 Holly Wobma1,2
Holly Wobma1,2 Lauren A. Henderson1,2
Lauren A. Henderson1,2 Christine N. Duncan1,3,4
Christine N. Duncan1,3,4 Susan E. Prockop1,3,4
Susan E. Prockop1,3,4 Adrienne G. Randolph1,5
Adrienne G. Randolph1,5 Barbara A. Degar1,3,4
Barbara A. Degar1,3,4 Leslie E. Lehmann1,3,4
Leslie E. Lehmann1,3,4 Susanne H. C. Baumeister1,3,4*
Susanne H. C. Baumeister1,3,4*- 1Harvard Medical School, Boston, MA, United States
- 2Division of Immunology, Boston Children’s Hospital, Boston, MA, United States
- 3Division of Hematology-Oncology, Boston Children’s Hospital, Boston, MA, United States
- 4Department of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, MA, United States
- 5Division of Critical Care, Boston Children’s Hospital, Boston, MA, United States
Hemophagocytic lymphohistiocytosis (HLH) describes a severe, hyperinflammatory syndrome that can originate from diverse etiologies, often requiring critical care level management. Primary HLH, initially described in the 1940s, derives from genetic defects that result in uncontrolled immune activation. Although chemotherapy and immunosuppressive agents can temporarily quell inflammation, allogeneic hematopoietic cell transplantation (HCT) is the only curative option. In 2025, HCT is indicated for primary HLH and some etiologies of secondary HLH but remains challenging due to both disease and transplant-related inflammation. Additionally, new cellular therapy approaches to treat malignancy, such as chimeric antigen receptor T cells, can trigger a spectrum of hyperinflammatory complications. Herein, we review the pathophysiology, diagnosis, and evolving management approaches of primary and secondary HLH, ultimately informing our management of hyperinflammation in the setting of new cell therapies.
Introduction
Inflammation is the natural response to injury or infection. It also requires tight regulation. When there is an imbalance between the level of stimulus and the magnitude and duration of response, hyperinflammation ensues. The quintessential hyperinflammatory disorder is hemophagocytic lymphohistiocytosis (HLH). HLH describes a clinical syndrome of fever, cytopenias, hepatosplenomegaly, hyperferritinemia and organ dysfunction, which is often life threatening, requiring critical care management (1). Key HLH pathophysiology involves dysregulated T cells, natural killer (NK) cells, and macrophage/myeloid cells, resulting in excessive cytokine production.
In healthy individuals, activation of antigen presenting cells (APCs) via pattern recognition receptors facilitates antigen-presentation, production of proinflammatory cytokines, and recruitment and activation of cytotoxic CD8+ T-lymphocytes and NK cells. CD8+ T cells and NK cells proliferate and release inflammatory cytokines such as interferon gamma (IFN-γ) (2, 3). This inflammatory milieu further increases the antigen presentation capacity of APCs and stimulates innate immune cells. One of the mechanisms by which CD8+ T cells and NK cells eliminate infected, autoreactive, or malignant cells, is granule-mediated cytotoxicity. This process involves delivery of cytotoxic granule contents into the immune synapse and perforin facilitated entry into the target cell, inducing target cell apoptosis. Once a successful immune response has been orchestrated and target cells are eliminated, the immune response is downregulated in a controlled fashion. CD8+ T cells eliminate activated APCs, NK cells can induce apoptosis of activated APCs and T cells, and ultimately, most involved immune cells undergo a programmed cell death, with only a small number of memory T cells remaining. When this process becomes dysregulated, there is overproduction of IFN-γ, a central cytokine in HLH pathophysiology.
HLH is separated into ‘primary’ and ‘secondary’ forms, although as discussed below, is often multifactorial. Primary HLH derives from pathogenic genetic variants in granule-mediated cytotoxicity or inflammasome regulation that result in an escalating hyperinflammatory feedback loop and ineffective contraction of the immune response. Granule-mediated cytotoxicity defects result in prolonged synapse time, overproduction of cytokines, accumulation of APCs to stimulate T cell inflammation and defective APC and T-cell clearance. In inflammasome regulation defects, this inflammatory cycle is triggered by excessive production of inflammasome-dependent cytokines.
In secondary HLH, a predominant underlying etiology (e.g., autoimmune disease, malignancy, infection, administration of Immune Effector Cells (IEC)) in the setting of additional modifiers (e.g., infection, genetic variants) may yield similar functional defects in immune regulation. This model, in which multiple factors converge to surpass a ‘threshold’ for development of HLH, is schematized in Figure 1 (4). Shared terminal pathophysiology may be marked by the presence of activated, Th1 polarized HLADR+CD38hi CD8+ T cells (5–7), which (amongst other lymphocytes) are the producers of IFN-γ that is characteristic of HLH-related inflammation (8).
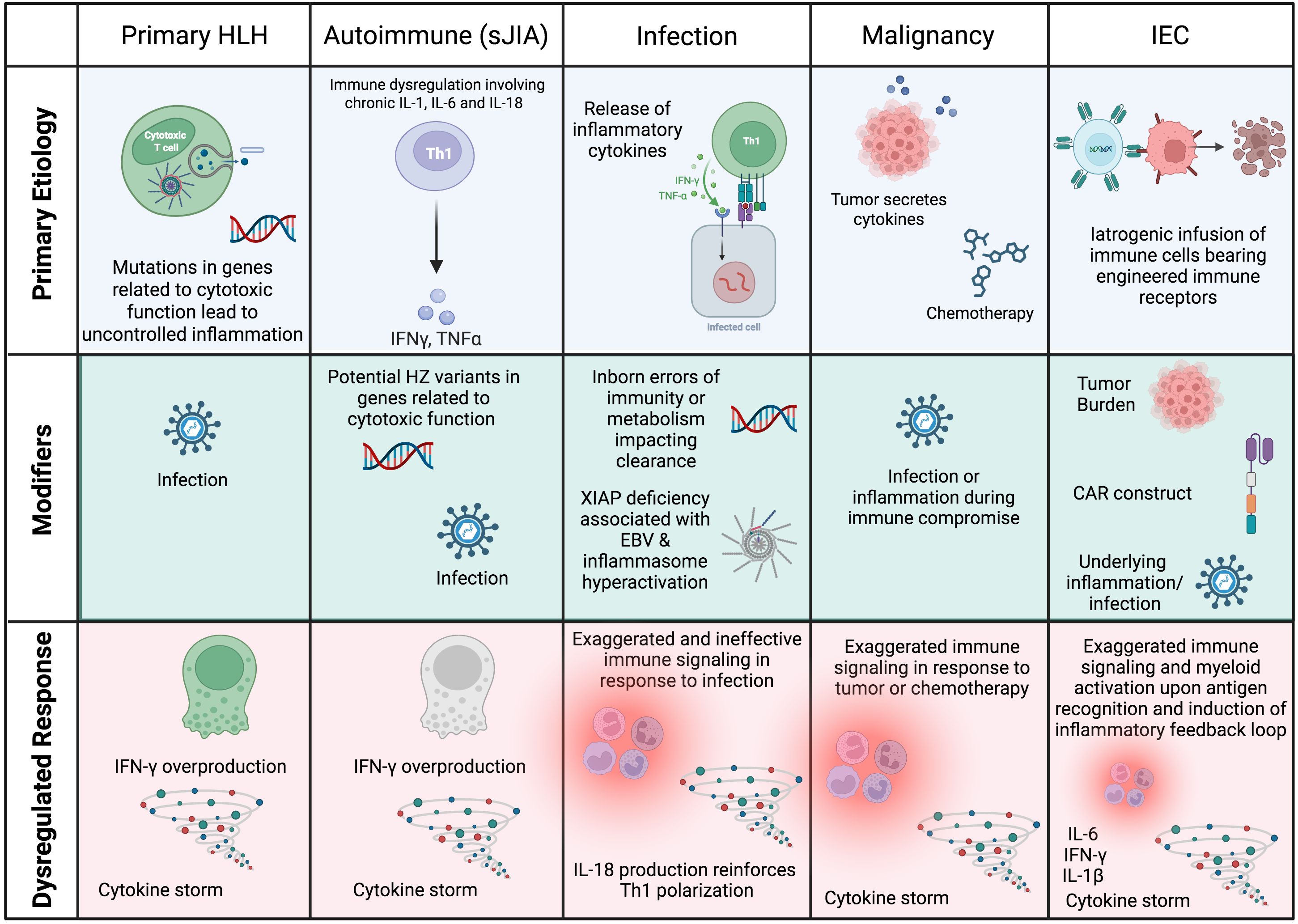
Figure 1. Schematic of the root causes of HLH and HLH-like syndromes in different settings. In most cases, HLH is multifactorial, with a predominating underlying cause that manifests as HLH when additional modifiers or ‘hits’ converge to result in a dysregulated immune response. CAR, chimeric antigen receptor; HSCs, hematopoietic stem cells; HZ, heterozygous; IEC, Immune Effector Cell. Created in BioRender.
Medical management targeting cytokine production and hyperactive immune cells may quell HLH and prevent its recurrence. However, when there is a strong genetic predisposition and/or a chronic underlying disorder, allogeneic hematopoietic cell transplantation (HCT) may be required for definitive treatment. The longstanding experience of cell therapy physicians in HLH management has been instrumental. Additionally, given the illness acuity of patients with HLH and HLH-like syndromes, physicians caring for these patients almost always include those in critical care medicine. Intensivists must balance treating multi-organ dysfunction and immune dysregulation in the context of a patient’s primary disorder. This results in overlapping but unique multidisciplinary management considerations in collaboration with HCT/Cellular therapy, Oncology, Neurology, Rheumatology, Immunology, and Infectious Disease specialists. Here we describe the different causes of HLH and HLH-like syndromes, as well as the role of medical management and HCT.
Primary HLH
Classification and pathophysiology
Primary HLH was initially described in the mid-1900s in a series of case reports (9). Infants would develop fevers, cytopenias, hepatosplenomegaly, and neurological features, and within weeks, succumb to disease. Post-mortem bone marrow analysis showed the characteristic finding of ‘hemophagocytosis’ – macrophages (histiocytes) phagocytosing blood cells. This syndrome eventually became known as HLH. The Histiocyte Society initially developed criteria for the diagnosis of HLH in children based on clinical and laboratory criteria for the HLH-94 trial (10). These criteria were subsequently refined for the HLH-2004 trial to include molecular and/or clinical criteria (11). Most recently, revised criteria were proposed in which functional assays in the setting of consistent clinical features can also be used to diagnose HLH (Table 1) (12). Additional laboratory tests may support the diagnosis of HLH and be useful for disease activity monitoring. These include detection of the IFN-γ induced protein CXCL9 (since IFN-γ itself is difficult to detect in the bloodstream) (8, 13) as well as HLADR+CD38hi CD8+ T cells, although the latter is not available as a certified laboratory test.

Table 1. 2024 revised HLH criteria (12).
Primary HLH results from inborn errors of immunity (IEIs) and is estimated to describe 12% of cases of HLH in pediatrics (14). In 1999, perforin (PRF1) mutations were found to cause HLH in a subset of patients (15). Perforin is key to the cytotoxic function of CD8+ T cells and NK cells, enabling them to appropriately contract an inflammatory response. Soon thereafter, mutations in other genes (STX11, STXBP2, UNC13D) with related functions in the cytotoxic response were implicated in HLH, resulting in a nomenclature of familial HLH (FHL), where ‘familial’ implied inherited, and a corresponding number implied a specific genetic defect in granule mediated cytotoxicity (e.g., FHL2 is HLH due to a perforin defect). As genetic variants driving HLH can be de novo, and there is now a larger range of pathways implicated, it is now more common to use the term “primary HLH” and to specify the gene mutation (if known) (16) (Table 2). For example, several IEIs that do not affect cytotoxic function but instead regulate the inflammasome (e.g. NLRC4, CDC42) have HLH as a common manifestation (17–19). In these disorders, overactive inflammasome signaling drives activation of caspase-1 and its cleavage products IL-1β and IL-18, which can promote a hyperinflammatory cascade (20). Lastly, IEIs that predispose to poor control of Epstein Barr virus (EBV) infection may increase the risk for HLH and will be discussed in a later section. There may also be an overlap, as XIAP deficiency exhibits both altered inflammasome regulation and increased susceptibility to EBV infection (21).
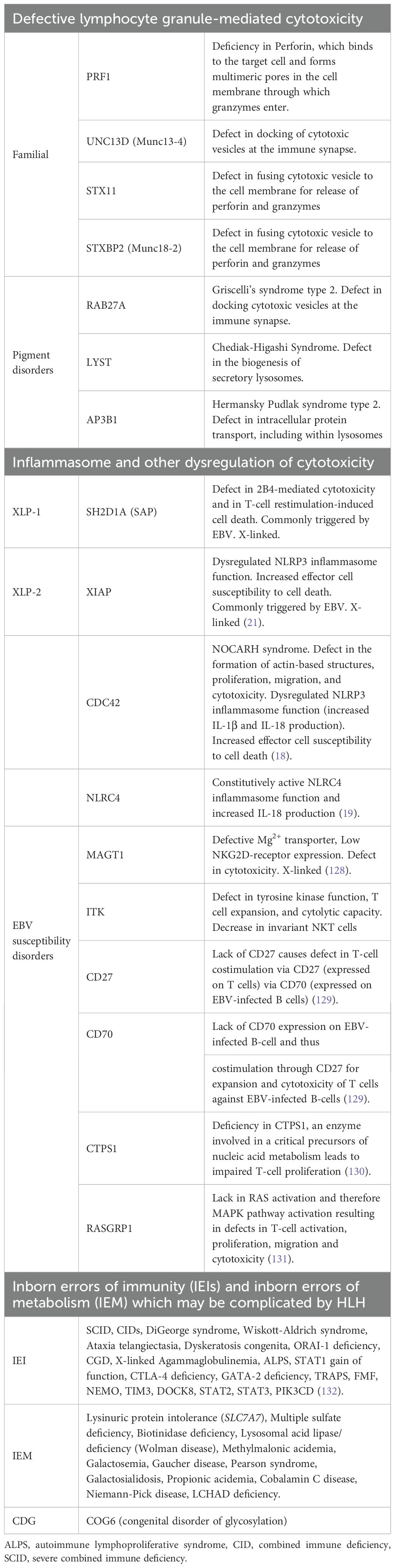
Table 2. Primary HLH (based on an underlying genetic defects).
Medical treatment of primary HLH
Early attempts to treat classic ‘familial’ HLH included adrenocorticotropic hormone, splenectomy, exchange transfusion, and steroids (1). There was also use of chemotherapeutic agents. One agent in particular – etoposide – was selected due to its use in monocytic leukemias suggesting a potential ability to subdue macrophage activation. Indeed, early studies with etoposide were highly promising (22). Ironically, the mechanism of its benefit was different than predicted. Etoposide is a topoisomerase II inhibitor, blocking repair of double stranded DNA breaks, triggering apoptotic cell death. This was shown to be true of activated, cytotoxic CD8+ T cells in murine models of HLH (23). Thus, etoposide eliminates a hyperactive cell population in HLH that would have otherwise been eliminated had there not been a genetic defect in cytotoxic function. In addition to etoposide, intrathecal methotrexate (anti-metabolite) was added to regimens for central nervous system (CNS) coverage, as there is CNS involvement in 30-73% of patients with primary HLH (24). As dexamethasone has better CNS penetrance than other forms of corticosteroids, it emerged as the steroid of choice. Calcineurin inhibitors – particularly cyclosporine – were added to protocols to block NFAT-mediated production of cytokines such as IL-2, TNF-α, and IFN-γ. Finally, allogeneic HCT was determined to be necessary for cure.
This constellation of therapies was the foundation of the HLH-94 protocol, used in a large international pediatric trial. Etoposide, dexamethasone, and intrathecal methotrexate were used as an 8-week induction regimen, followed by intermittent etoposide, dexamethasone, and twice daily cyclosporine as a bridge to HCT (25). In a long-term follow up study, 29% of patients died before HCT, and for those that underwent HCT, the 5-year survival was 66%. There was a trend for improved HCT outcomes in patients with better disease control at the time of HCT. Early deaths were reported in patients who developed jaundice, edema, and renal dysfunction, suggestive of veno-occlusive disease (VOD). Even with the 20-30% mortality rate pre- and post-HCT, outcomes in this study were significantly improved from historic regimens. As a follow up, the HLH-2004 study started cyclosporine during induction, but this did not improve outcomes, and there were concerns regarding the side effects of cyclosporine (26).
In the modern era, etoposide and dexamethasone still serve as the foundation of pre-HCT induction therapy in primary HLH (27). Due to the risk of myelosuppression (and associated infection risk) as well as secondary malignancy (albeit rare) with etoposide, alternative agents have been explored. For example, emapalumab (IFN-γ antagonist) is now FDA approved for patients with primary HLH who have had a refractory, recurrent, or progressive course or intolerance to conventional therapy (28). Other options include alemtuzumab, a monoclonal antibody against CD52 (targets T cells, B cells, and macrophages) and ruxolitinib (janus kinase (JAK)1/2 inhibitor) (29). In practice, these agents are not mutually exclusive with etoposide and can be used sequentially or in conjunction. For example, as will be discussed below, one of the major barriers to HCT for HLH is the high rate of graft failure, particularly with reduced intensity conditioning (RIC) regimens. This may be due to a detrimental effect of IFN-γ on donor stem cells that cannot compete with residual recipient cells in the bone marrow that are ‘better acclimated’ to high-level IFN-γ environments (30). Bridging emapalumab in patients previously treated with etoposide may address this challenge and improve donor chimerism (30).
Allogeneic HCT for primary HLH
Identifying the best conditioning regimen for HCT in HLH has been historically difficult. Initial transplants with busulfan (Bu)-based myeloablative conditioning (MAC) had high rates of fatal VOD, likely due to the hepatotoxicity of Bu in patients with pre-existing liver dysfunction from HLH. This motivated lower dose Bu or RIC regimens such as fludarabine/melphalan/alemtuzumab (Flu/Mel/Alem). While the toxicity of RIC regimens was lower, they had high rates of mixed chimerism and graft failure, with chimerism levels of at least 20-30% thought to be necessary to protect against HLH recurrence (31). A large study using the Center for International Blood and Marrow Transplant Research registry compared four different commonly used conditioning regimens, adjusted for year of transplant (32). These included Bu/Cyclophosphamide (Cy), Bu/Flu, Flu/Mel/Alem, and Flu/Mel/Alem/Thiotepa (TT). The primary outcome was event free survival (EFS) – i.e., survival without primary or secondary graft failure. While overall survival (OS) was similar between regimens, rates of graft failure were substantially higher in the Flu/Mel/Alem regimen, corresponding to the lowest EFS of 44% at 5 years. Addition of thiotepa (an alkylator) to this regimen substantially reduced graft failure, improved EFS, and was associated with a low rate of VOD, establishing Flu/Mel/Alem/TT as the currently recommended regimen.
As previously mentioned, CNS involvement occurs in a substantial fraction of patients with primary HLH, and in some forms of secondary HLH. Clinical symptoms may include seizures, altered mental status, and/or encephalopathy, with delayed or insufficient treatment risking permanent motor and cognitive deficits. Thus, early identification is crucial. As not all patients have clinical symptoms at onset, screening MRI and lumbar puncture are recommended (24). In addition to CNS inflammation being part of HLH, there is an entity of CNS-restricted HLH, often driven by similar pathogenic variants but more difficult to diagnose given the lack of systemic features and inflammatory markers (33). Compared to the classic systemic form of primary HLH, the onset of CNS-restricted HLH occurs later – at a median age of 6.5 years in one report (33). The differential diagnosis includes CNS vasculitis, acute disseminated encephalomyelitis, acute necrotizing encephalopathy, or CNS involvement of an underlying condition. Diffuse white matter and/or cerebellar involvement is commonly seen on imaging, and pleocytosis, elevated protein, and elevated neopterin may be seen in the cerebrospinal fluid (CSF). There are no consensus guidelines on treatment of CNS-restricted HLH but dexamethasone with or without intrathecal methotrexate may be considered, as can etoposide and emapalumab. Regardless of medical treatment choice, the definitive treatment of CNS-restricted HLH remains HCT, ideally as soon as feasible to avoid irreversible neurologic damage (34, 35).
Summary and future directions
Therapeutic approaches for primary HLH, including first line and HCT regimens, are likely to continue to evolve, as will supportive care measures. Our group has recently considered pre-emptive (prior to HCT) tracheostomy for small patients considered to be at high risk for respiratory complications. While not part of any established guidelines, this has been beneficial for respiratory support through the acute transplant period. In parallel, ex vivo autologous hematopoietic stem cell-based gene therapy for genetically defined HLH is being developed. The first trial is opening in France for patients with UNC13D mutations (NCT06736080). While this approach has great potential, challenges include the harvesting of stem cells from critically ill patients, the requirement for therapy while the genetically manipulated product is manufactured, and uncertainty about which conditioning regimen will be required and tolerated to facilitate engraftment of the genetically manipulated product.
HLH secondary to rheumatic disease
Classification and pathophysiology
HLH secondary to rheumatic disease is referred to as macrophage activation syndrome (MAS), with an estimated 10% of cases of HLH/MAS attributable to this cause in children (14). By far, the rheumatic disease most associated with MAS in pediatrics is systemic juvenile idiopathic arthritis (sJIA), also known as Still’s disease. Systemic JIA presents uniquely from other subclasses of JIA with daily fevers, rash, lymphadenopathy, and potentially MAS [in up to 14% of cases (36)]. This systemic inflammation results from an autoinflammatory component to the disease pathophysiology, with disease-specific cytokine elevation, including IL-1 (37), IL-18 (38), and IL-6 (39). While arthritis is part of historic disease definitions (40), not all patients have arthritis at presentation, and with modern therapy, the development of arthritis may even be avoided (41). Patients are thus recognized as having sJIA based on other consistent disease features, including MAS, as well as sJIA-suggestive biomarkers described below.
The definition of MAS in sJIA is unique from primary HLH, with classification criteria shown in Table 3 (42). The high incidence of MAS is hypothesized to result from elevations in IL-18 during active disease (43), which augments IFN-γ-production by cycling lymphocytes (6). These include HLADR+CD38hi CD8+ T cells as well as CD4+ cells and NK cells, which have been found to be highly glycolytic due to activation of mTORC1 signaling. mTORC1 is a metabolic sensor that can drive cell proliferation and determine function. This mTORC1 signaling was also found in activated monocytes in murine models of sJIA and MAS, potentially coordinating the inflammatory response (44). In addition to IL-18 and IFN-γ signaling in sJIA-MAS, an elevated type I interferon signature has been described that may further prime pathogenic T and NK cells and augment IL-18 production (6). As type I interferons are commonly induced by viral infection, this may explain infection as a trigger of sJIA-MAS. Lastly, it has also been observed that heterozygous variants in primary HLH related genes are enriched in patients with sJIA who develop MAS (45) (Figure 1).

Table 3. Classification of MAS in sJIA (2016 criteria) (42).
Medical treatment of sJIA and sJIA-MAS
The American College of Rheumatology recommendation (46) for first line treatment of sJIA is IL-1 inhibition (anakinra, canakinumab) or IL-6 inhibition (tocilizumab) (47–50). Use of JAK inhibitors in more severe cases, is increasingly common (51, 52). As plasma IL-1 levels are not reflective of IL-1 secretion, due to greater production in the tissues and binding to a natural decoy receptor in the blood (53), IL-18 – a related cytokine – is instead monitored. In active sJIA, IL-18 levels usually range from 104 to 105 pg/mL, which can help identify sJIA from other febrile disorders (38). During episodes of MAS, IL-18 levels >105 pg/mL are often observed, and IFN-γ related biomarkers (e.g. CXCL9) become significantly elevated (39).
Unlike in primary HLH, there are few dedicated trials or consensus guidelines for treatment of MAS (14). To create a framework for approaching MAS/HLH diagnosis and treatment, our center previously developed an Evidence Based Guideline (EBG) based on expert participation from diverse specialties at Boston Children’s Hospital, using a consensus-based process (54, 55). This led to treatment recommendations that include consideration of anakinra, intravenous immune globulin, calcineurin inhibitors, and high dose steroids for MAS. Since the implementation of this EBG, there has been updated evidence for use of JAK inhibitors (for other etiologies of HLH) (29) and emapalumab (recently FDA approved for MAS) (56), and the EBG has been updated accordingly. Use of etoposide, although described and beneficial for some patients with sJIA-MAS, is generally not recommended due to associated myelosuppression but could be considered in collaboration with Oncologists for patients with highly refractory disease (57, 58). A 2025 review on treatment evidence for MAS is provided (57). Importantly, while systemic reviews and single center guidelines are important reference tools, centers should adapt recommendations to local resources and patient specific needs. For example, general dosing guidelines may not be appropriate for patients with significant organ dysfunction or in the setting of potential drug-drug interactions (e.g. liver toxicity and/or antifungal prophylaxis with ‘azoles’ for ruxolitinib). Infection prophylaxis should be considered but is not detailed in this review. Lastly, with new advances in the field, guidelines will eventually become outdated.
Autologous and allogeneic HCT for sJIA and sJIA-MAS
Beyond treating individual episodes of MAS, there is also a history of HCT for sJIA, although transplant strategies and indications have evolved over time. Initially, in the late 1990s when there were no sJIA targeted biologics, autologous HCT was explored for patients with refractory arthritis. The premise was that high dose chemotherapy (usually ATG and Cy ± Flu or low-dose total body irradiation) could reset the immune system, and pre-collected hematopoietic stem cells would then be infused to restore hematopoiesis with subsequent production of naïve, clonally diverse T cells in the thymus. Intrinsic to this approach was ex vivo T cell depletion of the stem cell graft to avoid reinfusing dysregulated T cells. In a multicenter cohort of 34 patients with arthritis unresponsive to methotrexate and at least one other therapy, 53% of patients had a complete response, 18% a partial response, and 21% had no response by an average of 29 months of follow up (59). Prolonged depression of CD4+ T cells was common after transplant. Three patients developed fatal MAS triggered by infection, prompting an international consensus to reduce the degree of T cell depletion and improve disease control ahead of transplantation (60). Given the transplant related mortality of 9% and the emergence of IL-1 and IL-6 targeted biologics by the mid-2000s, autologous HCT became uncommon.
With new biologic agents curtailing the development of chronic arthritis, morbidity from sJIA transitioned from arthritis to recurrent MAS and potentially fatal lung disease (sJIA-LD), comprised of interstitial involvement and/or pulmonary hypertension (61). One of the earliest reports of sJIA-LD had a 5-year survival of only 42% (61). Due to increased screening, patients with less severe lung disease are now being identified, improving survival statistics (62). Risk factors for lung disease include young age of sJIA onset, HLA-DRB1*15 positivity, congenital heart disease, chronic IL-18 signaling, recurrent MAS, and trisomy 21 (Figure 2) (63–66). The latter two factors have a causal explanation – an IFN-γ signature has been found in the bronchoalveolar lavage fluid of patients with lung disease suggesting a role in its pathogenesis (67). MAS is largely IFN-γ driven, and in trisomy 21, there is increased interferon signaling due to triplicate copies of a receptor for IFN-γ (IFNGR2) (68). Due to concern for disease relapse and MAS flare with autologous HCT, the field shifted to allogeneic HCT for recurrent MAS ± lung disease as the main transplant indications. There have been two retrospective cohort studies in the last 10 years. The first included 11 patients with treatment-refractory sJIA with 5/11 having a history of MAS (69). All patients received a reduced toxicity conditioning (RTC) regimen of Flu/Mel/Alem or Flu/Treosulfan/Alem. Viral infections after transplant were common and 1/11 patients died of fungemia in the setting of graft-vs-host-disease treatment. At last follow-up, 9/10 surviving patients were in complete remission (no symptoms, off immunosuppression), while one patient had a flare of arthritis and an MAS-like episode. The second report included 13 patients with sJIA-LD – 5 with an oxygen requirement prior to transplant (70). Patients received RIC or RTC regimens, most commonly Flu/Mel/Alem/TT (akin to that used in primary HLH). Four patients died – two from cytomegalovirus infection, one from intracranial hemorrhage during asymptomatic SAR-CoV2 infection, and one from progressive sJIA-LD. All 9 surviving patients were in complete remission off immune suppression at the time of last follow up, with previously oxygen-dependent patients coming off oxygen (70).
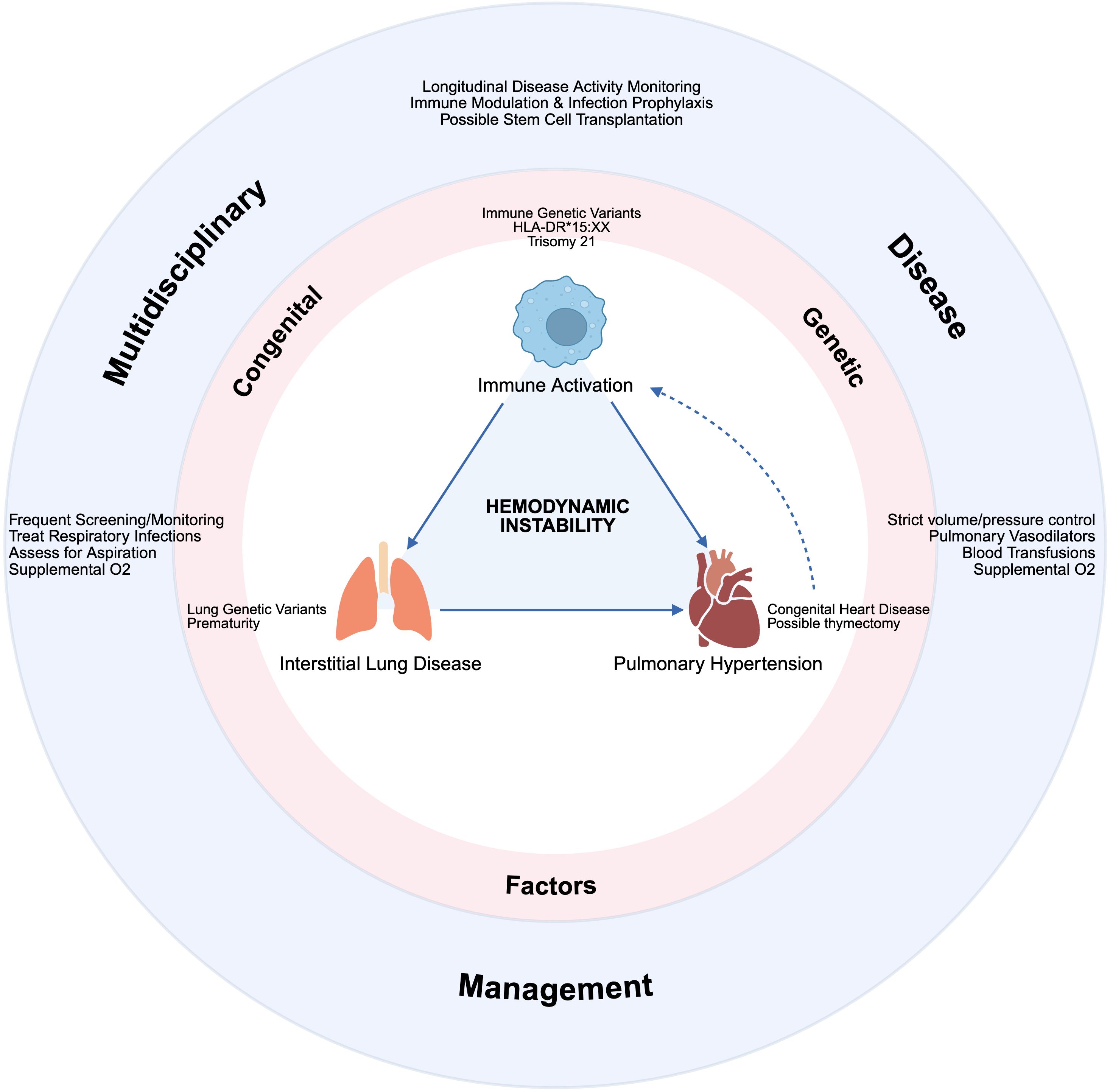
Figure 2. Schematic of the multi-organ involvement in sJIA and MAS, often requiring critical care management in collaboration with rheumatology, pulmonology, cardiology, and stem cell transplantation. Created in BioRender.
Summary and future directions
In summary, the pediatric rheumatic disease most associated with MAS is sJIA due to baseline activation of pathways that can trigger MAS-related inflammation. Medical management with small molecules and biologic agents may successfully control these episodes in some patients, but for those with recurrent MAS and/or lung disease, allogeneic HCT may be considered to halt a potentially fatal disease trajectory and achieve drug free remission. As transplant-related mortality remains 12.5-25% (higher for those with lung disease), careful patient selection is required, and there is currently no consensus on when this should be pursued.
HLH secondary to infection
Classification and pathophysiology
Infections are the most common trigger of HLH in children, both in primary and secondary cases (57% of cases overall) (14). Viral, bacterial, parasitic, and fungal infections have been linked to HLH in both immune competent and immune compromised hosts. Work-up for suspected HLH should therefore include comprehensive evaluation in consultation with an infectious disease expert. As HLH is an uncommon complication of infection overall, a genetic evaluation should be strongly considered to look for predisposing pathogenic variants.
The most common trigger of infection associated HLH is EBV. EBV is a herpesvirus that primarily infects B cells through their CD21 and CD35 receptors and establishes latency. Initial EBV infection is asymptomatic in most individuals, and an estimated >90% of adults are latently infected (71). Clinical sequelae can range from ‘infectious mononucleosis’ (e.g., fever, sore throat, splenomegaly) to EBV-related malignancy or HLH.
A recent study by Liu et al. elucidates why EBV has a higher predilection for triggering HLH (72). In this study, serum and peripheral blood mononuclear cells were compared amongst pediatric patients with non-EBV viral illness, EBV infection, and known HLH. Relative to other viral illnesses, acute EBV infection led to increases in IL-18 and IL-27 (which contribute to Th1 cell polarization) as well as CXCL9. As with other etiologies of HLH, there was also expansion of HLADR+CD38hi CD8+ T cells, which corresponded to the ‘atypical lymphocytes’ often observed in acute EBV infection (72). Overall, these data suggest that the response to acute EBV infection includes production of cytokines and populations of T cells that are hallmarks of HLH.
Medical treatment of infection-driven HLH
Treatment of infection-driven HLH involves targeting the underlying infection in addition to standard HLH directed therapies. For EBV driven HLH, addition of a B cell targeting agent like rituximab has been shown to facilitate elimination of the EBV reservoir (73). Anakinra is safe in the setting of infection (was initially investigated for patients with sepsis) (74, 75) but is not always sufficient to control EBV-HLH (76). In a single-arm phase II trial, 52 pediatric patients with a new diagnosis of HLH (diverse etiologies but malignancy excluded) received ruxolitinib as first-line therapy, with a complete response of 58.3% in the subset with EBV-HLH (29). Lastly, emapalumab may be used as an adjunctive agent to chemotherapy for cases that are particularly severe (77, 78). More specific guidelines for EBV-HLH will require prospective studies. However, one important consideration is whether a patient is at risk for recurrence.
Allogeneic HCT for CAEBV and EBV-predisposing IEIs
Allogeneic HCT may be needed to resolve EBV viremia and its associated complications. For example, there are several IEIs (due to loss-of-function variants in XIAP, SAP, CD27, CD70, ITK, and MAGT1) associated with EBV-HLH and EBV-triggered lymphoma (79) that impair the ability of the immune system to clear EBV infection. There is also an entity known as Chronic Active EBV (CAEBV), which describes persistent EBV viremia without a known IEI. CAEBV has clear geographic associations and has mainly been described in East Asia and in Central America. As with some IEIs, a characteristic of CAEBV is that EBV infection is found in other lymphoid and myeloid lineages – thus not restricted to the B cell compartment (Table 4). A 2024 study by Wang et al. provides a potential explanation (80). In this study, bone marrow and peripheral blood samples were obtained from patients with CAEBV, supporting infection of hematopoietic stem cells, which then differentiate to become EBV infected myeloid and lymphoid cells. While the circumstances under which they become infected with EBV are still unknown, this finding supports allogeneic HCT as the only curative option. There is no standardized treatment regimen for CAEBV, but a combination of immune targeted therapy (e.g. steroids, cyclosporine, PD1 inhibition) and/or chemotherapy is often used to control disease ahead of HCT (81, 82). In a large Japanese registry study of 102 patients with CAEBV that underwent allogeneic HCT, worse outcomes were associated with elevated soluble IL2 receptor at the time of transplant (suggesting poor control of inflammation) and use of radiotherapy in conditioning regimens (83). In a smaller study, MAC and RIC regimens were compared (84). The EFS at 3 years was 54.5 ± 15.0% in the MAC group and 85.0 ± 8.0% for the RIC group, supporting use of the latter.

Table 4. Diagnostic criteria for chronic active EBV (81).
HLH associated with malignancy
Classification and pathophysiology
HLH may be seen secondary to hematologic malignancies, which is a substantial challenge and major driver of HLH in adult patients (14) but observed in children more rarely (85). HLH may occur at diagnosis and be driven by the malignancy (M-HLH) or during chemotherapy (Ch-HLH). Ch-HLH or “on-therapy” HLH should raise high concern for a concomitant infection. Limited data on the frequency and management of malignancy associated HLH in pediatrics is available, although malignancy is estimated as the underlying etiology in approximately 5% of HLH cases (14). In one retrospective analysis of 29 pediatric and young adult patients, HLH was considered triggered by the malignancy in 21 patients with T- (n=12) or B-cell neoplasms (n=7), with EBV infection present as a co-trigger in 5 patients. The remaining 8 patients experienced HLH during chemotherapy (Ch-HLH) mainly for acute leukemias (86).
The diagnosis of M-HLH is frequently difficult to make, particularly when the underlying malignancy is yet undiagnosed. The diagnosis of M-HLH should be considered in a patient with a biopsy-proven malignancy who has clinical, and laboratory features consistent with HLH and no alternative cause of hyperinflammation. Different criteria and scoring systems have been used in adult patients, including the MD Anderson Criteria for M-HLH, H Score, and modification to the HLH-2004 criteria (87). Additionally, an Optimized HLH Inflammatory index (OHI) comprising soluble CD25 >3900 U/mL and Ferritin >1000ng/mL was developed to diagnose M-HLH more accurately and predict mortality in adult patients (88).
Medical treatment of malignancy associated HLH
Current treatment practices for M-HLH are largely based on expert opinion, given the absence of randomized clinical trials and heterogeneity of patient populations. Treatment is centered on the initiation of malignancy-directed therapy as soon as safely feasible and utilizing anti-inflammatory therapies to control hyperinflammation. In a pediatric cohort, the median overall survival (OS) was 1.2 years in M-HLH and 0.9 years in Ch-HLH (86). Given this was a small retrospective series, the superiority of prioritizing malignancy-directed vs. HLH-directed therapies could not be determined for the M-HLH cohort. In the Ch-HLH cohort, interventions ranged from postponement of chemotherapy to the use of etoposide-containing regimens (86). Adult M-HLH may be managed with an etoposide-based regimen that closely resembles the HLH-94 protocol, consisting of 8 weeks of treatment with dexamethasone 10mg/m2/d, subsequently tapered, or a dose-reduced regimen in patients with advanced age or significant comorbidities (89). A single-arm Phase 2 trial evaluated the safety and efficacy of a dose adjusted EPOCH regimen (Etoposide, Prednisolone, Vincristine, Cyclophosphamide, Doxorubicin) with or without rituximab in patients with previously untreated Non-Hodgkin Lymphoma-associated HLH to simultaneously address the HLH and underlying lymphoma. While this regimen led to an overall response rate (ORR) of 80.7% and 5-year OS of 73.1% in patients with B-cell lymphomas, those with T/NK lymphomas had dismal outcomes, with ORR of 13.8% and 1 year OS of 3.4% (90). A two-tiered approach of using etoposide and corticosteroids and, if needed, a salvage regimen of doxorubicin, etoposide, and methylprednisolone, to control the HLH with subsequent initiation of appropriate chemotherapy is frequently favored. Allogeneic HCT or cellular therapy in patients with malignancy associated HLH are employed only if this is indicated for the underlying malignancy.
HLH associated with IEC therapy
In contrast to HLH-syndromes in which an underlying predisposition and trigger need to be rapidly diagnosed and controlled, novel immunotherapeutic strategies purposefully trigger and employ the inflammatory capacities of the immune system to eradicate malignancies (91), and more recently, to address autoimmune conditions (92–94). IEC products can be generated by genetic modification of autologous or allogeneic T or NK cells to express Chimeric Antigen Receptors (CARs) that target cell-surface antigens. Similarly, genetically modified T cell receptors target intracellular antigens (TCR-T cells). With a growing number of FDA approved CAR T cell products and hundreds of active clinical trials for an expanding number of disease indications, we will focus on hyperinflammatory sequelae of CAR T cells below.
CRS
The earliest recognized hyperinflammatory complication of CAR T cell therapy was Cytokine Release Syndrome (CRS) (95). The hallmarks of CRS are fever, cardiopulmonary dysfunction, and other (hepatic, renal, gastrointestinal) organ dysfunction. Laboratory abnormalities include elevations in ferritin and IL-6, and coagulopathy, resembling those of HLH/MAS. CAR T cell activation leads to proliferation, secretion of proinflammatory cytokines, and activation of the host immune system including myeloid cells, thereby inducing an inflammatory feedback loop. In patients receiving 4-1BB costimulated CD19-CAR T cells, elevations in IFN-γ, IL-6, IL-8, sIL-2R, sgp130, MCP1, MIP1α, MIP1β, and GM-CSF as well as higher ferritin (>10,000) and CRP were significantly associated with severe (Grade 4-5) CRS compared to those with mild (Grade 0-3) CRS (96). Risk factors for the severity of CRS include cell dose, as well as recipient characteristics such as high tumor burden (97). Additionally, pre-infusion inflammation, peak expansion kinetics, and characteristics of the CAR itself are important (98) such as the transmembrane domain, costimulatory domain (e.g. CD28>41BB for CRS) (99) and affinity of the binder to target (less CRS with lower affinity binder) (100). Thus far, patients receiving CAR T cells for autoimmune indications have not been shown to be at increased risk for CRS, and rates and severity of CRS appear to be low (101). However, patients can experience localized inflammation in previously affected tissues, which is often mild and self resolves or resolves with steroid therapy (referred to as localized immune effector cell associated-toxicity syndrome or LICATS) (102).
The use of the monoclonal antibody tocilizumab revolutionized CRS management by interrupting the proinflammatory signaling cascade via IL-6 blockade without impacting CAR T cell efficacy (95, 103). While corticosteroids were initially avoided so as not to impair function and durability of CAR T cells, targeted use of corticosteroids during peak inflammation or prophylactically in high-risk patients has not been found to adversely impact efficacy (104).
ICANS
A related hyperinflammatory syndrome, IEC-associated neurotoxicity syndrome (ICANS), was recognized as a distinct entity that could overlap with CRS or occur following its resolution, and thus distinct grading systems and management algorithms were developed (105). Symptoms may range from mild confusion, headaches, tremors, and word finding difficulties to focal deficits, seizures, coma, and cerebral edema. Particularly in patients with prior CNS involvement or pre-existing neurological deficits, documentation of a patient’s baseline neurological exam and, if indicated, baseline MRI/imaging prior to CAR T administration is critical. Management guidelines suggest expeditious workup with neuroimaging, lumbar puncture, and EEG, in consultation with a Neurology team. Preclinical studies have identified IL-1 and IL-6 as key mediators in CRS and ICANS. However, while IL-6 blockade ameliorates CRS, only IL-1 blockade abrogates both CRS and ICANS (106–108). Initially treated supportively, management of ICANS currently includes corticosteroids, tocilizumab (if co-occurring with CRS, although there are concerns about enhancing IL-6 levels in the CSF due to non-CSF permeability of tocilizumab) and, if refractory, anakinra, intrathecal chemotherapy, and CSF pressure relief (109–111).
IEC-HS
Most recently, a separate entity, IEC-associated hemophagocytic lymphohistiocytosis-like syndrome (IEC-HS), was described following the experience with CD22-CAR T cell therapies (112). This syndrome has substantial overlap with the clinical and laboratory features of CRS and may be difficult to distinguish; however, it typically presents with delayed onset or following resolution of initial CRS and is not responsive to conventional CRS management with tocilizumab. Although the frequency was as high as 40.4% in a trial of 59 patients infused with CD22-CAR T cell products (112), other CD22-CAR T cell trials reported lower rates at 18.75% (113). A broad consensus definition (Table 5), grading system, and management recommendations have since been developed by the American Society for Transplantation and Cellular Therapy (114). With increasing recognition, it has also been described with other CAR T cell products such as those targeting CD19 (115) and in investigational CAR T cell products for solid tumors (116). Onset with resolving/resolved CRS or a worsening inflammatory response after initial improvement with CRS therapy is one of the most common manifestations of IEC-HS. Pre-infusion NK cell lymphopenia and higher bone marrow T cell:NK cell ratio may predispose to IEC-HS at least in the setting of CD22-CAR T cell therapy, possibly leading to a deficiency in regulating CD8+ T cell hyperactivation and CAR T cell contraction (112). Free IL-18 was differentially elevated in patients with CRS and IEC-HS compared to those with CRS alone (117) and may warrant investigation of agents targeting this pathway such as tadekinig alfa, a human recombinant IL-18-binding protein, which is not currently FDA approved. High pre-CAR T cell disease burden, a high level of pre-infusion inflammation based on ferritin and C-reactive protein, as well as lower platelet and neutrophil counts were associated with the development of IEC-HS in the context of the commercial CD19-CAR T cell product tisagenlecleucel (115). Germline genetic variants associated with HLH (118) and clonal hematopoiesis (119) may also contribute. Murine studies with perforin-deficient CAR T cells aimed at providing mechanistic insight, recapitulated elevated IL-1β and IL-18 levels and biphasic late CAR T cell expansion, which is characteristic of IEC-HS in humans (120). However, whether the pathophysiology underlying IEC-HS is distinct from or on the spectrum of CRS, and the identification of unifying risk factors, remains an area of active investigation. Similarly, the optimal therapeutic approach remains to be defined. Anakinra is recommended as first-line therapy given its favorable side-effect profile and short half-life allowing dose titration. Corticosteroids should be incorporated for moderate or higher disease severity or maintained in patients already receiving them for CRS. Given the importance of the IFN-γ signaling pathway in IEC-HS, as in other HLH-like conditions, ruxolitinib or emapalumab (used more frequently in pediatrics) are being employed for high grade/progressive IEC-HS (114, 121). Although the effect on durable CAR T cell function remains to be clarified in clinical studies, preclinical models predict that in hematologic malignancies, IFN-γ blockade can reduce macrophage activation without compromising CAR T cell function (122, 123). In contrast, IFN-γR signaling was shown to play a critical role in the susceptibility of solid tumors such as glioblastoma to CAR T cell mediated killing (124). This highlights the need for judicious use of these agents and further investigation to elucidate their impact mechanistically. Given the high rate of morbidity and mortality associated with IEC-HS, prompt diagnosis and aggressive management should be instituted.
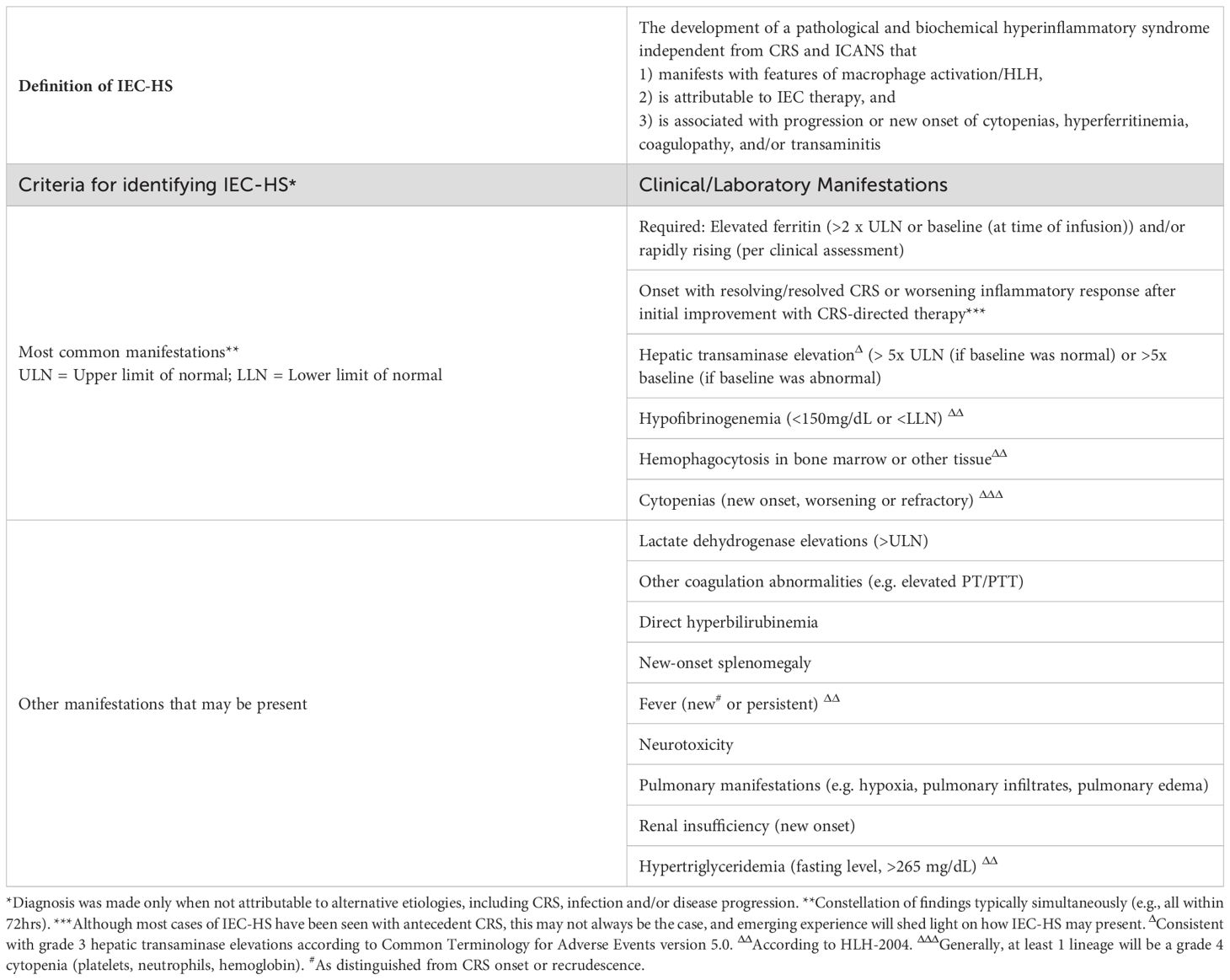
Table 5. Diagnostic criteria for IEC-associated HLH-like syndrome (114).
Diagnostic considerations in the critical care setting
Although dramatic advances have been made in the recognition, diagnostic workup, and treatment of HLH since the recognition of primary HLH in the 1950s and secondary HLH in the late 1970s, this life-threatening disorder remains underdiagnosed (125). A high degree of suspicion is required to expeditiously initiate diagnostic pathways for primary and secondary HLH syndromes and embark on appropriate treatment strategies, with consideration of the underlying etiology. All critically ill patients should have complete blood count testing, and multilineage cytopenias in a febrile patient should increase suspicion for HLH and prompt a ferritin to be sent if not already. Transaminitis, coagulopathy, and hepatosplenomegaly may also increase suspicion. If the ferritin is significantly elevated, this may trigger more specific markers of HLH to be sent such as CXCL9. The clinical context of fever and hyperinflammation is also important, as a patient with a history of sJIA, for example, is more likely to have sJIA-MAS, and a patient with symptoms of EBV may prompt consideration of EBV-HLH (vs. sepsis). Meanwhile, patients with positive blood cultures or suspicious sources for bacterial inoculation increase the relative suspicion for sepsis.
If initial workup raises suspicion for primary or secondary HLH, further diagnostic testing should be undertaken in consultation with rheumatologists, oncologists, immunologists, infectious disease specialists, neurologists (if any neurologic symptoms or deficits are present), and stem cell transplant/cell therapy physicians. Workup should extend beyond classic familial HLH or EBV susceptibility disorders, given that HLH may present as the first manifestation of a diverse spectrum of IEI, often with an unrecognized history of infections (126). Diagnostic workup and timely management should be rapidly instituted to prevent morbidity and mortality, even if diagnostic certainty is not yet achieved or the underlying genetic defect remains under investigation. Considerations for the multidisciplinary workup of HLH and HLH-like syndromes are summarized in Table 6.
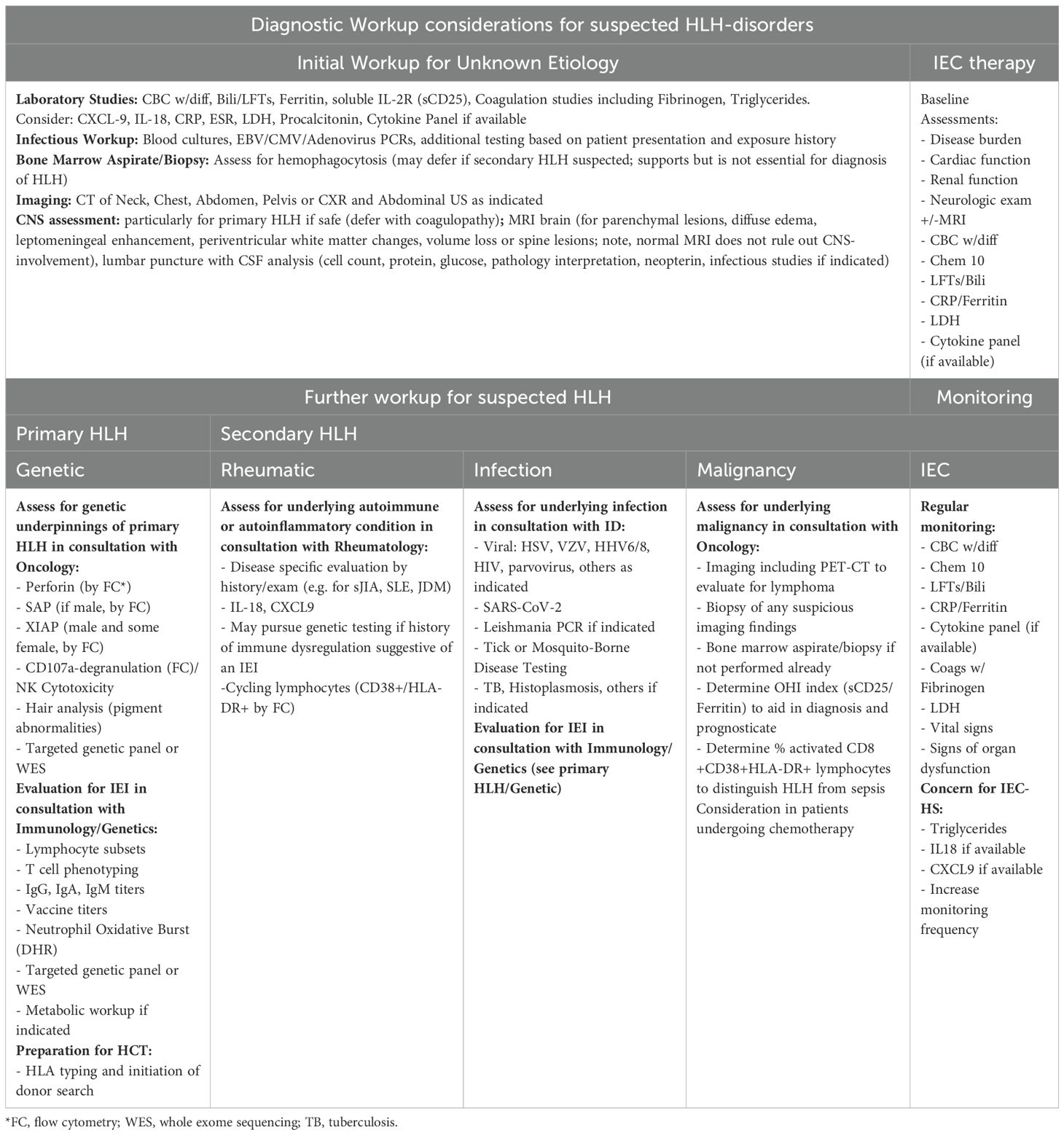
Table 6. Diagnostic considerations for suspected HLH-disorders.
Management considerations in the critical care setting
The overarching principles of treatment include gaining control of the hyperinflammatory feedback loop with immunosuppressive, cytokine-modulating, and chemotherapeutic agents (Figure 3, Table 7, Figure 4), providing supportive care, and treating the primary etiology as well as modifying factors as soon as safely feasible (127). Table 8 summarizes management considerations based on underlying etiology. In patients with EBV-driven HLH, EBV-directed therapy including rituximab to target CD20+ reservoirs of EBV should be employed. In patients with primary HLH, HLA-typing and donor workup should be undertaken to proceed to an allogeneic HCT once HLH is optimally controlled and HLH-directed therapy has been tapered to continuation therapy. Allogeneic HCT is also frequently indicated as the definitive therapy for IEIs presenting with HLH (often manifesting with an infectious trigger), CAEBV, and patients with recurrent MAS from sJIA, with similar goals of having controlled infection/inflammation at the time of transplant and potentially the use of reduced toxicity conditioning regimens to limit transplant related morbidity and mortality.
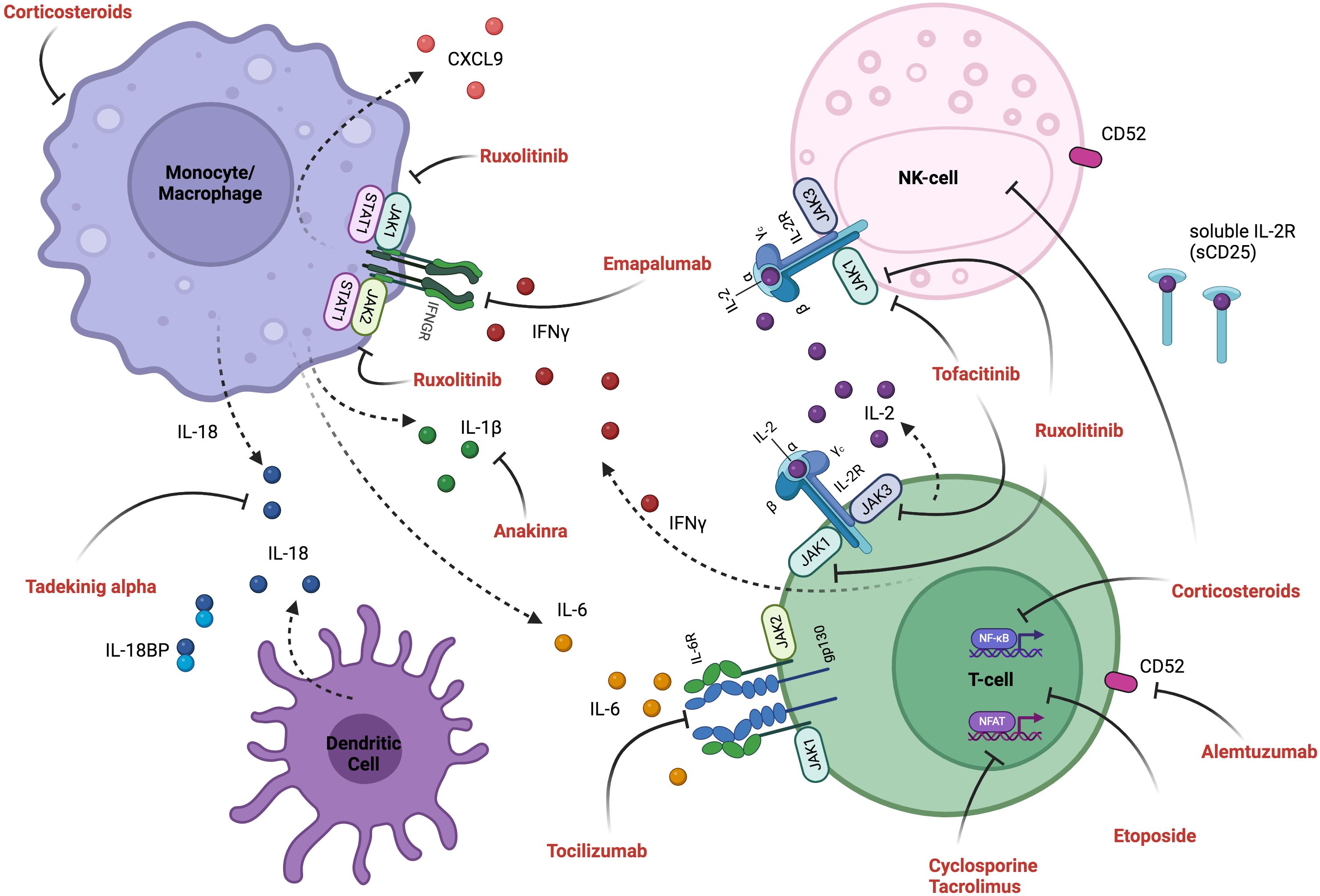
Figure 3. Inflammatory pathways and mechanism of therapeutic interventions. Activated T-cells produce pro-inflammatory cytokines (including IL-2, IFN-γ, TNF-α and GM-CSF), and their interaction with activated myeloid cells (producing IL-6, IL-18, IL-1β and CXCL9) induces an inflammatory feedback loop, which can be targeted therapeutically. See Figure 4 for detailed explanation of targeted therapeutics and Table 7 for cytokine descriptions. Created in BioRender.

Table 7. Cytokine signaling in HLH.
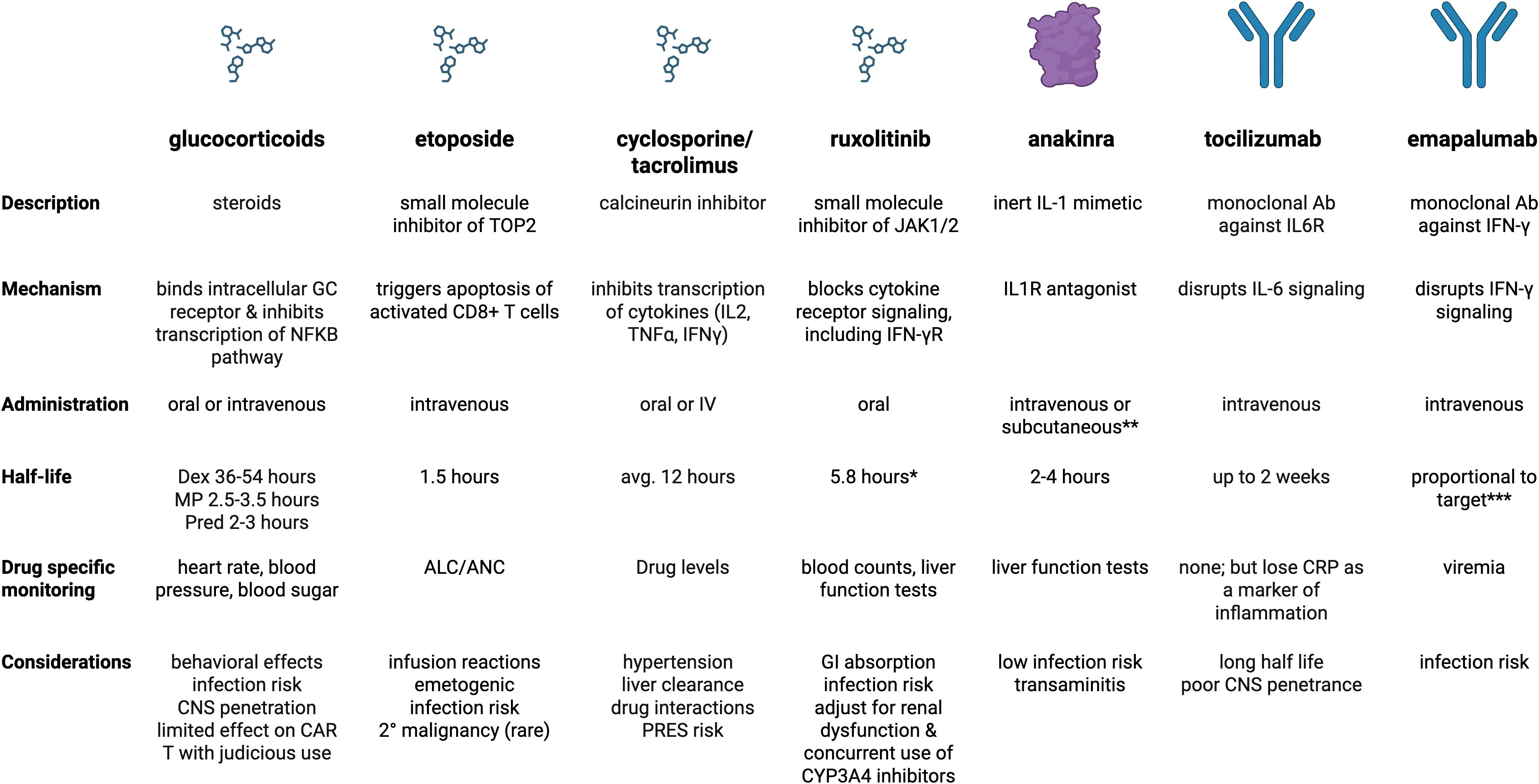
Figure 4. Summary of medications core to management of primary and secondary HLH. *includes ruxolitinib and active metabolites **subcutaneous form used more for outpatient management of sJIA ***clearance is proportional to total serum IFN-γ concentration when it is > 10,000 pg/mL (constant elimination if less). Created in BioRender. Dex, dexamethasone; MP, methylprednisolone; Pred, prednisone

Table 8. Management considerations for suspected or confirmed HLH-disorders.
In patients undergoing IEC therapies, standard institutional algorithms should be employed to monitor for the evolution of HLH-like toxicities. While the diagnosis and management of CRS and ICANS is distinct from the more recently described entity of IEC-HS, there is substantial overlap in presentation, diagnostic parameters, and management. CRS typically occurs more proximal to the IEC infusion and is managed with tocilizumab as the first-line agent, with addition of corticosteroids for high-grade persistent or recurrent symptoms. Anakinra and emapalumab, amongst other agents, have been used for refractory cases. The management of ICANS focuses on the use of corticosteroids, anakinra in refractory cases, and the addition of intrathecal hydrocortisone for severe, refractory situations. In contrast, IEC-HS timing is distinct and presents during improvement in CRS or after CRS resolution. IFN-γ plays a key role, and in addition to anakinra and/or corticosteroids, ruxolitinib or emapalumab may be employed.
Conclusion
In summary, HLH and HLH-like syndromes represent life-threatening entities in the pediatric setting and frequently require multidisciplinary critical care management with involvement by intensivists, rheumatology, immunology, oncology, infectious disease, and pediatric cell therapists. While the underlying etiology and the type and intensity of triggers required to produce the clinical HLH/HLH-like syndrome varies, these different entities ultimately converge to produce very similar clinical manifestations. Despite very distinct defects in the underlying immunologic pathophysiology, there is substantial overlap in the cytokines involved in the terminal pathways of the inflammatory feedback loop and the available therapeutics to mitigate the resulting toxicity. In this review, we aimed to outline differentiating features and common pathways and provide a framework for the management of these complex patients in the pediatric cell therapy and critical care setting.
Author contributions
HW: Conceptualization, Writing – original draft, Writing – review & editing. LH: Writing – review & editing. CD: Writing – review & editing. SP: Writing – review & editing. AR: Writing – review & editing. BD: Writing – review & editing. LL: Writing – review & editing. SB: Conceptualization, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
LH received salary support from CARRA. LH has received investigator-initiated research grants from BMS and consulting fees from Sobi and Pfizer. SP: Receives support for the conduct of clinical trials through Boston Children’s Hospital from Atara Biotherapeutics, Pierre Fabre and Cabaletta. Inventor of IP related to development of third party viral specific T cells program with all rights assigned to Memorial Sloan Kettering Cancer Center. Consulting Atara Biotherapeutics, Ensoma, HEOM, Pierre Fabre. DSMB Stanford University and NYBC. Equity share Regatta Biotherapy. AR has been paid as a scientific advisor to Thermo Fisher and Inotrem, Inc. She receives royalties from UpToDate Inc for editorial duties. SB's spouse is a former employee of Takeda and holds Takeda and AstraZeneca stock and stock options.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr. (1983) 140:221–30. doi: 10.1007/BF00443367
2. Jordan MB. Hemophagocytic lymphohistiocytosis: A disorder of T cell activation, immune regulation, and distinctive immunopathology. Immunol Rev. (2024) 322:339–50. doi: 10.1111/imr.13298
3. Nigrovic PA. Macrophage activation syndrome. Arthritis Rheumatol. (2024) 77(4):367–79. doi: 10.1002/art.43052
4. Brisse E, Wouters CH, and Matthys P. Advances in the pathogenesis of primary and secondary haemophagocytic lymphohistiocytosis: differences and similarities. Br J Haematol. (2016) 174:203–17. doi: 10.1111/bjh.14147
5. Nguyen TH, Kumar D, Prince C, Martini D, Grunwell JR, Lawrence T, et al. Frequency of HLA-DR(+)CD38(hi) T cells identifies and quantifies T-cell activation in hemophagocytic lymphohistiocytosis, hyperinflammation, and immune regulatory disorders. J Allergy Clin Immunol. (2024) 153:309–19. doi: 10.1016/j.jaci.2023.07.008
6. Huang Z, Brodeur KE, Chen L, Du Y, Wobma H, Hsu EE, et al. Type I interferon signature and cycling lymphocytes in macrophage activation syndrome. J Clin Invest. (2023) 133(22):e165616. doi: 10.1172/JCI165616
7. Chaturvedi V, Marsh RA, Zoref-Lorenz A, Owsley E, Chaturvedi V, Nguyen TC, et al. T-cell activation profiles distinguish hemophagocytic lymphohistiocytosis and early sepsis. Blood. (2021) 137:2337–46. doi: 10.1182/blood.2020009499
8. Jordan MB, Hildeman D, Kappler J, and Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. (2004) 104:735–43. doi: 10.1182/blood-2003-10-3413
9. Farquhar JW and Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child. (1952) 27:519–25. doi: 10.1136/adc.27.136.519
10. Henter JI, Samuelsson-Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. (2002) 100:2367–73. doi: 10.1182/blood-2002-01-0172
11. Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48:124–31. doi: 10.1002/pbc.21039
12. Henter JI, Sieni E, Eriksson J, Bergsten E, Hed Myrberg I, Canna SW, et al. Diagnostic guidelines for familial hemophagocytic lymphohistiocytosis revisited. Blood. (2024) 144:2308–18. doi: 10.1182/blood.2024025077
13. Lee PY, Schulert GS, Canna SW, Huang Y, Sundel J, Li Y, et al. Adenosine deaminase 2 as a biomarker of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Ann Rheum Dis. (2020) 79:225–31. doi: 10.1136/annrheumdis-2019-216030
14. Shakoory B, Geerlinks A, Wilejto M, Kernan K, Hines M, Romano M, et al. The 2022 EULAR/ACR points to consider at the early stages of diagnosis and management of suspected haemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS). Arthritis Rheumatol. (2023) 75:1714–32. doi: 10.1002/art.42636
15. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. (1999) 286:1957–9. doi: 10.1126/science.286.5446.1957
16. Gadoury-Levesque V, Dong L, Su R, Chen J, Zhang K, Risma KA, et al. Frequency and spectrum of disease-causing variants in 1892 patients with suspected genetic HLH disorders. Blood Adv. (2020) 4:2578–94. doi: 10.1182/bloodadvances.2020001605
17. Canna SW and Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. (2020) 135:1332–43. doi: 10.1182/blood.2019000936
18. Lam MT, Coppola S, Krumbach OHF, Prencipe G, Insalaco A, Cifaldi C, et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J Exp Med. (2019) 216:2778–99. doi: 10.1084/jem.20190147
19. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. (2014) 46:1140–6. doi: 10.1038/ng.3089
20. Guo H, Callaway JB, and Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
21. Rigaud S, Fondaneche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. (2006) 444:110–4. doi: 10.1038/nature05257
22. Ambruso DR, Hays T, Zwartjes WJ, Tubergen DG, and Favara BE. Successful treatment of lymphohistiocytic reticulosis with phagocytosis with epipodophyllotoxin VP 16-213. Cancer. (1980) 45:2516–20. doi: 10.1002/1097-0142(19800515)45:10<2516::AID-CNCR2820451008>3.0.CO;2-V
23. Johnson TS, Terrell CE, Millen SH, Katz JD, Hildeman DA, and Jordan MB. Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J Immunol. (2014) 192:84–91. doi: 10.4049/jimmunol.1302282
24. Horne A, Wickstrom R, Jordan MB, Yeh EA, Naqvi A, Henter JI, et al. How to treat involvement of the central nervous system in hemophagocytic lymphohistiocytosis? Curr Treat Options Neurol. (2017) 19:3. doi: 10.1007/s11940-017-0439-4
25. Trottestam H, Horne A, Arico M, Egeler RM, Filipovich AH, Gadner H, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. (2011) 118:4577–84. doi: 10.1182/blood-2011-06-356261
26. Ehl S, Astigarraga I, von Bahr Greenwood T, Hines M, Horne A, Ishii E, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH steering committee of the histiocyte society. J Allergy Clin Immunol Pract. (2018) 6:1508–17. doi: 10.1016/j.jaip.2018.05.031
27. Jordan MB, Allen CE, Weitzman S, Filipovich AH, and McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. (2011) 118:4041–52. doi: 10.1182/blood-2011-03-278127
28. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. (2020) 382:1811–22. doi: 10.1056/NEJMoa1911326
29. Zhang Q, Zhao YZ, Ma HH, Wang D, Cui L, Li WJ, et al. A study of ruxolitinib response-based stratified treatment for pediatric hemophagocytic lymphohistiocytosis. Blood. (2022) 139:3493–504. doi: 10.1182/blood.2021014860
30. Verkamp B, Jodele S, Sabulski A, Marsh R, Kieser P, and Jordan MB. Emapalumab therapy for hemophagocytic lymphohistiocytosis before reduced-intensity transplantation improves chimerism. Blood. (2024) 144:2625–36. doi: 10.1182/blood.2024025977
31. Hartz B, Marsh R, Rao K, Henter JI, Jordan M, Filipovich L, et al. The minimum required level of donor chimerism in hereditary hemophagocytic lymphohistiocytosis. Blood. (2016) 127:3281–90. doi: 10.1182/blood-2015-12-684498
32. Marsh RA, Hebert K, Kim S, Dvorak CC, Aquino VM, Baker KS, et al. Comparison of hematopoietic cell transplant conditioning regimens for hemophagocytic lymphohistiocytosis disorders. J Allergy Clin Immunol. (2022) 149:1097–104.e2. doi: 10.1016/j.jaci.2021.07.031
33. Blincoe A, Heeg M, Campbell PK, Hines M, Khojah A, Klein-Gitelman M, et al. Neuroinflammatory disease as an isolated manifestation of hemophagocytic lymphohistiocytosis. J Clin Immunol. (2020) 40:901–16. doi: 10.1007/s10875-020-00814-6
34. Benson LA, Li H, Henderson LA, Solomon IH, Soldatos A, Murphy J, et al. Pediatric CNS-isolated hemophagocytic lymphohistiocytosis. Neurol Neuroimmunol Neuroinflamm. (2019) 6:e560. doi: 10.1212/NXI.0000000000000560
35. Li H, Benson LA, Henderson LA, Solomon IH, Kennedy AL, Soldatos A, et al. Central nervous system-restricted familial hemophagocytic lymphohistiocytosis responds to hematopoietic cell transplantation. Blood Adv. (2019) 3:503–7. doi: 10.1182/bloodadvances.2018027417
36. Shimizu M. Macrophage activation syndrome in systemic juvenile idiopathic arthritis. Immunol Med. (2021) 44:237–45. doi: 10.1080/25785826.2021.1912893
37. Pascual V, Allantaz F, Arce E, Punaro M, and Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. (2005) 201:1479–86. doi: 10.1084/jem.20050473
38. Takahara T, Shimizu M, Nakagishi Y, Kinjo N, and Yachie A. Serum IL-18 as a potential specific marker for differentiating systemic juvenile idiopathic arthritis from incomplete Kawasaki disease. Rheumatol Int. (2015) 35:81–4. doi: 10.1007/s00296-014-3059-2
39. Put K, Avau A, Brisse E, Mitera T, Put S, Proost P, et al. Cytokines in systemic juvenile idiopathic arthritis and haemophagocytic lymphohistiocytosis: tipping the balance between interleukin-18 and interferon-gamma. Rheumatol (Oxford). (2015) 54:1507–17. doi: 10.1093/rheumatology/keu524
40. Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. (2004) 31:390–2.
41. Nigrovic PA. Review: is there a window of opportunity for treatment of systemic juvenile idiopathic arthritis? Arthritis Rheumatol. (2014) 66:1405–13. doi: 10.1002/art.38615.
42. Ravelli A, Minoia F, Davi S, Horne A, Bovis F, Pistorio A, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: A european league against rheumatism/American college of rheumatology/Paediatric rheumatology international trials organisation collaborative initiative. Arthritis Rheumatol. (2016) 68:566–76. doi: 10.1002/art.39332
43. Shimizu M, Nakagishi Y, Inoue N, Mizuta M, Ko G, Saikawa Y, et al. Interleukin-18 for predicting the development of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Clin Immunol. (2015) 160:277–81. doi: 10.1016/j.clim.2015.06.005
44. Huang Z, You X, Chen L, Du Y, Brodeur K, Jee H, et al. mTORC1 links pathology in experimental models of Still’s disease and macrophage activation syndrome. Nat Commun. (2022) 13:6915. doi: 10.1038/s41467-022-34480-6
45. Correia Marques M, Rubin D, Shuldiner EG, Datta M, Schmitz E, Gutierrez Cruz G, et al. Enrichment of rare variants of hemophagocytic lymphohistiocytosis genes in systemic juvenile idiopathic arthritis. Arthritis Rheumatol. (2024) 76:1566–72. doi: 10.1101/2024.03.13.24304215
46. Onel KB, Horton DB, Lovell DJ, Shenoi S, Cuello CA, Angeles-Han ST, et al. 2021 American college of rheumatology guideline for the treatment of juvenile idiopathic arthritis: therapeutic approaches for oligoarthritis, temporomandibular joint arthritis, and systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken). (2022) 74:521–37. doi: 10.1002/acr.24853
47. Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. (2011) 70:747–54. doi: 10.1136/ard.2010.134254
48. Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. (2012) 367:2396–406. doi: 10.1056/NEJMoa1205099
49. De Benedetti F, Brunner HI, Ruperto N, Kenwright A, Wright S, Calvo I, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. (2012) 367:2385–95. doi: 10.1056/NEJMoa1112802
50. Onel KB, Horton DB, Lovell DJ, Shenoi S, Cuello CA, Angeles-Han ST, et al. 2021 American college of rheumatology guideline for the treatment of juvenile idiopathic arthritis: therapeutic approaches for oligoarthritis, temporomandibular joint arthritis, and systemic juvenile idiopathic arthritis. Arthritis Rheumatol. (2022) 74:553–69. doi: 10.1002/art.42037
51. Gillard L, Pouchot J, Cohen-Aubart F, Kone-Paut I, Mouterde G, Michaud M, et al. JAK inhibitors in difficult-to-treat adult-onset Still’s disease and systemic-onset juvenile idiopathic arthritis. Rheumatol (Oxford). (2023) 62:1594–604. doi: 10.1093/rheumatology/keac440
52. Fang Z, Wang D, Ge J, Zhao Y, Lian H, Ma H, et al. Ruxolitinib-based regimen in children with autoimmune disease or autoinflammatory disease-related haemophagocytic lymphohistiocytosis. Br J Haematol. (2025) 206:215–23. doi: 10.1111/bjh.19803
53. Garlanda C, Riva F, Bonavita E, Gentile S, and Mantovani A. Decoys and regulatory “Receptors” of the IL-1/toll-like receptor superfamily. Front Immunol. (2013) 4:180. doi: 10.3389/fimmu.2013.00180
54. Taylor ML, Hoyt KJ, Han J, Benson L, Case S, Chandler MT, et al. An evidence-based guideline improves outcomes for patients with hemophagocytic lymphohistiocytosis and macrophage activation syndrome. J Rheumatol. (2022) 49:1042–51. doi: 10.3899/jrheum.211219
55. Halyabar O, Chang MH, Schoettler ML, Schwartz MA, Baris EH, Benson LA, et al. Calm in the midst of cytokine storm: a collaborative approach to the diagnosis and treatment of hemophagocytic lymphohistiocytosis and macrophage activation syndrome. Pediatr Rheumatol Online J. (2019) 17:7. doi: 10.1186/s12969-019-0309-6
56. De Benedetti F, Grom AA, Brogan PA, Bracaglia C, Pardeo M, Marucci G, et al. Efficacy and safety of emapalumab in macrophage activation syndrome. Ann Rheum Dis. (2023) 82:857–65. doi: 10.1136/ard-2022-223739
57. Baldo F, Erkens RGA, Mizuta M, Rogani G, Lucioni F, Bracaglia C, et al. Current treatment in macrophage activation syndrome worldwide: a systematic literature review to inform the METAPHOR project. Rheumatol (Oxford). (2025) 64:32–44. doi: 10.1093/rheumatology/keae391
58. Horne A, von Bahr Greenwood T, Chiang SCC, Meeths M, Bjorklund C, Ekelund M, et al. Efficacy of moderately dosed etoposide in macrophage activation syndrome-hemophagocytic lymphohistiocytosis. J Rheumatol. (2021) 48:1596–602. doi: 10.3899/jrheum.200941
59. Brinkman DM, de Kleer IM, ten Cate R, van Rossum MA, Bekkering WP, Fasth A, et al. Autologous stem cell transplantation in children with severe progressive systemic or polyarticular juvenile idiopathic arthritis: long-term follow-up of a prospective clinical trial. Arthritis Rheumatol. (2007) 56:2410–21. doi: 10.1002/art.22656
60. De Kleer IM, Brinkman DM, Ferster A, Abinun M, Quartier P, van der Net J, et al. Autologous stem cell transplantation for refractory juvenile idiopathic arthritis: analysis of clinical effects, mortality, and transplant related morbidity. Ann Rheum Dis. (2004) 63:1318–26. doi: 10.1136/ard.2003.017798
61. Saper VE, Chen G, Deutsch GH, Guillerman RP, Birgmeier J, Jagadeesh K, et al. Emergent high fatality lung disease in systemic juvenile arthritis. Ann Rheum Dis. (2019) 78:1722–31. doi: 10.1136/annrheumdis-2019-216040
62. Huang Y, Sompii-Montgomery L, Patti J, Pickering A, Yasin S, Do T, et al. Disease course, treatments, and outcomes of children with systemic juvenile idiopathic arthritis-associated lung disease. Arthritis Care Res (Hoboken). (2024) 76:328–39. doi: 10.1002/acr.25234
63. Schulert GS, Yasin S, Carey B, Chalk C, Do T, Schapiro AH, et al. Systemic juvenile idiopathic arthritis-associated lung disease: characterization and risk factors. Arthritis Rheumatol. (2019) 71:1943–54. doi: 10.1002/art.41073
64. Wobma H, Bachrach R, Farrell J, Chang MH, Day-Lewis M, Dedeoglu F, et al. Development of a screening algorithm for lung disease in systemic juvenile idiopathic arthritis. ACR Open Rheumatol. (2023) 5:556–62. doi: 10.1002/acr2.11600
65. Wobma H, Arvila SR, Taylor ML, Lam KP, Ohashi M, Gebhart C, et al. Incidence and risk factors for eosinophilia and lung disease in biologic-exposed children with systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken). (2023) 75:2063–72. doi: 10.1002/acr.25129
66. Aires BP, Wobma H, Samad A, Chandler MT, Chang MH, Dedeoglu F, et al. Severe features of systemic juvenile idiopathic arthritis in patients with congenital heart disease. J Rheumatol. (2024) 51:811–7. doi: 10.3899/jrheum.2024-0180
67. Gao DK, Salomonis N, Henderlight M, Woods C, Thakkar K, Grom AA, et al. IFN-gamma is essential for alveolar macrophage-driven pulmonary inflammation in macrophage activation syndrome. JCI Insight. (2021) 6(17):e147593. doi: 10.1172/jci.insight.147593
68. Araya P, Waugh KA, Sullivan KD, Nunez NG, Roselli E, Smith KP, et al. Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc Natl Acad Sci U S A. (2019) 116:24231–41. doi: 10.1073/pnas.1908129116
69. MFS J, Ladomenou F, Carpenter B, Chandra S, Sedlacek P, Formankova R, et al. Allogeneic hematopoietic stem cell transplantation for severe, refractory juvenile idiopathic arthritis. Blood Adv. (2018) 2:777–86. doi: 10.1182/bloodadvances.2017014449
70. Matt MG, Drozdov D, Bendstrup E, Glerup M, Hauge EM, Masmas T, et al. Allogeneic haematopoietic stem-cell transplantation for children with refractory systemic juvenile idiopathic arthritis and associated lung disease: outcomes from an international, retrospective cohort study. Lancet Rheumatol. (2024) 7(4):e243–51. doi: 10.1016/S2665-9913(24)00275-3
71. Dunmire SK, Verghese PS, and Balfour HH Jr. Primary Epstein-Barr virus infection. J Clin Virol. (2018) 102:84–92. doi: 10.1016/j.jcv.2018.03.001
72. Liu M, Brodeur KE, Bledsoe JR, Harris CN, Joerger J, Weng R, et al. Features of hyperinflammation link the biology of Epstein-Barr virus infection and cytokine storm syndromes. J Allergy Clin Immunol. (2024) 155(4):1346–56.e9. doi: 10.1016/j.jaci.2024.11.029
73. Chellapandian D, Das R, Zelley K, Wiener SJ, Zhao H, Teachey DT, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol. (2013) 162:376–82. doi: 10.1111/bjh.12386
74. Fisher CJ Jr., Slotman GJ, Opal SM, Pribble JP, Bone RC, Emmanuel G, et al. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: a randomized, open-label, placebo-controlled multicenter trial. Crit Care Med. (1994) 22:12–21. doi: 10.1097/00003246-199401000-00008
75. Fisher CJ Jr., Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. (1994) 271:1836–43. doi: 10.1001/jama.1994.03510470040032
76. Eloseily EM, Weiser P, Crayne CB, Haines H, Mannion ML, Stoll ML, et al. Benefit of anakinra in treating pediatric secondary hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. (2020) 72:326–34. doi: 10.1002/art.41103
77. Zhu W, Zhou F, Song Y, Zhou S, Du F, Zhu Q, et al. Low-dose emapalumab combined with chemotherapy for adult patients with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Transpl Immunol. (2024) 88:102162. doi: 10.1016/j.trim.2024.102162
78. Wang W, Yuan X, Yu L, and Pei F. Emapalumab as a therapeutic intervention for Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis: A case series. Med (Baltimore). (2024) 103:e39880. doi: 10.1097/MD.0000000000039880
79. Marsh RA. Epstein-barr virus and hemophagocytic lymphohistiocytosis. Front Immunol. (2017) 8:1902. doi: 10.3389/fimmu.2017.01902
80. Wang J, Su M, Wei N, Yan H, Zhang J, Gong Y, et al. Chronic active Epstein-Barr virus disease originates from infected hematopoietic stem cells. Blood. (2024) 143:32–41. doi: 10.1182/blood.2023021074
81. Kawada JI, Ito Y, Ohshima K, Yamada M, Kataoka S, Muramatsu H, et al. Updated guidelines for chronic active Epstein-Barr virus disease. Int J Hematol. (2023) 118:568–76. doi: 10.1007/s12185-023-03660-5
82. Chen R, Lin Q, Zhu Y, Shen Y, Xu Q, Tang H, et al. Sintilimab treatment for chronic active Epstein-Barr virus infection and Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in children. Orphanet J Rare Dis. (2023) 18:297. doi: 10.1186/s13023-023-02861-9
83. Yamamoto M, Sato M, Onishi Y, Sasahara Y, Sano H, Masuko M, et al. Registry data analysis of hematopoietic stem cell transplantation on systemic chronic active Epstein-Barr virus infection patients in Japan. Am J Hematol. (2022) 97:780–90. doi: 10.1002/ajh.26544
84. Kawa K, Sawada A, Sato M, Okamura T, Sakata N, Kondo O, et al. Excellent outcome of allogeneic hematopoietic SCT with reduced-intensity conditioning for the treatment of chronic active EBV infection. Bone Marrow Transplant. (2011) 46:77–83. doi: 10.1038/bmt.2010.122
85. Lofstedt A, Jadersten M, Meeths M, and Henter JI. Malignancy-associated hemophagocytic lymphohistiocytosis in Sweden: incidence, clinical characteristics, and survival. Blood. (2024) 143:233–42. doi: 10.1182/blood.2023020715
86. Lehmberg K, Sprekels B, Nichols KE, Woessmann W, Muller I, Suttorp M, et al. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br J Haematol. (2015) 170:539–49. doi: 10.1111/bjh.13462
87. Tamamyan GN, Kantarjian HM, Ning J, Jain P, Sasaki K, McClain KL, et al. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: Relation to hemophagocytosis, characteristics, and outcomes. Cancer. (2016) 122:2857–66. doi: 10.1002/cncr.30084
88. Zoref-Lorenz A, Murakami J, Hofstetter L, Iyer S, Alotaibi AS, Mohamed SF, et al. An improved index for diagnosis and mortality prediction in Malignancy-associated hemophagocytic lymphohistiocytosis. Blood. (2022) 139:1098–110. doi: 10.1182/blood.2021012764
89. La Rosee P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. (2019) 133:2465–77. doi: 10.1182/blood.2018894618
90. Liang JH, Wang L, Zhu HY, Qian J, Liao H, Wu JZ, et al. Dose-adjusted EPOCH regimen as first-line treatment for non-Hodgkin lymphoma-associated hemophagocytic lymphohistiocytosis: a single-arm, open-label, phase II trial. Haematologica. (2020) 105:e29–32. doi: 10.3324/haematol.2019.220301
91. Cappell KM and Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. (2023) 20:359–71. doi: 10.1038/s41571-023-00754-1
92. Mougiakakos D, Kronke G, Volkl S, Kretschmann S, Aigner M, Kharboutli S, et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med. (2021) 385:567–9. doi: 10.1056/NEJMc2107725
93. Mackensen A, Muller F, Mougiakakos D, Boltz S, Wilhelm A, Aigner M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. (2022) 28:2124–32. doi: 10.1038/s41591-022-02017-5
94. Wobma H, Chang JC, and Prockop SE. Releasing our model T - chimeric antigen receptor (CAR) T-cells for autoimmune indications. Curr Opin Rheumatol. (2024) 37(2):128–35. doi: 10.1097/BOR.0000000000001062
95. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. (2014) 124:188–95. doi: 10.1182/blood-2014-05-552729
96. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. (2016) 6:664–79. doi: 10.1158/2159-8290.CD-16-0040
97. Locke FL, Rossi JM, Neelapu SS, Jacobson CA, Miklos DB, Ghobadi A, et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. (2020) 4:4898–911. doi: 10.1182/bloodadvances.2020002394
98. Brudno JN and Kochenderfer JN. Current understanding and management of CAR T cell-associated toxicities. Nat Rev Clin Oncol. (2024) 21:501–21. doi: 10.1038/s41571-024-00903-0
99. Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med. (2020) 26:270–80. doi: 10.1038/s41591-019-0737-3
100. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. (2019) 25:1408–14. doi: 10.1038/s41591-019-0549-5
101. Vanni KMM, McCarter KR, Wang X, Duffy C, Cruz JPD, Wobma H, et al. Safety of CAR T-cell therapy for cancer in pre-existing autoimmune or inflammatory disease: a retrospective comparative cohort study. Lancet Rheumatol. (2025) 7:e226–e9. doi: 10.1016/S2665-9913(24)00402-8
102. Hagen M, Muller F, Wirsching A, Kharboutli S, Spoerl S, Dusing C, et al. Local immune effector cell-associated toxicity syndrome in CAR T-cell treated patients with autoimmune disease: an observational study. Lancet Rheumatol. (2025) 7(6):e424–33. doi: 10.1016/S2665-9913(25)00091-8
103. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
104. Gardner RA, Ceppi F, Rivers J, Annesley C, Summers C, Taraseviciute A, et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood. (2019) 134:2149–58. doi: 10.1182/blood.2019001463
105. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. (2019) 25:625–38. doi: 10.1016/j.bbmt.2018.12.758
106. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, and Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. (2018) 24:731–8. doi: 10.1038/s41591-018-0041-7
107. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
108. Morris EC, Neelapu SS, Giavridis T, and Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. (2022) 22:85–96. doi: 10.1038/s41577-021-00547-6
109. Santomasso BD, Nastoupil LJ, Adkins S, Lacchetti C, Schneider BJ, Anadkat M, et al. Management of immune-related adverse events in patients treated with chimeric antigen receptor T-cell therapy: ASCO guideline. J Clin Oncol. (2021) 39:3978–92. doi: 10.1200/JCO.21.01992
110. Ragoonanan D, Khazal SJ, Abdel-Azim H, McCall D, Cuglievan B, Tambaro FP, et al. Diagnosis, grading and management of toxicities from immunotherapies in children, adolescents and young adults with cancer. Nat Rev Clin Oncol. (2021) 18:435–53. doi: 10.1038/s41571-021-00474-4
111. Diorio C, Vatsayan A, Talleur AC, Annesley C, Jaroscak JJ, Shalabi H, et al. Anakinra utilization in refractory pediatric CAR T-cell associated toxicities. Blood Adv. (2022) 6:3398–403. doi: 10.1182/bloodadvances.2022006983
112. Lichtenstein DA, Schischlik F, Shao L, Steinberg SM, Yates B, Wang HW, et al. Characterization of HLH-like manifestations as a CRS variant in patients receiving CD22 CAR T cells. Blood. (2021) 138:2469–84. doi: 10.1182/blood.2021011898
113. Schultz LM, Jeyakumar N, Kramer AM, Sahaf B, Srinagesh H, Shiraz P, et al. CD22 CAR T cells demonstrate high response rates and safety in pediatric and adult B-ALL: Phase 1b results. Leukemia. (2024) 38:963–8. doi: 10.1038/s41375-024-02220-y
114. Hines MR, Knight TE, McNerney KO, Leick MB, Jain T, Ahmed S, et al. Immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome. Transplant Cell Ther. (2023) 29:438.e1– e16. doi: 10.1016/j.jtct.2023.03.006
115. McNerney KO, Si Lim SJ, Ishikawa K, Dreyzin A, Vatsayan A, Chen JJ, et al. HLH-like toxicities predict poor survival after the use of tisagenlecleucel in children and young adults with B-ALL. Blood Adv. (2023) 7:2758–71. doi: 10.1182/bloodadvances.2022008893
116. Ruemmele T, Macedo R, Stein MN, Chan HT, Mapara MY, Jacquemont CF, et al. Emapalumab for severe cytokine release syndrome in solid tumor CAR-T: a case report. Front Oncol. (2025) 15:1543622. doi: 10.3389/fonc.2025.1543622
117. Rocco JM, Inglefield J, Yates B, Lichtenstein DA, Wang Y, Goffin L, et al. Free interleukin-18 is elevated in CD22 CAR T-cell-associated hemophagocytic lymphohistiocytosis-like toxicities. Blood Adv. (2023) 7:6134–9. doi: 10.1182/bloodadvances.2023010708
118. Miller PG, Niroula A, Ceremsak JJ, Gibson CJ, Taylor MS, Birndt S, et al. Identification of germline variants in adults with hemophagocytic lymphohistiocytosis. Blood Adv. (2020) 4:925–9. doi: 10.1182/bloodadvances.2019001272
119. Miller PG, Sperling AS, Gibson CJ, Viswanathan K, Castellano C, McConkey M, et al. Contribution of clonal hematopoiesis to adult-onset hemophagocytic lymphohistiocytosis. Blood. (2020) 136:3051–5. doi: 10.1182/blood.2020008206
120. Ishii K, Pouzolles M, Chien CD, Erwin-Cohen RA, Kohler ME, Qin H, et al. Perforin-deficient CAR T cells recapitulate late-onset inflammatory toxicities observed in patients. J Clin Invest. (2020) 130:5425–43. doi: 10.1172/JCI130059
121. Schuelke MR, Bassiri H, Behrens EM, Canna S, Croy C, DiNofia A, et al. Emapalumab for the treatment of refractory cytokine release syndrome in pediatric patients. Blood Adv. (2023) 7:5603–7. doi: 10.1182/bloodadvances.2023010712
122. Manni S, Del Bufalo F, Merli P, Silvestris DA, Guercio M, Caruso S, et al. Neutralizing IFNgamma improves safety without compromising efficacy of CAR-T cell therapy in B-cell Malignancies. Nat Commun. (2023) 14:3423. doi: 10.1038/s41467-023-38723-y
123. Bailey SR, Vatsa S, Larson RC, Bouffard AA, Scarfo I, Kann MC, et al. Blockade or deletion of IFNgamma reduces macrophage activation without compromising CAR T-cell function in hematologic Malignancies. Blood Cancer Discov. (2022) 3:136–53. doi: 10.1158/2643-3230.BCD-21-0181
124. Larson RC, Kann MC, Bailey SR, Haradhvala NJ, Llopis PM, Bouffard AA, et al. CAR T cell killing requires the IFNgammaR pathway in solid but not liquid tumours. Nature. (2022) 604:563–70. doi: 10.1038/s41586-022-04585-5
125. Henter JI. Hemophagocytic lymphohistiocytosis. N Engl J Med. (2025) 392:584–98. doi: 10.1056/NEJMra2314005
126. Ricci S, Sarli WM, Lodi L, Canessa C, Lippi F, Dini D, et al. HLH as an additional warning sign of inborn errors of immunity beyond familial-HLH in children: a systematic review. Front Immunol. (2024) 15:1282804. doi: 10.3389/fimmu.2024.1282804
127. Hines MR, von Bahr Greenwood T, Beutel G, Beutel K, Hays JA, Horne A, et al. Consensus-based guidelines for the recognition, diagnosis, and management of hemophagocytic lymphohistiocytosis in critically ill children and adults. Crit Care Med. (2022) 50:860–72. doi: 10.1097/CCM.0000000000005361
128. Matsuda-Lennikov M, Biancalana M, Zou J, Ravell JC, Zheng L, Kanellopoulou C, et al. Magnesium transporter 1 (MAGT1) deficiency causes selective defects in N-linked glycosylation and expression of immune-response genes. J Biol Chem. (2019) 294:13638–56. doi: 10.1074/jbc.RA119.008903
129. Izawa K, Martin E, Soudais C, Bruneau J, Boutboul D, Rodriguez R, et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J Exp Med. (2017) 214:73–89. doi: 10.1084/jem.20160784
130. Martin E, Palmic N, Sanquer S, Lenoir C, Hauck F, Mongellaz C, et al. CTP synthase 1 deficiency in humans reveals its central role in lymphocyte proliferation. Nature. (2014) 510:288–92. doi: 10.1038/nature13386
131. Salzer E, Cagdas D, Hons M, Mace EM, Garncarz W, Petronczki OY, et al. RASGRP1 deficiency causes immunodeficiency with impaired cytoskeletal dynamics. Nat Immunol. (2016) 17:1352–60. doi: 10.1038/ni.3575
132. Aziz Bousfiha A, Jeddane L, Moundir A, Cecilia Poli M, Aksentijevich I, Cunningham-Rundles C, et al. The 2024 update of IUIS phenotypic classification of human inborn errors of immunity. J Hum Immunity. (2025) 1:e20250002. doi: 10.70962/jhi.20250002
133. Spolski R, Li P, and Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol. (2018) 18:648–59. doi: 10.1038/s41577-018-0046-y
134. Paliard X, de Waal Malefijt R, Yssel H, Blanchard D, Chretien I, Abrams J, et al. Simultaneous production of IL-2, IL-4, and IFN-gamma by activated human CD4+ and CD8+ T cell clones. J Immunol. (1988) 141:849–55. doi: 10.4049/jimmunol.141.3.849
135. Alspach E, Lussier DM, and Schreiber RD. Interferon gamma and its important roles in promoting and inhibiting spontaneous and therapeutic cancer immunity. Cold Spring Harb Perspect Biol. (2019) 11. doi: 10.1101/cshperspect.a028480
136. Tanaka T, Narazaki M, and Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
137. Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. (2011) 117:3720–32. doi: 10.1182/blood-2010-07-273417
138. Ricciotti E and FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. (2011) 31:986–1000. doi: 10.1161/ATVBAHA.110.207449
139. Landy E, Carol H, Ring A, and Canna S. Biological and clinical roles of IL-18 in inflammatory diseases. Nat Rev Rheumatol. (2024) 20:33–47. doi: 10.1038/s41584-023-01053-w
140. Nikiforow S and Duncan CN. Role of hematopoietic cell transplantation in pediatric and adult hemophagocytic lymphohistiocytosis-remaining unknowns and challenges. Hematol Oncol Clin North Am. (2025) 39(3):645–60. doi: 10.1016/j.hoc.2025.03.002
141. Baur K, Heim D, Beerlage A, Poerings AS, Kopp B, Medinger M, et al. Dasatinib for treatment of CAR T-cell therapy-related complications. J Immunother Cancer. (2022) 10(12):e005956. doi: 10.1136/jitc-2022-005956
142. Bajwa A, Zhao Q, Geer M, Lin C, Westholder J, Maakaron J, et al. Siltuximab for chimeric antigen receptor T-cell therapy-related CRS and ICANS: a multicenter retrospective analysis. Blood Adv. (2025) 9:170–5. doi: 10.1182/bloodadvances.2024013688
143. Shalabi H, Harrison C, Yates B, Calvo KR, Lee DW, and Shah NN. Intrathecal hydrocortisone for treatment of children and young adults with CAR T-cell immune-effector cell-associated neurotoxicity syndrome. Pediatr Blood Cancer. (2024) 71:e30741. doi: 10.1002/pbc.30741
144. Shah NN, Johnson BD, Fenske TS, Raj RV, and Hari P. Intrathecal chemotherapy for management of steroid-refractory CAR T-cell-associated neurotoxicity syndrome. Blood Adv. (2020) 4:2119–22. doi: 10.1182/bloodadvances.2020001626
Keywords: hemophagocytic lymphohistiocytosis, primary HLH, macrophage activation syndrome (MAS), systemic juvenile idiopathic arthritis (sJIA), CAEBV, immune effector cell associated HLH-like syndrome (IEC-HS), pediatric critical care, allogeneic hematopoietic cell transplant
Citation: Wobma H, Henderson LA, Duncan CN, Prockop SE, Randolph AG, Degar BA, Lehmann LE and Baumeister SHC (2025) Treating and triggering hyperinflammation: tackling hemophagocytic lymphohistiocytosis and HLH-like syndromes in the pediatric cell therapy and critical care setting. Front. Oncol. 15:1631557. doi: 10.3389/fonc.2025.1631557
Received: 19 May 2025; Accepted: 31 July 2025;
Published: 19 September 2025.
Edited by:
Jennifer Ann McArthur, St. Jude Children’s Research Hospital, United StatesReviewed by:
Hema Dave, Merck, United StatesE. Scott Halstead, Penn State University, United States
Claudio Pignata, University of Naples Federico II, Italy
Camille Keenan, Seattle Children’s Hospital, United States
Copyright © 2025 Wobma, Henderson, Duncan, Prockop, Randolph, Degar, Lehmann and Baumeister. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susanne H. C. Baumeister, c3VzYW5uZV9iYXVtZWlzdGVyQGRmY2kuaGFydmFyZC5lZHU=