 Duo Jin
Duo Jin Dong Zhang
Dong Zhang Qinyu Tang4*†
Qinyu Tang4*†- 1Comprehensive Laboratory, The First People’s Hospital of Changzhou, Changzhou, China
- 2Comprehensive Laboratory, The Third Affiliated Hospital of Soochow University, Changzhou, China
- 3The Ruminant Disease Early Warning, Prevention and Control Technology Innovation Team, Institute of Special Wild Economic Animals and Plants, Chinese Academy of Agricultural Sciences, Changchun, China
- 4Department of Dermatology, The Second People’s Hospital of Changzhou, the Third Affiliated Hospital of Nanjing Medical University, Changzhou, China
Neuroblastoma (NB) is a prevalent pediatric malignancy with poor clinical outcomes. As one of the most common childhood malignancies, it can arise in various locations along the sympathetic nervous system, complicating both fundamental studies and therapeutic approaches. The ARID1A protein has emerged as a pivotal regulator in the pathogenesis of diverse tumor types within oncology research. Recent studies have increasingly focused on the functional role of ARID1A in NB pathogenesis. As a tumor suppressor, ARID1A loss-of-function mutations enhance migratory and invasive capacities of NB cells through cell cycle dysregulation, thereby promoting tumor cell proliferation. At the molecular level, ARID1A functions as the core subunit of the BAF chromatin remodeling complex, critically regulating the proliferative behavior of tumor cells. Although research in this field remains at an early stage, it has established a solid foundation for elucidating NB pathogenesis, with promising implications for improving clinical outcomes and quality of life in affected children. This review summarizes the critical role of ARID1A in NB and explores emerging therapeutic strategies, with particular emphasis on targeted protein degradation approaches and immunotherapeutic interventions.
Introduction
Neuroblastoma (NB) is the fourth most common pediatric tumor, and affects 25–50 persons per million and accounts for 10-20% of pediatric malignancy related mortality (1). It can grow anywhere in the sympathetic nervous system since it affects neural crest-derived cells (2). NB may originate from multiple neural crest-derived cell types (e.g., neuroblasts, chromaffin cells, or mesenchymal-like cells) when genomic and epigenetic defects disrupt their normal differentiation during development (3). The precise moment and the specific cell or cells from which it arises remain uncertain and are still being investigated (4). The heterogeneity of NB is also reflected in the 5-year survival rate. Favorable NB, particularly low- and intermediate-risk cases, may spontaneously regress or differentiate without intervention, demonstrating 90-95% survival rates. Even with two novel therapies (anti-GD2 immunotherapy and tandem autologous stem cell transplantation), 40-50% of patients remain unresponsive to conventional treatments including surgery, chemotherapy, and immunotherapy (5, 6).
The BAF complex uses ATP hydrolysis to remodel nucleosomes and regulate gene expression (7). ARID1A serves as a core structural subunit of the BAF complex, mediating critical interactions with SMARCA4 and the base module to maintain complex integrity (8). Shuang He (8) et al. demonstrated that ARID1A deficiency disrupts BAF complex folding, significantly impairing nucleosome remodeling activity.
ARID1A recurrent mutations exhibit a broad tumor distribution, with particularly high prevalence in endometrioid and clear-cell ovarian cancers, gastric cancer, bladder cancer (9, 10) and neuroblastomas(~6%) (11, 12). Most ARID1A mutations are inactivating (frameshift or truncation) and distributed across the entire gene. These loss-of-function mutations are common in cancer, mainly including frameshifts, nonsense mutations, rearrangements, truncations, and splice-site defects (13). Emerging evidence indicates that ARID1A mutations serve as an independent prognostic biomarker, with loss-of-function variants significantly associated with reduced cancer-specific survival, increased recurrence rates (9) and diminished therapeutic response (14).
ARID1A has emerged as a critical tumor suppressor in cancer research. Although its role in NB remains poorly characterized, elucidating ARID1A-NB interactions may provide important insights into NB pathogenesis. This review summarizes current evidence on ARID1A in NB and discusses potential ARID1A-targeted therapeutic strategies.
Mutations in ARID1A cause increased migration and invasion in NB
The association between ARID1A and NB was first reported in 2001 by Takeuchi et al. (15), who identified amplified expression of a truncated ARID1A isoform (SMARCF1/B120) in NB. Their immunohistochemical analysis of 23 NB cases demonstrated detectable expression of this truncated protein. The B120 gene product (truncated ARID1A) showed significant cytoplasmic and nuclear expression in 4/23 NB cases. However, these findings received limited attention, and ARID1A/ARID1B were still considered novel neuroblastoma-associated genes as late as 2013. Genomic analyses (whole-genome sequencing, rearrangement mapping, and exome sequencing) identified ARID1A/ARID1B deletions or mutations in 8/71 pediatric NB cases, which correlated with early treatment failure and poorer survival (11). This study further analyzed the functional roles of ARID1A and ARID1B. Notably, ARID1B primarily exhibited deletion mutations, whereas ARID1A showed point mutations or compound mutations with biallelic deletions. In 2020, Flora Cimmino et al. (16) developed a novel NGS (next generation sequencing) approach to identify the special change in cDNA of NB patients, which included changes in the ARID1A gene. This role of ARID1A was further affirmed in 2023 by Zekiye Altun (17) et al. Through whole-exome sequencing and analysis using the cBioPortal cancer genomic database, their study identified ARID1A as one of two key high-risk genes.
C. Li (18) et al. demonstrated that ARID1A knockdown in SK-N-SH NB cells using shRNA significantly enhanced cell proliferation. Flow cytometry analysis confirmed cell cycle dysregulation, showing decreased G0/G1 phase and increased S phase populations. These results demonstrate that ARID1A knockdown disrupts cell cycle progression. Meanwhile, wound healing and transwell assays showed ARID1A silencing significantly increased cell migration rates and enhanced invasive capacity. Extensive studies have established ARID1A as a high-risk factor in NB pathogenesis. Multiple experimental approaches have consistently validated this finding. However, the molecular mechanisms underlying the regulation of ARID1A in NB remain unclear.
Chromosomal instability (CIN) is implicated in 90% of malignant tumors, with increasing severity correlating with worse prognosis (19). The 1p36 locus is among the most frequently deleted chromosomal regions across diverse malignancies, including neural, epithelial, and hematopoietic cancers. The 1p36 deletion exhibits a strong predilection for neurological tumors, with particularly high prevalence in NB (20). Notably, comprehensive genomic analyses reveal that approximately 70% of MYCN-driven NB cases harbor 1p36 deletions (21). MYCN gene amplification and overexpression represents one of the most prevalent high-risk genetic alterations in NB, occurring in approximately 25% of cases (22). Jesus García-López et al. (21) demonstrated through in vitro transformation assays using primary mouse neural crest cells (NCCs) that MYCN-driven carcinogenesis differentially transforms NCCs with ARID1A deletion compared to those with random 1p36 deletions, thereby identifying ARID1A as a MYCN-specific tumor suppressor. Hui Shi et al. (12) employed a zebrafish model [Tg(dβh:EGFP-MYCN)] to investigate the ARID1A-MYCN interaction in NB, wherein the dβh promoter drives EGFP-MYCN fusion gene expression for tumor visualization. ARID1A knock-down mutations were detected in early-onset tumors (5–13 weeks post-fertilization, wpf) but not in late-onset tumors (15–27 wpf). These results demonstrate that ARID1A loss-of-function advances the onset of MYCN-driven NB by 10–14 weeks in the Tg(dβh:EGFP-MYCN) model. Therefore, the deletion of ARID1A and overexpression of MYCN are synergistic in NB development and also imply a tumor suppressive role of ARID1A in NB (Figure 1 ARID1A acts as a tumor suppression gene of MYCN-driven NB.). Furthermore, RNA sequencing (RNA-seq) and Western blot analyses, combined with Transwell invasion assays, revealed that ARID1A depletion in MYCN-driven NB induces a phenotypic shift from the differentiated adrenergic (ADRN) state to a more aggressive mesenchymal (MES) state, as evidenced by altered expression of lineage-specific markers and enhanced tumor invasiveness (12, 23). In summary, these findings establish a critical functional interplay between ARID1A and MYCN in NB pathogenesis. Specifically, ARID1A acts as a tumor suppressor, and its loss exacerbates malignant progression by enhancing tumor cell invasiveness and metastatic potential. However, as a core subunit of the BAF chromatin remodeling complex, the mechanistic contribution of ARID1A within this complex to NB biology remains poorly understood.
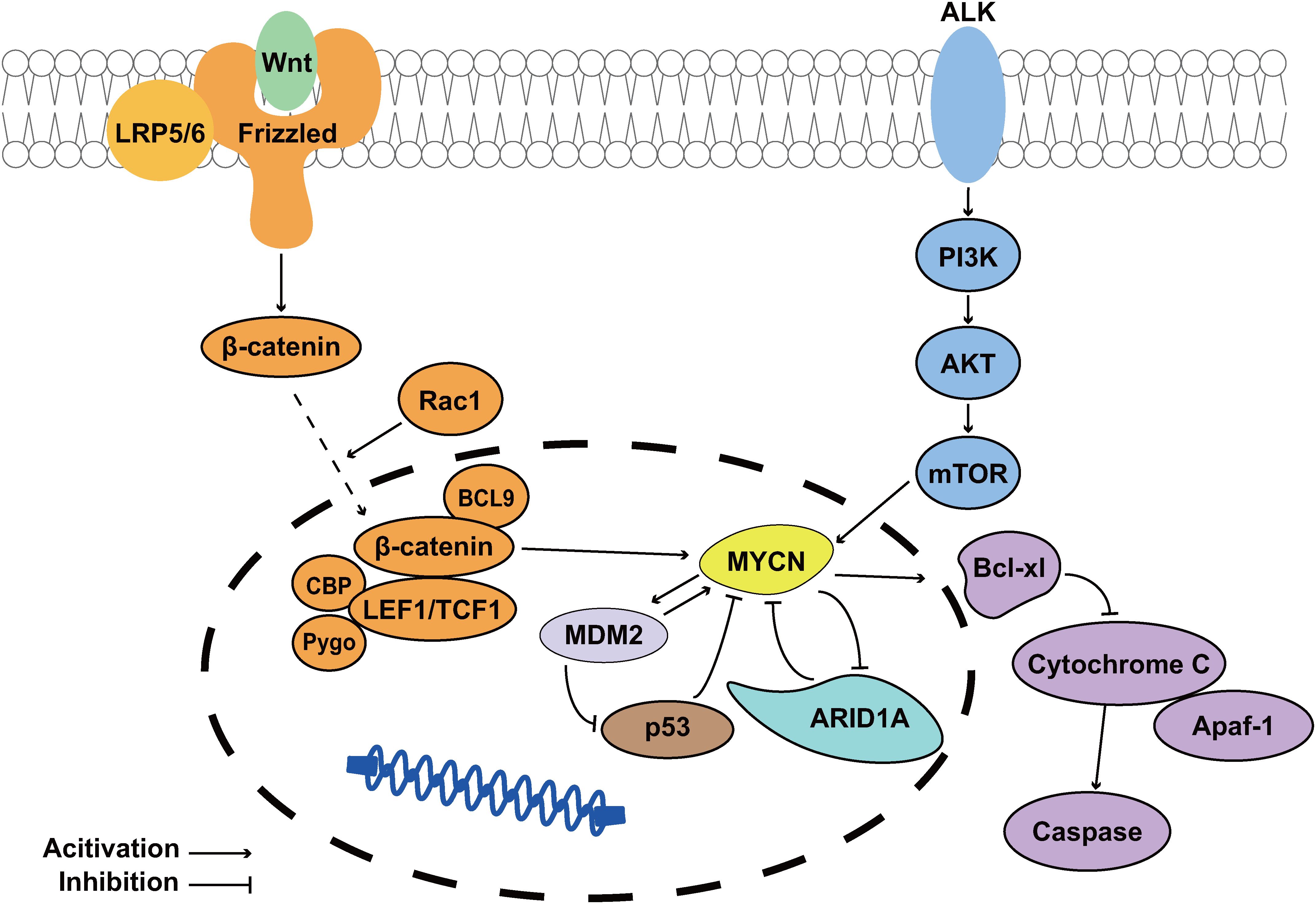
Figure 1. ARID1A acts as a tumor suppression gene of MYCN-driven NB. The PI3K/AKT/mTOR and Wnt signaling pathways upregulate MYCN expression, playing crucial roles in NB cell survival and drug resistance development. MDM2 suppresses the tumor suppressor p53 while simultaneously enhancing MYCN expression (24); The MYCN-encoded protein N-Myc transcriptionally upregulates Bcl-xL, thereby inhibiting apoptosis in NB cells (25, 26); Notably, ARID1A functions as a MYCN suppressor, and its loss promotes MYCN-driven NB pathogenesis.
Disruption of the BAF complex in which ARID1A resides inhibits NB proliferation
The BAF chromatin remodeling complex plays critical roles in tumorigenesis across multiple cancer types. As ARID1A is an essential subunit of this complex, an important question arises: To what extent can the functional consequences of ARID1A alterations be extrapolated to the ARID1A-containing BAF chromatin remodeling complex. Bieke Decaesteker et al. (27) found that SOX11 regulated 10 core proteins of the BAF complex, including ARID1A. Their findings indicate that the observed suppression of NB invasiveness and metastatic potential resulted from structural destabilization of the BAF complex, mediated through SOX11-dependent silencing of both its essential subunits and direct target ARID1A. This result differs from the effects observed with ARID1A as an individual component. Since SOX11 regulates 10 subunits of the BAF complex besides ARID1A, the disparity in conclusions might stem from strong alterations in the remaining subunits. It is also possible that the BAF complex, under the synergistic effect of multiple genes, exerts effects inconsistent with those of ARID1A alone. Alterations in the remaining 9 subunits could potentially lead to disruption of the integrity of the BAF complex and subsequently the balance of regulation between individual subunits.
It is hypothesized that this outcome is attributed to the disruption of the integrity of the BAF complex. Another study conducted in 2022 lends support to this hypothesis (28). Silencing the BAF-specific subunits ARID1A and ARID1B simultaneously reveals that NB cell proliferation depends significantly on the structural integrity of the BAF complex. In NB cells, this perturbation markedly reduced cyclin D1 protein levels and Rb phosphorylation, while flow cytometry analysis confirmed significant G1-phase accumulation with concomitant decreases in S and G2/M-phase populations. BAF complex disruption induced transcriptome-wide reprogramming in NB cells, altering cell cycle regulators and triggering G1-phase arrest, ultimately suppressing proliferation (Figure 2) (28). A 2023 study by Katerina Cermakova (29) et al. provided further evidence that the BAF complex is essential for maintaining adrenergic core regulatory circuitries (CRCs) in NB, primarily by preserving the open chromatin state of G1-phase enhancers and activating cell cycle-related genes. Inactivation of the BAF complex severely impairs the G1-to-S phase transition, leading to G1 arrest and ultimately cell death. These findings demonstrate that the BAF complex, as an integrated unit, differs markedly from ARID1A in both its mechanistic actions and phenotypic outcomes, underscoring the unique role of ARID1A in NB pathogenesis and highlighting its significance as a critical protein warranting further investigation.

Figure 2. Targeting of ARID1A results in disassembly of the BAF complex, affecting the cell cycle of NB cells. (A) Complete BAF Complex and normal cell cycle. (B) The disintegration of the BAF complex is accompanied by an abnormal cell cycle mainly characterized by the prolongation of the G1 phase.
While the architecture of the BAF complex has been partially characterized, structural insights into the ARID1A-deficient model remain elusive, while the structure of the BAF complex has been partially characterized, structural insights into the ARID1A-deficient model remain elusive. Furthermore, neither the standalone structure of ARID1A protein nor its interactive architectures with other proteins in NB have been resolved.Current structural data of the intact BAF complex lack complete information about ARID1A as well (8), fundamentally limiting our understanding of its mechanistic role. Future studies should employ cryo-EM and other structural biology approaches to determine how ARID1A deficiency alters the complex’s conformation and functional properties.
Future research directions at the therapeutic strategies in ARID1A-mutant NB
Most studies associate ARID1A loss with poor prognosis and tumor progression, recent studies suggest it may also increase susceptibility to specific therapies. Gene promoter hypermethylation correlated with reduced ARID1A expression in 86.4% of invasive ductal breast carcinomas (30). Decreased ARID1A immunoreactivity was associated with poorer prognosis in breast cancer patients. Both complete loss and partial reduction of ARID1A expression correlated with more aggressive tumor behavior. Furthermore, ARID1A protein expression served as an independent prognostic marker, with higher levels predicting better clinical outcomes (31).
Although ARID1A mutation is not an initiating event in NB, its deletion promotes disease progression and correlates with poor prognosis. Notably, immunoprecipitation assays using anti-ARID1B antibodies demonstrated that ARID1B-containing BAF complexes remain structurally intact in ARID1A-deficient NB cells (12). Consistent with prior studies demonstrating synthetic lethality between ARID1B and ARID1A, genetic depletion of ARID1B in ARID1A-deficient cells destabilizes the BAF complex and impairs cellular proliferation. These findings suggest ARID1B as a potential therapeutic target, though ARID1A loss itself remains clinically detrimental in NB patients. Recent advances in Proteolysis Targeting Chimeras (PROTACs) have demonstrated successful protein degradation strategies (32), including targeted disruption of the BAF complex. Notably, PROTAC-mediated degradation of the ATPase subunits has shown therapeutic efficacy in non-small cell lung cancer (33) (NSCLC) and prostate cancer (34). In NB therapeutic research, Adam D Durbin (35) et al. employed PROTAC technology to selectively degrade EP300 as a treatment strategy for MYCN-amplified NB. These findings provide a strong rationale for exploring ARID1B degradation in ARID1A-deficient NB, which may suppress tumor proliferation by destabilizing the BAF complex.
Beyond targeted protein degradation, emerging therapeutic strategies are now focusing on aberrantly expressed circular RNAs (circRNAs) in tumor cells. Aberrant expression of circRNAs has been implicated in nearly all cancer types, where they function as either oncogenes or tumor suppressors during tumorigenesis (36). In NB, circRNAs exhibit dual roles in tumor suppression and oncogenesis (37). Specifically, circARID1A has been shown to promote tumor cell proliferation and survival (23). Current therapeutic strategies targeting circRNAs include targeted degradation (38), immunotherapy (39), and RNA vaccine-based approaches to enhance treatment efficacy (40). However, research on circRNA-targeted therapies remains in its early stages, warranting further investigation.
Dysregulation at both the protein and RNA levels in tumor cells is fundamentally sustained by the tumor microenvironment (TME). TME not only provides structural support for tumor growth but also serves as a critical platform for complex cellular signaling interactions that collectively drive tumor proliferation and invasion. Within this context, immunotherapy has emerged as a promising strategy to enhance cancer treatment efficacy by modulating the immunosuppressive TME (41). Guangsheng Zhu (42) et al. conducted a large-scale clinical analysis of NSCLC patients and demonstrated that those harboring ARID1A/ARID1B mutations exhibited enhanced clinical responses to immune checkpoint blockade therapy, including PD-1/PD-L1 and CTLA-4 inhibitors. In NB treatment, immune checkpoint blockade therapy has been reported (43). Studies indicate that PD-L1 expression correlates with MYCN amplification, while ARID1A-mutated NB is closely associated with MYCN status. Immune checkpoint therapy may be particularly effective in ARID1A-mutated NB, provided these tumors exhibit PD-L1 expression. In a cohort of 31 NB patients analyzed by Shogo Zuo (44) et al., PD-L1 expression was detected in 11 cases (35%). These PD-L1-positive tumors may represent a promising target for immunotherapy. However, this study did not assess how many of the 11 PD-L1-positive cases harbored ARID1A mutations, nor whether such mutations would yield therapeutic outcomes similar to those observed in NSCLC. Thus, this potential treatment strategy remains theoretical at present.
The therapeutic potential of these approaches in NB may be further limited by poor drug delivery across the blood-brain barrier (45). To address these challenges, nanoparticle-based drug delivery systems (46, 47) and nanomedicines (48) have emerged as promising approaches. These technologies enhance tumor-specific drug accumulation, prolong drug retention, and minimize systemic toxicity (47). Integrating nanotechnology with existing therapies may thus provide a synergistic strategy to improve NB treatment outcomes.
Conclusions
ARID1A, a core subunit of the BAF chromatin remodeling complex, plays a pivotal role in NB pathogenesis. Studies have demonstrated that ARID1A synergizes with MYCN amplification to drive the transition of tumor cells from an adrenergic to a mesenchymal phenotype, thereby enhancing invasive and migratory capacities and accelerating disease progression. However, the role of ARID1A within the regulatory network of NB pathogenesis remains incompletely understood and warrants further investigation. Based on existing research findings, it is hypothesized that ARID1A may suppress MYCN transcription or inhibit its transcriptional activation function at the genetic level, thereby disrupting MYCN-driven signaling networks during NB initiation and progression. This proposed mechanism would position ARID1A as a tumor suppressor in this context. However, the specific involvement of ARID1A in MYCN-driven NB signaling networks has not yet been experimentally validated. Although the structures of the BAF complex and several related family members were resolved around 2020, the structural consequences of ARID1A deletion remain unknown. The lack of information on the conformational state of the ARID1A-deleted BAF complex and its associated structural rearrangements presents a critical knowledge gap, hindering mechanistic understanding at the molecular level.
Despite recent advances, several critical gaps remain in understanding the mechanistic role of ARID1A in NB. First, the cooperative interaction network between ARID1A and MYCN requires further elucidation, particularly regarding the functional involvement of the BAF complex in mediating their crosstalk. Second, therapeutic strategies targeting ARID1A-deficient NB remain underdeveloped. While several potential approaches have been proposed, most are still in the conceptual or preclinical stage, and their clinical efficacy awaits rigorous validation. Integrating clinical investigations with mechanistic studies will be essential to establish a solid theoretical foundation and identify actionable therapeutic pathways for ARID1A-driven NB.
Author contributions
QT: Writing – original draft, Writing – review & editing. DZ: Writing – original draft, Writing – review & editing. DJ: Writing – review & editing, Writing – original draft, Supervision.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Acknowledgments
I would like to express my deepest gratitude to The First People’s Hospital of Changzhou and The Second People’s Hospital of Changzhou for the tremendous support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yang R, Zheng S, and Dong R. Circulating tumor cells in neuroblastoma: Current status and future perspectives. Cancer Med. (2023) 12:7–19. doi: 10.1002/cam4.4893
2. Chung C, Boterberg T, Lucas J, Panoff J, Valteau-Couanet D, Hero B, et al. Neuroblastoma. Pediatr Blood Cancer. (2021) 68 Suppl 2:e28473. doi: 10.1002/pbc.28473
3. Ponzoni M, Bachetti T, Corrias MV, Brignole C, Pastorino F, Calarco E, et al. Recent advances in the developmental origin of neuroblastoma: an overview. J Exp Clin Cancer Res. (2022) 41:92. doi: 10.1186/s13046-022-02281-w
4. Sainero-Alcolado L, Bexelius TS, Santopolo G, Yuan Y, Liaão-Pons J, and Arsenian-Henriksson M. Defining Neuroblastoma: from origin to precision medicine. Neuro Oncol. (2024) 26:2174–92. doi: 10.1093/neuonc/noae152
5. Verhoeven BM, Mei S, Olsen TK, Gustafsson K, Valind A, and Lindström A. The immune cell atlas of human neuroblastoma. Cell Rep Med. (2022) 3:100657. doi: 10.1016/j.xcrm.2022.100657
6. Yoneda A. Role of surgery in neuroblastoma. Pediatr Surg Int. (2023) 39:177. doi: 10.1007/s00383-023-05459-1
7. Wilson BG and Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. (2011) 11:481–92. doi: 10.1038/nrc3068
8. He S, Wu Z, Tian Y, Yu Z, Yu J, Wang X, et al. Structure of nucleosome-bound human BAF complex. Science. (2020) 367:875–81. doi: 10.1126/science.aaz9761
9. Mathur R. ARID1A loss in cancer: Towards a mechanistic understanding. Pharmacol Ther. (2018) 190:15–23. doi: 10.1016/j.pharmthera.2018.05.001
10. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human Malignancy. Nat Genet. (2013) 45:592–601. doi: 10.1038/ng.2628
11. Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. (2013) 45:12–17. doi: 10.1038/ng.2493
12. Shi H, Tao T, Abraham BJ, Durbin AD, Zimmerman MW, Kadoch C, et al. ARID1A loss in neuroblastoma promotes the adrenergic-to-mesenchymal transition by regulating enhancer-mediated gene expression. Sci Adv. (2020) 6:eaaz3440. doi: 10.1126/sciadv.aaz3440
13. Jin F, Yang Z, Shao J, Tao J, Reissfelder C, Loges S, et al. ARID1A mutations in lung cancer: biology, prognostic role, and therapeutic implications. Trends Mol Med. (2023) 29:646–58. doi: 10.1016/j.molmed.2023.04.005
14. Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. (2018) 24:556–62. doi: 10.1038/s41591-018-0012-z
15. Takeuchi T, Nicole S, Misaki A, Furihata M, Iwata J, Sonobe H, et al. Expression of SMARCF1, a truncated form of SWI1, in neuroblastoma. Am J Pathol. (2001) 158:663–72. doi: 10.1016/S0002-9440(10)64008-4
16. Cimmino F, Lasorsa VA, Vetrella S, Iolascon A, and Capasso M. A targeted gene panel for circulating tumor DNA sequencing in neuroblastoma. Front Oncol. (2020) 10:596191. doi: 10.3389/fonc.2020.596191
17. Altun Z, Yuan H, Baran B, Aktas S, Sonmez EE, Kucuk C, et al. Whole-exome sequencing reveals genetic variants in low-risk and high-risk neuroblastoma. Gene. (2023) 860:147233. doi: 10.1016/j.gene.2023.147233
18. Li C, Xu ZL, Zhao Z, An Q, Wang L, Yu Y, et al. ARID1A gene knockdown promotes neuroblastoma migration and invasion. Neoplasma. (2017) 64:367–76. doi: 10.4149/neo_2017_307
19. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
20. Godfried MB, Veenstra M, Valent A, Sluis Pv, Voute PA, Versteeg R, et al. Lack of interstitial chromosome 1p deletions in clinically-detected neuroblastoma. Eur J Cancer. (2002) 38:1513–9. doi: 10.1016/S0959-8049(02)00137-5
21. Garcia-Lopez J, Wallace K, Otero JH, Olsen R, Wang YD, Finkelstein D, et al. Large 1p36 deletions affecting arid1a locus facilitate mycn-driven oncogenesis in neuroblastoma. Cell Rep. (2020) 30:454–64.e5. doi: 10.1016/j.celrep.2019.12.048
22. Huang M and Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med. (2013) 3:a014415. doi: 10.1101/cshperspect.a014415
23. Epp S, Chuah SM, and Halasz M. Epigenetic dysregulation in MYCN-amplified neuroblastoma. Int J Mol Sci. (2023) 24:17085. doi: 10.3390/ijms242317085
24. Zafar A, Wang W, Liu G, Wang X, Xian W, McKeon F, et al. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med Res Rev. (2021) 41:961–1021. doi: 10.1002/med.21750
25. Kang JH, Rychahou PG, Ishola TA, Qiao J, Evers BM, Chung DH, et al. MYCN silencing induces differentiation and apoptosis in human neuroblastoma cells. Biochem Biophys Res Commun. (2006) 351:192–7. doi: 10.1016/j.bbrc.2006.10.020
26. Otte J, Dyberg C, Pepich A, and Johnsen JI. MYCN function in neuroblastoma development. Front Oncol. (2020) 10:624079. doi: 10.3389/fonc.2020.624079
27. Decaesteker B, Louwagie A, Loontiens S, De Vloed F, Bekaert SL, Roels J, et al. SOX11 regulates SWI/SNF complex components as member of the adrenergic neuroblastoma core regulatory circuitry. Nat Commun. (2023) 14:1267. doi: 10.1038/s41467-023-36735-2
28. Jiménez C, Antonelli R, Nadal-Ribelles M, Devis-Jauregui L, Latorre P, Solé C, et al. Structural disruption of BAF chromatin remodeller impairs neuroblastoma metastasis by reverting an invasiveness epigenomic program. Mol Cancer. (2022) 21:175. doi: 10.1186/s12943-022-01643-4
29. Cermakova K, Tao L, Dejmek M, Sala M, Montierth MD, Chan YS, et al. Reactivation of the G1 enhancer landscape underlies core circuitry addiction to SWI/SNF. Nucleic Acids Res. (2024) 52:4–21. doi: 10.1093/nar/gkad1081
30. Zhang X, Sun Q, Shan M, Niu M, Liu T, Xia B, et al. Promoter hypermethylation of ARID1A gene is responsible for its low mRNA expression in many invasive breast cancers. PLoS One. (2013) 8:e53931. doi: 10.1371/journal.pone.0053931
31. Fontana B, Gallerani G, Salamon I, Pace I, Roncarati R, and Ferracin M. ARID1A in cancer: Friend or foe? Front Oncol. (2023) 13:1136248. doi: 10.3389/fonc.2023.1136248
32. Hu M, Zhou W, Wang Y, Yao D, Ye T, Yao Y, et al. Discovery of the first potent proteolysis targeting chimera (PROTAC) degrader of indoleamine 2,3-dioxygenase 1. Acta Pharm Sin B. (2020) 10:1943–53. doi: 10.1016/j.apsb.2020.02.010
33. Kotagiri S, Blazanin N, Xi Y, Han Y, Qudratullah M, Liang X, et al. Enhancer reprogramming underlies therapeutic utility of a SMARCA2 degrader in SMARCA4 mutant cancer. Cell Chem Biol. (2024) 31:2069–84.e9. doi: 10.1016/j.chembiol.2024.09.004
34. Xiao L, Parolia A, Qiao Y, Bawa P, Eyunni S, Mannan R, et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature. (2022) 601:434–9. doi: 10.1038/s41586-021-04246-z
35. Durbin AD, Wang T, Wimalasena VK, Zimmerman MW, Li D, Dharia NV, et al. EP300 selectively controls the enhancer landscape of MYCN-amplified neuroblastoma. Cancer Discov. (2022) 12:730–51. doi: 10.1158/2159-8290.CD-21-0385
36. Chen L and Shan G. CircRNA in cancer: Fundamental mechanism and clinical potential. Cancer Lett. (2021) 505:49–57. doi: 10.1016/j.canlet.2021.02.004
37. Wu K, Tan J, and Yang C. Recent advances and application value of circRNA in neuroblastoma. Front Oncol. (2023) 13:1180300. doi: 10.3389/fonc.2023.1180300
38. Karami Fath M, Pourbagher Benam S, Salmani K, Naderi S, Fahham Z, Ghiabi S, et al. Circular RNAs in neuroblastoma: Pathogenesis, potential biomarker, and therapeutic target. Pathol Res Pract. (2022) 238:154094. doi: 10.1016/j.prp.2022.154094
39. Zang J, Xiao L, Shi X, Liu S, Wang Y, Sun B, et al. Hsa_circ_0001479 accelerates tumorigenesis of gastric cancer and mediates immune escape. Int Immunopharmacol. (2023) 124:110887. doi: 10.1016/j.intimp.2023.110887
40. Li H, Peng K, Yang K, Ma W, Qi S, Yu X, et al. Circular RNA cancer vaccines drive immunity in hard-to-treat Malignancies. Theranostics. (2022) 12:6422–36. doi: 10.7150/thno.77350
41. Wu G, Pan B, Shi H, Yi Y, Zheng X, Ma H, et al. Neutrophils’ dual role in cancer: from tumor progression to immunotherapeutic potential. Int Immunopharmacol. (2024) 140:112788. doi: 10.1016/j.intimp.2024.112788
42. Zhu G, Shi R, Li Y, Zhang Z, Xu S, Chen C, et al. ARID1A, ARID1B, and ARID2 mutations serve as potential biomarkers for immune checkpoint blockade in patients with non-small cell lung cancer. Front Immunol. (2021) 12:670040. doi: 10.3389/fimmu.2021.670040
43. Nallasamy P, Chava S, Verma SS, Mishra S, Gorantla S, Coulter DW, et al. PD-L1, inflammation, non-coding RNAs, and neuroblastoma: Immuno-oncology perspective. Semin Cancer Biol. (2018) 52:53–65. doi: 10.1016/j.semcancer.2017.11.009
44. Zuo S, Sho M, Sawai T, Kanehiro H, Maeda K, Yoshida M, et al. Potential role of the PD-L1 expression and tumor-infiltrating lymphocytes on neuroblastoma. Pediatr Surg Int. (2020) 36:137–43. doi: 10.1007/s00383-019-04616-9
45. Xie J, Shen Z, Anraku Y, Kataoka K, and Chen X. Nanomaterial-based blood-brain-barrier (BBB) crossing strategies. Biomaterials. (2019) 224:119491. doi: 10.1016/j.biomaterials.2019.119491
46. Gao J, Wang WQ, Pei Q, Lord MS, and Yu HJ. Engineering nanomedicines through boosting immunogenic cell death for improved cancer immunotherapy. Acta Pharmacol Sin. (2020) 41:986–94. doi: 10.1038/s41401-020-0400-z
47. Xia D, Zhang X, Hao H, Jiang W, Chen C, Li H, et al. Strategies to prolong drug retention in solid tumors by aggregating Endo-CMC nanoparticles. J Control Release. (2023) 360:705–17. doi: 10.1016/j.jconrel.2023.07.006
Keywords: ARID1A, neuroblastoma, BAF complex, MYCN, treatment
Citation: Jin D, Zhang D and Tang Q (2025) Significance of ARID1A in neuroblastoma onset mechanism. Front. Oncol. 15:1624561. doi: 10.3389/fonc.2025.1624561
Received: 07 May 2025; Accepted: 25 August 2025;
Published: 10 September 2025.
Edited by:
Liam Chen, University of Minnesota, United StatesReviewed by:
Soraya Epp, University College Dublin, IrelandCopyright © 2025 Jin, Zhang and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Duo Jin, Y3p5eV9qZEAxNjMuY29t; Qinyu Tang, Y2hpbnl1a3RAMTYzLmNvbQ==
†These authors have contributed equally to this work