 Jingsong Mao
Jingsong Mao Qingxuan Gai
Qingxuan Gai Ming Zhong
Ming Zhong- Department of Oral Histopathology, School and Hospital of Stomatology, China Medical University, Shenyang, China
Background: Ameloblastoma is a benign but locally aggressive odontogenic tumor with frequent recurrence after conservative surgery. Evidence accumulated since 2010 implicates dysregulated non-coding RNAs (ncRNAs)—notably microRNAs (miRNAs) and circular RNAs (circRNAs)—as higher-order regulators of oncogenic signaling.
Objective: This study aimed to synthesize peer-reviewed mechanistic and translational evidence on ncRNA networks in ameloblastoma, with explicit grading by evidence tier and emphasis on druggable nodes.
Methods: We conducted a structured narrative search of PubMed, Scopus, and Web of Science (January 2010–May 31, 2025) using controlled terms for “ameloblastoma,” “microRNA,” “circRNA,” and key pathways (MAPK, PI3K–Akt–mTOR, Wnt/β-catenin, IL-33/STAT3; Hippo/YAP–TAZ considered contextually). Peer-reviewed studies with experimental validation in ameloblastoma were prioritized, while purely computational predictions and unrelated tumor entities were excluded.
Results: Across patient tissues, cell models, and limited in vivo studies, recurrent miRNA changes—i.e., loss of miR-524-5p, miR-141-3p, and miR-1-3p and gain of miR-29a-3p—converge on MAPK/ERK and PI3K–Akt–mTOR signaling. Loss of miR-524-5p derepresses IL-33/ST2, amplifying NF-κB/STAT3 and PI3K signaling (preclinical). miR-29a-3p targets CTNNBIP1 to reinforce Wnt/β-catenin (preclinical). miR-141-3p is anti-migratory and has been reported to upregulate NCAM1 in ameloblastoma models (preclinical). miR-1-3p restrains LAMP2-mediated autophagy (preclinical). Overexpressed circRNAs (e.g., circ-MAP3K7 and circ-HIPK3) can titrate tumor-suppressive miRNAs and sustain pathway activity (preclinical). No randomized clinical trials in ameloblastoma exist to date.
Conclusions: A coherent ncRNA network appears to maintain druggable signaling convergence in ameloblastoma. Translation will require multicenter validation of the ncRNA biomarkers, early-phase trials testing rational MAPK–mTOR combinations with ncRNA modulation, and jaw-targeted delivery approaches. Claims herein are limited to peer-reviewed, ameloblastoma-relevant evidence.
Graphical Abstract.
1 Introduction
1.1 Overview of ameloblastoma
Ameloblastoma is a benign but locally aggressive odontogenic epithelial tumor that arises from enamel–organ or dental–lamina remnants (1). The global incidence is estimated at ≈0.5–1.5 new cases per million person-years, and the lesion still represents ~1% of all jaw cysts and tumors. More than 80% occur in the posterior mandible (molar–ramus region). Maxillary lesions, although less common, often behave more aggressively due to the thin surrounding bone and the proximity to vital structures (2) (Table 1).
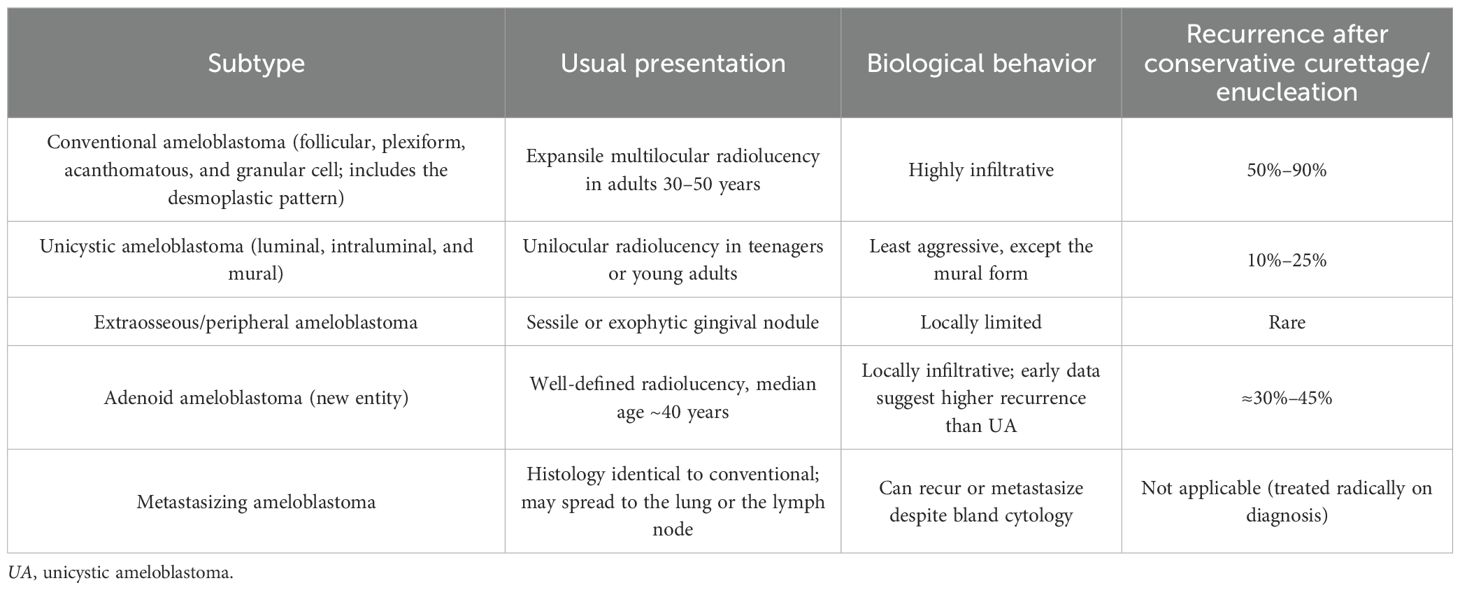
Table 1. Clinicopathologic subtypes recognized in WHO 2022 (2).
Although histologically benign, ameloblastoma can displace the teeth, perforate the cortical bone, and cause a marked facial asymmetry. Untreated tumors may attain a massive size. The standard of care is wide (≈1 cm) en bloc resection, where feasible. This reduces long-term recurrence to roughly 10%–15%, whereas curettage, marsupialization, or enucleation carries a several-fold higher risk (3).
Next-generation sequencing has uncovered site- and subtype-specific molecular alterations in odontogenic tumors: approximately 60%–70% of mandibular conventional and unicystic ameloblastomas harbor BRAF V600E mutations, while many maxillary cases exhibit activating SMO L412F/W535L mutations. Less frequent alterations involve PTCH1, KRAS, FGFR2, and other MAPK (mitogen-activated protein kinase) pathway genes (4). In contrast, adenoid ameloblastoma typically lacks BRAF mutations, but commonly carries CTNNB1 (β-catenin) exon 3 mutations, which underlie its distinctive cribriform ductal phenotype (5).
This review integrates studies retrieved through a structured search in PubMed, Scopus, and Web of Science, focusing on publications through May 2025. The search terms included “ameloblastoma,” “microRNA,” “circRNA,” “non-coding RNA,” and related combinations. Eligible sources were restricted to peer-reviewed original research and reviews reporting experimentally validated mechanisms or expression data in ameloblastoma. Purely computational predictions or unvalidated preprints were excluded.
1.2 miRNA and its role in cancer
MicroRNAs (miRNAs) are 19- to 24-nucleotide single-stranded RNAs loaded into the Argonaute-containing RISC complex by a typical Drosha–DGCR8→Exportin-5→Dicer pathway. The target transcripts’ 3′-UTR complementary sites coupled with the miRNA seed (nucleotides 2–8) cause translational repression or messenger RNA (mRNA) destruction. The human genome codes more than 2,700 mature miRNAs; combined, they regulate at least one-third of the protein-coding genes (6).
In oncogenesis, these miRNAs fit two primary functional groups: Their overexpression turns oncomiRs silent of tumor suppressors. Two such examples are miR-21 and miR-155, which downregulate PTEN or SHIP1, therefore boosting phosphatidyl-inositol 3-kinase (PI3K)–Akt signaling and supporting survival, invasion, and therapy resistance (7).
Loss of tumor-suppressive miRNAs releases oncogenic transcripts. Targeting ZEB1/2, the miR-200 family members impede the epithelial–mesenchymal transition (EMT). miR-34a (a direct p53 target) constrains MYC, MET, and BCL-2, while miR-1, miR-133, and miR-206 reduce the cell cycle progression and metabolism (8).
Cancer-associated miRNA dysregulation results from either gene amplification or deletion, promoter methylation, transcription factor alteration, defective miRNA-processing enzymes, or sequestration by competing endogenous RNAs (ceRNAs), including circular RNAs (circRNAs) and long non-coding RNAs (lncRNAs) (9). Small changes in abundance can produce significant phenotypic changes as individual miRNAs can simultaneously modify hundreds of transcripts within a signaling network. This makes miRNAs effective biomarkers and appealing therapeutic entry points (10).
1.3 Research background and significance
Studies on non-coding RNAs (ncRNAs) in ameloblastoma are still in their early years compared with oral squamous cancer. Fewer than 150 original research studies have examined the miRNA or circRNA expression in this tumor, while mechanistic dissection is extremely scarce. However, the scattered data formed a clear pattern: miR-29a-3p is consistently upregulated and drives invasion by stabilizing β-catenin and activating canonical Wnt signaling, while miR-524-5p, miR-141-3p, and miR-1-3p are downregulated. Loss of these miRNAs removes negative control over, respectively, the IL-33/ST2 inflammatory axis, the NCAM1-mediated motility, and the LAMP2-dependent autophagy. Multiple circRNAs—including circ_0007059, circ-HIPK3, and circ-FBXW7—act as “sponges,” binding tumor-suppressive miRNAs and, thus, indirectly amplifying the MAPK and PI3K/Akt/mTOR (mechanistic target of rapamycin) signaling cascades. On the genomic side, BRAF-mutant mandibular lesions and SMO-mutant maxillary lesions converge on downstream MAPK or Hedgehog activity. Both axes crosstalk with PI3K/Akt/mTOR, suggesting combinatorial vulnerabilities.
Nonetheless, the functions of miRNA and circRNA are not universally consistent across tumor types. For instance, miR-29a-3p, which is oncogenic in ameloblastoma, has been reported as tumor-suppressive in gastrointestinal and hematologic malignancies. Similarly, the expression and target selection of miR-141-3p vary in tissue-specific contexts. These discrepancies suggest that ncRNA networks may operate differently depending on the cellular background, microenvironmental factors, and the transcriptional landscapes, warranting careful contextual interpretation (11).
Adenoid ameloblastoma typically lacks BRAF V600E but harbors CTNNB1 exon 3 mutations with diffuse nuclear β-catenin activity, suggesting a distinct ncRNA–Wnt co-regulatory axis compared with classic ameloblastoma. Odontogenic keratocyst (OKC) is frequently linked to PTCH1/Hedgehog activation, and YAP/TAZ upregulation has been reported in keratocystic lesions, indicating Hippo–mechanotransduction involvement. These contrasts underscore the lesion-specific ncRNA wiring and support the novelty of ameloblastoma-focused ncRNA circuitry.
Radical excision remains the standard therapy, which often sacrifices mandibular continuity or maxillary integrity and requires complex reconstruction. Even then, micrometric epithelial islands left at the margin can seed recurrence years later. A precise molecular atlas that integrates miRNA, circRNA, and driver mutation data could transform management in three ways: biomarkers—saliva- or serum-based miRNA signatures may permit early detection, subtype stratification, and real-time monitoring for recurrence; targeted therapy—patients whose tumors are dependent on BRAF/MAPK or hyperactive Akt/mTOR signaling could receive pathway inhibitors, reducing the tumor volume preoperatively or controlling inoperable disease; and ncRNA therapeutics—synthetic miRNA mimics, anti-miRs, or circRNA disruptors delivered via tumor-targeted nanoparticles could restore critical regulatory circuits with far less morbidity than surgery (12).
However, the translation of these therapeutics faces critical barriers. The delivery of miRNA mimics or inhibitors to jaw lesions remains constrained by low vascular permeability, limited diffusion through mineralized matrix, rapid systemic clearance, and potential immune activation. Nanoparticles also risk off-target accumulation, and endosomal escape remains inefficient. Consequently, clinical success will require refined delivery systems with high tissue specificity and minimal systemic toxicity.
By weaving together a fragmented literature, the present review sets the stage for rational biomarker discovery, preclinical drug testing, and, ultimately, precision medicine trials capable of shifting ameloblastoma therapy from knife to molecule.
2 Literature selection criteria
To ensure comprehensiveness and scientific rigor, we employed a structured yet narrative approach to literature selection. The following criteria were applied.
2.1 Inclusion criteria
Publication date: Articles published between January 2010 and May 2025 in order to reflect the most current understanding of the ncRNA-related mechanisms in ameloblastoma and related tumors.
Article type: Peer-reviewed original research articles, systematic reviews, and meta-analyses published in indexed biomedical journals (e.g., PubMed, Scopus, and Web of Science).
Language: English-language publications only.
Scope: Studies focusing on ncRNAs (including miRNAs, circRNAs, and lncRNAs) and their involvement in ameloblastoma pathogenesis, therapeutic targeting, or molecular signaling pathways (e.g., PI3K/AKT/mTOR, MAPK, Wnt/β catenin, and Hippo).
Relevance to precision therapy: Studies providing molecular, preclinical, or translational evidence supporting the ncRNA-mediated regulation of key oncogenic or tumor suppressive pathways were prioritized.
2.2 Exclusion criteria
The following publications were excluded: non-peer-reviewed literature, including preprints, editorials, commentaries, and dissertations; articles focused exclusively on non-odontogenic tumors, unless a comparative framework was provided for ameloblastoma-related insights; studies lacking experimental validation (e.g., purely computational predictions without biological confirmation), unless cited for pathway modeling or hypothesis support; reports on ncRNAs unrelated to ameloblastoma or its analog signaling pathways, or where the ncRNA relevance was incidental or marginal; and duplicated data across multiple publications by the same research group, unless a significant update or extended findings were reported.
This framework ensured that the review maintained both depth and focus, capturing high-quality evidence while avoiding redundancy and irrelevant content.
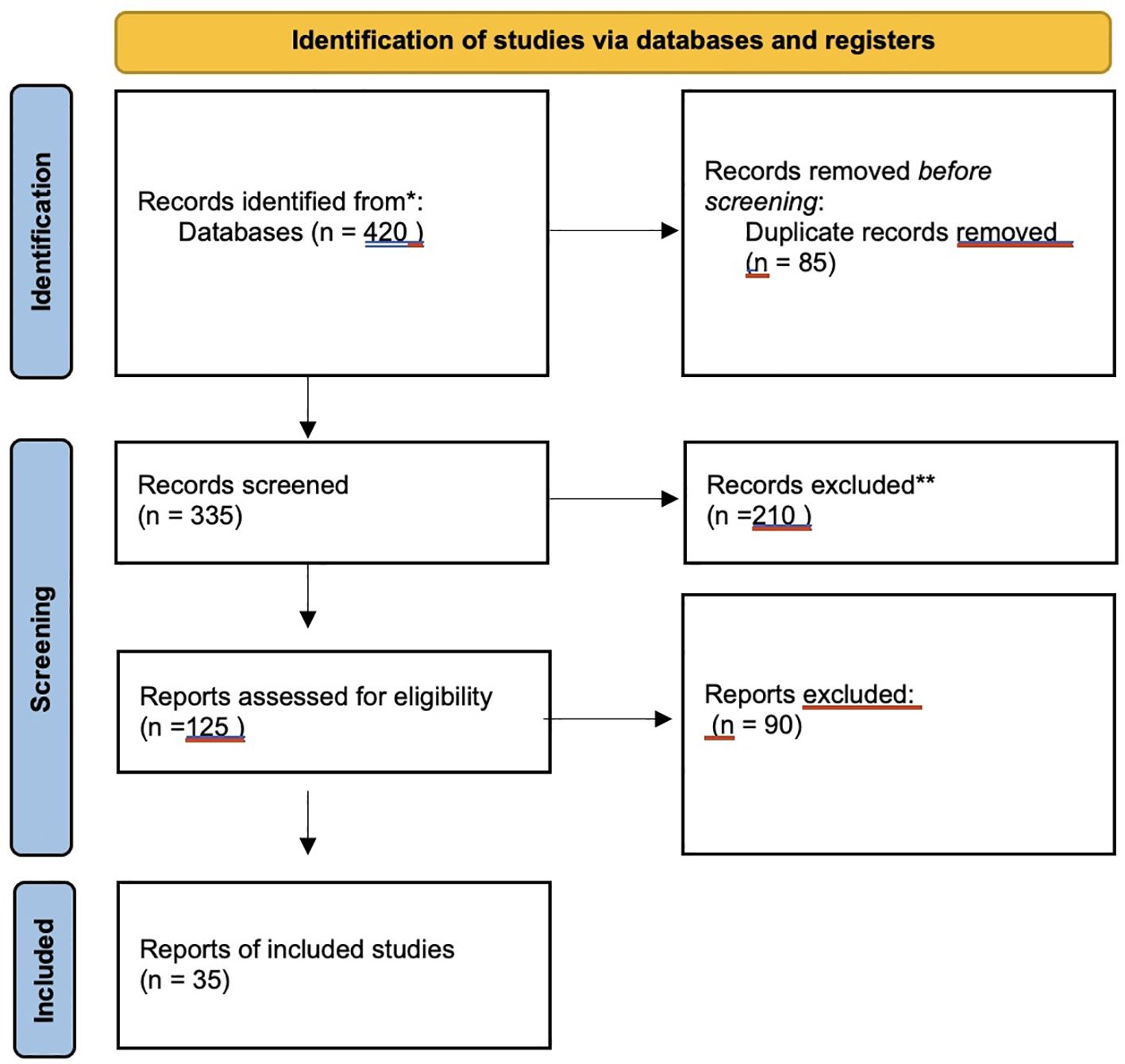
2.3 PRISMA flow diagram
3 Expression of miRNA in ameloblastoma
3.1 Expression and function of miR-524-5p
3.1.1 Expression characteristics of miR-524-5p in ameloblastoma
Across patient tissue and cell models, miR-524-5p is downregulated in ameloblastoma and directly targets IL-33. A reduced miR-524-5p is associated with a higher IL-33/ST2 signaling and a pro-inflammatory tone (luciferase validation and rescue experiments). To date, the evidence has been preclinical and ameloblastoma-specific.
Methylation profiling of the MIR525 promoter revealed dense CpG hypermethylation in approximately two-thirds of miR-524-5p-low tumors. Functionally, treatment of AM-1 cells with 5-aza-2′-deoxycytidine partially restores mature miR-524-5p and attenuates colony formation, confirming promoter methylation as a central silencing mechanism.
Concurrently, circRNA sequencing uncovered a dominant mandibular circRNA (circ-AB1) containing three conserved seed sites for miR-524-5p. Argonaute-2 immunoprecipitation assays verified substantial miR-524-5p sequestration, suggesting a ceRNA-like mechanism driving posttranscriptional repression (13).
Published data support IL-33 as a direct miR-524-5p target in ameloblastoma. Loss of miR-524-5p can augment NF-κB/STAT3 and PI3K signaling, consistent with a pro-inflammatory microenvironment. These findings are preclinical, and clinical validation and multicenter replication are needed (14, 15).
3.2 Effect of miR-29a-3p on ameloblastoma
3.2.1 Expression of miR-29a-3p in ameloblastoma
Published studies indicate that miR-29a-3p is upregulated in ameloblastoma. In cell models, gain- and loss-of-function experiments have supported enhanced migration and invasion via the targeting of CTNNBIP1 and the reinforcement of canonical Wnt/β-catenin signaling (16, 17).
3.2.2 miR-29a-3p promotes migration and invasion through the Wnt/β-catenin pathway
Functional studies have shown that miR-29a-3p suppresses CTNNBIP1, increases the β-catenin transcriptional activity and EMT markers, and enhances the motility in ameloblastoma cell models (18).
It is important to note that the regulatory efficacy of circRNAs as miRNA sponges is strongly influenced by their relative abundance and binding stoichiometry. Experimental models have indicated that effective derepression of β-catenin targets requires high circRNA-to-miRNA ratios and multiple seed matches. Inadequate circRNA levels or low site occupancy can fail to buffer the effects of miRNAs, suggesting that quantitative modeling is essential to validate the proposed ceRNA mechanisms (19).
Such findings underscore ncRNAs orchestrating a complex regulatory crosstalk among oncogenic pathways. For example, the miR-29a-3p-driven IRS-1 expression potentiates PI3K/Akt signaling, while miR-141-3p loss removes the repression of NCAM1, which in turn recruits Fyn–PI3K complexes. These events synchronize with the IL-33/ST2 axis influenced by miR-524-5p depletion. Together, these interlinked loops reinforce MAPK, Wnt/β-catenin, and mTOR activity, forming a resilient oncogenic network (18–20).
3.3 Regulation and effects of hsa-miR-141-3p on NCAM1
Hsa-miR-141-3p, a member of the miR-200 family that canonically reduces motility by imposing an epithelial phenotype, is the third node in the ameloblastoma miRNA network. Two convergent mechanisms for this depletion are shown by primary culture studies: a desmoplastic matrix rich in periostin down-modulates Dicer activity, hence further reducing miRNA maturation, and TGF-β stimulates ZEB1, which transcriptionally represses MIR141. NCAM1 thus escapes posttranscriptional surveillance since its 3′-UTR contains a high-affinity miR-141-3p seed site. Western blotting and immunostaining matched materials reveal a reciprocal pattern: low miR-141-3p corresponds with a thick membranous NCAM1 in tumor islands arranged along nerves and blood vessels. Functionally, the forced expression of miR-141-3p in AM-1 cells reduces the NCAM1 protein by more than 60%, attenuates FAK and paxillin phosphorylation, and slows the scratch-wound closure and Transwell migration by half (21), In ameloblastoma models, miR-141-3p exerts anti-migratory effects and has been reported to upregulate NCAM1. Its overexpression reduces migration in vitro. Thus, a reduced miR-141-3p may attenuate NCAM1 and facilitate motility. Linkage to PI3K/Akt is plausible, but remains preclinical (22).
3.4 Tumor heterogeneity and epigenetic architecture
Ameloblastoma exhibits substantial molecular heterogeneity beyond its histologic and anatomic classification. Genomic profiling has revealed distinct ncRNA expression profiles between BRAF^V600E-mutant mandibular tumors and SMO^L412F maxillary variants. In parallel, epigenetic modifications, such as MIR524 promoter methylation or Dicer downregulation, in periostin-rich microenvironments alter miRNA maturation. These changes result in differential regulatory control across spatial niches and tumor subclones. Such diversity complicates predictive modeling and therapeutic targeting, emphasizing the need for integrative single-cell and spatial transcriptomics in future ncRNA-based strategies.
3.5 The tumor-suppressive role of miR-1-3p
Across 80 tumors, quantitative RT-PCR places miR-1-3p among the five most downregulated genes. Its depletion is correlated with a bigger lesion size and a multilocular architecture. Transcriptional silencing seems to result from the disturbance of the serum response factor/myocardin enhancer complex in epithelial cells that have wandered far from their odontogenic root coupled with promoter hypermethylation (23). Loss of miR-1-3p sets the direct 3′-UTR target lysosomal membrane protein LAMP2 free from control. An elevated LAMP2 increases autophagic flux—shown by the conversion of LC3-I to LC3-II and the clearance of p62—allowing cancer cells to recycle nutrients under the hypoxic, nutrient-poor circumstances that define the growing mandibular marrow cavity. The reintroduction of miR-1-3p reduces the LAMP2 protein by over 70%, collapses autophagosome maturation, and generates the mitochondrial reactive oxygen species (ROS) buildup, leading to caspase-3-mediated death. These effects translate in vivo: orthotopic xenografts receiving a miR-1-3p mimic display reduced Ki-67 labeling, increased TUNEL positivity, and greatly slowed bone penetration. Crucially, pharmacologic autophagy inhibition with chloroquine duplicates the pro-apoptotic impact of miR-1-3p and shows additive growth restraint when combined with Akt/mTOR blockade, therefore highlighting a synthetic–lethal relationship between lysosomal turnover and the nutrient-sensing pathways already hyperactive through IL-33/ST2 and Wnt/β-catenin signaling (24). Driving the locally invasive phenotype of ameloblastoma, the miR-141-3p–NCAM1 and miR-1-3p–LAMP2 circuits together complete a trio of miRNA deficits that remove epithelial adhesion and metabolic and microenvironmental restrictions (25). However, it is worth noting that there is still a considerable distance between the above experiments and clinical practice (Table 2).
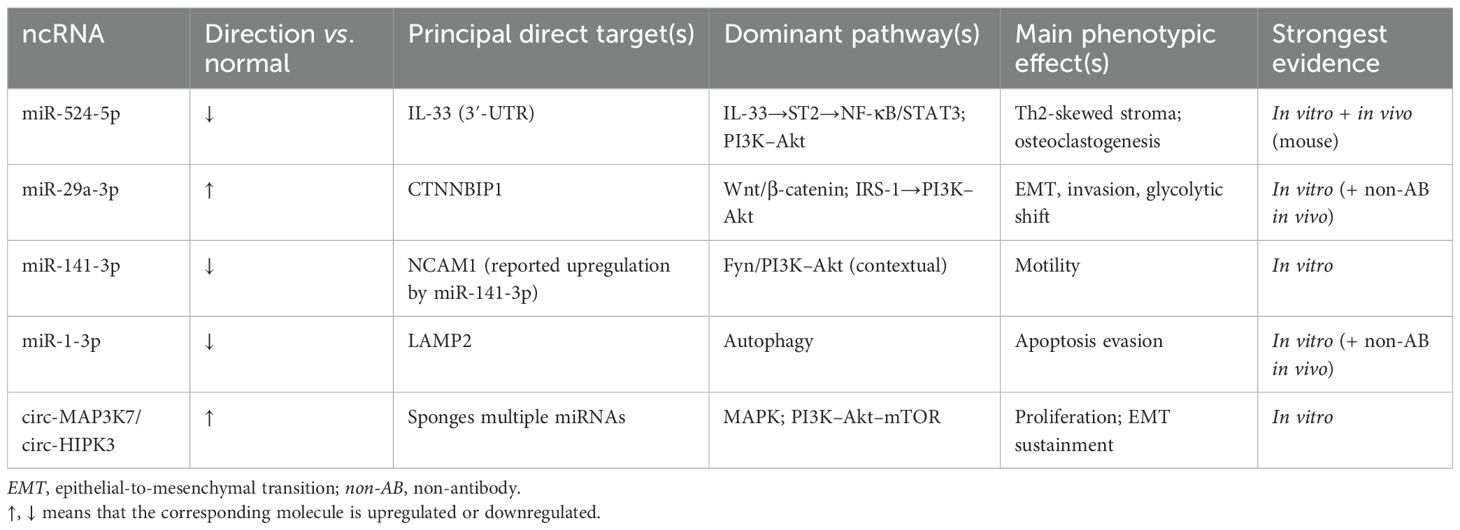
Table 2. Key non-coding RNAs (ncRNAs) implicated in ameloblastoma (26).
Fundamentally, miR-1-3p acts as a tumor suppressor in ameloblastoma models by repressing LAMP2 and autophagy. The reintroduction of miR-1-3p reduces LAMP2 and increases apoptosis in vitro.
4 Interaction between miRNAs and circRNAs in ameloblastoma
Figure 1 depicts the integrated regulatory network that controls ameloblastoma tumorigenesis through six major signaling pathways coordinated by the regulation of ncRNAs. The MAPK/ERK (extracellular signal-regulated kinase) signaling pathway (blue) processes growth factor signals via the RAS→RAF→MEK→ERK cascade, controlling proliferation, differentiation, and survival. The PI3K/Akt/mTOR signaling pathway (orange) regulates protein synthesis, survival, and metabolism through PI3K→PIP3→Akt→mTOR signaling. The Wnt/β-catenin signaling pathway (purple) controls stem cell maintenance, EMT, and transcription via Frizzled→β-catenin→TCF/LEF activation. The IL-33/STAT3 signaling pathway (green) mediates inflammatory responses through ST2→JAK→STAT3 signaling. The Hippo/YAP–TAZ signaling pathway (gold) regulates organ size and contact inhibition via MST1/2→LATS1/2→YAP/TAZ. The Hedgehog signaling pathway (purple) controls development and stem cell programs through Patched→SMO→GLI signaling. For pathway interconnections, the solid arrows represent direct protein–protein crosstalk mechanisms. RAS creates a central signaling hub linking the MAPK/ERK to the PI3K/Akt/mTOR pathways. GSK3β connects PI3K/Akt to Wnt/β-catenin through β-catenin phosphorylation control. YAP/β-catenin interaction bridges Hippo and Wnt signaling. Transcriptional enhanced associate domain (TEAD) transcription factors link Hippo/YAP–TAZ to MAPK/ERK gene expression programs. GLI1 connects Hedgehog to both the Wnt/β-catenin and PI3K/Akt pathways. STAT3 creates bidirectional communication with PI3K/Akt through transcriptional and metabolic mechanisms. T-cell factor (TCF)/lymphoid enhancer-binding factor (LEF) transcription factors mediate Wnt to IL-33/STAT3 connectivity. For the ncRNA regulatory architecture, the central tripartite hub represents the coordinated ncRNA control system comprising miRNAs, lncRNAs, and circRNAs. These ncRNAs regulate pathway activity through posttranscriptional control, epigenetic modification, and ceRNA networks. Red arrows indicate the ncRNA-mediated regulation affecting all pathways. The key regulatory ncRNAs include: oncogenic miRNAs—miR-21-5p targeting PTEN and PDCD4, miR-155 targeting APC and SOCS1, and miR-106b targeting p21 and LATS2; tumor suppressor miRNAs—miR-200 family targeting the EMT factors ZEB1/ZEB2 and miR-31-5p targeting YAP1; pathway-specific miRNAs—miR-324-5p and miR-125b targeting the Hedgehog components SMO and GLI1, respectively; regulatory lncRNAs—HOTAIR mediating chromatin remodeling, TUG1 functioning as an miR-31-5p sponge, and the tumor suppressor lncRNAs GAS5 and MEG3 activating p53 pathways; and circRNAs—oncogenic circPTEN promoting PI3K activation and tumor suppressor circITCH inhibiting Wnt signaling.
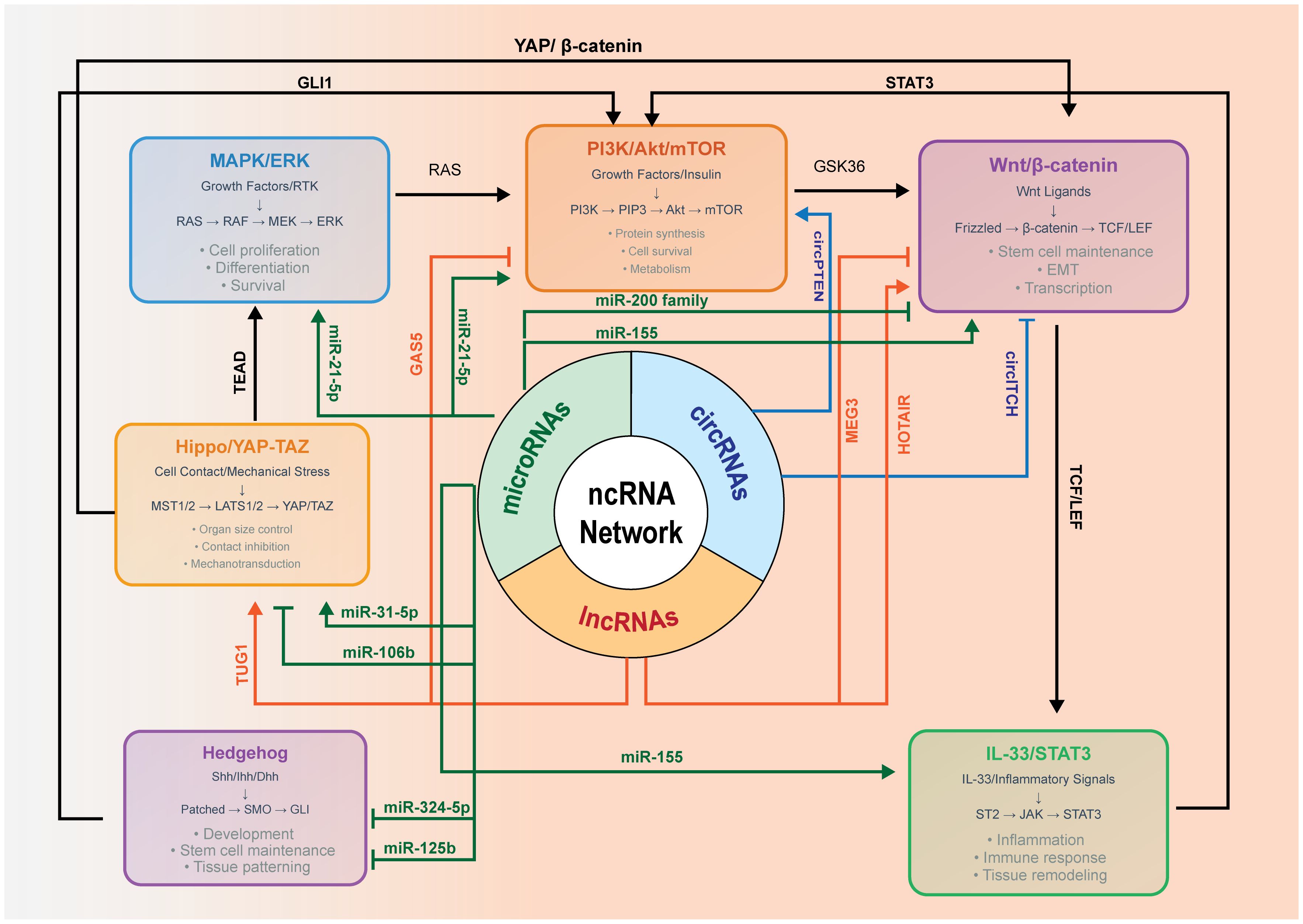
Figure 1. Non-coding RNA (ncRNA)-mediated crosstalk among MAPK/ERK, PI3K–Akt–mTOR, Wnt/β-catenin, IL-33/STAT3, and Hippo/YAP–TAZ in ameloblastoma.
Immunohistochemistry (IHC) indicates an elevated YAP expression in the ameloblastoma epithelium compared with the dental follicle, suggesting the involvement of the Hippo pathway in the tumor behavior. Mechanistically, YAP/TAZ integrates mechanical and growth factor cues and crosstalk with the Wnt/β-catenin and PI3K/mTOR programs, potentially influencing angiogenic outputs. Direct evidence in ameloblastoma remains limited. We therefore depict Hippo/YAP–TAZ as a contextual modulator and highlight it as a priority for targeted study.
4.1 Mechanism of action of circRNA
When a downstream splice donor joins an upstream splice acceptor, circRNAs result from a covalently closed loop impenetrable to exonuclease attack. By sequestering miRNAs, this topological stability enables circRNAs to accumulate to high copy number and to persist long enough to build ribonucleoprotein complexes that influence transcription, translation, and—most importantly—posttranscription. CircRNAs are appealing therapeutic substrates and regulatory molecules since their back-spliced junctions produce distinct sequence signatures that enable selective targeting using CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)–Cas (CRISPR-associated)-Cas13 or gapmer antisense technology regulation, as shown in Figure 2 (5).
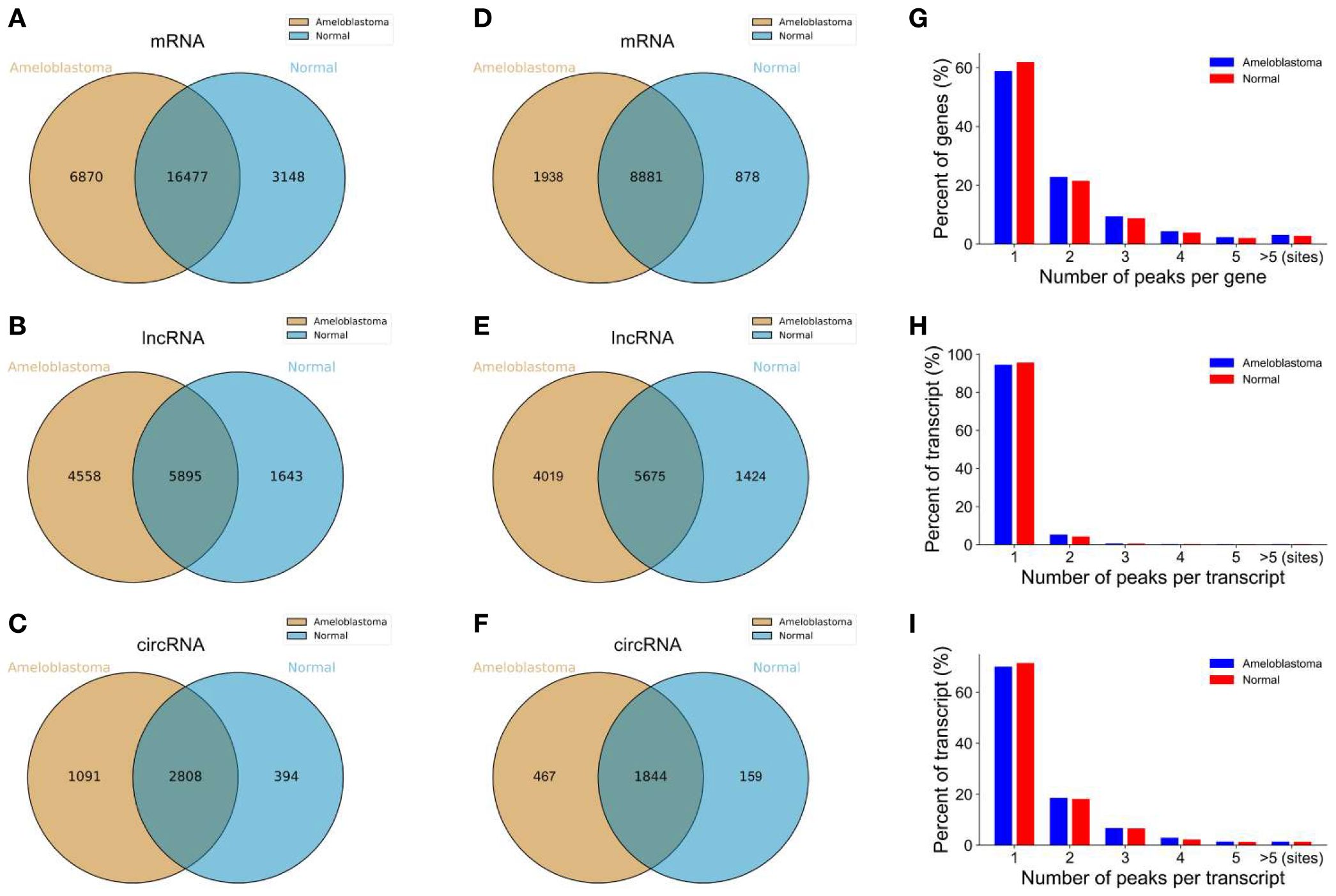
Figure 2. Overview of the m6A-modified peaks within messenger RNAs (mRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) in ameloblastoma and adjacent normal oral tissues. (A–C) Venn diagram depicting the overlapping and non-overlapping m6A peaks within the mRNAs, lncRNAs, and circRNAs between the two groups. (D–F) Venn diagram showing the differences and overlaps in the m6A-modified mRNAs, lncRNAs, and circRNAs between the two groups. (G–I) Number of m6A peaks per mRNA, lncRNA, and circRNA between the two groups (5).
4.1.1 Expression characteristics of circRNAs in ameloblastoma
More than 2,000 unique circRNAs were discovered by RiboMinus, RNase-R-enriched sequencing of solid/multicystic, unicystic, and desmoplastic tumors. Approximately 400 of these were differently expressed relative to healthy dental follicles (26). The global circRNA abundance increases in line with histological aggressiveness, with the most overrepresented circles relating to exonic regions of the genes previously underlined by oncogenic signaling, including HIPk3, SKA3, FBXW7, and MAP3K7. This enrichment makes a compelling case that the ameloblastoma transcriptional program is driven, not a by-product, by circRNA dysregulation (12).
4.1.2 The regulatory effect of circRNA on miRNA
Functionally, the main contribution of circRNAs to these cancers is their ceRNA sponging, modulating the miRNA dosage. Expanding substantially in mandibular lesions, circ-AB1 comprises three high-affinity seed matches for miR-524-5p and, in Argonaute pull-downs, catches approximately 40% of the residual miR-524-5p pool (5). Circ-HIPK3 binds miR-124 and miR-29b-1-5p, and circ-FBXW7 concentrates on miR-197-3p. Using CRISPR–Cas-Cas13, the degradation of each one of these circles releases its sequestered miRNAs, lowers their target gene expression, and alters the downstream signaling cascades, therefore proving a direct regulatory connection (27).
4.2 Interaction between miRNAs and circRNAs
The delivery of miRNA mimics or inhibitors to jaw lesions remains constrained by low vascular permeability, limited diffusion through mineralized matrix, rapid systemic clearance, and potential immune activation. Nanoparticles also risk off-target accumulation, and endosomal escape remains inefficient. Furthermore, immunogenicity remains a concern, particularly with the repeated administration of chemically modified oligonucleotides or lipid-based carriers, which may trigger innate immune responses. Tumor penetration is also hampered by the extracellular matrix density and the elevated interstitial pressure in ameloblastoma tissues. Delivery efficiency is further influenced by uptake heterogeneity among neoplastic cell populations, leading to variable therapeutic outcomes. Off-target effects remain a non-trivial risk, especially in closely related miRNA families with overlapping seed regions, necessitating rigorous in silico screening and in vivo validation. Consequently, clinical success will require refined delivery systems with high tissue specificity, minimal immunogenicity, and robust pharmacokinetic profiles that ensure durable and localized activity (28).
4.2.1 Examples of major miRNA–circRNA interactions
Whether a circRNA measurably “sponges” a miRNA is dependent on the relative copies and binding site count/affinity. Effective derepression generally requires high circRNA/miRNA ratios and multiple seed matches, and a low site occupancy often yields no phenotypic effect despite binding prediction. We therefore annotate the proposed ceRNA loops as hypotheses unless copy number and occupancy are demonstrated in ameloblastoma models.
Among the most powerful triplets is the axis circ-SKA3–miR-29a-3p–CTNNBIP1. An overexpressed circ-SKA3 sponges miR-29a-3p antagonists, releasing miR-29a-3p to silence CTNNBIP1 and to stabilize nuclear β-catenin, thereby boosting canonical Wnt signaling. Abundant circ-AB1 lowers the free miR-524-5p, lifting repression on IL-33 and its ST2 receptor to ignite the STAT3 and PI3K/Akt pathways in the surrounding stroma (28). Other circuits include circ-HIPK3–miR-124–FZD5, which dampens the Frizzled-mediated Wnt input, and circ-FBXW7–miR-197-3p–FBXW7, which reshapes the cell cycle control by modulating the SCF ubiquitin ligase activity. Although each triad is embedded in a wider continuum of crosstalk, it acts as an autonomous rheostat (29).
Corroborating the heightened Wnt tone predicted by these competing endogenous RNA loops, IHC and Western blot analyses have shown robust upregulation of the non-canonical ligand Wnt5a in ameloblastoma specimens, both the follicular and plexiform subtypes, and in AM-1 tumor cells compared with normal oral mucosa, tumor-adjacent tissues, and HaCaT keratinocytes (p < 0.05) (21). This selective enrichment of Wnt5a provides a tangible downstream read-out of circRNA/miRNA-driven Wnt pathway activation and highlights how aberrant ncRNA crosstalk simultaneously amplifies the canonical β-catenin and non-canonical Wnt5a cascades during ameloblastoma pathogenesis, as shown in Figure 3.
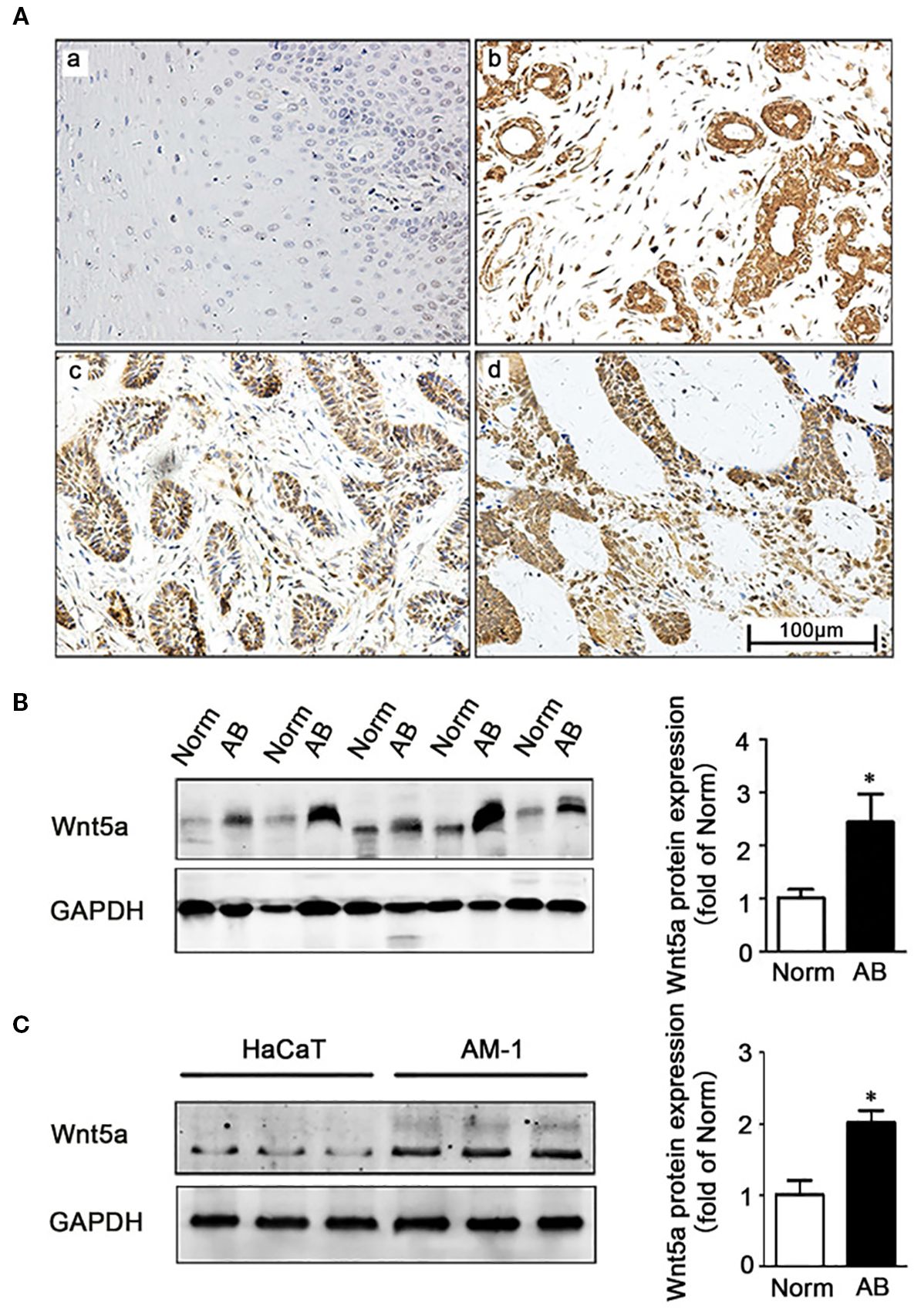
Figure 3. Expression of Wnt5a in ameloblastoma tissues and AM-1 cells. (A) Immunohistochemistry staining demonstrating Wnt5a protein expression in normal oral mucosa tissues (a) and in ameloblastoma tissues (b–d) [follicular type for (d, c) and plexiform type for (d)]. Scale bar, 100 µm. Magnification, ×200. (B) Western blot showing the Wnt5a protein expression in tumor-adjacent normal and ameloblastoma tissues. (C) Western blot showing the Wnt5a protein expression in HaCaT and AM-1 cell lines. *p < 0.05 (21).
4.2.2 Mechanism of the co-regulation of the MAPK signaling pathway by circRNAs and miRNAs
The main point of convergence for circRNA–miRNA co-regulation turns out to be MAPK signaling. Particularly high in recurrent lesions, circ-MAP3K7 has binding sites for miR-141-3p, miR-1-3p, and miR-542-3p, all of which typically reduce MAP3K7 or its adaptor TAB1. These miRNAs are titrated away when circ-MAP3K7 is plentiful. MAP3K7 autophosphorylates and the downstream ERK cascade stays constitutively active (29). Reiterating the pathway flux, loss of miR-181, miR-34a, or miR-125b brought on by other circRNA sponges releases the inhibitory pressure on RAS, MEK, and ERK itself. The activated ERK feeds back by raising the exon-skipping factor QKI, a facilitator of circRNA synthesis, hence generating a self-reinforcing loop that explains the increasing circRNA burden observed when tumors develop from a unicystic to a solid/multicystic type. The disruption of circ-MAP3K7 or similar sponges sensitizes three-dimensional tumors to MEK inhibitors and dual PI3K/mTOR inhibition, hence emphasizing circRNA targeting as a powerful complement to kinase-directed treatment in ameloblastoma (25).
5 The role of related signaling pathways in ameloblastoma
Less classical oncogene addiction drives ameloblastoma than the convergence of numerous nutrient- and stress-sensing cascades that collectively provide a metabolic excess, reduce apoptosis, and generate an aggressively osteolytic microenvironment. Among these, the PI3K–Akt–mTOR axis sits in the middle of a signaling nexus that integrates growth factor input from receptor tyrosine kinases, inflammatory ligands released by the tumor stroma, and the ncRNA derangements (25). Although the MAPK and Hedgehog pathways provide important initiating hits (e.g., BRAF^V600E in mandibular lesions and SMO-activating substitutions in maxillary tumors), durable expansion, marrow invasion, and postoperative recurrence correlate far more tightly with constitutive mTOR activity and its downstream transcriptional program (30).
5.1 The mTOR signaling pathway
5.1.1 The role of the mTOR pathway in ameloblastoma
Transcriptomic markers, phospho-proteomics, and IHC all point to mTOR complex 1 (mTORC1) being hyperactivated in the majority of mural unicystic lesions destined to recur and in over three-quarters of solid/multicystic tumors. The strong phosphorylation of mTOR at Ser2448 is complemented by the strong phosphorylation of its direct substrates, S6K1 and 4E-BP1, therefore suggesting increased cap-dependent translation and ribosomal biogenesis (31). Functional RNA sequencing (RNA-seq) has shown a characteristic mTORC1 transcriptional footprint—the upregulation of the TOP mRNAs encoding ribosomal proteins, translation elongation factors, and cholesterol biosynthesis enzymes. Lesions with the highest p-mTOR scores reveal the most extensive radiographic “soap bubble” growth, the deepest cortical perforations, and, at the tissue level, the highest Ki-67 indices. Especially diffuse p-mTOR staining mandibular tumors bearing BRAF^V600E is consistent with the MAPK–RSK crosstalk that cooperatively amplifies mTOR signaling from PI3K input (32).
Although less obvious histologically, the activation of mTOR complex 2 (mTORC2) has similarly important consequences. An active mTORC2 is indicated by the phosphorylation of Akt at Ser473, SGK1 at Ser422, and PKCα at Ser657. This corresponds with notable cortical bone erosion and suggests that an SGK-driven sodium transport and a PKC-mediated cytoskeletal remodeling increase the pressure resorption of the bone around the developing mass. Small interfering RNA (siRNA) knockdown of RICTOR, the defining subunit of mTORC2, lowered the Transwell invasion in AM-1 and A-T cell lines without appreciably changing proliferation, therefore highlighting a division of labor in which mTORC1 fuels biomass accumulation while mTORC2 coordinates the motility and cytoskeletal plasticity required for marrow invasion (33).
Confirming on-target pathway inhibition, orthotopic xenografts treated with rapamycin show a clearly reduced S6 ribosomal protein phosphorylation, small but considerable decreases in the tumor volume, and extensive central necrotic zones. Nevertheless, feedback stimulation of PI3K and ERK recovers Akt and 4E-BP1 phosphorylation within days, therefore highlighting the adaptive resilience of the network. Rapamycin by itself seldom causes persistent regression. In explant cultures, dual kinase inhibitors including BEZ-235 or the third-generation catalytic site inhibitor sapanisertib induce a deeper reduction of both mTORC1 and mTORC2, extend Akt dephosphorylation, and provide almost perfect cytostatic responses. Still, compensatory autophagy is a major escape route, and this fact makes the relationship between mTOR and miRNA control extremely relevant (34).
The above multiple IHC-based studies report the phosphorylation of mTORC1 targets (S6K1 and 4E-BP1) and Akt (Ser473/Thr308) in ameloblastoma tissues, consistent with PI3K–Akt–mTOR activation. These data are largely correlative and cross-sectional. Standardized prospective studies are needed (31–34). mTORC2 activity has been inferred from the phosphorylation of Akt (Ser473) and related substrates in some cohorts, with suggested links to invasion and motility; however, evidence remains heterogeneous and primarily descriptive. Rapalogs and catalytic site mTOR/PI3K inhibitors have shown preclinical activity in ameloblastoma cells/explants. Adaptive feedback and autophagy are recognized escape routes, underscoring the need for rational combinations. Claims of clinical readiness are avoided here.
5.1.2 Relationship between the mTOR pathway and miRNAs
Multiple layers of ncRNA regulation cross with the mTOR axis to generate a positive feed-forward circuit that stabilizes anabolic signaling. Through promoter hypermethylation and the activity of circRNA sponges produced from the HIPK3 and IGF1R loci, cancers of high grade continuously silence tumor-suppressive miR-199a-3p and miR-100, both direct repressors of the mTOR 3′-UTR. Their absence removes the main posttranscriptional brake on mTORC1. Conversely, oncomiRs specifically enriched in ameloblastoma strengthen the PI3K input: miR-21 and miR-155 decrease PTEN and SHIP1, while miR-29a-3p, through the β-catenin-mediated upregulation of IRS-1, increases the growth factor-dependent PI3K recruitment (35).
MiR-524-5p deficit produces perhaps the most remarkable ncRNA–mTOR interface. IL-33 escapes repression, hits the stromal receptor ST2, and starts a cytokine cascade that ends in PI3K–Akt activation in both the cancer epithelium and its peritumoral fibroblasts when this miRNA is reduced. The subsequent Akt activity phosphorylates and completely activates mTORC1, creating a loop linking microenvironmental inflammation to anabolic signaling. Concurrent with miR-1-3p loss, LAMP2 is lifted, enhancing autophagic flux. In the presence of mTORC1 inhibition by rapalogs, this autophagy surge provides the recycled amino acids needed to maintain minimum protein synthesis, hence reducing medication efficacy.
Restoring or imitating the repressed miRNAs greatly changes the pathway sensitivity. Transient transfection of miR-199a-3p or miR-100 decreases the mTOR protein by more than half, potentiates the anti-proliferative impact of everolimus, and greatly increases the cleaved caspase-3 levels in vitro. The analogous effects of a synthetic miR-1-3p mimic impede LAMP2-dependent autophagosome maturation, make cells metabolically fragile, and change rapamycin from a cytostatic to an apoptotic agent. Proof-of-concept ncRNA manipulation opens a hitherto resistant treatment window. In murine mandibular xenografts, nanoparticle co-delivery of the miR-1-3p mimic and everolimus delivers prolonged tumor reduction, complete abrogation of osteolytic craters, and little systemic toxicity (36).
A deliberate program of miRNA loss and oncomiR increase holds the mTOR network in ameloblastoma in a hyperactive state overall. Feedback loops resulting from inflammatory IL-33 signaling and β-catenin-driven IRS-1 tighten the grip on anabolic translation and angiogenic drive even more. Therapeutic success will thus depend on destroying these ncRNA inputs—either by reinstating the lost tumor-suppressive miRNAs, extinguishing oncomiRs, or selectively degrading their circRNA sponges—therefore enabling kinase inhibitors to exert uncompromised pressure on the metabolic engine that sustains tumor outgrowth and bone destruction (37).
5.2 Akt/mTOR signaling pathway
5.2.1 Expression and function of the Akt/mTOR cascade in ameloblastoma
Across available cohorts, the activation of Akt (Thr308/Ser473) has been reported by IHC/phospho-assays in ameloblastoma, with higher activity often observed at invasive fronts. Comprehensive phospho-proteomic prevalence figures remain to be established through multicenter studies (38, 39).
RTK amplification and adaptor loading: EGFR and IGF1R are overexpressed at the plasma membrane. The β-catenin-dependent transcription of IRS-1 and IRS-2 (secondary to miR-29a-3p gain) provides a dense shelf of p85-binding motifs that recruit class I PI3K to PIP2-rich rafts (40).
Loss of lipid phosphatase restraint: The coordinated overexpression of miR-21 and miR-155 reduces the PTEN and SHIP1 proteins by 60%–70%, allowing an unchecked PIP3 accumulation. PTEN immunoreactivity is particularly common in BRAF^V600E-positive mandibular tumors, which explains the frequent coexistence of MAPK and PI3K lesions in this anatomic subset (41).
Cytokine feed-forward: The depletion of miR-524-5p liberates IL-33. Binding to ST2 activates MyD88–IRAK4, which phosphorylates p110δ and further amplifies the PI3K flux. Inflammatory enhancement is most pronounced in tumors that show heavy desmoplastic reaction, where fibroblasts secrete additional IL-6 and CXCL12 that sustain long-range Akt signaling (42).
Once Akt is active, the phosphorylation of TSC2 (Thr1462) and PRAS40 (Thr246) eliminates the tonic brake on mTORC1, while mTORC2 concurrently phosphorylates SGK1 and PKCα, hence generating membrane ruffling and matrix metalloproteinase release. Confirming that mTORC1 reprograms translation toward biomass accumulation and amino acid scavenging, ribosome profiling has shown the preferential loading of mRNAs that produce ribosomal proteins, sterol regulatory element-binding proteins, and the glutamine transporter SLC1A5 (43). Metabolomics has also shown a threefold increase in lactate generation and a 40% increase in the pentose phosphate flux, alterations that reflect the increased fluorodeoxyglucose (FDG) absorption shown on PET-CT in fast-growing mandibular lesions. Under in vitro CRISPR–Cas deletion of RICTOR, Ser473-Akt is virtually eradicated, the traction force microscopy read-outs are half reduced, and marrow slice invasion is almost completely eliminated, therefore highlighting the motility license granted especially by mTORC2 (44).
5.2.2 Clinical significance of the Akt/mTOR pathway in ameloblastoma
Higher p-Akt/p-mTOR labeling has been associated with more aggressive radiographic and histologic features in some studies. The evidence is heterogeneous and largely single-center. No validated blood-based pharmacodynamic markers have been established for ameloblastoma (45).
Therapeutic efficacy hinges on both the breadth and the depth of pathway inhibition. While allosteric mTOR inhibitors such as sirolimus and everolimus transiently reduce S6K1 phosphorylation and modestly slow tumor growth, they leave 4E-BP1 largely unaffected and fail to disrupt the mTORC2–Akt feedback loops. Consequently, tumors frequently resume growth due to an IGF1R-driven rebound in Akt phosphorylation—a feedback effect paradoxically induced by rapalogs. In contrast, catalytic site mTOR inhibitors, such as sapanisertib and dactolisib, effectively dephosphorylate both S6K1 and 4E-BP1, leading to a near-complete cell cycle arrest in ameloblastoma explant cultures.
Nonetheless, resistance pathways persist. One major escape mechanism involves adaptive autophagy, which is driven by the derepression of LAMP2 following the loss of miR-1-3p regulation. This results in an accelerated lysosomal turnover and amino acid recycling, sustaining protein synthesis despite upstream inhibition. The co-administration of a miR-1-3p mimic with a catalytic mTOR inhibition suppresses this escape route. In addition, the introduction of an anti-miR-21 agent restores PTEN, downregulates PI3K, and curtails rebound Akt signaling. A triple combination therapy—sapanisertib + miR-1-3p mimic + anti-miR-21—has demonstrated >90% tumor regression, preserved inferior alveolar nerve function, eliminated osteolysis, and produced minimal systemic toxicity—an efficacy–toxicity profile that is currently unmatched by surgery alone (46).
Despite this, resistance remains a challenge. FGFR2-high clones can escape even dual PI3K/mTOR inhibition, with chronic Akt suppression inducing FGFR2 upregulation and activating STAT3. Preliminary studies have suggested that JAK1/2 inhibitors or a targeted FGFR2 blockade, administered in a rotating trimodal regimen, may prevent clonal expansion. The most promising strategy now under investigation involves the integration of ncRNA reprogramming—restoring miR-199a-3p, miR-100, and miR-1-3p while silencing miR-21, miR-29a-3p, and miR-155—with vertical kinase inhibition and intermittent autophagy suppression. These regimens are currently being evaluated in patient-derived organoid models and, if successful, may offer the first real therapeutic alternative to facially disfiguring surgery for high-risk ameloblastoma (46, 47) (Table 3).
6 Conclusion and future prospects
6.1 Integrated summary of key findings
6.1.1 ncRNA architecture that drives tumor biology
Today, ameloblastoma is well known as a disease driven by a tiny cadre of dysregulated miRNAs and their accompanying circRNA sponges. Loss of miR-524-5p, miR-141-3p, miR-1-3p, miR-199a-3p, and miR-100 eliminates the posttranscriptional brakes on IL-33/ST2, NCAM1, LAMP2, mTOR, and a number of downstream effectors. Concurrently, the upregulation of miR-29a-3p, miR-21, and miR-155 increases the Wnt/β-catenin flow, renders PTEN/SHIP1 inactive, and releases inflammatory cytokines. By sequestering the very miRNAs that would ordinarily temper MAPK, PI3K, and autophagy signaling, circRNAs—most notably circ-AB1, circ-SKA3, circ-HIPK3, circ-FBXW7, and circ-MAP3K7—further exacerbate this imbalance.
6.1.2 Pathway convergence and metabolic reprogramming
These ncRNA changes come together on a triad of kinase modules: Wnt/β-catenin, IL-33/STAT3, and the Akt/mTOR engine rather than being siloed. Together, this trio produces an anabolic phenotype distinguished by aerobic glycolysis, cap-dependent translation, increased pentose phosphate flow, and autophagy-assisted nutrition salvage. Explaining the osteolytic behavior of the lesion, the same circuitry upregulates matrix metalloproteinases and osteoclastogenic signals.
6.1.3 Biomarker implications
Early recurrence correlates with strong Ser473-Akt, Ser2448-mTOR, and nuclear β-catenin immunolabeling. Exosomal phospho-Akt, miR-29a-3p, and miR-21 mirror the intratumoral activity and diminish rapidly following successful pathway inhibition, thereby acting as real-time pharmacodynamic markers in the blood.
6.2 Unresolved challenges
6.2.1 Biological heterogeneity
There are subtype-specific differences between BRAF^V600E-positive mandibular tumors versus SMO-driven maxillary lesions. Solid/multicystic versus unicystic argues that a one-size-fits-all ncRNA catalogue is insufficient. Multicenter biobanks with single-cell and spatial omics annotation are required to capture the full spectrum of the epithelial, stromal, and immune states.
6.2.2 Delivery barriers
An efficient jaw-targeted delivery of miRNA mimics, antagomirs, or CRISPR–Cas13 constructs remains technically daunting. The dense mineral matrix, the hypoxic marrow niches, and the rich lymphatic drainage of the mandible demand bespoke nanoparticle or hydrogel platforms.
6.2.3 Preclinical model deficits
The current xenograft systems lack humanized immune and bone microenvironments, limiting accurate assessment of stroma-driven loops such as IL-33/ST2. Vascularized mandibular organoids and genetically engineered mouse models that recapitulate odontogenic lineage tracing are priorities.
6.3 Roadmap for future research and translation
6.3.1 Next-generation biomarkers
Longitudinal profiling of circulating miR-29a-3p, miR-21, and exosomal phospho-Akt in large patient cohorts should refine the risk algorithms for conservative surgery versus radical resection. Digital droplet PCR panels combining ncRNA and mutant cell-free DNA could detect micrometer-scale residuals months before radiographic relapse.
6.3.2 ncRNA-centric therapy design
LAMP2-dependent autophagy can be blocked by the therapeutic restoration of miR-524-5p, miR-141-3p, and miR-1-3p layered atop catalytic mTOR or dual PI3K/mTOR inhibitors, therefore transforming cytostasis into apoptosis. Parallel development of gapmer or Cas13 methods that degrade circ-MAP3K7 and circ-AB1 offers a pathway-wide derepression of many tumor-suppressive miRNAs.
6.3.3 Rational combination regimens
Adaptive laboratory evolution proposes that continuous Akt/mTOR suppression chooses for alternatives for FGFR2–STAT3. Early findings have shown that intermittent dosage schedules rotating catalytic mTOR inhibition, FGFR inhibition, and autophagy suppression outperform continuous monotherapy while protecting the normal mucosa. The development of these schedules calls for real-time liquid–biopsy feedback loops.
6.3.4 Bench to bedside pipelines
Concepts such as bone-targeted delivery systems for miRNA payloads and standardized Good Manufacturing Practice (GMP)-compliant synthesis pipelines remain largely at the proof-of-concept stage. Robust pharmacokinetics, biodistribution studies, and toxicology assessments are required to support future clinical translation.
6.3.5 Functional preservation metrics
Retention of mandibular continuity, occlusal performance, and face symmetry should all count toward success in radiographic regression. Molecular treatment will guarantee clinically significant effect by the co-development of outcome scales including bone density CT, electromyographic mastication indices, and patient-reported aesthetic scores.
Author contributions
JM: Supervision, Writing – original draft, Writing – review & editing, Conceptualization. QG: Writing – original draft. XB: Writing – original draft. MZ: Supervision, Conceptualization, Writing – review & editing, Funding acquisition, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was funded by National Natural Science Foundation of China (81072197, 81470758).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ghai S. Ameloblastoma: an updated narrative review of an enigmatic tumor. Cureus. (2022) 14:e27734. doi: 10.7759/CUREUS.27734
2. Soluk TM and Wright JM. The world health organization classification of odontogenic lesions: A summary of the changes of the 2022 (5th) edition. Turk Patoloji Derg. (2022) 38:168–84. doi: 10.5146/TJPATH.2022.01573
3. Vila S, Oster RA, James S, Morlandt AB, Powell KK, and Amm HM. A retrospective analysis of 129 ameloblastoma cases: clinical and demographical trends from a single institution. J Racial Ethn Health Disparities. (2025) 12:1612–20. doi: 10.1007/S40615-024-01993-3
4. Zlotogorski-Hurvitz A, Soluk TM, Passador-Santos F, Martins MVA, Salo T, Mauramo M, et al. Conceptual changes in ameloblastoma: Suggested re-classification of a “veteran” tumor. Oral Dis. (2022) 28:703–10. doi: 10.1111/ODI.13770
5. Niu X, Xu J, Liu J, Chen L, Qiao X, and Zhong M. Landscape of N6-methyladenosine modification patterns in human ameloblastoma. Front Oncol. (2020) 10:556497. doi: 10.3389/fonc.2020.556497
6. Sun Z, Shi K, Yang S, Liu J, Zhou Q, Wang G, et al. Effect of exosomal miRNA on cancer biology and clinical applications. Mol Cancer. (2018) 17:147. doi: 10.1186/s12943-018-0897-7
7. Liu L, Wang J, Khanabdali R, Kalionis B, Tai X, and Xia S. Circular RNAs: Isolation, characterization and their potential role in diseases. RNA Biol. (2017) 14:1715–21. doi: 10.1080/15476286.2017.1367886
8. Yoithapprabhunath TR, Srichinthu KK, Gupta D, Singh D, Pasupuleti S, and Nirmal RM. Effectiveness of molecular-targeted chemotherapy in ameloblastomas: A systematic review. Indian J Dent Res Off Publ Indian Soc Dent Res. (2022) 33:323–31. doi: 10.4103/IJDR.IJDR_456_22
9. Zhao Z, Xiong G, Wang C, and Cao W. From pathogenesis to precision medicine: Transformative advances in research and treatment of ameloblastoma. Cancer Lett. (2025) 612:217448. doi: 10.1016/J.CANLET.2025.217448
10. Gates JC, Clark AP, Cherkas E, Shreenivas AV, Kraus D, Danzinger N, et al. Genomic profiling and precision medicine in complex ameloblastoma. Head Neck. (2023) 45:816–26. doi: 10.1002/HED.27294
11. Conn VM, Chinnaiyan AM, and Conn SJ. Circular RNA in cancer. Nat Rev Cancer. (2024) 24:597–613. doi: 10.1038/s41568-024-00721-7
12. Huang X, Gu F, Zhao M, Huang W, Han W, Chen R, et al. Function and therapeutic potential of non-coding RNA in ameloblastoma. Onco Targets Ther. (2024) 31:643–53. doi: 10.2147/ott.s474038
13. Yang J, Liu J, Zhong M, Chen Y, Song M, and Du Y. Discoidin domain receptors: a promoter of the aggressive behavior of ameloblastomas. IUBMB Life. (2014) 66:292–9. doi: 10.1002/iub.1265
14. Ren MS, Wang J, Guo Y, and Zhong M. Expression and clinical significance of tumor-associated macrophages and microvessel density in ameloblastomas. Shanghai Kou Qiang Yi Xue Shanghai J Stoma-tol. (2012) 21:57–61. doi: 10.1016/j.surge.2013.07.003
15. Gao X, Wang G, Zhang YK, and Zhong M. Expression and mechanism of regulation of PP2A/Pr65 in ameloblastoma. Surg J R Coll Surg Edinb Irel. (2014) 12:129–33. doi: 10.1016/j.surge.2013.07.003
16. Chen L, Wang G, Qiao X, Wang X, Liu J, Niu X, et al. Downregulated miR-524-5p participates in the tumor microenvironment of ameloblastoma by targeting the interleukin-33 (IL-33)/suppression of tumorigenicity 2 (ST2) axis. Med Sci Monit Int Med J Exp Clin Res. (2020) 26:e921863. doi: 10.12659/msm.921863
17. Zhong M, Liu JD, Wang J, Liu J, Li L, Hou L, et al. Expression and significance of hTERT, c-fos and c-jun in ameloblastoma. Shanghai Kou Qiang Yi Xue Shanghai J Stomatol. (2006) 15:461–5.
18. Liu S, Liu D, Liu J, Liu J, and Zhong M. miR-29a-3p promotes migration and invasion in ameloblastoma via Wnt/β-catenin signaling by targeting catenin beta interacting protein 1. Head Neck. (2021) 43:3911–21. doi: 10.1002/HED.26888
19. Qi X, Zhang DH, Wu N, Xiao JH, Wang X, and Ma W. ceRNA in cancer: possible functions and clinical implications. J Med Genet. (2015) 52:710–8. doi: 10.1136/jmedgenet-2015-103334
20. Wei Z, Zhong M, Guo Y, Wang Y, Ren M, and Wang Z. Expression of β-catenin and AXIN2 in ameloblastomas. Contemp Oncol Poznan Pol. (2013) 17:250–6. doi: 10.5114/wo.2013.35278
21. Qiao X, Niu X, Shi J, Chen L, Wang X, Liu J, et al. Wnt5a regulates Ameloblastoma Cell Migration by modulating Mitochondrial and Cytoskeletal Dynamics. J Cancer. (2020) 11:5490–502. doi: 10.7150/jca.46547
22. Guan G, Niu X, Qiao X, Wang X, Liu J, and Zhong M. Upregulation of neural cell adhesion molecule 1 (NCAM1) by hsa-miR-141-3p suppresses ameloblastoma cell migration. Med Sci Monit Int Med J Exp Clin Res. (2020) 26:e923491. doi: 10.12659/msm.923491
23. Deng C, Huo M, Chu H, Zhuang X, Deng G, Li W, et al. Exosome circATP8A1 induces macrophage M2 polarization by regulating the miR-1-3p/STAT6 axis to promote gastric cancer progression. Mol Cancer. (2024) 23:49. doi: 10.1186/S12943-024-01966-4
24. Xu F, Xiao Q, Du WW, Wang S, and Yang BB. CircRNA: Functions, applications and prospects. Biomolecules. (2024) 14:1503. doi: 10.3390/biom14121503
25. Liu J, Qiao X, Liu J, and Zhong M. Identification of circ_0089153/miR-608/EGFR p53 axis in ameloblastoma via MAPK signaling pathway. Oral Dis. (2022) 28:756–70. doi: 10.1111/ODI.13788
26. Panda AC. Circular RNAs act as miRNA sponges. Adv Exp Med Biol. (2018) 1087:67–79. doi: 10.1007/978-981-13-1426-1_6
27. Zhong M, Li ZJ, Wang J, Yue YL, and Bao G. The study of the invasive biologic behaviour of ameloblastoma. Zhonghua Kouqiang Yixue Za zhi Chin J Stomatol. (2004) 39:45–8.
28. Al-Awsi GRL, Jasim SA, Fakri MY, Alhachami FR, Ziyadullaev S, Kandeel M, et al. The role of miRNA-128 in the development and progression of gastrointestinal and urogenital cancer. Future Oncol Lond Engl. (2022) 18:4209–31. doi: 10.2217/FON-2022-0574
29. Niu X, Huang B, Qiao X, Liu J, Chen L, and Zhong M. MicroRNA-1-3p suppresses Malignant phenotypes of ameloblastoma through down-regulating lysosomal associated membrane protein 2-mediated autophagy. Front Med. (2021) 8:670188. doi: 10.3389/FMED.2021.670188
30. Zhong M, Wang J, Gong YB, Li JC, Zhang B, and Hou L. Expression of HOXC13 in ameloblastoma. Zhonghua Kou Qiang Yi Xue Za Zhi Chin J Stomatol. (2007) 42:43–6.
31. Li N, Zhong M, and Song M. Expression of phosphorylated mTOR and its regulatory protein is related to biological behaviors of ameloblastoma. Int J Clin Exp Pathol. (2012) 5:660–7.
32. Li N, Sui J, Liu H, Zhong M, Zhang M, Wang Y, et al. Expression of phosphorylated Akt/mTOR and clinical significance in human ameloblastoma. Int J Clin Exp Med. (2015) 8:5236–44.
33. Ishaq Y, Ikram A, Alzahrani B, and Khurshid S. The Role of miRNAs, circRNAs and Their Interactions in Development and Progression of Hepatocellular Carcinoma: An Insilico Approach. Genes. (2022) 14:13. doi: 10.3390/GENES14010013
34. Uzuner E, Ulu GT, Gürler SB, and Baran Y. The role of miRNA in cancer: pathogenesis, diagnosis, and treatment. Methods Mol Biol Clifton NJ. (2022) 2257:375–422. doi: 10.1007/978-1-0716-1170-8_18
35. Sandiford OA, Moore CA, Du J, Boulad M, Gergues M, Eltouky H, et al. Human aging and cancer: role of miRNA in tumor microenvironment. Adv Exp Med Biol. (2018) 1056:137–52. doi: 10.1007/978-3-319-74470-4_9
36. May JM, Bylicky M, Chopra S, Coleman CN, and Aryankalayil MJ. Long and short non-coding RNA and radiation response: a review. Transl Res J Lab Clin Med. (2021) 233:162–79. doi: 10.1016/J.TRSL.2021.02.005
37. Sun TY, Li YQ, Zhao FQ, Sun HM, Gao Y, Wu B, et al. MiR-1-3p and miR-124-3p synergistically damage the intestinal barrier in the ageing colon. J Crohns Colitis. (2022) 16:656–67. doi: 10.1093/ECCO-JCC/JJAB179
38. Yu L, Wei J, and Liu P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin Cancer Biol. (2022) 85:69–94. doi: 10.1016/J.SEMCANCER.2021.06.019
39. Alves CL and Ditzel HJ. Drugging the PI3K/AKT/mTOR pathway in ER+ Breast cancer. Int J Mol Sci. (2023) 24:4522. doi: 10.3390/IJMS24054522
40. Peng Y, Wang Y, Zhou C, Mei W, and Zeng C. PI3K/Akt/mTOR pathway and its role in cancer therapeutics: are we making headway? Front Oncol. (2022) 12:819128. doi: 10.3389/FONC.2022.819128
41. Fattahi S, Amjadi-Moheb F, Tabaripou RR, Ashrafi GH, and Akhavan-Niaki H. PI3K/AKT/mTOR signaling in gastric cancer: Epigenetics and beyond. Life Sci. (2020) 262:118513. doi: 10.1016/j.lfs.2020.118513
42. Leiphrakpam PD and Are C. PI3K/Akt/mTOR signaling pathway as a target for colorectal cancer treatment. Int J Mol Sci. (2024) 25:3178. doi: 10.3390/IJMS25063178
43. Xu Z, Han X OD, Liu T, Li Z, Jiang G, et al. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl Microbiol Biotechnol. (2020) 104:575–87. doi: 10.1007/s00253-019-10257-8
44. Li Q, Li Z, Luo T, and Shi H. Targeting the PI3K/AKT/mTOR and RAF/MEK/ERK pathways for cancer therapy. Mol BioMed. (2022) 3:47. doi: 10.1186/S43556-022-00110-2
45. Shorning BY, Dass MS, Smalley MJ, and Pearson HB. The PI3K-AKT-mTOR pathway and prostate cancer: at the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci. (2020) 21:4507. doi: 10.3390/ijms21124507
46. Ersahin T, Tuncbag N, and Cetin-Atalay R. The PI3K/AKT/mTOR interactive pathway. Mol Biosyst. (2015) 11:1946–54. doi: 10.1039/c5mb00101c
Keywords: ameloblastoma, microRNAs, molecular targeted therapy, signaling pathway, non-coding RNA, precision therapy
Citation: Mao J, Gai Q, Bao X and Zhong M (2025) From miRNA sponges to mTOR blockades: mapping the multidimensional landscape of ameloblastoma pathogenesis and precision targeting. Front. Oncol. 15:1651236. doi: 10.3389/fonc.2025.1651236
Received: 21 June 2025; Accepted: 19 September 2025;
Published: 08 October 2025.
Edited by:
Lanrong Bi, Michigan Technological University, United StatesReviewed by:
Gaetano Santulli, Albert Einstein College of Medicine, United StatesVeenita Khare, University of California, San Diego, United States
Copyright © 2025 Mao, Gai, Bao and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Zhong, emhvbmdtaW5nX29yYWxAYWxpeXVuLmNvbQ==