 Prince Ahad Mir1*
Prince Ahad Mir1* Nishant Kumar2*
Nishant Kumar2* Sukesh K. Gupta3Anureet Kaur2Saeema Farooq4Gurpreet S. Sandhu5Anoop Kumar6
Sukesh K. Gupta3Anureet Kaur2Saeema Farooq4Gurpreet S. Sandhu5Anoop Kumar6- 1Department of Pharmacognosy and Phytochemistry, Khalsa College of Pharmacy, Amritsar, India
- 2Department of Pharmaceutics, Khalsa College of Pharmacy, Amritsar, India
- 3Department of Pharmacology, Amity Institute of Pharmacy, Amity University Kolkata, West Bengal, India
- 4Department of Pharmacognosy and Phytochemistry, University of Kashmir, Srinagar, Jammu and Kashmir, India
- 5Department of Pharmaceutical Analysis, Khalsa College of Pharmacy, Amritsar, India
- 6Department of Pharmacy, Dr KN Modi, Institute of Pharmaceutical Education and Research, Modinagar, Uttar Pradesh, India
Triple Negative Breast Cancer (TNBC) is a specific kind of breast cancer that is distinguished by the lack of expression of three specific receptors, namely human epidermal growth factor receptor 2 (HER2), estrogen receptor (ER), and progesterone receptor (PR) and are common in women under 40, especially among African American population or those with a BRCA1 genetic mutation. TNBC is characterized by its very aggressive behavior, elevated rates of recurrence, and restricted therapy alternatives in comparison to other subtypes of breast cancer. Chemotherapy is considered the most widely employed therapy against TNBC but experiences off-target toxicity due to its non-selectivity. Such a scenario led to the genetic profiling of the TNBC patients, which led to the identification of several targets and signaling pathways that can be considered as a therapeutic focus for the treatment of TNBC. In this review, we have compiled various therapeutic targets, including androgen receptor (AR) and PI3K/AKT/mTOR, Notch, Wnt/β-catenin, Hedgehog, and TGF-β signaling pathways, which are responsible for the progression of TNBC. In the current therapeutic landscape, the strategic targeting of key signaling pathways, coupled with the development of monoclonal antibody (mAb)-based immunotherapeutic interventions, has emerged as a promising and clinically relevant approach for the management of triple-negative breast cancer (TNBC). The mAbs reduce tumor development, modulate immune responses, and regulate the tumor microenvironment. This review summarizes their mechanisms, signaling pathway targets, clinical applications, and current therapeutic challenges.
Introduction
Triple-negative breast cancer (TNBC) is a very serious and complex form of breast carcinoma distinguished due to the lack of three prevalent receptors: human epidermal growth factor receptor 2 (HER2/neu), estrogen receptor (ER) and progesterone receptor (PR). The limited expression of receptors in this context hampers the efficacy of hormone-based and targeted treatments that are often used for various forms of breast carcinoma (1). TNBC constitutes around 10-15% of the total breast carcinoma cases, with a higher incidence seen in younger women, especially those of African-American ethnicity. The etiology of TNBC remains incompletely elucidated, while it is postulated to arise from a multifactorial interplay including lifestyle, environmental and genetic determinants (2). Consequently, patients with TNBC have limited treatment options, making it a particularly aggressive and difficult-to-treat form of breast cancer (3).
The prospects of TNBC may exhibit significant variability, contingent upon many variables such as the extent of the tumor, lymphatic participation, and the occurrence of distant metastases. TNBC has a propensity for a heightened incidence of early recurrence and distant metastases in contrast to other variants of breast carcinoma. Additionally, the lack of specific targeted therapies for TNBC makes it particularly challenging to manage and increases the risk of recurrence (4). Early detection and diagnosis of TNBC play a pivotal role in enhancing treatment results. Frequent breast self-examinations, mammographic screenings, and diagnostic breast examinations are necessary for detecting any abnormalities or changes in the breast tissue. If a suspicious lump or mass is detected, a biopsy is performed to confirm the diagnosis and determine the specific subtype of breast cancer (5). TNBC patients may also benefit from genetic screening to discover any inherent genetic alterations, like BRCA1 or BRCA2 alleles linked to a greater possibility of acquiring ovarian and breast cancer. Knowing the genetic profile of the tumor can help guide treatment decisions and identify potential targeted therapies or clinical trials (6).
Despite its aggressive nature, TNBC has shown some responsiveness to immunotherapy, like PD-1 and PD-L1 antagonists. Clinical investigations are under process to assess the potency of immunotherapy in TNBC, both as a standalone treatment and in combination with other therapeutic strategies (7).
In recent times, substantial advances have been made in the domain of targeted therapy, offering the potential for the management of TNBC. Precision medicine is a medical approach that emphasises the customization of therapies according to the specific features of a patient’s tumor. The proposed methodology entails the examination of the genetic and molecular composition of the tumor to discern distinct irregularities that may be effectively addressed via tailored therapeutic interventions (8, 9).
Clinical trials investigating novel treatment approaches, including targeted therapies and immunotherapies, are ongoing for TNBC. Researchers are exploring the potential of targeted agents that inhibit specific signaling pathways involved in TNBC growth and progression (10). Furthermore, researchers are investigating the potential of immunotherapies, including adoptive cell transfer and cancer vaccines, to exploit the immune system’s capabilities in identifying and eradicating TNBC cells (11). In addition to the advancements in treatment approaches, supportive care including strategies to manage the side effects of treatment, alleviate pain, provide psychological care, and immunostimulants plays a crucial role in managing disease and improving the quality of life (12, 13). In addition to therapeutic advances, monoclonal antibodies are expected to become integral components of personalized treatment regimens for TNBC, offering new hope for a historically underserved patient population (14, 15).
Signaling pathway for triple-negative breast cancer
TNBC lacks the molecular targets commonly used in the treatment of breast carcinoma, like hormone receptors and HER2. The signaling pathways implicated in TNBC are diverse and complex, involving multiple interconnecting networks (16–18). The crucial pathways implicated in TNBC include.
Phosphoinositide 3-kinase activation
TNBC is frequently associated with aberrant activation of the PI3K pathway. This pathway can be activated by several mechanisms, including (RTKs) receptor tyrosine kinases like IGF-1R (Insulin-like Growth Factor 1 Receptor) and EGFR (Epidermal Growth Factor Receptor) (19). These receptors, when activated by ligands, initiate a signaling cascade that leads to the recruitment and activation of PI3K. The activation of this process is essential in the advancement and growth of many types of malignancies, such as TNBC (20).
PI3K is a set of kinases that add a phosphate group to the hydroxyl group at the 3rd-position of phosphoinositides. This process converts phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-trisphosphate (PIP3) (21). Class I PI3Ks are most relevant in cancer, and they exist as heterodimers composed of a catalytic subunit (p110α, p110β, p110δ) and a regulatory subunit (p85α, p85β, p55γ, p55α, p50α) (22) (Figure 1). In TNBC, several RTKs are frequently overexpressed or mutated, leading to their constitutive activation. These RTKs, namely IGF-1R and EGFR, may trigger the activation of PI3K by binding to their ligands and beginning subsequent signaling pathways (23). Upon ligand binding to activated RTKs, PI3K is recruited to the cell membrane through interaction between the regulatory subunit and phosphorylated tyrosine residues on the RTK. This localization allows the catalytic subunit of PI3K to phosphorylate PIP2, generating PIP3 (24). PIP3 serves as a second messenger and recruits downstream signaling molecules to the membrane in the form of Akt and PDK1 to the plasma membrane (25, 26).
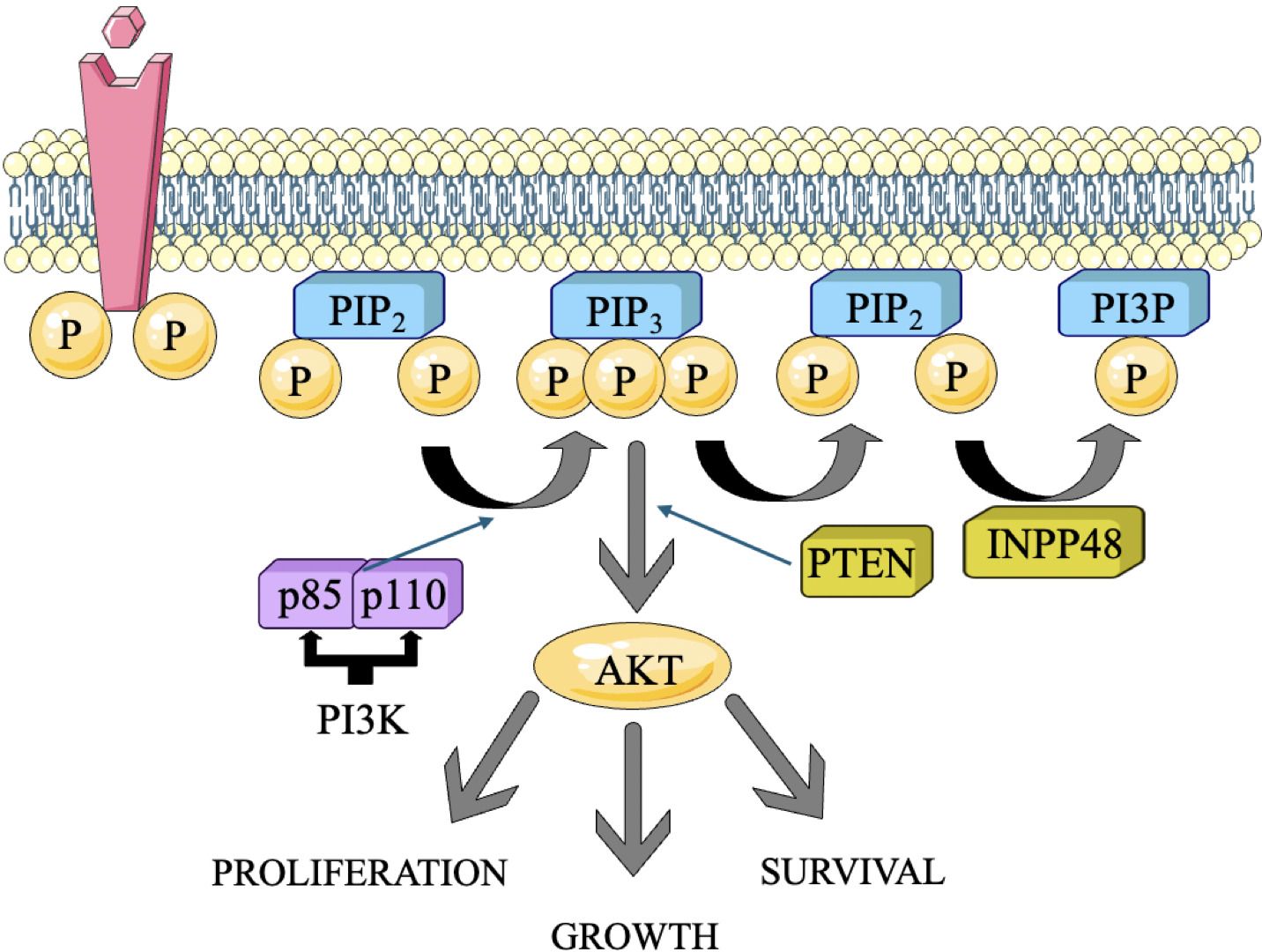
Figure 1. The PI3K pathway and the simplicity of its functions. The primary genomic abnormalities, including mutations and copy number variations (CNVs), identified in each gene in triple-negative breast cancer (TNBC) and their correlation with molecular subtypes (10).
Monoclonal antibodies against PI3K pathway
Monoclonal antibodies that directly activate PI3K in TNBC are not yet prevalent; however, some monoclonal antibodies influence PI3K expression indirectly by targeting immune checkpoints or alternative signaling networks (27). PI3K antagonists, when used in conjunction with these monoclonal antibodies, show potential for the treatment of TNBC by specifically targeting the pathway (28). Furthermore, investigations into innovative monoclonal antibodies that target specific PI3K isoforms represent a promising direction in the treatment of TNBC. Several monoclonal antibodies pertinent to TNBC and the phosphoinositide 3-kinase (PI3K) pathway are identified (29).
♣ Herceptin (Trastuzumab) a monoclonal antibody used in the treatment of HER2-positive breast cancer. It targets the HER2 receptor, inhibiting tumor growth and promoting apoptosis in cancer cells. Its efficacy has been established through numerous clinical trials, demonstrating improved outcomes in patients with this specific cancer subtype (30). Trastuzumab, that does not specifically target PI3K however, influences the PI3K/Akt cascade through downstream effects by inhibiting HER2 essential for triggering PI3K pathway (31). This treatment is primarily indicated for HER2-positive breast cancer; however, certain studies have investigated its potential application in TNBC, particularly in the HER2-negative subset that may exhibit significant signaling alterations (32).
♣ Margetuximab a monoclonal antibody targeting HER2, analogous to trastuzumab. It has demonstrated potential to influence PI3K signaling circuits (33). Preclinical findings indicate that margetuximab elicits a superior immune response relative to trastuzumab, potentially influencing PI3K-related downstream signaling in certain HER2-positive and even TNBC cells exhibiting HER2 overexpression (34).
♣ Anti-PD-L1 antibodies, such as Atezolizumab and Durvalumab, are utilized in therapeutic applications. While these antibodies do not directly activate the PI3K pathway, they regulate the immune reaction, and stimulating T cells that can indirectly affect PI3K signaling pathways (35). Immune checkpoint inhibitors are being utilized more frequently in TNBC, especially in conjunction with other treatments, including chemotherapy or targeted PI3K inhibitors (36).
♣ Bavituximab is a monoclonal antibody that targets phosphatidylserine, a molecule associated with the activation of PI3K/Akt signaling pathways. While this does not directly activate PI3K, it can indirectly affect the pathway, resulting in enhanced immunity against tumors and apoptosis in cancer cells (37).
♣ CureD3: Targeted PI3K isoforms. Monoclonal antibodies targeting specific isoforms of PI3K have been investigated in preclinical models that block the irregular PI3K pathway in cancers, like TNBC (29).
Protein kinase B activation
AKT, plays a pivotal part in the development and advancement of several types of malignancies, such as TNBC (38). It is a serine/threonine protein kinase that participates in several physiological functions, like cell survival, metabolism, growth, angiogenesis and proliferation. It operates after several signaling pathways, such as the PI3K/AKT pathway, which is often disrupted in TNBC (39). Activation of AKT in TNBC can occur through several mechanisms. One of the most common mechanisms is the overactivation of the PI3K pathway. Once activated, AKT phosphorylates and modulates the activity of numerous downstream effectors involved in cell survival and proliferation (40, 41) (Figure 2). The dysregulation of PI3K/AKT pathway can occur through various mechanisms, including genetic alterations, such as mutations in the PI3K catalytic subunit (PIK3CA) or loss of the tumor suppressor gene PTEN, which negatively regulates the pathway. Additionally, other signaling molecules, such as receptor tyrosine kinases (RTKs) or growth factor receptors, can activate AKT through the PI3K pathway (43).
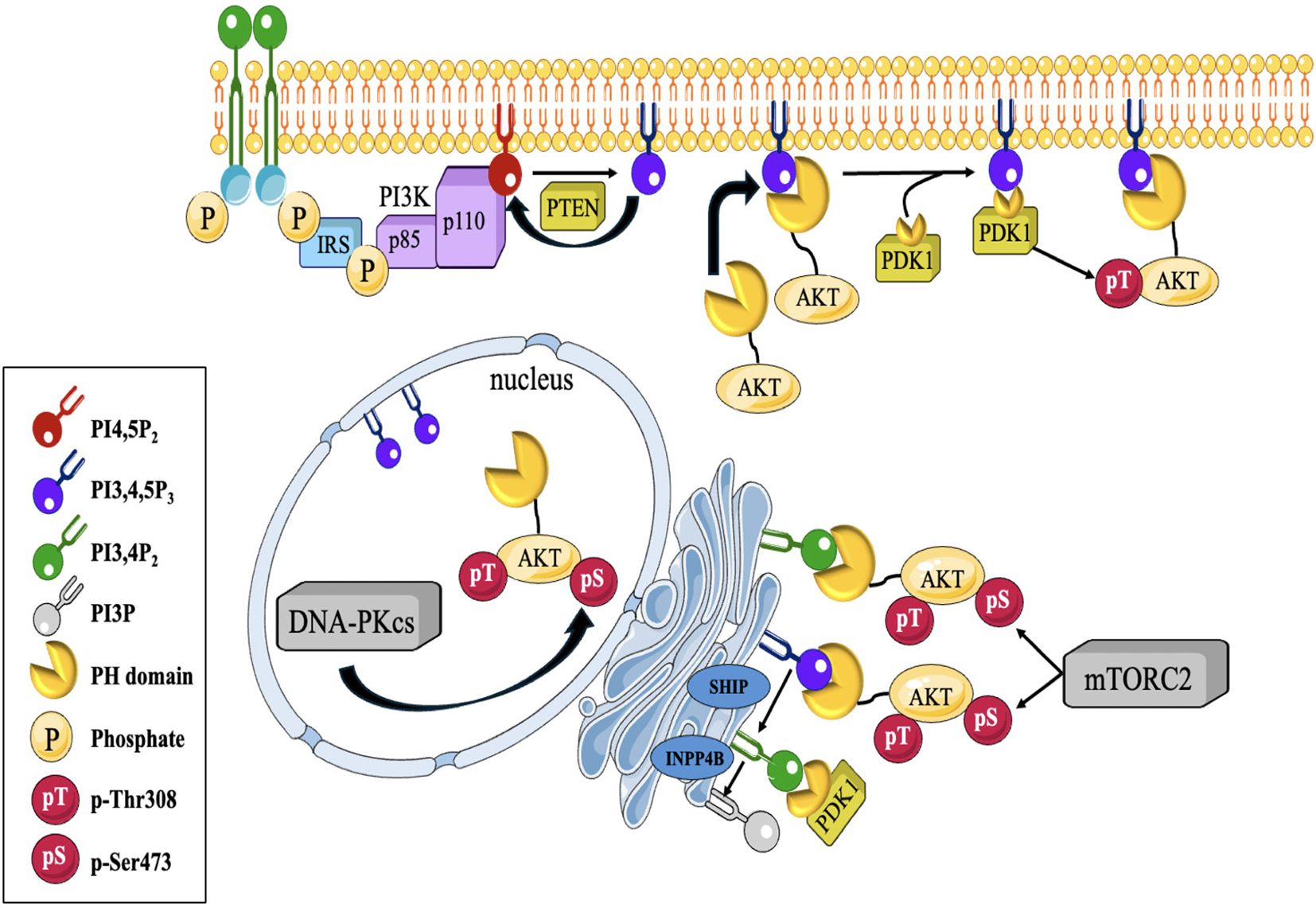
Figure 2. Depicts a diagrammatic illustration of compartmentalized Akt activation. PI(3,4,5)P3 synthesis at the cellular membrane is contingent upon the activation of PI3K by active RTKs and GPCRs downstream. Afterwards, Akt is brought to the cell membrane by attaching its PH- domain to the produced (PI(3,4,5)P3. Akt binds to PI (3,4,5)P3, which enables phosphorylation at T308 by PDK1, a PH-domain containing protein, and at S473 by mTORC2 or DNA-PKCS at different locations within the cell. Akt may also be triggered at endomembranes by attaching to P13,4P2, which is produced via a process catalyzed by SHIP from P1(3,4,5)P3. PTEN and INPP4B restrict Akt activation by regulating the levels of PI(3,4)P2 (42).
Monoclonal antibodies against AKT pathway
Akt, serves as a crucial regulator of numerous cellular processes, including survival, growth, proliferation, and metabolism. The Akt signaling pathway is often impaired in various cancers, such as TNBC, which contributes to chemoresistance, tumor progression, and unfavorable prognosis (42). Monoclonal antibodies (mAbs) that specifically activate Akt in TNBC are infrequent and not extensively investigated in contemporary treatments. Indirect stimulation or modulation of the Akt pathway represents a more common strategy in cancer therapy (44). Monoclonal antibodies may affect the Akt pathway via downstream mechanisms, either by inhibiting alternative signaling pathways (such as HER2 or immune checkpoint pathways) or by interfering with tumor dynamics (45).
♣ Anti-PD-L1 Antibodies: Atezolizumab (Tecentriq) and Durvalumab (Imfinzi) PD-L1 antibodies inhibit immune checkpoint signaling, thereby augmenting T-cell-mediated immune responses (46). Activated immune cells can influence tumor signaling pathways, such as Akt. PD-L1 inhibitors do not directly trigger Akt; however, they may enhance immune system activation, resulting in Akt suppression or indirectly influencing Akt in tumor cells (47).
♣ Antibodies targeting the Anti-Insulin-Like Growth Factor Receptor (IGF-1R): IGF-1R is a receptor that, upon activation, initiates multiple downstream signaling pathways, notably the PI3K/Akt pathway (48). Inhibition of IGF-1R signaling through monoclonal antibodies such as figitumumab (CP-751,871) may impede aberrant Akt activation in tumor cells, thereby potentially diminishing tumor growth. Monoclonal antibodies that target IGF-1R may indirectly influence Akt signaling by inhibiting tumor survival signals (48, 49).
♣ Anti-VEGF (Vascular Endothelial Growth Factor) agents Immunoglobulins: Bevacizumab (Avastin) inhibits the VEGF pathway, which plays a critical role in angiogenesis, the process of new blood vessel formation (50). This indirectly influence Akt activation in both tumor cells and endothelial cells (51). Bevacizumab has been utilized alongside chemotherapy for the treatment of TNBC. By inhibiting vascular endothelial growth factor (VEGF), it may obstruct certain survival signals that would typically activate Akt within the tumor microenvironment (52).
♣ Antibodies targeting Interleukin-6 (IL-6): Tocilizumab, also known as Actemra, is a monoclonal antibody that inhibits interleukin-6 (IL-6) receptors, utilized primarily in the treatment of autoimmune diseases such as rheumatoid arthritis and systemic juvenile idiopathic arthritis (53). Interleukin-6 (IL-6) is a cytokine that plays a role in inflammation and tumor progression and has the capacity to activate multiple signaling pathways, such as Akt (54). Tocilizumab indirectly modulate Akt signaling by blocking IL-6 receptor signaling and inhibiting the inflammatory microenvironment, which plays a crucial role in cancer progression (55).
♣ Combination therapies Numerous monoclonal antibodies are currently under investigation in conjunction with PI3K inhibitors or mTOR inhibitors to enhance targeting of the Akt pathway in TNBC (56). These combinations may indirectly modulate Akt signaling, leading to tumor cell apoptosis and diminished proliferation (57). Alpelisib (BYL719) a selective inhibitor of PI3Kα that, in conjunction with specific monoclonal antibodies (e.g., trastuzumab or immune checkpoint inhibitors), has the potential to diminish Akt activation in TNBC (58).
Mammalian target of rapamycin activation
mTOR is a protein kinase that plays a crucial role in cell growth, metabolism, and survival. It is a central regulator of various cellular processes, including protein synthesis, autophagy, and cell proliferation (38). Dysregulation of mTOR signaling has been implicated in the development and progression of several types of cancer, including TNBC. In TNBC, mTOR activation has been observed in a significant proportion of cases. Hyperactivation of mTOR signaling may arise from many pathways, such as the inactivation of adverse regulators like PTEN, and the stimulation of beneficial regulators like AKT and PI3K (59, 60). These modifications result in heightened mTOR activity, which then triggers the activation of the following pathways responsible for cell survival and development. mTOR signaling promotes cell cycle progression by regulating the translation of key proteins involved in cell growth and division. Enhanced mTOR activity in TNBC can contribute to uncontrolled cell proliferation, leading to tumor growth (61) (Figure 3). This activation can further promote cell development by suppressing apoptosis and also plays a pivotal role in the development of resistance to chemotherapy and targeted therapies in TNBC. Inhibiting mTOR signaling may sensitize TNBC cells to chemotherapy and enhance treatment response (63, 64). mTOR also regulates cellular metabolism, including glucose and lipid metabolism. In TNBC, mTOR activation can promote the uptake and utilization of nutrients, providing energy and resources for tumor growth (65).
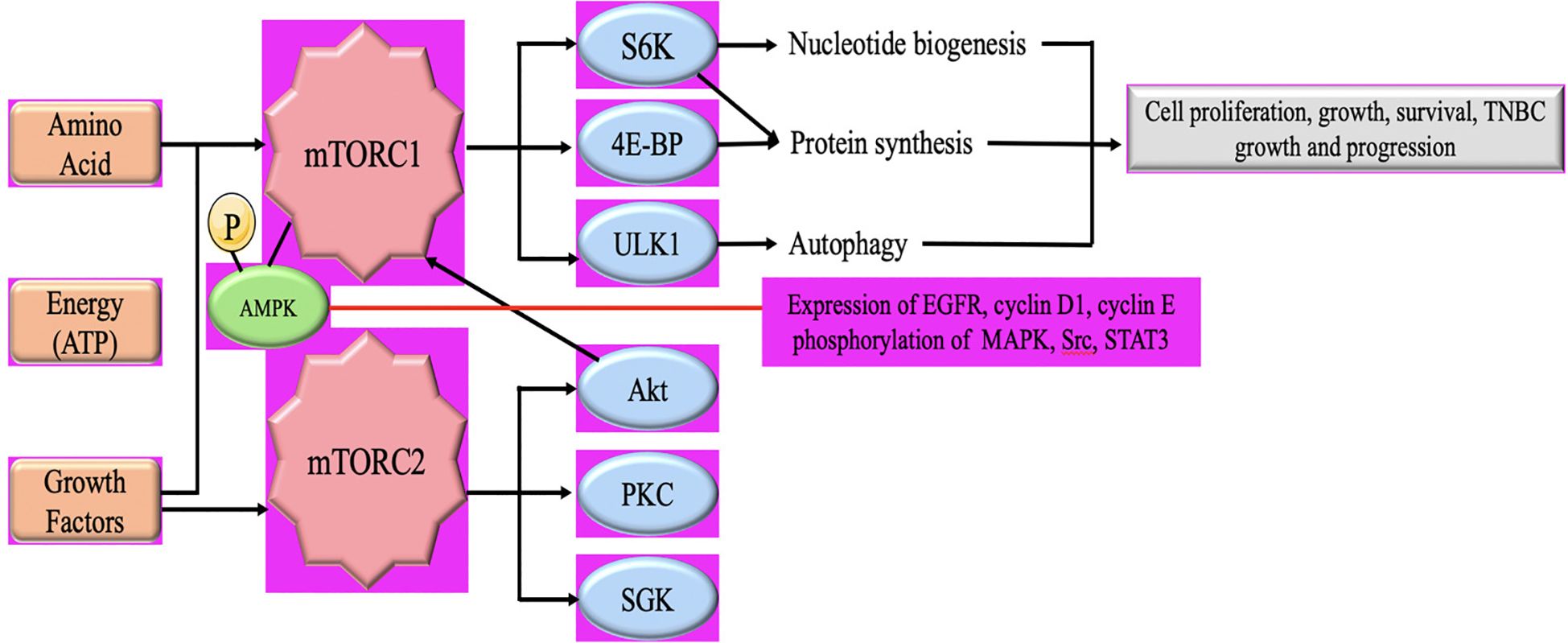
Figure 3. Schematic illustration of the AMPL/mTOR signaling pathway in TNBC Tumor growth and progression. mTORC1 comprises mTOR, mammalian lethal with sec-13 protein 8 (mLST8), and regulatory-associated protein of mammalian target of rapamycin (RAPTOR). mTORC1 is activated by growth factors, nutrients (amino acids), and cellular energy. mTORC2 consists of mTOR, mLST8, mammalian stress activated map kinase-interacting protein 1 (mSIN1) and rapamycin-insensitive companion of mTOR (RICTOR) and is activated by growth factors (62).
Monoclonal antibodies against mTOR pathway
The mTOR pathway comprises two complexes, mTORC1 and mTORC2, whose activity is meticulously regulated by several cellular signals, including growth hormones, nutrition, and stress indicators. Dysregulation of mTOR signaling has a significant role in carcinogenesis, making it a crucial therapeutic target, especially in TNBC (62). Currently, no monoclonal antibodies are authorized to directly activate mTOR in TNBC; however, some monoclonal antibodies influence mTOR signaling indirectly by targeting upstream pathways such as HER2, VEGF, PD-L1, and IGF-1R (66). These medicines can regulate mTOR activity, resulting in the suppression of tumor development. Nonetheless, mTOR inhibitors and combination treatments using monoclonal antibodies that target alternative pathways have shown potential in the treatment of TNBC via the modulation of mTOR signaling (67).
♣ Monoclonal Antibodies that Indirectly Modulate mTOR in TNBC:
Anti-PD-L1 Antibodies (e.g., Atezolizumab, Durvalumab): PD-L1 participates in immune evasion mechanisms, and the inhibition of PD-L1 with monoclonal antibodies such as atezolizumab (Tecentriq) and durvalumab (Imfinzi) may augment T-cell activation (68). Immune activation, including the stimulation of T-cell receptors (TCRs), may initiate many downstream signaling cascades, notably the PI3K/Akt/mTOR pathway (69). Although PD-L1 monoclonal antibodies are not intended to activate mTOR, they may indirectly augment immune responses that influence mTOR signaling inside the tumor microenvironment (70).
♣ Anti-IGF-1R antibodies: IGF-1R participates in the activation of the PI3K/Akt/mTOR signaling pathway. Monoclonal antibodies, such as figitumumab, inhibit the signaling pathway that results in mTOR activation. These monoclonal antibodies inhibit its aberrant activation, resulting in decreased cell survival and proliferation (71).
♣ Anti-VEGF antibodies: Inhibiting VEGF signaling using monoclonal antibodies such as bevacizumab (Avastin) might diminish the activation of downstream pathways, including the PI3K/Akt/mTOR pathway, since angiogenesis and tumor viability depend on these signaling mechanisms. Bevacizumab indirectly decreases mTOR by its impact on the tumor microenvironment (72).
Nuclear factor-κB activation
NF-κB, is a transcription protein that has a pivotal function in several physiological processes such as inflammation, immunological responses, cell proliferation, and longevity (73). In TNBC, NF-κB has been extensively studied because of its involvement in the development and progression of the disease. The activation of NF-κB in TNBC can occur through various mechanisms, and the most common pathway involves the canonical NF-κB pathway. Within this route, several triggers, like inflammatory mediators (e.g., TNF-α), growth factors, or pathogen-associated molecular patterns (PAMPs), initiate the signaling pathway that results in activation of κB kinase (IKK) complex (74, 75). The activated IKK complex phosphorylates the inhibitor of κB (IκB) proteins, which normally bind to NF-κB and keep it sequestered within the cytoplasm. The process of phosphorylating IκB results in its destruction, therefore releasing NF-κB to relocate to the nucleus. Upon entering the nucleus, NF-κB attaches itself to certain DNA sequences called κB sites, located in the promoter regions of target genes and stimulates the transcription of these genes (76). In TNBC, NF-κB activation can occur through various upstream signaling pathways, including those initiated by growth factor receptors (e.g., EGFR), cytokine receptors (e.g., TNF receptor), Toll-like receptors (TLRs), and DNA damage response pathways (77) (Figure 4). The transcriptional targets of NF-κB in TNBC include genes involved in cell survival (e.g., Bcl-2, Bcl-xL, cIAP1/2), cell proliferation (e.g., cyclin D1), angiogenesis (e.g., VEGF), invasion and metastasis (e.g., MMP-9, uPA), and inflammation (e.g., IL-6, IL-8, COX-2). By regulating the expression of these genes, NF-κB promotes tumor cell survival, proliferation, angiogenesis, and metastasis, and contributes to the development of an inflammatory microenvironment (79).
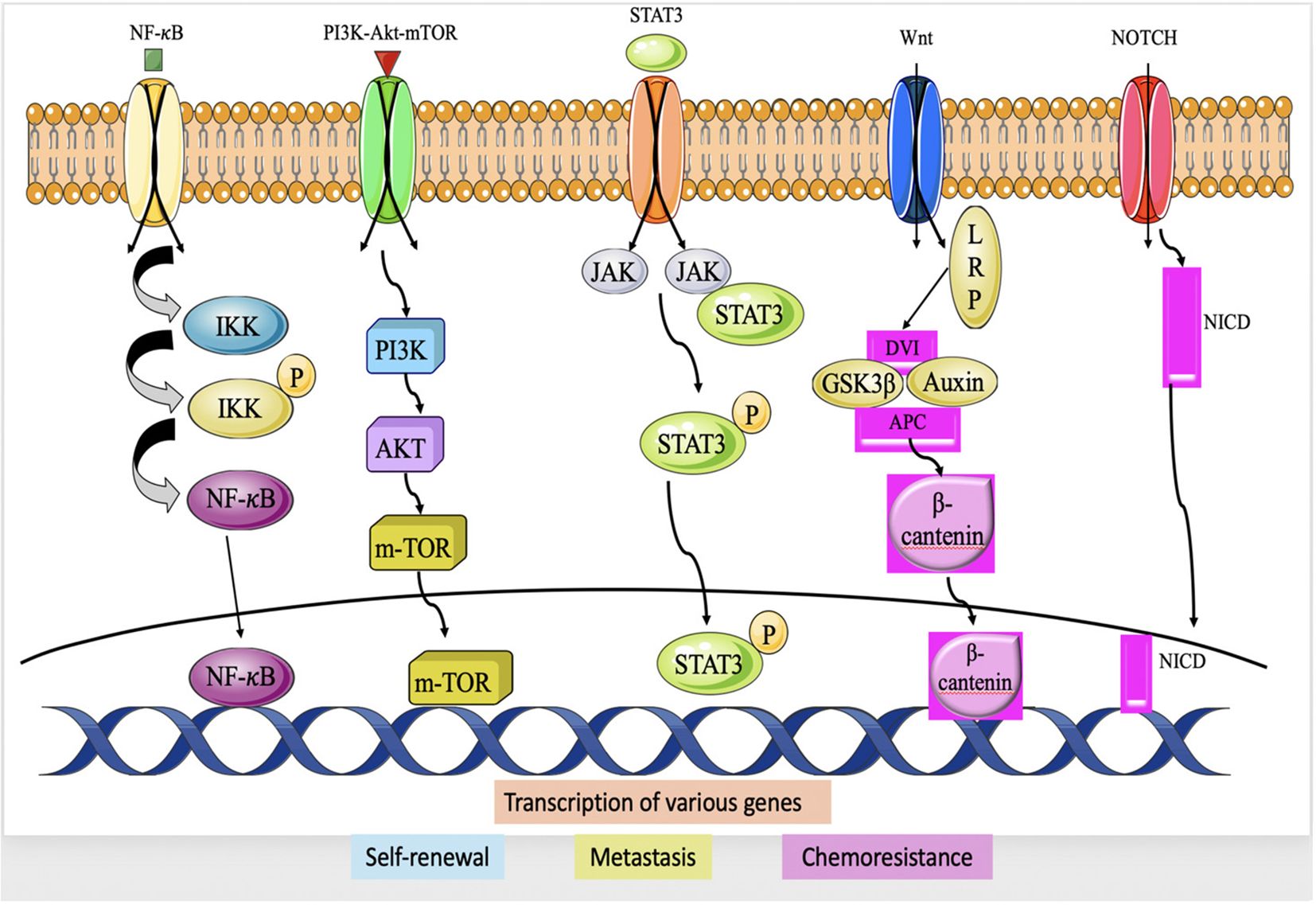
Figure 4. Major Signaling pathway in TNBC chemoresistance; IIK (IκB Kinase), NF-κB, PI3K, AKT, mTOR, JAK, STAT3, Dvl (Dishevlled) APC, GSK-3β, Notch intracellular domain (78).
Monoclonal antibodies against NF-κB pathway
Most treatment strategies concentrate on modifying immune responses or controlling inflammatory pathways that may indirectly influence NF-κB signaling in cancer cells and the tumor microenvironment (78). mAbs such as CD40 agonists, CD137 agonists, and TLR agonists are prospective immunomodulatory approaches that may affect NF-κB signaling via the activation of immune cells and the augmentation of anti-tumor immunity (80).
♣ CD40 Agonistic Monoclonal Antibodies: In TNBC, immune evasion is a considerable problem, and CD40 agonists are under investigation to enhance immunological responses, perhaps rendering tumors more vulnerable to immune cell-mediated eradication (81). CD40 is a costimulatory receptor on immune cells that activates NF-κB activation upon engagement. These antibodies are designed to activate CD40, hence initiating NF-κB signaling and further immune-activating pathways, including cytokine generation and T-cell activation. These reactions may indirectly stimulate NF-κB activation in immune cells (macrophages, dendritic cells) and tumor cells, hence augmenting anti-tumor immunity (82).
♣ CD137 (4-1BB) agonistic antibodies: CD137 is a costimulatory receptor present on T-cells, and its activation has been shown to initiate NF-κB signaling and T-cell activation (83). Agonistic antibodies such as Urelumab are under investigation in clinical studies to elicit immunological responses via NF-κB activation in T-cells. These antibodies indirectly activate NF-κB in cancer cells but may affect the tumor microenvironment, possibly altering NF-κB pathways in immune or cancer cells (84).
♣ TLR (Toll-Like Receptor) Agonists: TLRs are pattern recognition receptors that are crucial in immunological responses and in activation of NF-κB. TLR agonists, like Imiquimod, may stimulate NF-κB in immune cells such as dendritic cells and macrophages, hence enhancing the synthesis of pro-inflammatory cytokines (85). Although Imiquimod is not a monoclonal antibody, it exemplifies the activation of NF-κB via innate immunological signaling, which may be investigated in conjunction with monoclonal antibody treatment (86).
♣ Anti-DR5 (Death Receptor 5) Monoclonal Antibodies: Investigations into TRAIL (TNF-related apoptosis-inducing ligand) receptors and DR5 in TNBC indicates that agonistic antibodies may activate NF-κB in both immune and tumor cells, resulting in apoptosis and increased treatment sensitivity. Nonetheless, a significant portion of this research remains in the preclinical or experimental phase (87, 88).
♣ Targeting NF-κB Pathway Regulators (Inhibition of IκB Kinases and TAK1): In TNBC, dysregulated NF-κB activation fosters inflammation, immunological evasion, and metastasis (89). IKKβ and TAK1 serve as upstream regulators of the NF-κB signaling cascade. Inhibition of these pathways is being investigated to mitigate abnormal NF-κB activation in cancer (90).
Wnt/β-catenin pathway
The Wnt/β-catenin signaling system is of utmost importance in the genesis, growth, and spread of tumors in TNBC. The Wnt/β-catenin system is a ubiquitous signaling cascade that controls several physiological functions, such as stem cell maintenance, differentiation and cell proliferation. The fundamental constituents of the Wnt/β-catenin cascade are Frizzled receptors, Wnt ligands, and the intracellular protein β-catenin. When Wnt signaling is not present, β-catenin undergoes phosphorylation by a destruction complex composed of casein kinase 1 (CK1), adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK-3β) and axin that leads its destruction, hence maintaining low levels of β-catenin in the cytoplasm. The binding of Wnt ligands to Frizzled receptors initiates the signaling pathway that suppresses the expression of the destruction complex. Consequently, β-catenin builds up in the cytoplasm and moves into the nucleus (91). Within the nucleus, β-catenin engages with components of the T-cell factor/lymphoid promoter factor (TCF/LEF) family of transcription factors, resulting in the stimulation of certain genes responsible for cell division and viability (92).
In TNBC, aberrant activation of the Wnt/β-catenin pathway is commonly observed, contributing to tumor growth and progression. Several mechanisms can lead to the dysregulation of this pathway in TNBC, which include Wnt ligand overexpression (93). TNBC cells can produce excess Wnt ligands, which can activate the pathway in an autocrine manner. Changes or modifications in the genes that encode proteins participating in the Wnt/β-catenin cascade, like Axin1, β-catenin (CTNNB1) or APC might disturb the control of β-catenin and result in its stabilization and movement into the nucleus. (94, 95). Various signaling pathways, such as the PI3K/AKT pathway, can activate the Wnt/β-catenin pathway by inhibiting the activity of GSK-3β or promoting the nuclear translocation of β-catenin (96) (Figure 5).
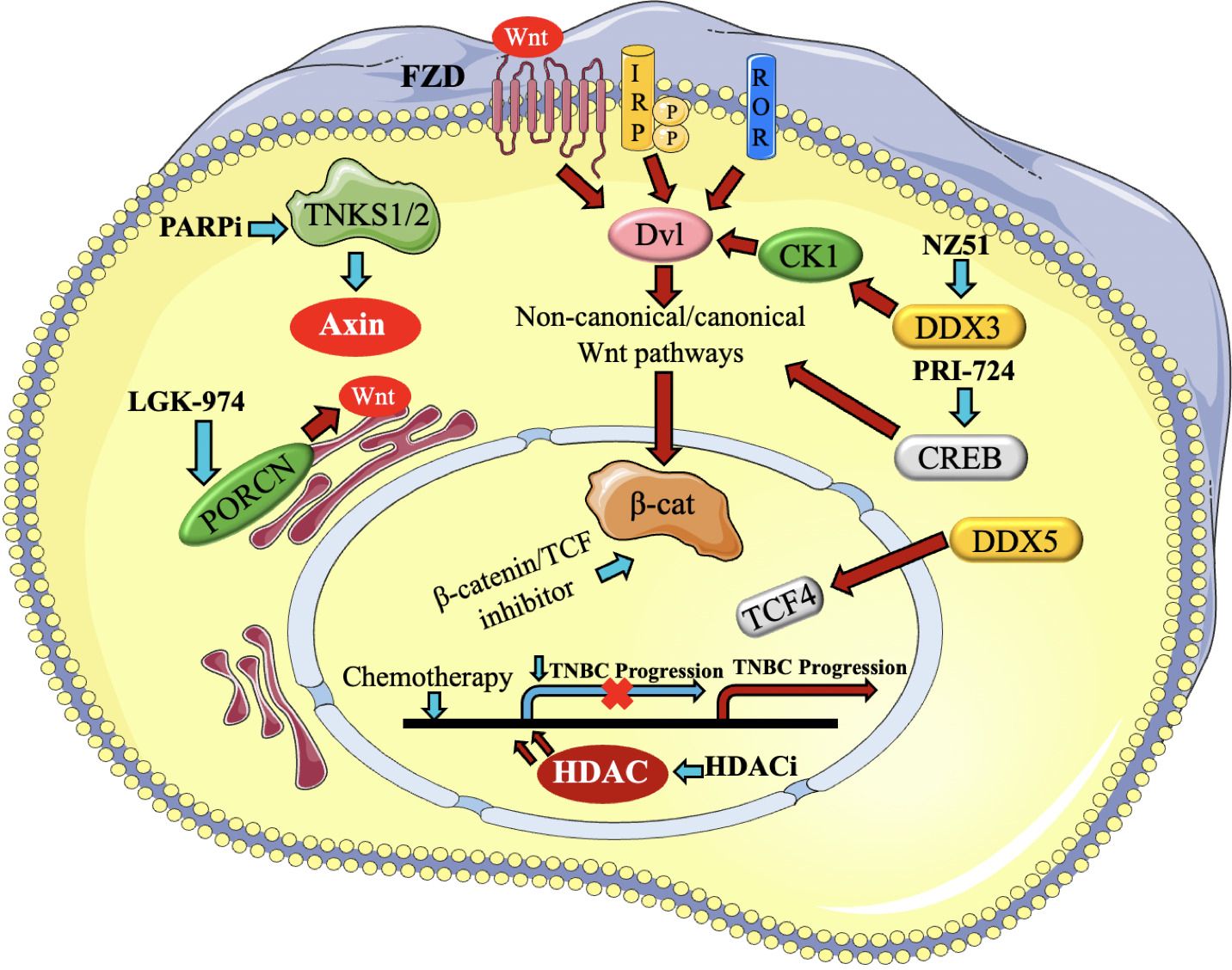
Figure 5. Presents a brief description of the regulators of Wnt signaling that contribute to the advancement of triple- negative breast cancer (TNBC) and the medicines that target them. The activation of canonical and non-canonical Wnt pathways occurs via Fzd, LRP, and ROR receptors. Blue arrows represent the reduction or blocking of Wnt regulators and routes, resulting in the impairment of Wnt target gene transcription, as represented by a yellow cross. On the other hand, red arrows reflect the stimulation of Wnt regulators and pathways (97).
The aberrant stimulation of the Wnt/β-catenin cascade in TNBC is associated with increased cell proliferation, decreased apoptosis, enhanced cancer stem cell properties, and resistance to chemotherapy. As a result, focusing on this sequence of events has become a possible treatment approach for TNBC (94). Experimental studies and preclinical models have explored the use of Wnt/β-catenin inhibitors, such as small molecule inhibitors or monoclonal antibodies, to suppress the pathway and inhibit tumor growth. Even though Wnt/β-catenin pathway is an attractive therapeutic target, further research is needed to fully understand its complexity and identify optimal strategies for intervention in TNBC (98, 99).
Monoclonal antibodies against Wnt/β-catenin pathway
The Wnt/β-Catenin pathway is often linked to cancer growth, metastasis, and chemoresistance; hence, most research has concentrated on blocking this system rather than activating it directly (97). Ongoing research is investigating monoclonal antibodies (mAbs) that may alter the Wnt/β-Catenin pathway, either indirectly or by targeting upstream components, to influence the tumor microenvironment or immune responses. mAbs specially engineered to activate Wnt/β-Catenin signaling in TNBC are few, with research mostly concentrating on the pathway’s suppression for therapeutic applications (100).
♣ Monoclonal Antibodies that May Influence Wnt/β-Catenin in TNBC
♣ Anti-LRP5/6 (Low-Density Lipoprotein Receptor-Related Protein 5/6) antibodies: LRP5 and LRP6 are co-receptors essential for the activation of Wnt/β-catenin signaling. Inhibiting the interaction between Wnt ligands and LRP5/6 generally suppresses Wnt signaling (101); however, agonistic antibodies directed at these co-receptors may activate Wnt/β-catenin signaling by promoting the formation of the Wnt receptor complex. The advancement of these medicines is mostly in the preclinical stage (102).
♣ GSK3β (Glycogen Synthase Kinase 3 Beta) inhibitors function by inhibiting the kinase GSK3β, which phosphorylates β-catenin, resulting in its destruction and subsequently suppressing Wnt/β-catenin signaling pathway (103). GSK3β inhibitors have been investigated for their potential to enhance β-catenin signaling, which may be used to influence tumor cell characteristics, including stemness and chemoresistance in TNBC (104). Nonetheless, GSK3β inhibitors remain mostly in preclinical or early-phase clinical trials.
Epidermal growth factor receptor pathway
EGFR is a member of the ErbB family of receptor tyrosine kinases, and its activation is primarily triggered by the binding of epidermal growth factor (EGF) or other ligands. In TNBC, EGFR is frequently overexpressed which is associated with several mechanisms that contribute to tumor progression and aggressiveness (105). EGFR signaling pathways stimulate cell proliferation and survival, promoting the growth and survival of TNBC cells. EGFR signaling amplifies the aggressive and spreading capabilities of TNBC cells and stimulates the development of matrix metalloproteinases (MMPs), that break down the extracellular matrix and accelerate the advancement and spread of malignant cells (106). EGFR signaling stimulates epithelial-mesenchymal transition (EMT), a mechanism that enables cancerous cells to adopt an enhanced mesenchymal phenotype, hence promoting infiltration and proliferation. It further enhances angiogenesis, by triggering the creation of pro-angiogenic substances including vascular endothelial growth factor (VEGF) (107) (Figure 6).
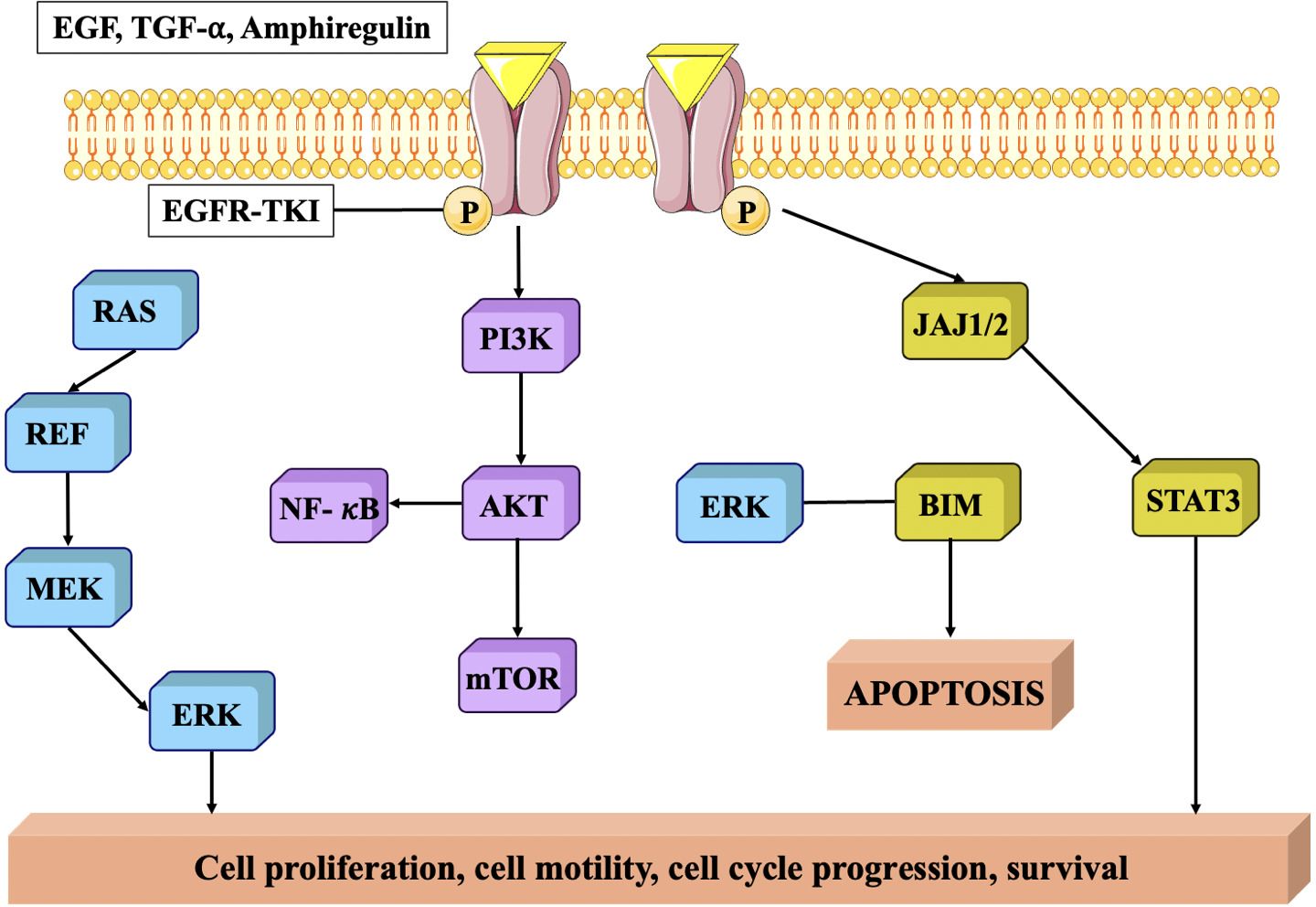
Figure 6. EGFR protein signaling pathway. EGFR is closely related to tumor cell proliferation, angiogenesis, tumor invasion, metastasis and inhibition of apoptosis (108).
In addition, overexpression of EGFR in TNBC has been linked to resistance to certain treatments. For example, it can activate pro-survival pathways that counteract the effects of chemotherapy drugs and can also interfere with the efficacy of targeted therapies, such as anti-HER2 treatments, by cross-talk between EGFR and HER2 signaling pathways (109, 110).
Monoclonal antibodies against EGFR pathway
The majority of monoclonal antibodies (mAbs) directed against EGFR in TNBC have focused on obstructing EGFR signaling to limit tumor proliferation.
♣ Monoclonal antibodies that modulate EGFR signaling in TNBC: EGFR-targeting bispecific antibodies have promise in TNBC, particularly because to the disease’s frequent immune resistance. Bispecific antibodies such as AMG 595 simultaneously target EGFR on tumor cells and CD3 on T cells, facilitating the closeness of immune cells to tumor cells. This medication may indirectly activate EGFR, leads to enhanced immune response via T cell activation having tumoricidal action (111).
♣ Combination therapies using EGFR-targeting monoclonal antibodies, such as Cetuximab or Panitumumab, are being integrated with immune checkpoint inhibitors (e.g., nivolumab, pembrolizumab) to augment the immune response. This strategy indirectly modify EGFR signaling while activating T cells via checkpoint inhibition in TNBC, resulting in enhanced tumor control (108).
♣ Anti-EGFRvIII Antibodies (Mutant EGFR Variant): EGFRvIII is a mutated variant of EGFR that is devoid of the extracellular ligand-binding domain and is often linked to more aggressive malignancies, such as TNBC. mAbs that target EGFRvIII induce receptor activation and subsequent signaling pathways. This constitute a strategy that alters the tumor microenvironment to counteract immune evasion or trigger apoptosis in cells harboring mutant EGFR (112).
JAK/STAT cascade
The JAK/STAT pathway is a signaling cascade that facilitates the transmission of extracellular signals to the cell nucleus, resulting in the expression of certain genes. The composition of this entity includes activator of transcription (STAT) proteins, signal transducer and Janus kinases (JAKs). Cytokines, which are tiny proteins involved in cell signaling, often trigger this signaling pathway (113). In TNBC, mediators like IFN-γ and IL-6 are often increased. The interaction between these mediators and their receptors initiates the expression of JAKs, resulting in the phosphorylation of STAT proteins. These proteins combine to form dimers and are exported to the nucleus, where they attach to certain DNA fragments called responder factors. The engagement of this molecule initiates the procedure of transcribing certain genes responsible for regulating a wide range of biological processes, such as differentiation, survival, development, and migration (114) (Figure 7). The JAK/STAT pathway in TNBC may stimulate tumor development by activating the transcription of genes related to cell cycle advancement, inhibition of programmed cell death, and formation of new blood vessels. Additionally, it can also regulate immunological responses (116). Activation of STAT3, stimulates the production of immunosuppressive substances such as programmed death-ligand 1 (PD-L1) and interleukin-10 (IL-10), which hinder the function of defence cells, namely cytotoxic T lymphocytes. This immune evasion mechanism can contribute to tumor immune escape and resistance to immunotherapies (117, 118).
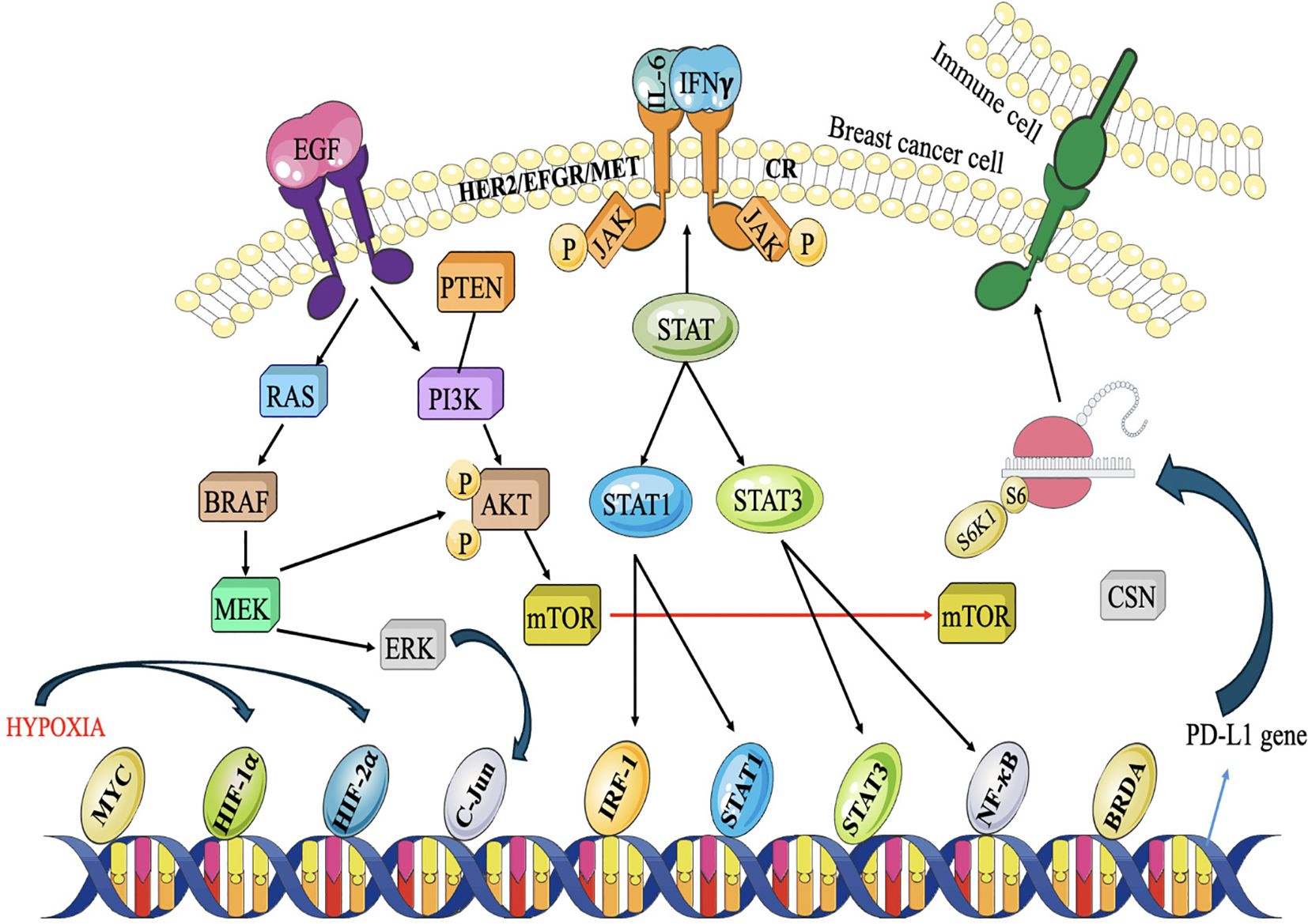
Figure 7. Akt, MAPK, and JAK/STAT pathways collaborate to inhibit the antitumor immune system. Under hy- poxic circumstances caused by Akt signaling, breast cancer cells produce PD-L1 to suppress T-cells. Three major signaling pathways are involved. RTKs stimulate the PI3K/Akt and MAPK pathways, whereas cytokine receptors (CR) are activated by cytokines released into the TME. JAK/STAT signaling is then initiated, recruiting STAT1/3 and other transcription factors to certain gene promoters, including the PD-L1 promoter. Proliferation and metas- tasis are triggered while anti-cancer immunity is deactivated (115).
Monoclonal antibodies against JAK/STAT signaling pathway
Although direct activation of JAK/STAT signaling with monoclonal antibodies is not a conventional method in the treatment of TNBC, the modulation of this pathway for therapeutic purposes is now a focus of ongoing study (119). Monoclonal antibodies and associated techniques that are currently under development are:
♣ Anti-IL-6 Receptor Antibodies (Indirect Modulation of JAK/STAT): IL-6 is a cytokine that stimulates the JAK/STAT3 system, via STAT3 phosphorylation, which facilitates tumor proliferation and immune evasion in TNBC (120). Tocilizumab, a monoclonal antibody targeting the IL-6 receptor, inhibits the interaction between IL-6 and its receptor, thereby obstructing STAT3 activation. Moreover, inhibiting IL-6 signaling may indirectly activate immunological responses by restoring immune cell function and thereby diminishing tumor immune evasion (115).
♣ Siltuximab an anti-IL-6 mAb that directly neutralizes IL-6, inhibiting its interaction with receptor and consequent JAK/STAT3 activation. Siltuximab generally inhibits IL-6-mediated signaling; which inadvertently stimulate immune system and surmount immunological resistance, resulting in enhanced therapeutic outcomes. This method is still under investigation in conjunction with combo medicines (121).
♣ Anti-GM-CSF monoclonal antibodies: GM-CSF, a cytokine that activates the JAK/STAT pathway and is involved in inflammatory processes and immune cell functionality (122). Gimsilumab is an antibody that targets GM-CSF and may influence JAK/STAT signaling by influencing the inflammatory milieu, including macrophages and dendritic cells, thus impacting the immunological suppression seen in TNBC. This may indirectly bolster the immune response, thereby aiding in the inhibition of tumor development (123).
♣ Anti-CD40 Monoclonal Antibodies (Indirect Modulation of JAK/STAT via Immune Activation). CD40 is a co-stimulatory receptor present on immune cells such as dendritic cells and macrophages. The activation of CD40 may initiate diverse immunological responses and result in the activation of JAK/STAT signaling in immune cells (124). CP-870,893 is an agonistic monoclonal antibody that targets CD40, hence promoting immunological activation and regulating the JAK/STAT pathway in immune cells indirectly (83). This method enhances the immune evasion often seen in TNBC by activating immunological responses and stimulating immune activation via JAK/STAT signaling in immune cells. CP-870,893 is under investigation in conjunction with other therapies to augment antitumor immunity (125, 126).
♣ Directly Targeting JAK1/2 with Monoclonal Antibodies (In Development): JAK1 and JAK2 are essential elements of the JAK/STAT signaling cascade. Oral inhibitors such as Ruxolitinib and Momelotinib are often used in hematologic malignancies, although mAbs that target JAK1/2 remain under development. These antibodies may influence the pathway in certain immunological settings or tumor microenvironments, thereby affecting tumor development or immune evasion seen in TNBC (127).
♣ Monoclonal Antibodies Targeting IL-4 and IL-13: IL-4 and IL-13 are cytokines capable of activating the JAK/STAT6 pathway, particularly within the Th2 immune response. Dupilumab is an antibody that inhibits IL-4 and IL-13 signaling, potentially affecting JAK/STAT signaling in the immune system indirectly. This regulate immune cell function and affect tumor immune evasion (128, 129).
Notch pathway
The Notch signaling cascade is strongly preserved and is vital in several biological activities, like determining cell destiny, promoting cell growth, facilitating cell specialisation, and ensuring cell survival (130). The Notch pathway is often disrupted in TNBC, resulting in aberrant signaling that plays a role in the pathogenesis and advancement of the illness. The disruption of Notch signaling in TNBC may arise from several causes, including genetic changes, modified expression levels of Notch receptors and ligands, and abnormal stimulation of subsequent signaling elements (131).
The fundamental constituents of the Notch pathway are Notch receptors (Notch1-4) and their ligands (Jagged1, Jagged2, Delta-like 1, 3, and 4). The Notch pathway is activated by a sequence of proteolytic cleavages. At first, Notch receptors are produced as a solitary polypeptide, subjected to proteolytic cleavage when a ligand binds to it. This process entails the consecutive fragmentation of the receptor by many enzymes, finally liberating the Notch intracellular domain (NICD). After being discharged, NICD moves to the nucleus and combines with other proteins, including co-activators, mastermind-like (MAML) and CSL (CBF1/RBPJκ, Su(H), Lag-1) to create a complex that activates certain genes (132, 133). These targeted genes affect several biological processes and pathways related to TNBC, including cell growth, longevity, EMT, vascular development, and stemness (134) (Figure 8).
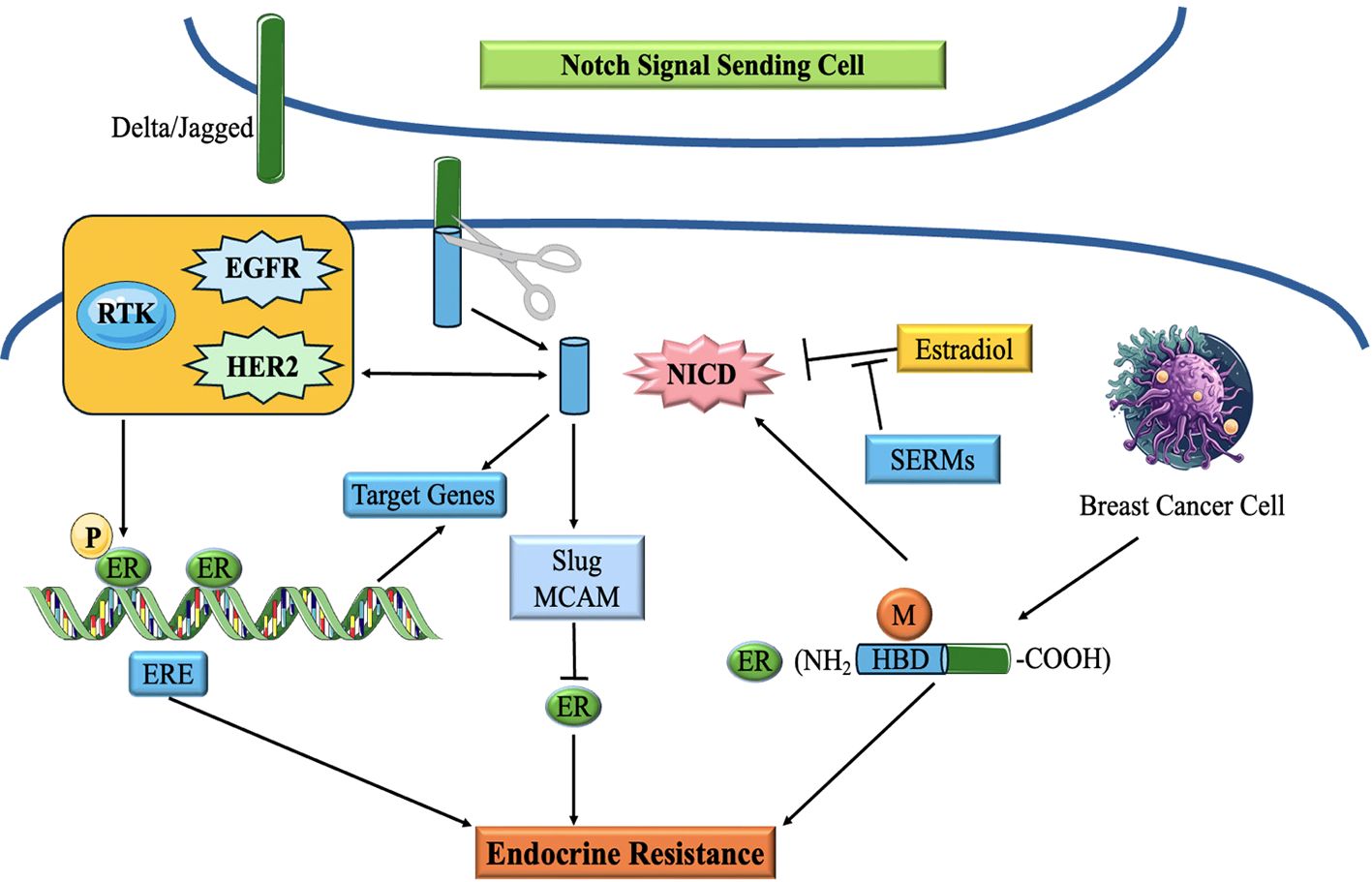
Figure 8. Schematic diagram of the correlation of Notch signaling pathway and ER in breast cancer. Notch Signaling modulates endocrine therapy by cooperating with ER in a complex network (135).
Notch signaling has been demonstrated to interact with various signaling systems implicated in TNBC, including the EGFR cascade and the Wnt/β-catenin system, hence amplifying its carcinogenic effects. Moreover, abnormal Notch signaling in TNBC has been linked to the initiation of EMT, a mechanism that facilitates tumor penetration and propagation (136, 137).
The activation of Notch signaling enhances the ability of cells to renew themselves and facilitates the development of tumor-initiating cells, also referred to as cancer stem cells (CSCs). The Notch pathway has a role in maintaining CSCs in TNBC by controlling the regulation of genes linked to stem cell characteristics, including Sox2, Nanog and Oct4 (138, 139). The Notch pathway in TNBC may resist conventional chemotherapy agents, such as paclitaxel and doxorubicin and regulate the production of drug efflux pumps, like ABC carriers, which decrease the number of chemotherapeutic drugs that build up within cells. Moreover, its activation can promote the survival of cancer cells by suppressing apoptosis and enhancing DNA repair mechanisms (140).
Monoclonal antibodies against Notch signaling pathway
Researches are being conducted on activating the Notch pathway through monoclonal antibodies, with greater emphasis on modulating tumor progression and immune responses rather than directly inducing Notch signaling for therapeutic benefits (135).
♣ Anti-Jagged1 Monoclonal Antibodies: Jagged1 serves as a ligand that activates Notch receptors, particularly Notch1, by interaction with the receptor’s extracellular domain (141). UHD 156, a monoclonal antibody that specifically targets Jagged1, obstructing its interaction with Notch receptors and hence inhibiting its activation leads to diminished tumor proliferation and stem cell properties (142).
♣ Anti-Delta-like Ligands (DLL) Monoclonal Antibodies: Delta-like 4 (DLL4) serves as a ligand that stimulates the Notch signaling pathway, particularly Notch1 and Notch4 (143). PF-06747775, a monoclonal antibody that specifically targets DLL4 and inhibits its capacity to activate Notch receptors, results in diminished Notch signaling. Researchers are also investigating mAbs with other medicines to enhance tumor sensitivity to therapy (144).
♣ Anti-Notch3 mAbs: Notch3 is significantly expressed in TNBC and is essential for the sustenance of cancer stem cells and tumor advancement (145). Investigations are underway about anti-Notch3 mAbs, which may diminish the proliferation and metastatic capacity of TNBC cells, particularly in cases resistant to standard treatment (146). Antibodies targeting Notch3 may also have the ability to influence immune responses inside the tumor microenvironment.
♣ Monoclonal antibodies directed against the Notch intracellular domain (NICD): Following ligand interaction with Notch receptors, the NICD undergoes cleavage and translocates to the nucleus, where it initiates the activation of target genes (147). Research is exploring methods to impede the activation of NICD, linked to tumor stem cell preservation, treatment resistance, and metastasis in TNBC (148).
♣ Notch Agonistic Antibodies (Under Investigation): Although the majority of current research emphasizes the inhibition of the Notch pathway in TNBC (149), there are continuing studies exploring agonistic mAbs that may stimulate Notch signaling in a regulated manner to promote the differentiation of cancer stem cells (150). By activating Notch receptors, these antibodies may compel tumor cells to differentiate, diminishing their stem-like characteristics and enhancing their susceptibility to treatment (151).
DNA damage response pathway
DDR is essential for preserving genomic integrity and inhibiting cancer formation. Approximately 15-20% of TNBC cases are associated with germline mutations in the BRCA1 gene. BRCA1 is an important DDR gene involved in DNA repair through homologous recombination (HR) (152). Mutations in BRCA1 impair its function, leading to defective DNA repair and increased genomic instability. These defects make TNBC cells more susceptible to further DNA damage and contribute to tumor development (153). Besides BRCA1 mutations, TNBC can exhibit other defects in HR DNA repair pathways, leading to a state of HRD. HRD causes a reduced capacity to restore DNA double-strand breaks (DSBs) and results in the buildup of genetic changes, which contributes to the development of TNBC tumors (154). TNBC tumors harbouring BRCA1 mutations or (HRD) are more susceptible to medicines that specifically target DNA repair proteins, like poly(ADP-ribose) polymerase (PARP) antagonists (155, 156) (Figure 9). DDR dysregulation, including defective DNA repair mechanisms, contributes to the accumulation of these genetic abnormalities which are believed to drive tumor progression and the development of aggressive phenotypes in TNBC (158).
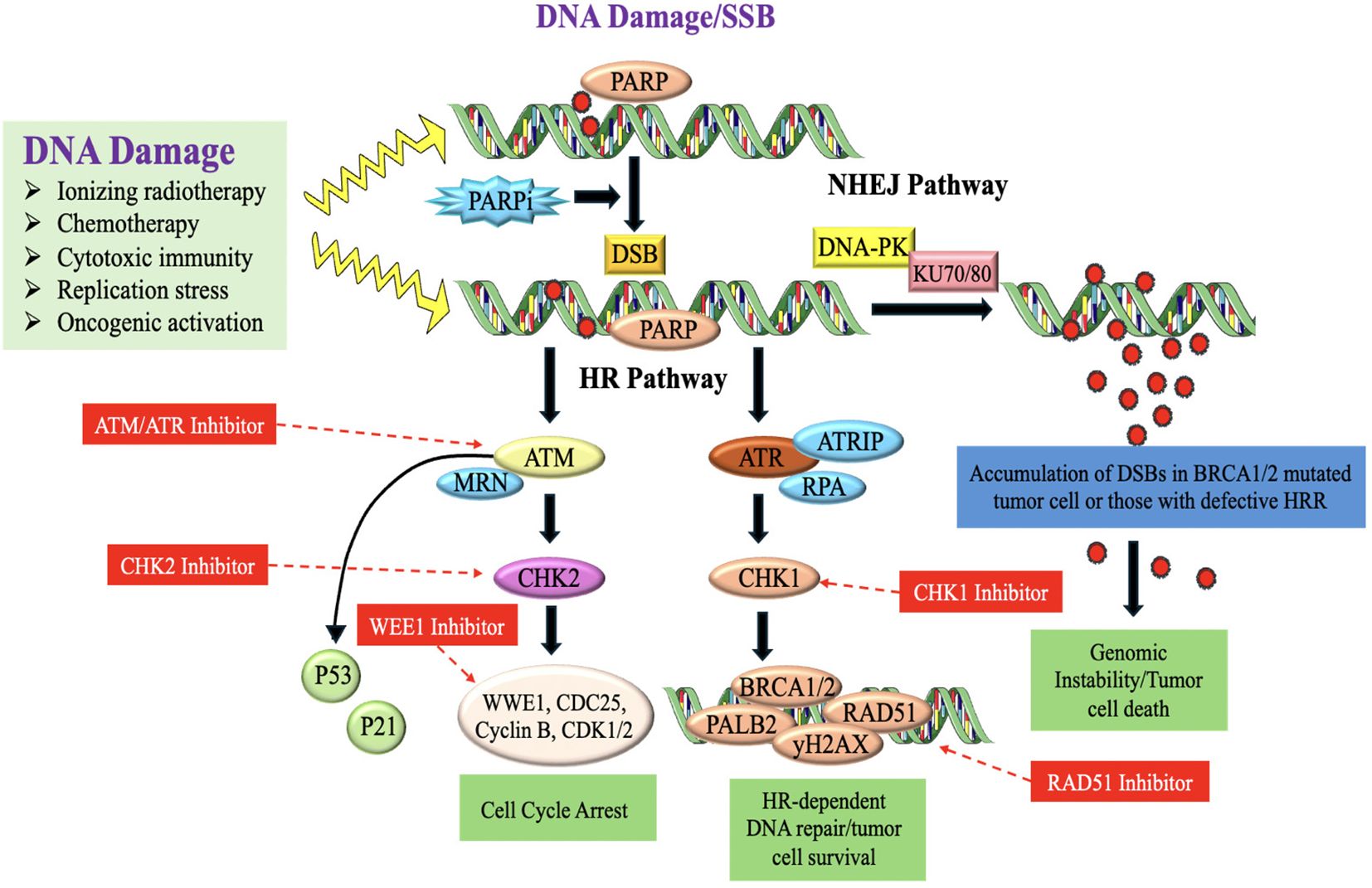
Figure 9. DNA-damaging therapies or endogenous replication dysfunction results in SSBs and DSBs which activate the DDR and repair signaling pathways (157).
Monoclonal antibodies against DNA damage response pathway
Activating the DDR in cancer treatment seeks to provoke genomic instability and death in tumor cells, especially in tumors with deficient DDR pathways (157). Research is continuing to elucidate how mAbs may influence the DDR pathway in the setting of TNBC. This involves targeting essential DDR proteins or augmenting DDR activity in tumor cells to provoke cell death or increase tumor sensitivity to radiation or chemotherapy. Certain medicines seek to stimulate DDR to surmount treatment resistance and induce apoptosis in tumor cells (159).
♣ Monoclonal Antibodies Directed Against DDR Pathways in TNBC:
Anti-ATM Monoclonal Antibodies: ATM is crucial for identifying DNA double-strand breaks and initiating subsequent repair mechanisms. Inhibition of ATM may enhance the susceptibility of cancer cells to DNA-damaging treatments such as radiation or chemotherapy (160). MedI-15 a developing monoclonal antibody that targets ATM, inhibiting its activity and possibly inducing genomic instability in cancers with abnormalities in DDR. Inhibition of ATM in TNBC, particularly in BRCA-deficient tumors, might augment the efficacy of chemotherapy or radiation by impairing repair mechanisms, eventually leading to cell death (161, 162).
♣ Anti-ATR Monoclonal Antibodies: ATR is an essential protein in the DDR system, particularly in the repair of single-strand breaks and the preservation of replication fork stability (163). Ceralasertib, principally a small molecule ATR inhibitor, is under investigation in conjunction with monoclonal antibodies to decrease ATR activity (164). This inhibition may elicit synthetic lethality in BRCA1/2-deficient TNBC by obstructing the repair of replication-associated DNA damage, hence rendering tumor cells more vulnerable to DNA-damaging therapies (165).
♣ Anti-PARP monoclonal antibodies enhance DNA damage response in BRCA-deficient TNBC by targeting PARP, which is important to repair single-strand DNA breaks. PARP inhibitors (PARPi), such as Olaparib and Veliparib, impede the repair of single-strand breaks, resulting in double-strand breaks that need BRCA1/2-mediated repair. In BRCA-deficient TNBC, PARP inhibition induces synthetic lethality, facilitating tumor cell apoptosis (166).
♣ Anti-BRCA Monoclonal Antibodies (Targeting DNA Repair Pathways): BRCA1/2 are essential components of homologous recombination (HR), a vital DNA repair process. mAbs directed against BRCA1/2 seek to alter the tumor’s capacity for DNA damage repair, hence enhancing vulnerability to chemotherapy and radiation. These drugs are being investigation in conjunction with other medicines to elicit synthetic lethality (157, 167).
♣ Anti-Checkpoint Kinase 1 (CHK1) Antibodies: CHK1 is a kinase that modulates the cell cycle checkpoint in reaction to DNA damage. Inhibition of CHK1 may result in cell cycle arrest and heightened DNA damage, hence amplifying the cytotoxic effects of chemotherapy and radiation. Prexasertib a CHK1 inhibitor that may be used in conjunction with mAbs aimed against DDR proteins to enhance the sensitivity of cancer cells (168).
Hedgehog pathway
The Hedgehog signaling system is essential in the development of TNBC. This signaling system is strongly preserved and controls a range of biological functions in embryonic development and tissue stability. Anomalous stimulation of this mechanism has been linked to the formation and advancement of numerous forms of malignancies, such as TNBC (169, 170). The Hedgehog pathway consists of numerous essential elements, which include Hedgehog ligands (Desert Hedgehog, Indian Hedgehog, and Sonic Hedgehog), transmembrane receptor Patched (PTCH), Smoothened (SMO), and downstream transcription factors belonging to the GLI family (171). When Hedgehog ligands are not present, PTCH prevents SMO from functioning, which leads to the suppression of Hedgehog pathway activity. When Hedgehog ligands connect to PTCH, SMO is no longer inhibited and moves to the main cilium, where it triggers downstream signaling processes (172).
In TNBC, dysfunctioning of the Hedgehog cascade is often linked with increased expression and secretion of Hedgehog ligands, such as Sonic Hedgehog. The activated Hedgehog signaling pathway promotes cancer stemness, cell proliferation, survival, angiogenesis, and metastasis, all of which contribute to TNBC pathogenesis. One of the key mechanisms by which the Hedgehog pathway promotes TNBC is through the activation of cancer stem cells (CSCs) (173, 174). In addition, the Hedgehog system stimulates angiogenesis, by increasing the production of pro-angiogenic proteins such as VEGF (134, 175). Angiogenesis is essential for the progression of tumors since it supplies oxygen and nutrients to support the development of the tumor mass and its spread to other parts of the body. This route also collaborates with other signaling mechanisms, including the Notch pathways and Wnt/β-catenin to facilitate the advancement of TNBC (176) (Figure 10).
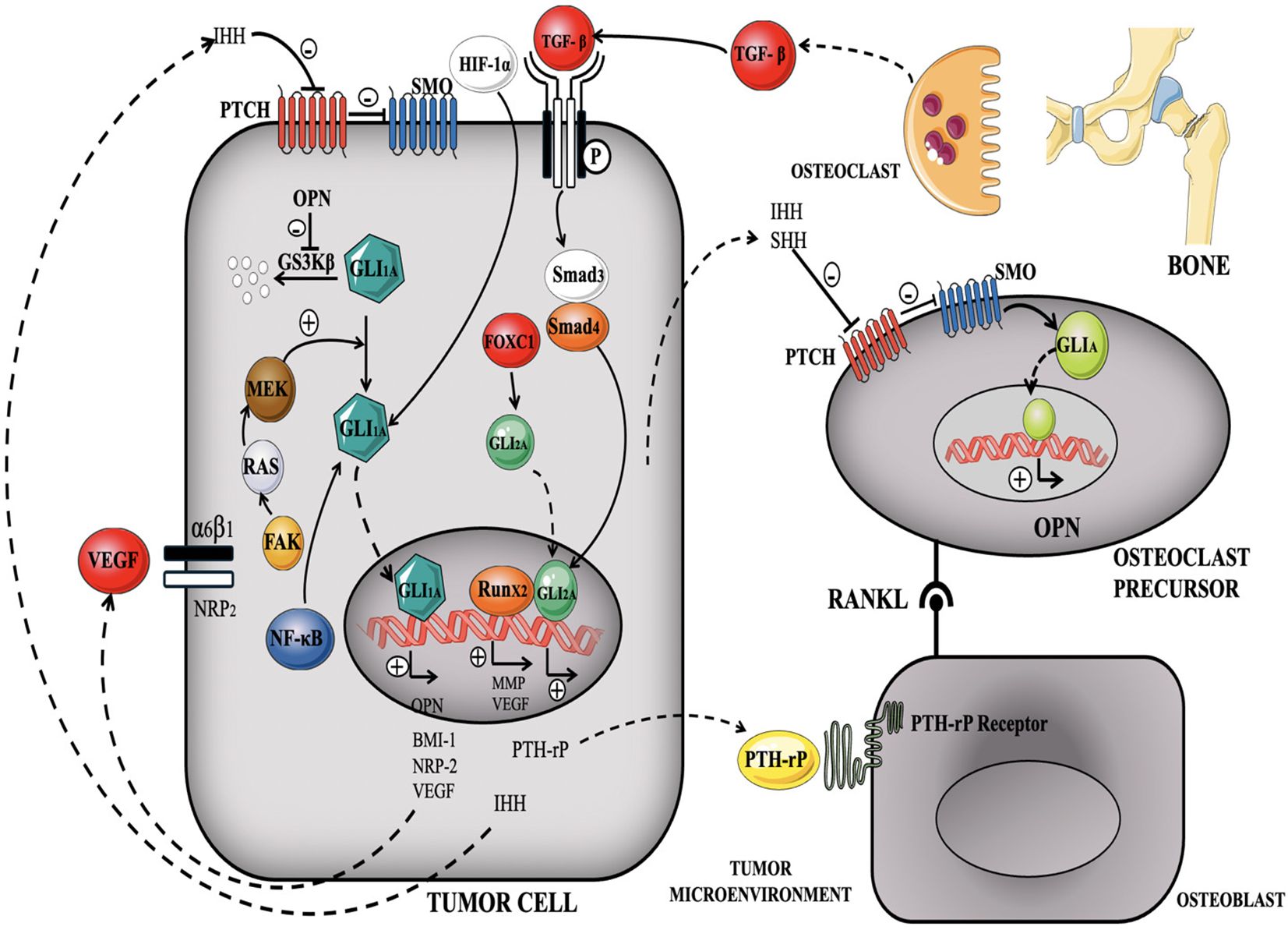
Figure 10. illustrates the triggering of Hedgehog signaling in TNBC, demonstrating the interaction between ligand-dependent and ligand-independent mechanisms (177).
Amplification of Hedgehog system increases cell cycle regulators, like Cyclin D1 and Cyclin E, resulting in elevated cell division and facilitating tumor development. Furthermore, this network promotes activation of anti-apoptotic proteins, including Bcl-xL and Bcl-2, which hamper apoptosis and enhance the survival of TNBC cells. Research has demonstrated that the amplification of the Hedgehog mechanism in TNBC is linked to the development of resistance to traditional chemotherapy medications by improving the production of drug efflux pumps, like ATP-binding cassette transporters. These pumps help remove chemotherapy medications from cancerous cells resulting in diminished efficacy (64, 178).
Monoclonal antibodies against hedgehog pathway
Monoclonal antibodies specially engineered to activate Hedgehog signaling in TNBC are less extensively investigated than inhibitory techniques (177). Research indicates that altering the system to augment or restore Hedgehog signaling may provide therapeutic promise in certain settings, such as overcoming drug resistance or stimulating the differentiation of cancer stem cells (179).
Vismodegib (GDC-0449): Vismodegib is a synthetic, small-molecule inhibitor of SMO, a key component of the hedgehog pathway. It binds and hinders SMO (a trans membrane protein), thus preventing systemic activation of the forward signaling. It results in suppression of Gli-1/2 transcriptional activation which ultimately leads to BCC tumor suppression proving the potential of vismodegib both as a tumoricidal and a tumoristatic candidate (180).
Sonidegib (LDE225): An additional SMO inhibitor now undergoing clinical studies to assess its effectiveness in TNBC. Studies have investigated the use of Sonidegib with chemotherapeutic drugs to address chemoresistance and inhibit tumor proliferation (181).
Anti-Patched1 (PTCH1) Antibodies: PTCH1 is a receptor that typically suppresses SMO in the absence of Hedgehog ligands. Upon binding of Hedgehog ligands to PTCH1, SMO is activated, resulting in downstream signaling. Anti-PTCH1 antibodies may alter the pathway by either obstructing PTCH1 activity to impede tumor growth or by targeting tumor cells that overexpress PTCH1 to possibly reinstate Hedgehog signaling (182).
Anti-GLI Transcription Factor Antibodies: GLI1 and GLI2 are the principal transcription factors that facilitate the ultimate activation of Hedgehog target genes. Anti-GLI antibodies inhibit GLI activation, hence obstructing the production of genes that promote carcinogenesis, chemoresistance, and the maintenance of cancer stem cells. These antibodies are under preliminary development and may be investigated in TNBC to inhibit tumor proliferation and enhance cellular sensitivity to treatment (183).
Anti-Hedgehog Ligand (Shh) Antibodies: Shh is a principal ligand that activates the Hedgehog pathway via its interaction with PTCH1. Anti-Shh mABs may inhibit this ligand’s capacity to activate the pathway, hence obstructing downstream signaling events that contribute to tumor growth and chemoresistance (184).
MET pathway
The MET pathway is primarily involved in cell growth, survival, motility, and invasion. It is activated by the hepatocyte growth factor (HGF), which binds to the MET receptor tyrosine kinase. When HGF binds to MET, it triggers a signaling cascade that leads to various cellular responses (185). The MET pathway in TNBC exhibits irregularity, which is associated with cancer growth and metastasis (Figure 11). The MET pathway enhances cellular proliferation and survival in TNBC (187). The signaling pathway triggers cell cycle progression, allowing cancer cells to grow and divide uncontrollably. It amplifies the capacity of TNBC cells to invade and spread, promoting cell migration, infiltration into neighbouring tissues, and the formation of metastatic tumors in organs like the lungs, liver, and bones (188). This signaling pathway controls the regulation of genes that are engaged in the course of EMT, which is linked with an upsurge in invasiveness and metastasis. The MET pathway is also crucial in facilitating new blood vessel formation. TNBC tumors rely on angiogenesis to receive adequate blood supply for their growth and survival. The MET pathway influences the production of pro-angiogenic factors, facilitating the formation of blood vessels within the tumor microenvironment (189).
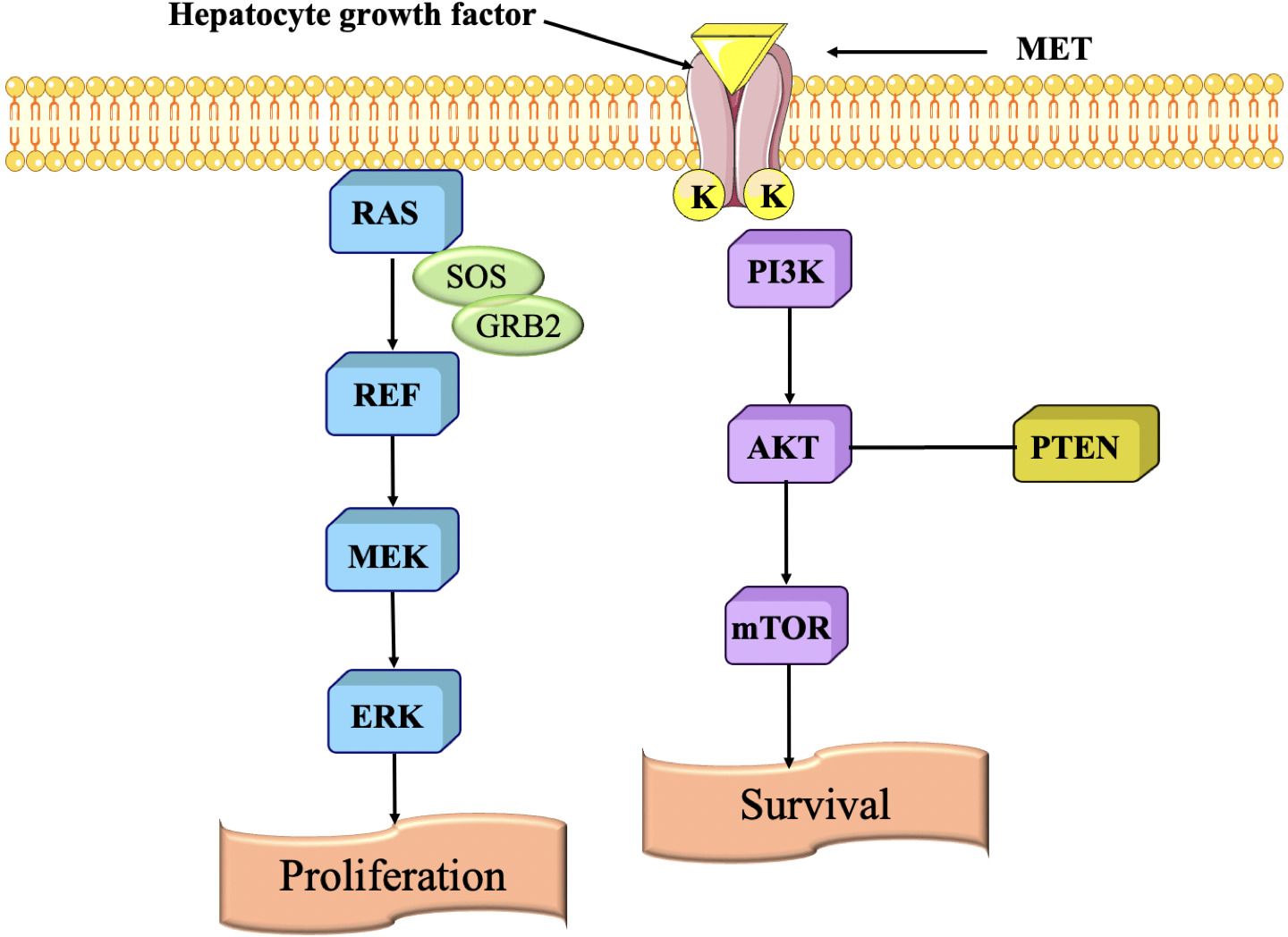
Figure 11. Signaling pathway are regulated by scatter factor (HGF) binding to MET resulting in activation of various pathways (186).
Monoclonal antibodies against MET pathway
Monoclonal antibodies that activate the MET pathway remain largely unexplored. The majority of research concentrates on obstructing MET signaling because of its involvement in facilitating tumor advancement and medication resistance (190).
♣ Monoclonal Antibodies Directed Against MET in TNBC
Inhibitory Anti-MET Monoclonal Antibodies: Although MET activation is often linked to carcinogenesis, mAbs that block MET (such as onartuzumab, savolitinib, and capmatinib) are now undergoing clinical trials for TNBC (186).
♣ Onartuzumab (MetMAb) a monoclonal antibody that targets the MET receptor, inhibiting the binding of its ligand HGF and obstructing MET activation. This antibody has been examined in early-phase clinical studies for MET-positive TNBC to evaluate its efficacy in inhibiting tumor development and chemoresistance. Onartuzumab is often used in conjunction with other medicines, including chemotherapy (191).
♣ Savolitinib (AZD6094) a MET inhibitor that obstructs the MET receptor. Clinical trials are under progress to assess its efficacy in inhibiting tumor proliferation and overcoming resistance in MET-driven TNBC (192).
♣ Anti-HGF Monoclonal Antibodies: Hepatocyte growth factor (HGF) serves as the ligand that activates MET, and anti-HGF monoclonal antibodies are under investigation to inhibit this activation (192). By inhibiting the binding of HGF to MET, these antibodies may impede tumor proliferation and invasion in MET-dependent malignancies such as TNBC (193).
HER2 signaling (Amplification)
“Although TNBC lacks HER2 amplification by definition, compensatory signaling through EGFR and other HER family members can activate downstream PI3K/AKT and MAPK pathways, functionally mimicking HER2-driven oncogenesis.” (194, 195). These systems control critical cellular functions, including cell cycle progression and cell survival (Figure 12). This pathway regulates key cellular processes involved in cancer progression, including cell cycle regulation and protein synthesis. The overexpression of HER2 can also lead to the dysregulation of the MAPK pathway. Stimulation of this route may enhance the growth, infiltration, and formation of new blood vessels in cells, hence promoting the possessive nature of the TNBC (106, 197).
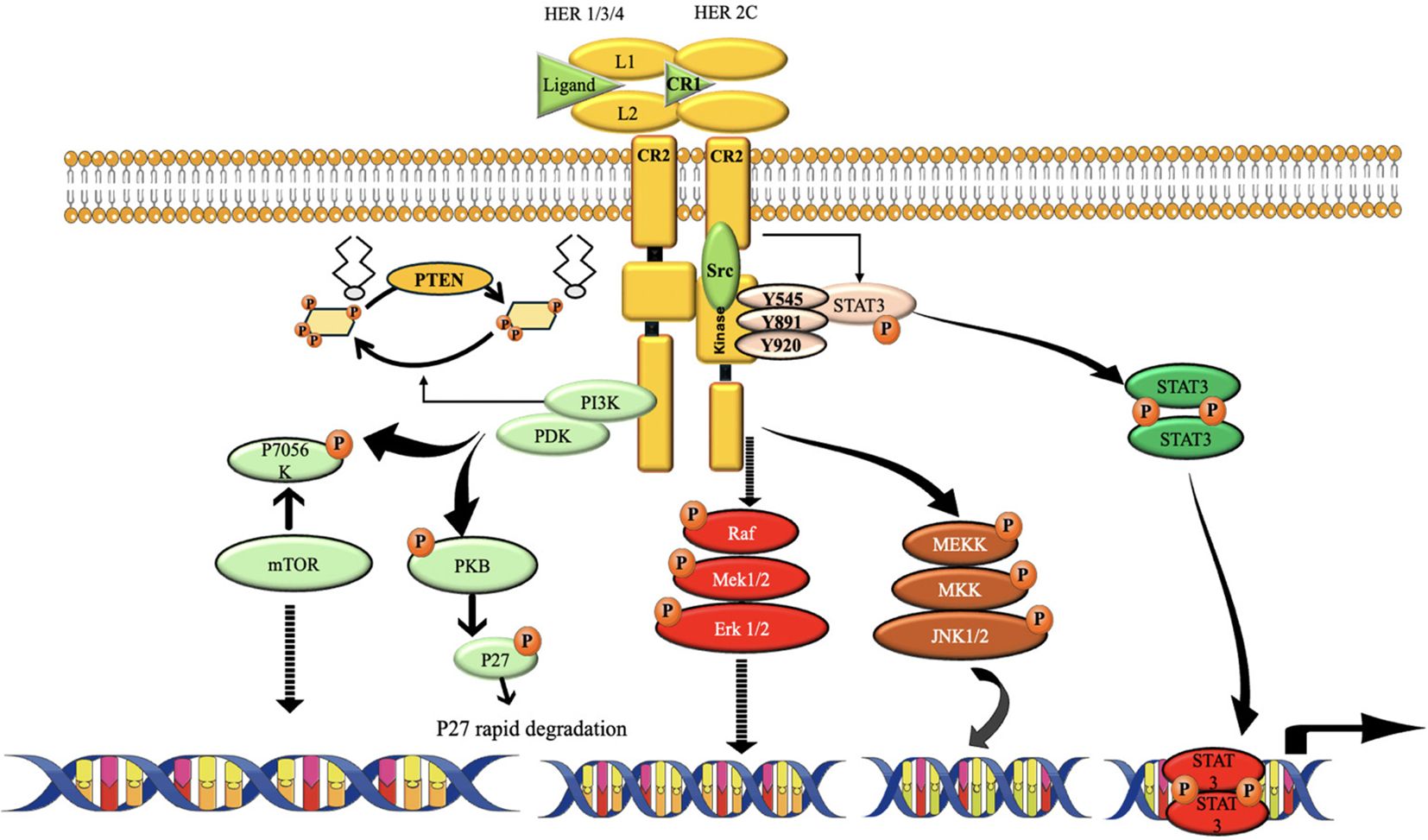
Figure 12. The route of HER2 signaling. The activation of HER2 requires the receptors to form homodimers or heterodimers. It promotes cell growth, proliferation, and survival, among other actions, by activating a wide variety of downstream cascades. One of the most extensively researched pathways triggered by HER2 is PI3K/Akt. The major positive regulator of cellular metabolism, mTOR, is activated when PI3k/Akt is. And via activating the Ras/Raf and MEK pathways, HER2 may promote the migration and development of cancer cells (196).
Monoclonal antibodies against HER2 pathway
While HER2-targeted medicines such as trastuzumab (Herceptin) are primarily used for HER2-positive breast cancer, novel mAbs and targeted therapies are being investigated for HER2-low or HER2-mutant TNBC to activate or modify HER2 signaling for therapeutic advantage. Certain techniques focus on targeting the HER2 receptor to elicit downstream signaling for specific results, such as differentiation or death in certain settings (198).
♣ Margetuximab (MGAH22) a recombinant monoclonal antibody that specifically targets HER2 and has enhanced immune-effector capabilities. It is intended to induce antibody-dependent cellular cytotoxicity (ADCC), resulting in the annihilation of malignant cells (196). Margetuximab is being evaluated for its efficacy in enhancing immune responses in HER2-positive tumors and may enhance HER2 signaling in HER2-low TNBC, facilitating immunogenic cell death and improving tumor detection by the immune system (196, 199).
♣ Pertuzumab (Perjeta) a monoclonal antibody that inhibits the dimerization domain of HER2, obstructing its interaction with other HER family receptors. This inhibits HER2 signaling and triggers apoptosis (200). It is often used in conjunction with Trastuzumab for HER2-positive malignancies. In HER2-low TNBC, Pertuzumab is being investigated to enhance immune response and induce apoptosis when used in conjunction with Trastuzumab and chemotherapy (198).
♣ HER2-Targeted Biologic Therapies with Modulatory Effects: Trastuzumab-Emtansine (T-DM1) is an antibody-drug conjugate (ADC) that integrates Trastuzumab with the cytotoxic compound emtansine. This medication is intended to provide chemotherapy specifically to tumor cells that express HER2. T-DM1, primarily used in HER2-positive breast cancer, is being investigated for its efficacy in HER2-low TNBC. It may activate HER2 signaling and enhance tumor cell apoptosis by selectively targeting cells with low HER2 expression (201).
♣ Sacituzumab Govitecan is an antibody-drug conjugate (ADC) that specifically targets Trop-2, a cell surface protein expressed in TNBC (202). Research is now being conducted to examine how HER2-targeting antibody-drug conjugates, such as Sacituzumab, may activate signaling pathways that potentially amplify their efficacy in HER2-low cancers (203). It is being evaluated in conjunction with HER2-targeted therapy to explore the potential for synergistic activation of signaling pathways and tumor suppression by concurrent targeting of HER2 and Trop-2 (204).
♣ Bispecific Antibodies: Blinatumomab (Blincyto) is a bispecific T-cell engager (BiTE) that attaches to CD3 on T cells and HER2 on tumor cells, therefore eliciting T-cell-mediated cytotoxicity (205). In HER2-low TNBC, Blinatumomab may augment the immune system’s capacity to identify and eliminate HER2-expressing tumor cells while activating HER2 signaling to promote tumor regression (206).
♣ Current and Upcoming Clinical Trials in HER2-Targeted Therapy for TNBC:
The IMpassion130 trial examines the efficacy of Atezolizumab (anti-PD-L1) in conjunction with nab-paclitaxel (chemotherapy) in HER2-negative TNBC, including HER2-low tumors in the subgroup analysis. The function of HER2 signaling in relation to immunotherapy is now under assessment in these individuals (207).
♣ The HER2CLIMB study: This clinical trial assesses Trastuzumab-Emtansine (T-DM1) in patients with HER2-low TNBC, investigating the efficacy of this HER2-targeted antibody-drug conjugate in tumors with reduced HER2 expression (208).
TGF-β (Transforming Growth Factor-beta) Pathway:
The TGF-β pathway is very influential in TNBC. This pathway is intricate and plays a role in several biological processes, such as cell proliferation, differentiation, death, and immunological control. The composition of this entity includes TGF-β receptors, TGF-β ligands, and downstream signaling effectors (209, 210). TGF-β ligands, namely TGF-β1, TGF-β2, and TGF-β3, are proteins that are released and attached to TGF-β receptors, which consist of type I and type II (TGF-βR) receptors. TGF-βRII phosphorylates and promotes TGF-βRI, which initiates the signaling cascade downstream (211). Once activated, TGF-βRI initiates a signaling cascade through the Smad proteins. Smad2 and Smad3 are phosphorylated by TGF-βRI and form a complex with Smad4 (212) (Figure 13). This intricate molecule moves into the nucleus, where it controls the process of transcribing certain genes. TGF-β functions as a tumor inhibitor in the first phases of breast cancer by impeding cell growth, triggering cell cycle arrest, and facilitating programmed cell death. Additionally, it contributes to the maintenance of tissue homeostasis. Nevertheless, at the later stages of TNBC, the TGF-β cascade may experience dysregulation, resulting in the elimination of its ability to control the tumor growth (214, 215).

Figure 13. Schematic representation of the TGF-β (Transforming growth factor-β/ SMAD (SMA and MAD related protein)-induced transcriptional response mediated by coactivators and corepressors. 'P' in yellow circles indicates phosphorylation. Arrows denote the addition of a modification or transfer of a protein complex and the dotted arrow represents the reverse of this. 'Ac' indicates acetylation (213).
Monoclonal antibodies against TGF-β pathway
Clinical studies are investigating mAbs that target the TGF-β pathway to either inhibit or regulate it, aiming to boost tumor suppression, immune activation, or improve the effectiveness of other treatments (216).
♣ Investigation of mAbs Targeting the TGF-β Pathway in TNBC
♣ Monoclonal Antibodies Against TGF-β: Fresolimumab (GC1008) a mAb targeting TGF-β that binds to all three isoforms (TGF-β1, TGF-β2, and TGF-β3), inhibiting their activation and signaling pathways (217). Fresolimumab seeks to restore normal immune function and diminish the tumor-promoting effects of TGF-β in cancer by inhibiting TGF-β signaling. In TNBC, Fresolimumab is undergoing evaluation with chemotherapy and immune checkpoint inhibitors to augment the immune system’s capacity to identify and eliminate tumor cells (213).
♣ M7824 is a bifunctional fusion protein that concurrently binds TGF-βRII and PD-L1. This combinatorial strategy obstructs TGF-β signaling and suppresses immunological checkpoint signaling (PD-L1), resulting in enhanced anti-tumor immunity (218). M7824 is being assessed in TNBC to enhance the immune response against the tumor while inhibiting the immune-evasive processes mediated by TGF-β (219).
♣ Trabedersen (AP 12009) is a TGF-β2-specific antisense oligonucleotide that targets the TGF-β2 gene at the RNA level, inhibiting the synthesis of TGF-β2 and mitigating its impact on tumor growth and immune suppression (220). It has been investigated for its capacity to inhibit TGF-β2 expression in TNBC, especially in instances when TGF-β2 significantly contributes to tumor spread and immune evasion (221).
Focal adhesion kinase pathway
FAK, a non-receptor tyrosine kinase, is essential for integrin-mediated signaling and the processes of cell adherence, growth migration, and longevity. FAK participates in signaling pathways that govern these activities, and its abnormal regulation has been linked to the development of several malignancies, such as TNBC (222, 223). FAK governs the generation and degradation of focal adhesions, which are dynamic structures that facilitate cell attachment to the extracellular matrix (ECM). FAK enhances cell migration by initiating downstream signaling pathways, such as the PI3K/Akt and MAPK pathways, resulting in heightened cell motility and invasion. (224). In TNBC, aberrant FAK signaling can enhance the migratory and invasive properties of cancer cells, facilitating metastasis. Its activation promotes cell survival and proliferation through various mechanisms. It can initiate pro-survival signaling pathways, such as the PI3K/Akt and STAT3, which play a role in promoting cell survival and preventing apoptosis (225) (Figure 14). FAK may also stimulate cancer development by facilitating angiogenesis, that provide nutrition and oxygen to the developing tumor. It also has been implicated in EMT regulation by activating transcription factors such as Snail, Slug, and Twist, which promote EMT-associated changes in cell morphology and behaviour (222). FAK signaling has been linked to resistance to many anti-cancer treatments, like targeted therapies and chemotherapy by promoting cell survival and reducing the efficacy of cytotoxic drugs. Additionally, FAK can modulate signaling pathways that mediate resistance to targeted therapies, such as HER2 inhibitors or PARP inhibitors (227).
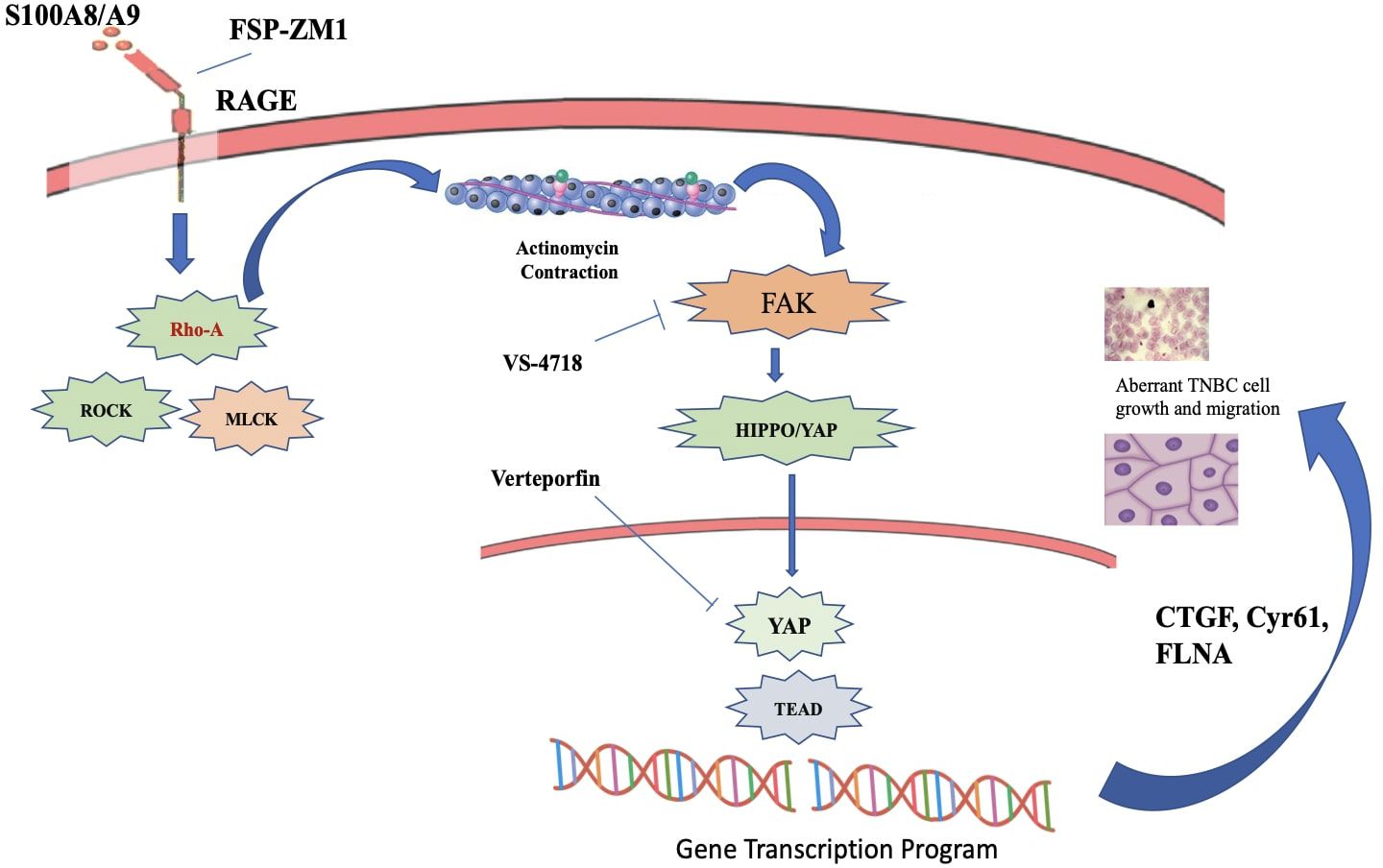
Figure 14. S100A8/A9-RAGE-FAK-YAP signaling in TNBC cells. A cartoon depicting the proposed S100A8/A9-RAGE-FAK-YAP transduction network in TNBC cells Targeting RAGE along with FAK/YAP-dependent transcriptional programs may disable the growth and migration of TNBC cells (226).
Pharmacotherapeutic therapies against FAK signaling pathway
Research on various pharmacotherapeutics therapies especially aimed at FAK signaling in TNBC is ongoing, with several strategies focusing on either directly targeting FAK or modifying pathways associated with FAK activation (228). Several therapies and methodologies under investigation that may influence FAK signaling in TNBC.
♣ Defactinib (VS-6063) a small molecule inhibitor that specifically inhibits the kinase activity of FAK and has been extensively researched in conjunction with other therapies, including immune checkpoint inhibitors and chemotherapy. It particularly inhibits FAK signaling and undermines the tumorigenic effects of FAK activation in cancer cells (229).
Signal transducer and activator of transcription 3 pathway
It is a transcription component that has a crucial function in the development of TNBC. The STAT3 pathway is activated by various cytokines, growth factors, and oncogenic signaling pathways (230). Upon activation, STAT3 relocates to the nucleus and controls the transcription of target genes associated with cell survival, proliferation, angiogenesis, immunological evasion, and metastasis (226).
The STAT3 pathway is often activated in TNBC, leading to abnormal gene expression that promotes cell cycle progression and suppresses apoptosis. It upregulates cyclin D1, cyclin-dependent kinase 4 (CDK4), and survivin, which promote cell proliferation and suppress cell death (116). STAT3 stimulates the production of pro-angiogenic substances, including fibroblast growth factor (FGF), interleukin-8 (IL-8) and VEGF (Figure 15). Additionally, it stimulates EMT and also hinders the body’s defences by impeding the activity of immune cells, including T cells and natural killer (NK) cells. It enhances the production of immunological checkpoint components, such as programmed death-ligand 1 (PD-L1), which helps in avoiding detection by the immune system (232).
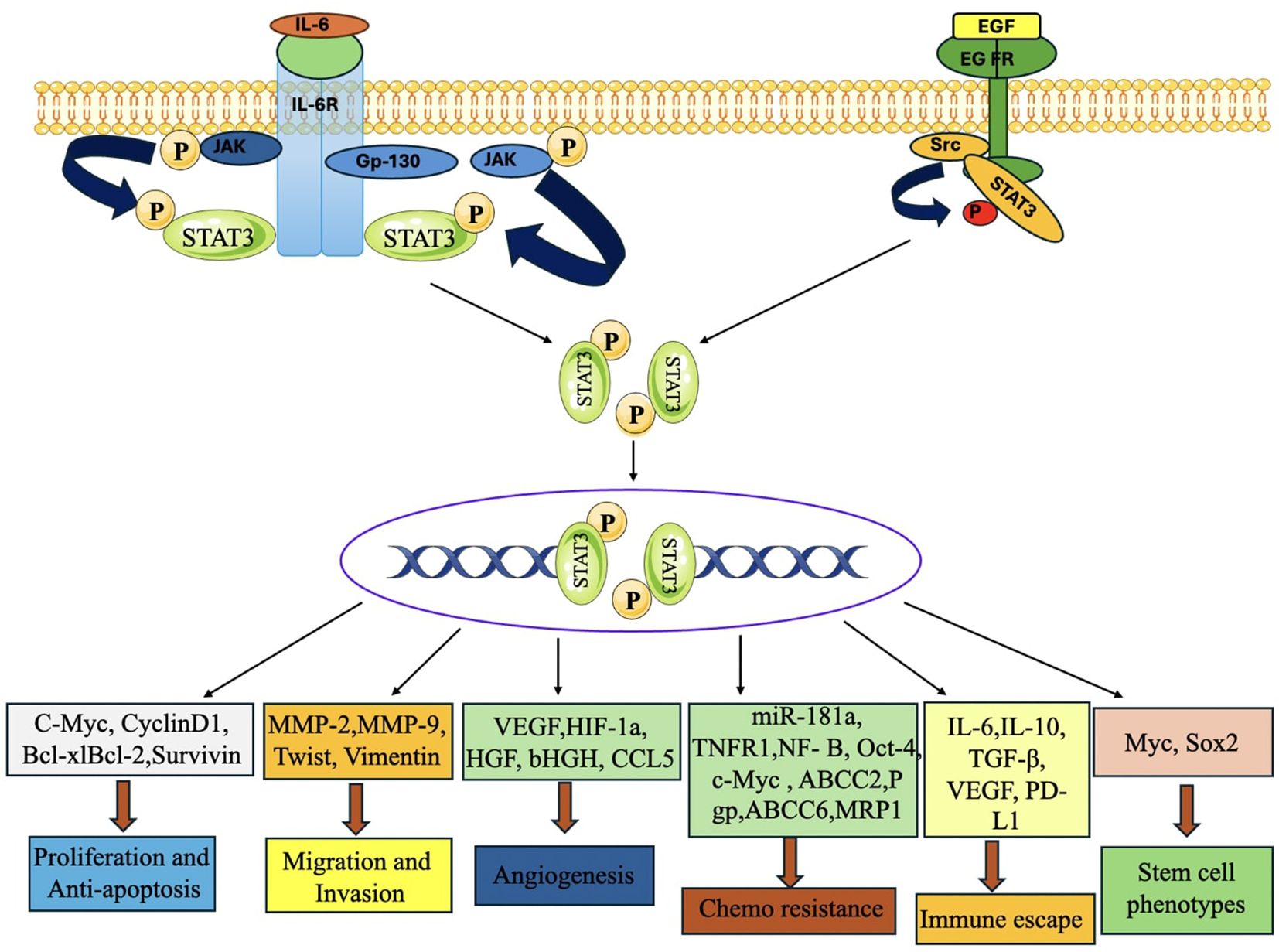
Figure 15. STAT3 signaling activation enhances tumor development, metastasis, chemoresistance, immunological evasion, and stemness in TNBC. When the upstream regulators are activated, STAT3 is phosphorylated, dimerized, and transported into the nucleus, where it activates the transcription of target genes that control cell proliferation, anti-apoptosis, migration, invasion, angiogenesis, chemoresistance, immune escape, stem cell phenotypes, and autophagy (231).
Activation of STAT3 has been linked to opposition to chemotherapy treatments via the promotion of drug efflux pumps, like P-glycoprotein (P-gp). These pumps decrease the buildup of pharmaceuticals within cells, hence contributing to resistance to therapy (233).
Combination strategies (monoclonal and non- monoclonal antibodies) against STAT3 pathway
Monoclonal antibodies that target the STAT3 pathway constitute a promising study domain for TNBC, especially since STAT3 plays a critical role in immune evasion and tumor advancement. Despite some medicines being in preliminary research phases, combination therapies using monoclonal antibodies and STAT3 inhibitors show significant promise in both preclinical and clinical studies (234).
♣ Monoclonal Antibodies Directed Against the STAT3 Pathway in TNBC.
The combination of N-803 (an IL-15 superagonist) with the anti-STAT3 mAb (CXXC5) is under investigation for its ability to target and inhibit the STAT3 pathway. N-803 promotes immunological activation, while CXXC5 inhibits the phosphorylation of STAT3. These drugs seek to inhibit STAT3 phosphorylation to reinstate immune surveillance, diminish cancer cell proliferation, and enhance tumor sensitivity to alternative treatments, such as immunotherapy (235).
♣ CDDO-Me, a synthetic STAT3 inhibitor, is now being evaluation for its ability to augment the effectiveness of mAb therapy, especially in metastatic TNBC (236). The objective is to diminish STAT3 activity, which often results in immune evasion and tumor persistence. This method remains in the preclinical phase, although it has potential as a combinatorial treatment with immune checkpoint inhibitors or other medicines targeting STAT3 suppression in TNBC (237).
♣ S3I-201 (STAT3 Inhibitor) Combined with Chemotherapy or Immunotherapy: S3I-201 a small molecule inhibitor of STAT3 has shown preclinical efficacy in many malignancies, including TNBC. S3I-201, a non-monoclonal antibody, directly inhibits STAT3 signaling by obstructing its phosphorylation and nuclear translocation (231). Researchers are evaluating this inhibitor with chemotherapy and immunotherapy (e.g., anti-PD-1/PD-L1 inhibitors) aiming to evaluate the safety, tolerability, and effectiveness of S3I-201, as well as its capacity to enhance the sensitivity of TNBC cells to other treatments, including mAbs (235).
Hippo pathway
The Hippo pathway is a well-preserved signaling route that regulates the size of organs and the growth of cells. This route has been linked to the development of many malignancies, such as TNBC (238). The disruption of the Hippo pathway has been recognized as a possible factor in the emergence and advancement of TNBC. The main mechanism of this route is controlled by a series of kinase reactions that modulate the function of transcriptional coactivator with PDZ-binding motif (TAZ) and Yes-associated protein (YAP) which are essential components of the process (239). When the Hippo pathway is activated, the upstream kinases, MST1/2 and LATS1/2, phosphorylate YAP and TAZ, causing them to be trapped in the plasma membrane and blocking their movement into the nucleus (Figure 16). Consequently, the process of gene transcription controlled by YAP/TAZ is suppressed (240).
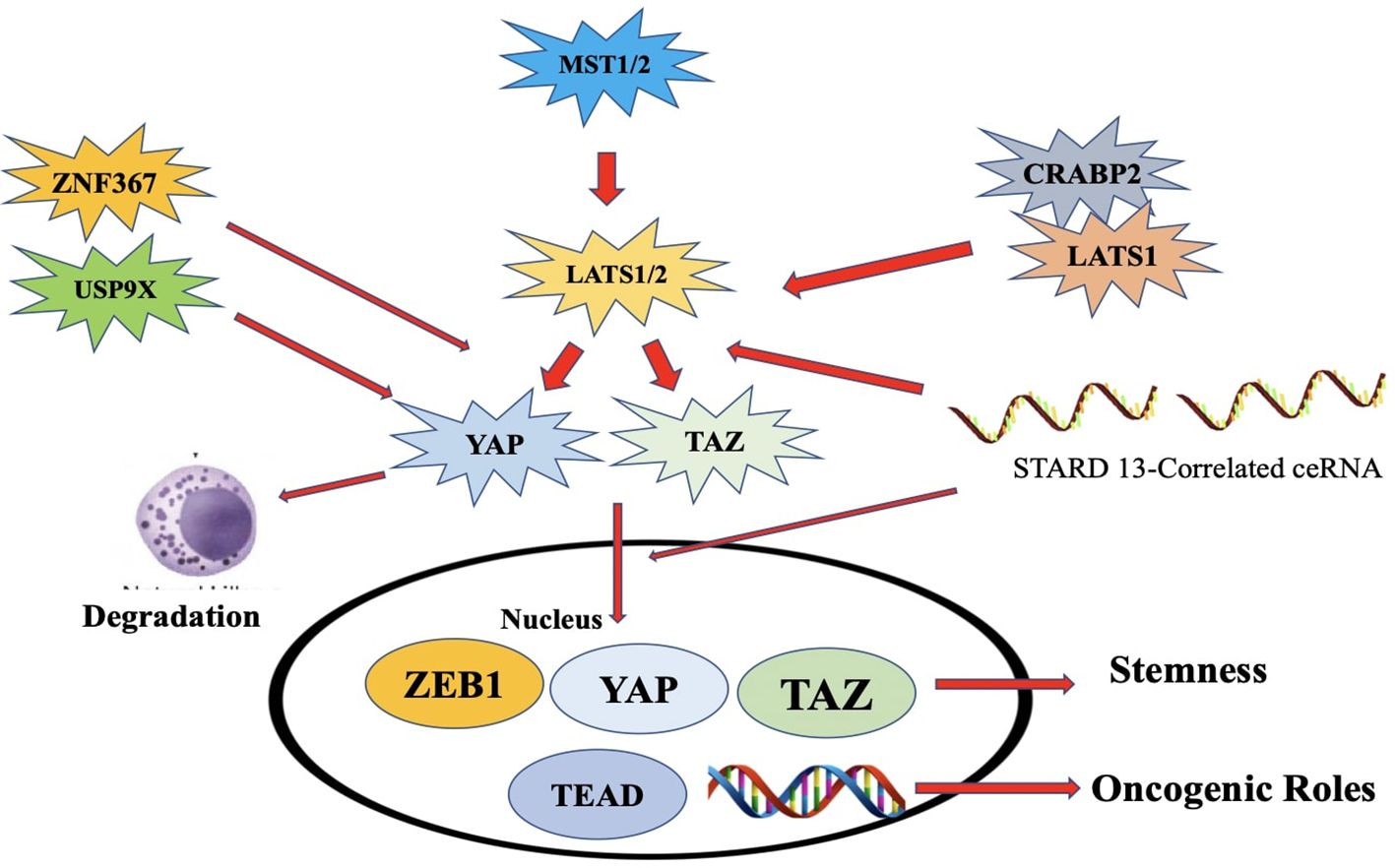
Figure 16. An illustration of the Hippo pathway's significance in ER+ breast cancer. Overexpression of ZNF367 promotes metastasis and stimulates Hippo/YAP signaling via blocking LATS. Overexpression of USP9x promotes cell proliferation by deubiquitinating and stabilizing YAP1.NE and EPI suppress breast cancer by rapidly phos-phorylating YAP and retaining it in the cytoplasm. ZEB1, a transcriptional activator, interacts with YAPI to in-crease transcription. The connection between LATS1 and CRABP2 decreases LATS1 ubiquitination, which sup-presses cell invasion. The STARD13-correlated ceRNA network controls TAZ distribution and may decrease the stem cell nature of breast cancer by upregulating LATS1/2 [248].
The Hippo pathway acts as a tumor suppressor by inhibiting the activity of YAP and TAZ, which are known to promote cell proliferation and survival. Dysregulation of the pathway, such as inactivating mutations or altered expression of its components, can result in YAP/TAZ activation, leading to uncontrolled cell growth and tumor formation (246). This Activation has also been associated with the initiation of EMT in TNBC cells, enhancing their capacity to move, infiltrate neighboring tissues, and spread to remote locations. Multiple studies have shown that stimulation of YAP in TNBC cells could have a role in making the cells resistant to chemotherapy. This resistance is believed to occur via the regulation of genes that take part in removing drugs from the cells, repairing DNA damage, and preventing cell death (247).
Combination strategies (monoclonal and non- monoclonal antibodies) against hippo pathway
Investigations into monoclonal antibodies that target the Hippo pathway remain in preliminary phases; nonetheless, several techniques are being explored to control this system in TNBC and other malignancies (248). Current Investigations on Monoclonal Antibodies Directed at the Hippo Pathway in TNBC includes:
♣ Monoclonal Antibodies Targeting YAP and TAZ: The development of anti-YAP/TAZ monoclonal antibodies, which directly target YAP/TAZ or block their nuclear activity, remains in the preliminary stages of research. These antibodies are designed to inhibit the interaction between YAP/TAZ and TEAD transcription factors, which facilitates the production of genes that promote cancer (249). Directly targeting YAP/TAZ with monoclonal antibodies is challenging owing to the intricate molecular relationships these proteins maintain with many biological components (246).
♣ TEAD Inhibitors and Monoclonal Antibodies: The connection between TEAD and YAP/TAZ is a primary therapeutic target. Monoclonal antibodies that inhibit the interaction between YAP/TAZ and TEAD are currently being studied. Verteporfin (a YAP-TEAD inhibitor) a tiny compound have shown some effectiveness in preclinical animals and are now advancing to clinical trials for TNBC. Researchers are now investigating the potential of engineered mAbs to disrupt this interaction with greater specificity and potency (244).
♣ Targeting MST1/2 Kinases: MST1 and MST2 serve as upstream regulators of the Hippo pathway, facilitating the phosphorylation and activation of LATS1/2 kinases, which then phosphorylate and inactivate YAP/TAZ. Activation of MST1/2 kinase reinstates the Hippo pathway and diminishes YAP/TAZ activity (241). Targeting MST1/2 may restore normal Hippo signaling in TNBC, thus inhibiting tumor growth and invasion. Although small compounds aimed at MST1/2 are being explored in preclinical investigations, mAbs for MST1/2 activation are not yet broadly accessible. The emphasis is on small chemical activators of MST1/2, which may subsequently facilitate the creation of specific antibodies (250). Researchers are exploring combination treatments including immune checkpoint inhibitors or chemotherapy to enhance the effectiveness of Hippo pathway inhibitors. They are also evaluating mAbs that target the Hippo pathway in conjunction with chemotherapy or immune checkpoint inhibitors (e.g., anti-PD-1/PD-L1, anti-CTLA-4) to improve the treatment response in TNBC. Combination therapy that simultaneously target YAP/TAZ activity and immune suppression may provide synergistic results for TNBC patients, who are often resistant to conventional treatments (242).
♣ mAbs aimed at the Hippo pathway in TNBC remain in preliminary development, with current research concentrating on YAP/TAZ inhibitors, TEAD inhibitors, and MST1/2 activators (239). Presently, there is a significant emphasis on using small compounds that target this route; however, monoclonal antibodies are being developed to possibly provide a more precise and enduring treatment (251).
Conclusion
TNBC exhibits the poorest prognosis of any kind of breast carcinoma in women. Many characteristics of TNBC contribute to its exceptional aggressiveness compared to other varieties of breast carcinoma. Therefore, comprehending the molecular signaling mechanism of TNBC in the tumor microenvironmnt by the implementation of interdisciplinary research would significantly enhance the diagnosis and treatment of TNBC. An extensive understanding of the genetic and proteomic mechanisms associated with TNBC may aid in the development of innovative treatment approaches and the effective design of clinical trials. The unfavorable clinical result of TNBC treatment may be attributed to cancer recurrence, metastasis, and death. Exploring several signaling pathways in depth and identifying the molecular targets for TNBC therapy is a valuable endeavour. Advancements in computer biology, together with breakthroughs in immunology, nanotechnology, and molecular biology, will provide early detection and customised therapy. Multiple studies are underway, and new targets are being identified, but they have yet to be successfully implemented. A Comprehensive table summarizing the key components of each signaling pathway and the corresponding potential therapeutic strategies is provided in Table 1. Scientists are motivated to continually investigate different signaling cascades to discover effective medications with a high rate of pathological complete response (pCR) for the treatment of TNBC. Presently, clinical trials are underway to assess the safety and effectiveness of several medications that target various routes in combination for patients with TNBC. The outcomes of these studies are currently pending.
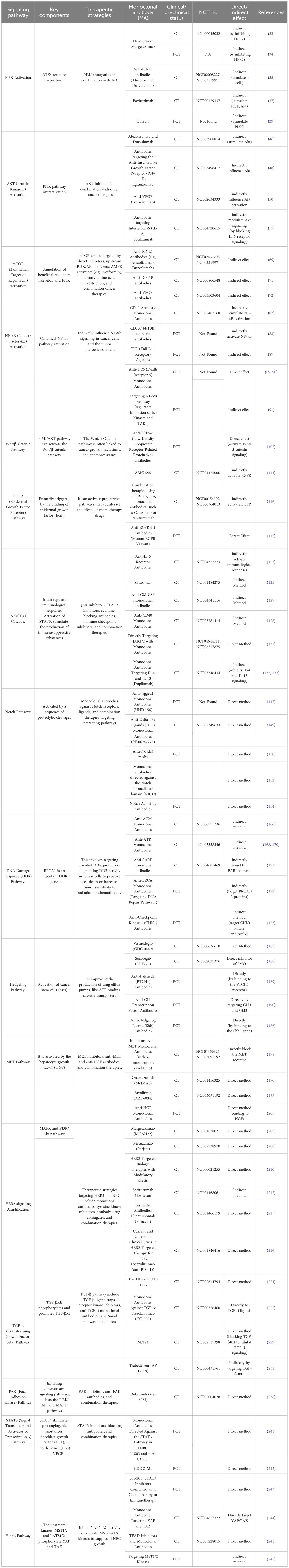
Table 1. Comprehensive table summarizing the key components of each signaling pathway and the corresponding potential therapeutic strategies.
Author contributions
PM: Writing – original draft, Investigation. NK: Writing – review & editing, Conceptualization. SG: Software, Writing – review & editing. AnuK: Writing – review & editing, Formal analysis. SF: Formal analysis, Data curation, Writing – review & editing. GS: Methodology, Writing – review & editing. AnoK: Software, Writing – review & editing.
Funding
The authors declare that no financial support was received for the research, and/or publication of this article.
Acknowledgments
The Authors would like to acknowledge the Director and Management of Khalsa College of Pharmacy, Amritsar, Punjab for providing all kinds of support for complying the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
This article has been corrected with minor changes. These changes do not impact the scientific content of the article.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
ADC: Antibody-drug conjugate
AKT: Protein Kinase B
APC: Adenomatous polyposis coli
AR: Androgen receptor
CDK4: Cyclin-dependent kinase 4
CK1: Casein kinase 1
CNVs: Copy number variations
COX-2: Cyclooxygenase-2
CSCs: Cancer stem cells
CTLA-4: Cytotoxic T-Lymphocyte-Associated Protein 4
DLL: Delta-like ligands
DSBs: Double-strand breaks
ECM: Extracellular matrix
EGF: Epidermal growth factor
EGFR: Epidermal Growth Factor Receptor
EMT: Epithelial-mesenchymal transition
ER: Estrogen receptor
EffNetb0: EfficientNet-b0
FAK: Focal adhesion kinase
FGF: Fibroblast growth factor
GSK3β: Glycogen Synthase Kinase 3 Beta
HGF: Hepatocyte growth factor
HER2: Human epidermal growth factor receptor 2
HRD: Homologous Recombination Deficiency
IFN-γ: Interferon-gamma
IGF-1R: Insulin-like Growth Factor 1 Receptor
IKKβ: Inhibitor of nuclear factor kappa-B kinase subunit beta
IL-6: Interleukin-6
IL-8: Interleukin-8
INPP4B: Inositol polyphosphate-4-phosphatase type II
JAK: Janus kinase
LRP: Low-density lipoprotein receptor-related protein
mAb: Monoclonal antibody
MMP-9: Matrix Metalloproteinase-9
MMPs: Metalloproteinases
mTOR: Mammalian Target of Rapamycin
NF-κB: Nuclear factor-κB
NICD: Notch intracellular domain
PAMPs: Pathogen-associated molecular patterns
PD-1: Programmed cell death protein 1
PDK1: Phosphoinositide-dependent protein kinase-1
PD-L1: Programmed death-ligand 1
PI3K: Phosphoinositide 3-kinase
PIK3CA: PI3K catalytic subunit
PIP2: Phosphatidylinositol 4,5-bisphosphate
PIP3: Phosphatidylinositol 3,4,5-trisphosphate
PR: Progesterone receptor
PTEN: Phosphatase and tensin homolog
ResizeMix: ResizeMix augmentation method
RNet18: Residual Network-18
RNet34: Residual Network-34
RNet50: Residual Network-50
RTKs: Receptor tyrosine kinases
SMO: Smoothened
STAT3: Signal Transducer and Activator of Transcription 3
TAK1: Transforming growth factor beta-activated kinase 1
TCF/LEF: T-cell factor/lymphoid promoter factor
TEAD: Transcriptional enhanced associate domian
TGF-β: Transforming growth factor-beta
TEAD: Transcriptional enhanced associate domain
TGF-β: Transforming growth factor-beta
TNBC: Triple negative breast cancer
TRAIL: TNF-related apoptosis-inducing ligand
VEGF: Vascular endothelial growth factor
YAP: Yes-associated protein
References
1. Dass SA, Tan KL, Selva Rajan R, Mokhtar NF, Mohd Adzmi ER, Wan Abdul Rahman WF, et al. Triple negative breast cancer: A review of present and future diagnostic modalities. Medicina. (2021) 57:62. doi: 10.3390/medicina57010062
2. Podo F, Buydens LMC, Degani H, Hilhorst R, Klipp E, Gribbestad IS, et al. Triple-negative breast cancer: present challenges and new perspectives. Mol Oncol. (2010) 4:209–29. doi: 10.1016/j.molonc.2010.04.006
3. Mahtani R, Kittaneh M, Kalinsky K, Mamounas E, Badve S, Vogel C, et al. Advances in therapeutic approaches for triple-negative breast cancer. Clin Breast Cancer. (2021) 21:383–90. doi: 10.1016/j.clbc.2020.12.011
4. Urru SAM, Gallus S, Bosetti C, Moi T, Medda R, Solli E, et al. Clinical and pathological factors influencing survival in a large cohort of triple-negative breast cancer patients. BMC Cancer. (2018) 18:1–11. doi: 10.1186/s12885-017-3969-y
5. Ludwick R and Gaczkowski T. Breast self-exams by teenagers: outcome of a teaching program. Cancer Nurs. (2001) 24:315–9. doi: 10.1097/00002820-200108000-00013
6. Afghahi A, Telli ML, and Kurian AW. Genetics of triple-negative breast cancer: Implications for patient care. Curr problems Cancer. (2016) 40:130–40. doi: 10.1016/j.currproblcancer.2016.09.007
7. Lazarus G, Audrey J, and Iskandar AWB. Efficacy and safety profiles of programmed cell death-1/programmed cell death ligand-1 inhibitors in the treatment of triple-negative breast cancer: A comprehensive systematic review. Oncol Rev. (2019) 13:161–9. doi: 10.4081/oncol.2019.425
8. Diamandis M, White NM, and Yousef GM. Personalized medicine: marking a new epoch in cancer patient management. Mol Cancer Res. (2010) 8:1175–87. doi: 10.1158/1541-7786.MCR-10-0264
9. Moscow JA, Fojo T, and Schilsky RL. The evidence framework for precision cancer medicine. Nat Rev Clin Oncol. (2018) 15:183–92. doi: 10.1038/nrclinonc.2017.186
10. Khan MA, Jain VK, Rizwanullah M, Ahmad J, and Jain K. PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: a review on drug discovery and future challenges. Drug Discov Today. (2019) 24:2181–91. doi: 10.1016/j.drudis.2019.09.001
11. Jia H, Truica CI, Wang B, Wang Y, Ren X, Harvey HA, et al. Immunotherapy for triple-negative breast cancer: Existing challenges and exciting prospects. Drug resistance updates. (2017) 32:1–15. doi: 10.1016/j.drup.2017.07.002
12. Gupta GK, Collier AL, Lee D, Hoefer RA, Zheleva V, Siewertsz van Reesema LL, et al. Perspectives on triple-negative breast cancer: current treatment strategies, unmet needs, and potential targets for future therapies. Cancers. (2020) 12:2392. doi: 10.3390/cancers12092392
13. Zaidi S, Hussain S, Verma S, Veqar Z, Khan A, Nazir SU, et al. Efficacy of complementary therapies in the quality of life of breast cancer survivors. Front Oncol. (2018) 7:326. doi: 10.3389/fonc.2017.00326
14. Abotaleb M, Kubatka P, Caprnda M, Varghese E, Zolakova B, and Zubor P. Chemotherapeutic agents for the treatment of metastatic breast cancer: An update. Biomedicine pharmacotherapy. (2018) 101:458–77. doi: 10.1016/j.biopha.2018.02.108
15. Satija A and Bhatnagar S. Complementary therapies for symptom management in cancer patients. Indian J palliative Care. (2017) 23:468. doi: 10.4103/IJPC.IJPC_100_17
16. Bosch A, Eroles P, Zaragoza R, Vina JR, and Lluch A. Triple-negative breast cancer: molecular features, pathogenesis, treatment and current lines of research. Cancer Treat Rev. (2010) 36:206–15. doi: 10.1016/j.ctrv.2009.12.002
17. Dawson S, Provenzano E, and Caldas C. Triple negative breast cancers: clinical and prognostic implications. Eur J Cancer. (2009) 45:27–40. doi: 10.1016/S0959-8049(09)70013-9
18. Sriroopreddy R and Sudandiradoss C. Integrative network-based approach identifies central genetic and transcriptomic elements in triple-negative breast cancer. Funct Integr Genomics. (2018) 18:113–24. doi: 10.1007/s10142-017-0579-3
19. Pascual J and Turner N. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann Oncol. (2019) 30:1051–60. doi: 10.1093/annonc/mdz133
20. Vara JáF, Casado E, Castro JD, Cejas P, Iniesta CB, and Baron MG. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. (2004) 30:193–204. doi: 10.1016/j.ctrv.2003.07.007
21. Rathinaswamy MK. Molecular basis for the regulation of phosphoinositide 3-kinase γ (PI3Kγ). Canada: Advances in Biological Regulation. (2021).
22. Fox M, Mott HR, and Owen D. Class IA PI3K regulatory subunits: p110-independent roles and structures. Biochem Soc Trans. (2020) 48:1397–417. doi: 10.1042/BST20190845
23. Hsu JL and Hung M-C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. (2016) 35:575–88. doi: 10.1007/s10555-016-9649-6
24. Butti R, Gunasekaran VP, Yadav AS, Kumar D, and Kundu GC. Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Mol Cancer. (2018) 17:1–18. doi: 10.1186/s12943-018-0797-x
25. Fayard E, Xue G, Parcellier A, Bozulic L, and Hemmings BA. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Phosphoinositide 3-kinase Health Dis. (2011) 1:31–56.
26. Lien EC, Dibble CC, and Toker A. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol. (2017) 45:62–71. doi: 10.1016/j.ceb.2017.02.007
27. You KS, Yi YW, Cho J, Park JS, and Seong YS. Potentiating therapeutic effects of epidermal growth factor receptor inhibition in triple-negative breast cancer. Pharmaceuticals. (2021) 14:589. doi: 10.3390/ph14060589
28. Zhang H-P, Jiang R-Y, Zhu J-Y, Sun K-N, Huang Y, Zhou H-H, et al. PI3K/AKT/mTOR signaling pathway: an important driver and therapeutic target in triple-negative breast cancer. Breast Cancer. (2024) 31:539–51. doi: 10.1007/s12282-024-01567-5
29. Fouqué A, Jean M, Weghe PVD, and Legembre P. Review of PI3K/mTOR inhibitors entering clinical trials to treat triple negative breast cancers. Recent patents anti-cancer Drug Discov. (2016) 11:283–96. doi: 10.2174/1574892811666160519113731
30. Modi S, Saura C, Yamashita T, Park YH, Kim S-B, Tamura K, et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. New Engl J Med. (2020) 382:610–21. doi: 10.1056/NEJMoa1914510
31. Maadi H, Soheilifar MH, Choi W-S, Moshtaghian A, and Wang Z. Trastuzumab mechanism of action; 20 years of research to unravel a dilemma. Cancers. (2021) 13:3540. doi: 10.3390/cancers13143540
32. Sukumar J, Gast K, Quiroga D, Lustberg M, and Williams N. Triple-negative breast cancer: promising prognostic biomarkers currently in development. Expert Rev Anticancer Ther. (2021) 21:135–48. doi: 10.1080/14737140.2021.1840984
33. Husna SMN and Wong KK. Margetuximab and trastuzumab deruxtecan: New generation of anti-HER2 immunotherapeutic agents for breast cancer. Mol Immunol. (2022) 152:45–54. doi: 10.1016/j.molimm.2022.10.005
34. Rugo HS, Im S-A, Cardoso F, Cortés J, Curigliano G, Musolino A, et al. Efficacy of margetuximab vs trastuzumab in patients with pretreated ERBB2-positive advanced breast cancer: a phase 3 randomized clinical trial. JAMA Oncol. (2021) 7:573–84. doi: 10.1001/jamaoncol.2020.7932
35. Córdova-Bahena L and Velasco-Velázquez MA. Anti-PD-1 and anti-PD-L1 antibodies as immunotherapy against cancer: A structural perspective. Rev investigación clínica. (2021) 73:8–16. doi: 10.24875/RIC.20000341
36. Zhang Z, Richmond A, and Yan C. Immunomodulatory properties of PI3K/AKT/mTOR and MAPK/MEK/ERK inhibition augment response to immune checkpoint blockade in melanoma and triple-negative breast cancer. Int J Mol Sci. (2022) 23:7353. doi: 10.3390/ijms23137353
37. Preetam S, Pandey A, Mishra R, Mohapatra G, Rath P, Malik S, et al. Phosphatidylserine: paving the way for a new era in cancer therapies. Materials Adv. (2024) 5:8384–403. doi: 10.1039/D4MA00511B
38. Massihnia D, Galvano A, Fanale D, Perez A, Castiglia M, Incorvaia L, et al. Triple negative breast cancer: shedding light onto the role of pi3k/akt/mtor pathway. Oncotarget. (2016) 7:60712. doi: 10.18632/oncotarget.10858
39. Jiang B-H and Liu L-Z. AKT signaling in regulating angiogenesis. Curr Cancer Drug Targets. (2008) 8:19–26. doi: 10.2174/156800908783497122
40. Alemi F, Sadigh AR, Malakoti F, Elhaei Y, Gaffari SH, Maleki M, et al. Molecular mechanisms involved in DNA repair in human cancers: An overview of PI3k/Akt signaling and PIKKs crosstalk. J Cell Physiol. (2022) 237:313–28. doi: 10.1002/jcp.30573
41. Song G, Ouyang G, and Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. (2005) 9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x
42. Hinz N and Jücker M. Distinct functions of AKT isoforms in breast cancer: a comprehensive review. Cell Communication Signaling. (2019) 17:1–29. doi: 10.1186/s12964-019-0450-3
43. McCubrey JA, Steelman LS, Chappell WH, Abrams S, Montalto G, Carvello M, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. (2012) 3:954. doi: 10.18632/oncotarget.652
44. Medina MA, Oza G, Sharma A, Arriaga LG, Hernández JMH, Rotello VM, et al. Triple-negative breast cancer: a review of conventional and advanced therapeutic strategies. Int J Environ Res Public Health. (2020) 17:2078. doi: 10.3390/ijerph17062078
45. Yip H and Papa A. Signaling pathways in cancer: therapeutic targets, combinatorial treatments, and new developments. Cells. (2021) 10:659. doi: 10.3390/cells10030659
46. Lee HT, Lee JY, Lim H, Lee SH, Moon YJ, Pyo HJ, et al. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci Rep. (2017) 7:5532. doi: 10.1038/s41598-017-06002-8
47. Lin X, Kang K, Chen P, Zeng Z, Li G, and Xiong W. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol Cancer. (2024) 23:108. doi: 10.1186/s12943-024-02023-w
48. Tognon CE and Sorensen PH. Targeting the insulin-like growth factor 1 receptor (IGF1R) signaling pathway for cancer therapy. Expert Opin Ther Targets. (2012) 16:33–48. doi: 10.1517/14728222.2011.638626
49. Lee J-S, Tocheny CE, and Shaw LM. The insulin-like growth factor signaling pathway in breast cancer: an elusive therapeutic target. Life. (2022) 12:1992. doi: 10.3390/life12121992
50. Itatani Y, Kawada K, Yamamoto T, and Sakai Y. Resistance to anti-angiogenic therapy in cancer—alterations to anti-VEGF pathway. Int J Mol Sci. (2018) 19:1232. doi: 10.3390/ijms19041232
51. Claesson-Welsh L and Welsh M. VEGFA and tumour angiogenesis. J Internal Med. (2013) 273:114–27. doi: 10.1111/joim.12019
52. Li Y, Zhang H, Merkher Y, Chen L, Liu N, Leonov S, et al. Recent advances in therapeutic strategies for triple-negative breast cancer. J Hematol Oncol. (2022) 15:121. doi: 10.1186/s13045-022-01341-0
53. Ohsugi Y. The immunobiology of humanized Anti-IL6 receptor antibody: From basic research to breakthrough medicine. J Trans Autoimmun. (2020) 3:100030. doi: 10.1016/j.jtauto.2019.100030
54. Yao X, Huang J, Zhong H, Shen N, Faggioni R, Fung M, et al. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther. (2014) 141:125–39. doi: 10.1016/j.pharmthera.2013.09.004
55. Sheppard M, Laskou F, Stapleton PP, Hadavi S, and Dasgupta B. Tocilizumab (actemra). Hum Vaccines immunotherapeutics. (2017) 13:1972–88. doi: 10.1080/21645515.2017.1316909
56. Kumari L, Mishra L, Patel P, Sharma N, Gupta G, Kurmi BD, et al. Emerging targeted therapeutic strategies for the treatment of triple-negative breast cancer. J Drug Targeting. (2023) 31:889–907. doi: 10.1080/1061186X.2023.2245579
57. He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW, et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal transduction targeted Ther. (2021) 6:425. doi: 10.1038/s41392-021-00828-5
58. Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, et al. A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER+/HER2– metastatic breast cancer. Clin Cancer Res. (2017) 23:26–34. doi: 10.1158/1078-0432.CCR-16-0134
59. Agostini D, Natalucci V, Baldelli G, Santi MD, Zeppa SD, Vallorani L, et al. New insights into the role of exercise in inhibiting mTOR signaling in triple-negative breast cancer. Oxid Med Cell Longevity. (2018) 2018:5896976. doi: 10.1155/2018/5896786
60. Cheung M and Testa JR. Diverse mechanisms of AKT pathway activation in human Malignancy. Curr Cancer Drug Targets. (2013) 13:234–44. doi: 10.2174/1568009611313030002
61. Cuyãs E, Faja BC, Joven J, and Menendez JA. Cell cycle regulation by the nutrient-sensing mammalian target of rapamycin (mTOR) pathway. In: Cell cycle control. Methods in Molecular Biology. (2014) 1170:113–144. doi: 10.1007/978-1-4939-0888-2_7
62. Szwed A, Kim E, and Jacinto E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol Rev. (2021) 101:1371–426. doi: 10.1152/physrev.00026.2020
63. Dong C, Wu J, Chen Y, Nie J, and Chen C. Activation of PI3K/AKT/mTOR pathway causes drug resistance in breast cancer. Front Pharmacol. (2021) 12:628690. doi: 10.3389/fphar.2021.628690
64. Nedeljković M and Damjanović A. Mechanisms of chemotherapy resistance in triple-negative breast cancer—how we can rise to the challenge. Cells. (2019) 8:957. doi: 10.3390/cells8090957
65. Sun X, Wang M, Wang M, Yu X, Guo J, Sun T, et al. Metabolic reprogramming in triple-negative breast cancer. Front Oncol. (2020) 10:428. doi: 10.3389/fonc.2020.00428
66. Panda VK, Mishra B, Mahapatra S, Swain B, Malhotra D, Saha S, et al. Molecular insights on signaling cascades in breast cancer: A comprehensive review. Cancers. (2025) 17:234. doi: 10.3390/cancers17020234
67. Mir MA, Qayoom H, Mehraj U, Nisar S, Bhat B, Wani NA, et al. Targeting different pathways using novel combination therapy in triple negative breast cancer. Curr Cancer Drug Targets. (2020) 20:586–602. doi: 10.2174/1570163817666200518081955
68. Zhu H, Du C, Yuan M, Fu P, He Q, Yang B, et al. PD-1/PD-L1 counterattack alliance: multiple strategies for treating triple-negative breast cancer. Drug Discov Today. (2020) 25:1762–71. doi: 10.1016/j.drudis.2020.07.006
69. Smith J. Role of PD-1/PD-L1 crosstalk on inhibition of T-cell activation and proliferation through blockade of PI3K/Akt/mTOR signaling pathway. Asia Pac J Surg Exp & Pathol. (2024) 1:49–56. doi: 10.32948/ajsep.2024.11.18
70. Quan Z, Yang Y, Zheng H, Zhan Y, Luo J, Ning Y, et al. Clinical implications of the interaction between PD-1/PD-L1 and PI3K/AKT/mTOR pathway in progression and treatment of non-small cell lung cancer. J Cancer. (2022) 13:3434. doi: 10.7150/jca.77619
71. Yap TA, Olmos D, Molife LR, and Bono JSD. Targeting the insulin-like growth factor signaling pathway: figitumumab and other novel anticancer strategies. Expert Opin investigational Drugs. (2011) 20:1293–304. doi: 10.1517/13543784.2011.602630
72. Wang L, Liu W-Q, Broussy S, Han B, and Fang H. Recent advances of anti-angiogenic inhibitors targeting VEGF/VEGFR axis. Front Pharmacol. (2024) 14:1307860. doi: 10.3389/fphar.2023.1307860
73. Mankan AK, Lawless MW, Gray SG, Kelleher D, and McManus R. NF-κB regulation: the nuclear response. J Cell Mol Med. (2009) 13:631–43. doi: 10.1111/j.1582-4934.2009.00632.x
74. Qiao Y, He H, Jonsson P, Sinha I, Zhao C, Wright KD, et al. AP-1 is a key regulator of proinflammatory cytokine TNFα-mediated triple-negative breast cancer progression. J Biol Chem. (2016) 291:5068–79. doi: 10.1074/jbc.M115.702571
75. Newton K and Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harbor Perspect Biol. (2012) 4:a006049. doi: 10.1101/cshperspect.a006049
76. Scheidereit C. IκB kinase complexes: gateways to NF-κB activation and transcription. Oncogene. (2006) 25:6685–705. doi: 10.1038/sj.onc.1209934
77. Wang W, Nag SA, and Zhang R. Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr medicinal Chem. (2015) 22:264–89. doi: 10.2174/0929867321666141106124315
78. Kim R, Emi M, and Tanabe K. Cancer cell immune escape and tumor progression by exploitation of anti-inflammatory and pro-inflammatory responses. Cancer Biol Ther. (2005) 4:924–33. doi: 10.4161/cbt.4.9.2101
79. Proença C, Freitas M, Ribeiro D, Rufino AT, Fernandes E, Oliveira JMPFD, et al. The role of flavonoids in the regulation of epithelial-mesenchymal transition in cancer: A review on targeting signaling pathways and metastasis. Medicinal Res Rev. (2023) 43:1878–945. doi: 10.1002/med.21966
80. Wittmann M. 37. Mainzer Allergie-und klinische Immunologie-Workshop–Frühjahrstagung der DGAKI; 21./22. März 2025. Allergologie. (2025) 48:100. doi: 10.5414/ALX02559
81. Marshall HT and Djamgoz MB. Immuno-oncology: emerging targets and combination therapies. Front Oncol. (2018) 8:315. doi: 10.3389/fonc.2018.00315
82. Mayes PA, Hance KW, and Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discov. (2018) 17:509–27. doi: 10.1038/nrd.2018.75
83. Baumeister SH, Freeman GJ, Dranoff G, and Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol. (2016) 34:539–73. doi: 10.1146/annurev-immunol-032414-112049
84. Jhajj H. Discovery and development of agonist antibodies for T cell receptors. University of Michigan. (2022). Available at: https://dx.doi.org/10.7302/6061
85. Schiller M, Metze D, Luger TA, Grabbe S, and Gunzer M. Immune response modifiers–mode of action. Exp Dermatol. (2006) 15:331–41. doi: 10.1111/j.0906-6705.2006.00414.x
86. Cao X, Chen J, Li B, Dang J, Zhang W, Zhong X, et al. Promoting antibody-dependent cellular phagocytosis for effective macrophage-based cancer immunotherapy. Sci Adv. (2022) 8:eabl9171. doi: 10.1126/sciadv.abl9171
87. Kundu M, Greer YE, Dine JL, and Lipkowitz S. Targeting TRAIL death receptors in triple-negative breast cancers: challenges and strategies for cancer therapy. Cells. (2022) 11:3717. doi: 10.3390/cells11233717
88. Yuan X, Gajan A, Chu Q, Xiong H, Wu K, Wu GS, et al. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev. (2018) 37:733–48. doi: 10.1007/s10555-018-9728-y
89. Guo Q, Jin Y, Lin M, Zeng C, and Zhang J. NF-κB signaling in therapy resistance of breast cancer: Mechanisms, approaches, and challenges. Life Sciences. (2024) 348:122684. doi: 10.1038/s41392-021-00762-6
90. Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol Sci. (2012) 33:522–30. doi: 10.1016/j.tips.2012.06.007
91. Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. (2012) 13:767–79. doi: 10.1038/nrm3470
92. MacDonald BT, Tamai K, and He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell. (2009) 17:9–26. doi: 10.1016/j.devcel.2009.06.016
93. King TD, Suto MJ, and Li Y. The wnt/β-catenin signaling pathway: A potential therapeutic target in the treatment of triple negative breast cancer. J Cell Biochem. (2012) 113:13–8. doi: 10.1002/jcb.23350
94. Merikhian P, Eisavand MR, and Farahmand L. Triple-negative breast cancer: Understanding Wnt signaling in drug resistance. Cancer Cell Int. (2021) 21:1–8. doi: 10.1186/s12935-021-02107-3
95. Bilir B, Kucuk O, and Moreno CS. Wnt signaling blockage inhibits cell proliferation and migration, and induces apoptosis in triple-negative breast cancer cells. J Trans Med. (2013) 11:1–12. doi: 10.1186/1479-5876-11-280
96. Zhang Y and Wang X. Targeting the Wnt/β-catenin signaling pathway in cancer. J Hematol Oncol. (2020) 13:1–16. doi: 10.1186/s13045-020-00990-3
97. Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal transduction targeted Ther. (2022) 7:3. doi: 10.1038/s41392-021-00762-6
98. Liu C, Takada K, and Zhu D. Targeting Wnt/β-catenin pathway for drug therapy. Med Drug Discov. (2020) 8:100066. doi: 10.1016/j.medidd.2020.100066
99. Krishnamurthy N and Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev. (2018) 62:50–60. doi: 10.1016/j.ctrv.2017.11.002
100. Patel S, Alam A, Pant R, and Chattopadhyay S. Wnt signaling and its significance within the tumor microenvironment: novel therapeutic insights. Front Immunol. (2019) 10:2872. doi: 10.3389/fimmu.2019.02872
101. Davidson G. LRPs in WNT signalling. In: Pharmacology of the WNT signaling System. Springer Cham. (2021) 269:45–73. doi: 10.1007/164_2021_526
102. Zhang X, Dong N, and Hu X. Wnt/β-catenin signaling inhibitors. Curr topics medicinal Chem. (2023) 23:880–96. doi: 10.2174/1568026623666230303101810
103. Tejeda-Muñoz N and Robles-Flores M. Glycogen synthase kinase 3 in Wnt signaling pathway and cancer. IUBMB Life. (2015) 67:914–22. doi: 10.1002/iub.1454
104. Vijay GV, Zhao N, Hollander PD, Toneff MJ, Joseph R, Pietila M, et al. GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. (2019) 21:1–14. doi: 10.1186/s13058-019-1125-0
105. Park HS, Jang MH, Kim EJ, Kim HJ, Lee HJ, Kim YJ, et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Modern Pathol. (2014) 27:1212–22. doi: 10.1038/modpathol.2013.251
106. Deepak K, Vempati R, Nagaraju GP, Dasari VR, Nagini S, Rao DN, et al. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol Res. (2020) 153:104683. doi: 10.1016/j.phrs.2020.104683
107. Wendt MK, Smith JA, and Schiemann WP. Transforming growth factor-β-induced epithelial–mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene. (2010) 29:6485–98. doi: 10.1038/onc.2010.377
108. Fremd C, Jaeger D, and Schneeweiss A. Targeted and immuno-biology driven treatment strategies for triple-negative breast cancer: current knowledge and future perspectives. Expert Rev Anticancer Ther. (2019) 19:29–42. doi: 10.1080/14737140.2019.1537785
109. Jutten B and Rouschop K. EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle. (2014) 13:42–51. doi: 10.4161/cc.27518
110. Panda M and Biswal BK. Cell signaling and cancer: a mechanistic insight into drug resistance. Mol Biol Rep. (2019) 46:5645–59. doi: 10.1007/s11033-019-04958-6
111. Sasada T, Azuma K, Ohtake J, and Fujimoto Y. Immune responses to epidermal growth factor receptor (EGFR) and their application for cancer treatment. Front Pharmacol. (2016) 7:405. doi: 10.3389/fphar.2016.00405
112. Ramani S, Samant S, and Manohar SM. The story of EGFR: from signaling pathways to a potent anticancer target. Future medicinal Chem. (2022) 14:1267–88. doi: 10.4155/fmc-2021-0343
113. Bharadwaj U, Kasembeli MM, Robinson P, and Tweardy DJ. Targeting janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: rationale, progress, and caution. Pharmacol Rev. (2020) 72:486–526. doi: 10.1124/pr.119.018440
114. Balogun E, Hoque M, Gong P, Killeen E, Green CJ, Foresti R, et al. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem J. (2003) 371:887–95. doi: 10.1042/bj20021619
115. Aliyu M, Zohora FT, Anka AU, Ali K, Maleknia S, Saffarioun M, et al. Interleukin-6 cytokine: An overview of the immune regulation, immune dysregulation, and therapeutic approach. Int Immunopharmacol. (2022) 111:109130. doi: 10.1016/j.intimp.2022.109130
116. Qin J-J, Yan L, Zhang J, and Zhang W-D. STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review. J Exp Clin Cancer Res. (2019) 38:1–16. doi: 10.1186/s13046-019-1206-z
117. Yaseen MM, Abuharfeil NM, Darmani H, and Daoud A. Mechanisms of immune suppression by myeloid-derived suppressor cells: the role of interleukin-10 as a key immunoregulatory cytokine. Open Biol. (2020) 10:200111. doi: 10.1098/rsob.200111
118. Li L, Yu R, Cai T, Chen Z, Lan M, Zou T, et al. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int Immunopharmacol. (2020) 88:106939. doi: 10.1016/j.intimp.2020.106939
119. Shao F, Pang X, and Baeg GH. Targeting the JAK/STAT signaling pathway for breast cancer. Curr medicinal Chem. (2021) 28:5137–51. doi: 10.2174/0929867328666201207202012
120. Huang B, Lang X, and Li X. The role of IL-6/JAK2/STAT3 signaling pathway in cancers. Front Oncol. (2022) 12:1023177. doi: 10.3389/fonc.2022.1023177
121. Mohamed AAH, Ahmed AT, Abdulmonem WA, Bokov DO, Shafie A, Al-Hetty HRAK, et al. Interleukin-6 serves as a critical factor in various cancer progression and therapy. Med Oncol. (2024) 41:182. doi: 10.1007/s12032-024-02422-5
122. Kumar A, Khani AT, Ortiz AS, and Swaminathan S. GM-CSF: a double-edged sword in cancer immunotherapy. Front Immunol. (2022) 13:901277. doi: 10.3389/fimmu.2022.901277
123. Tiwari H, Singh S, Sharma S, Gupta P, Verma A, Chattopadhaya A, et al. Deciphering the landscape of triple negative breast cancer from microenvironment dynamics and molecular insights to biomarker analysis and therapeutic modalities. Medicinal Res Rev. (2024) 45:817–41. doi: 10.1002/med.22090
124. Djureinovic D, Wang M, and Kluger HM. Agonistic CD40 antibodies in cancer treatment. Cancers. (2021) 13:1302. doi: 10.3390/cancers13061302
125. Owen KL, Brockwell NK, and Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers. (2019) 11:2002. doi: 10.3390/cancers11122002
126. Piechutta M and Berghoff AS. New emerging targets in cancer immunotherapy: the role of cluster of differentiation 40 (CD40/TNFR5). Esmo Open. (2019) 4:201–20. doi: 10.1136/esmoopen-2019-000510
127. Patel AA and Odenike O. The next generation of JAK inhibitors: an update on fedratinib, momelotonib, and pacritinib. Curr Hematologic Malignancy Rep. (2020) 15:409–18. doi: 10.1007/s11899-020-00596-z
128. Philips N, Samuel P, Samuel M, Perez G, Khundoker R, Alahmade G, et al. Interleukin-4 signaling pathway and effects in allergic diseases. Curr Signal Transduction Ther. (2018) 13:76–80. doi: 10.2174/1574362413666180319143151
129. Karo-Atar D, Bitton A, Benhar I, and Munitz A. Therapeutic targeting of the interleukin-4/interleukin-13 signaling pathway: in allergy and beyond. BioDrugs. (2018) 32:201–20. doi: 10.1007/s40259-018-0280-7
130. Kontomanolis EN, Kalagasidou S, Pouliliou S, Anthoulaki X, Georgiou N, Papamanolis V, et al. The notch pathway in breast cancer progression. Sci World J. (2018) 2018:2415489. doi: 10.1155/2018/2415489
131. Giuli M, Giuliani E, Screpanti I, Bellavia D, and Checquolo S. Notch signaling activation as a hallmark for triple-negative breast cancer subtype. J Oncol. (2019) 2019:8707053. doi: 10.1155/2019/8707053
132. Ehebauer M, Hayward P, and Martinez-Arias A. Notch signaling pathway. Science’s STKE. (2006) 2006:cm7–7. doi: 10.1126/stke.3642006cm7
133. Sharif A, Shaji A, Chammaa M, Pawlik E, and Fernandez-Valdivia R. Notch transduction in non-small cell lung cancer. Int J Mol Sci. (2020) 21:5691. doi: 10.3390/ijms21165691
134. Lee K-L, Kuo Y-C, Ho Y-S, and Huang Y-H. Triple-negative breast cancer: current understanding and future therapeutic breakthrough targeting cancer stemness. Cancers. (2019) 11:1334. doi: 10.3390/cancers11091334
135. Christopoulos PF, Gjolberg TT, Kruger S, Haraldsen G, Andersen JT, Sundlisaeter E, et al. Targeting the notch signaling pathway in chronic inflammatory diseases. Front Immunol. (2021) 12:668207. doi: 10.3389/fimmu.2021.668207
136. Li L, Li S, Qin X, Yang H, Wu C, Liu Y, et al. Notch signaling pathway networks in cancer metastasis: a new target for cancer therapy. Med Oncol. (2017) 34:1–10. doi: 10.1007/s12032-017-1039-6
137. Rota LM and Wood TL. Crosstalk of the insulin-like growth factor receptor with the Wnt signaling pathway in breast cancer. Front Endocrinol. (2015) 6:92. doi: 10.3389/fendo.2015.00092
138. Takebe N, Harris PJ, Warren RQ, and Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. (2011) 8:97–106. doi: 10.1038/nrclinonc.2010.196
139. Mahmoud R, Ordóñez-Morán P, and Allegrucci C. Challenges for triple negative breast cancer treatment: Defeating heterogeneity and cancer stemness. Cancers. (2022) 14:4280. doi: 10.3390/cancers14174280
140. McAuliffe SM, Morgan SL, Wyant GA, Tran LT, Muto KW, Chen YS, et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc Natl Acad Sci. (2012) 109:E2939–48. doi: 10.1073/pnas.1206400109
141. Pancewicz J, Niklinska W, and Eljaszewicz A. Anti-Jagged-1 immunotherapy in cancer. Adv Med Sci. (2022) 67:196–202. doi: 10.1016/j.advms.2022.04.001
142. MacLean MR, Walker OL, Arun RP, Fernando W, and Marcato P. Informed by cancer stem cells of solid tumors: advances in treatments targeting tumor-promoting factors and pathways. Int J Mol Sci. (2024) 25:4102. doi: 10.3390/ijms25074102
143. Wang YY, Wang WD, and Sun ZJ. Cancer stem cell-immune cell collusion in immunotherapy. Int J Cancer. (2023) 153:694–708. doi: 10.1002/ijc.34421
144. Kuhnert F, Kirshner JR, and Thurston G. Dll4-Notch signaling as a therapeutic target in tumor angiogenesis. Vasc Cell. (2011) 3:1–8. doi: 10.1186/2045-824X-3-20
145. Qayoom H, Wani NA, Alshehri B, and Mir MA. An insight into the cancer stem cell survival pathways involved in chemoresistance in triple-negative breast cancer. Future Oncol. (2021) 17:4185–206. doi: 10.2217/fon-2021-0172
146. Xie X, Lee J, Iwase T, Kai M, and Ueno NT. Emerging drug targets for triple-negative breast cancer: a guided tour of the preclinical landscape. Expert Opin Ther Targets. (2022) 26:405–25. doi: 10.1080/14728222.2022.2077188
147. Kumari L, Mishra L, Sharma Y, Chahar K, Kumar M, Patel P, et al. NOTCH signaling pathway: occurrence, mechanism, and NOTCH-directed therapy for the management of cancer. Cancer Biotherapy Radiopharmaceuticals. (2024) 39:19–34. doi: 10.1089/cbr.2023.0023
148. Jan A, Sofi S, Jan N, and Mir MA. An update on cancer stem cell survival pathways involved in chemoresistance in triple-negative breast cancer. Future Oncol. (2025) p:1–21. doi: 10.1080/14796694.2025.2461443
149. Rasool I, Bhat AH, Khurshid N, Andrabi I, Qadir Q, Ganie SA, et al. Navigating breast cancer complexity: Deciphering the cross talk of Wnt/β catenin and Notch pathways in development and diseases. (2024).
150. Zhdanovskaya N, Firrincieli M, Lazzari S, Pace E, Rossi PS, Felli MP, et al. Targeting notch to maximize chemotherapeutic benefits: Rationale, advanced strategies, and future perspectives. Cancers. (2021) 13:5106. doi: 10.3390/cancers13205106
151. Mollen EW, Lent J, Tjan-Heijen VCG, Boersma LJ, Miele L, Smidt ML, et al. Moving breast cancer therapy up a notch. Front Oncol. (2018) 8:518. doi: 10.3389/fonc.2018.00518
152. Engel C, Rhiem K, Hahnen E, Loibl S, Weber KE, Seiler S, et al. Prevalence of pathogenic BRCA1/2 germline mutations among 802 women with unilateral triple-negative breast cancer without family cancer history. BMC Cancer. (2018) 18:1–6. doi: 10.1186/s12885-018-4029-y
153. Deng C-X. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. (2006) 34:1416–26. doi: 10.1093/nar/gkl010
154. Ali RM, McIntosh SA, and Savage KI. Homologous recombination deficiency in breast cancer: Implications for risk, cancer development, and therapy. Genes Chromosomes Cancer. (2021) 60:358–72. doi: 10.1002/gcc.22921
155. Faraoni I and Graziani G. Role of BRCA mutations in cancer treatment with poly (ADP-ribose) polymerase (PARP) inhibitors. Cancers. (2018) 10:487. doi: 10.3390/cancers10120487
156. Bhattacharjee S and Nandi S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life. (2017) 69:929–37. doi: 10.1002/iub.1696
157. Jurkovicova D, Neophytou CM, Gašparović AČ, and Gonçalves AC. DNA damage response in cancer therapy and resistance: challenges and opportunities. Int J Mol Sci. (2022) 23:14672. doi: 10.3390/ijms232314672
158. Lee Y, Choi I, Kim J, and Kim K. DNA damage to human genetic disorders with neurodevelopmental defects. J Genet Med. (2016) 13:1–13. doi: 10.5734/JGM.2016.13.1.1
159. Nickoloff JA, Taylor L, Sharma N, and Kato TA. Exploiting DNA repair pathways for tumor sensitization, mitigation of resistance, and normal tissue protection in radiotherapy. Cancer Drug Resistance. (2021) 4:244. doi: 10.20517/cdr.2020.89
160. Karayazi Atici Ö, Urbanska A, Gopinathan SG, Boutillon F, Goffin V, Shemanko CS, et al. ATM is required for the prolactin-induced HSP90-mediated increase in cellular viability and clonogenic growth after DNA damage. Endocrinology. (2018) 159:907–30. doi: 10.1210/en.2017-00652
161. Clark CA and Yang ES. Harnessing DNA repair defects to augment immune-based therapies in triple-negative breast cancer. Front Oncol. (2021) 11:703802. doi: 10.3389/fonc.2021.703802
162. Albarakati N, Abdel-Fatah TMA, Doherty R, Russell R, Agarwal D, Moseley P, et al. Targeting BRCA1-BER deficient breast cancer by ATM or DNA-PKcs blockade either alone or in combination with cisplatin for personalized therapy. Mol Oncol. (2015) 9:204–17. doi: 10.1016/j.molonc.2014.08.001
163. Beyaert M. ATR signalling after DNA damage: from the discovery of a new target to the reevaluation of its function in primary chronic lymphocytic leukaemia cells. UCL-Université Catholique de Louvain (2017).
164. Salguero C, Valladolid C, Robinson HMR, Smith GCM, and Yap TA. Targeting ATR in cancer medicine. In: Targeting the DNA Damage Response for Cancer Therapy. Springer (2023). p. 239–83.
165. Mavroeidi D, Georganta A, Panagiotou E, Syrigos K, and Souliotis V. Targeting ATR Pathway in solid tumors: Evidence of improving therapeutic outcomes. Int J Mol Sci. (2024) 25:2767. doi: 10.3390/ijms25052767
166. Dibitetto D, Widmer CA, and Rottenberg S. PARPi, BRCA, and gaps: controversies and future research. Trends Cancer. (2024) 10:857–69. doi: 10.1016/j.trecan.2024.06.008
167. Clark CA and Yang ES. Therapeutic targeting of DNA damage repair in the era of precision oncology and immune checkpoint inhibitors. J Immunotherapy Precis Oncol. (2023) 6:31–49. doi: 10.36401/JIPO-22-15
168. Angius G, Tomao S, Stati V, Vici P, Bianco V, Tomao F, et al. Prexasertib, a checkpoint kinase inhibitor: from preclinical data to clinical development. Cancer chemotherapy Pharmacol. (2020) 85:9–20. doi: 10.1007/s00280-019-03950-y
169. Mustafa M, Abbas K, Alam M, Ahmad W, and Usmani N. Molecular pathways and therapeutic targets linked to triple-negative breast cancer (TNBC). Mol Cell Biochem. (2023) p:1–19.
170. Rangel MC, Bertolette D, Castro NP, Klauzinska M, Cuttitta F, Salomon DS, et al. Developmental signaling pathways regulating mammary stem cells and contributing to the etiology of triple-negative breast cancer. Breast Cancer Res Treat. (2016) 156:211–26. doi: 10.1007/s10549-016-3746-7
171. Cohen M. The hedgehog signaling network. Am J Med Genet Part A. (2003) 123:5–28. doi: 10.1002/ajmg.a.20495
172. Skoda AM, Simovic D, Karin V, Kardum V, Vranic S, Serman L, et al. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosnian J basic Med Sci. (2018) 18:8. doi: 10.17305/bjbms.2018.2756
173. Bhateja P, Cherian M, Majumder S, and Ramaswamy B. The hedgehog signaling pathway: a viable target in breast cancer? Cancers. (2019) 11:1126. doi: 10.3390/cancers11081126
174. Riobo-Del Galdo NA, Lara Montero A, and Wertheimer EV. Role of Hedgehog signaling in breast cancer: pathogenesis and therapeutics. Cells. (2019) 8:375. doi: 10.3390/cells8040375
175. Lin Y, Shao Y, Li J, Zhang W, Zheng K, Zheng X, et al. The hierarchical micro-/nanotextured topographies promote the proliferation and angiogenesis-related genes expression in human umbilical vein endothelial cells by initiation of Hedgehog-Gli1 signaling. Artif Cells Nanomedicine Biotechnol. (2018) 46:1141–51. doi: 10.1080/21691401.2018.1533845
176. Muz B, De la Puente P, Azab F, and Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia. (2015) 2015:83–92. doi: 10.2147/HP.S93413
177. Di Mauro C, Rosa R, D’Amato V, Ciciola P, Servetto A, Marciano R, et al. Hedgehog signalling pathway orchestrates angiogenesis in triple-negative breast cancers. Br J Cancer. (2017) 116:1425–35. doi: 10.1038/bjc.2017.116
178. O’Reilly EA, Gubbins L, Sharma S, Tully R, Guang MHZ, Weiner-Gorzel K, et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. (2015) 3:257–75. doi: 10.1016/j.bbacli.2015.03.003
179. de la Mare J-A, Contu L, Hunter MC, Moyo B, Sterrenberg JN, Dhamani KCH, et al. Breast cancer: current developments in molecular approaches to diagnosis and treatment. Recent Patents Anti-Cancer Drug Discov. (2014) 9:153–75. doi: 10.2174/15748928113086660046
180. Liu R, Yu Y, Wang Q, Zhao Q, Yao Y, Sun M, et al. Interactions between hedgehog signaling pathway and the complex tumor microenvironment in breast cancer: current knowledge and therapeutic promises. Cell Communication Signaling. (2024) 22:432. doi: 10.1186/s12964-024-01812-6
181. Ruiz-Borrego M, Jimenez B, Antolin S, Garcia-Saenz JA, Corral J, Jerez Y, et al. A phase Ib study of sonidegib (LDE225), an oral small molecule inhibitor of smoothened or Hedgehog pathway, in combination with docetaxel in triple negative advanced breast cancer patients: GEICAM/2012–12 (EDALINE) study. Investigational New Drugs. (2019) 37:98–108. doi: 10.1007/s10637-018-0614-9
182. Skóra B, Masicz M, Nowak P, Lachowska J, Soltysek P, Biskup J, et al. Suppression of sonic hedgehog pathway-based proliferation in glioblastoma cells by small-size silver nanoparticles in vitro. Arch Toxicol. (2023) 97:2385–98. doi: 10.1007/s00204-023-03552-x
183. Connell DR. Establishment of hedgehog pathway overexpression in osteosarcoma cell lines leading to an anti-apoptotic genotype which can be inhibited by the common anti-fungal and hedgehog pathway inhibitor, itraconazole. University of Illinois at Urbana-Champaign (2020).
184. Lospinoso Severini L, Ghirga F, Bufalieri F, Quaglio D, Infante P, Marcotullio LD, et al. The SHH/GLI signaling pathway: a therapeutic target for medulloblastoma. Expert Opin Ther Targets. (2020) 24:1159–81. doi: 10.1080/14728222.2020.1823967
185. Ilangumaran S, Villalobos-Hernandez A, Bobbala D, and Ramanathan S. The hepatocyte growth factor (HGF)–MET receptor tyrosine kinase signaling pathway: Diverse roles in modulating immune cell functions. Cytokine. (2016) 82:125–39. doi: 10.1016/j.cyto.2015.12.013
186. Gallo S, Folco CB, and Crepaldi T. The MET oncogene: An update on targeting strategies. Pharmaceuticals. (2024) 17:1473. doi: 10.3390/ph17111473
187. Cascione L, Gasparini P, Lovat F, Carasi S, Pulvirenti A, Ferro A, et al. Integrated microRNA and mRNA signatures associated with survival in triple negative breast cancer. PLoS One. (2013) 8:e55910. doi: 10.1371/journal.pone.0055910
188. Matthews HK, Bertoli C, and de Bruin RA. Cell cycle control in cancer. Nat Rev Mol Cell Biol. (2022) 23:74–88. doi: 10.1038/s41580-021-00404-3
189. Liu Z-L, Chen H-H, Zheng L-L, Sun L-P, and Shi L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduction Targeted Ther. (2023) 8:198. doi: 10.1038/s41392-023-01460-1
190. Rivas S, Marin A, Samtani S, Gonzalez-Feliu E, and Armisen R. MET signaling pathways, resistance mechanisms, and opportunities for target therapies. Int J Mol Sci. (2022) 23:13898. doi: 10.3390/ijms232213898
191. Morley R, Cardnas A, Hawkins P, Suzuki Y, Paton V, Phan S-C, et al. Safety of onartuzumab in patients with solid tumors: experience to date from the onartuzumab clinical trial program. PLoS One. (2015) 10:e0139679. doi: 10.1371/journal.pone.0139679
192. Moosavi F, Giovannetti E, Peters GJ, and Firuzi O. Combination of HGF/MET-targeting agents and other therapeutic strategies in cancer. Crit Rev Oncology/hematology. (2021) 160:103234. doi: 10.1016/j.critrevonc.2021.103234
193. Xia Y, Huang C, Zhong M, Zhong H, Ruan R, Xiong J, et al. Targeting HGF/c-MET signaling to regulate the tumor microenvironment: Implications for counteracting tumor immune evasion. Cell Communication Signaling. (2025) 23:46. doi: 10.1186/s12964-025-02033-1
194. Cheng Q, Chang JT, Geradts J, Neckers LM, Haystead T, Spector NL, et al. Amplification and high-level expression of heat shock protein 90 marks aggressive phenotypes of human epidermal growth factor receptor 2 negative breast cancer. Breast Cancer Res. (2012) 14:1–15. doi: 10.1186/bcr3168
195. Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, and Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther. (2013) 138:255–71. doi: 10.1016/j.pharmthera.2013.01.011
196. Wynn CS and Tang S-C. Anti-HER2 therapy in metastatic breast cancer: many choices and future directions. Cancer Metastasis Rev. (2022) 41:193–209. doi: 10.1007/s10555-022-10021-x
197. Swain SM, Shastry M, and Hamilton E. Targeting HER2-positive breast cancer: advances and future directions. Nat Rev Drug Discov. (2023) 22:101–26. doi: 10.1038/s41573-022-00579-0
198. Alasmari MM. A review of margetuximab-based therapies in patients with HER2-positive metastatic breast cancer. Cancers. (2022) 15:38. doi: 10.3390/cancers15010038
199. Ujlaky-Nagy L, Szöllősi J, and Vereb G. Disrupting EGFR–HER2 transactivation by pertuzumab in HER2-positive cancer: quantitative analysis reveals EGFR signal input as potential predictor of therapeutic outcome. Int J Mol Sci. (2024) 25:5978. doi: 10.3390/ijms25115978
200. Giuliano M, Hu H, Wang Y-C, Fu X, Nardone A, Herrera S, et al. Upregulation of ER signaling as an adaptive mechanism of cell survival in HER2-positive breast tumors treated with anti-HER2 therapy. Clin Cancer Res. (2015) 21:3995–4003. doi: 10.1158/1078-0432.CCR-14-2728
201. Seligson JM, Patron AM, Berger MJ, Harvey RD, and Seligson ND. Sacituzumab govitecan-hziy: An antibody-drug conjugate for the treatment of refractory, metastatic, triple-negative breast cancer. Ann Pharmacotherapy. (2021) 55:921–31. doi: 10.1177/1060028020966548
202. Nicolò E, Repetto M, Boscolo Bielo L, Tarantino P, and Curigliano G. Antibody-drug conjugates in breast cancer: what is beyond HER2? Cancer J. (2022) 28:436–45. doi: 10.1097/PPO.0000000000000629
203. Gao X, Bu T, Wang W, and Xu Y. Comparative analysis and future prospects of human epidermal growth factor receptor 2 (HER2) and trophoblast cell-surface antigen 2 (Trop-2) targeted antibody-drug conjugates in breast cancer treatment. Breast Cancer: Targets Ther. (2024) p:621–30.
204. Mercogliano MF, Bruni S, Mauro FL, and Schillaci R. Emerging targeted therapies for HER2-positive breast cancer. Cancers. (2023) 15:1987. doi: 10.3390/cancers15071987
205. Seung E, Xing Z, Wu L, Rao E, Cortez-Retamozo V, Ospina B, et al. A trispecific antibody targeting HER2 and T cells inhibits breast cancer growth via CD4 cells. Nature. (2022) 603:328–34. doi: 10.1038/s41586-022-04439-0
206. Popovic LS, Matovina-Brko G, Popovic M, Punie K, Cvetanovic A, Lambertini M, et al. Targeting triple-negative breast cancer: a clinical perspective. Oncol Res. (2023) 31:221. doi: 10.32604/or.2023.028525
207. Liang Y, Zhang P, Li F, Lai H, Qi T, Wang Y, et al. Advances in the study of marketed antibody-drug Conjugates (ADCs) for the treatment of breast cancer. Front Pharmacol. (2024) 14:1332539. doi: 10.3389/fphar.2023.1332539
208. Xu X, Zhang L, He X, Zhang P, Sun C, Xu X, et al. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem Biophys Res Commun. (2018) 502:160–5. doi: 10.1016/j.bbrc.2018.05.139
209. Jovanović B, Beeler JS, Pickup MW, Chytil A, Gorska AE, Ashby WJ, et al. Transforming growth factor beta receptor type III is a tumor promoter in mesenchymal-stem like triple negative breast cancer. Breast Cancer Res. (2014) 16:1–14. doi: 10.1186/bcr3684
210. Fujio K, Komai T, Inoue M, Morita K, Okamura T, Yamamoto K, et al. Revisiting the regulatory roles of the TGF-β family of cytokines. Autoimmun Rev. (2016) 15:917–22. doi: 10.1016/j.autrev.2016.07.007
211. Fanayan S, Firth SM, and Baxter RC. Signaling through the smad pathway by insulin-like growth factor-binding protein-3 in breast cancer cells: RELATIONSHIP TO TRANSFORMING GROWTH FACTOR-β1 SIGNALING. J Biol Chem. (2002) 277:7255–61. doi: 10.1074/jbc.M108038200
212. Chen H-S, Bai M-H, Zhang T, Li G-D, and Liu M. Ellagic acid induces cell cycle arrest and apoptosis through TGF-β/Smad3 signaling pathway in human breast cancer MCF-7 cells. Int J Oncol. (2015) 46:1730–8. doi: 10.3892/ijo.2015.2870
213. Li T, Wang X, Niu M, Wang M, Zhou J, Wu K, et al. Bispecific antibody targeting TGF-β and PD-L1 for synergistic cancer immunotherapy. Front Immunol. (2023) 14:1196970. doi: 10.3389/fimmu.2023.1196970
214. Siegel PM and Massagué J. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat Rev Cancer. (2003) 3:807–20. doi: 10.1038/nrc1208
215. Liu S, Ren J, and Ten Dijke P. Targeting TGFβ signal transduction for cancer therapy. Signal transduction targeted Ther. (2021) 6:8. doi: 10.1038/s41392-020-00436-9
216. Ramzan A, Yousaf MA, Rashid MU, Basheera S, and Malkani N. In-silico prediction of TGF-β1 non-synonymous variants and their impact on binding affinity to Fresolimumab. J Biomolecular Structure Dynamics. (2024) 42:12214–27. doi: 10.1080/07391102.2023.2268198
217. Dai X, Hua D, and Lu X. Roles of TGF-β in cancer hallmarks and emerging onco-therapeutic design. Expert Rev Mol Med. (2022) 24:e42. doi: 10.1017/erm.2022.37
218. Nedeljković M, Vuletić A, and Mirjačić Martinović K. Divide and conquer—Targeted therapy for triple-negative breast cancer. Int J Mol Sci. (2025) 26:1396. doi: 10.3390/ijms26041396
219. Schlingensiepen KH, Jaschinski F, Lang SA, Moser C, Geissler EK, Schlitt HJ, et al. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. (2011) 102:1193–200. doi: 10.1111/j.1349-7006.2011.01917.x
220. Sheikh KA, Amjad M, Irfan MT, Anjum S, Majeed T, Riaz MU, et al. Exploring TGF-β Signaling in cancer progression: prospects and therapeutic strategies. OncoTargets Ther. (2025) 2025:233–62. doi: 10.2147/OTT.S493643
221. Zhao X and Guan J-L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Advanced Drug delivery Rev. (2011) 63:610–5. doi: 10.1016/j.addr.2010.11.001
222. Rigiracciolo DC, Nohata N, Lappano R, Cirillo F, Talia M, Scordamaglia D, et al. IGF-1/IGF-1R/FAK/YAP transduction signaling prompts growth effects in triple-negative breast cancer (TNBC) cells. Cells. (2020) 9:1010. doi: 10.3390/cells9041010
223. Parsons JT, Martin KH, Sllack JK, Taylor JM, and Weed SA. Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene. (2000) 19:5606–13. doi: 10.1038/sj.onc.1203877
224. Rigiracciolo DC, Nohata N, Lappano R, Cirillo F, Talia M, Adame-Garcia SR, et al. Focal Adhesion Kinase (FAK)-Hippo/YAP transduction signaling mediates the stimulatory effects exerted by S100A8/A9-RAGE system in triple-negative breast cancer (TNBC). J Exp Clin Cancer Res. (2022) 41:1–25. doi: 10.1186/s13046-022-02396-0
225. Mohanty A, Pharaon RR, Nam A, Salgia S, Kulkarni P, Massarelli E, et al. FAK-targeted and combination therapies for the treatment of cancer: an overview of phase I and II clinical trials. Expert Opin investigational Drugs. (2020) 29:399–409. doi: 10.1080/13543784.2020.1740680
226. Singh S and Chakrabarti R. Consequences of EMT-driven changes in the immune microenvironment of breast cancer and therapeutic response of cancer cells. J Clin Med. (2019) 8:642. doi: 10.3390/jcm8050642
227. Perera DFTN. Targeting Focal Adhesion Kinase (FAK) in Triple Negative Breast Cancer (TNBC). (2020).
228. Kanno Y, Chen CY, Lee HL, Chiou JF, and Chen YJ. Molecular mechanisms of chemotherapy resistance in head and neck cancers. Front Oncol. (2021) 11:640392. doi: 10.3389/fonc.2021.640392
229. Fiori Lopes L, Guembarovski RL, Guembarovski AL, Kishima MO, Campos CZ, Oda JMM, et al. FOXP3 transcription factor: a candidate marker for susceptibility and prognosis in triple negative breast cancer. BioMed Res Int. (2014) 2014. doi: 10.1155/2014/341654
230. Carpenter RL and Lo H-W. STAT3 target genes relevant to human cancers. Cancers. (2014) 6:897–925. doi: 10.3390/cancers6020897
231. Wei C, Wang Y, and Li X. The role of Hippo signal pathway in breast cancer metastasis. OncoTargets Ther. (2018) 11:2185–93. doi: 10.2147/OTT.S157058
232. Mirzaei S, Gholami MH, Hashemi F, Zabolian A, Farahani MV, and Hushmandi K. Advances in understanding the role of P-gp in doxorubicin resistance: Molecular pathways, therapeutic strategies, and prospects. Drug Discov Today. (2022) 27:436–55. doi: 10.1016/j.drudis.2021.09.020
233. Adesoye T, Tripathy D, Hunt KK, and Keyomarsi K. Exploring novel frontiers: leveraging STAT3 signaling for advanced cancer therapeutics. Cancers. (2024) 16:492. doi: 10.3390/cancers16030492
234. Samad MA, Ahmad I, Hasan A, Alhashmi MH, Ayub A, Al-Abbasi FA, et al. STAT3 signaling pathway in health and disease. MedComm. (2025) 6:e70152. doi: 10.1002/mco2.70152
235. Ball MS, Bhandari R, Torres GM, Martyanov V, ElTanbouly MA, Archambault K, et al. CDDO-Me alters the tumor microenvironment in estrogen receptor negative breast cancer. Sci Rep. (2020) 10:6560. doi: 10.1038/s41598-020-63482-x
236. Battogtokh G, Obidiro O, and Akala EO. Recent developments in combination immunotherapy with other therapies and nanoparticle-based therapy for triple-negative breast cancer (TNBC). Cancers. (2024) 16:2012. doi: 10.3390/cancers16112012
237. Kang Z, Li S, Li Y, Song J, Peng Y, Chen Y, et al. Small molecular inhibitors and degraders targeting STAT3 for cancer therapy: An updated review (from 2022 to 2024). Chin Chem Lett. (2024), 110447.
238. Kyriazoglou A, Liontos M, Zakopoulou R, Kaparelou M, Tsiara A, Papatheodoridi AM, et al. The role of the Hippo pathway in breast cancer carcinogenesis, prognosis, and treatment: a systematic review. Breast Care. (2021) 16:6–15. doi: 10.1159/000507538
239. Xiao Y and Dong J. The hippo signaling pathway in cancer: A cell cycle perspective. Cancers. (2021) 13:6214. doi: 10.3390/cancers13246214
240. Piccolo S, Dupont S, and Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. (2014) 94:1287–312. doi: 10.1152/physrev.00005.2014
241. Jin X, Zhu L, Xiao S, Cui Z, Tang J, Yu J, et al. MST1 inhibits the progression of breast cancer by regulating the Hippo signaling pathway and may serve as a prognostic biomarker. Mol Med Rep. (2021) 23:383. doi: 10.3892/mmr.2021.12022
242. Thomas R, Al-Khadairi G, and Decock J. Immune checkpoint inhibitors in triple negative breast cancer treatment: promising future prospects. Front Oncol. (2021) 10:600573. doi: 10.3389/fonc.2020.600573
243. Li Q, Chen S, Wang X, Cai J, Huang H, Tang S, et al. Cisplatin-based combination therapy for enhanced cancer treatment. Curr Drug Targets. (2024) 25(7):473–91. doi: 10.2174/0113894501294182240401060343
244. Galan JA and Avruch J. MST1/MST2 protein kinases: regulation and physiologic roles. Biochemistry. (2016) 55:5507–19. doi: 10.1021/acs.biochem.6b00763
245. Crawford JJ, Bronner SM, and Zbieg JR. Hippo pathway inhibition by blocking the YAP/TAZ–TEAD interface: a patent review. Expert Opin Ther Pat. (2018) 28(12):867–73. doi: 10.1080/13543776.2018.1549226
246. Neophytou C, Boutsikos P, and Papageorgis P. Molecular mechanisms and emerging therapeutic targets of triple-negative breast cancer metastasis. Front Oncol. (2018) 8:31. doi: 10.3389/fonc.2018.00031
247. Li Q, Chen S, Wang X, Cai J, Huang H, Tang S, et al. Cisplatin-based combination therapy for enhanced cancer treatment. Curr Drug Targets. (2024) 25:473–91. doi: 10.2174/0113894501294182240401060343
248. Holden JK and Cunningham CN. Targeting the hippo pathway and cancer through the TEAD family of transcription factors. Cancers. (2018) 10:81. doi: 10.3390/cancers10030081
249. Crawford JJ, Bronner SM, and Zbieg JR. Hippo pathway inhibition by blocking the YAP/TAZ–TEAD interface: a patent review. Expert Opin Ther patents. (2018) 28:867–73. doi: 10.1080/13543776.2018.1549226
250. Marhelava K, Pilch Z, Bajor M, Graczyk-Jarzynka A, and Zagozdzon R. Targeting negative and positive immune checkpoints with monoclonal antibodies in therapy of cancer. Cancers. (2019) 11:1756. doi: 10.3390/cancers11111756
Keywords: TNBC, antitumor, BRCA1 or BRCA2, chemotherapy, signaling pathway
Citation: Mir PA, Kumar N, Gupta SK, Kaur A, Farooq S, Sandhu GS and Kumar A (2025) Therapeutic innovations in triple negative breast cancer: integrating molecular targeting and monoclonal antibody strategies. Front. Oncol. 15:1645438. doi: 10.3389/fonc.2025.1645438
Received: 11 June 2025; Accepted: 29 August 2025;
Published: 22 September 2025; Corrected: 24 September 2025.
Edited by:
Arunima Biswas, University of Kalyani, IndiaReviewed by:
Carla Van Den Berg, The University of Texas at Austin, United StatesZhenbo Tu, Beth Israel Deaconess Medical Center, United States
Copyright © 2025 Mir, Kumar, Gupta, Kaur, Farooq, Sandhu and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prince Ahad Mir, cHJpbmNlYWhhZEBrYXNobWlydW5pdmVyc2l0eS5uZXQ=; Nishant Kumar, bms1MDc0NDdAZ21haWwuY29t