 Rola H. Ali1,2*
Rola H. Ali1,2* Abdulaziz Hassan3
Abdulaziz Hassan3 Hussain H. Jarkhi2Abdullah Alshawish4Mohamad Almanabri4
Hussain H. Jarkhi2Abdullah Alshawish4Mohamad Almanabri4 Obada T. Alhalabi5
Obada T. Alhalabi5 Ahmad R. Alsaber6Nawal Y. Ali7Ehab Abdelnabi7
Ahmad R. Alsaber6Nawal Y. Ali7Ehab Abdelnabi7 Eiman M. A. Mohammed8Hiba Jama8Ammar Almarzooq8Zainab Alqallaf8Amir A. Ahmed8Shakir Bahzad8Stefan Hamelmann9
Eiman M. A. Mohammed8Hiba Jama8Ammar Almarzooq8Zainab Alqallaf8Amir A. Ahmed8Shakir Bahzad8Stefan Hamelmann9 Felix Sahm9Maryam Almurshed2
Felix Sahm9Maryam Almurshed2- 1Department of Pathology, College of Medicine, Kuwait University, Jabriya, Kuwait
- 2Department of Histopathology, Al Sabah Hospital, Shuwaikh, Kuwait
- 3Department of Diagnostic Radiology, Jaber Alahmad Hospital, South Surra, Kuwait
- 4Department of Neurosurgery, Jaber Alahmad Hospital, South Surra, Kuwait
- 5Department of Neurosurgery, Heidelberg University Hospital, Heidelberg, Germany
- 6Department of Management, College of Business and Economics, American University of Kuwait, Salmiya, Kuwait
- 7Department of Radiology, Ibn Sina Hospital, Shuwaikh, Kuwait
- 8Molecular Genetics Laboratory, Kuwait Cancer Control Center, Shuwaikh, Kuwait
- 9Department of Neuropathology, Heidelberg University Hospital, Heidelberg, Germany
Background: Prognostication in meningiomas has traditionally relied on histopathological grading, which has inherent limitations, including interobserver variability, intratumoral heterogeneity, and inconsistent correlation with clinical behavior. While molecular profiling enhances diagnostic precision and risk stratification, it is not yet routinely adopted in clinical practice. To date, no molecular data on meningiomas have been published from our country. This study aims to address this gap by characterizing the molecular landscape of meningiomas at our institution, incorporating insights from recent cIMPACT-NOW updates.
Methods: We retrospectively analyzed consecutive 131 meningiomas that underwent molecular sequencing at our institution between 2021 and 2023. Tumors were classified according to the latest WHO criteria. Next-generation sequencing (NGS) was performed using the Oncomine Comprehensive Assay, a targeted panel for solid tumors. Molecular findings were correlated with clinicopathological parameters.
Results: The cohort included 84 females and 47 males (median age: 51 years; range: 2–79). Tumor locations included the cerebral convexity (45.8%), skull base (38.2%), posterior fossa (3.1%), and spine (5.3%), with 7.6% being multifocal. CNS WHO grade 2 tumors were most common (58%), followed by grade 1 (35%) and grade 3 (7%). NF2 alterations (35%) were the most frequent, occurring across all grades but more prevalent in grades 2 and 3. Genotype (p = 0.004) and WHO grade (p = 0.002) were significantly associated with tumor location: NF2 alterations predominated in convexity and spine, while TRAKLS mutations (TRAF7, AKT1, KLF4, SMO) were enriched in lower-grade skull base tumors. High-risk homozygous CDKN2A/B deletions were identified in one grade 3 tumor, with hemizygous deletions, unexpectedly, in three grade 2 tumors.
Conclusion: This study provides regional insight into the molecular landscape of meningiomas in our population. While routine molecular profiling adds value to classification and prognostication, broader implementation may be limited by cost and panel coverage constraints.
1 Introduction
Meningioma is the most common primary intracranial neoplasm in adults, originating from arachnoid cap cells within the leptomeninges (1, 2). Grading of meningioma continues to rely primarily on histological criteria, as defined by the three-tier system in the current World Health Organization guidelines (WHO CNS5, 2021) (3). CNS WHO grade 1 meningiomas are generally slow-growing and associated with favorable outcomes, while CNS WHO grade 2 and 3 tumors demonstrate more aggressive behavior and increased risk of recurrence (4). However, recurrence remains a complex and nuanced challenge, as meningiomas can exhibit inconsistent behavior, even among grade 1 tumors (5, 6). This is further influenced by clinical factors, particularly the extent of surgical resection (7). Given these complexities, the incorporation of relevant molecular parameters that would reliably predict meningioma behavior has become increasingly important. The “Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy-Not Official WHO” (cIMPACT-NOW) has recently clarified the role of molecular markers in meningioma grading, emphasizing their potential clinical relevance (8).
Rapid advances in genomic profiling have uncovered distinct molecular subgroups of meningiomas, which correlate with unique clinical characteristics including tumor location, histological subtype, and WHO grade (9). While NF2 gene inactivation has long been recognized as a key driver of meningioma tumorigenesis (10, 11), more recent studies have identified non-NF2 alterations, particularly those involving the TRAKLS genes (TRAF7, AKT1, KLF4, SMO), as well as other genetic changes not otherwise classified (12, 13). In terms of clinical behavior, a few rare molecular alterations have been linked to aggressive meningioma biology, including oncogenic variants in the TERT promoter region and homozygous deletions of CDKN2A/CDKN2B (14, 15). These are frequently observed alongside NF2 alterations, reflecting a stepwise accumulation of genetic changes driving tumor progression (16–18). However, in the absence of these high-risk mutations, no reliable biomarkers currently exist to predict the risk of recurrence in the more prevalent lower-grade meningiomas. A growing body of literature supports the integration of genomic, epigenomic, and transcriptomic parameters into meningioma classification, offering enhanced predictive value and potential for personalized therapeutic approaches (19, 20). However, the widespread clinical implementation of such molecular tools remains constrained by cost, availability, and accessibility.
To complement histopathological evaluation and gain deeper insight into meningioma-associated molecular events, we implemented routine targeted sequencing for or all meningioma specimens at our laboratory beginning in 2021. In this study, we analyze data from 131 meningiomas diagnosed over the past three years, exploring the mutational landscape and its associations with clinicopathological parameters, with a focus on WHO grading refinements.
2 Materials and methods
2.1 Case selection
Meningioma cases diagnosed between 2021-2023 were retrospectively retrieved from the electronic records of the Department of Pathology at Sabah hospital, a major neuropathology site in Kuwait. Demographic and clinicopathological data were obtained from pathology reports and patient records.
2.2 Radiological and clinical data
Preoperative and postoperative magnetic resonance imaging (MRI) data were available for 105 (80%) and 118 (90%) patients, respectively. Tumor locations on MRI were categorized as follows: cerebral convexity (including falcine/parasagittal regions); skull base (including structures related to the sphenoid bone, ethmoid bone, and petroclival region in the posterior fossa); miscellaneous posterior fossa locations (e.g., cerebellopontine angle, tentorium cerebelli, cerebellar convexity); and spinal cord. Cases with ≥2 meningiomas at different locations were classified as multifocal. Other radiological parameters included tumor size, necrosis, cystic change, peri-tumoral brain edema, and parenchymal/skull invasion. Postoperative MRI was utilized to detect residual disease. Clinical follow-up data were obtained for 81 patients, and recurrence-free survival was calculated from the date of the first surgery to the first recurrence.
2.3 Histopathology review
Slides were re-evaluated by a neuropathologist according to the 2021 WHO Classification of CNS Tumors and the 2024 cIMPACT-NOW Update. Grade 2 was assigned based on any of the following criteria: ≥4 mitoses per 10 high-power fields (HPF), brain invasion, >50% chordoid or clear cell histology, or at least three of the following five morphological features: (i) hypercellularity, (ii) sheeting architecture, (iii) small cell change, (iv) prominent nucleoli, and (v) spontaneous necrosis. Grade 3 was assigned for tumors with ≥20 mitoses per 10 HPF, frank anaplasia, and/or molecular evidence of oncogenic TERT promoter variants or homozygous deletions of CDKN2A/CDKN2B. Immunohistochemical slides were reviewed as needed. Mitoses were manually counted in hotspots across 10 consecutive HPFs using a microscope with an objective lens providing a field area of 0.24 mm². The mitotic counts were adjusted to 1 mm², applying thresholds of ≥2.5 mitoses/mm² and ≥12.5 mitoses/mm² for grade 2 and grade 3 tumors, respectively.
2.4 Targeted next-generation sequencing
All sequencing and bioinformatics analyses were performed in-house at the Molecular Genetics Laboratory of the Kuwait Cancer Control Center. Formalin-fixed, paraffin-embedded (FFPE) tumor tissues were analyzed using next-generation sequencing (NGS) with the Oncomine Comprehensive Assay v3 (OCAv3; Thermo Fisher Scientific, USA) (21). This panel detects single-nucleotide variants, insertions/deletions, copy number variants, and gene fusions across 161 cancer-related genes (Supplementary Table 1), including meningioma-relevant genes listed in Table 1. DNA extraction was performed using the RecoverAll Total Nucleic Acid Isolation Kit, followed by library preparation with the Ion AmpliSeq Library Kit Plus (Thermo Fisher Scientific, USA). DNA concentration was measured using the Qubit 3.0 Fluorometer, which offers fluorometric quantification with high specificity for double-stranded DNA. Extraction from FFPE material was successful in the majority of cases; however, three specimens exhibited excessive nucleic acid degradation and failed downstream library preparation or sequencing quality control metrics, leading to their exclusion from analysis.
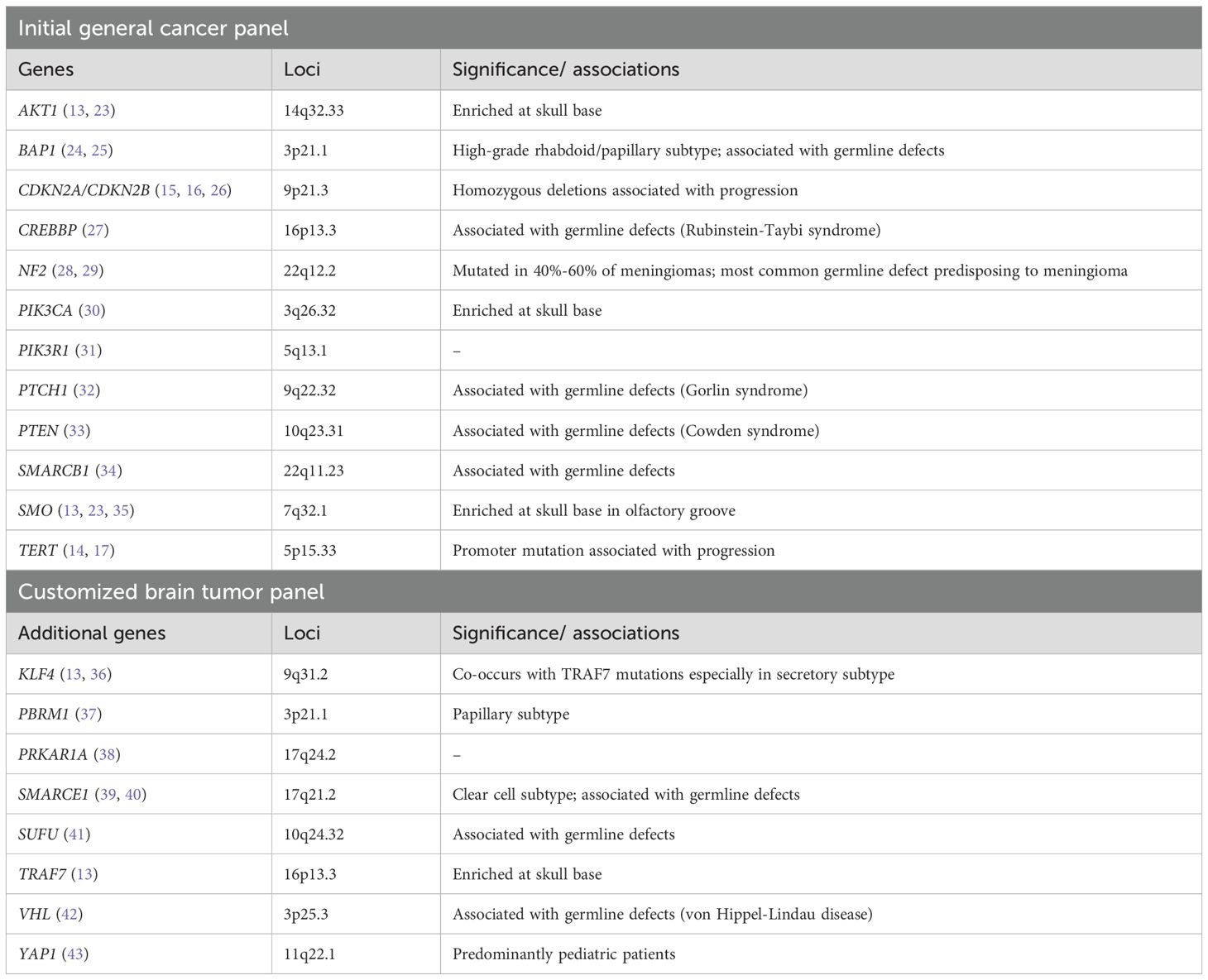
Table 1. Meningioma-relevant genes included in the NGS panels.
Sequencing was conducted on an Ion Torrent S5 XL platform, with data analyzed using the Ion Reporter™ Software (v5.10). Sequence reads were aligned to the human genome assembly GRCh37/hg19, achieving a minimum of 10 million total mapped reads. Variant calling was conducted with a minor allele frequency cutoff of 5% and a mean depth of ≥200x. To ensure sequencing reliability, stringent quality metrics were applied, including base quality scores (Q30 or higher), mapping quality (MQ ≥60), and uniformity of coverage (≥90%). Duplicate reads were filtered out when exceeding a 5% threshold. Variant calling further required a minimum of 20 supporting reads, an allele balance of at least 20%, and strand bias ≤10%. Annotations were obtained using databases such as ClinVar, COSMIC, and dbSNP, with pathogenicity assessed using tools like SIFT and PolyPhen-2. Subsequently, 24 mutation-negative tumors underwent further sequencing using a customized 201-gene NGS panel targeting brain tumor–relevant alterations (PANEL-NPHD2022A) performed on a NextSeq 500 instrument (Illumina) as previously described (22).
2.5 Statistical analysis
Descriptive statistics were used for continuous variables (mean, median, range, and standard deviation), as well as categorical variables (frequencies and graphical representations). Clinicopathological parameters were evaluated using univariate analysis: Pearson’s Chi-squared test for categorical variables and a two-sample independent t-test for continuous variables. A p-value <0.05 was considered statistically significant. The statistical analysis was conducted using JAMOVI Version 2.5.7.0.
3 Results
3.1 Clinicopathological findings
We analyzed 131 surgically resected meningiomas for genetic alterations from 2021 to 2023. Key clinicopathological features are summarized in Table 2. The cohort included 84 females and 47 males (female-to-male ratio ≈ 2:1), with a median age of 51 years (range 2–79) (Figure 1A). Three pediatric patients <18 years were identified. Most meningiomas were intracranial, with the cerebral convexity as the most common location (n=60, 45.8%), followed by skull base (n=50, 38.2%), miscellaneous posterior fossa locations (n=4, 3.1%), and spine (n=7, 5.3%). Ten (7.6%) were multifocal, including two syndromic neurofibromatosis type 2 (NF2) cases.
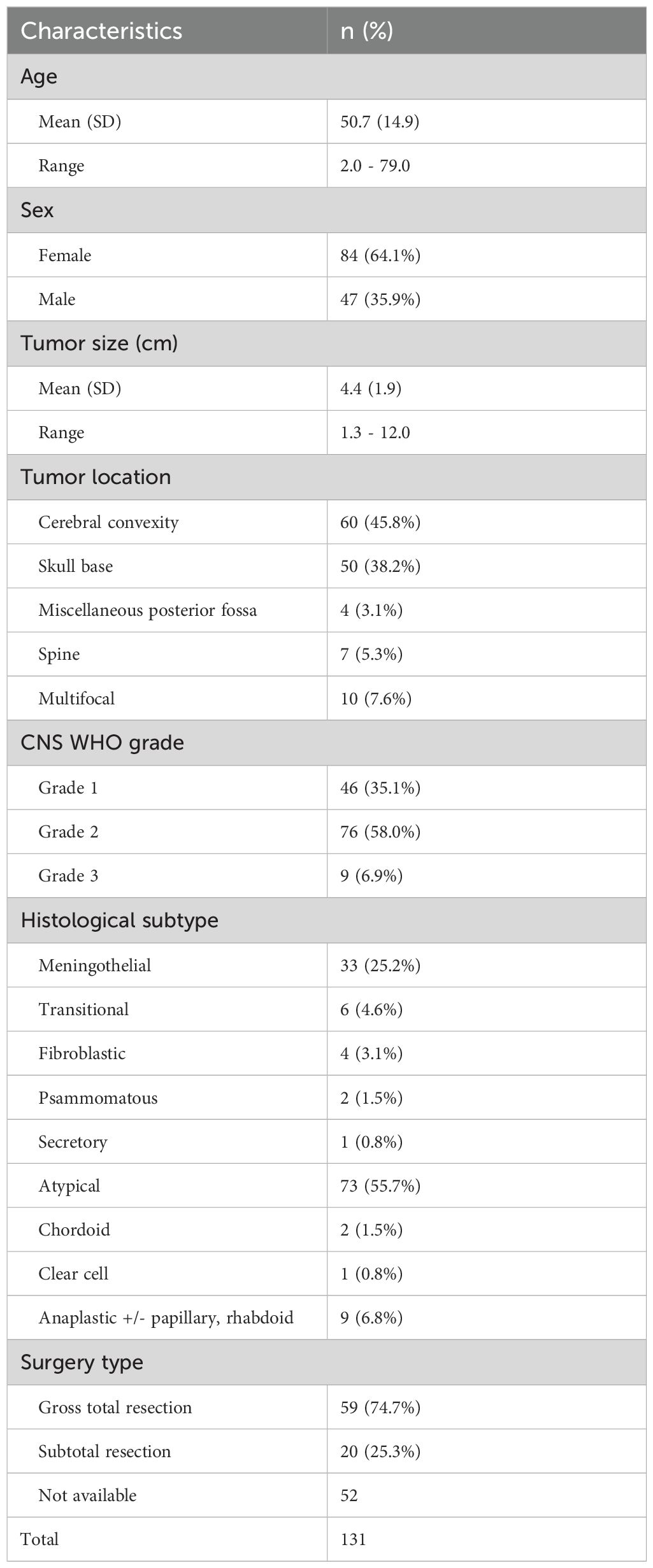
Table 2. Clinicopathological characteristics.
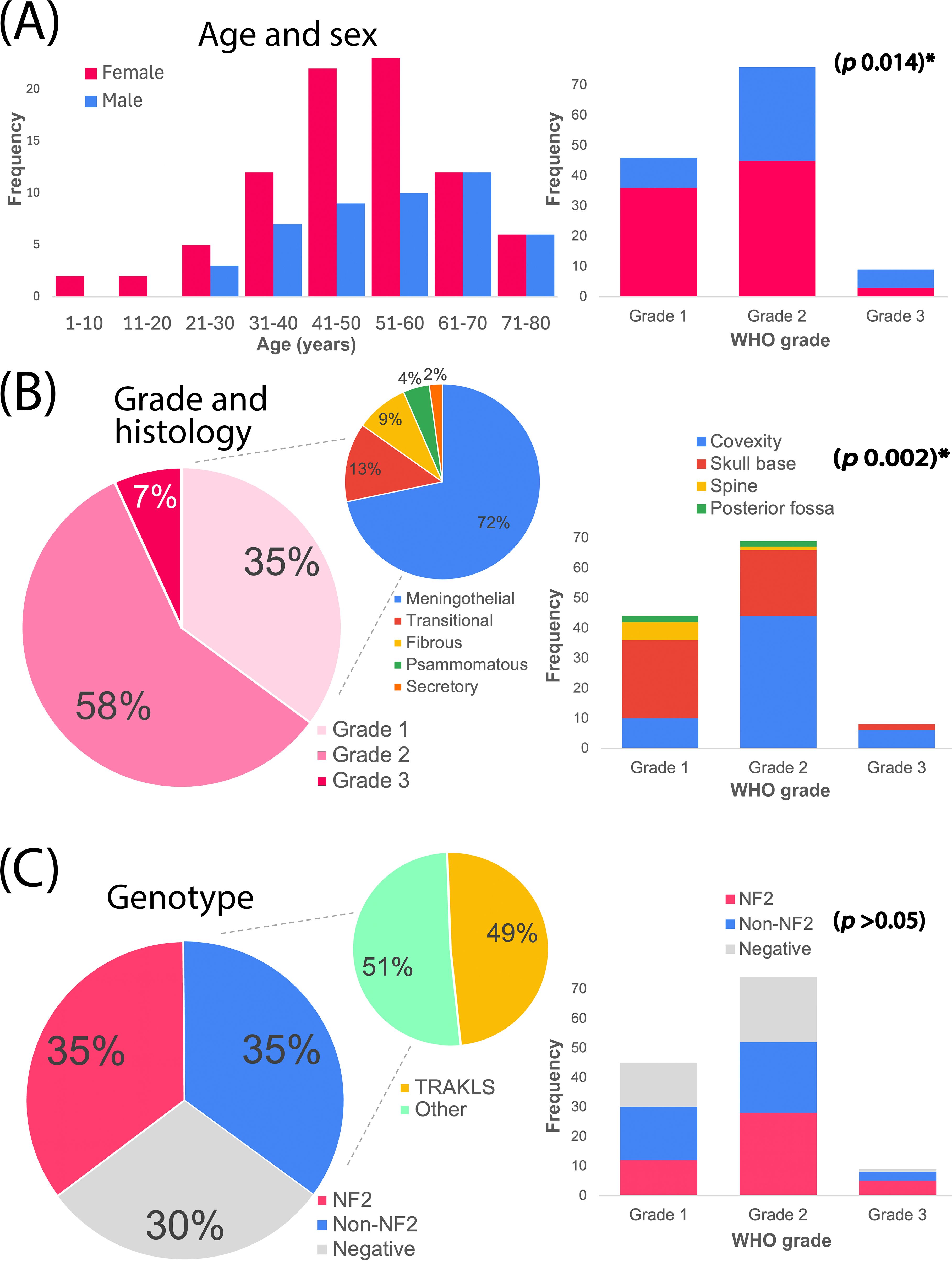
Figure 1. Overall characteristics of the meningioma cohort. (A) Age and sex distribution, alongside sex breakdown by WHO grade. (B) WHO histological grades and subtypes, alongside meningioma locations across grades. (C) Genotype groups, with genotype distribution across grades. TRAKLS= TRAF7, AKT1, KLF4, SMO alterations. *Statistically significant.
WHO histological grades were distributed as follows: 46 (35%) grade 1, 76 (58%) grade 2, and 9 (7%) grade 3 (Figure 1B). Grade 1 tumors included meningothelial (n=33), transitional (n=6), fibroblastic (n=4), psammomatous (n=2), and secretory (n=1) subtypes. Grade 2 tumors (largest group) comprised atypical meningiomas (n=73), chordoid (n=2), and clear-cell (n=1) variants. Grade 3 (anaplastic) meningiomas (n=9) included 3 cases with rhabdoid/papillary features.
Grade 2 was assigned based on morphological/brain invasion criteria in 37%, morphology alone in 26%, mitotic/morphological criteria in 19%, and all three criteria in 13% of tumors. Additionally, 4% were clear-cell or chordoid variants, and 1% exhibited isolated brain invasion (brain-invasive otherwise benign; BIOB). Grade 3 tumors were classified based on anaplasia +/- mitotic criteria (56%) or mitotic criteria alone (44%). The grades were determined following the conversion of mitotic counts from 10 high-powered fields (HPFs) to 1 mm², using mitotic cutoffs of ≥2.5/mm² and ≥12.5/mm² for grades 2 and 3, respectively. This adjustment changed the mitotic category in 31 cases, ultimately leading to the downgrading of 3 tumors to grade 1 and 7 tumors to grade 2 (Figure 2).
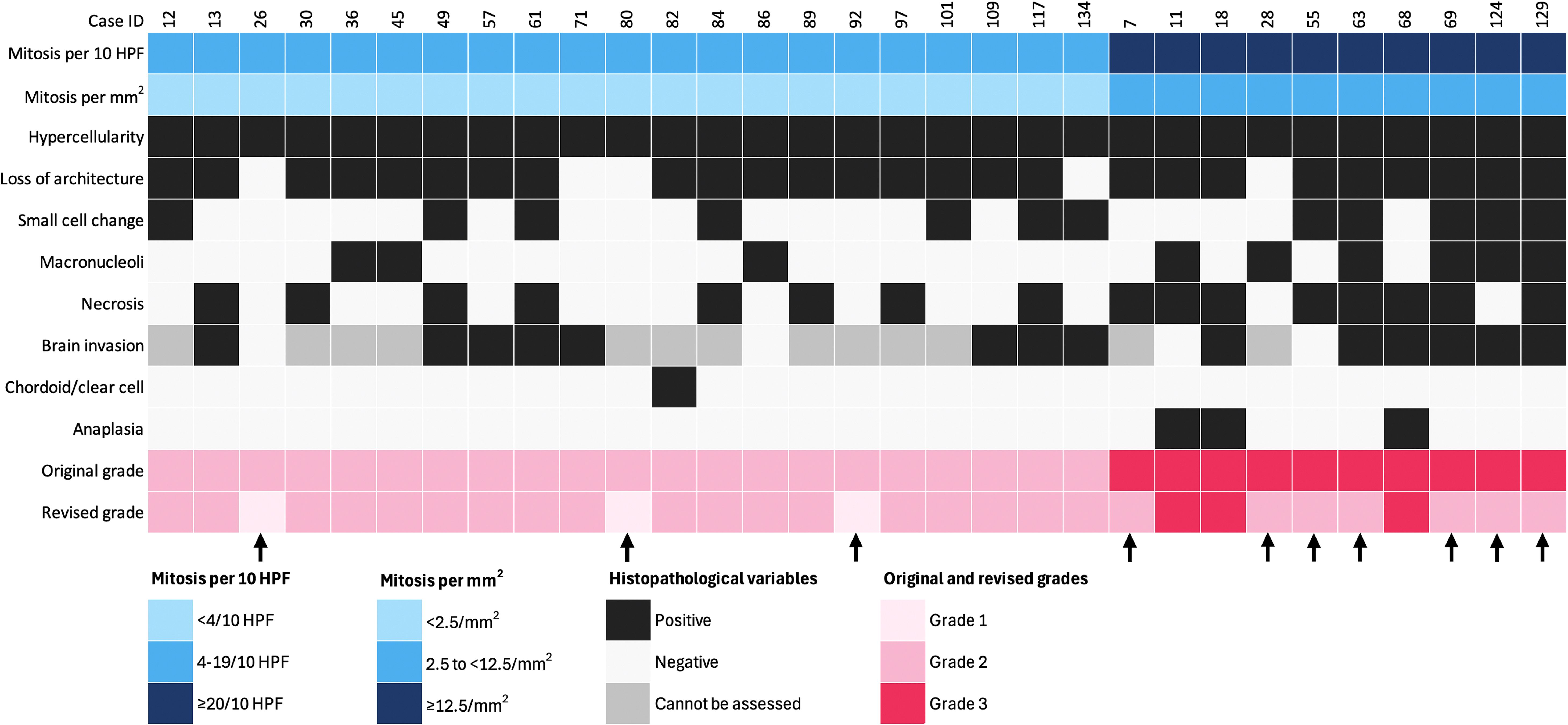
Figure 2. Impact of mitotic count adjustment on tumor grade in 31 cases (1 HPF = 0.24 mm2). Arrows indicate downgraded cases. Each column represents an individual patient, and each row corresponds to a specific variable.
3.2 Mutation analysis
Of the 128 successfully sequenced tumors, 115 (90%) were newly diagnosed (de novo), and 13 (10%) were recurrent. Genetic alterations were detected in 90 samples (70%), including 45 tumors (35%) with NF2 alterations, 45 (35%) with non-NF2 alterations, and 38 (30%) with no detectable alterations (Figure 1C). The annotated genetic variants and their allelic frequencies are provided in Supplementary Table 2.
NF2 alterations (n=45) were diverse, including frameshift deletions (n=13), in-frame deletion (n=1), nonsense mutations (n=15), frameshift insertions (n=6), splice site mutations (n=8), missense mutation (n=1), and heterozygous 22q12.2 loss (n=1) (Figure 3). The two syndromic NF2 tumors harbored a splice site mutation and a heterozygous 22q12.2 loss, respectively.
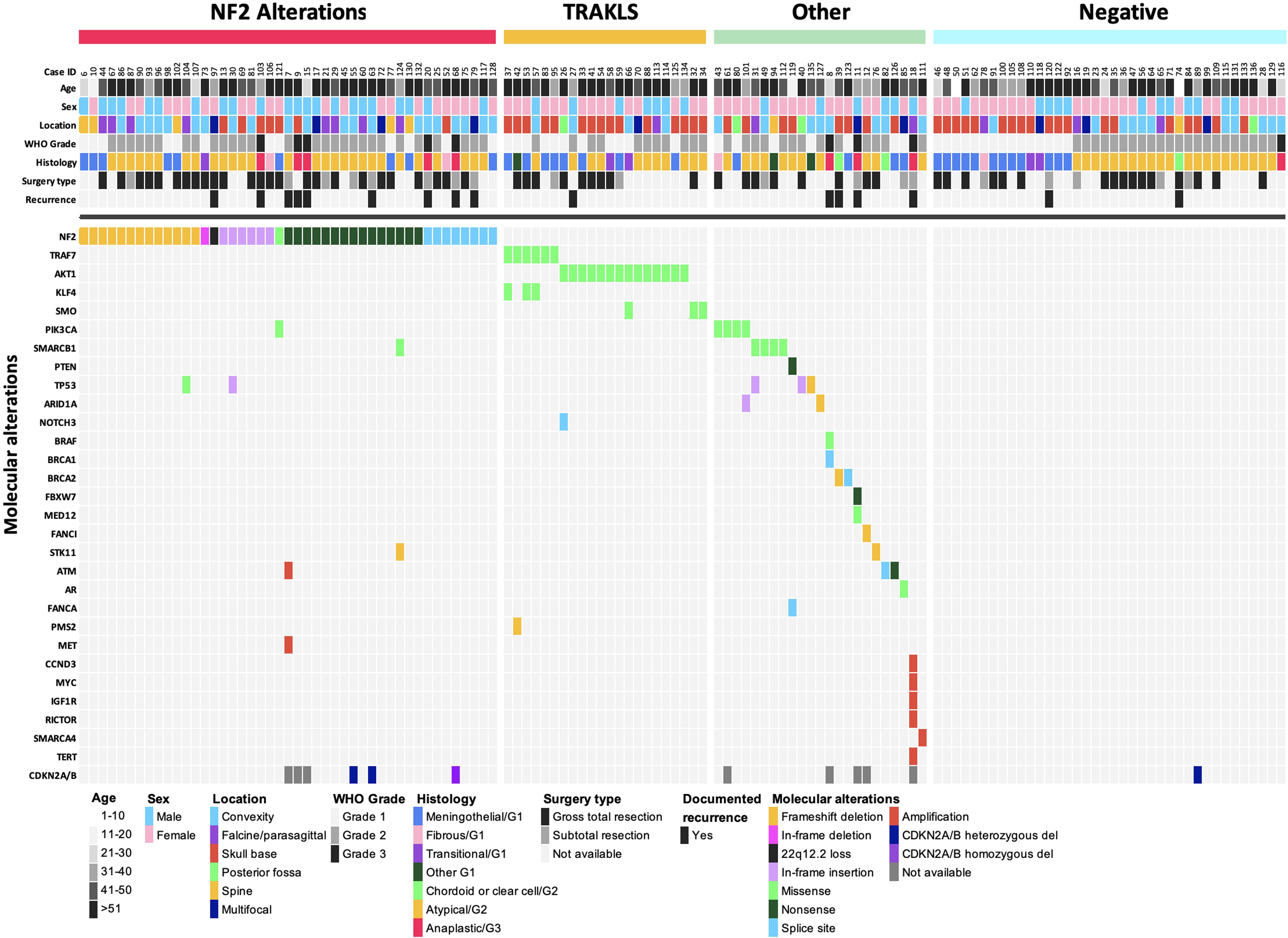
Figure 3. Oncoplot summary (n = 128). Each column represents an individual patient, and each row corresponds to a specific variable.
In the non-NF2 subgroup (n=45), a total 26 TRAKLS mutations were identified across 22 tumors, which were mutually exclusive of NF2 mutations. These included missense mutations in TRAF7 (n=6), AKT1 (n=14), KLF4 (n=3) and SMO (n=3). Recurrent variants were as follows: AKT1 p.E17K (n=12), KLF4 p.K409Q (n=3), and SMO p.L412F (n=2). TRAF7 mutations exhibited variability, often co-occurring with KLF4 p.K409Q. Other non-NF2 alterations were heterogenous involving SMARCB1, PIK3CA, TP53, and various other genes (Supplementary Table 2). NF2 co-mutations were observed with TP53 (n=2), PIK3CA (n=1), and SMARCB1/STK11 (n=1).
3.3 Clinicopathological and genotype correlations
Genotype correlated with tumor location (p = 0.004). NF2 alterations predominated in cerebral convexity (29/58; 50%) and spinal tumors (5/7; 71%), while TRAKLS mutations were enriched in skull base tumors (15/22; 68%) (Figure 4). Tumor size was significantly larger in the NF2 subgroup (mean 4.9 cm; p = 0.013). Qualitative radiological features (e.g., necrosis, edema, peritumoral brain edema, parenchymal/skull invasion) did not differ among genotypes.
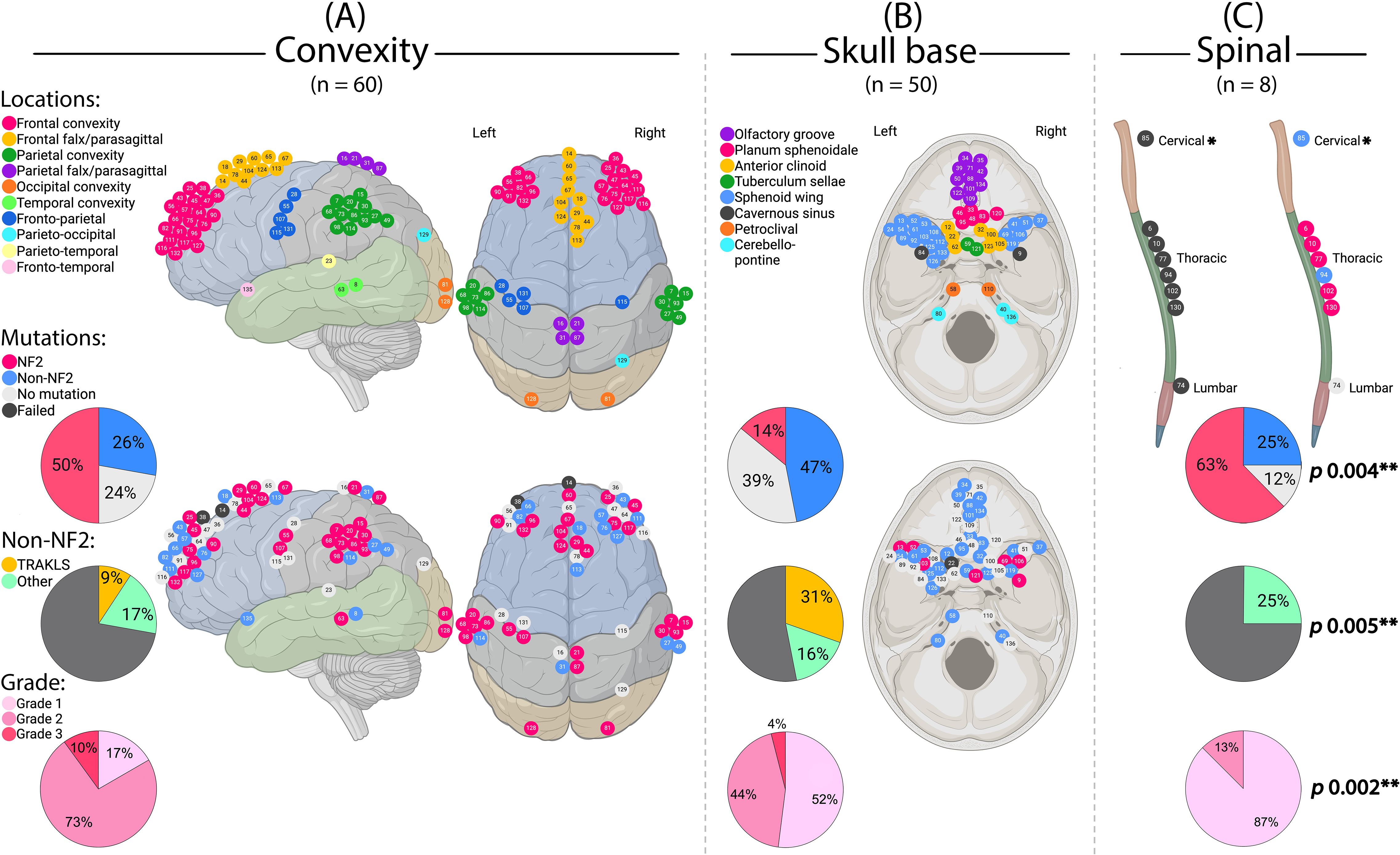
Figure 4. Meningioma location in relation to genotype and grade. (A) Cerebral convexity, sagittal and axial views. (B) Skull base, axial view. (C) Spinal cord. *This case also had another meningioma of the cavernous sinus. **Statistically significant. (Created by RHA with BioRender.com).
WHO grade also correlated with location (p = 0.002), with grade 1 tumors predominantly found at the skull base and higher grades at the convexity (Figure 4). Grade 3 tumors were more common in males (p = 0.014) and associated with necrosis (p = 0.002), cystic changes (p = 0.004), and perilesional edema (p = 0.002) on imaging.
NF2 alterations were more frequent in WHO grade 2 and 3 tumors (grade 1: 26%; grade 2: 37%; grade 3: 56%) (Figure 5). When combined, grade 2 and 3 tumors comprised 73% of the NF2 subgroup. In contrast, the TRAKLS subgroup included only grade 1 (50%) and grade 2 (50%) tumors, with no grade 3 cases. CDKN2A/B homozygous loss was found in one grade 3 NF2-mutated tumor, while hemizygous loss was detected in three grade 2 cases. No TERT promoter mutations were identified.
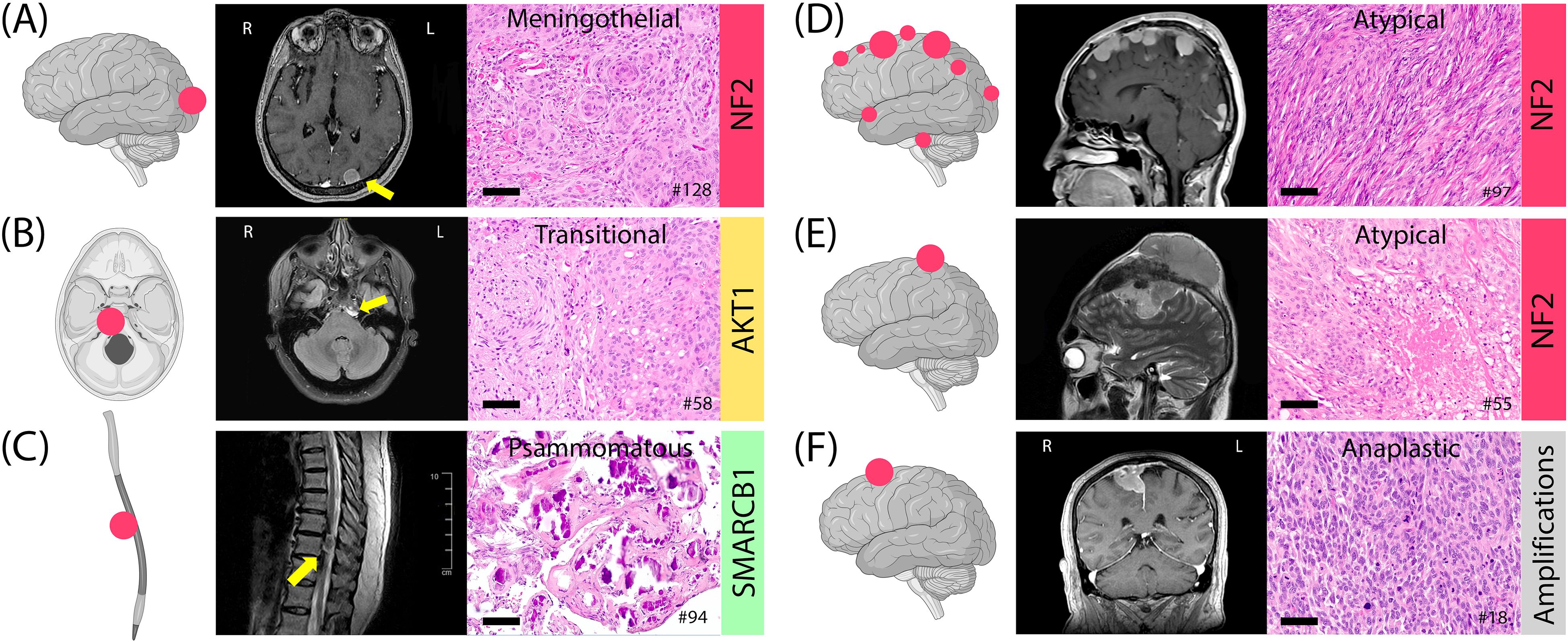
Figure 5. Examples of meningiomas with location, histology, MRI, and genotype. (A) Meningothelial meningioma, occipital convexity, grade 1. (B) Transitional meningioma, petroclival, grade 1. (C) Psammomatous meningioma, thoracic spine, grade 1. (D) Multifocal atypical meningiomas in NF2 syndrome with a heterozygous 22q12.2 loss, grade 2. (E) A 12-cm atypical meningioma, fronto-parietal convexity, with intra- and extra-axial components, grade 2. (F) Anaplastic meningioma with sarcoma-like histology, frontal parasagittal, grade 3. Histology images at 20x magnification; scale bars= 100µm.
Brain invasion was observed in 45 of 66 specimens containing brain parenchyma (40 grade 2 tumors and 5 grade 3 tumors). No molecular correlation with invasion was identified. One NF2-mutated BIOB tumor showed isolated brain invasion without mitotic activity or atypical morphological criteria.
Three pediatric cases were included: a spinal clear-cell meningioma (grade 2), an olfactory groove meningothelial meningioma (grade 1), and a sphenoid wing atypical meningioma (grade 2) with PTEN and FANCA pathogenic mutations (Figure 6). The latter case followed prior radiation therapy for medulloblastoma. The other two cases did not show detectable genetic alterations using the assay employed.
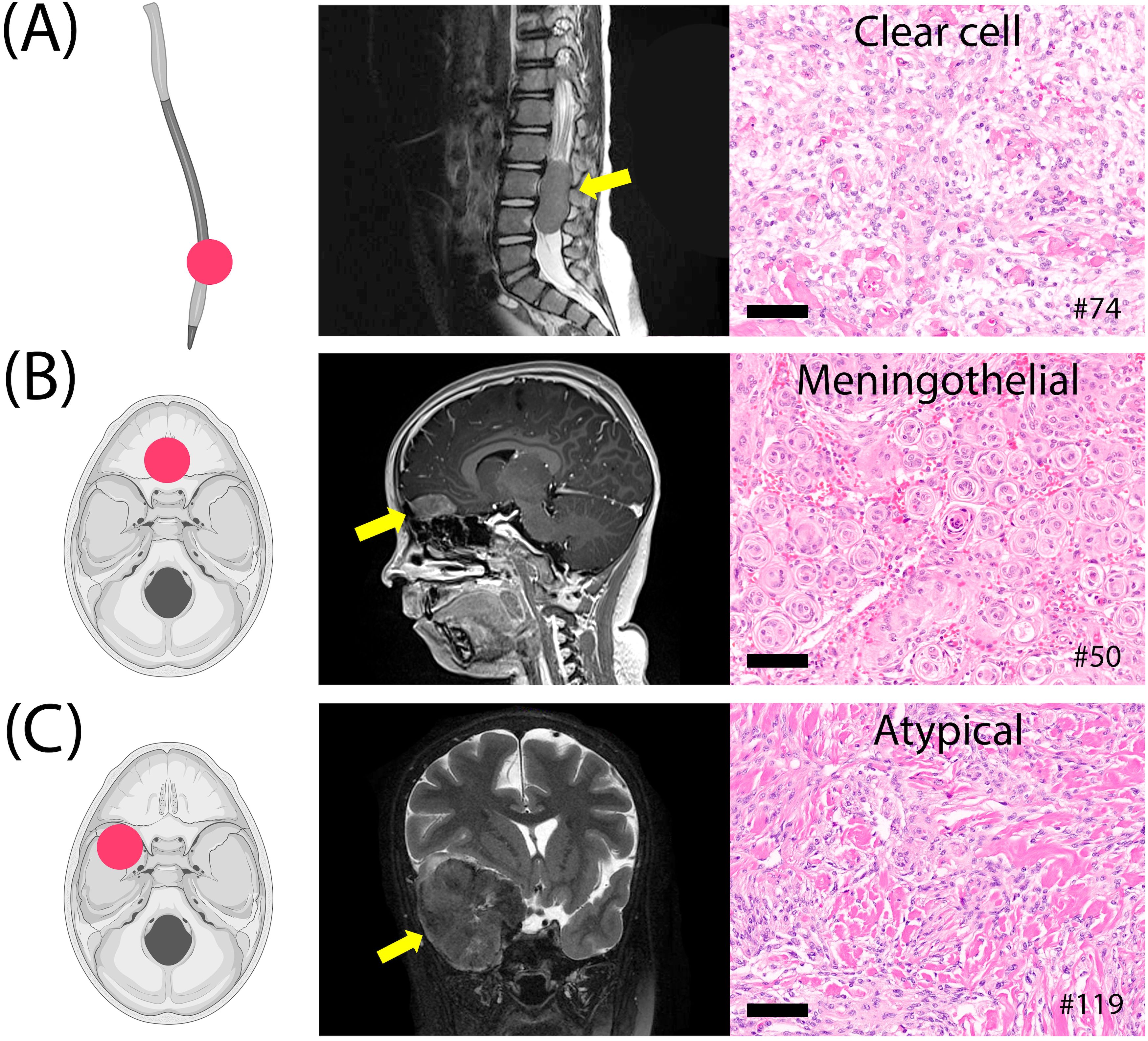
Figure 6. Three pediatric meningiomas. (A) Clear cell meningioma of the lumbar spine, grade 2. (B) Meningothelial meningioma of the olfactory groove, grade 1. (C) Atypical fibroblastic meningioma of the sphenoid wing, grade 2, post-radiation therapy. Histology images at 20x magnification; scale bars= 100µm.
Post-operative imaging (n=118) revealed residual tumors in 37 cases (31.4%), with significantly higher rates in subtotal resection (STR; 90%) compared to gross total resection (GTR; 17%) (p < 0.001). Residual disease was most common in grade 3 tumors (67%) but did not correlate with genotype. GTR was more frequently achieved in convexity and spinal tumors.
Clinical follow-up in 81 patients (median: 2.75 years) revealed recurrences in 16 cases (20%), significantly associated with WHO grade (p < 0.001): 89% in grade 3, 9% in grade 2, and 2% in grade 1 tumors. Among the recurrent cases, recurrence-free intervals ranged from 7.6 to 149.5 months (median: 46.6 months). Due to inconsistent follow-up documentation across the broader cohort, formal recurrence-free survival (RFS) analysis could not be performed. Recurrence rates did not differ significantly between genotype groups (p = 0.245), though the TRAKLS subgroup exhibited the lowest recurrence rate (4.5%). Syndromic NF2 patients experienced multiple recurrences requiring surgical reinterventions and gamma knife therapy. Among the 16 recurrent cases, 5 had undergone GTR, including 3 cases with no visible postoperative residual disease. All recurrent cases with GTR were grade 2 or 3 tumors. Tumor location showed no association with recurrence.
4 Discussion
Meningioma diagnostics has traditionally relied on histological criteria as the primary means of predicting outcomes, with the incorporation of molecular parameters being a more recent development. In this study, we present our analysis of 131 molecularly sequenced meningiomas, exploring the mutational profiles of the cohort and their associations with histopathological and clinical parameters. To our knowledge, this is the first comprehensive molecular profiling study of meningiomas conducted in Kuwait and one of the very few from the Middle East. While the genomic alterations identified align with previously reported patterns, the study offers significant regional value and contributes data from an underrepresented population. Notably, the median age of our cohort was 51 years, which is younger than the global median age of 66 years reported in the WHO Classification of CNS Tumours (3); whether this reflects demographic differences remains to be explored. In the context of evolving molecular classification frameworks, documenting institutional experiences, particularly from resource-limited settings, remains important for ensuring diagnostic consistency and guiding future research.
As anticipated, NF2 alterations represented the most frequent genetic abnormality. These were notably associated with convexity and spinal meningiomas, occurring across all grades but with enrichment in higher-grade tumors. While NF2 alterations are not direct predictors of prognosis or strictly correlated with WHO grading, they are linked to a more aggressive biological phenotype (11, 44). The NF2 mutational spectrum in our cohort was broad, with nonsense mutations and frameshift deletions being the most common, as described elsewhere (45). In contrast, TRAKLS alterations were more prevalent in lower-grade tumors at the skull base, in line with prior findings (46). The relatively high frequency of mutation-negative cases in our study (30%) can be attributed to the limited coverage of the NGS assay used, which did not capture several known meningioma-related alterations. Notably, a customized brain tumor–specific panel improved detection by 42% among 24 mutation-negative cases that were further tested. Histological subtype–specific mutations, such as KLF4/TRAF7 in secretory meningiomas (47, 48) and SMARCE1 in clear cell meningiomas (39, 40), were also not supported by our assay.
Homozygous deletions of CDKN2A/B and oncogenic TERT promoter mutations are well-established independent adverse prognostic factors and defining features of CNS WHO grade 3 meningiomas. In this study, homozygous CDKN2A/B loss was observed in a single grade 3 meningioma, while all histologically low-grade tumors retained intact CDKN2A/B and TERT loci. Interestingly, three grade 2 meningiomas displayed hemizygous CDKN2A/B loss, a finding that has been linked to poor outcomes in some studies (18, 49). Oncogenic TERT promoter variants, though rare, have been reported in lower-grade meningiomas (14). A recent study also identified significant discrepancies between histological grades and molecular profiles, highlighting the critical role of molecular screening in refining meningioma classification and prognostication (50). These findings reinforce the value of integrating routine molecular profiling to complement traditional histological grading and improve risk stratification. However, widespread adoption of routine meningioma genotyping in clinical practice remains constrained by cost-effectiveness concerns.
Three pediatric meningiomas were identified in this cohort: a grade 2 clear-cell meningioma in the spinal region, a grade 1 tumor in the olfactory groove with nasal extension, and a grade 2 sphenoid wing tumor that developed years after radiation therapy for medulloblastoma. Pediatric meningiomas are rare, comprising about 1% of all meningiomas, and have distinct clinicopathological and genetic features compared to adults (51). These include a lack of female preponderance (52), higher incidence of spinal and intraventricular localizations (52, 53), higher grade histopathological features (54, 55), and more aggressive histological subtypes such as clear-cell meningioma (56). While NF2 alterations are frequent in pediatric meningiomas, with a higher frequency of underlying NF2 syndrome (54), adult-type non-NF2 alterations are typically absent (57). Alternative alterations, such as those in YAP1, have been described (43, 58). DNA-methylation profiling of pediatric cases also differs from adults (57). No NF2 alterations were found in our pediatric cases, and we could not assess SMARCE1 mutations in the clear-cell meningioma due to limited NGS panel coverage. The post-radiation case showed mutations in PTEN and FANCA, though the significance of these remains uncertain (59).
Our cohort demonstrated a higher frequency of grade 2 meningiomas (58%) compared to grade 1 (35%), which exceeds what is typically reported in the literature (60). This discrepancy likely reflects cumulative factors beyond mitotic count thresholds or brain invasion alone. While we adopted recommended practices—such as full tissue processing and adjusting mitotic counts from 10 HPFs to mm²—we also observed a high frequency of subjective morphological features that contribute to grade 2 designation: hypercellularity in 84% (110/131), loss of architecture in 67% (88/131), necrosis in 37% (49/131), small cell change in 32% (42/131), and prominent nucleoli in 27% (36/131). Grade 2 was assigned based on combined morphological and brain invasion criteria in 37% of cases, morphology alone in 26%, morphology plus mitotic counts in 19%, and all three criteria in 13%. These findings may suggest a tendency toward overinterpretation of soft histological features which are inherently subjective. Nonetheless, we view this as a valuable institutional audit—offering insight into how WHO grading criteria are implemented in real-world diagnostic environments. While large-scale studies establish reference standards, single-institution experiences like ours remain essential for highlighting diagnostic variability, informing local quality assurance, and encouraging recalibration of histologic thresholds, particularly in settings where pathology remains the cornerstone of diagnosis in the absence of high-throughput molecular tools.
To mitigate potential overestimation, mitotic counts in our study—initially performed over 10 high-power fields (HPFs) using a 0.24 mm² field area—were standardized by converting to mitoses per mm², in line with recent cIMPACT-NOW recommendations (8) (61). This adjustment led to the downgrading of 10 cases: three from grade 2 to grade 1, and seven from grade 3 to grade 2. While such standardization is intended to improve grading reproducibility, it remains inconsistently applied across institutions and is not yet universally regulated. Whether these grading shifts reflect underlying biological behavior or merely technical variability remains uncertain and highlights the ongoing need for harmonized diagnostic criteria.
This study has several limitations. First, the cohort was heterogeneous, encompassing a broad range of age groups, anatomical locations (cranial convexity, skull base, spinal), and disease stages (both de novo and recurrent). Nonetheless, this heterogeneity was necessary to capture a comprehensive overview of the population’s characteristics. Second, although the commercial NGS panel provided broad oncologic coverage, it lacked certain meningioma-specific targets and did not assess large-scale chromosomal copy number alterations such as 1p and 22q loss, thereby limiting refined subclassification in some borderline tumors. Regarding clinical outcomes, the retrospective design and incomplete documentation prevented robust analysis of survival or progression-free intervals. This limitation was further compounded by structural challenges specific to our healthcare setting, including inconsistent follow-up, particularly among expatriate patients and patients who seek treatment abroad. Taken together, these constraints reflect the practical diagnostic realities faced by many institutions, particularly in settings where access to customized gene panels or high-throughput molecular tools remains limited.
5 Conclusion
Despite these limitations, our study presents a relatively large, well-characterized cohort and provides the first comprehensive molecular dataset on meningiomas from Kuwait. The findings are consistent with established genomic patterns while offering region-specific insights from an underrepresented population. Genotype and WHO histologic grade showed correlation with tumor location, while recurrence rates were more closely associated with grade and the extent of surgical resection, with no significant differences observed across molecular subtypes. Furthermore, our results highlight key limitations in conventional histopathologic assessment—particularly regarding mitotic thresholds and brain invasion criteria—underscoring the need for continued refinement of diagnostic and prognostic frameworks. We believe this work supports the broader implementation of molecular profiling in neuropathology and serves as a reference point for future investigations in similarly resource-limited contexts.
Data availability statement
The data presented in the study are deposited in the Figshare repository, at: https://figshare.com/articles/dataset/_b_Clinicopathological_and_Molecular_Data_of_131_Meningiomas_2021_2023_Kuwait_b_/29980738?file=57408235, under the DOI: 10.6084/m9.figshare.29980738.
Ethics statement
The studies involving humans were approved by Ethics Committee for Medical Research at the Ministry of Health, Kuwait (Ref: 1090, Study #2024/2585, dated July 10, 2024) and the Health Sciences Center Ethical Committee (Ref: 756, dated July 14, 2024). The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from Archival histopathology samples. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
RA: Methodology, Data curation, Conceptualization, Writing – review & editing, Supervision, Visualization, Writing – original draft, Project administration. AH: Writing – review & editing, Data curation. HHJ: Data curation, Writing – review & editing. AbA: Writing – review & editing, Data curation. MoA: Writing – review & editing, Data curation. OA: Resources, Writing – review & editing. AhA: Software, Formal analysis, Writing – review & editing. NA: Writing – review & editing, Data curation. EA: Writing – review & editing, Data curation. EM: Writing – review & editing, Resources, Investigation. HJ: Resources, Writing – review & editing, Investigation. AmA: Resources, Writing – review & editing, Investigation. ZA: Resources, Writing – review & editing, Investigation. AAA: Formal analysis, Writing – review & editing. SB: Writing – review & editing, Formal analysis. SH: Resources, Writing – review & editing. FS: Resources, Writing – review & editing. MaA: Writing – review & editing, Formal analysis, Supervision.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We are thankful to all contributors in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1648953/full#supplementary-material
References
1. Longstreth WT Jr, Dennis LK, McGuire VM, Drangsholt MT, and Koepsell TD. Epidemiology of intracranial meningioma. Cancer. (1993) 72:639–48. doi: 10.1002/1097-0142(19930801)72:3<639::aid-cncr2820720304>3.0.co;2-p
2. Whittle IR, Smith C, Navoo P, and Collie D. Meningiomas. Lancet. (2004) 363:1535–43. doi: 10.1016/S0140-6736(04)16153-9
3. Brat DJ, Ellison DW, Figarella-Branger D, Hawkins CE, Louis DN, Ng H, et al. WHO classification of tumours series. In: Central nervous system tumours, 5th ed, vol. 6. International Agency for Research on Cancer, Lyon (France. Available online at: https://tumourclassification.iarc.who.int/chapters/45.
4. Sughrue ME, Sanai N, Shangari G, Parsa AT, Berger MS, and McDermott MW. Outcome and survival following primary and repeat surgery for World Health Organization Grade III meningiomas. J Neurosurg. (2010) 113:202–9. doi: 10.3171/2010.1.JNS091114
5. Marciscano AE, Stemmer-Rachamimov AO, Niemierko A, Larvie M, Curry WT, Barker FG 2nd, et al. Benign meningiomas (WHO Grade I) with atypical histological features: correlation of histopathological features with clinical outcomes. J Neurosurg. (2016) 124:106–14. doi: 10.3171/2015.1.JNS142228
6. Lemée JM, Joswig H, Da Broi M, Corniola MV, Scheie D, Schaller K, et al. WHO grade I meningiomas: classification-tree for prognostic factors of survival. Neurosurg Rev. (2020) 43:749–58. doi: 10.1007/s10143-019-01117-0
7. Aizer AA, Bi WL, Kandola MS, Lee EQ, Nayak L, Rinne ML, et al. Extent of resection and overall survival for patients with atypical and Malignant meningioma. Cancer. (2015) 121:4376–81. doi: 10.1002/cncr.29639
8. Sahm F, Aldape KD, Brastianos PK, Brat DJ, Dahiya S, von Deimling A, et al. cIMPACT-NOW Update 8: Clarifications on molecular risk parameters and recommendations for WHO grading of meningiomas. Neuro Oncol. (2024) 27:319–30. doi: 10.1093/neuonc/noae170
9. Youngblood MW, Duran D, Montejo JD, Li C, Omay SB, Özduman K, et al. Correlations between genomic subgroup and clinical features in a cohort of more than 3000 meningiomas. J Neurosurg. (2020) 133:1345–54. doi: 10.3171/2019.8.JNS191266
10. Gutmann DH, Giordano MJ, Fishback AS, and Guha A. Loss of merlin expression in sporadic meningiomas, ependymomas and schwannomas. Neurology. (1997) 49:267–70. doi: 10.1212/wnl.49.1.267
11. Goutagny S, Yang HW, Zucman-Rossi J, Chan J, Dreyfuss JM, Park PJ, et al. Genomic profiling reveals alternative genetic pathways of meningioma Malignant progression dependent on the underlying NF2 status. Clin Cancer Res. (2010) 16:4155–64. doi: 10.1158/1078-0432.CCR-10-0891
12. Yuzawa S, Nishihara H, Yamaguchi S, Mohri H, Wang L, Kimura T, et al. Clinical impact of targeted amplicon sequencing for meningioma as a practical clinical-sequencing system. Mod Pathol. (2016) 29:708–16. doi: 10.1038/modpathol.2016.81
13. Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. (2013) 339:1077–80. doi: 10.1126/science.1233009
14. Sahm F, Schrimpf D, Olar A, Koelsche C, Reuss D, Bissel J, et al. TERT promoter mutations and risk of recurrence in meningioma. J Natl Cancer Inst. (2015) 108:djv377. doi: 10.1093/jnci/djv377
15. Boström J, Meyer-Puttlitz B, Wolter M, Blaschke B, Weber RG, Lichter P, et al. Alterations of the tumor suppressor genes CDKN2A (p16(INK4a)), p14(ARF), CDKN2B (p15(INK4b)), and CDKN2C (p18(INK4c)) in atypical and anaplastic meningiomas. Am J Pathol. (2001) 159:661–9. doi: 10.1016/S0002-9440(10)61737-3
16. Simon M, Park TW, Köster G, Mahlberg R, Hackenbroch M, Boström J, et al. Alterations of INK4a(p16-p14ARF)/INK4b(p15) expression and telomerase activation in meningioma progression. J Neurooncol. (2001) 55:149–58. doi: 10.1023/a:1013863630293
17. Goutagny S, Nault JC, Mallet M, Henin D, Rossi JZ, and Kalamarides M. High incidence of activating TERT promoter mutations in meningiomas undergoing Malignant progression. Brain Pathol. (2014) 24:184–9. doi: 10.1111/bpa.12110
18. Wach J, Basaran AE, Arlt F, Vychopen M, Seidel C, Barrantes-Freer A, et al. CDKN2A/B deletions are strongly associated with meningioma progression: a meta-analysis of individual patient data. Acta Neuropathol Commun. (2023) 11:189. doi: 10.1186/s40478-023-01690-y
19. Nassiri F, Liu J, Patil V, Mamatjan Y, Wang JZ, Hugh-White R, et al. A clinically applicable integrative molecular classification of meningiomas. Nature. (2021) 597:119–25. doi: 10.1038/s41586-021-03850-3
20. Maas SLN, Stichel D, Hielscher T, Sievers P, Berghoff AS, Schrimpf D, et al. Integrated molecular-morphologic meningioma classification: a multicenter retrospective analysis, retrospectively and prospectively validated. J Clin Oncol. (2021) 39:3839–52. doi: 10.1200/JCO.21.00784
21. Vestergaard LK, Oliveira DNP, Poulsen TS, Høgdall CK, and Høgdall EV. Oncomine Comprehensive Assay v3 vs. Oncomine Comprehensive Assay Plus. Cancers (Basel). (2021) 13:5230. doi: 10.3390/cancers13205230
22. Sahm F, Schrimpf D, Jones DT, Meyer J, Kratz A, Reuss D, et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol. (2016) 131:903–10. doi: 10.1007/s00401-015-1519-8
23. Brastianos PK, Horowitz PM, Santagata S, Jones RT, McKenna A, Getz G, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet. (2013) 45:285–9. doi: 10.1038/ng.2526
24. Shankar GM, Abedalthagafi M, Vaubel RA, Merrill PH, Nayyar N, Gill CM, et al. Germline and somatic BAP1 mutations in high-grade rhabdoid meningiomas. Neuro Oncol. (2017) 19:535–45. doi: 10.1093/neuonc/now235
25. Prasad RN, Gardner UG, Yaney A, Prevedello DM, Koboldt DC, Thomas DL, et al. Germline BAP1 mutation in a family with multi-generational meningioma with rhabdoid features: a case series and literature review. Front Oncol. (2021) 11:721712. doi: 10.3389/fonc.2021.721712
26. Sievers P, Hielscher T, Schrimpf D, Stichel D, Reuss DE, Berghoff AS, et al. CDKN2A/B homozygous deletion is associated with early recurrence in meningiomas. Acta Neuropathol. (2020) 140:409–13. doi: 10.1007/s00401-020-02188-w
27. Verstegen MJ, van den Munckhof P, Troost D, and Bouma GJ. Multiple meningiomas in a patient with Rubinstein-Taybi syndrome. Case report. J Neurosurg. (2005) 102:167–8. doi: 10.3171/jns.2005.102.1.0167
28. Kalamarides M, Niwa-Kawakita M, Leblois H, Abramowski V, Perricaudet M, Janin A, et al. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev. (2002) 16:1060–5. doi: 10.1101/gad.226302
29. Evans DG, Watson C, King A, Wallace AJ, and Baser ME. Multiple meningiomas: differential involvement of the NF2 gene in children and adults. J Med Genet. (2005) 42:45–8. doi: 10.1136/jmg.2004.023705
30. Abedalthagafi M, Bi WL, Aizer AA, Merrill PH, Brewster R, Agarwalla PK, et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro Oncol. (2016) 18:649–55. doi: 10.1093/neuonc/nov316
31. Dai J, Ma Y, Chu S, Le N, Cao J, and Wang Y. Identification of key genes and pathways in meningioma by bioinformatics analysis. Oncol Lett. (2018) 15:8245–52. doi: 10.3892/ol.2018.8376
32. Kijima C, Miyashita T, Suzuki M, Oka H, and Fujii K. Two cases of nevoid basal cell carcinoma syndrome associated with meningioma caused by a PTCH1 or SUFU germline mutation. Fam Cancer. (2012) 11:565–70. doi: 10.1007/s10689-012-9548-0
33. Yakubov E, Ghoochani A, Buslei R, Buchfelder M, Eyüpoglu IY, and Savaskan N. Hidden association of Cowden syndrome, PTEN mutation and meningioma frequency. Oncoscience. (2016) 3:149–55. doi: 10.18632/oncoscience.305
34. Christiaans I, Kenter SB, Brink HC, van Os TA, Baas F, van den Munckhof P, et al. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J Med Genet. (2011) 48:93–7. doi: 10.1136/jmg.2010.082420
35. Boetto J, Bielle F, Sanson M, Peyre M, and Kalamarides M. SMO mutation status defines a distinct and frequent molecular subgroup in olfactory groove meningiomas. Neuro Oncol. (2017) 19:345–51. doi: 10.1093/neuonc/now276
36. Tang H, Zhu H, Wang X, Hua L, Li J, Xie Q, et al. KLF4 is a tumor suppressor in anaplastic meningioma stem-like cells and human meningiomas. J Mol Cell Biol. (2017) 9:315–24. doi: 10.1093/jmcb/mjx023
37. Williams EA, Wakimoto H, Shankar GM, Barker FG 2nd, Brastianos PK, Santagata S, et al. Frequent inactivating mutations of the PBAF complex gene PBRM1 in meningioma with papillary features. Acta Neuropathol. (2020) 140:89–93. doi: 10.1007/s00401-020-02161-7
38. Clark VE, Harmancı AS, Bai H, Youngblood MW, Lee TI, Baranoski JF, et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat Genet. (2016) 48:1253–9. doi: 10.1038/ng.3651
39. Smith MJ, O’Sullivan J, Bhaskar SS, Hadfield KD, Poke G, Caird J, et al. Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat Genet. (2013) 45:295–8. doi: 10.1038/ng.2552
40. Smith MJ, Wallace AJ, Bennett C, Hasselblatt M, Elert-Dobkowska E, Evans LT, et al. Germline SMARCE1 mutations predispose to both spinal and cranial clear cell meningiomas. J Pathol. (2014) 234:436–40. doi: 10.1002/path.4427
41. Aavikko M, Li SP, Saarinen S, Alhopuro P, Kaasinen E, Morgunova E, et al. Loss of SUFU function in familial multiple meningioma. Am J Hum Genet. (2012) 91:520–6. doi: 10.1016/j.ajhg.2012.07.015
42. Governale LS, Vortmeyer AO, Zhuang Z, and Oldfield EH. Fibrous meningioma in a patient with von Hippel-Lindau disease: a genetic analysis. J Neurosurg. (2001) 95:1045–9. doi: 10.3171/jns.2001.95.6.1045
43. Sievers P, Chiang J, Schrimpf D, Stichel D, Paramasivam N, Sill M, et al. YAP1-fusions in pediatric NF2-wildtype meningioma. Acta Neuropathol. (2020) 139:215–8. doi: 10.1007/s00401-019-02095-9
44. Teranishi Y, Okano A, Miyawaki S, Ohara K, Ishigami D, Hongo H, et al. Clinical significance of NF2 alteration in grade I meningiomas revisited; prognostic impact integrated with extent of resection, tumour location, and Ki-67 index. Acta Neuropathol Commun. (2022) 10:76. doi: 10.1186/s40478-022-01377-w
45. Lee S, Karas PJ, Hadley CC, Bayley V JC, Khan AB, Jalali A, et al. The role of merlin/NF2 loss in meningioma biology. Cancers (Basel). (2019) 11:1633. doi: 10.3390/cancers11111633
46. Hao S, Huang G, Feng J, Li D, Wang K, Wang L, et al. Non-NF2 mutations have a key effect on inhibitory immune checkpoints and tumor pathogenesis in skull base meningiomas. J Neurooncol. (2019) 144:11–20. doi: 10.1007/s11060-019-03198-9
47. Reuss DE, Piro RM, Jones DT, Simon M, Ketter R, Kool M, et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol. (2013) 125:351–8. doi: 10.1007/s00401-013-1093-x
48. Najm P, Zhao P, Steklov M, Sewduth RN, Baietti MF, Pandolfi S, et al. Loss-of-function mutations in TRAF7 and KLF4 cooperatively activate RAS-like GTPase signaling and promote meningioma development. Cancer Res. (2021) 81:4218–29. doi: 10.1158/0008-5472.CAN-20-3669
49. Khan AB, English CW, Chen WC, Athukuri P, Bayley JC5, Brandt VL, et al. Even heterozygous loss of CDKN2A/B greatly accelerates recurrence in aggressive meningioma. Acta Neuropathol. (2023) 145:501–3. doi: 10.1007/s00401-023-02543-7
50. Roehrkasse AM, Peterson JEG, Fung KM, Pelargos PE, and Dunn IF. The discrepancy between standard histologic WHO grading of meningioma and molecular profile: a single institution series. Front Oncol. (2022) 12:846232. doi: 10.3389/fonc.2022.846232
51. Tauziède-Espariat A, Pfister SM, Mawrin C, and Sahm F. Pediatric meningiomas: A literature review and diagnostic update. Neurooncol Adv. (2023) 5:i105–11. doi: 10.1093/noajnl/vdac165
52. Dudley RWR, Torok MR, Randall S, Béland B, Handler MH, Mulcahy-Levy JM, et al. Pediatric versus adult meningioma: comparison of epidemiology, treatments, and outcomes using the Surveillance, Epidemiology, and End Results database. J Neurooncol. (2018) 137:621–9. doi: 10.1007/s11060-018-2756-1
53. Dash C, Pasricha R, Gurjar H, Singh PK, and Sharma BS. Pediatric intraventricular meningioma: A series of six cases. J Pediatr Neurosci. (2016) 11:193–6. doi: 10.4103/1817-1745.193356
54. Perry A, Giannini C, Raghavan R, Scheithauer BW, Banerjee R, Margraf L, et al. Aggressive phenotypic and genotypic features in pediatric and NF2-associated meningiomas: a clinicopathologic study of 53 cases. J Neuropathol Exp Neurol. (2001) 60:994–1003. doi: 10.1093/jnen/60.10.994
55. Toland A, McNulty SN, Pekmezci M, Evenson M, Huntoon K, Pierson CR, et al. Pediatric meningioma: a clinicopathologic and molecular study with potential grading implications. Brain Pathol. (2020) 30:1134–43. doi: 10.1111/bpa.12884
56. Tauziede-Espariat A, Parfait B, Besnard A, Lacombe J, Pallud J, Tazi S, et al. Loss of SMARCE1 expression is a specific diagnostic marker of clear cell meningioma: a comprehensive immunophenotypical and molecular analysis. Brain Pathol. (2018) 28:466–74. doi: 10.1111/bpa.12524
57. Kirches E, Sahm F, Korshunov A, Bluecher C, Waldt N, Kropf S, et al. Molecular profiling of pediatric meningiomas shows tumor characteristics distinct from adult meningiomas. Acta Neuropathol. (2021) 142:873–86. doi: 10.1007/s00401-021-02351-x
58. Schieffer KM, Agarwal V, LaHaye S, Miller KE, Koboldt DC, Lichtenberg T, et al. YAP1-FAM118B fusion defines a rare subset of childhood and young adulthood meningiomas. Am J Surg Pathol. (2021) 45:329–40. doi: 10.1097/PAS.0000000000001597
59. De Tommasi A, Occhiogrosso M, De Tommasi C, Cimmino A, Sanguedolce F, and Vailati G. Radiation-induced intracranial meningiomas: review of six operated cases. Neurosurg Rev. (2005) 28:104–14. doi: 10.1007/s10143-004-0366-1
60. Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. (2017) 19:v1–v88. doi: 10.1093/neuonc/nox158
Keywords: meningioma, molecular sequencing, NF2, WHO grade, cIMPACT-NOW
Citation: Ali RH, Hassan A, Jarkhi HH, Alshawish A, Almanabri M, Alhalabi OT, Alsaber AR, Ali NY, Abdelnabi E, Mohammed EMA, Jama H, Almarzooq A, Alqallaf Z, Ahmed AA, Bahzad S, Hamelmann S, Sahm F and Almurshed M (2025) Molecular and histopathological landscape of 131 meningiomas: a retrospective institutional study with insights from cIMPACT-NOW. Front. Oncol. 15:1648953. doi: 10.3389/fonc.2025.1648953
Received: 17 June 2025; Accepted: 11 August 2025;
Published: 29 August 2025.
Edited by:
Supriya Mallick, All India Institute of Medical Sciences, IndiaReviewed by:
Suvendu Purkait, All India Institute of Medical Sciences Bhubaneswar, IndiaPrashanth Giridhar, Tata Memorial Centre, India
Copyright © 2025 Ali, Hassan, Jarkhi, Alshawish, Almanabri, Alhalabi, Alsaber, Ali, Abdelnabi, Mohammed, Jama, Almarzooq, Alqallaf, Ahmed, Bahzad, Hamelmann, Sahm and Almurshed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rola H. Ali, cm9sYS5hbGlAa3UuZWR1Lmt3