 Mankgopo Kgatle1,2,3*
Mankgopo Kgatle1,2,3* Saidon Mbambara1,2,4
Saidon Mbambara1,2,4 Olalekan Fadebi1,2
Olalekan Fadebi1,2 Joseph Kabunda1,2
Joseph Kabunda1,2 Chimbabantu Kaoma1,2
Chimbabantu Kaoma1,2 Thobeka Dlangalala1,2
Thobeka Dlangalala1,2 Siphesihle Nxele1,2,5
Siphesihle Nxele1,2,5 Ndimo Modipane1,2
Ndimo Modipane1,2 Thato Serite1,2
Thato Serite1,2 Kgomotso Mokoala1,2
Kgomotso Mokoala1,2 Tivani Mashamba-Thompson5
Tivani Mashamba-Thompson5 Mike Sathekge1,2
Mike Sathekge1,2- 1Nuclear Medicine Research Infrastructure, Department of Basic and Translational Research, Pretoria, South Africa
- 2Department of Nuclear Medicine, University of Pretoria, Pretoria, South Africa
- 3Department of Medicine, University of Cape Town and Groote Schuur Hospital, Observatory, Cape Town, South Africa
- 4Department of Biomedical Sciences, Tropical Diseases Research Centre, Ndola, Zambia
- 5School of Health System and Public Health, Faculty of Health Sciences, University of Pretoria, Pretoria, South Africa
The overactivation of NRF2 (Nuclear factor erythroid 2-related factor 2) in female malignancies is an emerging field of study with significant implications for treatment efficacy. NRF2 plays a pivotal role in managing inflammation-induced oxidative stress, which is crucial components of the tumor microenvironment. Acting as a transcription factor and basic leucine zipper protein, it regulates the expression of various antioxidant genes that safeguard cells from oxidative stress and damage. While NRF2 activation is beneficial for the survival of normal cells, its overactivation in cancer cells can enhance tumor cell survival, proliferation, and resistance to treatments. Importantly, NRF2 has a dual context-dependent role, functioning as a tumor suppressor when transiently activated in normal cells to prevent carcinogenesis, but as an oncogene when persistently activated in established tumors. Understanding NRF2’s transcriptional alterations and developing targeted therapies could improve cancer management, prognosis and treatment outcomes, making it a promising target for precision oncology. This review aims to provide a comprehensive overview of NRF2 activation in female malignancies, including cervical, endometrial, ovarian, vaginal, vulvar and, breast cancers, and its association with chemoresistance, highlighting challenges and opportunities for developing more effective cancer treatments.
Introduction
Female cancers including breast and gynecological cancers significantly impact women’s health (1, 2). Gynecological cancers include cervical, endometrial (uterine), ovarian, vaginal, and vulvar cancers. Although not classified as a gynecological cancer, breast cancer also significantly impacts female reproductive health (3, 4). Both breast and gynecological cancers require early detection and specialized treatment to improve outcomes and survival rates. Treatment options vary depending on the type and stage of the cancer and may include surgery, radiation therapy, chemotherapy, and targeted therapies. However, treatment resistance remains a considerable hurdle.
These cancers can greatly affect reproductive health and overall well-being, presenting symptoms like abnormal bleeding, pelvic pain, and unusual discharge (1, 5). Late detection is common, particularly with ovarian cancer, which often manifests with nonspecific symptoms such as bloating and abdominal pain. These symptoms result from cancerous cells disrupting the normal functions of the female reproductive organs (6). The functioning of these organs relies heavily on the transcriptional regulation of various transcription factors that govern specific gene transcription necessary for cell growth, differentiation, and proper functioning (7). Any alterations in the activity or levels of these transcription factors can lead to disruptions in normal reproductive functions, potentially causing irregularities, infertility, infections, and diseases such as cancer (7). Improving early detection through regular screenings and increased symptom awareness is crucial for better treatment outcomes and patient prognosis.
Genetic alterations, such as mutations in BRCA1 and BRCA2 genes, play a critical role in the development, progression, and spread of breast and certain gynecological cancers. These genes are involved in DNA repair, and mutations can lead to heightened oxidative stress and genomic instability (8). Chronic inflammation contributes to cancer progression by fostering a microenvironment that activates multiple transcriptional regulation mechanisms. For example, inflammatory signaling pathways such as nuclear factor kappa B (NF-kB) and signal transducer and activator of transcription 3 (STAT3) regulate genes linked to cancer progression (9). Additionally, cytokines produced by inflammatory cells, including tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β), activate transcription factors that promote cell proliferation, survival, and angiogenesis. Additionally, epigenetic modifications, such as DNA methylation and histone changes, alter gene expression, and post-transcriptional processes affect mRNA stability and translation. These combined mechanisms significantly contribute to the onset, progression, and metastasis of female cancers (10–12).
NRF2 exhibits a dual role in cancer biology. In normal or pre-malignant cells, transient NRF2 activation acts tumor-suppressive by reducing ROS-induced DNA damage and carcinogenesis (13). Conversely, in established tumors, persistent NRF2 hyperactivation, often through KEAP1 mutations or epigenetic dysregulation, becomes oncogenic, promoting proliferation, metabolic reprogramming, and chemoresistance (13). This context dependency underpins the paradox of NRF2 as both ‘guardian’ and ‘driver’ of cancer, requiring careful therapeutic consideration, as illustrated in Figure 1. NRF2 not just as a redox regulator but as a driver of multiple cancer hallmarks, reinforcing its relevance in female cancers like breast, ovarian, and endometrial malignancies (13).
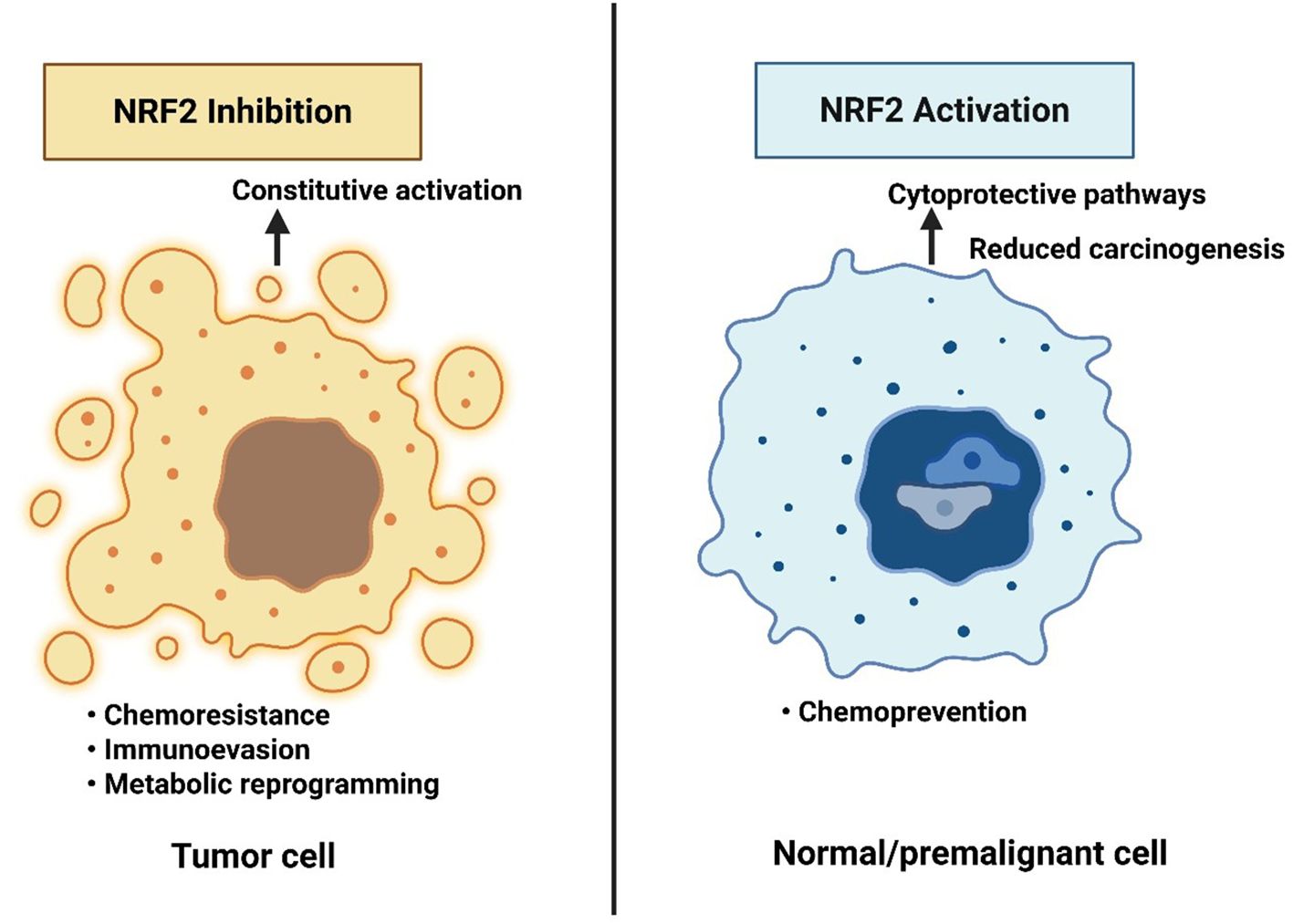
Figure 1. The NRF2 paradox in cancer therapy and prevention. Constitutive NRF2 activation in tumor cells promotes chemoresistance, immune evasion, and metabolic reprogramming, supporting malignant progression. In contrast, NRF2 activation in normal or premalignant cells enhances cytoprotective pathways and reduces carcinogenesis. This dual role highlights the therapeutic paradox: NRF2 inhibition may benefit established cancers, whereas NRF2 activation may serve as a chemopreventive strategy in high-risk but non-malignant contexts.
Overactivation of NRF2 may be linked to female cancers, some of which may be aggressive and exhibit distinct molecular features (14, 15). Mutations in KEAP1, hypoxic environment, along with epigenetic and post-translational alterations, lead to persistent NRF2 activation, enhancing cancer cells’ antioxidant capabilities and metabolic flexibility (16). This provides a survival advantage by neutralizing ROS and detoxifying chemotherapeutic agents. While transient NRF2 activation protects against carcinogenesis, its persistent activation may lead to malignant progression, increased aggressiveness, therapy resistance, and poor prognosis (17–19). NRF2 upregulates antioxidant and detoxifying genes, counteracting oxidative stress from chronic inflammation, influencing inflammatory responses, and maintaining cancer stem cells, which contribute to therapy resistance and tumor relapse (20–23).
This review aims to provide an overview of NRF2 activation in female malignancies and its association with chemoresistance, highlighting challenges and opportunities for developing more effective cancer treatments.
Activation of NRF2 and general implication in cancer
The NRF2 transcription factor plays a central role in regulating antioxidant and detoxification systems in response to oxidative and electrophilic stress (20–23). Reactive oxygen species (ROS), normally generated under homeostatic conditions, are essential signaling molecules that regulate proliferation, differentiation, and survival (19, 24). They also function as second messengers in extracellular signal transduction, coordinating communication within and between cells (19). However, excessive ROS and reactive nitrogen species (RNS) lead to oxidative stress, which fosters carcinogenesis by driving oncogene activation, inactivation of tumor-suppressor pathways, and mitochondrial dysfunction (19, 24).
The biological consequences of NRF2 activation are highly context-dependent. In normal physiology and early stages of carcinogenesis, NRF2 preserves redox balance, prevents ROS-induced mutagenesis, and thus acts as a tumor suppressor (22). By contrast, in established cancers, persistent NRF2 signaling is often hijacked by tumor cells to sustain anabolic metabolism, block apoptosis, and promote resistance to chemotherapy and radiotherapy (25). This duality underpins NRF2’s reputation as both a “protector” of normal tissue and an “enabler” of cancer progression (25).
At the molecular level, NRF2 controls the expression of genes that drive metabolic reprogramming and encode antioxidant and detoxifying enzymes. Under basal conditions, NRF2 is sequestered in the cytoplasm by its inhibitor KEAP1 (as illustrated in Figure 2), which forms a complex with the CUL3-based E3 ubiquitin ligase to target NRF2 for proteasomal degradation, thereby maintaining low NRF2 levels (26). Upon exposure to stressors such as ROS, modifications or mutations in KEAP1 disrupt this interaction, allowing NRF2 to escape degradation, accumulate in the nucleus, and dimerize with small MAF (sMAF) proteins (26). The NRF2-sMAF complex binds to antioxidant response elements (AREs) in target gene promoters, initiating transcriptional programs that enhance antioxidant defense, detoxification, and cellular repair.
Figure 2. Activation of NRF2 through ROS leads to the dissociation of NRF2 from the NRF2/CUL3/KEAP1 complex, allowing the NRF2 to translocate into the nucleus. In the nucleus, the NRF2 binds to the ARE, forming an NRF2/ARE complex and thereby regulating the transcription of its target genes. In its inactivated form, NRF2 levels are continuously kept low through proteasomal degradation in the cytoplasm. NRF2 activation pathway, showing KEAP1-NRF2 regulation, downstream antioxidant targets (GCLC, NQO1, HMOX1, TXNRD1, MRPs, GST), and potential pharmacological intervention points relevant to female cancers.
Key NRF2 target genes include glutamate-cysteine ligase catalytic subunit (GCLC), NAD(P)H quinone oxidoreductase 1 (NQO1), heme oxygenase-1 (HMOX1), UDP-glucuronosyltransferase (UGT), sulfiredoxin 1 (SRXN1), thioredoxin reductase 1 (TXNRD1), multidrug resistance-associated proteins (MRPs), and glutathione S-transferase (GST) (27). Together, these effectors underscore NRF2’s central role in maintaining cellular redox homeostasis and influencing cancer progression.
Key targets pathways for NRF2 activation
Glutamate-cysteine ligase catalytic subunit
Glutathione (GSH) is a critical tripeptide antioxidant involved in protein synthesis, DNA repair, and detoxification via conjugation reactions (28, 29). Its biosynthesis begins with the rate-limiting step catalyzed by γ-glutamylcysteine ligase (GCL), composed of a catalytic (GCLC) and a modifier (GCLM) subunit (30). GCLC links glutamate to cysteine, while GCLM enhances this efficiency (31). Oxidative stress strongly regulates GCL activity, which is essential for sustaining cellular GSH levels and protecting mitochondria from toxicity (32–34). In turn, GSH scavenges reactive oxygen species (ROS) and other free radicals, maintaining redox balance (32, 35).
Cysteine availability is the major constraint in GSH synthesis. It enters cells primarily as cystine via the cystine/glutamate antiporter xCT (SLC7A11) and is then reduced to cysteine by intracellular GSH or the NADPH-dependent thioredoxin system (36). Overexpression of xCT/SLC7A11 has been reported in triple-negative breast cancer (TNBC), where its activity fuels cysteine supply for GSH production; conversely, xCT inhibition reduces proliferation, partly through GSH depletion (37).
Mechanistically, NRF2 transcriptionally regulates GCL by binding to AREs in the promoter regions of the GCLC and GCLM genes, thereby enhancing glutathione biosynthesis and enabling cells to sustain redox homeostasis under oxidative stress. In cancer, particularly gynecological malignancies, dysregulation of the KEAP1-NRF2 axis results in persistent NRF2 activation, which upregulates GCL expression and promotes chemoresistance (38, 39). While some cancers exhibit reduced GCL activity and low glutathione levels, gynecological cancers such as endometrial and ovarian subtypes often display elevated GCLC expression, conferring metabolic flexibility and resistance to therapy. For instance, ARID1A-deficient ovarian cancers demonstrate synthetic lethality upon GCLC depletion, underscoring its therapeutic potential (31, 40).
Beyond regulating GCL, NRF2 also induces SLC7A11 (xCT) and GPX4, strengthening cellular defenses against ferroptosis and allowing tumor cells to evade iron-dependent cell death (15). In TNBC, mesenchymal stem-like cells exploit cystine uptake via xCT/SLC7A11 to activate NRF2, establishing antioxidant programs that extend beyond GSH synthesis. This cystine/NRF2/OSGIN1 axis promotes stemness, EMT, and survival, with NRF2-induced genes such as OSGIN1, SRXN1, and AKR1B10 linked to poor prognosis, highlighting a potential therapeutic vulnerability in aggressive TNBC (41).
Abnormal GSH levels are also linked to increased GCL expression or activity in cancers such as liver, lung, breast, and malignant mesothelioma, correlating with metastasis, chemoresistance, and poor prognosis (27). High GCLC expression has been consistently observed in multiple cancers, supporting its utility as a biomarker and therapeutic target (42). Additionally, NRF2-GCL deregulation has been reported in endometriotic lesions, where it contributes to growth and fibrosis (27). CRISPR knockout studies further demonstrate that GCLC is essential for the survival of acute myeloid leukemia (AML) cells but less critical for normal hematopoietic cells, highlighting its selective therapeutic potential (43, 44). Despite these insights, the expression and function of GCLC and GCLM in female cancers remain insufficiently defined, underscoring the need for further research.
NAD(P)H quinone oxidoreductase 1
NQO1 is one of the most robustly induced NRF2 targets. It is also a versatile, FAD-dependent flavoprotein that promotes 2-electron reductions of quinones and other substrates, reducing oxidative stress by minimizing ROS generation and thiol depletion through p53 tumor suppressor stabilization (45). Its expression is tightly linked to NRF2 activity and is often used as a surrogate marker of NRF2 pathway activation in cancer studies. NQO1 is highly inducible and regulated by the Keap1/Nrf2/ARE pathway (46). Its antioxidant function in combating oxidative stress is crucial, as evidenced by studies showing that NQO1 induction decreases susceptibility to oxidative stress, while its depletion increases it. NQO1 also stabilizes tumor suppressor p53 by preventing its degradation (46). Human polymorphisms suppressing NQO1 activity are linked to increased disease predisposition (47). NQO1 thus acts as a cytoprotective enzyme, providing antioxidant protection and regulating the degradation of specific proteins.
NQO1-deficient mice show lower p53 induction, reduced apoptosis, increased tumor susceptibility, and impaired NF-kB function. It has been demonstrated that the missense variant NQO1, found in 4-20% of the human population, results in no detectable NQO1 activity due to rapid degradation of the NQO1 P187S protein. Despite ongoing debates about the role of NQO1 in cancer, it has been found to be highly expressed in various cancers, including breast cancer, melanoma, lung cancer, cholangiocarcinoma, and pancreatic cancer (48–52). NQO1 homozygous individuals have a higher risk of benzene-induced hematotoxicity, acute nonlymphocytic leukemia, and other cancers, particularly leukemia. The variant also correlates with an increased risk of relapse or death in children treated for acute lymphoblastic leukemia and has been linked to increased breast cancer risk under specific conditions (53). Previous immunohistochemistry study has revealed that NQO1 plays a crucial role in the progression of gastric cancer, distinguishing it from non-cancer patients and healthy controls (49). Additionally, bioinformatics analysis of TCGA data also revealed significantly higher GCLC and GCLM expression in gastric adenocarcinoma tissues compared to normal tissues. Immunohistochemistry confirmed these findings, with 77% and 80% of gastric adenocarcinoma tissues showing positive staining for GCLC and GCLM, respectively, versus only 9% and 11% in adjacent normal tissues (54). The study concluded that dysregulated expression of GCLC and GCLM could promote tumorigenesis, presenting potential targets for valuable diagnostic or prognostic biomarker and therapeutic target.
Recent findings further underscore the oncogenic versatility of NQO1, particularly in gynecological malignancies. Elevated NQO1 expression in ovarian cancer has been shown to confer resistance to quinone-based chemotherapeutics and promote tumor survival, highlighting the dual role of NRF2 in cytoprotection and oncogenic adaptation (55). A study investigating ferroptosis, a regulated form of cell death driven by iron and lipid peroxidation, identified NQO1 as a key molecular target (56). Daphnetin (Daph), a natural compound, was found to directly bind and inhibit NQO1, thereby increasing oxidative stress and inducing ferroptosis in ovarian cancer cells. This inhibition significantly reduced cell proliferation and migration in vitro and in vivo (56). Notably, overexpression of NQO1 reversed these effects, confirming its protective role in ovarian cancer cell survival. These findings position NQO1 not only as a modulator of ferroptosis but also as a promising therapeutic target, especially in combination with agents like cisplatin.
Heme oxygenase-1
HMOX1 is a 32kDa key metabolic enzyme involved in the mammalian stress response (57). It has two major isoforms, HO-1 and HO-2, which degrade heme to carbon monoxide (CO), ferrous iron (Fe²+), and biliverdin (BV) (58, 59). HO-1 is inducible by stress factors like cadmium, hydrogen peroxide, hypoxia, and inflammation, while HO-2 is constitutively expressed (60, 61). Both isoforms localize to the endoplasmic reticulum but can redistribute under stress. Despite lacking heme groups, it plays essential roles, including catalyzing heme degradation with NADPH-Cytochrome P450 Reductase support. During injury healing, this process shifts heme to biliverdin and finally to bilirubin, facilitated by intramolecular hydroxylation of the heme molecule (62). HO-1 is found in many human tissues, especially the spleen, liver, and bone marrow, and plays a crucial role in iron homeostasis and protection against oxidative stress and inflammation (63). Notably, HMOX1 deficiency can be fatal, and this is evidenced by HO-1-deficient mice studies, which showed increased vulnerability to anemia, iron deposition, and oxidative stress-induced liver damage (64). Additionally, HO-1 influences bone metabolism and immune defense mechanisms, underlining its significance in various physiological processes.
The NRF2 also targets HMOX1 and breaks it down into beneficial products, and protects against various pathologies. NRF2 overactivation by early growth response 1 (EGR1) leads to increased HMOX1 transcription in breast cancer, providing a survival advantage to the cancer cells and promoting resistance to therapy (65). HMOX1 expression is notably higher in colon cancer cells compared to normal colonic epithelial cells across various hemin concentrations (66). However, in colorectal cancer tissues, HMOX1 expression is significantly lower than in adjacent non-neoplastic tissues (67). This suggests that while HMOX1 may provide cytoprotection within a certain range, excessive levels might contribute to tumor progression. Essentially, there’s a beneficial threshold where HMOX1 helps protect cells, but beyond this limit, it may act as an oncogene, promoting cancer development.
UDP-glucuronosyltransferase
Another NRF-2 targets, UGT, SRXN1 and TXNRD1, support the reduction of peroxiredoxins, aiding in the detoxification of harmful peroxides. UGTs are a superfamily of enzymes found in animals, plants, fungi, and bacteria. They add sugars from UDP-sugar donors to various lipophilic molecules, aiding in the detoxification and elimination of harmful substances (68). In mammals, the superfamily has four families: UGT1, UGT2, UGT3, and UGT8. UGT1 and UGT2 families, particularly, are involved in pharmacology and toxicology, conjugating glucuronic acid to make molecules water-soluble for excretion. Genetic variations in UGTs affect drug metabolism and disease susceptibility. UGT3 and UGT8, using different sugars, may primarily conjugate endogenous chemicals, impacting cellular functions and potentially influencing cancer and degenerative diseases (68).
In cancer, UGTs have a dual role. On a positive note, they safeguard normal cells by eliminating harmful substances, such as xenobiotics and endogenous compounds, through glucuronic acid conjugation (14). However, the overactivation of NRF2 upregulates these drug-metabolizing enzymes, leading to enhanced drug metabolism and clearance in cancers like lung, colorectal, breast, and prostate cancer. This rapid elimination of chemotherapy drugs reduces their effectiveness, enabling cancer cells to survive and proliferate (14). Consequently, higher doses of drugs are often required, which can increase toxicity and side effects for the patient.
Sulfiredoxin 1
A sulfiredoxin-1 is an antioxidant enzyme that catalyses the reduction of over-oxidized peroxiredoxins, restoring their active form (69). This reaction involves converting peroxiredoxin-(S-hydroxy-S-oxocysteine) to peroxiredoxin-(S-hydroxycysteine) using ATP and a thiol, producing ADP, phosphate, and a disulfide as by-products. Sulfiredoxins belongs to the thiol-based antioxidant system and can lower ROS under oxidative stress (70). They are also known by names such as Srx1 and peroxiredoxin-(S-hydroxy-S-oxocysteine) reductase. SRXN1 is essential for regulating various physiological processes like cell apoptosis, proliferation, invasion, and maintaining redox balance (71).
SRXN1 also plays a role in promoting carcinogenesis and protecting against ischemia/reperfusion injury in cardiac progenitor cells. When NRF2 is overactivated, it boosts the levels of SRXN1, which lowers ROS and helps cancer cells endure oxidative stress, thereby supporting their growth and survival (72). For instance, elevated levels of SRXN1 are associated with poor prognosis in several types of cancer, including non-small-cell lung cancer (NSCLC) and prostate cancer. Consequently, this leads to chemoresistance and radiotherapy resistance, thereby contributing to a more aggressive cancer phenotype with poorer patient prognosis. Another study has also identified SRXN1 as pro-tumorigenic in hepatocellular carcinoma (HCC) by influencing ROS signaling (73). Furthermore, SRXN1’s role in oxidoreductase activity suggested that its overexpression is critical in the tumorigenesis and progression of HCC (73).
Thioredoxin reductase 1
TXNRD1 gene encodes a pyridine nucleotide oxidoreductase that functions in selenium metabolism and oxidative stress protection by reducing thioredoxins and other substrates (74). The enzyme operates as a homodimer using FAD as a cofactor, with each subunit containing a selenocysteine (Sec) residue crucial for its catalytic activity. This residue is encoded by the UGA codon, typically a stop signal but recognized as Sec due to a common stem-loop structure in the 3’ UTR called the SECIS element (75). A thioredoxin-mediated reducing environment is essential for the effective DNA binding of redox-sensitive transcription factors like p53 and NF-κB. By binding ROS, thioredoxin protects against oxidative stress (76). Additionally, thioredoxin activates ribonucleotide reductase, inhibits apoptosis signal regulating kinase 1, and induces HIF-1 and vascular endothelial growth factor (VEGF), contributing to cancer traits such as increased proliferation, inhibited apoptosis, and angiogenesis.
TXNRD1 is a direct transcriptional target of NRF2, regulated through AREs located within its promoter region (77). Upon NRF2 activation, typically triggered by oxidative stress or KEAP1 disruption, the transcription factor translocates to the nucleus and induces TXNRD1 expression (26). This upregulation supports thioredoxin-mediated redox balance, selenium metabolism, and cellular protection against ROS-induced damage (41).
The cytoprotective function of TXNRD1 is particularly significant in cancer, where its overexpression is frequently observed across multiple malignancies, including lung, breast, and pancreatic cancers (78). In lung cancer, elevated TXNRD1 levels contribute to resistance against therapies such as cisplatin by maintaining low intracellular ROS levels, thereby shielding cancer cells from oxidative damage (78). In breast cancer, high TXNRD1 expression correlates with poor prognosis and increased metastatic potential, underscoring its role in tumor progression (79). Similarly, in pancreatic cancer, the thioredoxin system, of which TXNRD1 is a key component, enables cancer cells to evade oxidative stress-induced cell death (80). In non-small cell lung cancer (NSCLC), TXNRD1 overexpression, driven by NRF2 signaling, has been associated with tumor recurrence, adverse clinical outcomes, and chemoresistance, ultimately enhancing cancer cell survival (81).
Beyond its role as a downstream effector, TXNRD1 also engages in a dynamic feedback loop within the NRF2 regulatory network. When TXNRD1 levels are reduced, whether due to treatment-induced oxidative stress or post-translational modifications, compensatory NRF2 responses are triggered. These include increased nuclear accumulation of NRF2 and transcriptional activation of other ARE-regulated genes such as NQO1 and AOX1, aimed at restoring redox equilibrium (78). Consequently, TXNRD1 expression reflects both direct NRF2-mediated transcriptional control and adaptive feedback mechanisms responding to redox imbalance.
This dual regulatory role highlights the complexity of NRF2 signaling in cancer biology. Notably, the expression of NRF2 target genes may not consistently mirror pathway activation, particularly under therapeutic pressure or within heterogeneous tumor microenvironments. Such context-dependent modulation has important implications for interpreting TXNRD1 as a biomarker or therapeutic target in NRF2-driven malignancies.
Multidrug resistance-associated proteins
NRF2 upregulates MRPs, a family of transporters that move various molecular substrates, including drugs, across cell membranes, affecting their pharmacokinetics and toxicity (22, 82). MRPs are part of the ATP-binding cassette (ABCC) superfamily and are differentially expressed in various organs, including the liver, kidney, intestine, and blood-brain barrier. There are nine human MRPs, which transport a diverse array of endogenous and exogenous compounds and their conjugates (83, 84). MRP1 has a high affinity for leukotriene C4, MRP2 extrudes endogenous organic anions such as bilirubin glucuronide and certain anticancer agents, and MRP3 transports glutathione and glucuronate conjugates, as well as monoanionic bile acids (83, 84). MRP4 and MRP5 mediate the transport of cyclic nucleotides and confer resistance to some antiviral and anticancer nucleotide analogues, while MRP6 is involved in transporting glutathione conjugates and the cyclic pentapeptide BQ123; its deficiency leads to pseudoxanthoma elasticum (83, 84). MRPs play crucial roles in drug disposition and excretion, impacting drug toxicity and drug interactions, and understanding their functions and regulation can help minimize drug toxicity, prevent unfavorable interactions, and overcome drug resistance (85).
Increased levels of MRPs and NRF2 activation are linked to various cancers, including lung, colorectal, breast, and prostate cancers (86). Overexpression of MRPs (such as MRP1, MRP2, and MRP3), and high NRF2 activity enhance drug efflux in these cancers, leading to chemoresistance and poor response to treatments by effectively removing chemotherapy agents from cancer cells (22, 82). This dual role of MRPs and NRF2 complicates cancer treatment due to increased resilience and continued cancer cell proliferation (22, 82).
Glutathione S-transferase
GSTs are a family of Phase II detoxification enzymes that conjugate GSH to a variety of electrophilic compounds, both endogenous and exogenous (87). There are two main superfamilies of GSTs: membrane-bound microsomal and cytosolic. Microsomal GSTs, which form homo- and heterotrimers, are key in metabolizing leukotrienes and prostaglandins. Cytosolic GSTs, highly polymorphic, can be classified into six classes: α, μ, ω, π, θ, and ζ (87). The π and μ classes regulate cellular survival and death signals via interactions with JNK1 and ASK1, which are activated in response to cellular stress. GSTs are involved in developing resistance to chemotherapy agents through direct detoxification and inhibition of the MAP kinase pathway (87).
Continuous NRF2 overactivation exerts its transcriptional activities, upregulates GST (18). The GST family of enzymes detoxifies harmful compounds by conjugating them with GSH. This enhances cancer cells’ ability to detoxify ROS and other harmful compounds, leading to increased cancer cell proliferation, survival, and migration, thereby promoting tumor growth and metastasis (7). Furthermore, it increases cancer cells’ resistance to chemotherapy and radiation by enhancing their antioxidant and detoxifying capacities.
In summary, the GSH and the activity of various NRF2-regulated enzymes such as NQO1, HMOX1, SRXN1, TXNRD1, MRPs, and GST play crucial roles in cellular processes and maintaining cellular health.
The role of NRF2 activation in different female cancers
NRF2 activation in cervical cancer
Cervical cancer is one of the most prevalent cancers among women worldwide and originates in the cervical epithelial cells, which are divided into squamous and columnar epithelial cells (Figure 3, Table 1). These cells line the outer part (ectocervix) and the inner part (endocervix) of the cervix, the lower section of the uterus that connects to the vagina (92). The majority of cervical cancers are squamous cell carcinomas affecting the ectocervix. Adenocarcinoma, which affects the endocervix, is less common (93). The primary cause and significant risk factor for cervical cancer is infection with the human papillomavirus (HPV), which induces cellular changes that can lead to cancer development (94, 95).
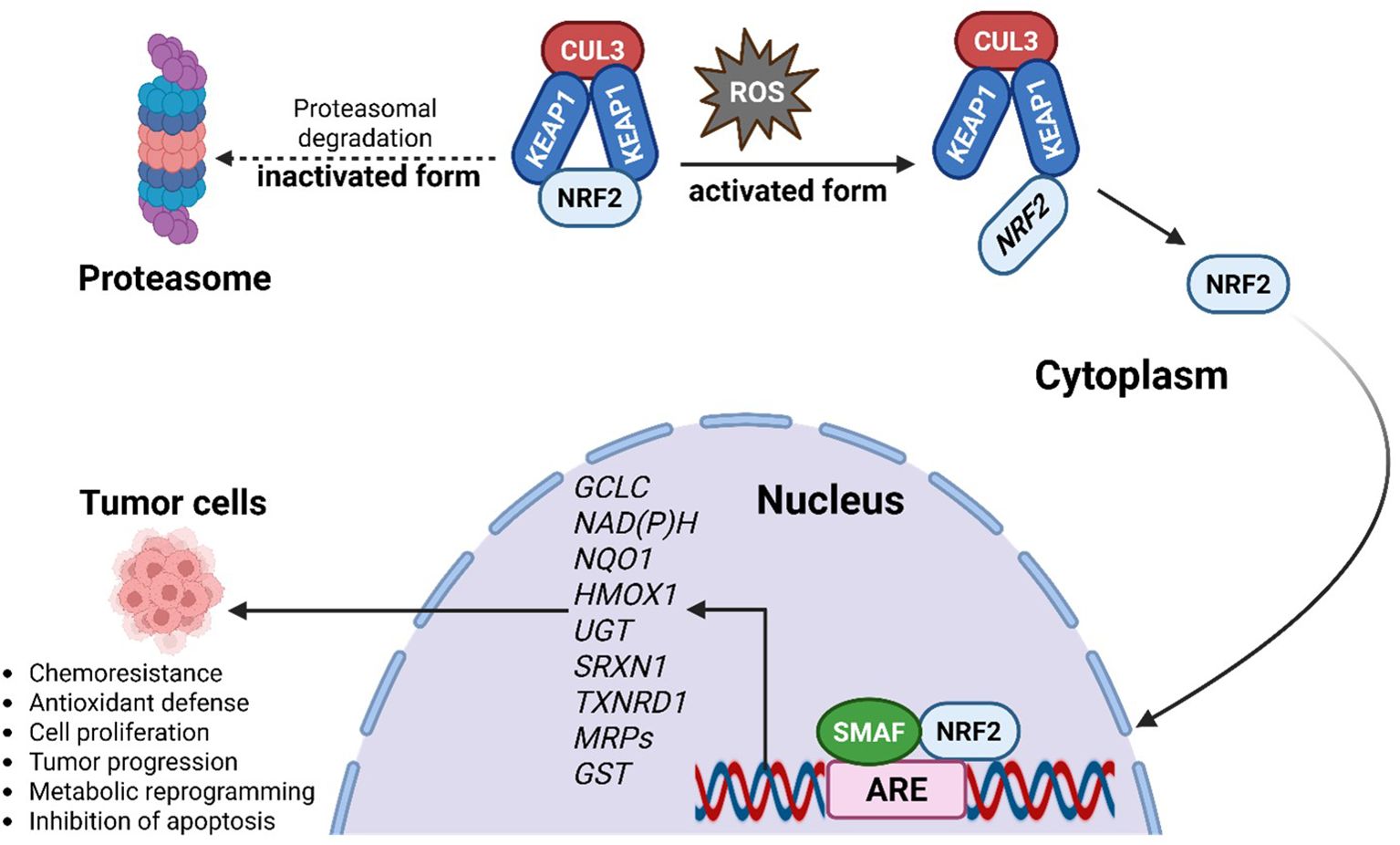
Figure 3. Distribution of female reproductive cancer cell types. Pink denotes gynecological malignancies; yellow marks breast cancer cells, which affect the reproductive system but are not considered gynecological.
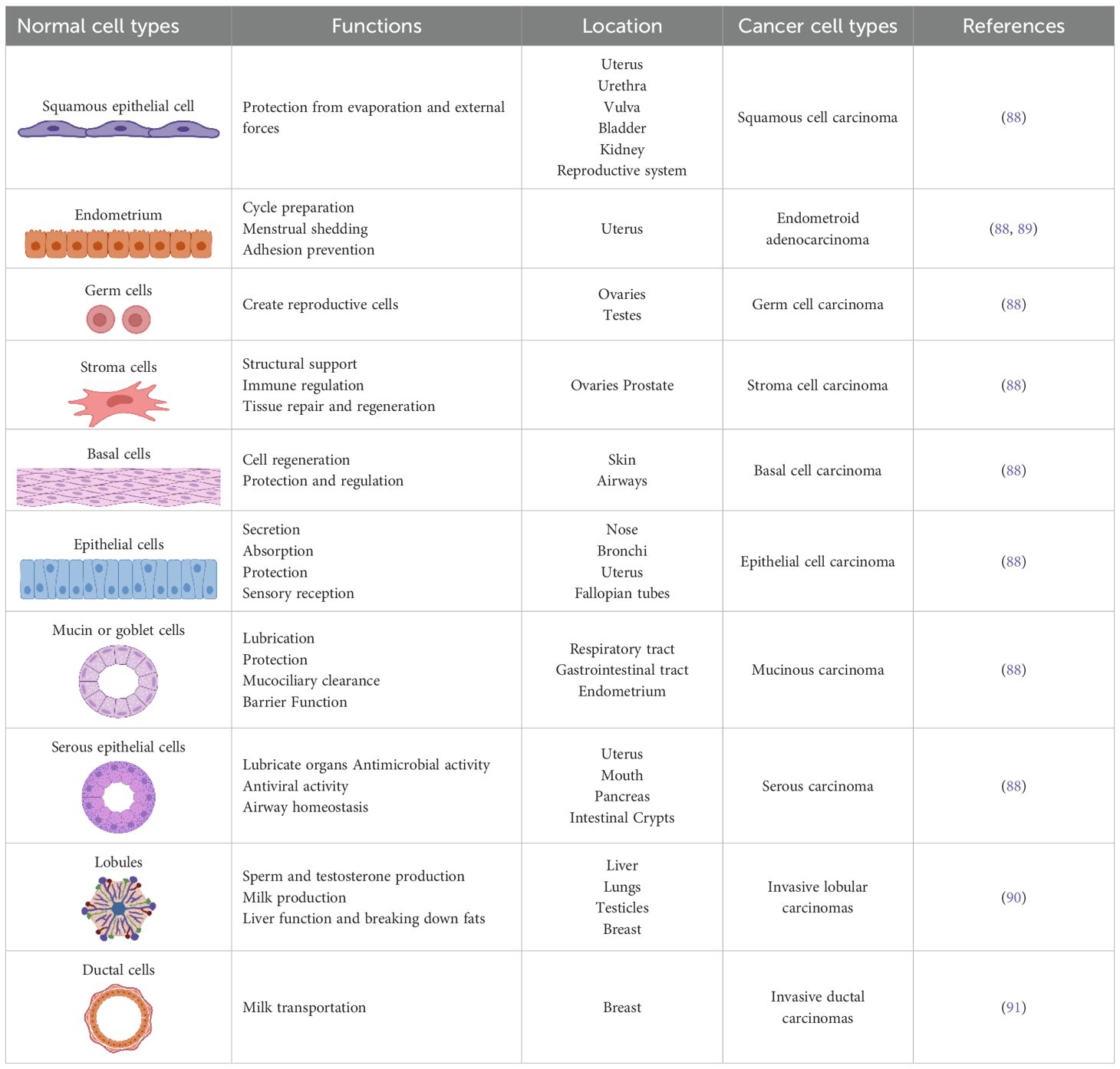
Table 1. The location and function of cell types affected by female cancers.
NRF2 activation plays a significant role in cervical cancer. In the early stages of cervical cancer development, NRF2 activation can prevent oxidative DNA damage and reduce the risk of mutations. NRF2-induced antioxidant genes, such as NQO1, HO-1, and GCLC, help mitigate oxidative stress, which is often elevated in cancer-prone tissues (14, 23). However, in advanced stages, persistent or aberrant activation of NRF2 can promote cancer cell survival, proliferation, and chemoresistance by enhancing antioxidant defenses (96). Studies have shown that cervical cancer tissues often exhibit increased nuclear NRF2 expression and decreased cytoplasmic KEAP1 expression. This imbalance leads to enhanced NRF2 activity, promoting cancer cell proliferation, inhibiting apoptosis, and increasing migration and invasion. Epigenetic changes, such as hypermethylation of the KEAP1 gene promoter, are also associated with increased NRF2 activity in cervical cancer (24, 96). Consequently, NRF2 may serve as a marker of poor prognosis and a potential therapeutic target in cervical cancer treatment (96).
There is considerable interplay between NRF2 activation and inflammatory pathways involving NF-κB and STAT3 (97). NF-κB, a key regulator of inflammation, can be activated by pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β (98). This activation typically leads to the production of ROS, which can, in turn, activate NRF2. In HPV-related cervical cancer, persistent infection may also induce chronic inflammation, further activating NF-κB and creating a pro-inflammatory environment (99). Consequently, NRF2 upregulates antioxidant and detoxifying genes, promoting cancer cell survival, proliferation, and resistance to therapy. STAT3, a transcription factor involved in cell proliferation, survival, and inflammation, can also be activated by cytokines like IL-6 and IL-1β. Once activated, STAT3 can induce the expression of NRF2, enhancing the cancer cells’ antioxidant and detoxifying capacity (18, 100–102). This interaction aids cancer cells in surviving oxidative stress and resisting therapy, contributing to tumor progression and chemoresistance. These pathways collectively create a more resilient and aggressive tumor microenvironment, posing challenges for effective cancer treatment. Understanding this crosstalk between NRF2, NF-κB, and STAT3 pathways can help in developing targeted therapies to disrupt these interactions and improve treatment outcomes for cervical cancer patients.
Targeting the NRF2 pathway in cervical cancer involves balancing its dual roles. In advanced cervical cancers with abnormal NRF2 activation, inhibitors like brusatol or ML385 can increase sensitivity to chemotherapy and radiation by reducing antioxidant defenses (14). Conversely, in early-stage cancer or pre-cancerous conditions, NRF2 activators such as sulforaphane may protect normal cells and prevent tumor initiation (14, 103). However, finding the right balance between activation and inhibition is challenging. Overactivation can promote tumor growth, while excessive inhibition may damage normal cells. Additionally, genetic and epigenetic variability among patients, resistance mechanisms, potential side effects, and the difficulties of translating preclinical findings to clinical applications further complicate the development of effective NRF2-targeted therapies. Addressing these challenges requires a nuanced approach, combining NRF2 inhibitors and activators with other therapeutic strategies to optimize treatment outcomes for cervical cancer patients.
NRF2 activation in uterine or endometrial cancer
Endometrial cancer, originating in the inner lining of the uterus, is classified into Type I and Type II. The most common Type I cancer is endometrioid adenocarcinoma, which originates in glandular cells (Figure 3). Type II cancers, which are non-estrogen-dependent, include serous, mixed, squamous cell, uterine, and adenosquamous carcinomas (Figure 3; Table 1). Serous and clear cell carcinomas, although less common, are more aggressive and have high recurrence rates (104). Mixed carcinoma involves a combination of different cancer cells, while squamous cell carcinoma and uterine carcinosarcoma are rare types originating from different cell types. Adenosquamous carcinoma includes both glandular and squamous cells.
The role of NRF2 in endometrial cancer is shaped by its interactions with oxidative stress, the tumor microenvironment, and metabolic reprogramming (23). In the early stages, NRF2 activation protects normal endometrial cells from oxidative damage and genotoxic stress, reducing the risk of malignant transformation (20). However, in advanced endometrial cancer, mutations in the KEAP1-NRF2 pathway or epigenetic changes can lead to persistent NRF2 activation (105). These alterations increase the antioxidant capacity of cancer cells, enabling them to thrive under oxidative stress and resist chemotherapy and radiotherapy by mitigating treatment-induced oxidative damage (105).
NRF2 activation is particularly observed in Type II endometrial cancers, especially serous and clear cell carcinomas, and is associated with increased aggressiveness, poor prognosis, and chemoresistance (104, 106). For instance, a study found that 89% of endometrial serous carcinoma (ESC) cases exhibited positive NRF2 expression, whereas only 28% of endometrioid carcinoma cases showed similar levels (107). Recognizing NRF2’s function as a transcription factor with known target genes, Beinse et al. investigated the mRNA levels of three such genes (NQO1, GCLC, and AKR1C3). They discovered that ESC shows low NQO1 expression, which might be due to an interaction between NRF2 and TP53, potentially contributing to the cancer’s aggressiveness (108).
Cell line studies indicated that SPEC-2 cells (derived from ESC) had higher NRF2 levels and were more resistant to chemotherapy than Ishikawa cells (derived from endometrioid carcinoma) (14). Silencing NRF2 in SPEC-2 cells significantly increased their sensitivity to cisplatin and paclitaxel, highlighting the potential of targeting NRF2 to overcome chemoresistance.
Immunohistochemical analysis has shown elevated levels of NRF2 in Type II variants of endometrial cancer tissues, linked to increased cancer aggressiveness. Specifically, NRF2 is significantly overexpressed in ESC tissues compared to other endometrial tumors and benign tissues, indicating a direct correlation between NRF2 levels and cancer aggressiveness (107) High NRF2 levels in these cancers also confer increased resistance of SPEC-2 cells to chemotherapy drugs like cisplatin and paclitaxel (107).
Strategies like downregulating NRF2 through siRNA transfection or overexpressing KEAP1 have shown potential in making resistant cancer cells more responsive to treatment. This resistance was mitigated by transiently transfecting NRF2 siRNA or stably overexpressing KEAP1 in both in vitro and xenograft models (107). NRF2 promotes the expression of genes involved in detoxification and antioxidant defense, helping cancer cells survive despite treatment. Compounds such as brusatol and ML385 are being studied to reduce NRF2 activity in cancers with constant activation (109). These inhibitors may lower antioxidant defenses, making cancer cells more vulnerable to oxidative damage and increasing their sensitivity to chemotherapy and radiotherapy (110). In pre-cancerous conditions or early-stage cancer, activating NRF2 with natural compounds like sulforaphane and curcumin might help protect normal cells from oxidative stress and inflammation (111). Therefore, targeting NRF2 could potentially improve the effectiveness of chemotherapy in endometrial cancers.
NRF2 activation in ovarian cancer
More than 95% of ovarian cancers are epithelial cell tumors (Figure 3; Table 1). These epithelial cells form the outer lining of the ovaries, which are the reproductive glands responsible for producing eggs and hormones like estrogen and progesterone (112). Ovarian cancer is especially dangerous because it is frequently diagnosed at a late stage (113). Genetic alterations are crucial in the development of ovarian cancer. While many ovarian cancers arise from random genetic mutations that build up over a person’s life, a significant portion (20-25%) is due to inherited genetic changes (23). For example, mutations in the BRCA1 and BRCA2 genes greatly elevate the risk of developing ovarian cancer (23).
Women with BRCA1 and BRCA2 mutations have a higher risk of developing breast and ovarian cancers, which tend to occur at younger ages (23, 114). The link between BRCA1 and BRCA2 mutations and NRF2 regulation lies in the increased oxidative stress caused by defective DNA repair mechanisms (23, 115). This oxidative stress can activate NRF2, promoting cancer cell survival and resistance to treatment (23, 115). Understanding the interplay between these genetic alterations and transcriptional regulation is crucial for developing targeted therapies for female cancers (23).
BRCA1 interacts with NRF2 to regulate antioxidant signaling by physically interacting with NRF2, promoting its stability and activation (116). NRF2 is a master regulator of antioxidant and cytoprotective genes that help both healthy and tumor cells cope with oxidative stress (117). The role of BRCA1 in regulating NRF2 activity has implications for the etiology and treatment of BRCA1-related cancers (116, 118).
NRF2 activation in ovarian cancer can have both positive and negative effects. Silencing NRF2 in ALDH1-enriched ovarian cancer cells significantly diminished their self-renewal capacity and stemness markers (119). This suggests that while NRF2 protects normal cells from oxidative damage, its overactivation in cancer cells may contribute to more aggressive tumor behavior. In early stages, NRF2 protects ovarian cells from oxidative stress and DNA damage due to ovulation-associated tissue damage and repair, potentially preventing cancer (43, 120). In advanced ovarian cancer, dysregulation of the Keap1-Nrf2 pathway leads to persistent NRF2 activation, promoting chemoresistance and immune evasion (120).
NRF2 and NQO1 were highly expressed in ovarian cancer and precancerous tissues compared to normal tissues, showing a positive correlation across lesion types (120, 121). NRF2 was present in both the nucleus and cytoplasm of ovarian cells, with its levels increasing as the cancer advanced (121).
Targeting NRF2 in ovarian cancer is challenging, but inhibitors like ML385 and brusatol are being investigated for cancers with abnormal NRF2 activation (122). These inhibitors enhance the sensitivity of cancer cells to chemotherapy by reducing their antioxidant defenses and increasing oxidative stress, making them more susceptible to apoptosis. Combining NRF2 inhibitors with conventional therapies, such as cisplatin and carboplatin, has shown promise in preclinical studies (122). It was suggested that targeting glutamate-cysteine ligase catalytic subunit (GCLC) could be an effective therapeutic strategy for ARID1A-deficient ovarian cancers (43). They identified GCLC as a synthetic lethal target in ARID1A-deficient ovarian and gastric cancers using CRISPR knockout technology. ARID1A mutations increase the sensitivity of these cancer cells to GCLC depletion, indicating that targeting GCLC could selectively attack cancer cells while sparing normal cells, thereby reducing the risk of excessive ROS buildup and DNA damage (43). Other genes that may be implicated in ovarian cancers include BRCA1 interacting protein C-terminal helicase 1 (BRIP1), MutL homolog 1 (MLH1), MutS homolog 2 (MSH2), MutS homolog 6 (MSH6), postmeiotic segregation increased 2 (PMS2), epithelial cell adhesion molecule (EPCAM), partner and localizer of BRCA2 (PALB2), RAD51 paralog C (RAD51C), and RAD51 paralog D (RAD51D) (123, 124).
NRF2 activation in vaginal cancer
Vaginal cancer is an uncommon type of cancer that affects the cells lining the vagina, the muscular canal connecting the cervix to the external genitalia (125, 126). It represents a small fraction (1-2%) of gynecological cancers, with the majority of cases being squamous cell carcinomas (Figure 3; Table 1), and is most frequently diagnosed in older women (127). Many instances of vaginal cancer are associated with DNA changes caused by HPV. For example, DNA damage response (DDR) mutations, which hinder the cell’s ability to repair DNA damage, have been identified through genomic sequencing. These mutations promote cancer growth and make cancer cells more dependent on specific repair pathways, such as homologous recombination repair (HRR), nucleotide excision repair (NER), and mismatch repair (MMR). The same mutations may also rely on NRF2 activation for DNA repair and survival (128).
Although the role of NRF2 activation in vaginal cancer remains largely unexplored, other transcription factors have been implicated in its pathogenesis. Studies have shown that P63, a transcription factor commonly associated with squamous epithelial cells, is overexpressed in vaginal tumors, indicating its involvement in vaginal squamous cell carcinoma in mouse models (129). Similarly, P53, an important transcription factor responsible for maintaining genome integrity by preventing the replication of damaged DNA, is frequently mutated in malignant vaginal tumors, highlighting its significance in tumor suppression and cancer progression (84, 130). Mutations in the P53 gene are often found in malignant vaginal tumors (131).
More insights can be drawn from cancers with similar histologic forms. Vaginal cancer predominantly presents as squamous cell carcinoma (SCC), which makes up approximately 90% of all cases (126). In other SCCs, such as those of the head, neck, and oesophagus, NRF2 is often constitutively activated, contributing to tumor progression and therapy resistance (132). For example, Hamad and colleagues demonstrated that NRF2 activation, in the absence of tumor suppressors p15 and p16, was essential for the development and progression of oral SCC in mouse models (133). Additionally, dysregulated NRF2 has been implicated in resistance to chemoradiotherapy in esophageal SCC (134, 135). Specifically, NRF2 was shown to enhance radiation resistance in esophageal SCC by upregulating the CaMKIIα gene, which promotes autophagy and facilitates cancer cell survival (136). Given these findings, it is plausible that NRF2 may play a similar role in vaginal squamous cell carcinoma by promoting tumor development and therapy resistance through similar molecular pathways.
Interestingly, research has revealed a potential link between NRF2 and P63, specifically its isoform Np63, which interacts with NRF2 to maintain cellular homeostasis and prevent tumor formation (137). Another study further demonstrated that NRF2 and P63 interact to promote keratinocyte proliferation in the epidermis. The presence of promoters and enhancers co-activated by both transcription factors suggests a coordinated role in epithelial renewal, which could extend to tumor development and progression in vaginal tissues (138). These findings suggest that NRF2, through its interactions with key transcription factors such as P63 and P53, may play an indirect role in vaginal cancer. Targeting NRF2 activity with inhibitors like brusatol and ML385 could be beneficial (139). This may enhance chemotherapy and radiation effectiveness by reducing antioxidant defenses and increasing oxidative stress in tumor cells (14). Combining these inhibitors with standard treatments could also improve outcomes by overcoming resistance mechanisms (14).
NRF2 activation in vulvar cancer
Most vulvar cancers are squamous cell carcinomas, originating in the thin, flat cells lining the vulvar surface (Figure 3; Table 1). These rare cancers develop in the external female genitalia, including the labia, clitoris, and vaginal opening (140). They primarily affect older women, though they can occur at any age (140). Vulvar cancer is relatively uncommon compared to breast and certain gynecological cancers, making early detection crucial for successful treatment (141). HPV infection is a significant risk factor, as it can cause genetic mutations that lead to cancerous changes in vulvar cells. Mutations in genes like TP53 and DNA repair genes can disrupt cell cycle regulation and impair the cell’s ability to repair DNA damage, resulting in genetic errors (142, 143). Additionally, epigenetic changes can contribute by silencing tumor suppressor genes or activating oncogenes, facilitating the progression of vulvar cancer (144).
Vulvar cancer, though rare, is aggressive with distinct molecular features implicating NRF2 activation in its development, progression, and treatment resistance (15). For instance, disruptions in the KEAP1-NRF2 regulatory axis are a primary mechanism of NRF2 dysregulation (145). KEAP1 mutations, frequently observed in vulvar squamous cell carcinoma, are associated with poor prognostic outcomes (146–148).
NRF2’s role in metabolic reprogramming contributes to vulvar cancer pathophysiology by driving gene expression in the pentose phosphate pathway and glutathione biosynthesis, providing cancer cells with metabolic flexibility for rapid proliferation (23). These shifts enable tumors to adapt to hypoxic conditions, presenting challenges in targeting NRF2-driven tumors (149).
NRF2 regulates cellular redox balance and interacts with pathways such as phosphoinositide 3-kinase/protein kinase B (PI3K/AKT), Notch, hypoxia-inducible factors (HIFs), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), myelocytomatosis oncogene (MYC), and forkhead box M1 (FOXM1), promoting tumor progression by enhancing EMT, angiogenesis, and immune evasion (18, 150–152). Elevated NRF2 levels are often associated with fewer immune cells in tumors, aiding cancer survival and spread (153). Although these interactions have not been specifically reported in vulvar cancer, they likely contribute to its aggressive nature and treatment resistance, warranting further investigation.
While targeting NRF2 presents challenges, it offers a promising avenue for improving treatment outcomes. Preclinical studies have explored small-molecule inhibitors that target NRF2 or its downstream effectors and agents that restore KEAP1 function to suppress NRF2 activity. These strategies show potential in reversing chemoresistance and sensitizing tumors to radiation (154). Combining NRF2 inhibitors with immune checkpoint blockade therapies may enhance anti-tumor immunity. Further research is needed to understand NRF2’s complex roles in vulvar cancer and identify biomarkers for personalized treatment.
NRF2 activation in breast cancer
Most breast cancers are invasive ductal carcinomas (Figure 3; Table 1), which are common and deadly, ranking fifth in cancer-related mortality worldwide (155). Breast cancer originates in the breast tissue, specifically in the ducts or lobules. Although not classified as a gynecological cancer, breast cancer significantly impacts female reproductive health. It is classified based on estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) status, which guide treatment (3, 4, 114). Risk factors include gender, age, hormonal status, genetic factors, and ionizing radiation (156). Prolonged estrogen exposure increases risk, especially with early menstruation and late menopause (156). Mutations in BRCA1 and BRCA2 genes, crucial for DNA repair, lead to breast cancer and genomic instability (4, 114, 116, 156).
NRF2 activation in breast cancer exhibits a dual role (24). In normal cells, NRF2 upregulates antioxidant and detoxifying genes, limiting oxidative DNA damage and preventing carcinogenesis, but in malignant cells it can promote survival, proliferation, and resistance to chemotherapy and radiotherapy (15). Somatic mutations, such as KEAP1 C23Y, impair NRF2 repression and drive its stabilization and persistent antioxidant signaling (157, 158). NRF2 also cross-talks with Notch pathways, contributing to invasion and therapy resistance (14, 65).
Importantly, NRF2 activity in breast cancer is modulated by sex-specific and hormonal influences. In ER-negative tumors, high NRF2 expression suppresses CXCL13 and limits proliferation, whereas in ER-positive tumors, low NRF2 levels increase CXCL13/CXCR5 expression, promoting invasion and metastasis (65). In BRCA1-deficient cells, ROS accumulation reflects a compromised NRF2 response, but reactivation of NRF2 restores survival. Estrogen protects BRCA1-deficient mammary cells from oxidative stress-induced death by activating NRF2 via the PI3K–AKT pathway (158). Consistently, NRF2–Keap1 signaling is more active in ER-, PR-, and HER2-positive tumors compared to TNBC (159).
Beyond breast cancer, female-specific vulnerabilities emerge in ovarian cancers associated with BRCA mutations, where disrupted NRF2-BRCA1 interactions compromise redox defenses and reveal unique therapeutic windows, reviewed in Li et al (14). These observations highlight that NRF2 regulation is not only context- but also sex-dependent, shaped by hormonal environment and receptor status. As reviewed by Adinolfi et al (17), NRF2 protects against tumor initiation but supports progression once malignancy is established. Thus, while NRF2 is a promising therapeutic target, interventions must be carefully tailored to the sex-specific and hormonal context to avoid unintended tumor-promoting effects (17).
NRF2 activation in breast cancer enhances the expression of Rho and its downstream proteins like Focal adhesion kinase 1 (FAK) and Modulator of volume-regulated anion channel current 1 (MLC), while suppressing estrogen-related receptor α (ERR1) (23). NRF2 can interact with BRCA1, enhancing its stability. In the absence of BRCA1, estrogen restores NRF2 activation, reducing ROS levels and protecting mammary gland cells in vitro (23). Continuous NRF2 signaling in cancer promotes malignant progression, therapy resistance, and poor clinical outcomes. Elevated NRF2 levels in breast cancer patients are linked to lower overall and disease-free survival. While NRF2 supports oncogenic signaling and cancer progression, it also plays a chemopreventive role in normal cells by suppressing ROS-induced DNA damage and carcinogenesis, highlighting its dual role (160, 161).
Therapeutic challenges and avenues
Emerging evidence indicates that the role of NRF2 in cancer extends far beyond redox regulation, presenting significant therapeutic challenges. Constitutive activation of NRF2, often resulting from KEAP1 mutations, facilitates immune evasion by suppressing interferon signaling, altering cytokine expression, and downregulating antigen-presentation pathways (13, 14). These changes foster immune-cold tumor microenvironments, particularly in cancers with limited T-cell infiltration. In parallel, NRF2 promotes metabolic reprogramming through enhanced glutaminolysis and pentose phosphate pathway activity, enabling tumor cells to withstand oxidative and metabolic stress (14). Its transcriptional regulation of drug efflux transporters and detoxification enzymes further contributes to broad-spectrum chemoresistance (14, 41).
In female-specific malignancies such as TNBC, platinum-resistant ovarian cancer and type II endometrial carcinoma, sustained NRF2 activation is consistently associated with poor outcomes, as illustrated in Figure 4 (38, 39). This effect is driven by upregulation of antioxidant genes (e.g., HO-1, NQO1, GCLC), which neutralize chemotherapy-induced ROS and suppress apoptotic signaling (14). These context-dependent effects complicate therapeutic targeting: while NRF2 inhibition may restore sensitivity to cytotoxic agents, it simultaneously risks compromising protective functions in normal tissues.
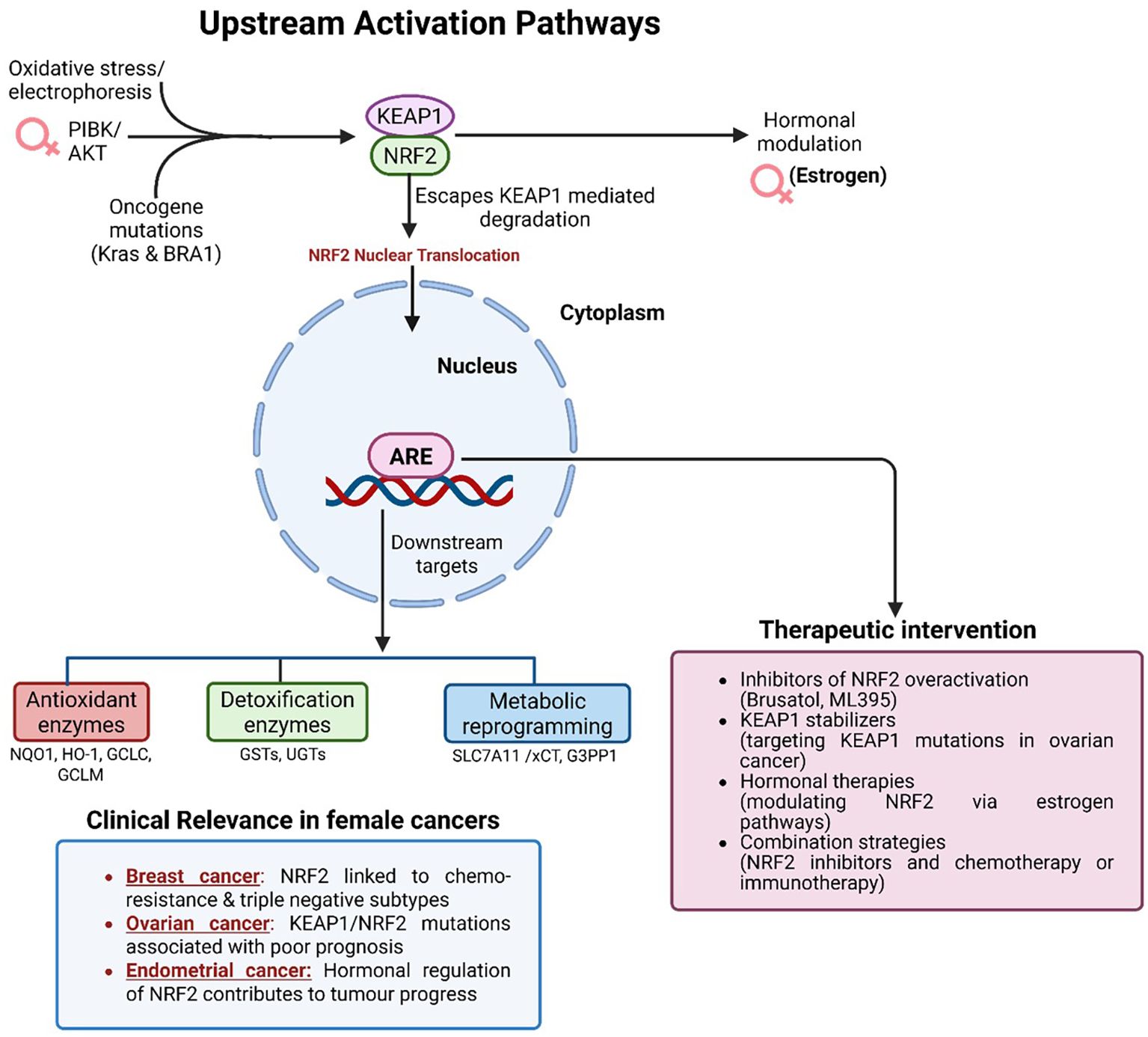
Figure 4. Upstream activation pathways and therapeutic targeting of NRF2 in female cancers. Oxidative stress, oncogenic mutations, and PI3K/AKT signaling disrupt KEAP1-mediated degradation, allowing NRF2 nuclear translocation and activation. Estrogen further modulates NRF2 activity in hormone-responsive cancers. In the nucleus, NRF2 drives expression of antioxidant, detoxification, and metabolic genes that promote chemoresistance, immune evasion, and tumor progression. Aberrant NRF2 activation is particularly relevant in triple-negative breast cancer, platinum-resistant ovarian cancer, and endometrial carcinoma. Therapeutic strategies include NRF2 inhibitors (Brusatol, ML385), KEAP1 stabilizers, hormonal therapies, and combination regimens with chemotherapy or immunotherapy.
Therapeutic strategies therefore require careful balancing of NRF2’s tumor-promoting versus cytoprotective roles. Selective modulation, ideally within genetically defined subgroups, represents a promising approach. Small-molecule inhibitors such as Brusatol (though its effects extend beyond NRF2, including global inhibition of protein synthesis) and ML385 have demonstrated efficacy in preclinical studies by suppressing NRF2-driven transcription and re-sensitizing tumors to chemotherapy and radiotherapy (Figure 4) (14, 17). Alternative strategies involve stabilizing KEAP1 or correcting KEAP1 loss-of-function mutations to re-establish upstream regulatory control (38).
Given the hormonal regulation of NRF2 in estrogen-responsive cancers, rational drug combinations are gaining attention. Integration of NRF2 inhibitors with endocrine therapies or immune checkpoint blockade may yield synergistic effects. Preclinical studies indicate that NRF2 inhibition enhances responsiveness to PD-1/PD-L1 inhibitors, particularly in poorly immunogenic tumors (15). These insights underscore the need for future clinical trials that incorporate NRF2 status as a predictive biomarker and test dual-modality regimens in stratified cohorts.
Synthetic lethality approaches further expand the therapeutic landscape. NRF2-overexpressing tumors display vulnerabilities to inhibitors of parallel survival pathways such as glutaminase and thioredoxin reductase (36, 40). Targeting these metabolic dependencies offers a precision-medicine strategy, particularly in genomically unstable or metabolically stressed tumors (37).
The expanding pipeline of NRF2 modulators highlights both opportunity and complexity. While small-molecule inhibitors like Brusatol and in Table 2 ML385 remain leading candidates for NRF2-addicted tumors, natural compounds such as sulforaphane and curcumin are under investigation for chemoprevention (14).
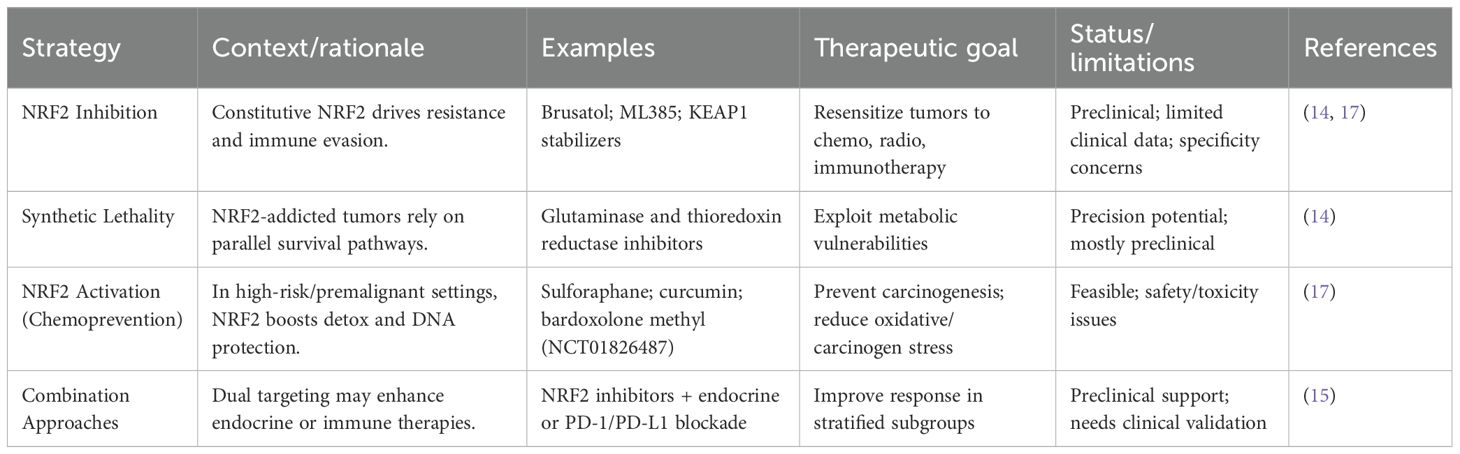
Table 2. Therapeutic strategies targeting NRF2: inhibition versus activation.
Conversely, systemic NRF2 activators (e.g., bardoxolone methyl, NCT01826487 in Table 2) illustrate translational interest but also raise safety concerns related to off-target toxicity (17). Importantly, this underscores the therapeutic paradox of NRF2 modulation: in established tumors, NRF2 inhibition may overcome resistance and restore sensitivity to therapy, whereas in premalignant or high-risk contexts, NRF2 activation may serve as a chemopreventive strategy by enhancing cellular defenses against carcinogens. The central challenge remains to achieve tumor-specific NRF2 inhibition without undermining its protective role in healthy tissues, and to harness NRF2 activation for prevention without inadvertently fueling tumor progression.
Positron emission tomography and radiotracers: potential in imaging NRF2
Current imaging modalities of female cancers and limitations
Positron emission tomography (PET) is a highly effective molecular imaging technique that enables the visualization and measurement of biological processes in living subjects (162). This method relies on radiotracers, which are molecules labelled with positron-emitting isotopes, allowing them to be detected and tracked within the body (163). The development of radiotracers targeting specific molecular pathways involved in cancer has significantly advanced early detection, precise staging, prediction of treatment responses, and monitoring of therapeutic effectiveness (164, 165).
18F-Fluorodeoxyglucose (18F-FDG) PET is a well-established imaging that plays a vital role in diagnosing, staging, treatment planning, and detecting recurrences in female cancers, particularly gynecological malignancies. When combined with computed tomography (FDG-PET/CT), it identifies increased glucose metabolism, a key characteristic of many cancers, allowing for precise tumor visualization and response assessment (Figure 5) (166, 167). FDG-PET/CT improves staging accuracy by detecting lymph node involvement and distant metastases, which are crucial for effective treatment planning (165, 168). Furthermore, it demonstrates high sensitivity in identifying recurrent disease, especially in patients with elevated tumor markers but no visible lesions on conventional imaging (165, 169).
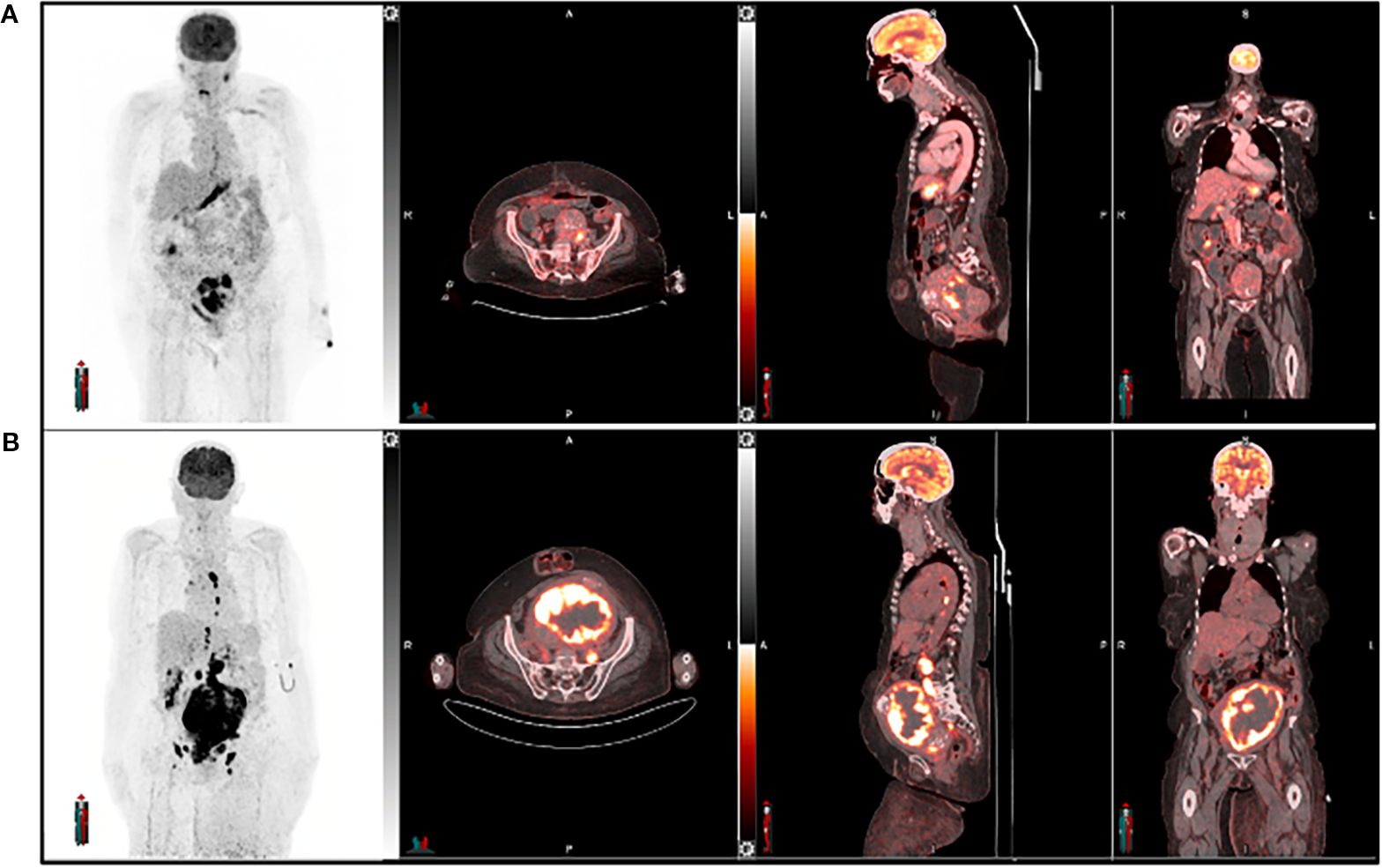
Figure 5. A 64-year-old female with endometrial cancer underwent [18F] FDG PET imaging for both diagnosis and management. The pre-treatment scan (A) was used for initial assessment, while the post-treatment scan (B) evaluated treatment response six weeks after therapy completion. Follow-up imaging revealed an increase in the size of the primary tumor and nodal metastatic lesions, along with heightened tracer uptake, suggesting disease progression or treatment failure.
Despite its benefits, FDG-PET/CT has several limitations. Certain low-grade tumors, such as mucinous ovarian cancers, exhibit low FDG uptake, leading to false-negative results (170). Additionally, metabolic changes induced by chemotherapy or radiotherapy can further influence FDG uptake, making treatment response assessment more challenging. Inflammatory conditions, including post-surgical changes, endometriosis, and pelvic inflammatory disease, can also cause false positives, complicating image interpretation (171–173). Furthermore, FDG-PET/CT has difficulty distinguishing viable tumors from inflammatory processes, assessing therapy resistance (including NRF2 status), and evaluating non-FDG-avid tumors (167, 174, 175). Moreover, 18F-FDG does not provide detailed insights into the molecular mechanisms driving tumor aggressiveness or treatment response. With the increasing shift toward personalized medicine and targeted cancer therapies, there is a growing demand for more specific PET tracers. These tracers could directly target receptors, signaling pathways, and molecular markers (e.g., 18F-FSPG) associated with tumor progression, metastasis, and therapy resistance. By offering a more precise and dynamic understanding of tumor biology, such advancements would enhance treatment monitoring, improve outcome prediction, and optimize therapy selection.
Potential imaging modalities through NRF2
Given NRF2’s pivotal role in cancer progression and resistance to various therapies, including chemotherapy, radiotherapy, immunotherapy, and KRAS G12C inhibitors, there is increasing interest in developing PET tracers to visualize NRF2 activation in vivo (176, 177). Although PET imaging of NRF2 is still in its early stages, several promising strategies have emerged, which can be broadly categorized into direct and indirect approaches. Direct strategies focus on imaging the NRF2 protein itself, while indirect methods target downstream effectors or metabolic consequences of NRF2 activation.
Direct imaging of NRF2 has proven challenging. One approach involves developing radiolabeled small-molecule inhibitors of NRF2. Several NRF2 inhibitors, such as brusatol and trigonelline, have been identified (174, 178), but creating radiolabeled versions with suitable properties for PET imaging has been difficult. Another direct strategy is the development of radiolabeled antibodies or antibody fragments targeting NRF2. While this approach offers high specificity, it faces significant challenges, including NRF2’s intracellular localization and the need for tracers to effectively cross the cell membrane (179).
Indirect imaging approaches provide valuable insights into tumor biology, especially when direct visualization of specific molecular targets is challenging. Tracers such as 18F-FDG and 68Ga-FAPI serve as surrogates for more specific biomarkers, offering indirect assessments of key tumor processes. For instance, in hypoxic tumor regions, where oxygen deprivation drives aggressive cancer behavior, 18F-FDG uptake increases due to enhanced glycolytic activity, making it a useful surrogate for hypoxia (180). Similarly, 68Ga-FAPI, which targets fibroblast activation protein (FAP), helps visualize tumor-associated stroma and its role in cancer progression, including hypoxia-induced changes in the tumor microenvironment (165). These surrogate markers are especially valuable when direct imaging of specific molecular pathways is challenging or not feasible. They play a crucial role in the advancing field of theragnostics, improving tumor characterization and optimizing treatment planning.
An indirect strategy for imaging NRF2 activity involves targeting the cystine/glutamate antiporter system xc-, which is upregulated by NRF2 activation (181). Under oxidative stress or antioxidant depletion, tumor cells enhance antioxidant production, such as GSH, to maintain redox balance, as illustrated in Figure 6 (117). This adaptive response is regulated by NRF2 and its negative regulator KEAP1, which influence transmembrane transporters like system xc- (182). System xc- facilitates cystine uptake in exchange for glutamate, with cystine serving as a precursor for GSH synthesis (183). Several PET tracers have been developed to assess system xc- activity, including 4-18F-fluoroglutamate (18F-FGlu) and (4S)-4-(3-18F-fluoropropyl)-L-glutamate (18F-FSPG) (184, 185). Additional tracers capable of imaging oxidative stress include [18F]5-fluoro aminosuberic acid (FASu), [¹¹C]ascorbic acid ([¹¹C]VitC), and [¹¹C]dehydroascorbic acid ([¹¹C]DHA) (186, 187). FASu, which targets system xc-, has demonstrated potential for imaging tumor redox status in breast cancer (188). Meanwhile, [¹¹C]VitC and [¹¹C]DHA track ROS and vitamin C uptake in NRF2-driven tumors, where accumulation is notably increased (189, 190).
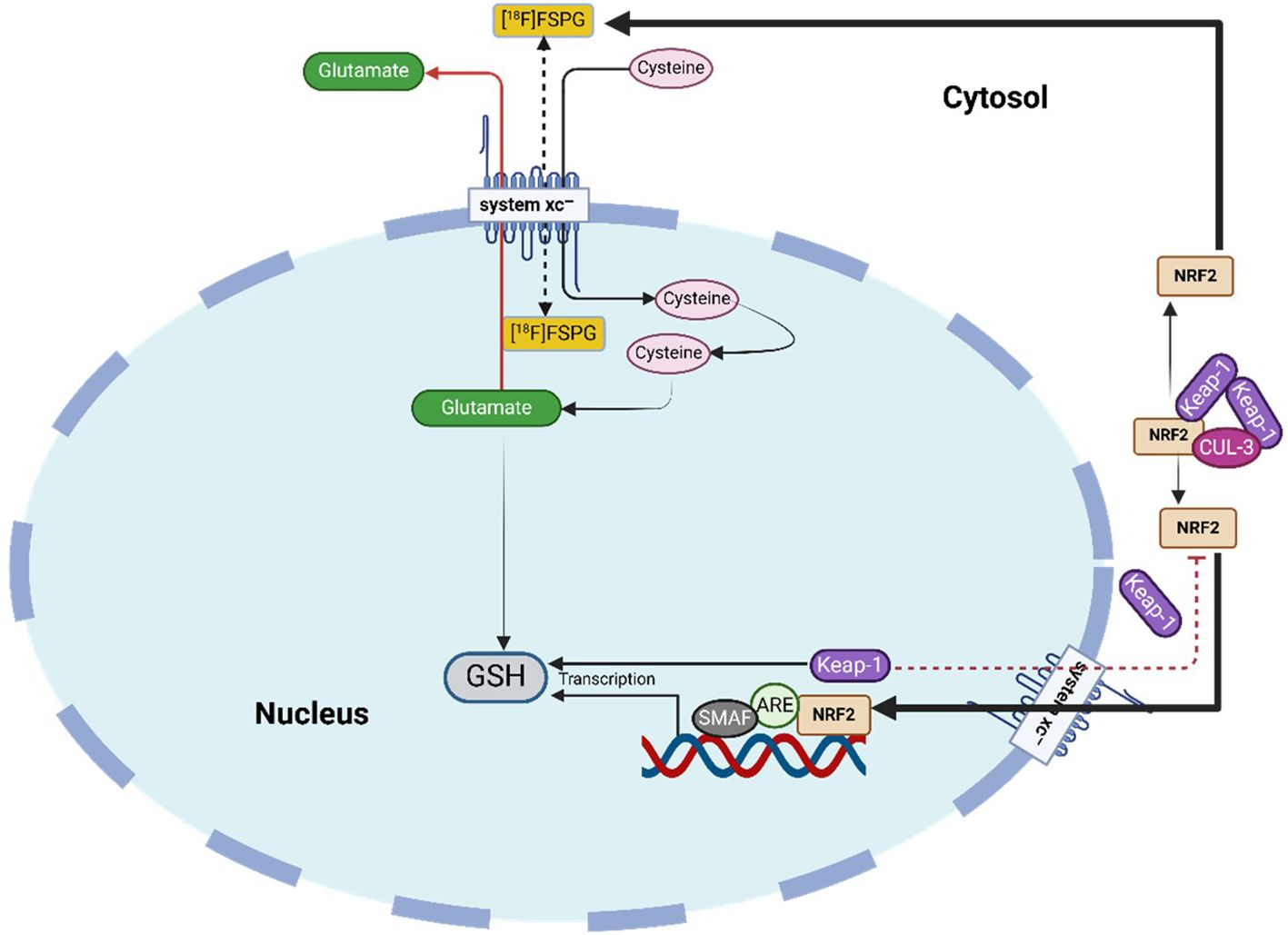
Figure 6. System xc- tracer uptake as an indirect marker of NRF2 activity and therapy response. NRF2 enhances system xc--mediated cystine import for GSH synthesis; oxidative stress from treatment can reduce uptake, while resistant tumors maintain high tracer retention.
18F-FSPG has demonstrated potential in both preclinical and clinical studies for imaging system xc- activity across various cancers, including lung, breast, ovarian, and head and neck cancers (191, 192). In NSCLC, 18F-FSPG uptake is associated with tumor aggressiveness and poor prognosis (174). Approximately one-third of NSCLC patients harbor NRF2/KEAP1 mutations, which contribute to poorer responses to standard therapies. The NRF2-regulated system xc- transporter supports GSH production, providing cancer cells with a survival advantage. 18F-FSPG PET enables the detection of NRF2 activation and related metabolic alterations, offering a promising tool for targeted therapeutic strategies (174). Tumors with elevated NRF2 activity exhibit increased xCT expression, higher GSH levels, and lower ROS accumulation. Targeting xCT with drugs such as HM30-tesirine has shown efficacy in overcoming therapy resistance in NRF2-driven NSCLC tumors (174). Non-invasive assessment of NRF2 activation via PET imaging could improve predictions of therapy resistance and inform the development of precision medicine approaches for resistant cancers. Furthermore, 18F-FSPG PET has been utilized to monitor treatment response to therapies designed to inhibit system xc- activity (112).
Conclusion and future perspectives
NRF2 is a paradoxical regulator in female cancers: while transient activation safeguards normal tissues against oxidative stress and carcinogen-induced damage, persistent hyperactivation supports tumor growth, immune evasion, and therapy resistance. This duality underscores the central translational challenge, defining when NRF2 activation should be preserved for chemoprevention and tissue protection, and when suppression is required to overcome tumor aggressiveness.
Pharmacological modulators highlight both the promise and complexity of NRF2-directed strategies. Inhibitors such as brusatol and ML385 can re-sensitize NRF2-addicted tumors, while natural activators like sulforaphane and curcumin hold chemopreventive potential. However, safety concerns from systemic activators (e.g., bardoxolone methyl) emphasize the need for selective and context-specific modulation that balances tumor suppression with preservation of physiological defenses. Hormonal regulation adds an additional layer, with estrogen-linked NRF2 activation in BRCA1-deficient contexts demonstrating the necessity of sex- and genotype-informed therapeutic design.
Emerging tools, particularly PET tracers such as 18F-FSPG, offer new opportunities to non-invasively monitor NRF2 activation, identify resistant tumors, and guide patient stratification. The integration of NRF2 imaging with molecular profiling could enable earlier detection, dynamic response monitoring, and refined precision therapies. Future advances will depend on multidisciplinary efforts to achieve context-dependent NRF2 modulation, ensuring that its protective benefits are harnessed while its tumor-promoting functions are contained, ultimately improving outcomes for women with breast, ovarian, and endometrial cancers.
Author contributions
MK: Conceptualization, Writing – original draft, Writing – review & editing. SM: Writing – original draft, Writing – review & editing. OF: Writing – original draft, Writing – review & editing. JK: Writing – original draft, Writing – review & editing. CK: Writing – original draft, Writing – review & editing. TD: Writing – original draft, Writing – review & editing. SN: Writing – original draft, Writing – review & editing. NM: Writing – original draft, Writing – review & editing. TS: Writing – original draft, Writing – review & editing. KM: Writing – original draft, Writing – review & editing. TM: Writing – original draft, Writing – review & editing. MS: Funding acquisition, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
Figures were created with BioRender.com (Agreement Reference Numbers: ZD27WGVYXU; RJ27WGUTH8; JC27WGUZ71 and XA27WGUL6Q).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor ZC declared a past co-authorship with the author MS.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. For editing and fixing grammatical errors.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Akazawa M and Hashimoto K. Artificial intelligence in gynecologic cancers: Current status and future challenges – A systematic review. Artif Intell Med. (2021) 120:102164. doi: 10.1016/j.artmed.2021.102164
2. Wahid M, Dar SA, Jawed A, Mandal RK, Akhter N, Khan S, et al. Microbes in gynecologic cancers: causes or consequences and therapeutic potential. Semin Cancer Biol. (2022). doi: 10.1016/j.semcancer.2021.07.013
3. Won KA and Spruck C. Triple−negative breast cancer therapy: Current and future perspectives (Review). Int J Oncol. (2020) 57:1245–61. doi: 10.3892/ijo.2020.5135
4. Yang H, Jenni S, Colovic M, Merkens H, Poleschuk C, Rodrigo I, et al. (18)F-5-fluoroaminosuberic acid as a potential tracer to gauge oxidative stress in breast cancer models. J Nucl Med. (2017) 58:367–73. doi: 10.2967/jnumed.116.180661
5. Ginsburg O, Bray F, Coleman MP, Vanderpuye V, Eniu A, Kotha SR, et al. The global burden of women’s cancers: a grand challenge in global health. Lancet. (2017) 389:847–60. doi: 10.1016/S0140-6736(16)31392-7
6. Jayson GC, Kohn EC, Kitchener HC, and Ledermann JA. Ovarian cancer. Lancet. (2014) 384:1376–88. doi: 10.1016/S0140-6736(13)62146-7
7. Vishnoi K, Viswakarma N, Rana A, and Rana B. Transcription factors in cancer development and therapy. Cancers (Basel). (2020) 12. doi: 10.3390/cancers12082296
8. Kristeleit RS, Miller RE, and Kohn EC. Gynecologic cancers: emerging novel strategies for targeting DNA repair deficiency. Am Soc Clin Oncol Educ Book. (2016) 36):e259–e68. doi: 10.1200/EDBK_159086
9. He G and Karin M. NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res. (2011) 21:159–68. doi: 10.1038/cr.2010.183
10. Holcakova J, Bartosik M, Anton M, Minar L, Hausnerova J, Bednarikova M, et al. New trends in the detection of gynecological precancerous lesions and early-stage cancers. Cancers. (2021) 13:6339. doi: 10.3390/cancers13246339
11. Watters AK, Seltzer ES, MacKenzie D, Young M, Muratori J, Hussein R, et al. The effects of genetic and epigenetic alterations of BARD1 on the development of non-breast and non-gynecological cancers. Genes. (2020) 11:829. doi: 10.3390/genes11070829
12. Keyvani V, Kheradmand N, Navaei ZN, Mollazadeh S, and Esmaeili S-A. Epidemiological trends and risk factors of gynecological cancers: an update. Med Oncol. (2023) 40:93. doi: 10.1007/s12032-023-01957-3
13. Pillai R, Hayashi M, Zavitsanou AM, and Papagiannakopoulos T. NRF2: KEAPing tumors protected. Cancer Discov. (2022) 12:625–43. doi: 10.1158/2159-8290.CD-21-0922
14. Lin L, Wu Q, Lu F, Lei J, Zhou Y, Liu Y, et al. Nrf2 signaling pathway: current status and potential therapeutic targetable role in human cancers. Front Oncol. (2023) 13:1184079. doi: 10.3389/fonc.2023.1184079
15. Kansanen E, Kuosmanen SM, Leinonen H, and Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. (2013) 1:45–9. doi: 10.1016/j.redox.2012.10.001
16. Zhang Y, Zhao H, and Li Y. Pleiotropic regulation of PGC-1α in tumor initiation and progression. Antioxidants Redox Signaling. (2024) 41:557–72. doi: 10.1089/ars.2023.0506
17. Adinolfi S, Patinen T, Deen AJ, Pitkänen S, Härkönen J, Kansanen E, et al. The KEAP1-NRF2 pathway: Targets for therapy and role in cancer. Redox Biol. (2023) 63:102726. doi: 10.1016/j.redox.2023.102726
18. He F, Ru X, and Wen T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci. (2020) 21:4777. doi: 10.3390/ijms21134777
19. Gjorgieva Ackova D, Maksimova V, Smilkov K, Buttari B, Arese M, and Saso L. Alkaloids as natural NRF2 inhibitors: Chemoprevention and cytotoxic action in Cancer. Pharmaceuticals. (2023) 16:850. doi: 10.3390/ph16060850
20. Tossetta G, Fantone S, Marzioni D, and Mazzucchelli R. Role of natural and synthetic compounds in modulating NRF2/KEAP1 signaling pathway in prostate cancer. Cancers. (2023) 15:3037. doi: 10.3390/cancers15113037
21. Ma JQ, Tuersun H, Jiao SJ, Zheng JH, Xiao JB, and Hasim A. Functional role of NRF2 in cervical carcinogenesis. PloS One. (2015) 10:e0133876. doi: 10.1371/journal.pone.0133876
22. Glorieux C, Enríquez C, González C, Aguirre-Martínez G, and Buc Calderon P. The multifaceted roles of NRF2 in cancer: friend or foe? Antioxidants. (2024) 13:70. doi: 10.3390/antiox13010070
23. Zimta AA, Cenariu D, Irimie A, Magdo L, Nabavi SM, Atanasov AG, et al. The role of nrf2 activity in cancer development and progression. Cancers (Basel). (2019) 11:1755. doi: 10.3390/cancers11111755
24. Pouremamali F, Pouremamali A, Dadashpour M, Soozangar N, and Jeddi F. An update of Nrf2 activators and inhibitors in cancer prevention/promotion. Cell Communication Signaling. (2022) 20:100. doi: 10.1186/s12964-022-00906-3
25. Rojo de la Vega M, Chapman E, and Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. (2018) 34:21–43. doi: 10.1016/j.ccell.2018.03.022
26. Ghareghomi S, Habibi-Rezaei M, Arese M, Saso L, and Moosavi-Movahedi AA. Nrf2 modulation in breast cancer. Biomedicines. (2022) 10:2668. doi: 10.3390/biomedicines10102668
27. Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. (2013) 2013:972913. doi: 10.1155/2013/972913
28. Meister A. Selective modification of glutathione metabolism. Science. (1983) 220:472–7. doi: 10.1126/science.6836290
29. Wu G, Lupton JR, Turner ND, Fang Y-Z, and Yang S. Glutathione metabolism and its implications for health. J Nutr. (2004) 134:489–92. doi: 10.1093/jn/134.3.489
30. Franklin CC, Backos DS, Mohar I, White CC, Forman HJ, and Kavanagh TJ. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol aspects Med. (2009) 30:86–98. doi: 10.1016/j.mam.2008.08.009
31. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. (2009) 30:42–59. doi: 10.1016/j.mam.2008.05.005
32. Rahman T, Hosen I, Islam MT, and Shekhar HU. Oxidative stress and human health. Adv biosci Biotechnol. (2012) 3:997–1019. doi: 10.4236/abb.2012.327123
33. Haddad JJ and Harb HL. l-γ-Glutamyl-l-cysteinyl-glycine (glutathione; GSH) and GSH-related enzymes in the regulation of pro-and anti-inflammatory cytokines: a signaling transcriptional scenario for redox (y) immunologic sensor (s)? Mol Immunol. (2005) 42:987–1014. doi: 10.1016/j.molimm.2004.09.029
34. Fernandez-Checa JC and Kaplowitz N. Hepatic mitochondrial glutathione: transport and role in disease and toxicity. Toxicol Appl Pharmacol. (2005) 204:263–73. doi: 10.1016/j.taap.2004.10.001
35. Griffith OW and Meister A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J Biol Chem. (1979) 254:7558–60. doi: 10.1016/S0021-9258(18)35980-5
36. Mandal PK, Seiler A, Perisic T, Kölle P, Banjac Canak A, Förster H, et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J Biol Chem. (2010) 285:22244–53. doi: 10.1074/jbc.M110.121327
37. Timmerman LA, Holton T, Yuneva M, Louie RJ, Padró M, Daemen A, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. (2013) 24:450–65. doi: 10.1016/j.ccr.2013.08.020
38. Tong X, Zhu X, Wang X, Xu Y, Huang P, Zhou L, et al. Oxidative stress-related KEAP1 and NRF2 genes contributed to the risk of epithelial ovarian cancer. Biochem Genet. (2025), 1–14. doi: 10.1007/s10528-025-11044-z
39. Cho HY, Kim K, Kim YB, Kim H, and No JH. Expression patterns of nrf2 and keap1 in ovarian cancer cells and their prognostic role in disease recurrence and patient survival. Int J Gynecol Cancer. (2017) 27:412–9. doi: 10.1097/IGC.0000000000000908
40. Ogiwara H, Takahashi K, Sasaki M, Kuroda T, Yoshida H, Watanabe R, et al. Targeting the vulnerability of glutathione metabolism in ARID1A-deficient cancers. Cancer Cell. (2019) 35:177–90.e8. doi: 10.1016/j.ccell.2018.12.009
41. Bottoni L, Minetti A, Realini G, Pio E, Giustarini D, Rossi R, et al. NRF2 activation by cysteine as a survival mechanism for triple-negative breast cancer cells. Oncogene. (2024) 43:1701–13. doi: 10.1038/s41388-024-03025-0
42. Zhang H, Limphong P, Pieper J, Liu Q, Rodesch CK, Christians E, et al. Glutathione-dependent reductive stress triggers mitochondrial oxidation and cytotoxicity. FASEB J. (2012) 26:1442–51. doi: 10.1096/fj.11-199869
43. Lin C-H, Vu JP, Yang C-Y, Sirisawad M, Chen C-T, Dao H, et al. Glutamate-cysteine ligase catalytic subunit as a therapeutic target in acute myeloid leukemia and solid tumors. Am J Cancer Res. (2021) 11:2911.
44. Lim J, Nakamura BN, Mohar I, Kavanagh TJ, and Luderer U. Glutamate cysteine ligase modifier subunit (Gclm) null mice have increased ovarian oxidative stress and accelerated age-related ovarian failure. Endocrinology. (2015) 156:3329–43. doi: 10.1210/en.2015-1206
45. Ross D and Siegel D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. (2021) 41:101950. doi: 10.1016/j.redox.2021.101950
46. Dinkova-Kostova AT and Talalay P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch Biochem Biophys. (2010) 501:116–23. doi: 10.1016/j.abb.2010.03.019
47. Wang M, Shen Y, Gao Y, Chen H, Duan F, Li S, et al. NQO1 polymorphism and susceptibility to ischemic stroke in a Chinese population. BMC Med Genomics. (2024) 17:219. doi: 10.1186/s12920-024-01992-7
48. Garate M, Wani AA, and Li G. The NAD(P)H:Quinone Oxidoreductase 1 induces cell cycle progression and proliferation of melanoma cells. Free Radic Biol Med. (2010) 48:1601–9. doi: 10.1016/j.freeradbiomed.2010.03.003
49. Lin L, Qin Y, Jin T, Liu S, Zhang S, Shen X, et al. Significance of NQO1 overexpression for prognostic evaluation of gastric adenocarcinoma. Exp Mol Pathol. (2014) 96:200–5. doi: 10.1016/j.yexmp.2013.12.008
50. Siegel D, Franklin WA, and Ross D. Immunohistochemical detection of NAD(P)H:quinone oxidoreductase in human lung and lung tumors. Clin Cancer Res. (1998) 4:2065–70. Available online at: https://aacrjournals.org/clincancerres/article/4/9/2065/7199/.
51. Buranrat B, Chau-in S, Prawan A, Puapairoj A, Zeekpudsa P, and Kukongviriyapan V. NQO1 expression correlates with cholangiocarcinoma prognosis. Asian Pac J Cancer Prev. (2012) 13 Suppl:131–6. Available online at: https://journal.waocp.org/article_27158.html.
52. Awadallah NS, Dehn D, Shah RJ, Russell Nash S, Chen YK, Ross D, et al. NQO1 expression in pancreatic cancer and its potential use as a biomarker. Appl Immunohistochem Mol Morphol. (2008) 16:24–31. doi: 10.1097/PAI.0b013e31802e91d0
53. Fagerholm R, Hofstetter B, Tommiska J, Aaltonen K, Vrtel R, Syrjäkoski K, et al. NAD (P) H: quinone oxidoreductase 1 NQO1* 2 genotype (P187S) is a strong prognostic and predictive factor in breast cancer. Nat Genet. (2008) 40:844–53. doi: 10.1038/ng.155
54. Da D, Pan Z, Zeng L, Dang Y, Dang C, Huang Y, et al. Glutamate-cysteine ligase catalytic and its modifier function as novel immunotargets in gastric adenocarcinoma. Asian J Surgery. (2023) 46:143–9. doi: 10.1016/j.asjsur.2022.02.005
55. Cui X, Li L, Yan G, Meng K, Lin Z, Nan Y, et al. High expression of NQO1 is associated with poor prognosis in serous ovarian carcinoma. BMC cancer. (2015) 15:244. doi: 10.1186/s12885-015-1271-4
56. Ma N, Zhang M, Hu J, Wei Z, and Zhang S. Daphnetin induces ferroptosis in ovarian cancer by inhibiting NAD (P) H: Quinone oxidoreductase 1 (NQO1). Phytomedicine. (2024) 132:155876. doi: 10.1016/j.phymed.2024.155876
57. Kikuchi G, Yoshida T, and Noguchi M. Heme oxygenase and heme degradation. Biochem Biophys Res Commun. (2005) 338:558–67. doi: 10.1016/j.bbrc.2005.08.020
58. Rücker H and Amslinger S. Identification of heme oxygenase-1 stimulators by a convenient ELISA-based bilirubin quantification assay. Free Radic Biol Med. (2015) 78:135–46. doi: 10.1016/j.freeradbiomed.2014.10.506
59. Huber WJ 3rd and Backes WL. Expression and characterization of full-length human heme oxygenase-1: the presence of intact membrane-binding region leads to increased binding affinity for NADPH cytochrome P450 reductase. Biochemistry. (2007) 46:12212–9. doi: 10.1021/bi701496z
60. Prawan A, Kundu JK, and Surh YJ. Molecular basis of heme oxygenase-1 induction: implications for chemoprevention and chemoprotection. Antioxid Redox Signal. (2005) 7:1688–703. doi: 10.1089/ars.2005.7.1688
61. Maines MD, Trakshel GM, and Kutty RK. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem. (1986) 261:411–9. doi: 10.1016/S0021-9258(17)42488-4
62. Spencer AL, Bagai I, Becker DF, Zuiderweg ER, and Ragsdale SW. Protein/protein interactions in the mammalian heme degradation pathway: heme oxygenase-2, cytochrome P450 reductase, and biliverdin reductase. J Biol Chem. (2014) 289:29836–58. doi: 10.1074/jbc.M114.582783
63. Consoli V, Sorrenti V, Grosso S, and Vanella L. Heme oxygenase-1 signaling and redox homeostasis in physiopathological conditions. Biomolecules. (2021) 11:589. doi: 10.3390/biom11040589
64. Poss KD and Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A. (1997) 94:10919–24. doi: 10.1073/pnas.94.20.10919
65. Kumar H, Kumar RM, Bhattacharjee D, Somanna P, and Jain V. Role of nrf2 signaling cascade in breast cancer: strategies and treatment. Front Pharmacol. (2022) 13. doi: 10.3389/fphar.2022.720076
66. Kodagoda Gamage SM, Cheng T, Lee KT-W, Dissabandara L, Lam AK-Y, and Gopalan V. Hemin, a major heme molecule, induced cellular and genetic alterations in normal colonic and colon cancer cells. Pathol - Res Practice. (2021) 224:153530. doi: 10.1016/j.prp.2021.153530
67. Li R, Wei R, Liu C, Zhang K, He S, Liu Z, et al. Heme oxygenase 1-mediated ferroptosis in Kupffer cells initiates liver injury during heat stroke. Acta Pharm Sin B. (2024) 14:3983–4000. doi: 10.1016/j.apsb.2024.05.007
68. Meech R, Hu DG, McKinnon RA, Mubarokah SN, Haines AZ, Nair PC, et al. The UDP-glycosyltransferase (UGT) superfamily: new members, new functions, and novel paradigms. Physiol Rev. (2019) 99:1153–222. doi: 10.1152/physrev.00058.2017
69. Mu B, Zeng Y, Luo L, and Wang K. Oxidative stress-mediated protein sulfenylation in human diseases: Past, present, and future. Redox Biol. (2024) 76:103332. doi: 10.1016/j.redox.2024.103332
70. Radzinski M, Oppenheim T, Metanis N, and Reichmann D. The Cys sense: thiol redox switches mediate life cycles of cellular proteins. Biomolecules. (2021) 11:469. doi: 10.3390/biom11030469
71. Wei J, Qiu D, Yang X, Wang J, Shi M, Sun L, et al. Unraveling the role of sulfiredoxin-1 in early-onset preeclampsia: A key player in trophoblast ferroptosis. J Reprod Immunol. (2024) 164:104273. doi: 10.1016/j.jri.2024.104273
72. Hammad M, Raftari M, Cesário R, Salma R, Godoy P, Emami SN, et al. Roles of oxidative stress and nrf2 signaling in pathogenic and non-pathogenic cells: A possible general mechanism of resistance to therapy. Antioxidants. (2023) 12:1371. doi: 10.3390/antiox12071371
73. Luo L, Yao X, Xiang J, Huang F, and Luo H. Identification of ferroptosis-related genes for overall survival prediction in hepatocellular carcinoma. Sci Rep. (2022) 12:10007. doi: 10.1038/s41598-022-14554-7
74. Shi W, Sun S, Liu H, Meng Y, Ren K, Wang G, et al. Guiding bar motif of thioredoxin reductase 1 modulates enzymatic activity and inhibitor binding by communicating with the co-factor FAD and regulating the flexible C-terminal redox motif. Redox Biol. (2024) 70:103050. doi: 10.1016/j.redox.2024.103050
75. Si C, Cao Y, and Zhang Y. Chapter Nine - SecMS analysis of selenoproteins with selenocysteine insertion sequence and beyond. In: Weerapana E, editor. Methods in enzymology, vol. 662. Cambridge, MA: Academic Press (2022). p. 227–40.
76. Seitz R, Tümen D, Kunst C, Heumann P, Schmid S, Kandulski A, et al. Exploring the thioredoxin system as a therapeutic target in cancer: mechanisms and implications. Antioxidants. (2024) 13:1078. doi: 10.3390/antiox13091078
77. Liang J, Wang S, Hu J, Hong X, Zhu M, Liu X, et al. Targeted inhibition of TXNRD1 prevents cartilage extracellular matrix degeneration by activating Nrf2 pathway in osteoarthritis. Biochem Biophys Res Commun. (2022) 635:267–76. doi: 10.1016/j.bbrc.2022.10.059
78. Yang R, Sun S, Zhang Q, Liu H, Wang L, Meng Y, et al. Pharmacological inhibition of TXNRD1 by a small molecule flavonoid butein overcomes cisplatin resistance in lung cancer cells. Biol Trace Elem Res. (2025) 203:1949–60. doi: 10.1007/s12011-024-04331-0
79. Cadenas C, Franckenstein D, Schmidt M, Gehrmann M, Hermes M, Geppert B, et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. (2010) 12:R44. doi: 10.1186/bcr2599
80. Cai LL, Ruberto RA, Ryan MJ, Eaton JK, Schreiber SL, and Viswanathan VS. Modulation of ferroptosis sensitivity by TXNRD1 in pancreatic cancer cells. BioRxiv. (2020) 2020-06:165647. doi: 10.1101/2020.06.25.165647
81. Delgobo M, Gonçalves RM, Delazeri MA, Falchetti M, Zandoná A, Nascimento das Neves R, et al. Thioredoxin reductase-1 levels are associated with NRF2 pathway activation and tumor recurrence in non-small cell lung cancer. Free Radic Biol Med. (2021) 177:58–71. doi: 10.1016/j.freeradbiomed.2021.10.020
82. Choi BH, Kim JM, and Kwak MK. The multifaceted role of NRF2 in cancer progression and cancer stem cells maintenance. Arch Pharm Res. (2021) 44:263–80. doi: 10.1007/s12272-021-01316-8
83. Wielinga PR, van der Heijden I, Reid G, Beijnen JH, Wijnholds J, and Borst P. Characterization of the MRP4- and MRP5-mediated transport of cyclic nucleotides from intact cells. J Biol Chem. (2003) 278:17664–71. doi: 10.1074/jbc.M212723200
84. Liu YH, Di YM, Zhou ZW, Mo SL, and Zhou SF. Multidrug resistance-associated proteins and implications in drug development. Clin Exp Pharmacol Physiol. (2010) 37:115–20. doi: 10.1111/j.1440-1681.2009.05252.x
85. Du T, Luo T, Wang J, Sun R, and Cai H. Role of MRPs transporters in pharmacokinetics and intestinal toxicity of irinotecan. Food Chem Toxicol. (2023) 182:114171. doi: 10.1016/j.fct.2023.114171
86. Wu S, Lu H, and Bai Y. Nrf2 in cancers: A double-edged sword. Cancer Med. (2019) 8:2252–67. doi: 10.1002/cam4.2101
87. Townsend DM and Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. (2003) 22:7369–75. doi: 10.1038/sj.onc.1206940
88. Prat J and Mutch DG. Pathology of cancers of the female genital tract including molecular pathology. Int J Gynaecol Obstet. (2018) 143 Suppl 2:93–108. doi: 10.1002/ijgo.12617
89. Yang HP, Wentzensen N, Trabert B, Gierach GL, Felix AS, Gunter MJ, et al. Endometrial cancer risk factors by 2 main histologic subtypes: the NIH-AARP Diet and Health Study. Am J Epidemiol. (2013) 177:142–51. doi: 10.1093/aje/kws200
90. Li C, Uribe Da, and Daling J. Clinical characteristics of different histologic types of breast cancer. Br J cancer. (2005) 93:1046–52. doi: 10.1038/sj.bjc.6602787
91. Makki J. Diversity of breast carcinoma: histological subtypes and clinical relevance. Clin Med Insights Pathol. (2015) 8:23–31. doi: 10.4137/CPath.S31563
92. Isham FSM. The Role of Micro-RNAs in Large Extracellular Vesicles Derived from Human Papillomavirus Associated Cervical Cancer Cells in the Cytokine Modulation of Dendritic Cells (Doctoral dissertation). New Zealand: University of Otago (2024).
93. Lu J, Na J, Li Y, Wang X, Wang J, and Han S. Gastric-type mucinous endocervical adenocarcinomas: A case report and literature review. Front Cell Infection Microbiol. (2022) 12:917009. doi: 10.3389/fcimb.2022.917009
94. Porter VL and Marra MA. The drivers, mechanisms, and consequences of genome instability in HPV-driven cancers. Cancers. (2022) 14:4623. doi: 10.3390/cancers14194623
95. Choi B, Na Y, Whang MY, Ho JY, Han M-R, Park S-W, et al. MGMT methylation is associated with human papillomavirus infection in cervical dysplasia: A longitudinal study. J Clin Med. (2023) 12:6188. doi: 10.3390/jcm12196188
96. Gu X, Mu C, Zheng R, Zhang Z, Zhang Q, and Liang T. The cancer antioxidant regulation system in therapeutic resistance. Antioxidants. (2024) 13:778. doi: 10.3390/antiox13070778
97. Ahsan H, Islam SU, Ahmed MB, and Lee YS. Role of Nrf2, STAT3, and Src as molecular targets for cancer chemoprevention. Pharmaceutics. (2022) 14:1775. doi: 10.3390/pharmaceutics14091775
98. Hariharan A, Hakeem AR, Radhakrishnan S, Reddy MS, and Rela M. The role and therapeutic potential of NF-kappa-B pathway in severe COVID-19 patients. Inflammopharmacology. (2021) 29:91–100. doi: 10.1007/s10787-020-00773-9
99. Fernandes JV, Fernandes T, de Azevedo JCV, Cobucci RNO, de Carvalho MGF, Andrade VS, et al. Link between chronic inflammation and human papillomavirus-induced carcinogenesis (Review). Oncol Lett. (2015) 9:1015–26. doi: 10.3892/ol.2015.2884
100. Chen X-W and Zhou S-F. Inflammation, cytokines, the IL-17/IL-6/STAT3/NF-κB axis, and tumorigenesis. Drug Design Dev Ther. (2015) 9:2941–6. doi: 10.2147/DDDT.S86396
101. Neganova M, Liu J, Aleksandrova Y, Klochkov S, and Fan R. Therapeutic influence on important targets associated with chronic inflammation and oxidative stress in cancer treatment. Cancers. (2021) 13:6062. doi: 10.3390/cancers13236062
102. Saha S, Buttari B, Panieri E, Profumo E, and Saso L. An overview of nrf2 signaling pathway and its role in inflammation. Molecules. (2020) 25:5474. doi: 10.3390/molecules25225474
103. Surh Y-J. Nrf2 paradox: Can cancer patients eat broccoli? Food Front. (2021) 2:25–8. doi: 10.1002/fft2.61
104. Gilyadova A, Ishchenko A, Babayan J, Avin M, Sekacheva M, and Reshetov I. Molecular genetic factors of risk stratification of lymph node metastasis in endometrial carcinoma. Cancers (Basel). (2024) 16:3560. doi: 10.3390/cancers16213560
105. Cheng W, Xu B, Zhang H, and Fang S. Lung adenocarcinoma patients with KEAP1 mutation harboring low immune cell infiltration and low activity of immune environment. Thorac Cancer. (2021) 12:2458–67. doi: 10.1111/1759-7714.14089
106. Ahtikoski AM, Kangas J, Salonen R, Puistola U, and Karihtala P. Cytoplasmic keap1 expression is associated with poor prognosis in endometrial cancer. Anticancer Res. (2019) 39:585–90. doi: 10.21873/anticanres.13151
107. Jiang T, Chen N, Zhao F, Wang XJ, Kong B, Zheng W, et al. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. (2010) 70:5486–96. doi: 10.1158/0008-5472.CAN-10-0713
108. Beinse G, Just PA, Rance B, Izac B, Letourneur F, Saidu NEB, et al. The NRF2 transcriptional target NQO1 has low mRNA levels in TP53-mutated endometrial carcinomas. PloS One. (2019) 14:e0214416. doi: 10.1371/journal.pone.0214416
109. Yan L, Hu H, Feng L, Li Z, Zheng C, Zhang J, et al. ML385 promotes ferroptosis and radiotherapy sensitivity by inhibiting the NRF2-SLC7A11 pathway in esophageal squamous cell carcinoma. Med Oncol. (2024) 41:309. doi: 10.1007/s12032-024-02483-6
110. Rai A, Patwardhan RS, Jayakumar S, Pachpatil P, Das D, Panigrahi GC, et al. Clobetasol propionate, a Nrf-2 inhibitor, sensitizes human lung cancer cells to radiation-induced killing via mitochondrial ROS-dependent ferroptosis. Acta Pharmacologica Sin. (2024) 45:1–14. doi: 10.1038/s41401-024-01233-8
111. Banna MN and Hasan S. Rochana MN. A review on phytochemical compounds used to treatment of breast cancer. Glob Acad J Pharm Drug Res. (2024) 6:1–11. doi: 10.36348/gajpdr.2024.v06i05.001
112. Sambasivan K, Tyrrell WE, Farooq R, Mynerich J, Edwards RS, Tanc M, et al. (18)F]FSPG-PET provides an early marker of radiotherapy response in head and neck squamous cell cancer. NPJ Imaging. (2024) 2:28. doi: 10.1038/s44303-024-00038-y
113. Pourbarkhordar V, Rahmani S, Roohbakhsh A, Hayes AW, and Karimi G. Melatonin effect on breast and ovarian cancers by targeting the pi3k/akt/mtor pathway. IUBMB Life. (2024) 76:1035–49. doi: 10.1002/iub.2900
114. Fadebi OO, Miya TV, Khanyile R, Dlamini Z, and Marima R. Long intergenic non-coding RNAs and BRCA1 in breast cancer pathogenesis: neighboring companions or nemeses? Non-Coding RNA. (2025) 11:9. doi: 10.3390/ncrna11010009
115. Uboveja A and Aird KM. Interplay between altered metabolism and DNA damage and repair in ovarian cancer. BioEssays. (2024) 46:2300166. doi: 10.1002/bies.202300166
116. Gorrini C, Baniasadi PS, Harris IS, Silvester J, Inoue S, Snow B, et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J Exp Med. (2013) 210:1529–44. doi: 10.1084/jem.20121337
117. Aboelella NS, Brandle C, Kim T, Ding Z-C, and Zhou G. Oxidative stress in the tumor microenvironment and its relevance to cancer immunotherapy. Cancers. (2021) 13:986. doi: 10.3390/cancers13050986
118. van der Wijst MGP, Brown R, and Rots MG. Nrf2, the master redox switch: The Achilles’ heel of ovarian cancer? Biochim Biophys Acta (BBA) - Rev Cancer. (2014) 1846:494–509. doi: 10.1016/j.bbcan.2014.09.004
119. Kim D, Choi BH, Ryoo IG, and Kwak MK. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. (2018) 9:896. doi: 10.1038/s41419-018-0903-4
120. Ding DN, Xie LZ, Shen Y, Li J, Guo Y, Fu Y, et al. Insights into the role of oxidative stress in ovarian cancer. Oxid Med Cell Longev. (2021) 2021:8388258. doi: 10.1155/2021/8388258
121. Osman N, Abd El-Maqsoud NMR, and El Gelany SAA. Correlation of NQO1 and nrf2 in female genital tract cancer and their precancerous lesions (Cervix, endometrium and ovary). World J Oncol. (2015) 6:364–74. doi: 10.14740/wjon931w
122. van der Wijst MG, Huisman C, Mposhi A, Roelfes G, and Rots MG. Targeting Nrf2 in healthy and Malignant ovarian epithelial cells: Protection versus promotion. Mol Oncol. (2015) 9:1259–73. doi: 10.1016/j.molonc.2015.03.003
123. Tomasova K, Cumova A, Seborova K, Horak J, Koucka K, Vodickova L, et al. DNA repair and ovarian carcinogenesis: impact on risk, prognosis and therapy outcome. Cancers (Basel). (2020) 12:1713. doi: 10.3390/cancers12071713
124. Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, et al. Frequency of mutations in individuals with breast cancer referred for 1 and 2 testing using next-generation sequencing with a 25-gene panel. Cancer. (2015) 121:25–33. doi: 10.1002/cncr.29010
125. Yildiz ED. Physiotherapy and Rehabilitation in Gynecologic Cancers. Cham: Springer International Publishing (2024) p. 1–29.
126. Kulkarni A, Dogra N, and Zigras T. Innovations in the management of vaginal cancer. Curr Oncol. (2022) 29:3082–92. doi: 10.3390/curroncol29050250
127. Li Z and Li Y. 6.29 - Bioactive Dietary Compounds and Epigenetics in Women’s Reproductive Cancers. In: Kenakin T, editor. Comprehensive Pharmacology. Elsevier, Oxford (2022). p. 595–610.
128. Li J, Xu C, and Liu Q. Roles of NRF2 in DNA damage repair. Cell Oncol (Dordr). (2023) 46:1577–93. doi: 10.1007/s13402-023-00834-5
129. Wang X, Praça MSL, Wendel JRH, Emerson RE, DeMayo FJ, Lydon JP, et al. Vaginal squamous cell carcinoma develops in mice with conditional arid1a loss and gain of oncogenic kras driven by progesterone receptor cre. Am J Pathol. (2021) 191:1281–91. doi: 10.1016/j.ajpath.2021.03.013
130. Marei HE, Althani A, Afifi N, Hasan A, Caceci T, Pozzoli G, et al. p53 signaling in cancer progression and therapy. Cancer Cell Int. (2021) 21:703. doi: 10.1186/s12935-021-02396-8
131. Egger EK, Condic M, Ralser DJ, Marinova M, Mustea A, Recker F, et al. The role of P16, P53, KI-67 and PD-L1 immunostaining in primary vaginal cancer. Cancers (Basel). (2023) 15:1046. doi: 10.3390/cancers15041046
132. Cloer EW, Goldfarb D, Schrank TP, Weissman BE, and Major MB. NRF2 activation in cancer: from DNA to protein. Cancer Res. (2019) 79:889–98. doi: 10.1158/0008-5472.CAN-18-2723
133. Hamad SH, Sellers RS, Wamsley N, Zolkind P, Schrank TP, Major MB, et al. NRF2 activation in trp53;p16-deficient mice drives oral squamous cell carcinoma. Cancer Res Commun. (2024) 4:487–95. doi: 10.1158/2767-9764.CRC-23-0386
134. Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote Malignancy. Proc Natl Acad Sci U S A. (2008) 105:13568–73. doi: 10.1073/pnas.0806268105
135. Kawasaki Y, Okumura H, Uchikado Y, Kita Y, Sasaki K, Owaki T, et al. Nrf2 is useful for predicting the effect of chemoradiation therapy on esophageal squamous cell carcinoma. Ann Surg Oncol. (2014) 21:2347–52. doi: 10.1245/s10434-014-3600-2
136. Xia D, Zhang X-R, Ma Y-L, Zhao Z-J, Zhao R, and Wang Y-Y. Nrf2 promotes esophageal squamous cell carcinoma (ESCC) resistance to radiotherapy through the CaMKIIα-associated activation of autophagy. Cell Biosci. (2020) 10:90. doi: 10.1186/s13578-020-00456-6
137. Napoli M, Deshpande AA, Chakravarti D, Rajapakshe K, Gunaratne PH, Coarfa C, et al. Genome-wide p63-target gene analyses reveal TAp63/NRF2-dependent oxidative stress responses. Cancer Res Commun. (2024) 4:264–78. doi: 10.1158/2767-9764.CRC-23-0358
138. Kurinna S, Seltmann K, Bachmann AL, Schwendimann A, Thiagarajan L, Hennig P, et al. Interaction of the NRF2 and p63 transcription factors promotes keratinocyte proliferation in the epidermis. Nucleic Acids Res. (2021) 49:3748–63. doi: 10.1093/nar/gkab167
139. Panieri E, Buha A, Telkoparan-Akillilar P, Cevik D, Kouretas D, Veskoukis A, et al. Potential applications of NRF2 modulators in cancer therapy. Antioxidants. (2020) 9:193. doi: 10.3390/antiox9030193
140. Olawaiye AB, Cuello MA, and Rogers LJ. Cancer of the vulva: 2021 update. Int J Gynecol Obstetrics. (2021) 155:7–18. doi: 10.1002/ijgo.13881
141. Pedrão PG, Guimarães YM, Godoy LR, Possati-Resende JC, Bovo AC, Andrade C, et al. Management of early-stage vulvar cancer. Cancers (Basel). (2022) 14:4184. doi: 10.3390/cancers14174184
142. Nooij LS, Ter Haar NT, Ruano D, Rakislova N, van Wezel T, Smit V, et al. Genomic characterization of vulvar (Pre)cancers identifies distinct molecular subtypes with prognostic significance. Clin Cancer Res. (2017) 23:6781–9. doi: 10.1158/1078-0432.CCR-17-1302
143. Williams EA, Werth AJ, Sharaf R, Montesion M, Sokol ES, Pavlick DC, et al. Vulvar squamous cell carcinoma: comprehensive genomic profiling of HPV+ Versus HPV- forms reveals distinct sets of potentially actionable molecular targets. JCO Precis Oncol. (2020) 4:647–61. doi: 10.1200/PO.19.00406
144. Trietsch MD, Nooij LS, Gaarenstroom KN, and van Poelgeest MI. Genetic and epigenetic changes in vulvar squamous cell carcinoma and its precursor lesions: a review of the current literature. Gynecol Oncol. (2015) 136:143–57. doi: 10.1016/j.ygyno.2014.11.002
145. Martins DBG, de Aguiar AC, de Araújo Barbosa FM, and Leitão GM. The dual role of NRF2 transcription factor in female cancer. IntechOpen. (2024), 65–110. doi: 10.5772/intechopen.111702
146. Tossetta G and Marzioni D. Targeting the NRF2/KEAP1 pathway in cervical and endometrial cancers. Eur J Pharmacol. (2023) 941:175503. doi: 10.1016/j.ejphar.2023.175503
147. Chen F, Xiao M, Hu S, and Wang M. Keap1-Nrf2 pathway: a key mechanism in the occurrence and development of cancer. Front Oncol. (2024) 14. doi: 10.3389/fonc.2024.1381467
148. Chakkittukandiyil A, Sajini DV, Karuppaiah A, and Selvaraj D. The principal molecular mechanisms behind the activation of Keap1/Nrf2/ARE pathway leading to neuroprotective action in Parkinson’s disease. Neurochem Int. (2022) 156:105325. doi: 10.1016/j.neuint.2022.105325
149. Bae T, Hallis SP, and Kwak MK. Hypoxia, oxidative stress, and the interplay of HIFs and NRF2 signaling in cancer. Exp Mol Med. (2024) 56:501–14. doi: 10.1038/s12276-024-01180-8
150. Toth RK and Warfel NA. Strange bedfellows: nuclear factor, erythroid 2-like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) in tumor hypoxia. Antioxidants. (2017) 6:27. doi: 10.3390/antiox6020027
151. Schiavello M, Gazzano E, Bergandi L, Silvagno F, Libener R, Riganti C, et al. Identification of redox-sensitive transcription factors as markers of Malignant pleural mesothelioma. Cancers. (2021) 13:1138. doi: 10.3390/cancers13051138
152. Park Y-H, Kim S-U, Kwon T-H, Kim J, Song I, Shin HJ, et al. Peroxiredoxin II promotes hepatic tumorigenesis through cooperation with Ras/Forkhead box M1 signaling pathway. Oncogene. (2016) 35:3503–13. doi: 10.1038/onc.2015.411
153. Oshi M, Angarita FA, Tokumaru Y, Yan L, Matsuyama R, Endo I, et al. High expression of NRF2 is associated with increased tumor-infiltrating lymphocytes and cancer immunity in ER-positive/HER2-negative breast cancer. Cancers (Basel). (2020) 12:3856. doi: 10.3390/cancers12123856
154. Yamamoto M, Kensler TW, and Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev. (2018) 98:1169–203. doi: 10.1152/physrev.00023.2017
155. Łukasiewicz S, Czeczelewski M, Forma A, Baj J, Sitarz R, and Stanisławek A. Breast cancer-epidemiology, risk factors, classification, prognostic markers, and current treatment strategies-an updated review. Cancers (Basel). (2021) 13:4287. doi: 10.3390/cancers13174287
156. Smolarz B, Nowak AZ, and Romanowicz H. Breast cancer-epidemiology, classification, pathogenesis and treatment (Review of literature). Cancers (Basel). (2022) 14:2569. doi: 10.3390/cancers14102569
157. Lau A, Villeneuve NF, Sun Z, Wong PK, and Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res. (2008) 58:262–70. doi: 10.1016/j.phrs.2008.09.003
158. Gorrini C, Gang BP, Bassi C, Wakeham A, Baniasadi SP, Hao Z, et al. Estrogen controls the survival of BRCA1-deficient cells via a PI3K–NRF2-regulated pathway. Proc Natl Acad Sci. (2014) 111:4472–7. doi: 10.1073/pnas.1324136111
159. Venugopalreddy Bovilla PM, Suma M, Natraj AS, and Madhunapantula SRV. Therapeutic potential of targeting nrf-2-keap-1 signaling in breast cancers. J Drug Des Res. (2017) 4:1038.
160. Xue D, Zhou X, and Qiu J. Emerging role of NRF2 in ROS-mediated tumor chemoresistance. Biomed Pharmacother. (2020) 131:110676. doi: 10.1016/j.biopha.2020.110676
161. Gall Trošelj K, Tomljanović M, Jaganjac M, Matijević Glavan T, Čipak Gašparović A, Milković L, et al. Oxidative stress and cancer heterogeneity orchestrate NRF2 roles relevant for therapy response. Molecules. (2022) 27:1468. doi: 10.3390/molecules27051468
162. Phelps ME. Positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci U S A. (2000) 97:9226–33. doi: 10.1073/pnas.97.16.9226
163. Ametamey SM, Honer M, and Schubiger PA. Molecular imaging with PET. Chem Rev. (2008) 108:1501–16. doi: 10.1021/cr0782426
164. Mankoff DA, Farwell MD, Clark AS, and Pryma DA. Making molecular imaging a clinical tool for precision oncology: A review. JAMA Oncol. (2017) 3:695–701. doi: 10.1001/jamaoncol.2016.5084
165. Mokoala K, Lawal I, Lengana T, Kgatle M, Giesel FL, Vorster M, et al. PSMA theranostics: science and practice. Cancers (Basel). (2021) 13:3904. doi: 10.3390/cancers13153904
166. Jadvar H, Alavi A, and Gambhir SS. 18F-FDG uptake in lung, breast, and colon cancers: molecular biology correlates and disease characterization. J Nucl Med. (2009) 50:1820–7. doi: 10.2967/jnumed.108.054098
167. Decazes P and Bohn P. Immunotherapy by immune checkpoint inhibitors and nuclear medicine imaging: current and future applications. Cancers. (2020) 12:371. doi: 10.3390/cancers12020371
168. Kitajima K, Doi H, Kanda T, Yamane T, Tsujikawa T, Kaida H, et al. Present and future roles of FDG-PET/CT imaging in the management of lung cancer. Japanese J Radiol. (2016) 34:387–99. doi: 10.1007/s11604-016-0546-2
169. Sironi S, Messa C, Mangili G, Zangheri B, Aletti G, Garavaglia E, et al. Integrated FDG PET/CT in patients with persistent ovarian cancer: correlation with histologic findings. Radiology. (2004) 233:433–40. doi: 10.1148/radiol.2332031800
170. Chung HH, Kang WJ, Kim JW, Park NH, Song YS, Chung JK, et al. Role of [18F]FDG PET/CT in the assessment of suspected recurrent ovarian cancer: correlation with clinical or histological findings. Eur J Nucl Med Mol Imaging. (2007) 34:480–6. doi: 10.1007/s00259-006-0260-x
171. Vranjesevic D, Filmont JE, Meta J, Silverman DH, Phelps ME, Rao J, et al. Whole-body (18)F-FDG PET and conventional imaging for predicting outcome in previously treated breast cancer patients. J Nucl Med. (2002) 43:325–9. Available online at: https://jnm.snmjournals.org/content/43/3/325.short.
172. Kinahan PE, Doot RK, Wanner-Roybal M, Bidaut LM, Armato SG, Meyer CR, et al. PET/CT assessment of response to therapy: tumor change measurement, truth data, and error. Transl Oncol. (2009) 2:223–30. doi: 10.1593/tlo.09223
173. Gomes Marin JF, Nunes RF, Coutinho AM, Zaniboni EC, Costa LB, Barbosa FG, et al. Theranostics in nuclear medicine: emerging and re-emerging integrated imaging and therapies in the era of precision oncology. RadioGraphics. (2020) 40:1715–40. doi: 10.1148/rg.2020200021
174. Greenwood HE, Barber AR, Edwards RS, Tyrrell WE, George ME, dos Santos SN, et al. Imaging NRF2 activation in non-small cell lung cancer with positron emission tomography. Nat Commun. (2024) 15:10484. doi: 10.1038/s41467-024-54852-4
175. Giammarile F, Castellucci P, Dierckx R, Estrada Lobato E, Farsad M, Hustinx R, et al. Non-FDG PET/CT in Diagnostic Oncology: a pictorial review. Eur J Hybrid Imaging. (2019) 3:20. doi: 10.1186/s41824-019-0066-2
176. Singh A, Boldin-Adamsky S, Thimmulappa RK, Rath SK, Ashush H, Coulter J, et al. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. (2008) 68:7975–84. doi: 10.1158/0008-5472.CAN-08-1401
177. Lee N, Choi JY, and Ryu YH. The development status of PET radiotracers for evaluating neuroinflammation. Nucl Med Mol Imaging. (2024) 58:160–76. doi: 10.1007/s13139-023-00831-4
178. Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. (2008) 29:1235–43. doi: 10.1093/carcin/bgn095
179. Cho HY, Gladwell W, Wang X, Chorley B, Bell D, Reddy SP, et al. Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am J Respir Crit Care Med. (2010) 182:170–82. doi: 10.1164/rccm.200907-1047OC
180. Dierckx RA and Van de Wiele C. FDG uptake, a surrogate of tumour hypoxia? Eur J Nucl Med Mol Imaging. (2008) 35:1544–9. doi: 10.1007/s00259-008-0758-5
181. Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. (2013) 18:522–55. doi: 10.1089/ars.2011.4391
182. Sasaki S, Negishi T, Tsuzuki T, and Yukawa K. Methylmercury-induced reactive oxygen species-dependent and independent dysregulation of MAP kinase-related signaling pathway in cultured normal rat cerebellar astrocytes. Toxicology. (2023) 487:153463. doi: 10.1016/j.tox.2023.153463
183. Bannai S and Tateishi N. Role of membrane transport in metabolism and function of glutathione in mammals. J Membr Biol. (1986) 89:1–8. doi: 10.1007/BF01870891
184. Koglin N, Mueller A, Berndt M, Schmitt-Willich H, Toschi L, Stephens AW, et al. Specific PET imaging of xC- transporter activity using a 18F-labeled glutamate derivative reveals a dominant pathway in tumor metabolism. Clin Cancer Res. (2011) 17:6000–11. doi: 10.1158/1078-0432.CCR-11-0687
185. Wardak M, Sonni I, Fan AP, Minamimoto R, Jamali M, Hatami N, et al. (18)F-FSPG PET/CT imaging of system x(C)(-) transporter activity in patients with primary and metastatic brain tumors. Radiology. (2022) 303:620–31. doi: 10.1148/radiol.203296
186. Park JY, Park SM, Lee TS, Lee SJ, Kim JY, Oh SJ, et al. Innovations in nuclear medicine imaging for reactive oxygen species: applications and radiopharmaceuticals. Antioxidants (Basel). (2024) 13:1254. doi: 10.3390/antiox13101254
187. Xiong F, Yang Y, Han Z, Zhang B, Kwak K, Wang P, et al. 18F-5-fluoro-aminosuberic acid PET/CT imaging of oxidative-stress features during the formation of DEN-induced rat hepatocellular carcinoma. Theranostics. (2025) 15:141–54. doi: 10.7150/thno.100467
188. Čolović M, Yang H, Merkens H, Colpo N, Bénard F, and Schaffer P. The effect of chirality on the application of 5-[18 F] fluoro-aminosuberic acid ([18 F] FASu) for oxidative stress imaging. Mol Imaging Biol. (2020) 22:873–82. doi: 10.1007/s11307-019-01450-2
189. Boutagy NE, Wu J, Cai Z, Zhang W, Booth CJ, Kyriakides TC, et al. In vivo reactive oxygen species detection with a novel positron emission tomography tracer, (18)F-DHMT, allows for early detection of anthracycline-induced cardiotoxicity in rodents. JACC Basic Transl Sci. (2018) 3:378–90. doi: 10.1016/j.jacbts.2018.02.003
190. McCormick PN, Greenwood HE, Glaser M, Maddocks ODK, Gendron T, Sander K, et al. Assessment of Tumor Redox Status through (S)-4-(3-[(18)F]fluoropropyl)-L-Glutamic Acid PET Imaging of System x(c) (-) Activity. Cancer Res. (2019) 79:853–63. doi: 10.1158/0008-5472.CAN-18-2634
191. Vermesh O and Mari Aparici C. Evaluation of 18F-FSPG versus 18F-FDG PET imaging in detecting Malignancy of solitary pulmonary nodules. J Nucl Med. (2022) 63:2595.
Keywords: NRF2 activation, oxidative stress, tumor progression, therapy resistance, gynecological cancers, targeted inhibitors, PET imaging, personalized treatment
Citation: Kgatle M, Mbambara S, Fadebi O, Kabunda J, Kaoma C, Dlangalala T, Nxele S, Modipane N, Serite T, Mokoala K, Mashamba-Thompson T and Sathekge M (2025) The implication of aberrant NRF2 activation in management of female cancers. Front. Oncol. 15:1579135. doi: 10.3389/fonc.2025.1579135
Received: 18 February 2025; Accepted: 13 October 2025;
Published: 17 November 2025.
Edited by:
Zamani Cele, Council for Scientific and Industrial Research (CSIR), South AfricaReviewed by:
Komal Ramani, Cedars Sinai Medical Center, United StatesHitesh Kumar, LCIT School of Pharmacy, India
Navkiran Kaur, Amity University, India
Copyright © 2025 Kgatle, Mbambara, Fadebi, Kabunda, Kaoma, Dlangalala, Nxele, Modipane, Serite, Mokoala, Mashamba-Thompson and Sathekge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mankgopo Kgatle, a2dhdGxlLm1hbmtnb3BvQGdtYWlsLmNvbQ==