 Philip K. Tan1,2
Philip K. Tan1,2 Timothy J. Martins3
Timothy J. Martins3 Pamela S. Becker4,5,6
Pamela S. Becker4,5,6 Robert J. Wechsler-Reya7,8
Robert J. Wechsler-Reya7,8 John Ross Crawford9,10,11*
John Ross Crawford9,10,11*- 1The Cure Starts Now Foundation, Cincinnati, OH, United States
- 2Yuvaan Tiwari Foundation, Atlanta, GA, United States
- 3Institute for Stem Cell and Regenerative Medicine, University of Washington School of Medicine, Seattle, WA, United States
- 4Department of Hematology and Hematopoietic Cell Transplantation, City of Hope National Medical Center, Duarte, CA, United States
- 5Department of Hematologic Malignancies Translational Science, City of Hope National Medical Center, Duarte, WA, United States
- 6Department of Medicine, University of Washington, Seattle, WA, United States
- 7Cancer Genome and Epigenetics Program, National Cancer Institute (NCI)-Designated Cancer Center, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA, United States
- 8Department of Neurology and Herbert Irving Comprehensive Cancer Center, Columbia University Medical Center, New York, NY, United States
- 9Rady Children’s Hospital, San Diego, CA, United States
- 10Children’s Hospital of Orange County, Orange, CA, United States
- 11Department of Pediatrics and Neurology, University of California Irvine, Irvine, CA, United States
Diffuse midline glioma (DMG) is a pediatric brain cancer that has a dismal prognosis with limited treatment options. We present the treatment course and outcome of an adolescent male diagnosed with a thalamic DMG carrying a histone H3.3 K27M (H3K27M) alteration. Tumor biopsies were taken at diagnosis for histological analysis, molecular profiling, and ex vivo drug sensitivity testing (DST). Seven months after diagnosis, the patient had recurrent/progressive disease after radiotherapy and an ineffective molecular-guided therapy based on tumor molecular profiling. The patient then started a novel functional precision medicine (FPM)-guided two-drug combination of disulfiram, based on the DST results of this drug on the patient’s tumor cells obtained at diagnosis, and ONC 201, the only drug that has advanced to a phase III clinical trial for H3K27M-DMG. Neuroimaging demonstrated a treatment response, and the patient lived for fifteen months after starting this personalized therapy. Disulfiram was discontinued after three months due to significant peripheral neuropathy. Our case describes the feasibility and limitations of using DST of patient-derived tumor cells to identify potentially effective personalized and novel therapies for DMG, which should be evaluated for efficacy and safety in formal N-of-1 clinical trials settings. We discuss the benefits and risks of this approach, particularly considering its use in children, adolescents, and young adults with pediatric brain cancers.
Introduction
Diffuse midline glioma (DMG) is a high-grade glioma that occurs mainly in children and more rarely in adults. It is the deadliest brain cancer in children and young adults with a life expectancy of about a year after diagnosis (1), although adults have a significantly longer life expectancy (2, 3). DMG is molecularly defined by a lysine-27 to methionine mutation in histone H3 (H3K27M) that leads to global hypomethylation of this residue and consequent epigenetic changes in gene expression patterns that are likely to promote oncogenesis (4, 5). The tumors occur within deep midline regions of the brain and spinal cord and are infiltrative in nature such that surgical resection is risky and often ineffective. There are no FDA-approved therapies and patient outcomes have not improved over decades. Radiation is the only standard of care therapy and is merely palliative, and clinical trials of chemotherapeutic and targeted agents after radiotherapy have been largely ineffective. Upon tumor progression, DMG patients have even fewer treatment options and a median life expectancy of only three months if no further treatments are pursued (6, 7).
Functional precision medicine (FPM) is a promising approach that utilizes drug sensitivity testing (DST) of patient-derived tumor cells either alone or in combination with molecular profiling as part of multi-omics testing to predict effective individualized treatment options (8–13). However, DST has not been widely applied to children and adolescents with pediatric brain cancer. This case describes an adolescent DMG patient whose tumor cells were subjected to ex vivo DST at diagnosis. Seven months later at the time of tumor progression, the patient started a personalized and experimental two-drug combination therapy of disulfiram, based on the DST results of this drug, and ONC201 (dordaviprone), the only drug that has advanced to a phase III clinical trial for H3K27M-DMG (14). This FPM-based therapy led to a tumor regression and the patient survived for 15 months after starting this treatment and for 22 months after diagnosis, although it also induced a peripheral neuropathy that reduced his quality of life and led to discontinuation of therapy. Our case highlights the challenges and opportunities of DST and serves as a basis for potential FPM-based clinical trials for patients with DMG.
Case description
An adolescent male presented with a 1-month history of fatigue and intractable vomiting. Examination was significant for obtundation, and right hemiparesis and neuroimaging revealed a left thalamic neoplasm with obstructive hydrocephalus and herniation. He underwent emergent external ventricular drain placement, fenestration of the septum pellucidum, and tumor biopsy. Histological staining and molecular analysis of the biopsies revealed the presence of the H3F3A K27M mutation along with mutations in TP53 (with accompanying loss of heterozygosity), PIK3CA, and a SMARCE1 mutation that was sub clonal (Table 1). Separately, the biopsied tumor cells were subjected to a feasibility study featuring multi-omics analysis, DNA methylation profiling, immunogenic potential analysis, and ex vivo DST with a customized panel of 175 FDA approved and investigational cancer drugs (15; John R. Crawford and Robert Wechsler-Reya, manuscript in preparation) at a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory at the University of Washington (16). Consistent with previously reported DST results on patient-derived DMG tumor cells, drugs with the most potency in viability assays included proteasome inhibitors such as marizomib and histone deacetylase (HDAC) inhibitors such as panobinostat (17, 18). Disulfiram also caused significant lethality of tumor cells, with an IC50 of 71 nanomolar and a tumor cell viability of just 2% at the maximal dose. Figure 1 shows the DST results of marizomib, as well as the results for other drugs that the patient used with his treatment plan. The Supplementary Table lists the ex vivo DST results for all the drugs that were evaluated.
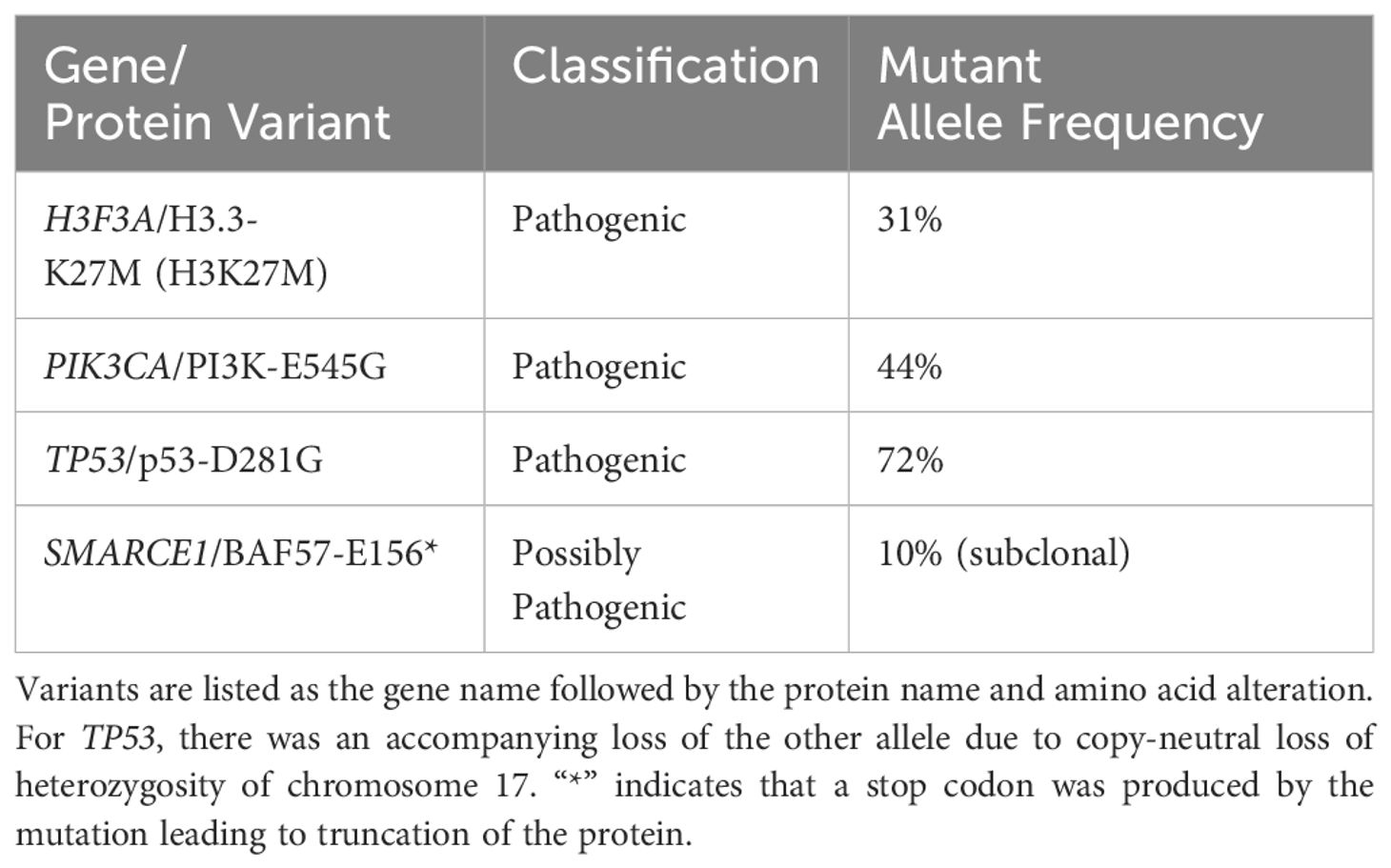
Table 1. Tumor molecular analysis, somatic alterations.
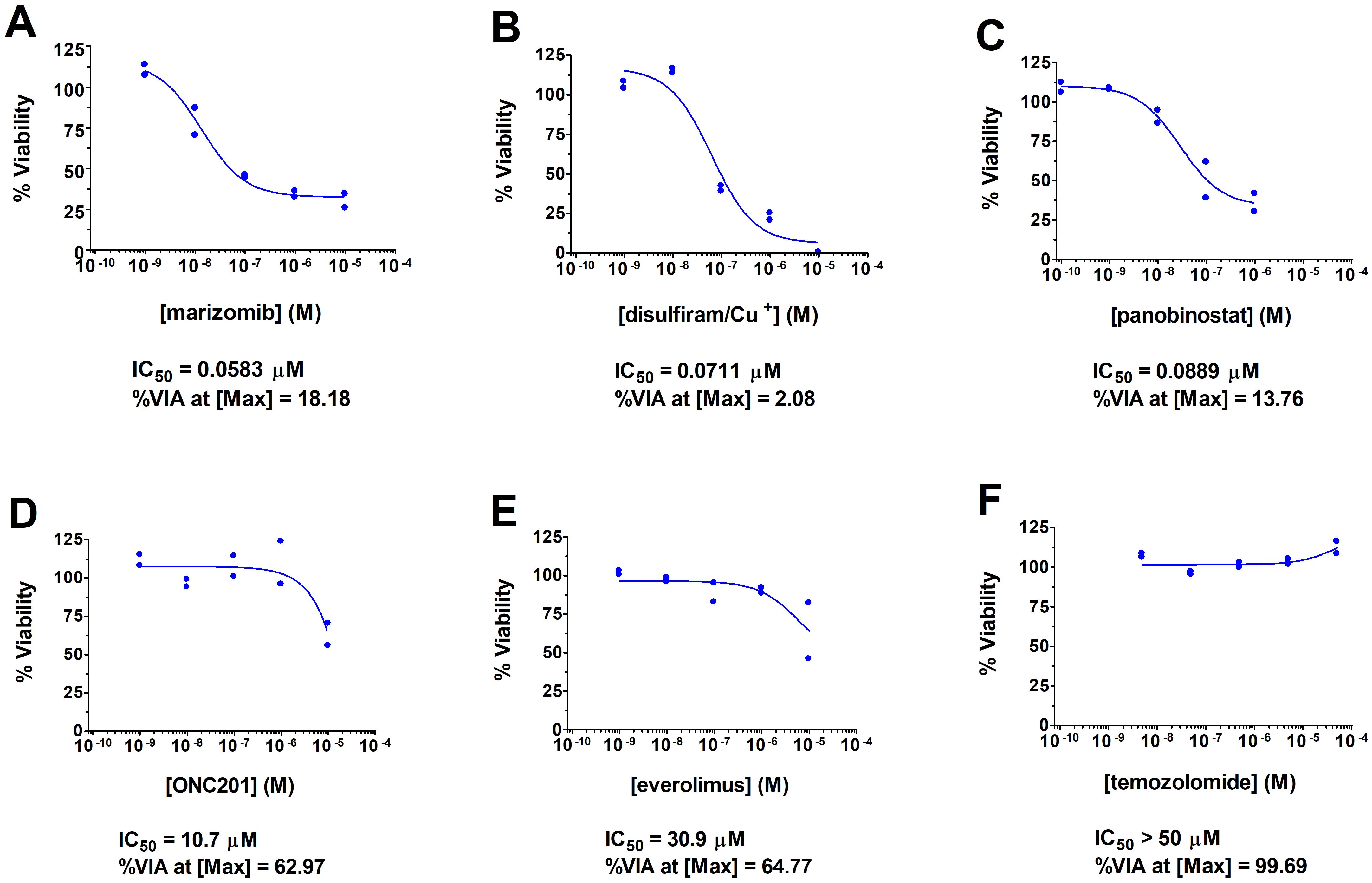
Figure 1. Summary of DST results for drugs used by the patient and for marizomib. DST curves are shown for marizomib (A), two other drugs with high potency that the patient used [disulfiram (B) and panobinostat (C)], and for three drugs with low potency that the patient used [ONC201 (D), everolimus (E), and temozolomide (F)]. Briefly, the freshly biopsied tumor cells were dispersed by enzymatic digestion, shipped on ice to a CLIA laboratory at the University of Washington (16), and on the next day were plated with each drug on opaque 6x384-well plates at 2000 cells per well. After a 3-day incubation, cell viability was measured using CellTiter Glo 2.0 (Promega Corp., Madison, WI, USA). Each drug was assayed at five concentrations in duplicate. XLFit from IBDS was used to generate dose-response curves using the 4-parameter logistic dose response model, tumor cell killing potency of each drug at the half-maximal concentration in micromolar, or IC50, and percentage of viable cells at the maximal concentration of each drug (%VIA at [Max]) as a measure of drug efficacy; the drug potency and efficacy values are shown below each dose response curve. Cu+, copper in the form of copper gluconate.
Figure 2 shows the course of treatment for the patient along with a graph of the area of the primary thalamic tumor for each magnetic resonance imaging (MRI), and the timing of molecular profiling and DST (lower left). Figure 3 presents representative post-gadolinium T1-weighted MRI images of the primary thalamic DMG tumor, shown in Figure 3A at diagnosis. Standard of care radiation therapy (proton therapy of 60 Gy over 30 cycles) from days 22–63 led to a continual reduction in tumor area based on imaging on days 88 and 149 (Figures 2, 3B, C). While in radiotherapy, the patient and his family expressed an interest in drug combination therapies, and since there are no FDA-approved therapies for DMG they were informed of the benefits and risks of using novel drug combinations, and they provided consent to use a molecular-guided combination drug therapy based on tumor molecular profiling obtained at diagnosis. On day 102 the patient started an individualized therapy consisting of panobinostat, everolimus, temozolomide, and hydroxychloroquine. An MRI on day 203 showed tumor progression (Figure 3D) and hence this combination therapy was deemed ineffective and was discontinued after 3.5 months.
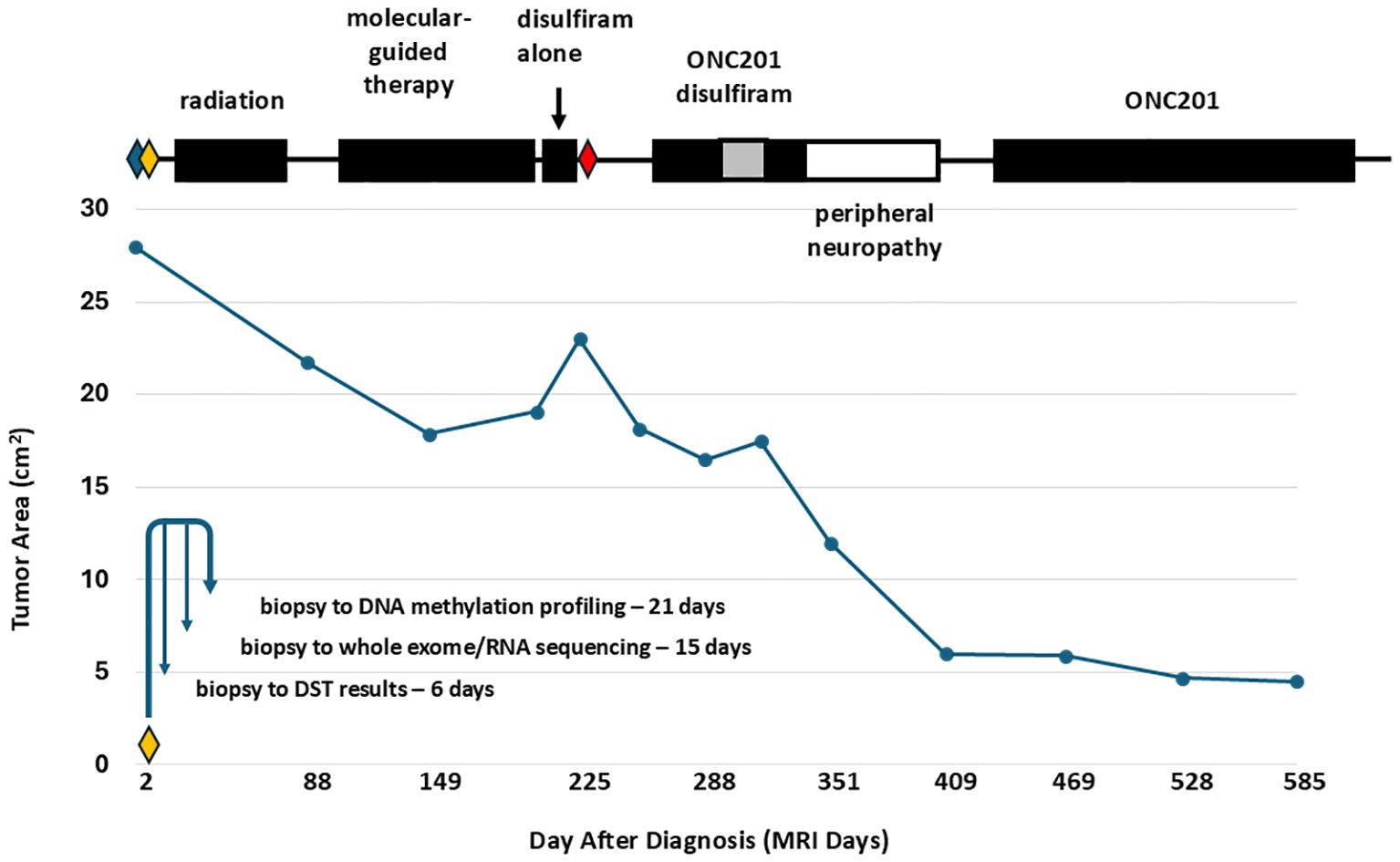
Figure 2. Treatment course and thalamic DMG tumor area measurements showing tumor regression with radiation therapy and with the FPM therapy of disulfiram and ONC201. The graph plots the measured area of the primary thalamic DMG tumor from each MRI. Above the graph, the time periods for the different treatments are shown in black boxes. Disulfiram was used at 250 mg QD, except during a time period at 250 mg BID (gray box). Surgeries are indicated by diamonds: blue diamond, external shunt placement and septostomy (day 1); yellow diamond, external shunt removal and third ventriculostomy (day 7); red diamond, internal shunt placement and septostomy (day 226). The lower left of the graph shows a timeline from tumor biopsy on the day 7 surgery to profiling including DST. The time period of the peripheral neuropathy is indicated as an open box. Standard of care radiation therapy led to tumor regression, observed as a reduction in tumor area on days 88 and 149. Following radiation therapy, an ineffective molecular-guided therapy with the drug combination temozolomide, panobinostat, everolimus, and hydroxychloroquine led to tumor progression, observed as an increase in tumor area on days 203 and 225. The FPM therapy of disulfiram alone, from days 210-224, disulfiram and ONC201, from days 258-333, and ONC201 alone led to a dramatic decrease in tumor area. The patient died on day 677 from treatment-resistant secondary tumors that were first visualized on day 255.
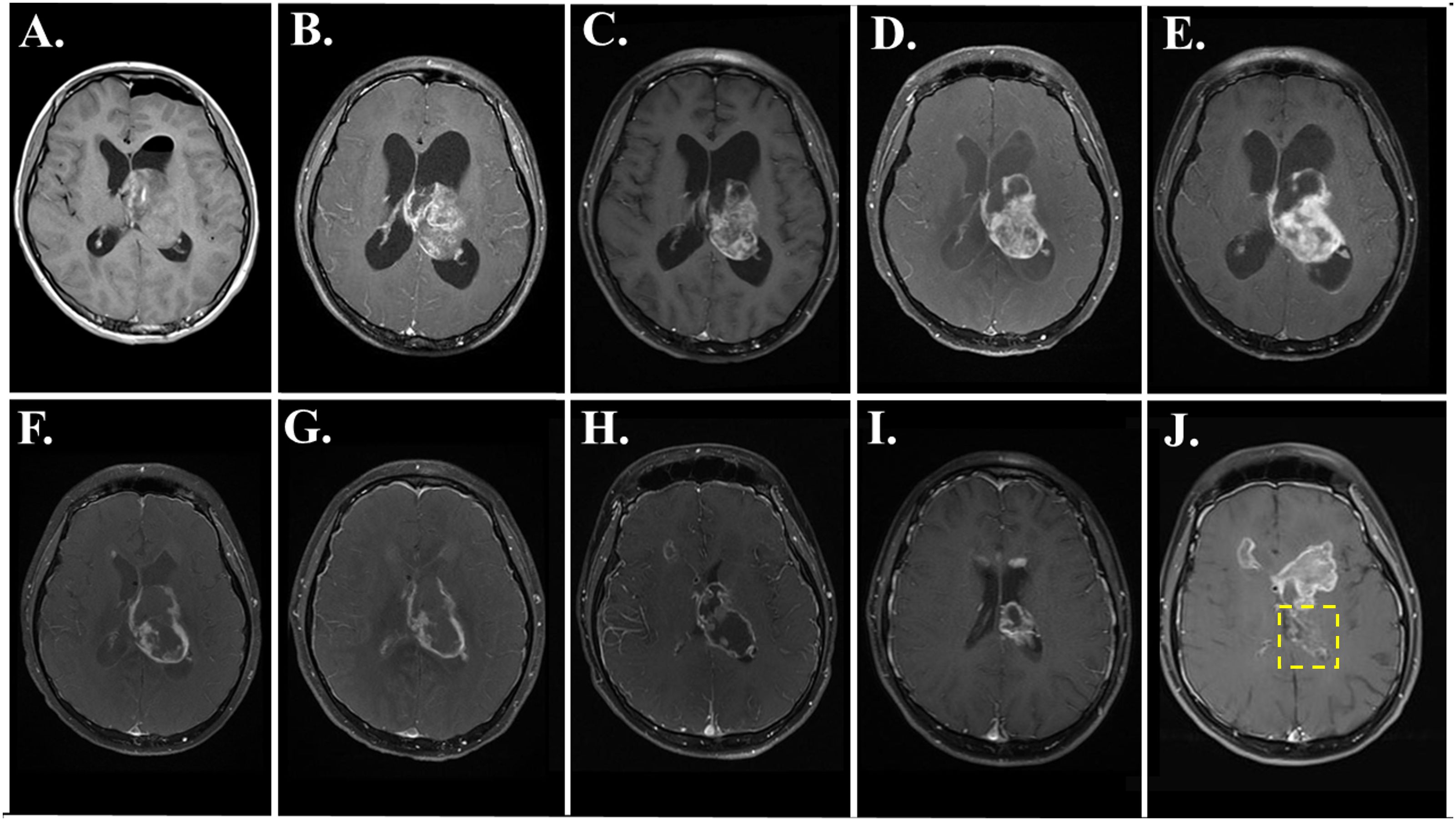
Figure 3. MRI images showing primary DMG tumor regression during and after FPM therapy with disulfiram and ONC201. T1-post gadolinium axial images are shown from the following days: (A) day 2; (B) day 88; (C) day 149; (D) day 203; (E) day 225; (F) day 255; (G) day 288; (H) day 351; (I) day 409; and (J) day 469. In J, the location of the primary tumor is within the yellow box, and secondary tumors are evident above the location of the primary tumor.
Based on the success of a feasibility study (15), the ex vivo DST results of the patient’s tumor cells were reviewed for the next treatment options as a salvage therapy, and the patient and family consented to a novel drug combination and FPM-based therapy of disulfiram and ONC201. Disulfiram, an FDA-approved drug for treating alcoholism that is in evaluation as a repurposed drug for treating high grade glioma and other cancers (19), resulted in high potency cell death in ex vivo DST (Figure 1). Meanwhile, ONC201 was at that time was available in Germany for off-trial use and serves as a potential molecular profiling therapy for DMG tumors carrying the H3K27M mutation (20, 21). Seven months after diagnosis on day 210, disulfiram was initiated alone at 250 milligrams (mg) once per day (QD). After 15 days, the disulfiram treatment was interrupted due to worsening hydrocephalus and an MRI on day 225 showed further tumor progression (Figure 3E). The patient then underwent another surgery for internal shunt placement, and did not receive any cancer therapies for over a month while in recovery. An MRI on day 255 at the end of this recovery period (Figure 3F) showed reduced hydrocephalus, and interestingly tumor regression from the previous month.
The patient then received the combination of disulfiram and German-sourced ONC201 starting on day 258. ONC201 was administered once per week at 680 mg, corresponding to 8.5 mg per kilogram of body weight. Disulfiram was initially administered at 250 mg QD for the first month, then increased to 250 mg twice per day (BID) for one month consistent with the dosing used in clinical trials for glioblastoma (22, 23) and then decreased to 250 mg QD for another 18 days. The disulfiram-ONC201 combination therapy was stopped on day 333 due to significant painful peripheral neuropathy. During this combination therapy, the tumor regressed continually month by month based on MRIs on days 288 (Figure 3G), and 351 (Figure 3H). After this therapy was stopped, MRIs on day 409 (Figure 3I) and day 469 (Figure 3J), with the primary tumor indicated within the yellow box) showed even further tumor regression.
The disulfiram-ONC201 combination therapy was stopped due to the onset of peripheral neuropathy featuring intense neuropathic pain in the feet and distal motor weakness/areflexia which led to inability to ambulate and subsequent hospitalization. MRI spine did not show any evidence of nerve root enhancement or cord signal abnormality. A lumbar puncture to assess for albuminocytologic dissociation was recommended but declined and the patient was treated for symptomatic neuropathic pain management and physical therapy. The painful neuropathy resolved after three months; however, the patient never regained his ability to ambulate due to persistent bilateral foot drop. After resolution of the neuropathy on day 433, the patient resumed ONC201 alone without disulfiram, and the peripheral neuropathy did not recur. Tumor progression appeared originating from the frontal horns of the right and left ventricles (first noticeable on day 225, Figure 3F, and prominent on day 469, Figure 3J) and the left cerebellar peduncle. The primary thalamic tumor never recurred, and the patient died of disease on day 677.
Discussion
Our case study demonstrates feasibility of ex vivo DST of the tumor cells from a DMG patient obtained at diagnosis, and a favorable treatment response of a primary thalamic DMG and extended survival of the patient to an off-trial N-of-1 FPM salvage therapy, consisting of a two-drug combination of disulfiram, a repurposed cancer drug (19) that was selected based on the DST results, and ONC201, an investigational drug for H3K27M-DMG currently being evaluated in phase III clinical trial setting (14). The patient started this therapy seven months after diagnosis, after radiotherapy and an ineffective molecular-guided therapy, when the tumor was recurrent and when the patient needed surgery for worsening hydrocephalus. Given the lack of clinical trial options available and at the request of the patient and family with informed consent, a novel personalized combination salvage therapy approach based on the DST results obtained at diagnosis was undertaken. After starting the FPM-guided therapy, the primary tumor regressed quickly and continuously in the following months, never recurred, and the patient survived for another fifteen months. This survival time compares favorably to recurrent DMG patients that lived for only three months when no further treatments were pursued or for 5–6 months after re-irradiation (6, 7). However, most DMG patients in survival studies were in children 18 years of age or younger. It is possible that the longer survival time of this adolescent is related to age-dependent biological differences between pediatric and adult DMG which may be responsible for improved survival reported in adults (2, 3). Ultimately, the treatment response in this case was mixed as secondary tumors in other areas of the brain that were initially visualized during this FPM therapy were treatment-resistant resulting in tumor progression and eventual death.
The FPM therapy was discontinued after three months due to peripheral neuropathy that was likely caused by disulfiram (24–28). Disulfiram-induced neuropathy is rare and dose dependent, and has been documented in clinical trials of glioblastoma patients where it was dosed at 500 mg QD or higher (22, 23), similar to the dosing used by this patient when the neuropathy first appeared. Disulfiram was shown to be safe in adolescents at a lower dose of 200 mg daily (29), and no studies have been reported in children. The combination of disulfiram and ONC201 has not been investigated, and therefore the mechanisms for the neuropathy due to a drug-drug interaction can only be speculative. The adverse event in this adolescent patient highlights the safety concerns of using uncharacterized drug combinations and treating adolescent and pediatric brain cancer patients with drugs that were developed for other indications in adults. After the neuropathy subsided, the patient declined to restart disulfiram at a lower dose due to the severity of neuropathic side effects. The family and patient also did not seek treatment using other drugs based on the DST results due to uncertainties with dosing for off-label use in an adolescent with DMG. Instead, they focused on treating the patient with ONC201 alone which does not cause peripheral neuropathy (30, 31), along with other palliative therapies.
Ex vivo DST of the patient’s tumor cells was part of a feasibility study for pediatric brain cancer patients that also included several multi-omics tumor profiling methods. Reliable DST results were obtained within one week, and all tumor profiling results were obtained within an average time of three weeks after tumor biopsy (15, Figure 2, lower left). Subsequently, these profiling methods have been used in an FPM clinical trial for patients with recurrent medulloblastoma (NCT05057702, PNOC027). Our patient’s tumor cells, in contrast to this clinical trial, were profiled just after diagnosis, and the DST results were not used to inform the molecular-guided therapy that the patient tried after surgery and standard of care radiotherapy. Therefore, it is important to note that the FPM therapy for this patient was initiated seven months after DST of the patient’s biopsied tumor cells and after prior therapies. Due to extensive tumor heterogeneity and adaptive resistance of high-grade gliomas, it is likely that time and prior therapies led to molecular changes in the tumor and hence resistance of the secondary tumors to the FPM therapy (32–36). Ideally, FPM therapy should be initiated immediately after DST testing, when the personalized therapy would likely be a better match with the DST results. The option of obtaining additional tissue for DST at the time of progression was not performed based upon patient/family and provider decision.
One must interpret the favorable treatment response with caution as we cannot know whether it was due to disulfiram, ONC201, or the drug combination. ONC201 has demonstrated promising early-stage clinical results including durable responses in a few individuals (30, 37–41). Disulfiram is approved for alcohol abuse and has been repurposed as a cancer therapy due to broad-spectrum anticancer activities (19), and has preclinical efficacy against DMG (42, 43). The efficacy of disulfiram on patients with DMG, however, is unknown, and its use on DMG patients has not been previously reported. The patient initially used disulfiram alone for only 15 days, and the subsequent MRI just 30 days later showed tumor regression (Figure 2, and compare Figures 3E, F), suggesting a favorable treatment response to disulfiram. In clinical trials, disulfiram demonstrated efficacy in a phase IIb trial for non-small cell lung cancer (44). However, there was no efficacy in a recent clinical trial for glioblastoma (45), The combination of disulfiram and ONC201 on the efficacy of any cancer, including potential synergistic effects, is unknown. Notably, in the absence of other treatment options, the patient and his family were willing to explore using ONC201 in a novel combination with another available and off-trial drug based on functional DST of the patient’s tumor cells.
Another limitation of this case report is extrapolating ex vivo DST results to in vivo drug efficacy. In this regard, DMG is challenging because there are no known effective individual or combination drug therapies, and drugs used for treating DMG also need to be brain penetrant. A starting point is using the drug potencies from ex vivo DST as a benchmark for potentially efficacious drug exposure. For example, ex vivo disulfiram had a high killing potency (IC50) of 0.071 μM against the patient’s tumor cells (Figure 1), and steady state plasma concentrations of disulfiram in patients reach levels that are up to ten times higher (46), suggesting that efficacious concentrations disulfiram could be achieved for this patient. Any brain penetrant drug with these features could be a candidate for an FPM therapy, and disulfiram was selected as a personalized therapy in part due to clinical trials in adult glioma that were ongoing at the time. However, there are other in vivo drug parameters to consider for potential efficacy. Disulfiram may have unfavorable metabolism and bioavailability that limits its efficacy (46, 47), perhaps explaining why disulfiram has failed in clinical trials for glioblastoma (23, 45). Meanwhile, ONC201 had an IC50 of around 10 μM against the patient’s tumor cells (Figure 1). Peak plasma concentrations of ONC201 reached a mean concentration of 12.2 μM when dosed in patients on two consecutive days per week (48). However, peak plasma concentrations were lower than 10 μM in most patients that were dosed once per week (31), as ONC201 was dosed for this patient, suggesting that this patient’s tumor may have been resistant to pharmacologically relevant concentrations of ONC201 and consistent with the lack of efficacy of ONC201 in clinical trials for some patients (38). However, ONC201 may have a higher anti-tumor efficacy in vivo through modulation of interactions between tumor cells with the nervous and immune systems (49, 50), interactions that may not occur in ex vivo DST. Therefore, when interpreting DST results for potential in vivo efficacy, it is important to understand the pharmacokinetics and mechanism of action of each drug of interest, as well as level of brain penetrance for treating brain cancers such as DMG.
Prior to the FPM therapy, the patient used an individualized molecular-guided therapy based on tumor genetic profiling (Table 1) consisting of temozolomide, panobinostat, everolimus, and hydroxychloroquine. At the time, everolimus and panobinostat were recommended for treating PIK3CA-mutated H3K27M-DMG (20), and temozolomide is a standard of care therapy for adult high-grade glioma (51). Unfortunately, this molecular-guided therapy proved ineffective. If the ex vivo DST results were used to inform therapeutic options at that time, they would have encouraged the use of panobinostat due to its high potency for killing the patient’s tumor cells (Figure 1) as well as preclinical efficacy in DMG (17, 18). Panobinostat was available at the time by accelerated FDA approval for treating multiple myeloma and was investigated in a phase I clinical trial for DMG (52), but has since been withdrawn due to inability of the provider to conduct a required post marketing trial (53). In contrast, the DST results would have discouraged the use of everolimus and temozolomide due to their low potency and lack of activity, respectively (Figure 1). Hydroxychloroquine was not examined in the DST. Meanwhile, tumor DNA methylation profiling that was concurrently done with DST for this patient (15, data not shown) would have also discouraged the use of temozolomide, which revealed an absence of methylation of the promoter for MGMT, encoding 6-O-methylguanine-DNA methyltransferase, that predicts temozolomide resistance (54). The ex vivo DST and tumor DNA methylation profiling results of this patient’s tumor cells may explain the failure of this molecular-guided therapy.
Another limitation to precision medicine for pediatric oncology patients is that childhood tumors have a 14-fold lower somatic mutation rate and hence fewer actionable mutations than adult cancers (55, 56). For this patient, H3K27M and TP53 mutations, which occur in 60% of H3K27M-DMG patients, are not directly actionable, while PI3KCA, which occurs in 15-20% of all DMG patients (57–59) and encodes phosphatidylinositol 3-kinase (PI3K), was the only actionable mutation. However, none of the FDA-approved PI3K inhibitors are brain-penetrant, and the investigational brain-penetrant inhibitor paxalisib was not available at the time. But even for adult cancer patients in large precision medicine studies, only 10% had beneficial treatment responses to personalized therapies based on tumor molecular profiling (60, 61). Similarly, a trial using molecular-guided therapies on nineteen DMG patients showed no clinical benefit (62). In contrast, certain FPM clinical trials yielded much better results. For example, 92% of patients who received personalized therapies informed by DST achieved stable disease or better (12). Exceptional treatment responses using FPM have also been reported in patients with pediatric cancer (8, 11).
Our case report promotes the wider use of DST for patients with DMG and other childhood brain cancers preferably in a clinical trial setting. For patients with DMG that have no FDA-approved treatment options, DST can be used to identify potentially effective investigational drugs. FPM for such patients should be used in N-of-1 clinical trials to match the results with the most appropriate therapy (63–65). The drugs used in DST should also be optimized for each tumor type. For example, for brain cancers, all brain-penetrant FDA-approved, investigational, and repurposed drugs should be included in the drug screen. Furthermore, various relevant drug combinations should also be evaluated in DST, including drug combinations in current clinical trials (e.g. ONC201 combined with paxalisib for DMG, clinical trial NCT05009992). FPM clinical trials are typically performed for patients with relapsed or progressive disease. However, for DMG patients we also support the use of DST upon initial biopsy or tumor resection, as was done for the patient in this study, to define FPM therapies for newly diagnosed patients. The inclusion of repurposed drugs such as disulfiram is important because these drugs are available and often inexpensive, thereby providing accessible off-label treatment options (66). However, in some cases these may be associated with significant dose limiting side effects as seen in our patient.
An expanded use of DST may provide valuable information that may lead to rapid identification of promising new therapies that would otherwise not be identified through conventional molecular tumor analysis. DST may be an option for pediatric brain tumors such as ependymoma that lack somatic driver mutations or for tumors such as DMG where there is no curative therapy. While our reported case demonstrates the feasibility of ex vivo DST in a single patient, many questions remain regarding the generalizability and mechanistic validation of the DST results. Ex-vivo testing of drug combinations from patient derived tumor cells is an attractive area of future study, but novel personalized combination therapies informed by ex vivo DST may also be associated with significant adverse events as was the case in our patient. DST together with pharmacologic predictive data of CNS penetrability (when available) and drug safety information should be explored further in the clinical trial setting.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Rady Children’s Hospital Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
PT: Writing – review & editing, Writing – original draft, Formal analysis. TM: Investigation, Project administration, Writing – original draft, Data curation, Writing – review & editing, Methodology. PB: Investigation, Writing – review & editing, Conceptualization, Writing – original draft, Formal analysis, Methodology, Validation. RW-R: Supervision, Formal analysis, Project administration, Writing – original draft, Conceptualization, Investigation, Methodology, Writing – review & editing, Data curation, Validation. JC: Data curation, Conceptualization, Writing – review & editing, Methodology, Supervision, Writing – original draft, Investigation, Formal analysis.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work is supported by a generous endowment from the Clayes Foundation to the Research Center for Neuro-Oncology and Genomics within the Rady Children’s Institute for Genomic Medicine. Critical infrastructure for the work was supported by a generous grant from Curebound (https://www.curebound.org). RJWR’s contributions were supported by R35 NS122339 from the National Institute for Neurological Disorders and Stroke (RJWR), William’s Superhero Fund and the McDowell Charity Trust.
Acknowledgments
The authors would like to thank the brave patients and families for allowing the exploration of novel approaches to identify personalized therapies that may lead to clinical trials and one day cures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1606575/full#supplementary-material
Supplementary Table 1 | Summary of compounds used in the ex-vivo drug screening of DMG.
Supplementary Methods | High-throughput drug screening (Clinical Laboratory Improvement Amendments–approved assay) The cell populations were analyzed after a 72-hour exposure to 5 customized drug concentrations (within the range of 5 pmol/L to 100 μmol/L) of each drug spanning 5 logs. After exposure viability was determined using CellTiter-Glo luminescent reagent (Promega) per manufacturer’s protocol, then the plates were analyzed with the EnVision Multilabel plate reader (Perkin Elmer). XLFit (IDBS), a Microsoft Excel Add-in, was used to analyze the data and generate dose–response curves based on standard 4- parameter logistic fit [i.e., fit = (A + (B/(1 + ((x/C)^D)))) where A and B equal minimum and maximum asymptotes, C equals IC50 and D equals slope]. The AUC values were calculated using the XLFit software utilizing minimum and maximum concentrations of drugs/compounds within the panel as the limits of the AUC calculation. For each plate, data were normalized to DMSO 100% viability and blank controls. The samples were not frozen but freshly, dissociated to single cell suspensions within hours from surgically removed tumors that were kept on ice prior to shipping. The drug sensitivity assay was initiated within 48 hours of surgery. Clinical Laboratory Improvement Amendments standards require that thawed cell line data be repeated for standardization of the assay every 6 months. The assay results were highly reproducible.
References
1. Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell. (2017) 32:520–37. doi: 10.1016/j.ccell.2017.08.017
2. Schulte JD, Buerki RA, Lapointe S, Molinaro AM, Zhang Y, Villanueva-Meyer JE, et al. Clinical, radiologic, and genetic characteristics of histone H3 K27M-mutant diffuse midline gliomas in adults. Neurooncol Adv. (2020) 2:vdaa142. doi: 10.1093/noajnl/vdaa142
3. Sim Y, McClelland AC, Choi K, Han K, Park YW, Ahn SS, et al. A comprehensive multicenter analysis of clinical, molecular, and imaging characteristics and outcomes of H3 K27-altered diffuse midline glioma in adults. J Neurosurg. (2025) 142:1307–18. doi: 10.3171/2024.8.JNS241180
4. Saratsis AM, Knowles T, Petrovic A, and Nazarian J. H3K27M mutant glioma: Disease definition and biological underpinnings. Neuro Oncol. (2024) 26:S92–S100. doi: 10.1093/neuonc/noad164
5. Al Sharie S, Abu Laban D, and Al-Hussaini M. Decoding diffuse midline gliomas: A comprehensive review of pathogenesis, diagnosis and treatment. Cancers (Basel). (2023) 15:4869. doi: 10.3390/cancers15194869
6. Lassaletta A, Strother D, Laperriere N, Hukin J, Vanan MI, Goddard K, et al. Reirradiation in patients with diffuse intrinsic pontine gliomas: The Canadian experience. Pediatr Blood Cancer. (2018) 65:e26988. doi: 10.1002/pbc.26988
7. Chavaz L, Janssens GO, Bolle S, Mandeville H, Ramos-Albiac M, Van Beek K, et al. Neurological symptom improvement after re-irradiation in patients with diffuse intrinsic pontine glioma: A retrospective analysis of the SIOP-E-HGG/DIPG project. Front Oncol. (2022) 12:1182994. doi: 10.3389/fonc.2023.1182994
8. Acanda de la Rocha AM, Berlow NE, Fader M, Coats ER, Saghira C, Espinal PS, et al. Feasibility of functional precision medicine for guiding treatment of relapsed or refractory pediatric cancers. Nat Med. (2024) 30:990–1000. doi: 10.1038/s41591-024-02848-4
9. Kornauth C, Pemovska T, Vladimer GI, Bayer G, Bergmann M, Eder S, et al. Functional precision medicine provides clinical benefit in advanced aggressive hematologic cancers and identifies exceptional responders. Cancer Discov. (2022) 12:372–87. doi: 10.1158/2159-8290.CD-21-0538
10. Malani D, Kumar A, Brück O, Kontro M, Yadav B, Hellesøy M, et al. Implementing a functional precision medicine tumor board for acute myeloid leukemia. Cancer Discov. (2022) 12:388–401. doi: 10.1158/2159-8290.CD-21-0410
11. Acanda de la Rocha AM, Fader M, Coats ER, Espinal PS, Berrios V, Saghira C, et al. Clinical utility of functional precision medicine in the management of recurrent/relapsed childhood rhabdomyosarcoma. JCO Precis Oncol. (2021) 5:1659–65. doi: 10.1200/PO.20.00438
12. Coffey DG, Cowan AJ, DeGraaff B, Martins TJ, Curley N, Green DJ, et al. High-throughput drug screening and multi-omic analysis to guide individualized treatment for multiple myeloma. JCO Precis Oncol. (2021) 5:602–12. doi: 10.1200/PO.20.00442
13. Swords RT, Azzam D, Al-Ali H, Lohse I, Volmar CH, Watts JM, et al. Ex-vivo sensitivity profiling to guide clinical decision making in acute myeloid leukemia: A pilot study. Leuk Res. (2018) 64:34–41. doi: 10.1016/j.leukres.2017.11.008
14. Arrillaga-Romany I, Lassman A, McGovern SL, Mueller S, Nabors B, van den Bent M, et al. ACTION: a randomized phase 3 study of ONC201 (dordaviprone) in patients with newly diagnosed H3 K27M-mutant diffuse glioma. Neuro Oncol. (2024) 26:S173–81. doi: 10.1093/neuonc/noae031
15. Crawford JR, Lo YY, Pagadala MS, Chapman OS, Sridhar S, Juarez EF, et al. TRLS-13. MULTI-OMICS AND FUNCTIONAL PRECISION MEDICINE FOR NEWLY DIAGNOSED AND RECURRENT PEDIATRIC CENTRAL NERVOUS SYSTEM TUMORS. Neuro Oncol. (2024) 26:iv47–8. doi: 10.1093/neuonc/noae064.166
16. Qin G, Dai J, Chien S, Martins TJ, Loera B, Nguyen QH, et al. Mutation patterns predict drug sensitivity in acute myeloid leukemia. Clin Cancer Res. (2024) 30:2659–71. doi: 10.1158/1078-0432.CCR-23-1674
17. Lin GL, Wilson KM, Ceribelli M, Stanton BZ, Woo PJ, Kreimer S, et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci Transl Med. (2019) 11:eaaw0064. doi: 10.1126/scitranslmed.aaw0064
18. Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. (2015) 21:555–9. doi: 10.1038/nm.3855
19. Zhong S, Liu S, Shi X, Zhang X, Li K, Liu G, et al. Disulfiram in glioma: Literature review of drug repurposing. Front Pharmacol. (2022) 13:933655. doi: 10.3389/fphar.2022.933655
20. Miklja Z, Pasternak A, Stallard S, Nicolaides T, Kline-Nunnally C, Cole B, et al. Molecular profiling and targeted therapy in pediatric gliomas: review and consensus recommendations. Neuro Oncol. (2019) 21:968–80. doi: 10.1093/neuonc/noz022
21. Duchatel RJ, Mannan A, Woldu AS, Hawtrey T, Hindley PA, Douglas AM, et al. Preclinical and clinical evaluation of German-sourced ONC201 for the treatment of H3K27M-mutant diffuse intrinsic pontine glioma. Neurooncol Adv. (2021) 3:vdab169. doi: 10.1093/noajnl/vdab169
22. Huang J, Campian JL, Gujar AD, Tran DD, Lockhart AC, DeWees TA, et al. A phase I study to repurpose disulfiram in combination with temozolomide to treat newly diagnosed glioblastoma after chemoradiotherapy. J Neurooncol. (2016) 128:259–66. doi: 10.1007/s11060-016-2104-2
23. Huang J, Campian JL, Gujar AD, Tsien C, Ansstas G, Tran DD, et al. Final results of a phase I dose-escalation, dose-expansion study of adding disulfiram with or without copper to adjuvant temozolomide for newly diagnosed glioblastoma. J Neurooncol. (2018) 138:105–11. doi: 10.1007/s11060-018-2775-y
24. Behan C, Lane A, and Clarke M. Disulfiram induced peripheral neuropathy: between the devil and the deep blue sea. Ir J Psychol Med. (2007) 24:115–16. doi: 10.1017/S0790966700010454
25. Karamanakos PN, Pappas P, Stephanou P, and Marselos M. Differentiation of disulfiram effects on central catecholamines and hepatic ethanol metabolism. Pharmacol Toxicol. (2001) 88:106–10. doi: 10.1034/j.1600-0773.2001.088002106.x
26. Palliyath SK, Schwartz BD, and Gant L. Peripheral nerve functions in chronic alcoholic patients on disulfiram: a six month follow up. J Neurol Neurosurg Psychiatry. (1990) 53:227–30. doi: 10.1136/jnnp.53.3.227
27. Frisoni GB and Di Monda V. Disulfiram neuropathy: a review (1971-1988) and report of a case. Alcohol. (1989) 24:429–37. doi: 10.1093/oxfordjournals.alcalc.a044938
28. Mokri B, Ohnishi A, and Dyck PJ. Disulfiram neuropathy. Neurology. (1981) 31:730–5. doi: 10.1212/wnl.31.6.730
29. Niederhofer H and Staffen W. Comparison of disulfiram and placebo in treatment of alcohol dependence of adolescents. Drug Alcohol Rev. (2003) 22:295–7. doi: 10.1080/0959523031000154436
30. Gardner SL, Tarapore RS, Allen J, McGovern SL, Zaky W, Odia Y, et al. Phase I dose escalation and expansion trial of single agent ONC201 in pediatric diffuse midline gliomas following radiotherapy. Neurooncol Adv. (2022) 4:vdac143. doi: 10.1093/noajnl/vdac143
31. Stein MN, Malhotra J, Tarapore RS, Malhotra U, Silk AW, Chan N, et al. Safety and enhanced immunostimulatory activity of the DRD2 antagonist ONC201 in advanced solid tumor patients with weekly oral administration. J Immunother Cancer. (2019) 7:136. doi: 10.1186/s40425-019-0599-8
32. Liu I, Jiang L, Samuelsson ER, Marco Salas S, Beck A, Hack OA, et al. The landscape of tumor cell states and spatial organization in H3-K27M mutant diffuse midline glioma across age and location. Nat Genet. (2022) 54:1881–94. doi: 10.1038/s41588-022-01236-3
33. Hoffman LM, DeWire M, Ryall S, Buczkowicz P, Leach J, Miles L, et al. Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol Commun. (2016) 4:1. doi: 10.1186/s40478-015-0269-0
34. Nikbakht H, Panditharatna E, Mikael LG, Li R, Gayden T, Osmond M, et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun. (2016) 7:11185. doi: 10.1038/ncomms11185
35. Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. (2013) 110:4009–14. doi: 10.1073/pnas.1219747110
36. Duchatel RJ, Jackson ER, Parackal SG, Kiltschewskij D, Findlay IJ, Mannan A, et al. PI3K/mTOR is a therapeutically targetable genetic dependency in diffuse intrinsic pontine glioma. J Clin Invest. (2024) 134:e170329. doi: 10.1172/JCI170329
37. Arrillaga-Romany I, Gardner SL, Odia Y, Aguilera D, Allen JE, Batchelor T, et al. ONC201 (Dordaviprone) in recurrent H3 K27M-mutant diffuse midline glioma. J Clin Oncol. (2024) 42:1542–52. doi: 10.1200/JCO.23.01134
38. Venneti S, Kawakibi AR, Ji S, Waszak SM, Sweha SR, Mota M, et al. Clinical efficacy of ONC201 in H3K27M-mutant diffuse midline gliomas is driven by disruption of integrated metabolic and epigenetic pathways. Cancer Discov. (2023) 13:2370–93. doi: 10.1158/2159-8290.CD-23-0131
39. Arrillaga-Romany I, Odia Y, Prabhu VV, Tarapore RS, Merdinger K, Stogniew M, et al. Biological activity of weekly ONC201 in adult recurrent glioblastoma patients. Neuro Oncol. (2020) 22:94–102. doi: 10.1093/neuonc/noz164
40. Chi AS, Tarapore RS, Hall MD, Shonka N, Gardner S, Umemura Y, et al. Pediatric and adult H3 K27M-mutant diffuse midline glioma treated with the selective DRD2 antagonist ONC201. J Neurooncol. (2019) 145:97–105. doi: 10.1007/s11060-019-03271-3
41. Hall MD, Odia Y, Allen JE, Tarapore R, Khatib Z, Niazi TN, et al. First clinical experience with DRD2/3 antagonist ONC201 in H3 K27M-mutant pediatric diffuse intrinsic pontine glioma: a case report. J Neurosurg Pediatr. (2019) 23:719–25. doi: 10.3171/2019.2.PEDS18480
42. Sharma M, Barravecchia I, Magnuson B, Ferris SF, Apfelbaum A, Mbah NE, et al. Histone H3 K27M-mediated regulation of cancer cell stemness and differentiation in diffuse midline glioma. Neoplasia. (2023) 44:100931. doi: 10.1016/j.neo.2023.100931
43. Meier S, Cantilena S, Niklison Chirou MV, Anderson J, Hargrave D, Salomoni P, et al. Alcohol-abuse drug disulfiram targets pediatric glioma via MLL degradation. Cell Death Dis. (2021) 12:785. doi: 10.1038/s41419-021-04078-9
44. Nechushtan H, Hamamreh Y, Nidal S, Gotfried M, Baron A, Shalev YI, et al. A phase IIb trial assessing the addition of disulfiram to chemotherapy for the treatment of metastatic non-small cell lung cancer. Oncologist. (2015) 20:366–7. doi: 10.1634/theoncologist.2014-0424
45. Werlenius K, Kinhult S, Solheim TS, Magelssen H, Löfgren D, Mudaisi M, et al. Effect of disulfiram and copper plus chemotherapy vs chemotherapy alone on survival in patients with recurrent glioblastoma: A randomized clinical trial. JAMA Netw Open. (2023) 6:e234149. doi: 10.1001/jamanetworkopen.2023.4149
46. Lanz J, Biniaz-Harris N, Kuvaldina M, Jain S, Lewis K, and Fallon BA. Disulfiram: mechanisms, applications, and challenges. Antibiotics (Basel). (2023) 12:524. doi: 10.3390/antibiotics12030524
47. Kang X, Jadhav S, Annaji M, Huang CH, Amin R, Shen J, et al. Advancing cancer therapy with copper/disulfiram nanomedicines and drug delivery systems. Pharmaceutics. (2023) 15:1567. doi: 10.3390/pharmaceutics15061567
48. Odia Y, Koschmann C, Vitanza NA, de Blank P, Aguilera D, Allen J, et al. Safety and pharmacokinetics of ONC201 (dordaviprone) administered two consecutive days per week in pediatric patients with H3 K27M-mutant glioma. Neuro Oncol. (2024) 26:S155–64. doi: 10.1093/neuonc/noae001
49. Jackson ER, Persson ML, Fish CJ, Findlay IJ, Mueller S, Nazarian J, et al. A review of current therapeutics targeting the mitochondrial protease ClpP in diffuse midline glioma, H3 K27-altered. Neuro Oncol. (2024) 26:S136–54. doi: 10.1093/neuonc/noad144
50. Jackson ER, Duchatel RJ, Staudt DE, Persson ML, Mannan A, Yadavilli S, et al. ONC201 in combination with paxalisib for the treatment of H3K27-altered diffuse midline glioma. Cancer Res. (2023) 83:2421–37. doi: 10.1158/0008-5472.CAN-23-0186
51. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
52. Monje M, Cooney T, Glod J, Huang J, Peer CJ, Faury D, et al. Phase I trial of panobinostat in children with diffuse intrinsic pontine glioma: A report from the Pediatric Brain Tumor Consortium (PBTC-047). Neuro Oncol. (2023) 25:2262–72. doi: 10.1093/neuonc/noad141
53. Federal Register. Secura bio, inc.; withdrawal of approval of new drug application for FARYDAK (Panobinostat) capsules, 10 milligrams, 15 milligrams, and 20 milligrams (2022). Available online at: https://www.federalregister.gov/documents/2022/03/24/2022-06182/secura-bio-inc-withdrawal-of-approval-of-new-drug-application-for-farydak-panobinostat-capsules-10 (Accessed February 24, 2025).
54. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. (2005) 352:997–1003. doi: 10.1056/NEJMoa043331
55. Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature. (2018) 555:321–7. doi: 10.1038/nature25480
56. Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. (2018) 555:371–6. doi: 10.1038/nature25795
57. Williams EA, Brastianos PK, Wakimoto H, Zolal A, Filbin MG, Cahill DP, et al. A comprehensive genomic study of 390 H3F3A-mutant pediatric and adult diffuse high-grade gliomas, CNS WHO grade 4. Acta Neuropathol. (2023) 146:515–25. doi: 10.1007/s00401-023-02609-6
58. Jovanovich N, Habib A, Head J, Hameed F, Agnihotri S, and Zinn PO. Pediatric diffuse midline glioma: Understanding the mechanisms and assessing the next generation of personalized therapeutics. Neurooncol Adv. (2023) 5:vdad040. doi: 10.1093/noajnl/vdad040
59. Stegat L, Eckhardt A, Gocke A, Neyazi S, Pohl L, Schmid S, et al. Integrated analyses reveal two molecularly and clinically distinct subtypes of H3 K27M-mutant diffuse midline gliomas with prognostic significance. Acta Neuropathol. (2024) 148:40. doi: 10.1007/s00401-024-02800-3
60. O’Dwyer PJ, Gray RJ, Flaherty KT, Chen AP, Li S, Wang V, et al. The NCI-MATCH trial: lessons for precision oncology. Nat Med. (2023) 29:1349–57. doi: 10.1038/s41591-023-02379-4
61. Haslam A, Kim MS, and Prasad V. Updated estimates of eligibility for and response to genome-targeted oncology drugs among US cancer patients, 2006-2020. Ann Oncol. (2021) 32:926–32. doi: 10.1016/j.annonc.2021.04.003
62. Kline C, Jain P, Kilburn L, Bonner ER, Gupta N, Crawford JR, et al. Upfront biology-guided therapy in diffuse intrinsic pontine glioma: therapeutic, molecular, and biomarker outcomes from PNOC003. Clin Cancer Res. (2022) 28:3965–78. doi: 10.1158/1078-0432.CCR-22-0803
63. Nikanjam M, Kato S, Sicklick JK, and Kurzrock R. At the right dose: personalised (N-of-1) dosing for precision oncology. Eur J Cancer. (2023) 194:113359. doi: 10.1016/j.ejca.2023.113359
64. Gouda MA, Buschhorn L, Schneeweiss A, Wahida A, and Subbiah V. N-of-1 trials in cancer drug development. Cancer Discov. (2023) 13:1301–9. doi: 10.1158/2159-8290.CD-22-1377
65. Kyr M, Svobodnik A, Stepanova R, and Hejnova R. N-of-1 trials in pediatric oncology: from a population-based approach to personalized medicine-A review. Cancers (Basel). (2021) 13:5428. doi: 10.3390/cancers13215428
Keywords: diffuse midline glioma, functional precision medicine, drug sensitivity testing, molecular guided therapy, pediatric brain cancer
Citation: Tan PK, Martins TJ, Becker PS, Wechsler-Reya RJ and Crawford JR (2025) Case Report: Application of ex-vivo drug sensitivity testing to identify personalized treatment options for an adolescent with diffuse midline glioma. Front. Oncol. 15:1606575. doi: 10.3389/fonc.2025.1606575
Received: 05 April 2025; Accepted: 07 July 2025;
Published: 07 August 2025.
Edited by:
Ashley Sloane Margol, Children’s Hospital of Los Angeles, United StatesReviewed by:
Shwetal Mehta, Barrow Neurological Institute (BNI), United StatesEvan Cantor, Connecticut Children’s Medical Center, United States
Copyright © 2025 Tan, Martins, Becker, Wechsler-Reya and Crawford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John Ross Crawford, Sm9obi5DcmF3Zm9yZEBjaG9jLm9yZw==