 Lu Wang
Lu Wang Weiwei Qiao1
Weiwei Qiao1 Yeqiong Zhang
Yeqiong Zhang Zhiwei Dong
Zhiwei Dong- 1Department of Diagnostics, Second School of Clinical Medicine, Binzhou Medical University, Yantai, Shandong, China
- 2Department of Infectious Diseases, Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong, China
- 3Department of Surgery, Faculty of Medicine, The Chinese University of Hong Kong, Hong Kong, Hong Kong SAR, China
Cholangiocarcinoma (CCA), a highly aggressive biliary tract malignancy, exhibits rising incidence rates and an extremely poor prognosis. Recent studies reveal that gut-liver axis dysregulation drives CCA progression through gut microbiota dysbiosis, bile acid (BA) metabolic disturbances, and immune microenvironment remodeling. Clinical evidence highlights significant alterations in the gut and biliary microbial composition of CCA patients, which correlate with tumor stage, vascular invasion, and survival outcomes. Dysregulated BA metabolism in CCA, characterized by accumulation of primary conjugated BAs, promotes tumor invasiveness via interaction with specific BA receptors and fosters an immunosuppressive microenvironment. Emerging therapeutic strategies include antibiotics for pathogenic microbiota modulation, probiotics for microbial homeostasis restoration, fecal microbiota transplantation, and BA pathway modulators. Future directions necessitate integrating synthetic biology (engineered microbiota), multi-omics, and artificial intelligence to develop precision therapies. Targeting the gut-liver axis offers novel therapeutic perspectives for CCA; however, clinical translation demands deeper mechanistic insights and standardized protocols to address challenges such as microbiota heterogeneity and receptor signaling duality.
1 Introduction
Cholangiocarcinoma (CCA), an aggressive malignant tumor of the bile ducts, exhibits distinct epidemiological trends across subtypes (1). CCA is anatomically classified as intrahepatic (iCCA) or extrahepatic (eCCA), with eCCA further subdivided into perihilar and distal subtypes based on their location relative to the cystic duct (2). Globally, CCA incidence is increasing, notably iCCA (3, 4), which is the second most common cause of primary liver cancer, after hepatocellular carcinoma (HCC) (5). The peak incidence of CCA ocures between 60 and 70 years, more commonly arising in males (6). While the risk factors for CCA subtypes are distinct, primary sclerosing cholangitis (PSC) is a well-established common risk factor for both iCCA and eCCA (7). The persistently poor prognosis (5-year survival <15%) largely stems from late-stage diagnosis, as the asymptomatic of disease onset often precludes timely therapeutic intervention (8). Current management of CCA remains clinically challenging. Surgical resection is the only curative option, which is limited to the majority patients (9). Patients with advanced unresectable or metastatic CCA face constrained therapeutic efficacy from systemic therapies, demonstrating median overall survival (OS) of 6-18 months (10, 11). Recent advancements incorporating chemoimmunotherapy regimens [e.g., gemcitabine/cisplatin combined with pembrolizumab (12)] and molecularly targeted agents [e.g., FGFR2 inhibitors synergized with cisplatin (13)] have broadened treatment paradigms. Nevertheless, clinical outcomes persist below expectations, with 12-month survival rates remaining under 40% in advanced-stage cohorts. Addressing the aggressive biology and molecular heterogeneity necessitates prioritized development of innovative neoadjuvant approaches leveraging multi-omics platforms to establish biomarker-directed therapeutic algorithms.
The gut-liver axis emerges as a master regulator of hepatobiliary disorders, governed by reciprocal signaling between hepatocytes, cholangiocytes, and intestinal microbiota (14, 15). This cellular triad drives disease pathogenesis through three interlinked mechanisms: microbiota-derived metabolite reprogramming, dysregulated bile acid (BA) enterohepatic cycling, and immune niche remodeling (16–18). Accumulating evidence establishes gut microbiota dysbiosis as a critical modulator of CCA pathogenesis and progression (19, 20). Pathobiont-derived metabolites translocate across the compromised intestinal barrier, activating hepatic Kupffer cells (KCs) to amplify pro-inflammatory cascades, thereby fueling hepatobiliary inflammation and fibrotic preconditioning (21). A critical component of this bidirectional interaction is the BA enterohepatic circulation. Bidirectional crosstalk emerges between microbial communities and BA metabolism, where altered BA composition and impaired excretion through BA specific receptor pathways perpetuate cholangitis-driven carcinogenesis (22). Concurrently, preclinical and clinical evidence converges to nominate the gut-liver axis as a high-value therapeutic frontier in CCA. This review will examine molecular mechanisms and clinical evidence linking the gut-liver axis to CCA progression. We will also evaluate dual targeting of gut microbiota and BA-associated signaling pathways as potential therapeutic strategies for CCA and propose the future research priorities and current translation challenges.
2 The gut-liver axis
The gut-liver axis orchestrates a bidirectional crosstalk network that coordinates environmental exposures, microbial dynamics, and host signaling to govern gastrointestinal homeostasis and disease progression (Figure 1). Mechanistically, disruption of this axis—characterized by intestinal barrier failure and microbial dysbiosis—promotes bacterial translocation into the biliary tract, where TLR activation and subsequent nuclear factor kappa-B(NF-κB) signaling drive immune-mediated cholangitis while impairing mucosal integrity (23). Notably, the gut epithelial tight junctions serve as a critical physical barrier against microbial invasion. Early perturbations of gut-liver axis homeostasis frequently originate from dietary insults or antibiotic-induced dysbiosis, which compromise tight junction integrity and initiate intestinal hyperpermeability (24). This breach facilitates systemic translocation of microbial metabolites—notably lipopolysaccharides (LPS)—from the portal circulation into hepatic tissues, triggering hepatic KCs to amplify pro-inflammatory cytokine production via coordinated activation of Toll-like receptor 4 (TLR4)/MyD88 signaling and IL-6/STAT3 signaling cascades. Such chronic inflammatory signaling drives both progressive hepatobiliary injury and direct oncogenic transformation through two interconnected mechanisms: (1) initiating oxidative DNA damage-induced genomic instability; and (2) activating dysregulated proliferative signaling pathways that bypass cell cycle checkpoints (14).

Figure 1. Gut–liver axis pathogenesis in cholangiocarcinoma and hepatocellular carcinoma.
Emerging clinical evidence highlights the pivotal role of genetically determined microbial alterations in destabilizing gut-liver axis, which accelerates steatohepatitis progression, exacerbates fibrogenesis, and ultimately fosters hepatocarcinogenesis (25–27). The pathogenic interplay between intestinal inflammation, gut microbiota alterations, and cholangiopathies is strikingly exemplified in PSC (28). In genetically predisposed individuals, gut-derived microbial components trigger cholangiocyte-specific immune activation, initiating or perpetuating biliary epithelial injury (29). Furthermore, gut dysbiosis critically impairs antitumor immune surveillance and propels the transition from chronic biliary disease to malignancy by orchestrating a pro-inflammatory microenvironment (19).
3 Gut microbiota and cholangiocarcinoma development
3.1 Gut microbiota and cholangiocarcinoma carcinogenesis
CCA patients exhibit significant gut microbiota dysbiosis, characterized by reduced microbial diversity, depletion of beneficial taxa (e.g., Faecalibacterium), and enrichment of pathobionts (e.g., Escherichia-Shigella), which collectively disrupt gut-liver axis homeostasis (Table 1). Of note, α-diversity alterations exhibit marked heterogeneity across CCA subtypes, with conflicting reports in extrahepatic and hilar variants (30). Emerging evidence on iCCA consistently demonstrate microbial dysbiosis accompanied by reduced α-diversity, likely reflecting tumor microenvironment-driven ecological pressures. In a study by Zhang et al., it was revealed that patients with iCCA exhibited significantly reduced abundances of beneficial gut bacteria Bacteroides, Faecalibacterium, and Roseburia, along with increased levels of pathogenic taxa Escherichia-Shigella and Subdoligranulum, compared to healthy controls (16). This study firstly unveiled that gut microbiota modulates glutamine metabolism to downregulate the ALK5/NOX1 axis, thereby inhibiting ferroptosis in CCA cells, offering a promising therapeutic strategy for precision treatment in iCCA. Moreover, Ito et al. reported enrichment of Enterobacteriaceae (harbouring the polyketide synthase carcinogenic island) in biliary tract cancer (BTC) faecal samples, suggesting its direct pro-carcinogenic role in biliary tract carcinogenesis (31). A case-control study and a cohort analysis found no significant difference in gut microbiota α-diversity between CCA patients and healthy controls (21, 32). Notably, within this cohort study, a machine learning-driven approach leveraging a random forest algorithm identified eight discriminative bacterial genera (e.g., Faecalibacterium, Klebsiella) to develop a tripartite microbial signature-based classifier, achieving exceptional diagnostic accuracy (AUC: 0.92–0.99) in effectively differentiating CCA from HCC (21). Furthermore, CCA patients exhibited increased abundance of Gram-negative bacteria (Enterobacteriaceae), correlating with elevated systemic inflammation markers (neutrophil-to-lymphocyte ratio, platelet-to-lymphocyte ratio). In contrast, Jia et al. revealed elevated α-diversity patterns in iCCA patients through a multi-omics approach integrating taxon-specific microbial biomarkers (notably Lactobacillus and Alloscardovia) with functional BA metabolism signatures (33). Despite established links between specific gut microbiota alterations (e.g., decreased Faecalibacterium, increased Escherichia-Shigella/Enterobacteriaceae) and CCA pathogenesis (16, 31, 33), interpretation remains constrained by methodological limitations. Predominantly small cohort sizes compromise statistical power and hinder validation/generalizability of findings, including biomarker classifiers or subtype-specific diversity patterns (e.g., the α-diversity heterogeneity observed) (16, 21, 31–33). Furthermore, reliance on observational designs impedes causal inference due to uncontrolled, dynamic confounders such as recent antibiotic use, dietary fluctuations, and variable underlying liver pathology (e.g., cirrhosis severity). Consequently, attributing dysbiosis directly to CCA versus confounders/disease consequences remains challenging. Future research necessitates large-scale, multi-center prospective cohorts with rigorous longitudinal monitoring of key confounders (antibiotics, diet, liver function) and robust causal inference methods..
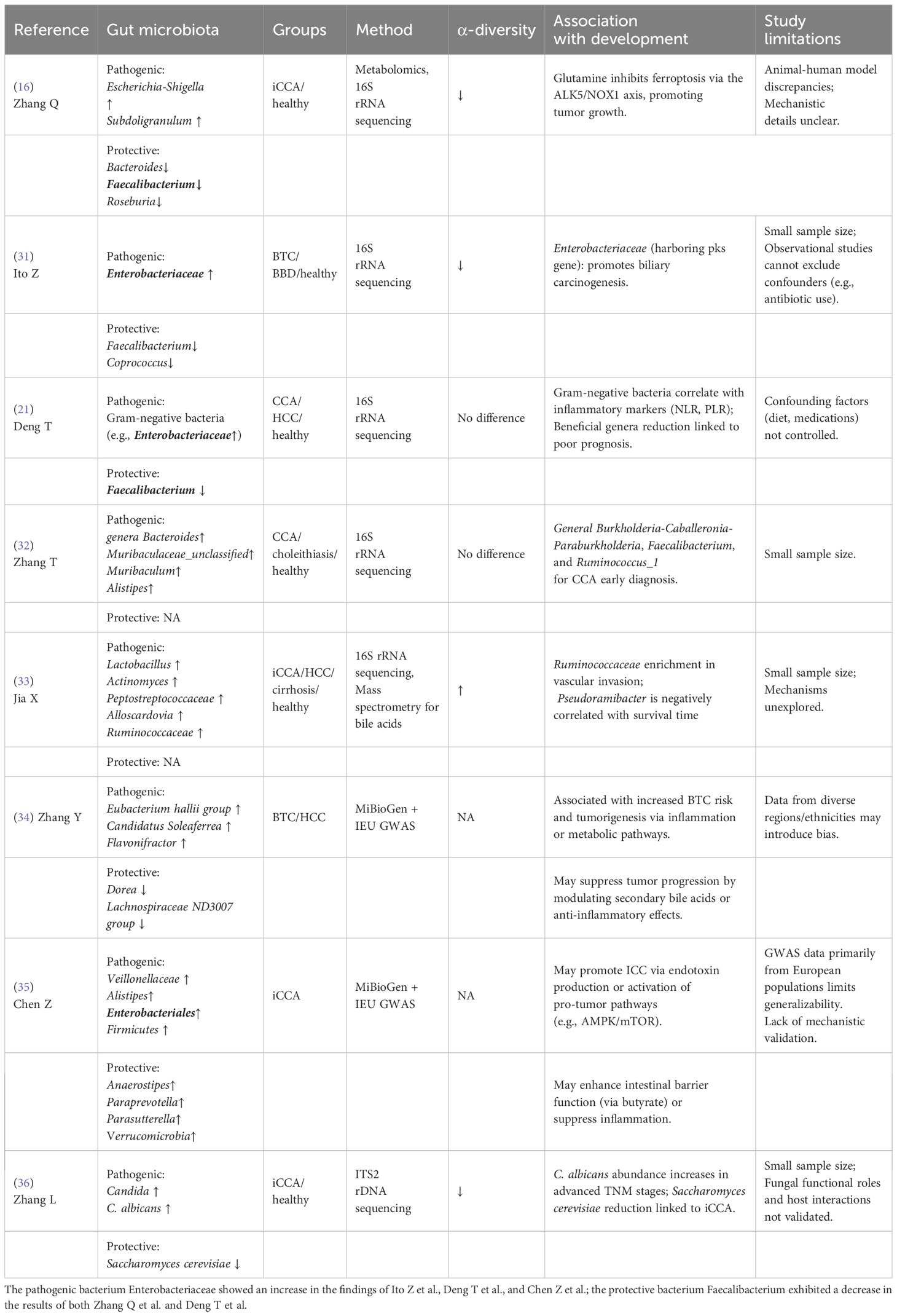
Table 1. Gut microbiota alterations in cholangiocarcinoma.
Mendelian randomization analyses leveraging MiBioGen gut microbiota genome-wide association study (GWAS) data have identified host genetic variants causally associated with microbial compositional changes. These analyses revealed that elevated abundances of Eubacterium hallii group, Candidatus Soleaferrea, and Flavonifractor confer increased BTC risk, while Dorea and Lachnospiraceae ND3007 demonstrated protective effects (34). Complementary Mendelian randomization studies further implicated Veillonellaceae, Alistipes, Enterobacteriales, and Firmicutes as iCCA-promoting taxa (35). Mechanistically, protective genera such as Anaerostipes, Paraprevotella, Parasutterella, and Verrucomicrobia enhance intestinal barrier integrity via butyrate production or exert anti-inflammatory activity, underscoring the dual diagnostic and therapeutic potential of gut microbiota in hepatobiliary malignancies. Notably, iCCA patients demonstrate gut mycobiota dysbiosis characterized by Candida spp. (particularly C. albicans) enrichment and depletion of beneficial Saccharomyces cerevisiae, with advanced TNM stages (III–IV) correlating strongly with pathogenic fungal predominance (36). These findings position gut microbiota dysbiosis as a driver of iCCA progression, warranting exploration of antifungal therapies (Supplementary Figure 1). However, critical gaps persist in addressing bacterial-fungal ecological interactions central to dysbiosis mechanisms and establishing experimental validation of causality between specific fungal taxa (e.g., C. albicans) and oncogenesis through animal models or in vitro systems.
While emerging evidence reveals subtype-specific gut microbial configurations differentiating CCA and healthy controls, current investigations remain constrained by geographically restricted cohorts. Future studies should prioritize mechanistic validation through germ-free animal models integrated with multi-ethnic validation cohorts—stratified into three principal groups: (1) Southeast Asian cohorts (≥40% allocation, bearing >50% of the global CCA burden), (2) European/North American cohorts (addressing industrial/environmental confounders), and (3) latent-risk diaspora cohorts. Crucially, ancestry-based principal component analysis with structured recruitment quotas must mitigate cryptic population substructure and allelic sampling biases, thereby ensuring robust control of geographic and migrant heterogeneity. This integrated methodology will enable rigorous delineation of causal host-microbiota interactions while controlling population stratification artifacts.
3.2 Gut microbiota and cholangiocarcinoma prognosis
Emerging evidence positions the gut microbiota as a pivotal determinant of CCA progression through multifaceted molecular mechanisms. Prognostically, tumor vascular invasion (VI) shows strong correlations with specific gut microbial alterations (37) (Supplementary Figure 1). VI-positive patients exhibit distinct dysbiotic patterns marked by Oscillospiraceae enrichment and depletion of beneficial taxa (Eubacteriaceae, Allobaculum, Pediococcus), patterns that independently predict reduced survival and elevated recurrence risks (33). Notably, advanced TNM stages (III-IV) in intrahepatic CCA show marked Candida albicans overgrowth in gut mycobiota, suggesting fungal dysbiosis as a potential progression biomarker (36). The microbial-immune interface further reveals VI-associated immunological shifts, with Ruminococcaceae-enriched cases displaying elevated IL-4 and suppressed IL-6 levels (21), highlighting microbiota-driven immune microenvironment remodeling.
Therapeutically, gut microbial signatures demonstrate predictive value for treatment responses across modalities. While Bacteroidetes enrichment (particularly Alistipes sp. Marseille-P5997) associates with improved anti-programmed cell death protein 1(PD-1) immunotherapy outcomes in biliary tract cancers (38), Proteobacteria dominance inversely correlates with Sintilimab-anlotinib combination efficacy in advanced cases (39). Mechanistically, microbial metabolites like cyclic dinucleotide c-di-AMP enhance radiotherapy-induced antitumor immunity through STING pathway activation (40), revealing novel microbiome-mediated therapeutic sensitization strategies. Nevertheless, critical knowledge gaps persist in delineating precise microbiota-immune crosstalk mechanisms and validating microbial biomarkers for clinical translation. Systematic investigation of host-microbiota metabolic interactions and standardized multi-omics approaches remain imperative to harness the full therapeutic potential of gut microbiome in CCA management.
3.3 Gut microbiota-derived metabolites and cholangiocarcinoma
The intestinal barrier serves as a critical interface that maintains microbial homeostasis through selective permeability, effectively restricting the translocation of exogenous and microbiota-derived molecules under physiological conditions (41). While inherently plastic adapts to physiological demands, this barrier exhibits vulnerability to age-related deterioration, environmental stressors, and pathological insults. Gut microbiota-derived metabolites, including those directly synthesized by bacteria or enzymatically transformed from dietary and host-derived molecules, exert oncogenic effects via bidirectional crosstalk with host signaling pathways. Dysbiosis disrupts intestinal barrier integrity, enabling systemic translocation of microbial components, including structural molecules (e.g., lipopolysaccharides, LPS) and metabolic byproducts (e.g., short-chain fatty acids (SCFAs)) (42–44). These translocated products, particularly endotoxins, access the liver via portal circulation, activating hepatic Kupffer cells (KCs) to initiate pro-inflammatory cytokine cascades (e.g., tumor factor alpha [TNF-α], IL-6) that perpetuate chronic inflammation, bile duct injury, and metabolic dysregulation (45, 46).
Elevated circulating LPS, a hallmark of Gram-negative bacterial dysbiosis, is clinically associated with chronic hepatobiliary diseases and malignancy progression. Impaired tight junction integrity (e.g., reduced Claudin-1/Occludin expression) facilitates LPS translocation to the liver (47). where it drives tumorigenesis via TLR4-mediated activation of oncogenic pathways (e.g., NF-κB, PI3K/AKT) (48, 49). Diet-induced metabolic perturbations exacerbate this cycle by promoting barrier dysfunction and endotoxemia, as evidenced in nonalcoholic fatty liver disease models (50, 51). In PSC models, dysbiosis-driven LPS translocation activates hepatic TLR4/MyD88 signaling, exacerbating cholangitis and fibrosis to establish a pro-carcinogenic niche (52). Patients with iCCA exhibit gut microbiota shifts marked by opportunistic pathogen enrichment (e.g., Veillonella, Klebsiella), with pathogen-derived LPS activating TLR4 on cholangiocytes and KCs to induce NF-κB-driven IL-6/TNF-α production, amplifying biliary injury and cholestasis (21). Mechanistically, LPS enhances tumor aggressiveness via METTL3-mediated PI3K/AKT pathway activation, promoting tumor cell migration and invasion (17). Additionally, LPS orchestrates immunosuppression in the tumor microenvironment (TME) by triggering TLR4-dependent hepatocyte C-X-C motif chemokine ligand (CXCL)1 secretion, which recruits polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) to dampen antitumor immunity (19). These findings collectively position the LPS/TLR4 axis as a therapeutic target, suggesting microbiota modulation and microenvironment reprogramming as viable strategies to combat iCCA progression.
3.4 Biliary microbiota and cholangiocarcinoma development
The biliary system harbors gut-associated microbes including Escherichia coli and Enterobacter spp., translocated via the gut-liver axis or establish local colonization (53). Systematic characterization by Carlo et al. revealed that biliary enrichment of E. coli and Klebsiella pneumoniae serves as an independent prognostic marker for reduced survival in pancreaticobiliary malignancies, highlighting their potential as diagnostic biomarkers (54). Notably, Alcaligenes faecalis demonstrates TME-specific colonization in eCCA, exhibiting diagnostic potential for early tumor detection and therapeutic monitoring (55). Contrasting with cholangitis-associated microbiotas, perihilar CCA exhibits a Gram-positive predominance featuring Enterococcus spp., which independently predicts portal vein thrombosis and multidrug resistance development (56). Furthermore, biliary colonization by Enterococcus or Candida spp. correlates strongly with clinical progression in PSC, solidifying microbial dysbiosis as a prognostic determinant (57). Critically, antibiotic-resistant strains such as Enterococcus faecalis and Enterobacter spp. in hepatic ducts directly exacerbate postoperative infection rates and mortality in eCCA, underscoring the clinical imperative for microbiota-guided perioperative management (58). These findings collectively establish biliary microbiota as both a driver of oncogenesis and a therapeutic target in CCA progression.
3.5 Oral microbiota and cholangiocarcinoma development
Comprising approximately 770 phylogenetically diverse species, the oral microbiota emerges as a pivotal regulator of systemic immunity and metabolic homeostasis with broad disease implications (59, 60). The functional crosstalk between oral and gut microbiotas has gained prominence given their collective influence on human pathophysiology (61), particularly through pathobiont translocation along the gut-liver axis during dysbiosis (62). Clinically, Rao and colleagues developed an oral microbiota-based diagnostic algorithm incorporating three bacterial biomarkers (Lautropia, Alloprevotella, and Actinomyces), achieving exceptional accuracy (AUC=0.981) in differentiating iCCA from HCC (63). A complementary Korean cohort study further demonstrated stratified oral microbial signatures across upper gastrointestinal malignancies, revealing significantly elevated α-diversity in oesophageal/gastric cancers compared to BTC and pancreatic cancer cohorts (64). Intriguingly, while distinct diagnostic microbial profiles were identified for oesophageal/gastric malignancies, BTC and pancreatic cancers lacked comparable signatures. This disparity suggests an anatomic microbial gradient along the gastrointestinal tract, where tumor proximity to the oral cavity correlates positively with both microbial dysbiosis severity and diagnostic biomarker detectability. While BTC-specific oral microbial biomarkers remain unidentified, these findings provide foundational evidence for the oncogenic involvement of the oral-gut-liver axis in BTC pathogenesis. Subsequent research must prioritize multi-institutional cohorts with longitudinal sampling to resolve spatiotemporal dynamics of microbial translocation and enable mechanistic dissection of tumor-promoting crosstalk within this axis.
4 Bile acid dysregulation in cholangiocarcinoma
The enterohepatic circulation maintains BA homeostasis through a hepatobiliary-intestinal loop, wherein hepatocyte-derived BAs undergo intestinal reabsorption followed by portal venous return to the liver (65, 66). Intrahepatic BA overload, while non-carcinogenic, drives cholangiocarcinogenesis via ductular hyperplasia, inflammatory niche formation, and impaired cytoprotective BA signaling (67). CCA development is strongly linked to chronic cholestasis, implicating prolonged exposure to elevated BA levels in promoting gastrointestinal carcinogenesis. Emerging evidence highlights BA metabolic reprogramming as a critical determinant of CCA heterogeneity and progression. The following will examine alterations in BA profiles in CCA, elucidates mechanisms through which BAs drive CCA pathogenesis, and evaluates their clinical implications.
4.1 Changes in bile acid profiles in cholangiocarcinoma
Serum total bile acid levels progressively escalate across the disease continuum from healthy controls to benign biliary disease (BBD) and CCA patients, correlating with biliary obstruction severity (68). Conjugated bile acids exhibit marked accumulation, particularly primary conjugated BAs (glycocholic acid [GCA], taurochenodeoxycholic acid [TCDCA]) showing marked increases in both serum and bile compared to BBD and healthy controls (69, 70) (Table 2). Paradoxically, secondary conjugated species (glycolithocholic acid [GLCA], glycoursodeoxycholic acid [GUDCA]) demonstrate malignant-specific reduction (71). Concurrently, primary unconjugated BAs (cholic acid [CA], chenodeoxycholic acid [CDCA]) accumulate due to cholestasis-induced excretion impairment (69), whereas secondary unconjugated derivatives (deoxycholic acid [DCA], lithocholic acid [LCA]) decline in plasma, reflecting gut microbial 7α-dehydroxylation defects (72). This conjugated/unconjugated BA ratio imbalance strongly correlates with suppressed bacterial deconjugation activity, establishing a pathogenic feedback loop between hepatic BA synthesis and gut microbial metabolic reprogramming that drives carcinogenesis. Thus, between-study heterogeneity in BA levels (particularly CCA vs BBD discrepancies) arises from methodological variables including biospecimen type (serum/bile/tissue), cohort heterogeneity, and gut microbiota-mediated BA metabolic variation. Critically, current BA metabolomic validation in CCA faces three critical gaps: (1) absence of multi-center harmonization in pre-analytical (sample collection/storage) and analytical (LC-MS) workflows; (2) deficient dynamic monitoring failing to track tumor progression/therapy-induced BA flux changes; and (3) inadequate pathological specificity against cirrhosis/cholangitis confounders. These limitations mandate prospective longitudinal cohorts with protocolized multi-omics integration to resolve CCA-specific BA dynamics.
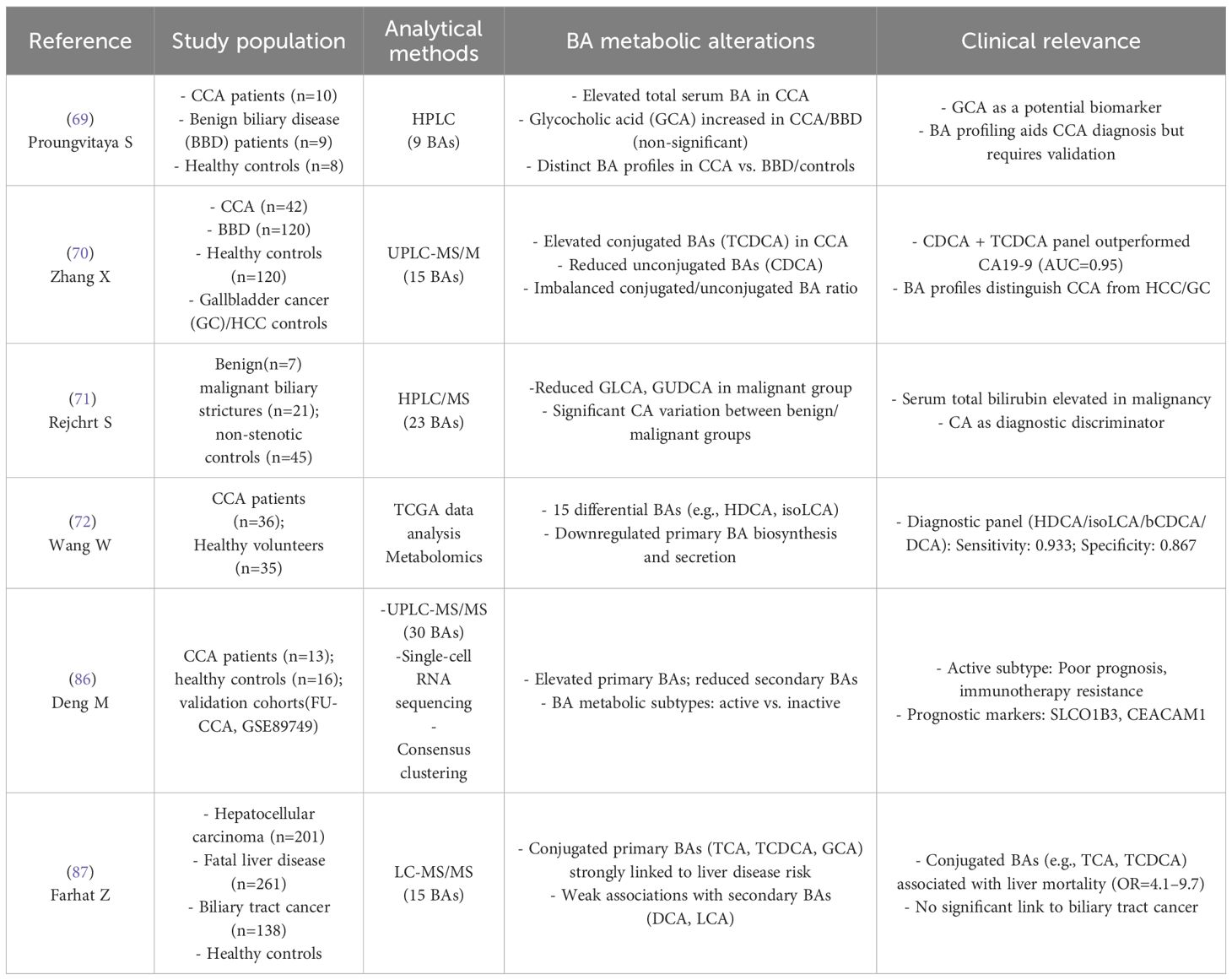
Table 2. Dysregulation of bile acid metabolism in cholangiocarcinoma.
4.2 Mechanisms of bile acid dysregulation driving CCA
Cholestasis-induced BA overload initiates a self-perpetuating pathogenic cycle central to CCA progression. Excessive BAs activate hepatocyte-cholangiocyte transdifferentiation via dual receptor systems: nuclear receptors (farnesoid X receptor, FXR) and membrane-bound receptors (Takeda G protein-coupled receptor 5, TGR5; sphingosine-1-phosphate receptor 2, S1PR2) (73, 74) (Figure 2). Sustained BA-receptor engagement generates a pro-tumorigenic niche marked by oxidative DNA damage (via reactive oxygen species [ROS] overproduction) and apoptotic resistance (through CD95 inactivation), thereby completing malignant transformation. This BA signalling duality manifests through distinct pathways: (1) Nuclear receptor modulation: Although frequently downregulated in CCA (84), FXR activation by CDCA or synthetic agonists (e.g., GW4064) exerts tumor-suppressive effects (75, 76). Mechanistically, FXR activation induces small heterodimer partner (SHP) expression, which suppresses STAT3 phosphorylation to downregulate Bcl-xL in BTC cells, ultimately triggering apoptosis and inhibiting proliferation (77–79). (2) Membrane receptor cascades: TGR5 promotes cholangiocyte proliferation through extracellular signal-regulated kinase (ERK)1/2 phosphorylation via a cascade comprising ROS generation, Src kinase activation, and epidermal growth factor receptor (EGFR) transactivation. Simultaneously, TGR5 triggers PKA-mediated CD95 serine/threonine phosphorylation that blocks death receptor signalling, conferring apoptotic resistance, with these dual mechanisms synergistically driving cholangiocarcinogenesis (80–82). S1PR2 axis: TCA-mediated S1PR2 activation triggers ERK1/2/Akt signalling through both G protein-dependent and EGFR-mediated transactivation pathways, subsequently inducing NF-κB activation and cyclooxygenase-2 (COX-2) upregulation. This signalling axis coordinately upregulates proinflammatory cytokines (IL-6, TNF-α), thereby establishing a self-sustaining inflammatory niche that promotes CCA progression (83–85).
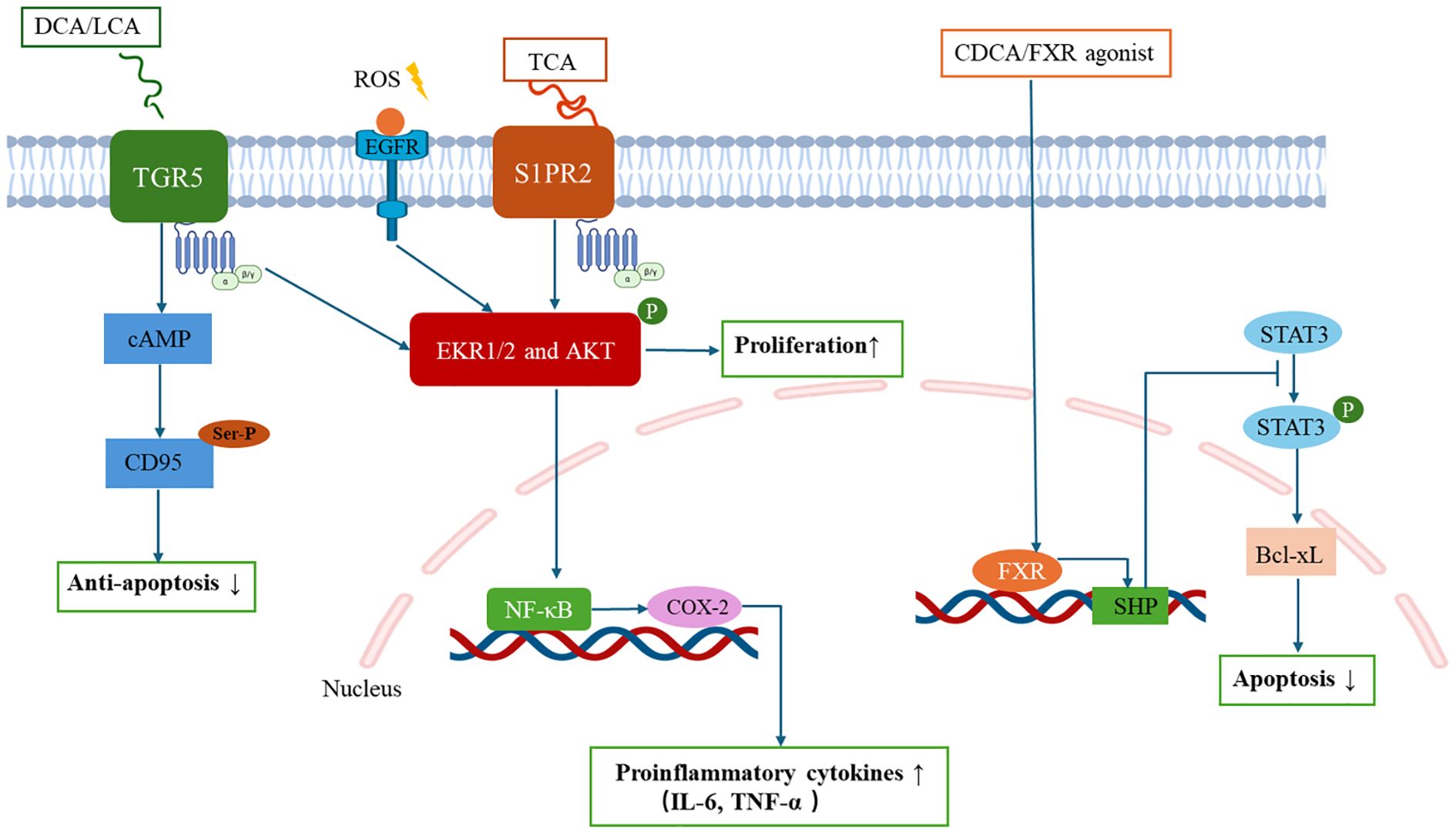
Figure 2. Bile acid receptor-mediated intracellular signaling pathways in cholangiocarcinoma cells.
BA signalling orchestrates TME immunosuppression and metastatic priming through macrophage polarization and T-cell functional suppression. S1PR2 activation by conjugated BAs upregulates COX-2/PGE2 signalling, which directly suppresses CD8+ cytotoxic T lymphocyte activity while promoting regulatory T cell infiltration, thereby establishing an immune-evasion niche conducive to tumor progression (83, 84). The functional duality of TGR5 pivots on disease stage-specific microenvironmental cues. In cholestatic conditions (e.g., early-stage PSC), TGR5 activation exerts hepatoprotective effects by mediating M2 macrophage polarization and suppressing NF-κB-mediated inflammation via cAMP-PKA signalling (52). Conversely, within the established TME of advanced CCA, TGR5 signalling shifts toward pro-tumorigenic activity by conferring apoptosis resistance and enhancing proliferation in malignant transformed biliary epithelial cells (81). The precise molecular mechanisms and pathological staging thresholds governing the transition of TGR5 from hepatoprotective to protumorigenic function remain incompletely characterized. Current evidence suggests this transition may correlate with progression of biliary intraepithelial neoplasia, advanced cirrhotic remodelling, sustained BA accumulation, and immune reprogramming. This receptor dichotomy underscores the delicate equilibrium between BA-mediated immune regulation and malignant transformation in hepatobiliary diseases. Elucidating these context-dependent switches represents a critical research priority for developing stage-adapted TGR5-targeted therapies in CCA.
4.3 Clinical significance of bile acid dysregulation in CCA
Clinical evidence highlights the critical association between dysregulated BA metabolism and the diagnosis, prognostic evaluation, and therapeutic strategies for CCA. BA profiling has emerged as a potential source of non-invasive biomarkers for CCA diagnosis (Table 2). Zhang et al. (70), developed a diagnostic model based on CDCA and TCDCA, which effectively distinguished CCA from BBD, gallbladder cancer, and HCC, demonstrating significantly superior specificity compared to the conventional biomarker CA19-9. Multi-omics studies further identified a quadruple diagnostic panel comprising hyodeoxycholic acid, isoLCA, bCDCA, and DCA, achieving 93.3% sensitivity and 86.7% specificity (72). Notably, serum GCA exhibits diagnostic potential but shows limited specificity in differentiating malignant from benign biliary disorders (69).
Metabolomic stratification studies demonstrate that BA metabolic activity serves as an independent prognostic determinant in CCA. Molecular subtyping based on BA metabolism features classifies CCA into metabolically active and inactive subgroups, where the active subtype exhibits significantly shorter OS and impaired immunotherapy response (86). These findings establish BA metabolic reprogramming as a mechanistic basis for precision oncology, providing a molecular classification framework for personalized therapeutic strategies. Intriguingly, while conjugated primary BAs (e.g., TCA, TCDCA) demonstrate strong correlation with HCC mortality, this association remains absent in BTCs, suggesting tumor-specific metabolic rewiring mechanisms (87). Overcoming these challenges necessitates collaborative multicenter studies that integrate machine learning pipelines to discriminate disease-relevant metabolic signatures from inter-individual physiological variability.
5 Gut microbiota-bile acid crosstalk in cholangiocarcinogenesis
The gut-liver axis establishes a bidirectional regulatory network wherein microbial BA metabolism and BA-mediated microbiota modulation cooperatively drive CCA progression through integrated metabolic and immune signaling (88). Gut commensals enzymatically transform primary BAs into secondary species (e.g., deconjugation, 7α-dehydroxylation), which in turn regulate microbial ecology and activate tumor-promoting pathways via receptor-mediated mechanisms (89, 90). This bidirectional crosstalk drives cholangiocarcinogenesis through chronic biliary inflammation, epigenetic reprogramming of cholangiocytes, and TME remodeling (Figure 3).
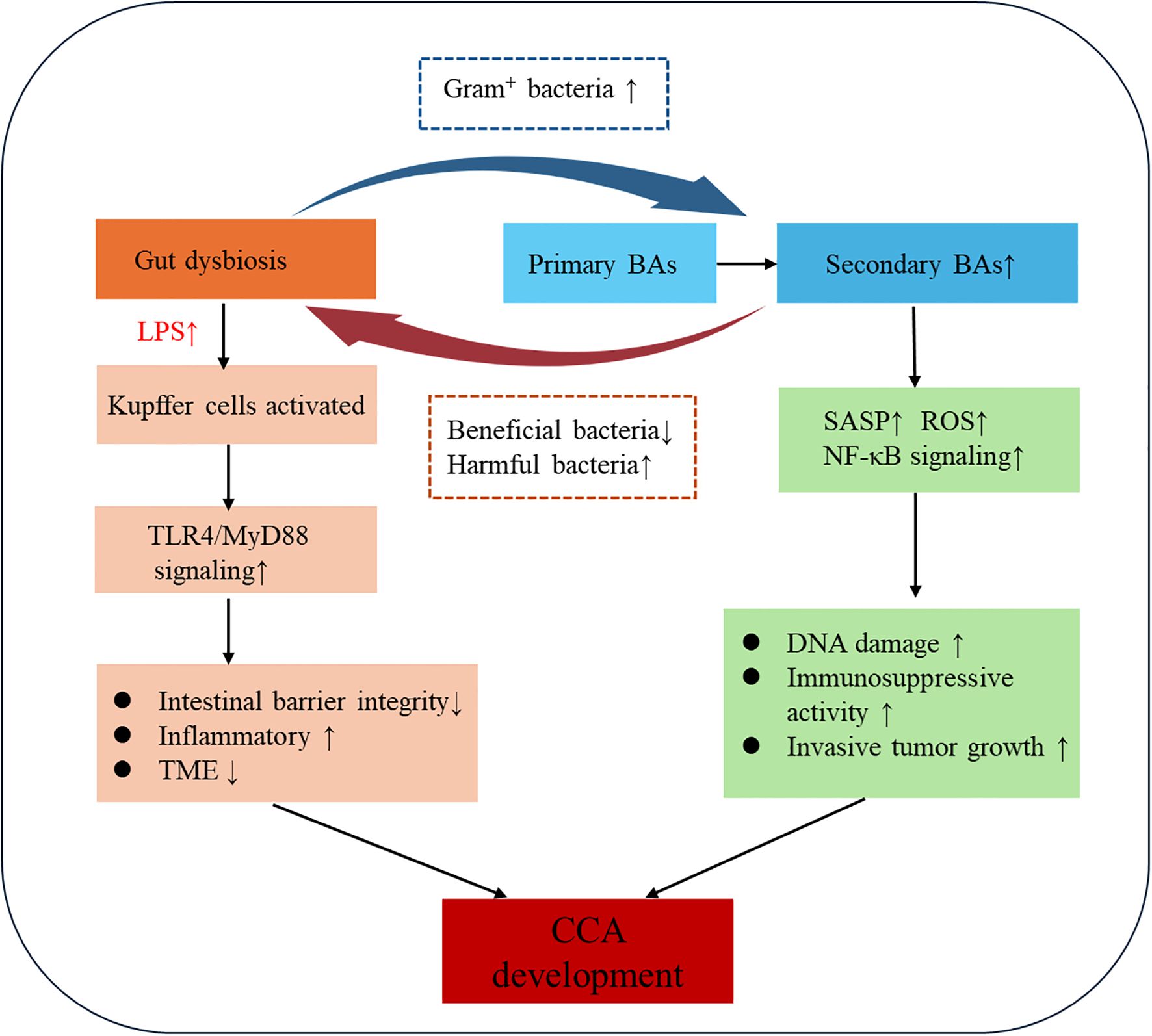
Figure 3. Mechanistic diagram of bile acid-microbiome-immune crosstalk in cholangiocarcinoma.
5.1 Microbial regulation of BA metabolism in CCA pathogenesis
Gut microbiota-derived enzymes convert primary BAs to tumorigenic secondary species (DCA, LCA) that activate oncogenic signalling cascades (91, 92). Clostridium scindens and other 7α-dehydroxylase-expressing species generate secondary BAs associated with diet-induced hepatocarcinogenesis models, where dysbiosis elevates circulating DCA. This hydrophobic secondary BA drives tumorigenesis by inducing senescence-associated secretory phenotype activation, ROS overproduction, and NF-κB-mediated inflammation via gut barrier disruption (93). Notably, DCA impairs hepatic CXCL16 secretion in liver sinusoidal endothelial cells, reducing CXCR6+ natural killer cell (NKT) recruitment and compromising antitumor immunity. Pharmacological depletion of Gram-positive 7α-dehydroxylase bacteria (e.g., vancomycin) attenuates secondary BA production and exerts chemopreventive effects in preclinical models (94). Furthermore, microbial bile salt hydrolases dynamically remodel the BA pool, facilitating intestinal absorption or further modification of BA (95). Therapeutic microbiota modulation demonstrates dual mechanisms: (1) Lactobacillus rhamnosus GG (LGG) activates FXR to reduce BA accumulation in cholestatic liver disease (96, 97); (2) Pediococcus pentosaceus Li05 enhances bile salt hydrolase activity while suppressing NLRP3 inflammasome activation in PSC (98, 99). These findings position gut microbiota as master regulators of BA homeostasis, linking microbial ecology to inflammation-driven carcinogenesis.
5.2 BA-mediated control of gut microbial dynamics in CCA
BAs critically shape gut microbiota composition and homeostasis through their chemical properties and signalling functions (67, 100). Primary BAs (e.g., CA, CDCA) exhibit surfactant-like properties that disrupt bacterial cell membrane integrity and interfere with DNA stability, thereby inhibiting pathogens such as Clostridium difficile, Salmonella spp., and Listeria spp. Notably, Gram-positive bacteria (Firmicutes) demonstrate heightened susceptibility to these antimicrobial effects compared to Gram-negative counterparts, a phenomenon attributable to structural differences in their cell wall architectures (101). Moreover, gut microbiota converts primary BAs into secondary BAs (e.g., DCA) through bile salt hydrolase and 7α-dehydroxylase activities. This biotransformation not only modulates BA toxicity and signalling activity but also reshapes microbial ecology via metabolic crosstalk. DCA selectively inhibits beneficial bacteria (e.g., Lactobacillus) while promoting the expansion of pathobionts such as Enterobacteriaceae, fostering a pro-inflammatory microenvironment (22).
Furthermore, BA signalling orchestrates gut microbial homeostasis through BA receptor-mediated pathways, predominantly FXR. Mechanistically, FXR activation stimulates the expression of antimicrobial peptides such as angiogenin-1, effectively curbing the expansion of pathobionts within the Proteobacteria phylum (102), while concurrently fortifying intestinal barrier function by upregulating tight junction proteins (e.g., occludin, ZO-1) (103). This dual regulatory axis not only maintains microbial equilibrium but also mitigates systemic inflammation triggered by bacterial endotoxin leakage. Future studies should delineate the mechanisms through which BA pool-mediated microbiota modulation governs host-microbial interactions.
6 Therapeutic translation: targeting the gut-liver axis in clinical practice
Gemcitabine/cisplatin combination therapy maintains its position as the standard first-line chemotherapeutic regimen for CCA, yet persistent limitations in objective response rates continue to challenge clinical outcomes. Emerging adjuvant strategies targeting the gut-liver axis show therapeutic promise, particularly given the pivotal role of gut microbiota dysbiosis in CCA pathogenesis. BA metabolism imbalance in specific CCA subtypes reveals a treatable weakness, positioning FXR/TGR5-targeted therapies as tailored approaches to fix cancer-driven metabolic defects in these patient groups. Therapeutically targeting this microbiota-metabolite-immune triad holds promise for intercepting CCA progression, advancing precision medicine through multimodal pathway modulation in CCA management (Table 3).

Table 3. Therapeutic strategies targeting the gut-liver axis in cholangiocarcinoma: molecular targets and mechanisms of action.
6.1 Targeting gut microbiota dysbiosis
Targeted microbial modulation—via antibiotics, probiotics, or faecal microbiota transplantation (FMT)—represents a potential therapeutic strategy for CCA by restoring microbiota homeostasis (104) (Table 4). These interventions mechanistically counteract dysbiosis-driven oncogenesis through pathogen depletion, intestinal barrier restoration, and suppression of protumorigenic signalling. However, translational progress remains limited, with current evidence largely confined to preclinical models.
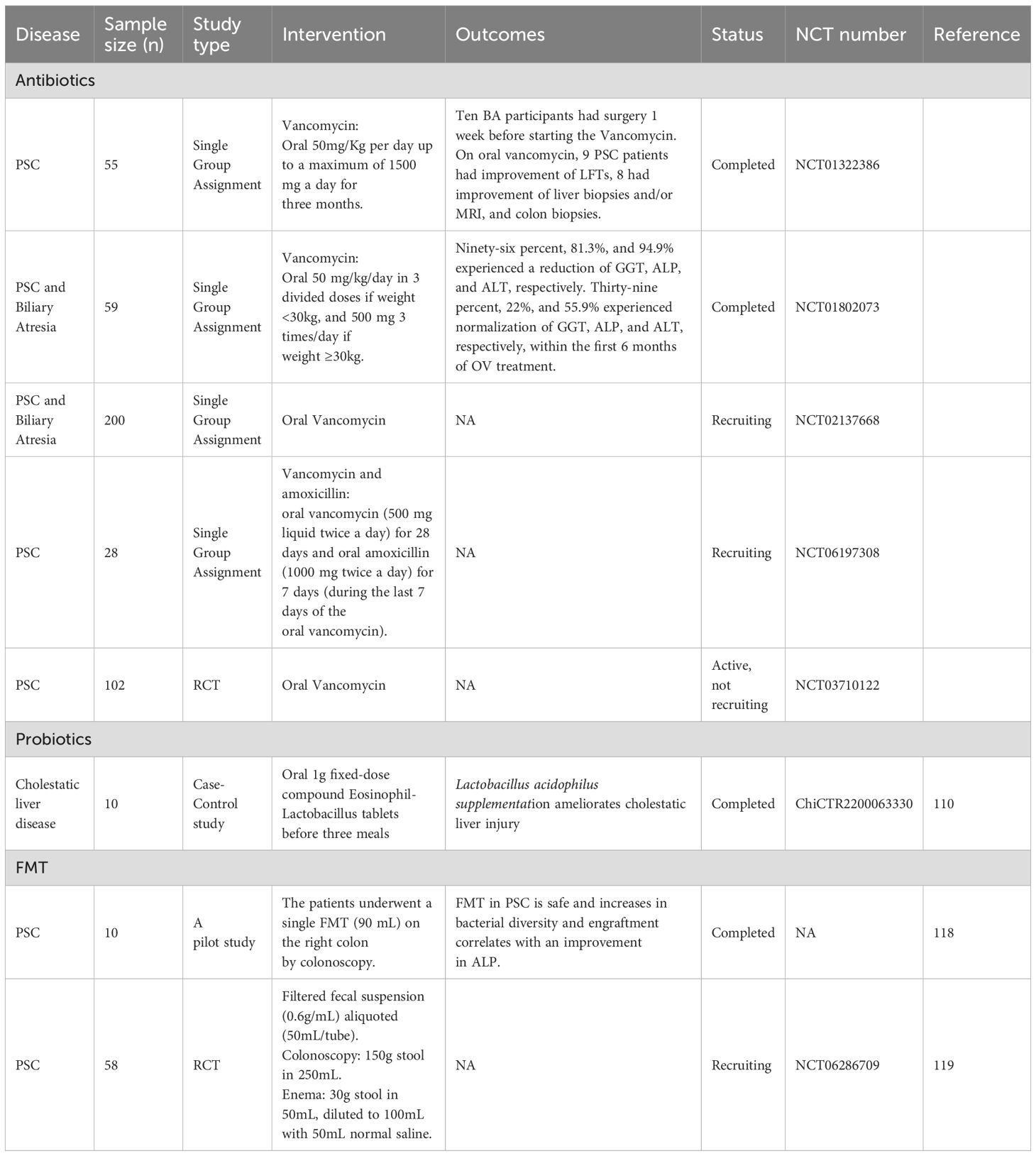
Table 4. Clinical trials of gut microbiota-targeted therapies for cholestatic liver disease.
6.1.1 Antibiotics
Preclinical studies in PSC model reveal that gut barrier dysfunction and dysbiosis facilitate hepatic bacterial/endotoxin accumulation. Klebsiella pneumoniae exacerbates bacterial translocation and TH17-driven hepatic inflammation, whereas metronidazole or vancomycin suppresses TH17 activation, demonstrating microbiota-targeted therapeutic potential (105). Gut dysbiosis further induces hepatocyte-derived CXCL1 to recruit CXCR2+ PMN-MDSCs, accelerating CCA progression. Neomycin, a Gram-negative-specific antibiotic, blocks this CXCL1-PMN-MDSC axis, validating the gut-liver-CCA interplay linking microbial imbalance, barrier integrity, and antitumor immunity (19). Notably, vancomycin depletes Gram-positive bacteria that convert primary to secondary BAs, exerting antitumor effects in murine liver cancer models by enhancing CXCL16-dependent hepatic NKT cell infiltration and suppressing tumor growth.
Clinically, multiple trials (NCT01322386, NCT01802073, NCT02137668) report improved liver biochemistry in PSC patients treated with oral vancomycin (106). A randomized controlled trial (NCT03710122) further explored the immunomodulatory effects of vancomycin by analyzing pro-inflammatory cytokines (e.g., TGF-β, IL-4). A pilot study (NCT06197308) evaluates FMT combined with vancomycin/amoxicillin dual therapy for PSC. Despite preclinical efficacy, antibiotic-based strategies remain unexplored in human CCA, necessitating multicenter trials to assess safety and efficacy.
Paradoxically, while antibiotics targeting pathogenic bacteria may enhance PD-1 inhibitor efficacy in hepatobiliary cancers (107), prolonged antibiotic use correlates with reduced survival in HCC patients receiving anti-PD-1 therapy, likely due to microbiota disruption (108). Thus, cautious antibiotic administration—combined with probiotics or FMT—is critical to preserve microbiota balance. These findings underscore antibiotics as a dual-edged therapeutic strategy, with immunomodulatory effects mediated through precise gut-liver-immune axis regulation.
6.1.2 Probiotics
Probiotic supplementation competitively colonizes gastrointestinal niches, mitigating intestinal dysbiosis by enhancing microbial homeostasis and inhibiting pathogenic bacterial adhesion. Emerging preclinical evidence highlights the therapeutic potential of Lactiplantibacillus species in CCA. For instance, LGG alleviates cholestasis by suppressing BA synthesis while enhancing excretion via activation of the intestinal FXR-FGF15 axis (96). Specific strains, including Lactiplantibacillus plantarum Lp 12 and Lp 355, exert dose-dependent antitumor effects against CCA by inducing tumor cell apoptosis and senescence through secreted metabolites such as peptidoglycans and exopolysaccharides (109). Synergistic interactions with gemcitabine further enhance antitumor responses while reducing chemotherapy doses and toxicity. Clinically, Lactobacillus acidophilus supplementation alleviates cholestatic liver injury through dual mechanisms: suppression of hepatic BA synthesis and enhancement of faecal BA excretion, as demonstrated in a registered case-control trial (ChiCTR2200063330) (110). Similarly, Pediococcus pentosaceus Li05 ameliorates PSC by activating the FXR-FGF15 axis, restoring BA homeostasis to attenuate hepatic inflammation and fibrosis (99). Traditional Chinese medicine formulations such as Si-Ni-San also modulate gut microbiota by promoting colonization of beneficial strains (e.g., Parabacteroides goldsteinii), thereby restoring microbial equilibrium and reinforcing intestinal barrier integrity (111).
Notably, probiotics may enhance immunotherapy efficacy. SCFAs derived from gut microbiota metabolism, including butyrate, exhibit anti-inflammatory and immunomodulatory properties. Specific taxa (e.g., Lachnospiraceae, Ruminococcaceae) enhance anti-PD-1 efficacy in BTC and HCC by mediating antitumor immunity through SCFAs and BA metabolic pathways (52). Lactobacillus rhamnosus Probio-M9 and polyphenol castalagin further potentiate anti-PD-1 responses by enriching immunotherapy-responsive bacterial taxa (e.g., Ruminococcaceae, Alistipes), offering strategies to overcome PD-1 resistance (112, 113).
Despite therapeutic promise, probiotics pose rare risks (114). Postoperative administration may lead to bacteraemia in immunocompromised patients, as reported in cases of Clostridium butyricum-associated bacteraemia following hepatic resection for biliary malignancies (115). Identified risk factors include intestinal barrier disruption, immunosuppression, and broad-spectrum antibiotic use. Clinicians should weigh benefits against potential adverse effects when prescribing probiotics. Collectively, microbiota-targeted interventions represent emerging strategies to optimize therapeutic outcomes in hepatobiliary cancers.
6.1.3 FMT
FMT, which involves transferring processed donor faecal material to restore intestinal microbial balance (116), modulates gut-liver axis dysbiosis and represents a potential therapeutic strategy for cholestatic liver diseases (117). Preliminary clinical studies in PSC patients demonstrate FMT safety, with associated increases in gut microbial diversity and reductions in serum alkaline phosphatase levels (118). A phase IIa randomized trial (NCT06286709) assesses the therapeutic potential of repeated-colonic FMT in patients with PSC-associated inflammatory bowel disease. Key translational objectives include identifying mucosal multi-omics signatures (metagenomic, metatranscriptomic, metabolomic) and immunophenotypic pathways linked to FMT response, aiming to elucidate microbiota-host crosstalk mechanisms driving clinical outcomes (119). Gut dysbiosis promotes intrahepatic HCC metastasis through neutrophil extracellular trap hyperactivation, while healthy donor FMT prolongs survival and reduces tumor burden in murine models by reprogramming the microbiota-NET axis (120). These findings underscore the therapeutic potential of microbial interventions in hepatobiliary malignancies. However, both preclinical and clinical investigations into FMT for CCA management remain in their infancy, underscoring an urgent need for mechanistic and translational studies to address this critical knowledge gap.
6.2 Targeting bile acid mediated signalling pathway
Metabolic dysregulation, characterized by activation of BA, fatty acid, and xenobiotic pathways, represents a hallmark of CCA (121). Emerging strategies aim to disrupt BA-driven oncogenic networks while preserving metabolic homeostasis. S1PR2 inhibitors (e.g., JTE-013) suppress tumor invasiveness in CCA preclinical models by inhibiting ERK/AKT/NF-κB signalling cascades (85). Similarly, TGR5 activation in CCA cells promotes tumor progression, its downregulation in cholangiocytes and tumor-associated macrophages attenuates inflammation through NF-κB and MAPK/ERK pathway inhibition (52, 122). Intriguingly, TGR5 signalling exhibits a stage-dependent duality: it exerts hepatoprotective effects during early cholestasis but becomes pro-tumorigenic in established CCA (123). This functional switch necessitates carefully timed therapeutic interventions. During the therapeutic window of biliary intraepithelial neoplasia, agonists like INT-777 may mitigate malignant transformation by restoring anti-inflammatory and epithelial barrier functions. Conversely, in advanced CCA characterized by CAF-rich microenvironments, antagonists such as SBI-115 can block pro-fibrotic and pro-metastatic signalling cascades, requiring stage-adapted regimens to optimize the therapeutic index.
FXR agonists (e.g., INT-747/OCA) demonstrate therapeutic potential in attenuating sepsis-induced cholestasis and intestinal injury. In cecal ligation and puncture -induced septic mice, INT-747 upregulates intestinal FXR/FGF15 signalling, preserving gut barrier integrity and reducing hepatic cholestasis (124). Clinically, OCA is approved for PSC treatment (125), with recent advances utilizing ROS-responsive nanoparticles for targeted OCA delivery to enhance efficacy (126). FXR expression was found to be markedly downregulated in samples of primary CCA (127, 128). OCA counteracts this deficit by restoring BA homeostasis via SHP/LRH-1-dependent pathways, suppressing CCA proliferation and migration (127). Critically, heterogeneous FXR expression across CCA subtypes directs subtype-specific therapeutic strategies. In chemotherapy-resistant iCCA models, the combination of FXR agonists (e.g., GW4064, CDCA) with standard chemotherapy (cisplatin/gemcitabine) potentiates anti-proliferative and pro-apoptotic responses (129). Conversely, pCCA and dCCA subtypes frequently exhibit low-to-moderate FXR levels (H-score <120), necessitating the priming use of epigenetic modulators (e.g., DNMT inhibitors) to restore agonist sensitivity (78, 130). Concerning carcinogenic risk, chronic FXR activation displays a complex tissue-selective duality: while systemic FXR loss promotes hepatocarcinogenesis and tumor-specific FXR silencing characterizes the HCC/CCA microenvironment (131), hyperactivation within specific gastrointestinal tissues may drive β-catenin-dependent oncogenesis (132). Therefore, the tissue-selective functional duality of FXR signalling necessitates rigorous long-term safety evaluation in forthcoming preclinical and clinical investigations.
Certain genetic mutations indirectly disrupt BA metabolism. For instance, frequent IDH1/2 mutations in iCCA drive 2-hydroxyglutarate accumulation, which epigenetically impairs cell differentiation and may suppress BA synthesis genes (e.g., CYP7A1) via DNA hypermethylation (133, 134). Ivosidenib (AG-120), a selective IDH1 inhibitor, demonstrated improved progression-free survival in the phase III ClarIDHy trial, becoming the first targeted therapy approved for IDH1-mutant CCA (135). HNF4A, a master regulator of hepatobiliary differentiation, directly modulates bile acid synthesis enzymes and transporters (e.g., BSEP/ABCB11) (136, 137). While HNF4A activation may suppress tumor progression by restoring BA homeostasis, direct targeting agents remain unavailable. Despite preclinical evidence supporting BA metabolic reprogramming as a therapeutic strategy for CCA, BA-targeted agents remain largely experimental with limited clinical translation. This gap necessitates large-scale trials and mechanistic studies to rigorously validate efficacy and safety, accelerating their clinical integration.
7 Future perspectives for gut-liver axis targeting in CCA
The gut-liver axis offers transformative potential for CCA therapy, yet clinical translation requires strategic prioritization to address biological complexity. We propose a phased framework (Figure 4) to accelerate clinical impact through defined milestones: Phase I establishes foundational biology through multi-omics validation. Phase II leverages these validated biomarkers to optimize therapeutic strategies using artificial intelligence (AI) -driven modeling of efficacy-toxicity landscapes. Phase III engineers precision interventions targeting CCA heterogeneity—notably cancer-associated fibroblast (CAF)-bile acid signaling inhibition or butyrate-producing microbiome modulation. This continuum from discovery to bioengineered solutions builds an individualized translational pipeline for CCA.
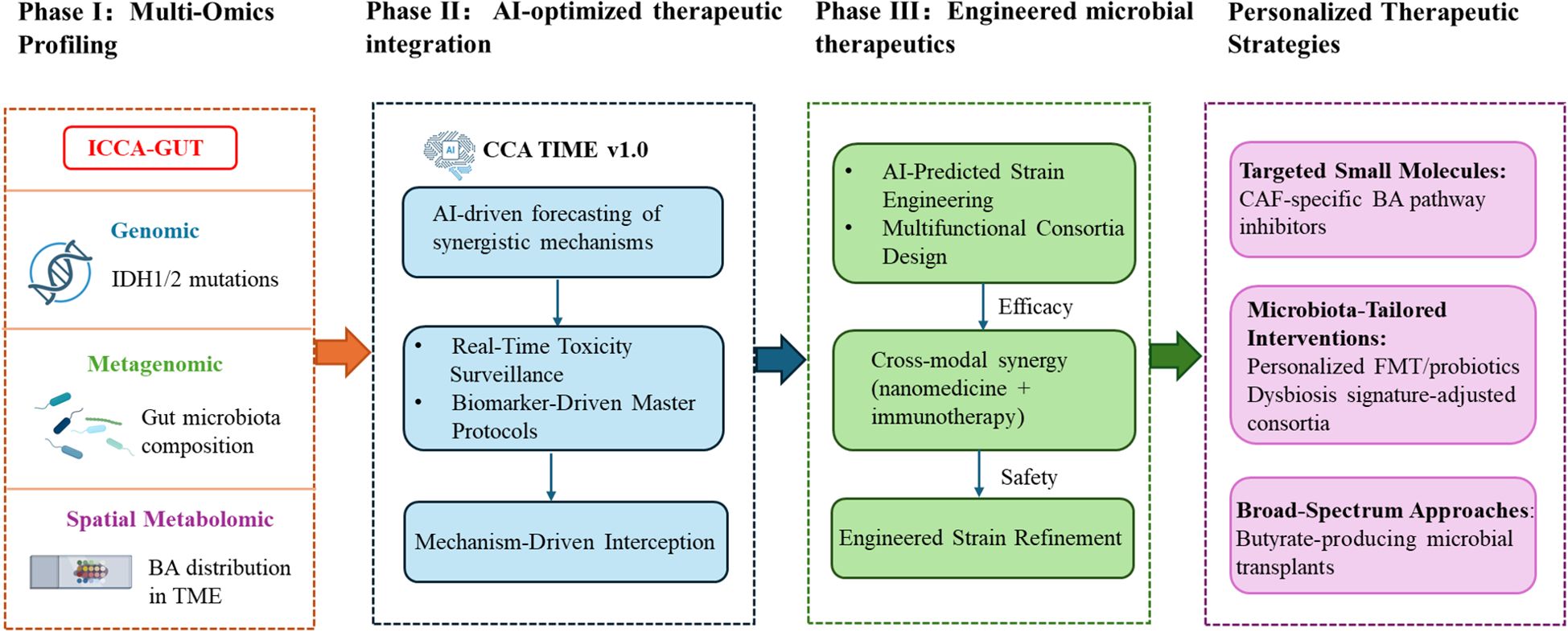
Figure 4. Multistage translational roadmap for gut-liver axis-targeted therapy in cholangiocarcinoma.
7.1 Multi-omics validation (phase I)
The pronounced heterogeneity of CCA necessitates therapeutic targeting based on robust molecular subtyping. Multi-omics approaches—encompassing genomics, metagenomics, and spatial metabolomics—systematically dissect the core mechanisms underlying gut-liver axis dysregulation, including critical microbiota-bile acid-host interactions. These approaches yield quantifiable and targetable biomarkers (e.g., dysbiosis subtypes, cancer-associated fibroblast BA dependency phenotypes) essential for subsequent therapeutic development phases. However, clinically applicable gut-liver axis biomarkers validated through large-cohort studies remain lacking. Phase I prioritizes the validation of foundational biomarkers through global consortia (ICCA-GUT), standardizing profiling to map BA distribution. Leveraging single-cell spatial transcriptomics will reveal cancer-associated fibroblast-specific BA dependencies. Cross-cancer comparative analyses will identify conserved vulnerabilities like pan-hepatobiliary butyrate deficiency. Machine learning frameworks will correlate IDH1/2status with microbiota-metabolome covariation, culminating in a validated dysbiosis subtype classifier (AUC>0.90). This establishes a closed-loop pipeline for refining precision medicine, from multi-omics discovery to mechanism-guided translation.
7.2 AI-optimized therapeutic integration (phase II)
Building on validated biomarkers, this phase utilizes AI platforms (CCA-TIME v1.0) to decode the multicellular interactome of tumor microenvironment, identifying therapeutic targets. The platform quantifies multidimensional efficacy-toxicity landscapes, forecasting synergistic interactions (e.g., probiotic-enhanced dendritic cell function augmenting anti-PD-1 responses) (138). Real-time toxicity monitoring flags risks like antibiotic-induced immunotherapy resistance. These insights guide master protocol trials: (1) Arm A targets dysbiosis index ≥5.2 tumors with FMT + anti-PD-1 to correct gut-liver axis dysregulation, and (2) Arm B addresses FXR-methylated subtypes via DNMT inhibitor + OCA combinations that destabilize chemotherapy-resistant microenvironments through cholesterol homeostasis modulation (121). This phase systematically converges microbial engineering, metabolic targeting, and AI-driven design to transition CCA management from empirical to mechanism-guided interception.
7.3 Engineered microbial therapeutics (phase III)
Synthetic biology tackles CCA heterogeneity through precision-engineered bacteria with logic-gated (“sense-decide-act”) capabilities, targeting dysregulation via tripartite mechanisms: microbiota modulation, BA metabolic intervention, and antitumor immunity activation (139). Crucially, this phase integrates outputs from prior stage: Phase I-derived single-cell metatranscriptomics enables AI-powered prediction of strain-engineered metabolic perturbations via hepatic flux balance analysis, informing designs like multifunctional consortia (BA regulation), Lactobacillus reuteri (IL-22-mediated mucosal repair), and attenuated Salmonella typhimurium (hypoxia-targeted gemcitabine delivery). Phase II-validated synergy models (e.g., butyrate-enhanced dendritic cell priming) guide strain-immunotherapy combinations. Convergence with nanomedicine (e.g., liver-tropic nanoparticles co-loaded with FXR agonists/probiotics) or immunotherapies (e.g., anti-PD-1/butyrate) amplifies therapeutic indices (126). Short-term priorities include strain optimization (kill switches, hypoxia-triggered gemcitabine) and ecological integration via butyrate-producing consortia for pan-hepatobiliary malignancies. The pivotal Phase III milestone requires demonstrating ≥30% reduction in CAF, validating engineered microbiota against desmoplastic CCA progression.
8 Persistent challenges and knowledge gaps
8.1 Mechanistic complexity and model limitations
Therapeutic development for CCA faces significant interspecies translational barriers. Murine models (e.g., KRAS/p53 KO) inadequately recapitulate human cholestatic pathophysiology due to species-specific BA metabolism—notably CYP8B1 deficiency—and the absence of key immune components, including Kupffer cells (140, 141). Further complexity arises from BA-specific receptor (e.g., TGR5) and metabolite (e.g., butyrate) exhibiting context-dependent dual roles (142), necessitating single-cell spatiotemporal resolution to dissect microenvironmental interactions. Biomarker standardization remains elusive, with analytical variability in quantifying >40 carcinogenic BA isoforms and microbial interference with BA gene signatures (e.g., CYP7A1/SLC10A2 ratios) confounding treatment predictions (70). Compounding these challenges, interindividual variability driven by genetic polymorphisms, geographic disparities in microbiota composition (e.g., Bacteroides abundance in Asian versus Western populations) (143, 144), and dietary influences (e.g., high-fat diet-induced TGR5 activation (93)) collectively modulate therapeutic responsiveness, underscoring the need for precision stratification models.
8.2 Safety-efficacy dilemmas in gut-liver axis therapeutic
The clinical translation of gut-liver axis modulators faces unresolved safety-efficacy dilemmas. Probiotic-related bacteremia, particularly septicemia caused by Clostridium butyricum, is predominantly concentrated in high-risk post-hepatectomy patients (e.g., with neutropenia or bilioenteric anastomoses), contrasting sharply with therapeutic benefits (145). This risk is mechanistically linked to intestinal barrier disruption (e.g., anastomotic leaks) facilitating bacterial translocation. Broad-spectrum antibiotics deplete oncobiotic taxa but concomitantly disrupt chemotherapy efficacy—for instance, by depleting folate-producing Faecalibacterium, thereby contributing to 5-FU resistance. This paradox highlights the critical need for optimizing this risk-benefit balance. Synthetic microbial therapies present significant biocontainment challenges. Engineered bacteria (e.g., utilizing the Escherichia coliNissle 1917 chassis) demonstrate concerning horizontal gene transfer frequencies (10-³–10-5 CFU/recipient via plasmids) in murine models, potentially facilitating dissemination of virulence genes (e.g., ctxfrom Vibrio cholerae) (146, 147). Furthermore, the limited standardization of FMT protocols and unclear pathogenic thresholds impede clinical reproducibility.
8.3 Multi-omics integration hurdles
Current static multi-omics approaches fail to capture the dynamic heterogeneity of CCA, particularly evolving microbiota-immune-metabolite interactions needed for adaptive therapy. While spatial metabolomics and AI enable microenvironment mapping, clinical translation is limited by inconsistent analytical standardization across workflows. The absence of closed-loop systems integrating longitudinal multi-omics data (e.g., IDH-mutation gut-liver signatures) with adaptive algorithms impedes progress from empirical treatment towards AI-driven precision oncology. Addressing this requires collaborative validation of predictive biomarkers through prospective trials and development of real-time monitoring framework.
9 Conclusions
The gut-liver axis plays a pivotal role in CCA pathogenesis through dysbiosis-driven microbial metabolite translocation, BA metabolic reprogramming, and immune microenvironment remodeling. Gut microbiota dysbiosis promotes pathogenic endotoxin influx, activating TLR/NF-κB signaling to fuel chronic inflammation and tumorigenesis. Concurrently, BA accumulation drives cholangiocyte proliferation and immunosuppression via FXR/TGR5/S1PR2 pathways, while bidirectional crosstalk between microbial BA metabolism and BA-mediated microbiota modulation perpetuates oncogenic signaling. Therapeutic strategies targeting this axis—antibiotics, probiotics, FMT, and BA receptor modulators—demonstrate potential to disrupt tumor-promoting circuits, particularly when integrated with immunotherapy. Future directions demand multi-omics-guided precision approaches, AI-powered combinatorial regimens, synthetic engineered microbiota to address heterogeneity and receptor duality. Overcoming challenges in mechanistic validation, clinical standardization, and microbiota-host metabolic interplay will be critical for translating gut-liver axis insights into effective CCA therapies.
Author contributions
LW: Writing – original draft, Writing – review & editing, Funding acquisition, Conceptualization. WQ: Conceptualization, Methodology, Writing – original draft, Funding acquisition. XZ: Methodology, Conceptualization, Writing – original draft. YZ: Supervision, Writing – review & editing. ZD: Writing – review & editing, Supervision.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by grants from the Shandong Provincial Natural Science Foundation (ZR2024QH660) and Shandong Provincial Natural Science Foundation (ZR2021MH259).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1646897/full#supplementary-material
Supplementary Figure 1 | Gut microbiota trajectories across advanced cholangiocarcinoma (TNM stage III–IV) and vascular invasion stratification.
Abbreviations
CCA, Cholangiocarcinoma; BA, bile acid; iCCA, intrahepatic CCA; eCCA, extrahepatic CCA; HCC, hepatocellular carcinoma; PSC, primary sclerosing cholangitis; KCs, Kupffer cells; NF-κB, nuclear factor kappa-B; LPS, lipopolysaccharides; SCFAs, short-chain fatty acids; TLR, Toll-like receptor; BTC, biliary tract cancer; GWAS, genome-wide association study; VI, vascular invasion; PD-1, programmed cell death protein 1; TNF-α, Tumor necrosis factor alpha; CXCL, C-X-C motif chemokine ligand; TME, tumor microenvironment; PMN-MDSCs, polymorphonuclear myeloid-derived suppressor cells; BBD, benign biliary disease; GCA, glycocholic acid; TCDCA, taurochenodeoxycholic acid; GLCA, glycolithocholic acid; GUDCA, glycoursodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid; FXR, farnesoid X receptor; TGR5, Takeda G protein-coupled receptor 5; S1PR2, sphingosine-1-phosphate receptor 2; ERK, extracellular signal-regulated kinase; EGFR, epidermal growth factor receptor; COX-2, cyclooxygenase-2; SHP, small heterodimer partner; NK cell, natural killer cell; LGG, Lactobacillus rhamnosus GG; FMT, fecal microbiota transplantation; AI, artificial intelligence; CAF, cancer-associated fibroblasts; ROS, reactive oxygen species.
References
1. Qurashi M, Vithayathil M, and Khan SA. Epidemiology of cholangiocarcinoma. Eur J Surg Oncol. (2025) 51:107064. doi: 10.1016/j.ejso.2023.107064
2. Welzel TM, McGlynn KA, Hsing AW, O’Brien TR, and Pfeiffer RM. Impact of classification of hilar cholangiocarcinomas (Klatskin tumors) on the incidence of intra- and extrahepatic cholangiocarcinoma in the United States. J Natl Cancer Inst. (2006) 98:873–5. doi: 10.1093/jnci/djj234
3. Vithayathil M and Khan SA. Current epidemiology of cholangiocarcinoma in Western countries. J Hepatol. (2022) 77:1690–8. doi: 10.1016/j.jhep.2022.07.022
4. Khan SA, Tavolari S, and Brandi G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. (2019) 39 Suppl 1:19–31. doi: 10.1111/liv.14095
5. Massarweh NN and El-Serag HB. Epidemiology of hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Cancer Control. (2017) 24:1073274817729245. doi: 10.1177/1073274817729245
6. Saha SK, Zhu AX, Fuchs CS, and Brooks GA. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist. (2016) 21:594–9. doi: 10.1634/theoncologist.2015-0446
7. Clements O, Eliahoo J, Kim JU, Taylor-Robinson SD, and Khan SA. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: A systematic review and meta-analysis. J Hepatol. (2020) 72:95–103. doi: 10.1016/j.jhep.2019.09.007
8. Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. (2020) 17:557–88. doi: 10.1038/s41575-020-0310-z
9. Marin JJG, Prete MG, Lamarca A, Tavolari S, Landa-Magdalena A, Brandi G, et al. Current and novel therapeutic opportunities for systemic therapy in biliary cancer. Br J Cancer. (2020) 123:1047–59. doi: 10.1038/s41416-020-0987-3
10. Bridgewater J, Lopes A, Palmer D, Cunningham D, Anthoney A, Maraveyas A, et al. Quality of life, long-term survivors and long-term outcome from the ABC-02 study. Br J Cancer. (2016) 114:965–71. doi: 10.1038/bjc.2016.64
11. Shroff RT, King G, Colby S, Scott AJ, Borad MJ, Goff L, et al. SWOG S1815: A phase III randomized trial of gemcitabine, cisplatin, and nab-paclitaxel versus gemcitabine and cisplatin in newly diagnosed, advanced biliary tract cancers. J Clin Oncol. (2025) 43:536–44. doi: 10.1200/jco-24-01383
12. Kelley RK, Ueno M, Yoo C, Finn RS, Furuse J, Ren Z, et al. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2023) 401:1853–65. doi: 10.1016/s0140-6736(23)00727-4
13. Goyal L, Meric-Bernstam F, Hollebecque A, Valle JW, Morizane C, Karasic TB, et al. Futibatinib for FGFR2-rearranged intrahepatic cholangiocarcinoma. N Engl J Med. (2023) 388:228–39. doi: 10.1056/NEJMoa2206834
14. Tilg H, Adolph TE, and Trauner M. Gut-liver axis: Pathophysiological concepts and clinical implications. Cell Metab. (2022) 34:1700–18. doi: 10.1016/j.cmet.2022.09.017
15. Herraez E, Romero MR, Macias RIR, Monte MJ, and Marin JJG. Clinical relevance of the relationship between changes in gut microbiota and bile acid metabolism in patients with intrahepatic cholangiocarcinoma. Hepatobiliary Surg Nutr. (2020) 9:211–4. doi: 10.21037/hbsn.2019.10.11
16. Zhang Q, Zhou J, Zhai D, Jiang Q, Yang M, and Zhou M. Gut microbiota regulates the ALK5/NOX1 axis by altering glutamine metabolism to inhibit ferroptosis of intrahepatic cholangiocarcinoma cells. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:167152. doi: 10.1016/j.bbadis.2024.167152
17. Ke J, Zhang CJ, Wang LZ, Xie FS, Wu HY, Li T, et al. Lipopolysaccharide promotes cancer cell migration and invasion through METTL3/PI3K/AKT signaling in human cholangiocarcinoma. Heliyon. (2024) 10:e29683. doi: 10.1016/j.heliyon.2024.e29683
18. Manieri E, Folgueira C, Rodríguez ME, Leiva-Vega L, Esteban-Lafuente L, Chen C, et al. JNK-mediated disruption of bile acid homeostasis promotes intrahepatic cholangiocarcinoma. Proc Natl Acad Sci U S A. (2020) 117:16492–9. doi: 10.1073/pnas.2002672117
19. Zhang Q, Ma C, Duan Y, Heinrich B, Rosato U, Diggs LP, et al. Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov. (2021) 11:1248–67. doi: 10.1158/2159-8290.Cd-20-0304
20. Lederer AK, Görrissen N, Nguyen TT, Kreutz C, Rasel H, Bartsch F, et al. Exploring the effects of gut microbiota on cholangiocarcinoma progression by patient-derived organoids. J Transl Med. (2025) 23:34. doi: 10.1186/s12967-024-06012-x
21. Deng T, Li J, He B, Chen B, Liu F, Chen Z, et al. Gut microbiome alteration as a diagnostic tool and associated with inflammatory response marker in primary liver cancer. Hepatol Int. (2022) 16:99–111. doi: 10.1007/s12072-021-10279-3
22. Jia W, Xie G, and Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol. (2018) 15:111–28. doi: 10.1038/nrgastro.2017.119
23. Wang X, Fang Y, Liang W, Cai Y, Wong CC, Wang J, et al. Gut-liver translocation of pathogen Klebsiella pneumoniae promotes hepatocellular carcinoma in mice. Nat Microbiol. (2025) 10:169–84. doi: 10.1038/s41564-024-01890-9
24. Leshem A, Liwinski T, and Elinav E. Immune-microbiota interplay and colonization resistance in infection. Mol Cell. (2020) 78:597–613. doi: 10.1016/j.molcel.2020.03.001
25. Schneider KM, Mohs A, Gui W, Galvez EJC, Candels LS, Hoenicke L, et al. Imbalanced gut microbiota fuels hepatocellular carcinoma development by shaping the hepatic inflammatory microenvironment. Nat Commun. (2022) 13:3964. doi: 10.1038/s41467-022-31312-5
26. Zhang X, Coker OO, Chu ES, Fu K, Lau HCH, Wang YX, et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut. (2021) 70:761–74. doi: 10.1136/gutjnl-2019-319664
27. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. (2019) 30:607. doi: 10.1016/j.cmet.2019.08.002
28. Giordano DM, Pinto C, Maroni L, Benedetti A, and Marzioni M. Inflammation and the gut-liver axis in the pathophysiology of cholangiopathies. Int J Mol Sci. (2018) 19:3003. doi: 10.3390/ijms19103003
29. Hov JR and Karlsen TH. The microbiota and the gut-liver axis in primary sclerosing cholangitis. Nat Rev Gastroenterol Hepatol. (2023) 20:135–54. doi: 10.1038/s41575-022-00690-y
30. Lederer AK, Rasel H, Kohnert E, Kreutz C, Huber R, Badr MT, et al. Gut microbiota in diagnosis, therapy and prognosis of cholangiocarcinoma and gallbladder carcinoma-A scoping review. Microorganisms. (2023) 11:2363. doi: 10.3390/microorganisms11092363
31. Ito Z, Koido S, Kato K, Odamaki T, Horiuchi S, Akasu T, et al. Dysbiosis of the fecal and biliary microbiota in biliary tract cancer. Cancers (Basel). (2022) 14:5379. doi: 10.3390/cancers14215379
32. Zhang T, Zhang S, Jin C, Lin Z, Deng T, Xie X, et al. A predictive model based on the gut microbiota improves the diagnostic effect in patients with cholangiocarcinoma. Front Cell Infect Microbiol. (2021) 11:751795. doi: 10.3389/fcimb.2021.751795
33. Jia X, Lu S, Zeng Z, Liu Q, Dong Z, Chen Y, et al. Characterization of gut microbiota, bile acid metabolism, and cytokines in intrahepatic cholangiocarcinoma. Hepatology. (2020) 71:893–906. doi: 10.1002/hep.30852
34. Zhang Y, Yang FJ, Jiang QR, Gao HJ, Song X, Zhu HQ, et al. Association between gut microbiota and hepatocellular carcinoma and biliary tract cancer: A mendelian randomization study. World J Clin Cases. (2024) 12:3497–504. doi: 10.12998/wjcc.v12.i18.3497
35. Chen Z, Shi W, Chen K, Lu C, Li X, and Li Q. Elucidating the causal association between gut microbiota and intrahepatic cholangiocarcinoma through Mendelian randomization analysis. Front Microbiol. (2023) 14:1288525. doi: 10.3389/fmicb.2023.1288525
36. Zhang L, Chen C, Chai D, Kuang T, Deng W, and Wang W. Alterations of gut mycobiota profiles in intrahepatic cholangiocarcinoma. Front Microbiol. (2022) 13:1090392. doi: 10.3389/fmicb.2022.1090392
37. Bartsch F, Baumgart J, Hoppe-Lotichius M, Schmidtmann I, Heinrich S, and Lang H. Visceral infiltration of intrahepatic cholangiocarcinoma is most prognostic after curative resection - Retrospective cohort study of 102 consecutive liver resections from a single center. Int J Surg. (2018) 55:193–200. doi: 10.1016/j.ijsu.2018.05.027
38. Mao J, Wang D, Long J, Yang X, Lin J, Song Y, et al. Gut microbiome is associated with the clinical response to anti-PD-1 based immunotherapy in hepatobiliary cancers. J Immunother Cancer. (2021) 9:e003334. doi: 10.1136/jitc-2021-003334
39. Jin S, Zhao R, Zhou C, Zhong Q, Shi J, Su C, et al. Feasibility and tolerability of sintilimab plus anlotinib as the second-line therapy for patients with advanced biliary tract cancers: An open-label, single-arm, phase II clinical trial. Int J Cancer. (2023) 152:1648–58. doi: 10.1002/ijc.34372
40. Li Z, Zhang Y, Hong W, Wang B, Chen Y, Yang P, et al. Gut microbiota modulate radiotherapy-associated antitumor immune responses against hepatocellular carcinoma Via STING signaling. Gut Microbes. (2022) 14:2119055. doi: 10.1080/19490976.2022.2119055
41. Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. (2009) 9:799–809. doi: 10.1038/nri2653
42. Blaak EE, Canfora EE, Theis S, Frost G, Groen AK, Mithieux G, et al. Short chain fatty acids in human gut and metabolic health. Benef Microbes. (2020) 11:411–55. doi: 10.3920/bm2020.0057
43. Bertani B and Ruiz N. Function and biogenesis of lipopolysaccharides. EcoSal Plus. (2018) 8:10. doi: 10.1128/ecosalplus.ESP-0001-2018
44. Fitzgerald KA and Kagan JC. Toll-like receptors and the control of immunity. Cell. (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
45. Pohl K, Moodley P, and Dhanda A. The effect of increasing intestinal short-chain fatty acid concentration on gut permeability and liver injury in the context of liver disease: A systematic review. J Gastroenterol Hepatol. (2022) 37:1498–506. doi: 10.1111/jgh.15899
46. Manilla V, Di Tommaso N, Santopaolo F, Gasbarrini A, and Ponziani FR. Endotoxemia and gastrointestinal cancers: insight into the mechanisms underlying a dangerous relationship. Microorganisms. (2023) 11:267. doi: 10.3390/microorganisms11020267
47. Xu M, Luo K, Li J, Li Y, Zhang Y, Yuan Z, et al. Role of intestinal microbes in chronic liver diseases. Int J Mol Sci. (2022) 23:12661. doi: 10.3390/ijms232012661
48. Killeen SD, Wang JH, Andrews EJ, and Redmond HP. Bacterial endotoxin enhances colorectal cancer cell adhesion and invasion through TLR-4 and NF-kappaB-dependent activation of the urokinase plasminogen activator system. Br J Cancer. (2009) 100:1589–602. doi: 10.1038/sj.bjc.6604942
49. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science. (2020) 368:973–80. doi: 10.1126/science.aay9189
50. Albillos A, de Gottardi A, and Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol. (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
51. Violi F, Cammisotto V, Bartimoccia S, Pignatelli P, Carnevale R, and Nocella C. Gut-derived low-grade endotoxaemia, atherothrombosis and cardiovascular disease. Nat Rev Cardiol. (2023) 20:24–37. doi: 10.1038/s41569-022-00737-2
52. Di Giorgio C, Urbani G, Marchianò S, Biagioli M, Bordoni M, Bellini R, et al. Liver GPBAR1 associates with immune dysfunction in primary sclerosing cholangitis and its activation attenuates cholestasis in abcb4-/- mice. Liver Int. (2025) 45:e16235. doi: 10.1111/liv.16235
53. Moon DK, Kang JS, Byun Y, Choi YJ, Lee HW, Jang JY, et al. Incidence of bactibilia and related factors in patients who undergo cholecystectomy. Ann Surg Treat Res. (2023) 104:10–7. doi: 10.4174/astr.2023.104.1.10
54. Di Carlo P, Serra N, D’Arpa F, Agrusa A, Gulotta G, Fasciana T, et al. The microbiota of the bilio-pancreatic system: a cohort, STROBE-compliant study. Infect Drug Resist. (2019) 12:1513–27. doi: 10.2147/idr.S200378
55. Di Carlo P, Serra N, Fasciana TMA, Giammanco A, D’Arpa F, Rea T, et al. Microbial profile in bile from pancreatic and extra-pancreatic biliary tract cancer. PloS One. (2024) 19:e0294049. doi: 10.1371/journal.pone.0294049
56. Bednarsch J, Czigany Z, Heij LR, Luedde T, van Dam R, Lang SA, et al. Bacterial bile duct colonization in perihilar cholangiocarcinoma and its clinical significance. Sci Rep. (2021) 11:2926. doi: 10.1038/s41598-021-82378-y
57. Zigmond E, Zecher BF, Bartels AL, Ziv-Baran T, Rösch T, Schachschal G, et al. Bile duct colonization with enterococcus sp. Associates with disease progression in primary sclerosing cholangitis. Clin Gastroenterol Hepatol. (2023) 21:1223–1232.e1223. doi: 10.1016/j.cgh.2022.09.006
58. Cammann S, Karabulut S, DeTemple DE, Oldhafer F, Kulik U, Schroeter A, et al. Antibiotic-resistant bacteria colonizing the bile duct are associated with increased morbidity and mortality after resection of extrahepatic cholangiocarcinoma. Surg Infect (Larchmt). (2022) 23:270–9. doi: 10.1089/sur.2021.117
59. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. (2012) 486:207–14. doi: 10.1038/nature11234
60. Hajishengallis G and Chavakis T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. (2021) 21:426–40. doi: 10.1038/s41577-020-00488-6
61. Kunath BJ, De Rudder C, Laczny CC, Letellier E, and Wilmes P. The oral-gut microbiome axis in health and disease. Nat Rev Microbiol. (2024) 22:791–805. doi: 10.1038/s41579-024-01075-5
62. Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, et al. Ectopic colonization of oral bacteria in the intestine drives T(H)1 cell induction and inflammation. Science. (2017) 358:359–65. doi: 10.1126/science.aan4526
63. Rao BC, Zhang GZ, Zou YW, Ren T, Ren HY, Liu C, et al. Alterations in the human oral microbiome in cholangiocarcinoma. Mil Med Res. (2022) 9:62. doi: 10.1186/s40779-022-00423-x
64. Oh S, Kim J, Shin CM, Lee HJ, Lee HS, and Park KU. Metagenomic characterization of oral microbiome signatures to predict upper gastrointestinal and pancreaticobiliary cancers: a case-control study. J Transl Med. (2025) 23:20. doi: 10.1186/s12967-024-05989-9
65. Chen MJ, Liu C, Wan Y, Yang L, Jiang S, Qian DW, et al. Enterohepatic circulation of bile acids and their emerging roles on glucolipid metabolism. Steroids. (2021) 165:108757. doi: 10.1016/j.steroids.2020.108757
66. Roberts MS, Magnusson BM, Burczynski FJ, and Weiss M. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin Pharmacokinet. (2002) 41:751–90. doi: 10.2165/00003088-200241100-00005
67. Lozano E, Sanchez-Vicente L, Monte MJ, Herraez E, Briz O, Banales JM, et al. Cocarcinogenic effects of intrahepatic bile acid accumulation in cholangiocarcinoma development. Mol Cancer Res. (2014) 12:91–100. doi: 10.1158/1541-7786.Mcr-13-0503
68. Sombattheera S, Proungvitaya T, Limpaiboon T, Wongkham S, Wongkham C, Luvira V, et al. Total serum bile acid as a potential marker for the diagnosis of cholangiocarcinoma without jaundice. Asian Pac J Cancer Prev. (2015) 16:1367–70. doi: 10.7314/apjcp.2015.16.4.1367
69. Proungvitaya S, Sombattheera S, Boonsiri P, Limpaiboon T, Wongkham S, Wongkham C, et al. Diagnostic value of serum bile acid composition patterns and serum glycocholic acid levels in cholangiocarcinoma. Oncol Lett. (2017) 14:4943–8. doi: 10.3892/ol.2017.6763
70. Zhang X, Yang Z, Shi Z, Zhu Z, Li C, Du Z, et al. Analysis of bile acid profile in plasma to differentiate cholangiocarcinoma from benign biliary diseases and healthy controls. J Steroid Biochem Mol Biol. (2021) 205:105775. doi: 10.1016/j.jsbmb.2020.105775
71. Rejchrt S, Hroch M, Repak R, Fejfar T, Douda T, Kohoutova D, et al. Investigation of 23 bile acids in liver bile in benign and Malignant biliary stenosis: A pilot study. Gastroenterol Res Pract. (2019) 2019:5371381. doi: 10.1155/2019/5371381
72. Wang W, Tian SL, Jin D, Liu B, Wang W, Chang H, et al. The role of bile acid subtypes in the diagnosis of cholangiocarcinoma. Asia Pac J Clin Oncol. (2022) 18:e163–72. doi: 10.1111/ajco.13588
73. Dai J, Wang H, Dong Y, Zhang Y, and Wang J. Bile acids affect the growth of human cholangiocarcinoma via NF-kB pathway. Cancer Invest. (2013) 31:111–20. doi: 10.3109/07357907.2012.762781
74. Amonyingcharoen S, Suriyo T, Thiantanawat A, Watcharasit P, and Satayavivad J. Taurolithocholic acid promotes intrahepatic cholangiocarcinoma cell growth via muscarinic acetylcholine receptor and EGFR/ERK1/2 signaling pathway. Int J Oncol. (2015) 46:2317–26. doi: 10.3892/ijo.2015.2939
75. Pellicciari R, Costantino G, Camaioni E, Sadeghpour BM, Entrena A, Willson TM, et al. Bile acid derivatives as ligands of the farnesoid X receptor. Synthesis, evaluation, and structure-activity relationship of a series of body and side chain modified analogues of chenodeoxycholic acid. J Med Chem. (2004) 47:4559–69. doi: 10.1021/jm049904b
76. Lv B, Ma L, Tang W, Huang P, Yang B, Wang L, et al. FXR acts as a metastasis suppressor in intrahepatic cholangiocarcinoma by inhibiting IL-6-induced epithelial-mesenchymal transition. Cell Physiol Biochem. (2018) 48:158–72. doi: 10.1159/000491715
77. Wang W, Zhan M, Li Q, Chen W, Chu H, Huang Q, et al. FXR agonists enhance the sensitivity of biliary tract cancer cells to cisplatin via SHP dependent inhibition of Bcl-xL expression. Oncotarget. (2016) 7:34617–29. doi: 10.18632/oncotarget.8964
78. Girisa S, Henamayee S, Parama D, Rana V, Dutta U, and Kunnumakkara AB. Targeting Farnesoid X receptor (FXR) for developing novel therapeutics against cancer. Mol Biomed. (2021) 2:21. doi: 10.1186/s43556-021-00035-2
79. Chen H, Zhu B, Zhao L, Liu Y, Zhao F, Feng J, et al. Allicin inhibits proliferation and invasion in vitro and in vivo via SHP-1-mediated STAT3 signaling in cholangiocarcinoma. Cell Physiol Biochem. (2018) 47:641–53. doi: 10.1159/000490019
80. Reich M, Klindt C, Deutschmann K, Spomer L, Häussinger D, and Keitel V. Role of the G protein-coupled bile acid receptor TGR5 in liver damage. Dig Dis. (2017) 35:235–40. doi: 10.1159/000450917
81. Reich M, Deutschmann K, Sommerfeld A, Klindt C, Kluge S, Kubitz R, et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation. Vivo vitro. Gut. (2016) 65:487–501. doi: 10.1136/gutjnl-2015-309458
82. Sato H, Macchiarulo A, Thomas C, Gioiello A, Une M, Hofmann AF, et al. Novel potent and selective bile acid derivatives as TGR5 agonists: biological screening, structure-activity relationships, and molecular modeling studies. J Med Chem. (2008) 51:1831–41. doi: 10.1021/jm7015864
83. Liu R, Li X, Qiang X, Luo L, Hylemon PB, Jiang Z, et al. Taurocholate induces cyclooxygenase-2 expression via the sphingosine 1-phosphate receptor 2 in a human cholangiocarcinoma cell line. J Biol Chem. (2015) 290:30988–1002. doi: 10.1074/jbc.M115.668277
84. Liu R, Zhao R, Zhou X, Liang X, Campbell DJ, Zhang X, et al. Conjugated bile acids promote cholangiocarcinoma cell invasive growth through activation of sphingosine 1-phosphate receptor 2. Hepatology. (2014) 60:908–18. doi: 10.1002/hep.27085
85. Wang Y, Aoki H, Yang J, Peng K, Liu R, Li X, et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology. (2017) 65:2005–18. doi: 10.1002/hep.29076
86. Deng M, Liu J, Zhang L, Lou Y, and Qiu Y. Identification of molecular subtypes based on bile acid metabolism in cholangiocarcinoma. BMC Cancer. (2024) 24:1313. doi: 10.1186/s12885-024-13081-0
87. Farhat Z, Freedman ND, Sampson JN, Falk RT, Koshiol J, Weinstein SJ, et al. A prospective investigation of serum bile acids with risk of liver cancer, fatal liver disease, and biliary tract cancer. Hepatol Commun. (2022) 6:2391–9. doi: 10.1002/hep4.2003
88. Perino A, Demagny H, Velazquez-Villegas L, and Schoonjans K. Molecular physiology of bile acid signaling in health, disease, and aging. Physiol Rev. (2021) 101:683–731. doi: 10.1152/physrev.00049.2019
89. Guan B, Tong J, Hao H, Yang Z, Chen K, Xu H, et al. Bile acid coordinates microbiota homeostasis and systemic immunometabolism in cardiometabolic diseases. Acta Pharm Sin B. (2022) 12:2129–49. doi: 10.1016/j.apsb.2021.12.011
90. Hofmann AF and Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. (2008) 65:2461–83. doi: 10.1007/s00018-008-7568-6
91. Portincasa P, Di Ciaula A, Garruti G, Vacca M, De Angelis M, and Wang DQ. Bile acids and GPBAR-1: dynamic interaction involving genes, environment and gut microbiome. Nutrients. (2020) 12:3709. doi: 10.3390/nu12123709
92. Thomas LA, Veysey MJ, Bathgate T, King A, French G, Smeeton NC, et al. Mechanism for the transit-induced increase in colonic deoxycholic acid formation in cholesterol cholelithiasis. Gastroenterology. (2000) 119:806–15. doi: 10.1053/gast.2000.16495
93. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. (2013) 499:97–101. doi: 10.1038/nature12347
94. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. (2018) 360:eaan5931. doi: 10.1126/science.aan5931
95. Wu N, Bayatpour S, Hylemon PB, Aseem SO, Brindley PJ, and Zhou H. Gut microbiome and bile acid interactions: mechanistic implications for cholangiocarcinoma development, immune resistance, and therapy. Am J Pathol. (2025) 195:397–408. doi: 10.1016/j.ajpath.2024.11.004
96. Liu Y, Chen K, Li F, Gu Z, Liu Q, He L, et al. Probiotic lactobacillus rhamnosus GG prevents liver fibrosis through inhibiting hepatic bile acid synthesis and enhancing bile acid excretion in mice. Hepatology. (2020) 71:2050–66. doi: 10.1002/hep.30975
97. Ren L, Song Q, Liu Y, Zhang L, Hao Z, and Feng W. Probiotic Lactobacillus rhamnosus GG prevents progesterone metabolite epiallaopregnanolone sulfate-induced hepatic bile acid accumulation and liver injury. Biochem Biophys Res Commun. (2019) 520:67–72. doi: 10.1016/j.bbrc.2019.09.103
98. Hu S, Tang B, Lu C, Wang S, Wu L, Lei Y, et al. Lactobacillus rhamnosus GG ameliorates triptolide-induced liver injury through modulation of the bile acid-FXR axis. Pharmacol Res. (2024) 206:107275. doi: 10.1016/j.phrs.2024.107275
99. Han S, Wang K, Shen J, Xia H, Lu Y, Zhuge A, et al. Probiotic Pediococcus pentosaceus Li05 Improves Cholestasis through the FXR-SHP and FXR-FGF15 Pathways. Nutrients. (2023) 15:4864. doi: 10.3390/nu15234864
100. Cabrera-Rubio R, Patterson AM, Cotter PD, and Beraza N. Cholestasis induced by bile duct ligation promotes changes in the intestinal microbiome in mice. Sci Rep. (2019) 9:12324. doi: 10.1038/s41598-019-48784-z
101. Collins SL, Stine JG, Bisanz JE, Okafor CD, and Patterson AD. Bile acids and the gut microbiota: metabolic interactions and impacts on disease. Nat Rev Microbiol. (2023) 21:236–47. doi: 10.1038/s41579-022-00805-x
102. Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. (2006) 103:3920–5. doi: 10.1073/pnas.0509592103
103. Xie XM, Zhang BY, Feng S, Fan ZJ, and Wang GY. Activation of gut FXR improves the metabolism of bile acids, intestinal barrier, and microbiota under cholestatic condition caused by GCDCA in mice. Microbiol Spectr. (2025) 13:e0315024. doi: 10.1128/spectrum.03150-24
104. Daniel N, Genua F, Jenab M, Mayén AL, Chrysovalantou Chatziioannou A, Keski-Rahkonen P, et al. The role of the gut microbiome in the development of hepatobiliary cancers. Hepatology. (2024) 80:1252–69. doi: 10.1097/hep.0000000000000406
105. Nakamoto N, Sasaki N, Aoki R, Miyamoto K, Suda W, Teratani T, et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat Microbiol. (2019) 4:492–503. doi: 10.1038/s41564-018-0333-1
106. Ali AH, Damman J, Shah SB, Davies Y, Hurwitz M, Stephen M, et al. Open-label prospective therapeutic clinical trials: oral vancomycin in children and adults with primary sclerosing cholangitis. Scand J Gastroenterol. (2020) 55:941–50. doi: 10.1080/00365521.2020.1787501
107. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. (2018) 359:91–7. doi: 10.1126/science.aan3706
108. Alshammari K, Alotaibi FM, Alsugheir F, Aldawoud M, Alolayan A, Algarni MA, et al. Antibiotic exposure concurrently with anti-PD1 blockade therapy reduces overall survival in patients with child-pugh class A advanced hepatocellular carcinoma. Cancers (Basel). (2023) 16:133. doi: 10.3390/cancers16010133
109. Duduyemi OP, Potapenko K, Limanska N, Kotsyuda S, Petriv N, Suo H, et al. Lactiplantibacillus plantarum inhibited the growth of primary liver cancer by inducing early apoptosis and senescence. vitro. Front Microbiol. (2024) 15:1451170. doi: 10.3389/fmicb.2024.1451170
110. Wu L, Zhou J, Zhou A, Lei Y, Tang L, Hu S, et al. Lactobacillus acidophilus ameliorates cholestatic liver injury through inhibiting bile acid synthesis and promoting bile acid excretion. Gut Microbes. (2024) 16:2390176. doi: 10.1080/19490976.2024.2390176
111. Li F, Han Q, Cai Y, Li Y, Yang Y, Li J, et al. Si-Ni-San ameliorates cholestatic liver injury by favoring P. goldsteinii colonization. J Ethnopharmacol. (2025) 337:118804. doi: 10.1016/j.jep.2024.118804
112. Gao G, Ma T, Zhang T, Jin H, Li Y, Kwok LY, et al. Adjunctive Probiotic Lactobacillus rhamnosus Probio-M9 Administration Enhances the Effect of Anti-PD-1 Antitumor Therapy via Restoring Antibiotic-Disrupted Gut Microbiota. Front Immunol. (2021) 12:772532. doi: 10.3389/fimmu.2021.772532
113. Messaoudene M, Pidgeon R, Richard C, Ponce M, Diop K, Benlaifaoui M, et al. A natural polyphenol exerts antitumor activity and circumvents anti-PD-1 resistance through effects on the gut microbiota. Cancer Discov. (2022) 12:1070–87. doi: 10.1158/2159-8290.Cd-21-0808
114. Zhang JW, Du P, Gao J, Yang BR, Fang WJ, and Ying CM. Preoperative probiotics decrease postoperative infectious complications of colorectal cancer. Am J Med Sci. (2012) 343:199–205. doi: 10.1097/MAJ.0b013e31823aace6
115. Shimura M, Mizuma M, Nakagawa K, Aoki S, Miura T, Takadate T, et al. Probiotic-related bacteremia after major hepatectomy for biliary cancer: a report of two cases. Surg Case Rep. (2021) 7:133. doi: 10.1186/s40792-021-01216-5
116. Keller JJ, Ooijevaar RE, Hvas CL, Terveer EM, Lieberknecht SC, Högenauer C, et al. A standardised model for stool banking for faecal microbiota transplantation: a consensus report from a multidisciplinary UEG working group. United Eur Gastroenterol J. (2021) 9:229–47. doi: 10.1177/2050640620967898
117. Zhao Y, Gong C, Xu J, Chen D, Yang B, Chen Z, et al. Research progress of fecal microbiota transplantation in liver diseases. J Clin Med. (2023) 12:1683. doi: 10.3390/jcm12041683
118. Allegretti JR, Kassam Z, Carrellas M, Mullish BH, Marchesi JR, Pechlivanis A, et al. Fecal microbiota transplantation in patients with primary sclerosing cholangitis: A pilot clinical trial. Am J Gastroenterol. (2019) 114:1071–9. doi: 10.14309/ajg.0000000000000115
119. Al-Shakhshir S, Quraishi MN, Mullish B, Patel A, Vince A, Rowe A, et al. FAecal micRobiota transplantation in primary sclerosinG chOlangitis (FARGO): study protocol for a randomised, multicentre, phase IIa, placebo-controlled trial. BMJ Open. (2025) 15:e095392. doi: 10.1136/bmjopen-2024-095392
120. Deng Z, Mei S, Ouyang Z, Wang R, Wang L, Zou B, et al. Dysregulation of gut microbiota stimulates NETs-driven HCC intrahepatic metastasis: therapeutic implications of healthy faecal microbiota transplantation. Gut Microbes. (2025) 17:2476561. doi: 10.1080/19490976.2025.2476561
121. Montal R, Sia D, Montironi C, Leow WQ, Esteban-Fabró R, Pinyol R, et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J Hepatol. (2020) 73:315–27. doi: 10.1016/j.jhep.2020.03.008
122. Li AD, Xie XL, Qi W, Wang WB, Ma JJ, Zhao DQ, et al. TGR5 promotes cholangiocarcinoma by interacting with mortalin. Exp Cell Res. (2020) 389:111855. doi: 10.1016/j.yexcr.2020.111855
123. Fiorucci S, Urbani G, Di Giorgio C, Biagioli M, and Distrutti E. Bile acids-based therapies for primary sclerosing cholangitis: current landscape and future developments. Cells. (2024) 13:1650. doi: 10.3390/cells13191650
124. Qian S, Su Z, Lin J, Hou Q, Wang X, Li Y, et al. Inhibition of Farnesoid-x-receptor signaling during abdominal sepsis by dysbiosis exacerbates gut barrier dysfunction. Cell Commun Signal. (2025) 23:236. doi: 10.1186/s12964-025-02224-w
125. Kowdley KV, Vuppalanchi R, Levy C, Floreani A, Andreone P, LaRusso NF, et al. A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J Hepatol. (2020) 73:94–101. doi: 10.1016/j.jhep.2020.02.033
126. Yao Q, Wang B, Yu J, Pan Q, Yu Y, Feng X, et al. ROS-responsive nanoparticle delivery of obeticholic acid mitigate primary sclerosing cholangitis. J Control Release. (2024) 374:112–26. doi: 10.1016/j.jconrel.2024.08.006
127. Erice O, Labiano I, Arbelaiz A, Santos-Laso A, Munoz-Garrido P, Jimenez-Agüero R, et al. Differential effects of FXR or TGR5 activation in cholangiocarcinoma progression. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1335–44. doi: 10.1016/j.bbadis.2017.08.016
128. Wang JP, Zhang MY, Luo M, Qin S, and Xia XM. Effect of FXR agonist GW4064 in the treatment of hilar cholangiocarcinoma in rats. Sci Rep. (2022) 12:18873. doi: 10.1038/s41598-022-23539-5
129. Di Matteo S, Nevi L, Costantini D, Overi D, Carpino G, Safarikia S, et al. The FXR agonist obeticholic acid inhibits the cancerogenic potential of human cholangiocarcinoma. PloS One. (2019) 14:e0210077. doi: 10.1371/journal.pone.0210077
130. Yang F, Huang X, Yi T, Yen Y, Moore DD, and Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. (2007) 67:863–7. doi: 10.1158/0008-5472.Can-06-1078
131. Yu J, Li S, Guo J, Xu Z, Zheng J, and Sun X. Farnesoid X receptor antagonizes Wnt/β-catenin signaling in colorectal tumorigenesis. Cell Death Dis. (2020) 11:640. doi: 10.1038/s41419-020-02819-w
132. Sun L, Cai J, and Gonzalez FJ. The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat Rev Gastroenterol Hepatol. (2021) 18:335–47. doi: 10.1038/s41575-020-00404-2
133. Ilyas SI, Affo S, Goyal L, Lamarca A, Sapisochin G, Yang JD, et al. Cholangiocarcinoma - novel biological insights and therapeutic strategies. Nat Rev Clin Oncol. (2023) 20:470–86. doi: 10.1038/s41571-023-00770-1
134. Sia D, Losic B, Moeini A, Cabellos L, Hao K, Revill K, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun. (2015) 6:6087. doi: 10.1038/ncomms7087
135. Abou-Alfa GK, Macarulla T, Javle MM, Kelley RK, Lubner SJ, Adeva J, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. (2020) 21:796–807. doi: 10.1016/s1470-2045(20)30157-1
136. Chen L, Vasoya RP, Toke NH, Parthasarathy A, Luo S, Chiles E, et al. HNF4 regulates fatty acid oxidation and is required for renewal of intestinal stem cells in mice. Gastroenterology. (2020) 158:985–999.e989. doi: 10.1053/j.gastro.2019.11.031
137. Beinsteiner B, Billas IML, and Moras D. Structural insights into the HNF4 biology. Front Endocrinol (Lausanne). (2023) 14:1197063. doi: 10.3389/fendo.2023.1197063
138. Marashi A, Hasany S, Moghimi S, Kiani R, Mehran Asl S, Dareghlou YA, et al. Targeting gut-microbiota for gastric cancer treatment: a systematic review. Front Med (Lausanne). (2024) 11:1412709. doi: 10.3389/fmed.2024.1412709
139. Landry BP and Tabor JJ. Engineering diagnostic and therapeutic gut bacteria. Microbiol Spectr. (2017) 5:10. doi: 10.1128/microbiolspec.BAD-0020-2017
140. Gåfvels M, Olin M, Chowdhary BP, Raudsepp T, Andersson U, Persson B, et al. Structure and chromosomal assignment of the sterol 12alpha-hydroxylase gene (CYP8B1) in human and mouse: eukaryotic cytochrome P-450 gene devoid of introns. Genomics. (1999) 56:184–96. doi: 10.1006/geno.1998.5606
141. Morgan RA. Human tumor xenografts: the good, the bad, and the ugly. Mol Ther. (2012) 20:882–4. doi: 10.1038/mt.2012.73
142. Liu H, Wang J, He T, Becker S, Zhang G, Li D, et al. Butyrate: A double-edged sword for health? Adv Nutr. (2018) 9:21–9. doi: 10.1093/advances/nmx009
143. Rehman A, Rausch P, Wang J, Skieceviciene J, Kiudelis G, Bhagalia K, et al. Geographical patterns of the standing and active human gut microbiome in health and IBD. Gut. (2016) 65:238–48. doi: 10.1136/gutjnl-2014-308341
144. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature. (2012) 486:222–7. doi: 10.1038/nature11053
145. DeFilipp Z, Bloom PP, Torres Soto M, Mansour MK, Sater MRA, Huntley MH, et al. Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. N Engl J Med. (2019) 381:2043–50. doi: 10.1056/NEJMoa1910437
146. Kim TH, Cho BK, and Lee DH. Synthetic biology-driven microbial therapeutics for disease treatment. J Microbiol Biotechnol. (2024) 34:1947–58. doi: 10.4014/jmb.2407.07004
Keywords: gut-liver axis, cholangiocarcinoma, gut microbiota, dysbiosis, bile salt, therapy, clinical translation
Citation: Wang L, Qiao W, Zhen X, Zhang Y and Dong Z (2025) Targeting the gut-liver axis in cholangiocarcinoma: mechanisms, therapeutic advances, and future directions. Front. Oncol. 15:1646897. doi: 10.3389/fonc.2025.1646897
Received: 14 June 2025; Accepted: 22 August 2025;
Published: 12 September 2025.
Edited by:
Jennifer M. Bailey-Lundberg, University of Nebraska Medical Center, United StatesCopyright © 2025 Wang, Qiao, Zhen, Zhang and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yeqiong Zhang, emhhbmd5cTUzQG1haWwuc3lzdS5lZHUuY24=; Zhiwei Dong, endkb25nQHN1cmdlcnkuY3Voay5lZHUuaGs=