 Renjie Xia1,2†
Renjie Xia1,2† Juan Liang3†Jianguo Ma2,4
Juan Liang3†Jianguo Ma2,4 Xiaoyu Du1,2Liangbin Ma1,2Xiongxiong Han2,4Yong Wang2,4Jianwei Qin2*
Xiaoyu Du1,2Liangbin Ma1,2Xiongxiong Han2,4Yong Wang2,4Jianwei Qin2* Long Yan2*
Long Yan2*- 1Department of Medicine, Northwest Minzu University, Lanzhou, China
- 2Department of Hepatobiliary Surgery, The 940th Hospital of Joint Logistic Support Force of Chinese People’s Liberation Army, Lanzhou, China
- 3Intensive Care Units, The 940th Hospital of Joint Logistic Support Force of Chinese People's Liberation Army, Lanzhou, China
- 4First School of Clinical Medicine, Gansu University of Chinese Medicine, Lanzhou, China
Immune checkpoint blockade (ICB), particularly targeting programmed cell death-1 (PD-1), has revolutionized cancer immunotherapy but remains limited by heterogeneous therapeutic responses and immune-related toxicities. This review systematically examines the integration of immune agonists—STING, TLR, CD40, and OX40 agonists—with PD-1 inhibitors to overcome resistance and amplify antitumor immunity. Nanoparticle delivery systems emerge as transformative platforms, addressing critical limitations of free agonists, including enzymatic degradation, off-target toxicity, and poor pharmacokinetics. By leveraging tunable physicochemical properties (e.g., size, surface charge, stimuli-responsive release), nanoparticles enhance tumor-specific accumulation, prolong agonist half-life, and synergize with PD-1 inhibitors to remodel immunosuppressive microenvironments. Preclinical and early clinical studies demonstrate combinatorial strategies achieving increases in T cell infiltration and enhancements in anti-angiogenic activity compared to monotherapies. However, translational challenges persist, including nanoparticle-induced immunotoxicity (ROS-mediated inflammation), manufacturing scalability hurdles, and interspecies discrepancies in murine models. Future directions emphasize personalized nanovaccines, supramolecular cytosolic delivery systems (e.g., Calix-STING), and biomarker-driven trials to optimize efficacy in advanced pancreatic, melanoma, and immunologically quiescent tumors. This work underscores the imperative for interdisciplinary collaboration to standardize nanoparticle design and clinical validation frameworks, ultimately bridging the gap between nanomedicine innovation and oncology practice.
1 Introduction
Malignant tumors remain globally challenging to cure. Conventional therapies face limitations: surgery risks organ damage and misses metastases; radiotherapy incompletely eradicates tumors; chemotherapy impairs immunity and quality of life (1). With advances in understanding tumor biology, immunotherapy has gained prominence as a therapeutic strategy. Immune checkpoint blockade (ICB) has demonstrated promise in patients with malignant tumors, particularly antibodies targeting programmed cell death-1 (PD-1) and cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), has demonstrated promise as a representative immunotherapy (2). Among these, PD-1 inhibitors comprise a diverse array of agents that have been extensively applied in immunotherapy for various malignancies.
However, the therapeutic efficacy of PD-1 inhibitors remains suboptimal in many malignancies. ‘Cold tumors’ exhibit limited T cell infiltration: immune-excluded types confine CD8+ T cells to margins, while immune-desert types lack intratumoral/peripheral CD8+ T cells (3). These phenotypes arise from low tumor mutational burden, diminished MHC-I expression, and reduced PD-L1 levels, compounded by immunosuppressive populations (e.g., TAMs, Tregs, MDSCs) (4). Consequently, cold tumors exhibit inherently defective antitumor immunity or ineffective T cell trafficking. As exemplified by glioblastoma (GBM)—a prototypical cold tumor—clinical trials of nivolumab or pembrolizumab failed to improve median overall survival, attributable to profound immunosuppression and spatial heterogeneity (5).
Beyond this, studies have demonstrated significant heterogeneity in treatment responses among colorectal cancer (CRC) subtypes, with PD-1 inhibitors exhibiting limited efficacy in microsatellite instability-low (MSI-L) CRC compared to improved outcomes in microsatellite instability-high (MSI-H) patients (6). PD-1 inhibitors have shown clinical benefits in only a subset of hepatocellular carcinoma (HCC) patients, while most exhibit primary resistance or rapidly develop acquired resistance (7). Furthermore, PD-1 inhibitor therapy in HCC frequently induces immune-related adverse events (irAEs), with an objective response rate (ORR) of 31% and grade 3/4 irAEs incidence of 37% observed in anti-PD-1/anti-CTLA-4 combination therapy for advanced HCC (8). Additionally, hepatotoxicity associated with PD-1 inhibitors poses a critical limitation to their long-term use. The incidence of drug-induced liver injury varies depending on agent type and dosage in HCC patients receiving PD-1 inhibitors, often manifesting as dose-dependent elevations in alanine aminotransferase (ALT) levels (9).
To enhance therapeutic efficacy while mitigating side effects and resistance without increasing PD-1 inhibitor dosage, researchers have pioneered combination therapies integrating immune agonists with ICB. Immune agonists activate the host immune system and amplify tumor immunogenicity, thereby reprogramming immunosuppressive microenvironments and promoting T cell infiltration in cold tumors— thereby potentiating PD-1 inhibitor activity.Immune agonists activate the host immune system and amplify tumor immunogenicity, thereby potentiating PD-1 inhibitor activity (10). This combinatorial strategy demonstrates potential for inhibiting HCC progression and reducing the incidence of PD-1 inhibitor resistance (11). Nevertheless, challenges such as suboptimal physicochemical stability and immune cell toxicity associated with free agonists persist. To address these limitations, nanoparticle-based delivery systems have been engineered for targeted agonist delivery, enabling precise modulation of immune cell function, reduced systemic toxicity, and specificity in immune cell activation (12). Consequently, the synergistic combination of immune agonist-loaded nanoparticles with PD-1 inhibitor immunotherapy is recognized as a novel therapeutic paradigm with significant translational potential in oncology.
2 Combinatorial immunotherapy: immune agonists and PD-1 inhibitors
Combinatorial Immunotherapy: Immune Agonists and PD-1 Inhibitors Current strategies to enhance PD-1 inhibitor efficacy include: Combination therapies with targeted agents and immune checkpoint inhibitors (ICB)、Adjunctive immune modulators、Cellular or cytokine therapies、Radiotherapy-ICB combinations (13). Among these approaches, immune agonists have demonstrated efficacy in remodeling the immunosuppressive tumor microenvironment (TME), thereby augmenting antitumor immunity and pre-conditioning the magnitude and duration of immune responses (14) (Figure 1).
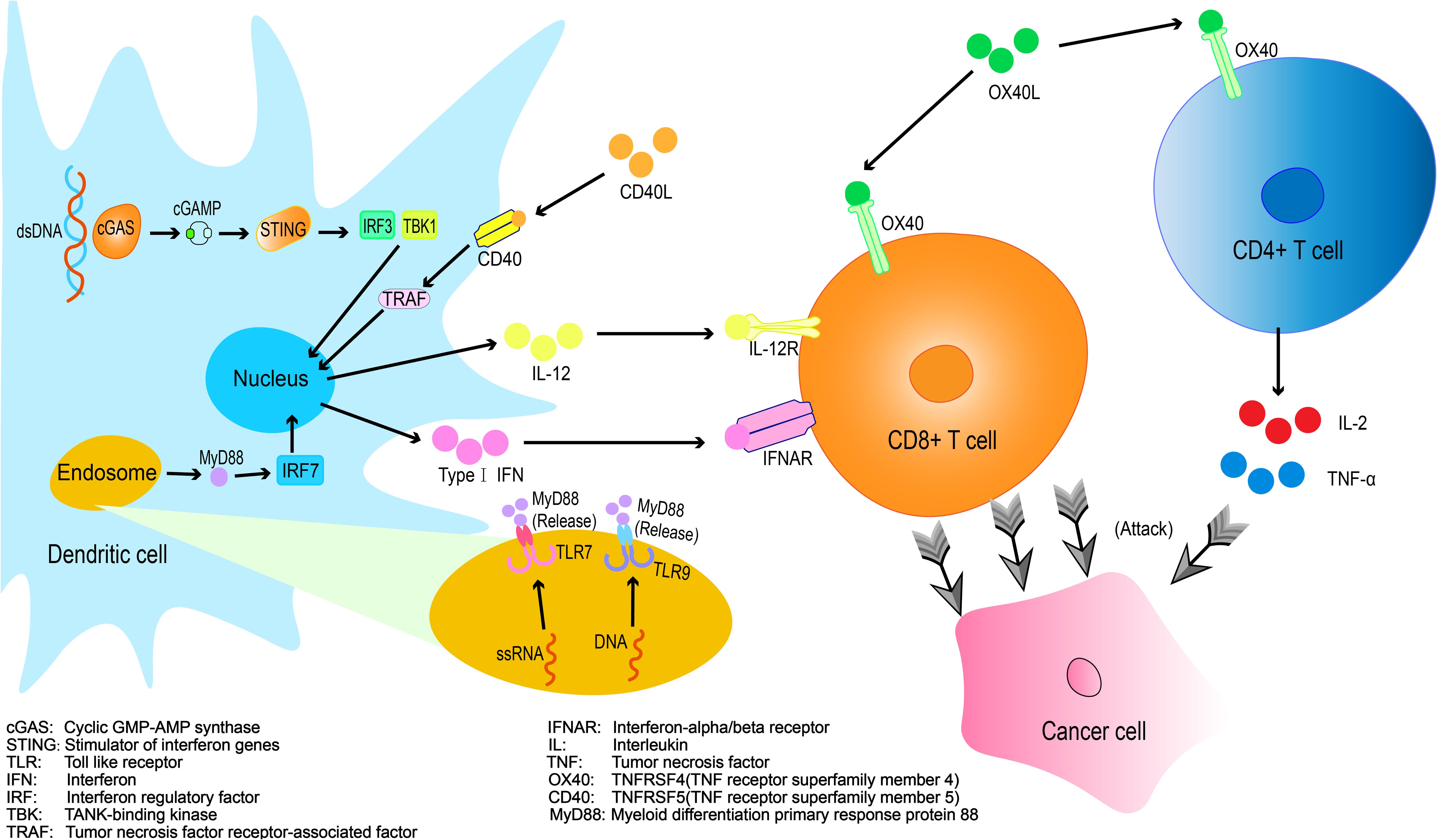
Figure 1. Mechanisms of immune agonists in enhancing T cell-mediated tumor killing.
2.1 Common immune agonists
2.1.1 STING agonists
The stimulator of interferon genes (STING) is a multifunctional immune adaptor protein that plays a critical role in antitumor immunity (15). This pathway enhances tumor immunogenicity by upregulating MHC-I expression and tumor antigen presentation required for CD8+ T cell recognition, while concurrently promoting B cell antibody production and amplifying the functionality of dendritic cell (DC) and T cell populations (16, 17).
The combination of STING agonists with immunotherapy has emerged as a novel cancer treatment modality (18). A Phase 1 trial evaluating the STING agonist MK-1454 combined with pembrolizumab in a mixed tumor cohort demonstrated partial responses in 3 head and neck squamous cell carcinoma (HNSCC), 2 anaplastic thyroid carcinoma (ATC), and 1 triple-negative breast cancer (TNBC) patient in the combination arm—with no responses in the MK-1454 monotherapy group (0% ORR). Treatment-related adverse events (TRAEs) were reported in both arms, with no treatment-related fatalities (19). A Phase Ib study demonstrated that the STING agonist MIW815 combined with spartalizumab exhibited a favorable safety profile but modest antitumor activity in patients with advanced/metastatic solid tumors or lymphomas, achieving an ORR of 10.4%. Pharmacokinetic analysis revealed rapid absorption of MIW815 at the intratumoral injection site, with systemic exposure increasing dose-proportionally and no observed drug accumulation. Future investigations should focus on optimizing drug formulation and delivery strategies to enhance intratumoral bioavailability and therapeutic efficacy (20). Mechanistically, cGAS-STING-activated M1 macrophages recruit T cells via STING-IRF3, but concurrent STING-IRF3–signal transducer and activator of transcription 1 (STAT1) activation induces immunosuppressive PD-L1 (21). The combination of STING agonists with PD-1 inhibitors synergistically activates the cGAS-STING pathway while blocking the PD-1/PD-L1 axis, offering a promising strategy to overcome monotherapy resistance in PD-1 inhibitor-based regimens.
2.1.2 TLR agonists
Toll-like receptors (TLRs), a class of pattern recognition receptor proteins, activate immune cells such as dendritic cells (DCs), T cells, and macrophages through agonist-specific recognition, playing a pivotal role in immunogenic signal transduction to promote antitumor immunity (22).
Investigators have developed combinatorial antitumor strategies integrating TLR agonists with PD-1 inhibitors. In CT26 murine models, monotherapy with the TLR7 agonist DSP-0509 achieved approximately 60% tumor growth inhibition (TGI), whereas combination therapy with anti-PD-1 antibodies enhanced TGI to >80%. Similarly, in 4T1 tumor-bearing mice, the combination regimen demonstrated significantly greater antitumor efficacy than either monotherapy. Critically, no significant reductions in body weight or other treatment-related adverse events were observed across experimental cohorts, indicating favorable tolerability profiles (23). In a Phase I/Ib trial evaluating the TLR-7 agonist LHC165 combined with spartalizumab for advanced solid tumors (including melanoma, HNSCC, and TNBC), efficacy analysis revealed an ORR of 4.8% (1/21) with monotherapy versus 8.3% (2/24) in the combination arm. Treatment discontinuation due to AEs occurred in 2 patients. Additionally, baseline biomarker analyses suggested that clinical responses to LHC165 may require a pre-existing immunologically hot TME (24). In contrast, the TLR9 agonist vidutolimod combined with PD-1 blockade demonstrated preliminary efficacy in PD-1 inhibitor-resistant melanoma patients with immunologically cold TMEs, achieving an ORR of 25% (11/44; including 7 partial responses and 4 complete responses) (25), providing critical context for patient stratification strategies. A study evaluating combined anti-PD-1 and TLR9 agonist immunotherapy in cardiac allograft-tolerant murine models demonstrated that intratumoral (i.t.) delivery achieved significantly greater suppression of tumor growth relative to systemic (i.p.) administration, without eliciting accelerated allograft rejection (26). Jiang et al. further demonstrated that the TLR9 agonist ODN1585 enhances CD8+ T cell-mediated antitumor immunity in colorectal cancer peritoneal metastasis by suppressing retinol metabolism in fibroblastic reticular cells (FRCs) and reducing Tim4+ peritoneal resident macrophage (PRM) populations, thereby potentiating anti-PD-1 therapy (27). These studies collectively demonstrate the promising therapeutic potential of TLR agonist/PD-1 inhibitor combination therapy in malignancies.
2.1.3 CD40 agonists
CD40, a co-stimulatory molecule predominantly expressed on APCs such as DCs, macrophages, and B cells, transmits activation signals via interaction with CD40L on T cells to promote their activation, proliferation, and cytokine secretion. CD40 agonist antibodies mimic CD40L binding to activate CD40 signaling, enhancing APC functionality and augmenting T cell-mediated antitumor immunity (28).
A phase II trial demonstrated that the CD40 agonist sotigalimab combined with nivolumab induced durable responses in PD-1 inhibitor-resistant melanoma patients, achieving an ORR of 15.2% (5/33 partial responses) with a median duration of response ≥26 months. Critically, no treatment discontinuations due to AEs were observed among evaluable patients (29). Preclinical studies further validated this strategy: the humanized CD40 agonist KHK2840 enhanced PD-1 inhibitor efficacy in syngeneic models, significantly augmenting Th1 cells in tumor-draining lymph nodes (TDLNs), elevating DC infiltration within tumors, and concomitantly increasing intratumoral CD8+ T cell populations in MC-38 murine colon carcinoma models (30). In the CT2A glioblastoma murine model, combination therapy employing CD40 agonists and PD-1 inhibitors significantly enhanced survival outcomes; Mechanistically, CD4+ T cells crucially maintain progenitor exhausted CD8+ T cell populations and their responsiveness to PD-1 blockade; CD40 agonism bypasses CD4+ T cell dysfunction, thereby potentiating PD-1 checkpoint therapy efficacy (31). Additionally, Laurence et al. showed CD40 agonism amplifies PD-1 inhibitor efficacy in iCCA models by expanding tumor-infiltrating CD4+/CD8+ T cell populations and enhancing their activation status (32). These findings confirm CD40 agonists enhance PD-1 inhibitor responses across malignancies.
2.1.4 OX40 agonists
OX40, a co-stimulatory molecule within the tumor necrosis factor receptor(TNFR) superfamily, delivers critical survival and differentiation signals that enhance T cell functionality, OX40 activation significantly amplifies the proliferation and cytokine production(including IL-2, IFN-γ, and TNF-α) in CD4+ and CD8+ T cells, augments effector functions such as granzyme B release and cytotoxic activity, promotes long-term memory cell generation, and modulates regulatory T cells (Tregs) by attenuating their immunosuppressive capacity or inducing Treg depletion (33).
In recent years, the combination of OX40 agonists with PD-1 inhibitors has emerged as a promising therapeutic strategy. Preclinical studies demonstrate that the OX40 agonist GSK3174998 synergizes with pembrolizumab to enhance antitumor immune responses in both in vitro and in vivo models, Clinically, this combination exhibits modest efficacy in immunologically ‘hot’ tumors (e.g., subsets of melanoma and NSCLC), yet demonstrates limited activity in ‘cold’ tumor microenvironments such as soft tissue sarcoma (STS) (34). This stark contrast underscores the imperative to define predictive TME biomarkers for refined patient stratification. In murine models of urothelial carcinoma, Alvim et al. reported that the triple combination of vascular-targeted photodynamic therapy (VTP), PD-1 inhibitors, and OX40 agonists achieved superior tumor growth suppression and prolonged survival compared to VTP or immunotherapy alone (35). Furthermore, Min et al. designed a phase II clinical trial (“R-ISV-RO”) integrating stereotactic radiotherapy, intratumoral injection of the RO adjuvant (a combination of TLR7 and OX40 agonists), and PD-1 blockade. Preclinical validation in urothelial carcinoma models demonstrated that triple-combination therapy (VTP + OX40 agonist + PD-1 inhibitor) significantly improved survival outcomes, achieving 60% survival at 60 days—markedly superior to monotherapy (VTP alone: 25%), dual therapies (VTP + PD-1 inhibitor: 31.25%; VTP + OX40 agonist: 20%), and untreated controls (p<0.001). This regimen significantly amplified antitumor immunity (36). Collectively, these findings provide a robust theoretical foundation for OX40/PD-1 inhibitor-based combination therapies.
2.2 Limitations of immune agonists
STING agonists face significant challenges in clinical translation due to their susceptibility to enzymatic degradation, short retention time, immunocyte toxicity, and inefficient cytosolic delivery (37); Additionally, they may induce epithelial barrier disruption, bronchial alveolar luminal dsDNA release, and cell death, leading to STING/type I IFN-dependent acute pulmonary inflammation (38). The STING pathway is also implicated in traumatic brain injury, spinal cord injury, subarachnoid hemorrhage, hypoxic-ischemic encephalopathy, and atherosclerosis (39, 40). Furthermore, STING agonists stimulate bone marrow-derived macrophages, triggering lipid peroxidation and cell death (41). TLR agonists are associated with adverse effects such as fever, chronic inflammation, and granuloma formation (42). In clinical trials, some TLR agonists exhibited high rates of injection site reactions (ISRs), including fever, chills, pain, and erythema (43). The pro-inflammatory cytokines and chemokines produced post-TLR activation may induce localized or systemic inflammation, contributing to irAEs (44, 45). CD40 agonists pose risks of cytokine release syndrome (CRS) and hepatotoxicity (46). Their poor tumor accumulation necessitates high doses, exacerbating toxicity while limiting efficacy (47). OX40 agonist/PD-1 inhibitor combinations demonstrate clinical responses in only a subset of patients (48). A trial evaluating the OX40 agonist MEDI0562 with immunotherapy reported frequent treatment-related adverse events (AEs) and dose-limiting toxicities (DLTs) requiring therapy discontinuation (49). Preclinical studies revealed that concurrent PD-1/OX40 agonism induces T cell apoptosis in the TME, impairing antitumor efficacy in TC-1 lung epithelial murine models, whereas delayed PD-1 inhibitor administration preserves OX40 agonist activity (50).To address these limitations, researchers have developed STING agonist-encapsulated immunostimulatory nanoparticles to enhance stability and prolong systemic retention (51, 52). Targeted delivery systems incorporating antibodies or peptides improve tumor-specific STING activation (53). Researchers have engineered nanoparticle-conjugated TLR7 agonist imiquimod to enhance therapeutic efficacy in mucosal tissues while significantly mitigating cutaneous irritation (54) (Table 1).
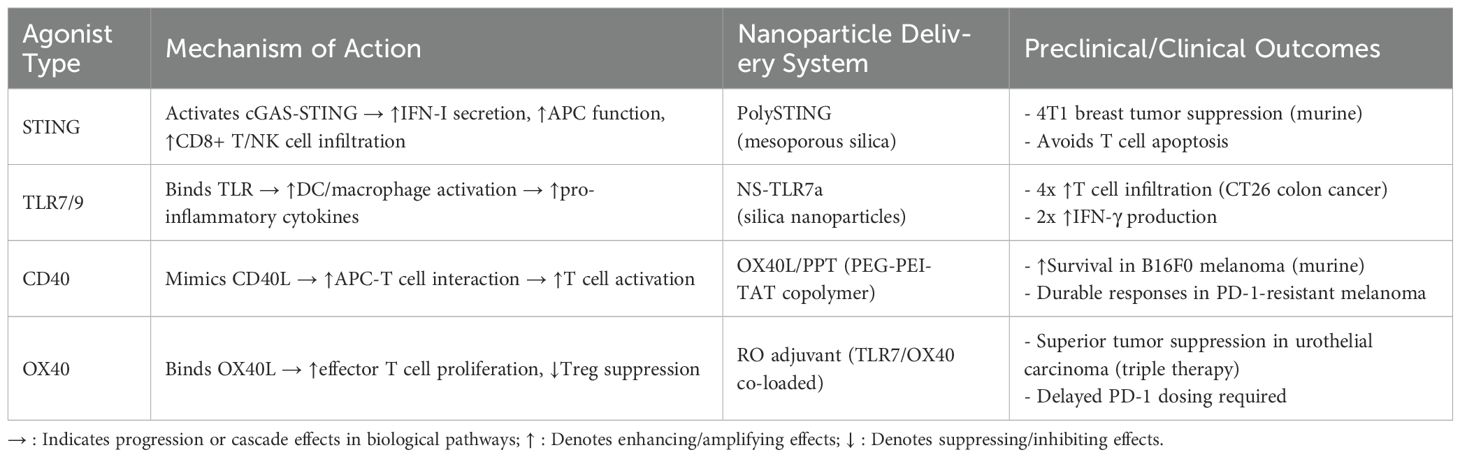
Table 1. Key immune agonists and their synergistic effects with pd-1 inhibitors.
To address off-target toxicity and improve tumor-specific delivery, key nanoplatform design strategies include: active targeting using antibody/peptide/aptamer ligands binding tumor or TME immune cell receptors; stimuli-responsive release (pH/enzyme/ROS/reduction-triggered) for site-specific agonist activation; localized administration (intratumoral injection/hydrogels) minimizing systemic exposure; and carrier optimization via biocompatible materials, size/surface charge tuning, and sustained-release profiles to lower peak toxicity. Immunotoxicity is governed by physicochemical in nanodelivery systems may enable precise targeting, toxicity mitigation, sustained release, and synergistic immune activation, offering comprehensive solutions to overcome the current shortcomings of immune agonists.
3 Current research status of immunostimulatory nanoparticles
3.1 Characteristics of immunostimulatory nanoparticles
Diverse nanosystems provide novel drug delivery strategies to improve cancer treatment. Nanoparticle (NP)-based delivery platforms can overcome biological barriers and enhance therapeutic drug accumulation at target sites (55). Additionally, nanoscale encapsulation not only reduces systemic toxicity but also improves tumor-specific drug delivery to malignant cells (56).
In multiple tumor models (MC38 colon cancer, B16F10 melanoma, Lewis lung cancer), immunostimulatory nanoparticles (PolySTING) demonstrated superior tumor suppression compared to free cGAMP. When combined with anti-PD-1 therapy, PolySTING further enhanced antitumor efficacy. Notably, PolySTING selectively targets myeloid cells—particularly DCs—while avoiding STING-mediated apoptosis in T cells, thereby improving therapeutic safety (57). Systemically administered drugs often fail to penetrate the dense desmoplastic stroma of pancreatic tumors, leading to vascular deposition, phagocytic clearance, and inefficient tumor cell delivery (58, 59). To address this, STING/TLR4 agonist-co-loaded immunostimulatory nanoparticles were engineered to target APC-rich perivascular regions in pancreatic ductal adenocarcinoma (PDAC), effectively reversing the immunosuppressive microenvironment and inducing synergistic antitumor effects (60). Studies confirmed that intravenously delivered nanoparticles are highly internalized by perivascular macrophages at tumor sites and retained intracellularly for prolonged periods (61). The APC-enriched perivascular niche in PDAC serves as an ideal immunotherapeutic target, as nanoparticle accumulation activates and expands APCs, enhancing tumor antigen processing. Furthermore, the tunable design of immunostimulatory nanoparticles allows optimization of agonist ratios (e.g., cdGMP/MPLA), significantly boosting functional synergy and IFN-β production in murine PDAC models (60).
The enhanced permeability and retention (EPR) effect—a phenomenon where leaky tumor vasculature permits nanoparticle extravasation and retention—has long been considered the primary mechanism of nanoparticle tumor accumulation (62). However, integrated analyses of murine models, human tumors, computational simulations, and advanced imaging techniques reveal that passive diffusion through vascular gaps accounts for <3% of nanoparticle delivery. Instead, >97% of nanoparticles enter tumors via active endothelial transcytosis (63). These findings highlight the need for deeper mechanistic investigations into nanoparticle delivery pathways.
3.2 Common types of immunostimulatory nanoparticles for tumor therapy
3.2.1 Metal ion-based nanoparticles
Manganese (Mn), an essential trace element, has been identified as an endogenous immunomodulator. Manganese ions (Mn²+) exhibit efficient cellular penetration, undergoing active uptake that results in significantly higher intracellular concentrations compared to extracellular levels, Under acidic conditions, MnO2 nanoparticles (MnO2 NPs) react with H2O2 to release Mn²+, which subsequently modulates intracellular oxidative stress and triggers diverse immune responses (64). Mechanistically, Mn²+ activate the cGAS-STING pathway, significantly augmenting type I interferon (IFN-I) secretion, promoting DC maturation, and enhancing the activation of CD8+ T cells and natural killer (NK) cells, with STING-deficient knockout models confirming pathway necessity; Furthermore, Phase I clinical trials demonstrated that Mn²+ combined with anti-PD-1 antibodies elicits promising therapeutic responses in patients with advanced metastatic solid tumors refractory to standard anticancer therapies (including failed combination chemotherapy, radiotherapy, or prior anti-PD-1 treatment) (n=22), achieving an objective response rate (ORR) of 45.5% (10/22) and a disease control rate (DCR) of 90.9% (20/22). These outcomes provide a robust foundation for clinical translation in malignancies including breast and ovarian cancers (65).
3.2.2 Silica-based nanoparticles
Silica-based materials, owing to their high drug-loading capacity and controlled release properties, serve as ideal carriers for STING/TLR agonists. Mesoporous silica nanoparticles (MSNs), surface-functionalized with positively charged TA molecules, enable recognition and uptake by immune cells (e.g., dendritic cells and macrophages), serving as an efficient delivery vehicle for primary immune cells (66). Chen et al. engineered cdG@RMSN-PEG-TA nanoparticles that critically protect c-di-GMP from extracellular enzymatic degradation while enabling sustained intracellular release. This sequential process robustly activates the STING pathway, ultimately promoting DC, macrophage, and T cell infiltration within the TME to significantly inhibit 4T1 breast tumor growth (67). Huang et al. developed TLR7 agonist (TLR7a)-loaded silica nanoparticles (NS-TLR7a), which stimulated TLR7 signaling to amplify immune responses. In CT26 colon cancer models, NS-TLR7a increased T cell infiltration by 4-fold, doubled IFN-γ production, and demonstrated synergistic efficacy with checkpoint inhibitors, showing promise for mismatch repair-proficient (MMRp) colorectal and other MMRp cancers (68).
3.2.3 Biocompatible organic nanoparticles
Low-toxicity biocompatible organic materials are widely employed for nanoparticle synthesis. Rakitina’s team engineered OX40L/PPT nanoparticles by conjugating OX40 ligand (OX40L) with a PEG-PEI-TAT copolymer (PPT). This platform enables charge-mediated complexation with plasmid DNA (pDNA), facilitating cellular uptake and permitting sustained intracellular pDNA release. The prolonged OX40 pathway activation drives persistent type I interferon production, thereby reversing tumor immunosuppression. Combined with PD-1 inhibitors, OX40L/PPT nanoparticles significantly improved survival rates in B16F0 melanoma and CT26 colon cancer models (69). Fu et al. engineered surface-modified nanocomplexes (aPD-1NCs&aOX40)@Gels that facilitate immune cell recognition and uptake. Leveraging redox-responsive properties, the platform enables rapid release of aOX40 followed by sustained release of aPD-1. This sequential release kinetics amplifies and prolongs immune activation. In B16F10 melanoma models, the nanocomplexes significantly enhanced infiltration of CD4+/CD8+ T cells, dendritic cells (DCs), and macrophages (70). Komura et al. engineered guanosine- and uridine-rich (GU-rich) hybrid hydrogels (RDgel) using DNA nanotechnology, which stabilized RNA and sustained TLR7/8 activation to amplify immunostimulatory activity (71).
3.2.4 Imiquimod-loaded nanosystems
To mitigate imiquimod toxicity while enhancing efficacy, researchers have developed imiquimod-loaded nanoparticles. Dias et al. fabricated NPImq via nanoprecipitation of preformed polymers, achieving 20-fold greater anti-angiogenic activity at low doses compared to free imiquimod; furthermore, NPImq treatment significantly reduced mean papilloma diameter versus tumor-induced and placebo groups (p < 0.05) (72). Gazzi et al. formulated imiquimod-loaded polymeric nanocapsules (PEC-NCimiq) embedded in pectin hydrogels, which exhibited superior skin penetration (*particularly in the dermis with 6.8 μg deposition vs 4.3 μg for PEC-imiq*), enhanced adhesion, and controlled release to minimize side effects (73). Frank et al. engineered imiquimod nanoemulsions (NEimiq) via spontaneous emulsification, suppressing SiHa cervical cancer clonogenicity by inducing apoptosis and autophagy (74). Lapteva et al. developed imiquimod-loaded nanogels using mPEG-hexPLA polymers and carboxymethyl cellulose (CMC), demonstrating enhanced tumor targeting and therapeutic efficacy (75).
3.2.5 Stimuli-responsive delivery systems
Researchers have successfully engineered pH-responsive delivery systems for immunostimulatory agonist-loaded nanoparticles that specifically target the TME. Song et al. synthesized epigallocatechin gallate (EGCG, a polyphenolic compound)- and R848 (TLR7/8 agonist)-loaded nanogels (E&R@NG) via polymerization, utilizing pH-sensitive DMAEP crosslinkers to enable acidic TME-triggered drug release. This pH-responsive immunostimulatory nanogel platform provides a synergistic immunotherapy strategy to address the limitations of immune checkpoint monotherapy (76). To overcome the poor tumor-targeting capability of CD40 agonists, Althobaiti et al. engineered low extracellular pH (pHe)-activated membrane-adhesive nanoliposomes encapsulating CD40a (pHTANL-CD40a). The system achieves pH-dependent membrane adhesion, selectively activating CD40 signaling in pancreatic tumors while minimizing systemic toxicity. *Critically, pHTANL-CD40a demonstrated significantly enhanced tumor growth inhibition (61.5% vs 27.3% for free CD40a; p<0.01)*. This approach holds significant potential to enhance the clinical efficacy of CD40 agonist/immune checkpoint inhibitor combinations in PDAC (77).
3.2.6 Lipid and ester-based nanoparticles
Atukorale et al. co-encapsulated the STING agonist cyclic diguanylate monophosphate(cdGMP) and TLR4 pathway agonist monophosphoryl lipid A(MPLA) within lipid nanoparticles, which synergized with PD-1 inhibitors to enhance tumor-clearing CD8+ T cell functionality in melanoma-bearing mice (78). STING/TLR4 co-loaded lipid nanoparticles (immuno-NPs) enable systemic delivery, maintaining payload stability while reducing systemic toxicity, and demonstrate potent efficacy in murine breast cancer models with 50-60% reduction in tumor weight versus vehicle controls (79). Studies further confirmed that STING agonist-loaded lipid nanoparticles (STING-LNPs) activate hepatic macrophages to produce type I interferons (IFN-I), which mobilize systemic NK cells—particularly PD-1+ NK cells—to overcome PD-1 inhibitor resistance in B16F10 melanoma lung metastasis models (80). Local intratumoral injection of CD137/IL-2Fc-anchored liposomes reduced systemic toxicity while amplifying antitumor efficacy in B16F10 melanoma, notably achieving complete tumor regression in 60%-70% of established tumors with no observed recurrence (81). Chang et al. developed a personalized autologous nanovaccine, CpG/cGAMP-hybrid liposome-mannose (C/G-HL-Man). This vaccine enriches in lymph nodes, promotes DC-mediated antigen cross-presentation, and activates tumor-specific cytotoxic T lymphocytes (CTLs); In B16F10 murine models, C/G-HL-Man combined with fenofibrate and PD-1 antibodies suppressed recurrent melanoma progression and extended survival (82). Hanson et al. engineered PEGylated lipid nanoparticles (NP-cdGMP) encapsulating cdGMP to enhance lymphatic trafficking and drainage lymph node (dLN) accumulation. Compared to free CDNs, NP-cdGMP exhibited superior systemic dissemination, dLN retention, and CD8+ T cell activation, significantly improving antitumor outcomes (83).
3.2.7 Innovative delivery strategies and engineered nanoadjuvants
Researchers designed NAcp@CD47 nanocapsules to co-deliver anti-CD47 antibodies and STING agonists into GBM tissues, where the anti-CD47 component blocks the CD47-SIRPα axis to enhance phagocytosis by macrophages and microglia, thereby reducing immunosuppression. Simultaneously, STING agonist activation promotes type-I interferon production, further amplifying antitumor immunity. This dual-action nanocapsule system reprograms the immunosuppressive microenvironment by polarizing microglia and macrophages toward M1-like tumor-associated macrophages (TAMs), ultimately reversing the ‘cold’ tumor phenotype (84). Biodegradable poly(β-amino ester) (PBAE) nanoparticles were engineered to facilitate cytosolic delivery of cyclic dinucleotides (CDNs), reducing cytotoxicity through physiological hydrolysis while enhancing nucleic acid binding efficiency, ultimately improving STING agonist-mediated cancer immunotherapy (85). Wang et al. developed c-di-AMP-loaded nanotubes (CDA-NT hydrogel) by electrostatically coupling the STING agonist with camptothecin (CPT) and the tumor-penetrating peptide iRGD. This system demonstrated potent antitumor efficacy across diverse murine tumor models, eliciting robust responses in GL-261 glioblastoma, CT26 colon carcinoma, and 4T1 breast cancer models; and induced durable immune memory. (86). Zhang et al. engineered self-assembling Ac-KLVFFAL-NH2 (KL-7) peptide nanotubes (PNT) for c-di-GMP delivery. In B16-F10 melanoma models, c-di-GMP-PNT significantly upregulated IFN-β, TNF-α, IL-6, and IL-1β expression, activated CD4+/CD8+ T cells, and enhanced tumor cell killing (87). To optimize CDN delivery, Wu’s team designed a calixarene-based supramolecular cytosolic delivery system (Calix-STING), which promoted melanoma regression, improved PD-1 inhibitor response rates, and established long-term immunological memory (88). Xian et al. chemically modified the non-nucleotide STING agonist MSA-2 with piperazine-functionalized hydrocarbon chains to synthesize nanoadjuvants. These agents robustly activated STING-mediated antitumor immunity in colorectal cancer models, reversed “cold” tumor phenotypes, and exhibited efficacy in triple-negative breast cancer, with further enhancement upon immune checkpoint inhibitor combination (89).
These studies collectively demonstrate that immunostimulatory nanoparticles amplify antitumor efficacy through enhanced antigen presentation, reversal of immunosuppressive microenvironments, and induction of memory T cell formation. By synergizing with chemotherapy, radiotherapy, and checkpoint inhibitors, these strategies establish multimodal therapeutic regimens that achieve durable antitumor immunity. Engineered agonist delivery systems represent a transformative paradigm for precision immunotherapy, particularly in advanced pancreatic cancer, melanoma, and immunologically “cold” tumors, underscoring their pivotal clinical value (Table 2).
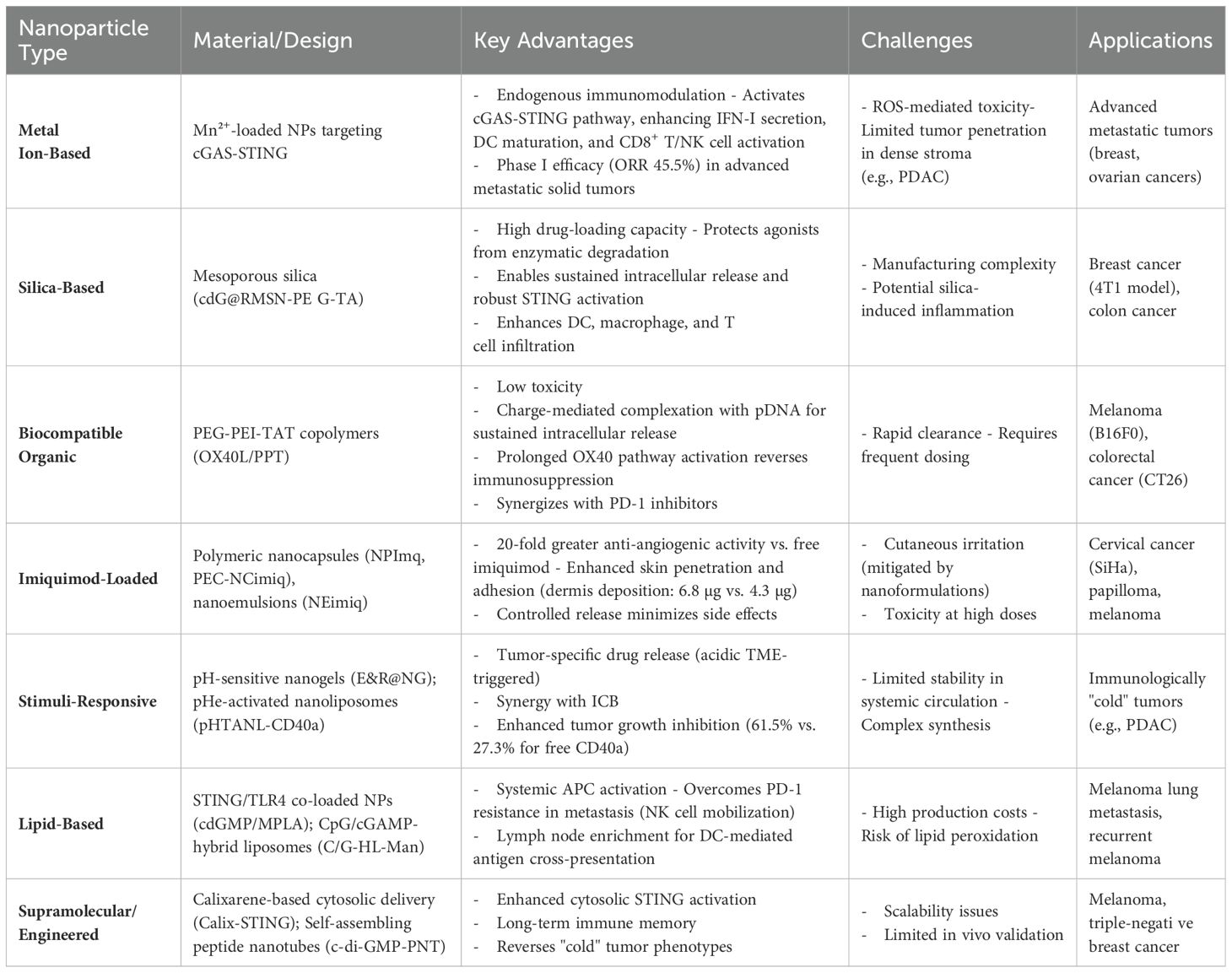
Table 2. Classification and characteristics of immunostimulatory nanoparticles.
4 Potential challenges of immunostimulatory nanoparticles
4.1 Immunogenicity and off-target toxicity
The immune system plays a pivotal role in maintaining physiological homeostasis, and any dysregulation may disrupt this equilibrium, potentially triggering pathological states (90). Distinguishing whether immunotoxicity originates primarily from nanocarrier components or encapsulated agonists is critical for targeted risk mitigation.
Metal oxide nanoparticles induce immunotoxicity via inflammation, oxidative stress, autophagy, and apoptosis pathways (91). Immunotoxicity is governed by physicochemical properties including size, morphology, surface chemistry, and composition. Nanoparticles can activate phagocytes via receptor interactions, leading to reactive oxygen species (ROS)-mediated cell death or tumorigenesis post-uptake (92). Interactions with immune cells (e.g., macrophages, dendritic cells, neutrophils) may further exacerbate inflammatory responses and systemic toxicity (93).
4.2 Complex manufacturing requirements
Nanoparticle production requires high-purity materials (polymers, lipids, albumin), incurring substantial costs that escalate at scale. Furthermore, the fabrication process requires precise control over particle size, morphology, and surface properties, rendering the scale-up from laboratory to industrial production a formidable challenge that demands rigorous quality control protocols (94).
4.3 Barriers to clinical translation
While immunodeficient murine models are useful for evaluating immunotherapy efficacy, they fail to recapitulate the complexity of human immunity, immunocompetent models (e.g., C57BL/6 mice) better approximate human antitumor responses but exhibit interspecies discrepancies in immune regulation. Additionally, murine TMEs oversimplify human stromal complexity, limiting preclinical predictability (95).
5 Conclusion
Immune agonists—targeting STING, TLR, CD40, and OX40 pathways—fundamentally potentiate PD-1 blockade efficacy by reprogramming immunosuppressive tumor microenvironments, converting immunologically “cold” tumors into T cell-inflamed niches through dendritic cell activation, enhanced antigen presentation, and cytotoxic lymphocyte recruitment. Nanoparticle delivery systems critically overcome inherent limitations of free agonists such as enzymatic degradation, off-target toxicity, and poor tumor accumulation. Through targeted cytosolic release exemplified by Calix-STING supramolecular carriers, ligand-directed tumor homing via antibody conjugation, and synergistic codelivery of dual agonists in STING/TLR4 nanoliposomes, nanoplatforms establish transformative synergy with immune agonists and PD-1 inhibitors. This integrated paradigm demonstrates compelling clinical translation: TLR9 agonist vidutolimod nanoparticles achieved 25% objective response in PD-1-resistant melanoma with immunologically cold phenotypes, while Mn²+-STING nanoformulations combined with anti-PD-1 elicited 45.5% response in therapy-refractory metastatic tumors. Similarly, CD40 agonist sotigalimab nanoparticles induced durable remission exceeding 26 months in 15.2% of PD-1 inhibitor-resistant melanoma patients. However, this review acknowledges limitations in comprehensively covering alternative PD-1 enhancement strategies such as bispecific antibodies and oncolytic viruses, which represent significant emerging approaches in the field. Critical translational barriers persist, including nanoparticle-induced immunotoxicity from ROS-mediated inflammation, manufacturing scalability hurdles for complex nanosystems, and interspecies discrepancies in murine models that limit clinical predictability. Future efforts should prioritize toxicity-mitigated combinatorial designs, standardized biomarker validation frameworks, and personalized nanovaccines for immunoquiescent malignancies. Ultimately, the strategic convergence of immune agonist pharmacology with precision nanodelivery establishes engineered nanoparticles as pivotal tools to overcome PD-1 resistance across diverse malignancies.
Author contributions
RX: Conceptualization, Writing – original draft. JL: Writing – original draft, Investigation. JM: Methodology, Writing – original draft. XD: Formal Analysis, Writing – original draft. LM: Writing – review & editing, Validation. XH: Project administration, Writing – review & editing. YW: Project administration, Writing – review & editing. JQ: Funding acquisition, Writing – review & editing, Resources. LY: Resources, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Gansu Provincial Natural Science Foundation (22JR5RA008), the Science and Technology Program of Lanzhou City (2024-9-152), and the Science and Technology Program of Lanzhou City (2024-9-131).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rui R, Zhou L, and He S. Cancer immunotherapies: advances and bottlenecks. Front Immunol. (2023) 14:1212476. doi: 10.3389/fimmu.2023.1212476
2. Sperandio RC, Pestana RC, Miyamura BV, and Kaseb AO. Hepatocellular carcinoma immunotherapy. Annu Rev Med. (2022) 73:267–78. doi: 10.1146/annurev-med-042220-021121
3. Khosravi GR, Mostafavi S, Bastan S, Ebrahimi N, Gharibvand RS, and Eskandari N. Immunologic tumor microenvironment modulators for turning cold tumors hot. Cancer Commun (Lond). (2024) 44:521–53. doi: 10.1002/cac2.12539
4. Liu YT and Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics. (2021) 11:5365–86. doi: 10.7150/thno.58390
5. Preddy I, Nandoliya K, Miska J, and Ahmed AU. Checkpoint: Inspecting the barriers in glioblastoma immunotherapies. Semin Cancer Biol. (2022) 86:473–81. doi: 10.1016/j.semcancer.2022.02.012
6. Yang W, Chang L, Guo Q, Chen J, Yu W, and Zhang W. Programmed cell death protein-1 inhibitors in the treatment of digestive system tumors in Chinese population: an observational study of effectiveness and safety. Ann Palliat Med. (2021) 10:9015–24. doi: 10.21037/apm-21-1827
7. Li Q, Han J, Yang Y, and Chen Y. PD-1/PD-L1 checkpoint inhibitors in advanced hepatocellular carcinoma immunotherapy. Front Immunol. (2022) 13:1070961. doi: 10.3389/fimmu.2022.1070961
8. Chuah S, Lee J, Song Y, Kim HD, Wasser M, Kaya NA, et al. Uncoupling immune trajectories of response and adverse events from anti-PD-1 immunotherapy in hepatocellular carcinoma. J Hepatol. (2022) 77:683–94. doi: 10.1016/j.jhep.2022.03.039
9. De Martin E, Michot JM, Rosmorduc O, Guettier C, and Samuel D. Liver toxicity as a limiting factor to the increasing use of immune checkpoint inhibitors. JHEP Rep. (2020) 2:100170. doi: 10.1016/j.jhepr.2020.100170
10. Xuan C and Hu R. Chemical biology perspectives on STING agonists as tumor immunotherapy. ChemMedChem. (2023) 18:e202300405. doi: 10.1002/cmdc.202300405
11. Hines JB, Kacew AJ, and Sweis RF. The development of STING agonists and emerging results as a cancer immunotherapy. Curr Oncol Rep. (2023) 25:189–99. doi: 10.1007/s11912-023-01361-0
12. Fan YN, Zhao G, Zhang Y, Ye QN, Sun YQ, Shen S, et al. Progress in nanoparticle-based regulation of immune cells. Med Rev (2021). (2023) 3:152–79. doi: 10.1515/mr-2022-0047
13. Hou K, Xu X, Ge X, Jiang J, and Ouyang F. Blockade of PD-1 and CTLA-4: A potent immunotherapeutic approach for hepatocellular carcinoma. Biofactors. (2024) 50:250–65. doi: 10.1002/biof.2012
14. Zhuang Q, Chao T, Wu Y, Wei T, Ren J, Cao Z, et al. Fluorocarbon modified chitosan to enable transdermal immunotherapy for melanoma treatment. Small. (2023) 19:e2303634. doi: 10.1002/smll.202303634
15. Amouzegar A, Chelvanambi M, Filderman JN, Storkus WJ, and Luke JJ. STING agonists as cancer therapeutics. Cancers (Basel). (2021) 13:2695. doi: 10.3390/cancers13112695
16. Mondal I, Das O, Sun R, Gao J, Yu B, Diaz A, et al. PP2Ac deficiency enhances tumor immunogenicity by activating STING-type I interferon signaling in glioblastoma. Cancer Res. (2023) 83:2527–42. doi: 10.1158/0008-5472.CAN-22-3382
17. Xiao Z, Chen H, Xu N, Chen Y, Wang S, and Xu X. MATR3 promotes liver cancer progression by suppressing DHX58-mediated type I interferon response. Cancer Lett. (2024) 604:217231. doi: 10.1016/j.canlet.2024.217231
19. Harrington KJ, Brody J, Ingham M, Strauss J, Cemerski S, Wang M, et al. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann Oncol. (2018) 29(Supplement 8):VIII712. Available online at: https://www.annalsofoncology.org/article/S0923-7534(19)50406-9/fulltext (Accessed January 13, 2025).
20. Meric-Bernstam F, Sweis RF, Kasper S, et al. Combination of the STING agonist MIW815 (ADU-S100) and PD-1 inhibitor spartalizumab in advanced/metastatic solid tumors or lymphomas: an open-label, multicenter, phase ib study. Available online at: https://aacrjournals.org/clincancerres/article/29/1/110/711972/Combination-of-the-STING-Agonist-MIW815-ADU-S100 (Accessed February 5, 2025).
21. Ma H, Kang Z, Foo TK, Shen Z, and Xia B. Disrupted BRCA1-PALB2 interaction induces tumor immunosuppression and T-lymphocyte infiltration in HCC through cGAS-STING pathway. Hepatology. (2023) 77:33–47. doi: 10.1002/hep.32335
22. Jeong S, Choi Y, and Kim K. Engineering therapeutic strategies in cancer immunotherapy via exogenous delivery of toll-like receptor agonists. Pharmaceutics. (2021) 13:1374. doi: 10.3390/pharmaceutics13091374
23. Ota Y, Nagai Y, Hirose Y, Hori S, Koga-Yamakawa E, Eguchi K, et al. DSP-0509, a systemically available TLR7 agonist, exhibits combination effect with immune checkpoint blockade by activating anti-tumor immune effects. Front Immunol. (2023) 14:1055671. doi: 10.3389/fimmu.2023.1055671
24. Curigliano G, Jimenez MM, Shimizu T, Keam B, Meric-Bernstam F, Rutten A, et al. A phase I trial of LHC165 single agent and in combination with spartalizumab in patients with advanced solid Malignancies. ESMO Open. (2024) 9:103643. doi: 10.1016/j.esmoop.2024.103643
25. Sullivan RJ. For whom the bell tolls? A toll-like receptor 9 agonist’s journey from vaccine adjuvant to promising agent in anti-PD-1-resistant melanoma. Cancer Discov. (2021) 11:2960. doi: 10.1158/2159-8290.CD-21-1226
26. Dang N, Waer M, Sprangers B, and Lin Y. Intratumoral immunotherapy with anti-PD-1 and TLR9 agonist induces systemic antitumor immunity without accelerating rejection of cardiac allografts. Am J Transplant. (2021) 21:60–72. doi: 10.1111/ajt.16105
27. Jiang T, Zhang H, Li Y, Jayakumar P, Liao H, Huang H, et al. Intraperitoneal injection of class A TLR9 agonist enhances anti-PD-1 immunotherapy in colorectal peritoneal metastases. JCI Insight. (2022) 7:e160063. doi: 10.1172/jci.insight.160063
28. Jimenez J, Amrute J, Ma P, Wang X, Dai R, and Lavine KJ. CD40 is an immune checkpoint regulator that potentiates myocardial inflammation through activation and expansion of CCR2 + macrophages and CD8 T-cells. bioRxiv. (2024). doi: 10.1101/2024.03.14.584418
29. Weiss SA, Sznol M, Shaheen M, Berciano-Guerrero MÁ, Couselo EM, Rodríguez-Abreu D, et al. A phase II trial of the CD40 agonistic antibody sotigalimab (APX005M) in combination with nivolumab in subjects with metastatic melanoma with confirmed disease progression on anti-PD-1 therapy. Clin Cancer Res. (2024) 30:74–81. doi: 10.1158/1078-0432.CCR-23-0475
30. Kobayashi C, Suzuki-Imaizumi M, Sakaguchi Y, Ishii T, Adachi M, Kaneda A, et al. The novel and potent CD40 agonist KHK2840 augments the antitumor efficacy of anti-PD-1 antibody and paclitaxel. Cancer Sci. (2024) 115:4008–20. doi: 10.1111/cas.16366
31. Khan SM, Desai R, Coxon A, Livingstone A, Dunn GP, Petti A, et al. Impact of CD4 T cells on intratumoral CD8 T-cell exhaustion and responsiveness to PD-1 blockade therapy in mouse brain tumors. J Immunother Cancer. (2022) 10:e005293. doi: 10.1136/jitc-2022-005293
32. Diggs LP, Ruf B, Ma C, Heinrich B, Cui L, Zhang Q, et al. CD40-mediated immune cell activation enhances response to anti-PD-1 in murine intrahepatic cholangiocarcinoma. J Hepatol. (2021) 74:1145–54. doi: 10.1016/j.jhep.2020.11.037
33. Redmond WL. Challenges and opportunities in the development of combination immunotherapy with OX40 agonists. Expert Opin Biol Ther. (2023) 23:901–12. doi: 10.1080/14712598.2023.2249396
34. Postel-Vinay S, Lam VK, Ros W, Bauer TM, Hansen AR, Cho DC, et al. First-in-human phase I study of the OX40 agonist GSK3174998 with or without pembrolizumab in patients with selected advanced solid tumors (ENGAGE-1). J Immunother Cancer. (2023) 11:e005301. doi: 10.1136/jitc-2022-005301
35. Alvim RG, Georgala P, Nogueira L, Somma AJ, Nagar K, Thomas J, et al. Combined OX40 agonist and PD-1 inhibitor immunotherapy improves the efficacy of vascular targeted photodynamic therapy in a urothelial tumor model. Molecules. (2021) 26:3744. doi: 10.3390/molecules26123744
36. Min L, Wang X, Chen A, Zhou Y, Ge Y, Dai J, et al. Design of a single-center, phase II trial to explore the efficacy and safety of ‘R-ISV-RO’ treatment in advanced tumors. Future Oncol. (2024) 20:1139–49. doi: 10.2217/fon-2023-0962
37. Huang C, Shao N, Huang Y, Chen J, Wang D, Hu G, et al. Overcoming challenges in the delivery of STING agonists for cancer immunotherapy: A comprehensive review of strategies and future perspectives. Mater Today Bio. (2023) 23:100839. doi: 10.1016/j.mtbio.2023.100839
38. Messaoud-Nacer Y, Culerier E, Rose S, Maillet I, Rouxel N, Briault S, et al. STING agonist diABZI induces PANoptosis and DNA mediated acute respiratory distress syndrome (ARDS). Cell Death Dis. (2022) 13:269. doi: 10.1038/s41419-022-04664-5
39. Hu X, Zhang H, Zhang Q, Yao X, Ni W, and Zhou K. Emerging role of STING signalling in CNS injury: inflammation, autophagy, necroptosis, ferroptosis and pyroptosis. J Neuroinflammation. (2022) 19:242. doi: 10.1186/s12974-022-02602-y
40. Pham PT, Fukuda D, Nishimoto S, Kim-Kaneyama JR, Lei XF, Takahashi Y, et al. STING, a cytosolic DNA sensor, plays a critical role in atherogenesis: a link between innate immunity and chronic inflammation caused by lifestyle-related diseases. Eur Heart J. (2021) 42:4336–48. doi: 10.1093/eurheartj/ehab249
41. Wu J, Liu Q, Zhang X, Wu X, Zhao Y, and Ren J. STING-dependent induction of lipid peroxidation mediates intestinal ischemia-reperfusion injury. Free Radic Biol Med. (2021) 163:135–40. doi: 10.1016/j.freeradbiomed.2020.12.010
42. Matthijs AMF, Auray G, Jakob V, García-Nicolás O, Braun RO, Keller I, et al. Systems immunology characterization of novel vaccine formulations for mycoplasma hyopneumoniae bacterins. Front Immunol. (2019) 10:1087. doi: 10.3389/fimmu.2019.01087
43. Rolfo C, Giovannetti E, Martinez P, McCue S, and Naing A. Applications and clinical trial landscape using Toll-like receptor agonists to reduce the toll of cancer. NPJ Precis Oncol. (2023) 7:26. doi: 10.1038/s41698-023-00364-1
44. Oboge H, Riitho V, Nyamai M, Omondi GP, Lacasta A, Githaka N, et al. Safety and efficacy of toll-like receptor agonists as therapeutic agents and vaccine adjuvants for infectious diseases in animals: a systematic review. Front Vet Sci. (2024) 11:1428713. doi: 10.3389/fvets.2024.1428713
45. Steeghs N, Hansen AR, Hanna GJ, Garralda E, Park H, Strauss J, et al. Manufacturing-dependent change in biological activity of the TLR4 agonist GSK1795091 and implications for lipid A analog development. Clin Transl Sci. (2022) 15:2625–39. doi: 10.1111/cts.13387
46. Bonnans C, Thomas G, He W, Jung B, Chen W, Liao M, et al. CD40 agonist-induced IL-12p40 potentiates hepatotoxicity. J Immunother Cancer. (2020) 8:e000624. doi: 10.1136/jitc-2020-000624
47. Caudell DL, Dugan GO, Babitzki G, Schubert C, Braendli-Baiocco A, Wasserman K, et al. Systemic immune response to a CD40 agonist antibody in nonhuman primates. J Leukoc Biol. (2024) 115:1084–93. doi: 10.1093/jleuko/qiae031
48. Hamid O, Chiappori AA, Thompson JA, Doi T, Hu-Lieskovan S, Eskens FALM, et al. First-in-human study of an OX40 (ivuxolimab) and 4-1BB (utomilumab) agonistic antibody combination in patients with advanced solid tumors. J Immunother Cancer. (2022) 10:e005471. doi: 10.1136/jitc-2022-005471
49. Goldman JW, Piha-Paul SA, Curti B, Pedersen KS, Bauer TM, Groenland SL, et al. Safety and tolerability of MEDI0562, an OX40 agonist mAb, in combination with durvalumab or tremelimumab in adult patients with advanced solid tumors. Clin Cancer Res. (2022) 28:3709–19. doi: 10.1158/1078-0432.CCR-21-3016
50. Shrimali RK, Ahmad S, Verma V, Zeng P, Ananth S, Gaur P, et al. Concurrent PD-1 blockade negates the effects of OX40 agonist antibody in combination immunotherapy through inducing T-cell apoptosis. Cancer Immunol Res. (2017) 5:755–66. doi: 10.1158/2326-6066.CIR-17-0292
51. Wu JJ, Zhao L, Hu HG, Li WH, and Li YM. Agonists and inhibitors of the STING pathway: Potential agents for immunotherapy. Med Res Rev. (2020) 40:1117–41. doi: 10.1002/med.21649
52. Petrovic M, Borchard G, and Jordan O. Considerations for the delivery of STING ligands in cancer immunotherapy. J Control Release. (2021) 339:235–47. doi: 10.1016/j.jconrel.2021.09.033
53. Garland KM, Sheehy TL, and Wilson JT. Chemical and biomolecular strategies for STING pathway activation in cancer immunotherapy. Chem Rev. (2022) 122:5977–6039. doi: 10.1021/acs.chemrev.1c00750
54. Scurtu LG, Jinga V, and Simionescu O. Fascinating molecular and immune escape mechanisms in the treatment of STIs (Syphilis, gonorrhea, chlamydia, and herpes simplex). Int J Mol Sci. (2022) 23:3550. doi: 10.3390/ijms23073550
55. Neetika, Sharma M, Thakur P, Gaur P, Rani GM, Rustagi S, et al. Cancer treatment and toxicity outlook of nanoparticles. Environ Res. (2023) 237:116870. doi: 10.1016/j.envres.2023.116870
56. Swartz MA, Hirosue S, and Hubbell JA. Engineering approaches to immunotherapy. Sci Transl Med. (2012) 4:148rv9. doi: 10.1126/scitranslmed.3003763
57. Wang J, Li S, Wang M, Wang X, Chen S, Sun Z, et al. STING licensing of type I dendritic cells potentiates antitumor immunity. bioRxiv. (2024). doi: 10.1101/2024.01.02.573934
58. Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. (2011) 6:815–23. doi: 10.1038/nnano.2011.166
59. Tanaka HY and Kano MR. Stromal barriers to nanomedicine penetration in the pancreatic tumor microenvironment. Cancer Sci. (2018) 109:2085–92. doi: 10.1111/cas.13630
60. Lorkowski ME, Atukorale PU, Bielecki PA, Tong KH, Covarrubias G, Zhang Y, et al. Immunostimulatory nanoparticle incorporating two immune agonists for the treatment of pancreatic tumors. J Control Release. (2021) 330:1095–105. doi: 10.1016/j.jconrel.2020.11.014
61. Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun. (2015) 6:8692. doi: 10.1038/ncomms9692
62. Ikeda-Imafuku M, Wang LLW, Rodrigues D, Shaha S, Zhao Z, and Mitragotri S. Strategies to improve the EPR effect: A mechanistic perspective and clinical translation. J Control Release. (2022) 345:512–36. doi: 10.1016/j.jconrel.2022.03.043
63. Sindhwani S, Syed AM, Ngai J, Kingston BR, Maiorino L, Rothschild J, et al. The entry of nanoparticles into solid tumours. Nat Mater. (2020) 19:566–75. doi: 10.1038/s41563-019-0566-2
64. Huang Y, Ruan Y, Ma Y, Chen D, Zhang T, Fan S, et al. Immunomodulatory activity of manganese dioxide nanoparticles: Promising for novel vaccines and immunotherapeutics. Front Immunol. (2023) 14:1128840. doi: 10.3389/fimmu.2023.1128840
65. Lv M, Chen M, Zhang R, Zhang W, Wang C, Zhang Y, et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. (2020) 30:966–79. doi: 10.1038/s41422-020-00395-4
66. Heidegger S, Gössl D, Schmidt A, Niedermayer S, Argyo C, Endres S, et al. Immune response to functionalized mesoporous silica nanoparticles for targeted drug delivery. Nanoscale. (2016) 8:938–48. doi: 10.1039/c5nr06122a
67. Chen YP, Xu L, Tang TW, Chen CH, Zheng QH, Liu TP, et al. STING activator c-di-GMP-loaded mesoporous silica nanoparticles enhance immunotherapy against breast cancer. ACS Appl Mater Interfaces. (2020) 12:56741–52. doi: 10.1021/acsami.0c16728
68. Huang CH, Mendez N, Echeagaray OH, Weeks J, Wang J, Yao S, et al. Immunostimulatory TLR7 agonist-nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Adv Ther (Weinh). (2020) 3:1900200. doi: 10.1002/adtp.201900200
69. Rakitina OA, Kuzmich AI, Bezborodova OA, Kondratieva SA, Pleshkan VV, Zinovyeva MV, et al. Non-viral-mediated gene transfer of OX40 ligand for tumor immunotherapy. Front Immunol. (2024) 15:1410564. doi: 10.3389/fimmu.2024.1410564
70. Fu Y, Huang Y, Li P, Wang L, Tang Z, Liu X, et al. Physical- and chemical-dually ROS-responsive nano-in-gel platforms with sequential release of OX40 agonist and PD-1 inhibitor for augmented combination immunotherapy. Nano Lett. (2023) 23:1424–34. doi: 10.1021/acs.nanolett.2c04767
71. Komura F, Okuzumi K, Takahashi Y, Takakura Y, and Nishikawa M. Development of RNA/DNA hydrogel targeting toll-like receptor 7/8 for sustained RNA release and potent immune activation. Molecules. (2020) 25:728. doi: 10.3390/molecules25030728
72. Dias MF, Figueiredo BCP, Teixeira-Neto J, Guerra MCA, Fialho SL, and Silva Cunha A. In vivo evaluation of antitumoral and antiangiogenic effect of imiquimod-loaded polymeric nanoparticles. BioMed Pharmacother. (2018) 103:1107–14. doi: 10.1016/j.biopha.2018.04.079
73. Gazzi RP, Frank LA, Onzi G, Pohlmann AR, and Guterres SS. New pectin-based hydrogel containing imiquimod-loaded polymeric nanocapsules for melanoma treatment. Drug Delivery Transl Res. (2020) 10:1829–40. doi: 10.1007/s13346-020-00805-5
74. Frank LA, Gazzi RP, Mello PA, Chaves P, Peña F, Beck RCR, et al. Anti-HPV nanoemulsified-imiquimod: A new and potent formulation to treat cervical cancer. AAPS PharmSciTech. (2020) 21:54. doi: 10.1208/s12249-019-1558-x
75. Lapteva M, Mignot M, Mondon K, Möller M, Gurny R, and Kalia YN. Self-assembled mPEG-hexPLA polymeric nanocarriers for the targeted cutaneous delivery of imiquimod. Eur J Pharm Biopharm. (2019) 142:553–62. doi: 10.1016/j.ejpb.2019.01.008
76. Song Q, Zhang G, Wang B, Cao G, Li D, Wang Y, et al. Reinforcing the combinational immuno-oncotherapy of switching “Cold” Tumor to “Hot” by responsive penetrating nanogels. ACS Appl Mater Interfaces. (2021) 13:36824–38. doi: 10.1021/acsami.1c08201
77. Althobaiti S, Parajuli P, Luong D, Sau S, Polin LA, Kim S, et al. Enhanced safety and efficacy profile of CD40 antibody upon encapsulation in pHe-triggered membrane-adhesive nanoliposomes. Nanomedicine (Lond). (2025) 20:155–66. doi: 10.1080/17435889.2024.2446008
78. Atukorale PU, Moon TJ, Bokatch AR, Lusi CF, Routhier JT, Deng VJ, et al. Dual agonist immunostimulatory nanoparticles combine with PD1 blockade for curative neoadjuvant immunotherapy of aggressive cancers. Nanoscale. (2022) 14:1144–59. doi: 10.1039/D1NR06577G
79. Atukorale PU, Raghunathan SP, Raguveer V, Moon TJ, Zheng C, Bielecki PA, et al. Nanoparticle encapsulation of synergistic immune agonists enables systemic codelivery to tumor sites and IFNβ-driven antitumor immunity. Cancer Res. (2019) 79:5394–406. doi: 10.1158/0008-5472.CAN-19-0381
80. Nakamura T, Sato T, Endo R, Sasaki S, Takahashi N, Sato Y, et al. STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. J Immunother Cancer. (2021) 9:e002852. doi: 10.1136/jitc-2021-002852
81. Kwong B, Gai SA, Elkhader J, Wittrup KD, and Irvine DJ. Localized immunotherapy via liposome-anchored Anti-CD137 + IL-2 prevents lethal toxicity and elicits local and systemic antitumor immunity. Cancer Res. (2013) 73:1547–58. doi: 10.1158/0008-5472.CAN-12-3343
82. Chang L, Fu S, Gao T, Sang X, Yang H, Liu X, et al. Regulating T-cell metabolic reprogramming and blocking PD-1 co-promote personalized postoperative autologous nanovaccines. Biomaterials. (2023) 297:122104. doi: 10.1016/j.biomaterials.2023.122104
83. Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, et al. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest. (2015) 125:2532–46. doi: 10.1172/JCI79915
84. Zhou Y, Guo Y, Chen L, Zhang X, Wu W, Yang Z, et al. Co-delivery of phagocytosis checkpoint and STING agonist by a Trojan horse nanocapsule for orthotopic glioma immunotherapy. Theranostics. (2022) 12:5488–503. doi: 10.7150/thno.73104
85. Wilson DR, Sen R, Sunshine JC, Pardoll DM, Green JJ, and Kim YJ. Biodegradable STING agonist nanoparticles for enhanced cancer immunotherapy. Nanomedicine. (2018) 14:237–46. doi: 10.1016/j.nano.2017.10.013
86. Wang F, Su H, Xu D, Dai W, Zhang W, Wang Z, et al. Tumour sensitization via the extended intratumoural release of a STING agonist and camptothecin from a self-assembled hydrogel. Nat BioMed Eng. (2020) 4:1090–101. doi: 10.1038/s41551-020-0597-7
87. Zhang Z, Liu J, Xiao M, Zhang Q, Liu Z, Liu M, et al. Peptide nanotube loaded with a STING agonist, c-di-GMP, enhance cancer immunotherapy against melanoma. Nano Res. (2023) 16:5206–15. doi: 10.1007/s12274-022-5102-z
88. Wu JJ, Chen FY, Han BB, Zhang HQ, Zhao L, Zhang ZR, et al. CASTING: A potent supramolecular strategy to cytosolically deliver STING agonist for cancer immunotherapy and SARS-CoV-2 vaccination. CCS Chem. (2022) 5:885–901. doi: 10.31635/ccschem.022.202201859
89. Xian S, Chen X, Ren S, Chen X, and Wang H. Ionizable STING-activating nanoadjuvants enhance tumor immunogenicity and potentiate immunotherapy efficacy in solid tumors. Cancer Res. (2024) 84:3044–57. doi: 10.1158/0008-5472.CAN-23-3511
90. Luebke R. Immunotoxicant screening and prioritization in the twenty-first century. Toxicol Pathol. (2012) 40:294–9. doi: 10.1177/0192623311427572
91. Bi J, Mo C, Li S, Huang M, Lin Y, Yuan P, et al. Immunotoxicity of metal and metal oxide nanoparticles: from toxic mechanisms to metabolism and outcomes. Biomater Sci. (2023) 11:4151–83. doi: 10.1039/d3bm00271c
92. Muhammad Q, Jang Y, Kang SH, Moon J, Kim WJ, and Park H. Modulation of immune responses with nanoparticles and reduction of their immunotoxicity. Biomater Sci. (2020) 8:1490–501. doi: 10.1039/c9bm01643k
93. Aljabali AA, Obeid MA, Bashatwah RM, Serrano-Aroca Á, Mishra V, Mishra Y, et al. Nanomaterials and their impact on the immune system. Int J Mol Sci. (2023) 24:2008. doi: 10.3390/ijms24032008
94. Chen C, Xu Y, Meng H, Bao H, Hu Y, Li C, et al. Nano-oncologic vaccine for boosting cancer immunotherapy: the horizons in cancer treatment. Nanomaterials (Basel). (2025) 15:122. doi: 10.3390/nano15020122
Keywords: immune agonists, PD-1 inhibitors, nanoparticle delivery systems, tumor immunotherapy, combinatorial therapy, immunotoxicity
Citation: Xia R, Liang J, Ma J, Du X, Ma L, Han X, Wang Y, Qin J and Yan L (2025) Research advances in immune agonists and their nanoparticles for enhancing the immunotherapeutic efficacy of PD-1 inhibitors in malignancies. Front. Oncol. 15:1618903. doi: 10.3389/fonc.2025.1618903
Received: 27 April 2025; Accepted: 31 July 2025;
Published: 26 August 2025.
Edited by:
Alicia Fernandez-Fernandez, Nova Southeastern University, United StatesReviewed by:
Hui-Yen Chuang, National Yang Ming Chiao Tung University, TaiwanCopyright © 2025 Xia, Liang, Ma, Du, Ma, Han, Wang, Qin and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Long Yan, bHp6eTk0MEAxNjMuY29t; Jianwei Qin, cWluamlhbndlaTk0MEAxNjMuY29t
†These authors have contributed equally to this work