 Jeries Kort
Jeries Kort Andrea Rivera3,4
Andrea Rivera3,4 Sindhuja Senigarapu
Sindhuja Senigarapu James J. Driscoll
James J. Driscoll- 1Division of Hematology and Oncology, Department of Medicine, University Hospitals Cleveland Medical Center, Cleveland, OH, United States
- 2Case Comprehensive Cancer Center, Case Western Reserve University, Cleveland, OH, United States
- 3Department of Medicine, Ponce Health Sciences University, Ponce, Puerto Rico
- 4Heart, Lung Blood Summer Program, Case Western Reserve University, Cleveland, OH, United States
- 5Department of Anesthesiology, University Hospitals Cleveland Medical Center, Cleveland, OH, United States
- 6Adult Hematologic Malignancies and Stem Cell Transplant Section, Seidman Cancer Center, University Hospitals Cleveland Medical Center, Cleveland, OH, United States
Multiple myeloma (MM) is a cancer of bone marrow plasma cells. A noteworthy ensemble of therapies has been introduced over the past quarter century that exert antimyeloma activities through diverse mechanisms and achieve durable disease control in many patients. The discovery that proteasome inhibitors (PIs) and immunomodulatory drugs (IMiDs) target specific plasma cell features that reflect disease biology and exert antimyeloma activity led to transformative changes in treatment algorithms. Recently, advances in immunotherapy have emerged and represent a promising option with the potential to capture immunologic memory and yield more durable responses in MM patients. Idecabtagene vicleucel and ciltacabtagene autoleucel are chimeric antigen receptor (CAR) T-cell immunotherapies that attach to the extracellular domain of the B-cell maturation antigen (BCMA) and have demonstrated significant response rates in heavily-treated patients. These agents are FDA-approved for relapsed and/or refractory (RR)MM patients previously treated with PIs, IMiDs, and CD38-directed monoclonal antibodies. Most patients who receive CAR T-cell therapy relapse after prolonged or brief remission, and a more thorough understanding of the resistance mechanisms following CAR T-cell infusion is needed. Bispecific antibodies (BsAbs) are engineered to simultaneously bind to both cancer and immune cells and trigger a direct tumor-specific cytotoxic response. BsAbs and CAR T-cells are major histocompatibility complex (MHC)-independent approaches to treat MM and do not require T-cell receptor (TCR) specificity. Agents that target BCMA and G protein-coupled receptor class C group 5 member D (GPRC5D) demonstrate impressive clinical responses, while early-phase trials targeting FcRH5 are promising. Here, we provide a comprehensive overview of their individual efficacy, adverse effects, and limitations that impact broader application.
1 Introduction
Multiple myeloma (MM) is a plasma cell (PC) malignancy that lacks a cure but can be effectively managed for many years in most patients (1–3). Patients diagnosed with MM in time relapse and become recalcitrant to current treatments (4, 5). Despite significant advances in treatment modalities, myeloma relapse is common and carries a poor prognosis (3–5). The incidence rate is 7/100,000 in the U.S with a median age of onset at 69 years (6). Monoclonal gammopathy of undetermined significance (MGUS) and smoldering MM (SMM) are premalignant PC proliferative disorders that are thought to universally precede the development of MM. MGUS affects ∼3% of individuals >50 years old, and MGUS patients usually do not advance to MM (7). SMM demonstrates high variability, with some cases closely resembling MGUS while others exhibit similarity and comparable risk of MM. Nearly 60% of SMM patients exhibit MM two years post-diagnosis depending on clinical and genetic factors (8). MM as well as the precursor disorders are relatively more common in those of African descent (9–11).
The majority of individuals diagnosed with MM are 65 to 75 years of age. MM is responsible for ~15% of hematological malignancies and ~22% of hematological malignancy-related deaths in the US. Globally, there is a rising trend in the incidence of MM, particularly in males and those >50 years of age, as well as individuals from high-income populations (12). An overall declining global trend of myeloma mortality was more evident in females. Lifestyle, diagnosis capacity, and treatment availability may be modified and improved to control the increasing trends that are observed populations at greater risk (10, 11).
Myeloma cells are predominantly detected in patient bone marrow (BM) but are additionally revealed in the circulatory system and at extramedullary sites, particularly at advanced stages of illness. Myeloma cells generally synthesize, fold, assemble and secrete monoclonal immunoglobulin (Ig) proteins (monoclonal protein, M protein). The IgG isotype accounts for ~50% for MM cases, IgA 20%, while the IgD and IgM phenotypes are observed less frequently (2% and 0.5%). Approximately ~20% of those diagnosed with myeloma secrete only monoclonal κ or λ free light chains (FLC) (light-chain myeloma or Bence Jones myeloma), while some have non- or hyposecretory disease (13–15).
Transformative advances in the treatment of MM over the past quarter century has led to a dramatic improvement in patient quality-of-life as well as progression free survival (PFS) and overall survival (OS) (Figure 1). A more precise mechanistic appreciation of myelomagenesis and disease features and the cellular, protein and genomic level has led to advances in diagnosis, prognosis, and response assessment, and tremendously informed the discovery of actionable targets and novel agents. Median OS for patients varies with age, eligibility for autologous stem cell transplant (ASCT), tumor cytogenetics, treatment depth and duration of response, and other factors (16). Data from randomized controlled trials using modern therapy show that the median survival in multiple myeloma is approximately 6 years (17). In the subset of patients eligible for ASCT, 4-year survival rates are more than 80% (18); the median OS among these patients is more than 8 years (19, 20). However, not all MM patients have benefited from the vast array of treatment options due to lack of timely access to treatment, inherently therapy refractory disease, and ineligibility to novel therapies. Hence, an improved understanding of timely and effective sequencing of currently approved MM drugs may maximize therapeutic efficacy.
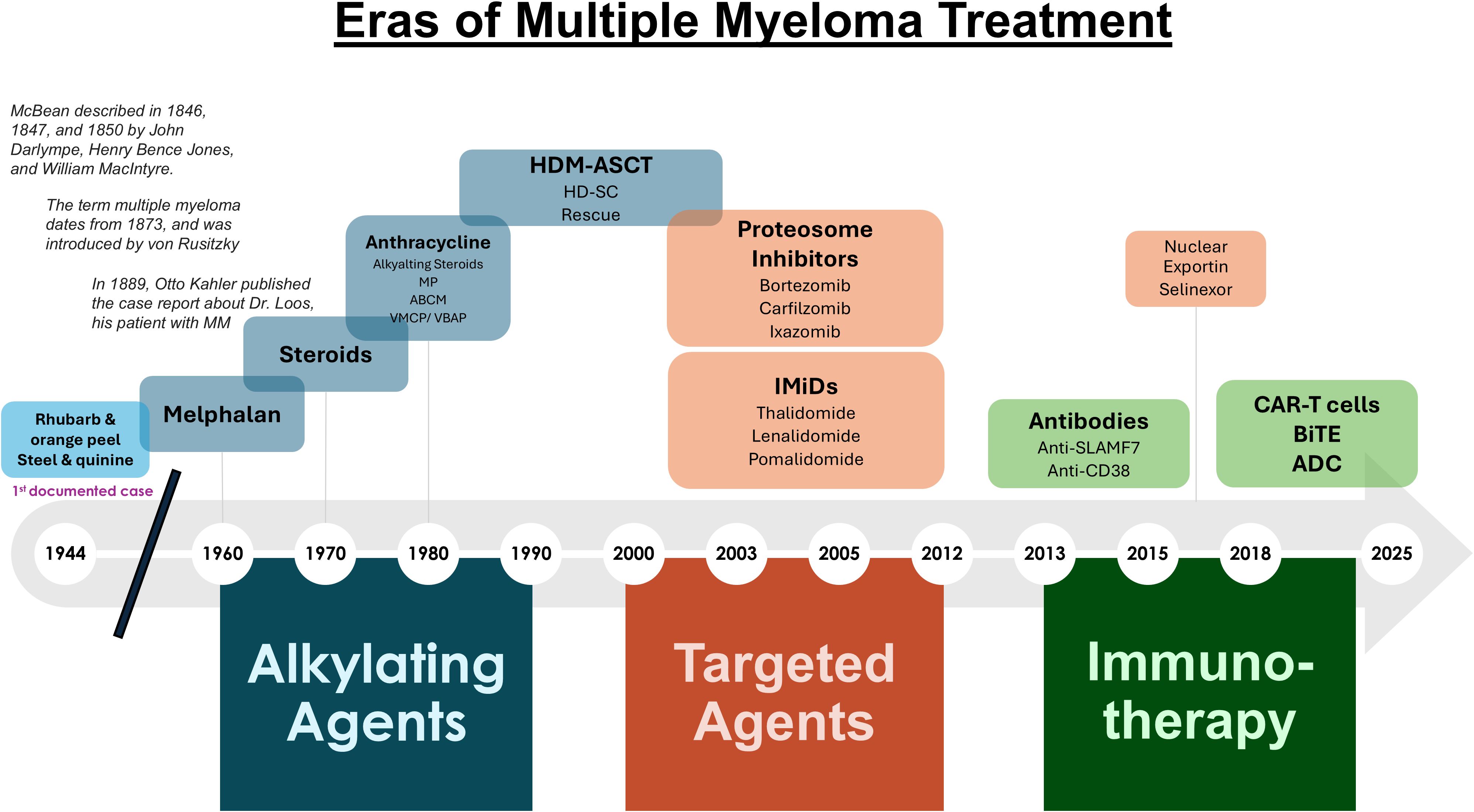
Figure 1. Eras of multiple myeloma treatment. Shown is a timeline of the agents used throughout the past two centuries to treat MM.
2 Earliest recognition on MM
The first widely recognized case of MM was reported in 1844 by Samuel Solly for a highly respectable tradesman from London named Mr. Thomas Alexander McBean (21, 22) Dr. William Macintyre documented attributes that resembled Bence Jones proteinuria. The chemical pathologist Henry Bence Jones confirmed the results of Macintyre (23). The disease was infrequently documented until 1889 with the case report by Kahler (24). The subject of Kahler survived for 8 years despite less than perfect chemotherapy. In 1922, Bayne-Jones and Wilson (25) identified two discrete groups of Bence Jones protein, but a connection between Bence Jones and myeloma serum proteins was not demonstrated until 1956 by Korngold and Lipari (26). The discovery that light chains from a serum IgG myeloma protein and the Bence Jones protein from the same patient’s urine were identical marked a significant breakthrough. Sarah Newbury was a 39-year-old that had developed fatigue, bone pain and fractures (21). At autopsy of the patient, her BM had been replaced by a red substance whose cells were remarkably similar to those found at the autopsy of Mr. McBean.
3 Early treatment of MM
Frontline therapy for newly diagnosed MM patients has improved markedly from the rhubarb pill and infusion of orange peel that was given in the 1840’s followed by phlebotomy and application of leeches later administered as “maintenance therapy” (Figure 1). In 1947, Alwall described that urethane reduced serum globulin levels, increased hemoglobin, eliminated proteinuria, and decreased BM PCs in a myeloma patient (27). The treatment approach remained the standard for nearly 15 years. Blokhin et al. in 1958 reported the benefit of sarcolysin (melphalan) in 3 of 6 patients with MM (28). Bergsagel et al. in 1962 reported significant improvement in 8 of 24 patients treated with melphalan (29). Holland et al. in 1966 administered urethane or placebo consisting of cherry and cola–flavored syrup to previously treated or untreated patients with promising results (30). Hoogstraten et al. found that melphalan followed yielded responses in 78% of patients that were either newly diagnosed (ND) or had previously treated disease (31).
A placebo-controlled double-blind trial demonstrated that prednisone as a single agent produced significant decreases in serum globulin and an increase in hematocrit but no difference in survival compared with a placebo (32). Two Cancer and Leukemia Group B (CALGB) protocols showed that single agent prednisone produced a 44% objective response rate (ORR) (33). Melphalan combined with prednisone (MelPred) was then established in a randomized trial of MM patients led by Alexanian et al, in which survival was 6 months longer with MP relative to Mel alone (34). In 1974, Lee et al. treated 36 MM patients with carmustine, cyclophosphamide, Mel, vincristine, and Pred (M-2 protocol) and reported that 60% had excellent subjective and objective responses (35). Case et al. later reported an 87% response rate in patients with MM given the M-2 protocol (36). High-dose chemotherapy with Mel 200 mg/m (2) (Mel200) became the conditioning regimen of choice with ASCT and consolidation after induction therapy for suitable patients, prolongs PFS and is recommended upfront for all eligible patients (18, 37).
4 Targeting the proteasome revolutionized multiple myeloma treatment
PCs are terminally differentiated B lymphocytes that dwell within the BM. A genotypically and phenotypically discrete population of long-lived PCs (LLPC) has the ability to live for extraordinarily extended periods in humans. Knowledge of LLPC biology impacts myelomagenesis and provides insights into disease treatment. Myeloma shares many tumor intrinsic survival programs as LLPCs. Most PCs are short-lived to limit antibody responses (38). Hence, MM cells are exquisitely sensitive to events that disrupt proteostasis (39–41). PCs differentiate from B lymphocytes to sustain antibody production, and as professional Ab secretors, PCs serve as a model system to dissect proteostasis and the stresses that high volume protein synthesis, folding and transport entail.
As early as the1940’s, it was recognized that all the constituents of an organism are in a constant state of chemical renewal (42). Schoenheimer developed the term “dynamic state of body constituents” to describe the state in which all constituents of the cell are continually degraded and replaced upon new biosynthesis. These thoughts led to the emergence of subsequent seminal discoveries that revealed the destruction of intracellular and cytosolic proteins within the cell is regulated with exquisite selectivity. Ciechanover, Hershko and Rose discovered, studied and revealed the discrete intricacies the ubiquitin (Ub)-mediated proteolytic pathway, a process where an enzymatic system tags unwanted protein substrates with the 76-amino acid polypeptide Ub (43). Ub-tagged proteins are then transported to the proteasome, a large multisubunit proteolytic complex, within which proteins are hydrolyzed to peptides and Ub is recycled. Myriad cellular processes, e.g., protein homeostasis and immunity, are regulated through the Ub pathway. A causal role for defects in the Ub-dependent proteolytic pathway have detected in a number of human diseases, e.g., cancer.
Proteasomes requisite components of the Ub-dependent protein degradation pathway and function as the catalytic core of the pathway to hydrolyze ubiquitinated protein substrates (44, 45). Proteasomes are extremely sophisticated complexes designed to perform the selective, efficient and processive hydrolysis of denatured, misfolded and redundant proteins (46). These structures are dynamic, tightly governed protein degrading, multicomponent complexes that are structurally and functionally conserved throughout evolution. Proteasomes consist of >30 distinct protein subunits and exhibit a MW of ~2.5 MDa. The barrel-shaped 20S proteasome core particle (CP) is formed by axial stacking of 4 heptameric rings: 2 identical inner β-rings, each formed by 7 distinct β-subunits (β1–7), and 2 identical outer α-rings each formed by 7 different α-subunits (α1–7). A 19S regulatory particle (RP) that recognizes proteins bearing a multi-Ub chain caps either or both end of the 20S CP (46, 47). Proteasomes collaborate with the Ub-conjugating enzymes to play a prominent role in the control of cellular activities by rapidly and unidirectionally catalyzing protein degradation. Constant, highly elevated rates of protein synthesis coupled with the need for highly efficient protein clearance mechanisms make MM cells exquisitely sensitive to the slightest perturbations in proteostasis. Hence, MM offers an opportunity to target this intrinsic survival vulnerability with the administration of PIs. Interestingly, endogenous PIs were also detected in mammalian cell lysates and shown to regulate proteasomal catalytic activities (48, 49). Carfilzomib and ixazomib were later developed as second-generation PIs and have increased patient response rates, survival and safety (50, 51). PIs are a cornerstone of current treatment approaches in MM. While survival outcomes have improved significantly over the past two decades for NDMM patients, elderly patients have not yielded the same magnitude of benefit as evidenced by higher rates of reported myeloma-related deaths in patients >75 years of age (52–54).
5 IMiDs
The development of the IMiDs which include thalidomide (Thal), lenalidomide (Len), and pomalidomide (Pom), has contributed significantly to these improved outcomes (55, 56). Len is widely used in the treatment of ND transplant-eligible and transplant-ineligible MM patients, in the maintenance setting post-transplant and in the relapsed/refractory setting, while Pom is currently utilized in the relapsed/refractory setting. IMiDs are a critical component of therapeutic combinations for all stages of the disease and are also used as a single-agent maintenance therapy after ASCT. At diagnosis, most patients are sensitive to IMiD-based combination therapy; however, ~5% are refractory and form an important group with difficult-to-manage disease (56). Patients who are initially sensitive eventually acquire resistance, and IMiD refractory states are associated with shorter PFS and OS in response to subsequent therapies. IMiDs demonstrate a multitude of activities, including anti-angiogenic, cytotoxic, and immunomodulatory. However, the more recent discoveries that the IMiDs bind to cereblon and thus regulate the ubiquitination of key transcription factors including IKZF1 and IKZF3, have provided greater insight into their mechanism of action (57). IMiDs have a direct impact on MM cells by functioning as a molecular glue, binding to a CRL4CRBN E3 ubiquitin ligase (cereblon) and leading to degradation of neo-substrates including ikaros (IKZF1) and aiolos (IKZF3). This results in downregulation of IRF4/MYC and MM cell death (58, 59).
6 Immunotherapies as a second revolution in multiple myeloma treatment
A number of recent strategies have been developed to revitalize a patient’s innate immune response towards cancer cells (Figures 2, 3). One of the earliest pieces of evidence of the role of immunotherapy in MM was the introduction of allogeneic (allo)-hematopoietic stem cell transplantation (HCT). HCT offers potential for long-term remission and possible curative outcome in a patient subset through potent graft-versus-myeloma (GVM) effects. Anti-tumor activity is generated through donor-derived alloreactive lymphocytes that target cancer cells, and additional benefit from donor lymphocyte infusions for post-transplant relapse (60). Allo-HCT is limited by harmful toxicities, e.g., graft-versus-host disease (GVHD), infections, combined with significant treatment-related mortality (TRM) (61). To broaden patient eligibility, reduced-intensity conditioning (RIC) regimens were evaluated which led to lower early morbidity countered by increased rates of disease relapse. GVM activity highlights the immune system’s remarkable ability to cure a subset of patients, pushing the role of immunotherapy. Additional modalities have subsequently been introduced to harness the host immune system, e.g., targeted monoclonal antibodies (mAbs), chimeric antigen receptor (CAR) targeting T-cells, and bispecific T-cell engagers (BiTEs).
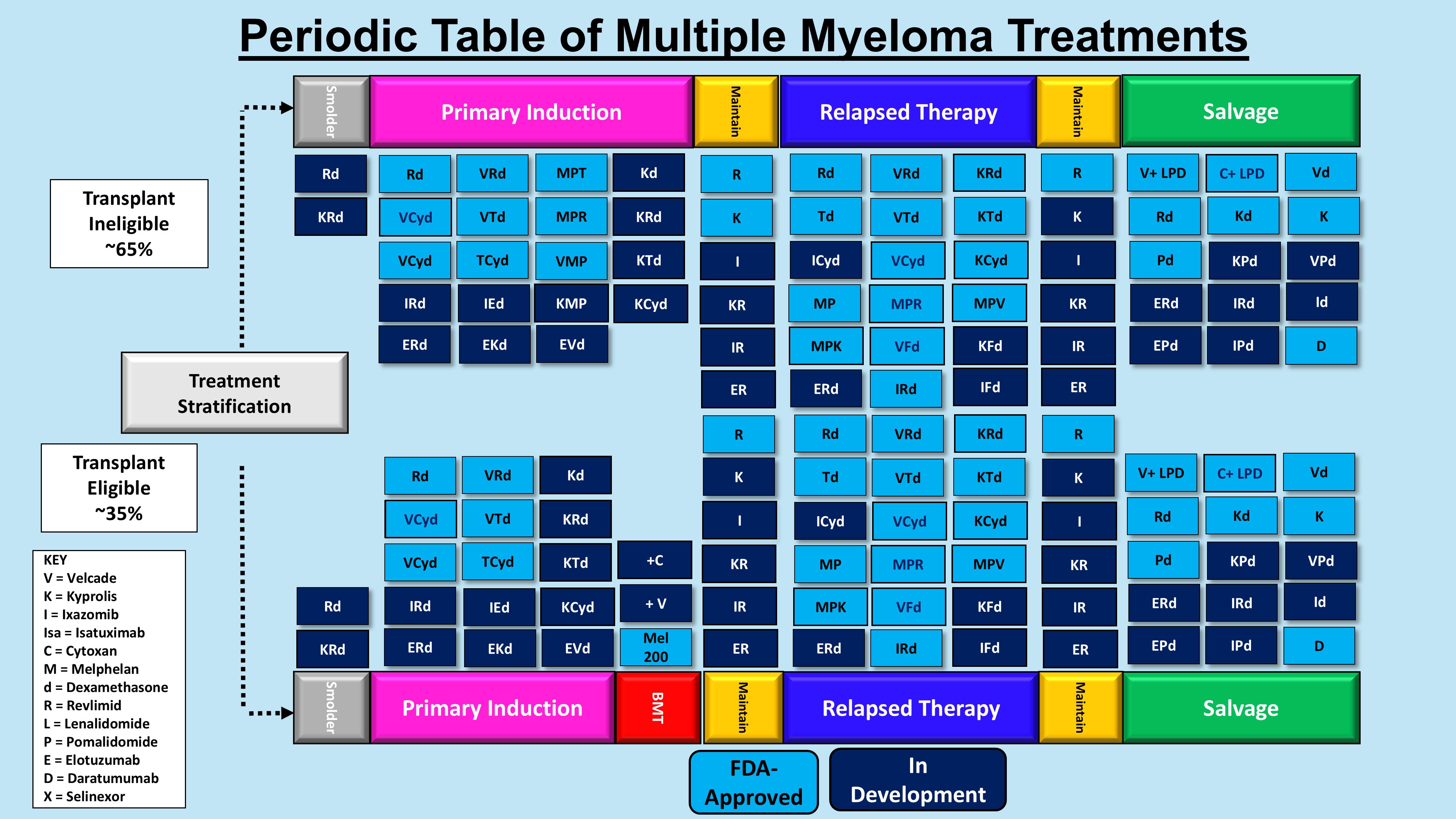
Figure 2. Periodic table of multiple myeloma treatments. Shown is a table of the agents used for transplant eligible and ineligible patients in the pre-immunotherapy era.
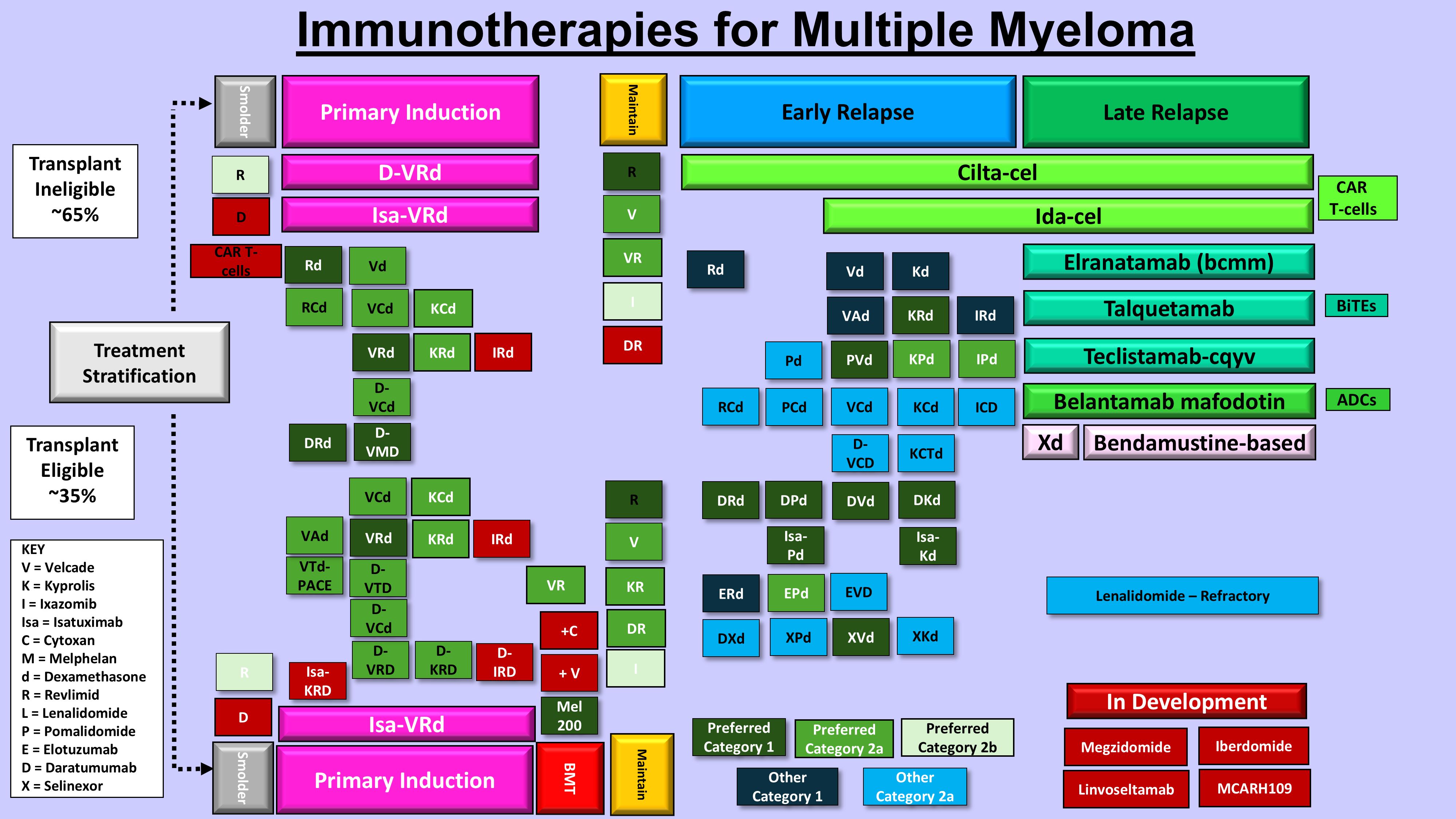
Figure 3. Immunotherapies for multiple myeloma treatment. Shown are the current FDA-approved and investigational immunotherapy agents for ND and RRMM.
6.1 Monoclonal antibodies in ND and RRMM
The US FDA has authorized three mAbs for the treatment of MM patients. Daratumumab (Dara) and isatuximab (Isa) both target CD38, while elotuzumab (Elo) targets the surface glycoprotein signaling lymphocytic activation molecule F7, SLAMF7/CS1, in the CD2 Ig superfamily (IgSF) subset (62–67). Elo directly activates NK cells and mediates ADCC through the CD16 pathway to yield antimyeloma activity. Anti-CD38 mAbs function through various mechanisms that include complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), and antibody-dependent cell-mediated phagocytosis (ADCP). CD38 mAbs can also induce programmed cell death, reduce mitochondrial transfer, inhibit adenosine production and adhesion molecule function, and regulate enzyme activity to induce the death of MM cells.
Dara was FDA-approved in 2016 as a breakthrough orphan drug based on two independent randomized trials in the relapsed setting. The FDA approved Dara-hyaluronidase-fihj (Darzalex Faspro) in combination with bortezomib, Len and Dex for induction and consolidation in patients with ASCT-eligible NDMM patients, based on the Perseus study (NCT03710603), which became the standard induction regimen for ND patients (68). The addition of Dara to a triplet induction regimen of VRd (bortezomib, Len and Dex) significantly improved PFS, and yielded an 84% 48-month PFS with minimal residual disease (MRD) negativity in transplant-eligible individuals.
The FDA has approved Isa-irfc<b> </b>(Sarclisa, Sanofi-Aventis) in combinations for patients with RRMM. Isatuximab-irfc<b> </b>has been authorized in combination with bortezomib, Len, and Dex (VRd) for NDMM, ASCT-ineligible individuals based on the IMROZ trial, which became the standard induction regimen for this patient population (69). Isatuximab added to VRd for transplant-ineligible patients enhanced MRD negativity and yielded a 48-month PFS rate ~70%. ICARIA-MM was a randomized, multicenter, open-label, phase 3 study of patients (aged ≥18 years) with RRMM who had received >2 previous lines of therapy (LOT), including Len and a PI, and had an ECOG PS of 0–2. Isa+POM/Dex resulted in a 6·9-month difference in median OS compared with POM/Dex and is a new standard of care for Len-refractory and PI-refractory/relapsed MM (65).
Elo has been approved by the FDA for use in combination with Len and Dex in patients with RRMM. Elo is effective as a single agent, as well as in combinations, supporting the use of Elo in NDMM patients (70, 71). ELOQUENT-1 was an open-label, multicenter, randomized, phase 3 trial conducted at 185 sites in 19 countries. Eligible patients were aged >18 years with ND, untreated, symptomatic myeloma and not candidates for high-dose therapy plus HSCT, and an Eastern Cooperative Oncology Group (ECOG) performance status of <2. Patients were randomly assigned (1:1) to receive Elo+Len/Dex or Len/Dex using an interactive voice response system, stratified by the International Staging System (ISS; stage I-II vs III), age (<75 years vs ≥75 years), and ECOG performance status (0 vs 1-2). In both treatment groups, patients received 25 mg Len orally on days 1–21 of each cycle and 40 mg Dex on days 1, 8, 15, and 22 of each cycle. The primary endpoint was PFS. Median PFS was not significantly different between the groups: 31·4 months (95% CI 26·2-36·8) in the Elo+Len/Dex group versus 29·5 months (23·5-34·3) in the Len/Dex group (HR 0·93, 95·71% CI 0·77-1·12; stratified log-rank p=0·44). The median follow-up was 70·6 months (IQR 35·1-79·2). The most common grade 3–4 treatment-related adverse event was neutropenia (64 [17%] of 371 vs 79 [21%] of 371). Study drug toxicity was the reported cause of death in five (1%) vs. four (1%) patients. Elo+Len/Dex did not significantly improve PFS vs. Len/Dex in patients with NDMM who are ineligible for HSCT.
In the phase III ELOQUENT 2 trial, the addition of Elo to Len/Dex (ERd) led to superior ORR, PFS and OS compared to Len/Dex in patients who had received 1–3 prior LOT. In the randomized phase II ELOQUENT 3 trial, the addition of Elo to Pom/Dex (EPd) led to superior ORR, PFS and OS compared to Pom/Dex in patients with RRMM, ~60% of which had only received ≤3 prior LOT. In ELOQUENT 2, patients were not previously treated with Dara and in ELOQUENT 3, <5% of patients were previously treated with Dara. Retrospective data suggest that the efficacy of Elo is diminished with prior Dara exposure (72). Current frontline treatment of MM often includes Len and Dara, with treatment administered until disease progression. There is no singular standard of care for patient’s refractory to these drugs at time of relapse. The results of a recent study suggest that EPd is an active and well-tolerated regimen in RRMM, even in real-world patients. Furthermore, EPd may be useful, especially in Dara-naïve patients (73).
6.2 CAR T-cells
In 1987, the immunologist Yoshikazu Kurosawa proposed the notion of a chimeric
T-cell receptor (TCR) which combined the antibody derived variable regions (VH/VL) with constant portions derived from the TCR (74). In 1989, Zelig Eshhar described a strategy to redirect T-cells to recognize antigens in a non-major histocompatibility complex (MHC)-restricted manner (75). Eshhar designed synthetic type I receptors that consisted of an extracellular Ag recognition, transmembrane, and an intracellular signaling domain to activate T-cells (76). Following transfection of these constructs, originally termed “T-bodies”, into a cytotoxic T-cell hybridoma, expression of a functional TCR was detected and the chimeric TCR exhibited the idiotope of the Sp6 anti-TNP Ab. The strategy provided T-cells with a non-MHC-restricted reaction to the given hapten. Transfectants specifically produced Il-2 and elicited cytotoxicity in response to TNP-bearing target cells.
Autologous T-cells were genetically modified to express a 2nd-generation CAR 19-28z, specific to the B-cell lineage CD19 and demonstrated efficacy in chemotherapy-refractory chronic lymphocytic leukemia (CLL) or relapsed B-cell acute lymphoblastic leukemia (ALL) patients (77). The short-term persistence of infused T-cells was enhanced by antecedent administration of cyclophosphamide and was inversely proportional to tumor burden within the peripheral blood. Patient T-cells were engineered to express not only CD19 but also costimulatory molecule CD28 joined with a CD3ζ chain as a 2nd generation CAR T-cell (78, 79). The 2nd-generation CAR T-cells were well tolerated, although there was notable cytokine release that correlated with burden of disease. Treated patients achieved tumor eradication and complete remission (CR).
Idecabtagene vicleucel (ide-cel) and ciltacabtagene autoleucel (cilta-cel) have been authorized by the US FDA for the treatment of MM patients following multiple prior LOT that included an IMiD, PI, and anti-CD38 monoclonal antibody (Table 1) (86, 93–101). The European Medicines Agency (EMA) likewise has endorsed these chimeric engineered T-cell therapies for MM patients following >3 prior LOT. While both cellular therapies target BCMA, they differ in their engineered TCR design, efficacy, and side effect profiles (Table 1). Ide-cel harbors a single BCMA-binding domain, while cilta-cel employs two distinct BCMA-binding regions to increase its binding affinity that may potentially contribute to higher response rates (86, 93–99). Ide-cel demonstrated efficacy in the KarMMa trial in which heavily pre-treated RRMM patients were enrolled. Ide-cel achieved an ORR of 73%, with 33% of patients achieving a CR and a median PFS of 8.8 months. However, relapses were frequent and underscored the need for more durable immune-based therapies and CAR T-cell designs. Cilta-cel achieved even more promising results in the Cartitude-1 trial yielding an ORR of 98% with 80% of patients deemed a stringent CR (sCR). Deep and durable responses were observed with cilta-cel to suggest it may offer long-term benefit. KarMMa-3 significantly improved PFS to 13.3 months with ide-cel to 4.4 months observed with standard regimens. Cartitude-4 demonstrated that cilta-cel achieved higher efficacy, with a PFS not yet reached and an ORR of 84%, including 71% of patients achieving MRD negativity (100, 101).
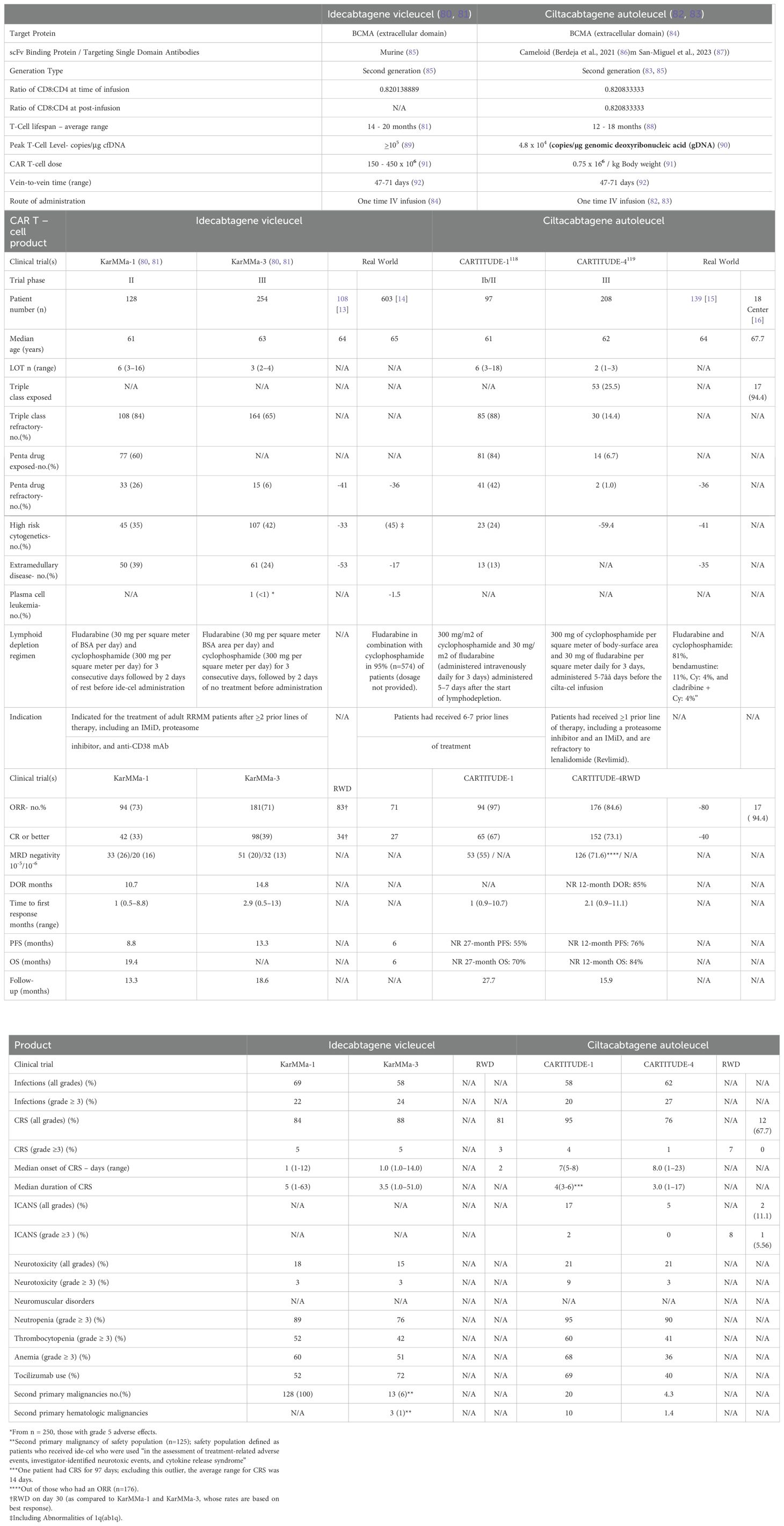
Table 1. Comparison of the FDA-approved CAR T-cells for the treatment of relapsed and/or refractory multiple myeloma.
A common side effect of CAR T-cell therapy is cytokine release syndrome (CRS), which can affect up to 90% of patients. A second complication of CAR T‐cell therapy is immune effector cell‐associated neurotoxicity syndrome (ICANS). Despite superior efficacy, cilta-cel carries a higher risk of neurotoxicity (102–105). ICANS commonly involves cognitive impairment that ranges from acute confusional states to severe encephalopathy. Although immune effector cell‐associated encephalopathy (ICE) scoring is a valid method to assess neurotoxicity, it does not generally capture subtle or early changes in status. In Cartitude-1, 95% of patients experienced CRS with 5% having grade >3 effects. Moreover, a concerning aspect of cilta-cel’s toxicity profile is the incidence of delayed neurotoxicity, e.g., parkinsonism and other movement disorders, which has been observed in up to 12% of patients. Neurotoxic effects can manifest weeks to months post-infusion and have prompted increased vigilance as well as specific mitigation strategies. Conversely, in the KarMMa study, patients exhibited a more manageable safety profile with CRS reported in 84% of patients following the infusion of ide-cel. Neurotoxicity was reported in 18% of patients, with severe cases occurring in ~3%. Acute anaphylaxis events and tumor lysis syndrome (TLS) also occur after CAR T-cell infusion. The relatively manageable neurotoxicity profile of ide-cel makes it an appealing option for the elderly and frail patient populations.
Real-world data have generally supported the results generated the pivotal trial for CAR T- cell therapies to treat MM, with both agents demonstrating high efficacy outside of the controlled setting (106). However, it is increasingly appreciated that patients that are enrolled on clinical trials with strict inclusion and exclusion criteria generally do not represent the average patient seen and treated in the non-academic or outpatient clinical practice. Real-world experiences highlight the challenges of managing the toxicities associated with CAR T-cells, particularly the neurotoxicity observed following cilta-cel. CAR T-cell dysfunction, tumor-intrinsic resistance, and an immunosuppressive tumor microenvironment (TME) remain obstacles that limit CAR T-cell efficacy (101–105). Also, CAR T-cell exhaustion caused by persistent Ag stimulation or tonic CAR signaling leads to gradual loss of function and reduced cytotoxic activity (106–108). Immunosuppressive cells and factors within the TME further also contribute to CAR T-cell exhaustion, exclusion from the tumor bed and loss of functionality.
6.3 Bispecific T-cell engagers
BiTes are a specific type of bispecific Ab designed to bridge T-cells and tumor cells, and, consequently, facilitate T-cell-mediated elimination of cancer cells (109–112). BiTEs have two distinct binding domains that can bind simultaneously to either two antigens or two epitopes (antigenic regions) of the same antigen. BiTEs typically bind to a tumor epitope as well as CD3 that is presented on autologous T-cells, leading to T-cell activation and tumor lysis. BiTEs can be considered as next generation mAbs that target antigenic epitopes (of the same or different proteins presented on the same or different cell types, e.g., tumor and T-cells), to elicit multiple downstream physiological or anti-tumor responses. Teclistamab and elranatamab (Elra) are each BiTEs that target BCMA on MM cells and also bind to CD3 presented on T-cells. Talquetamab targets G protein–coupled receptor, family C, group 5, member D (GPRC5D) expressed on MM cells and CD3 present on T-cells. Talquetamab may represent a viable target for patients resistant to BCMA-directed therapy. BiTes and Bispecifics are valuable not just as an alternative to ASCT but also have a role in transplant ineligible patients too.
Teclistamab is referred to as both a BiTE (specific T-cell Engager) and a BsAb (Bi specific Ab) because it functions as both (109). Teclistamab is a specific type of bispecific antibody that works by binding to two different targets: CD3 on T cells and BCMA on myeloma cells. in the phase 1 dose-defining portion of the study, teclistamab showed promising efficacy in patients with RRMM (109). Teclistamab is the first approved off-the-shelf BCMA×CD3 bispecific antibody for the treatment of patients (pts) with RRMM based on data from the pivotal phase 1/2 MajesTEC-1 study (NCT03145181/NCT04557098). MajesTEC-1 evaluated patients with RRMM after = 3 therapy LOT, including triple-class exposure to an IMiD, a PI, and anti-CD38 Ab, treated with teclistamab. Patients received weekly subcutaneous injection of teclistamab (1.5 mg/kg BW) after step-up doses of 0.06 mg and 0.3 mg/kg. An ORR of 63% was observed with 39.4% of patients achieving CR or better (109). Median PFS was 11.3 months and median DOR was 18.4 months. The FDA granted accelerated approval to teclistamab-cqyv (TECVAYLI; Janssen Biotech) for the treatment of adult patients with RRMM who have received >4 prior LOT. After ~2 y mFU, patients receiving teclistamab demonstrated deep and durable responses regardless of refractory status, with mPFS of 12.5 months and mDOR of 24 months (not reached in those achieving ≥CR). These long-term follow-up data support teclistamab as a safe and effective off-the-shelf BCMA bispecific therapy for pts with RRMM (110).
Elranatamab is a humanized BCMA-CD3 agent administered subcutaneously once weekly. MagnetisMM-3 (phase 2, NCT04649359) demonstrated that RRMM patients who had received >3 prior LOT and received Elra experienced an ORR of 61%, including 35% that achieved CR or better (111). The FDA also granted accelerated approval to elranatamab-bcmm (Elrexfio) for the treatment of patients with RRMM who have previously received >4 LOT, including a PI, an IMiD, and an anti-CD38 monoclonal antibody. The regulatory decision was based upon data which showed that Elra elicited an ORR of 57.7% (95% CI, 47.3%-67.7%) in 97 patients who were naïve to BCMA-directed therapy (110). The CR rate was 25.8%, VGPR rate of 25.8, and a PR rate of 6.2%. Notably, 82% of responders were estimated to continue to respond to treatment for >9 months.
Talquetamab targets the G protein–coupled receptor family C group 5 member D(GPRC5D). The FDA has also granted accelerated approval to talquetamab-tgvs (Talvey, Janssen Biotech, Inc.) for adults with RRMM who have received at least >4 LOT, including a PI, an IMiD, and an anti-CD38 monoclonal antibody (112–114). Efficacy was evaluated in MMY1001 (MonumenTAL-1) (NCT03399799, NCT4634552), a single-arm, open-label, multicenter study that included 187 patients who had previously received at least four prior systemic therapies. Patients received talquetamab-tgvs 0.4 mg/kg subcutaneously weekly, following two step-up doses in the first week of therapy, or talquetamab-tgvs 0.8 mg/kg subcutaneously biweekly (q 2 weeks), following three step-up doses, until disease progression or unacceptable toxicity. The main efficacy outcome measures were ORR and DOR as assessed IMWG criteria.
In MonumenTAL-1, talquetamab exhibited an impressive ORR of 70% in heavily pre-treated RRMM patients with a median PFS of 7.5 months. Based upon the distribution of the target antigen, a unique pattern of GPRC5D-associated AEs have been observed as well as T-cell redirection–associated AEs (115–117). GPRC5D-associated AEs included dermatologic, i.e., rash, non-rash, and nail toxicities, and oral AEs, i.e., dysgeusia, dysphagia, and dry mouth. Talquetamab, while effective, presents unique side effects related to its GPRC5D target, e.g., skin rashes and nail disorders. Infections are a critical issue, with serious cases reported in 30-40% of patients, necessitating rigorous monitoring and supportive care. The AEs underscore the need for careful patient selection. The incidence of CRS and ICANS were consistent with other T-cell redirection therapies. The incidence of high-grade infections was lower than that observed with BCMA-targeting Bispecific Abs, with less frequent use of intravenous immunoglobulin required. GPRC5D-associated AEs were mostly low grade and led to few discontinuations. Increased talquetamab exposure has led to a higher incidence of dysgeusia. Nutritional monitoring and appropriate supplementation were implemented and high caloric shakes were advised to ensure adequate nutritional intake and prevent weight loss. Oral toxicities and weight loss have beenreported as AEs in the MonumenTAL-1, MonumenTAL-2, TRIMM-2, and RedirecTT-1 studies.
The optimal benefit of BiTEs is hampered by an immunosuppressive TME, a hallmark of MM, which limits efficacy and by undesirable adverse events, especially CRS and severe infections (Table 2) (52, 80, 81, 118–122).BiTEs are also associated with significant adverse effects. CRS is a common toxicity, occurring in 60-75% of patients across all three BiTEs, though most cases are low grade. Neurotoxicity, generally mild to moderate, also remains a concern, particularly with elranatamab and teclistamab.
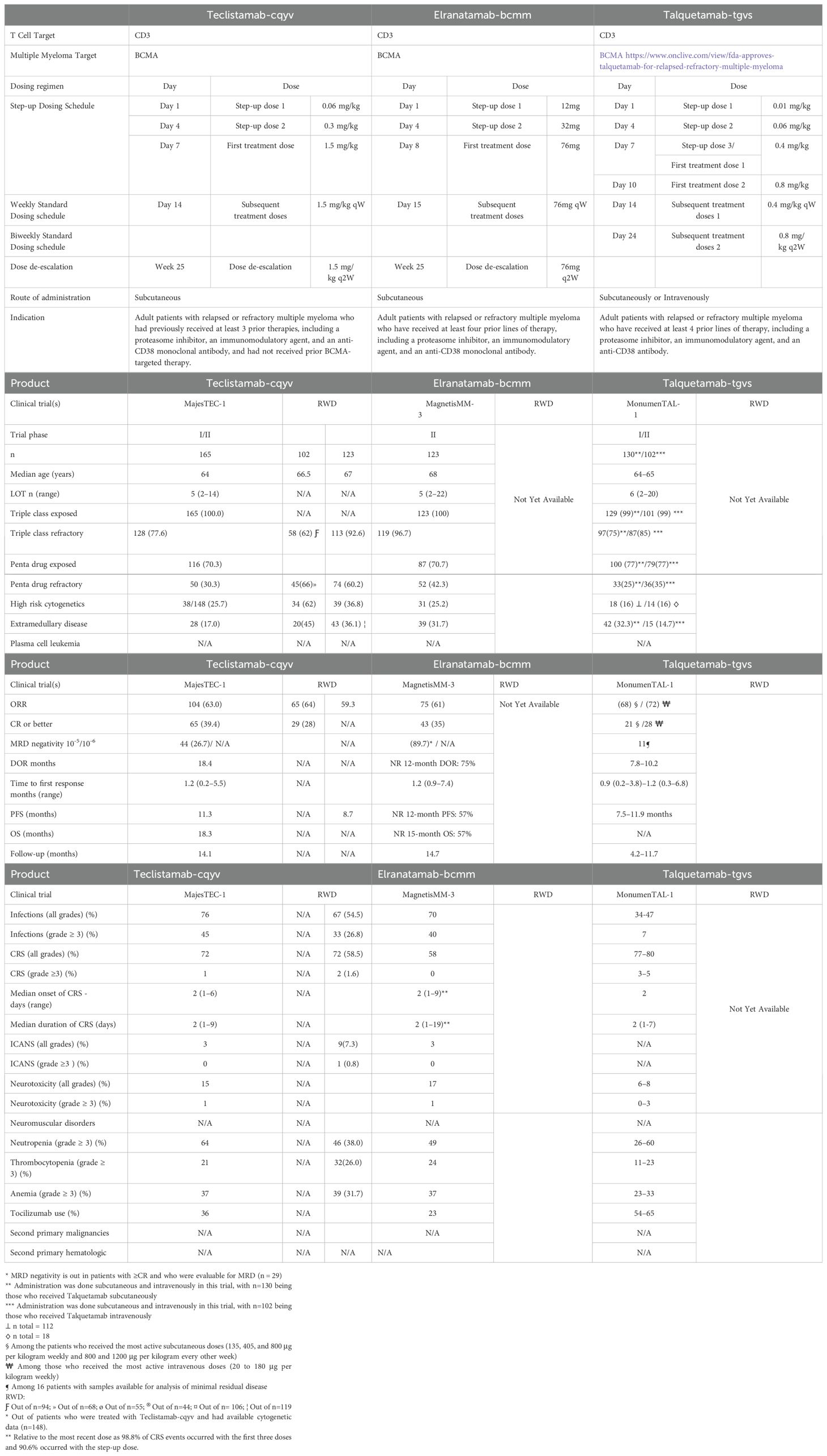
Table 2. Comparison of the FDA-approved BiTEs for the Treatment of Relapsed and/or Refractory Multiple Myeloma.
ASCT has been the standard of care patients with MM for the past three decades. However, high-risk patients still have poor outcomes. Many bispecific therapies are currently under investigation for the treatment of myeloma, e.g., blinatumomab, teclistamab, talquetamab and cevostamab, as well as CAR T-cell therapy using ide-cel and cilta-cel. With CAR-T and bispecific antibodies demonstrating deep and sustained remissions, the role of ASCT in the future treatment of myeloma has become a subject of debate, especially in the frail and elderly populations.
6.4 Antibody drug conjugates
ADCs are composed of a mAb that has been covalently linked to a cytotoxic chemotherapy drug, e.g., calicheamicin, monomethyl auristatin E, and microtubule inhibitor MMAF. The ADC is internalized by the tumor cell and then releases the cytotoxic drug, with the cancer cell to minimize damage to neighboring healthy cells. ADCs combine advantages of highly specific targeting ability and highly potent killing effect to achieve the accurate and efficient elimination of cancer cells. ADCs also improve the delivery of cytotoxic drugs to cancer cells and certain ADCs have exhibited improved efficacy over conventional chemotherapy. Belantamab mafodotin consists of a humanized IgG mAb linked to the microtubule-disrupting cytotoxic agent monomethyl auristatin, and targets BCMA (82–84). Initially approved for the treatment of RRMM, belantamab mafodotin was withdrawn from US and European markets following disappointing trial results.
DREAMM-1 and DREAMM-2 demonstrated promising activity of belantamab mafodotin in heavily pretreated RRMM populations (83, 85). DREAMM-1 reported an ORR of 60%, while DREAMM-2 exhibited an ORR of 30% at the approved dose of 2.5 mg/kg. The phase 3 DREAMM-3 study compared belantamab mafodotin to the combination of POM and Dex (Pd) and failed to demonstrate PFS or OS benefit. The phase 3 DREAMM-7 head-to-head trial evaluated the efficacy and safety of BVd triplet vs the standard of care triplet DVd, in patients with RRMM treated with ≥1 prior LOT. Belantamab mafodotin was dosed at 2.5mg/kg intravenously q3 weeks and BVd significantly improved PFS compared to DVd (37 vs. 13 months) but was associated with higher rates of serious AEs and drug discontinuation due to toxicity. DREAMM-8 investigated belantamab mafodotin with Pom and Dex (BPd) vs. bortezomib, Pom, and Dex (PVd). BPd improved PFS at 12 months (71% vs. 51%) but again exhibited higher rates of = grade 3 AEs, particularly ocular toxicity. Ocular toxicity is a significant AE associated with belantamab mafodotin caused by the accumulation of the cytotoxic payload in the cornea, leading to blurred vision, dry eyes, corneal ulceration, and, potentially, loss of vision. Ophthalmic monitoring is required before each dose. While ocular AEs can be managed with dose adjustments or temporary treatment holds, they remain a considerable burden for patients and healthcare providers. Other AEs of belantamab mafodotin include thrombocytopenia, nausea, pyrexia, infusion-related reactions, and fatigue.
7 Antimyeloma drugs in development
Cereblon E3 ligase modulatory drugs (CELMoDs) are an emerging new class of medications being studied in clinical trials for MM treatment (88, 89). CELMoDs build on the well-established platform of IMiDs and are designed not only target myeloma cells directly but also by engaging other immune cells. CELMoDs offer promise for those who have relapsed after treatment with IMiDs. Two promising orally-available CELMoDs in clinical trials are iberdomide and mezigdomide. Marizomib (MRZ) is a novel, irreversible proteasome inhibitor in clinical development for the treatment of relapsed or relapsed and refractory multiple myeloma (RRMM) (90). MRZ inhibits the 3 proteolytic activities of the 20S proteasome with specificity distinct from bortezomib and carfilzomib. Histone deacetylase (HDAC) inhibition improves the efficacy of proteasome inhibition for multiple myeloma but adds substantial toxicity. Preclinical models suggest that the observed synergy is due to the role of HDAC6 in mediating resistance to proteasome inhibition via the aggresome/autophagy pathway of protein degradation. A phase I/II trial of the HDAC6-selective inhibitor ricolinostat to define the safety, preliminary efficacy, and recommended phase II dose in combination with standard PI therapy (91). Patients with RRMM received oral ricolinostat on days 1–5 and 8–12 of each 21-day cycle. The ORR in combination with daily ricolinostat at ≥160 mg was 37%. The response rate to combination therapy among bortezomib-refractory patients was 14%. At the recommended phase II dose of ricolinostat of 160 mg daily, the combination with bortezomib and Dex is safe, well-tolerated, and active, suggesting that selective inhibition of HDAC6 is a promising approach to MM therapy.
There remain many challenges that warrant further development of CAR T structure and function as they relate to durability of response, T-cell exhaustion due to tonic signaling, immunogenicity, manufacturing-related limitations, and incidence of serious adverse events including cytokine release syndrome and immune effector cell associated neurotoxicity syndrome. D-domain based CAR T-cells are a new class of structurally and functionally distinct cell therapies that represent an alternative to conventional single-chain variable fragment based chimeric antigen receptor T-cells (92). Preclinical studies of a D-domain based targeting BCMA (CART-ddBCMA) have demonstrated effective anti-tumor response in both in vitro and tumor models. These studies currently support a first-in-human clinical study of CART-ddBCMA in MM patients. Durvalumab, a PD-L1 inhibitor, is being explored for its potential in treating MM. It works by blocking the PD-L1 protein, which helps cancer cells evade the immune system, thereby potentially enhancing the body’s ability to fight the disease (123). Durvalumab is often combined with other therapies, such
as like Len and Pom, and also with other targeted therapies like Dara.
8 Emerging combinations in myeloma therapy
Triplet regimens that consist of a PI, an IMiD, and Dex have been considered a standard of care for patients with NDMM based on RCTs demonstrating improved DOR and long-term PFS and OS compared with doublets (124). Quadruplet regimens have been investigated for NDMM in clinical trials evaluating a PI, IMiD, Dex, and anti-CD38 mAb. The results have shown improvement in increasing the DOR, including MRD negativity rates as well as PFS in patients with NDMM. The US FDA has approved a powerful four-drug combination therapy for frontline treatment of a regimen of Dara, bortezomib, Len and Dex for MM patients (68, 125–127). Disease progression in the first year post-QUADs is uncommon. Currently, there is a lack of high-level evidence to guide the management of transplant-eligible patients with NDMM who experience disease relapse after up-front quadruplet induction. Ravi et al. analyzed a large and mature institutional dataset of patients with NDMM treated with QUADs with the intent to proceed with ASCT (128). Similarly, a recent Mayo Clinic study reported that 6.7% of patients with NDMM had treatment refractoriness to initial induction therapy (129). Patients who did not respond to initial induction therapy had a poor median PFS of 4.2 months, compared to 50.8 months in patients who were able to achieve a complete response to initial induction therapy. Understanding the mechanisms of resistance to anti-myeloma therapy is an ongoing challenge. Efforts should focus on the early deployment of therapies with new mechanism of action for patients experiencing treatment failure after QUADs.
The FDA approved an injectable form of the drug Dara that includes hyaluronidase (Darzalex Faspro) given in combination with bortezomib, Len, and Dex for patients who are eligible for ASCT. The FDA has also approved isatuximab (Sarclisa) given with the same three drugs for patients newly diagnosed with MM who are ineligible for SCT. Because of the lengthy process required to treat a patient with CAR T-cells, bridging therapy (BT), administered after leukapheresis but prior to CAR T-cell infusion, has become an important component of safely administering CAR T therapy. Establishing a strategy for sequencing of T cell-redirecting therapies for RRMM also represents a pressing clinical need. The clinical and immunologic impact of bispecific T cell-engaging BsAb as BT to subsequent BCMA-directed CAR T-cell therapies in 52 patients with RRMM was recently evaluated. BsAbs were shown to be a potent and safe option for BT, achieving the highest ORR (100%) to BT compared with chemotherapy, anti-CD38, or anti-SLAMF7 antibody-based regimens (46%) (130).
9 Conclusions
The treatment paradigm for MM has evolved from non-specific agents to conventional chemotherapeutics to targeted agents generated based upon the biology of disease and more recently immunotherapy (41, 128, 131–133). Improved understanding of myelomagenesis and the biology of disease has given rise to new actionable targets for therapies that stimulate the immune system to effectuate long-lived tumor destruction (134, 135). Many of these pharmacologics are the pillars of present-day frontline treatment regimens, but are restricted by tight therapeutic indices, undesirable toxicities and significant AEs, combined with frequently acquired drug resistance.
Many challenges limit the therapeutic efficacy of CAR-T cells in hematological malignancies (86, 87, 96, 136–146). Barriers to effective CAR T-cell therapy include severe life-threatening toxicities, modest anti-tumor activity, antigen escape, restricted trafficking, and limited tumor infiltration. In addition, the host and tumor microenvironment interactions with CAR-T cells critically alter CAR-T cell function. Furthermore, a complex workforce is required to develop and implement these treatments. Lengthy waitlists to receive CAR T-cells introduce a pre-apheresis, patient selection bias. Post-apheresis patient dropout introduces a second layer of bias and further complicates the interpretation of treatment efficacy. Similar challenges also complicate the comparison of CAR T-cells with off-the-shelf agents, e.g., BsAbs and allogeneic CAR T-cell products (147–150). At present, both BsAbs and CAR T-cells are approved by the US FDA and are available as standard of care therapy. Certain patients may be better served by BsAbs that afford immediate access and a consequent reduced risk of disease progression or death while waiting for apheresis and CAR T-cell infusion. Patients who encounter manufacturing failure would have an even greater risk of compromised BM reserve and T-cell health and are more susceptible to cytopenia and life-threatening infections. Second, all patients in the pivotal KarMMA trial were required to be refractory to the last line of regimen before CAR T-cell therapy. However, in the study by Hansen et al, ~two-thirds of patients had refractory disease.
Selecting and sequencing immunotherapies in the myeloma setting presents a challenge, especially when lacking substantial historical data and necessitates the consideration of ECOG status, previous and potential AEs and disease resistance mechanisms. When sequencing BCMA-directed CAR T-cells and bispecific agents, clinicians must be aware of the effect that prior therapies may have on the efficacy of subsequent agent. Financial toxicity is another challenge since CAR T-cells and BiTEs are expensive and treatment decisions lead to healthcare disparities and raise concerns regarding long-term sustainability and access (86, 96, 137). Future myeloma care will undoubtedly continue to evolve over the next century to improve OS, access to care and quality of life. Ongoing research strives to identify targets that prevent or overcome drug resistance and advance precision oncology treatment approaches to tailors a patient’s care based on unique tumor and immune cell characteristics, e.g., mutations, genomics, T-cell profile. An approach focused on deciphering individual patient tumor genetics and MHC class I antigenic profiles will influence the parallel development of personalized (CAR) T-cell therapies and cancer vaccines.
Author contributions
JK: Writing – original draft, Writing – review & editing. AR: Writing – original draft, Writing – review & editing. SS: Writing – original draft, Writing – review & editing. JD: Conceptualization, Funding acquisition, Investigation, Software, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Research was supported by NIH R01 (5R01AI139141 to JD), University Hospitals Cleveland Medical Center/Seidman Cancer Center, and the Case Comprehensive Cancer Center.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cowan AJ, Green DJ, Kwok M, Lee S, Coffey DG, Holmberg LA, et al. Diagnosis and management of multiple myeloma: A review. JAMA. (2022) 327:464–77. doi: 10.1001/jama.2022.0003
2. Ludwig H, Novis Durie S, Meckl A, Hinke A, and Durie B. Multiple myeloma incidence and mortality around the globe; interrelations between health access and quality, economic resources, and patient empowerment. Oncologist. (2020) 25:e1406–13. doi: 10.1634/theoncologist.2020-0141
3. Malard F, Neri P, Bahlis NJ, Terpos E, Moukalled N, Hungria VTM, et al. Multiple myeloma. Nat Rev Dis Primers. (2024) 10:45. doi: 10.1038/s41572-024-00529-7
4. Mohty M, Facon T, Malard F, and Harousseau JL. A roadmap towards improving outcomes in multiple myeloma. Blood Cancer J. (2024) 14:135. doi: 10.1038/s41408-024-01115-6
5. Garfall AL. New biological therapies for multiple myeloma. Annu Rev Med. (2024) 29:75: 13–29. doi: 10.1146/annurev-med-050522-033815
6. Siegel RL, Miller KD, Wagle NS, and Jemal A. Cancer statistics, 2023. CA Cancer J Clin. (2023) 73:17–48. SEER. 2023. Cancer stat facts: myeloma. National Cancer Institute Surveillance, Epidemiology, and End Results Program. https://seer.cancer.gov/statfacts/html/mulmy.html. doi: 10.3322/caac.21763
6. Siegel RL, Miller KD, Wagle NS, and Jemal A. Cancer statistics, 2023. CA Cancer J Clin. (2023) 73:17–48. SEER. 2023. Cancer stat facts: myeloma. National Cancer Institute Surveillance, Epidemiology, and End Results Program. https://seer.cancer.gov/statfacts/html/mulmy.html. doi: 10.3322/caac.21763
7. Kyle RA, Larson DR, Therneau TM, Dispenzieri A, Kumar S, Cerhan JR, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. (2018) 378:241–49. doi: 10.1056/NEJMoa1709974
8. Mateos M-V, Kumar S, Dimopoulos MA, González-Calle V, Kastritis E, Hajek R, et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J. (2020) 10:102. doi: 10.1038/s41408-020-00366-3
9. Landgren O and Weiss BM. Patterns of monoclonal gammopathy of undetermined significance and multiple myeloma in various ethnic/racial groups: support for genetic factors in pathogenesis. Leukemia. (2009) 23:1691–97. doi: 10.1038/leu.2009.134
10. Siegel RL, Miller KD, Wagle NS, and Jemal A. Cancer statistics, 2023.CA. Cancer J Clin. (2023) 73:17–48. doi: 10.3322/caac.21763
11. Turesson I, Bjorkholm M, Blimark CH, Kristinsson S, Velez R, and Landgren O. Rapidly changing myeloma epidemiology in the general population: Increased incidence, older patients, and longer survival. Eur J haematology. (2018) 101:237–44. doi: 10.1111/ejh.2018.101.issue-2
12. Huang J, Chan SC, Lok V, Zhang L, Lucero-Prisno DE, Xu W, et al. The epidemiological landscape of multiple myeloma: a global cancer registry estimate of disease burden, risk factors, and temporal trends. Lancet Haematology. (2022) 9:e670–7. doi: 10.1016/S2352-3026(22)00165-X
13. Ouzzif Z, Eddair Y, Laassara W, El Maaroufi H, and Mahtat EM. Non-secretory multiple myeloma: A new observation and review of the literature. Cureus. (2024) 16:e54479. doi: 10.7759/cureus.54479
14. Migkou M, Avivi I, Gavriatopoulou M, Cohen YC, Fotiou D, Kanellias N, et al. Clinical characteristics and outcomes of oligosecretory and non-secretory multiple myeloma. Ann Hematology. (2020) 99:1251–5. doi: 10.1007/s00277-020-03984-w
15. Charliński G and Jurczyszyn A. Non-secretory multiple myeloma: diagnosis and management. Adv Clin Exp Med. (2022) 31:95–100. doi: 10.17219/acem/141455
16. Rajkumar SV. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. (2022) 97:1086–107. doi: 10.1002/ajh.26590
17. Durie BGM, Hoering A, Abidi MH, Rajkumar SV, Epstein J, Kahanic SP, et al. Bortezomib, Lenalidomide and Dexamethasone vs. Lenalidomide and Dexamethasone Induction Followed by Lenalidomide and Dexamethasone Maintenance in Patients with Newly Diagnosed Myeloma without Intent for Immediate Autologous Stem Cell Transplant: Results of the Randomised Phase III SWOG. Lancet. (2017) 389:519–27. doi: 10.1016/S0140-6736(16)31594-X
18. Attal M, Lauwers-Cances V, Hulin C, Leleu X, Caillot D, Escoffre M, et al. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N Engl J Med. (2017) 376:1311–20. doi: 10.1056/NEJMoa1611750
19. Goldschmidt H, Lokhorst HM, Mai EK, van der Holt B, Blau IW, Zweegman S, et al. Bortezomib before and after high-dose therapy in myeloma: long-term results from the phase III HOVON-65/GMMG-HD4 trial. Leukemia. (2018) 32:383–90. doi: 10.1038/leu.2017.211
20. Perrot A, Lauwers-Cances V, Cazaubiel T, Facon T, Caillot D, Clement-Filliatre L, et al. Early versus late autologous stem cell transplant in newly diagnosed multiple myeloma: long-term follow-up analysis of the IFM 2009 trial. Blood. (2020) 136:39. doi: 10.1182/blood-2020-134538
21. Solly S. Remarks on the pathology of mollities ossium; with cases. Med Chir Trans. (1844) 27:435–98. doi: 10.1177/095952874402700129
22. Kyle RA and Steensma DP. History of multiple myeloma. Recent Results Cancer Res. (2011) 183:3–23. doi: 10.1007/978-3-540-85772-3_1
23. Bence Jones H. Papers on clinical pathology. Lancet. (1847) 50:88–92. doi: 10.1016/S0140-6736(02)86528-X
24. Kahler O. Zur Symptomatologie des Multiplen Myeloms: Beobachtung von Albumosurie. Prague Med Wchnschr. (1889) 14:33.
25. Bayne-Jones S and Wilson DW. Immunological reactions of bence-jones proteins: I. Differences between bence-jones proteins and human serum proteins. Bull Johns Hopkins Hosp. (1922) 33:37–43.
26. Korngold L and Lipari R. Multiple-myeloma proteins. III. The antigenic relationship of Bence Jones proteins to normal gammaglobulin and multiple-myeloma serum proteins. Cancer. (1956) 9:262–72. doi: 10.1002/1097-0142(195603/04)9:2<262::aid-cncr2820090210>3.0.co;2-b
27. Alwall N. Urethane and stilbamidine in multiple myeloma: report on two cases. Lancet. (1947) 2:388–9. doi: 10.1016/s0140-6736(47)90375-9
28. Blokhin N, Larionov L, Perevodchikova N, Chebotareva L, and Merkulova N. Clinical experiences with sarcolysin in neoplastic diseases. Ann N Y Acad Sci. (1958) 68:1128–32. doi: 10.1111/j.1749-6632.1958.tb42675.x
29. Bergsagel DE, Sprague CC, Austin C, and Griffith KM. Evaluation of new chemotherapeutic agents in the treatment of multiple myeloma: IV. L-Phenylalanine mustard (NSC-8806). Cancer Chemother Rep. (1962) 21:87–99.
30. Holland JR, Hosley H, Scharlau C, Carbone PP, Frei E 3rd, Brindley CO, et al. A controlled trial of urethane treatment in multiple myeloma. Blood. (1966) 27:328–42.
31. Hoogstraten B, Sheehe PR, Cuttner J, Cooper T, Kyle RA, Oberfield RA, et al. Melphalan in multiple myeloma. Blood. (1967) 30:74–83.
32. Maas RE. A comparison of the effect of prednisone and a placebo in the treatment of multiple myeloma. Cancer Chemother Rep. (1962) 16:257–9.
33. Salmon SE, Shadduck RK, and Schilling A. Intermittent high-dose prednisone (NSC-10023) therapy for multiple myeloma. Cancer Chemother Rep. (1967) 51:179–87.
34. Alexanian R, Haut A, Khan AU, Lane M, McKelvey EM, Migliore PJ, et al. Treatment for multiple myeloma: combination chemotherapy with different melphalan dose regimens. JAMA. (1969) 208:1680–5. doi: 10.1001/jama.208.9.1680
35. Lee BJ, Sahakian G, Clarkson BD, and Krakoff IH. Proceedings: combination chemotherapy of multiple myeloma with alkeran, cytoxan, vincristine, prednisone, and BCNU. Cancer. (1974) 33:533–8. doi: 10.1002/1097-014233:2<533::aid-cncr2820330231>3.0.co;2-z
36. Case DC C.OMMAJ.R.X.X.X, Lee DJ 3rd, and Clarkson BD. Improved survival times in multiple myeloma treated with melphalan, prednisone, cyclophosphamide, vincristine and BCNU: M-2 protocol. Am J Med. (1977) 63:897–903. doi: 10.1016/0002-9343(77)90543-5
37. Richardson PG, Jacobus SJ, Weller EA, Hassoun H, Lonial S, Raje NS, et al. Triplet therapy, transplantation, and maintenance until progression in myeloma. New Engl J Med. (2022) 387:132–47. doi: 10.1056/NEJMoa2204925
38. Cenci S. The proteasome in terminal plasma cell differentiation. Semin Hematol. (2012) 49:215–22. doi: 10.1053/j.seminhematol.2012.04.005
39. Utley A, Lipchick B, Lee KP, and Nikiforov MA. Targeting multiple myeloma through the biology of long-lived plasma cells. Cancers (Basel). (2020) 12:2117. doi: 10.3390/cancers12082117
40. Ignatz-Hoover JJ, Murphy EV, and Driscoll JJ. Targeting proteasomes in cancer and infectious disease: A parallel strategy to treat Malignancies and microbes. Front Cell Infect Microbiol. (2022) 12:925804. doi: 10.3389/fcimb.2022.925804
41. Ignatz-Hoover JJ and Driscoll JJ. Therapeutics to harness the immune microenvironment in multiple myeloma. Cancer. Drug Resist. (2022) 5:647–61. doi: 10.20517/cdr.2022.23
43. The nobel prize in chemistry 2004. In: NobelPrize.org. Nobel prize outreach AB 2024. Stockholm, Sweden: The Nobel Foundation. Available at: https://www.nobelprize.org/prizes/chemistry/2004/summary (Accessed February 2025).
44. Driscoll JJ and Goldberg AL. The proteasome (multicatalytic protease) is a component of the 1500-kDa proteolytic complex which degrades ubiquitin-conjugated proteins. J Biol Chem. (1990) 286:4789–92. doi: 10.1016/S0021-9258(19)34041-4
45. Eytan E, Ganoth D, Armon T, and Hershko A. ATP-dependent incorporation of 20S protease into the 26S complex that degrades proteins conjugated to ubiquitin. Proc Natl Acad Sci U S A. (1989) 86:7751–5. doi: 10.1073/pnas.86.20.7751
46. Finley D and Prado MA. The proteasome and its network: engineering for adaptability. Cold Spring Harb Perspect Biol. (2020) 12:a033985. doi: 10.1101/cshperspect.a033985
47. Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci. (2009) 85:12–36. doi: 10.2183/pjab.85.12
48. Driscoll J and Goldberg AL. Skeletal muscle proteasome can degrade proteins in an ATP-dependent process that does not require ubiquitin. Proc Natl Acad Sci U S A. (1989) 86:787–91. doi: 10.1073/pnas.86.3.787
49. Driscoll J, Frydman J, and Goldberg AL. An ATP-stabilized inhibitor of the proteasome is a component of the 1500-kDa ubiquitin conjugate-degrading complex. Proc Natl Acad Sci U S A. (1992) 89:4986–90. doi: 10.1073/pnas.89.11.4986
50. Anderson KC. The 39th David A. Karnofsky lecture: Bench-to-Bedside translation of targeted therapies in multiple myeloma. J Clin Oncol. (2012) 30:445–52. doi: 10.1200/JCO.2011.37.8919
51. Anderson KC. Progress and paradigms in multiple myeloma. Clin Cancer Res. (2016) 22:5419–27. doi: 10.1158/1078-0432.CCR-16-0625
52. Kort J, Ignatz-Hoover J, and Driscoll JJ. Real-world evidence for autologous stem cell transplantation in elderly multiple myeloma patients. Transplant Cell Ther. (2025) 31:S437–8. doi: 10.1016/j.jtct.2025.01.672
53. Joseph NS, Gupta VA, Wyman S, Graiser M, Kaufman JL, Almaula D, et al. Benefits of autologous stem cell transplantation for elderly myeloma patients in the last quarter of life. Transplant Cell Ther. (2022) 28:75.e1–7. doi: 10.1016/j.jtct.2021.09.024
54. Driscoll JJ and Ignatz-Hoover J. Emerging strategies to manage relapsed and/or refractory Multiple Myeloma. Oncol (Williston Park NY). (2023) 37:166–7.
55. Holstein SA and McCarthy PL. Immunomodulatory drugs in multiple myeloma: mechanisms of action and clinical experience. Drugs. (2017) 77:505–20. doi: 10.1007/s40265-017-0689-1
56. Bird S and Pawlyn C. IMiD resistance in multiple myeloma: current understanding of the underpinning biology and clinical impact. Blood. (2023) 142:131–40. doi: 10.1182/blood.2023019637
57. Zamolodchikov D, Berk-Rauch HE, Oren DA, Stor DS, Singh PK, Kawasaki M, et al. Biochemical and structural analysis of the interaction between β-amyloid and fibrinogen. Blood. (2016) 128:1144–51. doi: 10.1182/blood-2016-03-705228
58. Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. (2014) 343:305–9. doi: 10.1126/science.1244917
59. Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, and McConkey M. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. (2014) 343:301–5. doi: 10.1126/science.1244851
60. Lokhorst MM, Schattenberg A, Cornelissen JJ, van Oers MH, Fibbe W, Russell I, et al. Donor lymphocyte infusions for relapsed multiple myeloma after allogeneic stem-cell transplantation: predictive factors for response and long-term outcome. JCO. (2000) 18:3031–7. doi: 10.1200/JCO.2000.18.16.3031
61. Greil C, Engelhardt M, Ihorst G, Schoeller K, Bertz H, Marks R, et al. Allogeneic transplantation of multiple myeloma patients may allow long-term survival in carefully selected patients with accepta ble toxicity and preserved quality of life. Haematologica. (2019) 104:370–9. doi: 10.3324/haematol.2018.200881
62. Xia C, Ribeiro M, Scott S, and Lonial S. Daratumumab: monoclonal antibody therapy to treat multiple myeloma. Drugs Today (Barc). (2016) 52:551–60. doi: 10.1358/dot.2016.52.10.2543308
63. Tzogani K, Penninga E, Schougaard Christiansen ML, Hovgaard D, Sarac SB, Camarero Jimenez J, et al. EMA review of daratumumab for the treatment of adult patients with multiple myeloma. Oncologist. (2018) 23:594–602. doi: 10.1634/theoncologist.2017-0328
64. Attal M, Richardson PG, Rajkumar SV, San-Miguel J, Beksac M, Spicka I, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): a randomised, multicentre, open-label, phase 3 study. Lancet. (2019) 394:2096–107. doi: 10.1016/S0140-6736(19)32556-5
65. Richardson PG, Perrot A, San-Miguel J, Beksac M, Spicka I, Leleu X, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): follow-up analysis of a randomised, phase 3 study. Lancet Oncol. (2022) 23:416–27. doi: 10.1016/S1470-2045(22)00019-5
66. Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. (2015) 373:621–31. doi: 10.1056/NEJMoa1505654
67. Collins SM, Bakan CE, Swartzel GD, Hofmeister CC, Efebera YA, Kwon H, et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunology Immunother. (2013) 62:1841–9. doi: 10.1007/s00262-013-1493-8
68. Sonneveld P, Dimopoulos MA, Boccadoro M, Quach H, Ho PJ, Beksac M, et al. Daratumumab, bortezomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. (2024) 390:301–13. doi: 10.1056/NEJMoa2312054
69. Facon T, Dimopoulos MA, Leleu XP, Beksac M, Pour L, Hájek R, et al. Isatuximab, bortezomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. (2024) 391:1597–609. doi: 10.1056/NEJMoa2400712
70. Jamil F, Shafqat M, Samuel S, Shah Z, Durer C, Durer S, et al. Efficacy and toxicity profile of elotuzumab for multiple myeloma: A systematic review and meta-analysis. Blood. (2018) 132 :5640. doi: 10.1182/blood-2018-99-117209
71. Dimopoulos MA, Richardson PG, Bahlis NJ, Grosicki S, Cavo M, Beksaç M, et al. Addition of elotuzumab to lenalidomide and dexamethasone for patients with newly diagnosed, transplantation ineligible multiple myeloma (ELOQUENT-1): an open-label, multicentre, randomised, phase 3 trial. Lancet Haematol. (2022) 9:e403–e414ccc. doi: 10.1056/NEJMoa1607751
72. Hoylman E, Brown A, Perissinotti AJ, Marini BL, Pianko M, Ye JC, et al. Optimal sequence of daratumumab and elotuzumab in relapsed and refractory multiple myeloma. Leukemia lymphoma. (2020) 61:691–8. doi: 10.1080/10428194.2019.1688324
73. Nakayama H, Aisa Y, Ito C, Sakurai A, and Nakazato T. The real-world outcomes of relapsed/refractory multiple myeloma treated with elotuzumab, pomalidomide, and dexamethasone. Hematol Rep. (2024) 16:593–602. doi: 10.3390/hematolrep16040058
74. Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived c regions. Biochem Biophys Res Commun. (1987) 149:960–8. doi: 10.1016/0006-291X(87)90502-X
75. Gross G, Waks T, and Eshhar Z. Expression of immunoglobulin-t-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
76. Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. (2003) 9:279–86. doi: 10.1038/nm827
77. Eshhar Z. Tumor-specific T-bodies: towards clinical application. Cancer Immunology Immunother. (1997) 45:131–6. doi: 10.1007/s002620050415
78. Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood J Am Soc Hematology. (2011) 118:4817–28. doi: 10.1182/blood-2011-04-348540
79. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Trans Med. (2013) 5:177ra38. doi: 10.1126/scitranslmed.3005930
80. Firestone R, Shekarkhand T, Patel D, Tan CRC, Hultcrantz M, Lesokhin AM, et al. Evaluating the efficacy of commercial teclistamab in relapsed refractory multiple myeloma patients with prior exposure to anti-BCMA therapies. J Clin Oncol. (2023) 41:8049. doi: 10.1200/JCO.2023.41.16_suppl.8049
81. Rodriguez-Otero P, van de Donk NWCJ, Pillarisetti K, Cornax I, Vishwamitra D, Gray K, et al. GPRC5D as a novel target for the treatment of multiple myeloma: a narrative review. Blood Cancer J. (2024) 14:24. doi: 10.1038/s41408-023-00966-9
82. Markham A. Belantamab mafodotin: first approval. Drugs. (2020) 80:1607–13. doi: 10.1007/s40265-020-01404-x
83. Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol. (2020) 21:207–21. doi: 10.1016/S1470-2045(19)30788-0
84. Trudel S and Stewart AK. A belantamab mafodotin revival in multiple myeloma therapy. . N Engl J Med. (2024) 391:461–2. doi: 10.1056/NEJMe2406799
85. Trudel S, Lendvai N, Popat R, Voorhees PM, Reeves B, Libby EN, et al. Antibody–drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: an update on safety and efficacy from dose expansion phase I study. Blood Cancer J. (2019) 9:37. doi: 10.1038/s41408-019-0196-6
86. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. (2021) 398:314–24. doi: 10.1016/S0140-6736(21)00933-8
87. San-Miguel J, Dhakal B, Yong K, Spencer A, Anguille S, Mateos MV, et al. Cilta-cel or standard care in lenalidomide-refractory multiple myeloma. New Engl J Med. (2023) 389:335–47. doi: 10.1056/NEJMoa2303379
88. Lonial S, Popat R, Hulin C, Jagannath S, Oriol A, Richardson PG, et al. Iberdomide plus dexamethasone in heavily pretreated late-line relapsed or refractory multiple myeloma (CC-220-MM-001): a multicentre, multicohort, open-label, phase 1/2 trial. Lancet Haematology. (2022) 9:e822–32. doi: 10.1016/S2352-3026(22)00290-3
89. Schütt J, Brinkert K, Plis A, Schenk T, and Brioli A. Unraveling the complexity of drug resistance mechanisms to T cell-engaging therapies and CELMoDs in multiple myeloma: a comprehensive review. Cancer Drug Resist. (2024) 7:26. doi: 10.20517/cdr.2024.39
90. Richardson PG, Zimmerman TM, Hofmeister CC, Talpaz M, Chanan-Khan AA, Kaufman JL, et al. Phase 1 study of marizomib in relapsed or relapsed and refractory multiple myeloma: NPI-0052–101 Part 1. Blood J Am Soc Hematology. (2016) 127:2693–700. doi: 10.1182/blood-2015-12-686378
91. Vogl DT, Raje N, Jagannath S, Richardson P, Hari P, Orlowski R, et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin Cancer research: an Off J Am Assoc Cancer Res. (2017) 23:3307. doi: 10.1158/1078-0432.CCR-16-2526
92. Frigault MJ, Bishop MR, Rosenblatt J, O’Donnell EK, Raje N, Cook D, et al. Phase 1 study of CART-ddBCMA for the treatment of subjects with relapsed and refractory multiple myeloma. Blood Adv. (2023) 7:768–77. doi: 10.1182/bloodadvances.2022007210
93. Ross S. Firestone, Sham Mailankody; Current use of CAR T cells to treat multiple myeloma. Hematol Am Soc Hematol Educ Program. (2023) 1:340–47. doi: 10.1182/hematology.2023000434
94. Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, et al. T-cell clones can be rendered specific for CD19: Toward the selective augmentation of the graft-versus-B–lineage leukemia effect. Blood J Am Soc Hematology. (2003) 101:1637–44. doi: 10.1182/blood-2002-07-1989
95. Mitra A, Barua A, Huang L, Ganguly S, Feng Q, and He B. From bench to bedside: the history and progress of CAR T cell therapy. Front Immunol. (2023) 14:1188049. doi: 10.3389/fimmu.2023.1188049
96. Munshi NC, Anderson LD Jr, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. (2021) 384:705–16. doi: 10.1056/NEJMoa2024850
97. Raje NS, Siegel DS, Jagannath S, Lonial S, Munshi NC, Moreau P, et al. Idecabtagene vicleucel (ide-cel, bb2121) in relapsed and refractory multiple myeloma: analyses of high-risk subgroups in the KarMMa Study. Blood. (2020) 136:37–8. doi: 10.1182/blood-2020-134319
98. Bar N, Diels J, van Sanden S, Mendes J, Hernando T, Cost T, et al. Comparative efficacy of ciltacabtagene autoleucel versus idecabtagene vicleucel in the treatment of patients with relapsed or refractory multiple myeloma previously treated with 2–4 prior lines of therapy using a matching-adjusted indirect comparison. Blood. (2023) 142 :2141. doi: 10.1182/blood-2023-182141
99. US Food and Drug Administration approves Bristol Myers Squibb’s and bluebird bio’s Abecma (idecabtagene vicleucel), the first anti-BCMA CAR T cell therapy for relapsed or refractory multiple myeloma. In: News release. Bristol Myers Squibb and Bluebird Bio. Available at: https://news.bms.com/news/details/2021/U.S.-Food-and-Drug-Administration-Approves-Bristol-Myers-Squibbs-and-bluebird-bios-Abecma-idecabtagene-vicleucel-the-First-Anti-BCMA-CAR-T-Cell-Therapy-for-Relapsed-or-Refractory-Multiple-Myeloma/default.aspx.
100. Usmani S, Patel K, Hari P, Berdeja J, Alsina M, Vij R, et al. KarMMa-2 cohort 2a: efficacy and safety of idecabtagene vicleucel in clinical high-risk multiple myeloma patients with early relapse after frontline autologous stem cell transplantation. Blood. (2022) 140:875–7. doi: 10.1182/blood-2022
101. Einsele H, Cohen AD, Delforge M, Hillengass J, Goldschmidt H, Weisel K, et al. Biological correlative analyses and updated clinical data of ciltacabtagene autoleucel (cilta-cel), a BCMA-directed CAR-T cell therapy, in lenalidomide (len)-refractory patients (pts) with progressive multiple myeloma (MM) after 1–3 prior lines of therapy (LOT): CARTITUDE-2, cohort A. J Clin Oncol. (2023) 40:8020. doi: 10.1200/JCO.2022.40.16_suppl
102. Rubin DB, Danish HH, Ali AB, Li K, LaRose S, Monk AD, et al. Neurological toxicities associated with chimeric antigen receptor T-cell therapy. Brain. (2019) 142:1334–48. doi: 10.1093/brain/awz053
103. Cohen AD, Parekh S, Santomasso BD, Pérez-Larraya JG, van de Donk NWCJ, Arnulf B, et al. Incidence and management of CAR-T neurotoxicity in patients with multiple myeloma treated with ciltacabtagene autoleucel in CARTITUDE studies. Blood Cancer J. (2022) 12:1–9. doi: 10.1038/s41408-022-00629-1
104. Hansen DK, Sidana S, Peres LC, Leitzinger CC, Shune L, Shrewsbury A, et al. Idecabtagene vicleucel for relapsed/refractory multiple myeloma: real-world experience from the myeloma CAR T consortium. J Clin Oncol. (2023) 41:2087–97. doi: 10.1200/JCO.22.01365
105. Hansen DK, Patel KK, Peres LC, Leitzinger CC, Shune L, Shrewsbury A, et al. Safety and efficacy of standard of care (SOC) ciltacabtagene autoleucel (Cilta-cel) for relapsed/refractory multiple myeloma (RRMM). J Clin Oncol. (2023) 41:8012. doi: 10.1200/JCO.2023.41.16_suppl.8012
106. Chakraborty R and Al Hadidi S. Intent matters: real-world applicability of idecabtagene vicleucel usage in the United States. J Clin Oncol. (2023) 41:3657–8. doi: 10.1200/JCO.23.00226
107. Van Oekelen O, Nath K, Mouhieddine TH, Farzana T, Aleman A, Melnekoff DT, et al. Interventions and outcomes of patients with multiple myeloma receiving salvage therapy after BCMA-directed CAR T therapy. Blood. (2023) 141:756–65. doi: 10.1182/blood.2022017848
108. Sales C, Anderson MA, Kuznetsova V, Rosenfeld H, Malpas CB, Roos I, et al. Patterns of neurotoxicity among patients receiving chimeric antigen receptor T-cell therapy: A single-centre cohort study. Eur J Neurol. (2024) 31:e16174. doi: 10.1111/ene.16174
109. Moreau P, Garfall AL, van de Donk NWCJ, Nahi H, San-Miguel JF, Oriol A, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. (2022) 387:495–505. doi: 10.1056/NEJMoa2203478
110. Garfall AL, Nooka AK, van de Donk NWCJ, Moreau P, Bhutani M, Oriol A, et al. Long-term follow-up from the phase 1/2 MajesTEC-1 trial of teclistamab in patients with relapsed/refractory multiple myeloma. JCO. (2024) 42:7540–0. doi: 10.1200/JCO.2024.42.16_suppl.7540
111. Lesokhin AM, Tomasson MH, Arnulf B, Bahlis NJ, Miles Prince H, Niesvizky R, et al. Elranatamab in relapsed or refractory multiple myeloma: phase 2 MagnetisMM-3 trial results. Nat Med. (2023) 29(9):2259–67. doi: 10.1038/s41591-023-02528-9
112. Chari A, Minnema MC, Berdeja JG, Oriol A, van de Donk NWCJ, Rodríguez-Otero P, et al. Talquetamab, a T-cell-redirecting GPRC5D bispecific antibody for multiple myeloma. N Engl J Med. (2022) 387:2232–44. doi: 10.1056/NEJMoa2204591
113. Liu L and Krishnan A. Talquetamab in multiple myeloma. Haematologica. (2024) 109:718–24. doi: 10.3324/haematol.2023.283931
114. Chari A. BCMA/GPRC5D/fcRH5—CARs/bsAbs—how to choose. In: SOHO annual meeting. Houston, TX (2023).
115. Mailankody S, Devlin SM, Landa J, Nath K, Diamonte C, Carstens EJ, et al. GPRC5D-targeted CAR T cells for myeloma. N Engl J Med. (2022) 387:1196–206. doi: 10.1056/NEJMoa2209900
116. Xia J, Li H, Yan Z, Zhou D, Wang Y, Qi Y, et al. Anti-G protein-coupled receptor, class C group 5 member D chimeric antigen receptor T cells in patients with relapsed or refractory multiple myeloma: a single-arm, phase II trial. J Clin Oncol. (2023) 41:2583–93. doi: 10.1200/JCO.22.01824
117. Zhang M, Wei G, Zhou L, Zhou J, Chen S, Zhang W, et al. GPRC5D CAR T cells (OriCAR-017) in patients with relapsed or refractory multiple myeloma (POLARIS): a first-in-human, single-centre, single-arm, phase 1 trial. Lancet Haematol. (2023) 10:e107–16. doi: 10.1016/S2352-3026(22)00372-6
118. Da Vià MC, Dietrich O, Truger M, Arampatzi P, Duell J, Heidemeier A, et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat Med. (2021) 27:616–9. doi: 10.1038/s41591-021-01245-5
119. Samur MK, Fulciniti M, Aktas Samur A, Bazarbachi AH, Tai YT, Prabhala R, et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat Commun. (2021) 12:868. doi: 10.1038/s41467-021-21177-5
120. Lee H, Ahn S, Maity R, LeBlay N, Ziccheddu B, Truger M, et al. Mechanisms of antigen escape from BCMA- or GPRC5D-targeted immunotherapies in multiple myeloma. Nat Med. (2023) 29:2295–306. doi: 10.1038/s41591-023-02491-5
121. Rana PS, Soler DC, and Kort J. and Driscoll JJ(2022), Targeting TGF-β signaling in the multiple myeloma microenvironment: Steering CARs and T cells in the right direction. Front Cell Dev Biol. (2022) 10:1059715. doi: 10.3389/fcell.2022.1059715
122. Rana PS, Murphy EV, Kort J, and Driscoll JJ. Road testing new CAR design strategies in multiple myeloma. Front Immunol. (2022) 13:957157. doi: 10.3389/fimmu.2022.957157
123. Frerichs KA, Verkleij CPM, Dimopoulos MA, Marin Soto JA, Zweegman S, Young MH, et al. Efficacy and safety of durvalumab combined with daratumumab in daratumumab-refractory multiple myeloma patients. Cancers (Basel). (2021) 13:2452. doi: 10.3390/cancers13102452
124. Dimopoulos MA, Moreau P, Terpos E, Mateos MV, Zweegman S, Cook G, et al. Multiple myeloma: EHA-ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Hemasphere. (2021) 5:e528. doi: 10.1097/HS9.0000000000000528
125. Moreau P, Hulin C, Perrot A, Arnulf B, Belhadj K, Benboubker L, et al. Maintenance with daratumumab or observation following treatment with bortezomib, thalidomide, and dexamethasone with or without daratumumab and autologous stem-cell transplant in patients with newly diagnosed multiple myeloma (CASSIOPEIA): an open-label, randomised, phase 3 trial. Lancet Oncol. (2021) 22:1378–90. doi: 10.1016/S1470-2045(21)00428-9
126. Voorhees PM, Sborov DW, Laubach J, Kaufman JL, Reeves B, Rodriguez C, et al. Addition of daratumumab to lenalidomide, bortezomib, and dexamethasone for transplantation-eligible patients with newly diagnosed multiple myeloma (GRIFFIN): final analysis of an open-label, randomised, phase 2 trial. Lancet Haematol. (2023) 10:e825–37. doi: 10.1016/S2352-3026(23)00217-X
127. Joseph NS, Kaufman JL, Gupta VA, Hofmeister CC, Dhodapkar MV, Boise LH, et al. Quadruplet therapy for newly diagnosed myeloma: comparative analysis of sequential cohorts with triplet therapy lenalidomide, bortezomib and dexamethasone (RVd) versus daratumamab with RVD (DRVd) in transplant-eligible patients. Blood Cancer J. (2024) 14:159. doi: 10.1038/s41408-024-01120-9
128. Ravi G, Bal S, Joiner L, Giri S, Sentell M, Hill T, et al. Subsequent therapy and outcomes in patients with newly diagnosed multiple myeloma experiencing disease progression after quadruplet combinations. Br J Haematol. (2024) 204:1300–6. doi: 10.1111/bjh.19303
129. Charalampous C, Goel U, Kapoor P, Binder M, Buadi FK, Cook J, et al. Outcomes of patients with primary refractory multiple myeloma in the era of triplet and quadruplet induction therapy. Blood Adv. (2023) 7:4371–80. doi: 10.1182/bloodadvances.2023009681
130. Bhaskar ST, Dholaria B, Savani BN, and Oluwole O. The evolving role of bridging therapy during CAR-T therapy. Clin Hematol Int. (2024) 6:9–16. doi: 10.46989/001c.116261
131. Driscoll JJ and Brailey M. Emerging small molecule approaches to enhance the antimyeloma benefit of proteasome inhibitors. Cancer Metastasis Rev. (2017) 36:585–98. doi: 10.1007/s10555-017-9698-5
132. Rana PS, Ignatz-Hoover JJ, and Driscoll JJ. Targeting proteasomes and the MHC class I antigen presentation machinery to treat cancer, infections and age-related diseases. Cancers (Basel). (2023) 15:5632. doi: 10.3390/cancers15235632
133. Rana PS, Ignatz-Hoover JJ, Kim BG, Malek E, Federov Y, Adams D, et al. HDAC6 inhibition releases HR23B to activate proteasomes, expand the tumor immuno-peptidome and amplify T-cell antimyeloma activity. Cancer Res Commun. (2024) 4:1517–32. doi: 10.1158/2767-9764
134. Rana PS, Ignatz-Hoover JJ, Guo C, Mosley AL, Malek E, Federov Y, et al. Mosley A et al, Immunoproteasome activation expands the MHC-class I immunopeptidome, unmasks neoantigens and enhances T-cell antimyeloma activity. Mol Cancer Ther. (2024) 23(12):1743–60. doi: 10.1158/1535-7163.MCT-23-0931
135. Mailankody S, Matous JV, Chhabra S, Liedtke M, Sidana S, Oluwole OO, et al. Allogeneic BCMA-targeting CAR T cells in relapsed/refractory multiple myeloma: phase 1 UNIVERSAL trial interim results. Nat Med. (2023) 29:422–9. doi: 10.1038/s41591-022-02182-7
136. Cohen AD, Mateos M-V, Cohen Y-C, Patel K, Cavo M, Nooka AK, et al. Efficacy and safety of cilta-cel in patients with progressive multiple myeloma after exposure to other BCMA-targeting agents. Blood. (2023) 141:219–30. doi: 10.1182/blood.2022015526
137. Rodriguez-Otero P, Ailawadhi S, Arnulf B, Patel K, Cavo M, Nooka AK, et al. Ide-cel or standard regimens in relapsed and refractory multiple myeloma. New Engl J Med. (2023) 388(11):1002–14. doi: 10.1056/NEJMoa2213614
138. Teoh PJ and Chng WJ. CAR T-cell therapy in multiple myeloma: more room for improvement. Blood Cancer J. (2021) 11:84. doi: 10.1038/s41408-021-00469-5
139. Simmons GL and Castaneda Puglianini O. T-cell-based cellular immunotherapy of multiple myeloma: current developments. Cancers (Basel). (2022) 14(3):101306. doi: 10.3390/cancers14174249
140. Jagannath S, Joseph N, Crivera C, Kharat A, Jackson CC, Valluri S, et al. Component costs of CAR-T therapy in addition to treatment acquisition costs in patients with multiple myeloma. Oncol Ther. (2023) 11:263–75. doi: 10.1007/s40487-023-00228-5
141. Wu W, Ding S, Mingming Z, Yuping Z, Sun X, Zhao Z, et al. Cost effectiveness analysis of CAR-T cell therapy for patients with relapsed/refractory multiple myeloma in China. J Med Econ. (2023) 26:701–9. doi: 10.1080/13696998.2023.2207742
142. Sanoyan DA, Kronig MN, Porret N, Wiedemann G, et al. Real-life experiences with CAR T-cell therapy with idecabtagene vicleucel (ide-cel) for triple-class exposed relapsed/refractory multiple myeloma patients. BMC Cancer. (2023) 23:345. doi: 10.1186/s12885-023-10824-3
143. Mann H and Comenzo RL. Evaluating the therapeutic potential of idecabtagene vicleucel in the treatment of multiple myeloma: evidence to date. Onco Targets Ther. (2022) 15:799–813. doi: 10.2147/OTT.S305429
144. Gong Z, et al. Adverse effects and non-relapse mortality of BCMA directed T cell therapies in multiple myeloma: an FAERS database study. Blood Cancer J. (2024) 14:36. doi: 10.1038/s41408-024-01023-9
145. Hansen DK, Sidana S, Peres L, Shune L, Sborov DW, Hashmi H, et al. Idecabtagene vicleucel (Ide-cel) chimeric antigen receptor (CAR) T-cell therapy for relapsed/refractory multiple myeloma (RRMM): Real-world experience. J Clin Oncol. (2022) 40(16):8042 doi: 10.1200/JCO.2022.40.16_suppl.8042
146. Lecat CSY, Taube JB, Wilson W, Carmichael J, Parrish C, Wallis G, et al. Defining unmet need following lenalidomide refractoriness: real-world evidence of outcomes in patients with multiple myeloma. Front Oncol. (2021) 11:703233. doi: 10.3389/fonc.2021.703233
147. Sidana S, Ahmed N, Akhtar OS, Heim M, Brazauskas R, Hansen DK, et al. Real world outcomes with idecabtagene vicleucel (Ide-cel) CAR-T cell therapy for relapsed/refractory multiple myeloma. Blood. (2023) 142:1027–7. doi: 10.1182/blood-2023-181762
148. Gill SK, Biran N, Phull P, Vesole DH, Siegel DS, and Parmar H. A real-world comparison of idecabtagene vicleucel and ciltacabtagene autoleucel CAR-T therapy: A single center experience for relapsed/refractory multiple myeloma. Blood. (2023) 142 :4717. doi: 10.1182/blood-2023-190583
149. Dima D, Sannareddy A, Ahmed N, Davis JA, Shaikh H, Mahmoudjafari Z, et al. Real-world safety and efficacy of teclistamab for patients with heavily pretreated relapsed-refractory multiple myeloma. Blood. (2023) 142:91. doi: 10.1182/blood-2023-180421
Keywords: myeloma, proteasome, immunotherapy, chemoresistance, CAR T cell, plasma cell
Citation: Kort J, Rivera A, Senigarapu S and Driscoll JJ (2025) Revolutions at the frontline of multiple myeloma treatment: lessons and challenges to finding a cure. Front. Oncol. 15:1578529. doi: 10.3389/fonc.2025.1578529
Received: 17 February 2025; Accepted: 22 May 2025;
Published: 20 June 2025.
Edited by:
Michael R. Shurin, University of Pittsburgh Medical Center, United StatesReviewed by:
Srinivas Devarakonda, The Ohio State University, United StatesGianfranco Lapietra, Josep Carreras Leukemia Research Institute (IJC), Spain
Copyright © 2025 Kort, Rivera, Senigarapu and Driscoll. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James J. Driscoll, SmFtZXMuRHJpc2NvbGxAVUhob3NwaXRhbHMub3Jn