 Tiantian Shan1
Tiantian Shan1 Xiaoying Li
Xiaoying Li- 1Department of Cardiology, The Affiliated Hospital of Qingdao University, Qingdao, China
- 2Department of Emergency, Central Hospital Affiliated Shandong First Medical University, Jinan, China
- 3Department of Emergency, Jinan Central Hospital, Jinan, China
- 4Medical College, Yangzhou University, Yangzhou, China
Invasive fibromatosis is a rare, locally invasive, benign fibrous tumor that mainly occurs in the anterior abdominal wall and limbs, with a lower incidence in the retroperitoneum. This article reports a case of retroperitoneal invasive fibromatosis in a 63-year-old male patient. The patient underwent resection for sigmoid colon cancer and was diagnosed with retroperitoneal invasive fibromatosis through enhanced CT, pathological examination and Whole Exome Sequencing one year after surgery. Given the rarity of postoperative surgery-related retroperitoneal invasive fibromatosis in patients with colorectal cancer, this article aims to provide a reference for clinical research on retroperitoneal invasive fibromatosis by reporting this case.
Introduction
Invasive fibromatosis, also known as desmoid-type fibromatosis (DF), is a rare, locally aggressive, benign fibrous tumor that originates from a mesenchymal cell line and is a subtype of mesenchymal tumor (1, 2). This tumor is a rare soft tissue sarcoma (STS), representing only 3% of all STSs and 0.03% of all tumors, with a high likelihood of recurrence without metastasis (3, 4). The local recurrence rate is high, ranging from 18% to 56% (5). DF usually occurs sporadically, but approximately 5% of cases are related to familial adenomatous polyposis (FAP); it most often occurs in the anterior abdominal wall and limbs and rarely occurs in the retroperitoneum (6, 7). The exact etiology is unclear. These tumors are rarely symptomatic, but when symptomatic, it is mainly due to the impact of the mass on the adjacent vasculature and organs. In most cases, they are found incidentally during imaging studies (7).
We report a case of a retroperitoneal invasive fibroma that was incidentally diagnosed. The patient is a 63-year-old man with a history of resection for sigmoid colon cancer who presented to the emergency department complaining of recurrent abdominal pain. In this case, we discuss the characteristics of this extremely rare tumor.
Case presentation
A 63-year-old man was found to have neoplastic polyps measuring approximately 2.2*1.4*1 cm in the sigmoid colon during routine gastrointestinal endoscopy. The polyps were removed via ESD. Pathology revealed moderately differentiated tubular adenocarcinoma with an SM invasion depth ≥ 1000 µm. The possibility of submucosal invasion was not excluded. Laparoscopic resection of colon cancer was performed at the Department of Gastroenterology, and approximately 24 cm of the sigmoid colon was removed. Pathology revealed that there was no atypical hyperplasia. Figure 1A shows the postoperative CT image.
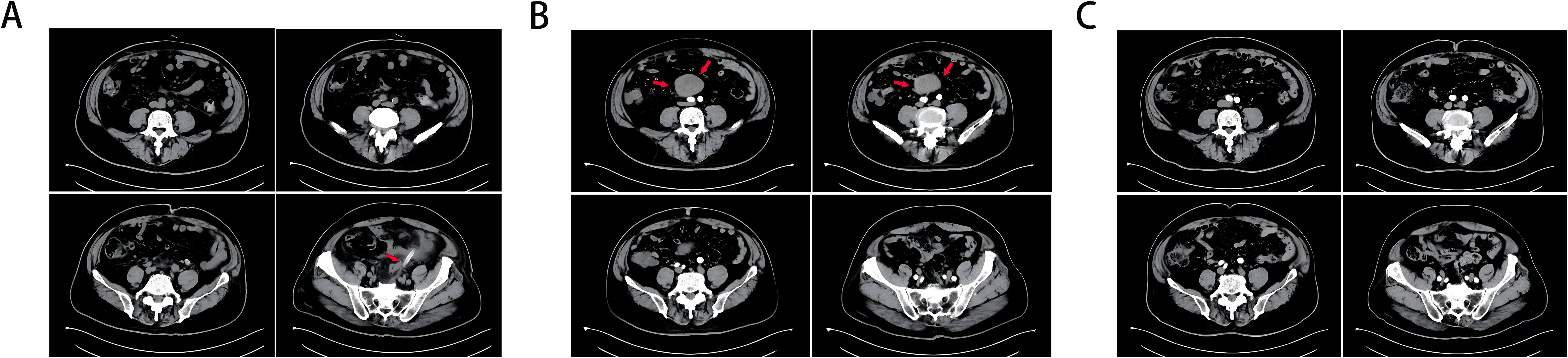
Figure 1. (A) CT taken 1 day after colon cancer surgery, and an arrow marks the drainage tube. (B) A retroperitoneal tumor (marked with an arrow) was found on intensive CT 1 year after colon cancer surgery, and there was vascular nourishment in the tumor. (C) Abdominal CT 3 years after resection of colon cancer and 2 years after resection of invasive fibroids, and no obvious abnormalities were found.
Within six months postsurgery (February–August 2021), the patient developed persistent symptoms, including increased stool frequency, loose stools, and weight loss. Gastrointestinal endoscopy performed in August 2021 revealed normal gastrointestinal mucosa without tumor recurrence, polyps, or anastomotic stricture. By December 2021, the patient’s bowel habits and stool morphology had normalized. In February 2022, the patient experienced lower abdominal discomfort and mild abdominal pain. Contrast-enhanced CT (Figure 1B) revealed a retroperitoneal mass with a vascular supply, suggesting possible tumor recurrence.
Since the possibility of tumor recurrence could not be ruled out, the patient underwent laparoscopic exploration and retroperitoneal tumor resection. The tumor (Figure 2A) was excised intraoperatively and sent for histopathological analysis. Under the microscope, spindle cell tumor-like hyperplasia was observed on HE stained sections, and the boundary with the surrounding adipose tissue was unclear. The results of immunohistochemical staining revealed the following: β-catenin (+), vimentin (+), Ki-67 (2%), SMA (-), ALK (-), MDM2 (-), CDK4 (-), CD34 (-), Bcl-2 (-), STAT-6 (-), S-100 (-), SOX-10 (-), CD117 (-), DoG-1 (-), and CK (-). The HE staining and immunohistochemical results suggested that the mass was invasive fibromatosis and a borderline tumor (Figure 2B).
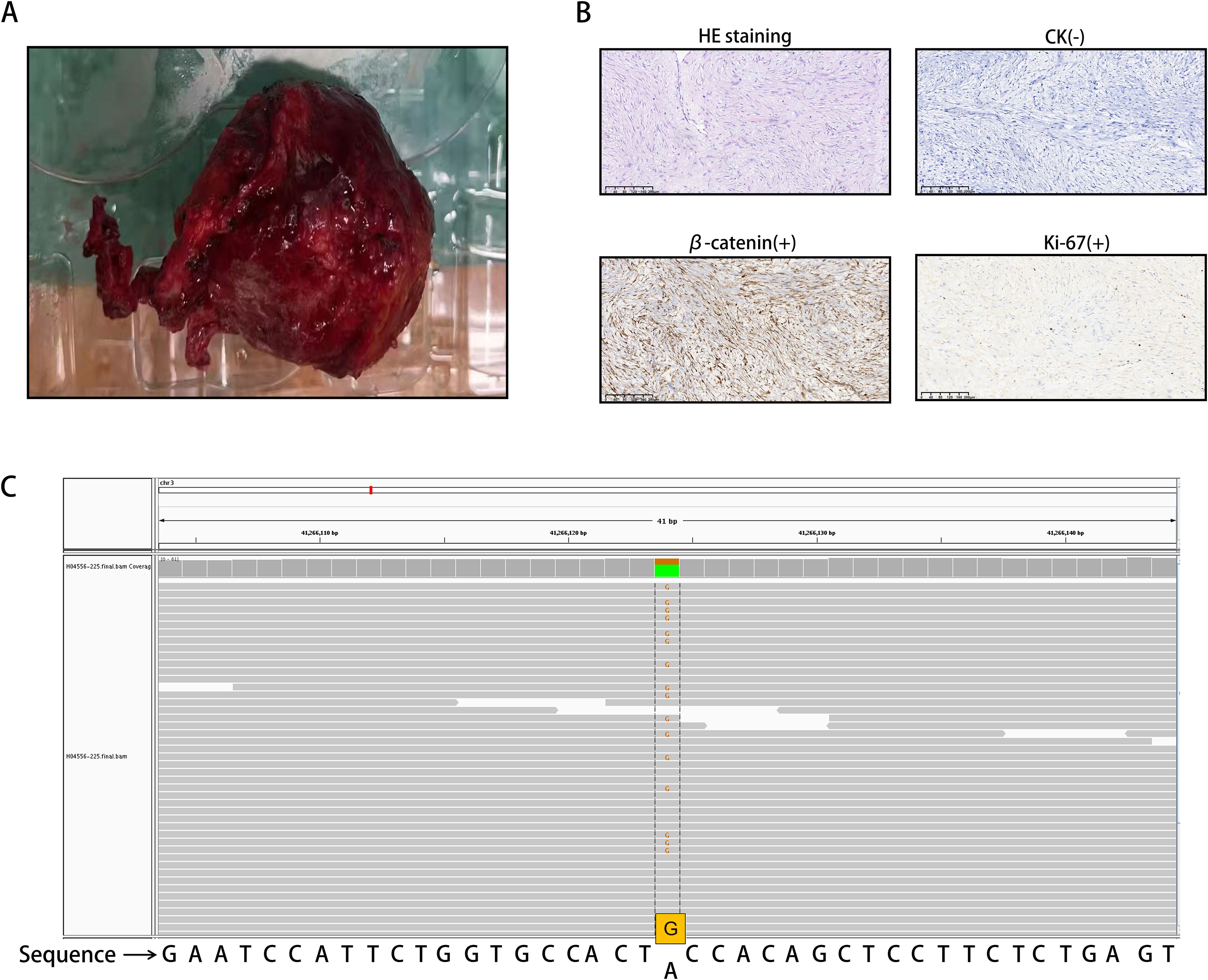
Figure 2. (A) Invasive fibroid surgery and excised tumor tissue. (B) Spindle cell neoplasia with ill-defined demarcation from surrounding adipose tissue on HE staining. The cells were sparse and dense and exhibited interstitial collagen degeneration, mucus degeneration, and high erythrocyte extravasation. The immunohistochemical staining results were as follows: Ki-67 (2%+), β-catenin (partial nucleus +), and CK (-). (C) The CTNNB1 gene mutation is located on chromosome 3, with mutation coordinates chr3:41266124 (hg19). The 121st nucleotide corresponding to exon 3 of the coding region changes from A to G (c.A121G). This was determined through the use of WES.
To confirm the diagnosis of tumor slices, whole exome sequencing (WES) was performed (Figure 2C). We found a CTNNB1 gene mutation on chromosome 3, with mutation coordinates chr3:41266124 (hg19). The 121st nucleotide corresponding to exon 3 of the coding region changes from A to G (c.A121G). This mutation is a missense mutation in all transcripts, causing the 41st amino acid of CTNNB1 encoded protein β-catenin to mutate from threonine to alanine (p.T41A). Based on the above results, the patient was diagnosed with DF.
The patient recovered uneventfully, with resolution of symptoms. During the 2-year follow-up period (March 2022–December 2024), the patient remained asymptomatic with normal bowel function, appetite, and weight restoration to preoperative levels. Serial abdominal CT scans (Figure 1C) confirmed that there was no recurrence of the retroperitoneal fibromatosis or colorectal carcinoma.
Discussion
Fibromatosis is a disease characterized by fibrous connective tissue hyperplasia and it can be divided into primary fibromatosis and invasive fibromatosis (8). Invasive fibromatosis, also known as desmoid-type fibromatosis (DF), is a type of interstitial neoplasm formed by adherent cells, fascia and aponeurosis. In terms of location, invasive fibromatosis can be divided into several groups: extraabdominal fibromatosis, which occurs in the shoulder (9), pelvis (10), chest (11) and neck muscles (12) and other muscle regions (13); intraabdominal fibromatosis, which involves the colon (14), small intestinal mesenteric connective tissue (15, 16), and retroperitoneal space (17); and fibromatosis, which can also occur in the abdominal wall (18) (Table 1). Two to six cases are newly diagnosed per million people worldwide every year, and more than 90% of DF cases are sporadic and associated with β-catenin gene (CTNNB1) mutations. A small number of DF patients are diagnosed with germline APC mutations, which present as familial adenomatous polyposis (FAP) (19). The incidence of DF in FAP patients is estimated to be 3–30% (20). However, approximately 8% of patients with sporadic DF have a family history of colon cancer, suggesting a genetic predisposition to both diseases (21).
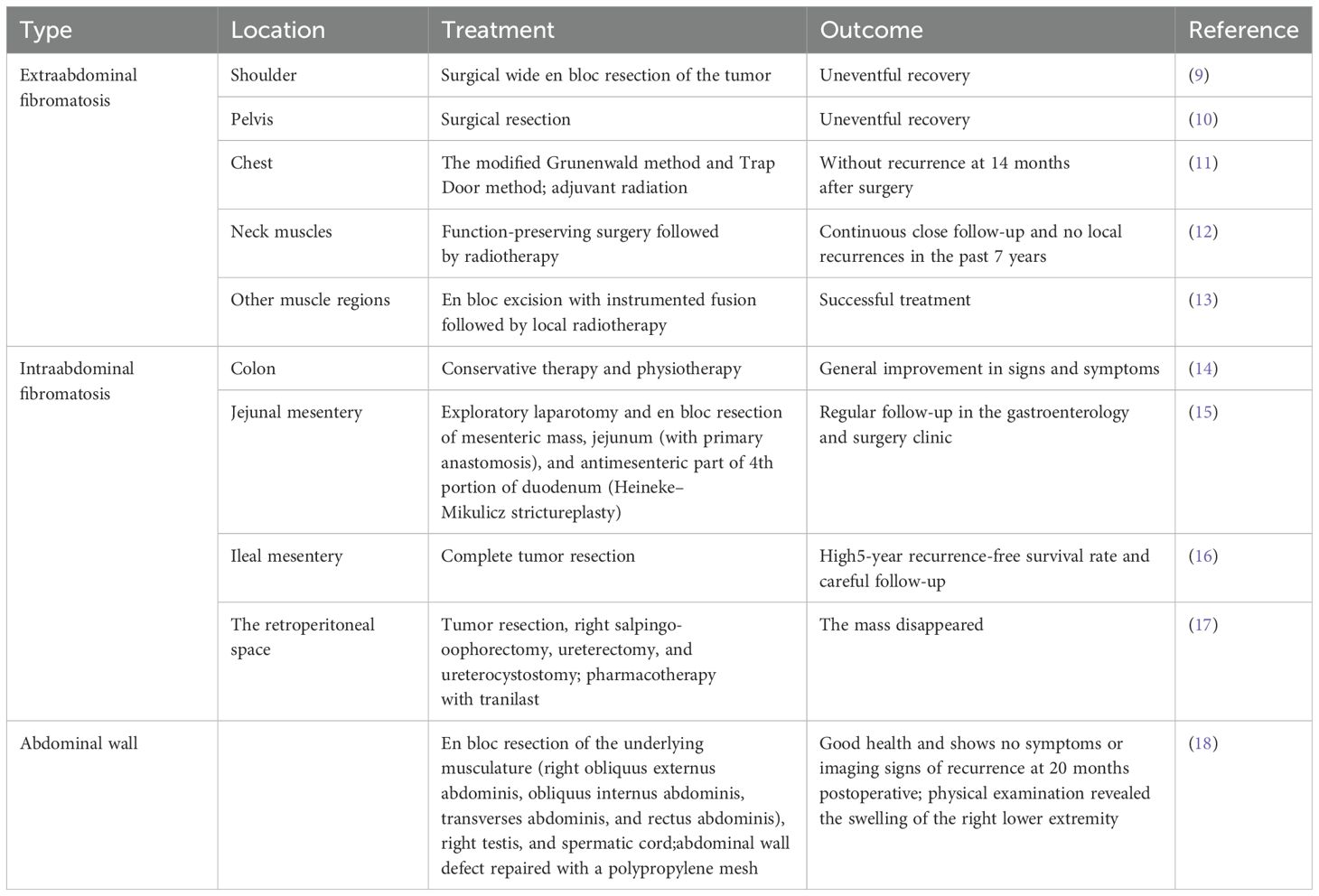
Table 1. Treatment and outcomes of invasive fibromatosis in different locations.
Computed tomography (CT) and magnetic resonance imaging (MRI) are the main imaging examination methods for detecting DF (22). With respect to the underlying mechanism of DF, the constitutive activation caused by the mutation of the β-catenin oncogene CTNNB1 (in sporadic cases) (23) or the germline activation of the adenomatous polyposis coli (APC) gene (in FAP patients) (24) will activate the Wnt pathway, thus preventing the degradation of cytosolic β-catenin. Therefore, dysregulation of the Wnt pathway plays a key role in the development of a variety of DFs. Moreover, the Wnt pathway is directly related to the pathogenesis of DF, but crosstalk with the Notch signaling pathway is crucial. Notch receptors are transmembrane proteins that play key roles in cell formation, differentiation, and apoptosis (22). In FAP patients, β-catenin-mediated upregulation of jagged-1/2 (jag-1), a notch-specific ligand, has been shown to activate Notch signaling (25). Therefore, the Wnt pathway and the Notch signaling pathway can interact with each other, leading to the occurrence of DF.
The etiology and pathogenesis of retroperitoneal invasive fibromatosis are not fully understood, but it has been reported that, in addition to the activation of abnormal Wnt signaling at the molecular level, which is mediated by the APC/β-catenin pathway and the activation of the Notch signaling pathway, it is related to trauma (including previous surgery), long-term estrogen use, pregnancy or puerperium (26). Studies have revealed that when DF occurs in the retroperitoneum, it usually has a clinical background of familial polyposis coli/familial adenomatous polyposis (FAP), colon cancer or Gardner syndrome, especially when the patient has a history of abdominal surgery (4). Postoperative development of a fibroma may be mistaken for tumor recurrence, especially when the tumor appears at the previous surgical site. It can be local or regional but never metastasizes. The incidence of this type of invasive fibroma is extremely low, and its etiology is unknown (27). Studies have revealed that fibromatosis tends to exhibit local invasive growth and recurrence. The growth is not constant and may eventually subside, but it will also grow rapidly in some periods. It is generally believed that the invasive growth of these tumors is triggered during surgery, particularly during abdominal surgery (27, 28), but the specific mechanism is not clear.
Owing to the rarity of this disease, the clinical diagnosis of DF is not simple, and the misdiagnosis rate can be as high as 30% to 40% (1). On CT images, fibromas usually appear as dense soft tissue masses with clear boundaries and uniform enhancement, but in some cases, they may appear more aggressive with unclear margins (29). Histological diagnosis usually requires the exclusion of other possible mesenchymal tumors, such as gastrointestinal stromal tumors or low-grade leiomyosarcomas (30), and fibromas are characterized by monoclonal fibroblast proliferation, manifested as small bundles of spindle cells in abundant fibrous stroma with low cell density and no malignant characteristics (2). Different types of soft tissue tumors have different immunohistochemical markers, in addition to differences in imaging and histology. Distinct immunophenotypic markers aid in differentiating these tumor types. For example, aggressive fibromatosis (a fibroblastic/myofibroblastic tumor) is characterized by diffuse cytoplasmic and nuclear β-catenin positivity along with vimentin and Ki-67 expression (31). Notably, other fibroblastic tumors lack nuclear β-catenin expression.
The Ki-67 index, widely used as a proliferation marker in clinical practice, reflects tumor cell proliferative activity through its expression in the G1, S, G2, and M phases (absent in the G0 phase), with higher values indicating greater malignant potential (32, 33). SMA and desmin positivity is typically indicative of leiomyoma (34). ALK positivity typically suggests an inflammatory myofibroblastic tumor (35), whereas liposarcoma (the most common soft tissue sarcoma) frequently harbors MDM2 and CDK4 amplifications in chromosomal region 12q13–15 (36). Coexpression of CD34, Bcl-2, CD99, and STAT-6 supports the diagnosis of solitary fibrous tumor (SFT) (37), whereas peripheral nerve sheath tumors are positive for S-100 and SOX-10 (38). Gastrointestinal stromal tumors (GISTs) typically express CD117 and DoG-1 (39). Carcinosarcoma, a biphasic malignancy containing both epithelial and mesenchymal components, is characterized by concurrent CK and vimentin expression (40).
In this case, MRI examination was declined by the patient. We comprehensively summarize the following immunohistochemical profile of this case: β-catenin (+), vimentin (+), Ki-67 (2%), SMA (-), ALK (-), MDM2 (-), CDK4 (-), CD34 (-), Bcl-2 (-), STAT-6 (-), S-100 (-), SOX-10 (-), CD117 (-), DoG-1 (-), and CK (-). This immunoprofile strongly supports the diagnosis of aggressive fibromatosis.
Every year, there are 2 to 6 newly diagnosed cases per million people worldwide, with over 90% of DF cases being sporadic and related to mutations in the β-catenin gene (CTNNB1). To demonstrate the relationship between tumor occurrence and gene mutations, tumor slices were sent for testing and subjected to WES. These results were interpreted based on the 2015 Gene Sequence Interpretation Guidelines and genome coordinates published by the American Society for Medical Genetics and Genomics (ACMG). We have detected a mutation in the CTNNB1 gene, which is located on chromosome 3 at 3p22.1. The coordinates of the gene mutation are located at chr3:41266124 (hg19), and the nucleotide at position 121 of the gene coding region has changed from A to G (c.A121G). Affects four transcripts, namely NM_001098209, NM_001098210, NM_001904, and NM_001330729. This mutation is a missense mutation in all transcripts, causing the 41st amino acid of the CTNNB1 encoded protein β-catenin to change from threonine to alanine (p.T41A), disrupting the GSK3 β phosphorylation site (a key site in the degradation signaling pathway), preventing the normal degradation of β-catenin, leading to abnormal protein accumulation and sustained activation of the Wnt signaling pathway, promoting cell proliferation, and ultimately leading to cancer. According to the ACMG classification, this mutation is defined as PP5 (pathogenicity, ClinVAR database) and PM2 (extremely low population frequency, consistent with carcinogenic mutation characteristics). Therefore, we believe that this disease can enrich our understanding of this type of tumor.
This type of tumor is difficult to treat, and many factors, such as age, tumor location and size, affect the prognosis. Among the many types of DFs, abdominal wall DFs have the best prognosis, followed by intra-abdominal DFs and limb DFs, which are known to have a greater risk of progression. Retroperitoneal invasive fibromas can be divided into three categories: asymptomatic resectable tumors, symptomatic resectable tumors, and unresectable and recurrent tumors. For asymptomatic patients, a “watch and wait” approach may be the most appropriate treatment, as such an approach can avoid unnecessary morbidity caused by surgery or radiotherapy. Surgical resection may be required to prevent disease progression, suggesting that a personalized, multidisciplinary approach to treatment is needed (22). For symptomatic retroperitoneal fibromas, their sensitivity to chemotherapy and radiotherapy is limited, and surgery is still the only curative treatment; however, studies have revealed that incomplete resection of DFs is associated with postoperative progression (41).
In the third category of unresectable and recurrent tumors, treatment options include antihormone therapy, NSAIDs, tyrosine kinase inhibitors, cytotoxic chemotherapy, radiotherapy, and close observation (42). Regardless of the type, this type of fibroma grows slowly, and patients often recover well. Although the prognosis of invasive fibromas after treatment is good, there is no reliable measure for preventing the development of an invasive fibroma after abdominal surgery.
In this case, the patient had a history of colon cancer and abdominal surgery. The invasive fibroma developed within a very short time after surgery. β-catenin showed characteristic nuclear positivity. Meanwhile, the WES results showed a mutation in the CTNNB1 gene. After laparoscopic resection, the patient recovered well without any symptoms or discomfort after the operation. Moreover, we followed the patient, and 2 years later, abdominal computed tomography (CT) revealed no recurrence of the retroperitoneal tumor or the colon cancer. The patient had returned to his preoperative weight.
Conclusion
Since invasive fibromas in the retroperitoneum are rare, it is essential to report any cases encountered. In this study, we present follow-up data for a patient who developed an invasive fibroma after colon cancer resection surgery. The CT image of the patient showed fibroma formation, with characteristic nuclear positivity of β-catenin immunohistochemistry. At the same time, WES results confirmed a mutation in the CTNNB1 gene. Retroperitoneal invasive fibromatosis is a medium-sized soft tissue tumor characterized by monoclonal fibroblast proliferation, and its clinical course is variable and unpredictable. However, few clinical trials have focused on such patients, and most studies are case series with relatively small numbers of patients. Therefore, more research on such tumors is needed.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
TS: Writing – original draft, Writing – review & editing. YZ: Conceptualization, Investigation, Writing – review & editing. YY: Writing – review & editing. ML: Conceptualization, Investigation, Writing – review & editing. XL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Shen C, Wang C, Yan J, He T, Zhou X, Ma W, et al. Clinicopathological characteristics, treatment, and survival outcomes of retroperitoneal desmoid-type fibromatosis: A single-institution experience in China. Med (Baltimore). (2019) 98:e18081. doi: 10.1097/MD.0000000000018081
2. Aidid F, Aichouni N, Afilal I, Abbou W, Jabi R, Miry N, et al. Retroperitoneal desmoid tumor in a patient with familial adenomatous polyposis: A case report. Radiol Case Rep. (2022) 17:2910–4. doi: 10.1016/j.radcr.2022.05.013
3. Meazza C, Bisogno G, Gronchi A, Fiore M, Cecchetto G, Alaggio R, et al. Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer. (2010) 116:233–40. doi: 10.1002/cncr.24679
4. Mishra DP and Rout SS. Desmoid tumors: A clear perspective or a persisting enigma? A case report and review of literature. World J Oncol. (2016) 7:21–7. doi: 10.14740/wjon961w
5. Peng PD, Hyder O, Mavros MN, Turley R, Groeschl R, Firoozmand A, et al. Management and recurrence patterns of desmoids tumors: a multi-institutional analysis of 211 patients. Ann Surg Oncol. (2012) 19:4036–42. doi: 10.1245/s10434-012-2634-6
6. Keusch CF and Bauer J. Mesenteric fibromatosis in Gardner’s syndrome. Mt Sinai J Med. (1989) 56:318–20.
7. El Charif MH, Tarhini H, Dushfunian D, Al Harake H, Khasawneh H, Abi Saad G, et al. Retroperitoneal desmoid-type fibromatosis: a case report. Ann Med Surg (Lond). (2023) 85:1258–61. doi: 10.1097/MS9.0000000000000491
8. Ferenc T, Sygut J, Kopczyński J, Mayer M, Latos-Bieleńska A, Dziki A, et al. Aggressive fibromatosis (desmoid tumors): definition, occurrence, pathology, diagnostic problems, clinical behavior, genetic background. Pol J Pathol. (2006) 57:5–15.
9. Limaiem F, Gharbi MA, Boujelbene N, Triki R, Romdhane KB, and Bouzidi R. Desmoid-type fibromatosis in an uncommon location: A case report of shoulder involvement misdiagnosed as rhabdomyosarcoma. Int J Surg Case Rep. (2024) 125:110508. doi: 10.1016/j.ijscr.2024.110508
10. Begum SA, et al. Surgical management of desmoid tumor of the female pelvis: A case report. Mymensingh Med J. (2016) 25:580–4.
11. Noda D, Abe M, Takumi Y, Anami K, Miyawaki M, Takeuchi H, et al. Resection and postoperative radiation therapy for desmoid fibromatosis of the chest wall in a young woman. Surg Case Rep. (2021) 7:28. doi: 10.1186/s40792-020-01006-5
12. Avinçsal Ö M, Shinomiya H, Otsuki N, Sasaki R, and Nibu KI. Successful management of aggressive fibromatosis of the neck: A case report. Balkan Med J. (2018) 35:278–81. doi: 10.4274/balkanmedj.2017.0509
13. Yogesh Kumar B, Vidyadhara S, and Vadhiraja BM. Pediatric recurrent aggressive spinal fibromatosis with progressive kyphosis and neurological deficits. J Orthop Surg (Hong Kong). (2019) 27:2309499019846618. doi: 10.1177/2309499019846618
14. Aljahdali NF, Alolah AA, Alghamdi AA, Alharthi FF, and Aljaziri NJ. Fibromatosis colli: A case report. Cureus. (2023) 15:e47308. doi: 10.7759/cureus.47308
15. Huang S, Shah JM, Quintero E, Xiao P, Asarian A, and Reddy M. Distal duodenal stricture secondary to mesenteric fibromatosis (Intra-abdominal desmoid tumor) of the jejunum. Case Rep Gastroenterol. (2024) 18:231–7. doi: 10.1159/000538489
16. Kuwabara H, Katayanagi S, Koganezawa I, Nakagawa M, Katsumata K, Tsuchida A, et al. Sporadic intra-abdominal desmoid tumor with a very unusual onset: two case reports. J Med Case Rep. (2021) 15:457. doi: 10.1186/s13256-021-03058-z
17. Ono H, Hori K, Tashima L, Tsuruta T, Nakatsuka SI, and Ito K. A case of retroperitoneal desmoid-type fibromatosis that involved the unilateral ureter after gynaecologic surgery. Int J Surg Case Rep. (2018) 47:30–3. doi: 10.1016/j.ijscr.2018.03.039
18. Zhao J, Cheng F, Yao Z, Zheng B, Niu Z, and He W. Surgical management of a giant desmoid fibromatosis of abdominal wall with vessels invasion in a young man: A case report and review of the literature. Front Surg. (2022) 9:851164. doi: 10.3389/fsurg.2022.851164
19. Kasper B, Ströbel P, and Hohenberger P. Desmoid tumors: clinical features and treatment options for advanced disease. Oncologist. (2011) 16:682–93. doi: 10.1634/theoncologist.2010-0281
20. Bertario L, Russo A, Sala P, Varesco L, Giarola M, Mondini P, et al. Multiple approach to the exploration of genotype-phenotype correlations in familial adenomatous polyposis. J Clin Oncol. (2003) 21:1698–707. doi: 10.1200/JCO.2003.09.118
21. Fiore M, Rimareix F, Mariani L, Domont J, Collini P, Le Péchoux C, et al. Desmoid-type fibromatosis: a front-line conservative approach to select patients for surgical treatment. Ann Surg Oncol. (2009) 16:2587–93. doi: 10.1245/s10434-009-0586-2
22. Mangla A, Agarwal N, and Schwartz G. Desmoid tumors: current perspective and treatment. Curr Treat Options Oncol. (2024) 25:161–75. doi: 10.1007/s11864-024-01177-5
23. Lazar AJ, Tuvin D, Hajibashi S, Habeeb S, Bolshakov S, Mayordomo-Aranda E, et al. Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol. (2008) 173:1518–27. doi: 10.2353/ajpath.2008.080475
24. Latchford A, Volikos E, Johnson V, Rogers P, Suraweera N, Tomlinson I, et al. APC mutations in FAP-associated desmoid tumours are non-random but not ‘just right’. Hum Mol Genet. (2007) 16:78–82. doi: 10.1093/hmg/ddl442
25. Rodilla V, Villanueva A, Obrador-Hevia A, Robert-Moreno A, Fernández-Majada V, Grilli A, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci U.S.A. (2009) 106:6315–20. doi: 10.1073/pnas.0813221106
26. Zou RQ, Liu F, and Li FY. Retroperitoneal desmoid-type fibromatosis: Challenges in the preoperative diagnosis and treatment. Asian J Surg. (2023) 46:1270–1. doi: 10.1016/j.asjsur.2022.08.059
27. Lewis JJ, Boland PJ, Leung DH, Woodruff JM, and Brennan MF. The enigma of desmoid tumors. Ann Surg. (1999) 229:866–72; discussion 872-3. doi: 10.1097/00000658-199906000-00014
28. Campara Z, Spasic A, Aleksic P, and Milev B. An aggressive retroperitoneal fibromatosis. Med Arch. (2016) 70:154–7. doi: 10.5455/medarh.2016.70.154-157
29. Teo HE, Peh WC, and Shek TW. Case 84: desmoid tumor of the abdominal wall. Radiology. (2005) 236:81–4. doi: 10.1148/radiol.2361031038
30. Lacka DE and Nasierowska-Guttmejer A. Fibromatosis - immunohistochemical evaluation, differential diagnosis from gastrointestinal tumors, and other mesenchymal tumours. Prz Gastroenterol. (2019) 14:79–85. doi: 10.5114/pg.2019.83429
31. Leithner A, Gapp M, Radl R, Pascher A, Krippl P, Leithner K, et al. Immunohistochemical analysis of desmoid tumours. J Clin Pathol. (2005) 58:1152–6. doi: 10.1136/jcp.2005.026278
32. Miller I, Min M, Yang C, Tian C, Gookin S, Carter D, et al. Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep. (2018) 24:1105–1112.e5. doi: 10.1016/j.celrep.2018.06.110
33. Kreipe H, Harbeck N, and Christgen M. Clinical validity and clinical utility of Ki67 in early breast cancer. Ther Adv Med Oncol. (2022) 14:17588359221122725. doi: 10.1177/17588359221122725
34. Campione E, Di Prete M, Costanza G, Saggini A, Agostinelli S, Terrinoni A, et al. Increased occurrence of cutaneous leiomyomas and dermatofibromas in patients with uterine leiomyomas without fumarate hydratase gene mutations. Dermatopathol (Basel). (2023) 10:231–43. doi: 10.3390/dermatopathology10030032
35. Zhao S, Liu W, Li S, Shi T, Chen Q, Li Q, et al. A case of simultaneously diagnosed lung adenocarcinoma and endobronchial inflammatory myofibroblastic tumor with two distinct types of ALK translocation. Cancer Res Treat. (2021) 53:601–6. doi: 10.4143/crt.2020.952
36. Ludwig MP, Galbraith MD, Eduthan NP, Hill AA, Clay MR, Tellez CM, et al. Proteasome inhibition sensitizes liposarcoma to MDM2 inhibition with nutlin-3 by activating the ATF4/CHOP stress response pathway. Cancer Res. (2023) 83:2543–56. doi: 10.1158/0008-5472.CAN-22-3173
37. Fonsêca TC, Agostini M, Paes JV, Roza ALOC, van Heerden WFP, Romañach MJ, et al. Solitary fibrous tumor of the mandible. Head Neck Pathol. (2024) 18:124. doi: 10.1007/s12105-024-01731-5
38. Zhou D, Xing X, Fan J, Zhang Y, Liu J, and Gong Y. PD-1/PD-L1 negative schwannoma mimicking obstructive bronchial Malignancy: A case report. Thorac Cancer. (2020) 11:2335–8. doi: 10.1111/1759-7714.13505
39. Hu D, Duan Y, Chen Y, Li B, Du Y, and Shi S. A case report of gastrointestinal stromal tumor of the duodenum. Am J Transl Res. (2022) 14:8279–85.
40. Wang Z, Jiang Z, Yang Y, Li J, Zhang C, and Liu Z. A case of tonsil sarcomatoid carcinoma. Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. (2020) 34:183–5. doi: 10.13201/j.issn.1001-1781.2020.02.021
41. Nuyttens JJ, Rust PF, Thomas CR Jr, and Turrisi AT 3rd. Surgery versus radiation therapy for patients with aggressive fibromatosis or desmoid tumors: A comparative review of 22 articles. Cancer. (2000) 88:1517–23. doi: 10.1002/(SICI)1097-0142(20000401)88:7<1517::AID-CNCR3>3.0.CO;2-9
Keywords: invasive fibromatosis, retroperitoneum, sigmoid colon cancer, imaging study, case report
Citation: Shan T, Zhang Y, Yao Y, Li M and Li X (2025) Retroperitoneal invasive fibromatosis after laparoscopic radical resection of colon cancer: a case report and literature review. Front. Oncol. 15:1582253. doi: 10.3389/fonc.2025.1582253
Received: 24 February 2025; Accepted: 27 June 2025;
Published: 17 July 2025.
Edited by:
Satvinder Singh Mudan, The London Clinic, United KingdomReviewed by:
Radu Minea, University of Southern California, United StatesKinan Mokbel, University of Exeter, United Kingdom
Copyright © 2025 Shan, Zhang, Yao, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoying Li, eWluZ2JhYnkxMDEwQDE2My5jb20=