1) inhibitors in combination with VEN/HMAs are being tested in R/R AML after initial VEN resistance. Pembrolizumab plus DAC ± VEN (NCT03969446), and Tislelizumab plus VEN/AZA (NCT06536959) are promising future therapeutic targets. Moreover, an ongoing open-label phase I/II study of Relatlimab (anti-LAG-3) plus Nivolumab (anti-PD1) in combination with AZA ± VEN for the treatment of patients with R/R or ND AML has demonstrated initial promising efficacy and manageable toxicity (130). Other agents currently tested with VEN/AZA as triplet regimens to overcome VEN resistance in AML are tagraxofusp, which targets CD123 (NCT05442216), the cyclin-dependent kinase 9 (CDK9) inhibitor QHRD107 (NCT06532058), the histone deacetylase (HDAC) inhibitor chidamide (NCT05305859) and the MDM2 inhibitor siremadlin (NCT05155709).
As more knowledge is established regarding the molecular pathways implicated in VEN resistance in AML patients, more targeted approaches will be applied and novel therapeutic avenues will be discovered. Extensive research is essential in order to decipher the aforementioned complex mechanisms of VEN response and mainly, resistance in AML.
Author contributions
MD: Conceptualization, Investigation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ABT-199, Venetoclax; BAK, Bcl-2 homologous antagonist-killer; BAX, Bcl-2-associated X Protein; BCL2, B-cell lymphoma 2; BCL-xL, B-cell lymphoma-extra large protein; BFL1, B-cell lymphoma-2-related protein A1; BH3, B-cell lymphoma-2 homology 3; BIM, B-cell lymphoma-2 interacting mediator; CDK9, Cyclin-dependent kinase 9; CR, Complete remission; CRc, Composite complete remission; [CRc, complete remission (CR) + CR with incomplete hematologic recovery (CRi)]; CRi, CR with incomplete hematologic recovery; CRISPR/Cas9, Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9; CTP, Cytidine triphosphate; dCTP, deoxycytidine triphosphate; DDX41 gene, Dead-box helicase 41 gene; DoR, duration of remission (response); EFS, Event-free survival; FLT3-ITD, FLT3 internal tandem duplication; HDAC, Histone deacetylase; HMA, Hypomethylating agent; HSCT, Hematopoietic stem cell transplantation; IVO, Ivosidenib; LSC, Leukemic stem cell; MAPK, Mitogen-activated protein kinases; MCL1, Myeloid cell leukemia sequence 1; MDM2, Murine double minute 2; MDS, Myelodysplastic syndrome; MPN, Myeloproliferative neoplasm; MRD, Measurable residual disease; NOXA (latin for damage), Phorbol-12-myristate-13-acetate-induced protein 1; NPM1, Nucleophosmin gene 1; OS, Overall survival; PUMA, P53-Upregulated modulator of apoptosis; RAS, Rat sarcoma gene; SPHK1, Sphingosine kinase 1; TEAEs, Treatment emergent adverse events; TLS, Tumor lysis syndrome; XPO1: Exportin 1.
References
1. Kantarjian H, Borthakur G, Daver N, DiNardo CD, Issa G, Jabbour E, et al. Current status and research directions in acute myeloid leukemia. Blood Cancer J. (2024) 14:163. doi: 10.1038/s41408-024-01143-2
2. DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. New Engl J Med. (2020) 383:617–29. doi: 10.1056/NEJMoa2012971
3. Pratz KW, Jonas BA, Pullarkat VA, Thirman MJ, Garcia JS, Fiedler W, et al. Long-term follow-up of the phase 3 viale-a clinical trial of venetoclax plus azacitidine for patients with untreated acute myeloid leukemia ineligible for intensive chemotherapy. Blood (ASH Annu Meeting Abstracts). (2022) 140:529–31. doi: 10.1182/blood-2022-158518
4. Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, et al. Relative mitochondrial priming of myeloblasts and normal hscs determines chemotherapeutic success in aml. Cell. (2012) 151:344–55. doi: 10.1016/j.cell.2012.08.038
5. Ong F, Kim K, and Konopleva MY. Venetoclax resistance: mechanistic insights and future strategies. Cancer Drug resistance (Alhambra Calif). (2022) 5:380–400. doi: 10.20517/cdr.2021.125
6. Wei Y, Cao Y, Sun R, Cheng L, Xiong X, Jin X, et al. Targeting bcl-2 proteins in acute myeloid leukemia. Front Oncol. (2020) 10:584974. doi: 10.3389/fonc.2020.584974
7. Niu J, Peng D, and Liu L. Drug resistance mechanisms of acute myeloid leukemia stem cells. Front Oncol. (2022) 12:896426. doi: 10.3389/fonc.2022.896426
8. Markouli M, Pagoni MN, and Diamantopoulos P. Bcl-2 inhibitors in hematological Malignancies: biomarkers that predict response and management strategies. Front Oncol. (2024) 14:1501950. doi: 10.3389/fonc.2024.1501950
9. Tiribelli M, Michelutti A, Cavallin M, Di Giusto S, Simeone E, Fanin R, et al. Bcl-2 Expression in Aml Patients over 65 Years: Impact on Outcomes across Different Therapeutic Strategies. J Clin Med. (2021) 10. doi: 10.3390/jcm10215096
10. Bogenberger JM, Delman D, Hansen N, Valdez R, Fauble V, Mesa RA, et al. Ex vivo activity of bcl-2 family inhibitors abt-199 and abt-737 combined with 5-azacytidine in myeloid Malignancies. Leuk Lymphoma. (2015) 56:226–9. doi: 10.3109/10428194.2014.910657
11. Tsao T, Shi Y, Kornblau S, Lu H, Konoplev S, Antony A, et al. Concomitant inhibition of DNA methyltransferase and bcl-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann Hematol. (2012) 91:1861–70. doi: 10.1007/s00277-012-1537-8
12. Sharon D, Cathelin S, Mirali S, Di Trani JM, Yanofsky DJ, Keon KA, et al. Inhibition of mitochondrial translation overcomes venetoclax resistance in aml through activation of the integrated stress response. Sci Transl Med. (2019) 11. doi: 10.1126/scitranslmed.aax2863
13. Jin S, Cojocari D, Purkal JJ, Popovic R, Talaty NN, Xiao Y, et al. 5-azacitidine induces noxa to prime aml cells for venetoclax-mediated apoptosis. Clin Cancer Res. (2020) 26:3371–83. doi: 10.1158/1078-0432.Ccr-19-1900
14. Hu X, Li L, Nkwocha J, Sharma K, Zhou L, and Grant S. Synergistic interactions between the hypomethylating agent thio-deoxycytidine and venetoclax in myelodysplastic syndrome cells. Hematol Rep. (2023) 15:91–100. doi: 10.3390/hematolrep15010010
15. Nguyen LXT, Troadec E, Kalvala A, Kumar B, Hoang DH, Viola D, et al. The bcl-2 inhibitor venetoclax inhibits nrf2 antioxidant pathway activation induced by hypomethylating agents in aml. J Cell Physiol. (2019) 234:14040–9. doi: 10.1002/jcp.28091
16. Kamachi K, Ureshino H, Watanabe T, Yoshida-Sakai N, Fukuda-Kurahashi Y, Kawasoe K, et al. Combination of a new oral demethylating agent, or2100, and venetoclax for treatment of acute myeloid leukemia. Cancer Res Commun. (2023) 3:297–308. doi: 10.1158/2767-9764.Crc-22-0259
17. Pollyea DA, DiNardo CD, Arellano ML, Pigneux A, Fiedler W, Konopleva M, et al. Impact of venetoclax and azacitidine in treatment-naïve patients with acute myeloid leukemia and idh1/2 mutations. Clin Cancer Res. (2022) 28:2753–61. doi: 10.1158/1078-0432.Ccr-21-3467
18. DiNardo CD, Tiong IS, Quaglieri A, MacRaild S, Loghavi S, Brown FC, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with aml. Blood. (2020) 135:791–803. doi: 10.1182/blood.2019003988
19. Fruchtman H, Avigan ZM, Waksal JA, Brennan N, and Mascarenhas JO. Management of isocitrate dehydrogenase 1/2 mutated acute myeloid leukemia. Leukemia. (2024) 38:927–35. doi: 10.1038/s41375-024-02246-2
20. Döhner H, Pratz KW, DiNardo CD, Wei AH, Jonas BA, Pullarkat VA, et al. Genetic risk stratification and outcomes among treatment-naive patients with aml treated with venetoclax and azacitidine. Blood. (2024) 144:2211–22. doi: 10.1182/blood.2024024944
21. Roboz GJ, DiNardo CD, Stein EM, de Botton S, Mims AS, Prince GT, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed idh1-mutant acute myeloid leukemia. Blood. (2020) 135:463–71. doi: 10.1182/blood.2019002140
22. Montesinos P, Recher C, Vives S, Zarzycka E, Wang J, Bertani G, et al. Ivosidenib and azacitidine in idh1-mutated acute myeloid leukemia. New Engl J Med. (2022) 386:1519–31. doi: 10.1056/NEJMoa2117344
23. Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ, et al. Isocitrate dehydrogenase 1 and 2 mutations induce bcl-2 dependence in acute myeloid leukemia. Nat Med. (2015) 21:178–84. doi: 10.1038/nm.3788
24. Stuani L, Sabatier M, Saland E, Cognet G, Poupin N, Bosc C, et al. Mitochondrial metabolism supports resistance to idh mutant inhibitors in acute myeloid leukemia. J Exp Med. (2021) 218. doi: 10.1084/jem.20200924
25. Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. New Engl J Med. (2012) 366:1079–89. doi: 10.1056/NEJMoa1112304
26. Boissel N, Nibourel O, Renneville A, Huchette P, Dombret H, and Preudhomme C. Differential prognosis impact of idh2 mutations in cytogenetically normal acute myeloid leukemia. Blood. (2011) 117:3696–7. doi: 10.1182/blood-2010-11-320937
27. Green CL, Evans CM, Zhao L, Hills RK, Burnett AK, Linch DC, et al. The prognostic significance of idh2 mutations in aml depends on the location of the mutation. Blood. (2011) 118:409–12. doi: 10.1182/blood-2010-12-322479
28. Zarnegar-Lumley S, Alonzo TA, Gerbing RB, Othus M, Sun Z, Ries RE, et al. Characteristics and prognostic impact of idh mutations in aml: A cog, swog, and ecog analysis. Blood Adv. (2023) 7:5941–53. doi: 10.1182/bloodadvances.2022008282
29. Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. New Engl J Med. (2005) 352:254–66. doi: 10.1056/NEJMoa041974
30. Issa GC, Bidikian A, Venugopal S, Konopleva M, DiNardo CD, Kadia TM, et al. Clinical outcomes associated with npm1 mutations in patients with relapsed or refractory aml. Blood Adv. (2023) 7:933–42. doi: 10.1182/bloodadvances.2022008316
31. Wei AH, Panayiotidis P, Montesinos P, Laribi K, Ivanov V, Kim I, et al. Long-term follow-up of viale-C in patients with untreated aml ineligible for intensive chemotherapy. Blood. (2022) 140:2754–6. doi: 10.1182/blood.2022016963
32. Wei AH and Roberts AW. Bcl2 inhibition: A new paradigm for the treatment of aml and beyond. Hemasphere. (2023) 7:e912. doi: 10.1097/hs9.0000000000000912
33. Chua CC, Roberts AW, Reynolds J, Fong CY, Ting SB, Salmon JM, et al. Chemotherapy and venetoclax in elderly acute myeloid leukemia trial (Caveat): A phase ib dose-escalation study of venetoclax combined with modified intensive chemotherapy. J Clin oncology: Off J Am Soc Clin Oncol. (2020) 38:3506–17. doi: 10.1200/jco.20.00572
34. Chua CC, Loo S, Fong CY, Ting SB, Tiong IS, Fleming S, et al. Final analysis of the phase 1b chemotherapy and venetoclax in elderly acute myeloid leukemia trial (Caveat). Blood Adv. (2025) 9:1827–35. doi: 10.1182/bloodadvances.2024014900
35. Zhang S, Qin F, Yang L, Xian J, Zou Q, Jin H, et al. Nucleophosmin mutations induce chemosensitivity in thp-1 leukemia cells by suppressing nf-Kb activity and regulating bax/bcl-2 expression. J Cancer. (2016) 7:2270–9. doi: 10.7150/jca.16010
36. Chin L, Wong CYG, and Gill H. Targeting and monitoring acute myeloid leukaemia with nucleophosmin-1 (Npm1) mutation. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24043161
37. Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, et al. Bcl-2, bcl-X(L) sequester bh3 domain-only molecules preventing bax- and bak-mediated mitochondrial apoptosis. Mol Cell. (2001) 8:705–11. doi: 10.1016/s1097-2765(01)00320-3
38. Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective bcl-2 inhibition by abt-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. (2014) 4:362–75. doi: 10.1158/2159-8290.Cd-13-0609
39. Rodríguez-Medina C, Stuckey R, Bilbao-Sieyro C, and Gómez-Casares MT. Biomarkers of response to venetoclax therapy in acute myeloid leukemia. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms25031421
40. Zhang W, Konopleva M, Burks JK, Dywer KC, Schober WD, Yang JY, et al. Blockade of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase and murine double minute synergistically induces apoptosis in acute myeloid leukemia via bh3-only proteins puma and bim. Cancer Res. (2010) 70:2424–34. doi: 10.1158/0008-5472.Can-09-0878
41. Arslan S, Zhang J, Dhakal P, Moran J, Naidoo N, Lombardi J, et al. Outcomes of therapy with venetoclax combined with a hypomethylating agent in favorable-risk acute myeloid leukemia. Am J Hematol. (2021) 96:E59–e63. doi: 10.1002/ajh.26057
42. Zhang K, Zhang X, Xu Y, Xue S, Qiu H, Tang X, et al. Efficacy of venetoclax combined with hypomethylating agents in young, and unfit patients with newly diagnosed core binding factor acute myeloid leukemia. Blood Cancer J. (2023) 13:155. doi: 10.1038/s41408-023-00928-1
43. Kadia TM, Reville PK, Borthakur G, Yilmaz M, Kornblau S, Alvarado Y, et al. Venetoclax plus intensive chemotherapy with cladribine, idarubicin, and cytarabine in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: A cohort from a single-centre, single-arm, phase 2 trial. Lancet Haematology. (2021) 8:e552–e61. doi: 10.1016/s2352-3026(21)00192-7
44. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of aml in adults: 2017 eln recommendations from an international expert panel. Blood. (2017) 129:424–47. doi: 10.1182/blood-2016-08-733196
45. Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, et al. Diagnosis and management of aml in adults: 2022 recommendations from an international expert panel on behalf of the eln. Blood. (2022) 140:1345–77. doi: 10.1182/blood.2022016867
46. Döhner H, Pratz KW, DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, et al. Eln risk stratification is not predictive of outcomes for treatment-naive ptients with acute myeloid leukemia treated with venetoclax and azacitidine. Blood. (2022) 140:1441–4. doi: 10.1182/blood-2022-169509
47. Bataller A, Bazinet A, DiNardo CD, Maiti A, Borthakur G, Daver NG, et al. Prognostic risk signature in patients with acute myeloid leukemia treated with hypomethylating agents and venetoclax. Blood Adv. (2024) 8:927–35. doi: 10.1182/bloodadvances.2023011757
48. Othman J, Tiong IS, O’Nions J, Dennis M, Mokretar K, Ivey A, et al. Molecular mrd is strongly prognostic in patients with npm1-mutated aml receiving venetoclax-based nonintensive therapy. Blood. (2024) 143:336–41. doi: 10.1182/blood.2023021579
49. Jimenez-Chillon C, Othman J, Taussig D, Jimenez-Vicente C, Martinez-Roca A, Tiong IS, et al. Venetoclax-based low intensity therapy in molecular failure of npm1-mutated aml. Blood Adv. (2024) 8:343–52. doi: 10.1182/bloodadvances.2023011106
50. Pratz KW, Jonas BA, Pullarkat V, Recher C, Schuh AC, Thirman MJ, et al. Measurable residual disease response and prognosis in treatment-naïve acute myeloid leukemia with venetoclax and azacitidine. J Clin Oncol. (2022) 40:855–65. doi: 10.1200/jco.21.01546
51. Döhner H, DiNardo CD, Appelbaum FR, Craddock C, Dombret H, Ebert BL, et al. Genetic risk classification for adults with aml receiving less-intensive therapies: the 2024 eln recommendations. Blood. (2024) 144:2169–73. doi: 10.1182/blood.2024025409
52. Gangat N, Karrar O, Iftikhar M, McCullough K, Johnson IM, Abdelmagid M, et al. Venetoclax and hypomethylating agent combination therapy in newly diagnosed acute myeloid leukemia: genotype signatures for response and survival among 301 consecutive patients. Am J Hematol. (2024) 99:193–202. doi: 10.1002/ajh.27138
53. Bataller A, Loghavi S, Gerstein Y, Bazinet A, Sasaki K, Chien KS, et al. Characteristics and clinical outcomes of patients with myeloid Malignancies and ddx41 variants. Am J Hematol. (2023) 98:1780–90. doi: 10.1002/ajh.27070
54. Weng G, Zhang Y, Yu G, Luo T, Yu S, Xu N, et al. Genetic characteristics predict response to venetoclax plus hypomethylating agents in relapsed or refractory acute myeloid leukemia. J Intern Med. (2023) 293:329–39. doi: 10.1111/joim.13581
55. Rahmani NE, Ramachandra N, Sahu S, Gitego N, Lopez A, Pradhan K, et al. Asxl1 mutations are associated with distinct epigenomic alterations that lead to sensitivity to venetoclax and azacytidine. Blood Cancer J. (2021) 11:157. doi: 10.1038/s41408-021-00541-0
56. Senapati J, Urrutia S, Loghavi S, Short NJ, Issa GC, Maiti A, et al. Venetoclax abrogates the prognostic impact of splicing factor gene mutations in newly diagnosed acute myeloid leukemia. Blood. (2023) 142:1647–57. doi: 10.1182/blood.2023020649
57. Shimony S, Garcia JS, Keating J, Chen EC, Luskin MR, Stahl M, et al. Molecular ontogeny underlies the benefit of adding venetoclax to hypomethylating agents in newly diagnosed aml patients. Leukemia. (2024) 38:1494–500. doi: 10.1038/s41375-024-02230-w
58. Waclawiczek A, Leppä AM, Renders S, Stumpf K, Reyneri C, Betz B, et al. Combinatorial bcl2 family expression in acute myeloid leukemia stem cells predicts clinical response to azacitidine/venetoclax. Cancer Discov. (2023) 13:1408–27. doi: 10.1158/2159-8290.Cd-22-0939
59. Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and biological correlates of response in a phase ii study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. (2016) 6:1106–17. doi: 10.1158/2159-8290.Cd-16-0313
60. Nwosu GO, Ross DM, Powell JA, and Pitson SM. Venetoclax therapy and emerging resistance mechanisms in acute myeloid leukaemia. Cell Death Dis. (2024) 15:413. doi: 10.1038/s41419-024-06810-7
61. Garciaz S, Saillard C, Hicheri Y, Hospital MA, and Vey N. Venetoclax in acute myeloid leukemia: molecular basis, evidences for preclinical and clinical efficacy and strategies to target resistance. Cancers. (2021) 13. doi: 10.3390/cancers13225608
62. Garciaz S, Hospital MA, Collette Y, and Vey N. Venetoclax resistance in acute myeloid leukemia. Cancers. (2024) 16. doi: 10.3390/cancers16061091
63. Konopleva MY. Mechanisms for resistance in aml insights into molecular pathways mediating resistance to venetoclax. Best Pract Res Clin haematology. (2021) 34:101251. doi: 10.1016/j.beha.2021.101251
64. Sango J, Carcamo S, Sirenko M, Maiti A, Mansour H, Ulukaya G, et al. Ras-mutant leukaemia stem cells drive clinical resistance to venetoclax. Nature. (2024) 636:241–50. doi: 10.1038/s41586-024-08137-x
65. Sturgeon CM, Wagenblast E, Izzo F, and Papapetrou EP. The crossroads of clonal evolution, differentiation hierarchy, and ontogeny in leukemia development. Blood Cancer Discov. (2025) 6:94–109. doi: 10.1158/2643-3230.Bcd-24-0235
66. Pei S, Pollyea DA, Gustafson A, Stevens BM, Minhajuddin M, Fu R, et al. Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discov. (2020) 10:536–51. doi: 10.1158/2159-8290.Cd-19-0710
67. Zhang Q, Riley-Gillis B, Han L, Jia Y, Lodi A, Zhang H, et al. Activation of ras/mapk pathway confers mcl-1 mediated acquired resistance to bcl-2 inhibitor venetoclax in acute myeloid leukemia. Signal Transduct Target Ther. (2022) 7:51. doi: 10.1038/s41392-021-00870-3
68. Chyla B, Daver N, Doyle K, McKeegan E, Huang X, Ruvolo V, et al. Genetic biomarkers of sensitivity and resistance to venetoclax monotherapy in patients with relapsed acute myeloid leukemia. Am J Hematol. (2018) 93:E202–5. doi: 10.1002/ajh.25146
69. Chatzilygeroudi T, Karantanos T, and Pappa V. Unraveling venetoclax resistance: navigating the future of hma/venetoclax-refractory aml in the nolecular era. Cancers. (2025) 17. doi: 10.3390/cancers17091586
70. Young AI, Timpson P, Gallego-Ortega D, Ormandy CJ, and Oakes SR. Myeloid cell leukemia 1 (Mcl-1), an unexpected modulator of protein kinase signaling during invasion. Cell Adh Migr. (2018) 12:513–23. doi: 10.1080/19336918.2017.1393591
71. Nechiporuk T, Kurtz SE, Nikolova O, Liu T, Jones CL, D’Alessandro A, et al. The tp53 apoptotic network is a primary mediator of resistance to bcl2 inhibition in aml cells. Cancer Discov. (2019) 9:910–25. doi: 10.1158/2159-8290.Cd-19-0125
72. Thijssen R, Diepstraten ST, Moujalled D, Chew E, Flensburg C, Shi MX, et al. Intact tp-53 function is essential for sustaining durable responses to bh3-mimetic drugs in leukemias. Blood. (2021) 137:2721–35. doi: 10.1182/blood.2020010167
73. Kim K, Maiti A, Loghavi S, Pourebrahim R, Kadia TM, Rausch CR, et al. Outcomes of tp53-mutant acute myeloid leukemia with decitabine and venetoclax. Cancer. (2021) 127:3772–81. doi: 10.1002/cncr.33689
74. Kuusanmäki H, Dufva O, Vähä-Koskela M, Leppä AM, Huuhtanen J, Vänttinen I, et al. Erythroid/megakaryocytic differentiation confers bcl-xl dependency and venetoclax resistance in acute myeloid leukemia. Blood. (2023) 141:1610–25. doi: 10.1182/blood.2021011094
75. Carter JL, Su Y, Qiao X, Zhao J, Wang G, Howard M, et al. Acquired resistance to venetoclax plus azacitidine in acute myeloid leukemia: in vitro models and mechanisms. Biochem Pharmacol. (2023) 216:115759. doi: 10.1016/j.bcp.2023.115759
76. Zhang H, Nakauchi Y, Köhnke T, Stafford M, Bottomly D, Thomas R, et al. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat Cancer. (2020) 1:826–39. doi: 10.1038/s43018-020-0103-x
77. Bisaillon R, Moison C, Thiollier C, Krosl J, Bordeleau ME, Lehnertz B, et al. Genetic characterization of abt-199 sensitivity in human aml. Leukemia. (2020) 34:63–74. doi: 10.1038/s41375-019-0485-x
78. Niu X, Zhao J, Ma J, Xie C, Edwards H, Wang G, et al. Binding of released bim to mcl-1 is a mechanism of intrinsic resistance to abt-199 which can be overcome by combination with daunorubicin or cytarabine in aml cells. Clin Cancer Res. (2016) 22:4440–51. doi: 10.1158/1078-0432.Ccr-15-3057
79. Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, et al. Anti-apoptotic mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. (2012) 26:120–5. doi: 10.1101/gad.182980.111
80. Xiang Z, Luo H, Payton JE, Cain J, Ley TJ, Opferman JT, et al. Mcl1 haploinsufficiency protects mice from myc-induced acute myeloid leukemia. J Clin Invest. (2010) 120:2109–18. doi: 10.1172/jci39964
81. Nachmias B, Aumann S, Haran A, and Schimmer AD. Venetoclax resistance in acute myeloid leukaemia-clinical and biological insights. Br J haematology. (2024) 204:1146–58. doi: 10.1111/bjh.19314
82. Tatarata QZ, Wang Z, and Konopleva M. Bcl-2 inhibition in acute myeloid leukemia: resistance and combinations. Expert Rev Hematol. (2024) 17:935–46. doi: 10.1080/17474086.2024.2429604
83. Zielonka K and Jamroziak K. Mechanisms of resistance to venetoclax in hematologic Malignancies. Adv Clin Exp Med. (2024) 33:1421–33. doi: 10.17219/acem/181145
84. Liu J, Chen Y, Yu L, and Yang L. Mechanisms of venetoclax resistance and solutions. Front Oncol. (2022) 12:1005659. doi: 10.3389/fonc.2022.1005659
85. Lin KH, Winter PS, Xie A, Roth C, Martz CA, Stein EM, et al. Targeting mcl-1/bcl-xl forestalls the acquisition of resistance to abt-199 in acute myeloid leukemia. Sci Rep. (2016) 6:27696. doi: 10.1038/srep27696
86. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. Abt-199, a potent and selective bcl-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. (2013) 19:202–8. doi: 10.1038/nm.3048
87. Walensky LD. Targeting bax to drug death directly. Nat Chem Biol. (2019) 15:657–65. doi: 10.1038/s41589-019-0306-6
88. Diepstraten ST, Anderson MA, Czabotar PE, Lessene G, Strasser A, and Kelly GL. The manipulation of apoptosis for cancer therapy using bh3-mimetic drugs. Nat Rev Cancer. (2022) 22:45–64. doi: 10.1038/s41568-021-00407-4
89. Brown FC, Wang X, Birkinshaw R, Chua CC, Morley T, Kasapgil S, et al. Acquired bcl2 variants associated with venetoclax resistance in acute myeloid leukemia. Blood Adv. (2025) 9:127–31. doi: 10.1182/bloodadvances.2024014446
90. Moujalled DM, Brown FC, Chua CC, Dengler MA, Pomilio G, Anstee NS, et al. Acquired mutations in bax confer resistance to bh3-mimetic therapy in acute myeloid leukemia. Blood. (2023) 141:634–44. doi: 10.1182/blood.2022016090
91. Nakao F, Setoguchi K, Semba Y, Yamauchi T, Nogami J, Sasaki K, et al. Targeting a mitochondrial E3 ubiquitin ligase complex to overcome aml cell-intrinsic venetoclax resistance. Leukemia. (2023) 37:1028–38. doi: 10.1038/s41375-023-01879-z
92. Springer MZ and Macleod KF. In brief: mitophagy: mechanisms and role in human disease. J Pathol. (2016) 240:253–5. doi: 10.1002/path.4774
93. Glytsou C, Chen X, Zacharioudakis E, Al-Santli W, Zhou H, Nadorp B, et al. Mitophagy promotes resistance to bh3 mimetics in acute myeloid leukemia. Cancer Discov. (2023) 13:1656–77. doi: 10.1158/2159-8290.Cd-22-0601
94. Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discov. (2019) 9:890–909. doi: 10.1158/2159-8290.Cd-19-0117
95. Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. (2018) 34:724–40.e4. doi: 10.1016/j.ccell.2018.10.005
96. Stevens BM, Jones CL, Pollyea DA, Culp-Hill R, D’Alessandro A, Winters A, et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat Cancer. (2020) 1:1176–87. doi: 10.1038/s43018-020-00126-z
97. Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, and Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. (2006) 5:219–34. doi: 10.1038/nrd1984
98. Xiao H, Zheng Y, Ma L, Tian L, and Sun Q. Clinically-relevant abc transporter for anti-cancer drug resistance. Front Pharmacol. (2021) 12:648407. doi: 10.3389/fphar.2021.648407
99. Ebner J, Schmoellerl J, Piontek M, Manhart G, Troester S, Carter BZ, et al. Abcc1 and glutathione metabolism limit the efficacy of bcl-2 inhibitors in acute myeloid leukemia. Nat Commun. (2023) 14:5709. doi: 10.1038/s41467-023-41229-2
100. Xu Y and Ye H. Progress in understanding the mechanisms of resistance to bcl-2 inhibitors. Exp Hematol Oncol. (2022) 11:31. doi: 10.1186/s40164-022-00283-0
101. Sullivan GP, Flanagan L, Rodrigues DA, and NC T. The path to venetoclax resistance is paved with mutations, metabolism, and more. Sci Transl Med. (2022) 14:eabo6891. doi: 10.1126/scitranslmed.abo6891
102. Naji NS, Sathish M, and Karantanos T. Inflammation and related signaling pathways in acute myeloid leukemia. Cancers. (2024) 16. doi: 10.3390/cancers16233974
103. Wang B, Reville PK, Yassouf MY, Jelloul FZ, Ly C, Desai PN, et al. Comprehensive characterization of ifnγ Signaling in acute myeloid leukemia reveals prognostic and therapeutic strategies. Nat Commun. (2024) 15:1821. doi: 10.1038/s41467-024-45916-6
104. Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The mcl1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. (2016) 538:477–82. doi: 10.1038/nature19830
105. Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E, et al. Discovery of mcl-1-specific inhibitor azd5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. (2018) 9:5341. doi: 10.1038/s41467-018-07551-w
106. Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, et al. Amg 176, a selective mcl1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. (2018) 8:1582–97. doi: 10.1158/2159-8290.Cd-18-0387
107. Wei AH, Roberts AW, Spencer A, Rosenberg AS, Siegel D, Walter RB, et al. Targeting mcl-1 in hematologic Malignancies: rationale and progress. Blood Rev. (2020) 44:100672. doi: 10.1016/j.blre.2020.100672
108. Bogenberger J, Whatcott C, Hansen N, Delman D, Shi CX, Kim W, et al. Combined venetoclax and alvocidib in acute myeloid leukemia. Oncotarget. (2017) 8:107206–22. doi: 10.18632/oncotarget.22284
109. Jonas BA, Hou JZ, Roboz GJ, Alvares CL, Jeyakumar D, Edwards JR, et al. A phase 1b study of venetoclax and alvocidib in patients with relapsed/refractory acute myeloid leukemia. Hematological Oncol. (2023) 41:743–52. doi: 10.1002/hon.3159
110. Baker A, Gregory GP, Verbrugge I, Kats L, Hilton JJ, Vidacs E, et al. The cdk9 inhibitor dinaciclib exerts potent apoptotic and antitumor effects in preclinical models of mll-rearranged acute myeloid leukemia. Cancer Res. (2016) 76:1158–69. doi: 10.1158/0008-5472.Can-15-1070
111. Barlaam B, Casella R, Cidado J, Cook C, De Savi C, Dishington A, et al. Discovery of azd4573, a potent and selective inhibitor of cdk9 that enables short duration of target engagement for the treatment of hematological Malignancies. J Med Chem. (2020) 63:15564–90. doi: 10.1021/acs.jmedchem.0c01754
112. Cidado J, Boiko S, Proia T, Ferguson D, Criscione SW, San Martin M, et al. Azd4573 is a highly selective cdk9 inhibitor that suppresses mcl-1 and induces apoptosis in hematologic cancer cells. Clin Cancer Res. (2020) 26:922–34. doi: 10.1158/1078-0432.Ccr-19-1853
113. Thomas D, Powell JA, Vergez F, Segal DH, Nguyen NY, Baker A, et al. Targeting acute myeloid leukemia by dual inhibition of pi3k signaling and cdk9-mediated mcl-1 transcription. Blood. (2013) 122:738–48. doi: 10.1182/blood-2012-08-447441
114. Akiyama H, Umezawa Y, Ishida S, Okada K, Nogami A, and Miura O. Inhibition of usp9x induces apoptosis in flt3-itd-positive aml cells cooperatively by inhibiting the mutant kinase through aggresomal translocation and inducing oxidative stress. Cancer Lett. (2019) 453:84–94. doi: 10.1016/j.canlet.2019.03.046
115. Lewis AC, Pope VS, Tea MN, Li M, Nwosu GO, Nguyen TM, et al. Ceramide-induced integrated stress response overcomes bcl-2 inhibitor resistance in acute myeloid leukemia. Blood. (2022) 139:3737–51. doi: 10.1182/blood.2021013277
116. Powell JA, Lewis AC, Zhu W, Toubia J, Pitman MR, Wallington-Beddoe CT, et al. Targeting sphingosine kinase 1 induces mcl1-dependent cell death in acute myeloid leukemia. Blood. (2017) 129:771–82. doi: 10.1182/blood-2016-06-720433
117. Brennan MS, Chang C, Tai L, Lessene G, Strasser A, Dewson G, et al. Humanized mcl-1 mice enable accurate preclinical evaluation of mcl-1 inhibitors destined for clinical use. Blood. (2018) 132:1573–83. doi: 10.1182/blood-2018-06-859405
118. Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, et al. Loss of mcl-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. (2013) 27:1365–77. doi: 10.1101/gad.215871.113
119. Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, et al. Deletion of mcl-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. (2013) 27:1351–64. doi: 10.1101/gad.215855.113
120. Pratz KW, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Döhner H, et al. Long-term follow-up of viale-A: venetoclax and azacitidine in chemotherapy-ineligible untreated acute myeloid leukemia. Am J Hematol. (2024) 99:615–24. doi: 10.1002/ajh.27246
121. Johnson IM, Karrar O, Rana M, Iftikhar M, Chen S, McCullough K, et al. Cardiac events in newly diagnosed acute myeloid leukaemia during treatment with venetoclax + Hypomethylating agents. Br J haematology. (2024) 204:1232–7. doi: 10.1111/bjh.19325
122. Wei AH, Montesinos P, Ivanov V, DiNardo CD, Novak J, Laribi K, et al. Venetoclax plus ldac for newly diagnosed aml ineligible for intensive chemotherapy: A phase 3 randomized placebo-controlled trial. Blood. (2020) 135:2137–45. doi: 10.1182/blood.2020004856
123. Venugopal S, Takahashi K, Daver N, Maiti A, Borthakur G, Loghavi S, et al. Efficacy and safety of enasidenib and azacitidine combination in patients with idh2 mutated acute myeloid leukemia and not eligible for intensive chemotherapy. Blood Cancer J. (2022) 12:10. doi: 10.1038/s41408-021-00604-2
124. Fathi AT, Issa GC, Wang ES, Erba H, Kaplan Altman J, Balasubramanian SK, et al. Ziftomenib combined with venetoclax/azacitidine in relapsed/refractory npm1-M or kmt2a-R acute myeloid leukemia: interim phase 1a results from komet-007. Blood. (2024) 144:2880–2. doi: 10.1182/blood-2024-199170
125. Issa GC, Aldoss I, Thirman MJ, DiPersio J, Arellano M, Blachly JS, et al. Menin inhibition with revumenib for kmt2a-rearranged relapsed or refractory acute leukemia (Augment-101). J Clin Oncol. (2025) 43:75–84. doi: 10.1200/jco.24.00826
126. Alawieh D, Cysique-Foinlan L, Willekens C, and Renneville A. Ras mutations in myeloid Malignancies: revisiting old questions with novel insights and therapeutic perspectives. Blood Cancer J. (2024) 14:72. doi: 10.1038/s41408-024-01054-2
127. Desikan SP, Ravandi F, Pemmaraju N, Konopleva M, Loghavi S, Jabbour EJ, et al. A phase ii study of azacitidine, venetoclax, and trametinib in relapsed or refractory acute myeloid leukemia harboring ras pathway-activating mutations. Acta haematologica. (2022) 145:529–36. doi: 10.1159/000525566
128. Konopleva MY, Dail M, Daver NG, Garcia JS, Jonas BA, Yee KWL, et al. Venetoclax and cobimetinib in relapsed/refractory aml: A phase 1b trial. Clin Lymphoma Myeloma Leuk. (2024) 24:364–74. doi: 10.1016/j.clml.2024.01.007
129. Daver NG, Dail M, Garcia JS, Jonas BA, Yee KWL, Kelly KR, et al. Venetoclax and idasanutlin in relapsed/refractory aml: A nonrandomized, open-label phase 1b trial. Blood. (2023) 141:1265–76. doi: 10.1182/blood.2022016362
130. Buecklein V, Magno G, Rausch C, Warm M, Emhardt AJ, Gottschlich A, et al. Aaron: an ongoing open-label phase I/ii study of relatlimab (Anti-lag-3) with nivolumab (Anti-pd-1) in combination with azacitidine ± Venetoclax for the treatment of patients with relapsed/refractory and non-fit patients with newly diagnosed acute myeloid leukemia - interim analysis. Cancer Res. (2025) 85:CT225. doi: 10.1158/1538-7445.AM2025-CT225
131. Short NJ, Daver N, Dinardo CD, Kadia T, Nasr LF, Macaron W, et al. Azacitidine, venetoclax, and gilteritinib in newly diagnosed and relapsed or refractory FLT3-Mutated AML. J Clin Oncol. (2024) 42:1499–508. doi: 10.1200/JCO.23.01911
132. Yilmaz M, Muftuoglu M, Dinardo CD, Kadia TM, Konopleva MY, Borthakur G, et al. Phase I/II study of quizartinib, venetoclax, and decitabine triple combination in FLT3-ITD mutated AML. Blood. (2023) 142:158–61. doi: 10.1182/blood-2023-186699
133. Lachowiez CA, Loghavi S, Zeng Z, Tanaka T, Kim YJ, Uryu H, et al. A Phase Ib/II study of ivosidenib with venetoclax ± azacitidine in IDH1-Mutated myeloid malignancies. Blood Cancer Discov. (2023) 4:276–93. doi: 10.1158/2643-3230.BCD-22-0205
Keywords: acute myeloid leukemia (AML), response, resistance, venetoclax (VEN), BCL2 (B-cell lymphoma 2) inhibition, azacitidine (AZA), hypomethylating agents (HMAs), MCL1 (myeloid cell leukemia sequence 1) overexpression
Citation: Diamantidis MD (2025) Factors affecting response and resistance to venetoclax in acute myeloid leukemia. Front. Oncol. 15:1577908. doi: 10.3389/fonc.2025.1577908
Received: 16 February 2025; Accepted: 29 August 2025;
Published: 15 September 2025.
Edited by:
Alisa Damnernsawad, Mahidol University, ThailandReviewed by:
Monika Kutyna, South Australian Health and Medical Research Institute (SAHMRI), AustraliaCopyright © 2025 Diamantidis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael D. Diamantidis, ZGlhbWFudGlkaXM3NkBnbWFpbC5jb20=
†ORCID: Michael D. Diamantidis, orcid.org/0000-0002-0041-5930