 Yasemin Gündoğdu1
Yasemin Gündoğdu1 Elif Şenocak Taşçı2†
Elif Şenocak Taşçı2† Leyla Özer3†Can Boynukara1
Leyla Özer3†Can Boynukara1 Recep Çeçen4
Recep Çeçen4 Arda Ulaş Mutlu5†İbrahim Yıldız2*†
Arda Ulaş Mutlu5†İbrahim Yıldız2*†- 1Department of Internal Medicine, Acıbadem MAA University, Istanbul, Türkiye
- 2Department of Medical Oncology, Acıbadem MAA University, Istanbul, Türkiye
- 3Department of Medical Oncology, Memorial Hospital, Istanbul, Türkiye
- 4Department of Internal Medicine, Faculty of Medicine, Istanbul University Cerrahpaşa, Istanbul, Türkiye
- 5Department of General Surgery, Acıbadem MAA University, Istanbul, Türkiye
Background: Sarcomas are rare mesenchymal tumors classified into soft tissue (STS) and bone sarcomas. Despite advances in treatment, the 5-year survival rate for metastatic disease remains low. There is still limited evidence regarding the use of next-generation sequencing (NGS).
Aim: To identify targetable genomic alterations that may play a crucial role in sarcoma treatment where therapeutic options are limited.
Study design: Methods: We conducted a retrospective; multicenter analysis of 81 patients diagnosed with STS and bone sarcomas who underwent NGS at Acıbadem Health Group Hospitals to investigate their mutation profiles and explore potential targeted therapies.
Results: Genomic profiling using four different NGS kits identified a total of 223 genomic alterations across the cohort. Genomic alterations were detectable in 90.1% of patients, with the most common types being copy number amplifications (26.9%) and deletions (24.7%). In addition, actionable mutations were identified in 22.2% of patients, rendering them eligible for FDA-approved targeted therapies. The most common alterations were found in TP53 (38%), RB1 (22%), and CDKN2A (14%) genes. Among the 79 patients with available microsatellite status data, all were microsatellite stable.
Conclusion: The high proportion of patients eligible for targeted therapies identified underscores the critical need to integrate NGS-derived genetic insights into clinical practice to improve survival rates and treatment outcomes through more tailored therapeutic approaches for each individual. NGS also led to a reclassification of diagnosis in four patients, demonstrating its utility not only in therapeutic decision-making but also as a powerful diagnostic tool.
Introduction
Sarcomas, a rare and heterogeneous group of tumors originating from mesenchymal cells, represent approximately 1% of adult malignancies and are broadly categorized into soft tissue sarcomas (STS) and bone sarcomas (1). With over 100 histological subtypes, these tumors exhibit distinct molecular and genetic alterations that significantly influence their biology, progression, and response to treatment (2). Despite advancements in treatment modalities, such as novel chemotherapeutics, immunotherapy, targeted therapies, and radiotherapy, the prognosis for sarcoma patients remains poor. Metastasis detection during follow-up after diagnosis remains high, with rates reported between 20% and 50% in various studies (3, 4).
Sarcomas can also be classified based on their genomic complexity into two major groups: simple karyotype (associated with translocations or specific activating mutations) and complex karyotype (marked by genomic heterogeneity and instability) (5). The identification and classification of sarcomas with complex karyotypes using conventional techniques, which often exhibit non-specific genomic alterations, present significant challenges (1). These complexities, coupled with the rarity of sarcomas, underscore the need for advanced tools to enhance diagnostic accuracy and optimize treatment strategies (6).
In recent years, the integration of comprehensive genomic profiling into clinical practice has begun to reshape standard care protocols, providing more personalized and precise therapeutic options (7). Among these innovations, next-generation sequencing (NGS) technologies have emerged as transformative tools in the diagnostic, prognostic, and therapeutic management of sarcomas. The 5th edition of the WHO classification of soft tissue and bone sarcomas, updated in 2020, highlights the significance of genetic mutations identified through NGS (2). NGS, with its advanced DNA and RNA sequencing capabilities, enables the simultaneous identification of multiple fusion mutations and previously unknown genetic alterations (8, 9). By offering a more detailed understanding of the genomic landscape of sarcomas, NGS facilitates targeted therapies tailored to specific mutational profiles (10).
However, despite its ability to identify numerous mutations through high-throughput sequencing, the clinical translation of these findings remains limited, with lower-than-expected rates of direct therapeutic benefit (11). Furthermore, tumors in patients receiving targeted therapies often develop resistance over time. In this context, NGS is a vital tool for uncovering resistance mechanisms and helping clinicians choose alternative treatment strategies, thereby advancing precision oncology in sarcomas.
In this study, we aim to investigate the genomic profiles of patients diagnosed with soft tissue and bone sarcomas using a comprehensive NGS-based approach and evaluate the potential impact of these findings in the management of sarcoma. As a referral center specialized in sarcoma treatment, we also seek to assess the role of NGS in improving diagnosis and treatment outcomes within our patient population.
Materials and methods
Study design and patient selection
In this multi-center, cross-sectional retrospective study (IRB number: 2024-4/150) patients diagnosed with soft tissue or bone sarcoma between 2017 and 2023 were analyzed. The study cohort included 81 adult patients diagnosed with STS (n=61) or bone sarcoma (n=20). Patients with gastrointestinal stromal tumors (GIST) and Kaposi sarcoma were excluded, as these tumors have distinct molecular characteristics and treatment approaches. Demographic and clinicopathological data were retrieved from electronic health records. All patients underwent either a biopsy or surgical resection, followed by standardized pathological examinations conducted by a team of sarcoma-specialized pathologists. The pathology specimens were consulted by the same specialized team if the patient is foreign.
Comprehensive molecular profiling
All participants underwent comprehensive molecular profiling at various points during their diagnosis or treatment using NGS tests (Foundation Medicine, Inc. [FoundationOne], Tempus, OncoDEEP (OncoDNA SA) and MI profile (Caris Inc.)). All the tests were performed in CLIA- regulated laboratories. Genomic alterations, such as insertions/deletions, copy number variations and structural rearrangements were recorded. Tumor mutation burden (TMB, an estimate of the number of mutations per megabase) and microsatellite instability (MSI) status were also documented when available. Variants of unknown significance were not included in the analysis. All sequencing was performed on tumor tissue without matched normal controls. As a result, the ability to definitively distinguish somatic from germline variants was limited. In tumor-only NGS analyses, variants with a variant allele frequency (VAF) greater than 50% were considered suspicious for possible germline origin. In addition, pathogenic variants occurring in well-known hereditary cancer predisposition genes (such as BRCA1/2, TP53, ATM, CHEK2) were also reviewed for potential germline significance. In such cases, or when a hereditary cancer syndrome was clinically suspected, confirmatory germline testing was subsequently performed using validated germline assays. Germline mutations were confirmed in two patients (BLM, TP53, ATM), followed by genetic counseling and family risk assessment. Actionable alterations were initially determined based on clinical annotations provided by the respective NGS platforms. Because most platforms used in this study are aligned with or directly integrated into the OncoKB evidence-level classification system, all actionable mutations were retrospectively reclassified according to OncoKB criteria. This framework incorporates FDA approval status, clinical guideline support, and the strength of supporting clinical or biological evidence. The NGS platforms used in this study differed in terms of gene content, sequencing depth, and variant calling pipelines. TMB and MSI values were interpreted using platform-specific thresholds and algorithms as provided in certified clinical reports. Given the differences in analytical design across platforms, direct standardization was not feasible.
Ethics
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Acıbadem MAA University (approval number: 2024-4/150, date: 14.03.2024). All methods and protocols were performed in accordance with the relevant guidelines and regulations.
Results
Patient characteristics
The study included 81 patients who underwent treatment for STS and bone sarcoma. The median age of patients was 45.2 years at diagnosis (range: 18-80) and there was slight female predominance (51.8%, n=42). Among these patients, 61 (75.3%) had STS while 20 (24.7%) were diagnosed with bone sarcoma. All patients had metastatic disease prior to performing NGS.
Undifferentiated pleomorphic sarcoma (UPS) was the most common STS subtype (22.7%), followed by leiomyosarcoma (16%) and synovial sarcoma (11.1%). The clinicopathological data of the patients is given in Table 1. The most frequently observed bone sarcomas were Ewing’s sarcoma (13.6%) and osteosarcoma (7.4%).
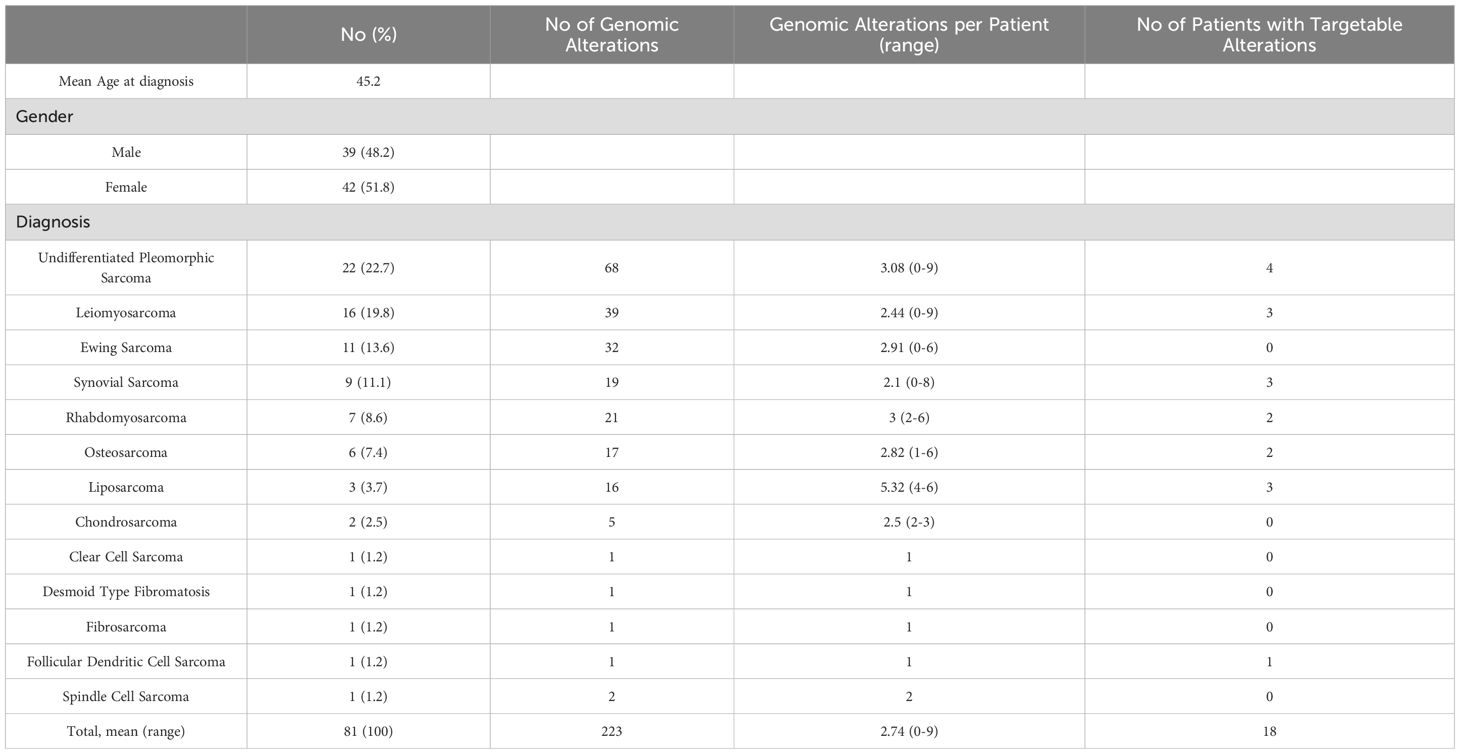
Table 1. Clinicopathological data of included patients (n=81).
Genomic alterations and instability
The four commercial NGS kits used were Tempus (Tempus Labs Inc.)(n:48), FoundationOne (Foundation Medicine Inc.)(n:24), OncoDEEP (OncoDNA SA)(n:6) and MI profile (Caris Inc.)(n:3). A total of 223 genomic alterations were identified across the cohort, with an average of 2.74 alterations per patient. Notably, at least one type of genomic alteration was present in 90.1% (n=73) of tumors. The remaining 8 did not possess any significant mutations according to the reports. The most frequently mutated genes were TP53 (38%, n=31), RB1 (22%, n=18) and CDKN2A (14%, n=12) followed by EWSR1 (13%, n= 11), CDKN2B (9%, n=8), MDM2 (8%, n= 7), PTEN (8%, n= 7), FRS2 (7%, n= 6) and MTAP (7%, n= 6) (Figure 1).
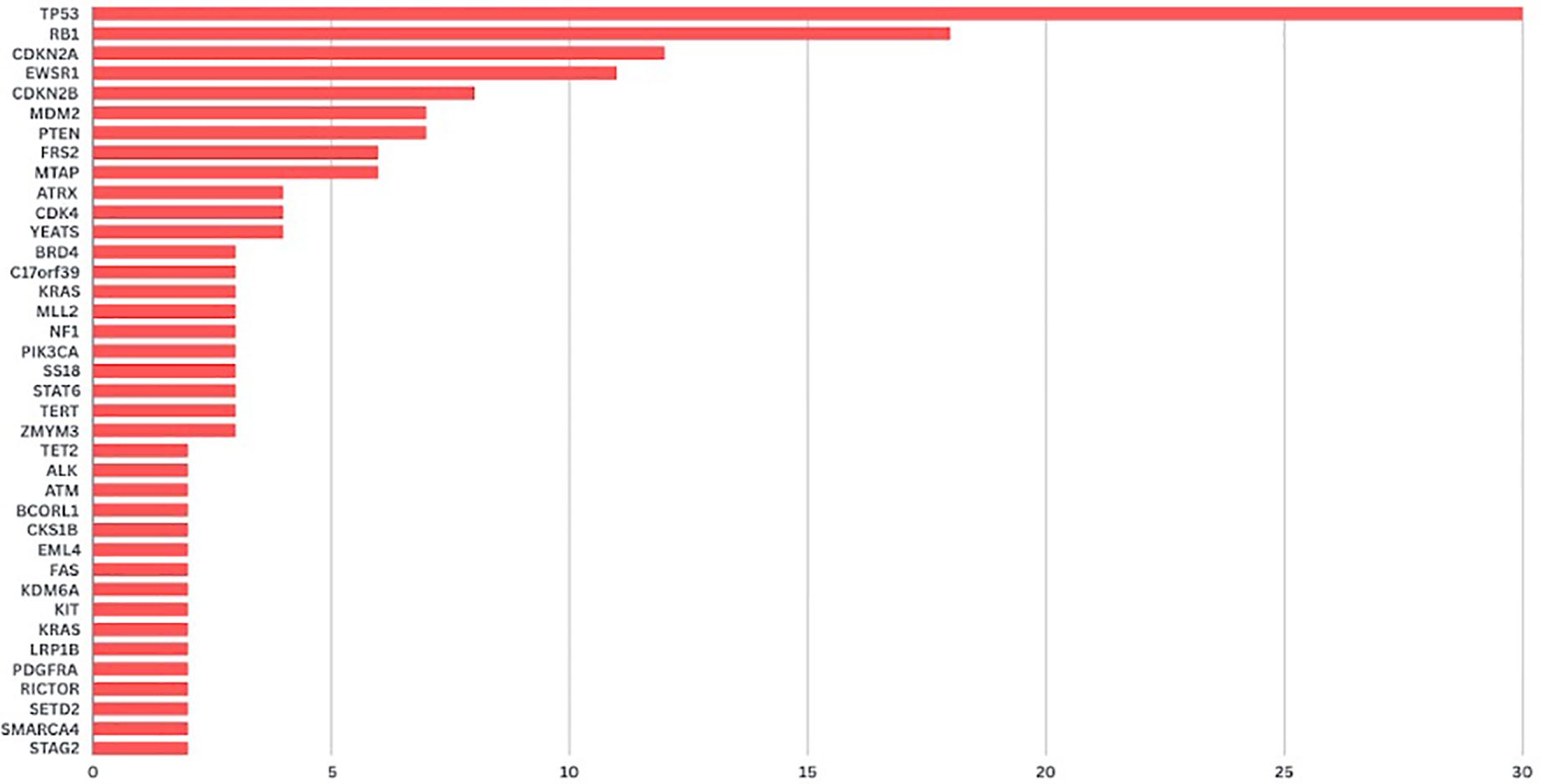
Figure 1. Number of genomic alterations detected in patients with sarcoma.
Upon functional analysis of genomic alterations, potentially targetable changes were identified in several key pathways. In the genomic stability regulation pathway, alterations were detected in TP53 (38%) and MDM2 (8%). In the cell cycle regulation pathway, mutations were observed in RB1 (22%), CDKN2A (14%), CDKN2B (9%), CDK4 (5%), and CDKN2A/B (1%). In the DNA repair pathway, alterations were found in RAD (1%). In the phosphoinositide-3 kinase (PI3K) pathway, mutations were identified in PTEN (8%), PIK3CA (4%), mTOR (1%), and RICTOR (2%). In the receptor tyrosine kinase pathway, alterations were detected in ALK (2%) and FGFR (4%).
Copy number amplifications were the most prevalent mutation type (26.9%), followed by copy number deletions (24.7%) (Figure 2). Chromosomal rearrangements were also notably present, particularly involving the EWSR1 gene in cases of Ewing’s sarcoma which is also a disease defining gene alteration.
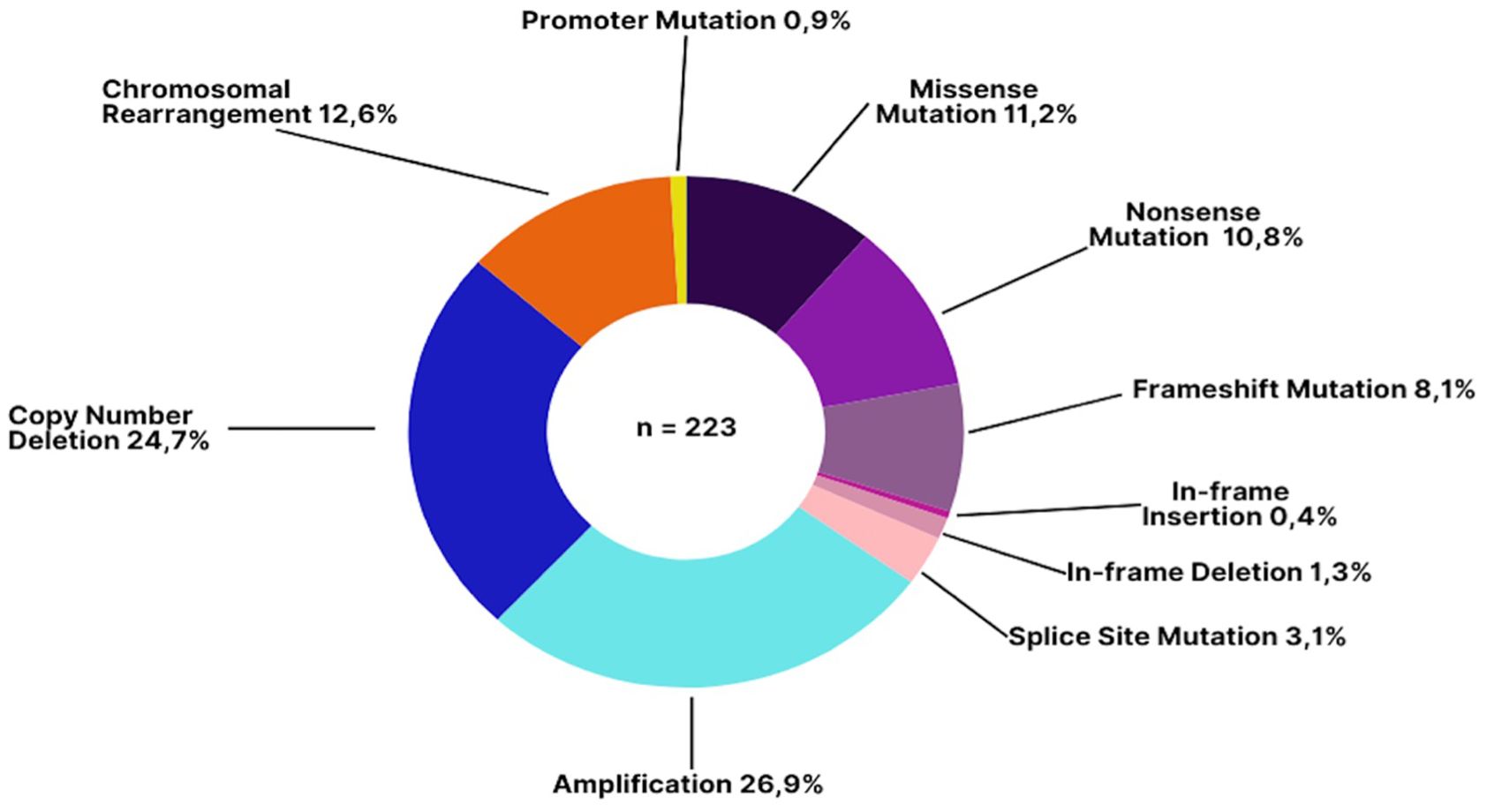
Figure 2. Types of genomic alterations detected in patients with sarcoma. The most common alteration types were amplifications (26.9%) and copy number deletions (24.7%), which are frequently implicated in oncogene activation and tumor suppressor loss, respectively. Chromosomal rearrangements (12.6%)—including translocations such as EWSR1 fusions—can have diagnostic utility in specific sarcoma subtypes. Missense (11.2%) and nonsense mutations (10.8%) often result in aberrant or truncated proteins, while frameshift mutations (8.1%) can dramatically alter protein function.
The types of genomic alterations detected in the most frequent genes are summarized in Figure 3. From a total of 31 TP53 alterations detected, 63.2% were loss of function mutations. Among the 18 patients with mutations in RB1, 61.1% had copy number variations. Of the 12 patients with alterations in CDKN2A, 83.3% had copy number variations.
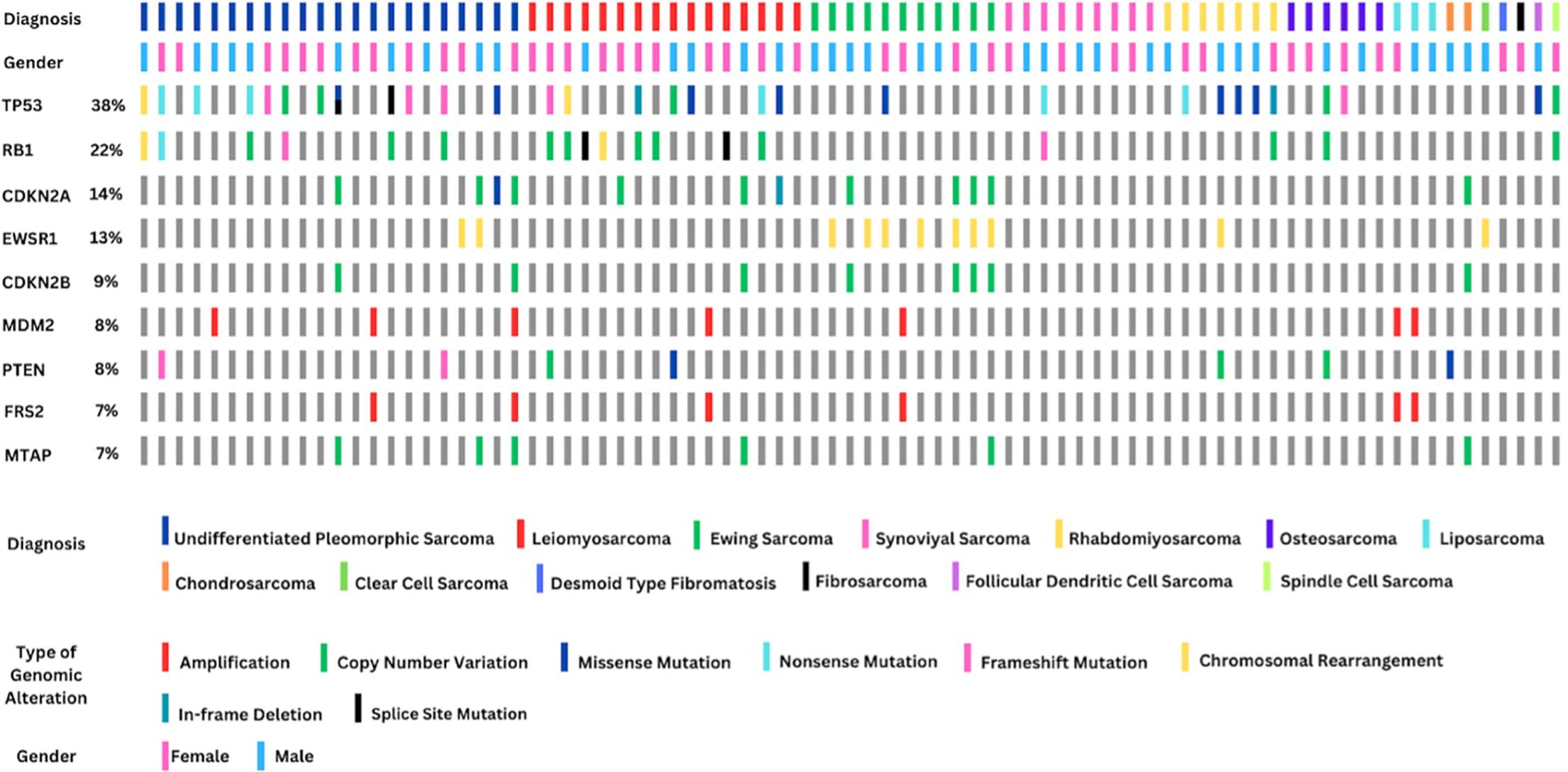
Figure 3. Genomic alteration summary of the most frequently altered genes across 81 sarcoma patients stratified by diagnosis, gender, and mutation type. The top row indicates histologic subtype, with undifferentiated pleomorphic sarcoma, leiomyosarcoma, and Ewing sarcoma being the most represented. Gender is shown in the second row. The remaining rows highlight alterations in the top 10 mutated genes, including TP53 (38%), RB1 (22%), and CDKN2A (14%). Mutations are color-coded by type (e.g., amplifications, copy number variations, missense, and chromosomal rearrangements). Notably, EWSR1 rearrangements (13%) serve as a diagnostic hallmark for Ewing sarcoma, while TP53 and RB1 mutations are common across several subtypes and may be prognostically relevant.
All genomic alterations involving the EWSR1 gene were observed to be chromosomal rearrangements. Among the 11 EWSR1-related translocations identified, seven were detected in patients diagnosed with Ewing sarcoma, while two were found in UPS, one in rhabdomyosarcoma, and one in a patient diagnosed with clear cell sarcoma. In 42% of the 11 patients histopathologically diagnosed with Ewing sarcoma, no EWSR1-related mutation was observed. A total of eight patients exhibited alterations in the CDKN2B gene, all of which were copy number losses. Notably, 50% of these patients were diagnosed with Ewing sarcoma. All seven MDM2 mutations identified were found to be copy number amplifications. Alterations in the MDM2 gene were detected in 66% of patients diagnosed with liposarcoma. Additionally, FRS2 amplification was observed in six patients, all of whom also had MDM2 amplification.
Microsatellite instability (MSI) analysis was performed in 79 patients, all of whom were classified as microsatellite stable (MSS), indicating a low likelihood of response to immunotherapy. Among these patients, the TMB ranged from 0 to 12.1 mut/Mb, with a mean TMB of 2.56 ± 2.31 mut/Mb. The analysis of TMB highlighted potential high mutation burdens in specific cases, notably in a UPS patient with a TMB of 12.1 mut/Mb, suggesting eligibility for immunotherapy.
Actionability
18 patients (22.2%) had an actionable mutation and were recommended therapeutic option by NGS testing reports. Only 10 patients were able to be treated with NGS-targeted therapy. No statistically significant differences were observed in overall mutation frequencies or in access to targeted therapies when stratified by histology, sex, or age group (p > 0.05).
Three patients had diagnosis-changing results in NGS. First patient’s histopathological diagnosis was UPS, according to the current WHO classification (2), tumors with CIC-DUX4 fusion are defined as CIC-rearranged sarcomas. Another example of a case where the pathological diagnosis contradicted the NGS profile was a patient with a histopathological diagnosis of UPS, in whom an EWSR1-ERG fusion was detected. Based on this finding, we concluded that the patient’s primary diagnosis was Ewing sarcoma. Third patient, whose histopathological diagnosis was defined as UPS but was found to have an EWSR1-ATF1 fusion, was ultimately determined to have a primary diagnosis of clear cell sarcoma. There was one instance in which the NGS-related findings were diagnosis-modifying. One patient diagnosed with rhabdomyosarcoma harbored PAX3-FOXO1 fusion mutation, which refined the diagnosis as alveolar rhabdomyosarcoma.
Two patients were identified with significant germline mutations. One patient with epithelioid fibrosarcoma had a germline BLM mutation, and another with UPS possessed TP53 and ATM mutations. These patients were further investigated for family risk assessment and were provided genetic counseling. The patient with germline BLM (p.W567 nonsense) mutation was diagnosed with Bloom syndrome and the patient with TP53 (p.R337C missense) and ATM (p.R1466 missense) mutations was diagnosed with Li-Fraumeni Syndrome (LFS). The molecular profiling had been life-changing for two patients.
Discussion
This study represents the most extensive investigation of unresectable sarcoma patients using comprehensive genomic profiling in Turkey, contributing significantly to the global understanding of sarcoma genomics. Sarcomas are characterized by significant genetic heterogeneity, necessitating precision medicine approaches for improved patient management. Our findings align with prior studies demonstrating that the vast majority of sarcomas harbor detectable genomic alterations, with TP53, RB1, and CDKN2A being the most frequently mutated genes (12). These alterations highlight the importance of cell cycle dysregulation in sarcomagenesis and may serve as potential therapeutic targets. Cole et al. also, in their analysis of 133 sarcoma patients using NGS, reported that the most common mutations were associated with cell cycle regulation (13).
The detection of genomic alterations in 90.1% of our cohort is consistent with previous international studies, which report a similarly high mutational diversity in sarcoma (12–16). However, only 22.2% of these alterations were deemed actionable with currently available FDA-approved therapies, underscoring the ongoing challenge of translating genomic findings into effective treatments, the gap between genomic discoveries and their clinical implementation. The presence of actionable mutations in a subset of patients suggests that precision oncology could play an increasingly critical role in sarcoma management, particularly for patients with limited treatment option (8). Still, NGS profiling had an impact on three patients’ diagnosis and potential prognostic trajectory and been life-changing for 2 patients who were diagnosed with familial syndromes after molecular profiling results.
Among the 81 patients in our study, 8 (9.9%) had no detectable genomic alterations, aligning with findings from Gusho et al. (10.3%) and other studies reporting at least one alteration in over 90% of cases (14–16). The predominant mutations in TP53 (38%), RB1 (22%), and CDKN2A (14%) were consistent with previous reports (16, 17). Additionally, PTEN and MDM2 mutations were identified as prognostic markers, where PTEN alterations were linked to reduced survival in STS (18). PTEN mutation was detected in 8% of cases, with loss-of-function mutations identified in 4 patients and copy number loss mutations in 3 patients. Notably, MDM2 and FRS2 co-occurrence (85.7%) was frequently observed, particularly in well-differentiated and dedifferentiated liposarcomas, where it serves as a key diagnostic marker (19, 20). Another notable genetic alteration identified through NGS was the HMGA2-DHX9 fusion. The HMGA2 mutation, previously described in certain sarcomas, particularly fibrosarcomas, has been associated with treatment resistance (21). To date, no cases of HMGA2-DHX9 fusion have been reported in sarcoma patients in the literature. Although its clinical significance remains unclear, the mutation is reported in a patient diagnosed with liposarcoma for the first time.
In our study, only 18 patients (22.2%) had actionable mutations with available FDA-approved targeted therapies. The frequency was found to be lower than the previous studies (8). In a recent large-scale analysis, clinically targetable mutations were identified in 31.7% of 7,494 sarcoma cases, where 81% of the cohort had STS (10). Differences in STS (75.3% in our study) distribution may contribute to this variation, as STS is known to harbor more targetable mutations and have broader therapeutic options. Some patients had multiple treatment options, resulting in a total of 34 targeted therapy recommendations. Given that sarcomas are rare and associated with poor prognosis, with survival estimates of 30% at two years (22), the identification of 34 treatment options through NGS in 81 patients underscores the significant role of precision medicine in clinical decision-making for this challenging malignancy. While 22.2% of patients in our cohort had actionable mutations, only 10 were able to receive targeted therapies. This gap underscores key challenges in translating molecular findings into clinical benefit. Barriers include limited access to approved targeted therapies for rare cancers such as sarcomas and lack of reimbursement for off-label use. Additionally, the absence of matched clinical trials and uncertainty among clinicians about the utility of targeted agents in off-label contexts may further limit implementation. Addressing these barriers is essential to fully harness the clinical potential of precision oncology in real-world settings.
Microsatellite instability analysis confirmed that all 79 patients with available data were MSS, consistent with the literature, which reports MSI in less than 1% of sarcomas (23). Consistent with large-scale studies reporting low TMB levels in sarcomas 10, our cohort exhibited a mean TMB of 2.56 ± 2.31 muts/megabase, further highlighting the limited benefit of immunotherapy in this population (24). Given the prevalence of translocation events—associated with relatively low genomic instability—in sarcomas, the observed low TMB levels were not unexpected.
This study underscores the importance of RNA-based NGS in detecting fusion-driven sarcomas, aiding in diagnostic accuracy and guiding treatment. For example, one patient was diagnosed with CIC-rearranged sarcoma based on the detection of a CIC-DUX4 fusion, a rare entity associated with poor prognosis and chemoresistance. Another case involved an EWSR1-ERG fusion in a patient initially diagnosed with UPS, ultimately leading to reclassification as Ewing sarcoma. In our cohort, the integration of genomic data led to a reclassification of initial histopathological diagnosis in four patients. These findings emphasize the diagnostic power of NGS, particularly in cases with ambiguous or overlapping histologic features (12, 25, 26). Such reclassifications can significantly alter clinical management, affecting both treatment strategy and prognosis. In a study done by Atiq et al. (27), a new amplicon-based targeted NGS assay was tested on the first 652 patients and the results also helped in changing or confirming diagnosis as well as identifying novel fusions. These findings support the routine inclusion of molecular profiling in the diagnostic workflow for challenging sarcoma cases.
Furthermore, the detection of significant germline mutations within the cohort suggests that NGS can play a valuable role in individual and familial risk management, providing essential information for genetic counseling and decision-making in cases suspected of hereditary sarcoma syndromes. Germline mutations were detected in two patients included in our study. One of the patients, diagnosed with Bloom syndrome, was enrolled in an intensive surveillance program for other potential cancers and was advised to undergo familial screening. In the other patient, a germline TP53 p.R337C missense mutation and a germline ATM p.R1466 missense mutation were identified. The patient was diagnosed with LFS, and due to the heterozygous ATM mutation, familial screening was recommended for both ataxia-telangiectasia and LFS, alongside cancer surveillance.
Germline mutations and associated hereditary syndromes are known to be more frequently detected in children and adolescents, with reported rates ranging from 4.6% to 10% (28). In our study, germline mutations associated with cancer predisposition were identified in 2.5% of cases, and the affected patients, aged 31 and 39, were relatively young. Approximately 10% of osteosarcomas are associated with hereditary cancer syndromes (29), and the increased familial risk of STS are shown with hereditary retinoblastoma, LFS, familial adenomatous polyposis, neurofibromatosis, tuberous sclerosis and Werner syndrome (30). STS should be evaluated clinically for personal history to be assessed for the possibility of cancer predisposition syndromes. Considering the potential benefits of early detection and treatment of accompanying findings, as well as the advantages of family screening for the healthcare system, evaluating germline mutations through NGS testing in young and middle-aged sarcoma patients may be a reasonable approach.
Despite its potential, NGS application must be tailored to specific sarcoma subtypes and clinical scenarios. Considering the rarity and unfavorable prognosis of sarcomas, integrating genomic profiling into multidisciplinary tumor boards may optimize clinical decision-making. While targeted and immunotherapy options remain limited, NGS serves as a valuable tool for patients with actionable mutations, refractory disease, or hereditary risk factors. Another emerging area of interest in precision oncology is the integration of molecular profiling with imaging characteristics, known as radiogenomics. This approach seeks to correlate imaging features (e.g., from MRI or CT) with underlying genomic alterations, potentially enabling non-invasive tumor characterization. In a study by Crombe et al. (31), underscoring the integration of radiomic and genomic data in STS, the tumors that appeared to evolve most rapidly on imaging scans not only enhanced proliferative activity but also exhibited immune response suppression. In sarcomas, radiogenomic analysis may help understand mutation status, guide personalized treatment, and monitor disease progression.
Limitations of this study include its retrospective design, relatively small cohort size, and the difficulty of capturing long-term treatment outcomes—particularly among international and second-opinion patients. Since the study spans cases diagnosed between 2017 and 2023, the evolving landscape of NGS technologies and the timing of regulatory approvals for targeted therapies may have influenced treatment decisions, especially in earlier cases where fewer therapeutic options were available. Another limitation is that genomic analysis was performed on the initial diagnostic biopsy or resected specimens, which may not reflect resistance mutations that developed under treatment pressure. Moreover, clinical outcome data for patients who received NGS-guided therapies were not consistently available, as many were externally referred and their follow-up occurred outside our institution. This limited our ability to assess the real-world impact of matched therapies. In addition, sequencing was performed without matched normal tissue, which restricts the ability to definitively distinguish somatic from germline variants. While tumor-only sequencing is widely accepted in clinical oncology, matched normal controls could enhance the precision of variant classification, particularly in patients with suspected hereditary cancer syndromes. Furthermore, in tumor-only sequencing, certain features can suggest possible germline origin. These include variants with high variant allele frequency (VAF >50%), pathogenic alterations in well-known hereditary cancer predisposition genes (e.g., BRCA1/2, TP53, ATM, CHEK2), recurrent detection of the same variant across multiple tumor samples in the same patient, and copy number patterns consistent with heterozygous germline deletions. Clinical context, such as young age at diagnosis or a strong family history of cancer, may further strengthen germline suspicion. However, these findings remain presumptive, and definitive classification always requires confirmation by independent germline testing and genetic counseling. Finally, a methodological limitation of this study is the use of multiple NGS platforms with varying gene panel sizes, sequencing depth, and bioinformatics pipelines. These differences may have affected the detection rates of certain alterations and limited the comparability of metrics such as TMB and MSI. Taken together, these factors highlight the broader challenges of implementing genomically guided therapy in sarcomas, where tumor evolution and clonal selection may result in molecular heterogeneity over time. Future studies incorporating dynamic tools such as serial biopsies or liquid biopsies may better capture resistance mechanisms and inform real-time treatment decisions.
Conclusions
Our study highlights the need to implement genetic information from molecular profiling with clinical insights to optimize the benefits of personalized medicine in sarcoma treatment. Although actionable mutations remain limited, NGS provides critical insights into tumor biology, refines diagnoses, and aids in treatment decision. This research sets the stage for a more targeted method of managing sarcomas, integrating genetic data with clinical practice to improve patient care.
Data availability statement
The data that support the findings of this study are available on request from the corresponding author ES.
Author contributions
YG: Investigation, Writing – original draft. EŞ: Writing – original draft, Writing – review & editing. LÖ: Data curation, Supervision, Writing – original draft. CB: Data curation, Formal analysis, Writing – original draft. RÇ: Data curation, Formal analysis, Writing – review & editing. AM: Writing – review & editing. İY: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial no support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. The WHO Classification of Tumours Editorial Board. WHO Classification of Tumours Soft Tissue and Bone Tumours. 5th. Lyon: IARC Press (2020).
3. Brown HK, Schiavone K, Gouin F, Heymann MF, and Heymann D. Biology of bone sarcomas and new therapeutic developments. Calcif Tissue Int. (2017) 102:174–95. doi: 10.1007/s00223-017-0372-2
4. Li RH, Zhou Q, Li AB, Zhang HZ, and Lin ZQ. A nomogram to predict metastasis of soft tissue sarcoma of the extremities. Med (Baltimore). (2020) 99:e20165. doi: 10.1097/MD.0000000000020165
5. Taylor BS, Barretina J, Maki RG, Antonescu CR, Singer S, and Ladanyi M. Advances in sarcoma genomics and new therapeutic targets. Nat Rev Cancer. (2011) 11:541–57. doi: 10.1038/nrc3087
6. Sbaraglia M, Bellan E, and Dei Tos AP. The 2020 WHO classification of soft tissue tumours: News and perspectives. Pathologica. (2020) 113:70–84. doi: 10.32074/1591-951X-213
7. Öfverholm I, Comstedt T, Bjerkehagen B, Stenman G, Jonsson M, Gylling B, et al. Comprehensive genomic profiling alters clinical diagnoses in a significant fraction of tumors suspicious of sarcoma. Clin Cancer Res. (2024) 30:2647–58. doi: 10.1158/1078-0432.CCR-24-0384
8. Lanic MD, Le Loarer F, Rainville V, Sater V, Viennot M, Beaussire L, et al. Detection of sarcoma fusions by a next-generation sequencing-based ligation-dependent multiplex RT-PCR assay. Mod Pathol. (2022) 35:649–63. doi: 10.1038/s41379-021-00980-x
9. Stark R, Grzelak M, and Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet. (2019) 20:631–56. doi: 10.1038/s41576-019-0150-2
10. Gounder MM, Agaram NP, Trabucco SE, Robinson V, Ferraro RA, Millis SZ, et al. Clinical genomic profiling in the management of patients with soft tissue and bone sarcoma. Nat Commun. (2022) 13:3406. doi: 10.1038/s41467-022-30496-0
11. Lucchesi C, Khalifa E, Laizet Y, Soubeyran I, Mathoulin-Pelissier S, Chomienne C, et al. Targetable alterations in adult patients with soft-tissue sarcomas: Insights for personalized therapy. JAMA Oncol. (2018) 4:1398–404. doi: 10.1001/jamaoncol.2018.0723
12. Nacev BA, Sanchez-Vega F, Smith SA, Antonescu CR, Rosenbaum E, Iafrate AJ, et al. Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. Nat Commun. (2022) 13:3405. doi: 10.1038/s41467-022-30453-x
13. Cote GM, He J, and Choy E. Next-generation sequencing for patients with sarcoma: A single center experience. Oncologist. (2018) 23:234–42. doi: 10.1634/theoncologist.2017-0290
14. Gusho CA, Weiss MC, Lee L, Gitelis S, Blank AT, Wang D, et al. The clinical utility of next-generation sequencing for bone and soft tissue sarcoma. Acta Oncol. (2022) 61:38–44. doi: 10.1080/0284186X.2021.1992009
15. Boddu S, Walko CM, Bienasz S, Brega N, Monga V, and Conley AP. Clinical utility of genomic profiling in the treatment of advanced sarcomas: a single-center experience. J Clin Oncol Precis Oncol. (2018) 2:1–8. doi: 10.1200/PO.18.00096
16. Groisberg R, Hong DS, Holla V, Janku F, Piha-Paul SA, Ravi V, et al. Clinical genomic profiling to identify actionable alterations for investigational therapies in patients with diverse sarcomas. Oncotarget. (2017) 8:39254–67. doi: 10.18632/oncotarget.16845
17. Lopes-Brás R, Lopez-Presa D, Esperança-Martins M, Melo-Alvim C, Gallego L, Costa L, et al. Genomic profiling of sarcomas: A promising weapon in the therapeutic arsenal. Int J Mol Sci. (2022) 23:14227. doi: 10.3390/ijms232214227
18. Pan M, Zhou MY, Jiang C, Li J, Yang M, Wu W, et al. PTEN pathogenic variants are associated with poor prognosis in patients with advanced soft tissue sarcoma. BJC Rep. (2024) 2:9. doi: 10.1038/s44276-023-00029-3
19. Kato S, Ross JS, Gay L, Dayyani F, Roszik J, Subbiah V, et al. Analysis of MDM2 amplification: Next-generation sequencing of patients with diverse Malignancies. JCO Precis Oncol. (2018) 2018:PO.17.00235. doi: 10.1200/PO.17.00235
20. Ricciotti RW, Baraff AJ, Jour G, Kyriss M, Wu Y, Liu Y, et al. High amplification levels of MDM2 and CDK4 correlate with poor outcome in patients with dedifferentiated liposarcoma: A cytogenomic microarray analysis of 47 cases. Cancer Genet. (2017) 218-219:69–80. doi: 10.1016/j.cancergen.2017.09.005
21. Mansoori B, Mohammadi A, Ditzel HJ, Duijf PHG, Khaze V, Gjerstorff MF, et al. HMGA2 as a critical regulator in cancer development. Genes (Basel). (2021) 12:269. doi: 10.3390/genes12020269
22. Lam SW, Kostine M, de Miranda NFCC, Schöffski P, Lee CJ, Morreau H, et al. Mismatch repair deficiency is rare in bone and soft tissue tumors. Histopathology. (2021) 79:509–20. doi: 10.1111/his.14377
23. Tazzari M, Bergamaschi L, De Vita A, Collini P, Barisella M, and Bertolotti A. Molecular determinants of soft tissue sarcoma immunity: Targets for immune intervention. Int J Mol Sci. (2021) 22:7518. doi: 10.3390/ijms22147518
24. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. (2017) 9:34. doi: 10.1186/s13073-017-0424-2
25. Abrahao-MaChado LF, Antunes B, Filippi RZ, Volc S, Boldrini E, Menezes WP, et al. Loss of MTAP expression is a negative prognostic marker in Ewing sarcoma family of tumors. biomark Med. (2018) 12:35–44. doi: 10.2217/bmm-2017-0152
26. Bui NQ, Przybyl J, Trabucco SE, Frampton G, Hastie T, and van de Rijn M. A clinico-genomic analysis of soft tissue sarcoma patients reveals CDKN2A deletion as a biomarker for poor prognosis. Clin Sarcoma Res. (2019) 9:12. doi: 10.1186/s13569-019-0122-5
27. Atiq MA, Balan J, Blackburn PR, Gross JM, Voss JS, Jin L, et al. SARCP, a clinical next-generation sequencing assay for the detection of gene fusions in sarcomas: A description of the first 652 cases. J Mol Diagn. (2025) 27:74–95. doi: 10.1016/j.jmoldx.2024.10.004
28. Alba-Pavón P, Alaña L, Gutierrez-Jimeno M, García-Rodríguez M, Moya-García AA, Perales S, et al. Identification of germline cancer predisposition variants in pediatric sarcoma patients from somatic tumor testing. Sci Rep. (2023) 13:2959. doi: 10.1038/s41598-023-29982-2
29. Calvert GT, Randall RL, Jones KB, Cannon-Albright L, Lessnick S, and Schiffman JD. At-risk populations for osteosarcoma: the syndromes and beyond. Sarcoma. (2012) 2012:152382. doi: 10.1155/2012/152382
30. Farid M and Ngeow J. Sarcomas associated with genetic cancer predisposition syndromes: A review. Oncologist. (2016) 21:1002–13. doi: 10.1634/theoncologist.2016-0079
Keywords: sarcoma, soft tissue, next-generation sequencing, molecular, targeted therapy
Citation: Gündoğdu Y, Şenocak Taşçı E, Özer L, Boynukara C, Çeçen R, Mutlu AU and Yıldız İ (2025) Next-generation sequencing-based genomic profiling of advanced soft tissue and bone sarcomas. Front. Oncol. 15:1627452. doi: 10.3389/fonc.2025.1627452
Received: 12 May 2025; Accepted: 17 September 2025;
Published: 03 October 2025.
Edited by:
Matthew J. Marton, MSD, United StatesReviewed by:
Eswari Dodagatta-Marri, University of California, San Francisco, United StatesKarthikeyan Murugesan, Foundation Medicine Inc., United States
Copyright © 2025 Gündoğdu, Şenocak Taşçı, Özer, Boynukara, Çeçen, Mutlu and Yıldız. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: İbrahim Yıldız, aWJyYWhpbS55aWxkaXpAYWNpYmFkZW0uY29t
†ORCID: Elif Şenocak Taşçı, orcid.org/0000-0002-1686-1628
Leyla Özer, orcid.org/0000-0003-4092-5051
Arda Ulaş Mutlu, orcid.org/0000-0001-7499-7155
İbrahim Yıldız, orcid.org/0000-0001-9094-7289