 Erika Sofia Torres-Narvaez1†
Erika Sofia Torres-Narvaez1† Daniel Felipe Mendivelso-González2†
Daniel Felipe Mendivelso-González2† Juan Andrés Artunduaga-Alvarado1
Juan Andrés Artunduaga-Alvarado1 Oscar Ortega-Recalde1,3*
Oscar Ortega-Recalde1,3*- 1Departamento de Morfología, Facultad de Medicina e Instituto de Genética, Universidad Nacional de Colombia, Bogotá, D.C, Colombia
- 2Department of Pathology, Instituto Nacional de Cancerología, Bogotá, D.C, Colombia
- 3Unidad de Biología Computacional y Analítica de datos, Biotecgen S.A.S., Bogotá, Colombia
Next-generation sequencing (NGS) technologies have revolutionized research and precision medicine in patients with cancer. Progress in this area has been accompanied by the development of efficient and robust bioinformatics methods along with computational resources able to handle the growing amount and complexity of sequencing data. Importantly, the implementation of such approaches has not been uniform around the globe and several regions, including Latin American countries, remain lagging behind in cancer genomics and precision oncology. Likewise, numerous studies have highlighted the complexity and particularities of such populations in terms of genetic background, healthcare systems and human and technological resources. In this review, we aim to describe current clinical applications of NGS-based tests, focusing on their bioinformatics analyses and implementation in Latin America. Furthermore, we describe several opportunities for development, perspectives, and challenges that face genomic data analysis in this geographical area. We expect this review to provide an up-to-date overview of cancer genomics and bioinformatics in Latin America, serving as a valuable resource for both local and international cancer researchers.
1 Introduction
Cancer is a complex and heterogeneous disease resulting from uncontrolled cell division, leading to abnormal growth and invasion. Currently, a strong body of evidence supports that cancer is predominantly a genetic disease, arising from mutations in genes associated with cell proliferation, survival, migration, and immune regulation (1). These mutations include a wide variety of DNA alterations ranging from point mutations to large genomic rearrangements and are considered critical for tumor development and progress. In addition to being a biologically complex disease, cancer has a devastating global impact in terms of social and economic burden (2, 3). Data from the latest report of the International Agency for Research on Cancer (IARC) showed that in 2022, 20 million new cases and 9.7 million deaths were reported worldwide (4). For Latin America, accounting for approximately 8.4% of the global population, 1.5 million cases and 750,000 deaths were recorded in the same year. Furthermore, model predictions suggest that by 2050 the number of new cases will increase to 35 million worldwide, principally driven by demographic transitions. This global burden has motivated intense research efforts and technological advancements in cancer diagnosis, treatment, and prevention, which in turn have significantly changed the course and prognosis of patients with this disease.
In the last two decades, next-generation sequencing (NGS) technologies, also known as massive parallel sequencing, have revolutionized the field of cancer genomics, providing unprecedented insights into cancer biology and accelerating the development of precision oncology (5). In addition to improving cancer treatment through targeted therapies, these techniques are currently used for molecular diagnosis, disease monitoring, and assessment of predictive biomarkers (6). Furthermore, NGS-based techniques can be employed to detect germline variants, important in hereditary cancer syndromes and pharmacogenomics; somatic variants, useful as tumor biomarkers; and transcriptomic profiles useful in clinical settings. Nowadays, several NGS platforms are available in the market, and significant improvements in cost-efficiency and accessibility have facilitated their adoption in healthcare around the globe (7). While promising, one of the main challenges concerning the implementation of NGS in clinical practice is data analysis (8). NGS studies produce a vast amount of raw data, which must be carefully processed and analyzed to ultimately generate a comprehensive report, useful to the clinical team and patient (8, 9). Data analysis is a non-trivial task and requires highly specialized personnel able to use and develop bioinformatics tools and strategies and correlate the findings with biological and clinical information. In addition, bioinformatics and computational methods are critical for the analysis of such amounts of data and are considered essential for the successful implementation of NGS in precision oncology. Finally, a robust computational infrastructure is required to process high-throughput sequencing data in a timely and effective manner.
The importance of bioinformatics in clinics is gaining momentum, nevertheless, key challenges remain to be addressed. Among these difficulties, we can highlight the fast-paced development of new methods, applications and technologies, the growing demand for genomic testing, limitations in human and computational resources, and gaps in knowledge amongst healthcare professionals. These challenges are particularly relevant for developing countries with limited healthcare resources, including most Latin American countries. Although not completely integrated into national healthcare systems, several groups and institutions in Latin America are using NGS and bioinformatics tools for clinical oncology. A quick search of cancer genomics studies in each Latin American country in the Scopus database, for example, showed 276 results with Brazil (86), Mexico (57), Colombia (34), Chile (26) and Peru (20) as the main contributors (Figure 1). Noteworthy, studies derived from genomic analyses have also stressed differences in genetic background and considerable heterogeneity amongst Latin American populations. Furthermore, these studies have also explored the feasibility, clinical relevance, and limitations that face the implementation of genomic analyses within routine cancer clinical care.
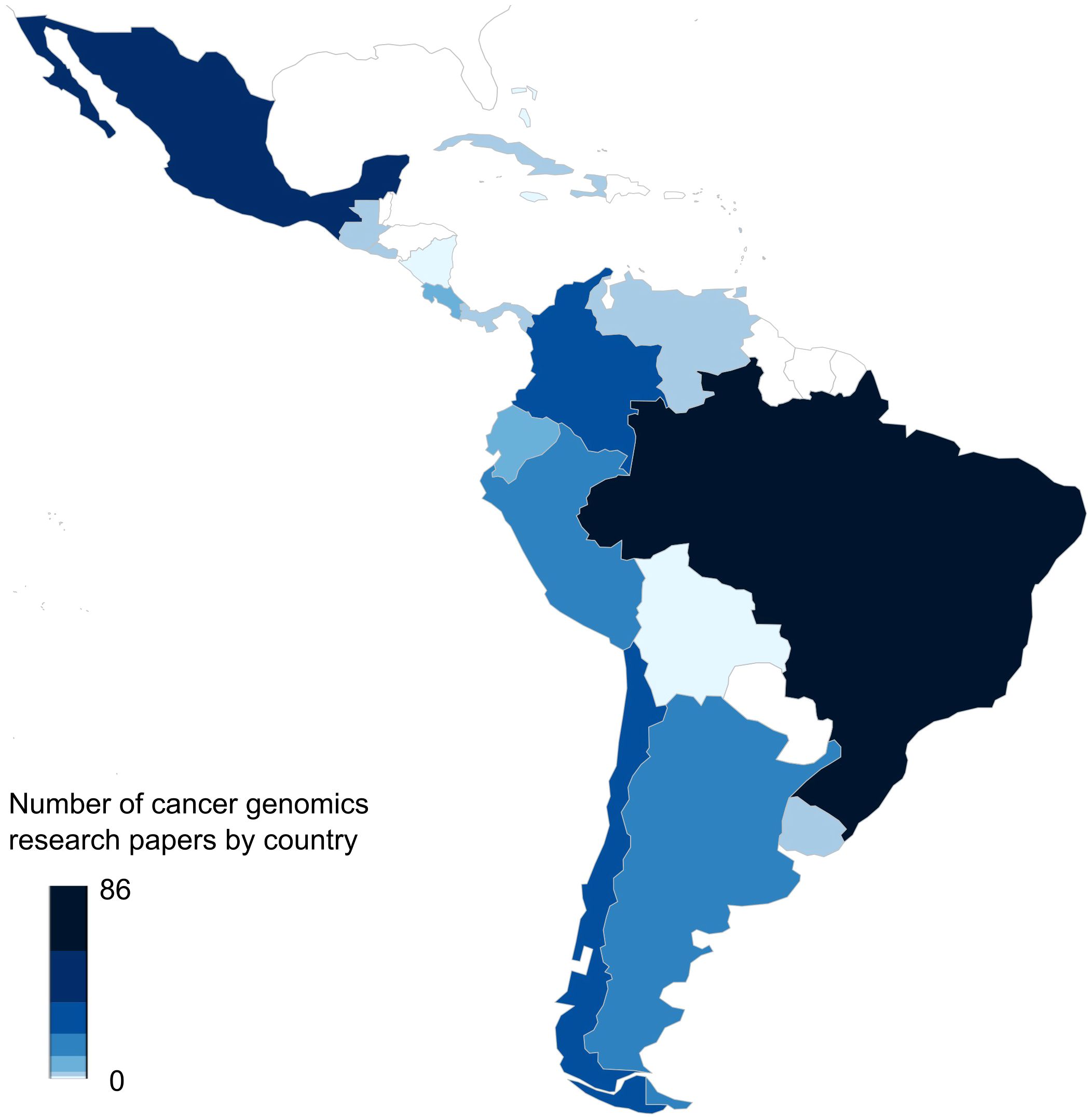
Figure 1. Number of publications in cancer genomics in each Latin American country. The figure shows the results of the frequencies of published papers found in the Scopus database until April 2025 about cancer genomics in each Latin American country. The map was generated using the rworldmap R package (v1.3-8) and the color bar represents the number of articles. The query used for each of the 33 countries was as follows: “TITLE-ABS-KEY(Genomics) OR TITLE-ABS-KEY(Transcriptomics) OR TITLE-ABS-KEY(Epigenomics) OR TITLE-ABS-KEY(Bioinformatics) AND TITLE-ABS-KEY(Neoplasms) OR TITLE-ABS-KEY(Tumor) OR TITLE-ABS-KEY(Neoplasm) OR TITLE-ABS-KEY(Tumors) OR TITLE-ABS-KEY(Neoplasia) OR TITLE-ABS-KEY(Neoplasias) OR TITLE-ABS-KEY(Cancer) OR TITLE-ABS-KEY(Cancers) OR TITLE-ABS-KEY(“Malignant Neoplasm”) OR TITLE-ABS-KEY(Malignancy) OR TITLE-ABS-KEY(Malignancies) OR TITLE-ABS-KEY(“Malignant Neoplasms”)”.
This review aims to present and discuss such clinical applications and, in a broader context, explore the challenges and opportunities of cancer genomics bioinformatics in the region. In the first part of this review, we will describe and illustrate examples of clinical applications of bioinformatics methodologies to study cancer genomics with special emphasis on the Latin American region. Next, we will focus on current challenges that hinder the successful implementation of bioinformatics platforms and propose possible solutions to address them. Finally, considering the vertiginous development of new technologies and bioinformatics approaches, we will present active areas of research that we consider will have a significant clinical impact in the near future.
2 Current clinical applications
NGS and bioinformatics tools are increasingly being used in the evaluation of cancer patients, bridging the gap between molecular data and oncology decision-making. Despite its relatively recent emergence, these tools have become increasingly available and utilized in clinical settings, particularly germline and somatic mutation testing and the analysis of transcriptomic profiles. These applications are reshaping cancer care worldwide, including the Latin American region, fostering precision medicine tailored to diverse populations.
2.1 Germline cancer testing
Germline cancer testing plays a critical role in identifying hereditary cancer syndromes, facilitating personalized preventive strategies such as enhanced surveillance, lifestyle modifications and prophylactic interventions (10). In addition, this strategy has important implications for patient screening, diagnosis, prognosis and treatment, which can be extended to other family members or communities. Currently, several guidelines and consensus include germline testing recommendations for specific tumors and high-risk patients (11, 12). Multiple recent studies have even explored the utility of universal germline cancer testing, this is cancer genetic testing for all cancer patients, providing strong evidence of its usefulness in clinical oncology and medical genetics (13–15). A pan-cancer study performed by Stadler et al., for example, analyzed 11,947 patients with advanced cancer, finding that 17% harbored likely pathogenic or pathogenic germline variants and 9% had a germline variant with therapeutic implications (13). Another study prospectively analyzed a cohort of 2,984 patients finding pathogenic variants in 13.3% of the cases, including 9.4% located in moderate- and high-penetrance cancer susceptibility genes (14). Furthermore, this study found that 28.3% of the patients with high-penetrance variants had modifications in their treatment based on their findings. These and other studies highlight the usefulness of germline cancer testing in patients with cancer and provide a strong foundation for the application of this approach in clinical practice.
Currently, several NGS-based strategies to identify germline variants associated with cancer are available, including targeted sequencing, exome sequencing and whole genome sequencing. These approaches vary in cost, diagnostic yield and analysis complexity, nevertheless, targeted sequencing, also known as gene-panel sequencing, is the most commonly used method to identify cancer-related variants in clinical settings (16). Any of these approaches involves a series of sequential steps that begin with a detailed clinical evaluation prior to ordering the test (17). This initial evaluation is not only critical to indicate opportunely the test but also for genetic counselling and interpretation purposes. In most cases, DNA is extracted from blood or saliva and sequencing library preparation is performed using standardized protocols, specific to the NGS platform to be employed (18). Regardless of the NGS technology used, sequencing raw data is primarily stored in FASTQ format and follows a standardized bioinformatics pipeline illustrated in Figure 2. First, the raw FASTQ files are trimmed to remove adaptors and low-quality bases and reads. Next, clean FASTQ files are mapped to a reference genome. The aligned reads are stored in a format known as SAM (Sequence Alignment/Map) which is commonly compressed in the binary format BAM (Binary Alignment Map). Next, variant calling is performed using a variant caller like GATK or DeepVariant incorporating bioinformatics best practices such as deduplication and recalibration (19, 20). Additionally, several bioinformatics tools allow the detection of copy number variants (CNV) using NGS data (21, 22). The variants obtained are stored in a format called VCF (Variant Call Format) and annotated. The next step involves a semi-automatic filtering of the variants identified. This process includes excluding variants based on allele frequencies, as those that are common in the general population are less likely to be associated with hereditary cancer syndromes, and using different sources of data such as bioinformatics predictions, functional analyses, genetic databases and family segregation information to prioritize and classify variants. Given the heterogeneity of data sources and variant interpretations, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) issued a guideline for classifying genetic variants using a five-point scale to assign pathogenicity in 2015 (23, 24). The scale ranges from benign (not disease causing) to pathogenic (disease causing), with intervening scores of likely benign, variant of uncertain significance (VUS) and likely pathogenic. Although several updates and alternatives have been proposed and implemented, the ACMG guidelines remain the most widely used classification system (25, 26). Nowadays, several bioinformatics companies offer automatic software and platforms to facilitate this process, nevertheless, it is important to highlight that given the clinical implications of these tests, all the steps and the generation of the final report should be supervised by a multidisciplinary team of physicians, geneticists, molecular biologists and bioinformaticians (27). In addition, similar to wet-lab protocols, oncology clinical practice guidelines firmly advocate for the validation of bioinformatics pipelines in local settings (28).
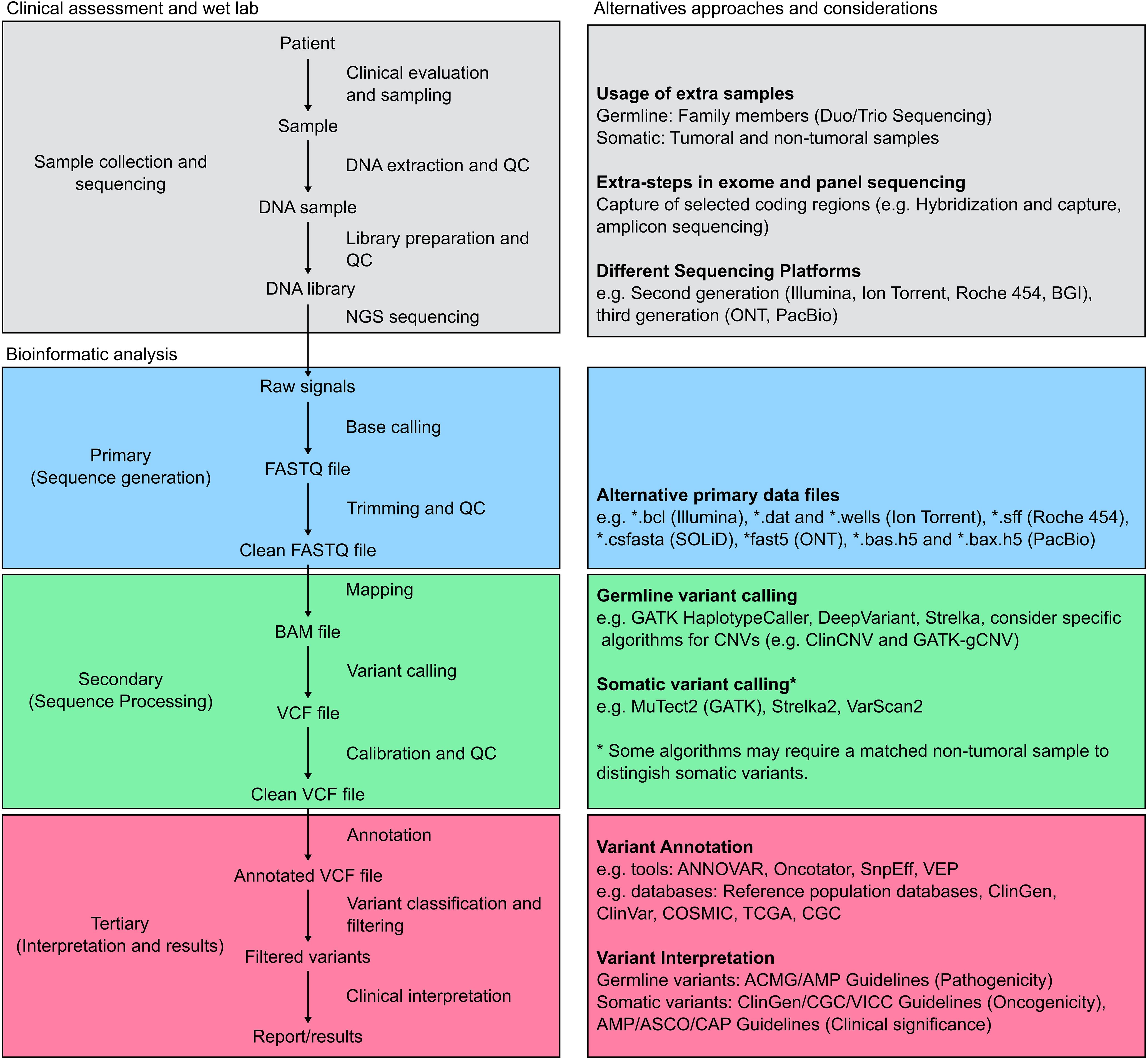
Figure 2. Standard pipeline for NGS analysis. The figure depicts a general pipeline for the analysis of NGS data along with complementary information. The grey boxes outline the process of clinical data and sample collection. Additional samples may be collected from family members and matched samples, and multiple sequencing approaches and platforms are currently available. The bioinformatic pipeline is then divided into three major steps: First, sequence generation (blue boxes), where raw sequencing data obtained from the equipment is converted into a sequence file format, most commonly FASTQ. Following trimming and quality control (QC), clean FASTQ files are mapped to a reference sequence and stored during the sequence processing step (green boxes). Different algorithms, optimized for germline or somatic variants, can then perform variant calling. Finally, variant annotation and interpretation (red boxes) are performed using a semi-automatic approach aiming to generate a clinical report or meaningful results according to the study goal.
The adoption of NGS platforms for germline cancer genetic testing in the Latin American region has been increasing over the last years. Interestingly, several studies have shown considerable differences between and within populations in this region. A recent study, for instance, analyzed 24,075 Latin American individuals undergoing testing for hereditary breast and ovarian cancer, finding that between 9.1% - 18.7% harbored pathogenic variants (29). This study included patients from Mexico, Central America, the Caribbean, South America, and US Hispanics reporting also a higher diagnostic yield in patients living in the Latin American region compared to US Hispanics. In another study, 403 individuals meeting the criteria for Hereditary Breast and Ovarian Cancer syndrome from Argentina, Colombia, Guatemala, Mexico, and Peru were analyzed for germline variants (30). The prevalence of pathogenic variants across these countries underscored the genetic heterogeneity of Latin American populations, with Argentina showing the highest prevalence at 25% and Colombia the lowest at 13%. Several examples of other studies in Latin American countries and their main results are presented in Table 1 (31–40). These results may stem from the complex genetic admixture in the region but also from differences in lifestyles and environmental factors, public health policies, and technical aspects (41). Regarding laboratory and bioinformatics practices in the Latin American region, these studies show significant variability. Some centers limit testing to specific genes, such as BRCA1 and BRCA2 for breast and ovarian cancer, while others employ NGS panels that include around 25 to >200 genes (34, 38). Remarkably, the guidelines and parameters used to define the clinical significance of genetic variants are not completely standardized across studies, potentially leading to inconsistencies in variant classification and difficulties in the comparison of results between centers and countries, creating challenges in establishing conclusions (42). On the other hand, bioinformatics methods and tools are not always presented in the studies and clinical reports, limiting their reproducibility and comparability. It is important to highlight that bioinformatics best practices and guidelines in clinical settings are increasingly relevant as these tests are more widely adopted worldwide (28). Finally, the genetic and bioinformatics “literacy” amongst clinical practitioners to interpret and use the results, a topic that will be discussed in more detail below, is a challenge probably underestimated in our region that significantly affects the utility of these tests (43).
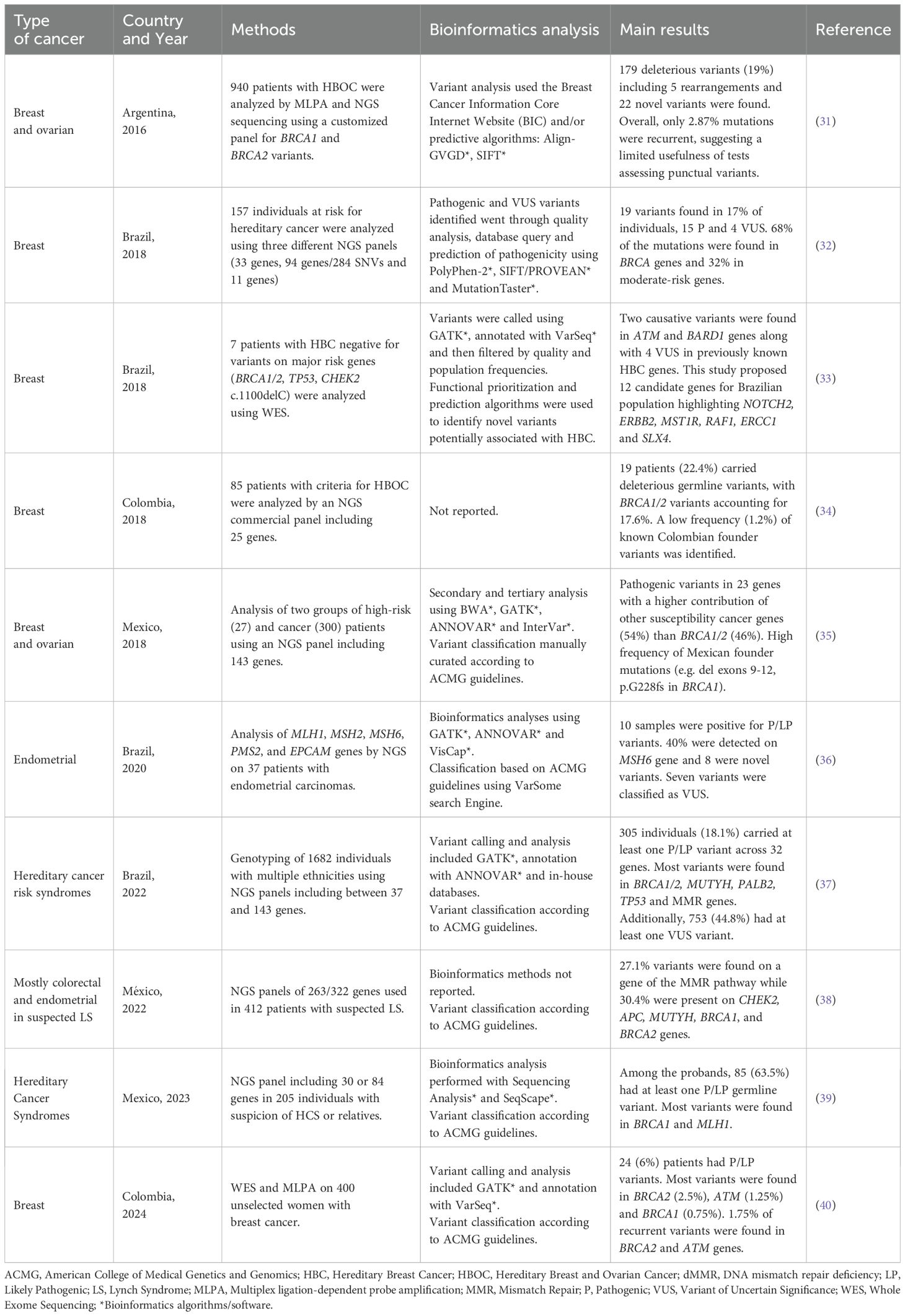
Table 1. Examples of germline NGS studies in patients with cancer from the Latin American region.
Despite the considerable advances in the implementation of hereditary cancer programs and genetic testing in Latin America, these have been heterogeneously implemented in different countries. This heterogeneity may be primarily due to structural differences in healthcare systems and limitations in human and economic resources (44). In Chile, for example, despite national recommendations advocating for universal genetic testing for patients with breast cancer, the country is far from achieving this objective. A study conducted by Acevedo et al. in two centers in Santiago, Chile during 2023 revealed that only 15% of patients with breast cancer meeting the criteria for genetic testing underwent this procedure (45). Furthermore, this study highlighted the disparities in access between private and public institutions. In Mexico, the public health system does not cover the costs of germline cancer testing and some studies mentioned the dependency on research projects to perform genetic testing (46). This approach possesses many challenges for the sustainability of germline cancer screening programs. In Colombia, germline testing is covered by the health insurer as it is considered a diagnostic procedure (47). In this country, a recent study by Sierra-Díaz et al., found that 6% of the Colombian women with unselected breast cancer had germline mutations in high-penetrance cancer susceptibility genes (40). Interestingly, the numerous challenges experienced in these countries have also generated opportunities to optimize resources and improve healthcare systems. For instance, one center in Mexico has successfully implemented a germline cancer testing service that includes telemedicine (46). This innovation enabled patients from rural and underserved areas to access genetic counseling and testing, effectively bridging a critical gap in genomic medicine. Similarly, the establishment of national cancer programs in countries such as Chile and Colombia has facilitated the gradual integration of germline screening programs in healthcare systems (48, 49). Notably, the development of robust and accessible cancer genomics programs has progressed more slowly than anticipated, highlighting the need for sustained efforts to overcome the existing barriers.
2.2 Somatic cancer testing
The identification of driver mutations in cancer genomes is considered one of the pillars of precision oncology (5). These mutations play a critical role in cancer development and are valuable biomarkers for diagnosis, prognosis and targeted therapy. In contrast to low-throughput molecular tests, NGS-based somatic cancer approaches can simultaneously analyze multiple gene regions and even the entire genome. Currently, leading organizations such as the European Society for Medical Oncology (ESMO), have included evidence-based recommendations for the use of tumor NGS in patients with advanced non-squamous non-small-cell lung cancer (NSCLC), prostate cancers, breast cancers, ovarian cancers, among other tumor types (50). These recommendations are based on multiple lines of evidence, for instance, a recent comprehensive review on the clinical impact of NGS tests for the management of advanced tumors showed that progression-free survival and overall survival among patients who received NGS-guided cancer treatment were significantly longer across multiple tumor types (51). Another large study assessing 109,695 patients with solid tumors found that among the most common cancer types, predictive, prognostic, and diagnostic markers were reported in 51.2% of tumor profiles, and 89.2% had genomic results that could inform guided therapies (52). While the decision to choose between different NGS test options may be challenging and relies on multiple factors including tumor biology characteristics, test availability, and cost-effectiveness, somatic cancer studies are critical to improving cancer care (53). Furthermore, numerous clinical trials matching specific genomic profiles to novel cancer therapies have shown that the growing knowledge gained through cancer research is continuously being integrated at this level, providing a powerful tool in translational medicine. Despite its relevance, one of the main bottlenecks of these approaches is the analysis and interpretation of the large amount of data generated through NGS, constituting a potential barrier to wide clinical adoption (54).
Several parallels can be drawn between germline and somatic cancer data analysis, nevertheless, the identification of somatic variants faces challenges. Regarding the tumor sample, DNA can be obtained from fresh samples, liquid biopsies, or formalin-fixed paraffin-embedded tissues (FFPE). Importantly, these samples may contain different amounts of genetic material and proportions of normal and tumor cells, described as the purity of the sample, which affects further analysis (6). Moreover, clonal evolution in cancer cells may result in genetic intratumoral heterogeneity, which is not always well represented in the sample taken and could lead to false negative results. In light of these confounding factors, it is suggested that clinical reports include sample quality parameters along with sequencing quality information (19).
Another important consideration is the correct distinction between germline and somatic variants. This is usually achieved through the direct comparison of tumor samples and patient-matched normal tissue samples, such as peripheral blood (19). When the study relies only on tumor samples, the origin can be inferred using variant allele frequencies (VAF), databases of recurrent germline variants and specific algorithms. While tumor-only studies are more cost-effective than matched tumor-normal sequencing, it carries inherent limitations. This is due to the potential misclassification of germline variants in population databases, variability in tumor purity, and differences in intratumor heterogeneity across specimens (55). Finally, to enhance sensitivity for detecting somatic variants, it is necessary to choose a specific sequencing depth, defined as the average number of aligned reads at a given genomic position. This will depend on the threshold defined to the limit of detection (LOD), tolerance for false positive/false negative results, and sequencing error rates (56). For example, increasing sequencing depth beyond standards used in germline studies is recommended for low-frequency somatic variants (19, 57). Although currently there is no consensus on the optimal sequencing depth in the context of somatic variants, some targeted somatic panels have recommended ranges between >500X for LOD of 5% to > 1000x for low tumor cellularity samples (56).
The bioinformatics analysis of NGS-based somatic cancer techniques follows similar steps to NGS germline techniques. A global overview of this pipeline is presented in Figure 2. Importantly, somatic variant calling remains a challenging task due to the cancer genome complexity and several bioinformatics tools have been specifically designed to optimize the identification of somatic variants, including MuTect2, Strelka2 and VarScan2 (58–60). These methods can integrate somatic and germline information to tackle biological and technical issues such as low VAF and low sample purity. Intriguingly, several studies comparing these tools have shown differences in performance, suggesting that the combination of techniques could maximize somatic variant discovery (61). Alternatively, when only tumor sequencing information is available, general or specific variant callers must be optimized to detect somatic variants, taking into account the potential issues previously mentioned (19, 62). The variants obtained from this step are then stored in VCF format for conducting tertiary analysis, including clinical interpretation and correlation. In addition to general and germline databases, numerous somatic and cancer-specific resources can be used to classify and interpret the findings, and somatic cancer reports often include more information about specific variants (63, 64).
Two concepts are particularly relevant in somatic cancer analyses and reports: oncogenicity and clinical significance. Oncogenicity, defined as “the pathogenicity of the variant in the context of a neoplastic disease”, is classified according to a joint consensus of the Clinical Genome Resource (ClinGen), the Cancer Genomics Consortium (CGC), and the Variant Interpretation for Cancer Consortium (VICC) (65). This system classifies the variants into 5 categories: oncogenic, likely oncogenic, variant of uncertain significance (VUS), likely benign, and benign, based on an evidence point system including population, functional and predictive data, cancer hotspots, and computational evidence. The evidence strength in each data type adds or subtracts points to the final score of each variant allowing its categorization. Second, clinical significance, defined as the variant’s impact on clinical care in terms of diagnosis, prognosis, and/or therapeutic biomarkers (55). Clinical significance is classified according to an evidence-based system proposed by a joint consensus of the AMP, the American Society of Clinical Oncology (ASCO), and the College of American Pathologists (CAP). This system uses different sources of information, including guidelines, FDA approvals, dedicated databases, and computational predictions, to classify variants in 4 Tiers: Tier I, variants with strong clinical significance; Tier II, variants with potential clinical significance; Tier III, variants with unknown clinical significance; and Tier IV, variants that are benign or likely benign. Given the growing amount of information and continuous updates related to clinical associations, knowledge databases are created to integrate the data. Examples of these efforts include the Cancer Genome Interpreter Cancer Biomarkers Database (CGI), Clinical Interpretation of Variants in Cancer (CIViC), Jackson Laboratory Clinical Knowledgebase (JAX-CKB), OncoKB and the Precision Medicine Knowledgebase (PMKB), among others (66–70). Harmonization of this data to obtain reproducible results using different tools is necessary to integrate and standardize the information included in the final clinical report (64).
The implementation of somatic NGS-based analyses in cancer research and clinical practice in Latin America has been slow but steady. By 2017, it was estimated that more than 221 NGS platforms were available in the region and 272 articles were reported to have Latin American authorship associations, with Brazil and Mexico as major contributors (71). Furthermore, the incorporation of NGS-based technologies has enabled the transition from specific mutation methods to comprehensive cancer genomics studies, allowing to detect novel clinical associations in these populations. A recent study performed by the CLICaP consortium, for example, analyzed the genomic landscape of primary resistance to Osimertinib among Hispanic patients with EGFR-mutant non-small cell lung cancer, showing that specific findings such as commutations, and the presence of the mutations EGFR p.T790M and p.L858R are associated with therapeutic responses and patient outcomes (72). Another recent study in Chile, suggested potential differences in driver mutations for Chilean patients with colon cancer when compared to cohorts with different ancestries (73). Given their epidemiological relevance, prostate (15%), breast (14%), colorectal (9%), lung (7%) and gastric (5%) cancers are amongst the principal focus of research and precision oncology initiatives in this region (74). Some examples of these studies and their conclusions are presented in Table 2 (75–83). These findings are particularly relevant in clinical oncology due to the underrepresentation of Latin American populations in precision medicine studies and databases. Interestingly, some studies have also focused on the design and validation of cost-effective NGS platforms optimized for our region (84). In addition to optimizing resources, these types of studies are remarkable in terms of technological appropriation and open source bioinformatics solutions.
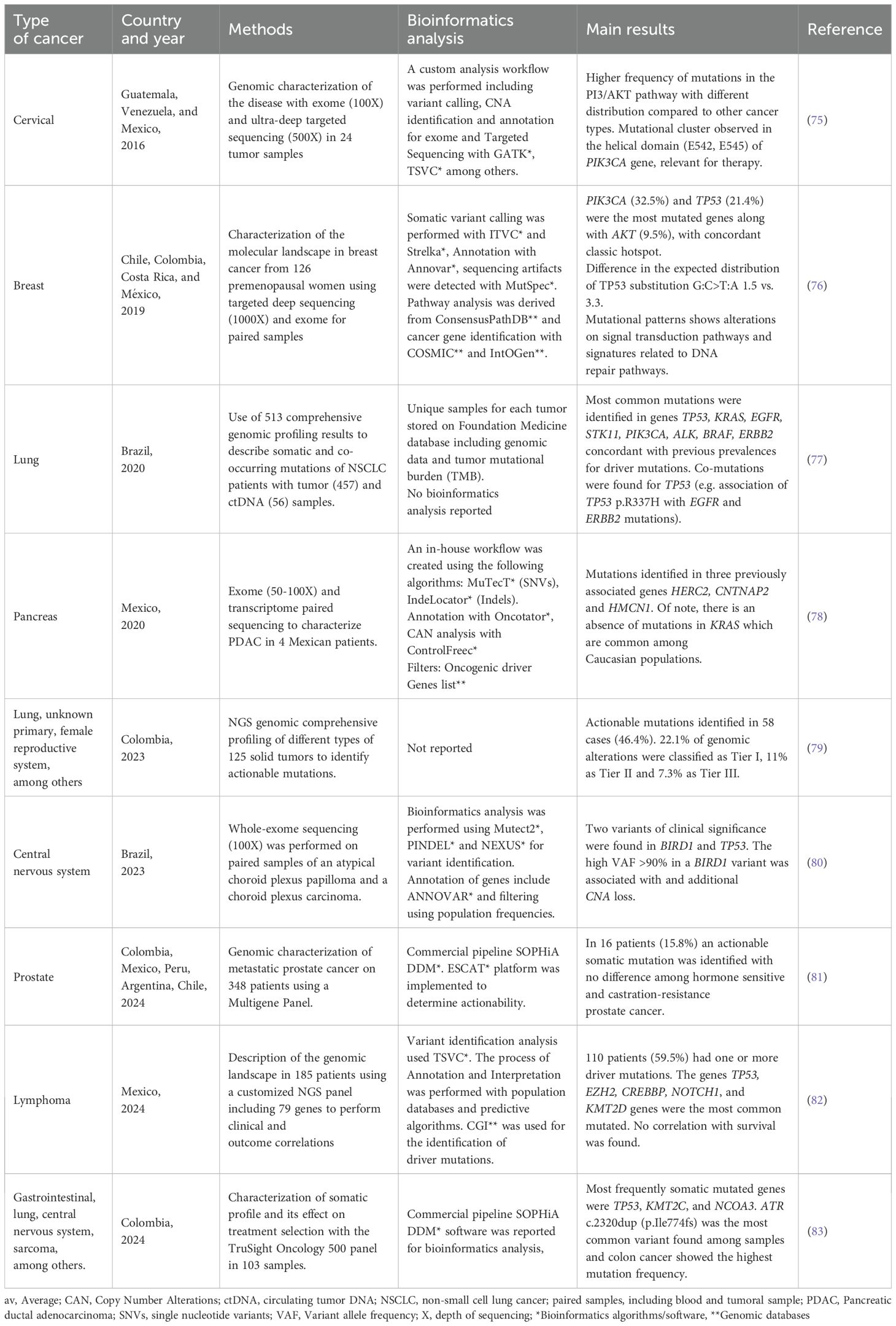
Table 2. Examples of somatic NGS studies in patients with cancer from the Latin American region.
Overall, diverse studies focused on somatic cancer studies in Latin America show high heterogeneity in technical and analytical aspects. Regarding mutation detection strategies, although multiple techniques are currently available, the transition to NGS-based technologies is accelerating (85). This transition is associated with increasing testing costs, nevertheless, several studies examining the cost-effectiveness of this approach suggest that robust analyses should be conducted in specific scenarios and that NGS-based tests are cost-effective in multiple clinical settings (86, 87). FFPE samples are the most common tissue analyzed due to their ease of storage and cost-efficiency, even though they are prone to DNA damage associated with the technique, time of storage and quality of the material and protocols (88). In this regard, it should be highlighted that high-quality materials and methods should be prioritized to optimize DNA recovery. Different NGS sequencing technologies, strategies and bioinformatics pipelines have been used in these analyses, including commercial and in-house gene panels and bioinformatics workflows. Despite the importance of quality parameters, these are not always included in clinical reports and studies. Similarly, there is still a large heterogeneity in the implementation of the oncogenicity and clinical significance parameters. Finally, the impact of these tests on clinical decisions has been rarely explored in our region. Given their importance in clinical practice, it is expected that somatic cancer studies will become standard-of-care in oncology and will dramatically improve the outcome of patients with cancer (50, 89).
2.3 Transcriptomic profiles
The transcriptome is the entire set of expressed RNA in a particular cell or population of cells at a specific time point. In contrast to the genome, which is considerably more stable in time, the transcriptome is highly dynamic, responding to environmental stimuli and endogenous cues (90). In cancer, gene expression studies have been critical to understanding tumor biology and in clinical practice (91, 92). Historically, these methods include Northern blotting and reverse transcriptase quantitative polymerase chain reaction (RT-qPCR). RT-qPCR, for example, can be used to detect specific gene fusions and quantify the expression of a limited number of genes (93). Although highly sensitive and specific, these techniques only allow the assessment of a determined and reduced number of transcripts or alterations. Later, the introduction of expression microarray enabled the analysis of a considerably larger number of genes, expanding the potential use of transcriptomic data in clinics (94). Currently, several commercial platforms, for example, Oncotype DX™ (Genomic Health), MammaPrint™ (Agendia) and EndoPredict™ (Myriad Genetics) offer gene expression-based analyses for clinical purposes (95–97). Despite their importance, the clinical usage of transcriptomic techniques remains limited due to several factors, including performance in different clinical settings complexity of the analyses, uncertain clinical interpretation and cost-effectiveness (98–100).
As a result of multiple technological and computational advances, NGS of RNA (RNA-seq) has been consolidated as a robust and versatile method for the analysis of tumor transcriptomes. In contrast to DNA sequencing, RNA-seq is primarily a qualitative and quantitative method (101). On the one hand, it allows the detection of isoforms, variants, aberrant splicing and gene fusions. On the other hand, it can be used to accurately measure gene expression levels, resulting in a robust and unbiased approach to studying the transcriptome and indirectly, the genome. Several types of RNA-seq are currently available, nevertheless, whole transcriptome RNA-seq (WTS) and targeted RNA-seq are the methods more commonly used for clinical purposes (102). WTS is a nonselective technique optimal for the discovery of new biomarkers and obtaining a complete picture of the transcriptome and being used for the detection of novel gene fusions, assessment of VUS and molecular characterization of transcriptomic profiles. Although versatile, the main setbacks of this technique are the quality requirements of the sample to be assessed, the sequencing depth to detect lower abundance events, and the costs and complexity of data analysis. Some of these limitations can be fixed by limiting the number of transcripts to be assessed through targeted RNA-seq. This method involves the selection and sequencing of specific transcripts of interest, reducing costs, making analyses more simple, and increasing the sequencing depth of informative events. As expected, the main setback of this approach is the inability to assess genes or events outside the targeted panel. In addition, multiomic approaches, integrating, for example, genomic and transcriptomic sequencing, have emerged as powerful tools to understand tumor complexity and ultimately improve cancer care (103).
The process of RNA-seq begins with the isolation of RNA from the tumor sample and library preparation. These steps, along with sample collection, are critical for obtaining high-quality data and have been extensively discussed in previous reviews (104, 105). An important point about RNA-seq data analysis is that there is not an optimal bioinformatics pipeline for all applications and scenarios in which this method can be used, therefore, these steps should be optimized accordingly (106). Overall, three major phases can be distinguished. First, a pre-analytical phase, which includes an adequate experimental and sequencing design. Once sequencing is performed, this phase includes raw reads quality control and other steps to ensure that data quality is appropriate, for example, read and alignment quality or assessment of batch effects. The second phase, or core analysis, begins with mapping reads to a reference genome, transcriptome or alternative database. This step may include quantitation of transcripts, transcript discovery and differential expression analysis. Finally, an advanced analysis phase can be performed according to the study goals. This phase may include data visualization, gene-fusion discovery, data interpretation, and integration with other techniques and data, including DNA sequencing and clinical information. It should be highlighted that in comparison to DNA-seq, RNA-seq data analysis is less standardized and might be more challenging, particularly in clinical scenarios.
Recently, numerous studies have shown the utility of RNA-seq in clinical settings, including pediatric low-grade glioma, acute lymphoblastic leukemia and breast cancer (92, 107–109). Hardin et al., for example, conducted a multicenter study analyzing 125 samples of patients with low-grade glioma using RNA-seq (107). Interestingly, the authors found that in addition to detecting genomic alterations previously found by other techniques, RNA-seq identified driver mutations not previously detected in 27 cases, 81% of them classified as actionable. Another study by Pleasance et al. combined whole genome and transcriptome sequencing analysis (WGTA) to study 570 patients with advanced or metastatic cancer of diverse etiologies (92). In this study, the authors identified clinically actionable targets for 83% of patients, of whom 37% received WGTA-informed treatments, and 46% of them resulted in clinical benefit. Remarkably, RNA-seq data was highly informative, being useful in 67% of WGTA-informed treatments. These studies highlight the transformative potential of integrating this technique into current cancer diagnostic methods to enhance patient outcomes.
In Latin America, the implementation of RNA-seq and expression arrays for analyzing solid tumors is gaining relevance, particularly in academic settings and specialized reference centers. These techniques have facilitated the identification of specific biomarkers and the molecular characterization of prevalent cancers in the region, including breast, lung and gastric cancers. Given its clinical relevance, multiple studies have focused on understanding the biological landscape of breast tumors in Latin American women (110–112). Romero-Cordoba et al., for instance, provided a detailed genomic and transcriptomic characterization of 204 breast tumors in Hispanic and Mexican women, contrasting their genomic context with patients from African, African American, Asian, and European ancestries and revealing unique molecular features in local populations (111). Another study conducted by Llera et al. used expression arrays to analyze a multi-country cohort of 1071 breast cancer patients as part of the Molecular Profile of Breast Cancer Study (MPBCS), identifying intrinsic subtypes using thePAM50 classification system and revealing similarities and differences in this cohort when compared to other studies (110). Other examples of studies in this and other cancer types are presented in Table 3 (110–116). In addition to solid tumors, RNA-seq has gained importance in the study of hematological malignancies, including non-Hodgkin lymphomas (NHL) and acute lymphoblastic leukemia (ALL). The Epidemiology of Lymphomas in Latin America (ELLA) cohort study, led by the “Grupo de Estudio Latinoamericano de Linfoproliferativos” (GELL), exemplifies these efforts by investigating the genomic and immunologic landscapes of NHL subtypes and developing prognostic models tailored to regional populations (117). Similarly, a retrospective multicenter cohort study by the Mexican Inter-Institutional Group for the Identification of the Causes of Childhood Leukemia (MIGICCL) utilized RNA-seq to analyze 49 children with ALL (114). This study identified a high prevalence of recurrent and novel gene fusions, including DUX4 and CRLF2 alterations, which correlated with poor outcomes, highlighting disparities in survival compared to global cohorts. Together, these initiatives demonstrate how transcriptomic analysis is unveiling critical molecular insights, advancing precision oncology, and addressing unique challenges in both solid and hematological malignancies across Latin America.
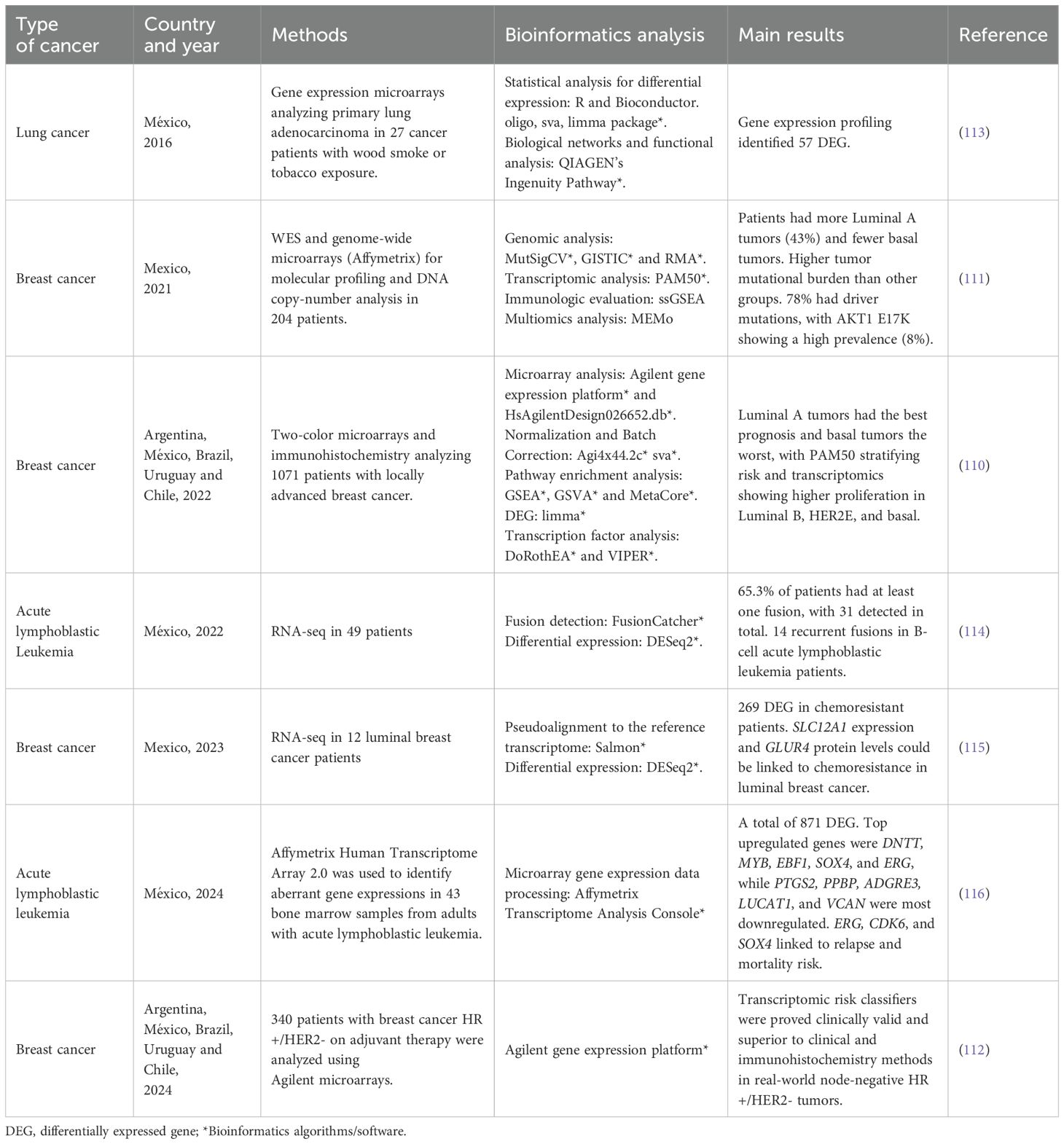
Table 3. Examples of transcriptomic studies in patients with cancer from the Latin American region.
3 Challenges and opportunities
Despite the transformative potential of cancer genomics in oncology practice and research, several challenges impede their widespread adoption in healthcare. Among these, limited human and computational resources hinder equitable access to genomic technologies, especially in low- and middle-income countries. Similarly, the lack of integration of genomic information into clinical practice and healthcare systems may slow its adoption. Given the growing importance of this field in medical practice, numerous opportunities have also emerged to tackle these challenges. Collaborative genomics initiatives and consortiums, for example, are powerful tools to optimize resource usage and reduce costs. Likewise, medical education and training are key to equipping healthcare professionals with the knowledge and skills needed for genomic-driven care. In this section, we will explore these topics in more depth and provide emphasis on the Latin American region.
3.1 Human and computational resources
The growing amount of cancer genomics data raises the need for specialized solutions able to provide efficient and reliable bioinformatics services to the clinical community. This increasing demand for bioinformatics solutions has long been recognized and plays an important role in current clinical settings (118). In order to provide these services, different resources should be considered, including computing platforms (infrastructure), algorithms and software, and human resources. Each of these levels has experienced significant progress and has accelerated the implementation of cancer genomics into the clinics.
At the computational infrastructure level, demands for handling, storing and analyzing massive amounts of genomic data can be solved on distributed high-performance computer (HPC) systems. HPC can be defined as a technology that uses supercomputers or computer networks, named clusters or grids, to process massive datasets and solve complex tasks at high speeds (119). Additionally, cloud storage and computing have emerged as powerful tools useful for data sharing and demand-driven computation, reducing costs, facilitating access and collaborations, and surpassing geographical and infrastructure barriers (120).
A second major area of concern includes software and algorithm development. These tools and methods are critical to optimizing data analysis and interpretation and constitute subjects of intense development and research. In variant calling, for example, new insights using artificial intelligence (AI)-based tools such as DeepVariant, developed by Google, and DRAGEN, developed by Illumina, have shown promising results (121, 122). Other efforts have been focused on developing software to improve interoperability and management of databases, this includes relational databases and standardization of genomic data for precision medicine. As an example, the Genomic Data Commons (GDC) platform developed by the National Cancer Institute (NCI) integrates renowned cancer genomics datasets, processing the data in reproducible bioinformatics pipelines and democratizing access to cancer genomics data (123). Other initiatives include the creation of APIs (Application Programming Interfaces) such as Beacon by GA4GH, developed to aid data sharing without compromising personal and sensitive information (124).
Last but not least, bioinformatics services require experts in the field not only to develop these tools but also to implement bioinformatics protocols and best practices, troubleshoot, and provide advice on the clinical interpretation of genomic data. Some large-scale projects, such as data integration between ICGC and TCGA in the Pancancer Analysis of Whole Genomes (PCAWG) study, for instance, required almost 1,300 researchers to complete bioinformatics tasks like consolidation of histopathological data, uniform data processing, variant calling, and quality control of somatic and germline variants from 2,600 cancer samples (125). Importantly, there is a critical need for bioinformatics expertise in healthcare and research, and although multiple solutions in education and training have been proposed to mitigate this shortage, several challenges remain to be addressed (126, 127).
Latin American countries have faced significant challenges in research and development, principally derived from insufficient and discontinuous funding that led to difficulties in maintaining and updating the necessary infrastructure, software and training for researchers to keep up with new technologies. These technological challenges arise from the need for standardization, the deployment of structured databases and the acquisition of advanced equipment, among others. Also, bioinformatics education and training are still considered an important challenge in Latin America and other low- to middle-income countries (LMICs) (128). In this regard, Argentina, Brazil, Chile, Colombia and Mexico have relatively advanced bioinformatics programs compared to other countries in Latin America and have shown successful results in integrating this field into basic and applied research (128–131). On the other hand, new computing paradigms related to decentralized and low-cost infrastructures, such as cloud computing, are alternatives to optimize available resources without compromising quality (120). Additionally, cooperative databases and open-source software and algorithms have brought opportunities to conduct high-quality research and offer cutting-edge bioinformatics services. Finally, international consortia and initiatives, explored below, have shown to be effective in optimizing the usage of these resources. Given the growing importance of cancer genomics bioinformatics in oncology, we highlight the importance of the allocation of resources and investment to improve the outcomes of patients with cancer.
3.2 National and international cancer genomics initiatives and consortiums
Advances in NGS and bioinformatics technologies have accelerated the generation of cancer genomics data, which in turn has been critical to understanding its molecular basis and the development of targeted therapies. Importantly, the number of patients/samples and the scale of projects aimed at studying such associations impose significant challenges in terms of large-scale patient recruitment, sample processing, data collection, and data analysis in a timely and resourceful manner. Cooperation arises as a powerful solution to tackle these problems under the figure of multicenter initiatives and consortia. In the scientific community, a consortium is defined as the association of a multidisciplinary group of scientists from diverse institutions and/or countries that collaborate on research efforts to achieve a common goal (132). In practical terms, these networks have contributed to the pooling of information, the development and validation of tools and the subsequent analyses in multicentric projects. Furthermore, consortia have served as a platform for training on genomic research and promotion of institutional infrastructure for data collection, analysis and sharing. One of the most significant examples of collaborative research worldwide was the Human Genome Project dedicated to establishing a standard sequence of the human genome by the International Human Genome Sequencing Consortium (133). Another illustrative example of recent large-scale cooperative efforts is the 100,000 Genomes Project, a British initiative aimed at sequencing the whole genome of patients from the United Kingdom National Health Service (NHS) affected by rare diseases and cancer (134). Importantly, these ambitious projects highlight the importance of interinstitutional and governmental cooperation to strengthen research capacities and optimize resources.
This kind of approach is particularly relevant in cancer genomics as it advances towards massive genomic and clinical data (GCD) analyses and fast-paced technological developments. Importantly, the integration of such data across different centers and institutions is necessary to achieve statistical power and obtain robust results in considerably shorter times, accelerating the implementation of precision oncology solutions (135). Global initiatives, such as the International Cancer Genome Consortium (ICGC) and Project GENIE (Genomics Evidence Neoplasia Information Exchange), created by the American Association for Cancer Research (AACR), are examples of relevant consortia in this area (136, 137). The ICGC was launched in 2008 as a large-scale collaborative effort to characterize genomic abnormalities among different cancer types using genomic, transcriptomic and epigenomic information (136). In 2019 the ICGC data portal contained data from 84 worldwide cancer projects, from 20,000 contributors and 77 million somatic mutations, including data from the Cancer Genome Atlas (TCGA) and the Sanger Cancer Genome projects. While the original web portal was available until June 2024, data remains available to researchers and the ICGC has advanced to a new phase, the ARGO (Accelerating Research in Genomic Oncology) project, an international initiative aimed at analyzing specimens from 100,000 cancer patients worldwide (136). Similarly, Project GENIE is a large international consortium aimed at catalyzing the sharing of GCD, enabling precision cancer medicine research (138). Launched in 2015, they have sequenced 214,487 samples from 184,988 patients and 18 contributing institutions until 2024. Remarkably, the utility of such initiatives is not limited to academic and research activities, but also to improving decision-making in oncology clinical practice.
In Latin American countries similar initiatives have been built over the years in an effort to ensure the representativity of such populations. Despite these advances, a clear underrepresentation of several ethnic groups, including Latin Americans, has been evidenced in medical and cancer genomics (139, 140). In addition to providing valuable information to fill this gap, these initiatives have successfully addressed multiple challenges, including limited funding, infrastructure and human resources (141). Most Latin American consortia are focused on studying specific cancer types and are funded and run by governmental and international organizations; examples of such efforts are presented in Table 4. Among some successful examples of such efforts, we can highlight the PRECAMA project, a large multicenter case-control study aimed at advancing the prevention and management of breast cancer in premenopausal Latin American women (142). This study is coordinated by the International Agency for Research on Cancer (IARC), enrolling patients from Chile, Colombia, Costa Rica, Mexico, and Brazil, and implementing a multidisciplinary approach that combines genetics, genomics, and metabolomics with lifestyle factors. Another example is the Latin American Consortium for Lung Cancer Research (CLICaP - “Consorcio Latinoamericano para la Investigación del Cáncer de Pulmón”), an initiative launched in 2011 by a network of Latin American oncologists to improve and promote clinical and translational research in lung cancer (143, 144). For 2021, this consortium included more than 75 researchers from most Latin American countries and has had a considerable impact on access to funding, coordination of multicenter research, number and quality of publications, and development of clinical guidelines adapted to a local context. Finally, another interesting example of collaboration in the region is the Brazilian Hereditary Tumors Study Group (145). Founded in 2003, the group initially published updates on hereditary cancer in Brazil with the mission of improving teaching and research into hereditary cancer and encouraging national and international collaboration. In 2007, numerous researchers and groups from other South American countries became interested in participating, widening its outreach and becoming the Study Group on Hereditary Tumors (GETH). Several publications and active participation and interaction of GETH members reflect the importance of local efforts to promote collaborations and partnerships (146, 147).
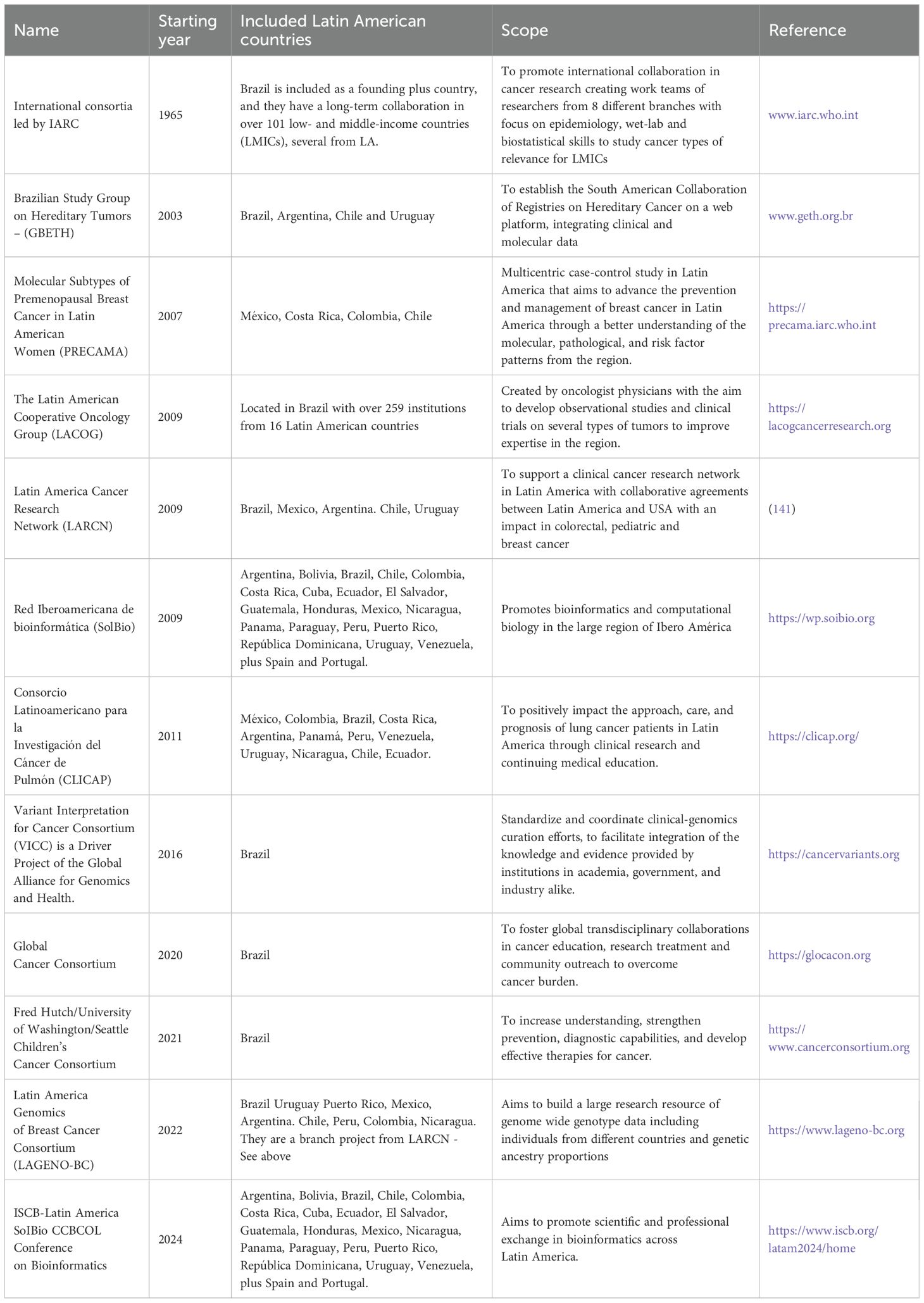
Table 4. Examples of initiatives and consortia in cancer genomics with participation of Latin American countries.
Regarding bioinformatics infrastructure initiatives, these have been essential to improve capacity building in other countries. For example, worldwide-known networks such as ELIXIR or BBMRI-ERIC offer several resources to support computational capabilities and maintain data repositories and biobank data (148, 149). They have also developed training resources for scientists/developers and created guidelines to allow interoperability between data and centers. In Latin America there are societies such as the CABANA Network, UNU BIOLAC and groups affiliated with SolBio (Iberoamerican Society for Bioinformatics) focused on accelerating the implementation of bioinformatics through training programs and research collaborations (150–152). However, in Latin America, data repositories are not interoperable between institutions, and there are no organized initiatives to share and store biological information. Latin America should walk toward an integrated and organized infrastructure to improve its role in global research (153).
3.3 Integration into the clinical practice and healthcare systems
There is a growing interest in genomic approaches in cancer research and clinical oncology. Over the past years, this interest has been translated into remarkable progress in cancer genomics worldwide, including Latin America, with Argentina, Brazil, Chile and Mexico leading the way in the region (154). The implementation of such approaches in clinical practice has been driven by numerous institutions, governmental actors, initiatives and consortia (155). While the importance of this integration is highlighted by the increasing usage of genomic biomarkers in oncology, several challenges regarding cost-benefit, clinical usefulness and precarious healthcare systems remain to be solved (42). Furthermore, these approaches have introduced new considerations such as increasing costs, the privacy of genomic data, and data sharing and harmonization, which will be increasingly important in the near future (156).
In some Latin American countries, cancer genomics profiling is currently part of the clinical practice to predict therapy response and identify relevant genetic variants (157). In Mexico, for example, the Genomic Diagnostic Laboratory, established at the National Institute of Genomic Medicine, offers comprehensive genetic testing services, analyzing genes linked to hereditary conditions and cancer predisposition syndromes (42). In Chile, a promising 25-gene NGS somatic panel called TumorSec™ has been recently developed and validated (84). This panel was designed to detect actionable mutations in tumors common in Latin America, including breast, colorectal, gastric, ovarian, pancreatic, and gallbladder cancers. This assay incorporates an automated bioinformatics analysis aiming to facilitate the implementation of precision medicine in Latin America by providing a cost-efficient alternative to multiple non-NGS assays and larger and more expensive NGS panels. In Colombia, cancer genomics research is currently centralized, with major medical centers such as the National Cancer Institute implementing NGS panels for the assessment of germline and somatic variants in cancer patients (158). Centers like these have assembled multidisciplinary teams of pathologists, geneticists and bioinformaticians to analyze genomic data, focusing on highly prevalent neoplasia such as breast, lung and colorectal cancer. The implementation of NGS technologies has unlocked new research opportunities, enabling a deeper understanding of the molecular epidemiology of these cancers and facilitating comparative studies with other populations.
Despite the advancements, the implementation of genomic cancer bioinformatics in Latin America is significantly constrained by multifaceted challenges, including pronounced geographical disparities that disadvantage rural areas, a concentration of healthcare professionals and technology in urban centers, and limited funding in the public sector. The high costs of targeted therapies and restricted availability of genomic platforms further deepen inequities in cancer treatment (159). Importantly, the region has been slower to adopt genomic technologies for routine use compared to other parts of the world. Significant challenges in funding and research infrastructure may explain this slow-paced adoption (154). In addition, navigating intricate and often inconsistent regulatory frameworks across countries can delay clinical trial approvals and implementation, hindering the region’s integration into global research efforts. The result is a complex landscape where precision oncology remains largely inaccessible to significant portions of the Latin American population, particularly in underserved and rural communities (160).
On the other hand, Latin America has not developed unified standards or guidelines to assess NGS technologies or bioinformatic procedures in healthcare and currently employs references from the US or Europe (e.g. AMP/ASCO/CAP guidelines, ESMO Scale for Clinical Actionability of molecular Targets - ESCAT). In general, most studies and laboratories have incorporated US guidelines, a trend also observed in clinical oncology in countries such as Brazil, Mexico, and Colombia (47, 161, 162). Interestingly, a growing number of studies aimed at strengthening research collaborations with European countries and organizations may change this trend (146, 163). With the growing importance of these technologies in healthcare in the region, it is worthwhile to develop and implement guidelines and protocols adapted to local settings and evidence-based, ideally based on transnational collaborations and with support of scientific societies and governmental agencies.
Other important barriers include insufficient funding for science and technology, expensive imported equipment, lack of local infrastructure, and a shortage of trained professionals. In fact, many institutions must send samples abroad for analysis, increasing costs and limiting flexibility. Some private laboratories, particularly in larger cities, offer advanced medical technologies as a service to hospitals that may not have these capabilities in-house. This arrangement allows smaller or less equipped hospitals to access cutting-edge diagnostic and treatment options without having to invest in expensive equipment themselves. Language barriers also exist, as many genomic analysis tools and educational programs are in English. To address these issues, experts recommend increasing regional collaboration, closer partnerships between hospitals and universities, improving government funding, developing local capacity, and creating resources in local languages (164). Similarly, several authors have highlighted the increasing importance of integrating genomic data into electronic health records, which is still incipient even in developed countries and requires an active effort to store, analyze, and share data relevant to oncologists and cancer researchers.
3.4 Medical education and training
NGS technologies and bioinformatics techniques have accelerated the adoption of cancer genomics in clinical oncology, allowing clinicians to deliver personalized treatments and improve diagnostics. However, one of the main challenges in this process is the knowledge translation of genomic information into clinical care by healthcare professionals. This issue is highly relevant as large amounts of new information and rapid technological advances are continuously transforming our understanding of cancer biology and its treatment. In order to face this challenge, numerous medical education training programs have highlighted the importance of the acquisition of abilities to obtain, understand, process, and use genomic information for cancer care-related decision-making, a concept termed cancer genomics literacy (43, 165). Numerous studies have explored this aspect among physicians and healthcare professionals. Ha et al., for example, analyzed 21 studies, 9 focused on cancer care, assessing three types of knowledge among the participants: awareness (general knowledge or perception), how-to (practical knowledge about the application) and principle knowledge (understanding of the theoretical principles) (43). Overall, the authors found that physicians’ knowledge about cancer genomics is limited. Interestingly, genomic literacy varied among specialty, location, years of practice, and type of genomic test, but even for oncologists, who felt more confident to communicate and interpret genomic results, an important percentage (~30%) did not feel confident with their knowledge about genomic tests. Another recent study also identified limited genomics training among physicians as an important barrier to the implementation of precision medicine in routine healthcare (166). Interestingly, this study found that 41% of physicians reported a lack of training to identify appropriate genetic tests and interpret their results. Another study in the UK, including approximately 10% of the country’s oncologists, found that 38.7% of them did not receive formal training in genomics, and 92.7% identified a need for additional genomics training (167). These studies highlight the urgent need to improve cancer genomics education among healthcare professionals.
Experts have proposed several solutions to enhance medical education and training in cancer genomics. The incorporation of genomic modules in undergraduate programs and specialized genomic training programs has shown successful results in bridging knowledge gaps in healthcare professionals and providing tools for continuous learning (168). In bioinformatics, an interesting approach focused on developing and delivering specialized workshops and courses for trainers (train-the-trainers) has been important in addressing the shortage of professionals in this area and could be applied to other emerging fields (127). Educational initiatives must also align with efforts to address infrastructure deficits. These efforts are particularly relevant in the Latin American context, where financial and healthcare resources are scarce, and the number of cancer genomics professionals is insufficient to meet the increasing demands. Furthermore, educational efforts must prioritize equity and accessibility, allowing professionals from different backgrounds to receive high-quality training and sustained mentorship (169). Finally, training programs should empower professionals to influence healthcare policies and promote regional collaborations (170). These strategies improve sustainability over time, stimulating the harmonization of standards, the implementation of good practices, and facilitating data sharing. Noteworthy, all these potential solutions require a substantial investment in education and training for clinicians at the undergraduate, graduate, and postgraduate levels, nevertheless, they constitute one of the backbones for future precision oncology.
Despite the limitations, several Latin American initiatives have been established to improve genomics education among healthcare professionals. The Latin American School of Human and Medical Genetics (ELAG) created by the Latin American Network of Human Genetics (RELAGH) in 2005, for example, has trained over 800 young researchers and professionals from 17 countries, emphasizing ethics and interdisciplinary collaboration in genomics (171). Similarly, organizations such as SOLFAGEN (Latin American Society of Pharmacogenomics and Personalized Medicine) and RELIVAF (Latin American Network for Implementation and Validation of Pharmacogenomic Clinical Guidelines) advocate for genomics and pharmacogenomics training opportunities to integrate genomic data into clinical practice (172, 173). Regarding policy advocacy, health organizations such as the Pan American Health Organization (PAHO) and the Americas Health Foundation (AHF) have emphasized the importance of creating genomic policies to improve access to precision oncology and shape public health strategies (160, 174). Additionally, leveraging existing groups and networks, such as RELAGH and national genetics and oncology societies, to strengthen educational programs and policies has shown to be useful in democratizing cancer genomics services and fostering collaborations (175). We strongly believe that education and knowledge are the basis for consolidating cancer genomics worldwide and should be prioritized in our region.
4 Future perspectives
Cancer genomics is a promising field poised to transform cancer diagnosis, prognosis and treatment through the integration of cutting-edge technologies into clinical practice. In addition to current approaches, we anticipate that novel and exciting emerging areas will have increasing importance in clinical oncology (Figure 3). Translational medicine, for example, plays a pivotal role in bridging the gap between genomic discoveries and clinical applications, accelerating the development of personalized targeted therapies. Multiomics and integrative approaches are crucial for gaining a holistic understanding of tumor biology and are increasingly valuable in tumor classification and biomarker identification. Single-cell sequencing offers unprecedented insights into intratumoral heterogeneity and tumor evolution, enabling highly tailored treatment strategies. Additionally, artificial intelligence is revolutionizing data analysis, enhancing biomarker discovery and advancing precision oncology. This section will briefly introduce and discuss these topics.
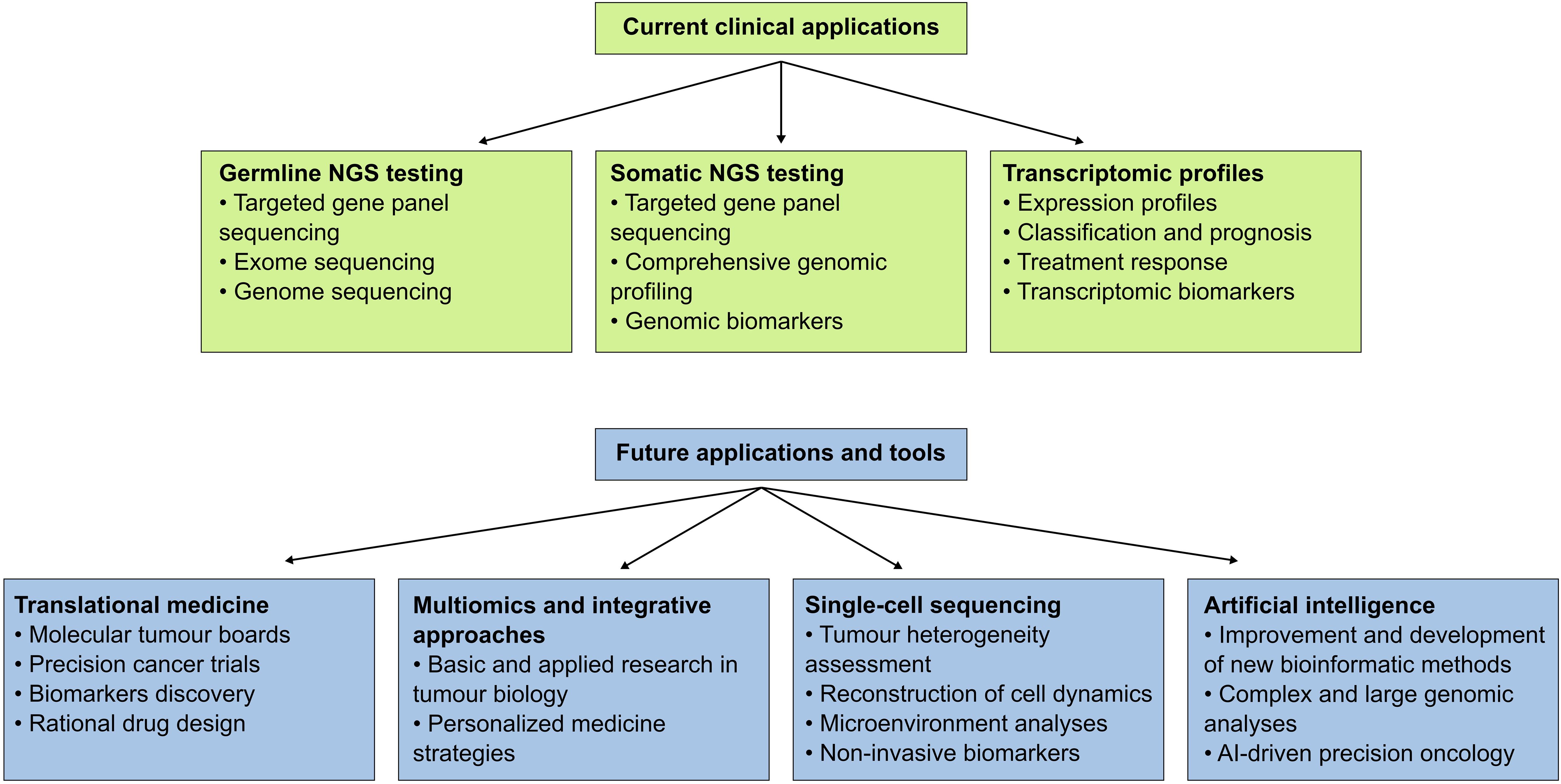
Figure 3. Current and future applications of NGS-based techniques in precision oncology and cancer research. Cancer genomics and bioinformatics are increasingly adopted in clinical settings; current applications include germline and somatic tests and transcriptomic profiles. Novel applications and tools in these areas are expected to improve cancer care and patient outcomes.
4.1 Translational medicine in cancer
The ultimate goal of translational medicine in oncology is the development and application of new treatments, technologies and insights to improve cancer care and, ultimately, patient and population outcomes (176). Molecular data, including genomic information, has been critical in this process by enabling personalized treatments, improving early detection, and advancing targeted therapies in oncology. Furthermore, cancer genomics has also accelerated drug discovery and revealed mechanisms of resistance, improving treatment effectiveness (177). Among these transformative technologies, NGS has revolutionized cancer genomics by allowing comprehensive profiling of tumors at a scale and resolution previously unattainable. In addition to the current applications previously stated, novel NGS-based techniques promise improvements in cancer prevention and treatment. Numerous studies, for example, have identified the additive effect of multiple genetic variants associated with increased risk for tumors, underscoring the potential for polygenic risk scores to improve screening and prevention strategies (178). On the other hand, non-invasive tools such as NGS in liquid biopsies may facilitate early cancer detection and monitoring, while insights into tumor heterogeneity and evolution may guide adaptive therapy strategies (179). Similarly, the development and implementation of rapid and affordable sequencing methods and bioinformatics solutions have the potential to transform cancer research and clinical care (180).
The establishment of molecular tumor boards (MTB), which bring together multidisciplinary teams to discuss and interpret genomic data for individual patients, gathering laboratory experts and clinicians, has been a remarkable effort to accelerate the implementation of NGS in clinical practice (181). These boards play a crucial role in the implementation of precision medicine by ensuring that genomic insights inform diagnosis and treatment decisions. For instance, the Moffitt Cancer Center’s experience with its Molecular Tumor Board illustrates how such collaborative frameworks can effectively translate genomic findings into actionable clinical strategies (182). In line with these observations, other studies have shown the importance of these groups in selecting appropriate antitumor agents and guide therapeutic decisions, particularly for advanced and recurrent malignancies (183, 184). The integration of genomic data into clinical workflows also prioritizes the development of robust bioinformatics tools and databases that can effectively catalog and interpret the vast array of genomic alterations found in tumors. Resources such as GENIE, ARGO, JAX-CKB, and My Cancer Genome, among many others, provide clinicians with critical information on somatic mutations and their therapeutic implications, thereby facilitating the selection of appropriate clinical trials and targeted therapies (70, 138, 185, 186).
Despite their importance, MTB are not broadly implemented in oncology care across the Latin American region. A limited number of centers have begun to include MTB in the analysis of difficult cases. For example, an MTB in the Alexander Fleming Institute in Argentina was launched in December 2019 and by 2021 they have attended 32 challenging cases of different cancer types. Remarkably, for 87.5% cases a potentially actionable alteration was identified, from this group 47% received an approved or off-label treatment recommendation (187). Another example is the development of a virtual MTB strategy by the Foundation for Clinical and Applied Cancer Research (FICMAC) in Bogota, Colombia. This effort gave oncologists from different locations in Colombia the opportunity to submit clinical and laboratory records to a group of physicians including clinical oncologist, biologist, geneticist, pathologist and clinical study coordinators. Of the 146 patients included between 2020 – 2021, 53.1% received treatment recommendations based on genomic profile analyses (188). Even though MTB recommendations are not mandatory or broadly implemented, these reports are examples of the clinical potential of multidisciplinary expert panels to improve cancer care through precision oncology and cancer genomics.
Several translational cancer studies have been conducted in the Latin American region, with Argentina, Brazil, and Mexico emerging as leading contributors (189). A study conducted in Brazil, for example, successfully established a novel cervical cancer cell line derived from Brazilian individuals (190). Researchers performed whole-exome sequencing (WES) on these cell lines and applied advanced bioinformatics tools for comprehensive analysis. Interestingly, the authors identified potential new targetable biomarkers specific to the Brazilian population. Another multinational study among different Latin American countries analyzed the prognostic value of transcriptomic analyses in a large cohort of patients with locally advanced breast cancer, finding specific expression patterns and providing novel insights into new therapeutic approaches and precision oncology (110). Several other studies focused on preclinical and clinical models, population differences and international partnerships are illustrative of the potential of this approach (189). Given the complexity and costs associated with these studies, it is important that governments and private actors prioritize in a steady and active manner such efforts to optimize the potential clinical impact.
4.2 Multiomics and integrative approaches
In recent years, multiple technological advancements have shifted the paradigm of cancer research towards multi-omics analyses (191). These technologies enable a comprehensive and unbiased integration of multiple high-dimensional datasets, including genomic, epigenomic, transcriptomic, proteomic, and metabolomic data, among others. This comprehensive and integrated approach can characterize the multilayer intersections between different data types, creating an extensive understanding of biological profiles from cancerous tissue or individual cells of tumors and patients. In cancer research and clinical settings, having orthogonal data streams provides a more robust and accurate picture of biological systems, which have been particularly useful in tumor classification and the development and assessment of novel predictive and therapeutic models (192, 193). Furthermore, conclusions obtained from multi-omics studies have provided novel and valuable insights into tumor pathophysiology and treatment resistance (191, 194). This kind of integrated information bridges the gap between genotype and phenotype, revealing how genetic and molecular perturbations translate into observable traits and clinical outcomes.
Despite the significant advances, there are multiple challenges and limitations to be addressed in this field. Data integration and harmonization, meaning combining different datasets to maximize their compatibility and comparability, remains a critical aspect of complex data analysis (195). This issue becomes critical in cancer research, where different formats, dynamic ranges and analytical or experimental errors may vary considerably between patients and omic data. In order to tackle this problem, mathematical, statistical and computational methods, such as machine learning and deep learning, have been implemented to improve the analysis of large volumes of high-dimensional datasets (194, 196). Most of these techniques are based on statistical modeling, classification and feature selection methods. Some of the most successful algorithms in overcoming the difficulties mentioned above use a method termed robust network-based penalized estimation, examples include ENET (Elastic net) and LASSO (Least Absolute Shrinkage and Selection Operator), which have been useful in identifying gene expression regulators, biomarkers and relationships between functional levels (197). Importantly, it should be highlighted that the performance of each algorithm or model depends on the biological characteristics of the samples and the specific aims of the study.
By integrating multiple molecular datasets, multiomic approaches have shown substantial promise in cancer subtyping, enabling the identification of tumor subgroups with unique biological features previously not identified (191). In these approaches, data clustering has proven particularly valuable in tumors such as lung, breast, and gastric cancer. For example, recent studies have revealed the important role of the KEAP1/NFE2L2 axis in lung cancer, dysregulation of cellular signaling pathways in gastric cancer and metabolic shifts in breast cancer subtypes (198–200). Other multiomic studies have contributed to the identification of prognostic and predictive biomarkers, such as MMP11 (Matrix metalloproteinase 11) and APOBEC, and real-time monitoring of treatment responses through liquid biopsies in patients with lung cancer (191, 201). Furthermore, the identification of dysregulated pathways, such as HER2 signaling in breast cancer and MAPK in gastric cancer, has facilitated the development of targeted therapies (202, 203). In addition to deepening our understanding of cancer initiation and development, this integrative characterization of tumors accelerates the development of innovative therapeutic interventions tailored to the complexity of individual cancers.
Extending these contributions, multi-omics approaches are now paving the way for the development of novel therapies and personalized cancer treatments. Emerging technologies like spatial multi-omics and single-cell multi-omics may strengthen these efforts by analyzing tumor heterogeneity and tumor microenvironments, identifying spatially regulated biomarkers and refining drug delivery strategies (204, 205). Furthermore, other integrative frameworks such as pharmacogenomics and epitranscriptomics are advancing personalized treatments by tailoring therapies to individual genetic and molecular profiles, predicting responses and mitigating resistance (194). As multi-omics continues evolving and improving, its capacity to unravel the intricate biological underpinnings of cancer remains promising in transforming cancer drug discovery and precision oncology, ensuring that treatments are optimized for each patient’s unique molecular profile. Remarkably, there is a considerable gap between basic research and clinical practice and most multiomic approaches are limited to research settings worldwide. Multiomic research is still incipient in most Latin American countries, nevertheless, given its increasing importance in oncology, significant growth is expected in the near future. Overcoming the challenges associated with its translation and implementation into clinical practice requires coordinated efforts to enhance education, standardize practices, and improve accessibility to multi-omics technologies, ensuring that all patients benefit from the potential of these innovative approaches.
4.3 Single-cell sequencing
Advances in single-cell sequencing (sc-seq) technologies have enabled cancer researchers to uncover cellular heterogeneity, and tumor microenvironment and dynamics in an unprecedented manner (206). In addition to characterizing the molecular state of each cell within a tumor, these techniques allow the analysis of large cell populations, making them powerful tools to identify rare cell types and dissect the molecular features of cancerous and adjacent noncancerous cells. Interesting initiatives such as the Human Tumor Atlas Network (HTAN) and the Human Cell Atlas, for instance, have aimed to use these methods to better characterize the molecular features of human cancers (207, 208). Also, recently, a growing number of studies have shown the utility of these techniques in translational oncology. Pellechia et al., for example, demonstrated the feasibility of anticancer drug response prediction at the single-cell level using computational and in vitro approaches (209). Other studies have been focused on the identification of cancer biomarkers related to patient outcomes and response to immunotherapy using sc-seq data (210, 211). Altogether, these findings suggest that sc-seq could be a promising approach in future precision oncology.
Several techniques currently enable the isolation and sequencing of single cells or nuclei (212). In addition to fresh tissues, recent protocols allow the processing of frozen and FFPE samples (213). Also, novel methods such as single-cell and spatial multiomics are increasingly available and are transforming the study of cancer biology (214). Similar to other high-throughput technologies, data analysis of sc-seq experiments is challenging and remains an important bottleneck that has motivated the development of novel computational methods (215). Standardized bioinformatics pipelines involve preprocessing of reads, quality control, gene count matrix generation and data normalization to obtain datasets compatible with downstream analyses such as clustering, cell annotation and cell-cell communication inference (216). Importantly, data collection and processing remain demanding steps that hinder a wider implementation of these methods in research and clinical settings.
In Latin America, these technologies are still incipient, and several additional barriers have limited their adoption, including poor distribution channels for equipment and reagents, lack of specialized expertise and limited number of core research facilities (217). Remarkably, several international initiatives led by research institutions such as Wellcome Connecting Science and the Human Cell Atlas have conducted hands-on laboratory and bioinformatics training courses and workshops in Latin American countries in an effort to support global scientific equity (218, 219). Likewise, NGS implementation in routine clinical settings in Latin America, we anticipate that sc-seq will be increasingly relevant in the study of patients with cancer.
4.4 Artificial intelligence
In recent years, Artificial Intelligence (AI) has become an emergent technology able to significantly disrupt many fields of research and medical practice. The main goal of AI is the development of computer systems capable of performing tasks that typically require human intelligence, such as learning, reasoning, problem-solving, and language understanding (220). In cancer genomics, the integration of AI tools is proving highly useful in managing and interpreting the vast amounts of data generated by NGS platforms and related technologies. One relevant example is the data analysis in big data cancer projects such as the TCGA (221). Since its creation, the TCGA has produced approximately 2.5 petabytes of data, enabling researchers to analyze and uncover novel patterns in disease behavior that could significantly influence cancer outcomes. Recently, numerous studies have shown the potential of AI tools in analyzing such massive amounts of information, illustrating the potential that these techniques offer (222, 223).
Given the versatility of AI-based techniques, these have been used in basic and translational cancer genomics research (221, 224). Complex and challenging tasks such as the identification of mutational signatures in cancer genomics data, integrative multiomic analyses, reconstruction of intratumoral heterogeneity using bulk sequencing, and single-cell genomic analyses are examples of successful applications of these techniques (225–228). Furthermore, common and critical bioinformatics tasks have also adopted AI techniques showing promising results. Different AI-based approaches, for example, have been used to improve variant calling in tumor samples (229, 230). On the other hand, numerous state-of-the-art bioinformatics tools used to predict variant pathogenicity, such as REVEL, spliceAI and AlphaMissense, employ AI methods such as random forest and deep neural networks (231–233). One of the main advantages of these approaches is the capacity to incorporate datasets with a large number of features or covariates to improve specific tasks such as classification, regression or clustering. Other tools have even moved further, allowing variant interpretation and prioritization, for example, VarChat and CancerVar (234, 235). It should be noted that although these tools are promising and have enormous potential, they have several limitations and therefore must be supervised by healthcare and bioinformatics professionals.
In Latin America, several research groups have begun utilizing AI in cancer genomics. The increasing accessibility of NGS platforms has facilitated the generation of tumor-related genomic data, encouraging the deployment of AI models. Recent studies by López-Cortés and other researchers from Ecuador and Spain, for example, have used numerous machine learning methods to identify cancer-driving proteins and targeted drugs using multi-omics data (236, 237). In Colombia, the national project GLORIA aims to integrate histology-genomic analysis using deep-learning models with Latin American population data (238). A team from Brazil and the Dominican Republic compared the performance of different deep learning autoencoders for cancer subtype detection using multiomics data from TCGA datasets, demonstrating their potential for predicting patient subgroups and survival profiles, and identifying differentially expressed genes (239). In Mexico, a team used single-cell RNA sequencing data to train deep neural networks for detecting myeloid malignancies in Fanconi anemia patients (240). Their models identified malignant cells with high accuracy, revealing their origin in specific blood cell precursors and signs of immune evasion. While these studies are still emerging, they highlight a growing interest in leveraging AI to address region-specific challenges in cancer genomics (241). We strongly believe that beyond the limitations of implementing AI and other cutting-edge technologies in our region, they constitute powerful tools in cancer genomics and precision oncology.
5 Conclusions
Recent years have seen novel and important insights into cancer genomics and precision oncology. This progress is largely due to advances in NGS and bioinformatics techniques and has profoundly transformed how we care for and treat patients with cancer. Currently, a number of NGS-based methods are available in the clinic. These methods include germline and somatic testing, as well as transcriptomic studies, and allow the identification of diagnostic, prognostic, and therapeutic cancer biomarkers. Despite the importance of these studies in regional settings, Latin America remains lagging in the implementation of these technologies compared to countries with advanced healthcare systems. In addition, bioinformatics analyses remain critical yet challenging. In Latin American groups several groups have conducted NGS-based studies showing the feasibility and constraints of these methods. We identified challenges that limit its wider adoption, including limited technological and human resources, difficulties in the integration of genomic information into clinical practice and healthcare systems, and insufficient education and training in cancer genomics. We highlight the importance of national and international collaborations as well as the consolidation of multidisciplinary groups, such as molecular tumor boards, to accelerate the implementation of these techniques and translation of cancer basic research into clinical practice. While these challenges are not limited to the Latin American region, several local factors exacerbate them and we noticed considerable differences in the adoption of NGS techniques among different countries. Finally, given the rapid technological advances in these areas, we highlight the importance of emerging technologies such as the growing implications of transitional medicine in cancer, multiomic approaches, sc-seq and AI-driven data analysis. Altogether, these current methods and future directions have tremendous potential to accelerate the implementation of precision oncology and ultimately improve the outcome and quality of life of patients with cancer in the Latin American region and worldwide.
Author contributions
ET-N: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. DM-G: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. JA-A: Conceptualization, Investigation, Visualization, Writing – original draft, Writing – review & editing. OO-R: Conceptualization, Funding acquisition, Investigation, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the HERMES project 61945 of the Universidad Nacional de Colombia, and Biotecgen S.A.S.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, et al. A compendium of mutational cancer driver genes. Nat Rev Cancer. (2020) 20:555–72. doi: 10.1038/s41568-020-0290-x
2. Hofmarcher T, Lindgren P, Wilking N, and Jönsson B. The cost of cancer in Europe 2018. Eur J Cancer. (2020) 129:41–9. doi: 10.1016/j.ejca.2020.01.011
3. Chen S, Cao Z, Prettner K, Kuhn M, Yang J, Jiao L, et al. Estimates and projections of the global economic cost of 29 cancers in 204 countries and territories from 2020 to 2050. JAMA Oncol. (2023) 9:465–72. doi: 10.1001/jamaoncol.2022.7826
4. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
5. Hussen BM, Abdullah ST, Salihi A, Sabir DK, Sidiq KR, Rasul MF, et al. The emerging roles of NGS in clinical oncology and personalized medicine. Pathol Res Pract. (2022) 230:153760. doi: 10.1016/j.prp.2022.153760
6. Berger MF and Mardis ER. The emerging clinical relevance of genomics in cancer medicine. Nat Rev Clin Oncol. (2018) 15:353–65. doi: 10.1038/s41571-018-0002-6
7. Phillips KA, Douglas MP, Wordsworth S, Buchanan J, and Marshall DA. Availability and funding of clinical genomic sequencing globally. BMJ Glob Health. (2021) 6:1–8. doi: 10.1136/bmjgh-2020-004415
8. Singer J, Irmisch A, Ruscheweyh H-J, Singer F, Toussaint NC, Levesque MP, et al. Bioinformatics for precision oncology. Brief Bioinform. (2019) 20:778–88. doi: 10.1093/bib/bbx143
9. Cortés-Ciriano I, Gulhan DC, Lee JJ-K, Melloni GEM, and Park PJ. Computational analysis of cancer genome sequencing data. Nat Rev Genet. (2022) 23:298–314. doi: 10.1038/s41576-021-00431-y
10. Singh DN, Daripelli S, Elamin Bushara MO, Polevoy GG, and Prasanna M. Genetic testing for successful cancer treatment. Cureus. (2023) 15:e49889. doi: 10.7759/cureus.49889
11. Daly MB, Pal T, Berry MP, Buys SS, Dickson P, Domchek SM, et al. Genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2021) 19:77–102. doi: 10.6004/jnccn.2021.0001
12. Marmolejo DH, Wong MYZ, Bajalica-Lagercrantz S, Tischkowitz M, and Balmaña J. extended ERN-GENTURIS Thematic Group 3. Overview of hereditary breast and ovarian cancer (HBOC) guidelines across Europe. Eur J Med Genet. (2021) 64:104350. doi: 10.1016/j.ejmg.2021.104350
13. Stadler ZK, Maio A, Chakravarty D, Kemel Y, Sheehan M, Salo-Mullen E, et al. Therapeutic implications of germline testing in patients with advanced cancers. J Clin Oncol. (2021) 39:2698–709. doi: 10.1200/JCO.20.03661
14. Samadder NJ, Riegert-Johnson D, Boardman L, Rhodes D, Wick M, Okuno S, et al. Comparison of universal genetic testing vs guideline-directed targeted testing for patients with hereditary cancer syndrome. JAMA Oncol. (2021) 7:230–7. doi: 10.1001/jamaoncol.2020.6252
15. Rezoug Z, Totten SP, Szlachtycz D, Atayan A, Mohler K, Albert S, et al. Universal genetic testing for newly diagnosed invasive breast cancer. JAMA Netw Open. (2024) 7:e2431427. doi: 10.1001/jamanetworkopen.2024.31427
16. Hall MJ, Forman AD, Pilarski R, Wiesner G, and Giri VN. Gene panel testing for inherited cancer risk. J Natl Compr Canc Netw. (2014) 12:1339–46. doi: 10.6004/jnccn.2014.0128
17. Stoffel EM and Carethers JM. Current approaches to germline cancer genetic testing. Annu Rev Med. (2020) 71:85–102. doi: 10.1146/annurev-med-052318-101009
18. Hu T, Chitnis N, Monos D, and Dinh A. Next-generation sequencing technologies: An overview. Hum Immunol. (2021) 82:801–11. doi: 10.1016/j.humimm.2021.02.012
19. Koboldt DC. Best practices for variant calling in clinical sequencing. Genome Med. (2020) 12:91. doi: 10.1186/s13073-020-00791-w
20. Poplin R, Chang P-C, Alexander D, Schwartz S, Colthurst T, Ku A, et al. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol. (2018) 36:983–7. doi: 10.1038/nbt.4235
21. Moreno-Cabrera JM, Del Valle J, Castellanos E, Feliubadaló L, Pineda M, Brunet J, et al. Evaluation of CNV detection tools for NGS panel data in genetic diagnostics. Eur J Hum Genet. (2020) 28:1645–55. doi: 10.1038/s41431-020-0675-z
22. Yao R, Yu T, Qing Y, Wang J, and Shen Y. Evaluation of copy number variant detection from panel-based next-generation sequencing data. Mol Genet Genomic Med. (2019) 7:e00513. doi: 10.1002/mgg3.513
23. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
24. Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. (2020) 22:245–57. doi: 10.1038/s41436-019-0686-8
25. Pejaver V, Byrne AB, Feng B-J, Pagel KA, Mooney SD, Karchin R, et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. (2022) 109:2163–77. doi: 10.1016/j.ajhg.2022.10.013
26. Houge G, Bratland E, Aukrust I, Tveten K, Žukauskaitė G, Sansovic I, et al. Comparison of the ABC and ACMG systems for variant classification. Eur J Hum Genet. (2024) 32:858–63. doi: 10.1038/s41431-024-01617-8
27. Horgan D, Curigliano G, Rieß O, Hofman P, Büttner R, Conte P, et al. Identifying the steps required to effectively implement next-generation sequencing in oncology at a national level in europe. J Pers Med. (2022) 12:1–27. doi: 10.3390/jpm12010072
28. Naito Y, Aburatani H, Amano T, Baba E, Furukawa T, Hayashida T, et al. Clinical practice guidance for next-generation sequencing in cancer diagnosis and treatment (edition 2.1). Int J Clin Oncol. (2021) 26:233–83. doi: 10.1007/s10147-020-01831-6
29. Ossa Gomez CA, Achatz MI, Hurtado M, Sanabria-Salas MC, Sullcahuaman Y, Chávarri-Guerra Y, et al. Germline pathogenic variant prevalence among latin american and US hispanic individuals undergoing testing for hereditary breast and ovarian cancer: A cross-sectional study. JCO Glob Oncol. (2022) 8:1–12. doi: 10.1200/GO.22.00104
30. Oliver J, Quezada Urban R, Franco Cortés CA, Díaz Velásquez CE, Montealegre Paez AL, Pacheco-Orozco RA, et al. Latin american study of hereditary breast and ovarian cancer LACAM: A genomic epidemiology approach. Front Oncol. (2019) 9:1429. doi: 10.3389/fonc.2019.01429
31. Solano AR, Cardoso FC, Romano V, Perazzo F, Bas C, Recondo G, et al. Spectrum of BRCA1/2 variants in 940 patients from Argentina including novel, deleterious and recurrent germline mutations: impact on healthcare and clinical practice. Oncotarget. (2017) 8:60487–95. doi: 10.18632/oncotarget.10814
32. de Souza Timoteo AR, Gonçalves AÉMM, Sales LAP, Albuquerque BM, de Souza JES, de Moura PCP, et al. A portrait of germline mutation in Brazilian at-risk for hereditary breast cancer. Breast Cancer Res Treat. (2018) 172:637–46. doi: 10.1007/s10549-018-4938-0
33. Torrezan GT, de Almeida FGDSR, Figueiredo MCP, Barros BD de F, de Paula CAA, Valieris R, et al. Complex landscape of germline variants in Brazilian patients with hereditary and early onset breast cancer. Front Genet. (2018) 9:161. doi: 10.3389/fgene.2018.00161
34. Cock-Rada AM, Ossa CA, Garcia HI, and Gomez LR. A multi-gene panel study in hereditary breast and ovarian cancer in Colombia. Fam Cancer. (2018) 17:23–30. doi: 10.1007/s10689-017-0004-z
35. Quezada Urban R, Díaz Velásquez CE, Gitler R, Rojo Castillo MP, Sirota Toporek M, Figueroa Morales A, et al. Comprehensive analysis of germline variants in mexican patients with hereditary breast and ovarian cancer susceptibility. Cancers (Basel). (2018) 10:1–19. doi: 10.3390/cancers10100361
36. Rosa RCA, Santis JO, Teixeira LA, Molfetta GA, Dos Santos JTT, Ribeiro VDS, et al. Lynch syndrome identification in a Brazilian cohort of endometrial cancer screened by a universal approach. Gynecol Oncol. (2020) 159:229–38. doi: 10.1016/j.ygyno.2020.07.013
37. de Oliveira JM, Zurro NB, Coelho AVC, Caraciolo MP, de Alexandre RB, Cervato MC, et al. The genetics of hereditary cancer risk syndromes in Brazil: a comprehensive analysis of 1682 patients. Eur J Hum Genet. (2022) 30:818–23. doi: 10.1038/s41431-022-01098-7
38. Padua-Bracho A, Velázquez-Aragón JA, Fragoso-Ontiveros V, Nuñez-Martínez PM, Mejía Aguayo M de la L, Sánchez-Contreras Y, et al. A previously unrecognized molecular landscape of lynch syndrome in the mexican population. Int J Mol Sci. (2022) 23:1–19. doi: 10.3390/ijms231911549
39. Pérez-Ibave DC, Garza-Rodríguez ML, Noriega-Iriondo MF, Flores-Moreno SM, González-Geroniz MI, Espinoza-Velazco A, et al. Identification of germline variants in patients with hereditary cancer syndromes in northeast Mexico. Genes (Basel). (2023) 14:1–11. doi: 10.3390/genes14020341
40. Sierra-Díaz DC, Morel A, Fonseca-Mendoza DJ, Bravo NC, Molano-Gonzalez N, Borras M, et al. Germline mutations of breast cancer susceptibility genes through expanded genetic analysis in unselected Colombian patients. Hum Genomics. (2024) 18:68. doi: 10.1186/s40246-024-00623-7
41. Borda V, Loesch DP, Guo B, Laboulaye R, Veliz-Otani D, French JN, et al. Genetics of Latin American Diversity Project: Insights into population genetics and association studies in admixed groups in the Americas. Cell Genomics. (2024) 4:100692. doi: 10.1016/j.xgen.2024.100692
42. Alvarez-Gomez RM, de la Fuente-Hernandez MA, Herrera-Montalvo L, and Hidalgo-Miranda A. Challenges of diagnostic genomics in Latin America. Curr Opin Genet Dev. (2021) 66:101–9. doi: 10.1016/j.gde.2020.12.010
43. Ha VTD, Frizzo-Barker J, and Chow-White P. Adopting clinical genomics: a systematic review of genomic literacy among physicians in cancer care. BMC Med Genomics. (2018) 11:18. doi: 10.1186/s12920-018-0337-y
44. Strasser-Weippl K, Chavarri-Guerra Y, Villarreal-Garza C, Bychkovsky BL, Debiasi M, Liedke PER, et al. Progress and remaining challenges for cancer control in Latin America and the Caribbean. Lancet Oncol. (2015) 16:1405–38. doi: 10.1016/S1470-2045(15)00218-1
45. Acevedo F, Walbaum B, Camus M, Manzor M, Muñiz S, Medina L, et al. Access disparities and underutilization of germline genetic testing in Chilean breast cancer patients. Breast Cancer Res Treat. (2023) 199:363–70. doi: 10.1007/s10549-023-06909-z
46. Chávarri-Guerra Y, Rodríguez-Olivares JL, Ramírez-González A, Moreno-Mirón JM, Lagunas-Medina A, Peñafort-Zamora JC, et al. Addressing the need for genetic cancer risk assessment in Mexico: From establishment of a formal program to delivery innovation and expansion. Genet Med Open. (2024) 2:101874. doi: 10.1016/j.gimo.2024.101874
47. Mantilla WA, Sanabria-Salas MC, Baldion AM, Sua LF, Gonzalez DM, and Lema M. NGS in lung, breast, and unknown primary cancer in Colombia: A multidisciplinary consensus on challenges and opportunities. JCO Glob Oncol. (2021) 7:1012–23. doi: 10.1200/GO.21.00046
48. Marcelain K, Selman-Bravo C, García-Bloj B, Bustamante E, Fernández J, Gaete F, et al. Avances y desafíos locales en el diagnóstico molecular de tumores sólidos: una mirada sanitaria hacia la oncología de precisión en Chile. Rev Med Chil. (2023) 151:1344–60. doi: 10.4067/s0034-98872023001001344
49. Vergara - Dagobeth E, Suárez-Causado A, and Gómez-Arias RD. Plan Control del cáncer en Colombia 2012-2021. Un análisis formal. Gerencia y Políticas Salud. (2017) 16:6–18. doi: 10.11144/Javeriana.rgps16-33.pccc
50. Mosele F, Remon J, Mateo J, Westphalen CB, Barlesi F, Lolkema MP, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO Precision Medicine Working Group. Ann Oncol. (2020) 31:1491–505. doi: 10.1016/j.annonc.2020.07.014
51. Gibbs SN, Peneva D, Cuyun Carter G, Palomares MR, Thakkar S, Hall DW, et al. Comprehensive review on the clinical impact of next-generation sequencing tests for the management of advanced cancer. JCO Precis Oncol. (2023) 7:e2200715. doi: 10.1200/PO.22.00715
52. Heilmann AM, Riess JW, McLaughlin-Drubin M, Huang RSP, Hjulstrom M, Creeden J, et al. Insights of clinical significance from 109 695 solid tumor tissue-based comprehensive genomic profiles. Oncologist. (2024) 29:e224–36. doi: 10.1093/oncolo/oyad251
53. Hicks JK, Howard R, Reisman P, Adashek JJ, Fields KK, Gray JE, et al. Integrating somatic and germline next-generation sequencing into routine clinical oncology practice. JCO Precis Oncol. (2021) 5:884–95. doi: 10.1200/PO.20.00513
54. Field MA. Bioinformatic challenges detecting genetic variation in precision medicine programs. Front Med (Lausanne). (2022) 9:806696. doi: 10.3389/fmed.2022.806696
55. Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the association for molecular pathology, american society of clinical oncology, and college of american pathologists. J Mol Diagn. (2017) 19:4–23. doi: 10.1016/j.jmoldx.2016.10.002
56. Petrackova A, Vasinek M, Sedlarikova L, Dyskova T, Schneiderova P, Novosad T, et al. Standardization of sequencing coverage depth in NGS: recommendation for detection of clonal and subclonal mutations in cancer diagnostics. Front Oncol. (2019) 9:851. doi: 10.3389/fonc.2019.00851
57. Zverinova S and Guryev V. Variant calling: Considerations, practices, and developments. Hum Mutat. (2022) 43:976–85. doi: 10.1002/humu.24311
58. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. (2013) 31:213–9. doi: 10.1038/nbt.2514
59. Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, and Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. (2012) 28:1811–7. doi: 10.1093/bioinformatics/bts271
60. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. (2012) 22:568–76. doi: 10.1101/gr.129684.111
61. Garcia-Prieto CA, Martínez-Jiménez F, Valencia A, and Porta-Pardo E. Detection of oncogenic and clinically actionable mutations in cancer genomes critically depends on variant calling tools. Bioinformatics. (2022) 38:3181–91. doi: 10.1093/bioinformatics/btac306
62. Little P, Jo H, Hoyle A, Mazul A, Zhao X, Salazar AH, et al. UNMASC: tumor-only variant calling with unmatched normal controls. NAR Cancer. (2021) 3:zcab040. doi: 10.1093/narcan/zcab040
63. Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, and Forbes SA. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer. (2018) 18:696–705. doi: 10.1038/s41568-018-0060-1
64. Wagner AH, Walsh B, Mayfield G, Tamborero D, Sonkin D, Krysiak K, et al. A harmonized meta-knowledgebase of clinical interpretations of somatic genomic variants in cancer. Nat Genet. (2020) 52:448–57. doi: 10.1038/s41588-020-0603-8
65. Horak P, Griffith M, Danos AM, Pitel BA, Madhavan S, Liu X, et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genet Med. (2022) 24:986–98. doi: 10.1016/j.gim.2022.01.001
66. Tamborero D, Rubio-Perez C, Deu-Pons J, Schroeder MP, Vivancos A, Rovira A, et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. (2018) 10:25. doi: 10.1186/s13073-018-0531-8
67. Griffith M, Spies NC, Krysiak K, McMichael JF, Coffman AC, Danos AM, et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet. (2017) 49:170–4. doi: 10.1038/ng.3774
68. Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: A precision oncology knowledge base. JCO Precis Oncol. (2017) 2017:1–16. doi: 10.1200/PO.17.00011
69. Yu Y, Wang Y, Xia Z, Zhang X, Jin K, Yang J, et al. PreMedKB: an integrated precision medicine knowledgebase for interpreting relationships between diseases, genes, variants and drugs. Nucleic Acids Res. (2019) 47:D1090–101. doi: 10.1093/nar/gky1042
70. Patterson SE, Liu R, Statz CM, Durkin D, Lakshminarayana A, and Mockus SM. The clinical trial landscape in oncology and connectivity of somatic mutational profiles to targeted therapies. Hum Genomics. (2016) 10:4. doi: 10.1186/s40246-016-0061-7
71. Torres Á, Oliver J, Frecha C, Montealegre AL, Quezada-Urbán R, Díaz-Velásquez CE, et al. Cancer genomic resources and present needs in the latin american region. Public Health Genomics. (2017) 20:194–201. doi: 10.1159/000479291
72. Chamorro DF, Cardona AF, Rodríguez J, Ruiz-Patiño A, Arrieta O, Moreno-Pérez DA, et al. Genomic landscape of primary resistance to osimertinib among hispanic patients with EGFR-mutant non-small cell lung cancer (NSCLC): results of an observational longitudinal cohort study. Target Oncol. (2023) 18:425–40. doi: 10.1007/s11523-023-00955-9
73. Tapia-Valladares C, Valenzuela G, González E, Maureira I, Toro J, Freire M, et al. Distinct driver pathway enrichments and a high prevalence of TSC2 mutations in right colon cancer in Chile: A preliminary comparative analysis. Int J Mol Sci. (2024) 25:1–18. doi: 10.3390/ijms25094695
74. Piñeros M, Laversanne M, Barrios E, Cancela M de C, de Vries E, Pardo C, et al. An updated profile of the cancer burden, patterns and trends in Latin America and the Caribbean. Lancet regional Health Americas. (2022) 13:1–14. doi: 10.1016/j.lana.2022.100294
75. Lou H, Villagran G, Boland JF, Im KM, Polo S, Zhou W, et al. Genome analysis of latin american cervical cancer: frequent activation of the PIK3CA pathway. Clin Cancer Res. (2015) 21:5360–70. doi: 10.1158/1078-0432.CCR-14-1837
76. Olivier M, Bouaoun L, Villar S, Robitaille A, Cahais V, Heguy A, et al. Molecular features of premenopausal breast cancers in Latin American women: Pilot results from the PRECAMA study. PloS One. (2019) 14:e0210372. doi: 10.1371/journal.pone.0210372
77. Mascarenhas E, Gelatti AC, Araújo LH, Baldotto C, Mathias C, Zukin M, et al. Comprehensive genomic profiling of Brazilian non-small cell lung cancer patients (GBOT 0118/LACOG0418). Thorac Cancer. (2021) 12:580–7. doi: 10.1111/1759-7714.13777
78. Sanchez P, Espinosa M, Maldonado V, Barquera R, Belem-Gabiño N, Torres J, et al. Pancreatic ductal adenocarcinomas from Mexican patients present a distinct genomic mutational pattern. Mol Biol Rep. (2020) 47:5175–84. doi: 10.1007/s11033-020-05592-3
79. Cifuentes C, Lombana M, Vargas H, Laguado P, Ruiz-Patiño A, Rojas L, et al. Application of comprehensive genomic profiling-based next-generation sequencing assay to improve cancer care in a developing country. Cancer Control. (2023) 30:10732748231175256. doi: 10.1177/10732748231175256
80. Garcia FA de O, Evangelista AF, Mançano BM, Moreno DA, Berardinelli GN, de Paula FE, et al. Genomic profile of two Brazilian choroid plexus tumors by whole-exome sequencing. Cold Spring Harb Mol Case Stud. (2023) 9:1–12. doi: 10.1101/mcs.a006245
81. Angel M, Freile B, Rodriguez A, Cayol F, Manneh Kopp R, Rioja P, et al. Genomic landscape in prostate cancer in a latin american population. JCO Glob Oncol. (2024) 10:e2400072. doi: 10.1200/GO.24.00072
82. Candelaria M, Cerrato-Izaguirre D, Gutierrez O, Diaz-Chavez J, Aviles A, Dueñas-Gonzalez A, et al. Characterizing the mutational landscape of diffuse large B-cell lymphoma in a prospective cohort of mexican patients. Int J Mol Sci. (2024) 25:1–13. doi: 10.3390/ijms25179328
83. López Rivera JJ, Rueda-Gaitán P, Rios Pinto LC, Rodríguez Gutiérrez DA, Gomez-Lopera N, Lamilla J, et al. Advancing cancer care in Colombia: results of the first in situ implementation of comprehensive genomic profiling. J Pers Med. (2024) 14:1–17. doi: 10.3390/jpm14090975
84. Salvo M, González-Feliú E, Toro J, Gallegos I, Maureira I, Miranda-González N, et al. Validation of an NGS panel designed for detection of actionable mutations in tumors common in latin america. J Pers Med. (2021) 11:1–17. doi: 10.3390/jpm11090899
85. Urbina-Jara LK, Rojas-Martinez A, Martinez-Ledesma E, Aguilar D, Villarreal-Garza C, and Ortiz-Lopez R. Landscape of germline mutations in DNA repair genes for breast cancer in latin america: opportunities for PARP-like inhibitors and immunotherapy. Genes (Basel). (2019) 10:2–24. doi: 10.3390/genes10100786
86. Payne K, Gavan SP, Wright SJ, and Thompson AJ. Cost-effectiveness analyses of genetic and genomic diagnostic tests. Nat Rev Genet. (2018) 19:235–46. doi: 10.1038/nrg.2017.108
87. Christofyllakis K, Bittenbring JT, Thurner L, Ahlgrimm M, Stilgenbauer S, Bewarder M, et al. Cost-effectiveness of precision cancer medicine-current challenges in the use of next generation sequencing for comprehensive tumour genomic profiling and the role of clinical utility frameworks (Review). Mol Clin Oncol. (2022) 16:21. doi: 10.3892/mco.2021.2453
88. Steiert TA, Parra G, Gut M, Arnold N, Trotta J-R, Tonda R, et al. A critical spotlight on the paradigms of FFPE-DNA sequencing. Nucleic Acids Res. (2023) 51:7143–62. doi: 10.1093/nar/gkad519
89. Gomez HL, Bargallo-Rocha JE, Billinghurst RJ, Núñez De Pierro AR, Coló FA, Gil LLB, et al. Practice-changing use of the 21-gene test for the management of patients with early-stage breast cancer in latin america. JCO Glob Oncol. (2021) 7:1364–73. doi: 10.1200/GO.21.00008
90. Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, et al. Human genomics. The human transcriptome across tissues and individuals. Science. (2015) 348:660–5. doi: 10.1126/science.aaa0355
91. Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science. (2017) 357:1–11. doi: 10.1126/science.aan2507
92. Pleasance E, Bohm A, Williamson LM, Nelson JMT, Shen Y, Bonakdar M, et al. Whole-genome and transcriptome analysis enhances precision cancer treatment options. Ann Oncol. (2022) 33:939–49. doi: 10.1016/j.annonc.2022.05.522
93. Tong Y-Q, Zhao Z-J, Liu B, Bao A-Y, Zheng H-Y, Gu J, et al. New rapid method to detect BCR-ABL fusion genes with multiplex RT-qPCR in one-tube at a time. Leuk Res. (2018) 69:47–53. doi: 10.1016/j.leukres.2018.04.001
94. Narrandes S and Xu W. Gene expression detection assay for cancer clinical use. J Cancer. (2018) 9:2249–65. doi: 10.7150/jca.24744
95. Cardoso F, van’t Veer LJ, Bogaerts J, Slaets L, Viale G, Delaloge S, et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. New Engl J Med. (2016) 375:717–29. doi: 10.1056/NEJMOA1602253
96. Sparano JA, Gray RJ, Makower DF, Pritchard KI, Albain KS, Hayes DF, et al. Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. N Engl J Med. (2018) 379:111–21. doi: 10.1056/NEJMoa1804710
97. Dubsky P, Filipits M, Jakesz R, Rudas M, Singer CF, Greil R, et al. EndoPredict improves the prognostic classification derived from common clinical guidelines in ER-positive, HER2-negative early breast cancer. Ann Oncol. (2013) 24:640–7. doi: 10.1093/annonc/mds334
98. Jahn SW, Bösl A, Tsybrovskyy O, Gruber-Rossipal C, Helfgott R, Fitzal F, et al. Clinically high-risk breast cancer displays markedly discordant molecular risk predictions between the MammaPrint and EndoPredict tests. Br J Cancer. (2020) 122:1744–6. doi: 10.1038/s41416-020-0838-2
99. Tsimberidou AM, Fountzilas E, Bleris L, and Kurzrock R. Transcriptomics and solid tumors: The next frontier in precision cancer medicine. Semin Cancer Biol. (2022) 84:50–9. doi: 10.1016/j.semcancer.2020.09.007
100. Lin VTG and Yang ES. The pros and cons of incorporating transcriptomics in the age of precision oncology. J Natl Cancer Inst. (2019) 111:1016–22. doi: 10.1093/JNCI/DJZ114
101. Reuther J, Roy A, and Monzon FA. Transcriptome sequencing (RNA-seq). Genomic Appl Pathology: Second Edition 2019. (2019), 33–49. doi: 10.1007/978-3-319-96830-8_4
102. Dickson BC and Swanson D. Targeted RNA sequencing: A routine ancillary technique in the diagnosis of bone and soft tissue neoplasms. Genes Chromosomes Cancer. (2019) 58:75–87. doi: 10.1002/gcc.22690
103. Menyhárt O and Győrffy B. Multi-omics approaches in cancer research with applications in tumor subtyping, prognosis, and diagnosis. Comput Struct Biotechnol J. (2021) 19:949–60. doi: 10.1016/j.csbj.2021.01.009
104. Hrdlickova R, Toloue M, and Tian B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip Rev RNA. (2017) 8:1–17. doi: 10.1002/wrna.1364
105. Hong M, Tao S, Zhang L, Diao L-T, Huang X, Huang S, et al. RNA sequencing: new technologies and applications in cancer research. J Hematol Oncol. (2020) 13:166. doi: 10.1186/s13045-020-01005-x
106. Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, et al. A survey of best practices for RNA-seq data analysis. Genome Biol. (2016) 17:13. doi: 10.1186/s13059-016-0881-8
107. Hardin EC, Schmid S, Sommerkamp A, Bodden C, Heipertz A-E, Sievers P, et al. LOGGIC Core BioClinical Data Bank: Added clinical value of RNA-Seq in an international molecular diagnostic registry for pediatric low-grade glioma patients. Neuro Oncol. (2023) 25:2087–97. doi: 10.1093/neuonc/noad078
108. Brown LM, Lonsdale A, Zhu A, Davidson NM, Schmidt B, Hawkins A, et al. The application of RNA sequencing for the diagnosis and genomic classification of pediatric acute lymphoblastic leukemia. Blood Adv. (2020) 4:930–42. doi: 10.1182/bloodadvances.2019001008
109. Brueffer C, Vallon-Christersson J, Grabau D, Ehinger A, Häkkinen J, Hegardt C, et al. Clinical value of RNA sequencing-based classifiers for prediction of the five conventional breast cancer biomarkers: A report from the population-based multicenter Sweden cancerome analysis network-breast initiative. JCO Precis Oncol. (2018) 2:1–18. doi: 10.1200/PO.17.00135
110. Llera AS, Abdelhay ESFW, Artagaveytia N, Daneri-Navarro A, Müller B, Velazquez C, et al. The transcriptomic portrait of locally advanced breast cancer and its prognostic value in a multi-country cohort of latin american patients. Front Oncol. (2022) 12:835626. doi: 10.3389/fonc.2022.835626
111. Romero-Cordoba SL, Salido-Guadarrama I, Rebollar-Vega R, Bautista-Piña V, Dominguez-Reyes C, Tenorio-Torres A, et al. Comprehensive omic characterization of breast cancer in Mexican-Hispanic women. Nat Commun. (2021) 12:2245. doi: 10.1038/s41467-021-22478-5
112. Alves da Quinta D, Rocha D, Retamales J, Giunta D, Artagaveytia N, Velazquez C, et al. Closing the gap: prognostic and predictive biomarker validation for personalized care in a Latin American hormone-dependent breast cancer cohort. Oncologist. (2024) 29:e1701–13. doi: 10.1093/oncolo/oyae191
113. Ortega-Gómez A, Rangel-Escareño C, Molina-Romero C, Macedo-Pérez EO, Avilés-Salas A, Lara-García A, et al. Gene-expression profiles in lung adenocarcinomas related to chronic wood smoke or tobacco exposure. Respir Res. (2016) 17:42. doi: 10.1186/s12931-016-0346-3
114. Barcenas-Lopez DA, Nuñez-Enriquez JC, Gomez-Romero LL, Tovar-Romero HA, Jimenez-Hernandez E, Medina-Sanson A, et al. Whole transcriptome analysis in mexican children with high-risk acute lymphoblastic leukemia: A preliminary report from the migiccl. Blood. (2022) 140:6365–6. doi: 10.1182/blood-2022-168167
115. Justo-Garrido M, López-Saavedra A, Alcaraz N, Cortés-González CC, Oñate-Ocaña LF, Caro-Sánchez CHS, et al. Association of SLC12A1 and GLUR4 ion transporters with neoadjuvant chemoresistance in luminal locally advanced breast cancer. Int J Mol Sci. (2023) 24:1–16. doi: 10.3390/ijms242216104
116. Cruz-Miranda GM, Olarte-Carrillo I, Bárcenas-López DA, Martínez-Tovar A, Ramírez-Bello J, Ramos-Peñafiel CO, et al. Transcriptome analysis in mexican adults with acute lymphoblastic leukemia. Int J Mol Sci. (2024) 25:1–15. doi: 10.3390/ijms25031750
117. Beltran B, Torres M, Villela L, Idrobo H, von Glasenapp A, Valvert F, et al. Epidemiology of lymphomas in latin america. JCO Glob Oncol. (2024) 10:64–5. doi: 10.1200/GO-24-57000
118. Mulder NJ, Adebiyi E, Adebiyi M, Adeyemi S, Ahmed A, Ahmed R, et al. Development of bioinformatics infrastructure for genomics research. Glob Heart. (2017) 12:91–8. doi: 10.1016/j.gheart.2017.01.005
119. Behl T, Kaur I, Sehgal A, Singh S, Bhatia S, Al-Harrasi A, et al. Bioinformatics accelerates the major tetrad: A real boost for the pharmaceutical industry. Int J Mol Sci. (2021) 22:1–28. doi: 10.3390/ijms22126184
120. Langmead B and Nellore A. Cloud computing for genomic data analysis and collaboration. Nat Rev Genet. (2018) 19:208–19. doi: 10.1038/nrg.2017.113
121. Behera S, Catreux S, Rossi M, Truong S, Huang Z, Ruehle M, et al. Comprehensive genome analysis and variant detection at scale using DRAGEN. Nat Biotechnol. (2024). doi: 10.1038/s41587-024-02382-1
122. Betschart RO, Thiéry A, Aguilera-Garcia D, Zoche M, Moch H, Twerenbold R, et al. Comparison of calling pipelines for whole genome sequencing: an empirical study demonstrating the importance of mapping and alignment. Sci Rep. (2022) 12:21502. doi: 10.1038/s41598-022-26181-3
123. Zhang Z, Hernandez K, Savage J, Li S, Miller D, Agrawal S, et al. Uniform genomic data analysis in the NCI Genomic Data Commons. Nat Commun. (2021) 12:1226. doi: 10.1038/s41467-021-21254-9
124. Rambla J, Baudis M, Ariosa R, Beck T, Fromont LA, Navarro A, et al. Beacon v2 and Beacon networks: A “lingua franca” for federated data discovery in biomedical genomics, and beyond. Hum Mutat. (2022) 43:791–9. doi: 10.1002/humu.24369
125. ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature. (2020) 578:82–93. doi: 10.1038/s41586-020-1969-6
126. Işık EB, Brazas MD, Schwartz R, Gaeta B, Palagi PM, van Gelder CWG, et al. Grand challenges in bioinformatics education and training. Nat Biotechnol. (2023) 41:1171–4. doi: 10.1038/s41587-023-01891-9
127. Attwood TK, Blackford S, Brazas MD, Davies A, and Schneider MV. A global perspective on evolving bioinformatics and data science training needs. Brief Bioinform. (2019) 20:398–404. doi: 10.1093/bib/bbx100
128. Ras V, Carvajal-López P, Gopalasingam P, Matimba A, Chauke PA, Mulder N, et al. Challenges and considerations for delivering bioinformatics training in LMICs: perspectives from pan-african and latin american bioinformatics networks. Front Educ (Lausanne). (2021) 6:710971. doi: 10.3389/feduc.2021.710971
129. Armenta-Medina D, Díaz de León-Castañeda C, and Valderrama-Blanco B. Bioinformatics in Mexico: A diagnostic from the academic perspective and recommendations for a public policy. PloS One. (2020) 15:e0243531. doi: 10.1371/journal.pone.0243531
130. Restrepo S, Pinzón A, Rodríguez-R LM, Sierra R, Grajales A, Bernal A, et al. Computational biology in Colombia. PloS Comput Biol. (2009) 5:e1000535. doi: 10.1371/journal.pcbi.1000535
131. Rocha M, Massarani L, Souza SJde, and Vasconcelos ATRde. The past, present and future of genomics and bioinformatics: A survey of Brazilian scientists. Genet Mol Biol. (2022) 45:e20210354. doi: 10.1590/1678-4685-GMB-2021-0354
132. Nicol D, Nielsen J, and Archer M. Data access arrangements in genomic research consortia. Sci Rep. (2024) 14:21685. doi: 10.1038/s41598-024-72653-z
133. International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. (2004) 431:931–45. doi: 10.1038/nature03001
134. Turnbull C, Scott RH, Thomas E, Jones L, Murugaesu N, Pretty FB, et al. The 100 000 Genomes Project: bringing whole genome sequencing to the NHS. BMJ. (2018) 361:k1687. doi: 10.1136/bmj.k1687
135. International Cancer Genome Consortium, Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, et al. International network of cancer genome projects. Nature. (2010) 464:993–8. doi: 10.1038/nature08987
136. Zhang J, Bajari R, Andric D, Gerthoffert F, Lepsa A, Nahal-Bose H, et al. The international cancer genome consortium data portal. Nat Biotechnol. (2019) 37:367–9. doi: 10.1038/s41587-019-0055-9
137. AACR Project GENIE Consortium. AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. (2017) 7:818–31. doi: 10.1158/2159-8290.CD-17-0151
138. Pugh TJ, Bell JL, Bruce JP, Doherty GJ, Galvin M, Green MF, et al. AACR project GENIE: 100,000 cases and beyond. Cancer Discov. (2022) 12:2044–57. doi: 10.1158/2159-8290.CD-21-1547
139. García-Cárdenas JM, Indacochea A, Pesantez-Coronel D, Terán-Navas M, Aleaga A, Armendáriz-Castillo I, et al. Toward equitable precision oncology: monitoring racial and ethnic inclusion in genomics and clinical trials. JCO Precis Oncol. (2024) 8:e2300398. doi: 10.1200/PO.23.00398
140. Corpas M, Pius M, Poburennaya M, Guio H, Dwek M, Nagaraj S, et al. Bridging genomics’ greatest challenge: The diversity gap. Cell Genomics. (2025) 5:100724. doi: 10.1016/j.xgen.2024.100724
141. Investigators of the US–Latin America Cancer Research Network. Translational cancer research comes of age in Latin America. Sci Transl Med. (2015) 7:319fs50. doi: 10.1126/scitranslmed.aad5859
142. Romieu I, Biessy C, Torres-Mejía G, Ángeles-Llerenas A, Sánchez GI, Borrero M, et al. Project profile: a multicenter study on breast cancer in young women in Latin America (PRECAMA study). Salud Publica Mex. (2019) 61:601. doi: 10.21149/10466
143. Arrieta O, Zatarain-Barrón ZL, Cardona AF, Corrales L, Martin C, and Cuello M. Uniting latin america through research: how regional research can strengthen local policies, networking, and outcomes for patients with lung cancer. Am Soc Clin Oncol Educ Book. (2022) 42:1–7. doi: 10.1200/EDBK_349951
144. Arrieta O, Zuluaga J, Cardona AF, Rojas L, Ordóñez-Reyes C, Wagner-Gutiérrez N, et al. CLICaP Guideline for the diagnosis, treatment, and follow-up of locally advanced non-small cell lung cancer. Medicina (B Aires). (2024) 46:596–625. doi: 10.56050/01205498.2386
145. Rossi BM, Sarroca C, Vaccaro C, Lopez F, Ashton-Prolla P, Ferreira F de O, et al. The development of the study of hereditary cancer in south america. Genet Mol Biol. (2016) 39:166–7. doi: 10.1590/1678-4685-GMB-2014-0366
146. Della Valle A, Rossi BM, Palmero EI, Antelo M, Vaccaro CA, López-Kostner F, et al. A snapshot of current genetic testing practice in Lynch syndrome: The results of a representative survey of 33 Latin American existing centres/registries. Eur J Cancer. (2019) 119:112–21. doi: 10.1016/j.ejca.2019.07.017
147. Vaccaro CA, López-Kostner F, Adriana DV, Palmero EI, Rossi BM, Antelo M, et al. From colorectal cancer pattern to the characterization of individuals at risk: Picture for genetic research in Latin America. Int J Cancer. (2019) 145:318–26. doi: 10.1002/ijc.31920
148. Durinx C, McEntyre J, Appel R, Apweiler R, Barlow M, Blomberg N, et al. Identifying ELIXIR core data resources. F1000Res. (2016) 5:2422. doi: 10.12688/F1000RESEARCH.9656.2
149. Mayrhofer MT, Holub P, Wutte A, and Litton JE. BBMRI-ERIC: Ein neuer Zugang zu Biobanken: Von Menschen für Menschen. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. (2016) 59:379–84. doi: 10.1007/S00103-015-2301-8
150. Daza C. Scientific research and training in biotechnology in Latin America and the Caribbean: the UNU/BIOLAC experience. Electronic J Biotechnol. (1998) 1:17–8. doi: 10.4067/S0717-34581998000200005
151. Campos-Sánchez R, Willis I, Gopalasingam P, López-Juárez D, Cristancho M, and Brooksbank C. CABANA model 2017–2022: research and training synergy to facilitate bioinformatics applications in Latin America. Front Educ (Lausanne). (2024) 9:1358620. doi: 10.3389/FEDUC.2024.1358620
152. De Las Rivas J, Bonavides-Martínez C, and Campos-Laborie FJ. Bioinformatics in Latin America and SoIBio impact, a tale of spin-off and expansion around genomes and protein structures. Brief Bioinform. (2019) 20:390–7. doi: 10.1093/BIB/BBX064
153. Valdés E and Lecaros JA. Biobanks and data interoperability in Latin America: engendering high-quality evidence for the global research ecosystem. Front Med (Lausanne). (2024) 11:1481891/BIBTEX. doi: 10.3389/FMED.2024.1481891/BIBTEX
154. Lozada-Martinez ID, Lozada-Martinez LM, Cabarcas-Martinez A, Ruiz-Gutierrez FK, Aristizabal Vanegas JG, Amorocho Lozada KJ, et al. Historical evolution of cancer genomics research in Latin America: a comprehensive visual and bibliometric analysis until 2023. Front Genet. (2024) 15:1327243. doi: 10.3389/fgene.2024.1327243
155. Calderón-Aparicio A and Orue A. Precision oncology in Latin America: current situation, challenges and perspectives. Ecancermedicalscience. (2019) 13:920. doi: 10.3332/ecancer.2019.920
156. Stark Z, Dolman L, Manolio TA, Ozenberger B, Hill SL, Caulfied MJ, et al. Integrating genomics into healthcare: A global responsibility. Am J Hum Genet. (2019) 104:13–20. doi: 10.1016/j.ajhg.2018.11.014
157. Perenguez M, Ramírez-Montaño D, Candelo E, Echavarria H, and de la Torre A. Genomic medicine: perspective of the challenges for the implementation of preventive, predictive, and personalized medicine in latin america. Curr Pharmacogenomics Person Med. (2024) 21:51–7. doi: 10.2174/0118756921304274240819071740
158. Manotas MC, Rivera AL, and Sanabria-Salas MC. Variant curation and interpretation in hereditary cancer genes: An institutional experience in Latin America. Mol Genet Genomic Med. (2023) 11:e2141. doi: 10.1002/mgg3.2141
159. Pinto JA, Saravia CH, Flores C, Araujo JM, Martínez D, Schwarz LJ, et al. Precision medicine for locally advanced breast cancer: frontiers and challenges in Latin America. Ecancermedicalscience. (2019) 13:896. doi: 10.3332/ecancer.2019.896
160. de Castilla EMR, Mayrides M, González H, Vidangossy F, Corbeaux T, Ortiz N, et al. Implementing precision oncology in Latin America to improve patient outcomes: the status quo and a call to action for key stakeholders and decision-makers. Ecancermedicalscience. (2024) 18:1653. doi: 10.3332/ecancer.2024.1653
161. Werneck Da Cunha I, De Almeida Coudry R, Petaccia De Macedo M, Augusto E, Pereira De Assis C, Stefani S, et al. A call to action: molecular pathology in Brazil. Surg Exp Pathol. (2021) 4:1–27. doi: 10.1186/S42047-021-00096-1
162. Hernandez-Martinez J-M, Guijosa A, Flores-Estrada D, Cruz-Rico G, Turcott J, Hernández-Pedro N, et al. Real-world survival outcomes in non–small cell lung cancer: the impact of genomic testing and targeted therapies in a latin american middle-income country. JCO Glob Oncol. (2024) 10:1–12. doi: 10.1200/GO-24-00338
163. van Schooten TS, Derks S, Jiménez-Martí E, Carneiro F, Figueiredo C, Ruiz E, et al. The LEGACy study: a European and Latin American consortium to identify risk factors and molecular phenotypes in gastric cancer to improve prevention strategies and personalized clinical decision making globally. BMC Cancer. (2022) 22:646. doi: 10.1186/s12885-022-09689-9
164. Calero-Cáceres W and Balcázar JL. Leveraging genomic surveillance for public health: insights from Latin America. Lancet Microbe. (2024) 5:100919. doi: 10.1016/S2666-5247(24)00159-9
165. Hurle B, Citrin T, Jenkins JF, Kaphingst KA, Lamb N, Roseman JE, et al. What does it mean to be genomically literate?: National Human Genome Research Institute Meeting Report. Genet Med. (2013) 15:658–63. doi: 10.1038/gim.2013.14
166. Schaibley VM, Ramos IN, Woosley RL, Curry S, Hays S, and Ramos KS. Limited genomics training among physicians remains a barrier to genomics-based implementation of precision medicine. Front Med (Lausanne). (2022) 9:757212. doi: 10.3389/fmed.2022.757212
167. Tutika RK, Bennett JA, Abraham J, Snape K, Tatton-Brown K, Kemp Z, et al. Mainstreaming of genomics in oncology: a nationwide survey of the genomics training needs of UK oncologists. Clin Med (Lond). (2023) 23:9–15. doi: 10.7861/clinmed.2022-0372
168. Wilson Sayres MA, Hauser C, Sierk M, Robic S, Rosenwald AG, Smith TM, et al. Bioinformatics core competencies for undergraduate life sciences education. PloS One. (2018) 13:e0196878. doi: 10.1371/journal.pone.0196878
169. Jjingo D, Mboowa G, Sserwadda I, Kakaire R, Kiberu D, Amujal M, et al. Bioinformatics mentorship in a resource limited setting. Brief Bioinform. (2022) 23:1–8. doi: 10.1093/bib/bbab399
170. Nisselle A, Terrill B, Janinski M, Metcalfe S, and Gaff C. Ensuring best practice in genomics education: A scoping review of genomics education needs assessments and evaluations. Am J Hum Genet. (2024) 111:1508–23. doi: 10.1016/j.ajhg.2024.06.005
171. Giugliani R, Baldo G, Vairo F, Lujan Lopez M, and Matte U. The Latin American School of Human and Medical Genetics: promoting education and collaboration in genetics and ethics applied to health sciences across the continent. J Community Genet. (2015) 6:189–91. doi: 10.1007/s12687-015-0230-8
172. Programa iberoamericano de ciencia Y tecnología para el desarrollo. Madrid, Spain: CYTED. (2025). Available at: https://cyted.org/RELIVAF.
173. Sociedad Latinoamericana de Farmacogenómica y Medicina Personalizada. SOLFAGEM. (2025). Available at: https://solfagem.com/.
174. Santos M, Coudry RA, Ferreira CG, Stefani S, Cunha IW, Zalis MG, et al. Increasing access to next-generation sequencing in oncology for Brazil. Lancet Oncol. (2019) 20:20–3. doi: 10.1016/S1470-2045(18)30822-2
175. Johnson DL, Korf BR, Ascurra M, El-Kamah G, Fieggen K, de la Fuente B, et al. “Preparing the workforce for genomic medicine: International challenges and strategies.,”. In: Kumar D, editor. Genomic medicine skills and competencies. 125 London Wall, London EC2Y 5AS, United Kingdom: Academic Press (2022). p. 131–9. doi: 10.1016/B978-0-323-98383-9.00008-4
176. Doroshow JH and Kummar S. Translational research in oncology–10 years of progress and future prospects. Nat Rev Clin Oncol. (2014) 11:649–62. doi: 10.1038/nrclinonc.2014.158
177. Khoury MJ, Clauser SB, Freedman AN, Gillanders EM, Glasgow RE, Klein WMP, et al. Population sciences, translational research, and the opportunities and challenges for genomics to reduce the burden of cancer in the 21st century. Cancer Epidemiol Biomarkers Prev. (2011) 20:2105–14. doi: 10.1158/1055-9965.EPI-11-0481
178. Jia G, Lu Y, Wen W, Long J, Liu Y, Tao R, et al. Evaluating the utility of polygenic risk scores in identifying high-risk individuals for eight common cancers. JNCI Cancer Spectr. (2020) 4:pkaa021. doi: 10.1093/jncics/pkaa021
179. Ren F, Fei Q, Qiu K, Zhang Y, Zhang H, and Sun L. Liquid biopsy techniques and lung cancer: diagnosis, monitoring and evaluation. J Exp Clin Cancer Res. (2024) 43:96. doi: 10.1186/s13046-024-03026-7
180. Banerjee S, Booth CM, Bruera E, Büchler MW, Drilon A, Fry TJ, et al. Two decades of advances in clinical oncology - lessons learned and future directions. Nat Rev Clin Oncol. (2024) 21:771–80. doi: 10.1038/s41571-024-00945-4
181. Luchini C, Lawlor RT, Milella M, and Scarpa A. Molecular tumor boards in clinical practice. Trends Cancer. (2020) 6:738–44. doi: 10.1016/j.trecan.2020.05.008
182. Knepper TC, Bell GC, Hicks JK, Padron E, Teer JK, Vo TT, et al. Key lessons learned from moffitt’s molecular tumor board: the clinical genomics action committee experience. Oncologist. (2017) 22:144–51. doi: 10.1634/theoncologist.2016-0195
183. Miller RW, Hutchcraft ML, Weiss HL, Wu J, Wang C, Liu J, et al. Molecular tumor board-assisted care in an advanced cancer population: results of a phase II clinical trial. JCO Precis Oncol. (2022) 6:e2100524. doi: 10.1200/PO.21.00524
184. Hönikl LS, Lange S, Butenschoen VM, Delbridge C, Meyer B, Combs SE, et al. The role of molecular tumor boards in neuro-oncology: a nationwide survey. BMC Cancer. (2024) 24:108. doi: 10.1186/s12885-024-11858-x
185. Holt ME, Mittendorf KF, LeNoue-Newton M, Jain NM, Anderson I, Lovly CM, et al. My cancer genome: coevolution of precision oncology and a molecular oncology knowledgebase. JCO Clin Cancer Inform. (2021) 5:995–1004. doi: 10.1200/CCI.21.00084
186. Accelerating research in genomic oncology. ICGC ARGO. (2025). Available at: https://www.icgc-argo.org.
187. Angel MO, Pupareli C, Soule T, Tsou F, Leiva M, Losco F, et al. Implementation of a molecular tumour board in LATAM: The impact on treatment decisions for patients evaluated at Instituto Alexander Fleming, Argentina. Ecancermedicalscience. (2021) 15:1–9. doi: 10.3332/ECANCER.2021.1312
188. Sotelo-Rodríguez C, Vallejo-Ardila D, Ruiz-Patiño A, Chamorro DF, Rodríguez J, Moreno-Pérez DA, et al. Molecular tumor board improves outcomes for hispanic patients with advanced solid tumors. JCO Glob Oncol. (2024) 10:1–10. doi: 10.1200/GO.23.00011
189. Cancela MB, Dinardi M, Aschero R, Zugbi S, Chantada G, Baroni L, et al. The importance of basic and translational research in caring for children with Malignant solid tumors in Latin America. Rev Panamericana Salud Pública. (2024) 48:e48. doi: 10.26633/RPSP.2024.48
190. Rosa MN, Evangelista AF, Leal LF, De Oliveira CM, Silva VAO, Munari CC, et al. Establishment, molecular and biological characterization of HCB-514: a novel human cervical cancer cell line. Sci Rep. (2019) 9:1913. doi: 10.1038/s41598-018-38315-7
191. Chakraborty S, Sharma G, Karmakar S, and Banerjee S. Multi-OMICS approaches in cancer biology: New era in cancer therapy. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:167120. doi: 10.1016/j.bbadis.2024.167120
192. Ektefaie Y, Yuan W, Dillon DA, Lin NU, Golden JA, Kohane IS, et al. Integrative multiomics-histopathology analysis for breast cancer classification. NPJ Breast Cancer. (2021) 7:147. doi: 10.1038/s41523-021-00357-y
193. Sammut S-J, Crispin-Ortuzar M, Chin S-F, Provenzano E, Bardwell HA, Ma W, et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature. (2022) 601:623–9. doi: 10.1038/s41586-021-04278-5
194. Heo YJ, Hwa C, Lee G-H, Park J-M, and An J-Y. Integrative multi-omics approaches in cancer research: from biological networks to clinical subtypes. Mol Cells. (2021) 44:433–43. doi: 10.14348/molcells.2021.0042
195. Cheng C, Messerschmidt L, Bravo I, Waldbauer M, Bhavikatti R, Schenk C, et al. A general primer for data harmonization. Sci Data. (2024) 11:152. doi: 10.1038/s41597-024-02956-3
196. Flores JE, Claborne DM, Weller ZD, Webb-Robertson B-JM, Waters KM, and Bramer LM. Missing data in multi-omics integration: Recent advances through artificial intelligence. Front Artif Intell. (2023) 6:1098308. doi: 10.3389/frai.2023.1098308
197. Hernández-Lemus E and Ochoa S. Methods for multi-omic data integration in cancer research. Front Genet. (2024) 15:1425456. doi: 10.3389/fgene.2024.1425456
198. Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell. (2020) 182:200–225.e35. doi: 10.1016/j.cell.2020.06.013
199. Mun D-G, Bhin J, Kim S, Kim H, Jung JH, Jung Y, et al. Proteogenomic characterization of human early-onset gastric cancer. Cancer Cell. (2019) 35:111–124.e10. doi: 10.1016/j.ccell.2018.12.003
200. Krug K, Jaehnig EJ, Satpathy S, Blumenberg L, Karpova A, Anurag M, et al. Proteogenomic landscape of breast cancer tumorigenesis and targeted therapy. Cell. (2020) 183:1436–1456.e31. doi: 10.1016/j.cell.2020.10.036
201. Chen Y-J, Roumeliotis TI, Chang Y-H, Chen C-T, Han C-L, Lin M-H, et al. Proteogenomics of non-smoking lung cancer in east asia delineates molecular signatures of pathogenesis and progression. Cell. (2020) 182:226–244.e17. doi: 10.1016/j.cell.2020.06.012
202. Chen L, Liu C-C, Zhu S-Y, Ge J-Y, Chen Y-F, Ma D, et al. Multiomics of HER2-low triple-negative breast cancer identifies a receptor tyrosine kinase-relevant subgroup with therapeutic prospects. JCI Insight. (2023) 8:1–18. doi: 10.1172/jci.insight.172366
203. Li S, Yu W, Xie F, Luo H, Liu Z, Lv W, et al. Neoadjuvant therapy with immune checkpoint blockade, antiangiogenesis, and chemotherapy for locally advanced gastric cancer. Nat Commun. (2023) 14:8. doi: 10.1038/s41467-022-35431-x
204. Wörheide MA, Krumsiek J, Kastenmüller G, and Arnold M. Multi-omics integration in biomedical research - A metabolomics-centric review. Anal Chim Acta. (2021) 1141:144–62. doi: 10.1016/j.aca.2020.10.038
205. Lee J, Hyeon DY, and Hwang D. Single-cell multiomics: technologies and data analysis methods. Exp Mol Med. (2020) 52:1428–42. doi: 10.1038/s12276-020-0420-2
206. Lim B, Lin Y, and Navin N. Advancing cancer research and medicine with single-cell genomics. Cancer Cell. (2020) 37:456–70. doi: 10.1016/j.ccell.2020.03.008
207. Rood JE, Maartens A, Hupalowska A, Teichmann SA, and Regev A. Impact of the human cell atlas on medicine. Nat Med. (2022) 28:2486–96. doi: 10.1038/s41591-022-02104-7
208. Rozenblatt-Rosen O, Regev A, Oberdoerffer P, Nawy T, Hupalowska A, Rood JE, et al. The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell. (2020) 181:236–49. doi: 10.1016/j.cell.2020.03.053
209. Pellecchia S, Viscido G, Franchini M, and Gambardella G. Predicting drug response from single-cell expression profiles of tumours. BMC Med. (2023) 21:476. doi: 10.1186/s12916-023-03182-1
210. Xue Y, Friedl V, Ding H, Wong CK, and Stuart JM. Single-cell signatures identify microenvironment factors in tumors associated with patient outcomes. Cell Rep Methods. (2024) 4:100799. doi: 10.1016/j.crmeth.2024.100799
211. Kang J, Lee JH, Cha H, An J, Kwon J, Lee S, et al. Systematic dissection of tumor-normal single-cell ecosystems across a thousand tumors of 30 cancer types. Nat Commun. (2024) 15:4067. doi: 10.1038/s41467-024-48310-4
212. Jovic D, Liang X, Zeng H, Lin L, Xu F, and Luo Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin Transl Med. (2022) 12:e694. doi: 10.1002/ctm2.694
213. Trinks A, Milek M, Beule D, Kluge J, Florian S, Sers C, et al. Robust detection of clinically relevant features in single-cell RNA profiles of patient-matched fresh and formalin-fixed paraffin-embedded (FFPE) lung cancer tissue. Cell Oncol (Dordrecht Netherlands). (2024) 47:1221–31. doi: 10.1007/s13402-024-00922-0
214. Baysoy A, Bai Z, Satija R, and Fan R. The technological landscape and applications of single-cell multi-omics. Nat Rev Mol Cell Biol. (2023) 24:695–713. doi: 10.1038/s41580-023-00615-w
215. Heumos L, Schaar AC, Lance C, Litinetskaya A, Drost F, Zappia L, et al. Best practices for single-cell analysis across modalities. Nat Rev Genet. (2023) 24:550–72. doi: 10.1038/s41576-023-00586-w
216. Lim J, Chin V, Fairfax K, Moutinho C, Suan D, Ji H, et al. Transitioning single-cell genomics into the clinic. Nat Rev Genet. (2023) 24:573–84. doi: 10.1038/s41576-023-00613-w
217. Boakye Serebour T, Cribbs AP, Baldwin MJ, Masimirembwa C, Chikwambi Z, Kerasidou A, et al. Overcoming barriers to single-cell RNA sequencing adoption in low- and middle-income countries. Eur J Hum Genet. (2024) 32:1206–13. doi: 10.1038/s41431-024-01564-4
218. Majumder PP, Mhlanga MM, and Shalek AK. The Human Cell Atlas and equity: lessons learned. Nat Med. (2020) 26:1509–11. doi: 10.1038/s41591-020-1100-4
219. Genome Research Limited. Wellcome Connecting Science courses and conferences – Inspiring Science Transforming Careers . Available online at: https://coursesandconferences.wellcomeconnectingscience.org/ (Accessed February 24, 2025).
220. Helm JM, Swiergosz AM, Haeberle HS, Karnuta JM, Schaffer JL, Krebs VE, et al. Machine learning and artificial intelligence: definitions, applications, and future directions. Curr Rev Musculoskelet Med. (2020) 13:69–76. doi: 10.1007/s12178-020-09600-8
221. Danishuddin, Khan S, and Kim JJ. From cancer big data to treatment: Artificial intelligence in cancer research. J Gene Med. (2024) 26:e3629. doi: 10.1002/jgm.3629
222. Liñares-Blanco J, Pazos A, and Fernandez-Lozano C. Machine learning analysis of TCGA cancer data. PeerJ Comput Sci. (2021) 7:e584. doi: 10.7717/peerj-cs.584
223. Sánchez-Marqués R, García V, and Sánchez JS. A data-centric machine learning approach to improve prediction of glioma grades using low-imbalance TCGA data. Sci Rep. (2024) 14:17195. doi: 10.1038/s41598-024-68291-0
224. Perez-Lopez R, Reis-Filho JS, and Kather JN. A framework for artificial intelligence in cancer research and precision oncology. NPJ Precis Oncol. (2023) 7:43. doi: 10.1038/s41698-023-00383-y
225. Lal A, Liu K, Tibshirani R, Sidow A, and Ramazzotti D. De novo mutational signature discovery in tumor genomes using SparseSignatures. PloS Comput Biol. (2021) 17:e1009119. doi: 10.1371/journal.pcbi.1009119
226. Satas G, Zaccaria S, El-Kebir M, and Raphael BJ. DeCiFering the elusive cancer cell fraction in tumor heterogeneity and evolution. Cell Syst. (2021) 12:1004–1018.e10. doi: 10.1016/j.cels.2021.07.006
227. Argelaguet R, Arnol D, Bredikhin D, Deloro Y, Velten B, Marioni JC, et al. MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol. (2020) 21:111. doi: 10.1186/s13059-020-02015-1
228. Del Giudice M, Peirone S, Perrone S, Priante F, Varese F, Tirtei E, et al. Artificial intelligence in bulk and single-cell RNA-sequencing data to foster precision oncology. Int J Mol Sci. (2021) 22:1–18. doi: 10.3390/ijms22094563
229. Hao Y, Xuei X, Li L, Nakshatri H, Edenberg HJ, and Liu Y. RareVar: A framework for detecting low-frequency single-nucleotide variants. J Comput Biol. (2017) 24:637–46. doi: 10.1089/cmb.2017.0057
230. Jeon H, Ahn J, Na B, Hong S, Sael L, Kim S, et al. AIVariant: a deep learning-based somatic variant detector for highly contaminated tumor samples. Exp Mol Med. (2023) 55:1734–42. doi: 10.1038/s12276-023-01049-2
231. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. (2016) 99:877–85. doi: 10.1016/j.ajhg.2016.08.016
232. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. (2019) 176:535–548.e24. doi: 10.1016/j.cell.2018.12.015
233. Cheng J, Novati G, Pan J, Bycroft C, Žemgulytė A, Applebaum T, et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science. (2023) 381:eadg7492. doi: 10.1126/science.adg7492
234. De Paoli F, Berardelli S, Limongelli I, Rizzo E, and Zucca S. VarChat: the generative AI assistant for the interpretation of human genomic variations. Bioinformatics. (2024) 40:1–4. doi: 10.1093/bioinformatics/btae183
235. Li Q, Ren Z, Cao K, Li MM, Wang K, and Zhou Y. CancerVar: An artificial intelligence-empowered platform for clinical interpretation of somatic mutations in cancer. Sci Adv. (2022) 8:eabj1624. doi: 10.1126/sciadv.abj1624
236. López-Cortés A, Cabrera-Andrade A, Echeverría-Garcés G, Echeverría-Espinoza P, Pineda-Albán M, Elsitdie N, et al. Unraveling druggable cancer-driving proteins and targeted drugs using artificial intelligence and multi-omics analyses. Sci Rep. (2024) 14:19359. doi: 10.1038/s41598-024-68565-7
237. López-Cortés A, Cabrera-Andrade A, Vázquez-Naya JM, Pazos A, Gonzáles-Díaz H, Paz-Y-Miño C, et al. Prediction of breast cancer proteins involved in immunotherapy, metastasis, and RNA-binding using molecular descriptors and artificial neural networks. Sci Rep. (2020) 10:8515. doi: 10.1038/s41598-020-65584-y
238. Mosquera-Zamudio A, Gomez-Suarez M, Sprockel J, Riaño-Moreno JC, Janssen EAM, Pantanowitz L, et al. Globalization of a telepathology network with artificial intelligence applications in Colombia: The GLORIA program study protocol. J Pathol Inform. (2024) 15:100394. doi: 10.1016/j.jpi.2024.100394
239. Franco EF, Rana P, Cruz A, Calderón VV, Azevedo V, Ramos RTJ, et al. Performance comparison of deep learning autoencoders for cancer subtype detection using multi-omics data. Cancers (Basel). (2021) 13:1–17. doi: 10.3390/cancers13092013
240. Flores-Mejía LA, Siliceo-Portugal P, Figueroa UJ, Quezada IG, Barcenas Martínez BT, Sosa D, et al. Prediction of myeloid Malignant cells in Fanconi anemia using machine learning. bioRxiv (2024), 1–30. doi: 10.1101/2023.12.31.573791
Keywords: cancer genomics, bioinformatics, precision oncology, next-generation sequencing (NGS), Latin America
Citation: Torres-Narvaez ES, Mendivelso-González DF, Artunduaga-Alvarado JA and Ortega-Recalde O (2025) Cancer genomics and bioinformatics in Latin American countries: applications, challenges, and perspectives. Front. Oncol. 15:1584178. doi: 10.3389/fonc.2025.1584178
Received: 26 February 2025; Accepted: 12 June 2025;
Published: 09 July 2025.
Edited by:
Sharon R. Pine, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Davide Dalfovo, University of Trento, ItalyMatteo Pallocca, National Research Council (CNR), Italy
Copyright © 2025 Torres-Narvaez, Mendivelso-González, Artunduaga-Alvarado and Ortega-Recalde. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oscar Ortega-Recalde, b3NjYXIub3J0ZWdhQGJpb3RlY2dlbi5jb20uY28=
†These authors have contributed equally to this work and share first authorship