 Xiaocui Liu1,2,3
Xiaocui Liu1,2,3 Xuefeng Kan1,2,3*
Xuefeng Kan1,2,3*- 1Department of Radiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Hubei Provincial Clinical Research Center for Precision Radiology and Interventional Medicine, Wuhan, China
- 3Hubei Province Key Laboratory of Molecular Imaging, Wuhan, China
N6-methyladenosine (m6A) is a reversible mRNA modification that plays important roles in malignant tumor processes. m6A modification has emerged as a significant research focus. Studies on the functions and mechanisms of m6A and its regulatory factors across various tumors have grown increasingly comprehensive and in-depth. Accumulating evidence has demonstrated that m6A modifications and their associated regulatory proteins can serve as biomarkers for cancer treatment and prognosis. Consequently, there has been a surge in research on the development and application of m6A regulatory factor inhibitors, particularly regarding their efficacy and mechanisms in tumor therapy. These advancements not only enhance the understanding of their therapeutic potential in diverse cancers but also facilitate their integration with existing treatments, accelerating the design of more effective, specific, and selective inhibitors. Such efforts hold promise for advancing m6A-targeted pharmaceutical development and promoting clinical applications. This review summarizes small-molecule and peptide inhibitors of m6A regulators for malignant tumors.
1 Introduction
N6-methyladenosine (m6A) specifically refers to the methylation occurring at the nitrogen-6 position of adenine, which is one of RNA modifications. This modification predominantly targets the conserved RRACH sequence (R = A/G, H = A/C/U) and is mainly enriched near the termination codon and 3’ untranslated region (3’UTR) (1, 2). RNA m6A methylation involves modifications in both coding mRNAs and non-coding RNAs (ncRNAs), with the latter category encompassing constitutive ncRNAs and regulatory ncRNAs (3–8). M6A influences the fate of modified RNA molecules by altering mRNA structure, maturation, stability, splicing, transport, localization, translation, degradation (9, 10), and the processing of miRNAs (11) or lncRNAs (12), as well as mediating RNA-protein interactions (13). In eukaryotes, m6A is the most abundant and conserved form of mRNA methylation modification (14). First discovered in mammalian mRNAs in 1974 (15), m6A has become a central focus of RNA epigenetics research, particularly with the advent of advanced sequencing technologies. These advancements have established m6A as the most extensively studied RNA methylation modification, especially in the context of cancer, where research on m6A modification of mRNAs has gained significant momentum (16).
m6A has a widespread impact on the normal physiological and biochemical processes of the body, as well as the occurrence and development of diseases, including metabolism, growth and development, viral infections, chronic diseases, and tumors (17–26). m6A and its regulatory proteins have emerged as critical diagnostic, therapeutic and prognostic biomarkers for cancers. Abnormal m6A modification levels are implicated in the initiation and development of various tumors in multiple systems throughout the body. This post-transcriptional modification exerts either oncogenic or tumor-suppressive effects by modulating m6A levels in mRNAs of involved genes, as well as by activating downstream signaling pathways (27). Aberrant expression of m6A regulators promotes growth, proliferation, migration, invasion, and metastasis of malignancies through multiple mechanisms (28–30). These include inducing tumor cell cycle arrest, inhibiting apoptosis and differentiation, disrupting cancer stem cell self-renewal, altering the tumor microenvironment, and enhancing immune evasion, all of which contribute to tumorigenesis and progression (31–36). Furthermore, the abnormality not only leads to the occurrence of radiotherapy or chemotherapy resistance (37–39), but also devastates the effectiveness of immunotherapy (40–43), ultimately influencing patient survival and prognosis.
The roles and mechanisms of m6A-related regulators in various tumors have been extensively studied and systematically summarized. In recent years, numerous studies have highlighted the potential of inhibitors targeting m6A regulatory proteins, offering promising benefits for cancer treatment. Despite these advancements, a comprehensive review specifically addressing the research progress of m6A inhibitors in cancer has been lacking. This review seeks to provide a comprehensive summary of the current research on the efficacy and mechanisms of all m6A regulatory factor inhibitors with demonstrated cellular activity in tumors. It aims to serve as a valuable reference for expanding the application of m6A-targeted inhibitors, integrating them with existing therapeutic strategies, and overcoming resistance to chemotherapy. Furthermore, this review describes the potential for m6A-targeted pharmaceuticals to cooperate with new materials and convert into clinical practice, offering new prospects for the treatment of incurable cancers.
2 Overview of m6A methylation regulatory factor
m6A modification is a dynamic and reversible process, which is regulated by three distinct types of proteins, including Writers, Erasers, and Readers, as shown in Figure 1. Writers are m6A methyltransferases that responsible for catalyzing RNA methylation; Erasers are m6A demethylases that remove methyl groups from RNA; Readers are a group of m6A-specific binding proteins that selectively recognize and interpret m6A modifications (40, 44–46).
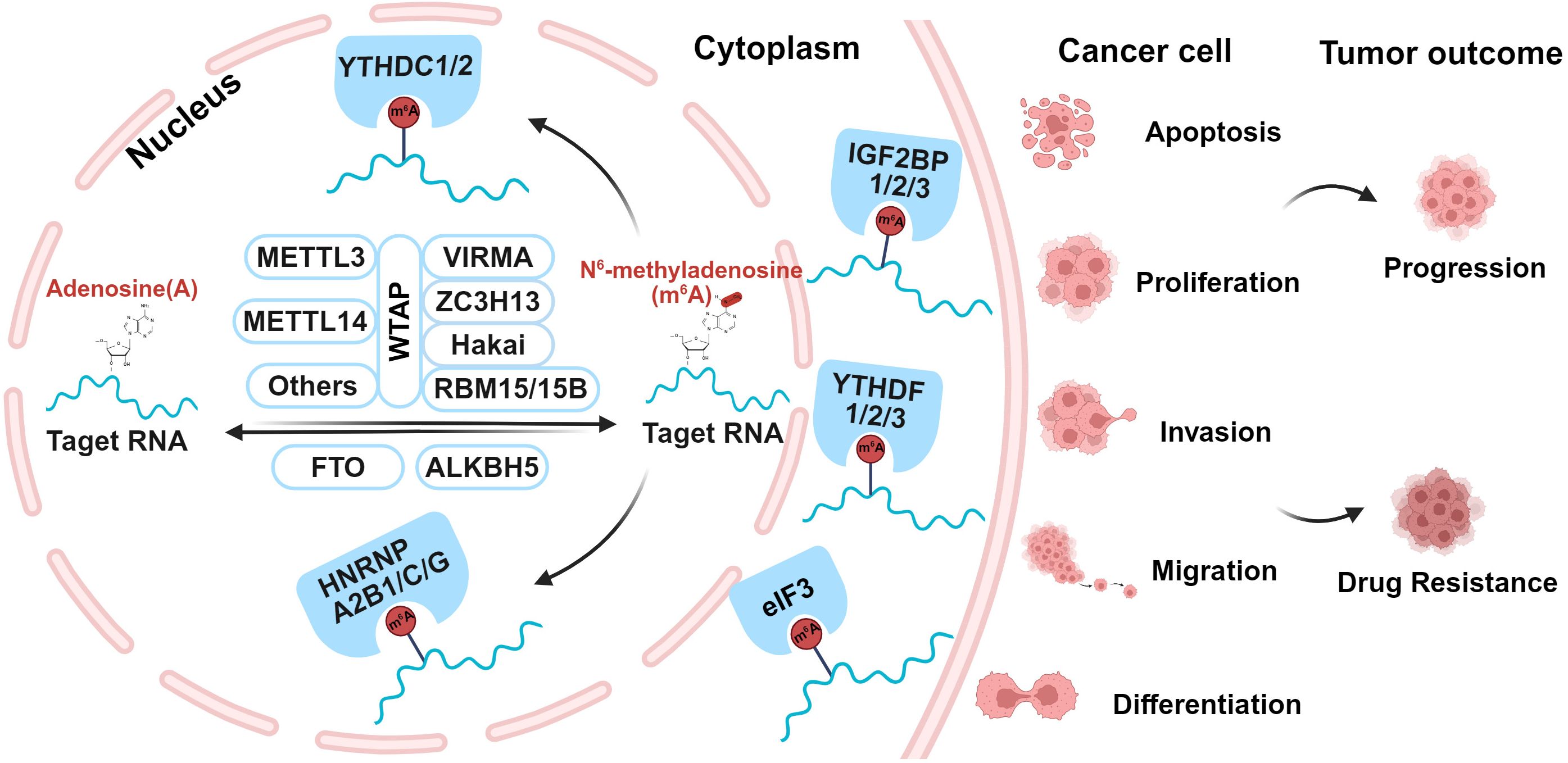
Figure 1. Schematic diagram of the mechanism and function of m6A and its regulatory proteins.
2.1 m6A methyltransferase
The m6A methyltransferases, known as “writers”, can be classified into three major categories: methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), and Wilms tumor 1-associated protein (WTAP). Other members include METTL5/16, RBM15, RBM15B, ZC3H13, ZCCHC4, CBLL1, VIRMA, and Hakai (47). Together, these proteins form the N6-methyltransferase complex (MTC), which utilizes S-adenosylmethionine (SAM) as a methyl donor to catalyze m6A methylation at the nitrogen-6 position of adenine in RNA (48). The MTC operates through the coordinated interaction of two core proteins, METTL3 and METTL14. They form a stable METTL3/14 heterodimer, serving as the catalytic core of the complex, while additional regulatory subunits provide structural support and fine-tune its activity (49). Both METTL3 and METTL14 belong to the S-adenosyl-L-methionine-dependent methyltransferase superfamily and bind either SAM or S-adenosylhomocysteine at the catalytic site. METTL3 contains an activated methyltransferase structural domain, which serves as the catalytic core to transfer the methyl group from SAM to adenosine (50). This domain is capable of catalyzing m6A modification of mRNA. In contrast, METTL14 lacks catalytic activity but specifically facilitates the recognition of METTL3 and the binding of MTC to the RNA substrates, thereby enhancing the catalytic activity of METTL3 (51). WTAP integrates and interacts with the METTL3/14 complex, regulating the localization of the MTC by recruiting it to nuclear speckles enriched with pre-mRNA processing factors. It maintains the catalytic activity of m6A methyltransferase, and also adjusts the recruitment of MTC to target mRNAs (52). Although METTL3, METTL14, and WTAP constitute the major components of the MTC, the contributions of other members should not be overlooked. VIRMA induces regioselective methylation by recruiting the catalytic major components, and mediates preferential methylation of mRNA (53). ZC3H13 regulates and promotes nuclear localization of MTC by facilitating nuclear translocation of WTAP, VIRMA and Hakai (54). Additionally, it serves a regulatory role as a bridge between RBM15 and WTAP (55). RBM15/15B interacts with the MTC and guides its recruitment to specific RNA sites, facilitating the deposition of m6A modifications (56). Hakai stabilizes the major components of MTC (57). These activities illustrate the diverse roles of m6A methyltransferase with diverse components in RNA regulation and cellular processes.
2.2 m6A demethylase
The m6A demethylases, also called “ Erasers “, include fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5), both of which belong to the ALKB protein family of Fe2+/α-ketoglutarate-dependent dioxygenases (58). They remove m6A modifications by catalyzing the hydroxylation of their target bases, thereby reducing RNA methylation levels and effectively reversing the methylation process (58, 59). FTO oxidizes m6A to unstable intermediates, including N6-hydroxy-methyl-adenosine (hm6A) and N6-formyl-adenosine (f6A), ultimately converting them into normal adenosine (A), thereby carrying out its demethylation function (60, 61). FTO primarily targets pre-mRNAs, influencing alternative splicing and 3’UTR processing (62). Though FTO can demethylate both mRNA m6A and cap m6Am in the cytoplasm, its primary demethylation activity occurs in the nucleus. In contrast, ALKBH5 localizes to nuclear speckles and specifically demethylates nuclear RNA substrates (63, 64). It directly converts m6A to A and catalyzes the release of f6A via the R130/K132/Y139 triad (60). ALKBH5-mediated erasure of m6A is required for proper splicing and the generation of longer 3’UTR mRNAs (65), ultimately affecting overall mRNA output (66). These activities underscore the critical roles of FTO and ALKBH5 in regulating RNA methylation dynamics and their subsequent impact on RNA metabolism.
2.3 m6A methylation recognition proteins
Readers, also referred to as methylation effectors or methylated reading proteins, specifically recognize and bind to m6A modifications. This diverse group primarily includes the YT521-B homology (YTH) domain family, insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs), HNRNPs, and eIF3 (67). Additionally, it encompasses BP1, G3BP2, PRRC2A, RBMX, FMR1, ELAVL1, G3BP1, and G3BP2 (68). Among them, YTH domain family consists of YTH domain family proteins 1–3 (YTHDF1–3) and YTH domain-containing protein 1–2 (YTHDC1–2). IGF2BPs are made up of IGF2BP1-3, also known as IMP1-3 (69). Meanwhile, HNRNPs encompass HNRNPC, HNRNPG, and HNRNPA2B1. Readers recognize m6A-modified bases through distinct regulatory mechanisms and subsequently bind to m6A-containing mRNAs, influencing their fate. By selectively recognizing m6A modification sites, these proteins play various roles in regulating key RNA processes, including splicing, translation, transport, and degradation. YTHDF1–3 specifically recognize m6A-modified mRNAs primarily in the cytoplasm. YTHDF1 induces the further translation of m6A-labeled transcripts into proteins (70–72), while YTHDF2 leads to mRNA decay (73, 74). YTHDF3 exhibits versatile roles in regulating RNA processing, translation and decay, with its binding to YTHDF1 to promote translation and YTHDF2 to facilitate degradation (75, 76). Unlike YTHDF1-3, YTHDC1–2 primarily function in the nucleus. YTHDC1 is involved in modulating mRNA selective splicing (77, 78), mediating mRNA nuclear export (79), accelerating the decline of certain transcripts (80). YTHDC2 contributes to mRNA stabilization and translation (81, 82). The IGF2BP1–3 proteins stabilize the target mRNAs and enhance their translation efficiency (83). HNRNPC and HNRNPG primarily interact with ncRNAs and also bind m6A-modified mRNAs, facilitating their splicing (45, 84). HNRNPA2B1 benefits the maturation, transport, and metabolism of mRNA, as well as regulating lncRNA expression (85, 86). The eIF3 protein binds to m6A modification sites at the 5’UTR of mRNA, facilitating translation through a novel mechanism of eIF3-driven initiation that operates independently of cap-binding factors (87). All above highlight the diverse regulatory roles of m6A readers in RNA biology and their impact on cellular processes.
3 METTL3 inhibitors
3.1 Advancements in classification, mechanisms, and therapeutic potential
Research on m6A methyltransferase regulatory factor inhibitors, as shown in Table 1, has predominantly centered on the core protein, METTL3. From one perspective, METTL3 inhibitors can be categorized into nucleoside and non-nucleoside compounds, with the latter further subdivided into allosteric and competitive inhibitors. Nucleoside compounds, mainly comprising adenosine and its analogs, are characterized by their poor selectivity, like neofenobufenacin (88). The allosteric inhibitor CDIBA, which features an indole core structure, binds non-competitively and reversibly to the METTL3-METTL14 enzyme complex, and compound 43n optimized from CDIBA exhibits significant antiproliferative efficacy against acute myeloid leukemia (AML) cells (89). However, an increasing number of studies are now focusing on competitive inhibitors.
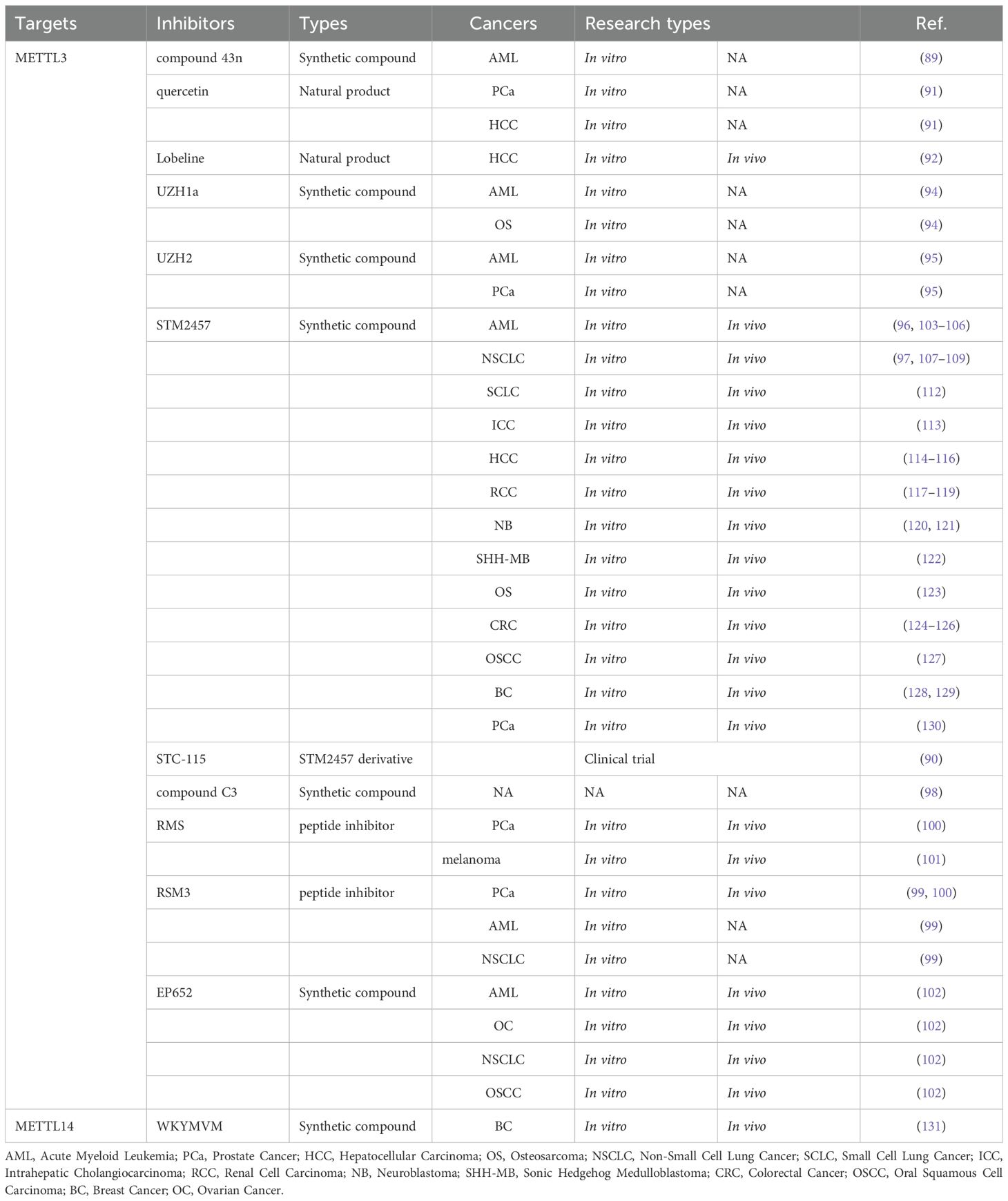
Table 1. Summary of m6A methyltransferase inhibitors.
From a complementary perspective, investigations into METTL3 inhibitors have primarily concentrated on four key domains: the application of natural pharmaceutical compounds, clinical evaluation of synthetic small-molecule drugs, the amalgamation of extensive datasets with computational predictive modeling, and the identification of drug targets and associated pathways (90). Quercetin, a natural compound, has been recognized as the pioneering METTL3 inhibitor. Studies showed that it exerts anti-proliferative effects by reducing global m6A levels in prostate cancer (PCa) and hepatocellular carcinoma (HCC) cell lines (91). Lobeline, another naturally derived inhibitor, was initially identified through screening of a natural product library (92). Research demonstrated that lobeline significantly enhances HCC chemosensitivity to Lenvatinib and effectively reverses therapeutic resistance, resulting in tumor inhibition (92). Mechanistically, it is linked to the regulation of UBE3B expression and function (92). Moreover, studies confirmed that many natural drug components regulating m6A methylation activity fall into categories like polyphenols, flavonoids, alkaloids, anthraquinones, and terpenoids (93).
In a series of studies involving protein crystallography, biochemical binding assays, and cellular experiments, Elena V. et al. (94) identified and characterized UZH1a, a small-molecule METTL3 inhibitor with selectivity, cell permeability, and potency. It effectively reduces the mRNA m6A/A ratio in various cell lines, including osteosarcoma and AML cells. Building on this, Dolbois et al. optimized a METTL3 hit compound using medicinal chemistry and protein crystallography techniques, ultimately leading to the development of lead compound UZH2, and it specifically inhibits intracellular METTL3 activity and significantly decreases the m6A levels in AML and PCa cell lines (95). STM2457 represents a breakthrough in METTL3 inhibition as the first bioavailable compound. This highly selective and potent inhibitor demonstrates both cellular activity and oral bioavailability, making it the most extensively characterized and clinically promising METTL3 inhibitor to date (96). STM2457 (C25H28N6O2; MW: 444.53 g/mol; CAS No. 2499663-01-1) is usually supplied as a stable solid powder (97). Since its initial discovery, it has undergone comprehensive pharmacological evaluation, exhibiting potent METTL3-targeting efficacy across various solid and hematological tumors. The STM2457 derivative STC-15 is the first clinical candidate oral drug targeting METTL3. Currently, a Phase I clinical trial (NCT05584111) is underway to assess STC-15 in patients with advanced solid tumors (90). This marks a major milestone in targeted cancer therapy with the development of METTL3-specific inhibitors as a new therapeutic strategy.
Additionally, Yang et al. (98) employed a series of rigorous and sophisticated methods to screen and identify the compound 2-(4-hydroxyphenyl)-5-[3-[2-(4-hydroxyphenyl)-1,3-dioxoisoindol-5-yl] oxyphenoxy] isoindole-1,3-dione (C3). which exhibits potent and selective inhibitory activity against METTL3. Though its efficacy has not yet to be further validated through preclinical studies, this discovery provides a novel perspective for the development of METTL3 inhibitors.
Unlike small molecule inhibitors, peptide inhibitors were innovatively developed. The representative peptide inhibitor RM3 exhibits significant antitumor properties by inhibiting METTL3 activity and promoting its degradation (99). Based on this, a stable peptide inhibitor named RSM3 was designed and confirmed its antitumor efficacy in various malignancies, including PCa, AML, and NSCLC (99). Furthermore, RM3 and RSM3 effectively inhibit PCa progression, where METTL3/m6A/RRBP1 plays a promoting role (100). Similarly, RM3 exerts anti-tumor effects on melanoma cells (101). Moreover, combining RM3 with anti-PD-1 shows a comparably greater anti-tumor efficacy (101). These findings lay the foundation for developing small peptide inhibitors of METTL3 and also provide new cancer therapy for binding peptide-based drugs with immune checkpoint inhibitors (ICIs).
Recently, Dutheuil et al. (102) discovered and optimized a new effective METTL3 small molecule inhibitor, EP652. In in vivo models of AML and ovarian cancer (OC), EP652 significantly inhibited cancer progression. This discovery opens new avenues for therapeutic strategies in both solid and hematologic tumors.
3.2 Applications of METTL3 inhibitor STM2457 in cancers
3.2.1 Acute myeloid leukemia
METTL3 serves as an oncogene in AML, thereby promoting AML progression (103, 104). Two primary mechanisms have been identified (1): Hyperactivation of METTL3 evokes AML cell proliferation and inhibits differentiation by promoting the translation of oncogenic targets such as c-MYC, BCL2, and PTEN (103); (2) CAATT enhancer binding protein Zeta (CEBPZ) recruits METTL3 to the transcription start site, leading to the enhanced translation of oncogenes SP1 and SP2 (104). Yankova et al. indicated that STM2457 lessened AML growth rate and induced differentiation and apoptosis, resulting in anti-leukemic outcomes (96). Similarly, Li et al. demonstrated that METTL3 can promote m6A modification of ITGA4 mRNA, thereby enhancing the mRNA stability and prolonging its half-life (105). This leads to the upregulation of ITGA4 protein expression, which subsequently promotes the homing and implantation of AML cells. Furthermore, AML cells may interact with the bone marrow microenvironment and finally provoke chemoresistance in AML (105, 106). However, STM2457 can significantly turn around the homing and implantation of AML cells, thus reversing drug resistance and improving efficacy (105). These findings highlight the potential of STM2457 as a promising therapeutic agent in combating AML and overcoming chemoresistance.
3.2.2 Lung cancer
The heterogeneity of non-small cell lung cancer (NSCLC) promotes its drug resistance, and PD-L1 expression suppresses its immunotherapy. STM2457 effectively inhibits the progression of NSCLC by targeting the translational regulatory mechanisms of METTL3 and its interplay with PD-L1 (97). STM2457 not only overcomes the heterogeneity of tumors to achieve inhibition of tumor progression, but also regulates the expression of PD-L1, thereby expanding the benefits of immunotherapy. Additionally, STM2457 can inhibit the METTL3/m6A/SRPK1 axis, thereby preventing glycolysis in lung adenocarcinoma (107). Generally speaking, METTL3 promotes glycolysis via increasing the m6A modification of SRPK1, thereby facilitating tumor progression. Furthermore, the combination of STM2457 and MAT2A inhibitors displays good synergistic anti-tumor effectiveness (108). Notably, the METTL3 inhibitor specifically targets the PI3K/AKT signaling axis. Additionally, the combination of STM2457 with paclitaxel (PTX) or carboplatin demonstrates significantly enhanced synergistic anti-tumor efficacy in NSCLC (109). The mechanism involves the reduction of ABCC2 expression in an m6A-YTHDF1-dependent manner.
In contrast to NSCLC, small cell lung cancer (SCLC) presents formidable therapeutic challenges characterized by its aggressive biological behavior, high malignant potential, and drug-resistant susceptibility (110, 111). The elevated expression of METTL3 mRNA and protein levels in SCLC individuals predicts poor prognosis (112). METTL3 reduces the m6A level of DCP2 mRNA, accelerating its degradation and suppressing DCP2 expression (112). This process activates Pink1-Parkin-mediated mitochondrial autophagy, maintaining cellular homeostasis and promoting chemotherapy resistance in SCLC (112). These findings highlight STM2457’s potential to target similar mechanisms in both NSCLC and SCLC, offering a promising strategy to overcome drug resistance and improve treatment outcomes in these aggressive lung cancers.
3.2.3 Intrahepatic cholangiocarcinoma
The overexpression of METTL3 in intrahepatic cholangiocarcinoma (ICC) patients is associated with poor prognosis (113). The upregulation of METTL3 promotes m6A modification of IFIT2 mRNA, reducing its stability and triggering its degradation. This leads to the downregulation of IFIT2 expression, which in turn facilitates tumor progression (113). However, the METTL3 inhibitor STM2457 effectively suppresses ICC by inhibiting cell proliferation, migration, and invasion, as well as inducing apoptosis in ICC cells (113). These findings show the potential of STM2457 in improving ICC patient treatment outcomes.
3.2.4 Hepatocellular carcinoma
METTL3 overexpression is strongly linked to poor prognosis and resistance to oxaliplatin (OXA) in HCC individuals (114). Mechanistically, the METTL3/TRIM21/G6PD pathway works well in promoting drug resistance of HCC (114). Notably, STM2457 reverses the drug resistance and enhances the therapeutic efficacy in HCC (114). In another study, abundant m6A was discovered in the core enzymes of SSP (PSPH, PSAT1, and PHGDH), and the METTL3/m6A/IGF2BP3 axis works in modulating associated mRNA stability and activating SSP (115). Furthermore, HCC cells with resistance to sorafenib depend on m6A to regulate the SSP pathway and reactive oxygen species (ROS) levels (115). Importantly, inhibiting SSP with STM2457 increases ROS accumulation, impairs HCC growth, and enhances the sensitivity of HCC to sorafenib (115). Additionally, STM2457 targets the METTL3/m6A/BMI1/RNF2 signaling pathway to suppress malignant growth (116). These studies provide more promising therapeutic ideas for the treatment of HCC.
3.2.5 Renal cell carcinoma
Elevated METTL3 expression in advanced RCC patients equals to poor prognosis (117). RCC tends to generate a hypoxic microenvironment due to rapid growth (118). Hypoxia upregulates METTL3 via HIF-1α, promoting m6A-modified PLOD2 mRNA translation, ultimately resulting in RCC migration and invasion (117, 119). Significantly, STM2457 targets the METTL3-PLOD2 axis, effectively reducing tumor growth and metastasis. These findings highlight STM2457 as a promising therapeutic agent for advanced RCC.
3.2.6 Neuroblastoma
On the one hand, STM2457 inhibits the proliferation of adrenal neuroblastoma and promotes a differentiated phenotype, subsequently restraining neuroblastoma growth (120). Additionally, the study preliminarily explored the role of differentiation-related gene expression in this process. On the other hand, KAP1, in conjunction with YTHDC1 and METTL3, forms a complex responsible for preserving the stability of MYCN mRNA (121). Consequently, STM2457 downregulates MYCN to achieve an anti-tumor effect in neuroblastoma. These findings provide a foundation for further mechanistic investigations and the development of potential therapeutic methods.
3.2.7 Sonic hedgehog medulloblastoma
High expression of METTL3 means poor prognosis in patients with Sonic Hedgehog medulloblastoma (SHH-MB) (122). The over-expression of METTL3 leads to persistent m6A hypermethylation of PTCH1 and GLI2, thereby promoting proliferation and suppressing cell death (122). However, STM2457 inhibits METTL3, thereby reducing tumor progression. Which presents a promising therapy for SHH-MB.
3.2.8 Osteosarcoma
Elevated expression of METTL3 incurs an increase in the overall m6A modification level of the oncogene ZBTB7C mRNA, eventually promoting osteosarcoma progression (123). Notably, STM2457 decreases m6A modification levels and weaken ZBTB7C protein abundance, leading to osteosarcoma growth inhibition. Furthermore, the increased METTL3 and ZBTB7C levels in human osteosarcoma tissues were confirmed, highlighting their potential as therapeutic targets and STM2457 as a promising drug.
3.2.9 Colorectal cancer
Yu et al. (124) discovered that STM2457 exhibits significant anti-tumor activity in CRC cells and tissues. Mechanistically, the inhibition of METTL3 reduces the m6A level of asparagine synthetase (ASNS) mRNA, thereby downregulating ASNS expression and exerting anti-tumor effects. However, Zhou et al. (125) found that m6A modification enhances mitochondrial fusion via RRM2B/GSH/OPA1 pathway, and STM2457 prevent the above fusion in order to prohibit CRC progression. Additionally, the combination of STM2457 with an anti-PD-1 antibody can strengthen the expression of interferon-gamma and granzyme B, thereby increasing the cytotoxic activity of T cells and boosting the anti-tumor immunity against colon cancer, as well as melanoma (126). These studies offer novel insights into combination therapy.
3.2.10 Oral squamous cell carcinoma
STM2457 combined with anlotinib could effectively prevent OSCC (127). Mechanistically, this combination significantly downregulates the expression of epidermal growth factor receptor (EGFR), suppresses the stemness properties and epithelial-mesenchymal transition (EMT) process. This combined therapeutic remedy may bring new insights for the clinically treatment of OSCC.
3.2.11 Breast cancer
A study (128) revealed a progression mechanism in triple-negative breast cancer (TNBC), where POP1 targets m6A to destabilize CDKN1A mRNA. The downregulation of CDKN1A accelerates TNBC progression. Notably, STM2457 not only reverses this situation but also increases the sensitivity of TNBC to PTX. Another study (129) about the endoplasmic reticulum (ER) discovered a new mechanism where XBP1s enhances the expression of METTL3/METTL14 and increases its m6A modification. In turn, the elevated m6A modification facilitates reticulophagy (ER-phagy) by stabilizing the mRNA of CALCOCO1 and p62, ultimately supporting cell survival under PTX-induced stress in BC. Further investigation showed that PTX could mediate ER stress and up-regulate m6A modification for ER-phagy. Importantly, experiments on BC declared remarkable therapeutic efficacy through combining STM2457 and PTX. These evidences bring novel approaches for PTX resistance in BC.
3.2.12 Prostate cancer
Recent studies (130) highlighted the therapeutic potential of targeting m6A modifications in PCa. Notably, STM2457 effectively reduces m6A levels in PCa cells, thereby impairing their proliferative, invasive, migratory, and stemness properties. Moreover, STM2457 demonstrates significant tumor-suppressive effects in animal models. Mechanistically, the IGFBP3/AKT axis acts as a key mediator of its biological effects. Additionally, STM2457 exhibits synergistic anti-PCa activity when combined with the PARP inhibitor olaparib. Undoubtedly, these findings provide new insights into the treatment and combination therapy of PCa.
4 METTL14 inhibitors
As one of the core components of methyltransferase, there have been few reports on METTL14 inhibitors. Recently, Liu et al. (131) identified a novel small molecule inhibitor, WKYMVM (Trp-LysTyr-Met-Val-Met-NH2), through screening. The study showed that METTL14 can increase the expression of E2F1 in BC, thereby promoting its resistance to CDK4/6 inhibitor therapy. However, the inhibitor WKYMVM can reverse the resistance of BC cells to CDK4/6 inhibitor treatment, restoring their sensitivity. This breakthrough paves the way for the development and exploration of METTL4 inhibitors.
5 FTO inhibitors
All demethylase inhibitors are shown in Table 2. FTO was the first RNA m6A demethylase to be discovered (64). To date, more than 10 FTO inhibitors have been identified, and the therapeutic efficacy of these inhibitors has been evaluated across various cancers. FTO inhibitors can be classified into three distinct categories based on their mechanism of action (1): metal ion-chelating inhibitors that disrupt the catalytic center, (2) 2-oxoglutarate(2-OG) analogs that compete with the essential cofactor, and (3) selective m6A-competitive inhibitors that specifically target the substrate-binding site (132). Targeting FTO can suppress tumor growth, enhance tumor immunotherapy, and mitigate tumor drug resistance (32). The development of FTO inhibitors provides additional therapeutic solutions for cancer treatment.
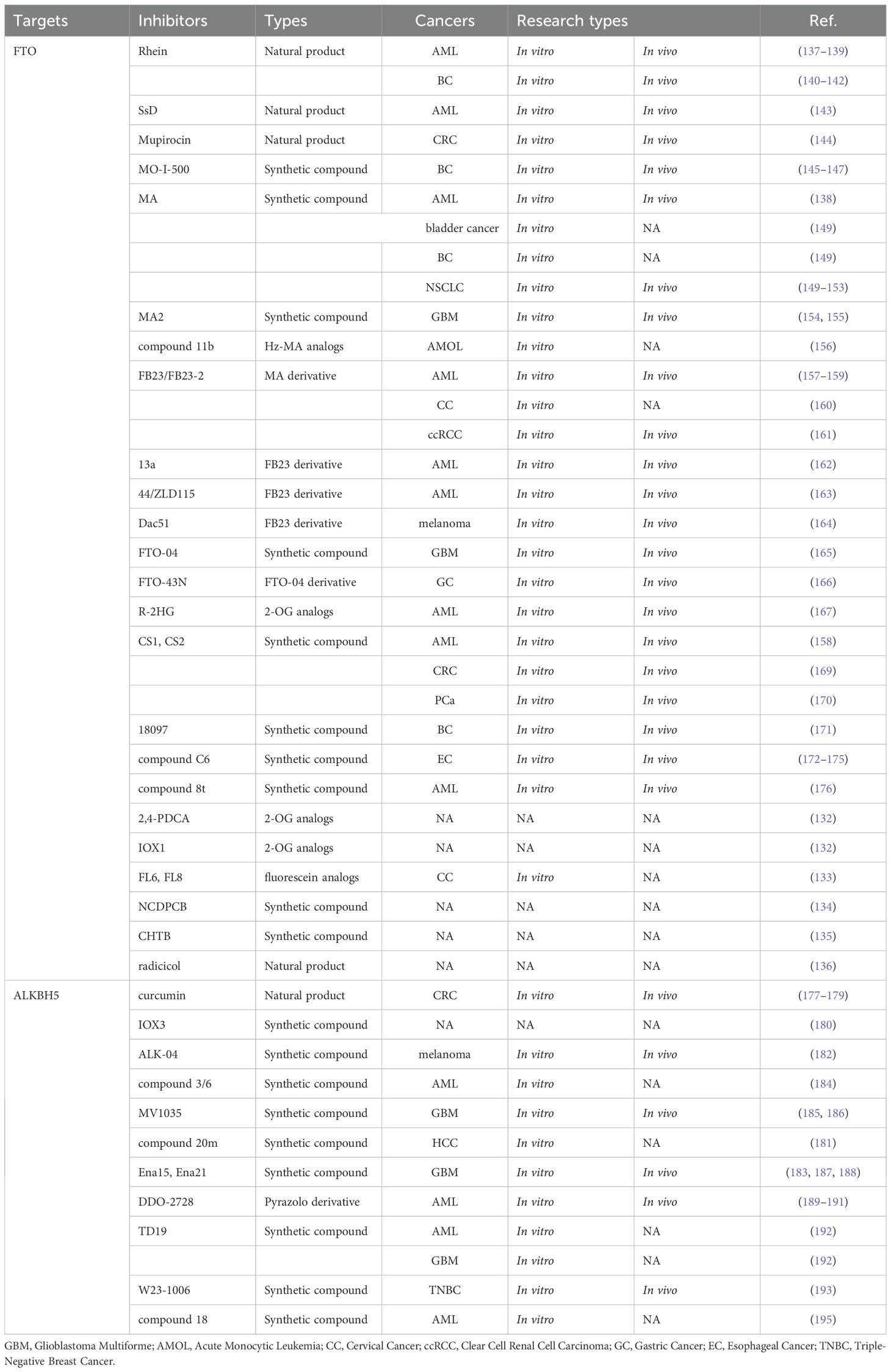
Table 2. Summary of m6A demethylase inhibitors.
5.1 FTO inhibitors without preclinical studies
Aik et al. identified two FTO inhibitors, 2,4-PDCA and IOX1, from the 2-OG analogs (132). Additionally, fluorescein and its derivatives act as bifunctional molecules via selectively inhibiting FTO demethylation while simultaneously labeling FTO proteins. Among fluorescein analogs, FL6 and FL8 perform superior cell permeability and effectiveness in treated cervical cancer HeLa cells (133). N-(5-chloro-2,4-dihydroxyphenyl)-1-phenylcyclobutanecarboxamide (NCDPCB) was identified as another potential FTO inhibitor (134). Similarly, 4-chloro-6-(6′-chloro-7′-hydroxy-2′,4′,4′-trimethyl-chroman-2′-yl) benzene-1,3-diol (CHTB) functions as a small-molecule FTO inhibitor (135). Furthermore, Wang et al. discovered that the natural compound radicicol potently inhibits FTO demethylation in a dose-dependent manner (136). All these inhibitors bind to the same site on FTO as MA. While their cancer inhibitory effects have not yet been fully validated through preclinical studies, these discoveries provide valuable insights into the structure and properties of FTO inhibitors. These foundational knowledges pave the way for the development of more potent, specific, and selective FTO inhibitors with enhanced therapeutic potential.
5.2 Natural products of FTO inhibitors
Rhubarbic acid (Rhein), a natural compound, was identified as the first bioactive competitive FTO inhibitor (137). This potent inhibitor with cellular activity binds to FTO’s active site, blocking m6A substrate recognition (137). Therefore, the FTO-m6A axis has emerged as a novel marker characterizing leukemia cell heterogeneity and a broad defense mechanism for its resistance to tyrosine kinase inhibitors (TKIs) (138). And when combined with TKIs, Rhein effectively eradicates drug-resistant leukemia cells and suppresses tumor growth in AML mouse models (138). Mechanistically, Rhein targets FTO to disrupt the AKT/mTOR signaling pathway (139). These findings highlight Rhein’s potential as a therapeutic agent in targeting FTO and overcoming drug resistance in leukemia.
Through suppressing activation of PTEN/PI3K/AKT/mTOR and MAPK/ERK pathways, which are induced by vascular endothelial growth factor and endothelial growth factor, Rhein effectively inhibits the growth of BC cells (140). Additionally, Rhein exhibits antitumor effects on MCF-7 cells with high HER2 expression, mechanistically involved in caspase-9-mediated apoptosis and ROS-mediated activation via NF-kB/P53 signaling pathway (141). Another study further evaluated Rhein’s anti-tumor function on BC cells and tissues through the combination of Rhein and atezolizumab (142). Mechanistically, they remarkably elevate the CD8+ T-cell infiltration, serum TNFα and IL-6 levels, apoptotic factors, and the Bax/Bcl2 mRNA level (142). These findings emphasize Rhein’s potential as a complementary agent to enhance the efficacy of immunotherapy in BC.
Saikosaponin-D (SsD), a triterpenoid saponin compound extracted from Bupleurum species, demonstrates significant anti-tumor activity. Sun et al. (143) reported that SsD effectively inhibits AML cells in both in vitro and in vivo models. Mechanistically, SsD directly targets FTO, leading to an increase in overall RNA m6A methylation levels. This increased methylation level reduces the stability of downstream gene transcripts and inhibits associated signaling pathways, thereby exerting a potent anti-tumor effect. Furthermore, the study uncovered that SsD can overcome FTO/m6A-mediated leukemia resistance to TKIs, making it a promising candidate for addressing drug resistance in AML therapy.
Mupirocin is also a natural FTO inhibitor. Qiao et al. (144) demonstrated that it effectively suppresses CRC cell growth through a ferroptosis-dependent manner. Moreover, FTO promotes the expression of SLC7A11 and GPX4, thereby reducing cancer cell ferroptosis in animal models and in vitro. These findings offer valuable insights for the design of novel therapeutic approaches for CRC.
As inhibitors, natural products often lack target specificity, prompting increasing research interest in the exploration of synthetic small-molecule inhibitors.
5.3 Synthetic compounds of FTO inhibitors
MO-I-500, a synthetic ascorbic acid analog, has been shown to inhibit FTO activity effectively. FTO exerts a tumor-promoting role in BC. Highly expressed FTO in human BC tissues significantly facilitates tumor progression (145, 146). The mechanism involves Bcl-2 nineteen kilodalton interacting protein 3 (BNIP3), a pro-apoptotic gene (147). FTO decreases m6A modification level of BNIP3 mRNA, resulting in a downregulation of BNIP3 expression. This enables FTO to exert its pro-tumorigenic effects (146). By targeting FTO, MO-I-500 exhibits anti-proliferative effects on BC cells, highlighting its potential as a therapeutic compound in BC treatment (145).
Meclofenamic acid (MA) and its derivative are substrate-competitive inhibitors of FTO with cellular activity. MA is a highly selective inhibitor, and competitively inhibits the binding of FTO to m6A-containing nucleic acids (148). Preliminary in vitro experiments have demonstrated that MA can inhibit the proliferation of BC, bladder cancer, and NSCLC cells (149). Additionally, BCRP and MRP-7 trigger Gefitinib (GE) resistance during tumor therapy process (150, 151). Therefore, inhibiting BCRP and MRP-7 function or reducing their expression could serve as promising strategies to overcome GE resistance in NSCLC (152). The combination of MA and GE significantly downregulated BCRP and MRP7 expression in GE-resistant cells by increasing m6A modification of MYC (153). This drug combination also inhibits GE resistance by accelerating apoptosis, inhibiting the EGFR downstream pathway, and promoting GE accumulation in cancer cells (153). Furthermore, the application of MA, when combined with TKIs in leukemia management exhibits comparable clinical efficacy to Rhein-based therapies (138). MA2, an acetyl derivative of MA, is another highly selective inhibitor of FTO (148). MA2 significantly inhibits the growth and self-renewal of glioblastoma stem cells (GSCs), thereby suppressing GSCs-induced tumor formation and prolonging the survival time of an animal model (154). MYC/miRNA/MXI1 feedback loop plays a crucial role in glioma proliferation and tumorigenesis. Among them, MYC suppresses MXI1 expression through related miRNA, while MXI1 represses MYC expression via binding to its promoter. Meanwhile, FTO works through stabilizing MYC transcripts. MA2 inhibits FTO activity, thereby disrupting this oncogenic feedback loop and exerting anti-tumor effects. Additionally, MA2 enhances the effectiveness of temozolomide (TMZ) in suppressing glioma cell proliferation (155), highlighting its potential as a therapeutic agent in glioblastoma treatment.
According to previous FTO inhibitors, Prakash et al. utilized a fragment-merging strategy to design and synthesize compound 11b, a hybrid analog combining key fragments of Hz and MA (156). This Hz-MA hybrid analog is a highly effective, selective, and cell-active FTO inhibitor (156). Treatment with Hz-MA in acute monocytic leukemia cells significantly reduces cell viability. Mechanistically, Hz-MA treatment induced upregulation of MYC oncogene expression while simultaneously suppressing RARA transcriptional activity (156). These findings position Hz-MA as a promising candidate for targeted leukemia therapy.
Firstly, two promising FTO inhibitors, FB23 and FB23-2, was designed through structural optimization. These compounds directly bind to FTO and selectively inhibit its m6A demethylase activity (157). FB23, an MA-derived inhibitor, shows more potent inhibition of FTO enzyme activity than MA. And FB23-2, a further optimized analog of FB23, exhibits superior cell permeability. FTO overexpression exerts oncogenic function in AML (158). Thus FB23–2 exerts anti-leukemic effects through multiple mechanisms: inhibiting AML cell proliferation, inducing differentiation and apoptosis, thereby significantly improving survival outcomes in leukemic mouse models (157). Tarullo et al. obtained similar anti-tumor effects against AML (159). However, Wang et al. confirmed the anticancer effect of FB23–2 in cervical cancer, with its mechanism involving the mRNA and protein levels of DIRAS family GTPase 1 (DIRAS1) (160). Additionally, Xu et al. found that FTO is significantly up-regulated in clear cell renal cell carcinoma (ccRCC), where it functions as an oncogene (161). This leads to the reduced level of m6A modification associated with autophagy, and predicts a poor prognosis for patients (161). Mechanistically, the FTO upregulation reduces m6A levels and destabilizes SIK2 mRNA through an m6A-IGF2BP2-dependent pathway, leading to decreased SIK2 expression (161). As a result, ccRCC exhibits enhanced autophagy, proliferation, migration, and invasion. Importantly, FB23–2 inhibits tumor growth to reverse the above malignant processes and prolong survival in animal models (161). Based on previous inhibitors, FB23 analogs (13a and 44/ZLD115) came into light. It was reported that 13a exhibits strong anti-proliferative effects in AML cells and improves survival outcomes in animal models (162). Mechanistically, 13a upregulates ASB2 and RARA expression while downregulating MYC (162). Similarly, 44/ZLD115, a tetracyclic benzoic acid derivative with a flexible basic side chain, significantly upregulates RARA and downregulates MYC to realize its anti-leukemic activity (163). Another promising analog, Dac51, shares a similar FTO-binding mode with FB23 but exhibits greater potency. Dac51 stabilizes FTO while simultaneously blocking FTO-mediated immune evasion, synergizing with ICIs to enhance anti-tumor efficacy (164). Dac51 remodels the tumor microenvironment by attenuating glycolytic metabolism and promoting CD8+ T cell infiltration, leading to significant anti-tumor effects in melanoma (164). Additionally, combining Dac51 with ICIs improves therapeutic outcomes (164). These findings exhibit the versatility and potential of FB23-derived inhibitors across a variety of malignancies.
FTO-02 and FTO-04, two potent and highly selective FTO competitive inhibitors, was developed by structural design, synthesis, and biochemical evaluation (165). The research demonstrated that FTO-04 effectively prevented neurosphere growth in patient-derived GSCs, and both the m6A and m6Am levels were elevated in patients after FTO-04 treatment, which ultimately led to the suppression of glioblastoma (165). In another study, through rational design and optimization, a new class of inhibitor oxaprostanes derived from FTO-04 came into birth (166). Among these, FTO-43N emerged as a lead compound, effectively elevating m6A and m6Am levels in gastric cancer cells, ultimately exerting anti-cancer effects by regulating the Wnt/PI3K-Akt signaling pathway (166). These findings highlight the potential of oxaprostanes, particularly FTO-43N, as promising therapeutic drugs for FTO-targeted cancer therapy.
R-2HG, a 2-OG competitive inhibitor, has demonstrated broad antileukemic effects both in vitro and in vivo. Su et al. (167) showed that R-2HG suppresses the proliferation and survival of leukemia cells while promoting cell cycle arrest and apoptosis. Mechanistically, R-2HG targets the FTO/m6A/MYC/CEBPA signaling pathway to inhibit the malignancy of leukemia cells with high FTO expression. By disrupting this pathway, R-2HG effectively reduces the oncogenic potential of FTO-overexpressing leukemia cells, making it a promising therapeutic agent for leukemia treatment.
CS1 and CS2, potent and selective FTO inhibitors, was identified through a series of screening and validation analyses (168). Studies revealed FTO’s significant role and potential mechanism in prohibiting tumor stem cell self-renewal and immune evasion. CS1 and CS2 selectively bind and occupy the catalytic pocket of FTO, preventing m6A-modified oligonucleotides from entering the catalytic pocket, thereby inhibiting FTO-mRNA binding and reducing MYC/CEBPA while increasing RARA/ASB2 expression (158, 167). Conversely, uninhibited FTO reduced RARA and ASB2 protein expression and suppressed all-trans retinoic acid (ATRA)-induced leukemia cell differentiation (158). Phan et al. found that CS1 inhibits CRC progression through the downregulation of several signaling pathways, including the expression of ERG, KRAP, PDE4B and SLC38A2 (169). In addition, CS1 can be applicable as a single alternative drug or in combination to overcome medicine resistance to 5-FU-based treatments for CRC (169). Moreover, Garg et al. demonstrated that PCa cells express higher levels of FTO than normal cells, and CS1-mediated inhibition of FTO performs significant anti-PCa function (170). These findings underscore the versatility of CS1 and CS2 as promising therapeutic methods against multiple cancers, offering potential for targeted therapy and overcoming drug resistance.
18077 and 18097 was designed to directly inhibit FTO demethylase activity with potency and cellular activity. 18097 has a significant inhibitory effect on the colony formation of BC cells (171). Additionally, 18097 exhibits strong anti-tumor activity in vivo, effectively inhibiting the progression of BC and lung metastases (171). Moreover, 18097 was found to metabolically inhibit cancer progression by regulating the key downstream effector cytokine signaling 1(SOCS1) in the P53 pathway (171). This finding makes 18097 a promising candidate for BC therapy targeting FTO.
1,2,3-Triazoles, an important category of nitrogen-containing heterocyclic compounds, possess strong anticancer activity (172). A class of 1,2,3-triazole-pyridine hybrid compounds containing pentafluorobenzoyl groups was designed as FTO inhibitors. Among them, compound C6 showed the strongest inhibitory effect, making it a highly efficient and low-toxicity oral anticancer agent (173). Increased expression of FTO in esophageal cancer tissues is associated with poor clinical prognosis (174). FTO plays an oncogenic role in esophageal cancer, promoting cell proliferation and migration (175). Compound C6 targets FTO to display anti-tumor activity against esophageal cancer via the inhibition of the EMT pathway. Furthermore, C6 regulates the PI3K/AKT signaling pathway, enhancing its inhibitory effect on esophageal cancer progression (173). These findings pave the way for esophageal cancer treatment.
Recently, Liang et al. identified a series of novel and effective FTO inhibitors with an acylhydrazone scaffold, among which compound 8t is a potent representative (176). Compound 8t can inhibit the proliferative activity of AML cells both in vivo and in vitro, laying the foundation for further research on effective cancer inhibitors.
6 ALKBH5 inhibitors
ALKBH5, the second m6A demethylase to be identified, has m6A as its only known catalytic substrate (66). There are few ALKBH5 inhibitors studied to date, especially natural compounds. As a phenolic compound extracted from turmeric root, Curcumin has been shown to reduce the expression of ALKBH5. Then curcumin enhances the translation of tumor necrosis factor receptor-associated factor 4 (TRAF4) and promotes the binding of TRAF4 to the m6A methyl-recognizing enzyme, thereby improving the efficiency of m6A methylation modification (177). Additionally, curcumin facilitates the conversion of microtubule-associated protein LC3-I to LC3-II, upregulates Beclin-1, and induces autophagy in CRC cells. These actions reduce cancer stem cell production and re-sensitize drug-resistant cells to chemotherapeutic agents such as 5-FU and OXA (178, 179). These findings highlight curcumin’s potential as a therapeutic agent targeting ALKBH5-mediated pathways in cancer treatment.
Natural products often lack selectivity, which is why an increasing number of studies are focused on developing selective small molecule inhibitors with targeted competency. Aik et al. unexpectedly discovered that the small molecule inhibitor IOX3 can covalently inhibit ALKBH5 during their study of the structure and function of ALKBH5 (180). Unfortunately, the application of this inhibitor in cancer has not been further explored. Fang et al. (181) obtained an effective compound, 5-hydroxy-1-(3-(trifluoromethyl) phenyl)-1H-pyrazole-3-carboxylic acid (20m), through a series of complex methods. They demonstrated that 20m can inhibit the demethylation process of m6A in HCC HepG2 cells. Additionally, they validated the efficacy, selectivity, and biological activity of 20m as an ALKBH5 inhibitor. However, further in vivo and in vitro validation is lacking.
ALK-04, a small-molecule inhibitor of ALKBH5, was identified through screening using the X-ray crystal structure and structure-activity relationship of the ALKBH5 protein. In preclinical studies, ALK-04 demonstrated significant therapeutic potential by synergistically reducing melanoma tumor growth when combined with GVAX/anti-PD-1 immunotherapy via MCT4/SLC16A3 axis (182). This finding turns ALK-04 into a promising candidate for enhancing the efficacy of immunotherapy in melanoma treatment.
ALKBH5, which is highly expressed in GSCs, significantly promotes tumor formation and proliferation (183). A study selected two compounds, 2-[(1-hydroxy-2-oxo-2-phenylethyl) sulfanyl] acetic acid (compound 3) and 4- (184)-1,2-diazinane-3,6-dione (compound 6), and validated their antiproliferative activity in various leukemia cell lines (184). All above lay the foundation for the development of selective ALKBH5 inhibitors. Malacrida et al. provided further evidence about MV1035, an ALKBH5-targeted inhibitor (185). They found that MV1035 inhibits the migration and invasion of glioblastoma cells. In a follow-up study, they reaffirmed these findings and demonstrated that MV1035, when combined with TMZ, significantly reduces cell viability and sphere formation in glioblastoma cells (186). Notably, MV1035 not only decreases glioblastoma migration and invasion but also overcomes TMZ resistance, making it a promising candidate for glioblastoma therapy (186). These studies highlight the therapeutic potential of ALKBH5 inhibitors, particularly MV1035, in addressing the challenges of glioblastoma progression and drug resistance.
By high-throughput screening, Takahashi et al. (187) identified two novel small-molecule inhibitors, Ena15 and Ena21, which exhibit non-competitive or competitive inhibition of 2-OG. The tumor-initiating cell GSCs are highly self-renewing, resistant to conventional treatments, and responsible for tumor recurrence because of sustained tumor growth (188). ALKBH5 reduces the m6A methylation level of FOXM1 transcripts, thereby enhancing FOXM1 expression. Where FOXM1 plays a critical role in regulating GSC proliferation, self-renewal, and tumorigenicity (183). Ena15 and Ena21 effectively inhibit cell proliferation in glioblastoma multiforme (GBM) cell lines by downregulating FOXM expression (187). These findings highlight the potential of Ena15 and Ena21 as therapeutic agents targeting ALKBH5-mediated pathways in glioblastoma.
Through structure-based virtual screening and optimization, DDO-2728, a potent and selective pyrazolo[1,5-a] pyrimidine derivative, was identified to directly bind to ALKBH5 (189). Unlike 2-hydroxyglutarate analogs, DDO-2728 selectively inhibits the demethylase activity of ALKBH5 without affecting FTO. Overexpression of ALKBH5 plays a tumorigenic role in AML by regulating the stability of the prognosis-related oncogene TACC3 mRNA (190). DDO-2728 inhibits ALKBH5, resulting in increased m6A modification levels in AML cells, decreased TACC3 mRNA stability, and induction of apoptosis and cell cycle arrest. These antiproliferative effects significantly suppress tumor growth (189). Furthermore, Fei et al. (191) developed and synthesized the covalent ALKBH5 inhibitor DDO-02267 via exploring the protein structure of ALKBH5 and incorporating a salicylaldehyde warhead into a noncovalent small molecule ligand. With the high selectivity and specificity for ALKBH5, DDO-02267 enhances m6A levels and modulates the ALKBH5-AXL signal pathway in AML tissues and cells. These findings highlight the therapeutic potential of ALKBH5 inhibitors in AML.
Recently, researchers developed a selective covalent inhibitor of ALKBH5, TD19, which shows good biological activity (192). And it exhibits significant anticancer efficacy in AML and GBM cells (192). Besides, some other researchers developed a more efficient novel covalent inhibitor, namely W23-1006, which exhibits selectivity and biological activity (193). Experiments about TNBC indicated that W23–1006 covalently inhibits ALKBH5, increasing the m6A level of fibronectin 1 (FN1) mRNA, thereby reducing the expression of FN1 and bringing overall tumor suppression both in vivo and in vivo (193). Notably, FN1 is a key gene that promotes the EMT process in tumors (194). More recently, investigators identified a series of maleimide-derived small molecule inhibitors targeting ALKBH5, with compound 18 emerging as a key candidate after optimization (195). AML cell assays further confirmed its antiproliferative activity, while comprehensive evaluations highlighted its potential as a lead compound for ALKBH5 inhibitor development (195). These findings offer fresh insights and novel strategies for advancing anticancer therapeutics.
7 Inhibitors of IGF2BPs
As is shown in Table 3, IGF2BPs share similar structures and functions. Their N-terminal contains two RNA recognition motifs (RRM1-2), while their C-terminal houses four KH homology domains (KH1-4) (196). Notably, IGF2BPs, particularly IGF2BP1 and IGF2BP3, are recognized as oncofetal proteins. These regulatory proteins, which are transiently expressed during embryonic development and subsequently silenced in most adult tissues, undergo tumor-specific reactivation and play pivotal roles in cancer progression and metastasis (33). Aberrant expression of IGF2BPs is associated with a wide range of tumorigenic processes. By regulating the transcripts of several oncogenes such as KRAS, MYC, PTEN, and MDR1, IGF2BPs contribute to numerous malignant behaviors, including enhanced cell proliferation, invasion, stemness, drug-resistance, and immune evasion (196, 197). These properties underline their significance as key players in tumor progress and potential therapeutic targets.
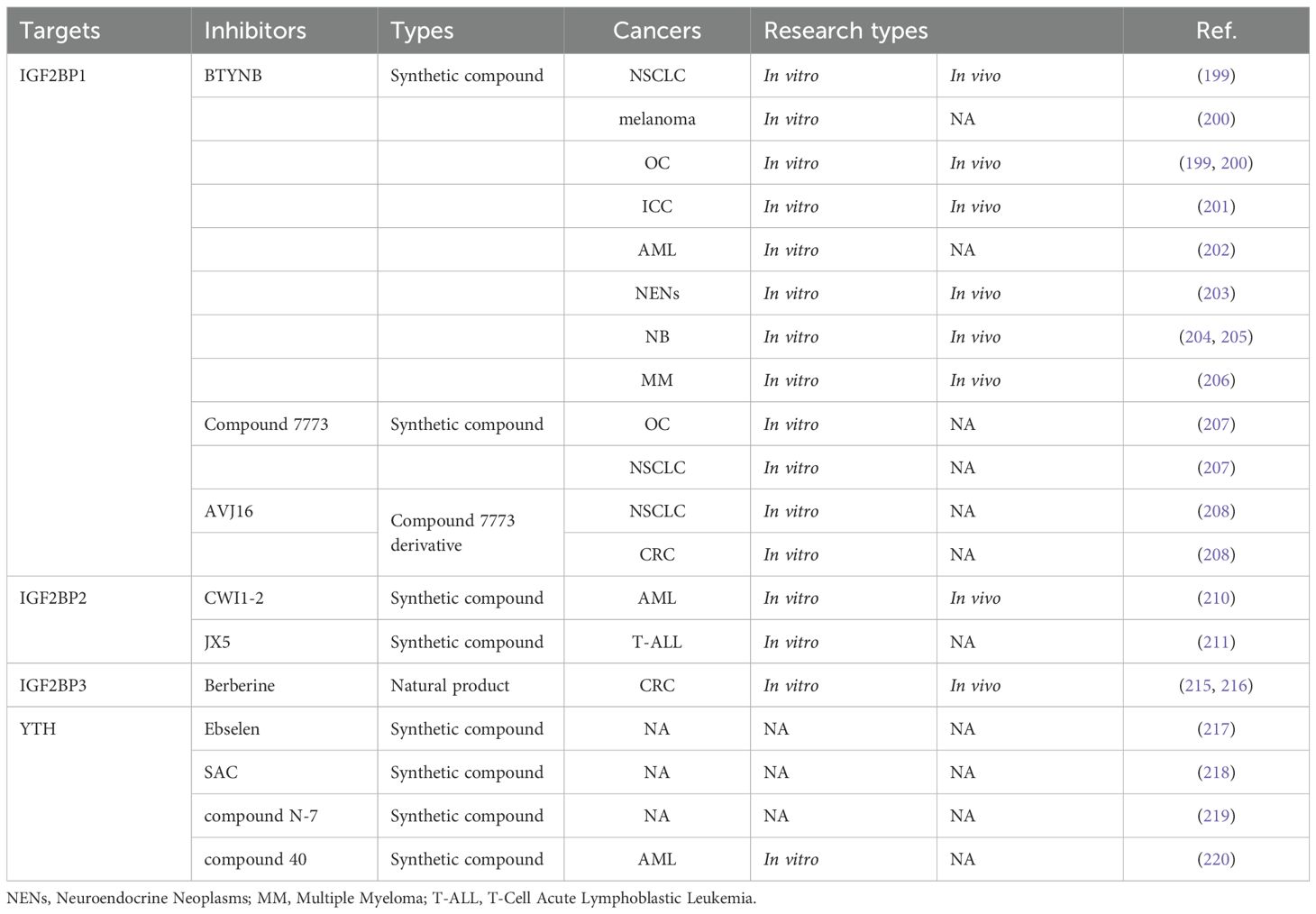
Table 3. Summary of m6A methylation recognition protein inhibitors.
7.1 IGF2BP1 inhibitors
IGF2BP1, also known as ZBP1, VICKZ1, CRD-BP, or IMP1, is an RNA-binding protein with six RNA-binding domains. It plays a key role in tumorigenesis by regulating the translation, stability, localization, and selective splicing of its target mRNAs (198). 2-[(5-bromo-2-thienyl) methylene] amino benzamide (BTYNB) is a structure-specific small molecule inhibitor of IGF2BP1. In OC and NSCLC cell lines and mouse models, BTYNB disrupts E2F-driven gene expression and then controls tumor growth (199). In OC and melanoma cells with high IMP1 expression, BTYNB effectively decreases intracellular c-MYC mRNA and protein levels, thereby suppressing tumor proliferation (200). In ICC cells and tissues, the overexpression of IGF2BP1 promotes growth and inhibits senescence by regulating the c-MYC/p16 axis (201). Additionally, IGF2BP1 induces tumor metastasis by activating the ZIC2/PAK4/AKT/MMP2 axis (201). Fortunately, BTYNB exerts critical antitumor effects in ICC models (201). Furthermore, Jamal et al. showed that BTYNB induces leukemia cell death and cell cycle arrest by upregulating BAK and p21, and promotes differentiation by modulating associated genes such as ITGAM, ZFPM1, and KLF5 (202). Sperling et al. demonstrated that BTYNB suppresses the IGF2BP1/MYC/EZH2 axis’s pro-proliferative effects on neuroendocrine neoplasms by blocking IGF2BP1 binding to MYC mRNA (203). Biegel et al. indicated that BTYNB strengthens neuroblastoma cells’ sensitivity to chemotherapy (204). Hagemann et al. suggested that IGF2BP1/MYCN serves as a poor prognostic factor for high-risk neuroblastoma (205). They also discovered that BTYNB can counter the synergistic pro-carcinogenic effects of IGF2BP1/MYCN by inhibiting IGF2BP1 in vivo (205). Xu et al. established a novel IGF2BP1/CDC5L pathogenic axis in multiple myeloma (MM), where BTYNB effectively suppresses the expression of CDC5L, thereby inhibiting MM progression (206). These findings highlight BTYNB as a promising therapeutic agent for IGF2BP1-driven cancers, with broad applicability across divorce cancers.
Compound 7773 was identified as a potent IGF2BP1 inhibitor through FP-based high-throughput screening (HTS) (207). It was shown to directly bind to intracellular IGF2BP1 and block its interaction with target mRNAs, including Kras (207). As a result, Kras mRNA and protein levels were reduced, leading to suppression of pERK signaling (207). Moreover, compound 7773 demonstrated significant inhibitory effects on OC and lung adenocarcinoma cells, suggesting its potential for targeting IGF2BP1-related pathways in various cancers.
AVJ16 was optimized from the lead compound 7773, which was experimentally shown to be a more potent, selective, and safer small-molecule inhibitor of IGF2BP1 (208). AVJ16 exhibits significant anti-cancer effects on NSCLC and CRC cell lines by the downregulation of Kras mRNA and protein (208). Which further confirms its potential as an effective therapeutic agent against cancers with elevated IGF2BP1 expression.
7.2 IGF2BP2 inhibitors
IGF2BP2 was validated as a potential anticancer target through various in vitro and in vivo experiments. KH structural domains, especially the KH3–4 double structural domain, are essential for the binding of IGF2BP2 to m6A-modified RNAs (83). HTS was used to identify 10 potential inhibitors of IGF2BP2/RNA interactions, mainly divided into two categories: benzamidobenzoic acid and ureidothiophene (209). These compounds demonstrated target-specific activity and promising anti-proliferative effects, establishing a framework for developing optimized IGF2BP2 inhibitors with enhanced potency and selectivity against IGF2BP2-driven cancers.
CWI1-2, a potent small molecule inhibitor, was developed to bind directly to IGF2BP2 and competitively inhibit its interaction with target transcripts (210). Elevated IGF2BP2 in AML portends adverse clinical outcomes. IGF2BP2 drives AML progression by m6A-dependently controlling glutamine metabolic regulators (MYC/GPT2/SLC1A5), sustaining leukemic stem cell self-renewal (210). Inhibition of IGF2BP2 with CWI1–2 demonstrates favorable antileukemic effects (210). Additionally, combining CWI1–2 with drugs like doxycycline hyclate or homoharringtonine enhances therapeutic efficacy on AML (210). These results position CWI1–2 as a promising therapeutic agent, both alone and in combination therapies targeting IGF2BP2 in AML.
IGF2BP2 is highly expressed in T-cell acute lymphoblastic leukemia (T-ALL), and directly binds to the T-ALL oncogene NOTCH1 in an m6A-dependent manner, thereby stabilizing its mRNA and promoting tumorigenesis (211). Furthermore, IGF2BP2 reduces apoptosis induced by chemotherapeutic agents such as acitretin, vincristine, vannamei, or dexamethasone, contributing to chemoresistance in T-ALL (211). Importantly, the IGF2BP2 inhibitor JX5 not only effectively increases apoptosis but also alleviates chemotherapy resistance, highlighting its potential for treating T-ALL (211).
7.3 IGF2BP3 inhibitors
IGF2BP3 acts as an oncogene and a reliable independent prognostic biomarker in CRC (212, 213). IGF2BP3 is markedly overexpressed in colon cancer, where it regulates the cell cycle and angiogenesis by recognizing m6A modifications of target mRNAs, thereby facilitating tumor growth (214). Berberine (BBR), an isoquinoline alkaloid derived from coptis chinensis, exhibits a wide range of pharmacological activities (215). BBR inhibits CRC cell proliferation via the down-regulation of IGF2BP3 (215). Further research demonstrated that BBR targets the degradation of IGF2BP3 through TRIM21-mediated ubiquitination (216). These findings show the therapeutic potential of BBR as an IGF2BP3 inhibitor, offering a promising strategy for treating CRC.
8 YTH domain family inhibitors
Though no m6A-recognizing protein inhibitor with antitumor activity has been identified to date. Organoselenium compound ebselen was reported to be a first-in-class inhibitor of the YTHDF m6A-binding domain, which binds and interacts with YTHDF (217). This interaction disrupts the ability of YTHDF recognizing and binding m6A-modified mRNA. Additionally, researchers found a series of structural analogs of ebselen, which are suitable for the development of new inhibitors. Their findings pave the way for developing effective inhibitors targeting m6A-recognizing proteins.
Salvianolic acid C (SAC) was identified as a selective small molecule inhibitor of YTHDF1, exhibiting a competitive inhibitory effect (218). A new pan-inhibitor, N-7, was developed to target YTHDF1-3, YTFDC1, and YTFDC2 (219). Although the in vitro inhibitory activity of SAC and N-7 has been confirmed, their application in cancers has not yet been realized. Recently, researchers designed a selective and potent ligand of YTHDC1, known as compound 40, which demonstrates significant anti-proliferative activity across various AML cell lines (220). All above lay the foundation for further refinement and promotion of potent inhibitors.
9 Therapeutic promise, clinical limitations, and future perspectives of m6A regulatory protein inhibitors
m6A regulatory factor inhibitors, as emerging anti-tumor agents targeting RNA epigenetic modifications, exhibit multiple therapeutic advantages in oncology. First, their high selectivity enables precise targeting of cancer cells, enhancing treatment accuracy (193, 221). Second, due to their target specificity, most inhibitors demonstrate minimal toxicity to critical organs such as the heart, liver, and kidneys at effective doses, resulting in a relatively broad therapeutic window (222). Third, by disrupting the dynamic balance of oncogenic RNAs, these inhibitors open novel avenues for investigating cancer-related molecular mechanisms (223, 224). Fourth, m6A-associated inhibitors have been shown to remodel immune cell functions and alter the tumor microenvironment, thereby potentiating anti-tumor immune responses (225–228). Fifth, they can overcome resistance to conventional therapies and exhibit synergistic effects when used in combination regimens (223, 229, 230).
Nonetheless, the application of m6A-related inhibitors also presents certain limitations. Foremost among them is the context-dependent function of m6A regulatory proteins, which may exhibit diametrically opposing roles in different tumor types—leading to unpredictable therapeutic outcomes or even pro-tumorigenic effects. For instance, ALKBH5 functions as an oncogene in TNBC but acts as a tumor suppressor in gastric cancer; thus, its inhibition may suppress TNBC metastasis while potentially promoting GC progression (193, 231). Moreover, beyond their roles in tumorigenesis, m6A regulatory proteins are intricately involved in physiological processes such as hematopoiesis and neurogenesis (232). Insufficient target specificity, or off-target effects among protein family members, may impair normal cellular function and cause toxicity concerns. For example, though YTHDF1 inhibition showed anti-leukemic activity, it also impaired the self-renewal capacity of normal hematopoietic stem cells (229). In addition, m6A regulatory factor inhibitors face similar physicochemical limitations as other small-molecule agents, including poor aqueous solubility, metabolic instability, and inadequate cell permeability, which collectively hinder the potential of transferring into drugs (223, 233). Finally, due to mechanistic complexity and the involvement of non-enzymatic functions, single-agent inhibition is sometimes insufficient to fully suppress oncogenic signaling pathways (18).
To address these challenges, researchers are actively exploring next-generation drug design strategies. These include enhancing selectivity, improving drug delivery systems, and identifying rational combination therapies. For selectivity, dual-target inhibitors (e.g., FTO/ALKBH5 inhibitors) and proteolysis-targeting chimeras (PROTACs) degradation technology have been proposed as potential solutions to increase specificity (48). In terms of drug delivery, researchers aim to optimize drug delivery systems to overcome pharmacokinetic limitations (93). Tan et al. (234) designed a hydrogel loaded with STM2457, which adheres to the surgical margins of CRC patients when injected. The hydrogel slowly and continuously releases the inhibitor, inhibiting METTL3 to suppress m6A methylation in CRC. This inhibition leads to increased expression of CXCL9 and CXCL10, effectively recruiting infused CAR-NK cells to prevent postoperative recurrence of CRC. Li et al. (235) developed a novel nano-delivery system, PLGA-STM-TAT, consisting of PLGA (poly lactic-co-glycolic acid) nanoparticles loaded with the METTL3 inhibitor STM2457 and the cell-penetrating peptide TAT. This nanoparticle-loaded drugs can modulate m6A methylation and target EphA2 through selectively accumulating in gastric cancer tissues. The system not only significantly enhances drug delivery but also inhibits cancer cell proliferation by downregulating key oncogenes, c-MYC and BRD4. For combination strategies, investigators seek to explore novel combination therapies to maximize efficacy and overcome resistance. Chen et al. (225) innovatively combined the FTO inhibitor CS2 with the anti-PD-1 body for the treatment of HCC, demonstrating favorable efficacy in both in vitro and in vivo experiments. Mechanistically, glycoprotein non-metastatic melanoma protein B (GPNMB), a downstream target of FTO, serves as an oncogenic factor in HCC. Tumor cell-derived extracellular vesicles encapsulate GPNMB and deliver it to surface-associated receptors on CD8+ T cells, thereby suppressing their immune activity. However, CS2, by targeting the FTO/m6A/GPNMB axis, significantly enhances the therapeutic efficacy of anti-PD-1 body. This study provides insight into the potential combination of m6A modification-related inhibitors with ICIs for improved therapeutic strategies.
Based on the previous inhibitor targets, as shown in Figure 2 (by Figdraw), these innovative approaches integrating m6A-associated inhibitors provide novel therapeutic strategies for cancers prone to residual disease, metastasis, and recurrence, representing a promising direction for future cancer treatment development.
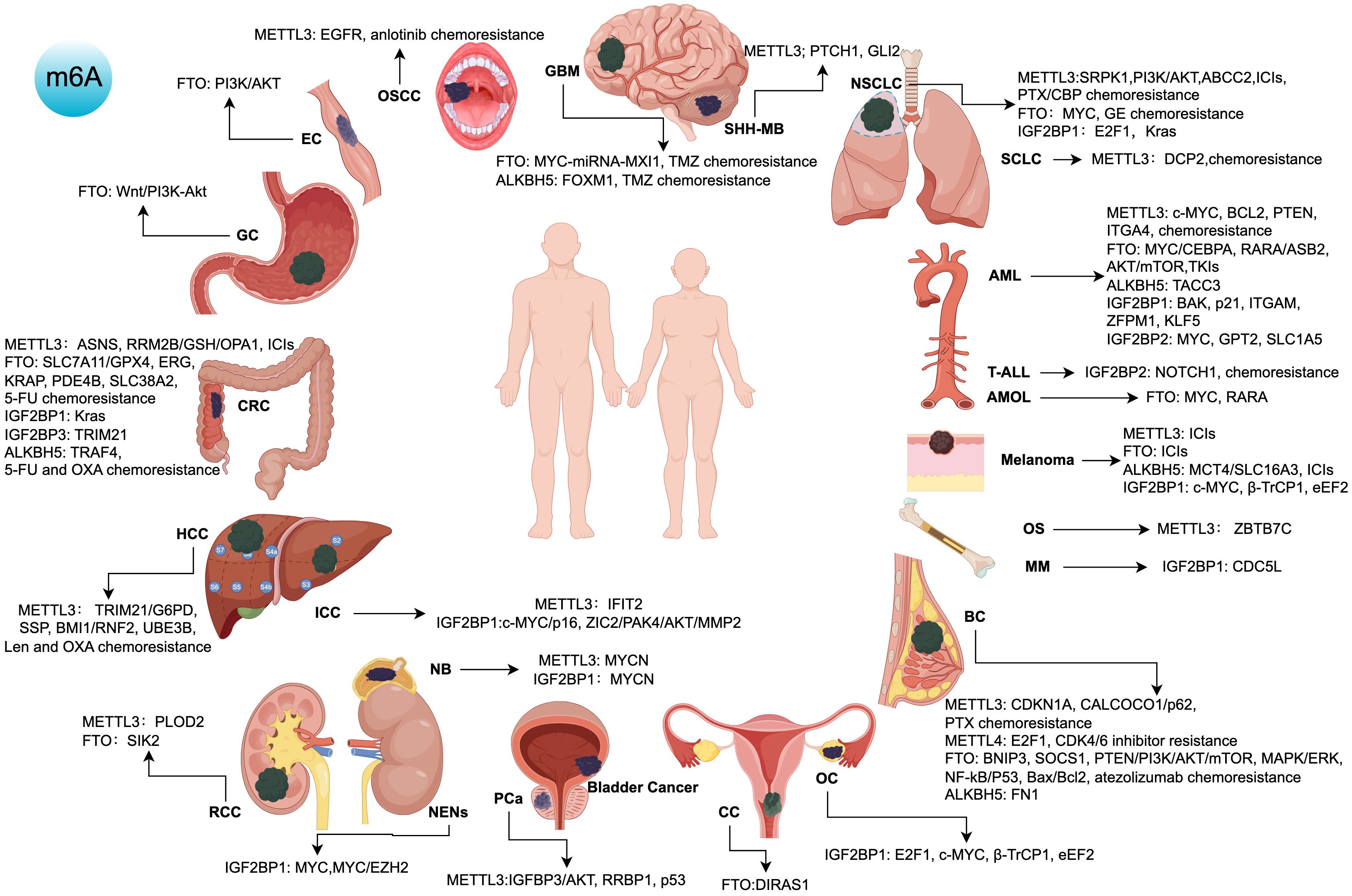
Figure 2. Targets, signaling pathways, and roles of m6A regulatory protein inhibitors in various cancers. ICIs, immune checkpoint inhibitors; TKIs, tyrosine kinase inhibitors; 5-FU, 5-Fluorouracil; OXA, Oxaliplatin; Len, Lenvatinib; PTX, Paclitaxel; CBP, Carboplatin; GE, Gefitinib; TMZ, Temozolomide; AML, Acute Myeloid Leukemia; PCa, Prostate Cancer; HCC, Hepatocellular Carcinoma; OS, Osteosarcoma; NSCLC, Non-Small Cell Lung Cancer; SCLC, Small Cell Lung Cancer; ICC, Intrahepatic Cholangiocarcinoma; RCC, Renal Cell Carcinoma; NB, Neuroblastoma; SHH-MB, Sonic Hedgehog Medulloblastoma; CRC, Colorectal Cancer; OSCC, Oral Squamous Cell Carcinoma; BC, Breast Cancer; OC, Ovarian Cancer; GBM, Glioblastoma Multiforme; AMOL, Acute Monocytic Leukemia; CC, Cervical Cancer; GC, Gastric Cancer; EC, Esophageal Cancer; NENs, Neuroendocrine Neoplasms; MM, Multiple Myeloma; T-ALL, T-Cell Acute Lymphoblastic Leukemia.
Despite the formidable hurdles, m6A-associated inhibitors remain a promising frontier in cancer therapy owing to their unique mechanisms—such as modulating oncogenic RNA stability, reshaping the immune microenvironment, and altering metabolic reprogramming. Notably, the clinical translation of m6A regulatory factor inhibitors remains to face substantial obstacles. Though a growing body of preclinical research supports their potential, only a few candidates have advanced into clinical trials. Issues such as potential toxicity to normal cells, insufficient understanding of resistance mechanisms, and a lack of reliable biomarkers to identify responsive patient populations all contribute to the hurdles facing clinical development. These gaps also highlight critical directions for future investigations aimed at optimizing m6A-targeted therapies.
10 Conclusion
In conclusion, although m6A regulatory protein inhibitors exhibit a duality of therapeutic potential and clinical risks, m6A and its regulatory proteins remain a prominent research focus, particularly in oncology. With advancing research, m6A regulators have become increasingly recognized as therapeutic targets across various cancers. Targeted inhibition of m6A regulatory proteins demonstrates promising therapeutic potential in suppressing tumor growth, enhancing chemosensitivity, and promoting cancer immunotherapy. In this review, we systematically summarize all reported inhibitors targeting m6A regulators, providing comparative analyses of their antitumor mechanisms and efficacy across various cancers. Additionally, we critically evaluate their complementary roles in overcoming chemotherapy resistance and enhancing immunotherapy effect. Notably, this review highlights innovative therapeutic approaches utilizing novel drug delivery systems for these inhibitors. Additionally, this review summarizes next-generation drug design strategies for m6A-associated inhibitors and their clinical translation challenges. These findings establish a foundation for developing novel cancer therapies and bring good news to patients with currently incurable malignancies.
Author contributions
XL: Writing – original draft, Conceptualization, Data curation. XK: Writing – review & editing, Conceptualization, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by grants of National Natural Science Foundation of China (No. 82372069) and the Outstanding Youth Foundation of Hubei Province, China (2023AFA107).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
METTL5: Methyltransferase-like 5
METTL16: Methyltransferase-like 16
RBM15: RNA Binding Motif Protein 15
RBM15B: RNA Binding Motif Protein 15B
ZC3H13: Zinc Finger CCCH-Type Containing 13
ZCCHC4: Zinc Finger CCHC-Type Containing 4
CBLL1: Cbl Proto-Oncogene Like 1
VIRMA: Vir Like m6A Methyltransferase Associated
HNRNPs: Heterogeneous Nuclear Ribonucleoproteins
eIF3: Eukaryotic Translation Initiation Factor 3
G3BP2: GTPase-Activating Protein SH3 Domain-Binding Protein 2
PRRC2A: Proline-Rich Coiled-Coil 2A
RBMX: RNA Binding Motif Protein X-Linked
FMR1: Fragile X Messenger Ribonucleoprotein 1
ELAVL1: ELAV Like RNA Binding Protein 1
G3BP1: GTPase-Activating Protein SH3 Domain-Binding Protein 1
IMP1: Insulin-like Growth Factor 2 mRNA-Binding Protein 1
IMP2: Insulin-like Growth Factor 2 mRNA-Binding Protein 2
IMP3: Insulin-like Growth Factor 2 mRNA-Binding Protein 3
RRBP1: Ribosome Binding Protein 1
c-MYC: MYC Proto-Oncogene
BCL2: B-Cell Lymphoma 2
PTEN: Phosphatase and Tensin Homolog
SP1: Specificity Protein 1
SPP2: Secreted Phosphoprotein 2
ITGA4: Integrin Subunit Alpha 4
SRPK1: Serine/Arginine-Rich Protein-Specific Kinase 1
PI3K: Phosphatidylinositol 3-Kinase
AKT: Serine/Threonine Kinase
ABCC2: ATP-Binding Cassette Subfamily C Member 2
DCP2: mRNA Decapping Enzyme 2
IFIT2: Interferon-Induced Protein with Tetratricopeptide Repeats 2
TRIM21: Tripartite Motif-Containing Protein 21
G6PD: Glucose-6-Phosphate Dehydrogenase
PSPH: Phosphoserine Phosphatase
PSAT1: Phosphoserine Aminotransferase 1
PHGDH: Phosphoglycerate Dehydrogenase
BMI1: B Lymphoma Mo-MLV Insertion Region 1 Homolog
RNF2: Ring Finger Protein 2
HIF-1α: Hypoxia-Inducible Factor 1 Subunit Alpha
PLOD2: Procollagen-Lysine,2-Oxoglutarate 5-Dioxygenase 2
KAP1: KRAB Associated Protein 1
MYCN: MYCN Proto-Oncogene
PTCH1: Patched 1
GLI2: GLI Family Zinc Finger 2
ZBTB7C: Zinc Finger and BTB Domain Containing 7C
RRM2B: Ribonucleotide Reductase Regulatory TP53 Inducible Subunit M2B
OPA1: Mitochondrial Dynamin Like GTPase
POP1: Processing of Precursors 1
CDKN1A: Cyclin Dependent Kinase Inhibitor 1A
XBP1s: X-Box Binding Protein 1 Spliced
CALCOCO1: Calcium Binding and Coiled-Coil Domain 1
mTOR: Mechanistic Target of Rapamycin Kinase
MAPK: Mitogen-Activated Protein Kinase
ERK: Extracellular Signal-Regulated Kinase
NF-kB: Nuclear Factor Kappa B
P53: Tumor Protein P53
Bax: BCL2 Associated X
SLC7A11: Solute Carrier Family 7 Member 11
GPX4: Glutathione Peroxidase 4
BCRP: ATP Binding Cassette Subfamily G Member 2
MRP-7: Multidrug Resistance Protein Mrp-7
MYC: Proto-Oncogene
EGFR: Epidermal Growth Factor Receptor
MXI1: MAX Interactor 1
RARA: Retinoic Acid Receptor Alpha
DIRAS1: DIRAS Family GTPase 1
SIK2: Salt Inducible Kinase 2
ASB2: Ankyrin Repeat and SOCS Box Containing 2
CEBPA: CCAAT Enhancer Binding Protein Alpha
ERG: ETS Transcription Factor ERG
KRAP: SUGP1 (SURP And G-Patch Domain Containing 1)
PDE4B: Phosphodiesterase 4B
SLC38A2: Solute Carrier Family 38 Member 2
TRAF4: TNF Receptor Associated Factor 4
LC3-I: Microtubule Associated Protein 1 Light Chain 3
LC3-II: Microtubule Associated Protein 1 Light Chain 3
MCT4: Solute Carrier Family 16 Member 3
FOXM1: Forkhead Box M1
TACC3: Transforming Acidic Coiled-Coil Containing Protein 3
FN1: Fibronectin 1
KRAS: Proto-Oncogene
MDR1: ATP Binding Cassette Subfamily B Member 1
ZBP1: Z-DNA Binding Protein 1
VICKZ1: IGF2BP1 (Insulin Like Growth Factor 2 mRNA Binding Protein 1)
CRD-BP: IGF2BP1 (Insulin Like Growth Factor 2 mRNA Binding Protein 1)
p16: Cyclin Dependent Kinase Inhibitor 2A
ZIC2: Zic Family Member 2
PAK4: p21-Activated Kinase 4
MMP2: Matrix Metallopeptidase 2
BAK: BCL2 Antagonist/Killer 1
ITGAM: Integrin Subunit Alpha M
ZFPM1: Zinc Finger Protein FOG Family Member 1
KLF5: Kruppel Like Factor 5
IGF2BP1: Insulin Like Growth Factor 2 mRNA Binding Protein 1
IGF2BP2: Insulin Like Growth Factor 2 mRNA Binding Protein 2
IGF2BP3: Insulin Like Growth Factor 2 mRNA Binding Protein 3
EZH2: Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit
CDC5L: Cell Division Cycle 5 Like
GPT2: Glutamic–Pyruvic Transaminase 2
SLC1A5: Solute Carrier Family 1 Member 5
NOTCH1: Notch Receptor 1
CXCL9: C-X-C Motif Chemokine Ligand 9
CXCL10: C-X-C Motif Chemokine Ligand 10
BRD4: Bromodomain Containing 4
GPNMB: Glycoprotein Nmb
References
1. Harper JE, Miceli SM, and Roberts RJ. Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res. (1990) 18:5735–41. JL. M. doi: 10.1093/nar/18.19.5735
2. Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, et al. A majority of m6A residues are in the last exons, allowing the potential for 3' UTR regulation. Genes Dev. (2015) 29:2037–53. doi: 10.1101/gad.269415.115
3. Liu Y, Leng P, Liu Y, Guo J, and Zhou H. Crosstalk between methylation and ncRNAs in breast cancer: therapeutic and diagnostic implications. Int J Mol Sci. (2022) 23:15759. doi: 10.3390/ijms232415759
4. Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G, et al. The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. (2019) 12:121. doi: 10.1186/s13045-019-0805-7
5. Li G, Ma L, He S, Luo R, Wang B, Zhang W, et al. WTAP-mediated m(6)A modification of lncRNA NORAD promotes intervertebral disc degeneration. Nat Commun. (2022) 13:1469. doi: 10.1038/s41467-022-28990-6
6. Wang J, Tan L, Yu X, Cao X, Jia B, Chen R, et al. lncRNA ZNRD1-AS1 promotes Malignant lung cell proliferation, migration, and angiogenesis via the miR-942/TNS1 axis and is positively regulated by the m(6)A reader YTHDC2. Mol Cancer. (2022) 21:229. doi: 10.1186/s12943-022-01705-7
7. Wei W, Sun J, Zhang H, Xiao X, Huang C, Wang L, et al. Circ0008399 interaction with WTAP promotes assembly and activity of the m(6)A methyltransferase complex and promotes cisplatin resistance in bladder cancer. Cancer Res. (2021) 81:6142–56. doi: 10.1158/0008-5472.CAN-21-1518
8. Boccaletto P, Stefaniak F, Ray A, Cappannini A, Mukherjee S, Purta E, et al. MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res. (2022) 50:D231–D5. doi: 10.1093/nar/gkab1083
9. Chen XY, Zhang J, and Zhu JS. The role of m(6)A RNA methylation in human cancer. Mol Cancer. (2019) 18:103. doi: 10.1186/s12943-019-1033-z
10. Boo SH and Kim YK. The emerging role of RNA modifications in the regulation of mRNA stability. Exp Mol Med. (2020) 52:400–8. doi: 10.1038/s12276-020-0407-z
11. Alarcon CR, Lee H, Goodarzi H, Halberg N, and Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. (2015) 519:482–5. doi: 10.1038/nature14281
12. Zhu L, Zhu Y, Han S, Chen M, Song P, Dai D, et al. Impaired autophagic degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in gastric cancer. Cell Death Dis. (2019) 10:383. doi: 10.1038/s41419-019-1585-2
13. Liu N, Dai Q, Zheng G, He C, Parisien M, and Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. (2015) 518:560–4. doi: 10.1038/nature14234
14. Wang S, Lv W, Li T, Zhang S, Wang H, Li X, et al. Dynamic regulation and functions of mRNA m6A modification. Cancer Cell Int. (2022) 22:48. doi: 10.1186/s12935-022-02452-x
15. Desrosiers R and Friderici K. Identification of methylated nucleosides in messenger RNA from novikoff hepatoma cells. Proc Natl Acad Sci USA. (1974) 71:3971–5. F. R. doi: 10.1073/pnas.71.10.3971
16. Lan Q, Liu PY, Haase J, Bell JL, Huttelmaier S, and Liu T. The critical role of RNA m(6)A methylation in cancer. Cancer Res. (2019) 79:1285–92. doi: 10.1158/0008-5472.CAN-18-2965
17. Yang C, Hu Y, Zhou B, Bao Y, Li Z, Gong C, et al. The role of m(6)A modification in physiology and disease. Cell Death Dis. (2020) 11:960. doi: 10.1038/s41419-020-03143-z
18. Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. (2021) 6:74. doi: 10.1038/s41392-020-00450-x
19. Weng H and Huang H. N6-methyladenosine RNA modification in normal and Malignant hematopoiesis. Adv Exp Med Biol. (2023) 1442:105–23. doi: 10.1007/978-981-99-7471-9_7
20. Zhao Y, Shi Y, Shen H, and Xie W. m(6)A-binding proteins: the emerging crucial performers in epigenetics. J Hematol Oncol. (2020) 13:35. doi: 10.1186/s13045-020-00872-8
21. Gao JF and Zhang L. The role of N6-methyladenosine (m6A) in eye diseases. Mol Biol Rep. (2021) 48:6145–50. doi: 10.1007/s11033-021-06596-3
22. Chen Y, Miao L, Lin H, Zhuo Z, and He J. The role of m6A modification in pediatric cancer. Biochim Biophys Acta Rev Cancer. (2022) 1877:188691. doi: 10.1016/j.bbcan.2022.188691
23. Hong J, Xu K, and Lee JH. Biological roles of the RNA m6A modification and its implications in cancer. Exp Mol Med. (2022) 54:1822–32. doi: 10.1038/s12276-022-00897-8
24. Deng X, Qing Y, Horne D, Huang H, and Chen J. The roles and implications of RNA m6A modification in cancer. Nat Rev Clin Oncol. (2023) 20:507–26. doi: 10.1038/s41571-023-00774-x
25. Huang C, Zhang K, Guo Y, Shen C, Liu X, Huang H, et al. The crucial roles of m6A RNA modifications in cutaneous cancers: Implications in pathogenesis, metastasis, drug resistance, and targeted therapies. Genes Diseases. (2023) 10:2320–30. doi: 10.1016/j.gendis.2022.03.006
26. Kisan A and Chhabra R. Modulation of gene expression by YTH domain family (YTHDF) proteins in human physiology and pathology. J Cell Physiol. (2023) 238:5–31. doi: 10.1002/jcp.v238.1
27. Zeng C, Huang W, Li Y, and Weng H. Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J Hematol Oncol. (2020) 13:117. doi: 10.1186/s13045-020-00951-w
28. Liu ZH, Ma P, He Y, Zhang YF, Mou Z, Fang T, et al. The mechanism and latest progress of m6A methylation in the progression of pancreatic cancer. Int J Biol Sci. (2025) 21:1187–201. doi: 10.7150/ijbs.104407
29. Zhao J, Li GY, Lu XY, Zhu LR, and Gao Q. Landscape of m(6)A RNA methylation regulators in liver cancer and its therapeutic implications. Front Pharmacol. (2024) 15:1376005. doi: 10.3389/fphar.2024.1376005
30. Hara T, Meng S, Arao Y, Saito Y, Inoue K, Rennie S, et al. Recent advances in noncoding RNA modifications of gastrointestinal cancer. Cancer Sci. (2025) 116:8–20. doi: 10.1111/cas.v116.1
31. Zhang F, Liu H, Duan M, Wang G, Zhang Z, Wang Y, et al. Crosstalk among m(6)A RNA methylation, hypoxia and metabolic reprogramming in TME: from immunosuppressive microenvironment to clinical application. J Hematol Oncol. (2022) 15:84. doi: 10.1186/s13045-022-01304-5
32. Li Y, Su R, Deng X, and Chen Y. FTO in cancer: functions, molecular mechanisms and therapeutic implications. Adv Trends Cancer. (2022) 8:598–614. doi: 10.1016/j.trecan.2022.02.010
33. Zhu TY, Hong LL, and Ling ZQ. Oncofetal protein IGF2BPs in human cancer: functions, mechanisms and therapeutic potential. biomark Res. (2023) 11:62. doi: 10.1186/s40364-023-00499-0
34. Yin H, Zhang X, Yang P, Zhang X, Peng Y, Li D, et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun. (2021) 12:1394. doi: 10.1038/s41467-021-21514-8
35. Lin W, Chen L, Zhang H, Qiu X, Huang Q, Wan F, et al. Tumor-intrinsic YTHDF1 drives immune evasion and resistance to immune checkpoint inhibitors via promoting MHC-I degradation. Nat Commun. (2023) 14:265. doi: 10.1038/s41467-022-35710-7
36. Boulias K and Greer EL. Biological roles of adenine methylation in RNA. Nat Rev Genet. (2023) 24:143–60. doi: 10.1038/s41576-022-00534-0
37. Liu Z, Zou H, Dang Q, Xu H, Liu L, Zhang Y, et al. Biological and pharmacological roles of m(6)A modifications in cancer drug resistance. Mol Cancer. (2022) 21:220. doi: 10.1186/s12943-022-01680-z
38. Wei J, Yin Y, Zhou J, Chen H, Peng J, Yang J, et al. METTL3 potentiates resistance to cisplatin through m(6) A modification of TFAP2C in seminoma. J Cell Mol Med. (2020) 24:11366–80. doi: 10.1111/jcmm.v24.19
39. Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X, et al. RNA m(6) A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. EMBO J. (2020) 39:e103181. doi: 10.15252/embj.2019103181
40. Zeng L, Huang X, Zhang J, Lin D, and Zheng J. Roles and implications of mRNA N(6) -methyladenosine in cancer. Cancer Commun (Lond). (2023) 43:729–48. doi: 10.1002/cac2.12458
41. Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. (2019) 10:2782. doi: 10.1038/s41467-019-10669-0
42. Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, et al. m(6) A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. (2020) 39:e104514. doi: 10.15252/embj.2020104514
43. Luo P, Li S, and Long X. N6-methyladenosine RNA modification in PD-1/PD-L1: Novel implications for immunotherapy. Biochim Biophys Acta Rev Cancer. (2023) 1878:188873. doi: 10.1016/j.bbcan.2023.188873
44. Shi H, Wei J, and He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. (2019) 74:640–50. doi: 10.1016/j.molcel.2019.04.025
45. Zaccara S, Ries RJ, and Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. (2019) 20:608–24. doi: 10.1038/s41580-019-0168-5
46. Esteve-Puig R, Bueno-Costa A, and Esteller M. Writers, readers and erasers of RNA modifications in cancer. Cancer Lett. (2020) 474:127–37. doi: 10.1016/j.canlet.2020.01.021
47. Li N, Wei X, Dai J, Yang J, and Xiong S. METTL3: a multifunctional regulator in diseases. Mol Cell Biochem. (2025) 480:3429–54. doi: 10.1007/s11010-025-05208-z
48. Feng G, Wu Y, Hu Y, Shuai W, Yang X, Li Y, et al. Small molecule inhibitors targeting m(6)A regulators. J Hematol Oncol. (2024) 17:30. doi: 10.1186/s13045-024-01546-5
49. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. (2014) 10:93–5. doi: 10.1038/nchembio.1432
50. Corbeski I, Vargas-Rosales PA, Bedi RK, Deng J, Coelho D, Braud E, et al. The catalytic mechanism of the RNA methyltransferase METTL3. Elife. (2024) 12:RP92537. doi: 10.7554/eLife.92537
51. Wang P, Doxtader KA, and Nam Y. Structural basis for cooperative function of mettl3 and mettl14 methyltransferases. Mol Cell. (2016) 63:306–17. doi: 10.1016/j.molcel.2016.05.041
52. Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. (2014) 24:177–89. doi: 10.1038/cr.2014.3
53. Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA mediates preferential m(6)A mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. (2018) 4:10. doi: 10.1038/s41421-018-0019-0
54. Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Mol Cell. (2018) 69:1028–38 e6. doi: 10.1016/j.molcel.2018.02.015
55. Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. (2018) 32:415–29. doi: 10.1101/gad.309146.117
56. Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. (2016) 537:369–73. doi: 10.1038/nature19342
57. Bawankar P, Lence T, Paolantoni C, Haussmann IU, Kazlauskiene M, Jacob D, et al. Hakai is required for stabilization of core components of the m(6)A mRNA methylation machinery. Nat Commun. (2021) 12:3778. doi: 10.1038/s41467-021-23892-5
58. Gao Z, Zha X, Li M, Xia X, and Wang S. Insights into the m(6)A demethylases FTO and ALKBH5: structural, biological function, and inhibitor development. Cell Biosci. (2024) 14:108. doi: 10.1186/s13578-024-01286-6
59. Jaafar C and Aguiar RCT. Dynamic multilayered control of m(6)A RNA demethylase activity. Proc Natl Acad Sci U S A. (2024) 121:e2317847121. doi: 10.1073/pnas.2317847121
60. Toh JDW, Crossley SWM, Bruemmer KJ, Ge EJ, He D, Iovan DA, et al. Distinct RNA N-demethylation pathways catalyzed by nonheme iron ALKBH5 and FTO enzymes enable regulation of formaldehyde release rates. Proc Natl Acad Sci U S A. (2020) 117:25284–92. doi: 10.1073/pnas.2007349117
61. Fu Y, Jia G, Pang X, Wang RN, Wang X, Li CJ, et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun. (2013) 4:1798. doi: 10.1038/ncomms2822
62. Bartosovic M, Molares HC, Gregorova P, Hrossova D, Kudla G, and Vanacova S. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3'-end processing. Nucleic Acids Res. (2017) 45:11356–70. doi: 10.1093/nar/gkx778
63. Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, et al. Differential m(6)A, m(6)A(m), and m(1)A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol Cell. (2018) 71:973–85 e5. doi: 10.1016/j.molcel.2018.08.011
64. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. (2011) 7:885–7. doi: 10.1038/nchembio.687
65. Tang C, Klukovich R, Peng H, Wang Z, Yu T, Zhang Y, et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3'-UTR mRNAs in male germ cells. Proc Natl Acad Sci U S A. (2018) 115:E325–E33. doi: 10.1073/pnas.1717794115
66. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015
67. Cai Y, Wang Y, Mao B, You Q, and Guo X. Targeting insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) for the treatment of cancer. Eur J Med Chem. (2024) 268:116241. doi: 10.1016/j.ejmech.2024.116241
68. Wu R, Li A, Sun B, Sun JG, Zhang J, Zhang T, et al. A novel m(6)A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. (2019) 29:23–41. doi: 10.1038/s41422-018-0113-8
69. Shen J and Ding Y. Multifaceted roles of insulin−like growth factor 2 mRNA binding protein 2 in human cancer (Review). Mol Med Rep. (2025) 31:75. doi: 10.3892/mmr.2025.13441
70. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. (2015) 161:1388–99. doi: 10.1016/j.cell.2015.05.014
71. Liu T, Wei Q, Jin J, Luo Q, Liu Y, Yang Y, et al. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. (2020) 48:3816–31. doi: 10.1093/nar/gkaa048
72. Han B, Yan S, Wei S, Xiang J, Liu K, Chen Z, et al. YTHDF1-mediated translation amplifies Wnt-driven intestinal stemness. EMBO Rep. (2020) 21:e49229. doi: 10.15252/embr.201949229
73. Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. (2016) 7:12626. doi: 10.1038/ncomms12626
74. Li M, Zhao X, Wang W, Shi H, Pan Q, Lu Z, et al. Ythdf2-mediated m(6)A mRNA clearance modulates neural development in mice. Genome Biol. (2018) 19:69. doi: 10.1186/s13059-018-1436-y
75. Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, et al. Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res. (2017) 27:444–7. doi: 10.1038/cr.2017.10
76. Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. (2017) 27:315–28. doi: 10.1038/cr.2017.15
77. Roundtree IA and He C. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Trends Genet. (2016) 32:320–1. doi: 10.1016/j.tig.2016.03.006
78. Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell. (2016) 61:507–19. doi: 10.1016/j.molcel.2016.01.012
79. Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. (2017) 6:e31311. doi: 10.7554/eLife.31311
80. Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, et al. S-adenosylmethionine synthesis is regulated by selective N(6)-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. (2017) 21:3354–63. doi: 10.1016/j.celrep.2017.11.092
81. Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. (2017) 27:1115–27. doi: 10.1038/cr.2017.99
82. Mao Y, Dong L, Liu XM, Guo J, Ma H, Shen B, et al. m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat Commun. (2019) 10:5332. doi: 10.1038/s41467-019-13317-9
83. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. (2018) 20:285–95. doi: 10.1038/s41556-018-0045-z
84. Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, and Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. (2017) 45:6051–63. doi: 10.1093/nar/gkx141
85. Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, and Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. (2015) 162:1299–308. doi: 10.1016/j.cell.2015.08.011
86. Wu B, Su S, Patil DP, Liu H, Gan J, Jaffrey SR, et al. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat Commun. (2018) 9:420. doi: 10.1038/s41467-017-02770-z
87. Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5' UTR m(6)A promotes cap-independent translation. Cell. (2015) 163:999–1010. doi: 10.1016/j.cell.2015.10.012
88. Bedi RK, Huang D, Eberle SA, Wiedmer L, Sledz P, and Caflisch A. Small-molecule inhibitors of METTL3, the major human epitranscriptomic writer. ChemMedChem. (2020) 15:744–8. doi: 10.1002/cmdc.202000011
89. Lee JH, Kim S, Jin MS, and Kim YC. Discovery of substituted indole derivatives as allosteric inhibitors of m(6) A-RNA methyltransferase, METTL3–14 complex. Drug Dev Res. (2022) 83:783–99. doi: 10.1002/ddr.21910
90. Shao C, Han Y, Huang Y, Zhang Z, Gong T, Zhang Y, et al. Targeting key RNA methylation enzymes to improve the outcome of colorectal cancer chemotherapy (Review). Int J Oncol. (2024) 64:17. doi: 10.3892/ijo.2023.5605
91. Du Y, Yuan Y, Xu L, Zhao F, Wang W, Xu Y, et al. Discovery of METTL3 small molecule inhibitors by virtual screening of natural products. Front Pharmacol. (2022) 13:878135. doi: 10.3389/fphar.2022.878135
92. Zhao L, Ma H, Jiang Y, Li Y, Qiao L, Chen Y, et al. Identification of an m6A natural inhibitor, lobeline, that reverses lenvatinib resistance in hepatocellular tumors. J Nat Prod. (2024) 87:1983–93. doi: 10.1021/acs.jnatprod.4c00406
93. Deng LJ, Deng WQ, Fan SR, Chen MF, Qi M, Lyu WY, et al. m6A modification: recent advances, anticancer targeted drug discovery and beyond. Mol Cancer. (2022) 21:52. doi: 10.1186/s12943-022-01510-2
94. Moroz-Omori EV, Huang D, Kumar Bedi R, Cheriyamkunnel SJ, Bochenkova E, Dolbois A, et al. METTL3 inhibitors for epitranscriptomic modulation of cellular processes. ChemMedChem. (2021) 16:3035–43. doi: 10.1002/cmdc.202100291
95. Dolbois A, Bedi RK, Bochenkova E, Muller A, Moroz-Omori EV, Huang D, et al. 1,4,9-triazaspiro[5.5]undecan-2-one derivatives as potent and selective METTL3 inhibitors. J Med Chem. (2021) 64:12738–60. doi: 10.1021/acs.jmedchem.1c00773
96. Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. (2021) 593:597–601. doi: 10.1038/s41586-021-03536-w
97. Xiao H, Zhao R, Meng W, and Liao Y. Effects and translatomics characteristics of a small-molecule inhibitor of METTL3 against non-small cell lung cancer. J Pharm Anal. (2023) 13:625–39. doi: 10.1016/j.jpha.2023.04.009
98. Yang J, He Y, Kang Y, Shen L, Zhang W, Yan Y, et al. Virtual screening and molecular docking: discovering novel METTL3 inhibitors. ACS Med Chem Lett. (2024) 15:1491–9. doi: 10.1021/acsmedchemlett.4c00216
99. Li Z, Feng Y, Han H, Jiang X, Chen W, Ma X, et al. A stapled peptide inhibitor targeting the binding interface of N6-adenosine-methyltransferase subunits METTL3 and METTL14 for cancer therapy. Angew Chem Int Ed Engl. (2024) 63:e202402611. doi: 10.1002/anie.202402611
100. Feng Y, Li Z, Zhu J, Zou C, Tian Y, Xiong J, et al. Stabilization of RRBP1 mRNA via an m(6)A-dependent manner in prostate cancer constitutes a therapeutic vulnerability amenable to small-peptide inhibition of METTL3. Cell Mol Life Sci. (2024) 81:414. doi: 10.1007/s00018-024-05418-6
101. Han H, Li Z, Feng Y, Song H, Fang Z, Zhang D, et al. Peptide degrader-based targeting of METTL3/14 improves immunotherapy response in cutaneous melanoma. Angew Chem Int Ed Engl. (2024) 63:e202407381. doi: 10.1002/anie.202407381
102. Dutheuil G, Oukoloff K, Korac J, Lenoir F, El Bousmaqui M, Probst N, et al. Discovery, optimization, and preclinical pharmacology of EP652, a METTL3 inhibitor with efficacy in liquid and solid tumor models. J Med Chem. (2025) 68:2981–3003. doi: 10.1021/acs.jmedchem.4c02225
103. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. (2017) 23:1369–76. doi: 10.1038/nm.4416
104. Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. (2017) 552:126–31. doi: 10.1038/nature24678
105. Li M, Ye J, Xia Y, Li M, Li G, Hu X, et al. METTL3 mediates chemoresistance by enhancing AML homing and engraftment via ITGA4. Leukemia. (2022) 36:2586–95. doi: 10.1038/s41375-022-01696-w
106. Wang A and Zhong H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology. (2018) 23:729–39. doi: 10.1080/10245332.2018.1486064
107. Wang A, Zeng Y, Zhang W, Zhao J, Gao L, Li J, et al. N(6)-methyladenosine-modified SRPK1 promotes aerobic glycolysis of lung adenocarcinoma via PKM splicing. Cell Mol Biol Lett. (2024) 29:106. doi: 10.1186/s11658-024-00622-5
108. Xuan YF, Lu S, Ou YJ, Bao XB, Huan XJ, Song SS, et al. The combination of methionine adenosyltransferase 2A inhibitor and methyltransferase like 3 inhibitor promotes apoptosis of non-small cell lung cancer cells and produces synergistic anti-tumor activity. Biochem Biophys Res Commun. (2024) 716:150011. doi: 10.1016/j.bbrc.2024.150011
109. Zhang R, Chen P, Wang Y, Zeng Z, Yang H, Li M, et al. Targeting METTL3 enhances the chemosensitivity of non-small cell lung cancer cells by decreasing ABCC2 expression in an m(6)A-YTHDF1-dependent manner. Int J Biol Sci. (2024) 20:4750–66. doi: 10.7150/ijbs.97425
110. Hamilton G, Hochmair MJ, and Stickler S. Overcoming resistance in small-cell lung cancer. Expert Rev Respir Med. (2024) 18:569–80. doi: 10.1080/17476348.2024.2388288
111. Solta A, Ernhofer B, Boettiger K, Megyesfalvi Z, Heeke S, Hoda MA, et al. Small cells - big issues: biological implications and preclinical advancements in small cell lung cancer. Mol Cancer. (2024) 23:41. doi: 10.1186/s12943-024-01953-9
112. Sun Y, Shen W, Hu S, Lyu Q, Wang Q, Wei T, et al. METTL3 promotes chemoresistance in small cell lung cancer by inducing mitophagy. J Exp Clin Cancer Res. (2023) 42:65. doi: 10.1186/s13046-023-02638-9
113. Xu QC, Tien YC, Shi YH, Chen S, Zhu YQ, Huang XT, et al. METTL3 promotes intrahepatic cholangiocarcinoma progression by regulating IFIT2 expression in an m(6)A-YTHDF2-dependent manner. Oncogene. (2022) 41:1622–33. doi: 10.1038/s41388-022-02185-1
114. Jin X, Lv Y, Bie F, Duan J, Ma C, Dai M, et al. METTL3 confers oxaliplatin resistance through the activation of G6PD-enhanced pentose phosphate pathway in hepatocellular carcinoma. Cell Death Differ. (2025) 32:466–79. doi: 10.1038/s41418-024-01406-2
115. Chan FF, Kwan KK, Seoung DH, Chin DW, Ng IO, Wong CC, et al. N6-Methyladenosine modification activates the serine synthesis pathway to mediate therapeutic resistance in liver cancer. Mol Ther. (2024) 32:4435–47. doi: 10.1016/j.ymthe.2024.10.025
116. Wu T, Chen W, Zhang J, Ma W, and Liu N. METTL3-mediated m6A modification regulates the polycomb repressive complex 1 (PRC1) components BMI1 and RNF2 in hepatocellular carcinoma cells. Mol Cancer Res. (2025) 23:190–201. doi: 10.1158/1541-7786.MCR-24-0362
117. Cao C, Ma Q, Huang X, Li A, Liu J, Ye J, et al. Targeted demethylation of the PLOD2 mRNA inhibits the proliferation and migration of renal cell carcinoma. Front Mol Biosci. (2021) 8:675683. doi: 10.3389/fmolb.2021.675683
118. Chen Y, He Y, Li Z, Zhang N, Zhou C, He X, et al. METTL3 facilitates renal cell carcinoma progression by PLOD2 m(6)A-methylation under prolonged hypoxia. Cell Death Dis. (2024) 15:62. doi: 10.1038/s41419-023-06411-w
119. Xu Q, Kong N, Zhao Y, Wu Q, Wang X, Xun X, et al. Pan-cancer analyses reveal oncogenic and immunological role of PLOD2. Front Genet. (2022) 13:864655. doi: 10.3389/fgene.2022.864655
120. Pomaville M, Chennakesavalu M, Wang P, Jiang Z, Sun HL, Ren P, et al. Small-molecule inhibition of the METTL3/METTL14 complex suppresses neuroblastoma tumor growth and promotes differentiation. Cell Rep. (2024) 43:114165. doi: 10.1016/j.celrep.2024.114165
121. Yang Y, Zhang Y, Chen G, Sun B, Luo F, Gao Y, et al. KAP1 stabilizes MYCN mRNA and promotes neuroblastoma tumorigenicity by protecting the RNA m(6)A reader YTHDC1 protein degradation. J Exp Clin Cancer Res. (2024) 43:141. doi: 10.1186/s13046-024-03040-9
122. Zhang ZW, Teng X, Zhao F, Ma C, Zhang J, Xiao LF, et al. METTL3 regulates m(6)A methylation of PTCH1 and GLI2 in Sonic hedgehog signaling to promote tumor progression in SHH-medulloblastoma. Cell Rep. (2022) 41:111530. doi: 10.1016/j.celrep.2022.111530
123. An X, Wu W, Yang L, Dong J, Liu B, Guo J, et al. ZBTB7C m6A modification incurred by METTL3 aberration promotes osteosarcoma progression. Transl Res. (2023) 259:62–71. doi: 10.1016/j.trsl.2023.04.005
124. Yu Y, Hai Y, Zhou H, Bao W, Hu X, Gao Y, et al. METTL3 inhibition suppresses cell growth and survival in colorectal cancer via ASNS downregulation. J Cancer. (2024) 15:4853–65. doi: 10.7150/jca.96760
125. Zhou J, Zhang H, Zhong K, Tao L, Lin Y, Xie G, et al. N6-methyladenosine facilitates mitochondrial fusion of colorectal cancer cells via induction of GSH synthesis and stabilization of OPA1 mRNA. Natl Sci Rev. (2024) 11:nwae039. doi: 10.1093/nsr/nwae039
126. Wu K, Li S, Hong G, Dong H, Tang T, Liu H, et al. Targeting METTL3 as a checkpoint to enhance T cells for tumour immunotherapy. Clin Transl Med. (2024) 14:e70089. doi: 10.1002/ctm2.v14.11
127. Liu L, Zhao T, Zheng S, Tang D, Han H, Yang C, et al. METTL3 inhibitor STM2457 impairs tumor progression and enhances sensitivity to anlotinib in OSCC. Oral Dis. (2024) 30:4243–54. doi: 10.1111/odi.14864
128. Zhang C, Wang S, Lu X, Zhong W, Tang Y, Huang W, et al. POP1 Facilitates Proliferation in Triple-Negative Breast Cancer via m6A-Dependent Degradation of CDKN1A mRNA. Res (Wash D C). (2024) 7:0472. doi: 10.34133/research.0472
129. Wang J, Fan P, Shen P, Fan C, Zhao P, Yao S, et al. XBP1s activates METTL3/METTL14 for ER-phagy and paclitaxel sensitivity regulation in breast cancer. Cancer Lett. (2024) 596:216846. doi: 10.1016/j.canlet.2024.216846
130. Chen X, Wang M, Wang H, Yang J, Li X, Zhang R, et al. METTL3 inhibitor suppresses the progression of prostate cancer via IGFBP3/AKT pathway and synergizes with PARP inhibitor. BioMed Pharmacother. (2024) 179:117366. doi: 10.1016/j.biopha.2024.117366
131. Liu C, Fan D, Sun J, Li G, Du R, Zuo X, et al. Inhibition of METTL14 overcomes CDK4/6 inhibitor resistance driven by METTL14-m6A-E2F1-axis in ERalpha-positive breast cancer. J Nanobiotechnology. (2025) 23:3. doi: 10.1186/s12951-024-03021-2
132. Aik W, Demetriades M, Hamdan MK, Bagg EA, Yeoh KK, Lejeune C, et al. Structural basis for inhibition of the fat mass and obesity associated protein (FTO). J Med Chem. (2013) 56:3680–8. doi: 10.1021/jm400193d
133. Wang T, Hong T, Huang Y, Su H, Wu F, Chen Y, et al. Fluorescein derivatives as bifunctional molecules for the simultaneous inhibiting and labeling of FTO protein. J Am Chem Soc. (2015) 137:13736–9. doi: 10.1021/jacs.5b06690
134. He W, Zhou B, Liu W, Zhang M, Shen Z, Han Z, et al. Identification of A novel small-molecule binding site of the fat mass and obesity associated protein (FTO). J Med Chem. (2015) 58:7341–8. doi: 10.1021/acs.jmedchem.5b00702
135. Qiao Y, Zhou B, Zhang M, Liu W, Han Z, Song C, et al. A novel inhibitor of the obesity-related protein FTO. Biochemistry. (2016) 55:1516–22. doi: 10.1021/acs.biochem.6b00023
136. Wang R, Han Z, Liu B, Zhou B, Wang N, Jiang Q, et al. Identification of natural compound radicicol as a potent FTO inhibitor. Mol Pharm. (2018) 15:4092–8. doi: 10.1021/acs.molpharmaceut.8b00522
137. Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. (2012) 134:17963–71. doi: 10.1021/ja3064149
138. Yan F, Al-Kali A, Zhang Z, Liu J, Pang J, Zhao N, et al. A dynamic N(6)-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. (2018) 28:1062–76. doi: 10.1038/s41422-018-0097-4
139. Zhang S, Zhou L, Yang J, Lu J, Tao L, Feng Y, et al. Rhein exerts anti-multidrug resistance in acute myeloid leukemia via targeting FTO to inhibit AKT/mTOR. Anticancer Drugs. (2024) 35:597–605. doi: 10.1097/CAD.0000000000001608
140. Fernand VE, Losso JN, Truax RE, Villar EE, Bwambok DK, Fakayode SO, et al. Rhein inhibits angiogenesis and the viability of hormone-dependent and -independent cancer cells under normoxic or hypoxic conditions in vitro. Chem Biol Interact. (2011) 192:220–32. doi: 10.1016/j.cbi.2011.03.013
141. Chang CY, Chan HL, Lin HY, Way TD, Kao MC, Song MZ, et al. Rhein induces apoptosis in human breast cancer cells. Evid Based Complement Alternat Med. (2012) 2012:952504. doi: 10.1155/2012/952504
142. Shen Z, Zhu B, Li J, and Qin L. Rhein augments antiproliferative effects of atezolizumab based on breast cancer (4T1) regression. Planta Med. (2019) 85:1143–9. doi: 10.1055/a-1012-7034
143. Sun K, Du Y, Hou Y, Zhao M, Li J, Du Y, et al. Saikosaponin D exhibits anti-leukemic activity by targeting FTO/m(6)A signaling. Theranostics. (2021) 11:5831–46. doi: 10.7150/thno.55574
144. Qiao Y, Su M, Zhao H, Liu H, Wang C, Dai X, et al. Targeting FTO induces colorectal cancer ferroptotic cell death by decreasing SLC7A11/GPX4 expression. J Exp Clin Cancer Res. (2024) 43:108. doi: 10.1186/s13046-024-03032-9
145. Singh B, Kinne HE, Milligan RD, Washburn LJ, Olsen M, and Lucci A. Important role of FTO in the survival of rare panresistant triple-negative inflammatory breast cancer cells facing a severe metabolic challenge. PloS One. (2016) 11:e0159072. doi: 10.1371/journal.pone.0159072
146. Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. (2019) 18:46. doi: 10.1186/s12943-019-1004-4
147. Chourasia AH and Macleod KF. Tumor suppressor functions of BNIP3 and mitophagy. Autophagy. (2015) 11:1937–8. doi: 10.1080/15548627.2015.1085136
148. Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. (2015) 43:373–84. doi: 10.1093/nar/gku1276
149. Kovala-Demertzi D, Dokorou V, Primikiri A, Vargas R, Silvestru C, Russo U, et al. Organotin meclofenamic complexes: Synthesis, crystal structures and antiproliferative activity of the first complexes of meclofenamic acid - novel anti-tuberculosis agents. J Inorg Biochem. (2009) 103:738–44. doi: 10.1016/j.jinorgbio.2009.01.014
150. Yang Y, Ji N, Cai CY, Wang JQ, Lei ZN, Teng QX, et al. Modulating the function of ABCB1: in vitro and in vivo characterization of sitravatinib, a tyrosine kinase inhibitor. Cancer Commun (Lond). (2020) 40:285–300. doi: 10.1002/cac2.12040
151. Cui Q, Cai CY, Gao HL, Ren L, Ji N, Gupta P, et al. Glesatinib, a c-MET/SMO dual inhibitor, antagonizes P-glycoprotein mediated multidrug resistance in cancer cells. Front Oncol. (2019) 9:313. doi: 10.3389/fonc.2019.00313
152. Sakamoto S, Sato K, Takita Y, Izumiya Y, Kumagai N, Sudo K, et al. ABCG2 C421A polymorphisms affect exposure of the epidermal growth factor receptor inhibitor gefitinib. Invest New Drugs. (2020) 38:1687–95. doi: 10.1007/s10637-020-00946-x
153. Chen H, Jia B, Zhang Q, and Zhang Y. Meclofenamic acid restores gefinitib sensitivity by downregulating breast cancer resistance protein and multidrug resistance protein 7 via FTO/m6A-demethylation/c-myc in non-small cell lung cancer. Front Oncol. (2022) 12:870636. doi: 10.3389/fonc.2022.870636
154. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. (2017) 18:2622–34. doi: 10.1016/j.celrep.2017.02.059
155. Xiao L, Li X, Mu Z, Zhou J, Zhou P, Xie C, et al. FTO inhibition enhances the antitumor effect of temozolomide by targeting MYC-miR-155/23a cluster-MXI1 feedback circuit in glioma. Cancer Res. (2020) 80:3945–58. doi: 10.1158/0008-5472.CAN-20-0132
156. Prakash M, Itoh Y, Fujiwara Y, Takahashi Y, Takada Y, Mellini P, et al. Identification of potent and selective inhibitors of fat mass obesity-associated protein using a fragment-merging approach. J Med Chem. (2021) 64:15810–24. doi: 10.1021/acs.jmedchem.1c01107
157. Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. (2019) 35:677–91 e10. doi: 10.1016/j.ccell.2019.03.006
158. Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N 6 -methyladenosine RNA demethylase. Cancer Cell. (2017) 31:127–41. doi: 10.1016/j.ccell.2016.11.017
159. Tarullo M, Fernandez Rodriguez G, Iaiza A, Venezia S, Macone A, Incocciati A, et al. Off-target inhibition of human dihydroorotate dehydrogenase (hDHODH) highlights challenges in the development of fat mass and obesity-associated protein (FTO) inhibitors. ACS Pharmacol Transl Sci. (2024) 7:4096–111. doi: 10.1021/acsptsci.4c00533
160. Wang YY, Ye LH, Zhao AQ, Gao WR, Dai N, Yin Y, et al. M6A modification regulates tumor suppressor DIRAS1 expression in cervical cancer cells. Cancer Biol Ther. (2024) 25:2306674. doi: 10.1080/15384047.2024.2306674
161. Xu Y, Zhou J, Li L, Yang W, Zhang Z, Zhang K, et al. FTO-mediated autophagy promotes progression of clear cell renal cell carcinoma via regulating SIK2 mRNA stability. Int J Biol Sci. (2022) 18:5943–62. doi: 10.7150/ijbs.77774
162. Liu Z, Duan Z, Zhang D, Xiao P, Zhang T, Xu H, et al. Structure-activity relationships and antileukemia effects of the tricyclic benzoic acid FTO inhibitors. J Med Chem. (2022) 65:10638–54. doi: 10.1021/acs.jmedchem.2c00848
163. Xiao P, Duan Z, Liu Z, Chen L, Zhang D, Liu L, et al. Rational design of RNA demethylase FTO inhibitors with enhanced antileukemia drug-like properties. J Med Chem. (2023) 66:9731–52. doi: 10.1021/acs.jmedchem.3c00543
164. Liu Y, Liang G, Xu H, Dong W, Dong Z, Qiu Z, et al. Tumors exploit FTO-mediated regulation of glycolytic metabolism to evade immune surveillance. Cell Metab. (2021) 33:1221–33 e11. doi: 10.1016/j.cmet.2021.04.001
165. Huff S, Tiwari SK, Gonzalez GM, Wang Y, and Rana TM. m(6)A-RNA demethylase FTO inhibitors impair self-renewal in glioblastoma stem cells. ACS Chem Biol. (2021) 16:324–33. doi: 10.1021/acschembio.0c00841
166. Huff S, Kummetha IR, Zhang L, Wang L, Bray W, Yin J, et al. Rational design and optimization of m(6)A-RNA demethylase FTO inhibitors as anticancer agents. J Med Chem. (2022) 65:10920–37. doi: 10.1021/acs.jmedchem.1c02075
167. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. (2018) 172:90–105 e23. doi: 10.1016/j.cell.2017.11.031
168. Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. (2020) 38:79–96 e11. doi: 10.1016/j.ccell.2020.04.017
169. Phan T, Nguyen VH, Su R, Li Y, Qing Y, Qin H, et al. Targeting fat mass and obesity-associated protein mitigates human colorectal cancer growth in vitro and in a murine model. Front Oncol. (2023) 13:1087644. doi: 10.3389/fonc.2023.1087644
170. Garg R, Melstrom L, Chen J, He C, and Goel A. Targeting FTO suppresses pancreatic carcinogenesis via regulating stem cell maintenance and EMT pathway. Cancers (Basel). (2022) 14:5919. doi: 10.3390/cancers14235919
171. Xie G, Wu XN, Ling Y, Rui Y, Wu D, Zhou J, et al. A novel inhibitor of N (6)-methyladenosine demethylase FTO induces mRNA methylation and shows anti-cancer activities. Acta Pharm Sin B. (2022) 12:853–66. doi: 10.1016/j.apsb.2021.08.028
172. Dobie C, Montgomery AP, Szabo R, Yu H, and Skropeta D. Synthesis and biological evaluation of selective phosphonate-bearing 1,2,3-triazole-linked sialyltransferase inhibitors. RSC Med Chem. (2021) 12:1680–9. doi: 10.1039/D1MD00079A
173. Qin B, Bai Q, Yan D, Yin F, Zhu Z, Xia C, et al. Discovery of novel mRNA demethylase FTO inhibitors against esophageal cancer. J Enzyme Inhib Med Chem. (2022) 37:1995–2003. doi: 10.1080/14756366.2022.2098954
174. Cui Y, Zhang C, Ma S, Li Z, Wang W, Li Y, et al. RNA m6A demethylase FTO-mediated epigenetic up-regulation of LINC00022 promotes tumorigenesis in esophageal squamous cell carcinoma. J Exp Clin Cancer Res. (2021) 40:294. doi: 10.1186/s13046-021-02096-1
175. Liu S, Huang M, Chen Z, Chen J, Chao Q, Yin X, et al. FTO promotes cell proliferation and migration in esophageal squamous cell carcinoma through up-regulation of MMP13. Exp Cell Res. (2020) 389:111894. doi: 10.1016/j.yexcr.2020.111894
176. Liang X, Huang Y, Ren H, Liu Q, Chen L, Zhao J, et al. Discovery of novel RNA demethylase FTO inhibitors featuring an acylhydrazone scaffold with potent antileukemia activity. J Med Chem. (2025) 68:2742–63. doi: 10.1021/acs.jmedchem.4c02076
177. Chen Y, Wu R, Chen W, Liu Y, Liao X, Zeng B, et al. Curcumin prevents obesity by targeting TRAF4-induced ubiquitylation in m(6) A-dependent manner. EMBO Rep. (2021) 22:e52146. doi: 10.15252/embr.202052146
178. Weng W and Goel A. Curcumin and colorectal cancer: An update and current perspective on this natural medicine. Semin Cancer Biol. (2022) 80:73–86. doi: 10.1016/j.semcancer.2020.02.011
179. Su P, Yang Y, Wang G, Chen X, and Ju Y. Curcumin attenuates resistance to irinotecan via induction of apoptosis of cancer stem cells in chemoresistant colon cancer cells. Int J Oncol. (2018) 53:1343–53. doi: 10.3892/ijo.2018.4461
180. Aik W, Scotti JS, Choi H, Gong L, Demetriades M, Schofield CJ, et al. Structure of human RNA N(6)-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res. (2014) 42:4741–54. doi: 10.1093/nar/gku085
181. Fang Z, Mu B, Liu Y, Guo N, Xiong L, Guo Y, et al. Discovery of a potent, selective and cell active inhibitor of m(6)A demethylase ALKBH5. Eur J Med Chem. (2022) 238:114446. doi: 10.1016/j.ejmech.2022.114446
182. Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U S A. (2020) 117:20159–70. doi: 10.1073/pnas.1918986117
183. Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. (2017) 31:591–606 e6. doi: 10.1016/j.ccell.2017.02.013
184. Selberg S, Seli N, Kankuri E, and Karelson M. Rational design of novel anticancer small-molecule RNA m6A demethylase ALKBH5 inhibitors. ACS Omega. (2021) 6:13310–20. doi: 10.1021/acsomega.1c01289
185. Malacrida A, Rivara M, Di Domizio A, Cislaghi G, Miloso M, Zuliani V, et al. 3D proteome-wide scale screening and activity evaluation of a new ALKBH5 inhibitor in U87 glioblastoma cell line. Bioorg Med Chem. (2020) 28:115300. doi: 10.1016/j.bmc.2019.115300
186. Malacrida A, Di Domizio A, Bentivegna A, Cislaghi G, Messuti E, Tabano SM, et al. MV1035 overcomes temozolomide resistance in patient-derived glioblastoma stem cell lines. Biol (Basel). (2022) 11:70. doi: 10.3390/biology11010070
187. Takahashi H, Hase H, Yoshida T, Tashiro J, Hirade Y, Kitae K, et al. Discovery of two novel ALKBH5 selective inhibitors that exhibit uncompetitive or competitive type and suppress the growth activity of glioblastoma multiforme. Chem Biol Drug Des. (2022) 100:1–12. doi: 10.1111/cbdd.v100.1
188. Biserova K, Jakovlevs A, Uljanovs R, and Strumfa I. Cancer stem cells: significance in origin, pathogenesis and treatment of glioblastoma. Cells. (2021) 10:621. doi: 10.3390/cells10030621
189. Wang YZ, Li HY, Zhang Y, Jiang RX, Xu J, Gu J, et al. Discovery of pyrazolo[1,5-a]pyrimidine derivative as a novel and selective ALKBH5 inhibitor for the treatment of AML. J Med Chem. (2023) 66:15944–59. doi: 10.1021/acs.jmedchem.3c01374
190. Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, et al. RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self-renewal in acute myeloid leukemia. Cell Stem Cell. (2020) 27:64–80 e9. doi: 10.1016/j.stem.2020.04.009
191. Fei WL, Wang YZ, Feng QL, Li CT, Jiang RX, Zhang SD, et al. Discovery of the salicylaldehyde-based compound DDO-02267 as a lysine-targeting covalent inhibitor of ALKBH5. Eur J Med Chem. (2025) 284:117183. doi: 10.1016/j.ejmech.2024.117183
192. Lai GQ, Li Y, Zhu H, Zhang T, Gao J, Zhou H, et al. A covalent compound selectively inhibits RNA demethylase ALKBH5 rather than FTO. RSC Chem Biol. (2024) 5:335–43. doi: 10.1039/D3CB00230F
193. Yang X, Huang K, Wu XN, Zhang C, Sun Y, Gao Y, et al. Discovery of a novel selective and cell-active N(6)-methyladenosine RNA demethylase ALKBH5 inhibitor. J Med Chem. (2025) 68:4133–47. doi: 10.1021/acs.jmedchem.4c01542
194. Tao M, Li X, He L, Rong X, Wang H, Pan J, et al. Decreased RNA m6A methylation enhances the process of the epithelial mesenchymal transition and vasculogenic mimicry in glioblastoma. Am J Cancer Res. (2022) 12:893–906.
195. Liang L, Fei W, Wang Y, Zhang Z, You Q, and Guo X. Discovery of maleimide derivatives as m(6)A demethylase ALKBH5 inhibitors. Bioorg Med Chem. (2025) 120:118083. doi: 10.1016/j.bmc.2025.118083
196. Bell JL, Wachter K, Muhleck B, Pazaitis N, Kohn M, Lederer M, et al. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell Mol Life Sci. (2013) 70:2657–75. doi: 10.1007/s00018-012-1186-z
197. Du QY, Zhu ZM, and Pei DS. The biological function of IGF2BPs and their role in tumorigenesis. Invest New Drugs. (2021) 39:1682–93. doi: 10.1007/s10637-021-01148-9
198. Degrauwe N, Suvà ML, Janiszewska M, and Riggi N. IMPs: an RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. (2016) 30:2459–74. I. S. doi: 10.1101/gad.287540.116
199. Muller S, Bley N, Busch B, Glass M, Lederer M, Misiak C, et al. The oncofetal RNA-binding protein IGF2BP1 is a druggable, post-transcriptional super-enhancer of E2F-driven gene expression in cancer. Nucleic Acids Res. (2020) 48:8576–90. doi: 10.1093/nar/gkaa653
200. Mahapatra L, Andruska N, Mao C, Le J, and Shapiro DJ. A novel IMP1 inhibitor, BTYNB, targets c-myc and inhibits melanoma and ovarian cancer cell proliferation. Transl Oncol. (2017) 10:818–27. doi: 10.1016/j.tranon.2017.07.008
201. Xiao P, Meng Q, Liu Q, Lang Q, Yin Z, Li G, et al. IGF2BP1-mediated N6-methyladenosine modification promotes intrahepatic cholangiocarcinoma progression. Cancer Lett. (2023) 557:216075. doi: 10.1016/j.canlet.2023.216075
202. Jamal A, Hassan Dalhat M, Jahan S, Choudhry H, and Imran Khan M. BTYNB, an inhibitor of RNA binding protein IGF2BP1 reduces proliferation and induces differentiation of leukemic cancer cells. Saudi J Biol Sci. (2023) 30:103569. doi: 10.1016/j.sjbs.2023.103569
203. Sperling F, Misiak D, Huttelmaier S, Michl P, and Griesmann H. IGF2BP1 promotes proliferation of neuroendocrine neoplasms by post-transcriptional enhancement of EZH2. Cancers (Basel). (2022) 14:2121. doi: 10.3390/cancers14092121
204. Biegel JM, Dhamdhere M, Gao S, Gowda CP, Kawasawa YI, and Spiegelman VS. Inhibition of the mRNA-binding protein IGF2BP1 suppresses proliferation and sensitizes neuroblastoma cells to chemotherapeutic agents. Front Oncol. (2021) 11:608816. doi: 10.3389/fonc.2021.608816
205. Hagemann S, Misiak D, Bell JL, Fuchs T, Lederer MI, Bley N, et al. IGF2BP1 induces neuroblastoma via a druggable feedforward loop with MYCN promoting 17q oncogene expression. Mol Cancer. (2023) 22:88. doi: 10.1186/s12943-023-01792-0
206. Xu J, Wang Y, Ren L, Li P, and Liu P. IGF2BP1 promotes multiple myeloma with chromosome 1q gain via increasing CDC5L expression in an m(6)A-dependent manner. Genes Dis. (2025) 12:101214. doi: 10.1016/j.gendis.2024.101214
207. Wallis N, Oberman F, Shurrush K, Germain N, Greenwald G, Gershon T, et al. Small molecule inhibitor of Igf2bp1 represses Kras and a pro-oncogenic phenotype in cancer cells. RNA Biol. (2022) 19:26–43. doi: 10.1080/15476286.2021.2010983
208. Singh A, Singh V, Wallis N, Abis G, Oberman F, Wood T, et al. Development of a specific and potent IGF2BP1 inhibitor: A promising therapeutic agent for IGF2BP1-expressing cancers. Eur J Med Chem. (2024) 263:115940. doi: 10.1016/j.ejmech.2023.115940
209. Dahlem C, Abuhaliema A, Kessler SM, Krohler T, Zoller BGE, Chanda S, et al. First small-molecule inhibitors targeting the RNA-binding protein IGF2BP2/IMP2 for cancer therapy. ACS Chem Biol. (2022) 17:361–75. doi: 10.1021/acschembio.1c00833
210. Weng H, Huang F, Yu Z, Chen Z, Prince E, Kang Y, et al. The m(6)A reader IGF2BP2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell. (2022) 40:1566–82 e10. doi: 10.1016/j.ccell.2022.10.004
211. Feng P, Chen D, Wang X, Li Y, Li Z, Li B, et al. Inhibition of the m(6)A reader IGF2BP2 as a strategy against T-cell acute lymphoblastic leukemia. Leukemia. (2022) 36:2180–8. doi: 10.1038/s41375-022-01651-9
212. Wei Q, Zhou H, Zhong L, Shi L, Liu J, Yang Q, et al. IMP3 expression in biopsy specimens as a diagnostic biomarker for colorectal cancer. Hum Pathol. (2017) 64:137–44. doi: 10.1016/j.humpath.2017.03.013
213. Xu W, Sheng Y, Guo Y, Huang Z, Huang Y, Wen D, et al. Increased IGF2BP3 expression promotes the aggressive phenotypes of colorectal cancer cells in vitro and vivo. J Cell Physiol. (2019) 234:18466–79. doi: 10.1002/jcp.v234.10
214. Yang Z, Wang T, Wu D, Min Z, Tan J, and Yu B. RNA N6-methyladenosine reader IGF2BP3 regulates cell cycle and angiogenesis in colon cancer. J Exp Clin Cancer Res. (2020) 39:203. doi: 10.1186/s13046-020-01714-8
215. Zhang Y, Liu X, Yu M, Xu M, Xiao Y, Ma W, et al. Berberine inhibits proliferation and induces G0/G1 phase arrest in colorectal cancer cells by downregulating IGF2BP3. Life Sci. (2020) 260:118413. doi: 10.1016/j.lfs.2020.118413
216. Gui Z, Li J, Li J, Li X, Chen L, Ma Z, et al. Berberine promotes IGF2BP3 ubiquitination by TRIM21 to induce G1/S phase arrest in colorectal cancer cells. Chem Biol Interact. (2023) 374:110408. doi: 10.1016/j.cbi.2023.110408
217. Micaelli M, Dalle Vedove A, Cerofolini L, Vigna J, Sighel D, Zaccara S, et al. Small-molecule ebselen binds to YTHDF proteins interfering with the recognition of N (6)-methyladenosine-modified RNAs. ACS Pharmacol Transl Sci. (2022) 5:872–91. doi: 10.1021/acsptsci.2c00008
218. Zou Z, Wei J, Chen Y, Kang Y, Shi H, Yang F, et al. FMRP phosphorylation modulates neuronal translation through YTHDF1. Mol Cell. (2023) 83:4304–17 e8. doi: 10.1016/j.molcel.2023.10.028
219. Wang CH and Zhou H. Discovery of a new inhibitor for YTH domain-containing m(6)A RNA readers. RSC Chem Biol. (2024) 5:914–23. doi: 10.1039/D4CB00105B
220. Zalesak F, Nai F, Herok M, Bochenkova E, Bedi RK, Li Y, et al. Structure-based design of a potent and selective YTHDC1 ligand. J Med Chem. (2024) 67:9516–35. doi: 10.1021/acs.jmedchem.4c00599
221. Cun Y, Guo W, Ma B, Okuno Y, and Wang J. Decoding the specificity of m6A RNA methylation and its implication in cancer therapy. Mol Ther. (2024) 32:2461–9. doi: 10.1016/j.ymthe.2024.05.035
222. Zhou C, Zhang Y, Shi SM, Yin D, Li XD, Shi YH, et al. FTO downregulation-mediated m6A modification resulting in enhanced hepatocellular carcinoma invasion. Cell Biosci. (2025) 15:58. doi: 10.1186/s13578-025-01395-w
223. Bai Y, Li K, Peng J, and Yi C. m(6)A modification: a new avenue for anti-cancer therapy. Life Med. (2023) 2:lnad008. doi: 10.1093/lifemedi/lnad008
224. Yu F, Wei J, Cui X, Yu C, Ni W, Bungert J, et al. Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic Acids Res. (2021) 49:5779–97. doi: 10.1093/nar/gkab415
225. Chen A, Zhang VX, Zhang Q, Sze KM, Tian L, Huang H, et al. Targeting the oncogenic m6A demethylase FTO suppresses tumourigenesis and potentiates immune response in hepatocellular carcinoma. Gut. (2024) 74:90–102. doi: 10.1136/gutjnl-2024-331903
226. Liu F, Liu Q, Li X, Wang Y, Cao R, Zhang S, et al. m6A epitranscriptomic modification in hepatocellular carcinoma: implications for the tumor microenvironment and immunotherapy. Front Immunol. (2025) 16:1538658. doi: 10.3389/fimmu.2025.1538658
227. Lu Z, Lyu Z, Dong P, Liu Y, and Huang L. N6-methyladenosine RNA modification in stomach carcinoma: Novel insights into mechanisms and implications for diagnosis and treatment. Biochim Biophys Acta Mol Basis Dis. (2025) 1871:167793. doi: 10.1016/j.bbadis.2025.167793
228. Gao L, Gao J, He J, Fan W, Che X, Wang X, et al. Identification of m6A methyltransferase-related WTAP and ZC3H13 predicts immune infiltrates in glioblastoma. Sci Rep. (2025) 15:4412. doi: 10.1038/s41598-025-88671-4
229. Zhou X, Li C, Chen T, Li W, Wang X, and Yang Q. Targeting RNA N6-methyladenosine to synergize with immune checkpoint therapy. Mol Cancer. (2023) 22:36. doi: 10.1186/s12943-023-01746-6
230. Zhao Z, Han L, and Ge Q. Targeting YTHDF2 impacts the epitranscriptome and overcomes tumor therapy resistance. Trends Cell Biol. (2025) 35:177–9. doi: 10.1016/j.tcb.2025.02.003
231. Zheng Z, Lin F, Zhao B, Chen G, Wei C, Chen X, et al. ALKBH5 suppresses gastric cancer tumorigenesis and metastasis by inhibiting the translation of uncapped WRAP53 RNA isoforms in an m6A-dependent manner. Mol Cancer. (2025) 24:19. doi: 10.1186/s12943-024-02223-4
232. Livneh I, Moshitch-Moshkovitz S, Amariglio N, Rechavi G, and Dominissini D. The m6A epitranscriptome: transcriptome plasticity in brain development and function. Nat Rev Neurosci. (2019) 21:36–51. doi: 10.1038/s41583-019-0244-z
233. Dong Z, Huang Y, Xia W, Liao Y, and Yang CG. A patenting perspective of fat mass and obesity associated protein (FTO) inhibitors: 2017-present. Expert Opin Ther Pat. (2025) 35:533–42. doi: 10.1080/13543776.2025.2477482
234. Tan Z, Tian L, Luo Y, Ai K, Zhang X, Yuan H, et al. Preventing postsurgical colorectal cancer relapse: A hemostatic hydrogel loaded with METTL3 inhibitor for CAR-NK cell therapy. Bioact Mater. (2025) 44:236–55. doi: 10.1016/j.bioactmat.2024.10.015
Keywords: RNA modification, N6-methyladenosine, inhibitors, molecular targets, cancer treatment
Citation: Liu X and Kan X (2025) Small-molecule and peptide inhibitors of m6A regulators. Front. Oncol. 15:1629864. doi: 10.3389/fonc.2025.1629864
Received: 16 May 2025; Accepted: 07 July 2025;
Published: 01 August 2025.
Edited by:
Qun Chen, University of Oklahoma, United StatesReviewed by:
Xiangting Wang, University of Science and Technology of China, ChinaRais Ahmad Ansari, Nova Southeastern University, United States
Copyright © 2025 Liu and Kan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuefeng Kan, eGtsaXVsYW5nMTMxNEAxNjMuY29t