 Elena Diana Chiru
Elena Diana Chiru Lina Sojak2
Lina Sojak2 Marcus Vetter
Marcus Vetter Cvetka Grašič Kuhar
Cvetka Grašič Kuhar- 1University Center for Hematology and Oncology, Cantonal Hospital Baselland, Liestal, Switzerland
- 2Basel Medical University, Basel, Switzerland
- 3Department of Medical Oncology, Institute of Oncology Ljubljana, Ljubljana, Slovenia
- 4Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
Breast cancer (BC) remains the most prevalent cancer among women worldwide, with hormone receptor-positive (HR+) and human epidermal growth factor receptor 2-negative (HER2-) subtypes representing approximately 75% of cases. Endocrine therapy (ET) has been foundational in HR+ BC treatment, significantly reducing recurrence and mortality rates, yet resistance to ET remains a critical challenge, particularly in the metastatic setting. Recent treatment advancements—especially CDK4/6 inhibitors (CDK4/6i) in first-line therapy, have reshaped management of HR+ HER2- mBC. Additionally, novel agents like selective estrogen receptor degraders (SERDs) and proteolysis-targeting chimeras (PROTACs), have proven to be effective against ER-resistance inducing mutations, such as ESR1, while poly ADP-ribose polymerase inhibitors (PARPi) showed targeted benefit in BRCA-mutated tumors. In breast cancer expressing AKT/PIK3CA pathway alterations, drugs like alpelisib, capivasertib, and inavolisib have recently been approved, demonstrating improved PFS in this specific patient population. Recent developments of antibody-drug conjugates (ADCs) have also extended therapeutic options to previously labeled HER2-negative tumors, with drugs like trastuzumab deruxtecan (T-DXd) demonstrating efficacy in newly emerged HER2-low and HER2-ultralow pathologic subgroups, extending median overall survival to almost 2 years. Most of these drugs have paved the way for personalized medicine and opened questions around optimal sequencing of ET and application of combination therapies, which continue to be investigated through clinical trials. This review seeks to highlight current and emerging treatment strategies addressing ET resistance to improve survival outcomes for HR+ mBC patients, emphasizing the need for personalized approaches.
Introduction
With approximately 2.3 million new cases in 2022, breast cancer (BC) represents 25% of all cancer cases diagnosed in women worldwide. It accounts for nearly 12% of all cancer cases globally and 7% of cancer-related deaths annually (1). Despite recent treatment advances, most of these fatalities are due to development of therapy-resistant metastatic disease (2).
Most breast tumors are hormone-dependent, driven by estrogen dependent factors of growth and proliferation. Therefore, incidence of BC is approximately 150 times higher in women than in men, with a rapid increase in incidence observed in premenopausal women. Age-standardized incidence rates of premenopausal breast cancer are predominantly higher in high-income countries, while the incidence of postmenopausal BC has been rising significantly in countries undergoing economic and social transitions (3), possibly as a consequence of overlapping reduced prevention, increased prevalence of co-morbidities in older patients of transitioning countries and aging factors. Menopause experienced 10 years earlier than median age (52 years old) reduces the risk of BC by 35%. Similarly, other key moments in hormone development, such as age at menarche and timing of pregnancy play pivotal roles in BC occurrence. In a similar way, men who develop BC first experience gynecomastia, indicating an underlying endocrine disbalance, as 90% of male breast tumors are hormone receptor positive (HR+) (3, 4).
Assessment of HR status – estrogen receptor (ER) and progesterone receptor (PR), as well as human epidermal receptor 2 (HER2) status, and proliferation index (Ki67) is essential for diagnostic workup, molecular classification and risk stratification. The most commonly accepted subtypes have been determined based on immunohistochemical expression of HR and HER2, and have been classified into luminal A-like (HR+/HER2-, low Ki-67 expression), luminal B HER2-negative (HR+/HER2-, PR- or higher Ki67 expression), luminal B-like HER2+ (ER+/HER+, any level of Ki67, PR-/+), HER2 enriched (HR-, HER2+) and triple-negative or basal-like (HR-/HER2-) (5, 6).
In early stages of HR+/HER2- BC, endocrine therapy (ET) is the standard management, having proven efficacy in lowering mortality rate by one third (7). ET options have evolved from selective estrogen receptor modulator (SERM) tamoxifen to aromatase inhibitors (AIs). In a metanalysis conducted by the Early Breast Cancer Trialists Collaborative Group (EBCTCG) in 2011, 5 years of tamoxifen therapy in ER-positive BC patients lowered recurrence rates by half in the first 4 years (RR 0.53) and by one-third in years 5-9 (RR 0.68), with a sustained 30% reduction in BC mortality over 15 years (RR 0.71 in years 0-4, RR 0.66 in years 5-9, RR 0.68 in years 10-14) (8). Results from a EBCTCG metanalysis in postmenopausal patients, showed that 5 years of aromatase inhibitor (AI) treatment significantly reduced recurrence rates by about 30% when compared to tamoxifen (RR 0.70, 95% CI 0.64–0.77) and led to a 10-year BC mortality reduction of about 15% (RR 0.86, 95% CI 0.80–0.94). AIs are associated with fewer endometrial cancers, and are a better treatment alternative in patients with higher risk of thromboembolic events and those at risk of endometrial cancer. However, they pose a higher risk of bone fractures when compared to tamoxifen (9).
In premenopausal women on ovarian suppression and ET, the rate of BC recurrence was lower for women receiving AI than for women assigned to tamoxifen (RR 0.79, 95% CI 0.69–0.90, p=0.0005). The main benefit was seen in years 0–4 (RR 0.68), leading to a 3.2% reduction of recurrence at 5 years (10). However, in these younger patients no effect on mortality rate was observed.
History
Benefits of ET in BC management have been acknowledged since the end of the 19th century, when British surgeon George Beatson introduced the practice of oophorectomy to reduce soft tissue metastases in cases of advanced BC (11). However, it wasn’t until Clara Szego’s discovery of the ER pathway and its mediators, that ET became an established BC treatment option (12).
Although, antiestrogen strategies can now provide significant disease control, development of ET resistance constitutes one of the main challenges to date and is associated with late recurrence events (>5 years) (13).
Risk of recurrence is clinically assessed through features such as tumor size and lymph node status, and guides therapy decision-making. An earlier metanalysis conducted by the EBCTCG in HR+/HER2- patients showed in tumors harboring more than 3 positive lymph nodes, a distant recurrence risk at 5 years of 3%, in those with 1–3 nodes 2%, and in node negative tumors ≥ 20 mm 1%, while node-negative tumors smaller than 20 mm had a risk of 0.5-1% (13). Moreover, in an effort to guide de-escalation of CHT application, multigene prognostic assays such as Oncotype DX, MammaPrint, EndoPredict and Prosigna, have emerged in the past two decades to inform about risk of recurrence in early stage HR+ BC and have been incorporated into international oncologic guidelines to refine risk stratification, replacing clinical risk factors (14–17).
In the metastatic setting, however, first-line treatment outcomes do not appear to differ significantly between patients receiving chemotherapy (CHT) and those treated with ET alone (18). Moreover, patients with HR+ mBC are more likely to than those with early-stage BC to express an actionable mutation, specifically in the mitogen-activated protein kinases, such as MAPK/ERK (37% vs. 22%, respectively) and homologous recombinant deficiency (HRD) pathways (22% vs. 10%, respectively) (19).
Since 2012, a variety of ET-based combination treatments—typically using AIs or fulvestrant as the backbone—have become available. The first was the approval of the mTOR inhibitor everolimus, followed by the introduction of CDK4/6 inhibitors: palbociclib (2015), ribociclib (2018), and abemaciclib (2018). Subsequent approvals included PI3K inhibitors such as alpelisib (2019) (20), and more recently inavolisib (21), as well as AKT inhibitors, such as capivasertib (22). Additionally, PARP inhibitors—olaparib (2018) and talazoparib (2023)—have been approved for patients with germline BRCA1 or BRCA2 mutations (19) – see Tables 1a, b.
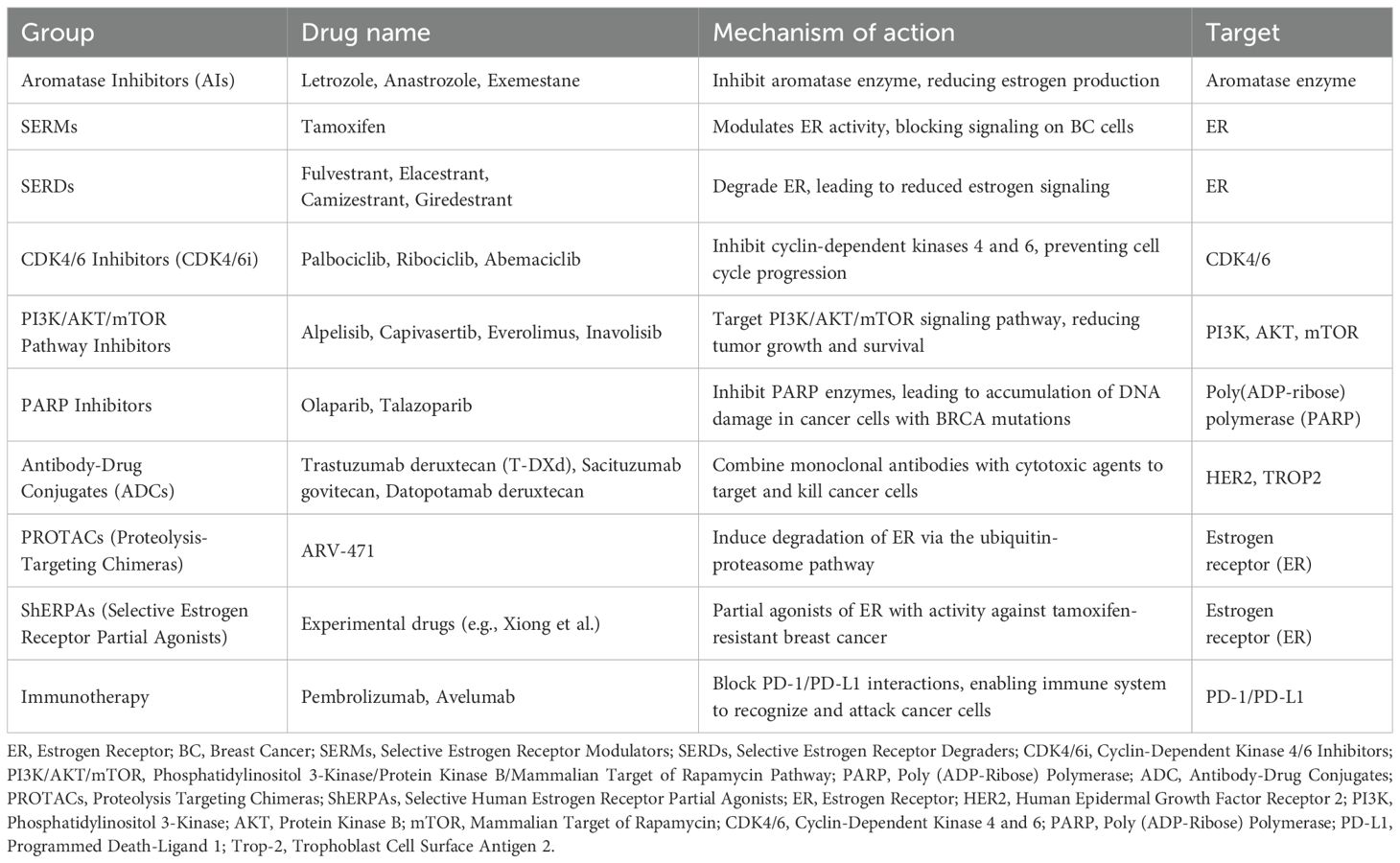
Table 1a. Therapeutic agents in metastatic breast cancer grouped by mechanism and target.
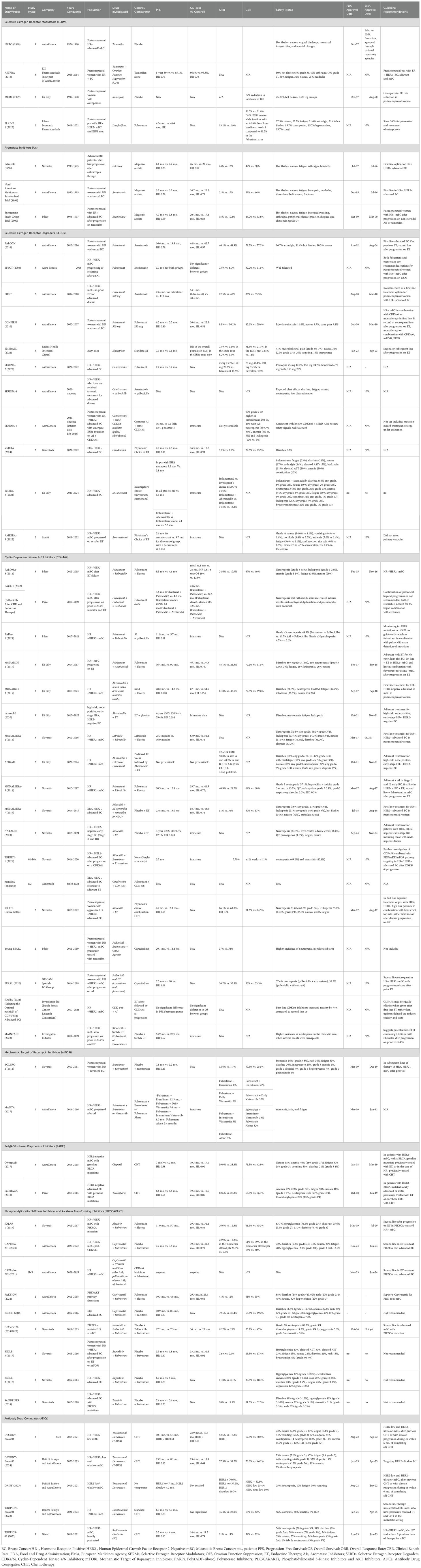
Table 1b. List of clinical trials and drugs presented in this paper, grouped by class.
In HR+ mBC, ET is better tolerated than CHT, and better tumor control is achieved with ET than with CHT. According to current ESMO guidelines, the preferred first-line approach in the metastatic setting is ET in combination therapy with CDK4/6i. More recently, this has also become standard in early-stage high risk BC patients (23). While tamoxifen remains a viable option for premenopausal patients with childbearing potential, non-steroidal AIs such as anastrozole and letrozole are generally preferred due to their superior effectiveness in achieving disease control. Moreover, they are even more potent when combined with CDK4/6i, although they seem to be directly related to the development of estrogen receptor 1 (ESR1) mutations.
In case of disease progression, several treatment options should be considered based on the presence of targetable mutations, including ESR1. To overcome this resistance mechanism, newer selective estrogen receptor degraders (SERD) such as elacestrant (24) and other novel antiestrogens such as complete estrogen receptor antagonists (CERANs), selective estrogen receptor covalent antagonists (SERCAs), and proteolysis targeting chimera (PROTACs) have been evaluated in clinical studies (25, 26).
In those cases where targeted treatment is not available, combination with aromatase inhibitors and everolimus still remain an option (27) (ESMO Metastatic Breast Cancer Living Guidelines, 2023) (Figure 1).
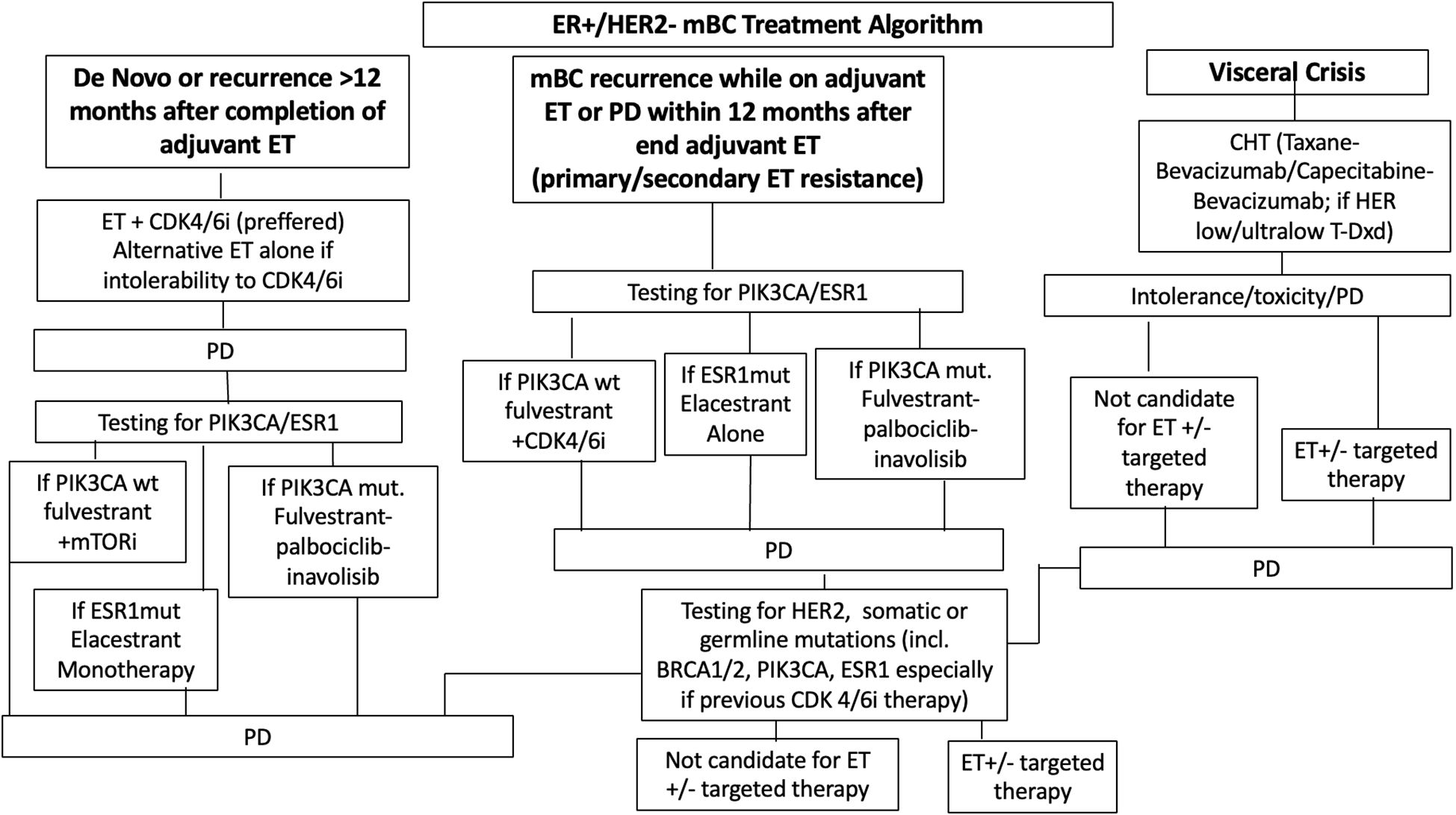
Figure 1. Treatment paradigm for hormone receptor positive (HR+) metastatic breast cancer. ET, endocrine therapy; CDK4/6i, cyclin-dependent kinase 4/6 inhibitor; AI, aromatase inhibitor; CHT, chemotherapy; PD, progressive diseases.
For women with rapidly progressing disease or visceral crisis, CHT continues to be the preferred strategy, with anthracyclines and taxanes being the most used agents (28). However, recent advances have delivered more modern CHT options, such as antibody-drug conjugates (ADCs). Trastuzumab deruxtecan (T-DXd) has emerged as a key agent for patients with HER2-low disease (defined as IHC 1+ or 2+ with ISH <2), while sacituzumab govitecan is playing an increasingly important role in later-line settings (27) (Figure 1).
Beyond tumor-specific risk factors, therapy selection is becoming increasingly personalized taking into account factors such as age, menopausal status, performance status, comorbidities, and prior treatment history. Emerging combination and sequencing strategies are gaining attention, aiming to de-escalate CHT while extending progression-free survival (PFS).
Supporting this approach, recent findings from the RIGHT Choice trial showed that first-line ribociclib plus ET improved median PFS when compared with combination CHT, had comparable overall response rates (ORR) and lower rates of adverse events in HR+/HER2– advanced BC (29).
Main treatment paradigm for HR+ mBC is illustrated in Figure 1.
Pharmacologic landscape of HR+/HER2− mBC
Aromatase inhibitors
Aromatase inhibitors (AIs) are designed to block the activity of CYP19A1, an aromatase enzyme belonging to the cytochrome P450 family, which catalyzes the conversion of androgens to estrogens in peripheral tissues. These tissues—such as adipose tissue, skin, and muscle—lie outside the gonads and become the primary sites of estrogen synthesis in postmenopausal women. By suppressing this peripheral conversion, AIs effectively lower circulating levels of estradiol and limit estrogen-driven tumor growth.
Among them, third-generation non-steroidal AIs (letrozole and anastrozole) are commonly used in combination with CDK4/6i (30) in first-line treatment for HR+ mBC. In later treatment lines, the steroidal AI exemestane is often paired with the mTOR inhibitor everolimus (24), particularly in the absence of visceral crisis or imminent organ failure that would necessitate chemotherapy.
Classification of common hormonal agents is illustrated in Figure 2.
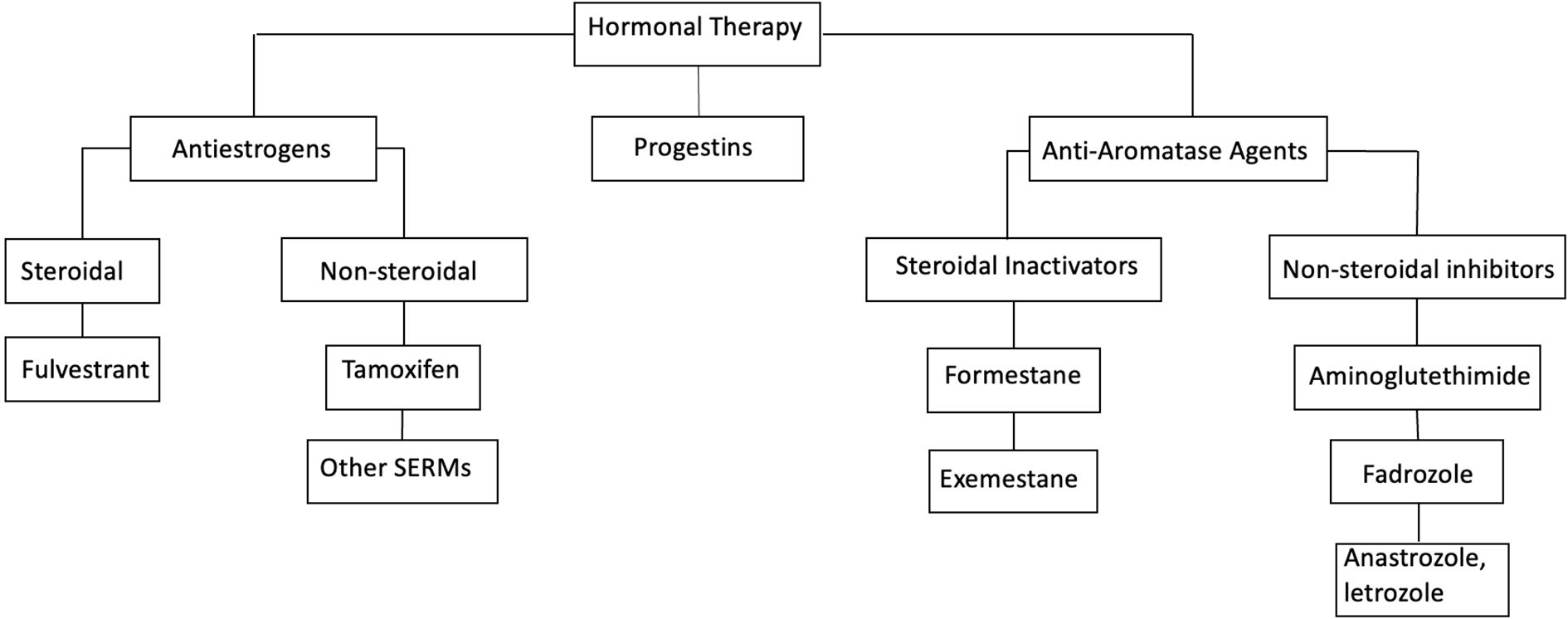
Figure 2. Classification of hormonal therapy agents. SERM, selective estrogen receptor modulators.
Significant structural differences allow for different mechanism of actions among steroidal and non-steroidal AIs, making these drugs lack cross-resistance, and supporting their individual use in different circumstances. As such, exemestane binds to the androgen substrate anchoring site of the aromatase enzyme and permanently inactivates it, while non-steroidal AIs block the peripheral conversion of androstenedione to estrone through reversible covalent bonding with the heme moiety of the aromatase – see Table 2. All third-generation AIs inhibit about 97-99% of peripheral aromatization (31).
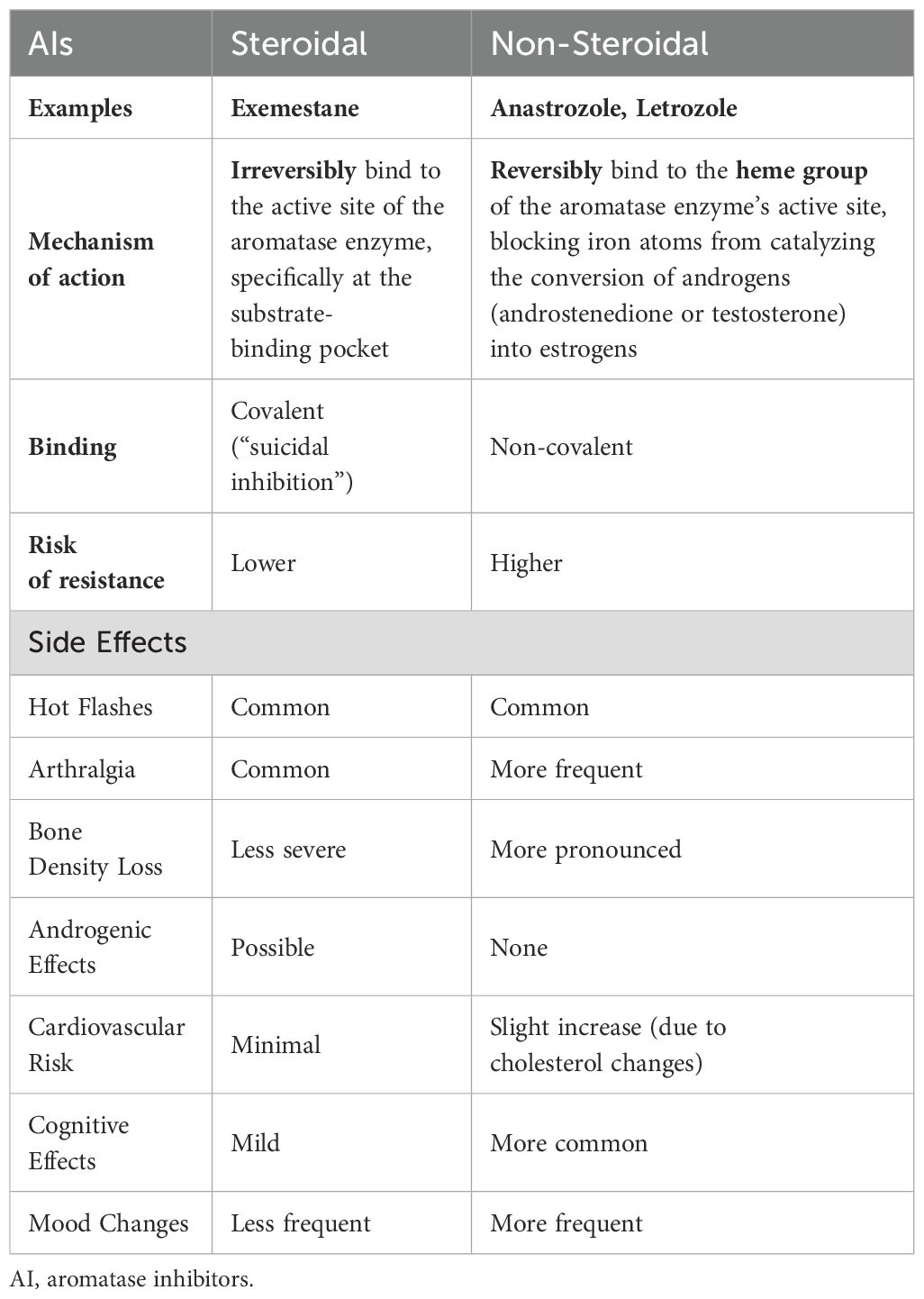
Table 2. Classification and comparison of steroidal vs. non-steroidal aromatase inhibitors (AIs, their mechanism of action, resistance and side effects).
Mechanism of action of aromatase inhibitors is illustrated in Figure 3.
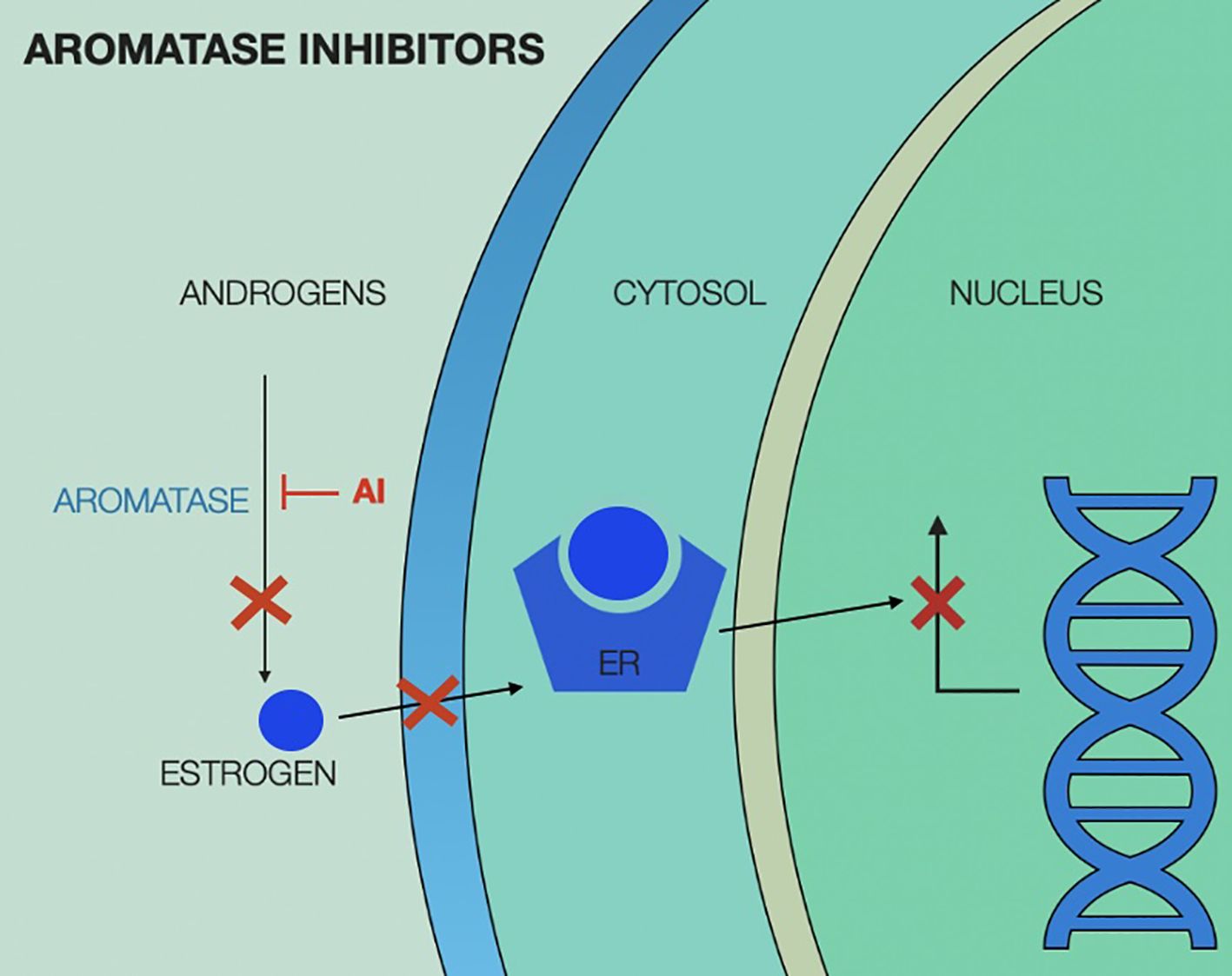
Figure 3. Aromatase inhibitors (AI) mechanism of action, showing interruption of estrogen production in the peripheral tissue, leading to no estrogen translocating in the cytosol and therefore lack of estrogen-estrogen receptor complex to activate nuclear transcription factors. ER, estrogen receptor; AI, aromatase inhibitor.
However, while AI monotherapy can achieve a median PFS of 1 to 4 years, when used in first line treatment for mBC, its efficacy declines significantly in later lines, with median PFS dropping to approximately 2 to 6 months in the second-line setting. In some cases, this seems to be due to resistance to AIs, linked to mutations of the ligand-binding domain of the ESR1 (32), which were first discovered in 1997. These mutations result in a constitutively active estrogen receptor that can signal independently of estrogen, rendering AI-induced estrogen depletion ineffective (33). The importance of ESR1 mutations in mBC development has not been discovered until 2013, when genomic sequencing of circulating tumor cells (CTC) in patients with metastatic disease allowed for its identification as a key factor of ET resistance (34–36).
According to current guidelines, endocrine resistance is classified as either primary or secondary. Primary endocrine resistance refers to relapse during the first 2 years of adjuvant ET or progression within the first 6 months of first-line ET for mBC, in the absence of prior ET in the metastatic setting. Secondary resistance is defined as relapse after 2 years of adjuvant therapy or within one year after completion of ET for early BC or progression after 6 months of ET in the metastatic setting (5).
ET resistant mutations, such as ESR1 are usually found in patients with secondary endocrine resistance (27).
In patients who received AI in the neoadjuvant setting, ESR1 mutations are found in 1.5%-7%, whereas in those who received AI for recurrent BC after previous adjuvant ET, it ranges from 4% to 5%. The highest prevalence of ESR1 mutations was documented in patients who have previously received AI for mBC, where it was found to range between 20%-40%, while in ET naïve mBC patients, ESR1 mutations were present in 1% of cases, suggesting that these mutations are primarily induced by AIs (32). Moreover, resistance is influenced by the choice of drug, with a specific 30-fold reduction in binding affinity for tamoxifen and a 40-fold reduction for fulvestrant, necessitating higher dosages. Resistance also appears to be driven by specific ESR1 mutations, with the Y537S mutation associated with a worse prognosis compared to D538G (37).
In metastatic breast tumors harboring ESR1 mutations, selective estrogen receptor modulators (SERMs) and selective estrogen receptor covalent antagonists (SERCAs) seem to work better than AI.
With regards to combination therapies, in addition to CDK4/6 inhibitors, tyrosine kinase inhibitors (TKIs) have been studied in combination with AIs for HR+ mBC. While clinical trials have shown some benefit, their use is currently limited to HER2-positive disease (38).
Selective estrogen receptor modulators
Tamoxifen is the most extensively evaluated SERM, with antagonistic effects in the breast but predominantly agonistic effects in the endometrial and liver tissue. Tamoxifen binds to the ER, leads to homodimerization of the complex, and translocation to the nucleus, where it blocks binding of co-activators and promotes binding of co-repressors, blocking transcription of activation factor 2 domain (AF2), but not of AF1. This explains its partial agonistic effects in the uterus (39). Tamoxifen has been a cornerstone of treatment in mBC since the 1970s, following the discovery of its ability to stop tumor growth, preserve bone density, and lower serum cholesterol. Long-term follow-up from early clinical trials demonstrated improved 15-year outcomes, with average annual reductions in the risk of relapse and mortality by 12% and 9%, respectively (11). In metastatic disease the median duration of response to tamoxifen therapy is 9–12 months, with an overall response rate of 30–40%. Response rates are even higher in patients with soft tissue metastases (35%) compared to those with visceral (29%) or bone metastases (25%) (11).
Notably, even in early stages HR+ BC, ET with tamoxifen or AI for 5–10 years were until recently, standard treatment. Front-line therapy with AIs has demonstrated a 3.6% reduction in 10-year recurrence risk and a 2.1% improvement in OS compared to tamoxifen. However, AIs are particularly advantageous for patients with advanced-stage (node-positive), high-grade, HER2-positive, or highly proliferative BC and are the preferred choice for lobular cancers. As a result, an upfront AI or a sequential approach—starting with tamoxifen for 2–5 years followed by an AI—were long considered the standard of care, achieving a 2% reduction in recurrence risk and a 1.5% decrease in risk of mortality when compared to tamoxifen alone (40).
In premenopausal women with stage I–III BC, the ASTRRA trial showed that addition of ovarian function suppression (OFS) to tamoxifen led to a 5-year PFS rate of 91.1%, compared to 87.5% with tamoxifen alone (HR 0.69, 95% CI 0.48–0.97, p=0.033). This benefit was sustained at the 8-year follow-up, when PFS rate reached 85.4% in the tamoxifen plus OFS group versus 80.2% in the tamoxifen monotherapy group (HR 0.67, 95% CI 0.51–0.87) (41). Despite these findings, no significant OS benefit was observed (96.5% in the OFS group vs. 95.3% in the tamoxifen group, HR 0.78, 95% CI 0.49–1.25) (42).
Common side effects of tamoxifen include hot flashes and vaginal bleeding, but more serious side effects, such as thromboembolic events (3–4%), must also be considered. Despite its well-established benefits, tamoxifen use is associated with a significantly elevated risk of endometrial cancer, with reported incidence rates of 2.20 per 1000 woman-years, compared to 0.71 per 1000 woman-years observed with placebo (28).
Raloxifene, a SERM with a favorable risk profile and lacking the agonistic effects of tamoxifen, demonstrated no significant benefit in a heavily pre-treated population. However, a clinical benefit rate of 33% was observed when combining partial responses with prolonged stable disease in 21 patients with HR+ mBC (43). Currently, raloxifene has limited applications in the mBC setting.
Mechanisms of action of SERMs are illustrated in Figure 4.
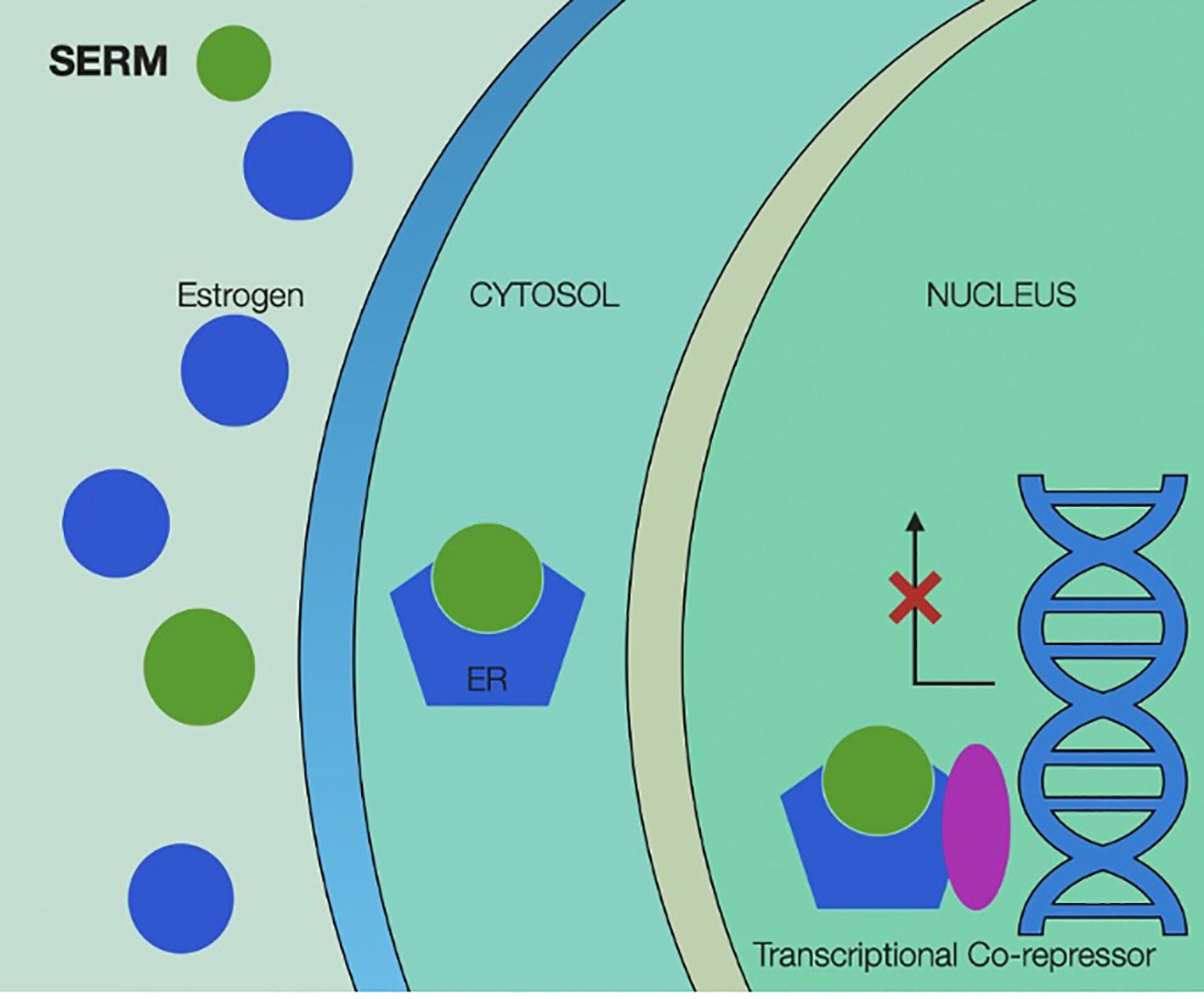
Figure 4. Selective estrogen receptor modulators’ mechanism of action, showing binding of the selective modulator by estrogen receptor and activating transcriptional co-repressors inside the cancer cell’s nucleus. SERM, selective estrogen receptor modulators; ER, estrogen receptor.
Among newer SERMs, the oral agent lasofoxifene has been evaluated in the phase II ELAINE 1 trial in ESR1-mutated, endocrine therapy–resistant mBC following progression on AI plus CDK4/6 inhibitors. While differences in PFS and response rates compared to fulvestrant did not reach statistical significance, lasofoxifene was associated with a more pronounced reduction in circulating ESR1 mutant allele fraction (82.9% drop from baseline at week 8 compared to 61.5% in the fulvestrant arm), suggesting potential biological activity warranting further investigation (44).
Selective estrogen receptor degraders
Fulvestrant is a SERD approved for the treatment of HR+ BC following progression on SERM and AI. It binds the ligand-binding domain of ERα, inhibiting conformational changes at both AF-1 and AF-2, and preventing co-activators recruitment. Because the complex is unstable, it degrades, leading to pure ER antagonism. Intramuscular injection of fulvestrant every 4 weeks reduces ER expression in a dose-depending fashion. Common side effects are menopause-like symptoms (28).
Benefit of fulvestrant was established at the beginning of the century when it was directly compared to AI in treatment of postmenopausal women with mBC or locally advanced BC progressing after ET (45, 46). Subsequent trials, such as FIRST and FALCON, demonstrated the benefits of a higher fulvestrant dose (500 mg versus 250 mg) with a loading dose strategy as first-line treatment in ET-naïve mBC, showing superiority over AIs (47). Loading dose strategies were later indirectly evaluated in two clinical trials, along with possible additional combinations – SoFEA (48) and EFFECT (48). Notably, the SoFEA trial found no PFS benefit with the addition of anastrozole to fulvestrant compared to fulvestrant alone (4.4 months [95% CI 3.4–5.4] vs. 4.8 months [3.6–5.5], respectively) or exemestane monotherapy (3.4 months [3.0–4.6]) (49). However, both trials confirmed efficacy of fulvestrant modality of application with 500 mg intramuscularly on day 0, 250 mg on days 14, 28, and 250 mg every 28 days thereafter.
This was later reinforced through the CONFIRM trial, which showed that fulvestrant 500 mg led to a significant improvement in median OS to 26.4 months compared to 22.3 months with the 250 mg dose (HR, 0.81; 95% CI, 0.69–0.96; p=0.02) (50). It is important to note that these studies included patients who had progressed on prior non-steroidal AIs and had acquired endocrine resistance. In contrast, the S0226 evaluated fulvestrant in ET–naïve postmenopausal patients with mBC and showed improved outcomes with the combination of fulvestrant and anastrozole. In this population, median PFS was 15.0 months with combination therapy compared to 13.5 months with anastrozole alone (HR 0.81, p = 0.007), and extended to 16.7 vs. 12.7 months in patients who were entirely endocrine-naïve. Overall survival was also prolonged (49.8 vs. 42.0 months; HR 0.82, p = 0.03), with the most pronounced benefit observed in tamoxifen-naïve patients (52.2 vs. 40.3 months; HR 0.73). No significant OS advantage was seen in those previously treated with tamoxifen (51).
In ESR1-mutated BC a combined analysis of the SoFEA and EFECT trials further demonstrated higher OS at one year with fulvestrant (80%) vs. exemestane (62%) (p=0.04). In contrast, among patients without ESR1 mutations, 1-year OS was similar between the two treatments (81% for fulvestrant vs. 79% for exemestane, p=0.69). Additionally, the EFECT trial reported no significant difference in response rate or clinical benefit between fulvestrant and exemestane in an unselected metastatic cohort. These findings suggest a survival advantage for fulvestrant in ESR1-mutated disease, while both agents appear equally effective in wild-type cases (51, 52). More recently, the Chinese FRIEND trial further supported the superiority of fulvestrant over exemestane, demonstrating improved PFS (8.5 vs. 5.6 months, p=0.014), ORR (19.5% vs. 6%, p=0.017), and time to treatment failure (8.4 vs. 5.5 months, p=0.008). However, in this trial, no significant difference in efficacy was observed between ESR1-mutated and ESR1 wild-type HR+ mBC (53).
Fulvestrant-based combination therapies, especially with CDK4/6i, have been evaluated in multiple trials, which will be explored in the following sections.
To address the limitations of fulvestrant and provide improved bioavailability and efficacy compared to AIs, particularly in somatic ESR1-mutated BC, novel oral SERDs are currently under development. Among these, elacestrant has shown promising PFS results (54) and it is approved for clinical use since 2023.
Elacestrant is a nonsteroidal oral SERD that degrades ER-alpha in a proteasome dose-dependent manner. It also inhibits estradiol-dependent, ER-directed gene transcription, thereby suppressing tumor growth in heavily pretreated BC, including both non-mutated and ESR1-mutated cases (55).
Recent findings from the EMERALD trial have demonstrated the efficacy of elacestrant in HR+ mBC, particularly within the ESR1 mutated breast tumors. The trial reported improved 1-year PFS in all patients (HR 0.70, 95% CI 0.55–0.88, p=0.002), including those with ESR1-mutated tumors (HR 0.55, 95% CI 0.39–0.77, p=0.005), without significant limiting side (54) effects (54).
Additionally, the SERENA-2 trial demonstrated the efficacy of camizestrant, a next-generation selective estrogen receptor degrader (ngSERD), in patients with advanced HR+ HER2- BC who had progressed or relapsed after ≤1 line of ET and ≤1 line of CHT in the advanced setting, without prior exposure to fulvestrant or other SERDs. The primary endpoint was PFS, comparing camizestrant at 75 mg and 150 mg daily to fulvestrant 500 mg. Median PFS was 7.2 months (90% CI, 3.7–10.9) with camizestrant 75 mg, 7.7 months (90% CI, 5.5–12.9) with 150 mg, and 3.7 months (90% CI, 2.0–6.0) with fulvestrant. Both camizestrant doses significantly reduced the risk of progression versus fulvestrant, with HRs of 0.59 (90% CI, 0.42–0.82; p=0.017) for 75 mg and 0.64 (90% CI, 0.46–0.89; p=0.009) for 150 mg (56).
Patients previously treated with CDK4/6 inhibitors had poorer outcomes, with PFS of 5.5 months (HR 0.49) in the camizestrant 75 mg plus fulvestrant group, 3.8 months (HR 0.68) with 150 mg, and 2.1 months with fulvestrant alone. Among patients with ESR1 mutations, PFS was 6.3 months (HR 0.33) with 75 mg of camizestrant, 9.2 months (HR 0.655) with 150 mg, and 2.2 months with fulvestrant (56, 57).
Most recently, the SERENA-6 phase III trial, presented at ASCO 2025, demonstrated that camizestrant, when administered at 75 mg daily in combination with a CDK4/6 inhibitor to patients with detectable emergent ESR1 mutations during ongoing AI plus CDK4/6i therapy, significantly improved outcomes. The median PFS was 16.0 months with camizestrant versus 9.2 months with continued AI (HR 0.44; 95% CI, 0.31–0.60; p<0.00001). Camizestrant also substantially prolonged time to global health status deterioration (23.0 vs. 6.4 months; HR 0.53; 95% CI, 0.33–0.82; p<0.001) (58), positioning it as a promising early intervention strategy upon molecular progression. Notably, SERENA-6 is the first global registrational study to validate ctDNA-guided treatment intensification based on ESR1 mutation emergence, highlighting its potential to redefine endocrine sequencing.
More recently, results from the phase III EMBER-3 trial were also published. This study investigated the oral SERD imlunestrant in patients with ER-positive, HER2-negative mBC who progressed on prior AI therapy, with or without CDK4/6 inhibitors. Patients were randomized to imlunestrant, standard ET, or imlunestrant plus abemaciclib. In the ESR1-mutant subgroup, imlunestrant significantly improved PFS compared to standard ET (5.5 vs. 3.8 months; P<0.001). In the overall population, PFS was similar between imlunestrant and standard ET (5.6 vs. 5.5 months; HR 0.87; P=0.12). However, the combination of imlunestrant with abemaciclib yielded superior PFS (9.4 vs. 5.5 months; HR 0.57; P<0.001), regardless of ESR1 status. Imlunestrant alone was well tolerated, with fewer grade ≥3 adverse events than the combination arm (59). These findings support imlunestrant as a promising endocrine backbone, particularly in ESR1-mutant mBC.
Other SERDs such as giradestrant and amcenestrant, evaluated in the acelERA and AMEERA-3 studies respectively, showed no benefit over traditional ET (60), despite previous promising findings. Giradestrant was initially evaluated as monotherapy (30 mg orally daily) versus combination therapy with palbociclib in postmenopausal women with HR+ mBC, demonstrating an ORR of 38% in the combination arm compared to 20% in the monotherapy arm. Paired biopsies from 21 patients taken before and during treatment revealed downregulation of ER, PR, Ki67, and ER pathway activity, along with a significant reduction in ESR1 ctDNA mutations detected in 94% of patients after 4 weeks of therapy (26).
Combination trials involving giradestrant and palbociclib versus letrozole and palbociclib, as well as giradestrant and everolimus versus exemestane and everolimus, are currently ongoing. Additionally, a randomized umbrella trial is evaluating giradestrant in various combination with ipatasertib, inavolisib, everolimus, and samuraciclib (26).
Mechanisms of action of SERDs are illustrated in Figure 5.
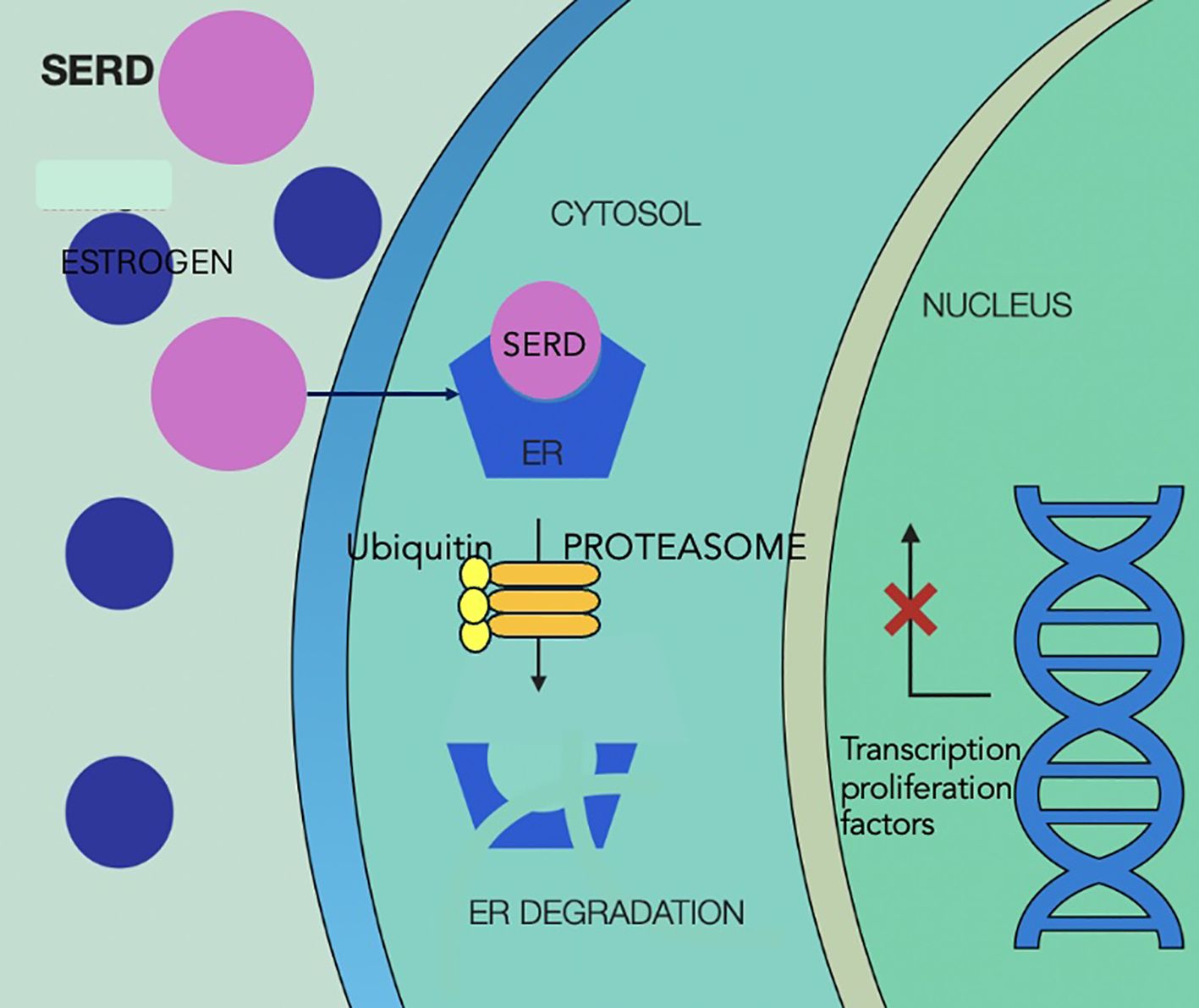
Figure 5. Selective estrogen receptor degraders mechanism of action, showing competitive binding of SERD to the estrogen receptor in the cytoplasm, displacing estrogen and preventing receptor activation. SERD binding leads to conformational change in the receptor, making it a target for ubiquitin-mediated degradation via the proteasome pathway, reducing levels of estrogen receptor and estrogen-driven gene expression in the nucleus, which ultimately leads to less activation of transcription factors and inhibition of tumor proliferation. SERD, selective estrogen receptor degraders; ER, estrogen receptor.
CDK4/6 inhibitors
The introduction of CDK4/6 inhibitors has been one of the most transformative advances in the treatment of HR+/HER2– mBC over the past decade, leading to significant survival benefits. Palbociclib, ribociclib, and abemaciclib—routinely used in combination with ET—now represent the standard first-line approach for most patients with HR+/HER2– mBC (61).
CDK4/6 forms active complexes with D-type cyclins, driving hyperphosphorylation of the retinoblastoma (Rb) protein. This inactivates Rb, leading to the release of E2F transcription factors that promote cell cycle progression from G1 to S phase. Dysregulation of this pathway is common in cancer, facilitating uncontrolled cellular proliferation (61). Estrogen induces expression of cyclin D and promotes CDK4/6 activity in HR+ BC. Therefore, estrogen depletion is essential for cell-cycle arrest.
CDK4/6 inhibitors are currently approved in combination with ET for HR+ mBC, with abemaciclib additionally approved as monotherapy.
For endocrine-sensitive cases, AI-based combinations were evaluated in PALOMA-2, MONALEESA-2, and MONARCH-3, reporting hazard ratios for progression of 0.54, 0.55, and 0.58, respectively, with median PFS gains of 9–13 months. Specifically, median PFS was 24.8 months with palbociclib plus letrozole compared to 14.5 months with placebo plus letrozole in PALOMA-2 (62), and 25.3 months with ribociclib plus letrozole versus 16 months with placebo plus (61) letrozole in the MONALEESA-2 study (63). Abemaciclib also showed significant benefit, achieving a PFS of 28.18 months in combination with anastrozole/letrozole vs. 14.76 months in the placebo plus anastrozole/letrozole group (64).
Regarding OS ribociclib is the only CDK4/6 inhibitor to show a statistically significant OS benefit in the first-line setting. In the MONALEESA-2 trial, ribociclib plus letrozole achieved a median OS of 63.9 months (95% CI, 52.4–71.0) versus 51.4 months (95% CI, 47.2–59.7) with placebo plus letrozole (HR 0.76; 95% CI, 0.63–0.93; p=0.008) (65). In contrast, the PALOMA-2 trial failed to demonstrate a significant OS advantage for palbociclib plus letrozole compared to placebo (53.9 vs. 51.2 months; HR 0.956; p=0.3378) (62). Similarly, the MONARCH-3 trial, with 8 years of follow-up, reported a non-significant OS improvement of 13.1 months for abemaciclib plus AI (66.8 vs. 53.7 months; HR 0.804; p=0.0664) (66).
In endocrine-resistant disease, CDK4/6i were investigated in combinations with fulvestrant-based ET. Importantly, primary endocrine resistance does not preclude clinical benefit from subsequent endocrine-based combinations. Several studies, including PALOMA-3, MONARCH 2 and MONALEESA-3, have demonstrated that CDK4/6 inhibitors such as palbociclib, abemaciclib and ribociclib, when combined with fulvestrant, retain efficacy in both primary and secondary endocrine-resistant HR+/HER2− mBC.
In PALOMA-3, the combination of palbociclib and fulvestrant significantly improved PFS compared to fulvestrant plus placebo in HR+/HER2– mBC patients who had progressed on prior ET. Median PFS was 9.5 months versus 4.6 months (HR 0.46; p<0.0001), and objective response rate (ORR) was notably higher with palbociclib (24.6% vs. 10.9%). Importantly, this benefit was independent of PIK3CA mutation status (67).
Similarly, MONARCH-2 evaluated abemaciclib in combination with fulvestrant in a chemotherapy-naïve population with progression on prior ET. Abemaciclib significantly prolonged PFS to 16.4 months compared to 9.3 months with placebo (HR 0.553; p<0.001), with an ORR of 48.1% versus 21.3%, respectively — representing a gain of over 7 months and more than doubling response rates (68).
In MONALEESA-3, ribociclib was assessed alongside fulvestrant in a broader population that included both treatment-naïve and previously treated patients. The combination achieved a median PFS of 20.5 months versus 12.8 months with placebo (HR 0.593; p<0.001). This benefit was consistent across subgroups, including treatment-naïve patients (HR 0.577) and those with prior ET for mBC (HR 0.565). ORR was also higher with ribociclib (40.9%) compared to placebo (28.7%) (69).
Collectively, these findings established all three CDK4/6 inhibitors—palbociclib, abemaciclib, and ribociclib—as key components of ET-based therapy for both endocrine sensitive and endocrine resistant HR+/HER2– mBC.
The clinical benefit of CDK4/6 inhibitors extends beyond postmenopausal, endocrine-sensitive and -resistant settings. In pre- and perimenopausal patients with HR+/HER2– mBC, the combination of ribociclib with a non-steroidal AI and ovarian suppression (goserelin) has also demonstrated significant efficacy. In this population, the MONALEESA-7 trial reported a median OS of 58.7 months with ribociclib versus 48.0 months with placebo (HR 0.76; 95% CI, 0.61–0.96), with 4-year OS rates of 60% and 50%, respectively (70). PFS similarly improved from 13.0 to 23.8 months (HR 0.55; p<0.0001) (71).
Additional evidence supporting the use of CDK4/6 inhibitors in younger patients comes from the Young PEARL trial, a phase II randomized study comparing palbociclib plus ET and ovarian suppression to capecitabine in premenopausal women with HR+/HER2– mBC. Median PFS was 19.5 months (90% CI, 14.3–22.2) in the palbociclib arm versus 14.0 months (90% CI, 11.7–18.7) in the capecitabine arm (HR 0.74; 90% CI, 0.57–0.98; p =0.036) (72), confirming the efficacy of CDK4/6i in this subgroup with a more favorable toxicity profile.
The ABIGAIL trial further explored the role of abemaciclib in premenopausal mBC patients with visceral disease. Patients were randomized to receive either abemaciclib plus ET (letrozole or fulvestrant) or an induction regimen of paclitaxel followed by abemaciclib plus ET. The primary endpoint was met, with a 12-week objective response rate (ORR) of 59% in the abemaciclib plus ET arm compared to 40% in the chemotherapy-sequenced arm (OR 2.12; 95% CI, 1.13–3.96; p=0.019) (73), supporting the efficacy of endocrine-based regimens even in the presence of aggressive disease biology.
Despite the synergy observed with ET combinations, abemaciclib remains the only CDK4/6 inhibitor approved as monotherapy. This indication was based on the MONARCH 1 trial, which evaluated abemaciclib in heavily pretreated, CDK4/6i-naïve patients with HR+/HER2– mBC (65). This trial reported an ORR of 19.7% and a median PFS of 6 months (74). Notably, all participants in this study were CDK4/6i-naïve (61). Subsequent data suggest that abemaciclib retains efficacy even after prior CDK4/6i exposure: in a multicenter cohort, patients previously treated with palbociclib and ET achieved a median OS of 17.2 months and PFS of 5.3 months with abemaciclib, administered either alone or in combination with ET (75).
As briefly mentioned earlier, CDK4/6i were also validated against CHT in the phase II RIGHT Choice trial, which compared ribociclib plus ET to physician’s choice of CHT in 223 patients with HR+ mBC experiencing rapid disease progression. With a median follow-up of 24.1 months, the trial demonstrated nearly a 12-month advantage in median PFS for the ribociclib arm compared to CHT (24 months vs. 12.3 months; HR 0.54; 95% CI, 0.36–0.79; p=0.007). ORR were comparable between the groups, with 65.2% for ribociclib and 60% for CHT, although ribociclib showed a slightly longer time to response (29).
These findings are consistent with an earlier phase II study in premenopausal HR+ mBC patients who relapsed or progressed on tamoxifen therapy. This trial compared palbociclib plus ET to capecitabine and demonstrated a median PFS of 20.1 months in the palbociclib arm versus 14.4 months in the capecitabine arm (HR 0.659; 95% CI, 0.437–0.994; p=0.0235) (76).
However, findings from the phase III PEARL trial, which compared palbociclib plus fulvestrant or exemestane to capecitabine in AI-resistant HR+ mBC, showed no efficacy advantage in patients with ESR1 mutations, despite the more favorable toxicity profile of the endocrine-based regimens. These results underscore the importance of individualized treatment sequencing, taking into account both the agent’s mechanism of action and the tumor’s molecular profile (77).
This issue has increasingly been recognized as crucial for managing therapy resistance and optimizing patient outcomes. The PADA-1 trial provided compelling evidence supporting a molecularly guided approach by demonstrating that early intervention based on the detection of rising ESR1 mutations in circulating tumor DNA (bESR1mut) significantly prolonged PFS. Patients who switched from AI plus palbociclib to fulvestrant plus palbociclib upon bESR1mut detection—before radiographic progression—achieved a median PFS of 11.9 months compared to 5.7 months in those who continued AI and palbociclib (HR 0.61; p=0.004). Importantly, this trial underscored the feasibility and clinical benefit of guiding treatment decisions using molecular dynamics rather than waiting for standard radiologic progression, offering a paradigm shift in the timing and duration of therapy in HR+/HER2– mBC (78).
In contrast, the TRINITI-1 and PACE studies explored rational combination strategies for patients progressing after CDK4/6i therapy, with a focus on overcoming endocrine resistance through additional pathway targeting or immune modulation.
TRINITI-1 evaluated a triplet regimen of ribociclib, everolimus, and exemestane in heavily pretreated HR+/HER2– mBC patients. Notably, those with wild-type ESR1 and PIK3CA tumors derived greater benefit (median PFS 9.9 vs. 3.2 months), underscoring the role of molecular context in response to targeted combinations (79).
The PACE trial tested the addition of avelumab to fulvestrant with or without palbociclib in the post-CDK4/6i setting. Although PFS was not significantly improved, the triplet arm (avelumab + fulvestrant + palbociclib) showed the most favorable OS outcomes (42.5 months), suggesting potential benefit of immunotherapy-enhanced endocrine strategies in a subset of resistant patients (80).
Together, these studies highlight two complementary but distinct approaches in managing CDK4/6i resistance: one centered on molecular surveillance and dynamic treatment switching (e.g., PADA-1), and the other exploring post-progression therapeutic intensification through rational combinations (e.g., TRINITI-1, PACE). While these strategies focus on optimizing outcomes once resistance has developed, another critical question is whether all patients benefit equally from early CDK4/6i use in the first-line setting.
Addressing this, the phase III SONIA trial examined the sequencing of CDK4/6 inhibition in endocrine-sensitive HR+/HER2– mBC. Specifically, it evaluated whether initiating treatment with a CDK4/6 inhibitor plus an AI provided superior benefit compared to deferring CDK4/6i until after progression on AI monotherapy. The results showed no statistically significant time to second progression (PFS2) advantage with upfront CDK4/6i use (31.0 vs. 26.8 months; HR 0.87; p=0.10), despite increased toxicity and higher treatment burden (81). These findings question the universal application of first-line CDK4/6i and highlight the importance of tailoring treatment initiation to individual patient context.
Building on this, several studies have evaluated the effectiveness of continuing or reintroducing CDK4/6 inhibitors after progression. The phase III postMONARCH trial demonstrated that abemaciclib plus fulvestrant reduced the risk of disease progression by 27% (HR 0.66; p=0.01), with benefits observed across subgroups, including patients with ESR1 or PIK3CA mutations (57, 82). Likewise, the phase II MAINTAIN trial showed that switching the endocrine partner while continuing CDK4/6 inhibition with ribociclib significantly improved PFS, with a median of 5.29 months versus 2.76 months for placebo (HR 0.57; p=0.006). Improved 6- and 12-month PFS rates further supported this strategy (83).
Given the variability in trial outcomes, the clinical value of continuing CDK4/6 inhibition beyond progression remains a subject of ongoing debate. Emerging evidence suggests that the distinct pharmacologic and biological profiles of individual CDK4/6 inhibitors may shape patterns of resistance and efficacy. This is exemplified by the divergent results seen in the adjuvant setting: ribociclib demonstrated a significant benefit in the phase III NATALEE trial (83), while palbociclib failed to improve outcomes in both the PALLAS (84) and PENELOPE-B (85) trials. Additionally, abemaciclib has distinguished itself with single-agent activity and favorable results in the nextMONARCH trial when combined with tamoxifen, reinforcing its potential utility in later lines (86). These differences underscore the need for a more nuanced, agent-specific approach to sequencing and rechallenge strategies, ideally guided by molecular profiling and individual tumor biology.
Ongoing trials, including ELAINE 3 and CAPItello-292, as well as the recently published INAVO-120, aim to provide further insights into optimal sequencing and resistance management for CDK4/6-based therapy, but will be discussed in a separate section.
mTOR inhibitors
The PI3K/AKT/mTOR pathway plays a critical role in both genomic and non-genomic ER signaling and is frequently upregulated in endocrine-resistant HR+/HER2– mBC. This has led to the investigation of mTOR inhibitors as a means to restore endocrine sensitivity. Early efforts focused on temsirolimus, which, in a subgroup of patients under 65, demonstrated a PFS benefit when combined with letrozole (median PFS 9 vs. 5.6 months; HR 0.75; p=0.009) (38).
Subsequently, everolimus—an oral allosteric mTOR inhibitor with a superior pharmacological profile—was evaluated in several pivotal trials. The BOLERO-4 phase II study established everolimus combined with letrozole as an active first-line strategy in ET-naïve HR+/HER2– mBC, reporting a median PFS of 22 months, though with considerable toxicity (stomatitis, diarrhea, weight loss, anemia) (87). In the endocrine-resistant setting, BOLERO-2 confirmed the efficacy of everolimus with exemestane versus placebo (median PFS 7.8 vs. 3.2 months; HR 0.45; p<0.0001) however, without having shown an overall survival benefit (24). Similarly, other trials demonstrated clinical benefit with everolimus combined with tamoxifen or fulvestrant, with TAMRAD study reporting a 6-month clinical benefit rate of 61% versus 42% with tamoxifen alone (88) while the MANTA trial reported a PFS of 12.3 months for everolimus combined with fulvestrant and 7.6 months for vistusertib combined with fulvestrant (88).
The MIRACLE phase II trial extended this concept to premenopausal patients, showing that everolimus plus letrozole (with goserelin) significantly improved median PFS (19.4 vs. 12.9 months; HR 0.64; p=0.008) and clinical benefit rate (72.7% vs. 47.5%) compared to letrozole alone. Interestingly, patients who crossed over to everolimus after progression still gained a median PFS of 5.5 months, emphasizing potential utility even in delayed sequencing (89).
More recently, the role of mTOR inhibition post-CDK4/6i progression has garnered attention. A real-world cohort study involving 161 patients treated with everolimus plus various ETs after CDK4/6i failure (primarily exemestane or fulvestrant) demonstrated a median PFS of 6.0 months, with longer benefit observed in patients with prior CDK4/6i exposure ≥18 months (8.7 months), no visceral disease (8.0 months), or who were chemotherapy-naïve in the metastatic setting (7.2 months) (90). These findings are particularly relevant when contrasted with endocrine monotherapy post-CDK4/6i, where median PFS typically ranges from 2.8 to 4.8 months, suggesting that everolimus-based combinations may offer a clinically meaningful alternative in selected patients.
Despite this promise, not all studies were positive. A phase I/IIa study of exemestane, everolimus, and palbociclib in CDK4/6i-pretreated HR+/HER2– mBC patients failed to meet its primary endpoint of clinical benefit rate, achieving just 18.8%, although median OS reached 24.7 months. Multi-omic profiling in this study revealed resistance mechanisms including ESR1, HER2, and BRAF alterations with high RTK/MAPK pathway activity, hinting at molecular subsets more or less likely to benefit (91).
Together, this body of evidence underscores the importance of biologically guided therapy selection. The activity of everolimus appears preserved even after CDK4/6i exposure, especially in patients without visceral crisis or prior chemotherapy, and its utility may be enhanced in the presence of certain molecular aberrations. Future strategies will likely rely on integrative biomarker-driven approaches to define the optimal placement of mTOR inhibitors in the evolving treatment landscape of endocrine-resistant HR+/HER2– mBC.
Phosphoinositide 3 kinase inhibitors
Aberrant activation of the PI3K/AKT/mTOR pathway is a hallmark of endocrine resistance in HR+/HER2– advanced breast cancer (BC). PI3K mutations, which lead to constitutive pathway activation, are identified in approximately 28%–46% of cases and are consistently associated with a poorer prognosis (92).
Although mTOR inhibitors like everolimus have demonstrated clinical efficacy, their therapeutic potential is limited by compensatory feedback loops that reactivate upstream signaling. One such mechanism involves the IGF-1R–mediated phosphorylation of insulin receptor substrates IRS1 and IRS2 by activated S6K, which reactivates AKT signaling and promotes metabolic adaptation through increased translocation of glucose transporter 4 (GLUT4) to the plasma membrane (38). This facilitates enhanced glucose uptake and accelerates tumor cell metabolism, potentially contributing to the hyperglycemia, ketosis, and insulin dependency observed in some patients receiving mTOR-targeted therapies (93).
To counteract these feedback-driven escape mechanisms, targeted agents directed at upstream nodes of the pathway have been developed. These include isoform-specific PI3K inhibitors, pan-PI3K inhibitors, dual PI3K/mTOR inhibitors, and AKT inhibitors. The latter are broadly categorized into two mechanistic classes: ATP-competitive inhibitors, such as alpelisib, capivasertib, and ipatasertib, which directly inhibit kinase activity at the ATP-binding site; and allosteric inhibitors, such as MK-2206 and miransertib, which bind the pleckstrin homology domain of AKT, preventing its translocation to the plasma membrane and subsequent activation by upstream kinases (94).
Several pan-PI3K inhibitors have been evaluated in combination with ET to overcome resistance in HR+/HER2– mBC particularly in patients harboring PIK3CA mutations. Buparlisib and pictilisib were the most extensively studied agents in this class, with buparlisib advancing to two pivotal phase III trials—BELLE-2 and BELLE-3 (94).
The BELLE-2 trial enrolled patients with AI-resistant disease and prior exposure to CHT. Buparlisib (100 mg daily) combined with fulvestrant modestly improved median PFS compared to fulvestrant plus placebo (6.9 vs. 5.0 months; HR 0.78, 95% CI 0.67–0.89; p=0.00021). Importantly, in the subset of patients with confirmed PIK3CA mutations, the benefit was slightly more pronounced (6.8 vs. 4.0 months; HR 0.76, 95% CI 0.60–0.97; p=0.014) (95).
BELLE-3 extended these findings to a more heavily pretreated population—postmenopausal women with prior progression on everolimus. Here, buparlisib plus fulvestrant resulted in a median PFS of 3.9 months compared to 1.8 months with fulvestrant alone (HR 0.67, 95% CI 0.53–0.84; p=0.00030) (96).
Despite the moderate improvements in PFS (ranging from 2 to 3 months), the clinical use of buparlisib is limited by its unfavorable toxicity profile. Reported side effects include elevated liver enzymes, hyperglycemia, hypertension, and fatigue, with more severe adverse events such as pleural effusion, dyspnea, and significant liver toxicity observed in 22% of patients in the buparlisib group, compared to 16% in the placebo group (96). However, despite the statistically significant gains, the clinical translation of buparlisib has been hindered by a challenging toxicity profile. Treatment-related adverse events—including transaminitis, hyperglycemia, hypertension, and fatigue—were frequent, with grade ≥3 events such as pleural effusion and liver toxicity occurring in 22% of patients receiving buparlisib, compared to 16% in the placebo arm (96). These safety concerns, coupled with modest efficacy, ultimately limited its therapeutic development and redirected focus toward isoform-selective PI3K inhibitors with more favorable tolerability.
Given the toxicity and limited therapeutic window of pan-PI3K inhibitors, attention has shifted toward isoform-specific agents, particularly those targeting the PI3Kα isoform, which is most frequently mutated in HR+/HER2– BC. Alpelisib, a selective PI3Kα inhibitor, was evaluated in the pivotal phase III SOLAR-1 trial. Among patients with PIK3CA-mutant tumors, the combination of alpelisib and fulvestrant significantly improved PFS compared to fulvestrant alone (11.0 vs. 5.7 months; HR 0.65, 95% CI 0.50–0.85; p<0.001) (92). Although the trial did not meet the prespecified threshold for overall survival (OS) benefit in the overall mutant cohort (39.3 vs. 31.4 months; HR 0.86; p=0.15), a clinically meaningful OS gain was observed in the subset of patients with visceral disease, particularly those with lung and/or liver metastases (37.2 vs. 22.8 months; HR 0.68; 95% CI 0.46–1.00) (92).
Further support for alpelisib’s biological activity was provided by a phase II study of alpelisib monotherapy, which demonstrated that early reductions in PIK3CA mutations detected in circulating tumor DNA (ctDNA) were strongly associated with improved outcomes. Patients with a molecular response by week 8 experienced longer PFS (HR 0.24, 95% CI 0.083–0.67; p=0.0065), and similar associations were seen in those with detectable ESR1 mutations at baseline (HR 0.22, 95% CI 0.078–0.60; p=0.003) (92, 97).
These findings highlight the potential utility of ctDNA as an early predictive biomarker for response to PI3K inhibition, particularly in endocrine-resistant disease.
Despite its efficacy, alpelisib’s clinical use is constrained by a notable toxicity profile. In the SOLAR-1 study, grade ≥3 hyperglycemia occurred in 36.6% of patients, while rash and diarrhea were reported in 9.9% and 6.7%, respectively (98). These adverse events require early intervention and may limit the use of alpelisib in frail patients or those with pre-existing metabolic comorbidities.
Similarly, in the SANDPIPER trial, the PI3K inhibitor taselisib was evaluated in women with recurrent or progressive BC, including those with metastatic disease following treatment with an AI. Patients were randomized to receive either taselisib or placebo in combination with fulvestrant. The taselisib group demonstrated a PFS improvement of 2 months (7.4 months [95% CI, 7.26–9.07] vs. 5.4 months [95% CI, 3.68–7.29]; HR 0.70, 95% CI 0.56–0.89; p=0.0037). However, serious adverse events were noted in the taselisib arm compared to the placebo arm (32.0% vs. 8.9%) (99).
Considering the toxicity limitations observed with alpelisib and taselisib, attention has expanded toward AKT inhibition as a complementary strategy to disrupt the PI3K/AKT/mTOR axis. Capivasertib, a selective ATP-competitive pan-AKT inhibitor, has demonstrated notable antitumor activity in HR+/HER2– breast cancer across various clinical settings. Early-phase data from the BEECH trial, which combined capivasertib with paclitaxel in endocrine-resistant mBC, did not yield significant improvements in PFS or tolerability. However, this may have been confounded by CHT-related toxicities and the absence of molecular stratification (100).
More encouraging results emerged from endocrine-based combinations. In the phase II FAKTION trial, capivasertib plus fulvestrant significantly improved median PFS (10.3 vs. 4.8 months; HR 0.56; p=0.0023) and OS (29.3 vs. 23.4 months; HR 0.66; p=0.0035) compared to fulvestrant alone. The benefit was particularly pronounced in patients with PI3K/AKT/PTEN pathway alterations, where median PFS reached 12.8 months versus 4.6 months (HR 0.44; p=0.0014), and median OS extended to 38.9 months compared to 20.0 months with placebo (HR 0.46; p=0.0047) (22).
While capivasertib was generally well tolerated, treatment-related grade 3/4 toxicities such as hyperglycemia (20–24%), diarrhea (14–17%), and rash (11–16%) were not uncommon, underscoring the importance of adverse event monitoring.
Building on these findings, the phase III CAPItello-291 trial confirmed the benefit of AKT inhibition in a more heterogeneous, heavily pretreated population. In this study, capivasertib plus fulvestrant doubled PFS compared to placebo (7.2 vs. 3.6 months; HR 0.60; p<0.001), with even greater efficacy in patients harboring AKT-pathway aberrations (7.3 vs. 3.1 months; HR 0.50; p<0.001) (101).
These results firmly establish capivasertib as a clinically meaningful option, especially in the post-CDK4/6i setting. The ongoing CAPItello-292 trial is now evaluating capivasertib in triplet therapy with palbociclib and fulvestrant, aiming to clarify its role earlier in the treatment sequence.
Most recently, the selective PI3K inhibitor inavolisib has garnered significant attention following the positive findings of the phase III INAVO120 trial. In patients with PIK3CA-mutant, HR+/HER2– advanced breast cancer, the addition of inavolisib to palbociclib and fulvestrant substantially extended PFS to 17.2 months compared to 7.3 months with placebo. Objective response rates were also markedly improved (62.7% vs. 28.0%), and according to the most recent ASCO announcement there was also a significant improvement in the OS with 34 months in the experimental, inavolisib arm vs. 27 months in the placebo arm (102). Importantly, the safety profile was favorable relative to alpelisib, with lower rates of grade ≥3 hyperglycemia (5.6%) and minimal occurrences of stomatitis or diarrhea. Neutropenia remained the most common high-grade toxicity (80.2%), consistent with palbociclib-related myelosuppression (21). Treatment discontinuation due to adverse events was infrequent (6.8%), suggesting that inavolisib may represent a more tolerable and efficacious next-generation alternative to alpelisib for patients with endocrine-resistant, PIK3CA-mutated disease (21, 94).
Taken together, these data underscore the growing importance of biomarker-guided inhibition of the PI3K/AKT pathway in HR+/HER2– mBC. With multiple agents now demonstrating efficacy in genomically enriched populations, future therapeutic strategies will likely hinge on refined molecular profiling to optimize the timing, combination, and sequencing of targeted therapies.
Antibody drug conjugates
In HR+ mBC that has become resistant to endocrine and targeted therapies, single-agent CHT remains a standard treatment option. However, its efficacy is modest, with median PFS typically ranging from 6 to 7 months.
Antibody-drug conjugates (ADCs) have emerged as a transformative therapeutic strategy, consisting of monoclonal antibodies directed against tumor-associated antigens, conjugated to cytotoxic payloads. This design allows for the selective delivery of chemotherapy agents to tumor cells expressing the target antigen (103). In BC, the first ADC to gain regulatory approval was ado-trastuzumab emtansine (T-DM1) in 2013 for HER2+ disease, followed by fam-trastuzumab deruxtecan (T-DXd, DS-8201) in 2022 (104).
The therapeutic efficacy of ADCs is influenced by several factors, beyond the payload itself, including the drug to antibody ratio, the level of antigen expression on tumor cells, and quite critically, the nature of the linker (105). Linkers can be either cleavable or non-cleavable, determining the mechanism of intracellular drug release. Non-cleavable linkers require internalization and lysosomal degradation of the ADC for payload release within the targeted cell. In contrast, cleavable linkers, such as those used in T-DXd, enable payload release in response to specific intracellular conditions, such as acidic pH or enzymatic activity (105). Notably, cleavable payloads can diffuse beyond the initially targeted cell, exerting cytotoxic effects on neighboring tumor cells with lower or heterogeneous antigen expression — a phenomenon known as the bystander effect (106). This has been a central hypothesis underlying the evaluation of ADCs in tumors with low or heterogeneous target expression.
In light of these insights, HER2 classification was redefined in 2021 to encompass a broader continuum of HER2 expressions, including HER2-low and ultra-low categories. HER2-0 is now defined by a complete absence of staining in infiltrating tumor cells, HER2 ultra-low by ≤10% of cells with faint or weak membrane staining and HER2-low by an IHC score of 1+ or 2+ with a negative in situ hybridization (ISH) result.
This reclassification has significantly expanded the therapeutic landscape for BC, with HER-2 low tumors comprising almost 70% of cases, previously considered HER2-negative tumors, now showing clinical benefit from agents such as T-DXd.
Subsequent genomic studies have supported the clinical distinction of HER2-low tumors. In a large sequencing study of 1,039 HER2- mBC, 47% were reclassified as HER2-low and 53% as HER2-0. While HER2-low tumors exhibited a slightly higher ERBB2 copy count compared to HER2-0 (2.05 vs. 1.79), no significant differences were observed in mutational burden or broader genomic alterations, suggesting that HER2-low may not represent a distinct molecular subtype (107). Longitudinal studies of matched primary and recurrent tumor samples further highlight the dynamic nature of HER-2 expression. HER2-low status was identified in 34.2% of primary tumors, rising to 37.3% in recurrent lesions. Notably, about 38% of patients exhibited shifts in HER2 expression over time, most frequently transitioning between HER2–0 and HER2-low (108).
What truly supported this paradigm shift were its clinical implications, as evidenced by the DESTINY-Breast 04 and DESTINY-Breast 06 trials, which demonstrated that patients with low and ultra-low HER2 expression can benefit from HER2-directed therapy.
In the DESTINY-Breast04, a pivotal phase III trial, 557 patients with HER2-low mBC were enrolled, of whom 494 (88.7%) were HR+ tumors and 63 (11.3%) HR-. Among patients with HR+ tumors, T-DXd significantly improved median PFS to 10.1 months compared to 5.4 months with CHT (HR 0.51; p<0.001) and extended OS to 23.9 months compared to 17.5 months (HR 0.64; p=0.003). Across the entire cohort, median PFS and OS were also superior with T-DXd: 9.9 months versus 5.1 months (HR 0.50; p<0.001), and 23.4 months versus 16.8 months (HR 0.64; p=0.001), respectively. The ORR in HR+ patients reached 52.6% with T-DXd compared to 16.3% with CHT. Grade 3 or higher adverse events occurred in 52.6% of T-DXd-treated patients versus 67.4% with CHT, with T-DXd being associated with interstitial lung disease (ILD) or pneumonitis in 12.1% of patients (109). Similarly, results were favorable in patients without previous CHT for metastatic disease, in both the HER2-low and ultralow categories, as illustrated through the DESTINY-Breast 06 phase 3 trial. In the HER2-low group, T-DXd significantly extended median PFS to 13.2 months compared to 8.1 months with CHT (HR 0.62; p<0.001), while in the ultralow category median PFS was similar 13.2 months vs. 8.3 months in the physician’s choice of chemotherapy (HR 0.72). However, OS data is still immature. Grade 3 or higher adverse events occurred in 52.8% of T-DXd patients versus 44.4% in the CHT group, with ILD or pneumonitis reported in 11.3% of T-DXd patients (110).
The DAISY phase 2 trial further provided mechanistic insights into the differential efficacy of T-DXd across HER2 expression levels. ORR was highest in HER2-overexpressing tumors (70.6%) with a median PFS of 11.1 months, while HER2-low and HER2–0 tumors demonstrated ORRs of 37.5% and 29.7%, and PFS of 6.7 and 4.2 months, respectively. Tumor shrinkage in target lesions reflected this gradient, with median reductions of 57% in HER2-positive, 25% in HER2-low, and 12.5% in HER2-0 cohorts, respectively (111).
Beyond DESTINY-Breast04 and DESTINY-Breast06, additional trials have reinforced the versatility of T-DXd across distinct subpopulations in advanced BC. The DESTINY-Breast07 trial evaluated T-DXd alone or in combination with pertuzumab in the first-line setting for HER2-positive mBC. Both treatment arms demonstrated high objective response rates (77–82%) and sustained clinical benefit, with 12-month PFS rates nearing 90% in the combination arm (112). These findings support the feasibility of T-DXd-based doublet regimens and inform the design of ongoing first-line studies such as DESTINY-Breast09.
The DESTINY-Breast12 trial addressed a critical unmet need by prospectively assessing the efficacy of T-DXd in patients with HER2-positive mBC and brain metastases—a population frequently underrepresented in clinical trials. T-DXd showed meaningful intracranial activity, with a 12-month PFS of 61.6% and central nervous system (CNS)-specific PFS of 58.9% in patients with brain metastases. In patients without CNS involvement, the systemic ORR reached 62.7% (113). All these findings support the use of T-DXd regardless of the presence or activity of brain metastases.
Collectively, these trials underscore the expanding therapeutic footprint of T-DXd—from HER2-low and ultralow hormone receptor-positive disease to HER2-positive tumors with CNS involvement—highlighting its potential as a foundational agent across molecular and clinical subtypes in advanced BC.
Beyond HER2-directed therapy, ADCs have shown promise in other molecular contexts. In heavily pretreated patients, sacituzumab govitecan (SG), an ADC targeting trophoblast cell-surface antigen-2 (Trop-2) and linked via a hydrolysable linker to SN-38, demonstrated robust activity in pretreated HR+, HER2− mBC patients. Internalization is not required, as hydrolysis can occur in the tumor microenvironment, contributing to the bystander effect (104).
The TROPiCS-02 trial showed a significant OS benefit with SG compared to CHT (median 14.4 vs. 11.2 months; HR 0.79; p=0.020), with consistent efficacy across different levels of Trop-2 expression. The ORR was also superior with SG (21% vs. 14%; OR 1.63; p=0.035), and the safety profile remained manageable (114).
Datopotamab deruxtecan (Dato-DXd), a novel Trop-2 directed ADC using the same cleavable linker and topoisomerase I inhibitor payload as T-DXd, is currently under investigation. In the TROPION-Breast01 trial, Dato-DXd significantly improved PFS (6.9 vs. 4.9 months; HR 0.63, p<0.0001) and demonstrated a higher ORR (36.4% vs. 22.9%) compared to CHT in patients who experienced disease progression on ET and had undergone up to two prior lines of CHT in the metastatic setting. Notably, Dato-DXd was associated with fewer grade ≥3 treatment-related adverse events (20.8% vs. 44.7%), suggesting an improved safety profile over conventional cytotoxic regimens (115).
Poly ADP-ribose polymerase inhibitors
In BRCA-mutated BC, loss of homologous recombination repair renders tumor cells unable to effectively repair double-strand DNA breaks. PARP inhibitors (PARPi) capitalize on this vulnerability by blocking the repair of single-strand breaks, through inhibition of PARP enzymes. During DNA replication, these unresolved single-strand breaks are converted into double-strand breaks, which accumulate and lead to cell death in the context of BRCA deficiency. This forms the mechanistic basis for PARPi as a targeted therapy for BC with DNA repair deficiencies (115).
In HR+ mBC patients with germline BRCA1 or BRCA2 mutations, PARPi such as olaparib and talazoparib, have demonstrated clinical efficacy in pivotal trials. The OlympiAD trial reshowed that olaparib significantly improved PFS over CHT (7.0 months vs. 4.2 months) in patients with HER2- germline BRCA-mutated mBC, including HR+ subtypes. In first-line settings, olaparib extended median OS to 22.6 months versus 14.7 months with CHT (HR = 0.55; 95% CI, 0.33–0.95), with 3 year survival rates of 40.8% compared to 12.8% (116). Similarly, the phase 3 EMBRACA trial demonstrated that talazoparib significantly prolonged median PFS of 8.6 months compared to 5.6 months with CHT (HR = 0.54; 95% CI, 0.41–0.71; p < 0.0001) and achieved a higher ORR of 62.6% versus 27.2%. While talazoparib was associated with increased hematologic toxicity, particularly grade 3–4 anemia (55% vs. 38%), it was linked to improved quality of life and delayed symptom deterioration compared to CHT (117).
Following these findings, PARPi are currently recommended as second-line therapy after progression on CDK4/6i and ET in patients with BRCA germline mutations. Given the significant OS advantage associated with CDK4/6i, these agents are generally prioritized before PARP inhibition, as no OS benefit was observed with PARPi alone (40). Nevertheless, the optimal sequencing of therapies continues to be a topic of active debate among clinicians and researchers.
PROteolysis TArgeting Chimeras
PROteolysis Targeting Chimeras (PROTACs), alongside lysosome targeting chimeras (LYTACs), represent an innovative class of targeted protein degradation therapies. These heterobifunctional molecules consist of two ligands: one that binds the protein of interest, and one that recruits E3 ubiquitin ligase, connected by a flexible linker. The formation of this ternary complex promotes polyubiquitination of the target protein, leading to its proteasomal degradation. Unlike traditional inhibitors, PROTACs eliminate rather than inhibit their targets, offering a strategy to overcome resistance driven by receptor overexpression or mutation.
Since their conceptualization in 2001, PROTACs targeting ER for BC have advanced considerably. ARV-471 (vepdegestrant), a novel oral ER-targeting PROTAC, recently received FDA fast-track designation for monotherapy in endocrine-resistant mBC (118), based on results from the NCT04072952 and the VERITAC clinical trials.
In the phase 1b NCT04072952 trial, vepdegestrant combined with palbociclib demonstrated a clinical benefit rate of 63.0% (95% CI, 47.5%-76.8%) for HR+ mBC. Subgroup analyses revealed even higher activity in ESR1-mutant tumors (CBR 72.4%) versus ESR1 wild-type (CBR 53.3%). Median PFS was 11.2 months (95% CI, 8.2-16.5) overall, extending to 13.7 months (95% CI, 8.2–not reached) in ESR1-mutant patients and 11.1 months (95% CI, 2.8-19.3) in ESR1 wild-type patients (119).
In the monotherapy VERITAC study which enrolled heavily pretreated ER+/HER2− mBC patients, ARV-471–200 mg QD achieved a CBR of 37.1% (95% CI: 21–55) overall and 47.4% (95% CI: 24–71) in those with mutant ESR1. Median PFS was 3.5 months (95% CI: 1.8–7.8). The safety profile was favorable, with most adverse events being grade 1–2 and including fatigue (40%), hot flushes (17%), and nausea (14%), supporting its advancement to phase III evaluation (120).
Just presented at ASCO 2025, the results of the pivotal phase III randomized, open-label trial comparing vepdegestrant (200 mg) with fulvestrant further underscore its clinical potential. In this study, 624 patients with ER+/HER2− advanced BC previously treated with a CDK4/6 inhibitor and up to two lines of ET were randomized. Among the 270 patients harboring ESR1 mutations, vepdegestrant significantly improved median PFS to 5.0 months (95% CI, 3.7–7.4) versus 2.1 months (95% CI, 1.9–3.5) with fulvestrant (HR 0.58; 95% CI, 0.43–0.78; P<0.001). In the overall population, median PFS was 3.8 months vs. 3.6 months (HR 0.83; 95% CI, 0.69–1.01; P=0.07). Grade ≥3 adverse events were reported in 23.4% of patients in the vepdegestrant group and in 17.6% of those receiving fulvestrant, with low rates of treatment discontinuation (2.9% vs. 0.7%) (121).
Mechanism of action of PROTACs are illustrated in Figure 6.
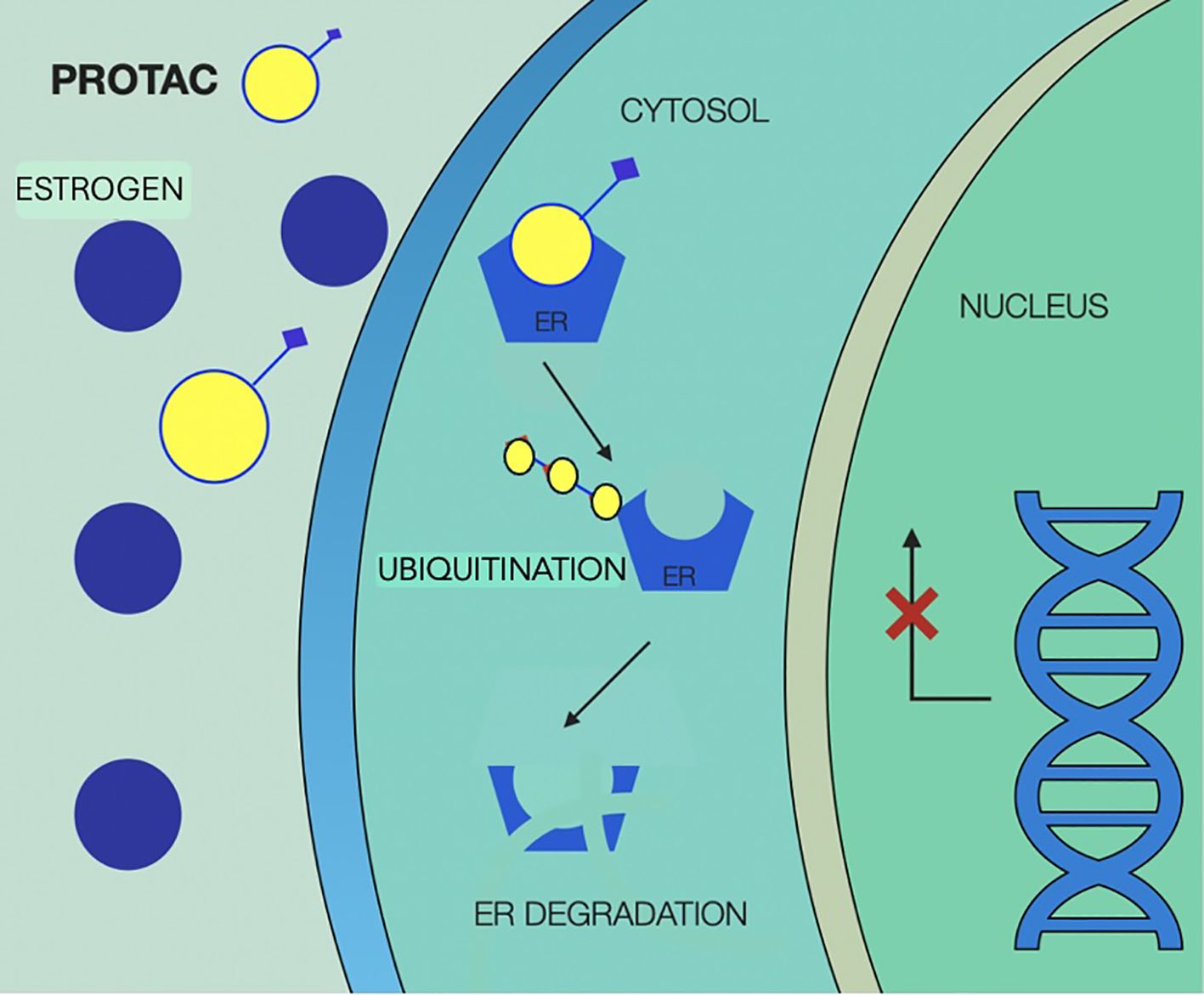
Figure 6. Mechanism of action of Proteolysis Targeting Chimera agents (PROTACs), illustrating selective degradation of the estrogen receptor (ER). PROTAC molecules simultaneously bind ER and recruit an E3 ubiquitin ligase, forming a ternary complex that leads to polyubiquitination of the receptor. This post-translational modification marks ER for proteasomal degradation, resulting in the complete elimination of both ligand-bound and mutant receptor species. PROTAC, proteolysis targeting chimera; ER, estrogen receptor.
Complete estrogen receptor antagonists
The complete estrogen receptor antagonists (CERANs) are a distinct class of small molecules designed to comprehensively inhibit both activation domains, AF1 and AF2, of the ER. Their mechanism involves degradation of ER, functional silencing through co-repressors recruitment, such as nuclear receptor co-repressor (N-CoR) binding to AF1, and consequently inhibition of ER-driven transcription and tumor proliferation (118).
Among agents in this category, OP-1250 has emerged as an orally bioavailable CERAN with selective ER degradation (SERD) activity. In preclinical studies, OP-1250 effectively inhibited and degraded both wild-type and mutant ER. Early clinical evaluation in a phase I/II dose-escalation and expansion trial showed promising results. At the recommended phase II dose, OP-1250 achieved in heavily pretreated HR+ mBC patients, an ORR of 18% and a CBR of 38% (26, 118).
CERANs mechanism of action is illustrated in Figure 7.
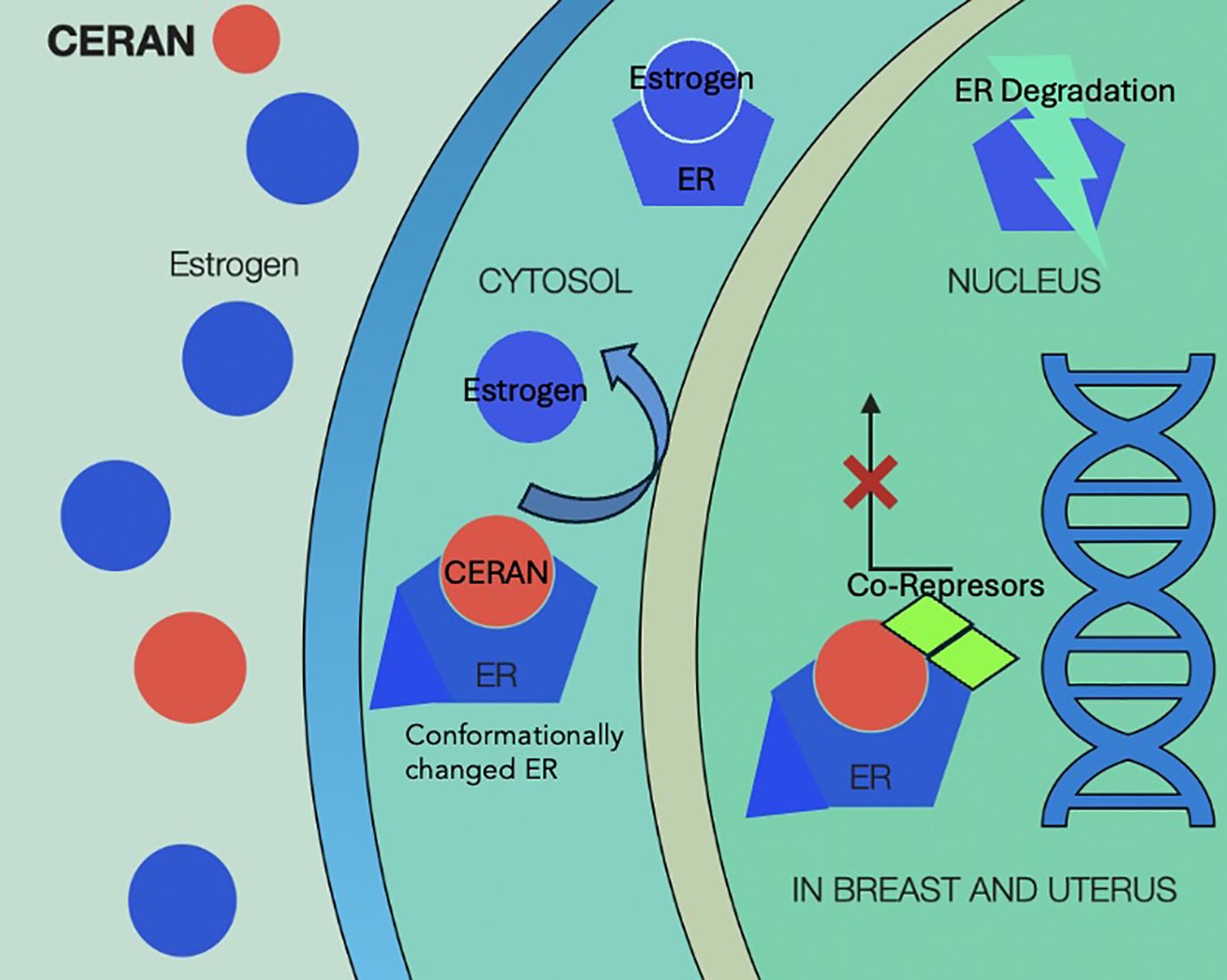
Figure 7. Mechanism of action of complete estrogen receptor antagonists (CERANs) showing competitive binding to the estrogen receptor (ER) in the cytosol, displacement of endogenous estrogen, and inhibition of ER activation. Upon CERAN binding, the receptor fails to undergo appropriate conformational change, leading to co-repressor recruitment, nuclear translocation blockade, or transcriptional silencing. This results in ER degradation and suppression of estrogen-driven gene expression in target tissues. CERAN, complete estrogen receptor antagonist; ER, estrogen receptor.
Selective estrogen receptor covalent antagonists
Selective Estrogen Receptor Covalent Antagonists (SERCAs) are an innovative class of ER targeting agents that inactivate ER by covalently binding to cysteine 530 (C530), a non-conserved residue within the ligand-binding pocket of ER alpha. This mechanism effectively antagonizes both wild-type and mutant ER receptors, including clinically relevant ESR1 mutations associated with endocrine resistance (26).
H3B-5942 was the first-in-class experimental oral compound with high selectivity for C530 demonstrating superior antitumor activity to fulvestrant in preclinical BC xenograft models with both ESR1 Y37S mutation and ESR1 wild type. Research around this agent paved the way for development of H3B-6545, a next generation SERCA currently under clinical investigation in multiple trials for ET-resistant HR+ BC (clinical trials: NCT03250676, NCT04568902, NCT04288089) (118). In a phase I/II trial H3B-6545 demonstrated an objective response rate (ORR) of 16.4% and a clinical benefit rate (CBR) of 39.7% in a heavily pretreated population (n=94), most of whom had prior CDK4/6 inhibitor exposure. Median PFS was 3.8 months, with responses enriched in patients with visceral disease and ESR1 mutations. Adverse events included bradycardia (asymptomatic in 35%, grade ≥2 in 5%), gastrointestinal symptoms, cytopenias, and renal function changes. Serious adverse events occurred in 21% of patients, leading to treatment discontinuation in 13% (26).
H3B-6545 is also being evaluated in combination with palbociclib in patients with HR+ mBC after ≥2 prior treatment lines (NCT04288089), further supporting its potential as a next-generation endocrine backbone in resistant disease.
Mechanism of action of SERCAs are illustrated in Figure 8.
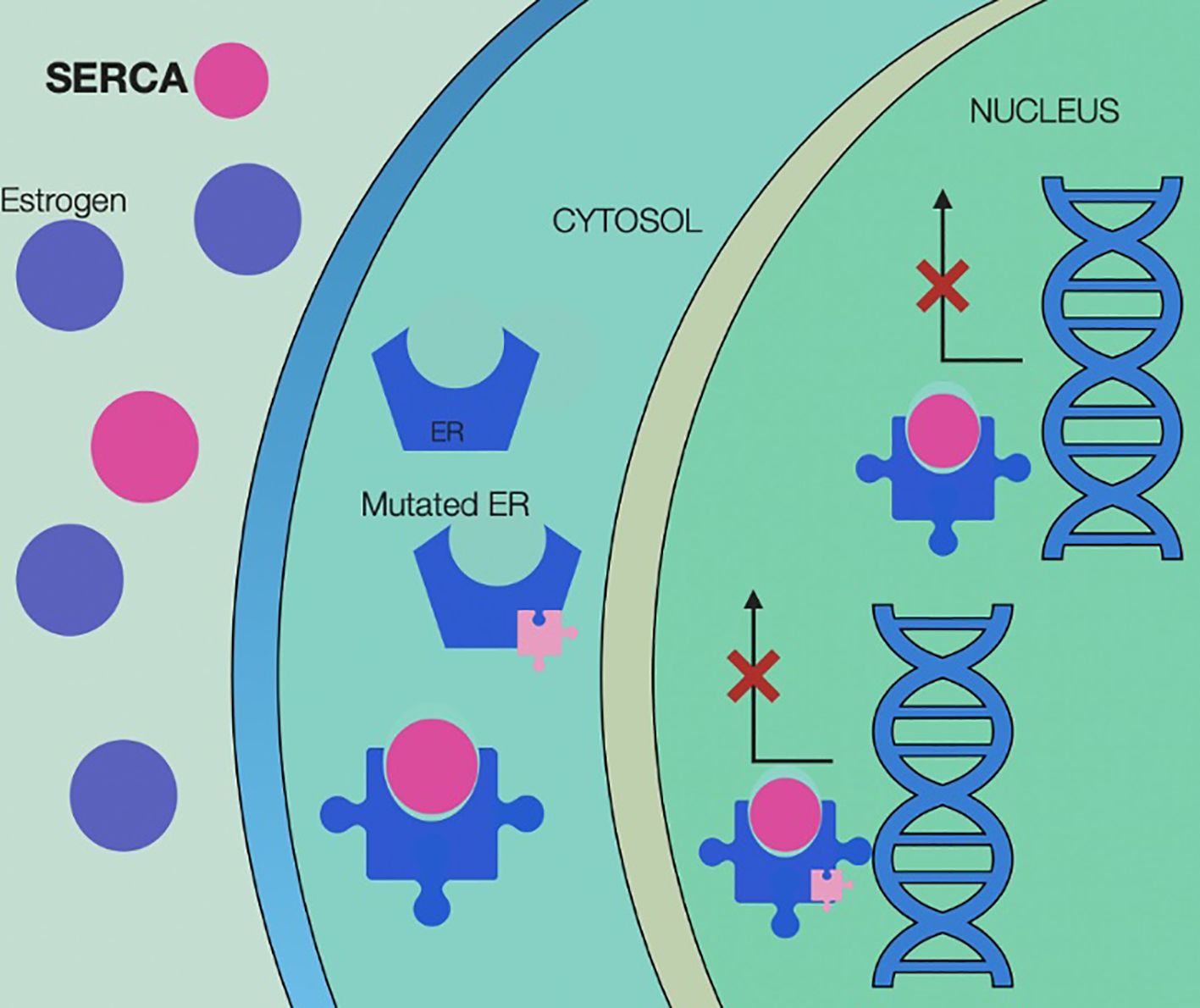
Figure 8. Selective estrogen receptor covalent antagonists (SERCA) mechanism of action, illustrating irreversible inhibition of both wild-type and mutant estrogen receptors (ER) through covalent binding at cysteine 530 within the ligand-binding domain. By forming a covalent bond, SERCAs prevent receptor activation, block estrogen binding, and disrupt transcriptional activity—even in the presence of activating ESR1 mutations. This results in sustained receptor inactivation, suppression of estrogen-driven gene expression, and inhibition of tumor cell proliferation. SERCA, Selective Estrogen Receptor Covalent Antagonists; ER, estrogen receptor.
Selective Human Estrogen Receptor Partial Agonist
Selective Human Estrogen Receptor Partial Agonists (ShERPAs) represent a novel class of benzothiophene-derived compounds engineered to selectively modulate ER signaling with partial agonist activity. These agents exbibit nanomolar potency in BC cell lines and are designed to mimic estradiol activity by binding ER in the nucleus, promoting its translocation to extranuclear compartments, and ultimately disrupting proliferative signaling. In preclinical models, several ShERPAs have shown significant antitumor activity, including in xenografts of endocrine-independent tamoxifen-resistant BC (122). Specifically, the compounds BMI-135 and TTC-352 have emerged as leading drugs within their category. Notably, TTC-352 has progressed to a phase I clinical trial for HR+ BC resistant to ET, demonstrating a favorable safety profile alongside early signs of antitumor efficacy (118).
Future directions
Although development of immune checkpoint inhibition strategies (ICI) targeting tumor-infiltrating lymphocytes (TILs) within the tumor microenvironment has revolutionized cancer treatment, patients with HR+/HER2- BC have derived limited benefit. This is primarily due to the unique pathophysiological characteristics of the tumor microenvironment in HR+/HER2- BC, which is characterized by low levels of TILs, reduced expression of HLA class I molecules (123), and the recruitment of immunosuppressive cells such as macrophages and regulatory T cells (Tregs). These factors render HR+/HER2- BC less responsive to conventional immunotherapies and are associated with a poorer prognosis (124).
However, ICI targeting the programmed death receptor 1 (PD-1) pathway, along with other immunotherapeutic approaches such as CTLA-4 monoclonal antibodies (e.g., ipilimumab), have been explored in HR+ BC, with several clinical trials still underway.
In the KEYNOTE-028 study, pembrolizumab achieved an ORR of 12% in heavily pretreated PD-L1-positive HR+/HER2− BC patients, albeit with prolonged response durations in some cases. These findings highlight the challenges of achieving significant clinical benefit with ICIs, especially in heavily pretreated HR+/HER2- BC populations (125). Similarly, avelumab demonstrated an ORR of only 3% in a broader mBC population, including 43% with HR+/HER2− disease, again underscoring the limitations of ICIs in this group (126).
Preclinical data suggest that CDK4/6 inhibitors may sensitize tumors to ICIs by reducing Treg populations, downregulating PD-1 expression, enhancing antigen presentation, and promoting cytotoxic T cell activity. This has spurred clinical investigations into ICI–CDK4/6i combinations, sometimes paired with ET (20). A phase 1b trial evaluating abemaciclib plus pembrolizumab—with or without anastrozole—was terminated early due to hepatotoxicity and pneumonitis, despite preliminary antitumor activity (127). Conversely, a small open-label trial with palbociclib, pembrolizumab, and letrozole demonstrated encouraging responses with 31% CR and 25% PR among patients who received pembrolizumab upfront. When pembrolizumab was added later in the treatment sequence, the CR and PR rates increased to 50% (20, 128).
These findings underscore the need for further research to validate the potential benefits of ICIs combination therapy in HR+/HER2- BC.
Beyond immunotherapy, proteasome inhibitors such as bortezomib are being reevaluated in HR+/HER2− BC for their ability to destabilize ER signaling and enhance the efficacy of ET (129).
Antiprogestins—including selective progesterone receptor antagonists like onapristone—have also emerged as potential options in endocrine-resistant disease. Targeting additional signaling pathways beyond ER remains an important strategy to overcome endocrine resistance. Notably, ER activation can occur independently of ligand binding through the Notch signaling axis, leading to ERα dysregulation and sustained transcriptional activity even in the absence of estrogen. Inhibiting the Notch pathway has therefore emerged as a promising approach to restore ET sensitivity. Similarly, overactivation of AURKA contributes to ERα destabilization and downregulation, further driving resistance. The AURKA inhibitor alisertib is currently under investigation in this context. Another emerging strategy involves modulation of the JAK-STAT pathway via the lymphocyte adaptor protein LNK, which plays a role in hormone receptor signaling and immune cross-talk. Together, these pathways—Notch, AURKA, and JAK/STAT—represent rational and biologically distinct targets under exploration for restoring hormone sensitivity in resistant HR+ breast cancers (130).
Artificial intelligence (AI) is poised to accelerate progress across several domains in HR+/HER2− BC. In drug discovery, AI-based platforms have been utilized to predict the binding affinity of SERDs and to identify synergistic combinations with CDK4/6i by integrating pharmacogenomic and transcriptomic datasets. In prognostication, AI-enhanced digital pathology tools—such as those developed by PathAI and Paige—have demonstrated the ability to predict recurrence risk and treatment response in ER+ BC by analyzing H&E-stained histology slides in conjunction with molecular profiles (131). For instance, a study introduced the BRACE marker, derived from AI-based assessment of tumor heterogeneity in H&E images, which effectively stratified early-stage luminal/HER2-negative BC patients for distant metastasis-free survival and BC-specific survival, showing comparable prediction accuracy to traditional prognostic indices (132). Additionally, circulating tumor DNA (ctDNA) analysis, interpreted through AI algorithms, has enabled earlier detection of ESR1 mutations and prediction of endocrine resistance. Notably, the PADA-1 trial utilized ctDNA monitoring to detect ESR1 mutations, allowing for timely therapeutic interventions (133). In therapeutic delivery, AI algorithms are now being piloted to optimize CDK4/6 inhibitor dosing based on patient-specific hematologic trends and toxicity profiles, reducing the incidence of treatment interruptions and improving adherence. Collectively, these advances support the integration of AI as a transformative tool for precision oncology in HR+/HER2− mBC, with the potential to refine prognostic models, enhance therapeutic targeting, and personalize treatment delivery.
Discussion
The therapeutic landscape for HR+/HER2- mBC has evolved markedly with the integration of targeted therapies and next-generation endocrine strategies. While CDK4/6i combined with ET remain foundational in first-line treatment, resistance – particularly via ESR1 mutations – continues to drive disease progression and poses a major clinical challenge.
The EMERALD trial established the efficacy of oral selective estrogen receptor degraders in this space. Elacestrant significantly improved PFS over standard endocrine therapy in patients with ESR1 mutations, leading to its regulatory approval. Building on this, the EMBER-3 phase III trial demonstrated that imlunestrant, another oral SERD, achieved a statistically significant progression-free survival benefit in both ESR1-mutant and wild-type populations following prior CDK4/6 inhibitor exposure, while also offering a favorable safety profile.
Most recently, findings from the SERENA-6 trial showed compelling data on camizestrant. This biomarker-driven, ctDNA-guided study showed that early intervention with camizestrant—upon detection of rising ESR1 mutations prior to radiologic progression—delayed disease progression compared to standard AI continuation. This represents the first phase III evidence supporting a molecular response–adapted strategy in HR+ HER2- mBC and positions camizestrant as a viable option not only for resistant disease but potentially in mutation-interceptive protocols.
In parallel, the INAVO120 trial has reaffirmed the clinical value of targeting the PI3K/AKT pathway. In patients with PIK3CA-mutant tumors, the addition of inavolisib to palbociclib and fulvestrant nearly doubled median PFS versus placebo, with manageable toxicity. These findings validate a biomarker-driven approach post-CDK4/6i failure and support the sequencing of targeted pathway inhibitors in genomically defined subgroups.
The therapeutic horizon is also expanding with ADCs. Initially positioned in later-line settings, agents like T-DXd and sacituzumab govitecan are now being evaluated—and in some cases approved—for earlier lines of treatment, including post-CDK4/6i and even in the first line setting for select high-risk populations. Their impressive activity in HER2-low and Trop-2–positive disease may soon redefine standard treatment sequences.
Efforts to enhance immunotherapy efficacy are also ongoing, though the immunologically “cold” tumor microenvironment typical of HR+, HER2-negative tumors remains a barrier. Trials combining checkpoint inhibitors with CDK4/6i or ET have yielded mixed results, but certain histologies, such as lobular carcinoma, may harbor enhanced sensitivity.
Finally, AI is emerging as a disruptive tool across drug discovery, prognostic modeling, and treatment personalization. AI-enabled pathology markers such as BRACE, ctDNA analytics for ESR1 dynamics, and predictive dosing algorithms for CDK4/6i are early examples of how machine learning is transforming care in this setting. These technologies offer unprecedented granularity in treatment adaptation, accelerating the shift toward dynamic, biomarker-informed precision oncology.
Conclusion
The management of HR+ HER2− mBC is undergoing a paradigm shift, driven by biomarker-informed treatment selection, adaptive therapy strategies, and integration of emerging technologies. Oral SERDs such as elacestrant, imlunestrant, and camizestrant are reshaping endocrine resistance management, with SERENA-6 demonstrating the feasibility of ctDNA-guided early intervention. The INAVO120 trial highlights the value of targeting the PI3K/AKT pathway in genomically defined subgroups, while ADCs are gaining traction beyond second line use and are being positioned for earlier implementation.
As combination approaches evolve, incorporating ET, specific pathway inhibitors, ADCs, and immunotherapies into rational regimens will be key. AI will play an increasingly central role in optimizing these strategies—through drug discovery, real-time monitoring, and personalized dosing—to achieve maximum clinical benefit.
Ultimately, future advances in HR+ mBC will rely on the convergence of molecular diagnostics, translational research, and AI-enabled tools to deliver precision oncology solutions that will not only overcome resistance but will be proactively designed to prevent it.
Author contributions
EC: Writing – review & editing, Methodology, Data curation, Visualization, Writing – original draft, Conceptualization. LS: Data curation, Project administration, Resources, Writing – review & editing. JL: Data curation, Methodology, Investigation, Writing – review & editing. MV: Investigation, Methodology, Validation, Writing – review & editing, Supervision, Software, Project administration, Data curation, Conceptualization. CK: Supervision, Writing – review & editing, Methodology.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Loscalzo J, Fauci A, Kasper D, Hauser S, Longo D, Jameson JL, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21841
2. Quaglino E and Cavallo F. Breast cancer stem cell antigens as targets for immunotherapy. Semin Immunol. (2020) 47:101386. doi: 10.1016/j.smim.2020.101386
3. Rakha EA and Ellis IO. Modern classification of breast cancer: should we stick with morphology or convert to molecular profile characteristics. Anatomic Pathol. (2011) 18:255–67. doi: 10.1097/PAP.0b013e318220f5d1
4. Loibl S, André F, Bachelot T, Barrios CH, Bergh J, Burstein HJ, et al. Harrison’s Principles of Internal Medicine. New York: McGraw-Hill (2015).
5. ESMO. Breast cancer guidelines. (2024). European Society for Medical Oncology. Available online at: https://www.esmo.org/guidelines/esmo-clinical-practice-guidelines-breast-cancer (Accessed May 2, 2025).
6. Early Breast Cancer Trialists' Collaborative Group (EBCTCG), Davies C, Godwin J, Gray R, Clarke M, Cutter D, Darby S, et al. Early breast cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. (2024) 35:159–82. doi: 10.1016/j.annonc.2023.11.016
7. Herzog SK and Fesl C. ESR1 mutations and therapeutic resistance in metastatic breast cancer: progress and remaining challenges. Br J Cancer. (2022) 126:174–86. doi: 10.1038/s41416-021-01564-x
8. Gnant M, Pfeiler G, Dubsky PC, Hubalek M, Greil R, Jakesz R, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. (2011) 378:771–84. doi: 10.1016/S0140-6736(11)60993-8
9. Gnant M, et al. Adjuvant denosumab in breast cancer (ABCSG-18): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet. (2015) 386:433–43. doi: 10.1016/S0140-6736(15)60995-3
10. Bradley R, Braybrooke J, Gray R, Hills RK, Liu Z, Pan H, et al. Aromatase inhibitors versus tamoxifen in premenopausal women with oestrogen receptor-positive early-stage breast cancer treated with ovarian suppression: a patient-level meta-analysis of 7030 women from four randomised trials. Lancet Oncol. (2022) 23:382–92. doi: 10.1016/S1470-2045(21)00758-0
11. Campos SM and Winer EP. Hormonal therapy in postmenopausal women with breast cancer. Oncology. (2003) 64:289–99. doi: 10.1159/000070284
12. Levin ER. Plasma membrane estrogen receptors. Trends Endocrinol Metab. (2009) 20:477–82. doi: 10.1016/j.tem.2009.06.004
13. Battisti NML and Sundararajan IE. Preventing late recurrence in hormone receptor-positive early breast cancer: a review. Eur J Cancer. (2022) 168:53–64. doi: 10.1016/j.ejca.2022.05.028
14. Fitzal F, Filipits M, Rudas M, Greil R, Dietze O, Samonigg H, et al. The genomic expression test EndoPredict is a prognostic tool for identifying risk of local recurrence in postmenopausal endocrine receptor-positive, HER2-negative breast cancer patients randomised within the prospective ABCSG 8 trial. Br J Cancer. (2015) 112:1405–10. doi: 10.1038/bjc.2015.98
15. Sparano JA and Paik S. Development of the 21-gene assay and its application in clinical practice and clinical trials. J Clin Oncol. (2008) 26:721–8. doi: 10.1200/JCO.2007.15.1068
16. Cardoso F, van't Veer LJ, Bogaerts J, Slaets L, Viale G, Delaloge S, et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med. (2016) 375:717–29. doi: 10.1056/NEJMoa1602253
17. Matikas A, Foukakis T, Swain S, and Bergh J. Avoiding over- and undertreatment in patients with resected node-positive breast cancer with the use of gene expression signatures: are we there yet? Ann Oncol. (2019) 30:1044–50. doi: 10.1093/annonc/mdz126
18. Jacquet E, Lardy-Cléaud A, Pistilli B, Franck S, Cottu P, Delaloge S, et al. Endocrine therapy or chemotherapy as first-line therapy in hormone receptor-positive HER2-negative metastatic breast cancer patients. Eur J Cancer. (2018) 90:93–101. doi: 10.1016/j.ejca.2017.11.003
19. Shah AN, Metzger O, Bartlett CH, Liu Y, Huang X, Cristofanilli M, et al. Hormone receptor-positive/human epidermal growth receptor 2-negative metastatic breast cancer in young women: emerging data in the era of molecularly targeted agents. Oncologist. (2020) 25:900–8. doi: 10.1634/theoncologist.2019-0622
20. McAndrew NP and Finn RS. Management of ER positive metastatic breast cancer. Semin Oncol. (2020) 47:270–7. doi: 10.1053/j.seminoncol.2020.07.001
21. Turner NC, Im SA, Saura C, Juric D, Loibl S, Kalinsky K, et al. Inavolisib-based therapy in PIK3CA-mutated advanced breast cancer. N Engl J Med. (2024) 391:1584–96. doi: 10.1056/NEJMoa2401972
22. Turner NC, Oliveira M, Howell SJ, Dalenc F, Cortes J, Gomez Moreno HL, et al. Capivasertib in hormone receptor-positive advanced breast cancer. N Engl J Med. (2023) 388:2058–70. doi: 10.1056/NEJMoa2215022
23. Hortobagyi GN, Lacko A, Sohn J, Cruz F, Ruiz Borrego M, Manikhas A, et al. A phase III trial of adjuvant ribociclib plus endocrine therapy vs endocrine therapy alone in patients with HR+/HER2- early breast cancer: final invasive disease-free survival results from the NATALEE trial. Ann Oncol. (2024) 35:S7534. doi: 10.1016/annonc/ann.2024.7534
24. Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. (2012) 366:520–9. doi: 10.1056/NEJMoa1109653
25. Cao LQ, Sun H, Xie Y, Patel H, Bo L, Lin H, et al. Therapeutic evolution in HR+/HER2- breast cancer: from targeted therapy to endocrine therapy. Front Pharmacol. (2024) 15:1340764. doi: 10.3389/fphar.2024.1340764
26. Patel R, Klein P, Tiersten A, and Sparano JA. An emerging generation of endocrine therapies in breast cancer: a clinical perspective. NPJ Breast Cancer. (2023) 9:6. doi: 10.1038/s41523-023-00549-1
27. ESMO. Metastatic breast cancer living guidelines (2023). European Society for Medical Oncology. Available online at: https://www.esmo.org/guidelines/breast-cancer (Accessed May 2, 2025).
28. Kaklamani VG and Gradishar WJ. Endocrine therapy in the current management of postmenopausal estrogen receptor-positive metastatic breast cancer. Oncologist. (2017) 22:507–17. doi: 10.1634/theoncologist.2016-0471
29. Lu YS, Mahidin EIBM, Azim H, Eralp Y, Yap YS, Im SA, et al. Final results of RIGHT choice: ribociclib plus endocrine therapy versus combination chemotherapy in premenopausal women with clinically aggressive hormone receptor-positive/HER2-negative advanced breast cancer. J Clin Oncol. (2024) 42:2812–21. doi: 10.1200/JCO.23.00214
30. Wang X, Zhao S, Xin Q, Zhang Y, Wang K, Li M, et al. Recent progress of CDK4/6 inhibitors’ current practice in breast cancer. Cancer Gene Ther. (2024) 31:1283–91. doi: 10.1038/s41417-023-00631-w
31. Glück S. Exemestane as first-line therapy in postmenopausal women with recurrent or metastatic breast cancer. Am J Clin Oncol. (2010) 33:314–9. doi: 10.1097/COC.0b013e31819e3bd0
32. Brett JO, Spring LM, Bardia A, and Wander SA. ESR1 mutation as an emerging clinical biomarker in metastatic hormone receptor-positive breast cancer. Breast Cancer Res. (2021) 23:85. doi: 10.1186/s13058-021-01428-3
33. Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. (2013) 45:1439–45. doi: 10.1038/ng.2822
34. Gibson WJ, Hoivik EA, Halle MK, Taylor-Weiner A, Cherniack AD, Berg A, et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat Genet. (2016) 48:848–55. doi: 10.1038/ng.3602
35. Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61–70. doi: 10.1038/nature11412
36. Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, et al. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. (2014) 345:216–20. doi: 10.1126/science.1253533
37. Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell. (2018) 33:173–86. doi: 10.1016/j.ccell.2017.12.010
38. Aggelis V and Johnston SRD. Advances in endocrine-based therapies for estrogen receptor-positive metastatic breast cancer. Drugs. (2019) 79:1849–66. doi: 10.1007/s40265-019-01200-3
39. Adamo V, Iorfida M, Montalto E, Festa V, Garipoli C, Scimone A, et al. Overview and new strategies in metastatic breast cancer (MBC) for treatment of tamoxifen-resistant patients. Ann Oncol. (2007) 18:53–7. doi: 10.1093/annonc/mdm272
40. Loibl S, Poortmans P, Morrow M, Denkert C, and Curigliano G. Breast cancer. Lancet. (2021) 397:1750–69. doi: 10.1016/S0140-6736(20)32381-3
41. Kim HA, Lee JW, Nam SJ, Park BW, Im SA, Lee ES, et al. Adding ovarian suppression to tamoxifen for premenopausal breast cancer: A randomized phase III trial. J Clin Oncol. (2020) 38:434–43. doi: 10.1200/JCO.19.01683
42. Baek SY, Noh WC, Ahn SH, Kim HA, Ryu JM, Kim SI, et al. Adding ovarian suppression to tamoxifen for premenopausal women with hormone receptor-positive breast cancer after chemotherapy: an 8-year follow-up of the ASTRRA trial. J Clin Oncol. (2023) 41:4864–71. doi: 10.1200/JCO.23.00665
43. Gradishar W, Glusman J, Lu Y, Vogel C, Cohen FJ, Sledge GW, et al. Effects of high dose raloxifene in selected patients with advanced breast carcinoma. Cancer. (2000) 88:2047–53. doi: 10.1002/(SICI)1097-0142(20000501)88:9<2047::AID-CNCR27>3.0.CO;2-O
44. Goetz MP, Bagegni NA, Batist G, Brufsky A, Cristofanilli MA, Damodaran S, et al. Lasofoxifene versus fulvestrant for ER+/HER2– metastatic breast cancer with an ESR1 mutation: results from the randomized, phase II ELAINE 1 trial. Ann Oncol. (2023) 34:1141–51. doi: 10.1016/j.annonc.2023.06.013
45. Osborne CK, Pippen J, Jones SE, Parker LM, Ellis M, Come S, et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J Clin Oncol. (2002) 20:3386–95. doi: 10.1200/JCO.2002.11.023
46. Howell A, Robertson JF, Quaresma Albano J, Aschermannova A, Mauriac L, Kleeberg UR, et al. Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol. (2002) 20:3396–403. doi: 10.1200/JCO.2002.10.093
47. Shafaee MN and Emens MJ. Fulvestrant in management of hormone receptor-positive metastatic breast cancer. Future Oncol. (2018) 14:1789–800. doi: 10.2217/fon-2018-0120
48. Chia S, Gradishar W, Mauriac L, Bines J, Amant F, Federico M, et al. Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J Clin Oncol. (2008) 26:1664–70. doi: 10.1200/JCO.2007.13.5564
49. Johnston SR, Kilburn LS, Ellis P, Dodwell D, Cameron D, Hayward L, et al. Fulvestrant plus anastrozole or placebo versus exemestane alone after progression on non-steroidal aromatase inhibitors in postmenopausal patients with hormone-receptor-positive locally advanced or metastatic breast cancer (SoFEA): a composite, multicentre, phase 3 randomised trial. Lancet Oncol. (2013) 14:989–98. doi: 10.1016/S1470-2045(13)70383-4
50. Di Leo A, Jerusalem G, Petruzelka L, Torres R, Bondarenko IN, Khasanov R, et al. Final overall survival: fulvestrant 500 mg vs 250 mg in the randomized CONFIRM trial. J Natl Cancer Inst. (2014) 106:djt337. doi: 10.1093/jnci/djt337
51. Mehta RS, Barlow WE, Albain KS, Vandenberg TA, Dakhil SR, Tirumali NR, et al. Overall survival with fulvestrant plus anastrozole in metastatic breast cancer. N Engl J Med. (2019) 380:1226–34. doi: 10.1056/NEJMoa1804693
52. Turner NC, Swift C, Kilburn L, Fribbens C, Beaney M, Garcia-Murillas I, et al. ESR1 mutations and overall survival on fulvestrant versus exemestane in advanced hormone receptor-positive breast cancer: A combined analysis of the phase III soFEA and EFECT trials. Clin Cancer Res. (2020) 26:5172–7. doi: 10.1158/1078-0432.CCR-20-1021
53. Wang J, Cai L, Song Y, Sun T, Tong Z, Teng Y, et al. Clinical efficacy of fulvestrant versus exemestane as first-line therapies for Chinese postmenopausal oestrogen-receptor positive /human epidermal growth factor receptor 2 -advanced breast cancer (FRIEND study). Eur J Cancer. (2023) 184:73–82. doi: 10.1016/j.ejca.2023.01.027
54. Bardia A, Cortés J, Bidard FC, Neven P, Garcia-Sáenz J, Aftimos P, et al. Elacestrant (oral selective estrogen receptor degrader) versus standard endocrine therapy for estrogen receptor-positive, HER2-negative advanced breast cancer: results from the randomized phase III EMERALD trial. J Clin Oncol. (2022) 40:3246–56. doi: 10.1200/JCO.21.01392
55. Garcia-Fructuoso I and González-Billalabeitia E. Integrating new oral selective oestrogen receptor degraders in the breast cancer treatment. Curr Opin Oncol. (2022) 34:635–42. doi: 10.1097/CCO.0000000000000866
56. Bidard FC, Kaklamani VG, Neven P, Streich G, Montero AJ, Forget F, et al. Camizestrant, a next-generation oral SERD, vs fulvestrant in post-menopausal women with advanced ER-positive HER2-negative breast cancer: results of the randomized, multi-dose phase 2 SERENA-2 trial. J Clin Oncol. (2022) 40:3246–56. doi: 10.1200/JCO.22.01528
57. Helwick C. Phase III postMONARCH trial: switching to abemaciclib may improve outcomes after disease progression. ASCO Post. (2024) 25:1424–39. doi: 10.1016/S1470-2045(24)00387-5
58. Cookson F and Gradishar WJ. Camizestrant reduced the risk of disease progression or death by 56% in patients with advanced HR-positive breast cancer with an emergent ESR1 tumor mutation in SERENA-6 Phase III trial. Business Wire. (2025). Available at: https://www.businesswire.com/news/home/20250601316083/en/Camizestrant-reduced-the-risk-of-disease-progression-or-death-by-56-in-patients-with-advanced-HR-positive-breast-cancer-with-an-emergent-ESR1-tumor-mutation-in-SERENA-6-Phase-III-trial (Accessed June 2, 2025).
59. Jhaveri KL, Neven P, Casalnuovo ML, Kim S-B, Tokunaga E, Aftimos P, et al. Imlunestrant with or without abemaciclib in advanced breast cancer. N Engl J Med. (2025) 392:1189–202. doi: 10.1056/NEJMoa241085
60. Jhaveri KL, Neven P, Casalnuovo ML, Kim SB, Tokunaga E, Aftimos P, et al. Update breast cancer 2023 part 2 – advanced-stage breast cancer. Geburtshilfe Frauenheilkd. (2023) 83:664–72. doi: 10.1055/a-2085-9525
61. Lux MP, Hartkopf AD, Fehm TN, Welslau M, Müller V, Schütz F, et al. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. Lancet. (2020) 395:817–27. doi: 10.1016/S0140-6736(20)30165-3
62. Finn RS, Slamon DJ, Crown JP, Winer EP, Hortobagyi GN, Lee HJ, et al. Overall survival with first-line palbociclib plus letrozole versus placebo plus letrozole in women with ER+/HER2– advanced breast cancer: Analyses from PALOMA-2. J Clin Oncol. (2022) 40:817–27. doi: 10.1200/JCO.22.00354
63. Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch Shimon S, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. (2016) 375:1738–48. doi: 10.1056/NEJMoa1609709
64. Johnston S, Toi M, O’Shaughnessy J, Rastogi P, Campone M, Neven P, et al. MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer. (2019) 5:5. doi: 10.1038/s41523-019-0097-4
65. Hortobagyi GN, Turner NC, Yap YS, Bittoni A, Aftimos P, Aktas B, et al. Overall survival with ribociclib plus letrozole in advanced breast cancer. N Engl J Med. (2022) 386:942–50. doi: 10.1056/NEJMoa2119114
66. Goetz MP, Harbeck N, Tripathy D, Spring LM, Shah N, Ruiz Borrego M, et al. Abemaciclib plus a nonsteroidal aromatase inhibitor as initial therapy for HR+, HER2- advanced breast cancer: final overall survival results of MONARCH 3. Ann Oncol. (2024) 35:718–27. doi: 10.1016/j.annonc.2024.03.011
67. Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. (2016) 17:425–39. doi: 10.1016/S1470-2045(15)00613-0
68. Sledge GW Jr., Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2– advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. (2017) 35:2875–84. doi: 10.1200/JCO.2017.73.7585
69. Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, HER2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol. (2018) 36:2465–72. doi: 10.1200/JCO.2018.78.9901
70. Lu YS, Krop I, Sohn J, Kaufman PA, Blackwell KL, Wang DY, et al. Updated overall survival of ribociclib plus endocrine therapy versus endocrine therapy alone in pre- and perimenopausal patients with HR+/HER2– advanced breast cancer in MONALEESA-7: a phase III randomized clinical trial. Clin Cancer Res. (2022) 28:851–9. doi: 10.1158/1078-0432.CCR-21-3094
71. Tripathy D, Im SA, Colleoni M, Franke F, Bardia A, Harbeck N, et al. Ribociclib plus endocrine therapy for premenopausal women with hormone receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol. (2018) 19:904–15. doi: 10.1016/S1470-2045(18)30292-4
72. Ahn HK, Im SA, Guerrero-Zotano A, Ruiz-Borrego M, Tripathy D, Sohn J, et al. Palbociclib plus endocrine therapy versus capecitabine in premenopausal women with hormone receptor-positive, HER2-negative metastatic breast cancer (Young-PEARL): overall survival analysis of a randomised, open-label, phase 2 study. Lancet Oncol. (2025) 26:343–54. doi: 10.1016/S1470-2045(24)00023-6
73. de la Haba Rodriguez J, Cejalvo JM, Gómez P, Daver N, Acevedo A, Oliver L, et al. ABIGAIL: Randomized phase II study of abemaciclib plus endocrine therapy with or without a short course of induction paclitaxel in patients with HR+/HER2– advanced breast cancer with aggressive disease criteria. Ann Oncol. (2024) 35:1215–6. doi: 10.1016/j.annonc.2024.04.021
74. Dickler MN, Tolaney SM, Rugo HS, Neven P, Johnston S, Goetz MP, et al. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR+/HER2– metastatic breast cancer. Clin Cancer Res. (2017) 23:5218–24. doi: 10.1158/1078-0432.CCR-17-0754
75. Wander SA, Payne R, Locklair D, Krop I, Chu L, Partridge A, et al. Clinical outcomes with abemaciclib after prior CDK4/6 inhibitor progression in breast cancer: a multicenter experience. J Natl Compr Canc. Netw. (2021) 19:1–8. doi: 10.6004/jnccn.2020.7651
76. Park YH, Im SA, Sohn J, Lee KS, Hwang J, Kim SW, et al. Palbociclib plus exemestane with gonadotropin-releasing hormone agonist versus capecitabine in premenopausal women with hormone receptor-positive, HER2-negative metastatic breast cancer (KCSG-BR15-10): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. (2019) 20:1750–9. doi: 10.1016/S1470-2045(19)30548-4
77. Martín M, Rodríguez CA, Ruiz-Borrego M, Im SA, Masuda N, Takahashi M, et al. Palbociclib in combination with endocrine therapy versus capecitabine in hormone receptor-positive, HER2-negative, aromatase inhibitor-resistant metastatic breast cancer: a phase III randomised controlled trial—PEARL. Ann Oncol. (2021) 32:488–99. doi: 10.1016/j.annonc.2021.01.068
78. Bidard FC, Simone C, Pierga JY, Pol B, Mansi L, Lotz JP, et al. Switch to fulvestrant and palbociclib versus no switch in advanced breast cancer with rising ESR1 mutation during aromatase inhibitor and palbociclib therapy (PADA-1): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. (2022) 23:1367–77. doi: 10.1016/S1470-2045(22)00498-2
79. Bardia A, Hurvitz SA, Mayer IA, Tolaney SM, Isakoff SJ, Forero A, et al. Phase I/II trial of exemestane, ribociclib, and everolimus in women with HR+/HER2– advanced breast cancer after progression on CDK4/6 inhibitors (TRINITI-1). Clin Cancer Res. (2021) 27:4177–85. doi: 10.1158/1078-0432.CCR-21-0096
80. Mayer EL, Liu MC, Gandhi L, O’Shaughnessy J, Abramson VG, Winer EP, et al. Palbociclib after CDK4/6i and endocrine therapy (PACE): a randomized phase II study of fulvestrant, palbociclib, and avelumab for endocrine pre-treated ER+/HER2– metastatic breast cancer. J Clin Oncol. (2023) 41 Suppl:16. doi: 10.1200/JCO.2023.41.16_suppl.LBA1004
81. Sonke GS, van Diest P, Johnston SR, Loibl S, Turner NC, Piccart M, et al. Primary outcome analysis of the phase 3 SONIA trial (BOOG 2017-03) on selecting the optimal position of CDK4/6 inhibitors for patients with HR+/HER2– advanced breast cancer. J Clin Oncol. (2023) 41:2050–60. doi: 10.1200/JCO.2023.41.17_suppl.LBA1001
82. Kalinsky K, Han HS, Conlin AK, Tolaney SM, Ro-Engel CE, Vamos EP, et al. Abemaciclib plus fulvestrant vs fulvestrant alone for HR+, HER2– advanced breast cancer following progression on a prior CDK4/6 inhibitor plus endocrine therapy: primary outcome of the phase 3 postMONARCH trial. J Clin Oncol. (2024) 42:LBA1001. doi: 10.1200/JCO.2024.42.17_suppl.LBA1001
83. Slamon D, Fasching PA, Hurvitz SA, Macneill FA, Mayer IA, Rubovits S, et al. Ribociclib plus endocrine therapy in early breast cancer. N Engl J Med. (2024) 390:1080–91. doi: 10.1056/NEJMoa2309491
84. Gnant M, Rugo H, Bhattacharya S, Fazli L, Glitza-Oliva IC, Madi A, et al. Adjuvant palbociclib for early breast cancer: The PALLAS trial results (ABCSG-42/AFT-05/BIG-14-03). J Clin Oncol. (2022) 40:282–93. doi: 10.1200/JCO.21.01556
85. Loibl S, Turner NC, Ro J, Neven P, Im SA, Hillman DW, et al. Palbociclib for residual high-risk invasive HR-positive and HER2-negative early breast cancer: the Penelope-B trial. J Clin Oncol. (2021) 39:1518–30. doi: 10.1200/JCO.20.09299
86. Mittal A, Arun B, Barcelona T, Montgomery LL, Ito Y, Puhalla S, et al. Filling the gap after CDK4/6 inhibitors: novel endocrine and biologic treatment options for metastatic hormone receptor-positive breast cancer. Cancers (Basel). (2023) 15:2015. doi: 10.3390/cancers15072015
87. O'Shaughnessy J, Thaddeus Beck J, and Royce M. Everolimus plus endocrine therapy for postmenopausal women with estrogen receptor-positive, HER2-negative advanced breast cancer: a clinical trial. JAMA Oncol. (2018) 4:977–84. doi: 10.1001/jamaoncol.2018.0262
88. O’Shaughnessy J, Beck JT, and Royce M. Everolimus-based combination therapies for HR+, HER2– metastatic breast cancer. Cancer Treat Rev. (2018) 71:204–14. doi: 10.1016/j.ctrv.2018.01.008
89. Fan Y, Sun T, Shao Z, Zhang Q, Ouyang Q, Tong Z, et al. Effectiveness of adding everolimus to the first-line treatment of advanced breast cancer in premenopausal women who experienced disease progression while receiving selective estrogen receptor modulators: a phase 2 randomized clinical trial. JAMA Oncol. (2021) 7:e213428. doi: 10.1001/jamaoncol.2021.3428
90. Sánchez-Bayona R, Lopez de Sa A, Jerez Gilarranz Y, Sanchez de Torre A, Alva M, Echavarria I, et al. Everolimus plus endocrine therapy beyond CDK4/6 inhibitor progression for HR+/HER2– advanced breast cancer: a real-world evidence cohort. Breast Cancer Res Treat. (2024) 206:551–9. doi: 10.1007/s10549-023-07083-3
91. Gómez Tejeda Zañudo J, Barroso-Sousa R, Jain E, Jin Q, Li T, Buendia-Buendia JE, et al. Exemestane plus everolimus and palbociclib in metastatic breast cancer: clinical response and genomic/transcriptomic determinants of resistance in a phase I/II trial. Nat Commun. (2024) 15:2446. doi: 10.1038/s41467-024-40119-2
92. André F, Cruelos EM, Juric D, Loibl S, Campone M, Mayer IA, et al. Alpelisib plus fulvestrant for PIK3CA-mutated, HR-positive, HER2-negative advanced breast cancer: final overall survival results from SOLAR-1. Ann Oncol. (2021) 32:208–17. doi: 10.1016/j.annonc.2020.10.015
93. Ladraa S, Zerbib L, Bayard C, Fraissenon A, Venot Q, Morin G, et al. PIK3CA gain-of-function mutation in adipose tissue induces metabolic reprogramming with Warburg-like effect and severe endocrine disruption. Sci Adv. (2022) 8:eade7823. doi: 10.1126/sciadv.ade7823
94. Andrikopoulou A, Chatzinikolaou S, Panourgias E, Kaparelou M, Liontos M, Dimopoulos M-A, et al. The emerging role of capivasertib in breast cancer. Breast. (2022) 62:157–67. doi: 10.1016/j.breast.2022.06.009
95. Baselga J, Im S-A, Iwata H, Cortés J, De Laurentiis M, Jiang Z, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, HR-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. (2017) 18:904–16. doi: 10.1016/S1470-2045(17)30313-6
96. Di Leo A, Johnston S, Lee KS, Ciruelos E, Lønning PE, Janni W, et al. Buparlisib plus fulvestrant in postmenopausal women with HR-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. (2018) 19:87–100. doi: 10.1016/S1470-2045(17)30787-2
97. Rugo HS, André F, Yamashita T, Cerda H, Toledano I, Stemmer SM, et al. Time course and management of key adverse events during the randomized phase III SOLAR-1 study of PI3K inhibitor alpelisib plus fulvestrant in patients with HR-positive advanced breast cancer. Ann Oncol. (2020) 31:1001–10. doi: 10.1016/j.annonc.2020.03.300
98. Dent S, Cortés J, Im YH, Diéras V, Harbeck N, Krop IE, et al. Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor-positive, PIK3CA-mutant, HER2-negative, advanced breast cancer: the SANDPIPER trial. Ann Oncol. (2021) 32:197–207. doi: 10.1016/j.annonc.2020.11.006
99. Turner NC, Alarcón E, Armstrong AC, Philco M, López Chuken YA, Sablin MP, et al. BEECH: a dose-finding run-in followed by a randomized phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with ER-positive advanced or metastatic breast cancer, and in a PIK3CA mutant sub-population. Ann Oncol. (2019) 30:774–80. doi: 10.1093/annonc/mdz121
100. Howell SJ, Casbard A, Carucci M, Cox C, Butler R, Alchami F, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. (2020) 21(3):345–57. doi: 10.1016/S1470-2045(19)30817-4
101. Turner NC, Im SA, Saura C, Juric D, Loibl S, Kalinsky K, et al. INAVO120: Phase III trial final overall survival (OS) analysis of first-line inavolisib/placebo + palbociclib + fulvestrant in patients with PIK3CA-mutated, HR-positive, HER2-negative, endocrine-resistant advanced breast cancer. J Clin Oncol. (2025) 42:1003. doi: 10.1200/JCO.2025.42.16_suppl.1003
102. Barroso-Sousa R and Tolaney SM. Clinical development of new antibody-drug conjugates in breast cancer: to infinity and beyond. BioDrugs. (2021) 35:159–74. doi: 10.1007/s40259-021-00462-6
103. Stecklein SR and Perez EA. Last but not least: antibody-drug conjugates in hormone receptor-positive metastatic breast cancer. Ann Oncol. (2020) 31:1594–6. doi: 10.1016/j.annonc.2020.07.006
104. Nakada T, Sugihara K, Jikoh T, Abe Y, and Agatsuma T. The latest research and development into the antibody-drug conjugate, [fam-] trastuzumab deruxtecan (DS-8201a), for HER2 cancer therapy. Chem Pharm Bull (Tokyo). (2019) 67:173–85. doi: 10.1248/cpb.c18-00986
105. Martín M, Pandiella A, Vargas-Castrillón E, Díaz-Rodríguez E, Iglesias-Hernangómez T, Martínez Cano C, et al. Trastuzumab deruxtecan in breast cancer. Crit Rev Oncol Hematol. (2024) 187:104355. doi: 10.1016/j.critrevonc.2024.104355
106. Tarantino P, Gandini S, and Sparano T. Navigating the HER2-low paradigm in breast oncology: new standards, future horizons. Cancer Discov. (2022) 12:2026–30. doi: 10.1158/2159-8290.CD-21-1713
107. Miglietta F, Griguolo G, Bottosso M, Giarratano T, Lo Mele M, Fassan M, et al. Evolution of HER2-low expression from primary to recurrent breast cancer. NPJ Breast Cancer. (2021) 7:137. doi: 10.1038/s41523-021-00345-4
108. Modi S, Jacot W, Yamashita T, Sohn J, Vidal M, Tokunaga E, et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N Engl J Med. (2022) 387:9–20. doi: 10.1056/NEJMoa2203690
109. Bardia A, Hu X, Dent R, Yonemori K, Barrios CH, O'Shaughnessy JA, et al. Trastuzumab deruxtecan after endocrine therapy in metastatic breast cancer. N Engl J Med. (2024) 198:104355. doi: 10.1056/NEJMoa2401167
110. Mosele F, Deluche E, Lusque A, Le Bescond L, Filleron T, Pradat Y, et al. Trastuzumab deruxtecan in metastatic breast cancer with variable HER2 expression: the phase 2 DAISY trial. Nat Med. (2023) 29:2110–20. doi: 10.1038/s41591-023-02571-z
111. André F, Hamilton EP, Loi S, Anders CK, Schmid P, Stroyakovskiy D, et al. DESTINY-Breast07: Dose-expansion interim analysis of T-DXd monotherapy and T-DXd + pertuzumab in patients with previously untreated HER2+ mBC. J Clin Oncol. (2024) 42:1009. doi: 10.1200/JCO.2024.42.16_suppl.1009
112. Harbeck N, Ciruelos E, Jerusalem G, Müller V, Niikura N, Viale G, et al. Trastuzumab deruxtecan in HER2-positive advanced breast cancer with or without brain metastases: a phase 3b/4 trial. Nat Med. (2024) 30:3717–27. doi: 10.1038/s41591-024-03027-z
113. Rugo HS, Bardia A, Marmé F, Cortés J, Schmid P, Loirat D, et al. Overall survival with sacituzumab govitecan in HR-positive/HER2-negative metastatic breast cancer (TROPiCS-02): a randomised, open-label, multicentre, phase 3 trial. Lancet. (2023) 402:1423–33. doi: 10.1016/S0140-6736(23)00727-3
114. Bardia A, Jhaveri K, Im SA, Pernas S, De Laurentiis M, Wang S, et al. Datopotamab deruxtecan versus chemotherapy in previously treated inoperable/metastatic hormone receptor-positive, HER2-negative breast cancer: primary results from TROPION-Breast01. J Clin Oncol. (2024) 42:00920. doi: 10.1200/JCO.2023.42.16_suppl.LBA1002
115. Zheng F, Zhang Y, Chen S, Weng X, Rao Y, Fang H, et al. Mechanism and current progress of poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed Pharmacother. (2020) 123:109661. doi: 10.1016/j.biopha.2019.1091
116. Robson ME, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. OlympiAD extended follow-up for overall survival and safety: olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Eur J Cancer. (2023) 184:39–47. doi: 10.1016/j.ejca.2023.01.026
117. Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee KH, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. (2018) 379:753–63. doi: 10.1056/NEJMoa1802905
118. Iacopetta D, Ceramella J, Baldino N, Sinicropi MS, Catalano A, et al. Targeting breast cancer: an overlook on current strategies. Int J Mol Sci. (2023) 24:3643. doi: 10.3390/ijms24043643
119. Seymour C. Vepdegestrant plus palbociclib sustains efficacy in advanced ER+ breast cancer. OncLive (2024). Available online at: https://www.onclive.com/view/vepdegestrant-plus-palbociclib-sustains-efficacy-in-advanced-er-breast-cancer (Accessed March 24, 2024).
120. Hurvitz SA, Schott AF, Ma C, Hamilton E, Nanda R, Zahrah G, et al. VERITAC update: Phase II study of ARV-471, a PROteolysis TArgeting Chimera (PROTAC) estrogen receptor (ER) degrader in ER+/HER2– advanced breast cancer. ESMO Open. (2023) 8:101394. doi: 10.1016/j.esmoop.2023.101394
121. Campone M, De Laurentiis M, Jhaveri K, Hu X, Ladoire S, Patsouris A, et al. Vepdegestrant, a PROTAC estrogen receptor degrader, in advanced breast cancer. N Engl J Med. (2025) 392:1123–35. doi: 10.1056/NEJMoa2402059
122. Xiong R, Patel HK, Gutgesell LM, Zhao J, Delgado Rivera L, Pham TND, et al. Selective human estrogen receptor partial agonists (ShERPAs) for tamoxifen-resistant breast cancer. J Med Chem. (2016) 59:219–37. doi: 10.1021/acs.jmedchem.5b01057
123. Goldberg JE, Pastorello RG, Vallius T, Davis J, Cui YX, Agudo J, et al. The immunology of hormone receptor positive breast cancer. Front Immunol. (2021) 12:674192. doi: 10.3389/fimmu.2021.674192
124. Debien V, De Caluwé A, Wang X, Piccart-Gebhart M, Tuohy VK, Romano E, et al. Immunotherapy in breast cancer: an overview of current strategies and perspectives. NPJ Breast Cancer. (2023) 9:27. doi: 10.1038/s41523-023-00502-2
125. Rugo HS, Delord JP, Im SA, Ott PA, Piha-Paul SA, Bedard PL, et al. Safety and antitumor activity of pembrolizumab in patients with estrogen receptor-positive/HER2-negative advanced breast cancer. Clin Cancer Res. (2018) 24:2804–11. doi: 10.1158/1078-0432.CCR-17-3458
126. Dirix LY, Takacs I, Jerusalem G, Nikolinakos P, Arkenau HT, Forero-Torres A, et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN Solid Tumor study. Breast Cancer Res Treat. (2018) 167:671–86. doi: 10.1007/s10549-017-4537-6
127. Rugo HS, Kabos P, Beck JT, Guy J, Wildiers H, Sevillano E, et al. Abemaciclib in combination with pembrolizumab for HR+, HER2– metastatic breast cancer: Phase 1b study. NPJ Breast Cancer. (2022) 8:15. doi: 10.1038/s41523-022-00368-1
128. Yuan Y, Lee JS, Yost SE, Frankel PH, Ruel C, Egelston CA, et al. Phase I/II trial of palbociclib, pembrolizumab and letrozole in patients with hormone receptor-positive metastatic breast cancer. Eur J Cancer. (2021) 154:11–20. doi: 10.1016/j.ejca.2021.06.018
129. Thaler S, Thiede G, Hengstler JG, Schad A, Schmidt M, Sleeman JP, et al. The proteasome inhibitor bortezomib (Velcade) as potential inhibitor of estrogen receptor-positive breast cancer. Int J Cancer. (2015) 137:686–97. doi: 10.1002/ijc.29418
130. Raheem F, Karikalan SA, Batalini F, El Masry A, Mina L, et al. Metastatic ER+ breast cancer: mechanisms of resistance and future therapeutic approaches. Int J Mol Sci. (2023) 24:16198. doi: 10.3390/ijms242016198
131. Soliman A, Li Z, and Parwani AV. Artificial intelligence’s impact on breast cancer pathology: a literature review. Diagn Pathol. (2024) 19:38. doi: 10.1186/s13000-024-01322-2
132. Wahab N, Toss M, Miligy IM, Jahanifar M, Atallah NM, Lu W, et al. AI-enabled routine H&E image-based prognostic marker for early-stage luminal breast cancer. NPJ Precis Oncol. (2023) 7:122. doi: 10.1038/s41698-023-00393-0
Keywords: hormone receptor positive breast cancer, endocrine resistance, metastatic breast cancer, CDK4/6 inhibitors, aromatase inhibitors
Citation: Chiru ED, Sojak L, Landin J, Vetter M and Grašič Kuhar C (2025) Pharmacologic management of HR+/HER2− mBC: a clinically oriented review. Front. Oncol. 15:1596634. doi: 10.3389/fonc.2025.1596634
Received: 19 March 2025; Accepted: 08 August 2025;
Published: 08 September 2025.
Edited by:
Ozgur Kutuk, Sabancı University, TürkiyeReviewed by:
Simone Nardin, Università Degli Studi del Piemonte Orientale, ItalySimon Udovica, Klinik Ottakring, Austria
Copyright © 2025 Chiru, Sojak, Landin, Vetter and Grašič Kuhar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Diana Chiru, ZWxlbmEtZGlhbmEuY2hpcnVAa3NibC5jaA==
†These authors have contributed equally to this work