 Zhenjiang Pan
Zhenjiang Pan Jing Bao
Jing Bao Shepeng Wei
Shepeng Wei- 1Department of Neurosurgery, Shidong Hospital, Yangpu District, Shanghai, China
- 2Shidong Hospital, University of Shanghai for Science and Technology, Shanghai, China
Intracranial ependymomas are glial tumors arising from the ependymal lining of the ventricular system, most commonly affecting young children (median age: 5 years), though they can occur across all age groups. Typically located in the posterior fossa, they account for fewer than 10% of pediatric central nervous system neoplasms and show a slight male predominance. Clinical symptoms vary by location, with posterior fossa tumors often causing hydrocephalus-related signs, and supratentorial lesions presenting with seizures or focal deficits. The 2021 WHO CNS5 classification integrates histologic, anatomic, and molecular features, distinguishing prognostically significant subgroups such as posterior fossa group A (PFA) and supratentorial ZFTA-fusion ependymomas. Diagnosis requires histologic confirmation, aided by MRI and cerebrospinal fluid analysis, with dissemination present in up to 10% of cases at diagnosis. Maximal safe surgical resection is the cornerstone of treatment. Children over one year with grade 2 or 3 tumors typically receive adjuvant focal radiotherapy, while chemotherapy is used to delay irradiation in infants or after subtotal resection. Disseminated disease may require craniospinal irradiation or systemic therapy. Despite multimodal treatment, prognosis remains guarded. Ten-year overall survival ranges from 50% to 75%, influenced by extent of resection, molecular subtype, and age. This review synthesizes current knowledge of ependymoma pathogenesis, classification, diagnosis, and therapy, highlighting the growing role of molecular profiling and the importance of specialized, multidisciplinary care.
1 Introduction
Ependymomas are glial tumors thought to originate from radial glial cells in the subventricular zone, typically adjacent to the ependymal lining of the ventricular system (1–3). They predominantly occur in the posterior fossa near the fourth ventricle or within the intramedullary spinal cord, with rare occurrences in the cerebral parenchyma outside the posterior fossa and exceptional cases outside the central nervous system. This review focuses on the clinical presentation and management of intracranial ependymomas.
Ependymomas are glial tumors that arise from cells lining the ventricular system of the brain and spinal cord. Although they represent a relatively small proportion of central nervous system (CNS) tumors overall, they pose a significant clinical challenge due to their location, potential for recurrence, and molecular heterogeneity. In pediatric populations, intracranial ependymomas account for approximately 5–10% of all primary CNS tumors, with the posterior fossa being the most common site of origin (1–4). In contrast, adults more frequently develop spinal ependymomas, although intracranial variants are not uncommon (1, 3, 5).
Historically, classification and prognostication relied heavily on histopathological grading and anatomic location. However, recent advances in molecular diagnostics have revolutionized the understanding and management of these tumors. The 2021 World Health Organization (WHO) Classification of Tumors of the Central Nervous System (CNS5) emphasizes integrated diagnoses based on a combination of histology, molecular markers, and anatomical site (6, 7). This paradigm shift has led to the recognition of distinct molecular subgroups with divergent clinical behaviors and outcomes, such as posterior fossa group A (PFA), group B (PFB), and supratentorial ZFTA- or YAP1-fused tumors (8–10).
In this review, we aim to summarize contemporary approaches to the diagnosis and treatment of intracranial ependymomas, highlighting the critical role of molecular characterization in guiding clinical decision-making. We also explore ongoing controversies, emerging therapies, and directions for future research in this evolving field.
2 Epidemiology
Intracranial ependymomas predominantly affect young children, with a slight male predominance (3, 4). The median age at diagnosis is 5 years, with 25–40% of cases occurring before age 2 (5–7). In adults, most cases present before age 40. Ependymomas account for less than 10% of central nervous system tumors in children and young adults but represent approximately 25% of primary spinal cord tumors (8, 9). Spinal ependymomas typically arise between ages 30 and 40 and are more prevalent in patients with NF2-related schwannomatosis. Subependymomas, often incidental or identified at autopsy, primarily affect middle-aged and older men (3, 4).
3 Pathology
3.1 Histology and classification
In the World Health Organization (WHO) classification of central nervous system (CNS) tumors, ependymal tumors are categorized by anatomic location, histology, and molecular characteristics. The 2021 revision (5th edition, CNS5) introduces several new entities and subgroups defined by molecular genetic features (3, 10).
3.1.1 Ependymomas
Ependymomas may arise throughout the ventricular system and spinal canal, most commonly in the fourth ventricle and spinal cord. They are typically well circumscribed and may show calcification, hemorrhage, or cystic change. Ependymal rosettes, though not always present, are a diagnostic hallmark. Histologic variants—including papillary, clear cell, and tanycytic—lack distinct clinical relevance.
Traditionally classified as classic or anaplastic based on cellularity and mitotic activity, ependymomas have shown inconsistent correlation between histologic grade and prognosis. Some studies, such as the Children’s Oncology Group ACNS0121 phase II trial (11), suggest worse outcomes with anaplastic (grade 3) tumors, but findings are not uniform, particularly in retrospective analyses (12). Tumor heterogeneity and interobserver variability may underlie this inconsistency (13).
Reflecting improved understanding of molecular subgroups, the WHO CNS5 classification abandons the classic–anaplastic distinction and incorporates histology, location, and molecular features (10). Histologic grading remains an integral part of diagnosis and prognostication, although its predictive power is increasingly supplemented by molecular classification (14–18).
3.1.2 Subependymoma
Subependymomas are rare WHO grade 1 tumors typically located in the fourth or lateral ventricles of adults (3). They have a benign histologic appearance, featuring a coarse fibrillar matrix, uniform nuclei in clusters, microcysts, and occasionally calcifications or hemorrhage.
3.1.3 Myxopapillary ependymoma
Myxopapillary ependymomas arise almost exclusively in the conus medullaris and filum terminale. Molecular differences have been noted between adult and pediatric cases (19). Under the WHO CNS5 classification, they are designated as grade 2, reflecting a recurrence risk similar to conventional spinal ependymomas (3, 10, 20–23).
3.2 Molecular groups
Epigenomic and transcriptomic analyses have identified at least nine distinct molecular entities of ependymoma, each with unique demographics and clinical behavior (33). Further “omics” studies reveal additional heterogeneity within posterior fossa group A (PFA) (34) and group B (PFB) (35) tumors. Mapping of enhancer landscapes may provide a basis for targeted drug development across molecular subtypes (36).
Genomic alterations in ependymomas vary by anatomic site (16–18, 24–36). Comprehensive genomic and epigenetic profiling has defined distinct molecular subgroups (24, 37–39). Emerging classifications continue to evolve and require prospective validation to establish their prognostic and therapeutic relevance (40).
3.2.1 Posterior fossa ependymoma group A
PFA ependymomas, an aggressive molecular subgroup, primarily affect infants and young children (2, 12, 41, 42). These tumors exhibit a CpG island methylator phenotype and transcriptional silencing of polycomb repressive complex 2 (PRC2), leading to downregulation of differentiation genes (43). Chromosomal alterations—particularly 1q gain, which is found in 15–20% of newly diagnosed cases—are associated with poor outcomes. Combined 1q gain and 6q loss occurs less frequently but may define an ultra–high-risk subgroup. These alterations are detected in up to 60% of recurrent cases (11, 12, 15, 16, 18, 24, 28, 44–46).
Loss of nuclear H3K27me3 expression—a hallmark of PFA tumor cells (32)—was first described in H3 K27-altered diffuse midline gliomas (47). Both tumor types appear to share a PRC2-inhibitory oncogenic mechanism involving peptidyl PRC2 inhibitors (48, 49). Immunohistochemical absence of H3K27me3 offers a reliable, clinically applicable marker for identifying PFA tumors (31, 50).
3.2.2 Posterior fossa ependymoma group B
PFB ependymomas predominantly affect older children and adults and are linked to a more favorable prognosis (38, 41).
3.2.3 Supratentorial ependymoma with ZFTA fusion
Most supratentorial ependymomas (70–80%) harbor an oncogenic fusion between ZFTA (formerly C11orf95) on chromosome 11 and a partner gene—most commonly RELA, a key effector of NF-κB signaling (51, 52). ZFTA fusions are typically identified by fluorescence in situ hybridization (FISH), although RNA sequencing and next-generation sequencing (NGS) are also widely used in molecular diagnostic workflows. Although retrospective studies have linked these fusions to poor prognosis (51), prospective data have challenged this association (11, 12, 53, 54), warranting caution in clinical interpretation. Additional prognostic markers are under investigation. Biallelic CDKN2A loss has emerged as a candidate marker of poor outcome in ZFTA-fusion tumors, though validation in prospective cohorts is needed (45, 55, 56).
3.2.4 Supratentorial ependymoma with YAP1 fusion
YAP1 fusion tumors represent a small fraction of supratentorial ependymomas and are predominantly observed in infants. This subgroup may exhibit a more favorable prognosis compared with other ependymoma subtypes (12, 45), though prospective validation is required.
3.2.5 Supratentorial ependymoma without ZFTA or YAP1 fusion
A minority of supratentorial ependymomas lack ZFTA or YAP fusions and display clinical and molecular heterogeneity, underscoring the need for larger studies to clarify their biology (57, 58).
4 Clinical features
4.1 Presenting signs and symptoms
Clinical presentation varies by tumor location:
4.1.1 Increased intracranial pressure
Posterior fossa ependymomas commonly cause obstructive hydrocephalus, leading to headache, nausea, vomiting, ataxia, vertigo, and papilledema. Cranial nerve palsies—particularly of nerves VI to X—are frequent, and brainstem invasion may occur.
4.1.2 Seizures and focal deficits
Supratentorial ependymomas often present with seizures or focal deficits, such as hemiparesis, due to mass effect and peritumoral edema.
4.1.3 Myelopathy and radiculopathy
Spinal ependymomas cause symptoms from tract or nerve root involvement, with findings determined by tumor location along the cord.
4.1.4 Leptomeningeal disease
Cerebrospinal fluid (CSF) dissemination is present in fewer than 5% of patients at diagnosis (12, 53, 59). Both infratentorial and supratentorial tumors may spread, with clinical presentations ranging from minimal symptoms to multifocal deficits involving cranial nerves or the cauda equina. Although earlier studies linked spinal seeding to higher histologic grade, recent data suggest that prognosis is more closely associated with molecular subgroup.
4.2 Anatomic location
The fourth ventricle is the most frequent site of intracranial ependymomas, often extending into the subarachnoid space and occasionally encasing the medulla and upper cervical cord. Supratentorial tumors may be intraventricular (typically in the lateral ventricles) or parenchymal. Ependymoma locations vary by age:
4.2.1 Children
Approximately 90% of ependymomas in children are intracranial, with 75% in the posterior fossa and 10% in the spinal cord (5, 6).
4.2.2 Adults
In adults, about 65% are spinal, 25% infratentorial, and 10% supratentorial (60).
5 Diagnosis
Diagnosis of ependymoma requires histologic confirmation but is often suspected preoperatively based on imaging, tumor location, and patient age. Because gross total resection is central to management—and appropriate for most differential diagnoses in children—diagnosis is typically made during resection. Biopsy is reserved for cases with diagnostic uncertainty or high surgical risk.
5.1 Neuroimaging appearance
On magnetic resonance imaging (MRI), posterior fossa ependymomas typically appear hypointense on T1-weighted and hyperintense on T2-weighted or proton density sequences, with prominent gadolinium enhancement. Extension into the foramen of Luschka is common. These tumors often obstruct the fourth or supratentorial ventricles, causing hydrocephalus; however, peritumoral edema is uncommon. Restricted diffusion may be present. Supratentorial ZFTA-fused ependymomas frequently have a large cystic component with nodular areas showing restricted diffusion (61, 62).
On computed tomography (CT), ependymomas are usually hyperdense with homogeneous enhancement and may contain cysts or calcifications. Calcifications in a fourth ventricle mass suggest ependymoma but are not pathognomonic. Subependymomas appear as nonenhancing, well-circumscribed intraventricular nodules, isodense on CT and typically isointense on T1 and hyperintense on T2 MRI sequences (63).
5.2 Extent of disease evaluation
All patients with suspected or confirmed ependymoma should undergo contrast-enhanced MRI of the brain and entire spine, along with cerebrospinal fluid (CSF) analysis. Spinal or leptomeningeal dissemination occurs in up to 10% of cases, though some reports suggest a lower incidence at diagnosis (12, 53, 59). CSF cytology is recommended for staging in classic, anaplastic, and myxopapillary ependymomas when imaging suggests spread.
Preoperative lumbar puncture is preferred, as postoperative samples may be contaminated by surgical debris. However, obstructive hydrocephalus often precludes lumbar puncture at presentation. In such cases, CSF sampling should be deferred for 10–14 days postoperatively to allow debris clearance (64). Although dissemination is rare, it significantly affects management and prognosis. Notably, about one-third of cases are identified solely by CSF cytology (53, 65). Given the potential for cytologic misinterpretation, a second CSF sample is advised to confirm isolated positive results.
6 Surgical resection
The primary treatment for suspected ependymoma is maximal safe resection.
6.1 Extent of resection
Ependymomas often arise in the posterior fossa, adjacent to cranial nerves and the brainstem, making resection challenging. Nonetheless, extent of resection is a key determinant of oncologic outcome and survival, underscoring the importance of optimal initial surgery. Referral to centers with pediatric oncologic neurosurgical expertise is strongly recommended. While terminology may vary, resection extent is generally classified as follows:
6.1.1 Gross total resection
Achieved when postoperative MRI reveals no residual enhancing or nonenhancing tumor, and the surgeon confirms complete removal intraoperatively.
6.1.2 Near total resection
Near-total resection refers to minimal residual tumor on postoperative MRI (typically <5 mm) or no visible disease, despite intraoperative evidence of tumor adherence to critical structures such as the brainstem or cranial nerves that prevents complete resection. For prognostic and therapeutic purposes, these cases are generally treated similarly to gross total resections.
6.1.3 Subtotal resection
Subtotal resection—defined by residual tumor on postoperative MRI and encompassing terms such as partial or incomplete resection—has not been evaluated in randomized trials. Given the overwhelming observational evidence supporting gross total resection, randomizing patients to subtotal resection would now be considered ethically impermissible. However, observational studies consistently associate gross total resection with lower local recurrence and improved long-term survival compared to partial resection (66–69). Most deaths in prospective studies are due to local recurrence, which is often difficult to control.
Brainstem-invasive tumors present particular surgical challenges, and outcomes are poorer with incomplete resection. As a result, many centers use preradiation chemotherapy and consider second-look surgery when residual tumor appears safely resectable.
6.2 Complications
Common complications following posterior fossa ependymoma resection include:
6.2.1 Cerebellar ataxia
Ataxia may emerge or worsen postoperatively. Injury to the lateral cerebellar hemispheres causes limb dysmetria, while midline involvement leads to gait ataxia.
6.2.2 Lower cranial nerve injury
Tumors in the cerebellopontine angle can damage lower cranial nerves, resulting in hemifacial weakness, dysarthria, dysphagia, or hearing loss. Infants may experience functional recovery after resection.
6.2.3 Posterior fossa syndrome
Cerebellar mutism, or posterior fossa syndrome, is a well-recognized complication, particularly when the superior or middle cerebellar peduncles are involved (70–73).
7 Postoperative therapy
7.1 Intracranial ependymoma, grade 2 or 3
Ependymomas are infiltrative primary brain tumors capable of CNS dissemination. Management typically includes maximal safe resection, followed by radiotherapy and, in some cases, chemotherapy—tailored to patient age, tumor location, and extent of resection (algorithm 1) (39, 74, 75). At present, treatment is not stratified by molecular subgroup.
Given the complexity of care, referral to specialized centers is strongly advised, particularly for infants with subtotal resections or patients with positive cerebrospinal fluid cytology. A treatment algorithm based on age, resection extent, and craniospinal dissemination is presented in Figure 1.
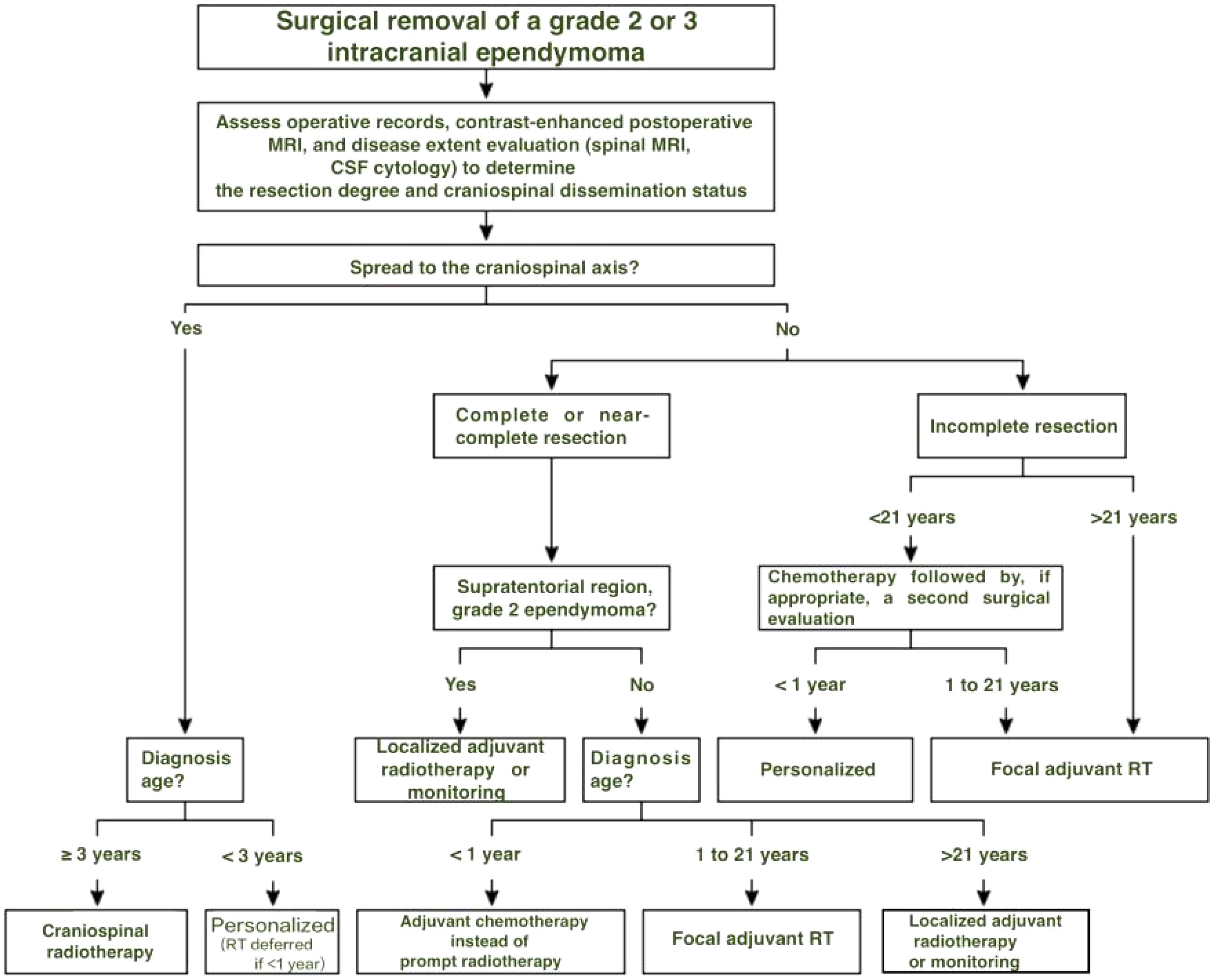
Figure 1. Treatment decision algorithm following surgical removal of grade 2 or 3 intracranial ependymoma. In children under 1 year, radiotherapy is generally deferred due to neurodevelopmental risks. Chemotherapy is used to delay irradiation whenever feasible, including in cases with craniospinal dissemination.
7.1.1 Gross total resection, age 1 to 21 years
Postoperative management of children older than one year with gross or near-total resection of intracranial grade 2 or 3 ependymomas continues to evolve. Radiotherapy alone remains standard in most cases (74–77). Preliminary data from the COG ACNS0831 trial suggest a possible benefit of maintenance chemotherapy after RT in selected patients, though final results are pending (78).
An exception includes children with supratentorial grade 2 tumors who achieve gross total resection; these patients may have favorable outcomes with observation alone. This approach was evaluated in one arm of the ACNS0831 trial, with outcomes not yet reported.
7.1.1.1 Adjuvant focal RT
In children over 1 year of age with grade 2 or 3 ependymomas, gross or near-total resection is typically followed by conformal focal radiotherapy (RT). Although prophylactic cranial irradiation was once standard, focal RT is now preferred, as most recurrences are local and PCI has not shown a survival benefit (5, 6, 79, 80). Broader radiation fields are reserved for patients with confirmed dissemination by imaging or CSF cytology. Limiting irradiated brain volume improves quality of life by reducing the risks of neurocognitive decline, stroke, and secondary malignancies.
Conformal RT targets the tumor bed with a margin while sparing healthy brain tissue. Standard doses range from 54 to 59.4 Gy; observational studies show no added benefit with higher doses after gross total resection (81), though escalation may be warranted for residual disease. Optimal margin size is under investigation, with trials such as ACNS0121 and ACNS0831 evaluating 1.0 cm and 0.5 cm clinical target volumes, respectively.
In the COG ACNS0121 phase II trial, 281 children with gross or near-total resection of grade 2 or 3 ependymomas (excluding completely resected supratentorial grade 2 tumors) received postoperative focal RT. At a median follow-up of 7.9 years, five-year event-free and overall survival rates were 69% and 86%, respectively. For patients with near-total or macroscopic resection (stratum 3), EFS and OS were 67% and 83%; for those with microscopic gross total resection (stratum 4), rates were 70% and 88% (11).
7.1.1.2 Post-RT chemotherapy
Post-radiotherapy (RT) chemotherapy for ependymoma remains investigational pending mature data from the completed COG ACNS0831 trial. This phase III trial randomized children aged 1–21 years with newly diagnosed grade 2 or 3 ependymomas after gross-total or near-total resection to focal RT alone or RT with maintenance chemotherapy (vincristine, cisplatin, cyclophosphamide, and etoposide) (78). Preliminary results from 325 patients (median age, 4.9 years) at a median follow-up of 3.5 years demonstrated a trend towards improved event-free survival (EFS) with RT plus chemotherapy (78% vs. 72%; hazard ratio [HR], 0.73; 90% CI, 0.51–1.06). This benefit was statistically significant in a subgroup with complete or near-complete resections (81% vs. 71%, P = 0.03). Overall survival (OS) data remain unavailable.
Adherence to chemotherapy was suboptimal; 27% of the chemotherapy-assigned patients received RT alone, mostly due to refusal. An as-treated analysis, excluding patients who did not receive chemotherapy and some in the RT-alone arm who missed RT, showed improved EFS with chemotherapy (80% vs. 71%; HR, 0.58; 95% CI, 0.36–0.94), though this result is prone to bias. Final results of ACNS0831, including molecular subgroup outcomes, are pending. The ongoing SIOP-EP-II trial (NCT02265770), comparing adjuvant chemotherapy to observation post-resection and RT, will provide further clarity. Currently, adjuvant chemotherapy after RT is not recommended due to insufficient evidence of benefit.
7.1.2 Subtotal resection, age 1 to 21 years
Incompletely resected grade 2 or 3 ependymomas are associated with inferior progression-free and overall survival compared to gross total resection. For these high-risk patients, current practice includes a short postoperative chemotherapy course, followed by second-look surgery when feasible, and then conformal radiotherapy (RT) (5, 12, 82). Active agents include cisplatin, carboplatin, cyclophosphamide, and etoposide, with greater efficacy seen in multidrug regimens (56, 77, 83–88). One to four cycles of chemotherapy are typically administered postoperatively, followed by MRI to reassess resectability. Patients with safely removable residual disease undergo second debulking prior to focal RT.
RT dosing mirrors that used in completely resected tumors, though higher doses may be appropriate for residual macroscopic disease. This multimodal strategy is supported by both single- and multicenter studies (5, 11, 77, 82, 89). The most robust prospective data come from stratum 2 of the COG ACNS0121 trial, which enrolled 64 patients with incomplete resection. All received chemotherapy, second-look surgery when feasible (achieved in 39%), and focal RT. At a median follow-up of 7.9 years, event-free and overall survival were 37% and 70%, respectively (11). Chemotherapy consisted of two cycles of carboplatin, cyclophosphamide, etoposide, and vincristine over seven weeks.
Post-RT chemotherapy appears to confer no additional benefit. In the ACNS0831 trial, patients who achieved complete response through induction chemotherapy or second-look surgery had similar outcomes whether treated with RT alone or RT plus maintenance chemotherapy (78). No benefit was observed in those with residual disease post-induction.
7.1.3 Children <1 year of age
Although adjuvant radiotherapy (RT) is the standard of care for most ependymoma patients, its use in infants is limited due to concerns about developmental toxicity (42). For children under 1 year, adjuvant chemotherapy is recommended to delay RT. Those with residual tumor after chemotherapy may be considered for second-look surgery and should ideally be managed at tertiary care centers. In children over 1 year, the benefits of focal RT generally outweigh the risks. Chemotherapy in lieu of RT for patients aged 1 to 3 should be restricted to clinical protocols.
No randomized trials directly compare immediate postoperative RT with chemotherapy followed by deferred RT. However, several cooperative group studies have explored this approach in young children:
● In a cohort of 41 children under age 3 (one with disseminated disease), multiagent chemotherapy followed resection. Regimens included vincristine, methotrexate, and cyclophosphamide alternating with cisplatin and etoposide, or a shorter vincristine–etoposide–cyclophosphamide protocol (90). Twenty-nine experienced local progression (median, 9 months). Of 13 survivors, six avoided RT. Five-year progression-free survival (PFS), event-free survival (EFS), and overall survival (OS) were 27%, 26%, and 37%, respectively, with no significant cognitive differences between those who received or avoided RT.
● In a study of 73 children under 5 without dissemination, seven chemotherapy cycles followed maximal resection (84). At a median follow-up of 5 years, four-year OS was 59%, and five-year EFS was 22%. RT was required for recurrence in 49% at a median of 15 months.
● Among 89 children aged 3 years or younger treated with resection and chemotherapy, 80 had nondisseminated disease; 50 relapsed (91). At 6-year median follow-up, five-year EFS and OS were 42% and 63%, respectively, and RT was avoided in 42%.
High-dose chemotherapy with autologous stem cell rescue remains investigational. In one pilot study, five children under age 3 with anaplastic ependymoma (most with residual or disseminated disease) were treated with this approach. All reached age 3 without RT; only one experienced progression at a median follow-up of 45 months (92).
The long-term neurocognitive consequences of various treatment strategies in infants remain poorly characterized and represent a critical area for future research.
7.1.4 Patients with disseminated disease
Leptomeningeal dissemination or spinal seeding from intracranial ependymoma portends a poor prognosis, though patients with ZFTA fusion–positive tumors may experience prolonged survival (53). Management should be individualized. Given the limited sensitivity of cerebrospinal fluid cytology in ependymoma, repeat sampling 10 to 14 days after surgery is recommended to confirm initial findings. Referral to a tertiary center is advisable when feasible.
Postoperative craniospinal irradiation (CSI) is generally indicated but carries greater developmental risk than focal radiotherapy. In children younger than three years, CSI is typically deferred in favor of chemotherapy, with or without focal radiation.
7.1.5 Areas of uncertainty and ongoing trials
•Observation after gross total resection of supratentorial grade 2 tumors.
Retrospective data suggest that adjuvant radiotherapy (RT) may be omitted in selected patients with completely resected supratentorial grade 2 ependymomas, though this approach remains controversial (69, 93–95). As most recurrences are local, deferring RT aims to avoid early toxicities, with salvage surgery and RT reserved for recurrence. In the COG ACNS0121 phase II trial, 11 pediatric patients managed with observation alone had a five-year progression-free survival (PFS) of 61%, and all were alive at five years (11). However, the small sample size limited conclusions. While North American trials such as ACNS0831 have guided much of current practice, the ongoing European SIOP-Ependymoma II trial is expected to provide critical data on the role of post-radiotherapy chemotherapy and observation in stratified patient groups.
•Chemotherapy alone in children over 1 year.
The role of primary chemotherapy in children over 1 year remains undefined. As modern conformal radiation techniques reduce long-term toxicities, replacing RT with chemotherapy should be limited to clinical trials.
7.1.6 Adults, age >21 years
Intracranial ependymoma is rare in adults, and no randomized trials guide management. Treatment relies on retrospective studies and extrapolation from pediatric protocols. As in children, the extent of initial resection is the strongest predictor of outcome, and maximal safe resection is recommended for all patients (95–98). Postoperative focal radiotherapy (RT) is indicated for localized grade 3 (anaplastic) tumors and for residual disease after resection of grade 2 tumors (74, 75, 99, 100). Craniospinal RT is reserved for disseminated disease confirmed by imaging or CSF cytology (99). Adjuvant chemotherapy offers no proven benefit in adults.
The benefit of adjuvant focal RT after gross total resection of grade 2 ependymomas remains uncertain. Current guidelines from the National Comprehensive Cancer Network (NCCN) and the European Association for Neuro-Oncology (EANO) recommend observation, supported by retrospective data showing no survival advantage with RT in this setting (75, 99, 101, 102). Nonetheless, some advocate postoperative RT even after complete resection—particularly when microscopic residual disease is suspected—citing its tolerability, the limited efficacy of salvage options, and the risks of recurrence, especially in the posterior fossa.
7.2 Other ependymal tumors
Subependymomas are often incidental findings in older adults and typically require no treatment unless symptomatic or enlarging. When resection is indicated, complete removal of large, symptomatic tumors is usually curative, underscoring their more indolent course relative to other ependymomas (103). Radiotherapy is generally reserved for unresectable or progressive lesions.
8 Follow-up and monitoring
Although most recurrences in pediatric ependymoma occur within five years of diagnosis, late progression can occur, supporting surveillance beyond this window (104).
8.1 Surveillance
Imaging practices vary, but a strategy consistent with the Children’s Oncology Group (COG) ACNS0831 trial is commonly adopted:
Brain MRI: Every 3–4 months for the first 3 years post-treatment, every 6 months from years 3 to 5, and annually for an additional 2–5 years thereafter in survivorship care.
Spine MRI: Performed annually with brain MRI, if symptoms suggest spinal involvement, or if brain imaging shows progression.
The RAPNO group recommends more frequent spine imaging (e.g., every other brain MRI), and combined brain/spine studies at each interval for patients with metastatic disease or 1q gain (64), though these guidelines remain unvalidated and primarily research-focused.
8.2 Survivorship
Long-term survivors of childhood CNS tumors are at risk for neurocognitive deficits, focal neurologic impairments, hearing loss, endocrine and growth disturbances, radiation necrosis, vasculopathy, and second malignancies (42, 105–112). Morbidity in adult survivors often includes fatigue, pain, numbness, and sleep disruption (113). The COG offers long-term follow-up guidelines for these patients (114).
9 Recurrent disease
The long-term prognosis for patients with recurrent ependymoma is poor, with most succumbing within years of relapse despite extended palliation. First recurrence is local in approximately two-thirds of cases, metastatic (within the CNS) in about 20%, or combined in 10–15% (115, 116). Although various treatment options exist, patients and caregivers should be informed of the dismal long-term outlook following recurrence or progression after surgery and radiotherapy (RT). Thoughtful treatment selection can optimize palliation and quality of life, though no single approach is standard or proven effective (117).
Outside clinical trials, management should be individualized, considering age, original and recurrent disease location, metastasis, prior therapy, and functional status. Beyond experimental treatments, several strategies have been employed:
9.1 Surgery
Aggressive resection may offer effective palliation in select cases. Chemotherapy, with or without RT, may reduce residual or recurrent tumor to facilitate reresection, aiming to delay progression and death (83, 86, 118).
9.2 Radiation
Reirradiation of recurrent ependymomas may improve outcomes, achieving salvage in select cases (117, 119–123). Options include stereotactic radiosurgery (SRS), focal fractionated reirradiation, or craniospinal irradiation (CSI), tailored to recurrence location, patient age, and extent (119, 124). Focal reirradiation carries a risk of disseminated metastases, with local recurrence eventually affecting most patients. In a large retrospective series of 101 patients undergoing aggressive repeat resection and reirradiation (median 27 months after initial RT), median progression-free survival (PFS) and overall survival (OS) from the start of second RT were 27 and 75 months, respectively (123). Tumor progression occurred in 57 patients (56%) after focal RT or CSI with boost, with local failure contributing in 35 (61%). Outcomes were best in patients with distant-only failure post-initial RT, no anaplasia at recurrence, and subsequent CSI with boost reirradiation. The completed phase II RERTEP trial (NCT02125786) investigated this approach, with results pending.
9.3 Chemotherapy
Conventional chemotherapy may provide symptomatic relief in recurrent ependymoma, though no standard regimen exists for children (99). Agents with the most activity—based on adjuvant or neoadjuvant data—include cisplatin, carboplatin, cyclophosphamide, and etoposide. In a series of 28 adults with recurrent or progressive intracranial disease, objective responses were observed in six patients, with better outcomes in those receiving platinum-based regimens (125). Modest activity has also been reported with oral etoposide, nitrosoureas, temozolomide, and fluorouracil (99, 126–131). However, responses are generally short-lived, with rapid disease progression following chemotherapy alone.
9.4 Molecular targets
Emerging molecular targets in ependymoma include EGFR (18), VEGF, and various epigenetic and metabolic regulators (132–136). In adults, temozolomide combined with lapatinib (a HER2/EGFR inhibitor) has shown activity in recurrent disease. A phase II trial involving 50 adults with recurrent intracranial or spinal ependymoma reported a median progression-free survival (PFS) of 7.8 months and overall survival (OS) of 2.25 years, with two complete and six partial responses; many experienced symptom stabilization or improvement (137). In a series of eight adults, six had partial responses to bevacizumab, with a median time to progression of six months (138), although other retrospective studies suggest limited efficacy (139).
In children, treatment remains investigational. A trial of bevacizumab and irinotecan in 13 children with recurrent or progressive ependymoma yielded no objective responses (median time to progression, 2.2 months) (140). Similarly, a phase II study of bevacizumab plus lapatinib in 24 children showed no benefit (141).
10 Prognosis
10.1 Children
Despite surgery and adjuvant therapy, pediatric intracranial ependymomas carry a poor long-term prognosis, with 10-year overall survival (OS) ranging from 50% to 75% (11, 77, 104, 142). Several factors influence disease-free survival following treatment (143):
10.1.1 Extent of resection
Local recurrence accounts for approximately 80% of failures, underscoring the importance of maximal safe resection (66, 100, 144–146). Gross total resection is consistently associated with better outcomes, though benefit varies (5, 66, 100, 144). Still, up to 40% of children relapse or die within 10 years (104).
10.1.2 Tumor location
Data on survival by location are mixed. Some studies report better outcomes for supratentorial tumors (5, 142, 147); others favor infratentorial lesions (100). Cervical spinal involvement is linked to poorer survival due to increased risk of distant relapse (148).
10.1.3 Age
Older children fare better than infants, who often present with infratentorial PFA tumors and face delayed radiotherapy due to concerns over developmental toxicity (146).
10.1.4 Histologic grade
The prognostic significance of histologic grade remains debated. Some studies associate grade 3 tumors with worse outcomes (11, 142, 149), but grading inconsistencies limit definitive conclusions (12).
10.1.5 Molecular genetic groups
Molecular subgrouping may surpass histology in prognostic precision, with emerging classifications refining risk stratification.
10.1.6 Additional molecular alterations
Chromosome 1q gain is associated with adverse outcomes in PFA tumors, confirmed by retrospective and prospective studies (15, 18, 24, 28, 44). Loss of 6q, especially when co-occurring with 1q gain, may define an ultra–high-risk subgroup (44, 46). In ZFTA fusion–positive tumors, CDKN2A deletion may also predict poor prognosis, though prospective validation is needed (45, 55, 56).
10.2 Adults
Intracranial ependymoma is uncommon in adults, and outcomes data remain limited. Two large European series (222 patients total) and one from a U.S. center (123 patients) reported 5-year survival rates of 67% to 85% and 10-year survival of 50% to 77% (96, 97, 150). Multivariate analyses associated poorer outcomes with high-grade histology, subtotal resection (151), tumor location (150, 152), and a Karnofsky Performance Status of 80 or lower (97). The detrimental effect of incomplete resection was corroborated in a population-based SEER study including both adults and children (68). A recent 2025 comprehensive review emphasizes the growing importance of molecularly guided management in adults, suggesting improved prognostic accuracy with molecular subgrouping, despite continued challenges due to disease heterogeneity and the rarity of cases in adults (153).
11 Summary of enhanced WHO classification of ependymomas
To consolidate the clinical, molecular, and prognostic features of ependymomas as classified under the enhanced WHO framework, Table 1 provides a comprehensive summary of key subtypes across supratentorial and infratentorial regions, integrating data discussed throughout this review.
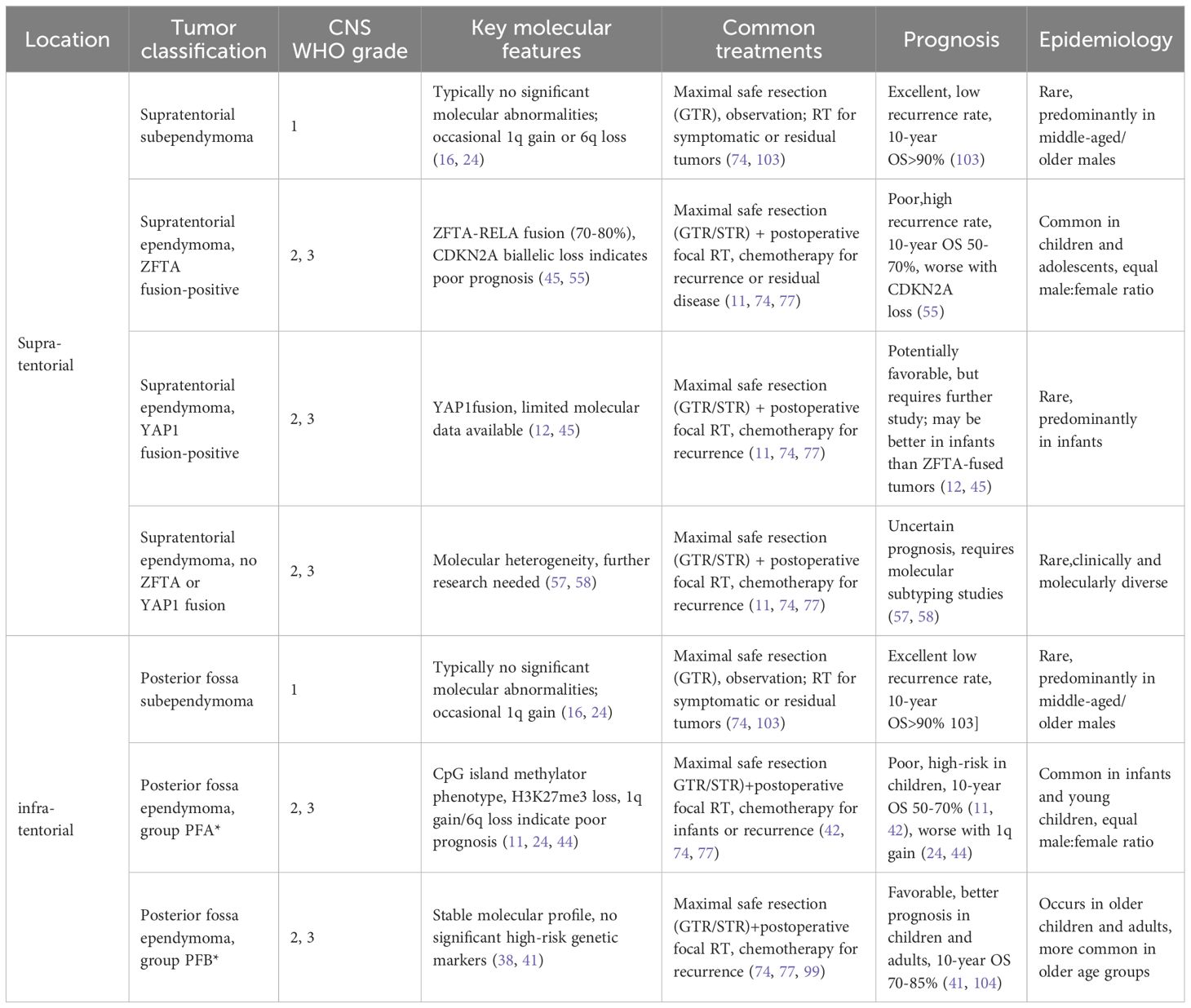
Table 1. Enhanced WHO classification of ependymomas: clinical and molecular features.
12 Conclusion
Intracranial ependymomas predominantly arise in early childhood (median age: 5 years), while spinal ependymomas are more frequently observed in adults. The spectrum includes subependymoma (WHO grade 1), myxopapillary ependymoma (WHO grade 2), and classic ependymoma localized to the supratentorial, posterior fossa, or spinal compartments. The traditional distinction between grade 2 and grade 3 tumors is increasingly being superseded by molecular stratification. Clinical symptoms vary by tumor location: posterior fossa lesions commonly present with headache, nausea, ataxia, and papilledema secondary to hydrocephalus.
Diagnosis requires histopathological confirmation and is guided by imaging features, tumor location, and patient age. Staging with spinal MRI and cerebrospinal fluid analysis is essential, as dissemination is present in approximately 10% of cases at diagnosis.
Maximal safe surgical resection remains the cornerstone of initial therapy, with gross total resection as the goal; however, proximity to eloquent structures such as the brainstem may preclude complete excision, necessitating referral to specialized pediatric neurosurgical centers. Adjuvant focal radiotherapy (RT) is the standard of care for children aged 1–21 years with WHO grade 2 or 3 ependymomas following gross or near-total resection, except in two key scenarios: children aged 1–3 years, where chemotherapy is administered within clinical protocols to defer RT, and patients with supratentorial grade 2 tumors, for whom observation may be appropriate. In cases of subtotal resection, management typically includes chemotherapy, second-look surgery (when feasible), and RT. Infants under 1 year are treated with chemotherapy to delay RT, while patients with disseminated disease require craniospinal irradiation or systemic chemotherapy. In adults, focal RT is commonly employed for grade 3 tumors or incompletely resected grade 2 lesions.
Surveillance should continue for 7–10 years post-treatment. Despite multimodal management, the 10-year overall survival rate for pediatric patients remains between 50% and 70%.
Author contributions
JB: Writing – original draft, Visualization, Data curation, Resources, Formal analysis, Conceptualization, Project administration, Writing – review & editing, Methodology, Investigation, Validation. ZP: Writing – original draft, Formal analysis, Project administration, Visualization, Data curation, Software, Methodology, Investigation, Conceptualization, Writing – review & editing, Resources, Validation. SW: Visualization, Project administration, Resources, Data curation, Formal analysis, Validation, Writing – original draft, Supervision, Conceptualization, Writing – review & editing, Methodology, Investigation, Software.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Shuangshoti S, Rushing EJ, Mena H, Olsen C, and Sandberg GD. Supratentorial extraventricular ependymal neoplasms: a clinicopathologic study of 32 patients. Cance. (2005) 103:2598. doi: 10.1002/cncr.v103:12
2. Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cel. (2005) 8:323. doi: 10.1016/j.ccr.2005.09.001
3. WHO Classification of Tumours Editorial Board. Central Nervous System Tumours. Int Agency Res Cance. (2021).
4. Louis DN, Ohgaki H, Wiestler OD, and Cavenee WK. WHO Classification of Tumours of the Central Nervous System. 4th ed. New York, NY, USA: International Agency for Research on Cancer (2016).
5. Massimino M, Miceli R, Giangaspero F, Boschetti L, Modena P, Antonelli M, et al. Final results of the second prospective AIEOP protocol for pediatric intracranial ependymoma. Neuro Onco. (2016) 18:1451. doi: 10.1093/neuonc/now108
6. Merchant TE, Li C, Xiong X, Kun LE, Boop FA, and Sanford RA. Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study. Lancet Onco. (2009) 10:258. doi: 10.1016/S1470-2045(08)70342-5
7. Chamberlain MC. Ependymomas. Curr Neurol Neurosci Re. (2003) 3:193. doi: 10.1007/s11910-003-0078-x
8. Price M, Neff C, Nagarajan N, Kruchko C, Waite KA, Cioffi G, et al. CBTRUS statistical report: american brain tumor association & NCI neuro-oncology branch adolescent and young adult primary brain and other central nervous system tumors diagnosed in the United States in 2016-2020. Neuro Onco. (2024) 26:iii1. doi: 10.1093/neuonc/noae047
9. Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2016-2020. Neuro Onco. (2023) 25:iv1. doi: 10.1093/neuonc/noad149
10. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Onco. (2021) 23:1231. doi: 10.1093/neuonc/noab106
11. Merchant TE, Bendel AE, Sabin ND, Burger PC, Shaw DW, Chang E, et al. Conformal radiation therapy for pediatric ependymoma, chemotherapy for incompletely resected ependymoma, and observation for completely resected, supratentorial ependymoma. J Clin Onco. (2019) 37:974. doi: 10.1200/JCO.18.01765
12. Upadhyaya SA, Robinson GW, Onar-Thomas A, Orr BA, Billups CA, Bowers DC, et al. Molecular grouping and outcomes of young children with newly diagnosedependymoma treated on the multi- institutional SJYC07 trial. Neuro Onco. (2019) 21:1319. doi: 10.1093/neuonc/noz069
13. Ellison DW, Kocak M, Figarella-Branger D, Felice G, Catherine G, Pietsch T, et al. Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J Negat Results Biome. (2011) 10:7. doi: 10.1186/1477-5751-10-7
14. Milde T, Hielscher T, Witt H, Kool M, Mack SC, Deubzer HE, et al. Nestin expression identifies ependymoma patients with poor outcome. Brain Patho. (2012) 22:848. doi: 10.1111/j.1750-3639.2012.00600.x
15. Godfraind C, Kaczmarska JM, Kocak M, Dalton J, Wright KD, Sanford RA, et al. Distinct disease-risk groups in pediatric supratentorial and posterior fossa ependymomas. Acta Neuropathol. (2012) 124:247. doi: 10.1007/s00401-012-0981-9
16. Carter M, Nicholson J, Ross F, Crolla J, Allibone R, Balaji V, et al. Genetic abnormalities detected in ependymomas by comparative genomic hybridisation. Br J Cancer. (2002) 86:929. doi: 10.1038/sj.bjc.6600180
17. Dyer S, Prebble E, Davison V, Davies P, Ramani P, Ellison D, et al. Genomic imbalances in pediatric intracranial ependymomas define clinically relevant groups. Am J Patho. (2002) 161:2133. doi: 10.1016/S0002-9440(10)64491-4
18. Mendrzyk F, Korshunov A, Benner A, Toedt G, Pfister S, Radlwimmer B, et al. Identification of gains on 1q and epidermal growth factor receptor overexpression as independent prognostic markers in intracranial ependymoma. Clin Cancer Re. (2006) 12:2070. doi: 10.1158/1078-0432.CCR-05-2363
19. Barton VN, Donson AM, Kleinschmidt-DeMasters BK, Birks DK, Handler MH, and Foreman NK. Unique molecular characteristics of pediatric myxopapillary ependymoma. Brain Patho. (2010) 20:560. doi: 10.1111/j.1750-3639.2009.00333.x
20. Akyurek S, Chang EL, Yu TK, et al. Spinal myxopapillary ependymoma outcomes in patients treated with surgery and radiotherapy at M. D. Anderson Cancer Center. J Neuroonco. (2006) 80:177. doi: 10.1007/s11060-006-9169-2
21. Merchant TE, Kiehna EN, Thompson SJ, Heideman R, Sanford RA, and Kun LE. Pediatric low-grade and ependymal spinal cord tumors. Pediatr Neurosur. (2000) 32:30. doi: 10.1159/000028894
22. Agbahiwe HC, Wharam M, Batra S, Cohen K, and Terezakis SA. Management of pediatric myxopapillary ependymoma: the role of adjuvant radiation. Int J Radiat Oncol Biol Phy. (2013) 85:421. doi: 10.1016/j.ijrobp.2012.05.001
23. Al-Halabi H, Montes JL, Atkinson J, Farmer JP, and Freeman CR. Adjuvant radiotherapy in the treatment of pediatric myxopapillary ependymomas. Pediatr Blood Cancer. (2010) 55:639. doi: 10.1002/pbc.22614
24. Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cel. (2015) 27:728. doi: 10.1016/j.ccell.2015.04.002
25. Rousseau A, Idbaih A, Ducray F, Crinière E, Fèvre-Montange M, Jouvet A, et al. Specific chromosomal imbalances as detected by array CGH in ependymomas in association with tumor location, histological subtype and grade. J Neurooncol. (2010) 97:353. doi: 10.1007/s11060-009-0039-6
26. Suarez-Merino B, Hubank M, Revesz T, Harkness W, Hayward R, Thompson D, et al. Microarray analysis of pediatric ependymoma identifies a cluster of 112 candidate genes including four transcripts at 22q12. 1-q13.3. Neuro Onco. (2005) 7:20. doi: 10.1215/S1152851704000596
27. Huang B, Starostik P, Schraut H, Krauss J, Sörensen N, and Roggendorf. W. Human ependymomas reveal frequent deletions on chromosomes 6 and 9. Acta Neuropatho. (2003) 106:357. doi: 10.1007/s00401-003-0739-5
28. Korshunov A, Witt H, Hielscher T, Benner A, Remke M, Ryzhova M, et al. Molecular staging of intracranial ependymoma in children and adults. J Clin Onco. (2010) 28:3182. doi: 10.1200/JCO.2009.27.3359
29. Wani K, Armstrong TS, Vera-Bolanos E, Raghunathan A, Ellison D, Gilbertson R, et al. A prognostic gene expression signature in infratentorial ependymoma. Acta Neuropathol. (2012) 123:727. doi: 10.1007/s00401-012-0941-4
30. Yang I, Nagasawa DT, Kim W, Spasic M, Trang A, Lu DC, et al. Chromosomal anomalies and prognostic markers for intracranial and spinal ependymomas. J Clin Neurosc. (2012) 19:779. doi: 10.1016/j.jocn.2011.11.004
31. Panwalkar P, Clark J, Ramaswamy V, Hawes D, Yang F, Dunham C, et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. (2017) 134:705. doi: 10.1007/s00401-017-1752-4
32. Bayliss J, Mukherjee P, Lu C, Jain SU, Chung C, Martinez D, et al. Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci Transl Me. (2016) 8:366ra161. doi: 10.1126/scitranslmed.aah6904
33. Mack SC and Taylor MD. Put away your microscopes: the ependymoma molecular era has begun. Curr Opin Onco. (2017) 29:443. doi: 10.1097/CCO.0000000000000411
34. Pajtler KW, Wen J, Sill M, Lin T, Orisme W, Tang B, et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. (2018) 136:211. doi: 10.1007/s00401-018-1877-0
35. Cavalli FMG, Hübner JM, Sharma T, Luu B, Sill M, Zapotocky M, et al. Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol. (2018) 136:227. doi: 10.1007/s00401-018-1888-x
36. Mack SC, Pajtler KW, Chavez L, Okonechnikov K, Bertrand KC, Wang X, et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Natur. (2018) 553:101. doi: 10.1038/nature25169
37. Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Natur. (2010) 466:632. doi: 10.1038/nature09173
38. Witt H, Mack SC, Ryzhova M, Bender S, Sill M, Isserlin R, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cel. (2011) 20:143. doi: 10.1016/j.ccr.2011.07.007
39. Pajtler KW, Mack SC, Ramaswamy V, Smith CA, Witt H, Smith A, et al. The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol. (2017) 133:5. doi: 10.1007/s00401-016-1643-0
40. Ellison DW, Aldape KD, Capper D, Fouladi M, Gilbert MR, Gilbertson RJ, et al. cIMPACT-NOW update 7: advancing the molecular classification of ependymal tumors. Brain Patho. (2020) 30:863. doi: 10.1111/bpa.12866
41. Ramaswamy V, Hielscher T, Mack SC, Lassaletta A, Lin T, Pajtler KW, et al. Therapeutic impact of cytoreductive surgery and irradiation of posterior fossa ependymoma in the molecular era: A retrospective multicohort analysis. J Clin Onco. (2016) 34:2468. doi: 10.1200/JCO.2015.65.7825
42. Zapotocky M, Beera K, Adamski J, Laperierre N, Guger S, Janzen L, et al. Survival and functional outcomes of molecularly defined childhood posterior fossa ependymoma: Cure at a cost. Cance. (2019) 125:1867. doi: 10.1002/cncr.31995
43. Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stütz AM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Natur. (2014) 506:445. doi: 10.1038/nature13108
44. Baroni LV, Sundaresan L, Heled A, Coltin H, Pajtler KW, Lin T, et al. Ultra high-risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro Onco. (2021) 23:1360. doi: 10.1093/neuonc/noab034
45. Pohl LC, Leitheiser M, Obrecht D, Mynarek M, Rutkowski S, Pajtler KW, et al. Molecular characteristics and improved survival prediction in a cohort of 2023 ependymomas. Acta Neuropathol. (2024) 147:24. doi: 10.1007/s00401-023-02674-x
46. Donson AM, Bertrand KC, Riemondy KA, Gao D, Zhuang Y, Sanford B, et al. Significant increase of high-risk chromosome 1q gain and 6q loss at recurrence in posterior fossa group A ependymoma: A multicenter study. Neuro Onco. (2023) 25:1854. doi: 10.1093/neuonc/noad096
47. Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Scienc. (2013) 340:857. doi: 10.1126/science.1232245
48. Hübner JM, Müller T, Papageorgiou DN, Mauermann M, Krijgsman O, Kouwenhoven MCM, et al. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Onco. (2019) 21:878. doi: 10.1093/neuonc/noz058
49. Jain SU, Do TJ, Lund PJ, Rashoff AQ, Diehl KL, Cieslik M, et al. PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat Commu. (2019) 10:2146. doi: 10.1038/s41467-019-09981-6
50. Chapman RJ, Ghasemi DR, Andreiuolo F, Zschernack V, Mynarek M, Hasselblatt M, et al. Optimizing biomarkers for accurate ependymoma diagnosis, prognostication, and stratification within International Clinical Trials: A BIOMECA study. Neuro Onco. (2023) 25:1871. doi: 10.1093/neuonc/noad055
51. Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y, et al. C11orf95-RELA fusions drive oncogenic NF-kB signalling in ependymoma. Natur. (2014) 506:451. doi: 10.1038/nature13109
52. Pietsch T, Wohlers I, Goschzik T, Dreschmann V, Denkhaus D, Dörner E, et al. Supratentorial ependymomas of childhood carry C11orf95-RELA fusions leading to pathological activation of the NF-kB signaling pathway. Acta Neuropathol. (2014) 127:609. doi: 10.1007/s00401-014-1264-4
53. Benesch M, Mynarek M, Witt H, Warmuth-Metz M, Pietsch T, Bison B, et al. Newly diagnosed metastatic intracranial ependymoma in children: frequency, molecular characteristics, treatment, and outcome in the prospective HIT series. Oncologis. (2019) 24:e921. doi: 10.1634/theoncologist.2018-0489
54. Ng CH, Obrecht D, Wells O, Davies BR, Warren M, De S, et al. A multi-institutional retrospective pooled outcome analysis of molecularly annotated pediatric supratentorial ZFTA-fused ependymoma. Neurooncol Ad. (2023) 5:vdad057. doi: 10.1093/noajnl/vdad057
55. Jünger ST, Andreiuolo F, Mynarek M, Dörner E, Zur Mühlen A, Rutkowski S, et al. CDKN2A deletion in supratentorial ependymoma with RELA alteration indicates a dismal prognosis: a retrospective analysis of the HIT ependymoma trial cohort. Acta Neuropathol. (2020) 140:405. doi: 10.1007/s00401-020-02169-z
56. Massimino M, Barretta F, Modena P, Witt H, Minasi S, Pfister SM, et al. Second series by the Italian Association of Pediatric Hematology and Oncology of children and adolescents with intracranial ependymoma: an integrated molecular and clinical characterization with a long-term follow-up. Neuro Onco. (2021) 23:848. doi: 10.1093/neuonc/noaa257
57. Zschernack V, Jünger ST, Mynarek M, Rutkowski S, Garre ML, Ebinger M, et al. Supratentorial ependymoma in childhood: more than just RELA or YAP. Acta Neuropathol. (2021) 141:455. doi: 10.1007/s00401-020-02260-5
58. Fukuoka K, Kanemura Y, Shofuda T, Fukushima S, Yamashita S, Narushima D, et al. Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol Commu. (2018) 6:134. doi: 10.1186/s40478-018-0630-1
59. Fangusaro J, Van Den Berghe C, Tomita T, Rajaram V, Habiby R, Heller G, et al. Evaluating the incidence and utility of microscopic metastatic dissemination as diagnosed by lumbar cerebro-spinal fluid (CSF) samples in children with newly diagnosed intracranial ependymoma. J Neurooncol. (2011) 103:693. doi: 10.1007/s11060-010-0448-6
60. McGuire CS, Sainani KL, and Fisher PG. Incidence patterns for ependymoma: a surveillance, epidemiology, and end results study. J Neurosur. (2009) 110:725. doi: 10.3171/2008.9.JNS08117
61. Pagès M, Pajtler KW, Puget S, Castel D, Boddaert N, Tauziède-Espariat A, et al. Diagnostics of pediatric supratentorial RELA ependymomas: integration of information from histopathology, genetics, DNA methylation and imaging. Brain Patho. (2019) 29:325. doi: 10.1111/bpa.12664
62. Nowak J, Jünger ST, Huflage H, Seidel C, Hohm A, Vandergrift LA, et al. MRI phenotype of RELA-fused pediatric supratentorial ependymoma. Clin Neuroradio. (2019) 29:595. doi: 10.1007/s00062-018-0704-2
63. Maiuri F, Gangemi M, Iaconetta G, Signorelli F, and Del Basso De Caro M. Symptomatic subependymomas of the lateral ventricles. Rep eight cases. Clin Neurol Neurosur. (1997) 99:17. doi: 10.1016/S0303-8467(96)00554-9
64. Lindsay HB, Massimino M, Avula S, Davies R, Hargrave D, Grundy RG, et al. Response assessment in paediatric intracranial ependymoma: recommendations from the Response Assessment in Pediatric Neuro-Oncology (RAPNO) working group. Lancet Onco. (2022) 23:e393. doi: 10.1016/S1470-2045(22)00222-4
65. Moreno L, Pollack IF, Duffner PK, Geyer JR, Grill J, Massimino M, et al. Utility of cerebrospinal fluid cytology in newly diagnosed childhood ependymoma. J Pediatr Hematol Onco. (2010) 32:515. doi: 10.1097/MPH.0b013e3181d7adf5
66. Timmermann B, Kortmann RD, Kühl J, Meisner C, Dieckmann K, Pietsch T, et al. Combined postoperative irradiation and chemotherapy for anaplastic ependymomas in childhood: results of the German prospective trials HIT 88/89 and HIT 91. Int J Radiat Oncol Biol Phy. (2000) 46:287. doi: 10.1016/S0360-3016(99)00414-9
67. Rodriguez D, Cheung MC, Housri N, Quinones-Hinojosa A, Camphausen K, Koniaris LG, et al. Outcomes of malignant CNS ependymomas: an examination of 2408 cases through the Surveillance, Epidemiology, and End Results (SEER) database (1973-2005). J Surg Re. (2009) 156:340. doi: 10.1016/j.jss.2009.04.024
68. Amirian ES, Armstrong TS, Aldape KD, Gilbert MR, Scheurer ME, Bondy ML, et al. Predictors of survival among pediatric and adult ependymoma cases: a study using Surveillance, Epidemiology, and End Results data from 1973 to 2007. Neuroepidemiolog. (2012) 39:116. doi: 10.1159/000339320
69. Aizer AA, Ancukiewicz M, Nguyen PL, Shih HA, Loeffler JS, Oh KS, et al. Natural history and role of radiation in patients with supratentorial and infratentorial WHO grade II ependymomas: results from a population-based study. J Neurooncol. (2013) 115:411. doi: 10.1007/s11060-013-1237-9
70. Doxey D, Bruce D, Sklar F, Swift D, and Shapiro K. Posterior fossa syndrome: identifiable risk factors and irreversible complications. Pediatr Neurosur. (1999) 31:131. doi: 10.1159/000028848
71. McEvoy SD, Lee A, Poliakov A, Friedman S, Shaw D, Browd SR, et al. Longitudinal cerebellar diffusion tensor imaging changes in posterior fossa syndrome. NeuroImage Cli. (2016) 12:582. doi: 10.1016/j.nicl.2016.09.007
72. Liu JF, Dineen RA, Avula S, Chambers T, Dutta M, Jaspan T, et al. Development of a pre-operative scoring system for predicting risk of post-operative paediatric cerebellar mutism syndrome. Br J Neurosur. (2018) 32:18. doi: 10.1080/02688697.2018.1431204
73. Khan RB, Patay Z, Klimo P, Huang J, Kumar R, Boop FA, et al. Clinical features, neurologic recovery, and risk factors of postoperative posterior fossa syndrome and delayed recovery: a prospective study. Neuro Onco. (2021) 23:1586. doi: 10.1093/neuonc/noab030
74. Reni M. Guidelines for the treatment of adult intra-cranial grade II-III ependymal tumours. Forum (Genova). (2003) 13:90.
75. Rudà R, Reifenberger G, Frappaz D, Anon A, Anon A, Anon A, et al. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro Onco. (2018) 20:445. doi: 10.1093/neuonc/nox166
76. Merchant TE. Current clinical challenges in childhood ependymoma: A focused review. J Clin Onco. (2017) 35:2364. doi: 10.1200/JCO.2017.73.1265
77. Ritzmann TA, Chapman RJ, Kilday JP, Anon A, Anon A, Anon A, et al. SIOP Ependymoma I: Final results, long-term follow-up, and molecular analysis of the trial cohort - A BIOMECA Consortium Study. Neuro Onco. (2022) 24:936. doi: 10.1093/neuonc/noac012
78. Smith A, Onar-Thomas A, Ellison D, Anon A, Anon A, Anon A, et al. Phase III randomized trial of post-radiation chemotherapy in patients with newly diagnosed ependymoma ages 1 to 21 years. (Geneva, Switzerland: WHO Classification of Tumours of the Central Nervous System, WHO Press), (2020).
79. Merchant TE, Fouladi M, Anon A, Anon A, Anon A, Anon A, et al. Ependymoma: new therapeutic approaches including radiation and chemotherapy. J Neurooncol. (2005) 75:287. doi: 10.1007/s11060-005-6753-9
80. Taylor RE, Anon A, Anon A, Anon A, Anon A, and Anon A. Review of radiotherapy dose and volume for intracranial ependymoma. Pediatr Blood Cancer. (2004) 42:457. doi: 10.1002/pbc.10470
81. Rose ML, Sachdeva R, Mezgueldi Y, Anon A, Anon A, Anon A, et al. Systematic review and meta-analysis of adjuvant radiation dose for pediatric patients (≤22 years) with nonmetastatic intracranial ependymomas. Int J Radiat Oncol Biol Phy. (2025) 121:667. doi: 10.1016/j.ijrobp.2024.07.2335
82. Van Poppel M, Klimo P Jr, Dewire M, Anon A, Anon A, Anon A, et al. Resection of infantile brain tumors after neoadjuvant chemotherapy: the St. Jude experience. J Neurosurg Pediat. (2011) 8:251. doi: 10.3171/2011.6.PEDS11158
83. Siffert J, Allen JC, Anon A, Anon A, Anon A, Anon A, et al. Chemotherapy in recurrent ependymoma. Pediatr Neurosur. (1998) 28:314. doi: 10.1159/000028669
84. Grill J, Le Deley MC, Gambarelli D, Anon A, Anon A, Anon A, et al. Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: a multicenter trial of the French Society of Pediatric Oncology. J Clin Onco. (2001) 19:1288. doi: 10.1200/JCO.2001.19.5.1288
85. Chamberlain MC, Anon A, Anon A, Anon A, Anon A, and Anon A. Salvage chemotherapy for recurrent spinal cord ependymoma. Cance. (2002) 95:997. doi: 10.1002/cncr.10826
86. Gornet MK, Buckner JC, Marks RS, Anon A, Anon A, Anon A, et al. Chemotherapy for advanced CNS ependymoma. J Neurooncol. (1999) 45:61. doi: 10.1023/A:1006394407245
87. Bouffet E, Foreman N, Anon A, Anon A, Anon A, and Anon A. Chemotherapy for intracranial ependymomas. Childs Nerv Sys. (1999) 15:563. doi: 10.1007/s003810050544
88. Fouladi M, Gururangan S, Moghrabi A, Anon A, Anon A, Anon A, et al. Carboplatin-based primary chemotherapy for infants and young children with CNS tumors. Cance. (2009) 115:3243. doi: 10.1002/cncr.v115:14
89. Garvin JH Jr, Selch MT, Holmes E, Anon A, Anon A, Anon A, et al. Phase II study of pre-irradiation chemotherapy for childhood intracranial ependymoma. Children’s Cancer Group protocol 9942: a report from the Children’s Oncology Group. Pediatr Blood Cancer. (2012) 59:1183. doi: 10.1002/pbc.24274
90. Massimino M, Gandola L, Barra S, Anon A, Anon A, Anon A, et al. Infant ependymoma in a 10-year AIEOP experience with omitted or deferred radiotherapy. Int J Radiat Oncol Biol Phy. (2011) 80:807. doi: 10.1016/j.ijrobp.2010.02.048
91. Grundy RG, Wilne SA, Weston CL, Anon A, Anon A, Anon A, et al. Primary postoperative chemotherapy without radiotherapy for intracranial ependymoma in children: the UKCCSG/SIOP prospective study. Lancet Onco. (2007) 8:696. doi: 10.1016/S1470-2045(07)70208-5
92. Sung KW, Lim DH, Lee SH, Anon A, Anon A, Anon A, et al. Tandem high-dose chemotherapy and autologous stem cell transplantation for anaplastic ependymoma in children younger than 3 years of age. J Neurooncol. (2012) 107:335. doi: 10.1007/s11060-011-0745-8
93. Hukin J, Epstein F, Lefton D, Allen J, Anon A, Anon A, et al. Treatment of intracranial ependymoma by surgery alone. Pediatr Neurosur. (1998) 29:40. doi: 10.1159/000028683
94. Palma L, Celli P, Mariottini A, Anon A, Anon A, Anon A, et al. The importance of surgery in supratentorial ependymomas. Long-term survival in a series of 23 cases. Childs Nerv Sys. (2000) 16:170. doi: 10.1007/s003810050487
95. Rogers L, Pueschel J, Spetzler R, Anon A, Anon A, Anon A, et al. Is gross-total resection sufficient treatment for posterior fossa ependymomas? J Neurosur. (2005) 102:629. doi: 10.3171/jns.2005.102.4.0629
96. Reni M, Brandes AA, Vavassori V, Anon A, Anon A, Anon A, et al. A multicenter study of the prognosis and treatment of adult brain ependymal tumors. Cance. (2004) 100:1221. doi: 10.1002/cncr.v100:6
97. Metellus P, Barrie M, Figarella-Branger D, Anon A, Anon A, Anon A, et al. Multicentric French study on adult intracranial ependymomas: prognostic factors analysis and therapeutic considerations from a cohort of 152 patients. Brai. (2007) 130:1338. doi: 10.1093/brain/awm046
98. Hollon T, Nguyen V, Smith BW, Anon A, Anon A, Anon A, et al. Supratentorial hemispheric ependymomas: an analysis of 109 adults for survival and prognostic factors. J Neurosur. (2016) 125:410. doi: 10.3171/2015.7.JNS151187
99. National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncolog. . Available at: https://www.nccn.org/ (Accessed September 05, 2024).
100. Mansur DB, Perry A, Rajaram V, Anon A, Anon A, Anon A, et al. Postoperative radiation therapy for grade II and III intracranial ependymoma. Int J Radiat Oncol Biol Phy. (2005) 61:387. doi: 10.1016/j.ijrobp.2004.06.002
101. Metellus P, Guyotat J, Chinot O, Anon A, Anon A, Anon A, et al. Adult intracranial WHO grade II ependymomas: long-term outcome and prognostic factor analysis in a series of 114 patients. Neuro Onco. (2010) 12:976. doi: 10.1093/neuonc/noq047
102. Nuño M, Yu JJ, Varshneya K, Anon A, Anon A, Anon A, et al. Treatment and survival of supratentorial and posterior fossa ependymomas in adults. J Clin Neurosc. (2016) 28:24. doi: 10.1016/j.jocn.2015.11.014
103. Kweh BTS, Rosenfeld JV, Hunn M, and Tee JW. Tumor characteristics and surgical outcomes of intracranial subependymomas: a systematic review and meta-analysis. J Neurosur. (2022) 136:736. doi: 10.3171/2021.2.JNS204052
104. Marinoff AE, Ma C, Guo D, Anon A, Anon A, Anon A, et al. Rethinking childhood ependymoma: a retrospective, multi-center analysis reveals poor long-term overall survival. J Neurooncol. (2017) 135:201. doi: 10.1007/s11060-017-2568-8
105. Broniscer A, Ke W, Fuller CE, Anon A, Anon A, Anon A, et al. Second neoplasms in pediatric patients with primary central nervous system tumors: the St. Jude Children’s Research Hospital experience. Cance. (2004) 100:2246. doi: 10.1002/cncr.v100:10
106. Spiegler BJ, Bouffet E, Greenberg ML, Anon A, Anon A, Anon A, et al. Change in neurocognitive functioning after treatment with cranial radiation in childhood. J Clin Onco. (2004) 22:706. doi: 10.1200/JCO.2004.05.186
107. Duffner PK, Krischer JP, Horowitz ME, Anon A, Anon A, Anon A, et al. Second malignancies in young children with primary brain tumors following treatment with prolonged postoperative chemotherapy and delayed irradiation: A Pediatric Oncology Group study. Ann Neuro. (1998) 44:313. doi: 10.1002/ana.410440305
108. Garwicz S, Anderson H, Olsen JH, Anon A, Anon A, Anon A, et al. Second malignant neoplasms after cancer in childhood and adolescence: A population-based case-control study in the 5 Nordic countries. Int J Cancer. (2000) 88:672. doi: 10.1002/1097-0215(20001115)88:4<672::AID-IJC24>3.0.CO;2-N
109. Stavrou T, Bromley CM, Nicholson HS, Anon A, Anon A, Anon A, et al. Prognostic factors and secondary malignancies in childhood medulloblastoma. J Pediatr Hematol Onco. (2001) 23:431. doi: 10.1097/00043426-200110000-00008
110. Neglia JP, Friedman DL, Yasui Y, Anon A, Anon A, Anon A, et al. Second malignant neoplasms in five-year survivors of childhood cancer: Childhood Cancer Survivor Study. J Natl Cancer Ins. (2001) 93:618. doi: 10.1093/jnci/93.8.618
111. Devarahally SR, Severson RK, Chuba P, Anon A, Anon A, Anon A, et al. Second malignant neoplasms after primary central nervous system malignancies of childhood and adolescence. Pediatr Hematol Onco. (2003) 20:617. doi: 10.1080/08880010390243031
112. Bass JK, Hua CH, Huang J, Anon A, Anon A, Anon A, et al. Hearing loss in patients who received cranial radiation therapy for childhood cancer. J Clin Onco. (2016) 34:1248. doi: 10.1200/JCO.2015.63.6738
113. Armstrong TS, Vera-Bolanos E, Gilbert MR, Anon A, Anon A, Anon A, et al. Clinical course of adult patients with ependymoma: results of the Adult Ependymoma Outcomes Project. Cance. (2011) 117:5133. doi: 10.1002/cncr.v117.22
114. Landier W, Bhatia S, Eshelman DA, Anon A, Anon A, Anon A, et al. Development of risk-based guidelines for pediatric cancer survivors: The Children’s Oncology Group long-term follow-up guidelines from the Children’s Oncology Group Late Effects Committee and Nursing Discipline. J Clin Onco. (2004) 22:4979. doi: 10.1200/JCO.2004.11.032
115. Zacharoulis S, Ashley S, Moreno L, Anon A, Anon A, Anon A, et al. Treatment and outcome of children with relapsed ependymoma: a multi-institutional retrospective analysis. Childs Nerv Sys. (2010) 26:905. doi: 10.1007/s00381-009-1067-4
116. Desrousseaux J, Claude L, Chaltiel L, Anon A, Anon A, Anon A, et al. Respective roles of surgery, chemotherapy, and radiation therapy for recurrent pediatric and adolescent ependymoma: A national multicentric study. Int J Radiat Oncol Biol Phy. (2023) 117:404. doi: 10.1016/j.ijrobp.2023.04.008
117. Massimino M, Barretta F, Modena P, Anon A, Anon A, Anon A, et al. Treatment and outcome of intracranial ependymoma after first relapse in the 2nd AIEOP protocol. Neuro Onco. (2022) 24:467. doi: 10.1093/neuonc/noab230
118. Merchant TE. Current management of childhood ependymoma. Oncol (Williston Park. (2002) 16(5):629–42.
119. Merchant TE, Boop FA, Kun LE, Sanford RA, Anon A, Anon A, et al. A retrospective study of surgery and reirradiation for recurrent ependymoma. Int J Radiat Oncol Biol Phy. (2008) 71:87. doi: 10.1016/j.ijrobp.2007.09.037
120. Stafford SL, Pollock BE, Foote RL, Anon A, Anon A, Anon A, et al. Stereotactic radiosurgery for recurrent ependymoma. Cance. (2000) 88:870. doi: 10.1002/(SICI)1097-0142(20000215)88:4<870::AID-CNCR18>3.0.CO;2-I
121. Liu AK, Foreman NK, Gaspar LE, Anon A, Anon A, Anon A, et al. Maximally safe resection followed by hypofractionated re-irradiation for locally recurrent ependymoma in children. Pediatr Blood Cance. (2009) 52:804. doi: 10.1002/pbc.21982
122. Bouffet E, Hawkins CE, Ballourah W, Anon A, Anon A, Anon A, et al. Survival benefit for pediatric patients with recurrent ependymoma treated with reirradiation. Int J Radiat Oncol Biol Phy. (2012) 83:1541. doi: 10.1016/j.ijrobp.2011.10.039
123. Tsang DS, Burghen E, Klimo P Jr, Anon A, Anon A, Anon A, et al. Outcomes after reirradiation for recurrent pediatric intracranial ependymoma. Int J Radiat Oncol Biol Phy. (2018) 100:507. doi: 10.1016/j.ijrobp.2017.10.002
124. Tsang DS, Murray L, Ramaswamy V, Anon A, Anon A, Anon A, et al. Craniospinal irradiation as part of re-irradiation for children with recurrent intracranial ependymoma. Neuro Onco. (2019) 21:547. doi: 10.1093/neuonc/noy191
125. Brandes AA, Cavallo G, Reni M, Anon A, Anon A, Anon A, et al. A multicenter retrospective study of chemotherapy for recurrent intracranial ependymal tumors in adults by the Gruppo Italiano Cooperativo di Neuro-Oncologia. Cance. (2005) 104:143. doi: 10.1002/cncr.v104:1
126. Rudà R, Bosa C, Magistrello M, Anon A, Anon A, Anon A, et al. Temozolomide as salvage treatment for recurrent intracranial ependymomas of the adult: a retrospective study. Neuro Onco. (2016) 18:261. doi: 10.1093/neuonc/nov167
127. Komori K, Yanagisawa R, Miyairi Y, Anon A, Anon A, Anon A, et al. Temozolomide treatment for pediatric refractory anaplastic ependymoma with low MGMT protein expression. Pediatr Blood Cancer. (2016) 63:152. doi: 10.1002/pbc.25696
128. Wright KD, Daryani VM, Turner DC, Anon A, Anon A, Anon A, et al. Phase I study of 5-fluorouracil in children and young adults with recurrent ependymoma. Neuro Onco. (2015) 17:1620. doi: 10.1093/neuonc/nov181
129. Chamberlain MC, Anon A, Anon A, Anon A, Anon A, Anon A, et al. Recurrent intracranial ependymoma in children: salvage therapy with oral etoposide. Pediatr Neuro. (2001) 24:117. doi: 10.1016/S0887-8994(00)00249-6
130. Sandri A, Massimino M, Mastrodicasa L, Anon A, Anon A, Anon A, et al. Treatment with oral etoposide for childhood recurrent ependymomas. J Pediatr Hematol Onco. (2005) 27:486. doi: 10.1097/01.mph.0000181430.71176.b7
131. Adolph JE, Fleischhack G, Mikasch R, Anon A, Anon A, Anon A, et al. Local and systemic therapy of recurrent ependymoma in children and adolescents: short- and long-term results of the E-HIT-REZ 2005 study. Neuro Onco. (2021) 23:1012. doi: 10.1093/neuonc/noaa276
132. Michealraj KA, Kumar SA, Kim LJY, Anon A, Anon A, Anon A, et al. Metabolic regulation of the epigenome drives lethal infantile ependymoma. Cel. (2020) 181:1329. doi: 10.1016/j.cell.2020.04.047
133. Gojo J, Englinger B, Jiang L, Anon A, Anon A, Anon A, et al. Single-cell RNA-seq reveals cellular hierarchies and impaired developmental trajectories in pediatric ependymoma. Cancer Cel. (2020) 38:44. doi: 10.1016/j.ccell.2020.06.004
134. Arabzade A, Zhao Y, Varadharajan S, Anon A, Anon A, Anon A, et al. ZFTA-RELA dictates oncogenic transcriptional programs to drive aggressive supratentorial ependymoma. Cancer Discover. (2021) 11:2200. doi: 10.1158/2159-8290.CD-20-1066
135. Kupp R, Ruff L, Terranova S, Anon A, Anon A, Anon A, et al. ZFTA translocations constitute ependymoma chromatin remodeling and transcription factors. Cancer Discover. (2021) 11:2216. doi: 10.1158/2159-8290.CD-20-1052
136. Zheng T, Ghasemi DR, Okonechnikov K, Anon A, Anon A, Anon A, et al. Cross-species genomics reveals oncogenic dependencies in ZFTA/C11orf95 fusion-positive supratentorial ependymomas. Cancer Discover. (2021) 11:2230. doi: 10.1158/2159-8290.CD-20-0963
137. Gilbert MR, Yuan Y, Wu J, Anon A, Anon A, Anon A, et al. A phase II study of dose-dense temozolomide and lapatinib for recurrent low-grade and anaplastic supratentorial, infratentorial, and spinal cord ependymoma. Neuro Onco. (2021) 23:468. doi: 10.1093/neuonc/noaa240
138. Green RM, Cloughesy TF, Stupp R, Anon A, Anon A, Anon A, et al. Bevacizumab for recurrent ependymoma. Neurolog. (2009) 73:1677. doi: 10.1212/WNL.0b013e3181c1df34
139. Couec ML, Andre N, Thebaud E, Anon A, Anon A, Anon A, et al. Bevacizumab and irinotecan in children with recurrent or refractory brain tumors: toxicity and efficacy trends. Pediatr Blood Cancer. (2012) 59:34. doi: 10.1002/pbc.24066
140. Gururangan S, Fangusaro J, Young Poussaint T, Anon A, Anon A, Anon A, et al. Lack of efficacy of bevacizumab + irinotecan in pediatric recurrent ependymoma – a Pediatric Brain Tumor Consortium study. Neuro Onco. (2012) 14:1404. doi: 10.1093/neuonc/nos213
141. DeWire M, Fouladi M, Turner DC, Anon A, Anon A, Anon A, et al. Bevacizumab and lapatinib in children with recurrent/refractory ependymoma: A CERN study. J Neurooncol. (2015) 123:85. doi: 10.1007/s11060-015-1764-7
142. Jagtiani P, Karabacak M, Le C, Anon A, Anon A, Anon A, et al. Intracranial ependymomas in pediatric patients: patterns of care, disparities, and survival outcomes from the National Cancer Database. J Neurosurg Pediat. (2024) 34:495. doi: 10.3171/2024.5.PEDS2480
143. Pajtler KW, Pfister SM, and Kool M. Molecular dissection of ependymomas. Oncoscienc. (2015) 2:827. doi: 10.18632/oncoscience.v2i10
144. Healey EA, Barnes PD, Kupsky WJ, Anon A, Anon A, Anon A, et al. The prognostic significance of postoperative residual tumor in ependymoma. Neurosurger. (1991) 28:666. doi: 10.1227/00006123-199105000-00005
145. McLaughlin MP, Marcus RB Jr, Buatti JM, Anon A, Anon A, Anon A, et al. Ependymoma: results, prognostic factors and treatment recommendations. Int J Radiat Oncol Biol Phy. (1998) 40:845. doi: 10.1016/S0360-3016(97)00893-6
146. Bouffet E, Perilongo G, Canete A, and Massimino M. Intracranial ependymomas in children: a critical review of prognostic factors and a plea for cooperation. Med Pediatr Onco. (1998) 30:319.
147. Horn B, Heideman R, Geyer R, Anon A, Anon A, Anon A, et al. Risk factors in pediatric ependymoma: A multi-institutional retrospective study. J Pediatr Hematol Onco. (1999) 21:203. doi: 10.1097/00043426-199905000-00008
148. Shu HK, Sall WF, Maity A, Anon A, Anon A, Anon A, et al. Childhood intracranial ependymoma: twenty-year experience from a single institution. Cance. (2007) 110:432. doi: 10.1002/cncr.v110:2
149. De B, Khakoo Y, Souweidane MM, Anon A, Anon A, Anon A, et al. Patterns of relapse in children with localized intracranial ependymoma. J Neurooncol. (2018) 138:435. doi: 10.1007/s11060-018-2815-7
150. Armstrong TS, Vera-Bolanos E, Bekele BN, Anon A, Anon A, Anon A, et al. Adult ependymal tumors: Prognosis and the M. D. Anderson Cancer Center experience. Neuro Onco. (2010) 12:862. doi: 10.1093/neuonc/noq009
151. Vitanovics D, Balint K, Hanzely Z, Anon A, Anon A, Anon A, et al. Ependymoma in adults: surgery, reoperation, and radiotherapy for survival. Pathol Oncol Re. (2010) 16:93. doi: 10.1007/s12253-009-9194-5
152. Amirian ES, Armstrong TS, Gilbert MR, Scheurer ME, Anon A, Anon A, et al. Predictors of survival among older adults with ependymoma. J Neurooncol. (2012) 107:183. doi: 10.1007/s11060-011-0730-2
Keywords: intracranial ependymoma, molecular classification, gross total resection, radiotherapy, pediatric neuro-oncology
Citation: Pan Z, Bao J and Wei S (2025) Optimizing outcomes in intracranial ependymoma: a contemporary review. Front. Oncol. 15:1617169. doi: 10.3389/fonc.2025.1617169
Received: 24 April 2025; Accepted: 22 May 2025;
Published: 10 June 2025.
Edited by:
Brent T Harris, Georgetown University, United StatesReviewed by:
Timothy A Ritzmann, University of Nottingham, United KingdomCopyright © 2025 Pan, Bao and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shepeng Wei, ZHJfd2Vpc2hlcGVuZ0AxMjYuY29t