 Daniele Fanale1†
Daniele Fanale1† Lidia Rita Corsini1†Paola Piraino1†Erika Pedone1†
Lidia Rita Corsini1†Paola Piraino1†Erika Pedone1† Chiara Brando1
Chiara Brando1 Tancredi Didier Bazan Russo1Pietro Ferraro1Alisia Simone1Silvia Contino1Ornella Prestifilippo1Ugo Randazzo1Ambra Giurintano1Carla Ferrante Bannera1
Tancredi Didier Bazan Russo1Pietro Ferraro1Alisia Simone1Silvia Contino1Ornella Prestifilippo1Ugo Randazzo1Ambra Giurintano1Carla Ferrante Bannera1 Antonio Galvano1
Antonio Galvano1 Lorena Incorvaia1Gianfranco Pernice2
Lorena Incorvaia1Gianfranco Pernice2 Salvatore Vieni3Gianni Pantuso3
Salvatore Vieni3Gianni Pantuso3 Calogero Cipolla3Antonino Giulio Giannone4
Calogero Cipolla3Antonino Giulio Giannone4 Giuseppe Badalamenti1
Giuseppe Badalamenti1 Antonio Russo1*‡
Antonio Russo1*‡ Viviana Bazan5*‡
Viviana Bazan5*‡- 1Section of Medical Oncology, Department of Precision Medicine in Medical, Surgical and Critical Care (Me.Pre.C.C.), University of Palermo, Palermo, Italy
- 2U.O. Oncologia, Fondazione Istituto G. Giglio, Palermo, Italy
- 3Division of General and Oncological Surgery, Department of Precision Medicine in Medical, Surgical and Critical Care (Me.Pre.C.C.), University of Palermo, Palermo, Italy
- 4Pathology Unit, Department of Health Promotion Sciences, Maternal and Infant Care, Internal Medicine and Medical Specialties (PROMISE), University of Palermo, Palermo, Italy
- 5Department of Biomedicine, Neuroscience and Advanced Diagnostics, University of Palermo, Palermo, Italy
Endometrial carcinoma (EC) is one of the most common gynecological cancers showing a survival rate of 15-17% in the case of advanced disease. Based on the mutational burden and copy number alteration, EC is classified into four different molecular subgroups: POLE-mutated (ultramutated), microsatellite unstable (hypermutated), low copy number (endometrioid), and high copy number (serous-like). Despite the high tumor grading, the ultramutated subtype, accounting for about 8-10% of all ECs, showed favorable prognostic potential, enhanced immune response, and excellent clinical outcomes. Somatic POLE alterations have been found in 6-10% of ECs, whereas germline pathogenic variants have been reported only in 0.25-4% of cases. Germline POLE alterations are linked to genome instability and are associated with onset of hereditary tumors, including colorectal cancer and EC. Emerging data suggests that knowledge of POLE mutational status could be clinically important, as ultramutated ECs may be more likely to respond to immunotherapy. In this Review, we will investigate the role of germline/somatic POLE genetic alterations in EC, discussing the potential future theranostic applications and evaluating the benefit of performing a routine genetic testing, in order to adopt prevention and surveillance strategies in germline POLE mutation carriers.
1 Introduction
Endometrial carcinoma (EC) is one of the most common gynecological cancers with a steady increase in incidence worldwide, accounting for 6th most frequent cancer in women (1, 2). Approximately 382,000 new cases of EC and 90,000 deaths are diagnosed annually worldwide (2). In particular, the high EC incidence rate in North America and Western Europe may be associated with a high presence of lifestyle-related risk factors, such as obesity (3). Racial disparity and socioeconomic and geographical differences are important variables for EC-related incidence and mortality (4). Although most women show at an early-stage disease favorable prognosis, with 5-year overall survival (OS) of 81%, some patients present with advanced disease, and the 5-year OS for stages IVA and IVB is only 17% and 15%, respectively (5, 6).
EC may be classified based on main three aspects: pathogenetic, histopathological and molecular (7). In 1983, Bokhman (8) described two pathogenetic types of EC: type I, more common, favorable from a prognostic point of view, associated with obesity, metabolic syndrome and hyperestrogenism, and type II, unrelated to risk factors known at that time (9).
The histopathological classification of EC includes several forms: endometrioid and its variants, mucinous, serous, clear cell, neuroendocrine, mixed, undifferentiated, or dedifferentiated. In addition, there are also some carcinomas with mesenchymal differentiation, defined as carcinosarcomas (10). It is possible to highlight a correlation between the histopathological subtypes and pathogenetic types: type I tumors show an endometrioid histology in 70%-80% of cases; type II tumors may have serous, clear cell, or undifferentiated histology and are more clinically aggressive (10, 11).
For the first time, the Cancer Genome Atlas (TCGA), through a comprehensive genomic analysis, classified EC into molecular subgroups based on the mutational burden and copy number alterations (12). The updated classification allowed to identify four distinct subgroups: POLE-mutated (ultramutated), microsatellite unstable (hypermutated), low copy number (endometrioid), and high copy number (serous-like) (11, 13, 14) (Figure 1). Generally, endometrioid carcinomas occur in all molecular subgroups, instead serous carcinomas arise almost exclusively in the high copy number subgroup (10).
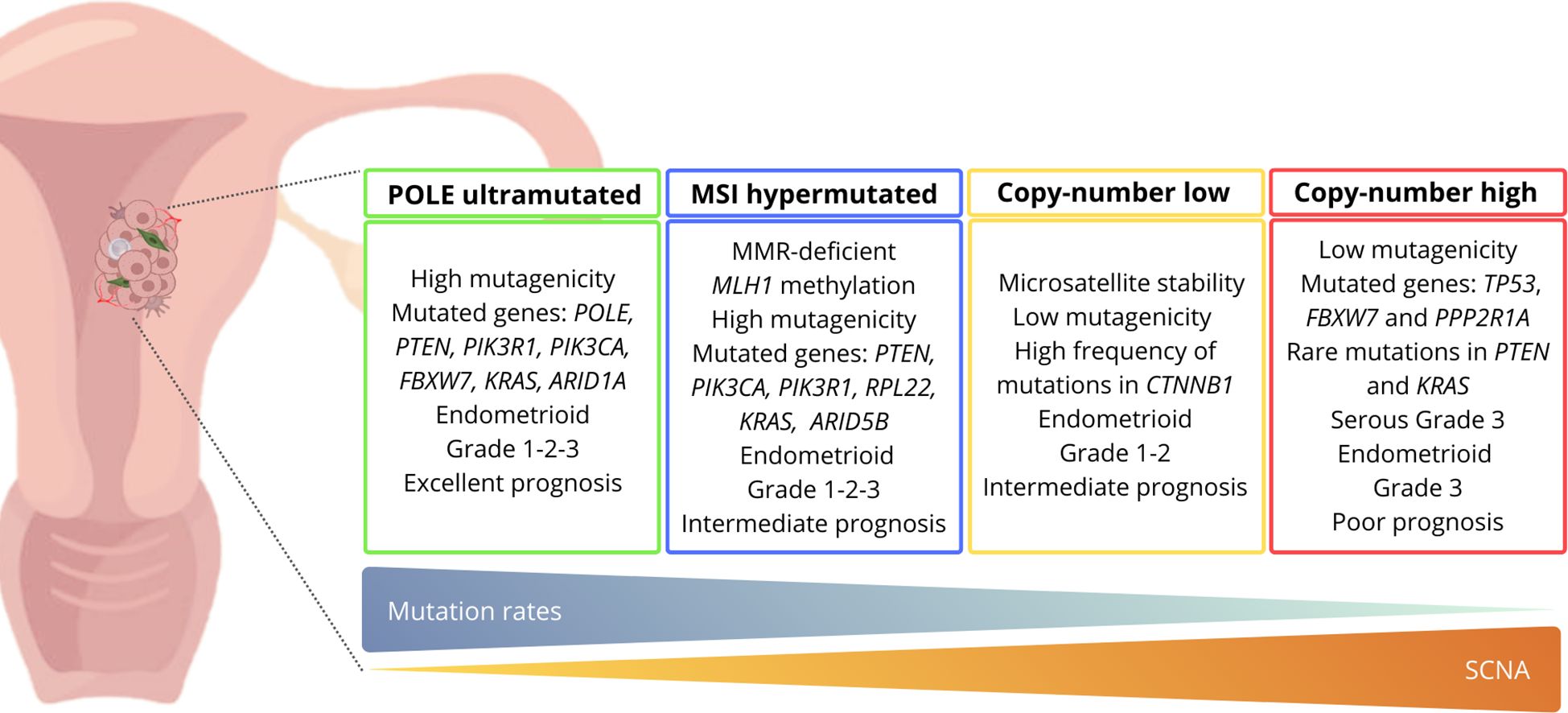
Figure 1. Molecular classification of endometrial cancer and related characteristics according to the TCGA criteria. ARID1A, AT-rich interaction domain 1A; ARID5B, AT-rich interaction domain 5B; CTNNB1, Catenin beta-1; FBXW7, F-box/WD repeat-containing protein 7; KRAS, Kirsten rat sarcoma viral oncogene homolog; MLH1, mutL homolog 1; MMR, mismatch repair; MSI, microsatellite instability; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase; PIK3R1, phosphoinositide-3-kinase regulatory subunit 1; POLE, DNA polymerase epsilon, catalytic subunit; PPP2R1A, protein phosphatase 2 scaffold subunit Aalpha; PTEN, phosphatase and tensin homolog; RPL22, ribosomal protein L22; SCNA, somatic copy number alterations; TP53, cellular tumor antigen 53.
Based on the TCGA classification, the Proactive Molecular Risk Classifier for Endometrial Cancer (ProMisE) used surrogate biomarkers to summarize the TCGA subtypes, in order to evaluate the utility of those classifiers in risk prediction (14).
Like ProMisE, Stelloo et al. (15), by means of subsequent immunohistochemistry (IHC) studies, classified EC into four subgroups: POLE-mutated, microsatellite instability (MSI) positive, p53 mutant, and no specific molecular profile (NSMP). The POLE-mutated and MSI-positive subgroups showed the most favorable outcomes (6, 10).
The use of these new integrated ‘histomolecular’ diagnostic entities offers the possibility to characterize EC in a different way and improve the risk assessment and treatment in patients, adding interesting diagnostic, prognostic, and therapeutic objectives (15, 16). The POLE-ultramutated subgroup is very promising as regards the perspectives of EC women (10, 11, 14).
Several studies have investigated the clinical impact of the detection of a POLE mutation in EC. Levine et al. (12), in a study published in 2013, found possible correlations between the presence of POLE alterations and therapeutic applications. Since then, much more attention has been paid to the prognostic and predictive role of POLE mutations in EC, as highlighted by the investigation by León‐Castillo et al. (17). Finally, the RUBY study provided important evidence on the role of POLE mutations in EC and their impact on the response to immunotherapy (18). All these studies, together with others, have allowed a better understanding of the biology of EC, providing an important contribution to risk stratification.
Therefore, in this Review, we will investigate the role of germline POLE genetic alterations in EC predisposition, discussing the potential future theranostic applications and evaluating the benefit of performing a routine genetic testing, in order to adopt prevention and surveillance strategies in patients harboring germline POLE pathogenic variants (PVs) and unaffected family members.
2 POLE: more than believed
2.1 POLE functions
POLE is involved in DNA replication process and has been recently recognized as an inherited gene able to predispose to cancer (19). POLE alterations are linked to genome instability and are associated with the prognosis and development and onset of tumors, including colorectal cancer (CRC) and EC, which results in an “ultramutated” phenotype (13, 20).
POLE, also known as POLE1, FILS and CRCS12, is located on the human chromosome 12q24.3. Its gene product, the largest subunit of Pol ϵ, contains 2286 amino acids and has a molecular weight of 262 kDa (21).
The POLE gene encodes the main catalytic subunit of the enzyme DNA polymerase ϵ, which synthesizes leading and lagging strands, during DNA replication in eukaryotes. Pol ϵ and Pol δ both belong to the DNA polymerase B family, which have polymerase and 3′-5′ exonuclease activities. Polymerase ϵ is believed to work in leading strand synthesis, although there is conflicting data on whether polymerase δ synthesizes both the leading and lagging strands, and that polymerase ϵ serves only in repair and proofreading activity (22, 23). The proofreading activity allows polymerase ϵ to recognize and remove incorrect nucleotides through its exonuclease activity, thus enabling high-fidelity DNA replication (24) (Figure 2a).
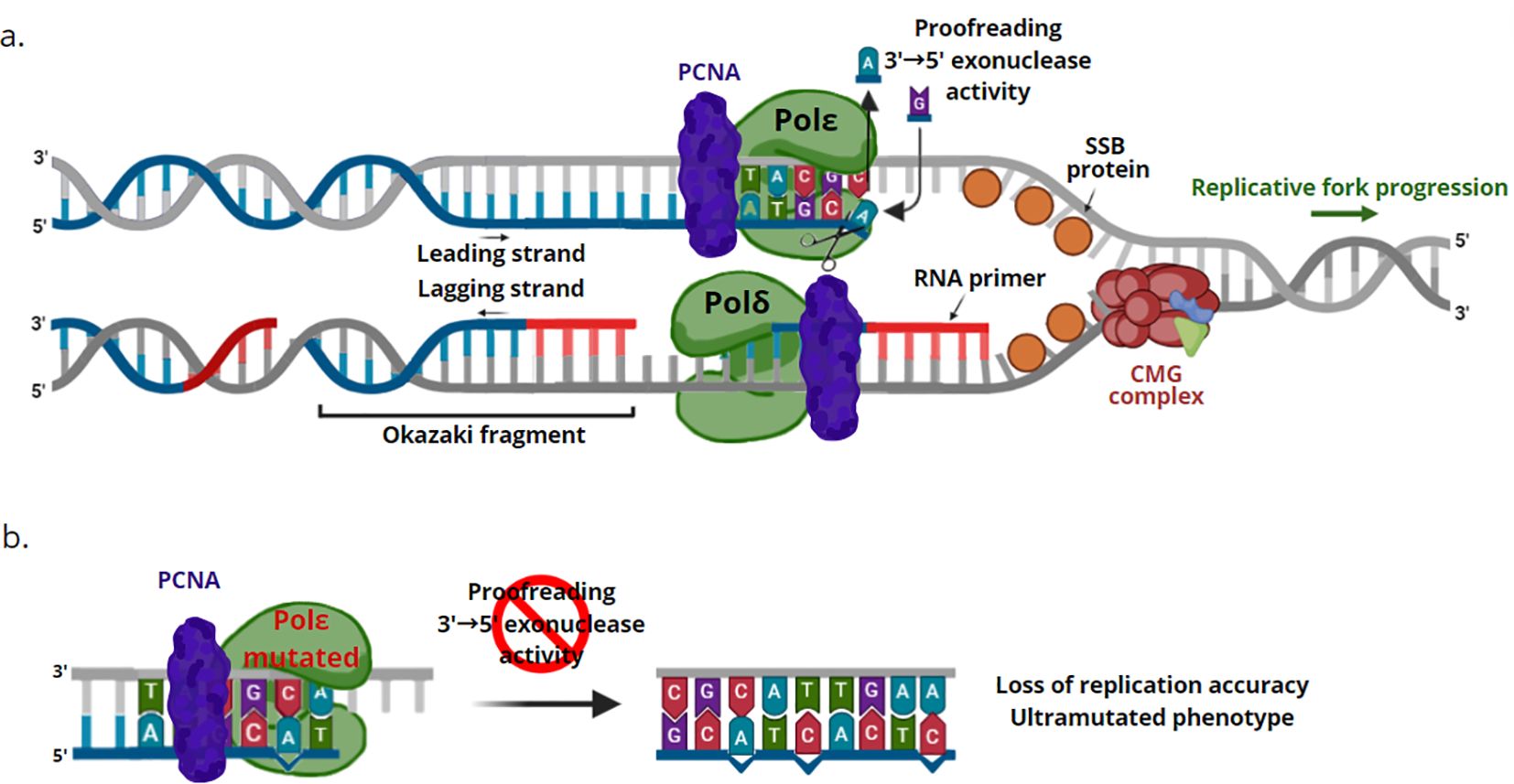
Figure 2. Functioning mechanism of DNA polymerase ϵ. (a) Proofreading activity. The neosynthesized DNA strand is indicated in blue, while the RNA primers are represented in red. Polymerase ϵ synthesizes the guide strand, continuously following the progression of the replicative fork. Polymerase δ uses the other strand as a template, moving in the opposite direction from the replicative fork, generating the lagging strand. The resulting Okazaki fragments contain around 200 nucleotides in human cells; for convenience, they are shown shorter in the figure. When an incorrect nucleotide is incorporated, the 3’ end of the newly synthesized strand moves from the polymerase active site to the 3’-5’ exonuclease domain (exonuclease proofreading). The incorrect nucleotide is removed, allowing the formation of the primer-template junction, which can bind the polymerase active site, permitting DNA synthesis to continue. (b) Pol ϵ deficiency. Genetic alterations which inactivate or suppress the exonuclease proofreading activity of the polymerase ϵ cause increased replicative error rates and an ultramutated phenotype. CMG complex, Cdc45-Mcm2-7-GINS complex; PCNA, proliferating cell nuclear antigen; POL δ, DNA polymerase δ; POL ϵ, DNA polymerase ϵ; SSB protein, single-stranded binding protein.
POLE alterations can occur in the polymerase exonuclease domain, within hotspot regions. These genetic alterations inactivate or suppress the 3′ to 5′ proofreading capabilities of the polymerase, causing a loss of replication fidelity, the occurrence of genomic instability, an increase in replicative error rates and consequently an ultramutated phenotype (11, 22) (Figure 2b).
The effects of individual mutations on exonuclease activity are likely to be distinct. Some mutations in exon 13, which hosts the exonuclease domain, can generate more than 8,000 neo-epitopes, favoring and determining a highly immunogenic microenvironment, as highlighted by an increase in the number of peri-tumor and tumor-infiltrating lymphocytes (TILs) and by a concomitant high expression of PD-1 or PD-L1 (25). Indeed, TILs express the PD-1 receptor, playing an important role in tumor immune escape (26). Also, POLE-mutated tumors demonstrated a high frequency of transversions from C to A (22). The presence of germline POLE PVs is a predisposing factor for EC and CRC, but also for ovarian cancer and brain tumors (21).
2.2 POLE alterations
The Cancer Genome Atlas (TCGA) project first allowed to identify the molecular subset of POLE-ultramutated EC, characterized by a favorable prognostic potential, enhanced immune response, and excellent clinical outcomes, despite the high tumor grading (11). The POLE-ultramutated subtype, accounting for about 8-10% of all ECs, is characterized by epsilon polymerase (POLE) exonuclease domain mutations (EDMs) with extremely high mutation rates (≥100 mutations/Mb) (19, 20). In particular, the pathogenicity of five most common POLE variants (P286R, V411L, S297F, A456P and S459F) has been shown. The five POLE mutation hotspots were associated with an elevated tumor mutational burden (TMB = 268 mut/Mb), which varied between different mutational hotspots (range 37.5-791.9 mut/Mb) and between tumors with identical hotspot mutations (e.g. P286R: 41.9-550.1 mut/Mb). POLE hotspot mutant ECs typically exhibited a high rate of C>A and T>G substitutions, whereas the percentage of C>G and indel substitutions were low (17).
A recent work by León‐Castillo et al. (17) showed that POLE-mutated ECs typically exhibit specific genomic alterations: a high prevalence of C>A substitutions (often greater than 20%), a low percentage of small insertion and deletion mutations (indels), and a very high TMB (>100 mut/Mb). In a study performed by the TCGA, 17 cancers classified as ultramutated had a POLE EDM, including the most frequent P286R (exon 9) and V411L (exon 13) substitutions (eight and five cases, respectively) and one case each of M444K, A456P, L424I, and S297F substitutions. Interestingly, 10 out of the 231 non-ultramutated ECs also harbored a POLE variant inside or outside the exonuclease domain (12).
In addition to the TCGA study, further investigations confirmed the prevalence of the five PVs and identified additional alterations having uncertain pathogenicity. In their study, Leòn-Castillo and colleagues developed a scoring system able to assess the pathogenicity of new POLE variants, on the basis of the presence or absence of genomic alterations associated with known POLE PVs. This scoring system was improved, assessing whether POLE alterations were recurrent in EC within the COSMIC or TCGA databases (17), since recurrent mutations are more likely to be pathogenic as they are responsible for tumor ultramutation (27). Therefore, the presence of “recurrence” was incorporated into the final scoring system related to pathogenicity, called the “POLE-score”. The “POLE-score” system allows to predict the pathogenicity of POLE mutations in EC, when its value is ≥ 4 points. Instead, POLE EDMs in EC with a “POLE score” ≤ 2 were classified as non-pathogenic, based on the absence of genomic alterations associated with the ultramutated phenotype. Finally, tumors with a score of 3 were classified as having a variant of uncertain significance (e.g., L424V, T278M, A428T and A465V). Furthermore, pragmatic guidelines for interpreting the clinical significance of POLE variants, where no complete genomic data is available, were provided, in order to evaluate the prognostic and therapeutic value associated with ultramutated-POLE ECs. The EC mutant subtype harboring POLE EDMs attracted more attention due to better results obtained in terms of survival (17).
2.2.1 Technique approaches for genetic testing
The most frequent somatic alterations detected in ECs, within the POLE proofreading domain, are P286R, V411L, and S459F, which have been identified in 67-92% of POLE-mutated tumors. The recommendations of the revised WHO classification system and ESMO guidelines for EC suggest to test the five POLE “hotspot” mutations by means of the Next-Generation Sequencing (NGS) and Sanger sequencing (28). In the work by Leòn Castillo et al. (17), these five hotspot PVs in the POLE gene were detected, with a frequency of 10.8%, in Formalin-Fixed Paraffin-Embedded (FFPE) samples from EC women, using droplet digital PCR (ddPCR). Approximately 30% of POLE variants identified by ddPCR were not detected by Sanger sequencing. The POLE mutations with low allele frequency are likely be undetected, due to the limit of detection of Sanger sequencing. Interestingly, the most common alterations detected by ddPCR were V411L (67%) and P286R (25%), whereas TCGA data analysis revealed that P286R was more common than V411L (17). Using ddPCR to detect POLE hotspot mutations could integrate the current ProMisE classification system, which involves immunohistochemistry to evaluate MMR status, followed by POLE genetic testing and immunohistochemistry analysis to evaluate the expression of p53 (29). Furthermore, Murali et al. (30) also suggested that POLE sequencing should be performed before immunohistochemistry evaluation for MMR status, since the expression loss for MMR proteins in a POLE-mutated case would be assigned to the dMMR (MMR deficiency) subgroup rather than POLE-positive subgroup. Some researchers suggested to perform the immunohistochemistry analysis and ddPCR simultaneously to evaluate the status of POLE, MMR, and p53, which could prevent misclassification and then be used for prognosis and treatment choice (17). Another innovative genetic analysis is represented by the OncoPanel test, reported in study by Veneris et al. (22). The OncoPanel test, developed at the Dana-Farber Cancer Institute, is a targeted NGS test, which, in addition to detecting somatic alterations, also allows to identify copy number variation and structural DNA rearrangements, and to perform mutational signature analysis. The current version of the OncoPanel test analyzes exon sequences of 447 genes involved in cancer biology and 191 regions of 60 genes for the rearrangement detection (22).
Mutational signature analysis is a unique aspect of this panel. Mutational signatures which can be detected with this approach include DNA damage patterns associated with exposure to UV light, tobacco and alkylating agent, and with apolipoprotein mRNA editing enzyme dysregulation B (APOBEC), MMR deficiency and impairment of POLE function. Each carcinogen or DNA repair defect leaves a characteristic “imprint” describing the mode in which DNA is altered. The POLE signature shows a strand distortion for C-to-A transitions at TpCpT positions and T-to-G mutations in the TpTpT genomic context. Currently, 30 mutational signatures have been characterized, the list of which it is possible to find in the Catalog of Somatic Mutations in Cancer (COSMIC) database. A specimen is considered to harbor a mutational signature if it is detected, so it is possible for a sample to be attributed with more than one (or no) signature (22).
2.3 POLE/TP53 correlations
The presence of secondary TP53 alterations has been reported in up to 42% of POLE-mutated EC cases (16). In the study carried out by Vermij et al. (16) a small subgroup of patients (3-5%) showed more than one classifying alteration (e.g., POLE-mut/dMMR EC, POLE-mut/p53abn EC, dMMR/p53abn EC or POLE-mut/dMMR-p53abn EC), which allowed to define these ECs as “multiple classifiers”.
Certainly, the combination of POLE-mut/p53abn EC is the most controversial, as the tumor harbors a favorable PV in the POLE domain of the exonuclease but also an unfavorable aberrant IHC expression of p53. In opposition to the excellent prognosis of POLE-mutated ECs, p53abn ECs are associated with poor clinical outcomes (16).
Molecular clustering of these “multiple classifier ECs” showed that POLE-mut/p53abn ECs clustered together with POLE-mut ECs without TP53 alterations. In addition, it was observed that p53-IHC, in these cases, often showed “subclonal” expression of mutant-like p53. This unusual expression pattern of p53 can be observed in POLE-mut and dMMR ECs, reflecting their genetic heterogeneity. Available survival data demonstrated that POLE-mut/p53abn ECs exhibit clinical outcomes comparable to POLE-mut EC without abnormal p53 expression. This would appear to indicate that TP53 mutations in these “multiple classifiers” are likely transient and do not affect clinical behavior, indicating that these cases should be classified and treated as POLE-mut ECs (16).
2.4 POLE/MSI correlations
POLE-mut ECs have been shown to be genetically distinct from MSI-high tumors (20). The most common alteration in MSI-H tumors is the frameshift/deletion mutation, whereas the most frequent variant in POLE-mutated tumors is the missense mutation. Tumors with dMMR/MSI-H or POLE mutations are usually associated with high TMB. MSI-H is mainly restricted to tumors in the 10–100 mut/Mb range, whereas TMB of POLE/POLD1 alterations can overcome 100 mut/Mb. POLE mutation and MSI were classified as ultra-high mutation phenotype and strong mutation phenotype, respectively (21).
Mismatch repair deficiency combined with loss of proofreading activity of the replication polymerase can produce DNA repair defects, resulting in an ultrahigh mutation with microsatellite stability (MSS) (31). TCGA considers POLE mutation and MSI as two important indicators of molecular typing. However, up to 30% of POLE-mutated tumors exhibit MMR deficiency or elevated MSI (21). Since universal screening for Lynch syndrome using MMR IHC has been widely used (32), MMR-deficient POLE EDM ECs may represent a confounding factor in the screening process (20). POLE mutations coexisting with MSI in EC are more likely to be non-exonuclease and non-pathogenic, although this is not an universal assumption. A pragmatic tool was developed in order to classify EC with somatic POLE mutations in clinical practice (17). The POLE score and presence or absence of MSI/dMMR can be used to stratify EC cases in POLE-mut, dMMR, or one of the other two TCGA subgroups depending on p53 status (33). The only presence of a POLE deleterious alteration is not sufficient to categorize tumors as “POLE-mut”, but the classification of ECs with combined POLE alteration and dMMR/MSI depends on the POLE score (17, 22).
2.5 Prognostic value of POLE in EC
POLE-mut ECs are characterized by a favorable prognosis. Several hypotheses have been proposed about the prognosis of POLE-mut ECs, the main one being that the extremely high mutational load may generate a major immune response (34). A less favored hypothesis is that intrinsically defective DNA repair related to POLE mutations makes tumor cells more susceptible to standard chemotherapy, although in vitro data showed platinum resistance rather than susceptibility. Another little-established hypothesis is that the abundance of mutations may result deleterious for cancer cells, reducing the their ability to proliferate or metastasize. However POLE-mutated tumors appear as widespread diseases, at high risk of relapse (22).
Li et al. (35) demonstrated that POLE mutations improve the prognosis of EC by regulating cellular glucose metabolism via AMF/AMFR signal transduction pathway.
On the one hand, the POLE PVs greatly affect patient prognosis, on the other hand, 3-6% of EC patients exhibit multiple molecular alterations, including POLE/p53 mutations and dMMR. These patients usually have a good prognosis, if they are carriers of a POLE PV (36).
An emerging link has been observed between high TMB and improved prognosis in cancer patients (37). Indeed, POLE-mut ECs exhibit higher immune infiltration and PD-1/PD-L1 expression, which may compensate for low survival risk caused by higher tumor grades in ultramutated POLE tumors, determining a favorable prognosis (38).
In 2020, the ESGO/ESTRO/ESP published their joint guidelines for the management of EC patients, incorporating findings of the TCGA to assess the prognosis in association with conventional and distinct clinic-pathological prognostic factors (tumor stage, grade and histotype, myometrial invasion or lymphovascular space invasion) in risk stratification of EC (39). However, several points remain to be clarified, since the prognostic value of the TCGA molecular group may vary depending on different EC histotypes (19).
3 Therapeutic approaches in POLE-mutated EC
3.1 POLE and therapy
The standard treatment for EC is surgery involving total hysterectomy and bilateral salpingo-oophorectomy, with lymph node evaluation (40). Adjuvant treatment often involves significant side effects and toxicities. Therefore, the objective of oncologists is to find an optimal patient selection approach, in order to reduce recurrence risk, improve survival, and avoid overtreatment side effects (41). The clinical and surgical histopathological features, which stratify patients into low-risk, low-intermediate, high-intermediate and high-risk groups, determine the need and type of adjuvant treatment (6, 42). Molecular classification allowed to classify EC into four subgroups: POLE-mutated, p53-abnormal, dMMR, and NSMP (no specific molecular profile). These four groups of ECs represent a promising clinical tool to assist in decision making regarding adjuvant treatment (43). Specifically, the POLE mutations are associated with a low relapse risk, probably due to the high mutational burden of highly immunogenic POLE-mutated ECs (44). This setting of patients will likely not require any adjuvant treatment in the early stages. Conversely, patients harboring p53-altered tumors have a high risk of recurrence and reduced survival, therefore require a chemotherapy treatment (6, 45).
When the molecular classification is known, according to the new guidelines of The European Society of Gynaecological Oncology (ESGO), adjuvant treatment is not recommended in stage I-II patients harboring POLE alteration, due to lack of benefit (7). Furthermore, advanced stage POLE-mutated patients (III-IVA) are not included in this category by the ESGO guidelines, due to data restrictions. These guidelines also suggest not performing the POLE mutational analysis for low-risk patients. Indeed, for patients who do not require adjuvant treatment based on clinic-pathological parameters, the knowledge of the POLE mutational status would has no impact on clinical management (6, 39).
Clinical trials, such as PORTEC-4a, RAINBO, CANSTAMP and TAPER, are playing a key role in the development of personalized treatments against EC, based on molecular and clinic-pathological features, leading to more tailored care strategies for patients (Table 1) (6, 50, 52, 53).
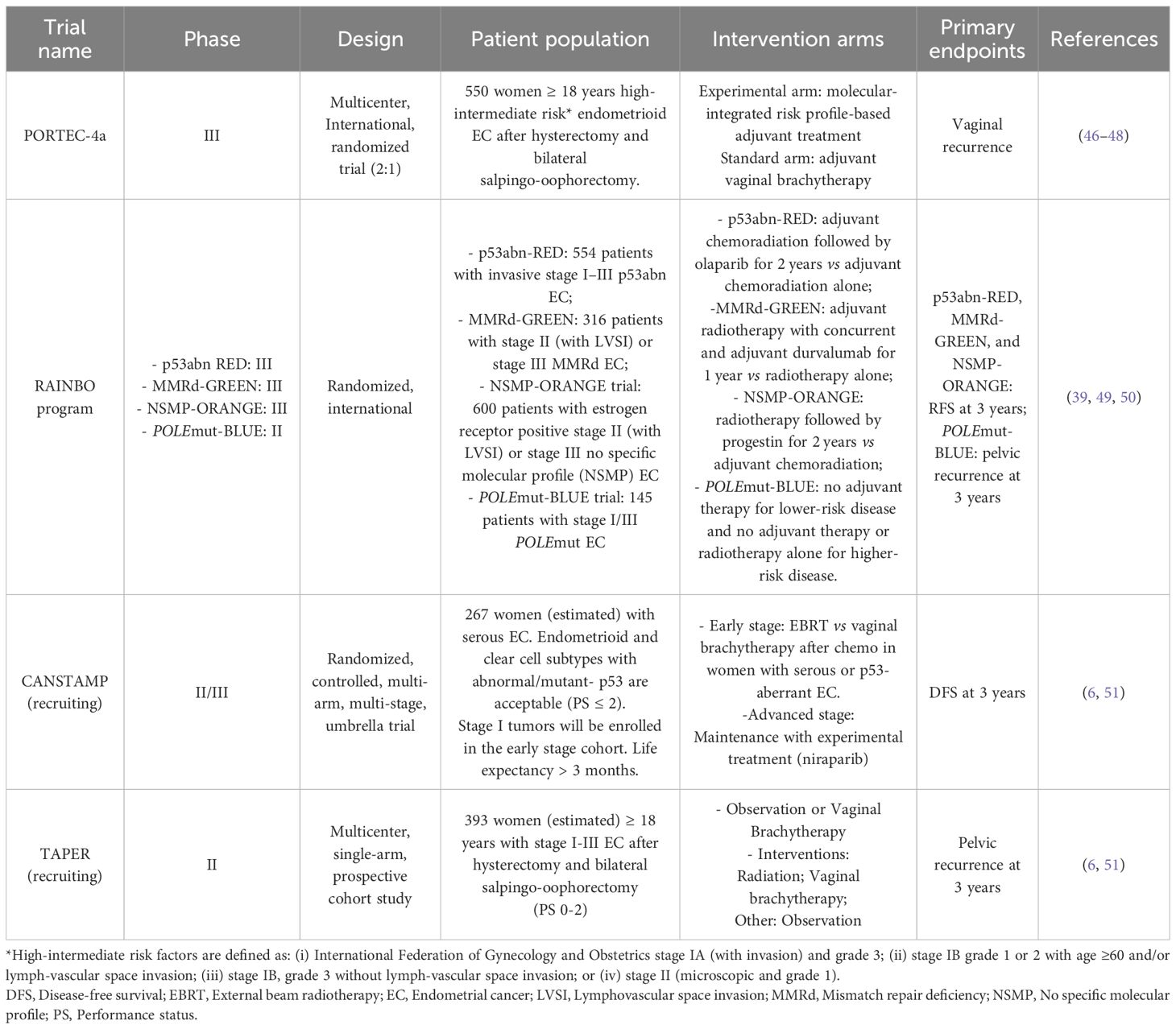
Table 1. Clinical trials regarding personalized treatments against EC based on molecular and clinic-pathological features.
POLE-mut EC showed favorable clinical outcomes, while retrospective studies revealed that avoiding adjuvant treatment is safe in this patient group. In addition, two prospective clinical trials, PORTEC-4 and TAPER, are underway to evaluate this aspect. The PORTEC-4a study will be the first to prospectively show the use of adjuvant therapy after combining molecular and clinic-pathological characteristics in EC (53). The RAINBO (Refining Adjuvant treatment iN endometrial cancer Based On molecular profile) study, instead, is an international TransPORTEC collaboration aimed at evaluating the molecular-based adjuvant treatment for high-risk EC women (6).
Furthermore, two additional studies carried out in Canada are examining specific molecular subtypes, such as the POLE and p53 status. The TAPER (Tailored Adjuvant therapy in POLE-mutated and p53 wildtype early-stage Endometrial cancer) is a multicenter, single-arm, prospective cohort study, which is specifically investigating treatment reductions in patients affected by early stage POLE-mutated/p53-wild-type EC with non-specific molecular profile (NSMP) (54). The second Canadian CANSTAMP trial (NCT04159155) is a randomized, controlled, multi-arm, multi-stage study evaluating the first-line treatment in serous or p53-mutated ECs (6, 55).
The finding of good clinical results in POLE-mut ECs highlights that this group of patients could be unnecessarily exposed to unwanted side effects, if subjected to radio- and/or chemotherapy (54). Therefore, the clinical practice, whereby many patients currently undergo adjuvant therapy, may represent an overtreatment, suggesting a reduction in treatment or perhaps no additional therapeutic option (19). Results from the ongoing PORTEC4a study will show whether this is a valid approach for patients with intermediate-risk POLE-mut EC. For high-risk POLE-mut ECs, the recently presented molecular characterization in the PORTEC-3 study is resulted highly informative (16, 56). POLE-mut tumors in the high-risk EC population showed an excellent prognosis when adjuvant treatment is restricted to radiotherapy, strongly suggesting that these patients do not benefit from adjuvant chemotherapy (16).
To date, treatment options for patients with advanced or recurrent EC are restricted. POLE-mut and dMMR ECs, due to their high mutational load, acquire high levels of neoantigens and TILs, making them interesting candidates for immunotherapy (57).
3.2 Immunotherapy as treatment option for metastatic and relapsed POLE-mutated EC
In general, EC women have a favorable prognosis, however patients with relapsed or metastatic disease have an overall survival (OS) poor (58). Emerging data suggests that knowledge of POLE status could be important in the context of relapse, as ultramutated tumors may be more likely to respond to immunotherapy (20, 59, 60).
Stasenko et al. (20) showed the effectiveness of immunotherapy agents in dMMR or MSI-H tumors. In particular, pembrolizumab, anti-PD-1 drug, has obtained the approval by the FDA for the treatment of these tumors. However, the role of immunotherapy in MSS tumors with POLE EDM and ultramutated phenotype is less clear (20, 61).
Although most POLE-mut ECs are MSS, preclinical data support the use of PD-1/PD-L1 inhibitors in this subgroup of ultramutated tumors, as the genomic instability present in tumors with a POLE EDM leads to a high number of neo-antigens and an increased quantity of TILs (62, 63). Several studies have reported the effective use of anti-PD-1 immune checkpoint inhibitors for the management of relapsed POLE-mutated EC (20, 64, 65).
Standard systemic treatment for metastatic disease includes hormone therapy for patients with low-grade, estrogen and progesterone receptor-positive tumors (66). Two randomized phase III clinical trials (KEYNOTE-775/NCT03517449 and ENGOT-EN9/LEAP-001/NCT03884101) focusing on the combinatorial use of lenvatinib and pembrolizumab compared to chemotherapy are currently recruiting (67, 68).
Finally, monotherapy with PARP and PD-1/PD-L1 inhibitors showed promising results (69, 70). Several phase II studies are evaluating whether combined therapy of PARP inhibition and PD-1/PD-L1 pathway inhibition is even more effective, as limited data have suggested an additive or even synergistic effect (69). The NCT02912572 study will include 70 patients with metastatic EC previously treated with at least one line of chemotherapy. The first cohort will include patients with dMMR and/or POLE mut EC treated with avelumab monotherapy. The second cohort will include patients with MSS without POLE mutation treated with either avelumab or talazoparib (58). The DOMEC study (NCT03951415), an ongoing multicenter, single-arm phase II study, will include 55 patients with metastatic EC to study the combination of olaparib and durvalumab, treatment response and recurrence-free survival (69, 71). Table 2 summarizes the main studies involving the use of immunotherapy treatments currently used or under evaluation in POLE-mutated EC.
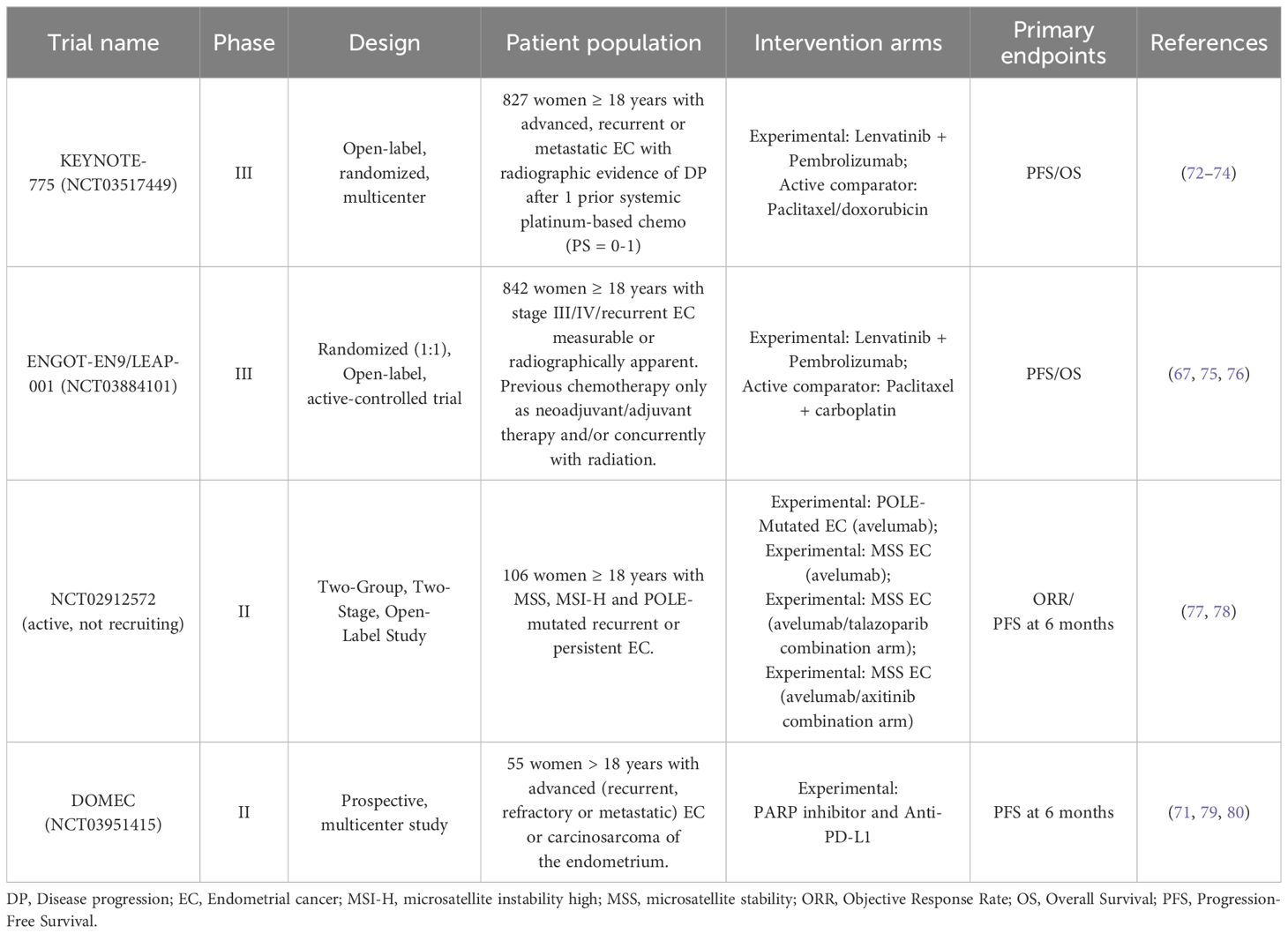
Table 2. Clinical trials involving immunotherapy treatments against POLE-mutated EC.
Ionizing radiations have been shown to induce genomic instability through direct and indirect damage to DNA, resulting in ROS production, which triggers chemical reactions with potentially lethal damage (81). The combination of radiotherapy with PARP inhibitors leads to the inhibition of DNA repair mechanisms in malignant cells, which contributes to promote the DNA damage and apoptosis (82, 83). Furthermore, radiations enhance tumor immunogenicity through the release of proinflammatory cytokines and chemokines via tumor antigen uptake and cross-presentation by dendritic cells (84). The combination of immunotherapy and radiations determines an alteration of the tumor microenvironment by intensifying the recruitment and infiltration of immune cells (58).
4 Prevention pathway: genetic counseling and screening
Genetic counseling is a procedure that analyzes personal and family history of patients to evaluate the presence of hereditary features. The objective of the genetic counseling is to identify potential correlations between the risk of developing a hereditary cancer syndrome and the presence of an underlying predisposing mutation, in order to increase awareness and possibility of adopting suitable screening strategies (13, 85).
Somatic POLE alterations have been found in 6-10% of ECs, but germline POLE PVs may also predispose to EC. However, germline POLE alterations in this tumor are rare, since they have been reported only in 0.25-4% of EC cases (13). Several reports support the hypothesis that germline PVs in POLE (exons 9-14) and POLD1 (exons 8-13), which are deleterious to polymerase proofreading function, predispose to EC (86–89). However, to date, due to the paucity of data in this regard, the risks of EC associated with POLD1/POLE PVs have yet to be estimated. Furthermore, though little is known about prognosis of germline PV carriers, somatic POLE alterations correlate with favorable clinical outcome in EC women.
Although the absolute risk of EC associated with germline POLD1/POLE PVs has not been established, Bellido et al. (89) published preliminary clinical management recommendations, suggesting a colonoscopy every 1–2 years and gastroduodenoscopy every 3 years, starting at the age of 20–25 years (the periodicity should be re-evaluated according to the findings), adding EC screening beginning at the age of 40 years for female carriers of germline POLD1 PVs.
The germline POLE/POLD1 genetic testing should be considered in the context of a strong family history of colorectal or endometrial cancer (two or more relatives) or a single relative with colorectal cancer or endometrial cancer <60 years (89).
The advent of new techniques, such as NGS, provided substantial advantages compared to single-gene testing. Therefore, cancer-related multiple genes should be tested in patients with suspect POLE associated tumor syndromes (13).
Although POLD1 and POLE have recently been suggested as genes predisposing to hereditary EC, absolute risk for EC and even other cancer types is very uncertain, therefore the POLD1/POLE genetic testing of unselected EC patients should be restricted to the research setting (90).
5 Discussion
Somatic POLE mutations have been detected in 6-10% of EC cases, whereas germline POLE alterations are very rare (0.25-4%), limiting the applicability of findings to a broader EC population (13). Although germline POLE genetic testing can enable the inclusion of mutation carriers in suitable and intensive preventive and surveillance pathways, its performing involves ethical and psychological implications for probands and family members, causing a state of potential anxiety or misinterpretation of cancer risk (90). Additionally, not all POLE mutations have an established clinical significance, as some variants of unknown significance (VUS) may complicate diagnosis and treatment decisions. Functional studies are often required to confirm pathogenicity (91). Another issue regards the access to comprehensive genetic testing (somatic and germline), which may not be uniformly available, increasing the variability (92). Lack of standardized protocols for testing and interpreting POLE variants could lead to inconsistent clinical application (91, 93). Despite data on long-term outcomes in germline mutation carriers are still limited (13), however, some evidence supports the integration of the routine POLE mutation testing in EC patients, especially useful for stratifying treatment and managing familial cancer risk.
POLE EDMs identify a subgroup of EC patients with an excellent prognosis. The use of this biomarker has been suggested to refine adjuvant treatment decisions, also helping to identify patients who would benefit from immune checkpoint inhibitors (59).
Despite high tumor grade, POLE-ultramutated ECs showed excellent clinical outcomes, strong immune response, potentially higher sensitivity to immunotherapy (20, 94, 95). The screening for POLE alterations is performed by sequencing the exonuclease domain (exons 9-14), but the relatively high cost of this analysis hinders the clinical use of this important biomarker. The evaluation of simple morphological, histological and immunohistochemical features (tumor type, grade, peritumoral lymphocytes, MLH1 and p53 expression) could help in the pre-screening phase and molecular characterization for identifying POLE EDMs in EC, increasing the likelihood from 7% to 33% that a mutation may be detected (15) (Figure 3, left and middle panels). Different therapeutic strategies are currently used or under evaluation for encouraging the development of targeted therapies and personalized treatments in women affected by POLE-mutated EC (60) (Figure 3, right panel). This could facilitate the use of this significant prognostic and predictive biomarker in routine clinical practice (96). Although POLE-ultramutated ECs show immune activation, not all cases respond to immune checkpoint inhibitors, as immune microenvironment and co-mutations may influence therapeutic outcomes (97, 98). Therefore, a potential overestimation of immunotherapy benefit is a problem that clinicians should take into account. Finally, most data supporting POLE as a biomarker are from retrospective studies or small cohorts. More prospective clinical trials are needed to validate POLE status as a predictor of immunotherapy response (92).
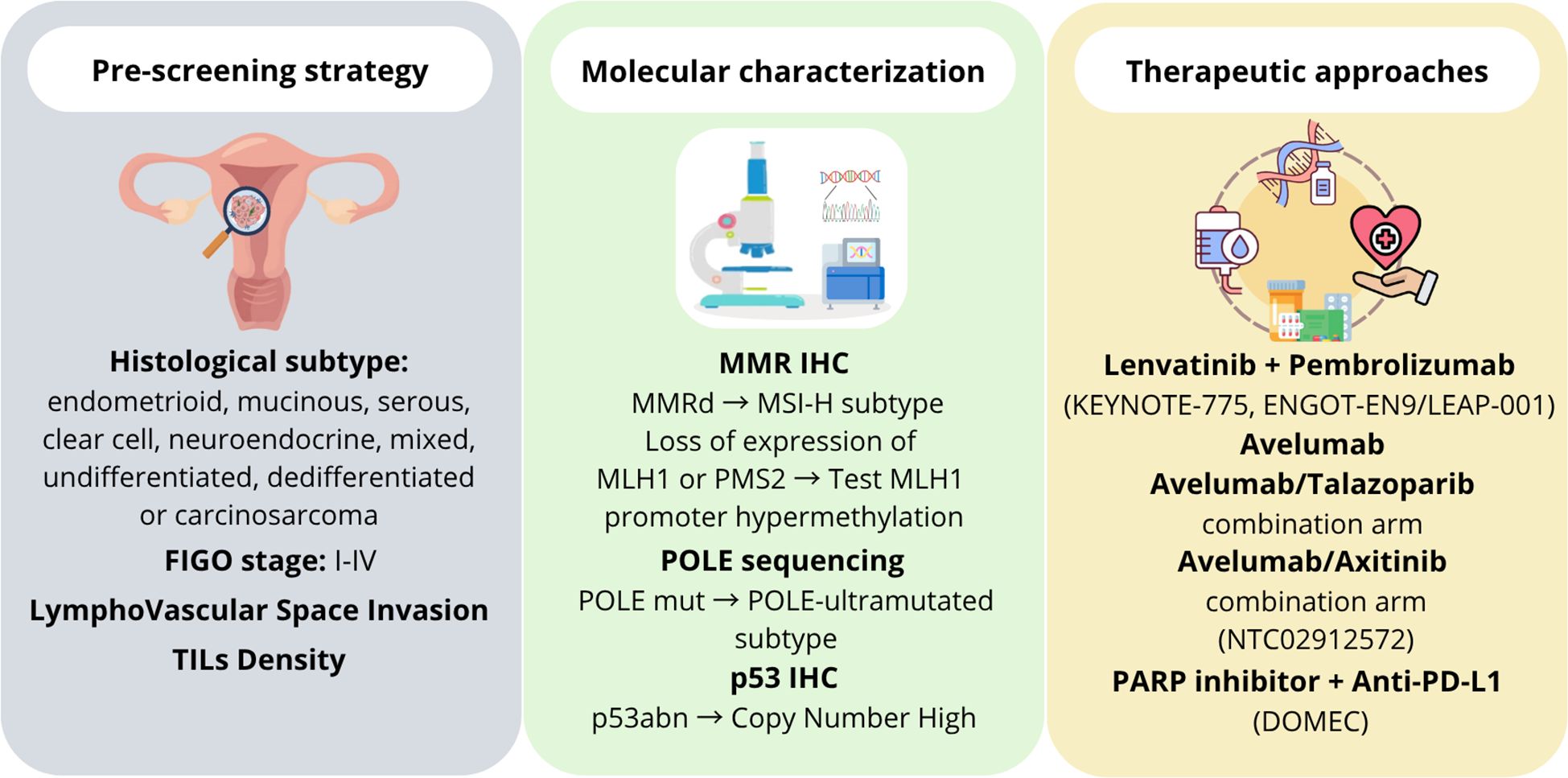
Figure 3. Schematic representation of theranostic approaches currently used in POLE-mutated endometrial cancer. The left panel shows the pre-screening strategies mainly based on morphological and histological features of endometrial cancer. The middle panel reports the approaches used for the molecular characterization. The right panel lists the main therapeutic strategies currently used or under evaluation against POLE-mutated endometrial cancer.
Author contributions
DF: Conceptualization, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing. LC: Conceptualization, Supervision, Validation, Writing – original draft, Writing – review & editing. PP: Conceptualization, Supervision, Validation, Writing – original draft, Writing – review & editing. EP: Conceptualization, Supervision, Validation, Writing – original draft, Writing – review & editing. CB: Writing – original draft, Writing – review & editing. TB: Writing – original draft, Writing – review & editing. PF: Writing – original draft, Writing – review & editing. AS: Writing – original draft, Writing – review & editing. SC: Writing – original draft, Writing – review & editing. OP: Writing – original draft, Writing – review & editing. UR: Writing – original draft, Writing – review & editing. AmG: Writing – original draft, Writing – review & editing. CF: Writing – original draft, Writing – review & editing. AG: Writing – original draft, Writing – review & editing. LI: Writing – original draft, Writing – review & editing. GP: Writing – original draft, Writing – review & editing. SV: Writing – original draft, Writing – review & editing. GP: Writing – original draft, Writing – review & editing. CC: Writing – original draft, Writing – review & editing. AnG: Writing – original draft, Writing – review & editing. GB: Writing – original draft, Writing – review & editing. AR: Conceptualization, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing. VB: Conceptualization, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. European Union - Next Generation EU, Mission 4, Component 2, Project SAMOTHRACE CUP B73C22000810001 (MUR, PNRR-M4C2, ECS_00000022).
Acknowledgments
Project Title: SiciliAn MicronanOTecH Research And Innovation CEnter “SAMOTHRACE” (MUR, PNRR-M4C2, ECS_00000022), spoke 3: Università degli Studi di Palermo, “S2-COMMs - Micro and Nanotechnologies for Smart & Sustainable Communities”.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, and Jemal A. Cancer statistics, 2021. CA: A Cancer J Clin. (2021) 71:7–33. doi: 10.3322/caac.21654
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, and Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
3. Makker V, MacKay H, Ray-Coquard I, Levine DA, Westin SN, Aoki D, et al. Endometrial cancer. Nat Rev Dis Primers. (2021) 7. doi: 10.1038/s41572-021-00324-8
4. Calle EE, Rodriguez C, Walker-Thurmond K, and Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. Adults. New Engl J Med. (2003) 348:1625–38. doi: 10.1056/NEJMoa021423
5. Siegel RL, Miller KD, and Jemal A. Cancer statistics, 2018. CA: A Cancer J Clin. (2018) 68:7–30. doi: 10.3322/caac.21442
6. Mitric C and Bernardini MQ. Endometrial cancer: transitioning from histology to genomics. Curr Oncol. (2022) 29:741–57. doi: 10.3390/curroncol29020063
7. Santoro A, Angelico G, Travaglino A, Inzani F, Arciuolo D, Valente M, et al. New pathological and clinical insights in endometrial cancer in view of the updated ESGO/ESTRO/ESP guidelines. Cancers. (2021) 13:2623. doi: 10.3390/cancers13112623
8. Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecologic Oncol. (1983) 15:10–7. doi: 10.1016/0090-8258(83)90111-7
9. Alexa M, Hasenburg A, and Battista MJ. The TCGA molecular classification of endometrial cancer and its possible impact on adjuvant treatment decisions. Cancers. (2021) 13:1478. doi: 10.3390/cancers13061478
10. Bell DW and Ellenson LH. Molecular genetics of endometrial carcinoma. Annu Rev Pathol: Mech Disease. (2019) 14:339–67. doi: 10.1146/annurev-pathol-020117-043609
11. Jumaah AS, Salim MM, Al-Haddad HS, McAllister KA, and Yasseen AA. The frequency of POLE-mutation in endometrial carcinoma and prognostic implications: a systemic review and meta-analysis. J Pathol Trans Med. (2020) 54:471–9. doi: 10.4132/jptm.2020.07.23
12. Levine DA. Integrated genomic characterization of endometrial carcinoma. Nature. (2013) 497:67–73. doi: 10.1038/nature12113
13. Magrin L, Fanale D, Brando C, Fiorino A, Corsini LR, Sciacchitano R, et al. POLE, POLD1, and NTHL1: the last but not the least hereditary cancer-predisposing genes. Oncogene. (2021) 40:5893–901. doi: 10.1038/s41388-021-01984-2
14. Patankar MS, Jumaah AS, Al-Haddad HS, McAllister KA, and Yasseen AA. The clinicopathology and survival characteristics of patients with POLE proofreading mutations in endometrial carcinoma: A systematic review and meta-analysis. PLoS One. (2022) 17:e0263585. doi: 10.1371/journal.pone.0263585
15. Stelloo E, Nout RA, Osse EM, Jürgenliemk-Schulz IJ, Jobsen JJ, Lutgens LC, et al. Improved risk assessment by integrating molecular and clinicopathological factors in early-stage endometrial cancer—Combined analysis of the PORTEC cohorts. Clin Cancer Res. (2016) 22:4215–24. doi: 10.1158/1078-0432.CCR-15-2878
16. Vermij L, Smit V, Nout R, and Bosse T. Incorporation of molecular characteristics into endometrial cancer management. Histopathology. (2019) 76:52–63. doi: 10.1111/his.14015
17. León-Castillo A, Britton H, McConechy MK, McAlpine JN, Nout R, Kommoss S, et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J Pathol. (2020) 250:323–35. doi: 10.1002/path.5372
18. Powell MA, Bjørge L, Willmott L, Novák Z, Black D, Gilbert L, et al. Overall survival in patients with endometrial cancer treated with dostarlimab plus carboplatin–paclitaxel in the randomized ENGOT-EN6/GOG-3031/RUBY trial. Ann Oncol. (2024) 35:728–38. doi: 10.1016/j.annonc.2024.05.546
19. Wu Q, Zhang N, and Xie X. The clinicopathological characteristics of POLE-mutated/ultramutated endometrial carcinoma and prognostic value of POLE status: a meta-analysis based on 49 articles incorporating 12,120 patients. BMC Cancer. (2022) 22. doi: 10.1186/s12885-022-10267-2
20. Stasenko M, Tunnage I, Ashley CW, Rubinstein MM, Latham AJ, Da Cruz Paula A, et al. Clinical outcomes of patients with POLE mutated endometrioid endometrial cancer. Gynecologic Oncol. (2020) 156:194–202. doi: 10.1016/j.ygyno.2019.10.028
21. Ma X, Dong L, Liu X, Ou K, and Yang L. POLE/POLD1 mutation and tumor immunotherapy. J Exp Clin Cancer Res. (2022) 41. doi: 10.1186/s13046-022-02422-1
22. Veneris JT, Lee EK, Goebel EA, Nucci MR, Lindeman N, Horowitz NS, et al. Diagnosis and management of a recurrent polymerase-epsilon (POLE)-mutated endometrial cancer. Gynecologic Oncol. (2019) 153:471–8. doi: 10.1016/j.ygyno.2019.03.247
23. Pursell ZF, Isoz I, Lundström E-B, Johansson E, and Kunkel TA. Yeast DNA polymerase ϵ Participates in leading-strand DNA replication. Science. (2007) 317:127–30. doi: 10.1126/science.1144067
24. Hussein YR, Weigelt B, Levine DA, Schoolmeester JK, Dao LN, Balzer BL, et al. Clinicopathological analysis of endometrial carcinomas harboring somatic POLE exonuclease domain mutations. Modern Pathol. (2015) 28:505–14. doi: 10.1038/modpathol.2014.143
25. Bellone S, Bignotti E, Lonardi S, Ferrari F, Centritto F, Masserdotti A, et al. Polymerase ϵ (POLE) ultra-mutation in uterine tumors correlates with T lymphocyte infiltration and increased resistance to platinum-based chemotherapy in vitro. Gynecologic Oncol. (2017) 144:146–52. doi: 10.1016/j.ygyno.2016.11.023
26. Fanale D, Dimino A, Pedone E, Brando C, Corsini LR, Filorizzo C, et al. Prognostic and predictive role of tumor-infiltrating lymphocytes (TILs) in ovarian cancer. Cancers. (2022) 14:4344. doi: 10.3390/cancers14184344
27. Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, et al. Comprehensive analysis of hypermutation in human cancer. Cell. (2017) 171:1042–56.e10. doi: 10.1016/j.cell.2017.09.048
28. Joe S, Lee M, Kang J, Kim J, Hong S-H, Lee SJ, et al. Enhanced risk stratification in early-stage endometrial cancer: integrating POLE through droplet digital PCR and L1CAM. Cancers. (2023) 15:4899. doi: 10.3390/cancers15194899
29. Talhouk A, McConechy MK, Leung S, Li-Chang HH, Kwon JS, Melnyk N, et al. A clinically applicable molecular-based classification for endometrial cancers. Br J Cancer. (2015) 113:299–310. doi: 10.1038/bjc.2015.190
30. Murali R, Delair DF, Bean SM, Abu-Rustum NR, and Soslow RA. Evolving roles of histologic evaluation and molecular/genomic profiling in the management of endometrial cancer. J Natl Compr Cancer Network. (2018) 16:201–9. doi: 10.6004/jnccn.2017.7066
31. Chung J, Maruvka YE, Sudhaman S, Kelly J, Haradhvala NJ, Bianchi V, et al. DNA polymerase and mismatch repair exert distinct microsatellite instability signatures in normal and Malignant human cells. Cancer Discov. (2021) 11:1176–91. doi: 10.1158/2159-8290.CD-20-0790
32. Fanale D, Corsini LR, Brando C, Dimino A, Filorizzo C, Magrin L, et al. Impact of different selection approaches for identifying lynch syndrome-related colorectal cancer patients: unity is strength. Front Oncol. (2022) 12. doi: 10.3389/fonc.2022.827822
33. Singh N, Piskorz AM, Bosse T, Jimenez-Linan M, Rous B, Brenton JD, et al. p53 immunohistochemistry is an accurate surrogate for TP53 mutational analysis in endometrial carcinoma biopsies. J Pathol. (2020) 250:336–45. doi: 10.1002/path.5375
34. van Gool IC, Eggink FA, Freeman-Mills L, Stelloo E, Marchi E, de Bruyn M, et al. POLE proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin Cancer Res. (2015) 21:3347–55. doi: 10.1158/1078-0432.CCR-15-0057
35. Li Y, Bian Y, Wang K, and Wan X-P. POLE mutations improve the prognosis of endometrial cancer via regulating cellular metabolism through AMF/AMFR signal transduction. BMC Med Genet. (2019) 20. doi: 10.1186/s12881-019-0936-2
36. Yu S, Sun Z, Zong L, Yan J, Yu M, Chen J, et al. Clinicopathological and molecular characterization of high-grade endometrial carcinoma with POLE mutation: a single center study. J Gynecologic Oncol. (2022) 33. doi: 10.3802/jgo.2022.33.e38
37. Aggarwal C, Ben-Shachar R, Gao Y, Hyun SW, Rivers Z, Epstein C, et al. Assessment of tumor mutational burden and outcomes in patients with diverse advanced cancers treated with immunotherapy. JAMA Network Open. (2023) 6:e2311181. doi: 10.1001/jamanetworkopen.2023.11181
38. Zhang J, An L, Zhou X, Shi R, and Wang H. Analysis of tumor mutation burden combined with immune infiltrates in endometrial cancer. Ann Trans Med. (2021) 9:551–. doi: 10.21037/atm-20-6049
39. Concin N, Matias-Guiu X, Vergote I, Cibula D, Mirza MR, Marnitz S, et al. ESGO/ESTRO/ESP guidelines for the management of patients with endometrial carcinoma. Int J Gynecologic Cancer. (2021) 31:12–39. doi: 10.1136/ijgc-2020-002230
40. Sonoda Y. Surgical treatment for apparent early stage endometrial cancer. Obstetrics Gynecol Science. (2014) 57:1. doi: 10.5468/ogs.2014.57.1.1
41. DeLeon MC, Ammakkanavar NR, and Matei D. Adjuvant therapy for endometrial cancer. J Gynecologic Oncol. (2014) 25:136. doi: 10.3802/jgo.2014.25.2.136
42. Kasius JC, Pijnenborg JMA, Lindemann K, Forsse D, van Zwol J, Kristensen GB, et al. Risk stratification of endometrial cancer patients: FIGO stage, biomarkers and molecular classification. Cancers. (2021) 13:5848. doi: 10.3390/cancers13225848
43. Yang Y, Wu SF, and Bao W. Molecular subtypes of endometrial cancer: Implications for adjuvant treatment strategies. Int J Gynecol Obstetrics. (2023) 164:436–59. doi: 10.1002/ijgo.14969
44. Casanova J, Duarte GS, da Costa AG, Catarino A, Nave M, Antunes T, et al. Prognosis of polymerase epsilon (POLE) mutation in high-grade endometrioid endometrial cancer: Systematic review and meta-analysis. Gynecologic Oncol. (2024) 182:99–107. doi: 10.1016/j.ygyno.2024.01.018
45. Tresa A, Sambasivan S, Rema P, Dinesh D, Sivaranjith J, Nair SP, et al. Clinical profile and survival outcome of endometrial cancer with p53 mutation. Indian J Surg Oncol. (2022) 13:580–6. doi: 10.1007/s13193-022-01523-9
46. Colombo N, Creutzberg C, Amant F, Bosse T, Gonzalez-Martin A, Ledermann J, et al. ESMO-ESGO-ESTRO consensus conference on endometrial cancer. Int J Gynecological Cancer. (2016) 26:2–30. doi: 10.1097/IGC.0000000000000609
47. ASTEC/EN.5 Study Group, Blake P SA, Orton J, Kitchener H, Whelan T, Lukka H, et al. Adjuvant external beam radiotherapy in the treatment of endometrial cancer (MRC ASTEC and NCIC CTG EN.5 randomised trials): pooled trial results, systematic review, and meta-analysis. Lancet. (2009) 373:137–46. doi: 10.1016/S0140-6736(08)61767-5
48. van den Heerik ASVM, Horeweg N, Nout RA, Lutgens LCHW, van der Steen-Banasik EM, Westerveld GH, et al. PORTEC-4a: international randomized trial of molecular profile-based adjuvant treatment for women with high-intermediate risk endometrial cancer. Int J Gynecological Cancer. (2020) 30:2002–7. doi: 10.1136/ijgc-2020-001929
49. Crosbie EJ, Kitson SJ, McAlpine JN, Mukhopadhyay A, Powell ME, and Singh N. Endometrial cancer. Lancet. (2022) 399:1412–28. doi: 10.1016/S0140-6736(22)00323-3
50. Consortium RR. Refining adjuvant treatment in endometrial cancer based on molecular features: the RAINBO clinical trial program. Int J Gynecologic Cancer. (2023) 33:109–17. doi: 10.1136/ijgc-2022-004039
51. Peng Y and Yang X. Molecular classification guides for the postoperative adjuvant therapy of early-stage endometrial carcinoma. Thermal Science. (2024) 28:2217–24. doi: 10.2298/TSCI2403217P
52. Jamieson A, Bosse T, and McAlpine JN. The emerging role of molecular pathology in directing the systemic treatment of endometrial cancer. Ther Adv Med Oncol. (2021) 13:175883592110359. doi: 10.1177/17588359211035959
53. Wortman BG, Bosse T, Nout RA, Lutgens LCHW, van der Steen-Banasik EM, Westerveld H, et al. Molecular-integrated risk profile to determine adjuvant radiotherapy in endometrial cancer: Evaluation of the pilot phase of the PORTEC-4a trial. Gynecologic Oncol. (2018) 151:69–75. doi: 10.1016/j.ygyno.2018.07.020
54. McAlpine JN, Chiu DS, Nout RA, Church DN, Schmidt P, Lam S, et al. Evaluation of treatment effects in patients with endometrial cancer and POLE mutations: An individual patient data meta-analysis. Cancer. (2021) 127:2409–22. doi: 10.1002/cncr.33516
55. Tabata J, Takenaka M, and Okamoto A. Molecular typing guiding treatment and prognosis of endometrial cancer. Gynecol Obstetrics Clin Med. (2023) 3:7–17. doi: 10.1016/j.gocm.2023.01.011
56. León-Castillo A, de Boer SM, Powell ME, Mileshkin LR, Mackay HJ, Leary A, et al. Molecular classification of the PORTEC-3 trial for high-risk endometrial cancer: impact on prognosis and benefit from adjuvant therapy. J Clin Oncol. (2020) 38:3388–97. doi: 10.1200/JCO.20.00549
57. Fanale D, Corsini LR, Scalia R, Brando C, Cucinella A, Madonia G, et al. Can the tumor-agnostic evaluation of MSI/MMR status be the common denominator for the immunotherapy treatment of patients with several solid tumors? Crit Rev Oncology/Hematology. (2022) 170:103597. doi: 10.1016/j.critrevonc.2022.103597
58. van den Heerik ASVM, Horeweg N, de Boer SM, Bosse T, and Creutzberg CL. Adjuvant therapy for endometrial cancer in the era of molecular classification: radiotherapy, chemoradiation and novel targets for therapy. Int J Gynecologic Cancer. (2021) 31:594–604. doi: 10.1136/ijgc-2020-001822
59. Madariaga A, Garg S, Tchrakian N, Dhani NC, Jimenez W, Welch S, et al. Clinical outcome and biomarker assessments of a multi-centre phase II trial assessing niraparib with or without dostarlimab in recurrent endometrial carcinoma. Nat Commun. (2023) 14. doi: 10.1038/s41467-023-37084-w
60. Wang L, Liu L, Huo D, and Zhang Y. A comprehensive analysis of immunotherapy in advanced endometrial cancer (Review). Oncol Letters. (2023) 27. doi: 10.3892/ol.2023.14210
61. Castellucci E, He T, Goldstein DY, Halmos B, and Chuy J. DNA polymerase ϵ Deficiency leading to an ultramutator phenotype: A novel clinically relevant entity. Oncologist. (2017) 22:497–502. doi: 10.1634/theoncologist.2017-0034
62. Howitt BE, Shukla SA, Sholl LM, Ritterhouse LL, Watkins JC, Rodig S, et al. Association of polymerase e–mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol. (2015) 1:1319. doi: 10.1001/jamaoncol.2015.2151
63. Eggink FA, Van Gool IC, Leary A, Pollock PM, Crosbie EJ, Mileshkin L, et al. Immunological profiling of molecularly classified high-risk endometrial cancers identifies POLE-mutant and microsatellite unstable carcinomas as candidates for checkpoint inhibition. OncoImmunology. (2017) 6:e1264565. doi: 10.1080/2162402X.2016.1264565
64. Johnson RL, Ganesan S, Thangavelu A, Theophilou G, de Jong D, Hutson R, et al. Immune checkpoint inhibitors targeting the PD-1/PD-L1 pathway in advanced, recurrent endometrial cancer: A scoping review with SWOT analysis. Cancers. (2023) 15:4632. doi: 10.3390/cancers15184632
65. Green AK, Feinberg J, and Makker V. A review of immune checkpoint blockade therapy in endometrial cancer. Am Soc Clin Oncol Educ Book. (2020) 40:238–44. doi: 10.1200/EDBK_280503
66. Mahdi H, Ray-Coquard I, Lorusso D, Mirza MR, Monk BJ, and Slomovitz B. Evolving treatment paradigms in metastatic or recurrent low-grade endometrial cancer: When is hormonal-based therapy the preferred option? Int J Gynecologic Cancer. (2023) 33:1675–81. doi: 10.1136/ijgc-2023-004454
67. Marth C, Tarnawski R, Tyulyandina A, Pignata S, Gilbert L, Kaen D, et al. Phase 3, randomized, open-label study of pembrolizumab plus lenvatinib versus chemotherapy for first-line treatment of advanced or recurrent endometrial cancer: ENGOT-en9/LEAP-001. Int J Gynecologic Cancer. (2022) 32:93–100. doi: 10.1136/ijgc-2021-003017
68. Aravantinou−Fatorou A, Andrikopoulou A, Liontos M, Fiste O, Georgakopoulou V, Dimopoulos M-A, et al. Pembrolizumab in endometrial cancer: Where we stand now (Review). Oncol Lett. (2021) 22. doi: 10.3892/ol.2021.13082
69. Post CCB, Westermann AM, Bosse T, Creutzberg CL, and Kroep JR. PARP and PD-1/PD-L1 checkpoint inhibition in recurrent or metastatic endometrial cancer. Crit Rev Oncology/Hematology. (2020) 152:102973. doi: 10.1016/j.critrevonc.2020.102973
70. Peyraud F and Italiano A. Combined PARP inhibition and immune checkpoint therapy in solid tumors. Cancers. (2020) 12:1502. doi: 10.3390/cancers12061502
71. Post CCB, Westermann AM, Boere IA, Witteveen PO, Ottevanger PB, Sonke GS, et al. Efficacy and safety of durvalumab with olaparib in metastatic or recurrent endometrial cancer (phase II DOMEC trial). Gynecologic Oncol. (2022) 165:223–9. doi: 10.1016/j.ygyno.2022.02.025
72. Brooks RA, Fleming GF, Lastra RR, Lee NK, Moroney JW, Son CH, et al. Current recommendations and recent progress in endometrial cancer. CA: A Cancer J Clin. (2019) 69:258–79. doi: 10.3322/caac.21561
73. Makker V, Colombo N, Casado Herráez A, Santin AD, Colomba E, Miller DS, et al. Lenvatinib plus pembrolizumab for advanced endometrial cancer. New Engl J Med. (2022) 386:437–48. doi: 10.1056/NEJMoa2108330
74. Makker V, Colombo N, Herráez AC, Monk BJ, Mackay H, Santin AD, et al. Lenvatinib plus pembrolizumab in previously treated advanced endometrial cancer: updated efficacy and safety from the randomized phase III study 309/KEYNOTE-775. J Clin Oncol. (2023) 41:2904–10. doi: 10.1200/JCO.22.02152
75. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
76. Sant M, Chirlaque Lopez MD, Agresti R, Sánchez Pérez MJ, Holleczek B, Bielska-Lasota M, et al. Survival of women with cancers of breast and genital organs in Europe 1999–2007: Results of the EUROCARE-5 study. Eur J Cancer. (2015) 51:2191–205. doi: 10.1016/j.ejca.2015.07.022
77. Konstantinopoulos PA, Gockley AA, Xiong N, Krasner C, Horowitz N, Campos S, et al. Evaluation of treatment with talazoparib and avelumab in patients with recurrent mismatch repair proficient endometrial cancer. JAMA Oncol. (2022) 8:1317. doi: 10.1001/jamaoncol.2022.2181
78. Lee EK, Xiong N, Krasner C, Polak M, Campos S, Wright AA, et al. Phase 2, two-stage study of avelumab and axitinib in patients with mismatch repair proficient recurrent or persistent endometrial cancer. Gynecologic Oncol. (2025) 198:1–8. doi: 10.1016/j.ygyno.2025.05.006
79. Miller D, Filiaci V, Fleming G, Mannel R, Cohn D, Matsumoto T, et al. Late-Breaking Abstract 1: Randomized phase III noninferiority trial of first line chemotherapy for metastatic or recurrent endometrial carcinoma: A Gynecologic Oncology Group study. Gynecologic Oncol. (2012) 125:771. doi: 10.1016/j.ygyno.2012.03.034
80. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord J-P, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair–deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. (2020) 38:1–10. doi: 10.1200/JCO.19.02105
81. Srinivas US, Tan BWQ, Vellayappan BA, and Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol. (2019) 25:101084. doi: 10.1016/j.redox.2018.101084
82. Li W-H, Wang F, Song G-Y, Yu Q-H, Du R-P, and Xu P. PARP-1: a critical regulator in radioprotection and radiotherapy-mechanisms, challenges, and therapeutic opportunities. Front Pharmacol. (2023) 14. doi: 10.3389/fphar.2023.1198948
83. Sun C, Chu A, Song R, Liu S, Chai T, Wang X, et al. PARP inhibitors combined with radiotherapy: are we ready? Front Pharmacol. (2023) 14. doi: 10.3389/fphar.2023.1234973
84. Derer A, Deloch L, Rubner Y, Fietkau R, Frey B, and Gaipl US. Radio-immunotherapy-induced immunogenic cancer cells as basis for induction of systemic anti-tumor immune responses – pre-clinical evidence and ongoing clinical applications. Front Immunol. (2015) 6. doi: 10.3389/fimmu.2015.00505
85. Cicero G, De Luca R, Dorangricchia P, Lo Coco G, Guarnaccia C, Fanale D, et al. Risk perception and psychological distress in genetic counselling for hereditary breast and/or ovarian cancer. J Genet Counseling. (2017) 26:999–1007. doi: 10.1007/s10897-017-0072-0
86. Valle L, Hernández-Illán E, Bellido F, Aiza G, Castillejo A, Castillejo M-I, et al. New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Hum Mol Genet. (2014) 23:3506–12. doi: 10.1093/hmg/ddu058
87. Elsayed FA, Kets CM, Ruano D, van den Akker B, Mensenkamp AR, Schrumpf M, et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur J Hum Genet. (2014) 23:1080–4. doi: 10.1038/ejhg.2014.242
88. Spier I, Holzapfel S, Altmüller J, Zhao B, Horpaopan S, Vogt S, et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer. (2015) 137:320–31. doi: 10.1002/ijc.29396
89. Bellido F, Pineda M, Aiza G, Valdés-Mas R, Navarro M, Puente DA, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med. (2016) 18:325–32. doi: 10.1038/gim.2015.75
90. Spurdle AB, Bowman MA, Shamsani J, and Kirk J. Endometrial cancer gene panels: clinical diagnostic vs research germline DNA testing. Modern Pathol. (2017) 30:1048–68. doi: 10.1038/modpathol.2017.20
91. Mur P, Viana-Errasti J, García-Mulero S, Magraner-Pardo L, Muñoz IG, Pons T, et al. Recommendations for the classification of germline variants in the exonuclease domain of POLE and POLD1. Genome Med. (2023) 15. doi: 10.1186/s13073-023-01234-y
92. Kögl J, Pan TL, Marth C, and Zeimet AG. The game-changing impact of POLE mutations in oncology—a review from a gynecologic oncology perspective. Front Oncol. (2024) 14. doi: 10.3389/fonc.2024.1369189
93. Valentine MC, Wong A, Chen L, Du F, Hughes AEO, Spencer DH, et al. A fully next-generation sequencing-based method of classifying molecular sub-types of endometrial cancer retains prognostic value and expands biomarker targets. Int J Gynecological Cancer. (2025) 35:100060. doi: 10.1016/j.ijgc.2024.100060
94. McConechy MK, Talhouk A, Leung S, Chiu D, Yang W, Senz J, et al. Endometrial carcinomas with POLE exonuclease domain mutations have a favorable prognosis. Clin Cancer Res. (2016) 22:2865–73. doi: 10.1158/1078-0432.CCR-15-2233
95. Mehnert JM, Panda A, Zhong H, Hirshfield K, Damare S, Lane K, et al. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J Clin Invest. (2016) 126:2334–40. doi: 10.1172/JCI84940
96. Van Gool IC, Ubachs JEH, Stelloo E, de Kroon CD, Goeman JJ, Smit VTHBM, et al. Blinded histopathological characterisation of POLE exonuclease domain-mutant endometrial cancers: sheep in wolf’s clothing. Histopathology. (2017) 72:248–58. doi: 10.1111/his.13338
97. Gargiulo P, Della Pepa C, Berardi S, Califano D, Scala S, Buonaguro L, et al. Tumor genotype and immune microenvironment in POLE-ultramutated and MSI-hypermutated Endometrial Cancers: New candidates for checkpoint blockade immunotherapy? Cancer Treat Rev. (2016) 48:61–8. doi: 10.1016/j.ctrv.2016.06.008
Keywords: endometrial cancer, exonuclease domain mutations, germline/somatic POLE variants, microsatellite instability, POLE, POLE-ultramutated subtype, prognosis
Citation: Fanale D, Corsini LR, Piraino P, Pedone E, Brando C, Bazan Russo TD, Ferraro P, Simone A, Contino S, Prestifilippo O, Randazzo U, Giurintano A, Ferrante Bannera C, Galvano A, Incorvaia L, Pernice G, Vieni S, Pantuso G, Cipolla C, Giannone AG, Badalamenti G, Russo A and Bazan V (2025) POLE-mutated endometrial cancer: new perspectives on the horizon? Front. Oncol. 15:1633260. doi: 10.3389/fonc.2025.1633260
Received: 22 May 2025; Accepted: 11 August 2025;
Published: 27 August 2025.
Edited by:
Mayumi Kobayashi Kato, National Cancer Centre, JapanReviewed by:
Eswari Dodagatta-Marri, University of California, San Francisco, United StatesDongbo Yang, The University of Chicago, United States
Copyright © 2025 Fanale, Corsini, Piraino, Pedone, Brando, Bazan Russo, Ferraro, Simone, Contino, Prestifilippo, Randazzo, Giurintano, Ferrante Bannera, Galvano, Incorvaia, Pernice, Vieni, Pantuso, Cipolla, Giannone, Badalamenti, Russo and Bazan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viviana Bazan, dml2aWFuYS5iYXphbkB1bmlwYS5pdA==; Antonio Russo, YW50b25pby5ydXNzb0B1c2EubmV0
†These authors have contributed equally to this work
‡These authors share last authorship