 Erin T. Hamanishi
Erin T. Hamanishi Derek Dang
Derek Dang Sriram Venneti2,3,4,5*
Sriram Venneti2,3,4,5*- 1Division of Pediatric Hematology, Oncology and Bone Marrow Transplant, Department of Pediatrics, University of Michigan Medical School, Ann Arbor, MI, United States
- 2Department of Pediatrics, University of Michigan Medical School, Ann Arbor, MI, United States
- 3Laboratory of Brain Tumor Metabolism and Epigenetics, Department of Pathology, University of Michigan Medical School, Ann Arbor, MI, United States
- 4Chad Carr Pediatric Brain Tumor Center, University of Michigan, Ann Arbor, MI, United States
- 5Department of Pathology, University of Michigan Medical School, Ann Arbor, MI, United States
Epigenetic modifications, particularly histone post-translational modifications (PTMs), are central to pediatric brain tumor pathogenesis, impacting chromatin structure, gene expression, and genomic stability. Disruptions in histone PTMs, especially lysine methylation and acetylation, arising due to histone mutations or aberrant enzyme modulation are critical drivers of oncogenesis. Lysine methylation, catalyzed by histone methyltransferases (KMTs), modulates chromatin interactions and gene expression through activation or repression, depending on the methylation state and the specific histone residue. Key enzymes, including histone methyltransferases and demethylases, and associated proteins exemplify the functions of writers, readers, and erasers in maintaining histone modification balance. Similarly, histone acetylation, a dynamic process regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), plays a crucial role in pediatric brain tumors. Alterations in these components lead to aberrant gene expression and tumorigenesis. Understanding these disrupted processes offers potential for targeted therapies to rewire oncogenic chromatin states and potentially improve patient outcomes.
1 Introduction
Pediatric brain cancers are the most common solid tumor in children, leading to significant morbidity and mortality (1). In contrast to many adult tumors, pediatric cancers are often characterized by a paucity of recurrent mutations and bear a relatively lower tumor mutational burden (2–4). In contrast, extensive research into pediatric brain tumors has revealed fundamental deregulation in chromatin biology and epigenomic alterations. These differences highlight the unique molecular landscape of pediatric malignancies, offering insights into how chromatin structure and gene regulation are disrupted in these cancers.
Within eukaryotic cells, DNA is organized into chromatin, a complex of DNA and proteins arranged into repeating units called nucleosomes. Each nucleosome consists of a core octamer of histone proteins around which approximately 146 base pairs of DNA are tightly wrapped, forming the fundamental unit of chromatin structure (5, 6). The core octamer comprises four core histones: H2A, H2B, H3, and H4 (7, 8) (Figure 1). Linker DNA connects these nucleosomes, and linker histone H1 binds to the nucleosome core at the sites where DNA enters and exits, contributing to chromatin structure and stability (11). Chromatin organization is dynamic and tightly regulated; modulation of chromatin structural changes plays a crucial role in regulating gene expression and maintaining genomic stability, underscoring the importance of chromatin’s dynamic nature in cellular function.
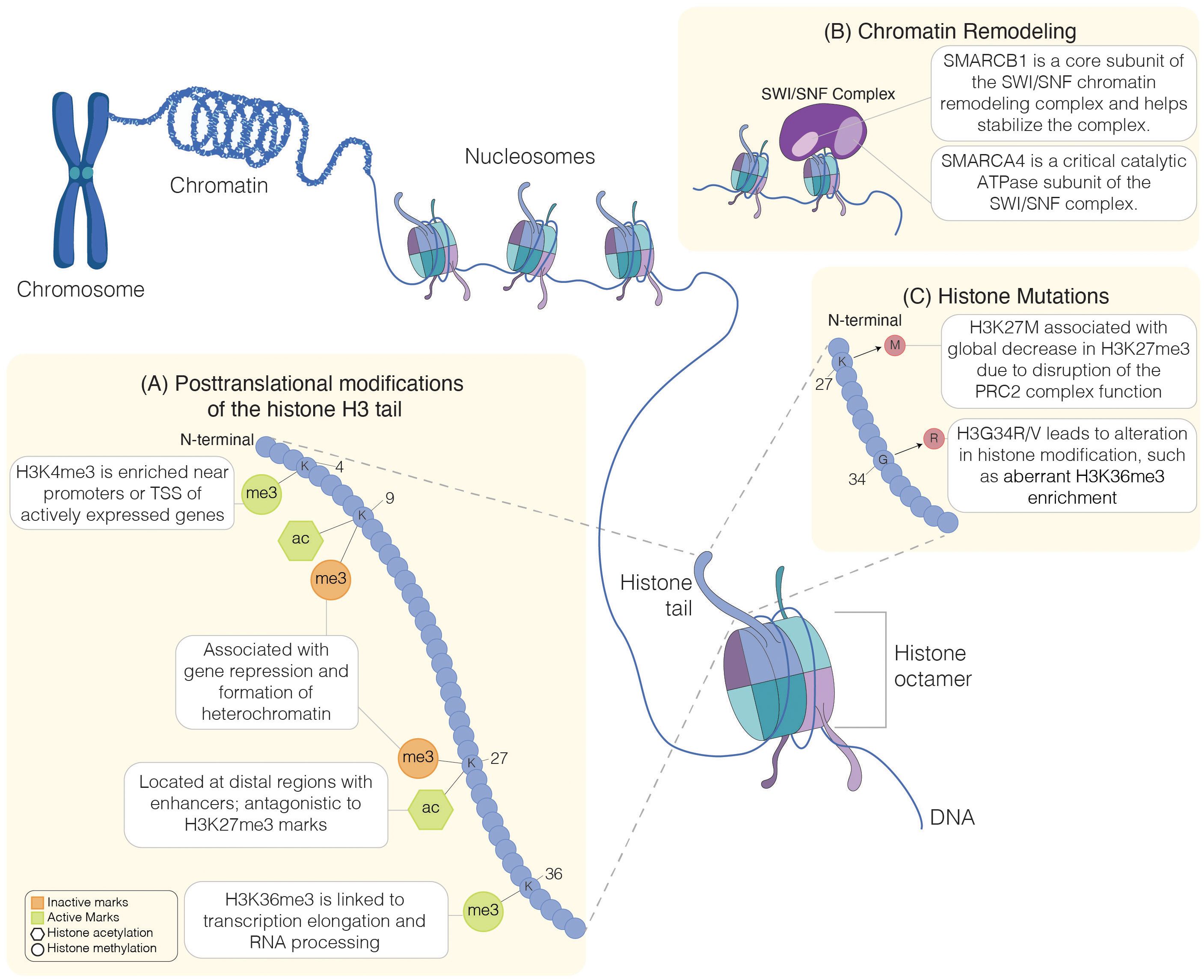
Figure 1. Regulation of chromatin structure. Schematic representation of chromatin organization. Chromatin is made up of DNA wound around histone proteins. Each histone complex comprises an octamer of histone protein, including H2A, H2B, H3, and H4 (7, 8). Chromatin remodeling, histone mutations, and histone modifications influence chromatin structure. Lysine-specific post-translational modifications of histone H3 tail (A) include methylation (me) and acetylation (ac). Histone modification can be inactive (orange symbols) or active (green symbols). Chromatin accessibility can be influenced by chromatin remodelers, such as the SWI/SNF complexes, which are composed of multiple subunits, such as the SMARCB1 subunit involved in the complex stabilization (9) or the catalytic subunit SMARCA4 (10) (B). Histone mutations (C), including H3K27M or H3G34R, interfere with histone modifications, leading to aberrant chromatin structure and accessibility. Chromosome, chromatin: Created in BioRender. Chung, C. (2025) https://BioRender.com/a87r173.
Disruption of chromatin regulation and structure can have profound effects, as alterations in chromatin due to mutations in histone post-translational modifications (PTMs) (Figure 1A), chromatin remodeling complexes (Figure 1B), histone proteins (Figure 1C), or DNA methylation can significantly impact gene expression. Such disruptions may lead to the aberrant activation of oncogenes or the silencing of tumor suppressor genes, both critical in cancer development (12). In the setting of cancer, sequencing efforts have revealed mutations in genes involved in chromatin organization and regulation (13, 14). Beyond cancer, disruptions in chromatin regulation are linked to various other diseases, including neurodegenerative disorders and developmental syndromes (15–17). These alterations can also impact normal cellular processes, such as DNA repair, replication, and cell differentiation, highlighting the essential role of chromatin in maintaining cellular and genomic integrity. Herein, we will focus on the impact of aberrant histone modifications underlying pediatric brain tumors. Understanding these disruptions provides crucial insights into disease mechanisms and identifies potential therapeutic targets for correcting these aberrant chromatin states.
2 Post-translational histone modifications
Histone PTMs, like other mechanisms of chromatin modifications, create changes in chromatin structure that can impact genomic stability and gene expression (18, 19). Diverse arrays of PTMs have been reported, with some of the most well-studied histone modifications reported on lysine residues in the N-terminal tail of H3 and H4 (20). The covalent modifications to histone residues consist primarily of methylation, acetylation, phosphorylation, and ubiquitination, although not limited to those mentioned. In the common paradigm of histone PTMs, distinct categories of proteins—referred to as “writers,” “readers,” and “erasers”—play crucial roles in regulating PTMs (21). Writers are enzymes that add specific PTMs to histones, such as methylation, acetylation, or phosphorylation. For instance, methyltransferases such as SET1 (histone-lysine N-methyltransferase, H3 lysine-4 specific - Saccharomyces cerevisiae) and EZH2 (enhancer of zeste 2) add methyl groups to lysine residues on histone tails (22–25). Similarly, histone acetyltransferases (HATs) can add acetyl groups to histone lysine residues (26–30). Readers are proteins that recognize and bind to these modifications, translating the histone code into functional outcomes by regulating gene expression. These include chromodomain-containing proteins like HP1 (Heterochromatin protein 1), which binds to methylated lysine residues (31, 32). Erasers are enzymes that remove or reverse these modifications. These include histone deacetylases (HDACs) or demethylases like KDM1A/LSD1 (33, 34). Together, these readers, writers, and erasers maintain the complex dynamic balance of histone modifications, essential for regulating gene expression, maintaining genomic stability, and coordinating cellular processes. Understanding how these chromatin-modifying processes are disrupted in pediatric brain tumors offers potential therapeutic targets for precision treatments to restore normal chromatin function and improve patient outcomes.
The methylation of basic residues, such as lysine, arginine, and histidine, is a well-studied process, and the methylation of lysine residues, specifically on histone H3 and H4, has been extensively reviewed (35). Lysine can be mono- (me1), di- (me2), or trimethylated (me3), whereas arginine can be mono- or di-methylated, for example. Unlike the function of other PTMs, histone methylation does not affect the charge of the histone residue; instead, it regulates interaction with chromatin-binding proteins, thus impacting gene expression. Histone methylation at lysine residues can be associated with either transcriptional activation or repression, depending on the methylation level or the lysine residue involved (35, 36). Generally, H3K4 or H3K36 methylation is associated with the activation of genes. For example, H3K4 methylation marks are enriched near transcription start sites (TSS), and H3K4me2/me3 are strongly associated with euchromatin formation and active transcription. Certain transcriptional coactivators and chromatin remodeling complexes, such as the SAGA complex, recognize this modification, leading to the recruitment of RNA polymerase II and gene transcription (37, 38). Global increase in H3K4me3 positively correlates with higher grade and poor survival in certain pediatric brain tumors, such as ependymomas (39) and pediatric high-grade gliomas (pHGG) (40). H3K36me3 marks are typically localized to bodies of active genes and are associated with transcription elongation and RNA processing (41). In G34-mutant diffuse hemispheric gliomas (DHG), H3K36me3 marks are decreased in cis; however, they have preserved global levels (42). On the other hand, trimethylation of H3K9 (H3K9me3) or H3K27 (H3K27me3) is associated with repression of gene expression (36). A decrease in global H3K27me3 is commonly noted in certain high-grade tumors, such as pediatric high-grade gliomas, and the dysregulation of H3K27me3 can contribute to the oncogenesis of these tumors (43, 44). In addition to histone methylation, acetylation of lysine residues in the histone tail influences chromatin structure and gene expression, generally activating transcription. Along with the global loss of H3K27me3 marks, there is a reciprocal gain of H3K27ac across the genome in pHGG (45). Together, alterations in histone modifications can drive tumorigenesis through aberrant regulation of chromatin structure and gene expression (Figure 1A).
Histone methylation occurs via the writer proteins known as histone methyltransferases. Lysine methyltransferases (KMTs) catalyze the methylation of lysine residues by donating a methyl group from S-Adenosyl-L-Methionine (SAM). Histone methylation was first reported in the 1960s. However, researchers began to pay significant attention to the role of histone methyltransferases in the early 2000s with the discovery of SUV39H1 (KMT1) and EZH2 (46–49). These enzymes contain SET (Su(var)3–9, enhancer of zeste, and Trithorax) domains (SET-domain protein methyltransferase family) that play critical roles in methylation of histone lysine residues (50). SUV39H1 (KMT1) and its homolog SUV39H2 (KMT1B) are involved in H3K9 methylation (H3K9me2/me3) (51). EZH2 mediates trimethylation of H3K27 (H3K27me3) and is a key component of the polycomb repressive complex 2 (PRC2) (52). These enzymes can facilitate a repressive chromatin state by silencing gene expression by H3K9me3 and H3K27me3 enrichment. The first histone demethylase, KDM1A/LSD, was described in 2004, which removes the methyl mark from H3K4 (33). Since then, an extensive family of KMTs has been characterized; many principal writers (KMTs) and erasers (KDMs) have been identified, often for specific residues (34). Together, the combined functions of KMTs and KDMs can orchestrate complex patterns of genomic methylation and demethylation to regulate multiple cellular functions.
3 Pediatric brain tumors
Pediatric brain tumors encompass a diverse spectrum of malignancies, each defined by distinct biological and clinical characteristics (Figures 2A, B). They most commonly arise in the hindbrain/posterior fossa (PF) region. This region of the brain houses critical structures, including the brainstem and cerebellum. Medulloblastomas (MB) are the most common brain tumor in children (1) and arise from the cerebellum. They are classified into four distinct subgroups: WNT, SHH, Group 3, and Group 4 (71, 72). Each subgroup is characterized by unique driver genetic events, proposed cell-of-origin, and distinct clinical implications (53, 73, 74), and are now further subdivided into several subtypes (54, 55, 75). Despite exhibiting a relative paucity of mutations across MB, alterations in epigenetic regulators and gene expression contribute to the heterogeneity and pathogenesis of MB.
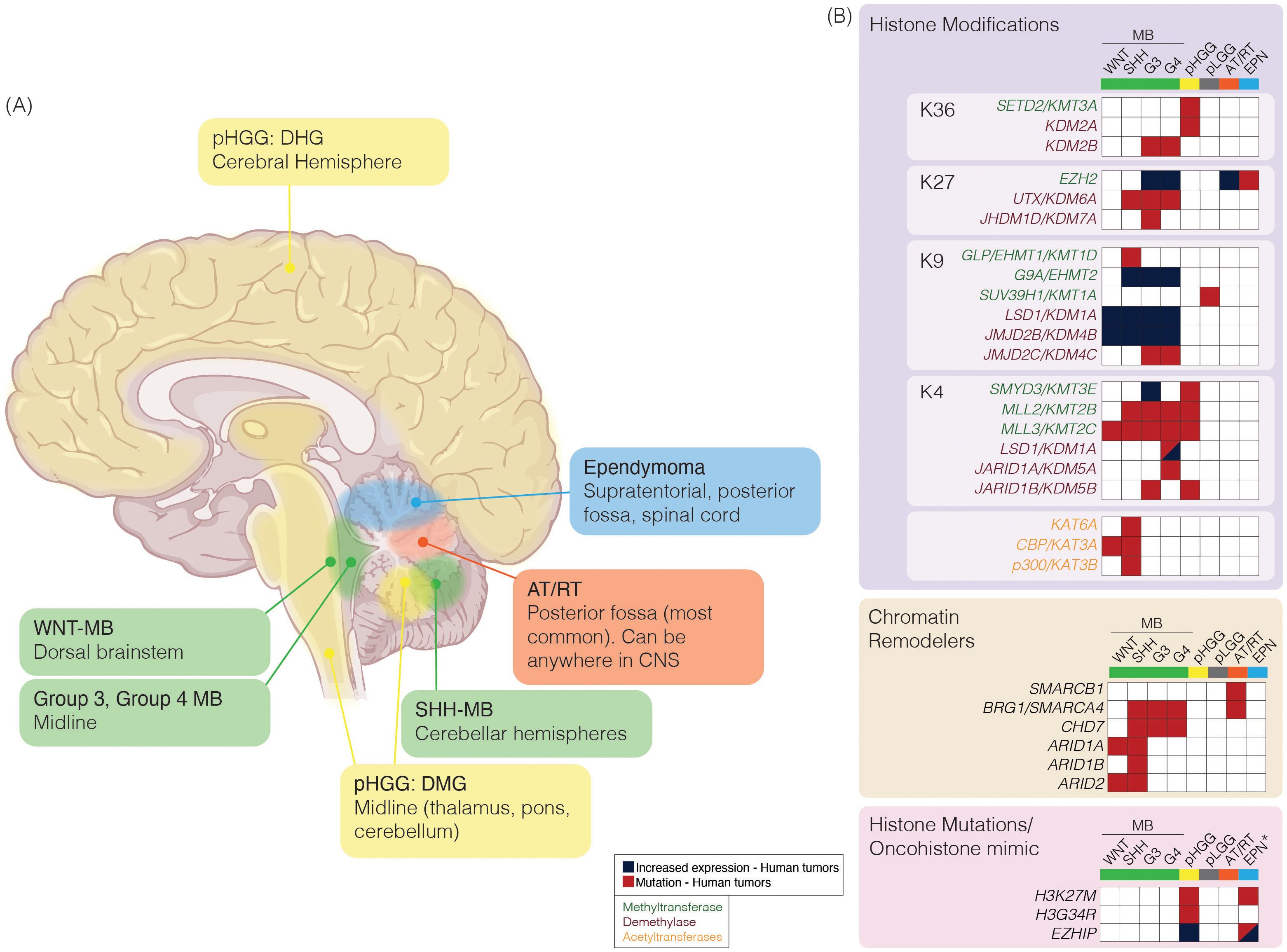
Figure 2. (A) Schematic of pediatric brain tumor location and characteristic epigenetic modifications. Pediatric brain tumors vary in anatomic location (left) and alterations in expression or mutations in epigenetic regulators (right). Medulloblastoma (MB; green) can be divided into subgroups based on clinical and molecular characteristics. Pediatric high-grade gliomas (pHGG; yellow) can be further characterized by characteristic histone mutations (pink box); diffuse hemispheric gliomas (DHG) are associated with H3G34R/V mutation, or diffuse midline glioma (DMG) are associated with H3K27M mutation. Atypical teratoid/rhabdoid tumor (AT/RT; orange) are most commonly located in the posterior fossa; however, it can be found anywhere in the CNS among pediatric patients. Ependymoma (EPN; blue) can be found in three anatomic compartments, supratentorial (ST), posterior fossa (PF) or spinal (SP). (B) Epigenetic alterations reported in at least one tumor sample from previously reported molecular characterization efforts in MB (53–59), pHGG (60–64), pediatric low-grade glioma (pLGG; grey) (65) or AT/RT (66–70). Mutations or alterations in expression among writers [histone methyltransferases (green text) or histone acetyltransferases (orange text)], erasers [histone demethylases (purple text)], chromatin remodelers, or histone mutations/oncohistone mimics are represented. *Increased expression or mutations in EZHIP reported, or H3K27M histone mutations reported specifically in PFA-EPN. Brain: Created in BioRender. https://BioRender.com/tmiyitf.
Other aggressive brain tumors arising in children and adolescents include high-grade gliomas, ependymomas, and atypical teratoid/rhabdoid tumors (AT/RT). High-grade gliomas are characterized by infiltrative growth and malignant behavior. Mutations leading to the amino acid substitutions at lysine 27 to methionine (collectively referred to as H3K27M) in the N-terminal tail of histone H3 were identified in H3-3A (previously called H3F3A) and H3-C2 (previously called HIST1H3B) (60, 76) (Figure 2B). Additionally, H3-3A mutations at glycine 34 to either arginine or valine (referred to as H3G34R/V) were noted and were mutually exclusive from H3K27M mutations (60). H3-3A encodes the non-canonical variant H3.3, while H3-C2 encodes canonical, cell cycle-couple H3.1 (77–79). Subsequent studies have shown rare H3K27M tumors involving H3-C3 (also encodes H3.1) (80, 81).
Ependymomas (EPN) exhibit variable biological behavior depending on their location within the central nervous system (82). Classification of EPN has further subdivided this disease entity into subgroups based on anatomical region and molecular characteristics (83). A majority of posterior fossa EPN tumors fall within the PF-A subgroup and are characterized by global hypomethylation of H3K27 (84–86). AT/RTs are highly aggressive and often present in very young children. They are driven by loss-of-function SMARCB1 gene alterations (66). SMARCB1 is a critical component of the chromatin remodeler SWI/SNF (SWItch/Sucrose Non-Fermentable) complex. The SWI/SNF complex regulates nucleosome incorporation and the eviction of genes and functions in tandem with histone PTM modifiers (87, 88). Rare loss-of-function mutations in SMARCA4, another SWI/SNF component, have been reported in AT/RT (67, 68). A comprehensive understanding of the epigenetic alterations in these tumors is essential for dissecting the mechanisms underpinning their pathogenesis and developing effective and targeted therapies (Figure 2B).
4 Post-translational histone modifications in the developing hindbrain
Pediatric brain tumors most commonly arise within the hindbrain/PF region. Histone modifications are key to brain development, as illustrated in the development of the hindbrain and cerebellum. Understanding this complex phenomenon may help understand how these pathways are deregulated in pediatric brain cancers. The cerebellum is a highly foliated structure composed of many different types of neurons and astrocytes. Accordingly, the development of the human cerebellum is a highly complex process that begins one month after conception and extends into the second postnatal year (89, 90). Many efforts have been made to establish the genetic and developmental programs that underlie cerebellar ontogenesis. These efforts led to the identification of two primary zones of neurogenesis in the developing cerebellum: the ventricular zone and the rhombic lip (91). The cerebellar ventricular zone gives rise to GABAergic cerebellar neuronal derivatives, including Purkinje cells, GABAergic cerebellar nuclei, Bergmann glial cells, and inhibitory interneurons (91–94). On the other hand, cerebellar glutamatergic derivatives, including granule neuron progenitors, glutamatergic cerebellar nuclei, and unipolar brush cells, arise from the rhombic lip (95). Although human cerebellar development research continues to be obfuscated by the lack of relevant tissues and the intrinsic discordance between human and murine cerebellar development (90), there is growing evidence that supports critical roles for epigenetics in the proper development of this vital hindbrain structure.
The expression of proliferation and differentiation genes is tightly regulated by epigenetic modifications during mammalian cerebellar development. This is particularly well-documented in granule neurons, the most common neuron in the brain (96). Granule neuron progenitor (GNPs) cells are born in the rhombic lip and migrate along the cerebellar anlage, where they eventually form the cerebellar granular layer. Bivalent chromatin are regions of DNA that are simultaneously marked by both activating H3K4me3 and repressive H3K27me3 (97). This dual enrichment of both repressive and activation marks keeps genes in a “poised” state that can be readily modulated. Bivalent chromatin is particularly important in embryonic stem cells, as it helps maintain their undifferentiated state by keeping developmental genes poised for activation when necessary (97). Bivalent chromatin plays an important role during the proliferative phase of nascent GNPs, and these cells maintain high levels of H3K4me3 at genes associated with cell cycling and suppress differentiation genes via increased H3K27me3 (98). Accordingly, knockout of Ezh2 in the developing mouse cerebellum decreases GNP and Purkinje cell proliferation (99). Histone modifications of genes that encode key effector proteins in signaling pathways can also affect GNP proliferation. The Sonic hedgehog (SHH) pathway is a key driver of granule neuron proliferation (100). SHH directly induces HDAC1, which in turn deacetylates gene regulatory regions of Gli2 and allows Gli2 to translocate to the nucleus and induce Gli1 expression (101). Gli1 promotes genes related to cell cycling and proliferation (101). Both HAT (Gcn5) and other HDAC (HDAC2, HDAC3) enzymes can also act directly on cell cycling gene programs to ensure proper GNP proliferation (102–104). ATP-dependent chromatin remodeler complexes, including Chd7 (99, 105), Snf2h (106), and npBAF (107), increase chromatin accessibility of pro-proliferation genes and regulate expression of genes that induce differentiation, such as reelin (105). Improper proliferation of GNPs caused by mutations in chromatin-remodeling enzymes causes cerebellar hypoplasia, underlying the importance of these complexes in the maintenance of adequate GNP pools (99, 105–107).
As GNPs stop proliferating and begin to differentiate into mature granule neurons, they exhibit simultaneous increase of H3K9me3 and H3K27me3 at cell cycling and proliferation genes and activating marks H3K4me3, H3K9ac, H3K14ac, H3K27ac at genes essential for synaptogenesis, ion channels, and cell adhesion (98, 108–111). Whereas HDAC1 activity induced GNP proliferation, the inhibition of HDAC1 via activation of pro-neurotrophin receptor p75NTR induces cell cycle arrest and subsequent neutrophin activity. These processes contribute to shifting cells towards differentiation (112). DNA methylation is also dynamically regulated during granule neuron differentiation. TET demethylases are highly expressed during this time and increase 5-hydroxymethylcytosine (5hmC) at genes associated with axonal guidance and ion channels (113). 5hmc is also present in differentiated Purkinje cells, suggesting an important role for this epigenetic mark among multiple cellular niches (114, 115). Concurrently, 5-methylcytosine (5mc) is increased at genes that promote proliferation and cell cycling (98). Chromatin-remodeling enzymes can also impact GNP differentiation. As GNPs mature, the nucleosome remodeling and deacetylase (NuRD) complex silences genes expressed in immature GNPs that induce ectopic early synaptogenesis, while its CHD4 subunit increases transcription of genes essential for dendrite pruning by inducing active chromatin (116). To summarize, histone tail modifying enzymes, DNA methylation modulators, and chromatin-remodeler complexes work in synchrony to modulate gene expression throughout cerebellar development which allows for proper GNP proliferation and differentiation.
5 Deregulated histone modifications in pediatric brain tumors
5.1 Oncohistones and oncohistone-like proteins
Recurrent somatic histone mutations in childhood brain tumors were discovered in 2012 through exome sequencing of pediatric glial tumors, including H3K27M and H3G34R/V mutations (Figure 1C) (60, 76). H3K27M and H3G34R/V can arise in differing age groups and within distinct anatomic regions of the brain. H3K27M gliomas arise mainly in younger children from the midline of the CNS and are collectively designated diffuse midline gliomas (DMGs) (71). These midline structures include the pons [where they are referred to as diffuse intrinsic pontine gliomas (DIPG)], thalamus, cerebellum, and spine (71). In contrast, H3G34R/V mutations are hemispheric, frequently observed in older children and adolescents, and are termed DHG H3-G34-mutant (71). These mutant histones, referred to as oncohistones, are associated with distinct gene expression profiles and global DNA methylation patterns (117).
H3K27M mutations occur most frequently in the non-canonical H3.3 (approximately 60-70%), whereas mutations targeting H3.1 or H3.2 are identified at a lower frequency in DMGs (118–120). H3.1 and H3.2 K27M tumors are located primarily in the brainstem, restricted to the pons, and often associated with younger age. In contrast, those harboring an H3.3 K27M are found across the midline, often in older children and adults (119, 121). In addition to the hallmark mutation, H3K27M, many DMGs harbor additional genetic alterations. The genetic mutations associated with H3.3 and H3.1/H3.2 vary. For example, H3.1 and H3.2 K27M DMGs are associated with activin A receptor type I (ACVR1) mutations. The gain of function mutations in ACVR1 leads to the hyperactivation of the bone morphogenic protein (BMP) signaling pathway (122). Meanwhile, H3.3 mutant variants are commonly linked with loss of function alterations in platelet-derived growth factor receptor A (PDGFRA), ultimately leading to activation of downstream PI3K/AKT/mTOR and Ras/Raf/MEK/ERK downstream signaling (63, 121–125).
H3K27M mutations cause global H3K27me3 reduction due to dominant negative effects on the PRC2 complex (43, 126, 127). Once PRC2 is recruited to specific sites in the genome, it spreads and deposits H3K27me3 in adjacent regions (128–130). H3K27M can bind to EZH2/PRC2 and inhibit this spreading function of PRC2, resulting in a global reduction in H3K27me3 levels (43, 131–133). Despite this global decrease in H3K27me3, these high-affinity PRC2 genomic loci retain H3K27me3 in these tumors (127, 131, 134–137). H3K27M mutations can reprogram other histone PTMs including H3K27ac (discussed below), H3K36me3, H3K4me3 and bivalent histone marks (138–148).
EZHIP protein (EZH Inhibitory Protein or Cxorf67 or Catacomb) is an oncohistone-like protein expressed in the testis and important for spermatogenesis (149). EZHIP is overexpressed in majority of childhood posterior fossa group-A (PFA) ependymomas, resulting in a global reduction of H3K27me3 levels (150). EZHIP contains a methionine residue at position 406 that enables EZHIP to phenotypically mimics H3K27M by binding to and inhibiting the function of EZH2/PRC2 complex (126, 131, 134, 135, 146, 149, 151, 152). H3K27ac is an activating mark that opposes H3K27me3 and localizes to gene promoters and enhancers. Global H3K27me3 reduction in H3K27M gliomas and PFA ependymomas is associated with a global increase in H3K27ac levels (153). Genomic distribution of H3K27ac in these tumors converges on several neuro-developmental related enhancers and super-enhancers contributing to aberrant differentiation tumor cell states (45, 133, 154–165). Rare population of H3-wildtype, low-H3K27me3 DMGs overexpress EZHIP, and similarly small percentage of PFAs harbor H3K27M mutations (64, 71). Because DMGs can harbor both H3K27M mutations and overexpress EZHIP in rare tumors, they are designated collectively as DMG, H3-K27-altered (71).
Like H3K27 mutant tumors, DHG H3-G34-mutant tumors are characterized by a H3-3A mutation targeting the N-terminal tail of the histone protein. However, unlike H3K27M, the H3G34 mutation leads to glycine replacement by arginine or valine (60, 121). H3G34 mutant tumors are characteristically located in the cerebral hemispheres, with a propensity for the temporoparietal hemispheres. These tumors are most prevalent in adolescents and young adults (12–35 years) (166). Khazaei et al. demonstrated that H3-G34 substitutions lead to distinct phenotypic outcomes affecting neurodevelopment by altering the epigenome (167). Specifically, H3-G34R mutations reduce H3K36me2 and H3K36me3 levels on the mutant histone tail, disrupting the recruitment and distribution of DNMT3A deposition and mCH methylation (167). In general, when compared to other pHGG, H3-G43-mutant tumors are hypomethylated (117, 139). Many H3-G34 mutant tumors also bear activating mutations in PDGFRA or abnormal PDGFRA activation through enhancer hijacking (168). Abnormal downstream signaling of PDGFRA increases cell survival and proliferation, promoting gliomagenesis (168, 169). They are also often associated with loss of function mutations in TP53 and ATRX (encoding a chromatin remodeling protein) (117, 121). In a genetically engineered mouse model, Atrx loss in the presence of H3.3G34R upregulates of HOX genes and inhibits differentiation pathways (170). The replacement of glycine by arginine or valine leads to decreased SET Domain Containing 2, Histone Lysine Methyltransferase (SETD2) activity, ultimately resulting in altered PTMs of nearby H3K36 and H3K27 (42, 171–175). These epigenetic state-driven gene expression profiles map to interneuron-like GABAergic states and can impact the tumor microenvironment (176). To gain deeper insight into the immunological landscape of H3-G34-altered tumors, Garcia-Fabiani et al. examined the epigenetic and transcriptomic reprogramming in H3.3-G34R tumors. They found that the H3-G34R mutation reshapes the tumor microenvironment, leading to upregulation of immune-related genes and activation of the JAK/STAT pathway (177).
Given the need to further characterize the histone mutational landscape in cancers, comprehensive sequencing efforts have identified histone mutations in various cancers, including mutations in both the tail and the globular domains (178–180). Analysis of cancer genomes across various age groups, including adults, adolescents, and young adults, reveals that approximately 11% carry mutations in histone-encoding genes (180). Among CNS tumors, there is an increased prevalence of histone mutations among pediatric, adolescent, and young adults (AYA) when compared to adults. Not surprisingly, many of these histone mutations identified in the pediatric and AYA population were H3 K27/G34 mutations; however, non-H3, core, and linker mutations were also identified in a variety of CNS malignancies, including atypical teratoid/rhabdoid tumor (ATRT), DMG, HGG, ependymoma, and MB (180). Although mutant oncohistones may be a hallmark of tumorigenesis, it is evident through extensive investigations that they do not act alone but instead in concert with other genetic alterations, leading to the oncogenesis of these brain tumors.
5.2 Aberrant histone modifications in pediatric brain tumors
5.2.1 Histone methylation
The activity and balance of specific KMTs and KDMs regulate the genomic enrichment of both repressive and activating histone marks. Disruption in the expression or function of these enzymes has been implicated in the pathogenesis of pediatric brain tumors. This is particularly evident in MB across many of the molecular subgroups. Molecular studies have identified numerous mutations that alter histone methylation patterns. Key alterations among KMTs in pediatric brain tumors include aberrant expression of EZH2, NSD1, SETD1A, SMYD3, MLL2 (KMT2D), and G9A (EHMT2) (40, 181–184). Conversely, mutations in KDMs are also frequently observed, for example amplification of JMJD family proteins such as JMJD2C (KDM4C), JMJD2B (KDM4B), and JMJD3 (KDM6B), as well as mutations in MYST3 and UTX (KDM6A) (181–184). In MB, mutations in KDM6A and MLL2 are associated with a loss of H3K27me3 (Dubuc et al., 2013). Additionally, global hypomethylation of H3K9 has been reported in approximately 40% of MB cases compared to normal brain tissue, and in vitro restoration of genes regulating H3K9 methylation results in a decrease in cell proliferation (54). Together, epigenetic dysregulation in pediatric brain cancers through altered histone methylation patterns plays a crucial role in the pathogenesis of these devastating tumors.
5.2.1.1 Histone methyltransferases
The most studied methyltransferase in pediatric brain tumors is EZH2, a critical component of PRC2. PRC2 is involved in differentiation, proliferation, and maintenance of cellular identity in pediatric brain cancers, including H3K27- altered DMG, PFA ependymomas, MB, and AT/RT (185). Despite the global reduction of H3K27me3, H3K27-altered DMGs and PFA ependymomas retain genomic H3K27me3 to repress gene expression at high-affinity PRC2 sites, including the CDKN2A/B locus encoding the senesce-associated protein p16. Inhibition of EZH2 in these tumors is therapeutic by lowering H3K27me3 at these sites, leading to increased gene expression, including p16 (132, 135, 186–189).
PRC2 is composed of four core subunits: the catalytic EZH2 (responsible for methylation of lysine 27), EED, SUZ12, and RBAp46/48, in addition to several additional axillary subunits (190). Recruitment of PRC2 through EED to H3K27me3 stimulates the catalytic activity of EZH2 (190). Complete loss of Eed acts as a tumor suppressor in murine MB models, whereas mosaic Eed mutations can promote tumor growth, highlighting the impact of PRC2 heterogeneity driving tumor growth in MB (191). Deletion of Eed destabilizes PRC2 (192), and deletion of Eed or Ezh2 in SHH-MB models is sufficient to promote the expression of genes typically suppressed by PRC2, specifically promoting myeloid differentiation and tumor progression (192).
MB tumors demonstrate elevated levels of EZH2 across groups, with the highest levels appreciated in groups 3 and 4 (56, 193). Subsequent alterations in genomic H3K27me3 can contribute to stem-like tumor cell states in groups 3 and 4 MB (191). EZH2 interacts with maternal embryonic leucine-zipper kinase (MELK), a member of the AMPK protein kinase family involved in the regulation of cell cycle and cellular function (194). Along with EZH2, MELK is frequently upregulated in MB and associated with reduced survival (195). MELK binds to and phosphorylates EZH2, thus working together to promote cancer stem-like cell proliferation and stemness (195).
Therapeutic inhibition of EZH2 in SHH-MB cells promotes tumor cell differentiation, impairs tumor growth and proliferation, and reduces stemness, suggesting that EZH2 represents a promising druggable target, which shows significantly reduced proliferation and impaired self-renewal in response to EZH2 inhibition (196–198). Human and mouse MB cells from the SHH-MB subgroup significantly reduced proliferation and impaired self-renewal in response to EZH2 inhibition (196, 197). Similarly, treatment with EZH2 inhibitors extended survival in SHH and Group 3 MB xenograft models (198). This suggests that EZH2 inhibition has promising evidence supporting reduced MB growth in a subset of MB in vitro and in vivo. However, inhibition of EZH2 in MB must be approached cautiously, as inhibition of EZH2 can lead to Gfi1 upregulation through epigenetic remodeling, promoting tumor progression in MYC-driven Group 3 MB (196, 199), supporting a nuanced role for EZH2 in MB.
In cancers, including MB, microRNAs (miRNAs) are implicated in cancer initiation and progression through their crucial role in regulating gene expression (200). MiRNAs can act as tumor suppressors or oncomiR. In MB, miRNA profiling identified several miRNAs with unique profiles, with only a few upregulated and the majority downregulated in MB (201–203). The relationship between miRNAs and the KMT, EZH2, is complex. For example, during development, miR-10 downregulates key midbrain markers, such as Otx2, and upregulates hindbrain markers, such as Gbx2 (204). In many cancer types, including some subgroups of MB, miR-10 family members are dysregulated (203, 205) leading to altered expression of their downstream targets. In group 3 MB, Otx2 is frequently overexpressed (206). When OTX2 is silenced in MB tumorsopheres, EZH2 and SUZ12 levels decreased, suggesting that OTX2 plays a role in the regulation of PRC2 (207). Alternatively, EZH2 can be regulated by specific miRNAs, such as miR-101-3p and miR-423-5p, which have been identified of negative regulators of EZH2 in MB (208). Another example is the exosomal miR-130b-3p, which functions as a tumor suppressor in vitro and in vivo in MB (209). Specifically, EZH2 was identified as a target gene of miR-101-3p in MB (208). In the absence of EZH2, the inhibitory effect of the exosomal miR-130b-3p is lost, suggesting that miR-130b-3p mediates MB progression and may be a potential therapeutic target for the treatment of pediatric MB.
In addition to MB, aberrant EZH2 expression has been reported in AT/RT (210). The absence of SMARCB1 protein in AT/RT promotes EZH2 expression (210, 211). Disruption of EZH2 via genetic or pharmacological inhibitors impairs cell growth, self-renewal and may potently sensitize ATRT cells to radiation therapy (69, 210). AT/RT tumors can also show a global increase in H3K37me3, independent of EZH2 expression (69). Although global increase in H3K27m3 is observed in ATRT, specific genomic regions were identified with higher H3K27ac occupancy in association with BET bromodomain-containing protein 4 (BET4) (212). In vitro and in vivo, combination therapy with EZH2 and BET4 inhibitors reduced cell proliferation and invasiveness (213). Interestingly, in SMARCB1-deficient AT/RT tumors, pharmacological inhibition of EZH2 induces the viral mimicry response (214).
Tazemetostat (EPZ-6438) is a potent and selective EZH2 inhibitor. Initial preclinical studies demonstrated that it induced apoptosis and differentiation in SMARCB1-deleted rhabdoid tumors in vitro and in vivo (215), with significant anti-tumor activity observed in rhabdoid tumor models, including AT/RT, but variable responses across other pediatric solid tumors (216). Subsequent preclinical studies demonstrated therapeutic potential in variety of pediatric CNS tumors, such as MB with wild type p53, AT/RT, or HGG, supporting further evaluation in pediatric brain tumors with EZH2 overexpression, though combination strategies may be needed to overcome resistance and intratumoral heterogeneity (198, 217). Encouraging preclinical evidence led to a phase 1 clinical trial (NCT02601937) examining tazemetostat monotherapy in pediatric patients with relapsed or refractory SMARCB1 (also known as INI1) negative tumors, such as malignant rhabdoid tumors (MRT), including AT/RT, or other SMARCB1-deficient tumors and synovial sarcoma. Among subjects with AT/RT, one had a complete response, and 5/21 had an objective response, with a 6.5-month duration of response (218). Additionally, in the phase 2 NCI-COG pediatric MATCH trial (NCT03213665), Arm C, tazemetostat was evaluated in a variety of pediatric tumors harboring EZH2 mutation or SMARCB1 or SMARCA4 loss (219). Within this trial, two pediatric CNS malignancies were represented, including AT/RT (n=8) and ependymoma (n=1), among other tumors, including MRT, epithelioid sarcoma, renal medullary carcinoma, hepatocellular carcinoma, Ewing sarcoma, and Langerhans cell histiocytosis (219). Although treatment with tazemetostat in this trial did not meet its primary efficacy endpoint, a diverse array of tumor diagnoses and molecular alterations were represented (219). Vejmelkova et al. reported the results of a small cohort of four pediatric patients with primary AT/RT treated with tazemetostat maintenance after the completion of upfront therapy with surgery, radiotherapy (older than 2 years), and chemotherapy. The most significant adverse effects were thrombocytopenia or other cytopenias requiring dose reduction (220). However, resistance to tazemetostat in patient-derived SMARCB1-deficient epithelioid sarcomas or rhabdoid tumors has been observed. Multiple factors can contribute to the development of resistance to EZH2 inhibitors. NSD1 is a histone H3 lysine 36 methyltransferase and was identified by CRISPR screens as a critical regulator of resistance to EZH2 inhibitors in AT/RT and extra-CNS rhabdoid tumors (221). Additionally, functional sequencing has uncovered distinct acquired mutations affecting the RB1/E2F axis that decouple EZH2-dependent differentiation from cell-cycle control, thus circumventing the drug’s intended mechanism (222). Beyond the intricacies of EZH2 inhibition, the broader landscape of histone modification offers additional avenues for therapeutic intervention.
G9a (euchromatic histone lysine methyltransferase 2, EHMT2) is another nuclear KMT belonging to the Su(var) 3–9 family that catalyzes histone H3K9 methylation (223, 224). G9a is higher in a subset of MB tumors, specifically those belonging to groups 3 and 4 (57). In conjunction with G9a, the repressor element-1 silencing transcription factor (REST) works to repress transcription by modifying chromatin through methylation of H3K9. In MB, increased expression of REST and, in turn, G9a activity leads to the subsequent downregulation of USP37 through methylation at its promoter region. USP37 is a component of the ubiquitin system implicated in the regulation of cell proliferation and reduced expression (225). Higher expression of G9a and REST both may be indicators of poor prognosis in this subset of MB (57, 225). Interestingly, higher levels of REST and USP7 (deubiquitylase) are reported in SHH-MB and are associated with increased LSD1/KDM1A expression (226). In vitro, cell migration was promoted by REST elevation in conjunction with elevated LSD1, and pharmacological inhibition of LSD1 decreased cell migration and viability (226). While REST is elevated in multiple MB subtypes, the distinct co-expression of opposing histone modifying enzymes like G9a and LSD1/KDM1A reveals that REST’s functional consequences are dependent on the specific epigenetic context, underscoring the complexity of its role in MB.
5.2.1.2 Histone demethylases
Like KMTs, the role of KDMs has been extensively studied in the context of cancer (227, 228). More specifically, in MB, multiple KDMs have been implicated with aberrant methylation patterns of H3K27 and H3K4 (56, 184). KDM1A, also known as lysine-specific histone demethylase 1A (LSD1), is a H3K4 specific KDM. It is overexpressed across all subgroups of MB (229). Lsd1/Kdm1a knockdown was associated with apoptosis and suppression of proliferation; similarly, using a KDM1A inhibitor, NCL-1, inhibited the growth of MB cells (229). Inhibition of LSD1 sensitized H3K27M gliomas to HDAC inhibitors, promoted differentiation pathways, and induced natural killer (NK) cell infiltration into the tumor microenvironment (148, 230). KDM6 subfamily members are involved in the demethylation of di- and trimethylated H3K27, and dysregulation of KDM6s plays an important role in various cancers (231). In MB, KDM6 plays a role in oncogenic processes and the tumor microenvironment (232). Mutations in both KDM6A/6B have been identified, and copy number loss of these KDM6 subfamily members, predominantly in group 4 MB (54, 183). Inhibition of the histone demethylases KDM6A and KDM6B by a pharmacological inhibitor, GSKJ4, increased global H3K27me3 in K27M-mutant brainstem gliomas, leading to reduced tumor growth, improved survival, and sensitized tumor cells to radiation therapy in preclinical models (233). Other KDMs that have been shown to have altered expression in MB include KDM3A (H3K9me2/me1 histone demethylase), KDM4C (H3K36me2/me3 histone demethylase), KDM5A/B (H3K4me2/me3 histone demethylases), and KDM7A (dual H3K9 and H3K27 histone demethylase) (54, 56).
Group 3 MB has the highest potential for metastasis (55). MYC amplification is considered one of the defining features of group 3 MB (54, 234), although it is thought not to be sufficient to promote tumorigenesis alone (235, 236). More recently, other pathways promoting metastasis were identified independent of MYC amplification. Prune exopolyphosphatase 1 (Prune-1) enhances the TGF-β pathway, with subsequent upregulation of Otx2 and Snail and downregulation of Pten (237). High expression of Prune-1 and Lsd1/Kdm1a are reported in group 3 MB, and Bibbò et al. suggest that LSD1/KDM1A is an epigenetic regulator of Prune-1 (238). In vitro, dual inhibition of PRUNE-1 and LSD1/KDM1A promoted differentiation and altered the tumor microenvironment in MB cells, identifying a potential therapeutic approach for group 3 MB tumors (238). Additionally, GFI1 and GFI1B work cooperatively with MYC to drive tumorigenesis in a subset of group 3 MB (239). Lee et al. (2019) demonstrated that Lsd1 physically interacts with Gfi1, and together, they are involved in the inhibition of genes involved in neuronal commitment and differentiation (240). The pharmacological inhibition of Lsd1 in Gfi1-driven MB in vitro and in vivo inhibits tumor cell growth and supports the idea that targeting Lsd1 may be an effective strategy for these tumors (240).
In cerebellar granular progenitor cells (CGPCs), the repressive PRC2 and G9A/G9A-like protein oppose the elevation of MLL4 and KDM7A activities to maintain REST homeostasis. However, alterations, either up- or downregulation in KDM7A activities, lead to dysregulation of REST homeostasis. Altered KDM7A expression in human SHH-MB leads to poor survival (241). This suggests that KDM7A may not act solely as a repressor or activator but may depend on the cellular context; therefore, targeting KDM7A in MB may require a nuanced approach to avoid unintended REST deregulation. This intricate interplay between KDM7A and REST homeostasis in SHH-MB exemplifies the challenge of targeting epigenetic regulators, such as KDMs and KMTs. The potential for unintended, system-wide effects due to the interconnected nature of epigenetic modifications highlights the necessity for comprehensive investigations to understand and mitigate these risks. This complexity extends beyond individual regulators. Disruption of global methylation patterns, or mutations in histone methyltransferases or demethylases, can cause alterations to normal cellular processes, contributing to oncogenesis by promoting uncontrolled cell growth and survival. Continued expansion of our understanding of the mechanisms regulating histone methylation in pediatric brain tumors, including the context-dependent roles of enzymes that regulate histone methylation will continue to provide insights into the biology of these cancers and help delineate potential and better treatment options for these devastating tumors.
5.2.2 Histone acetylation
Histone acetylation, like methylation, was one of the first histone PTMs described, and it is a dynamic process governed by the opposing actions of histone acetyl transferases (HATs) and histone deacetylases (HDACs) (47). The balance between acetylation and deacetylation is crucial for regulating various cellular processes, including gene expression, DNA repair, and cell cycle progression. Dysregulation of HATs and HDACs can lead to aberrant acetylation patterns.
5.2.2.1 Histone acetyl transferases
HATs add acetyl groups to lysine residues on histones lysine residues and are typically associated with open chromatin structure and enhanced gene expression (242). HATs serve as multifunctional transcriptional coactivators and facilitate acetylation of several histone lysine residues including H3K27 (243–246). H3K27ac is an activating mark associated with enhancers, super enhancers, and promoters. Pediatric brain tumors including MB, AT/RT, H3K27-altered DMG, H3-G34 DHG and ependymomas have distinct H3K27ac profiles that relate with epigenetic and tumor cell states (45, 66, 133, 148, 160, 161, 168, 176, 239, 247–253). HATs are classified into type A and B HATs. Type A HATs are subdivided into five families: the GNAT family, p300/CBP family, MYST family, basal TF family, and NRCF family (254). These proteins contain several homologous domains including the catalytic HAT domain and bromodomains (BRD) (243–246). In MB, somatic mutations involving the HATs CREBBP and EP300 (encoding CBP/KAT3A and P300, respectively) affecting histone acetylation regulation are observed across all subgroups (54, 56, 73, 182, 184). Germline mutations in CREBBP are associated with Rubinstein-Taybi syndrome (RTS), a neurodevelopmental disorder that predisposes individuals to CNS malignancies, including MB (255–257). Interestingly, during embryonic development, the loss of Crebbp in GNPs impairs cerebellar development, whereas postnatal loss of Crebbp synergizes with SHH signaling to enhance the growth of MB (258). Schoof et al. found that concurrent loss of CREBBP function and MYCN overexpression in neural stem cells resulted in the development of aggressive forebrain tumors, suggesting a critical mechanistic link between these two factors in oncogenesis (259). Several cancer cell lines, including MB subtypes, are sensitive to CBP/EP300 inhibitors. Shendy et al. (2024) demonstrated that A485 (HAT domain inhibitor) and CCS1477 (BRD domain inhibitor) have varying effects across tumor types, with Group 3 MB exhibiting particular sensitivity to BRD inhibition (260). Together, these findings underscore the importance of understanding the complex roles of HATs in pediatric brain cancers, as understanding their specific contributions to oncogenesis may reveal useful therapeutic vulnerabilities.
Dysregulation of other HATs are reported in MB. For example, the downregulation of the histone H4 lysine K16-specific acetyltransferase (MOF) and lower H4K16 acetylation in MB has been associated with lower survival rates (261, 262). H4K16ac is associated with DNA damage repair and gene expression (263). Although not explicitly reported for MB, the loss of global H4K16ac in concert with MOF may lead to pathogenesis via dysregulation of oncogenes or tumor suppressor genes, increased genomic instability, or dysregulation of cell cycle (263–265).
5.2.2.2 Histone deacetylases
In contrast to HATs, HDACs remove acetyl groups, which generally results in chromatin condensation and transcriptional repression (266). There are ~18 human HDACs, classified into five classes based on their sequence similarity to yeast deacetylases, domain composition, and dependence on specific cofactors (267). Alterations in HDAC expression have frequently been reported in pediatric brain cancers. In MB, increased HDAC2 expression is observed, especially in MYC-driven MB tumors (268, 269). Also, increased HDAC5 and HDAC9 expression is seen in a subset of MB and is associated with poor prognosis (270). In AT/RT, HDAC1 was reported to be differentially expressed, suggesting that HDAC inhibitors targeting HDAC1 may be beneficial compared to those with less specificity in young patients with ATRT (271).
Trichostatin A (TSA), the first reported potent and specific inhibitor of HDAC, led to alterations in cell proliferation and differentiation in vivo (272). Subsequently, vorinostat [also known as suberoylanilide hydroxamic acid (SAHA)], a Class I and Class IIb HDAC inhibitor, was shown to induce differentiation, growth arrest, and apoptosis in vitro (273, 274). Vorinostat effectively induces cell death in MB cell lines, patient-derived primary tumor cultures, and xenograft models, with minimal prohibitive toxicity observed in fibroblasts and animal models (4). In MB cell lines, the class I HDAC, HDAC2, is overexpressed in poor-prognosis subtypes, including SHH, Group 3, and Group 4, with MYC-amplified Group 3 cells demonstrating increased sensitivity to HDAC inhibition (269). Specifically, HDAC inhibition reduced metabolic activity and increased cell death in the MYC-amplified MB cells (269). Separately, high-throughput screening identified panobinostat (LBH-589), a pan-HDAC inhibitor, as highly effective against MYC-amplified Group 3 MB, with treatment decreasing MYC expression and inhibiting cell growth (275). To further elucidate the mechanism of HDAC inhibition in MYC-amplified MB, Ecker et al. (2020) investigated the interaction between MYC and HDAC2 and reported that HDAC inhibition disrupts the MYC-HDAC2 complex in MYC-amplified medulloblastoma, leading to reduced chromatin binding of MYC. This results in the downregulation of MYC-activated genes and the upregulation of MYC-repressed genes, effectively reversing the MYC-dependent transcriptional program and providing a therapeutic strategy for these MYC-amplified MB tumors (276).
A phase 1 trial in pediatric patients showed that Vorinostat was well-tolerated (277). However, despite promising preclinical and early clinical data, Vorinostat has shown limited efficacy in pediatric brain tumor clinical trials, including in DMGs (278). Ongoing trials are investigating Vorinostat in combination with other therapies for pediatric solid or CNS tumors (e.g., NCT02420613, NCT04308330, NCT06693284). Similarly, Panobinostat, another pan-HDAC inhibitor, suppressed MB leptomeningeal seeding in preclinical mouse models (279). Panobinostat was identified from a drug screening study to effectively kill H3K27M DMG cells (280). Panobinostat has been studied in several preclinical DMG models as monotherapy or in combination with various drugs (281–287). However, clinical trials of Panobinostat in DMGs have shown limited efficacy, although the drug was tolerated with expected toxicities, such as myelosuppression and diarrhea (288). Current Panobinostat trials are exploring blood-brain barrier disruption with focused ultrasound or direct intraventricular administration in DMG (NCT04804709, NCT04315064). Despite encouraging preclinical results and safety profiles, HDAC inhibitors have not demonstrated significant efficacy in pediatric brain tumors as monotherapy. Current strategies focus on combining HDAC inhibitors with cytotoxic chemotherapy, hypomethylating agents, or immunotherapy and on improving drug delivery across the blood-brain barrier. These approaches will be crucial for future clinical investigations. Overall, the role of HATs and HDACs in pediatric brain cancers highlights the therapeutic potential of both HAT and HDAC inhibitors.
6 Conclusion
The intricate landscape of pediatric brain cancers reveals a critical reliance on precise chromatin regulation and histone post-translational modifications. Unlike many adult tumors, pediatric malignancies do not bear a high burden of recurrent mutations, but rather aberrant epigenetic modifications. These disruptions, encompassing alterations in histone proteins, PTMs, and chromatin remodeling complexes, profoundly influence gene expression and contribute to oncogenesis. The diverse roles of histone modifications and the enzymes that regulate them—writers, readers, and erasers—underscore the complexity and importance of this field. By focusing on the impact of chromatin biology and modifications in pediatric brain tumors, we gain crucial insights into disease mechanisms and identify potential therapeutic targets for correcting aberrant chromatin states. Ultimately, a deeper understanding of these epigenetic vulnerabilities will pave the way for developing more effective and targeted therapies for these devastating childhood cancers.
Author contributions
EH: Writing – original draft, Writing – review & editing. DD: Writing – original draft, Writing – review & editing. SV: Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by Mark Foundation for Cancer Research (SV), National Institutes of Health Grant R01-NS110572 (SV), National Institutes of Health Grant R01-CA261926 (SV), National Institute of Health Grant R01-NS127799, Sontag Foundation (SV), Alex Lemonade Stand Foundation for Childhood Cancer (SV), Hyundai Hope on Wheels (SV), Mark Foundation for Cancer Research (SV), University of Michigan Chad Carr Pediatric Brain Tumor Center (EH, SV), ChadTough Defeat DIPG Foundation (SV), and National Institutes of Health Grant 5F31CA260935 (DD).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2016—2020. Neuro-Oncol. (2023) 25:iv1–iv99. doi: 10.1093/neuonc/noad149
2. Patel RR, Ramkissoon SH, Ross J, and Weintraub L. Tumor mutational burden and driver mutations: Characterizing the genomic landscape of pediatric brain tumors. Pediatr Blood Cancer. (2020) 67:e28338. doi: 10.1002/pbc.28338
3. Meléndez B, Campenhout CV, Rorive S, Remmelink M, Salmon I, and D’Haene N. Methods of measurement for tumor mutational burden in tumor tissue. Transl Lung Cancer Res. (2018) 7:661–7. doi: 10.21037/tlcr.2018.08.02
4. Noskova H, Kyr M, Pal K, Merta T, Mudry P, Polaskova K, et al. Assessment of tumor mutational burden in pediatric tumors by real-life whole-exome sequencing and in silico simulation of targeted gene panels: how the choice of method could affect the clinical decision? Cancers. (2020) 12:230. doi: 10.3390/cancers12010230
5. Kornberg RD. Chromatin structure: A repeating unit of histones and DNA. Science. (1974) 184:868–71. doi: 10.1126/science.184.4139.868
6. Luger K, Mäder AW, Richmond RK, Sargent DF, and Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. (1997) 389:251–60. doi: 10.1038/38444
7. Eickbush TH and Moudrianakis EN. The histone core complex: an octamer assembled by two sets of protein-protein interactions. Biochemistry. (1978) 17:4955–64. doi: 10.1021/bi00616a016
8. Arents G, Burlingame RW, Wang BC, Love WE, and Moudrianakis EN. The nucleosomal core histone octamer at 3.1 A resolution: a tripartite protein assembly and a left-handed superhelix. Proc Natl Acad Sci. (1991) 88:10148–52. doi: 10.1073/pnas.88.22.10148
9. Wang X, Lee RS, Alver BH, Haswell JR, Wang S, Mieczkowski J, et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat Genet. (2017) 49:289–95. doi: 10.1038/ng.3746
10. Wang W, Côté J, Xue Y, Zhou S, Khavari PA, Biggar SR, et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. (1996) 15:5370–82. doi: 10.1002/j.1460-2075.1996.tb00921.x
11. Fyodorov DV, Zhou B-R, Skoultchi AI, and Bai Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol. (2018) 19:192–206. doi: 10.1038/nrm.2017.94
12. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. (2022) 12:31–46. doi: 10.1158/2159-8290.cd-21-1059
13. Beck S, Bernstein BE, Campbell RM, Costello JF, Dhanak D, Ecker JR, et al. A blueprint for an international cancer epigenome consortium. A report from the AACR cancer epigenome task force. Cancer Res. (2012) 72:6319–24. doi: 10.1158/0008-5472.can-12-3658
14. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Dias Jr. LA, and Kinzler KW. Cancer genome landscapes. Science. (2013) 339:1546–58. doi: 10.1126/science.1235122
15. Berson A, Nativio R, Berger SL, and Bonini NM. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. (2018) 41:587–98. doi: 10.1016/j.tins.2018.05.005
16. Sharma H, Koirala S, Chew YL, and Konopka A. DNA damage and chromatin rearrangement work together to promote neurodegeneration. Mol Neurobiol. (2024) 62:1282–90. doi: 10.1007/s12035-024-04331-0
17. Nie Y, Song C, Huang H, Mao S, Ding K, and Tang H. Chromatin modifiers in human disease: from functional roles to regulatory mechanisms. Mol BioMed. (2024) 5:12. doi: 10.1186/s43556-024-00175-1
18. Kouzarides T. Chromatin modifications and their function. Cell. (2007) 128:693–705. doi: 10.1016/j.cell.2007.02.005
19. Allis CD and Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. (2016) 17:487–500. doi: 10.1038/nrg.2016.59
20. Strahl BD and Allis CD. The language of covalent histone modifications. Nature. (2000) 403:41–5. doi: 10.1038/47412
21. Biswas S and Rao CM. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur J Pharmacol. (2018) 837:8–24. doi: 10.1016/j.ejphar.2018.08.021
22. Herz H-M, Garruss A, and Shilatifard A. SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem Sci. (2013) 38:621–39. doi: 10.1016/j.tibs.2013.09.004
23. Malik S and Bhaumik SR. Mixed lineage leukemia: histone H3 lysine 4 methyltransferases from yeast to human. FEBS J. (2010) 277:1805–21. doi: 10.1111/j.1742-4658.2010.07607.x
24. Cao R and Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev. (2004) 14:155–64. doi: 10.1016/j.gde.2004.02.001
25. Sparmann A and van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. (2006) 6:846–56. doi: 10.1038/nrc1991
26. Brownell JE and Allis CD. An activity gel assay detects a single, catalytically active histone acetyltransferase subunit in Tetrahymena macronuclei. Proc Natl Acad Sci. (1995) 92:6364–8. doi: 10.1073/pnas.92.14.6364
27. Kleff S, Andrulis ED, Anderson CW, and Sternglanz R. Identification of a gene encoding a yeast histone H4 acetyltransferase (∗). J Biol Chem. (1995) 270:24674–7. doi: 10.1074/jbc.270.42.24674
28. Bannister AJ and Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. (1996) 384:641–3. doi: 10.1038/384641a0
29. Ogryzko VV, Schiltz RL, Russanova V, Howard BH, and Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. (1996) 87:953–9. doi: 10.1016/s0092-8674(00)82001-2
30. Phillips D. The presence of acetyl groups in histones. Biochem J. (1963) 87:258–63. doi: 10.1042/bj0870258
31. Schoelz JM and Riddle NC. Functions of HP1 proteins in transcriptional regulation. Epigenet Chromatin. (2022) 15:14. doi: 10.1186/s13072-022-00453-8
32. Musselman CA, Lalonde M-E, Côté J, and Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. (2012) 19:1218–27. doi: 10.1038/nsmb.2436
33. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. (2004) 119:941–53. doi: 10.1016/j.cell.2004.12.012
34. Hyun K, Jeon J, Park K, and Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. (2017) 49:e324–4. doi: 10.1038/emm.2017.11
35. Lanouette S, Mongeon V, Figeys D, and Couture J. The functional diversity of protein lysine methylation. Mol Syst Biol. (2014) 10:724. doi: 10.1002/msb.134974
36. Black JC, Van Rechem C, and Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. (2012) 48:491–507. doi: 10.1016/j.molcel.2012.11.006
37. Bian C, Xu C, Ruan J, Lee KK, Burke TL, Tempel W, et al. Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation. EMBO J. (2011) 30:2829–42. doi: 10.1038/emboj.2011.193
38. Li J, Xue X, Ruan J, Wu M, Zhu Z, and Zang J. Cloning, purification, crystallization and preliminary crystallographic analysis of the tandem tudor domain of Sgf29 from Saccharomyces cerevisiae. Acta Crystallogr Sect F. (2010) 66:902–4. doi: 10.1107/s1744309110016726
39. Lewis R, Li YD, Hoffman L, Hashizume R, Gravohac G, Rice G, et al. Global reduction of H3K4me3 improves chemotherapeutic efficacy for pediatric ependymomas. Neoplasia. (2019) 21:505–15. doi: 10.1016/j.neo.2019.03.012
40. Wadhwani N, Nayak S, Wang Y, Hashizume R, Jie C, Mania-Farnell B, et al. WDR82-mediated H3K4me3 is associated with tumor proliferation and therapeutic efficacy in pediatric high-grade gliomas. Cancers. (2023) 15:3429. doi: 10.3390/cancers15133429
41. Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, and Strahl BD. A novel domain in set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. (2005) 25:3305–16. doi: 10.1128/mcb.25.8.3305-3316.2005
42. Huang TY-T, Piunti A, Qi J, Morgan M, Bartom E, Shilatifard A, et al. Effects of H3.3G34V mutation on genomic H3K36 and H3K27 methylation patterns in isogenic pediatric glioma cells. Acta Neuropathol Commun. (2020) 8:219. doi: 10.1186/s40478-020-01092-4
43. Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. (2013) 340:857–61. doi: 10.1126/science.1232245
44. Venneti S, Garimella MT, Sullivan LM, Martinez D, Huse JT, Heguy A, et al. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. (2013) 23:558–64. doi: 10.1111/bpa.12042
45. Krug B, Jay ND, Harutyunyan AS, Deshmukh S, Marchione DM, Guilhamon P, et al. Pervasive H3K27 acetylation leads to ERV expression and a therapeutic vulnerability in H3K27M gliomas. Cancer Cell. (2019) 35:782–797.e8. doi: 10.1016/j.ccell.2019.04.004
46. Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun Z-W, Schmid M, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. (2000) 406:593–9. doi: 10.1038/35020506
47. Allfrey VG, Faulkner R, and Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci. (1964) 51:786–94. doi: 10.1073/pnas.51.5.786
48. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science. (2002) 298:1039–43. doi: 10.1126/science.1076997
49. Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. (2006) 439:871–4. doi: 10.1038/nature04431
50. Dillon SC, Zhang X, Trievel RC, and Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. (2005) 6:227. doi: 10.1186/gb-2005-6-8-227
51. Weirich S, Khella MS, and Jeltsch A. Structure, activity and function of the suv39h1 and suv39h2 protein lysine methyltransferases. Life. (2021) 11:703. doi: 10.3390/life11070703
52. Margueron R and Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. (2011) 469:343–9. doi: 10.1038/nature09784
53. Northcott PA, Shih DJH, Peacock J, Garzia L, Morrissy AS, Zichner T, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. (2012) 488:49–56. doi: 10.1038/nature11327
54. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. (2017) 547:311–7. doi: 10.1038/nature22973
55. Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. (2017) 31:737–754.e6. doi: 10.1016/j.ccell.2017.05.005
56. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. (2012) 488:43–8. doi: 10.1038/nature11213
57. Souza BK, Freire NH, Jaeger M, de Farias CB, Brunetto AL, Brunetto AT, et al. G9a/EHMT2 is a potential prognostic biomarker and molecular target in SHH medulloblastoma. NeuroMolecular Med. (2022) 24:392–8. doi: 10.1007/s12017-022-08702-5
58. Hendrikse LD, Haldipur P, Saulnier O, Millman J, Sjoboen AH, Erickson AW, et al. Failure of human rhombic lip differentiation underlies medulloblastoma formation. Nature. (2022) 609:1021–8. doi: 10.1038/s41586-022-05215-w
59. Asuthkar S, Venkataraman S, Avilala J, Shishido K, Vibhakar R, Veo B, et al. SMYD3 promotes cell cycle progression by inducing cyclin D3 transcription and stabilizing the cyclin D1 protein in medulloblastoma. Cancers. (2022) 14:1673. doi: 10.3390/cancers14071673
60. Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. (2012) 482:226–31. doi: 10.1038/nature10833
61. Fontebasso AM, Schwartzentruber J, Khuong-Quang D-A, Liu X-Y, Sturm D, Korshunov A, et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. (2013) 125:659–69. doi: 10.1007/s00401-013-1095-8
62. Petralia F, Tignor N, Reva B, Koptyra M, Chowdhury S, Rykunov D, et al. Integrated proteogenomic characterization across major histological types of pediatric brain cancer. Cell. (2020) 183:1962–1985.e31. doi: 10.1016/j.cell.2020.10.044
63. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. (2014) 46:444–50. doi: 10.1038/ng.2938
64. Castel D, Kergrohen T, Tauziède-Espariat A, Mackay A, Ghermaoui S, Lechapt E, et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol. (2020) 139:1109–13. doi: 10.1007/s00401-020-02142-w
65. Griesinger AM, Birks DK, Donson AM, Amani V, Hoffman LM, Waziri A, et al. Characterization of distinct immunophenotypes across pediatric brain tumor types. J Immunol. (2013) 191:4880–8. doi: 10.4049/jimmunol.1301966
66. Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. (2016) 29:379–93. doi: 10.1016/j.ccell.2016.02.001
67. Hasselblatt M, Gesk S, Oyen F, Rossi S, Viscardi E, Giangaspero F, et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/Rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol. (2011) 35:933–5. doi: 10.1097/pas.0b013e3182196a39
68. Hasselblatt M, Nagel I, Oyen F, Bartelheim K, Russell RB, Schüller U, et al. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. (2014) 128:453–6. doi: 10.1007/s00401-014-1323-x
69. Alimova I, Birks DK, Harris PS, Knipstein JA, Venkataraman S, Marquez VE, et al. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro-Oncol. (2013) 15:149–60. doi: 10.1093/neuonc/nos285
70. Frühwald MC, Hasselblatt M, Nemes K, Bens S, Steinbügl M, Johann PD, et al. Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro-Oncol. (2020) 22:1006–17. doi: 10.1093/neuonc/noz244
71. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncol. (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
72. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho Y-J, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. (2012) 123:465–72. doi: 10.1007/s00401-011-0922-z
73. Jones DTW, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. (2012) 488:100–5. doi: 10.1038/nature11284
74. Kool M, Korshunov A, Remke M, Jones DTW, Schlanstein M, Northcott PA, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. (2012) 123:473–84. doi: 10.1007/s00401-012-0958-8
75. Schwalbe EC, Lindsey JC, Nakjang S, Crosier S, Smith AJ, Hicks D, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol. (2017) 18:958–71. doi: 10.1016/s1470-2045(17)30243-7
76. Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. (2012) 44:251–3. doi: 10.1038/ng.1102
77. Maze I, Noh K-M, Soshnev AA, and Allis CD. Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat Rev Genet. (2014) 15:259–71. doi: 10.1038/nrg3673
78. Kardalinou E, Eick S, Albig W, and Doenecke D. Association of a human H1 histone gene with an H2A pseudogene and genes encoding H2B.1 and H3.1 histones. J Cell Biochem. (1993) 52:375–83. doi: 10.1002/jcb.240520402
79. Marzluff WF, Gongidi P, Woods KR, Jin J, and Maltais LJ. The human and mouse replication-dependent histone genes. Genomics. (2002) 80:487–98. doi: 10.1006/geno.2002.6850
80. Reisz Z, Pereira R, Nevis S, Mackay A, Bhaw L, Grabovska Y, et al. An exceptionally rare case of a diffuse midline glioma with concomitant H3.1 K27M and G34R mutations in the HIST1H3C (H3C3) gene. Acta Neuropathol Commun. (2025) 13:7. doi: 10.1186/s40478-024-01899-5
81. Kara B, Danyeli AE, Öztürk M, Ertan K, and Köksal Y. A pediatric bithalamic high grade glioma with concomitant H3K27M and EGFR mutations. Turk J Pediatr. (2022) 64:754–8. doi: 10.24953/turkjped.2021.1140
82. Lester A and McDonald KL. Intracranial ependymomas: molecular insights and translation to treatment. Brain Pathol. (2020) 30:3–12. doi: 10.1111/bpa.12781
83. Pajtler KW, Witt H, Sill M, Jones DTW, Hovestadt V, Kratochwil F, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. (2015) 27:728–43. doi: 10.1016/j.ccell.2015.04.002
84. Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stütz AM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. (2014) 506:445–50. doi: 10.1038/nature13108
85. Bayliss J, Mukherjee P, Lu C, Jain SU, Chung C, Martinez D, et al. Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci Transl Med. (2016) 8:366ra161. doi: 10.1126/scitranslmed.aah6904
86. Panwalkar P, Clark J, Ramaswamy V, Hawes D, Yang F, Dunham C, et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. (2017) 134:705–14. doi: 10.1007/s00401-017-1752-4
87. St Pierre R and Kadoch C. Mammalian SWI/SNF complexes in cancer: emerging therapeutic opportunities. Curr Opin Genet Dev. (2017) 42:56–67. doi: 10.1016/j.gde.2017.02.004
88. Mittal P and Roberts CWM. The SWI/SNF complex in cancer — biology, biomarkers and therapy. Nat Rev Clin Oncol. (2020) 17:435–48. doi: 10.1038/s41571-020-0357-3
89. Rakic P and Sidman RL. Histogenesis of cortical layers in human cerebellum, particularly the lamina dissecans. J Comp Neurol. (1970) 139:473–500. doi: 10.1002/cne.901390407
90. Haldipur P, Aldinger KA, Bernardo S, Deng M, Timms AE, Overman LM, et al. Spatiotemporal expansion of primary progenitor zones in the developing human cerebellum. Science. (2019) 366:454–60. doi: 10.1126/science.aax7526
91. Leto K, Arancillo M, Becker EBE, Buffo A, Chiang C, Ding B, et al. Consensus paper: cerebellar development. Cerebellum. (2016) 15:789–828. doi: 10.1007/s12311-015-0724-2
92. Sudarov A, Turnbull RK, Kim EJ, Lebel-Potter M, Guillemot F, and Joyner AL. Ascl1 genetics reveals insights into cerebellum local circuit assembly. J Neurosci. (2011) 31:11055–69. doi: 10.1523/jneurosci.0479-11.2011
93. Haldipur P, Dang D, and Millen KJ. Chapter 2 embryology. Handb Clin Neurol. (2018) 154:29–44. doi: 10.1016/b978-0-444-63956-1.00002-3
94. Leto K, Carletti B, Williams IM, Magrassi L, and Rossi F. Different types of cerebellar GABAergic interneurons originate from a common pool of multipotent progenitor cells. J Neurosci. (2006) 26:11682–94. doi: 10.1523/jneurosci.3656-06.2006
95. Englund C, Kowalczyk T, Daza RAM, Dagan A, Lau C, Rose MF, et al. Unipolar brush cells of the cerebellum are produced in the rhombic lip and migrate through developing white matter. J Neurosci. (2006) 26:9184–95. doi: 10.1523/jneurosci.1610-06.2006
96. Bandeira F, Lent R, and Herculano-Houzel S. Changing numbers of neuronal and non-neuronal cells underlie postnatal brain growth in the rat. Proc Natl Acad Sci. (2009) 106:14108–13. doi: 10.1073/pnas.0804650106
97. Mätlik K, Govek E-E, and Hatten ME. Histone bivalency in CNS development. Genes Dev. (2025) 39:428–44. doi: 10.1101/gad.352306.124
98. Yang Y, Yamada T, and Bonni A. “Epigenetic Regulation of the Cerebellum,” in Handbook of the cerebellum and cerebellar disorders (Springer, Cham) (2021), 409–28. doi: 10.1007/978-3-030-23810-0_110
99. Feng X, Juan AH, Wang HA, Ko KD, Zare H, and Sartorelli V. Polycomb Ezh2 controls the fate of GABAergic neurons in the embryonic cerebellum. Development. (2016) 143:1971–80. doi: 10.1242/dev.132902
100. Lewis PM, Gritli-Linde A, Smeyne R, Kottmann A, and McMahon AP. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev Biol. (2004) 270:393–410. doi: 10.1016/j.ydbio.2004.03.007
101. Canettieri G, Marcotullio LD, Greco A, Coni S, Antonucci L, Infante P, et al. Histone deacetylase and Cullin3–RENKCTD11 ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. (2010) 12:132–42. doi: 10.1038/ncb2013
102. Montgomery RL, Hsieh J, Barbosa AC, Richardson JA, and Olson EN. Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc Natl Acad Sci. (2009) 106:7876–81. doi: 10.1073/pnas.0902750106
103. Norwood J, Franklin JM, Sharma D, and D’Mello SR. Histone deacetylase 3 is necessary for proper brain development*. J Biol Chem. (2014) 289:34569–82. doi: 10.1074/jbc.m114.576397
104. Martínez-Cerdeño V, Lemen JM, Chan V, Wey A, Lin W, Dent SR, et al. N-Myc and GCN5 regulate significantly overlapping transcriptional programs in neural stem cells. PloS One. (2012) 7:e39456. doi: 10.1371/journal.pone.0039456
105. Whittaker DE, Riegman KLH, Kasah S, Mohan C, Yu T, Sala BP, et al. The chromatin remodeling factor CHD7 controls cerebellar development by regulating reelin expression. J Clin Invest. (2017) 127:874–87. doi: 10.1172/jci83408
106. Alvarez-Saavedra M, Repentigny YD, Lagali PS, Ram EVSR, Yan K, Hashem E, et al. Snf2h-mediated chromatin organization and histone H1 dynamics govern cerebellar morphogenesis and neural maturation. Nat Commun. (2014) 5:4181. doi: 10.1038/ncomms5181
107. Moreno N, Schmidt C, Ahlfeld J, Pöschl J, Dittmar S, Pfister SM, et al. Loss of smarc proteins impairs cerebellar development. J Neurosci. (2014) 34:13486–91. doi: 10.1523/jneurosci.2560-14.2014
108. Frank CL, Liu F, Wijayatunge R, Song L, Biegler MT, Yang MG, et al. Regulation of chromatin accessibility and Zic binding at enhancers in the developing cerebellum. Nat Neurosci. (2015) 18:647–56. doi: 10.1038/nn.3995
109. Pal S, Gupta R, Kim H, Wickramasinghe P, Baubet V, Showe LC, et al. Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res. (2011) 21:1260–72. doi: 10.1101/gr.120535.111
110. Song C-X, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. (2011) 29:68–72. doi: 10.1038/nbt.1732
111. Yang Y, Yamada T, Hill KK, Hemberg M, Reddy NC, Cho HY, et al. Chromatin remodeling inactivates activity genes and regulates neural coding. Science. (2016) 353:300–5. doi: 10.1126/science.aad4225
112. Zanin JP, Abercrombie E, and Friedman WJ. Proneurotrophin-3 promotes cell cycle withdrawal of developing cerebellar granule cell progenitors via the p75 neurotrophin receptor. eLife. (2016) 5:e16654. doi: 10.7554/elife.16654
113. Zhu X, Girardo D, Govek E-E, John K, Mellén M, Tamayo P, et al. Role of Tet1/3 genes and chromatin remodeling genes in cerebellar circuit formation. Neuron. (2016) 89:100–12. doi: 10.1016/j.neuron.2015.11.030
114. Kriaucionis S and Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science. (2009) 324:929–30. doi: 10.1126/science.1169786
115. Szulwach KE, Li X, Li Y, Song C-X, Wu H, Dai Q, et al. 5-hmC–mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. (2011) 14:1607–16. doi: 10.1038/nn.2959
116. Yamada T, Yang Y, Hemberg M, Yoshida T, Cho HY, Murphy JP, et al. Promoter decommissioning by the nuRD chromatin remodeling complex triggers synaptic connectivity in the mammalian brain. Neuron. (2014) 83:122–34. doi: 10.1016/j.neuron.2014.05.039
117. Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones DTW, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. (2012) 22:425–37. doi: 10.1016/j.ccr.2012.08.024
118. Buczkowicz P, Bartels U, Bouffet E, Becher O, and Hawkins C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol. (2014) 128:573–81. doi: 10.1007/s00401-014-1319-6
119. Castel D, Philippe C, Kergrohen T, Sill M, Merlevede J, Barret E, et al. Transcriptomic and epigenetic profiling of ‘diffuse midline gliomas, H3 K27M-mutant’ discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol Commun. (2018) 6:117. doi: 10.1186/s40478-018-0614-1
120. Khuong-Quang D-A, Buczkowicz P, Rakopoulos P, Liu X-Y, Fontebasso AM, Bouffet E, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. (2012) 124:439–47. doi: 10.1007/s00401-012-0998-0
121. Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell. (2017) 32:520–537.e5. doi: 10.1016/j.ccell.2017.08.017
122. Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset P-O, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet. (2014) 46:462–6. doi: 10.1038/ng.2950
123. Pajovic S, Siddaway R, Bridge T, Sheth J, Rakopoulos P, Kim B, et al. Epigenetic activation of a RAS/MYC axis in H3.3K27M-driven cancer. Nat Commun. (2020) 11:6216. doi: 10.1038/s41467-020-19972-7
124. Casillo SM, Gatesman TA, Chilukuri A, Varadharajan S, Johnson BJ, Premkumar DRD, et al. An ERK5-PFKFB3 axis regulates glycolysis and represents a therapeutic vulnerability in pediatric diffuse midline glioma. Cell Rep. (2024) 43:113557. doi: 10.1016/j.celrep.2023.113557
125. Koncar RF, Dey BR, Stanton A-CJ, Agrawal N, Wassell ML, McCarl LH, et al. Identification of novel RAS signaling therapeutic vulnerabilities in diffuse intrinsic pontine gliomas. Cancer Res. (2019) 79:4026–41. doi: 10.1158/0008-5472.can-18-3521
126. Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. (2016) 7:11316. doi: 10.1038/ncomms11316
127. Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DTW, Kool M, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. (2013) 24:660–72. doi: 10.1016/j.ccr.2013.10.006
128. Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stützer A, et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell. (2014) 53:49–62. doi: 10.1016/j.molcel.2013.10.030
129. Margueron R, Justin N, Ohno K, Sharpe ML, Son J, III WJD, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. (2009) 461:762–7. doi: 10.1038/nature08398
130. Oksuz O, Narendra V, Lee C-H, Descostes N, LeRoy G, Raviram R, et al. Capturing the onset of PRC2-mediated repressive domain formation. Mol Cell. (2018) 70:1149–1162.e5. doi: 10.1016/j.molcel.2018.05.023
131. Jain SU, Rashoff AQ, Krabbenhoft SD, Hoelper D, Do TJ, Gibson TJ, et al. H3 K27M and EZHIP impede H3K27-methylation spreading by inhibiting allosterically stimulated PRC2. Mol Cell. (2020) 80:726–735.e7. doi: 10.1016/j.molcel.2020.09.028
132. Mohammad F, Weissmann S, Leblanc B, Pandey DP, Højfeldt JW, Comet I, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. (2017) 23:483–92. doi: 10.1038/nm.4293
133. Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med. (2017) 23:493–500. doi: 10.1038/nm.4296
134. Harutyunyan AS, Krug B, Chen H, Papillon-Cavanagh S, Zeinieh M, Jay ND, et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat Commun. (2019) 10:1262. doi: 10.1038/s41467-019-09140-x
135. Jain SU, Do TJ, Lund PJ, Rashoff AQ, Diehl KL, Cieslik M, et al. PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat Commun. (2019) 10:2146. doi: 10.1038/s41467-019-09981-6
136. Fang D, Gan H, Cheng L, Lee J-H, Zhou H, Sarkaria JN, et al. H3.3K27M mutant proteins reprogram epigenome by sequestering the PRC2 complex to poised enhancers. eLife. (2018) 7:e36696. doi: 10.7554/elife.36696
137. Chan K-M, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. (2013) 27:985–90. doi: 10.1101/gad.217778.113
138. Chaouch A, Berlandi J, Chen CCL, Frey F, Badini S, Harutyunyan AS, et al. Histone H3.3 K27M and K36M mutations de-repress transposable elements through perturbation of antagonistic chromatin marks. Mol Cell. (2021) 81:4876–4890.e7. doi: 10.1016/j.molcel.2021.10.008
139. Siddaway R, Canty L, Pajovic S, Milos S, Coyaud E, Sbergio S-G, et al. Oncohistone interactome profiling uncovers contrasting oncogenic mechanisms and identifies potential therapeutic targets in high grade glioma. Acta Neuropathol. (2022) 144:1027–48. doi: 10.1007/s00401-022-02489-2
140. Furth N, Algranati D, Dassa B, Beresh O, Fedyuk V, Morris N, et al. H3-K27M-mutant nucleosomes interact with MLL1 to shape the glioma epigenetic landscape. Cell Rep. (2022) 39:110836. doi: 10.1016/j.celrep.2022.110836
141. Diehl KL, Ge EJ, Weinberg DN, Jani KS, Allis CD, and Muir TW. PRC2 engages a bivalent H3K27M-H3K27me3 dinucleosome inhibitor. Proc Natl Acad Sci. (2019) 116:22152–7. doi: 10.1073/pnas.1911775116
142. Silveira AB, Kasper LH, Fan Y, Jin H, Wu G, Shaw TI, et al. H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth. vivo. Acta Neuropathol. (2019) 137:637–55. doi: 10.1007/s00401-019-01975-4
143. Larson JD, Kasper LH, Paugh BS, Jin H, Wu G, Kwon C-H, et al. Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell. (2019) 35:140–155.e7. doi: 10.1016/j.ccell.2018.11.015
144. Harutyunyan AS, Chen H, Lu T, Horth C, Nikbakht H, Krug B, et al. H3K27M in gliomas causes a one-step decrease in H3K27 methylation and reduced spreading within the constraints of H3K36 methylation. Cell Rep. (2020) 33:108390. doi: 10.1016/j.celrep.2020.108390
145. An S, Camarillo JM, Huang TY-T, Li D, Morris JA, Zoltek MA, et al. Histone tail analysis reveals H3K36me2 and H4K16ac as epigenetic signatures of diffuse intrinsic pontine glioma. J Exp Clin Cancer Res. (2020) 39:261. doi: 10.1186/s13046-020-01773-x
146. Stafford JM, Lee C-H, Voigt P, Descostes N, Saldaña-Meyer R, Yu J-R, et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv. (2018) 4:eaau5935. doi: 10.1126/sciadv.aau5935
147. Yu J-R, LeRoy G, Bready D, Frenster JD, Saldaña-Meyer R, Jin Y, et al. The H3K36me2 writer-reader dependency in H3K27M-DIPG. Sci Adv. (2021) 7:eabg7444. doi: 10.1126/sciadv.abg7444
148. Anastas JN, Zee BM, Kalin JH, Kim M, Guo R, Alexandrescu S, et al. Re-programing chromatin with a bifunctional LSD1/HDAC inhibitor induces therapeutic differentiation in DIPG. Cancer Cell. (2019) 36:528–544.e10. doi: 10.1016/j.ccell.2019.09.005
149. Ragazzini R, Pérez-Palacios R, Baymaz IH, Diop S, Ancelin K, Zielinski D, et al. EZHIP constrains Polycomb Repressive Complex 2 activity in germ cells. Nat Commun. (2019) 10:3858. doi: 10.1038/s41467-019-11800-x
150. Pajtler KW, Wen J, Sill M, Lin T, Orisme W, Tang B, et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. (2018) 136:211–26. doi: 10.1007/s00401-018-1877-0
151. Hübner J-M, Müller T, Papageorgiou DN, Mauermann M, Krijgsveld J, Russell RB, et al. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro-Oncol. (2019) 21:noz058. doi: 10.1093/neuonc/noz058
152. Piunti A, Smith ER, Morgan MAJ, Ugarenko M, Khaltyan N, Helmin KA, et al. CATACOMB: An endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism. Sci Adv. (2019) 5:eaax2887. doi: 10.1126/sciadv.aax2887
153. Panwalkar P, Tamrazi B, Dang D, Chung C, Sweha S, Natarajan SK, et al. Targeting integrated epigenetic and metabolic pathways in lethal childhood PFA ependymomas. Sci Transl Med. (2021) 13:eabc0497. doi: 10.1126/scitranslmed.abc0497
154. Pun M, Pratt D, Nano PR, Joshi PK, Jiang L, Englinger B, et al. Common molecular features of H3K27M DMGs and PFA ependymomas map to hindbrain developmental pathways. Acta Neuropathol Commun. (2023) 11:25. doi: 10.1186/s40478-023-01514-z
155. Wiese M, Hamdan FH, Kubiak K, Diederichs C, Gielen GH, Nussbaumer G, et al. Combined treatment with CBP and BET inhibitors reverses inadvertent activation of detrimental super enhancer programs in DIPG cells. Cell Death Dis. (2020) 11:673. doi: 10.1038/s41419-020-02800-7
156. Nagaraja S, Quezada MA, Gillespie SM, Arzt M, Lennon JJ, Woo PJ, et al. Histone variant and cell context determine H3K27M reprogramming of the enhancer landscape and oncogenic state. Mol Cell. (2019) 76:965–980.e12. doi: 10.1016/j.molcel.2019.08.030
157. Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, Zhang L, et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell. (2017) 31:635–652.e6. doi: 10.1016/j.ccell.2017.03.011
158. Brien GL, Bressan RB, Monger C, Gannon D, Lagan E, Doherty AM, et al. Simultaneous disruption of PRC2 and enhancer function underlies histone H3.3-K27M oncogenic activity in human hindbrain neural stem cells. Nat Genet. (2021) 53:1221–32. doi: 10.1038/s41588-021-00897-w
159. Wang J, Huang TY-T, Hou Y, Bartom E, Lu X, Shilatifard A, et al. Epigenomic landscape and 3D genome structure in pediatric high-grade glioma. Sci Adv. (2021) 7:eabg4126. doi: 10.1126/sciadv.abg4126
160. Mack SC, Pajtler KW, Chavez L, Okonechnikov K, Bertrand KC, Wang X, et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature. (2018) 553:101–5. doi: 10.1038/nature25169
161. Okonechnikov K, Camgöz A, Chapman O, Wani S, Park DE, Hübner J-M, et al. 3D genome mapping identifies subgroup-specific chromosome conformations and tumor-dependency genes in ependymoma. Nat Commun. (2023) 14:2300. doi: 10.1038/s41467-023-38044-0
162. Liu I, Jiang L, Samuelsson ER, Salas SM, Beck A, Hack OA, et al. The landscape of tumor cell states and spatial organization in H3-K27M mutant diffuse midline glioma across age and location. Nat Genet. (2022) 54:1881–94. doi: 10.1038/s41588-022-01236-3
163. Gojo J, Englinger B, Jiang L, Hübner JM, Shaw ML, Hack OA, et al. Single-cell RNA-seq reveals cellular hierarchies and impaired developmental trajectories in pediatric ependymoma. Cancer Cell. (2020) 38:44–59.e9. doi: 10.1016/j.ccell.2020.06.004
164. Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, Mathewson ND, et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science. (2018) 360:331–5. doi: 10.1126/science.aao4750
165. Gillen AE, Riemondy KA, Amani V, Griesinger AM, Gilani A, Venkataraman S, et al. Single-cell RNA sequencing of childhood ependymoma reveals neoplastic cell subpopulations that impact molecular classification and etiology. Cell Rep. (2020) 32:108023. doi: 10.1016/j.celrep.2020.108023
166. Korshunov A, Capper D, Reuss D, Schrimpf D, Ryzhova M, Hovestadt V, et al. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol. (2016) 131:137–46. doi: 10.1007/s00401-015-1493-1
167. Khazaei S, Chen CCL, Andrade AF, Kabir N, Azarafshar P, Morcos SM, et al. Single substitution in H3.3G34 alters DNMT3A recruitment to cause progressive neurodegeneration. Cell. (2023) 186:1162–1178.e20. doi: 10.1016/j.cell.2023.02.023
168. Chen CCL, Deshmukh S, Jessa S, Hadjadj D, Lisi V, Andrade AF, et al. Histone H3.3G34-mutant interneuron progenitors co-opt PDGFRA for gliomagenesis. Cell. (2020) 183:1617–1633.e22. doi: 10.1016/j.cell.2020.11.012
169. Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. (2013) 73:6219–29. doi: 10.1158/0008-5472.can-13-1491
170. Abdallah AS, Cardona HJ, Gadd SL, Brat DJ, Powla PP, Alruwalli WS, et al. Novel genetically engineered H3.3G34R model reveals cooperation with ATRX loss in upregulation of Hoxa cluster genes and promotion of neuronal lineage. Neuro-Oncol Adv. (2023) 5:vdad003. doi: 10.1093/noajnl/vdad003
171. Jain SU, Khazaei S, Marchione DM, Lundgren SM, Wang X, Weinberg DN, et al. Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc Natl Acad Sci. (2020) 117:27354–64. doi: 10.1073/pnas.2006076117
172. Zhang Y, Shan C-M, Wang J, Bao K, Tong L, and Jia S. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci Rep. (2017) 7:43906. doi: 10.1038/srep43906
173. Nguyen AV, Soto JM, Gonzalez S-M, Murillo J, Trumble ER, Shan FY, et al. H3G34-mutant gliomas—A review of molecular pathogenesis and therapeutic options. Biomedicines. (2023) 11:2002. doi: 10.3390/biomedicines11072002
174. Fang J, Huang Y, Mao G, Yang S, Rennert G, Gu L, et al. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3–MutSα interaction. Proc Natl Acad Sci. (2018) 115:9598–603. doi: 10.1073/pnas.1806355115
175. Sweha SR, Chung C, Natarajan SK, Panwalkar P, Pun M, Ghali A, et al. Epigenetically defined therapeutic targeting in H3.3G34R/V high-grade gliomas. Sci Transl Med. (2021) 13:eabf7860. doi: 10.1126/scitranslmed.abf7860
176. Liu I, Cruzeiro GAV, Bjerke L, Rogers RF, Grabovska Y, Beck A, et al. GABAergic neuronal lineage development determines clinically actionable targets in diffuse hemispheric glioma, H3G34-mutant. Cancer Cell. (2024) 42:1528–1548.e17. doi: 10.1016/j.ccell.2024.08.006
177. Garcia-Fabiani MB, Haase S, Banerjee K, McClellan B, Zhu Z, Mujeeb A, et al. H3.3-G34R mutation-mediated epigenetic reprogramming leads to enhanced efficacy of immune stimulatory gene therapy in pediatric high-grade gliomas. bioRxiv. (2023) 2023:6. doi: 10.1101/2023.06.13.544658
178. Bagert JD, Mitchener MM, Patriotis AL, Dul BE, Wojcik F, Nacev BA, et al. Oncohistone mutations enhance chromatin remodeling and alter cell fates. Nat Chem Biol. (2021) 17:403–11. doi: 10.1038/s41589-021-00738-1
179. Nacev BA, Feng L, Bagert JD, Lemiesz AE, Gao J, Soshnev AA, et al. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature. (2019) 567:473–8. doi: 10.1038/s41586-019-1038-1
180. Bonner ER, Dawood A, Gordish-Dressman H, Eze A, Bhattacharya S, Yadavilli S, et al. Pan-cancer atlas of somatic core and linker histone mutations. NPJ Genom Med. (2023) 8:23. doi: 10.1038/s41525-023-00367-8
181. Haltom AR, Toll SA, Cheng D, Maegawa S, Gopalakrishnan V, and Khatua S. Medulloblastoma epigenetics and the path to clinical innovation. J Neuro-Oncol. (2020) 150:35–46. doi: 10.1007/s11060-020-03591-9
182. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC-H, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. (2011) 331:435–9. doi: 10.1126/science.1198056
183. Dubuc AM, Remke M, Korshunov A, Northcott PA, Zhan SH, Mendez-Lago M, et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. (2013) 125:373–84. doi: 10.1007/s00401-012-1070-9
184. Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. (2009) 41:465–72. doi: 10.1038/ng.336
185. Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, et al. Control of developmental regulators by polycomb in human embryonic stem cells. Cell. (2006) 125:301–13. doi: 10.1016/j.cell.2006.02.043
186. Dhar S, Gadd S, Patel P, Vaynshteyn J, Raju GP, Hashizume R, et al. A tumor suppressor role for EZH2 in diffuse midline glioma pathogenesis. Acta Neuropathol Commun. (2022) 10:47. doi: 10.1186/s40478-022-01336-5
187. Zhang Y, Dong W, Zhu J, Wang L, Wu X, and Shan H. Combination of EZH2 inhibitor and BET inhibitor for treatment of diffuse intrinsic pontine glioma. Cell Biosci. (2017) 7:56. doi: 10.1186/s13578-017-0184-0
188. Michealraj KA, Kumar SA, Kim LJY, Cavalli FMG, Przelicki D, Wojcik JB, et al. Metabolic regulation of the epigenome drives lethal infantile ependymoma. Cell. (2020) 181:1329–1345.e24. doi: 10.1016/j.cell.2020.04.047
189. Lummus SC, Donson AM, Gowan K, Jones KL, Vibhakar R, Foreman NK, et al. p16 Loss and E2F/cell cycle deregulation in infant posterior fossa ependymoma. Pediatr Blood Cancer. (2017) 64. doi: 10.1002/pbc.26656
190. Shi Y, Wang X, Zhuang Y, Jiang Y, Melcher K, and Xu HE. Structure of the PRC2 complex and application to drug discovery. Acta Pharmacol Sin. (2017) 38:963–76. doi: 10.1038/aps.2017.7
191. Yi J, Kim B, Shi X, Zhan X, Lu QR, Xuan Z, et al. PRC2 heterogeneity drives tumor growth in medulloblastoma. Cancer Res. (2022) 82:2874–86. doi: 10.1158/0008-5472.can-21-4313
192. Cleveland AH, Malawsky D, Churiwal M, Rodriguez C, Reed F, Schniederjan M, et al. PRC2 disruption in cerebellar progenitors produces cerebellar hypoplasia and aberrant myoid differentiation without blocking medulloblastoma growth. Acta Neuropathol Commun. (2023) 11:8. doi: 10.1186/s40478-023-01508-x
193. Smits M, van Rijn S, Hulleman E, Biesmans D, van Vuurden DG, Kool M, et al. EZH2-regulated DAB2IP is a medulloblastoma tumor suppressor and a positive marker for survival. Clin Cancer Res. (2012) 18:4048–58. doi: 10.1158/1078-0432.ccr-12-0399
194. Jung H, Seong H-A, and Ha H. Murine protein serine/threonine kinase 38 activates apoptosis signal-regulating kinase 1 via thr838 phosphorylation*. J Biol Chem. (2008) 283:34541–53. doi: 10.1074/jbc.m807219200
195. Liu H, Sun Q, Sun Y, Zhang J, Yuan H, Pang S, et al. MELK and EZH2 cooperate to regulate medulloblastoma cancer stem-like cell proliferation and differentiation. Mol Cancer Res. (2017) 15:1275–86. doi: 10.1158/1541-7786.mcr-17-0105
196. Cheng Y, Liao S, Xu G, Hu J, Guo D, Du F, et al. NeuroD1 dictates tumor cell differentiation in medulloblastoma. Cell Rep. (2020) 31:107782. doi: 10.1016/j.celrep.2020.107782
197. Miele E, Valente S, Alfano V, Silvano M, Mellini P, Borovika D, et al. The histone methyltransferase EZH2 as a druggable target in SHH medulloblastoma cancer stem cells. Oncotarget. (2017) 8:68557–70. doi: 10.18632/oncotarget.19782
198. Zhang H, Zhu D, Zhang Z, Kaluz S, Yu B, Devi NS, et al. EZH2 targeting reduces medulloblastoma growth through epigenetic reactivation of the BAI1/p53 tumor suppressor pathway. Oncogene. (2020) 39:1041–8. doi: 10.1038/s41388-019-1036-7
199. Vo BT, Li C, Morgan MA, Theurillat I, Finkelstein D, Wright S, et al. Inactivation of Ezh2 upregulates gfi1 and drives aggressive myc-driven group 3 medulloblastoma. Cell Rep. (2017) 18:2907–17. doi: 10.1016/j.celrep.2017.02.073
200. Leva GD, Garofalo M, and Croce CM. MicroRNAs in cancer. Annu Rev Pathol: Mech Dis. (2014) 9:287–314. doi: 10.1146/annurev-pathol-012513-104715
201. Ferretti E, Smaele ED, Po A, Marcotullio LD, Tosi E, Espinola MSB, et al. MicroRNA profiling in human medulloblastoma. Int J Cancer. (2009) 124:568–77. doi: 10.1002/ijc.23948
202. Mollashahi B, Aghamaleki FS, and Movafagh A. The roles of miRNAs in medulloblastoma: A systematic review. J Cancer Prev. (2019) 24:79–90. doi: 10.15430/jcp.2019.24.2.79
203. Bevacqua E, Farshchi J, Niklison-Chirou MV, and Tucci P. Role of microRNAs in the development and progression of the four medulloblastoma subgroups. Cancers. (2021) 13:6323. doi: 10.3390/cancers13246323
204. Jönsson ME, Wahlestedt JN, Åkerblom M, Kirkeby A, Malmevik J, Brattaas PL, et al. Comprehensive analysis of microRNA expression in regionalized human neural progenitor cells reveals microRNA-10 as a caudalizing factor. Development. (2015) 142:3166–77. doi: 10.1242/dev.122747
205. Li KK, Xia T, Ma FMT, Zhang R, Mao Y, Wang Y, et al. miR-106b is overexpressed in medulloblastomas and interacts directly with PTEN. Neuropathol Appl Neurobiol. (2015) 41:145–64. doi: 10.1111/nan.12169
206. Saulnier O, Zagozewski J, Liang L, Hendrikse LD, Layug P, Gordon V, et al. A group 3 medulloblastoma stem cell program is maintained by OTX2-mediated alternative splicing. Nat Cell Biol. (2024) 26:1233–46. doi: 10.1038/s41556-024-01460-5
207. Zagozewski J, Shahriary GM, Morrison LC, Saulnier O, Stromecki M, Fresnoza A, et al. An OTX2-PAX3 signaling axis regulates Group 3 medulloblastoma cell fate. Nat Commun. (2020) 11:3627. doi: 10.1038/s41467-020-17357-4
208. Xue P, Huang S, Han X, Zhang C, Yang L, Xiao W, et al. Exosomal miR-101-3p and miR-423-5p inhibit medulloblastoma tumorigenesis through targeting FOXP4 and EZH2. Cell Death Differ. (2022) 29:82–95. doi: 10.1038/s41418-021-00838-4
209. Huang S, Xue P, Han X, Zhang C, Yang L, Liu L, et al. Exosomal miR-130b-3p targets SIK1 to inhibit medulloblastoma tumorigenesis. Cell Death Dis. (2020) 11:408. doi: 10.1038/s41419-020-2621-y
210. Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho Y-J, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. (2010) 18:316–28. doi: 10.1016/j.ccr.2010.09.006
211. Kia SK, Gorski MM, Giannakopoulos S, and Verrijzer CP. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol Cell Biol. (2008) 28:3457–64. doi: 10.1128/mcb.02019-07
212. Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature. (2018) 555:321–7. doi: 10.1038/nature25480
213. Ishi Y, Zhang Y, Zhang A, Sasaki T, Piunti A, Suri A, et al. Therapeutic targeting of EZH2 and BET BRD4 in pediatric rhabdoid tumors. Mol Cancer Ther. (2022) 21:715–26. doi: 10.1158/1535-7163.mct-21-0646
214. Feng S, Marhon SA, Sokolowski DJ, D’Costa A, Soares F, Mehdipour P, et al. Inhibiting EZH2 targets atypical teratoid rhabdoid tumor by triggering viral mimicry via both RNA and DNA sensing pathways. Nat Commun. (2024) 15:9321. doi: 10.1038/s41467-024-53515-8
215. Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. (2012) 8:890–6. doi: 10.1038/nchembio.1084
216. Kurmasheva RT, Sammons M, Favours E, Wu J, Kurmashev D, Cosmopoulos K, et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. (2017) 64. doi: 10.1002/pbc.26218
217. Qi L, Lindsay H, Kogiso M, Du Y, Braun FK, Zhang H, et al. Evaluation of an EZH2 inhibitor in patient-derived orthotopic xenograft models of pediatric brain tumors alone and in combination with chemo- and radiation therapies. Lab Invest. (2022) 102:185–93. doi: 10.1038/s41374-021-00700-8
218. Chi SN, Bourdeaut F, Laetsch TW, Fouladi M, Macy ME, Makin GW, et al. Phase I study of tazemetostat, an enhancer of zeste homolog-2 inhibitor, in pediatric pts with relapsed/refractory integrase interactor 1-negative tumors. J Clin Oncol. (2020) 38:10525–5. doi: 10.1200/jco.2020.38.15_suppl.10525
219. Chi SN, Yi JS, Williams PM, Roy-Chowdhuri S, Patton DR, Coffey BD, et al. Tazemetostat for tumors harboring SMARCB1/SMARCA4 or EZH2 alterations: results from NCI-COG pediatric MATCH APEC1621C. JNCI: J Natl Cancer Inst. (2023) 115:1355–63. doi: 10.1093/jnci/djad085
220. Vejmelkova K, Pokorna P, Noskova K, Faustmannova A, Drabova K, Pavelka Z, et al. Tazemetostat in the therapy of pediatric INI1-negative Malignant rhabdoid tumors. Sci Rep. (2023) 13:21623. doi: 10.1038/s41598-023-48774-2
221. Drosos Y, Myers JA, Xu B, Mathias KM, Beane EC, Radko-Juettner S, et al. NSD1 mediates antagonism between SWI/SNF and polycomb complexes and is required for transcriptional activation upon EZH2 inhibition. Mol Cell. (2022) 82:2472–2489.e8. doi: 10.1016/j.molcel.2022.04.015
222. Kazansky Y, Cameron D, Mueller HS, Demarest P, Zaffaroni N, Arrighetti N, et al. Overcoming clinical resistance to EZH2 inhibition using rational epigenetic combination therapy. Cancer Discov. (2024) 14:965–81. doi: 10.1158/2159-8290.cd-23-0110
223. Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. (2002) 16:1779–91. doi: 10.1101/gad.989402
224. Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, et al. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. (2005) 19:815–26. doi: 10.1101/gad.1284005
225. Dobson THW, Hatcher RJ, Swaminathan J, Das CM, Shaik S, Tao R-H, et al. Regulation of USP37 expression by REST-associated G9a-dependent histone methylation. Mol Cancer Res. (2017) 15:1073–84. doi: 10.1158/1541-7786.mcr-16-0424
226. Callegari K, Maegawa S, Bravo-Alegria J, and Gopalakrishnan V. Pharmacological inhibition of LSD1 activity blocks REST-dependent medulloblastoma cell migration. Cell Commun Signal. (2018) 16:60. doi: 10.1186/s12964-018-0275-5
227. D’Oto A, Tian Q-W, Davidoff AM, and Yang J. Histone demethylases and their roles in cancer epigenetics. J Med Oncol Ther. (2016) 1:34–40. doi: 10.35841/medical-oncology.1.2.34-40
228. Tong D, Tang Y, and Zhong P. The emerging roles of histone demethylases in cancers. Cancer Metastasis Rev. (2024) 43:795–821. doi: 10.1007/s10555-023-10160-9
229. Pajtler KW, Weingarten C, Thor T, Künkele A, Heukamp LC, Büttner R, et al. The KDM1A histone demethylase is a promising new target for the epigenetic therapy of medulloblastoma. Acta Neuropathol Commun. (2013) 1:19. doi: 10.1186/2051-5960-1-19
230. Bailey CP, Figueroa M, Gangadharan A, Yang Y, Romero MM, Kennis BA, et al. Pharmacologic inhibition of lysine-specific demethylase 1 as a therapeutic and immune-sensitization strategy in pediatric high-grade glioma. Neuro-Oncol. (2020) 22:1302–14. doi: 10.1093/neuonc/noaa058
231. Hua C, Chen J, Li S, Zhou J, Fu J, Sun W, et al. KDM6 demethylases and their roles in human cancers. Front Oncol. (2021) 11:779918. doi: 10.3389/fonc.2021.779918
232. Yi J, Shi X, Xuan Z, and Wu J. Histone demethylase UTX/KDM6A enhances tumor immune cell recruitment, promotes differentiation and suppresses medulloblastoma. Cancer Lett. (2021) 499:188–200. doi: 10.1016/j.canlet.2020.11.031
233. Hashizume R, Smirnov I, Liu S, Phillips JJ, Hyer J, McKnight TR, et al. Characterization of a diffuse intrinsic pontine glioma cell line: implications for future investigations and treatment. J Neuro-Oncol. (2012) 110:305–13. doi: 10.1007/s11060-012-0973-6
234. Northcott PA, Jones DTW, Kool M, Robinson GW, Gilbertson RJ, Cho Y-J, et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer. (2012) 12:818–34. doi: 10.1038/nrc3410
235. Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell. (2012) 21:168–80. doi: 10.1016/j.ccr.2011.12.023
236. Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho Y-J, et al. An animal model of MYC-driven medulloblastoma. Cancer Cell. (2012) 21:155–67. doi: 10.1016/j.ccr.2011.12.021
237. Ferrucci V, de Antonellis P, Pennino FP, Asadzadeh F, Virgilio A, Montanaro D, et al. Metastatic group 3 medulloblastoma is driven by PRUNE1 targeting NME1–TGF-β–OTX2–SNAIL via PTEN inhibition. Brain. (2018) 141:1300–19. doi: 10.1093/brain/awy039
238. Bibbò F, Asadzadeh F, Boccia A, Sorice C, Bianco O, Saccà CD, et al. Targeting group 3 medulloblastoma by the anti-PRUNE-1 and anti-LSD1/KDM1A epigenetic molecules. Int J Mol Sci. (2024) 25:3917. doi: 10.3390/ijms25073917
239. Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. (2014) 511:428–34. doi: 10.1038/nature13379
240. Lee C, Rudneva VA, Erkek S, Zapatka M, Chau LQ, Tacheva-Grigorova SK, et al. Lsd1 as a therapeutic target in Gfi1-activated medulloblastoma. Nat Commun. (2019) 10:332. doi: 10.1038/s41467-018-08269-5
241. Swaminathan J, Maegawa S, Shaik S, Sharma A, Bravo-Alegria J, Guo L, et al. Cross-talk between histone methyltransferases and demethylases regulate REST transcription during neurogenesis. Front Oncol. (2022) 12:855167. doi: 10.3389/fonc.2022.855167
242. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. (1997) 389:349–52. doi: 10.1038/38664
243. Martínez-Balbás MA, Bannister AJ, Martin K, Haus-Seuffert P, Meisterernst M, and Kouzarides T. The acetyltransferase activity of CBP stimulates transcription. EMBO J. (1998) 17:2886–93. doi: 10.1093/emboj/17.10.2886
244. Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, Vassilev A, et al. HATs off Selective Synthetic Inhibitors of the Histone Acetyltransferases p300 and PCAF. Mol Cell. (2000) 5:589–95. doi: 10.1016/s1097-2765(00)80452-9
245. Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou M-M, et al. Structure and ligand of a histone acetyltransferase bromodomain. Nature. (1999) 399:491–6. doi: 10.1038/20974
246. Zucconi BE, Makofske JL, Meyers DJ, Hwang Y, Wu M, Kuroda MI, et al. Combination targeting of the bromodomain and acetyltransferase active site of p300/CBP. Biochemistry. (2019) 58:2133–43. doi: 10.1021/acs.biochem.9b00160
247. Huang T, Garcia R, Qi J, Lulla R, Horbinski C, Behdad A, et al. Detection of histone H3 K27M mutation and post-translational modifications in pediatric diffuse midline glioma via tissue immunohistochemistry informs diagnosis and clinical outcomes. Oncotarget. (2018) 9:37112–24. doi: 10.18632/oncotarget.26430
248. Ozawa T, Kaneko S, Szulzewsky F, Qiao Z, Takadera M, Narita Y, et al. C11orf95-RELA fusion drives aberrant gene expression through the unique epigenetic regulation for ependymoma formation. Acta Neuropathol Commun. (2021) 9:36. doi: 10.1186/s40478-021-01135-4
249. Sardar D, Chen H-C, Reyes A, Varadharajan S, Jain A, Mohila C, et al. Sox9 directs divergent epigenomic states in brain tumor subtypes. Proc Natl Acad Sci. (2022) 119:e2202015119. doi: 10.1073/pnas.2202015119
250. Hu X, Wu X, Berry K, Zhao C, Xin D, Ogurek S, et al. Nuclear condensates of YAP fusion proteins alter transcription to drive ependymoma tumourigenesis. Nat Cell Biol. (2023) 25:323–36. doi: 10.1038/s41556-022-01069-6
251. Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature. (2016) 530:57–62. doi: 10.1038/nature16546
252. Torchia J, Golbourn B, Feng S, Ho KC, Sin-Chan P, Vasiljevic A, et al. Integrated (epi)-genomic analyses identify subgroup-specific therapeutic targets in CNS rhabdoid tumors. Cancer Cell. (2016) 30:891–908. doi: 10.1016/j.ccell.2016.11.003
253. Arabzade A, Zhao Y, Varadharajan S, Chen H-C, Jessa S, Rivas B, et al. ZFTA–RELA dictates oncogenic transcriptional programs to drive aggressive supratentorial ependymoma. Cancer Discov. (2021) 11:2200–15. doi: 10.1158/2159-8290.cd-20-1066
254. Marmorstein R and Zhou M-M. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb Perspect Biol. (2014) 6:a018762. doi: 10.1101/cshperspect.a018762
255. Kool M, Jones DTW, Jäger N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. (2014) 25:393–405. doi: 10.1016/j.ccr.2014.02.004
256. Taylor MD, Mainprize TG, Rutka JT, Becker L, Bayani J, and Drake JM. Medulloblastoma in a child with rubenstein-taybi syndrome: case report and review of the literature. Pediatr Neurosurg. (2001) 35:235–8. doi: 10.1159/000050428
257. Bourdeaut F, Miquel C, Richer W, Grill J, Zerah M, Grison C, et al. Rubinstein–Taybi syndrome predisposing to non-WNT, non-SHH, group 3 medulloblastoma. Pediatr Blood Cancer. (2014) 61:383–6. doi: 10.1002/pbc.24765
258. Merk DJ, Ohli J, Merk ND, Thatikonda V, Morrissy S, Schoof M, et al. Opposing effects of CREBBP mutations govern the phenotype of rubinstein-taybi syndrome and adult SHH medulloblastoma. Dev Cell. (2018) 44:709–724.e6. doi: 10.1016/j.devcel.2018.02.012
259. Schoof M, Epplen GD, Walter C, Ballast A, Holdhof D, Göbel C, et al. The tumor suppressor CREBBP and the oncogene MYCN cooperate to induce Malignant brain tumors in mice. Oncogenesis. (2023) 12:36. doi: 10.1038/s41389-023-00481-3
260. Shendy NAM, Bikowitz M, Sigua LH, Zhang Y, Mercier A, Khashana Y, et al. Group 3 medulloblastoma transcriptional networks collapse under domain specific EP300/CBP inhibition. Nat Commun. (2024) 15:3483. doi: 10.1038/s41467-024-47102-0
261. Pfister S, Rea S, Taipale M, Mendrzyk F, Straub B, Ittrich C, et al. The histone acetyltransferase hMOF is frequently downregulated in primary breast carcinoma and medulloblastoma and constitutes a biomarker for clinical outcome in medulloblastoma. Int J Cancer. (2008) 122:1207–13. doi: 10.1002/ijc.23283
262. Taipale M, Rea S, Richter K, Vilar A, Lichter P, Imhof A, et al. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol Cell Biol. (2005) 25:6798–810. doi: 10.1128/mcb.25.15.6798-6810.2005
263. Krishnan V, Chow MZY, Wang Z, Zhang L, Liu B, Liu X, et al. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc Natl Acad Sci. (2011) 108:12325–30. doi: 10.1073/pnas.1102789108
264. Gupta A, Guerin-Peyrou TG, Sharma GG, Park C, Agarwal M, Ganju RK, et al. The mammalian ortholog of drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Mol Cell Biol. (2008) 28:397–409. doi: 10.1128/mcb.01045-07
265. Zhao L, Wang D-L, Liu Y, Chen S, and Sun F-L. Histone acetyltransferase hMOF promotes S phase entry and tumorigenesis in lung cancer. Cell Signal. (2013) 25:1689–98. doi: 10.1016/j.cellsig.2013.04.006
266. Seto E and Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. (2014) 6:a018713. doi: 10.1101/cshperspect.a018713
267. Park S-Y and Kim J-S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp Mol Med. (2020) 52:204–12. doi: 10.1038/s12276-020-0382-4
268. Park PC, Taylor MD, Mainprize TG, Becker LE, Ho M, Dura WT, et al. Transcriptional profiling of medulloblastoma in children. J Neurosurg. (2003) 99:534–41. doi: 10.3171/jns.2003.99.3.0534
269. Ecker J, Oehme I, Mazitschek R, Korshunov A, Kool M, Hielscher T, et al. Targeting class I histone deacetylase 2 in MYC amplified group 3 medulloblastoma. Acta Neuropathol Commun. (2015) 3:22. doi: 10.1186/s40478-015-0201-7
270. Milde T, Oehme I, Korshunov A, Kopp-Schneider A, Remke M, Northcott P, et al. HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin Cancer Res. (2010) 16:3240–52. doi: 10.1158/1078-0432.ccr-10-0395
271. Sredni ST, Halpern AL, Hamm CA, Bonaldo M de F, and Tomita T. Histone deacetylases expression in atypical teratoid rhabdoid tumors. Child’s Nerv Syst. (2013) 29:5–9. doi: 10.1007/s00381-012-1965-8
272. Yoshida M, Kijima M, Akita M, and Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. (1990) 265:17174–9. doi: 10.1016/s0021-9258(17)44885-x
273. Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci. (1998) 95:3003–7. doi: 10.1073/pnas.95.6.3003
274. Richon VM. Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br J Cancer. (2006) 95:S2–6. doi: 10.1038/sj.bjc.6603463
275. Pei Y, Liu K-W, Wang J, Garancher A, Tao R, Esparza LA, et al. HDAC and PI3K antagonists cooperate to inhibit growth of MYC-driven medulloblastoma. Cancer Cell. (2016) 29:311–23. doi: 10.1016/j.ccell.2016.02.011
276. Ecker J, Thatikonda V, Sigismondo G, Selt F, Valinciute G, Oehme I, et al. Reduced chromatin binding of MYC is a key effect of HDAC inhibition in MYC amplified medulloblastoma. Neuro-Oncol. (2020) 23:226–39. doi: 10.1093/neuonc/noaa191
277. Fouladi M, Park JR, Stewart CF, Gilbertson RJ, Schaiquevich P, Sun J, et al. Pediatric phase I trial and pharmacokinetic study of vorinostat: A children’s oncology group phase I consortium report. J Clin Oncol. (2010) 28:3623–9. doi: 10.1200/jco.2009.25.9119
278. Su JM, Kilburn LB, Mansur DB, Krailo M, Buxton A, Adekunle A, et al. Phase I/II trial of vorinostat and radiation and maintenance vorinostat in children with diffuse intrinsic pontine glioma: A Children’s Oncology Group report. Neuro-Oncol. (2021) 24:655–64. doi: 10.1093/neuonc/noab188
279. Phi JH, Choi SA, Kwak PA, Lee JY, Wang K-C, Hwang DW, et al. Panobinostat, a histone deacetylase inhibitor, suppresses leptomeningeal seeding in a medulloblastoma animal model. Oncotarget. (2017) 8:56747–57. doi: 10.18632/oncotarget.18132
280. Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily M-A, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. (2015) 21:555–9. doi: 10.1038/nm.3855
281. Fujita N, Bondoc A, Simoes S, Ishida J, Taccone MS, Luck A, et al. Combination treatment with histone deacetylase and carbonic anhydrase 9 inhibitors shows therapeutic potential in experimental diffuse intrinsic pontine glioma. Brain Tumor Pathol. (2024) 41:117–31. doi: 10.1007/s10014-024-00493-w
282. Jane EP, Reslink MC, Gatesman TA, Halbert ME, Miller TA, Golbourn BJ, et al. Targeting mitochondrial energetics reverses panobinostat- and marizomib-induced resistance in pediatric and adult high-grade gliomas. Mol Oncol. (2023) 17:1821–43. doi: 10.1002/1878-0261.13427
283. Chang W-I, Honeyman JN, Zhang J, Lin C, Sharma A, Zhou L, et al. Novel combination of imipridones and histone deacetylase inhibitors demonstrate cytotoxic effect through integrated stress response in pediatric solid tumors. Am J Cancer Res. (2023) 13:6241–55.
284. Meel MH, de Gooijer MC, Metselaar DS, Sewing ACP, Zwaan K, Waranecki P, et al. Combined therapy of AXL and HDAC inhibition reverses mesenchymal transition in diffuse intrinsic pontine glioma. Clin Cancer Res. (2020) 26:3319–32. doi: 10.1158/1078-0432.ccr-19-3538
285. Choi SA, Lee C, Kwak PA, Park C-K, Wang K-C, Phi JH, et al. Histone deacetylase inhibitor panobinostat potentiates the anti-cancer effects of mesenchymal stem cell-based sTRAIL gene therapy against Malignant glioma. Cancer Lett. (2019) 442:161–9. doi: 10.1016/j.canlet.2018.10.012
286. Hennika T, Hu G, Olaciregui NG, Barton KL, Ehteda A, Chitranjan A, et al. Pre-clinical study of panobinostat in xenograft and genetically engineered murine diffuse intrinsic pontine glioma models. PloS One. (2017) 12:e0169485. doi: 10.1371/journal.pone.0169485
287. Lin GL, Wilson KM, Ceribelli M, Stanton BZ, Woo PJ, Kreimer S, et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci Transl Med. (2019) 11. doi: 10.1126/scitranslmed.aaw0064
Keywords: epigenetics, histone modifications, histone methylation, histone acetylation, pediatric
Citation: Hamanishi ET, Dang D and Venneti S (2025) Aberrant histone modifications in pediatric brain tumors. Front. Oncol. 15:1587157. doi: 10.3389/fonc.2025.1587157
Received: 04 March 2025; Accepted: 21 May 2025;
Published: 10 June 2025.
Edited by:
Ashley Sloane Margol, Children’s Hospital of Los Angeles, United StatesReviewed by:
Joshua John Breunig, Cedars Sinai Medical Center, United StatesDebanjan Bhattacharya, University of Cincinnati, United States
Copyright © 2025 Hamanishi, Dang and Venneti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sriram Venneti, c3Zlbm5ldGlAbWVkLnVtaWNoLmVkdQ==