 Feilong Chen
Feilong Chen Lei Lyu1
Lei Lyu1 Shaofan Hu
Shaofan Hu Meng Wang
Meng Wang- 1Sports Medicine Key Laboratory of Sichuan Province, Expert Centre of Sichuan Province, Institute of Sports Medicine and Health, Chengdu Sport University, Chengdu, China
- 2Jinfeng Laboratory, Chongqing, China
- 3Department of Pathophysiology, College of High Altitude Military Medicine, Third Military Medical University (Army Medical University), Chongqing, China
- 4Institute for Cancer Medicine, School of Basic Medical Sciences, Southwest Medical University, Luzhou, Sichuan, China
Fibrosis, which is characterized by pathological extracellular matrix (ECM) accumulation impairing organ function, is governed primarily by dysregulated transforming growth factor-β (TGF-β)/Smad signalling. TGF-β1 triggers canonical (Smad2/3-dependent) and noncanonical pathways upon receptor binding, driving profibrotic processes such as fibroblast activation, epithelial–mesenchymal transition (EMT), excessive ECM production (e.g., collagen), and the suppression of matrix degradation. This pathway is central to organ-specific fibrogenesis: In liver fibrosis, it activates hepatic stellate cells (HSCs); in renal fibrosis, it promotes tubular injury and ECM deposition; in pulmonary fibrosis, it induces EMT/fibroblast transition in radiation/bleomycin models; in cardiac fibrosis, it mediates fibroblast activation in diabetic cardiomyopathy/atrial fibrillation via NPRC/TGIF1/USP mechanisms; and in skin fibrosis (e.g., scleroderma), it stimulates collagen overproduction, which is suppressed by osthole or mesenchymal stem cells. The TGF-β/Smad axis thus represents a pivotal therapeutic target. Future research should clarify tissue-specific regulatory networks and develop combinatorial antifibrotic strategies.
1 Introduction
Fibrosis is a pathological process characterized by persistent deposition of cells and mediators, leading to structural damage to and dysfunction of normal tissues and organs and significantly impacting patients’ physical and mental health and quality of life (1). Notably, the TGF-β/Smad signalling pathway plays a crucial role in cell biology and disease progression, primarily involving processes such as cell proliferation, growth, differentiation, apoptosis, and ECM regulation (2–4). In recent years, with increasing research, it has become apparent that the TGF-β/Smad pathway plays a pivotal role in the initiation and development of numerous diseases. Its involvement in fibrotic diseases, in particular, has garnered significant attention. Fibrosis in various organs, such as the lung (5), heart (6), kidney (7), and liver (8), is closely associated with aberrant activation of the TGF-β/Smad signalling pathway. For example, in diabetic cardiomyopathy (DCM) models, deficiency of natriuretic peptide receptor C (NPRC) alleviates cardiac fibrosis in diabetic mice by inhibiting the TGF-β/Smad pathway (6). Conversely, in pulmonary fibrosis, selpercatinib inhibits fibroblast proliferation, migration, activation, and ECM deposition by suppressing the TGF-β/Smad pathway, thereby attenuating the progression of lung fibrosis (9).
In this review, we summarize research advances concerning the TGF-β1/Smad signalling pathway, focusing on its role and associated mechanisms in various fibrotic diseases. Building upon this foundation, we analyse the commonalities and differences in the actions of TGF-β across different fibrotic diseases on the basis of the latest research findings and discuss current challenges and future research directions. In subsequent sections, we detail various molecules and regulatory mechanisms related to the TGF-β signalling pathway and analyse their potential roles in disease progression and therapy. In this review, we focus primarily on TGF-β1 and Smad2/3.
2 Current research status of TGF-β1
2.1 Classification of TGF-β family proteins
The TGF-β family comprises three principal isoforms (TGF-β1, -β2, and -β3) that function as pleiotropic cytokines (10, 11). Upon binding to specific cell surface receptors, TGF-β initiates distinct intracellular signalling cascades, which are classically categorized into Smad-dependent (canonical) and Smad-independent (noncanonical) pathways, with their activation and propagation being precisely regulated through multiple molecular mechanisms (12). The TGF-β receptor system consists of two transmembrane serine/threonine kinase receptors, type I (TβRI) and type II (TβRII) (13). The signalling cascade is initiated by ligand binding to TβRII, which subsequently recruits and phosphorylates TβRI to form an active receptor complex, thereby stimulating its kinase activity to propagate downstream signalling. Importantly, coreceptors, including endoglin (CD105) and betaglycan (TGFβR3), serve as critical modulators of TGF-β signal transduction through their regulatory functions in receptor complex assembly and signal amplification (14).
2.2 TGF-β1 biogenesis, activation and signal transduction
The generation, activation, and signal transduction of TGF-β1 constitute a sophisticated biological process. Initially, synthesized in the endoplasmic reticulum as a propeptide, TGF-β1 consists of an N-terminal latency-associated peptide (LAP) and a C-terminal mature TGF-β1 domain. Following biosynthesis, two propeptide monomers form homodimers via disulfide bonds before being transported to the Golgi apparatus (15). Within the Golgi, furin-mediated proteolytic cleavage separates the mature TGF-β1 domain from LAP, although these cleavage products remain noncovalently associated (16, 17). This results in the formation of the small latent complex (SLC), also called latent TGF-β1 (LTGF-β1), wherein the mature cytokine remains structurally sequestered and biologically inactive.
TGF-β1 is secreted through two distinct pathways: as an SLC associated with either GARP or LRRC33 or as a large latent complex (LLC) formed by binding to latent TGF-β-binding proteins (LTBPs), which subsequently anchor to the ECM via covalent interactions with ECM proteins such as fibronectin or fibrillin (18). The bioavailability and activity of TGF-β are tightly regulated by accessory molecules, collectively termed “TGF-β milieu molecules (19)”, including LTBPs (which anchor latent TGF-β to extracellular complexes), GARP (20)(which are expressed on Tregs, platelets, and endothelial cells (21–23)), and LRRC33 (which tethers TGF-β to the cell surface), as well as αvβ6 and αvβ8 integrins (which are critical for latent TGF-β activation). The activation of latent TGF-β1 is mediated by αvβ6 or αvβ8 integrin heterodimers, which bind the Arg-Gly-Asp (RGD) motif in LAP, generating mechanical tension that disrupts the constrained conformation and releases the active form (24).
TGF-β1 signalling pathway activation is initiated when the active ligand binds to TGF-β receptor type II (TGF-βRII) on target cells. This binding induces the phosphorylation and activation of TGF-β receptor type I (TGF-βRI). TGF-β1 signalling occurs through canonical (Smad-dependent) and noncanonical pathways (25). In the canonical pathway, activated TGF-βRI phosphorylates receptor-regulated Smads (R-Smads: Smad2 and Smad3), which then form a complex with Smad4. This complex translocates to the nucleus to regulate target gene transcription. Additionally, TGF-β1 utilizes multiple noncanonical pathways, including the PI3K/AKT/mTOR, MEK/ERKand ERK1/2, signalling cascades, to modulate diverse cellular functions (Figure 1) (25–29).
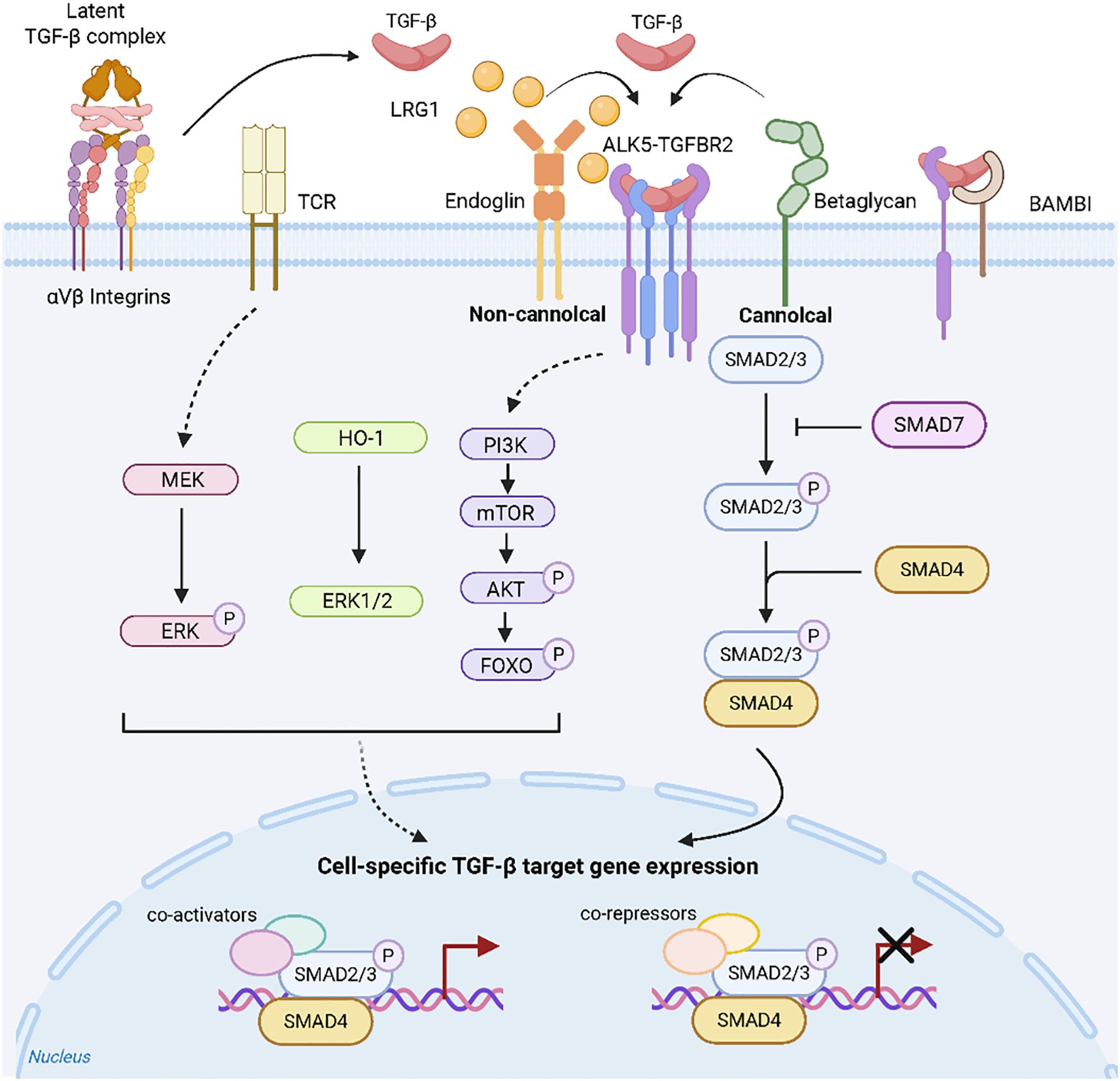
Figure 1. TGF-β signalling (25). The interaction of TGF-β with its specific receptors can initiate a series of intracellular pathways, which are categorized into Smad-dependent (canonical) and Smad-independent (non-canonical) signaling pathways, with their activation/transmission being finely regulated by various mechanisms.
These non-canonical pathways regulate fibrotic processes either independently or cooperatively with Smad signaling, as evidenced by organ-specific models.Notably, in canine myxomatous mitral valve disease, TGF-β1 activates the PI3K/AKT pathway to drive myofibroblast transdifferentiation (25).Similarly, during bleomycin-induced pulmonary fibrosis, sinomenine concurrently suppresses both canonical (TGF-β1/Smad3) and non-canonical (PI3K/Akt) pathways to attenuate fibrogenesis (30).Furthermore, in murine non-alcoholic fatty liver disease, triptolide activates AMPK signaling, thereby inhibiting TGF-β1 expression, improving lipid metabolism, and reducing hepatic fibrosis (31).Critically, in radiation-induced intestinal fibrosis, TGF-β1 orchestrates connective tissue growth factor (CTGF) expression through dual activation of Smad-dependent signaling and the Rho/ROCK axis (32).
3 Research status of Smad2/3
3.1 Classification, structure and function of Smad family proteins
3.1.1 Classification of Smad family proteins
The Smad protein family, comprising Mad proteins, Smad proteins, and their homologues (33, 34), serves as direct substrates for TGF-β family members. Although nine Smad proteins have been identified in total, only eight have been found in animals; these proteins can be functionally classified into three distinct subfamilies. First, receptor-regulated Smads (R-Smads), including Smad1, Smad2, Smad3, Smad5, and Smad8, are recognized by TβRI and function as downstream targets of TGF-β receptors. Notably, while Smad2 and Smad3 mediate TGF-β1 signalling, Smad1, Smad5, and Smad8 are phosphorylated by BMP receptors to transduce BMP - 7 signalling (35–37). Second, the common-mediator Smad (Co-Smad) subfamily consists solely of Smad4, which forms complexes with phosphorylated R-Smads in the cytoplasm before nuclear translocation to execute transcriptional regulation (38). Finally, the inhibitory Smads (I-Smads), comprising Smad6 and Smad7, function as negative regulators of the TGF-β signalling pathway (38).
3.1.2 Structure and function of Smads family proteins
Smad family proteins, with molecular weights ranging from 42 – 60 kDa, exhibit high evolutionary conservation across species and typically consist of approximately 500 amino acids. Structurally, these proteins contain two conserved domains: an N-terminal MH1 (Mad homology domain-1) and a C-terminal MH2 (Mad homology domain-2), connected by a variable-length proline-rich linker region. Importantly, the MH1 domain has dual functions: it suppresses MH2 domain activity in the unactivated state, but upon activation, it binds specific DNA sequences to regulate transcription (39). Furthermore, comparative analysis revealed distinct conservation patterns between these domains: while the MH2 domain is highly conserved across all Smad family members, the MH1 domain is conserved only in R-Smads and Co-Smads and is completely absent in I-Smads (39–41).
Smad family proteins play essential roles in diverse cellular signalling pathways by regulating gene expression through two distinct mechanisms. First, they can directly modulate target gene transcription by either activating or repressing transcriptional activity (42). Alternatively, they may indirectly regulate gene expression by interacting with DNA-binding factors to control the expression of associated genes or other transcriptional regulators (43). For example, Smad3/4 complexes can form heteromeric complexes with E2F4/DP1 dimers that subsequently bind to the TGF-β inhibitory element (TIE) in the c-Myc promoter to repress target gene expression (44–46). Notably, while Smad2 and Smad3 serve as direct downstream effectors of TGF-β1 to promote tissue fibrosis, Smad7 functions as a negative regulator to suppress TGF-β1-mediated fibrotic responses (47).
3.2 Structural and functional similarities and differences between Smad2 and Smad3
Smad2 and Smad3 differ in many aspects. In the following sections, we compare the structure, activity, transcription level and function of Smad2 and Smad3.
3.2.1 Similarities between Smad2 and Smad3
Smad2 and Smad3 exhibit significant structural and functional homology. Structurally, these proteins share 92% amino acid sequence identity in mice (48), with both containing conserved MH1 and MH2 domains connected by a proline-rich linker region. Functionally, as members of the R-Smad subfamily, they serve as downstream effectors of TGF-β1 signalling and demonstrate considerable functional redundancy (49, 50). Mechanistically, both proteins require TβRI-mediated phosphorylation before forming complexes with phosphorylated Smad4, a critical step for their subsequent nuclear translocation and transcriptional regulation (51–53). In terms of antioxidative stress, Smad2/3 can physically interact with Keap1 (Kelch-like ECH-associated protein 1) and its isoforms α and β through the EDGETSD and DLG motifs in the connection region between its MH1 and MH2 domains, mediating the expression of the Keap1–Nrf2 (nuclear factor E2-related factor 2, also called NFE2L2) pathway (54, 55).
3.2.2 The activation and transcriptional differences of Smad2 and Smad3
Notably, Smad2 and Smad3 exhibit distinct activation patterns, primarily due to differences in their phosphorylation mechanisms. Although both proteins undergo phosphorylation, this process requires multiple regulatory proteins that have differential effects on each Smad isoform. Specifically, studies have revealed that SARA (Smad anchor for receptor activation) deficiency significantly impairs Smad2 phosphorylation but has a minimal effect on Smad3 (56, 57).
Smad2 and Smad3 modulate transcriptional activity both by repressing target gene promoters and through regulation by accessory proteins. Critically, their DNA-binding capacity depends on structural differences within the MH1 domain: Smad3 lacks the TID (transcriptional inhibitory domain) and binds DNA directly, whereas Smad2 requires Smad4 interaction for DNA association owing to its inhibitory TID (58, 59).
3.2.3 Differences in function between Smad2 and Smad3
While both Smad2 and Smad3 are regulated by TGF-β1 signalling across multiple cell types, they exhibit significant functional divergence. Notably, during embryonic development, Smad2 expression is detectable at early stages, whereas Smad3 expression remains undetectable (60). Furthermore, genetic ablation studies have revealed differential impacts on viability: Smad2 knockout causes embryonic lethality, whereas Smad3-deficient mice survive postnatally but have markedly reduced lifespans (<6 months; minimum survival of 1 month) (61–63).
4 Cell regulatory function and mechanism of the TGF-β/Smad signalling pathway in fibrosis
4.1 Functions in cell proliferation and growth
Cellular proliferation, a fundamental biological process involving cell division, is distinct from cellular growth, which refers to an increase in cell size through protein synthesis, organelle biogenesis, and membrane expansion. Importantly, the TGF-β/Smad signalling pathway plays a critical role in negatively regulating both the proliferation and growth of normal cells. Mechanistically, the TGF-β-mediated inhibition of proliferation involves receptor activation of Smad2/Smad3, their subsequent complex formation with Smad4, their nuclear translocation, and the transcriptional repression of cell cycle-related genes. This regulatory mechanism is essential for maintaining tissue homeostasis and preventing aberrant proliferation. For example, in epithelial cells, the TGF-β–Smad axis suppresses proliferation by downregulating key cell cycle regulators (64). Furthermore, with respect to growth modulation, studies using human lung fibroblasts cultured in cystatin C (CST3)- and growth differentiation factor 15 (GDF15)-enriched media demonstrated that the inhibition of TGF-β/Smad signalling was correlated with reduced cellular proliferation (65).
4.2 Functions in cell differentiation and apoptosis
Importantly, the TGF-β/Smad signalling pathway critically regulates both cellular differentiation and apoptosis. Specifically, TGF-β promotes cell differentiation by activating Smad2/3 to modulate gene expression programs, as demonstrated by the psoralidin (PL)-induced differentiation of bone marrow mesenchymal stem cells into nucleus pulposus-like cells through this pathway (66). Conversely, during intervertebral disc degeneration, Grem1 accelerates disc cell apoptosis by inhibiting TGF-β-mediated Smad2/3 phosphorylation, thereby reducing inflammation-associated apoptotic events (67). In terms of apoptosis in myxoid mitral valve disease (MMVD), PI3K/AKT/mTOR signaling induced by TGF-β contributes to the pathogenesis of MMVD and plays a key role in the regulation of myofibroblast apoptosis (26).
4.3 Regulatory role in ECM
Fibrosis is characterized by the abnormal accumulation of extracellular matrix (ECM) components, such as collagen and fibronectin. The TGF-β signaling pathway plays a pivotal role in both physiological and pathological processes of tissue fibrosis (68). Mechanistically, TGF-β activation promotes the synthesis and deposition of ECM components—including collagens and fibronectin—through Smad2/3-mediated signalling (69). This regulatory function is particularly relevant to both tissue repair and fibrotic processes. Notably, in murine lung fibrosis models, ganoderic acid (GA) attenuates aberrant ECM accumulation by suppressing fibronectin expression and inhibiting hyperactivated TGF-β/Smad signalling (70). Conversely, during diabetic nephropathy, TGF-β drives pathological ECM deposition in glomerular mesangial cells via Smad-dependent pathways (71). Furthermore, activation of this pathway enhances ECM production and accelerates wound closure in diabetic foot ulcer mouse models (72). TGF-β1 can also be secreted in M2-type macrophages to promote myofibroblast proliferation, leading to ECM deposition (73). In the pathogenesis of idiopathic pulmonary fibrosis, the activation of TGF-β signaling pathway will accelerate the excessive production and deposition of ECM components, which will lead to the accumulation of fibrotic tissue. Functionally, the TGF-β/Smad signalling pathway serves as a multifunctional regulator of normal cellular physiology, maintaining tissue homeostasis through the precise coordination of proliferation, growth, differentiation, programmed cell death, and extracellular matrix turnover. However, dysregulation of this tightly controlled network frequently disrupts signalling equilibrium, thereby promoting pathological processes, including tumorigenesis and organ fibrosis. Notably, accumulating evidence reveals the dual regulatory nature of the pathway: it exerts tumour-suppressive effects during early carcinogenesis while paradoxically facilitating metastasis in advanced malignancies through mechanisms such as epithelial–mesenchymal transition promotion.
5 Regulatory role of the TGF-β/Smad signalling pathway in tissue fibrosis
Fibrosis represents a progressive pathological condition characterized by exaggerated wound-healing responses to persistent tissue injury across multiple organ systems. During this process, functional parenchymal cells are progressively replaced by excessive extracellular matrix deposition, ultimately compromising organ architecture and function. Notably, fibrogenesis affects diverse anatomical sites, such as cardiac and dermal tissues, beyond classical targets like lung, liver, and kidney. Critically, the TGF-β/Smad signalling pathway has been established as a central driver in numerous fibrotic disorders, where its aberrant activation correlates directly with increasing disease severity. Smad2 and Smad3, two primary downstream mediators of this pathway, exhibit high structural homology that confers partial functional redundancy, yet demonstrate distinct selectivity in their transcriptional targets (47, 74). Consequently, therapeutic strategies increasingly focus on targeting this pivotal pathway to attenuate pathological matrix accumulation.
5.1 Role of the TGF-β/Smad signalling pathway in hepatic fibrosis
The progression of hepatic fibrosis involves intricate interactions between hepatic stellate cells (HSCs) and various immune cells, particularly macrophages, within the liver. Extensive research has focused on elucidating the mechanisms underlying liver fibrosis, with particular emphasis on pharmacological interventions, signalling pathway modulation, and alterations in gene expression. Among these pathways, the TGF-β/Smad signalling pathway has been widely implicated in the pathological progression of liver fibrosis. Both in vitro and in vivo studies have demonstrated that astragaloside IV (75) and MOTS-c (mitochondrial open reading frame of the 12S rRNA type-c) (76) suppress HSCs activation and attenuate liver fibrosis by inhibiting the TGF-β1/Smad pathway through antioxidant stress mechanisms. Similarly, in CCL4-induced liver injury models, melatonin ameliorates hepatic fibrosis and improves liver function by downregulating TGF-β1/Smad signalling (77).
In nonalcoholic fatty liver disease (NAFLD), activation of the TGF-β1 signaling pathway leads to dysregulated hepatic lipid accumulation, thereby promoting the progression from a benign NAFLD condition to fibrosis, cirrhosis, and hepatic malignancies (78).Furthermore, (Pro)renin promotes HSCs activation and fibrosis in both human and murine HSCs, whereas its receptor knockdown TGF-β1/Smad3 pathway activity and mitigates fibrosis (79). Smad2 and Smad3 exhibit structural homology yet functional divergence. Multiple lines of evidence demonstrate that in HSCs, Smad2 overexpression downregulates collagen I and tissue inhibitor of metalloproteinase-1 (TIMP - 1) expression while augmenting matrix metalloproteinase-2 (MMP - 2) production. Conversely, Smad3 overexpression elicits diametrically opposed effects (80).Astaxanthin (ASTX) exerts antifibrotic effects by suppressing TGF-β1-induced mRNA and protein expression of α-smooth muscle actin (α-SMA) and procollagen alpha-1(I), thereby blocking TGF-β1 signal transduction and inhibiting Smad3 pathway activation in HSCs (81). Notably, the antifibrotic drug fluorofenidone (AKF-PD) inhibits HSCs autophagy via the TGF-β1/Smad pathway, thereby attenuating liver fibrosis (82). Moreover, studies on miRNAs have revealed that exosomal miR-342-3p derived from primary hepatic macrophages improves liver fibrosis by upregulating HPCAL1 (Hippocalcin-like protein 1) in HSCs, which subsequently inhibits TGF-β signalling (83).
5.2 Role of the TGF-β/Smad signalling pathway in renal fibrosis
Renal fibrosis, a hallmark of chronic kidney disease characterized by fibrotic alterations in the glomeruli and tubulointerstitium, frequently progresses to end-stage renal disease. Notably, dietary supplementation with eicosapentaenoic acid-enriched phospholipids (EPA-PLs) in spontaneously hypertensive rats suppresses TGF-β and Smad3 activation in renal tubular cells, enhances PI3K/AKT phosphorylation, diminishes the expression of proinflammatory cytokines (including IL - 1β and IL - 6), and consequently reduces tubulointerstitial fibrosis (84). Furthermore, research on ferroptosis in renal tubular epithelial cells (TECs) revealed a close link between its profibrotic mechanisms and the TGF-β/Smad pathway, and persistent ferroptosis effectively promotes fibrotic progression via this signalling axis (85). In experimental models of tubulointerstitial fibrosis, TGF-β1 ameliorates renal fibrotic progression through differential Smad-mediated regulation: Smad3 governs CTGF (connective tissue growth factor) and E-cadherin expression, while Smad2 modulates MMP - 2 expression. These findings demonstrate distinct target specificity of Smad2 versus Smad3 in tubulointerstitial fibrogenesis (86).
Moreover, therapeutic strategies targeting noncoding RNAs (such as miR-21, miR-145-5P, and miR-145-29b) have shown efficacy in mitigating nephritis and fibrosis in chronic kidney disease models, primarily through inhibition of the TGF-β1/Smad pathway (87). αKlotho, functioning as both an anti-fibrotic and anti-tumor agent, ameliorates impairment of renal structure and function in chronic kidney disease by inhibiting the binding of transforming growth factor-β (TGF-β) to its receptors (88). SIS3, a selective Smad3 inhibitor, directly suppresses Smad3-mediated collagen matrix expression and inhibits the accumulation of α-SMA-positive myofibroblasts in fibrotic kidneys. This blockade of endothelial-mesenchymal transition (EndMT) attenuates renal fibrosis and impedes disease progression (89). Lupus nephritis is a serious complication of systemic lupus erythematosus (SLE), driven by inflammation and fibrosis, which often leads to chronic kidney disease. Previous studies have shown that SPRY4-IT1 and TUG1 regulate TGF-β/Smad signaling to promote renal fibrosis in lupus nephritis (90). It is particularly important to note that the heart and kidneys are closely linked through the circulatory system. Primary dysfunction in one organ often leads to secondary dysfunction or injury in the other. These interactions shape cardiorenal syndrome, in which the activated fibrotic TGF-β1/Smad signaling pathway accelerates its pathological progression (91).
Macrophage subset differentiation is primarily categorized into classically activated M1 and alternatively activated M2 macrophages. In the kidney, persistent inflammation and prolonged release of factors like TGF-β lead to renal injury, ultimately resulting in renal fibrosis. Notably, studies have shown that macrophages are critically involved in renal fibrosis: within the kidney, M1 macrophages exert proinflammatory functions, and sustained inflammation coupled with the prolonged release of factors such as TGF-β drives progressive renal injury culminating in fibrosis. Conversely, M2 macrophages secrete TGF-β1 and fibroblast growth factor, promoting the proliferation of myofibroblasts and leading directly to ECM deposition (73). Similarly, studies on renal allograft rejection have demonstrated that METTL3 enhances the M2 macrophage-to-myofibroblast transition within this context by promoting the TGF-β1/Smad3 axis (92). Therefore, macrophages play a pivotal role in the pathogenesis and progression of renal fibrosis.
Type 2 diabetes (T2D) is a global health concern. Diabetic kidney disease in type 2 diabetes (T2DN) is one of the major microvascular complications of T2D and a leading cause of end-stage renal disease. TGF-β is a key regulator in renal fibrosis and mediates T2DN through its downstream Smad3-dependent mechanisms. Studies have demonstrated that treatment of prediabetic db/db mice (from 4 to 12 weeks of age) with SIS3, a Smad3 inhibitor, significantly reduced blood glucose levels and suppressed the elevation of serum creatinine, microalbuminuria, renal fibrosis, and inflammation, thereby substantially mitigating both T2D and T2DN. However, when db/db mice received late-stage SIS3 treatment during 8 – 16 weeks of age, although it inhibited the pathological progression of T2DN, it did not significantly improve T2D. This suggests that early intervention during the prediabetic stage may effectively prevent the development of both T2D and T2DN (93).
The signal transduction of TGF-β1 initiates upon its binding to the transforming growth factor beta type II receptor (TGFBR2), a constitutively active kinase (Figure 1). Subsequently, TGFBR2 phosphorylates and activates the type I receptor (TGFBRI), which then propagates downstream signaling through Smad2/3. Notably, the ubiquitin-proteasome system (UPS) serves as a critical mechanism for post-translational regulation. Of particular relevance, USP11— a key deubiquitinating enzyme—is significantly upregulated in fibrotic kidneys. Mechanistically, USP11 directly interacts with TGFBR2 and stabilizes it by counteracting its ubiquitin-mediated degradation via deubiquitination. Consequently, elevated TGFBR2 levels drive hyperactivation of the downstream Smad3/p53 pathway. This aberrant signaling cascade ultimately triggers renal tubular cell senescence and collectively exacerbates renal fibrosis progression (94).
Additionally, several investigations of natural compound extracts have revealed that their antifibrotic effects are mediated by the suppression of TGF-β/Smad signalling. For example, Ginkgo biloba leaf extract (EGb) ameliorates cisplatin-induced chronic renal interstitial fibrosis by downregulating the protein expression of TGF-β1, Smad2/3, and phosphorylated (p)-Smad2/3, thereby attenuating epithelial–mesenchymal transition in TECs (95). Similarly, ganoderic acid (GA) inhibits ECM deposition in the kidneys of unilateral ureteral obstruction (UUO) mice by suppressing fibronectin expression and preventing the overactivation of TGF-β/Smad signalling (69).
5.3 Role of the TGF-β/Smad signalling pathway in pulmonary fibrosis
Pulmonary fibrosis, a severe condition characterized by the replacement of normal lung architecture with excessive fibrous tissue, leads to significant impairment of lung function. Importantly, the TGF-β/Smad signalling pathway has been demonstrated to play a critical role in pulmonary fibrosis in diverse models, including those induced by radiation, perfluorooctanoic acid (PFOA), and bleomycin. Specifically, astilbin was shown to mitigate radiation-induced pulmonary fibrosis (RIPF) in both in vitro radiation injury models using murine lung epithelial cells (MLE - 12 and TC - 1) and in vivo RIPF models in C57BL/6J mice by inhibiting EMT through targeting the circPRKCE/TGF-β/Smad axis (96). Similarly, PFOA exposure induced pulmonary toxicity and fibrosis in adult male rats via activation of the TGF-β1/Smad signalling pathway (97). Furthermore, in bleomycin-induced mouse models, atractylodin (ATD) attenuated lung injury and fibrosis through the TGF-β/Smad pathway; concomitantly, ATD significantly suppressed both TGF-β1-induced EMT and fibroblast-to-myofibroblast transition in vitro (98). In the pathogenesis of pulmonary fibrosis, Forkhead box protein O3 (FOXO3) inhibits fibroblast activation and ECM deposition via its interaction with TGF-β1-induced Smad3, thereby attenuating idiopathic pulmonary fibrosis (IPF) (93, 99). Moreover, research into miRNAs underscores the importance of this pathway: for example, miR-326-mediated overexpression of nuclear factor I-B (NFIB) inhibits TGF-β-induced EMT and reverses pulmonary fibrosis (100), while miR-486-5p alleviates fibrosis in radiation-induced lung injury (RILI) by suppressing Smad2 and activating Akt (101).
5.4 Role of the TGF-β/Smad signalling pathway in cardiac fibrosis
Myocardial fibrosis, characterized by the excessive accumulation of ECM synthesized by cardiac fibroblasts (CFs), represents a common pathophysiological process in various cardiac diseases, including myocardial infarction (MI), hypertensive heart disease, and cardiomyopathies. Crucially, this pathological remodelling has detrimental effects on cardiac function. Two distinct types of myocardial fibrosis exist: reactive fibrosis and reparative (replacement) fibrosis. Importantly, the TGF-β1 signalling pathway is crucially involved in myocardial fibrosis and significantly influences disease progression and severity. In the context of MI, activation of the TGF-β/Smad signaling pathway can exert protective effects. For instance, in cardiac fibroblasts (CFs), overexpression of Carbonic Anhydrase III (CAIII) potentiates fibroblast activation via the Smad7-TGF-β/Smad2/3 signaling axis. Subsequent activation of these fibroblasts promotes cardiac wound healing (102). In contrast, the ubiquitin-like protein HLA-F adjacent transcript 10 (FAT10) mediates cardiac fibrosis through Smad3 in MI (103). Pulmonary arterial hypertension (PAH) is characterized by excessive proliferation and anti-apoptosis of pulmonary artery smooth muscle cells (PASMCs). Studies have shown that DJ - 1 alleviates DJ - 1-induced PASMCs injury by inhibiting TGFβ/Smad signaling pathway (104).
Mounting evidence shows that the TGF-β1 signalling pathway is a pivotal therapeutic target for cardiac fibrosis. For example, in the context of atrial fibrillation (AF), inhibition of the TGF-β1/Smad pathway mitigates angiotensin II (Ang II)-mediated fibrotic remodelling (105). Furthermore, studies utilizing human AF samples, TGF-β-treated human atrial endocardial endothelial cells (AEECs), and cardiac-specific TGF-β transgenic mice have demonstrated that miR-181b ameliorates AF fibrosis by suppressing the TGF-β-induced endothelial-to-mesenchymal transition (EndMT) (106). Similarly, research in human cardiac fibroblasts (HCFs) revealed that miR-452-5p regulates fibrotic progression under SCN5A deficiency by targeting the TGF-β/Smad axis (107). Moreover, investigations in diabetic cardiomyopathy (DCM) models have shown that the loss of natriuretic peptide receptor C (NPRC) in both DCM mice and patient myocardia upregulates TGF-β-induced factor homeobox 1 (TGIF1); consequently, TGIF1 inhibits Smad2/3 phosphorylation, thereby attenuating cardiac fibrosis and improving remodelling and function in diabetic mice (6).
Notably, the ubiquitin–proteasome system (UPS) plays a significant role in the development of myocardial fibrosis by regulating the degradation and synthesis of proteins involved in both the TGF-β-dependent and the TGF-β-independent profibrotic pathways. Specifically, members of the USP family can modulate this process by targeting signalling molecules and transcription factors critical for fibroblast proliferation and differentiation. For example, USP10 promotes fibrosis through the TGF-β/Smad signalling pathway by deubiquitinating Smad4; similarly (108), USP15 facilitates fibroblast activation and ECM production by activating the TGF-β/Smad pathway (109).
5.5 Role of the TGF-β/Smad signalling pathway in skin fibrosis
Skin fibrosis, characterized by excessive deposition and abnormal proliferation of the ECM, underlies various pathological conditions, including oral submucous fibrosis, scleroderma (systemic sclerosis, SSc), and scarring. Aberrant scar formation, including keloids and hypertrophic scars, is associated with a pathological, dysregulated chronic inflammatory wound healing process. The TGF-β/Smad signaling pathway represents the classic pathway regulating collagen synthesis in fibroblasts and myofibroblasts (78). Studies demonstrate that Zyxin effectively attenuates keloid formation through inhibitory regulation of the TGF-β signaling pathway (110). In support of this notion, osthole attenuates myofibroblast activity in oral submucous fibrosis by inhibiting the TGF-β/Smad2 pathway (111). Moreover, in a bleomycin (BLM)-induced SSc skin fibrosis mouse model generated by repeated subcutaneous injections, both adipose-derived mesenchymal stem cells (AMSCs) and their exosomes reduced dermal thickness and the collagen volume fraction, concurrently suppressing α-smooth muscle actin (α-SMA) and type III collagen (COL3A1) expression in skin tissue via the TGF-β1/Smad3 axis (112). Similarly, artesunate effectively mitigated hypertrophic scar formation in a rabbit ear model by inhibiting endothelial–mesenchymal transition and fibroblast activation through the downregulation of key proteins in the TGF-β/Smad signalling pathway (113).
5.6 Role of the TGF-β/Smad signalling pathway in fibrosis in other diseases
Fibrotic diseases are pathological processes affecting multiple organ systems. In addition to well-characterized fibrosis in organs such as the liver, heart, skin, and kidneys, other tissues, including the eyes and mammary glands, are also susceptible. For example, in a streptozotocin (STZ)-induced diabetic Sprague–Dawley (SD) rat model, Poldip2 overexpression contributes to diabetic retinal fibrosis via the TGF-β1/Smad signalling pathway (114). In contrast, in mouse corneal mechanical injury models, calcitonin gene-related peptide (CGRP) reduces TGF-β1 signalling and prevents TGF-β1-mediated stromal fibroblast activation and tissue fibrosis (115). Similarly, in triple-negative breast cancer (TNBC), PCK2 modulates Smad3 expression and phosphorylation by inhibiting TRIM67-mediated Smad3 ubiquitination; this action consequently enhances TGF-β-stimulated Smad3 activity and activates TGF-β/Smad3 signalling (116). Neuroblastoma (NB) is a cancer arising from neuroblasts. In the context of NB treatment, atypical protein kinase Cs (aPKCs) upregulate the Akt1/NF-κB and TGF-β pathways by binding to 14 - 3–3 and Smad2/3 (117). Regarding juvenile polyposis syndrome (JPS), a rare autosomal dominant disorder, genetic testing reveals aberrant Smad4 expression, which serves as a biomarker for rapid diagnosis of this disease (118). In order to facilitate an intuitive understanding of the main contents of Part 5, statistics are presented in the form of tables (Table 1).
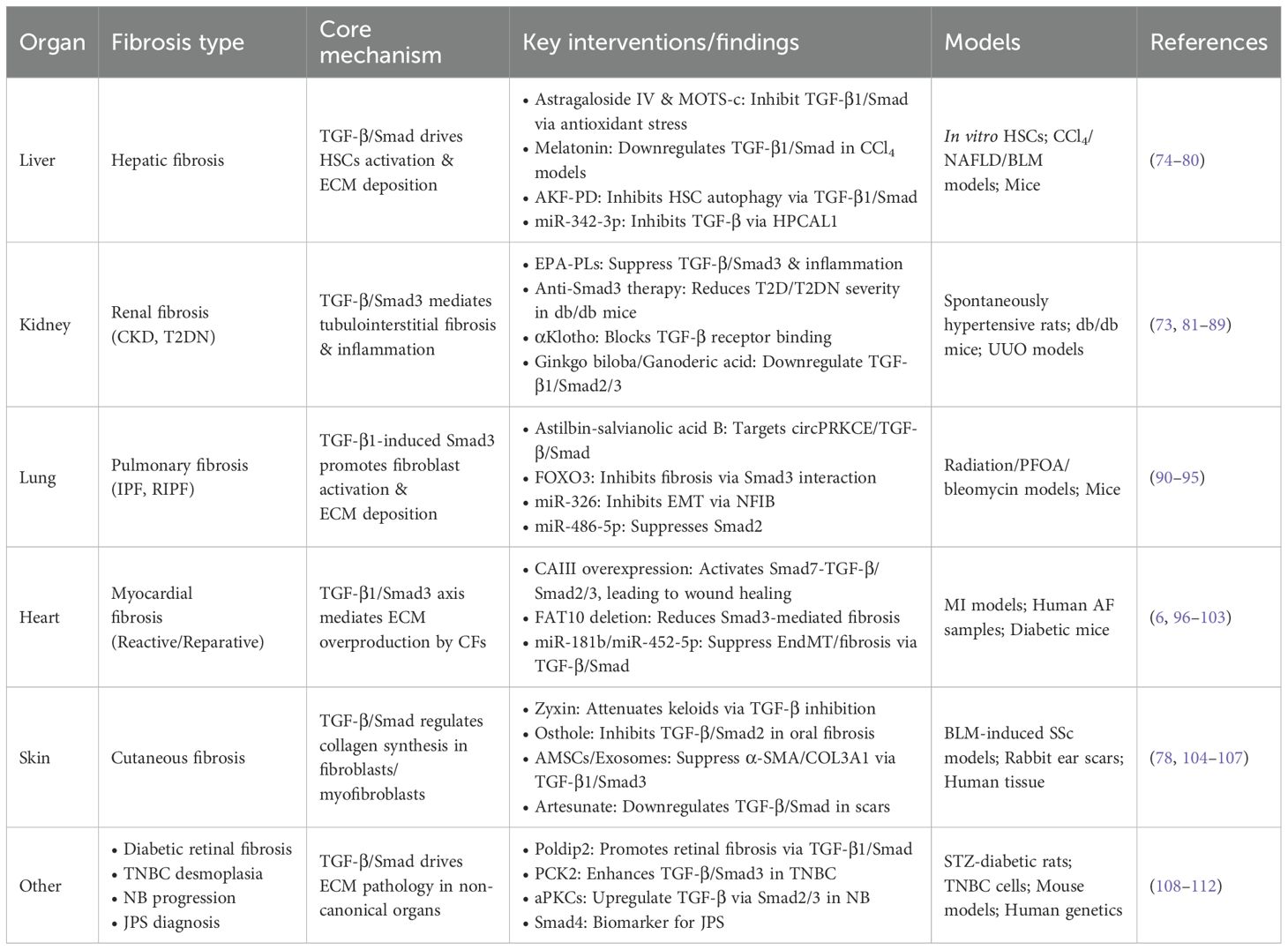
Table 1. Regulatory roles of TGF-β/Smad signaling pathway in organ fibrosis.
6 Discussion
Fibrotic diseases involve complex pathogenic mechanisms that frequently progress to organ failure, yet effective therapeutic interventions remain limited. Therefore, this review aims to elucidate the molecular mechanisms through which the TGF-β1/Smad signaling pathway drives fibrotic pathogenesis. Specifically, we focus on its core biological functions: (1) driving pathological cellular phenotypic transitions, (2) promoting excessive extracellular matrix (ECM) deposition, and (3) suppressing ECM degradation—collectively culminating in structural disruption and functional decline of affected organs. Ultimately, this work establishes a foundation for developing targeted therapeutic or prophylactic agents against this pivotal pathway. Importantly, numerous recent studies have focused on elucidating the pivotal role of the TGF-β/Smad signalling pathway in these disorders. Specifically, this pathway has been established as a key regulatory mechanism in multiple fibrotic conditions, including hepatic, pulmonary, cardiac, and renal fibrosis (Figure 2). Furthermore, oxidative stress and autophagy are closely involved in fibrosis pathogenesis. For example, The Keap1-Nrf2 pathway represents a canonical antioxidant response mechanism. Keap1 interacts with Smad2/3 proteins, and upon Nrf2-mediated antioxidant activation, TGF-β/Smad signaling is downregulated (54, 119). In experimental models of diabetic nephropathy, Mefunidone (MFD) treatment reduces ROS generation, suppresses TGF-β1/Smad pathway activation, and inhibits epithelial-mesenchymal transition (EMT) (120). Though the role of autophagy in fibrosis appears to be context-dependent, with both pro- and anti-fibrotic effects reported. Additionally, other signalling pathways contribute to fibrotic pathogenesis. Notably, the AMPKα/MMP9 axis attenuates skeletal muscle fibrosis, while PPAR-γ/NLRP3/NF-κB signaling mitigates pulmonary fibrosis progression (121, 122). Collectively, this evidence suggests that, therapeutic strategies targeting a single pathway may be insufficient, necessitating a comprehensive approach that considers the interplay of multiple signalling networks.
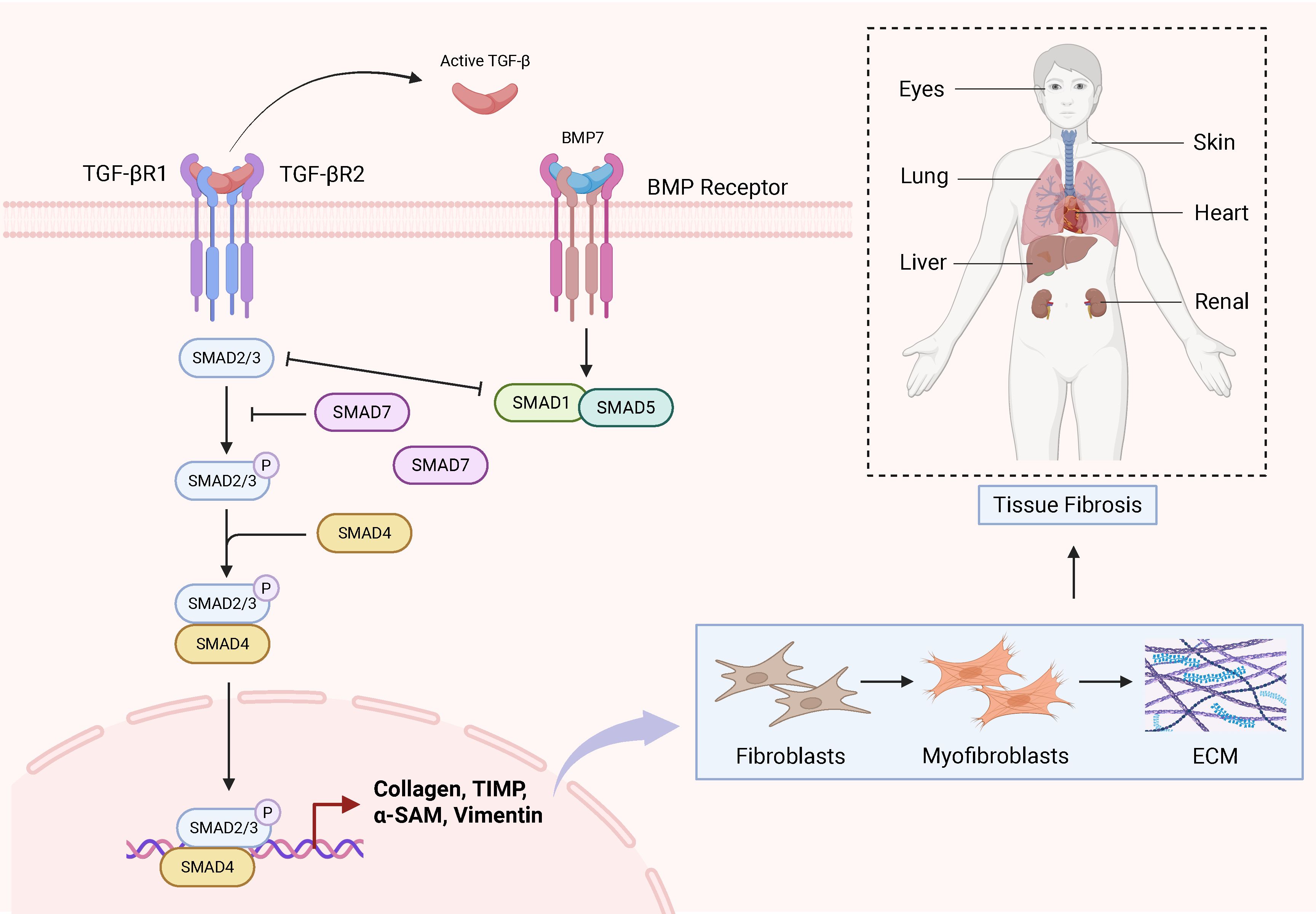
Figure 2. Schematic diagram of the TGF-β/Smad signalling pathway in fibrosis in various tissues.
In the future, promising research directions include first, further delineating the precise regulatory mechanisms of the TGF-β/Smad pathway to identify novel molecular targets; second, investigating its interactions with other pathways, such as those involving oxidative stress, to construct a more holistic map of fibrotic mechanisms (76); third, developing innovative therapeutic modalities, including extracellular vesicles (exosomes), targeted therapies, and phytomedicine formulations, aiming to enhance efficacy and minimize adverse effects (123–125); and finally, elucidating how environmental determinants (e.g., pollutants) influence fibrotic initiation/progression to inform preventive strategies (126, 127). Collectively, these approaches will advance our rapidly evolving understanding of fibrotic pathogenesis and accelerate the translation of novel interventions towards improved patient outcomes, while simultaneously presenting challenges in drug development—particularly regarding tissue specificity and safety profiles.
7 Conclusion
The TGF-β/Smad pathway is the master regulator of multi-organ fibrosis, driving pathological ECM accumulation via fibroblast activation, EMT, and suppressed matrix degradation. Key organ-specific mechanisms include: hepatic stellate cell activation (e.g.,inhibited by astragaloside IV/miR-342-3p); Smad3-mediated renal inflammation (e.g., attenuated by SIS3/αKlotho); TGF-β1-induced pulmonary fibroblast transition (e.g., targeted by miR-326/FOXO3); USP-dependent cardiac Smad deubiquitination; and dermal collagen overproduction (e.g., suppressed by osthole/exosomes). Therapeutic inhibition consistently attenuates fibrosis across models, validating its clinical potential. Future research must resolve tissue-specific regulatory divergence (e.g., Smad2 vs. Smad3) and develop combinatorial strategies (e.g., miRNA-phytomedicine hybrids) for precision antifibrotic therapy.
Author contributions
FC: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. LL: Investigation, Writing – review & editing. CX: Supervision, Writing – review & editing. YC: Writing – review & editing. SH: Methodology, Writing – review & editing. MW: Conceptualization, Writing – review & editing. ZA: Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by Natural Science Foundation of Sichuan (2024NSFSC1228), the China Postdoctoral Science Foundation (Certificate Number 2023M740381), the Postdoctoral Fellowship Program (Grade B) of China Postdoctoral Science Foundation under Grant Number GZB20240083, Key Laboratory of Sports Medicine of Sichuan Province (2025-A003).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Henderson NC, Rieder F, and Wynn TA. Fibrosis: from mechanisms to medicines. Nature. (2020) 587:555–66. doi: 10.1038/s41586-020-2938-9
2. Giarratana AO, Prendergast CM, Salvatore MM, and Capaccione KM. TGF-β signaling: critical nexus of fibrogenesis and cancer. J Trans Med. (2024) 22:594. doi: 10.1186/s12967-024-05411-4
3. Liu J, Jin J, Liang T, and Feng XH. To Ub or not to Ub: a regulatory question in TGF-β signaling. Trends Biochem Sci. (2022) 47:1059–72. doi: 10.1016/j.tibs.2022.06.001
4. Li K, Zhang Y, Zhao W, Wang R, Li Y, Wei L, et al. DPP8/9 inhibition attenuates the TGF-β1-induced excessive deposition of extracellular matrix (ECM) in human mesangial cells via Smad and Akt signaling pathways. Toxicol Lett. (2024) 395:1–10. doi: 10.1016/j.toxlet.2024.03.001
5. Zhao M, Wang M, Chen X, Gao Y, Chen Q, Wang L, et al. Targeting progranulin alleviated silica particles-induced pulmonary inflammation and fibrosis via decreasing Il-6 and Tgf-β1/Smad. J hazardous materials. (2024) 465:133199. doi: 10.1016/j.jhazmat.2023.133199
6. Meng L, Lu Y, Wang X, Cheng C, Xue F, Xie L, et al. NPRC deletion attenuates cardiac fibrosis in diabetic mice by activating PKA/PKG and inhibiting TGF-β1/Smad pathways. Sci Adv. (2023) 9:eadd4222. doi: 10.1126/sciadv.add4222
7. Wang Y, Li Y, Chen Z, Yuan Y, Su Q, Ye K, et al. GSDMD-dependent neutrophil extracellular traps promote macrophage-to-myofibroblast transition and renal fibrosis in obstructive nephropathy. Cell Death Dis. (2022) 13:693. doi: 10.1038/s41419-022-05138-4
8. Xiang D, Zou J, Zhu X, Chen X, Luo J, Kong L, et al. Physalin D attenuates hepatic stellate cell activation and liver fibrosis by blocking TGF-β/Smad and YAP signaling. Phytomedicine. (2020) 78:153294. doi: 10.1016/j.phymed.2020.153294
9. Li S, Liu Z, Jiao X, Gu J, Liu Z, Meng L, et al. Selpercatinib attenuates bleomycin-induced pulmonary fibrosis by inhibiting the TGF-β1 signaling pathway. Biochem Pharmacol. (2024) 225:116282. doi: 10.1016/j.bcp.2024.116282
10. Pelullo M, Zema S, Nardozza F, Checquolo S, Screpanti I, Bellavia D, et al. Wnt, notch, and TGF-β Pathways impinge on hedgehog signaling complexity: an open window on cancer. Front Genet. (2019) 10:711. doi: 10.3389/fgene.2019.00711
11. Culhaci N, Sagol O, Karademir S, Astarcioglu H, Astarcioglu I, Soyturk M, et al. Expression of transforming growth factor-beta-1 and p27Kip1 in pancreatic adenocarcinomas: relation with cell-cycle-associated proteins and clinicopathologic characteristics. BMC Cancer. (2005) 5:98. doi: 10.1186/1471-2407-5-98
12. Turati M, Mousset A, Issa N, Turtoi A, and Ronca R. TGF-β mediated drug resistance in solid cancer. Cytokine Growth factor Rev. (2023) 71-72:54–65. doi: 10.1016/j.cytogfr.2023.04.001
13. Massagué J and Sheppard D. TGF-β signaling in health and disease. Cell. (2023) 186:4007–37. doi: 10.1016/j.cell.2023.07.036
14. Yu L, Wan Q, Liu Q, Fan Y, Zhou Q, Skowronski AA, et al. IgG is an aging factor that drives adipose tissue fibrosis and metabolic decline. Cell Metab. (2024) 36:793–807.e5. doi: 10.1016/j.cmet.2024.01.015
15. Moreau JM, Velegraki M, Bolyard C, Rosenblum MD, and Li Z. Transforming growth factor-β1 in regulatory T cell biology. Sci Immunol. (2022) 7:eabi4613. doi: 10.1126/sciimmunol.abi4613
16. Dubois CM, Laprise MH, Blanchette F, Gentry LE, and Leduc R. Processing of transforming growth factor beta 1 precursor by human furin convertase. J Biol Chem. (1995) 270:10618–24. doi: 10.1074/jbc.270.18.10618
17. Dubois CM, Blanchette F, Laprise MH, Leduc R, Grondin F, and Seidah NG. Evidence that furin is an authentic transforming growth factor-beta1-converting enzyme. Am J Pathol. (2001) 158:305–16. doi: 10.1016/S0002-9440(10)63970-3
18. Kanzaki T, Olofsson A, Morén A, Wernstedt C, Hellman U, Miyazono K, et al. TGF-beta 1 binding protein: a component of the large latent complex of TGF-beta 1 with multiple repeat sequences. Cell. (1990) 61:1051–61. doi: 10.1016/0092-8674(90)90069-Q
19. Qin Y, Garrison BS, Ma W, Wang R, Jiang A, Li J, et al. A milieu molecule for TGF-β Required for microglia function in the nervous system. Cell. (2018) 174:156–71.e16. doi: 10.1016/j.cell.2018.05.027
20. Zimmer N, Trzeciak ER, Graefen B, Satoh K, and Tuettenberg A. GARP as a therapeutic target for the modulation of regulatory T cells in cancer and autoimmunity. Front Immunol. (2022) 13:928450. doi: 10.3389/fimmu.2022.928450
21. Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, and Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci USA. (2009) 106:13445–50. doi: 10.1073/pnas.0901944106
22. Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, and Unutmaz D. Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc Natl Acad Sci USA. (2009) 106:13439–44. doi: 10.1073/pnas.0901965106
23. Wang R, Wan Q, Kozhaya L, Fujii H, and Unutmaz D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PloS One. (2008) 3:e2705. doi: 10.1371/journal.pone.0002705
24. Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C, et al. Force interacts with macromolecular structure in activation of TGF-β. Nature. (2017) 542:55–9. doi: 10.1038/nature21035
25. Hong Q, Kim H, Cai GY, Chen XM, He JC, and Lee K. Modulation of TGF-β signaling new approaches toward kidney disease and fibrosis therapy. Int J Biol Sci. (2025) 21:1649–65. doi: 10.7150/ijbs.101548
26. Tang Q, Markby GR, Macnair AJ, Tang K, Tkacz M, Parys M, et al. TGF-β-induced PI3K/AKT/mTOR pathway controls myofibroblast differentiation and secretory phenotype of valvular interstitial cells through the modulation of cellular senescence in a naturally occurring in vitro canine model of myxomatous mitral valve disease. Cell proliferation. (2023) 56:e13435. doi: 10.1111/cpr.13435
27. Chen X, Ji Y, Liu R, Zhu X, Wang K, Yang X, et al. Mitochondrial dysfunction: roles in skeletal muscle atrophy. J Trans Med. (2023) 21:503. doi: 10.1186/s12967-023-04369-z
28. Muthusamy BP, Budi EH, Katsuno Y, Lee MK, Smith SM, Mirza AM, et al. ShcA Protects against Epithelial-Mesenchymal Transition through Compartmentalized Inhibition of TGF-β-Induced Smad Activation. PloS Biol. (2015) 13:e1002325. doi: 10.1371/journal.pbio.1002325
29. Kunnen SJ, Leonhard WN, Semeins C, Hawinkels L, Poelma C, Ten Dijke P, et al. Fluid shear stress-induced TGF-β/ALK5 signaling in renal epithelial cells is modulated by MEK1/2. Cell Mol Life sciences: CMLS. (2017) 74:2283–98. doi: 10.1007/s00018-017-2460-x
30. Yao F, Xu M, Dong L, Shen X, Shen Y, Jiang Y, et al. Sinomenine attenuates pulmonary fibrosis by downregulating TGF-β1/Smad3, PI3K/Akt and NF-κB signaling pathways. BMC pulmonary Med. (2024) 24:229. doi: 10.1186/s12890-024-03050-5
31. Huang R, Guo F, Li Y, Liang Y, Li G, Fu P, et al. Activation of AMPK by triptolide alleviates nonalcoholic fatty liver disease by improving hepatic lipid metabolism, inflammation and fibrosis. Phytomedicine. (2021) 92:153739. doi: 10.1016/j.phymed.2021.153739
32. Gervaz P, Morel P, and Vozenin-Brotons MC. Molecular aspects of intestinal radiation-induced fibrosis. Curr Mol Med. (2009) 9:273–80. doi: 10.2174/156652409787847164
33. Liu D, Black BL, and Derynck R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. (2001) 15:2950–66. doi: 10.1101/gad.925901
34. Das P, Inoue H, Baker JC, Beppu H, Kawabata M, Harland RM, et al. Drosophila dSmad2 and Atr-I transmit activin/TGFbeta signals. Genes Cells. (1999) 4:123–34. doi: 10.1046/j.1365-2443.1999.00244.x
35. Rubtsov YP and Rudensky AY. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat Rev Immunol. (2007) 7:443–53. doi: 10.1038/nri2095
36. Schmierer B and Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. (2007) 8:970–82. doi: 10.1038/nrm2297
37. Izzi L and Attisano L. Regulation of the TGFbeta signalling pathway by ubiquitin-mediated degradation. Oncogene. (2004) 23:2071–8. doi: 10.1038/sj.onc.1207412
38. Miyazawa K, Shinozaki M, Hara T, Hara T, Furuya T, and Miyazono K. Two major Smad pathways in TGF-beta superfamily signalling. Genes Cells. (2002) 7:1191–204. doi: 10.1046/j.1365-2443.2002.00599.x
39. Shi Y, Wang YF, Jayaraman L, Yang H, Massagué J, and Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-beta signaling. Cell. (1998) 94:585–94. doi: 10.1016/S0092-8674(00)81600-1
40. Cheng P, Wirka RC, Kim JB, Kim HJ, Nguyen T, Kundu R, et al. Smad3 regulates smooth muscle cell fate and mediates adverse remodeling and calcification of the atherosclerotic plaque. Nat Cardiovasc Res. (2022) 1:322–33. doi: 10.1038/s44161-022-00042-8
41. Ng JWK, Ong EHQ, Tucker-Kellogg L, and Tucker-Kellogg G. Deep learning for de-convolution of Smad2 versus Smad3 binding sites. BMC Genomics. (2022) 23:525. doi: 10.1186/s12864-022-08565-x
42. Itman C and Loveland KL. SMAD expression in the testis: an insight into BMP regulation of spermatogenesis. Dev Dyn. (2008) 237:97–111. doi: 10.1002/dvdy.21401
43. Lee MY, Lim HW, Lee SH, and Han HJ. Smad, PI3K/Akt, and Wnt-dependent signaling pathways are involved in BMP-4-induced ESC self-renewal. Stem Cells (Dayton Ohio). (2009) 27:1858–68. doi: 10.1002/stem.124
44. Siegel PM and Massagué J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. (2003) 3:807–21. doi: 10.1038/nrc1208
45. Chen CR, Kang Y, Siegel PM, and Massagué J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell. (2002) 110:19–32. doi: 10.1016/S0092-8674(02)00801-2
46. Yang J, Song K, Krebs TL, Jackson MW, and Danielpour D. Rb/E2F4 and Smad2/3 link survivin to TGF-beta-induced apoptosis and tumor progression. Oncogene. (2008) 27:5326–38. doi: 10.1038/onc.2008.165
47. Hu HH, Chen DQ, Wang YN, Feng YL, Cao G, Vaziri ND, et al. New insights into TGF-β/Smad signaling in tissue fibrosis. Chemico-biological Interact. (2018) 292:76–83. doi: 10.1016/j.cbi.2018.07.008
48. Shen X, Hu PP, Liberati NT, Datto MB, Frederick JP, and Wang XF. TGF-beta-induced phosphorylation of Smad3 regulates its interaction with coactivator p300/CREB-binding protein. Mol Biol Cell. (1998) 9:3309–19. doi: 10.1091/mbc.9.12.3309
49. Dunn NR, Koonce CH, Anderson DC, Islam A, Bikoff EK, and Robertson EJ. Mice exclusively expressing the short isoform of Smad2 develop normally and are viable and fertile. Genes Dev. (2005) 19:152–63. doi: 10.1101/gad.1243205
50. Liu Y, Festing M, Thompson JC, Hester M, Rankin S, El-Hodiri HM, et al. Smad2 and Smad3 coordinately regulate craniofacial and endodermal development. Dev Biol. (2004) 270:411–26. doi: 10.1016/j.ydbio.2004.03.017
51. Massagué J, Seoane J, and Wotton D. Smad transcription factors. Genes Dev. (2005) 19:2783–810. doi: 10.1101/gad.1350705
52. Derynck R and Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. (2003) 425:577–84. doi: 10.1038/nature02006
53. Schmierer B and Hill CS. Kinetic analysis of Smad nucleocytoplasmic shuttling reveals a mechanism for transforming growth factor beta-dependent nuclear accumulation of Smads. Mol Cell Biol. (2005) 25:9845–58. doi: 10.1128/MCB.25.22.9845-9858.2005
54. Chen F, Wang Q, Xiao M, Lou D, Wufur R, Hu S, et al. A novel crosstalk between Nrf2 and Smad2/3 bridged by two nuanced Keap1 isoforms with their divergent effects on these distinct family transcription factors. Free Radical Biol Med. (2024) 213:190–207. doi: 10.1016/j.freeradbiomed.2024.01.025
55. Chen F, Xiao M, Hu S, and Wang M. Keap1-Nrf2 pathway: a key mechanism in the occurrence and development of cancer. Front Oncol. (2024) 14:1381467. doi: 10.3389/fonc.2024.1381467
56. Sflomos G, Kostaras E, Panopoulou E, Pappas N, Kyrkou A, Politou AS, et al. ERBIN is a new SARA-interacting protein: competition between SARA and SMAD2 and SMAD3 for binding to ERBIN. J Cell Sci. (2011) 124:3209–22. doi: 10.1242/jcs.062307
57. Runyan CE, Schnaper HW, and Poncelet AC. The role of internalization in transforming growth factor beta1-induced Smad2 association with Smad anchor for receptor activation (SARA) and Smad2-dependent signaling in human mesangial cells. J Biol Chem. (2005) 280:8300–8. doi: 10.1074/jbc.M407939200
58. Lin HK, Bergmann S, and Pandolfi PP. Cytoplasmic PML function in TGF-beta signalling. Nature. (2004) 431:205–11. doi: 10.1038/nature02783
59. Faresse N, Colland F, Ferrand N, Prunier C, Bourgeade MF, and Atfi A. Identification of PCTA, a TGIF antagonist that promotes PML function in TGF-beta signalling. EMBO J. (2008) 27:1804–15. doi: 10.1038/emboj.2008.109
60. Faure S, Lee MA, Keller T, ten Dijke P, and Whitman M. Endogenous patterns of TGFbeta superfamily signaling during early Xenopus development. Dev (Cambridge England). (2000) 127:2917–31. doi: 10.1242/dev.127.13.2917
61. De Robertis EM, Larraín J, Oelgeschläger M, and Wessely O. The establishment of Spemann's organizer and patterning of the vertebrate embryo. Nat Rev Genet. (2000) 1:171–81. doi: 10.1038/35042039
62. Zhu Y, Richardson JA, Parada LF, and Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. (1998) 94:703–14. doi: 10.1016/S0092-8674(00)81730-4
63. Heyer J, Escalante-Alcalde D, Lia M, Boettinger E, Edelmann W, Stewart CL, et al. Postgastrulation Smad2-deficient embryos show defects in embryo turning and anterior morphogenesis. Proc Natl Acad Sci USA. (1999) 96:12595–600. doi: 10.1073/pnas.96.22.12595
64. Miyazono KI, Ito T, Fukatsu Y, Wada H, Kurisaki A, and Tanokura M. Structural basis for transcriptional coactivator recognition by SMAD2 in TGF-β signaling. Sci Signaling. (2020) 13(662):eabb9043. doi: 10.1126/scisignal.abb9043
65. Kim YI, Shin HW, Chun YS, Cho CH, Koh J, Chung DH, et al. Epithelial cell-derived cytokines CST3 and GDF15 as potential therapeutics for pulmonary fibrosis. Cell Death Dis. (2018) 9:506. doi: 10.1038/s41419-018-0530-0
66. Li S, Liu X, Nie Y, Yang L, Zhang C, Guo Y, et al. Psoralidin induced differentiation from adipose-derived stem cells to nucleus pulposus-like cells by TGF-β/smad signaling. Curr Mol Med. (2023) 23:688–97. doi: 10.2174/1566524022666220816165135
67. Chen S, Lei L, Li Z, Chen F, Huang Y, Jiang G, et al. Grem1 accelerates nucleus pulposus cell apoptosis and intervertebral disc degeneration by inhibiting TGF-β-mediated Smad2/3 phosphorylation. Exp Mol Med. (2022) 54:518–30. doi: 10.1038/s12276-022-00753-9
68. Ren LL, Miao H, Wang YN, Liu F, Li P, and Zhao YY. TGF-β as A master regulator of aging-associated tissue fibrosis. Aging Dis. (2023) 14:1633–50. doi: 10.14336/AD.2023.0222
69. Geng XQ, Ma A, He JZ, Wang L, Jia YL, Shao GY, et al. Ganoderic acid hinders renal fibrosis via suppressing the TGF-β/Smad and MAPK signaling pathways. Acta Pharmacol Sin. (2020) 41:670–7. doi: 10.1038/s41401-019-0324-7
70. Liang M, Si L, Yu Z, Ding H, Wang L, Chen X, et al. Intermittent hypoxia induces myofibroblast differentiation and extracellular matrix production of MRC5s via HIF-1α-TGF-β/Smad pathway. Sleep Breath. (2024) 28:291–300. doi: 10.1007/s11325-023-02889-y
71. Liu F, Cao Y, Zhang C, and Su H. Decreased DANCR contributes to high glucose-induced extracellular matrix accumulation in human renal mesangial cell via regulating the TGF-β/Smad signaling. FASEB J. (2023) 37:e22926. doi: 10.1096/fj.202300146R
72. Geng K, Ma X, Jiang Z, Gu J, Huang W, Wang W, et al. WDR74 facilitates TGF-β/Smad pathway activation to promote M2 macrophage polarization and diabetic foot ulcer wound healing in mice. Cell Biol Toxicol. (2023) 39:1577–91. doi: 10.1007/s10565-022-09748-8
73. Chung JY-F, Zhang Y-Y, Ji ZZ-Y, Tang T, Chen JY, Tang SCW, et al. immunodynamics_of_macrophages_in_renal_fibrosis. Integr Med Nephrol Androl. (2023) 10:e00001. doi: 10.1097/IMNA-D-23-00001
74. Tang PM, Zhang YY, Mak TS, Tang PC, Huang XR, Lan HY, et al. Transforming growth factor-β signalling in renal fibrosis: from Smads to non-coding RNAs. J Physiol. (2018) 596:3493–503. doi: 10.1113/JP274492
75. Yuan X, Gong Z, Wang B, Guo X, Yang L, Li D, et al. Astragaloside inhibits hepatic fibrosis by modulation of TGF-β1/smad signaling pathway. Evid Based Complement Alternat Med. (2018) 2018:3231647. doi: 10.1155/2018/3231647
76. Chen F, Li Z, Wang T, Fu Y, Lyu L, Xing C, et al. MOTS-c mimics exercise to combat diabetic liver fibrosis by targeting Keap1-Nrf2-Smad2/3. Sci Rep. (2025) 15:18460. doi: 10.1038/s41598-025-03526-2
77. Wang YR, Hong RT, Xie YY, and Xu JM. Melatonin ameliorates liver fibrosis induced by carbon tetrachloride in rats via inhibiting TGF-β1/smad signaling pathway. Curr Med Sci. (2018) 38:236–44. doi: 10.1007/s11596-018-1871-8
78. Zhang T, Wang XF, Wang ZC, Lou D, Fang QQ, Hu YY, et al. Current potential therapeutic strategies targeting the TGF-β/Smad signaling pathway to attenuate keloid and hypertrophic scar formation. Biomed Pharmacother. (2020) 129:110287. doi: 10.1016/j.biopha.2020.110287
79. Hsieh YC, Lee KC, Lei HJ, Lan KH, Huo TI, Lin YT, et al. (Pro)renin receptor knockdown attenuates liver fibrosis through inactivation of ERK/TGF-β1/SMAD3 pathway. Cell Mol Gastroenterol Hepatol. (2021) 12:813–38. doi: 10.1016/j.jcmgh.2021.05.017
80. Zhang L, Liu C, Meng XM, Huang C, Xu F, and Li J. Smad2 protects against TGF-β1/Smad3-mediated collagen synthesis in human hepatic stellate cells during hepatic fibrosis. Mol Cell Biochem. (2015) 400:17–28. doi: 10.1007/s11010-014-2258-1
81. Shen M, Zheng Y, Tu J, and Zhao F. Mechanism of astaxanthin-mediated TGF-β/SMAD signaling pathway in the activation of LX-2 cells and anti-hepatic fibrosis. ESRSA. (2025) 18(3):1641–55. doi: 10.1016/j.jrras.2025.101713
82. Peng X, Yang H, Tao L, Xiao J, Zeng Y, Shen Y, et al. Fluorofenidone alleviates liver fibrosis by inhibiting hepatic stellate cell autophagy via the TGF-β1/Smad pathway: implications for liver cancer. PeerJ. (2023) 11:e16060. doi: 10.7717/peerj.16060
83. Li W, Chen L, Zhou Q, Huang T, Zheng W, Luo F, et al. Liver macrophage-derived exosomal miRNA-342-3p promotes liver fibrosis by inhibiting HPCAL1 in stellate cells. Hum Genomics. (2025) 19:9. doi: 10.1186/s40246-025-00722-z
84. Shi HH, Zhang LY, Chen LP, Yang JY, Wang CC, Xue CH, et al. EPA-enriched phospholipids alleviate renal interstitial fibrosis in spontaneously hypertensive rats by regulating TGF-β Signaling pathways. Mar Drugs. (2022) 20(2):152. doi: 10.3390/md20020152
85. Chen Y, Dai Y, Huang Y, Zhang L, Zhang C, Gao H, et al. Inhibition of tubular epithelial cells ferroptosis alleviates renal interstitial fibrosis by reducing lipid hydroperoxides and TGF-β/Smad signaling. Cell Commun Signal. (2025) 23:81. doi: 10.1186/s12964-025-02068-4
86. Phanish MK, Wahab NA, Colville-Nash P, Hendry BM, and Dockrell ME. The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem J. (2006) 393:601–7. doi: 10.1042/BJ20051106
87. Gu YY, Liu XS, and Lan HY. Therapeutic potential for renal fibrosis by targeting Smad3-dependent noncoding RNAs. Mol therapy: J Am Soc Gene Ther. (2024) 32:313–24. doi: 10.1016/j.ymthe.2023.12.009
88. Wang Y and Zhao J. The protective function of αKlotho in chronic kidney disease: evidence and therapeutic implications. Integr Med Nephrol Androl. (2024) 11:e24–00021. doi: 10.1097/IMNA-D-24-00021
89. Li J, Qu X, Yao J, Caruana G, Ricardo SD, Yamamoto Y, et al. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes. (2010) 59:2612–24. doi: 10.2337/db09-1631
90. Abd-Elmawla MA, Zidan M, Elsabagh YA, Elfar N, and Radwan AF. Dissecting the role of SPRY4-IT1 and TUG1 in modulating miR-425/TGF-β/ Smad signaling in mediating renal fibrosis and inflammation in lupus nephritis: Novel biomarkers and therapeutic targets. Int Immunopharmacol. (2025) 162:115132. doi: 10.1016/j.intimp.2025.115132
91. Zhao BR, Hu XR, Wang WD, and Zhou Y. Cardiorenal syndrome: clinical diagnosis, molecular mechanisms and therapeutic strategies. Acta pharmacologica Sin. (2025) 46:1539–55. doi: 10.1038/s41401-025-01476-z
92. Yao Q, Zheng X, Zhang X, Wang Y, Zhou Q, Lv J, et al. METTL3 potentiates M2 macrophage-driven MMT to aggravate renal allograft fibrosis via the TGF-β1/smad3 pathway. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2025) 12:e2412123. doi: 10.1002/advs.202412123
93. He H, Wang H, Chen X, Zhong Y, Huang XR, Ma RC, et al. Treatment for type 2 diabetes and diabetic nephropathy by targeting Smad3 signaling. Int J Biol Sci. (2024) 20:200–17. doi: 10.7150/ijbs.87820
94. Ni JY, Wang X, Xie HY, Yang NH, Li JY, Sun XA, et al. Deubiquitinating enzyme USP11 promotes renal tubular cell senescence and fibrosis via inhibiting the ubiquitin degradation of TGF-β receptor II. Acta pharmacologica Sin. (2023) 44:584–95. doi: 10.1038/s41401-022-00977-5
95. Wei C, Zhang Y, Zhong X, Lu S, Zou X, Yang Y, et al. Ginkgo biloba leaf extract mitigates cisplatin-induced chronic renal interstitial fibrosis by inhibiting the epithelial-mesenchymal transition of renal tubular epithelial cells mediated by the Smad3/TGF-β1 and Smad3/p38 MAPK pathways. Chin Med. (2022) 17:25. doi: 10.1186/s13020-022-00574-y
96. Shi Z, Liu J, Qin J, Liang X, Ou X, Zhang T, et al. Astilbin Alleviates Radiation-Induced Pulmonary Fibrosis via circPRKCE Targeting the TGF-β/Smad7 Pathway to Inhibit Epithelial-Mesenchymal Transition. Biomedicines. (2025) 13(3):689. doi: 10.3390/biomedicines13030689
97. Elsheikh AA, Shalaby AM, Alabiad MA, Abd-Almotaleb NA, and Khayal EE. Perfluorooctanoic acid induced lung toxicity via TGF-β1/Smad pathway, crosstalk between airway hyperresponsiveness and fibrosis: withdrawal impact. Environ Sci pollut Res Int. (2025) 32:4989–5007. doi: 10.1007/s11356-025-36005-2
98. Hao M, Guan Z, Zhang Z, Ai H, Peng X, Zhou H, et al. Atractylodinol prevents pulmonary fibrosis through inhibiting TGF-β receptor 1 recycling by stabilizing vimentin. Mol therapy: J Am Soc Gene Ther. (2023) 31:3015–33. doi: 10.1016/j.ymthe.2023.08.017
99. Liu T, Zhang X, Yan X, Cheng L, Yan X, Zeng F, et al. Smad4 deficiency in S100A4(+) macrophages enhances colitis-associated tumorigenesis by promoting macrophage lipid metabolism augmented M2 polarization. Int J Biol Sci. (2024) 20:6114–29. doi: 10.7150/ijbs.98529
100. Pattnaik B, Negi V, Chaudhuri R, Desiraju K, Faizan MI, Akhtar A, et al. MiR-326-mediated overexpression of NFIB offsets TGF-β induced epithelial to mesenchymal transition and reverses lung fibrosis. Cell Mol Life sciences: CMLS. (2023) 80:357. doi: 10.1007/s00018-023-05005-1
101. Zhang WY, Wen L, Du L, Liu TT, Sun Y, Chen YZ, et al. S-RBD-modified and miR-486-5p-engineered exosomes derived from mesenchymal stem cells suppress ferroptosis and alleviate radiation-induced lung injury and long-term pulmonary fibrosis. J nanobiotechnol. (2024) 22:662. doi: 10.1186/s12951-024-02830-9
102. Su Y, Shi D, Xia G, Liu Y, Xu L, Dao L, et al. Carbonic Anhydrase 3 is required for cardiac repair post myocardial infarction via Smad7-Smad2/3 signaling pathway. Int J Biol Sci. (2024) 20:1796–814. doi: 10.7150/ijbs.91396
103. Chen C, Li X, Zhou T, Su Y, Yu B, Jin J, et al. Ubiquitin like protein FAT10 repressed cardiac fibrosis after myocardial ischemic via mediating degradation of Smad3 dependent on FAT10-proteasome system. Int J Biol Sci. (2023) 19:881–96. doi: 10.7150/ijbs.77677
104. Gao W, Shao R, Zhang X, Liu D, Liu Y, and Fa X. Up-regulation of caveolin-1 by DJ-1 attenuates rat pulmonary arterial hypertension by inhibiting TGFβ/Smad signaling pathway. Exp Cell Res. (2017) 361:192–8. doi: 10.1016/j.yexcr.2017.10.019
105. Zhang B, Hou J, Liu J, He J, Gao Y, Li G, et al. Hydrogen decreases susceptibility to AngII-induced atrial fibrillation and atrial fibrosis via the NOX4/ROS/NLRP3 and TGF-β1/Smad2/3 signaling pathways. PloS One. (2025) 20:e0310852. doi: 10.1371/journal.pone.0310852
106. Lai YJ, Tsai FC, Chang GJ, Chang SH, Huang CC, Chen WJ, et al. miR-181b targets semaphorin 3A to mediate TGF-β-induced endothelial-mesenchymal transition related to atrial fibrillation. J Clin Invest. (2022) 132(13):e142548. doi: 10.1172/JCI142548
107. Mushtaq I, Hsieh TH, Chen YC, Kao YH, and Chen YJ. MicroRNA-452-5p regulates fibrogenesis via targeting TGF-β/SMAD4 axis in SCN5A-knockdown human cardiac fibroblasts. iScience. (2024) 27:110084. doi: 10.1016/j.isci.2024.110084
108. Xie S, Xing Y, Shi W, Zhang M, Chen M, Fang W, et al. Cardiac fibroblast heat shock protein 47 aggravates cardiac fibrosis post myocardial ischemia-reperfusion injury by encouraging ubiquitin specific peptidase 10 dependent Smad4 deubiquitination. Acta Pharm Sin B. (2022) 12:4138–53. doi: 10.1016/j.apsb.2022.07.022
109. Tu L, Lin Z, Huang Q, et al. USP15 enhances the proliferation, migration, and collagen deposition of hypertrophic scar-derived fibroblasts by deubiquitinating TGF-βR1 in vitro. Plast Reconstr Surg. (2021) 148:1040–51. doi: 10.1097/PRS.0000000000008488
110. Huang Y, Zhao H, Zhang Y, Tang Y, Shi X, Jiang S, et al. Enhancement of zyxin promotes skin fibrosis by regulating FAK/PI3K/AKT and TGF-β Signaling pathways via integrins. Int J Biol Sci. (2023) 19:2394–408. doi: 10.7150/ijbs.77649
111. Yang PY, Hsieh PL, Yeh JC, Ho CT, Liao YW, Wei YL, et al. Osthole mitigates the myofibroblast properties in oral submucous fibrosis by suppressing the TGF-β/smad2 signaling pathway and NCK-AS1 expression. J Dental Sci. (2025) 20:911–8. doi: 10.1016/j.jds.2024.08.021
112. Xiao Y, Xiang Q, Wang Y, Huang Z, Yang J, Zhang X, et al. Exosomes carrying adipose mesenchymal stem cells function alleviate scleroderma skin fibrosis by inhibiting the TGF-β1/Smad3 axis. Sci Rep. (2025) 15:7162. doi: 10.1038/s41598-024-72630-6
113. Shang RY, Yang JC, Hu WG, Xiao R, Hu DS, Lin ZC, et al. Artesunate attenuates skin hypertrophic scar formation by inhibiting fibroblast activation and EndMT of vascular endothelial cells. Phytomedicine. (2025) 140:156498. doi: 10.1016/j.phymed.2025.156498
114. Ji Z, Lin S, Gui S, Gao J, Cao F, Guan Y, et al. Overexpressed poldip2 incurs retinal fibrosis via the TGF-β1/SMAD3 signaling pathway in diabetic retinopathy. Diabetes. (2024) 73:1742–55. doi: 10.2337/db23-1036
115. Zidan AA, Zhu S, Elbasiony E, Najafi S, Lin Z, Singh RB, et al. Topical application of calcitonin gene-related peptide as a regenerative, antifibrotic, and immunomodulatory therapy for corneal injury. Commun Biol. (2024) 7:264. doi: 10.1038/s42003-024-05934-y
116. Chang TM, Fang WY, Hsu HP, Chu PY, Jiang SS, Huang KW, et al. PCK2 promotes invasion and epithelial-to-mesenchymal transition in triple-negative breast cancer by promoting TGF-β/SMAD3 signaling through inhibiting TRIM67-mediated SMAD3 ubiquitination. Cancer Biol Ther. (2025) 26:2478670. doi: 10.1080/15384047.2025.2478670
117. Breedy S, Ratnayake WS, Lajmi L, Hill R, and Acevedo-Duncan M. 14-3–3 and Smad2/3 are crucial mediators of atypical-PKCs: Implications for neuroblastoma progression. Front Oncol. (2023) 13:1051516. doi: 10.3389/fonc.2023.1051516
118. Li H, Li J, Hu Y, Zhang R, Gu X, Wei Y, et al. FOXO3 regulates Smad3 and Smad7 through SPON1 circular RNA to inhibit idiopathic pulmonary fibrosis. Int J Biol Sci. (2023) 19:3042–56. doi: 10.7150/ijbs.80140
119. Chen F, Xiao M, Feng J, Wufur R, Liu K, Hu S, et al. Different inhibition of nrf2 by two keap1 isoforms α and β to shape Malignant behaviour of human hepatocellular carcinoma. Int J Mol Sci. (2022) 23(18):10342. doi: 10.3390/ijms231810342
120. Jiang Y, Xie F, Lv X, Wang S, Liao X, Yu Y, et al. Mefunidone ameliorates diabetic kidney disease in STZ and db/db mice. FASEB J. (2021) 35:e21198. doi: 10.1096/fj.202001138RR
121. Hussein ZA, Abu-Raghif AR, Tahseen NJ, Rashed KA, Shaker NS, and Fawzi HA. Vinpocetine alleviated alveolar epithelial cells injury in experimental pulmonary fibrosis by targeting PPAR-γ/NLRP3/NF-κB and TGF-β1/Smad2/3 pathways. Sci Rep. (2024) 14:11131. doi: 10.1038/s41598-024-61269-y
122. Huang Q, Chen J, Liao S, Long J, Fang R, He Y, et al. The SGLT2 inhibitor empagliflozin inhibits skeletal muscle fibrosis in naturally aging male mice through the AMPKα/MMP9/TGF-β1/Smad pathway. Biogerontology. (2024) 25:567–81. doi: 10.1007/s10522-024-10093-y
123. Wang W, Li Y, Zhang C, Zhou H, Li C, Cheng R, et al. Small Extracellular Vesicles from Young Healthy Human Plasma Inhibit Cardiac Fibrosis After Myocardial Infarction via miR-664a-3p Targeting SMAD4. Int J nanomed. (2025) 20:557–79. doi: 10.2147/IJN.S488368
124. Zuo R, Guo X, Song X, Gao X, Zhang J, Jiang S, et al. New uses of halofuginone to treat cancer. J Pharm Anal. (2025) 15:101080. doi: 10.1016/j.jpha.2024.101080
125. Yang Z, Chang Y, Zhou T, Sui W, Dai P, Wei Y, et al. Astragalus mongholicus bunge and Angelica sinensis botanical drug decoction mitigates lung inflammation through NOX4/TGF-β1/SMAD3 signaling. Front Pharmacol. (2025) 16:1565569. doi: 10.3389/fphar.2025.1565569
126. Liu H, Lai W, Nie H, Shi Y, Zhu L, Yang L, et al. PM(2.5) triggers autophagic degradation of Caveolin-1 via endoplasmic reticulum stress (ERS) to enhance the TGF-β1/Smad3 axis promoting pulmonary fibrosis. Environ Int. (2023) 181:108290. doi: 10.1016/j.envint.2023.108290
Keywords: fibrosis, TGF-β/Smad signalling pathway, ECM, miRNA, EMT
Citation: Chen F, Lyu L, Xing C, Chen Y, Hu S, Wang M and Ai Z (2025) The pivotal role of TGF-β/Smad pathway in fibrosis pathogenesis and treatment. Front. Oncol. 15:1649179. doi: 10.3389/fonc.2025.1649179
Received: 18 June 2025; Accepted: 18 August 2025;
Published: 03 September 2025.
Edited by:
Xiaoyong Yu, Shaanxi Provincial Hospital of Traditional Chinese Medicine, ChinaReviewed by:
Wenjing Wu, Guangdong Provincial People’s Hospital, ChinaMarwan Almoiliqy, University of Texas MD Anderson Cancer Center, United States
Eskandar Qaed, Lanzhou University, China
Güven Gürsoy, Faculty of Medicine. Mugla University, Türkiye
Copyright © 2025 Chen, Lyu, Xing, Chen, Hu, Wang and Ai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feilong Chen, Y2hlbmZlaWxvbmdAY2RzdS5lZHUuY24=