 Marwah M. Albakri
Marwah M. Albakri- 1Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, Taibah University, Madinah, Saudi Arabia
- 2Health and Life Research Center, Taibah University, Madinah, Saudi Arabia
TP53 mutations drive oncogenesis and therapeutic resistance in myelodysplastic syndromes (MDSs) and acute myeloid leukemia (AML), impairing p53-regulated functions such as apoptosis, immune surveillance, and genomic stability, leading to immune evasion and metabolic reprogramming. The tumor microenvironment in TP53-mutated MDS and AML fosters leukemic progression through cytokine dysregulation, altered metabolism, and immune suppression. Current therapies, including chemotherapy and hypomethylating agents, offer limited efficacy, resulting in poor overall survival rates for these high-risk patients. However, novel therapeutic approaches provide promising avenues, including MDM2 inhibitors, p53-reactivating agents, pathway-targeted inhibitors (Hedgehog, Wnt, NF-κB), immune modulation (checkpoint inhibitors, CAR-T therapy), metabolic interventions (fatty acid metabolism, glycolysis), and gene-editing technologies (CRISPR/Cas9, base editing). This review explores the mechanisms of immune dysfunction in TP53-mutated MDS and AML while highlighting emerging therapeutic strategies, emphasizing the integration of targeted, metabolic, and immune-modulating therapies as a transformative approach to improve patient outcomes.
1 Introduction
The TP53 gene encodes p53, a tumor suppressor crucial for cell cycle control, apoptosis, genomic integrity, metabolism, immune response, and inflammation. Mutation in TP53 disrupt these functions, resulting in uncontrolled proliferation, immune evasion, and therapy resistance (1–9). These mutations occur through direct gene mutations or via MDM2 overexpression, a negative regulator of p53 (10). These alterations contribute to genomic instability, impaired apoptosis, and therapy-resistant disease (11–13).
The prevalence of TP53 mutations varies across cancer types, with an estimated 50% incidence in all cancers (14). While these mutations are prevalent in solid tumors such as ovarian carcinoma (90%), they occur in less than 10% of hematologic malignancies such as AML (15). However, in therapy-related AML, the frequency rises to 50%, highlighting its role in disease progression and treatment resistance (16).
This review investigates the roles that TP53 mutations play in immunological dysregulation, tumor microenvironment changes, metabolic dysfunction, and treatment resistance. It integrates clinical and preclinical data from immunological, metabolic, and gene-editing methods to highlight potential treatment approaches to TP53-mutated MDS and AML.
2 Pathogenesis of TP53 mutations and clonal hematopoiesis in AML and MDS
TP53 regulates hematopoietic stem cell (HSC) quiescence, proliferation, and the induction of apoptosis, maintaining hematopoietic homeostasis. When TP53 is mutated, these functions are disrupted, leading to genomic instability, clonal expansion, and the transformation of HSCs into preleukemic stem cells, a key step in the progression to MDS or AML.
2.1 Clonal hematopoiesis and TP53 mutations
A major driver of TP53-mutated MDS and AML is clonal hematopoiesis, where a single mutated HSC clone expands due to a selective advantage (17). In CHIP, commonly seen in older individuals, TP53 mutations are among the most significant genetic abnormalities, greatly increasing the risk of progression to hematologic malignancies (18). These mutations promote self-renewal and clonal expansion, driving MDS or AML progression (19).
2.2 TP53 mutation types and their impact on disease progression
The TP53 mutations occurring through various genetic alterations include the following:
● Deletions: Loss of chromosome 17p, which contains TP53, leads to a monoallelic loss, often followed by mutations in the remaining allele, resulting in a complete LOF (20).
● Missense mutations: This is the most frequent type of TP53 mutation, leading to either dominant-negative effects (where mutant p53 inhibits wild-type function) or GOF mutations that promote oncogenic properties (2, 12–14).
● Truncating mutations: These result in premature stop codons, producing non-functional p53 proteins incapable of exerting tumor-suppressive effects.
Mutation hotspots (codons 175, 245, 248, 249, 273, and 282) correlate with poor AML prognosis (15). TP53 mutations are more frequent in older patients and those with prior chemotherapy exposure, supporting their role in therapy-related myeloid neoplasms (21).
2.3 TP53 as a distinct molecular entity
The World Health Organization 5th edition and the International Consensus Classification recognize TP53 mutations as a unique molecular entity (22, 23).
2.4 Molecular and clinical determinants of therapy in TP53-mutated MDS/AML
Effective treatment of TP53-mutated MDS and AML requires integrating molecular and clinical risk factors. The allelic status of TP53 mutations has significant prognostic implications. Patients with multi-hit (biallelic) TP53 mutations exhibit complex karyotyping, primary resistance to chemotherapy, and poor overall survival compared to those with monoallelic mutations (24, 25). Integrating allelic burden into prognostic models such as IPSS-M provides more accurate risk classification and informs intensity of treatment (25, 26). TP53 mutations co-occur with other genetic mutations, such as RUNX1 or ASXL1, which may further influence disease progression and therapeutic response (24, 27). Updated guidelines, including ELN 2022 and NCCN, now incorporate TP53 status (including allelic burden) into risk models to enable more personalized, risk-adapted treatment strategies. Patients with biallelic mutations or adverse co-mutations are often directed toward clinical trials or novel agents, while those with monoallelic TP53 mutations and good fitness may benefit from standard therapies followed by transplant (28–30).
Beyond molecular characteristics, clinical factors including patient age, comorbid conditions, and eligibility for hematopoietic stem cell transplantation remain central to risk assessment and treatment planning. Recent studies have shown that post-transplant survival in TP53-mutated MDS/AML is highly dependent on patient fitness and disease status, with poor outcomes in those with high comorbidity, poor performance, or residual disease at transplant (31, 32). These findings highlight the need to align treatment with both clinical fitness and molecular risk: fit patients may pursue intensive approaches like induction and transplant, while unfit patients may be directed to hypomethylating therapy or supportive care.
3 Mechanisms of immune evasion in TP53-mutated MDS
The role of p53 in tumor immune surveillance has been widely studied across various cancers; p53 enhances anti-tumor immunity by regulating cytokines and tumor recognition (33–36). However, these protective mechanisms are disrupted in TP53-mutated MDS and AML, enabling immune evasion and disease progression (4).
TP53 mutations contribute to genomic instability, leading to a higher mutation burden in tumor cells. Although TP53-mutant cancers generate neoantigens, TP53-mutated MDS/AML typically have lower tumor mutational burdens, suggesting immune evasion mechanisms hinder anti-tumor responses (37).
A key immune escape mechanism is impaired antigen presentation due to the downregulation of major histocompatibility complex class I (MHC I) molecules essential for presenting tumor antigens to cytotoxic T lymphocytes (CTLs) (38). The reduced expression of MHC I decreases the ability of the immune system to recognize and eliminate malignant cells, contributing to an immunosuppressive tumor microenvironment (TME) that facilitates leukemic progression. Additionally, TP53-mutated MDS and AML are characterized by cytokine dysregulation, immune suppression through regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), and metabolic reprogramming, including increased glycolysis and fatty acid metabolism. Beyond immune suppression, metabolic adaptations further reinforce the leukemic microenvironment, creating additional barriers to effective immune responses (Figure 1).
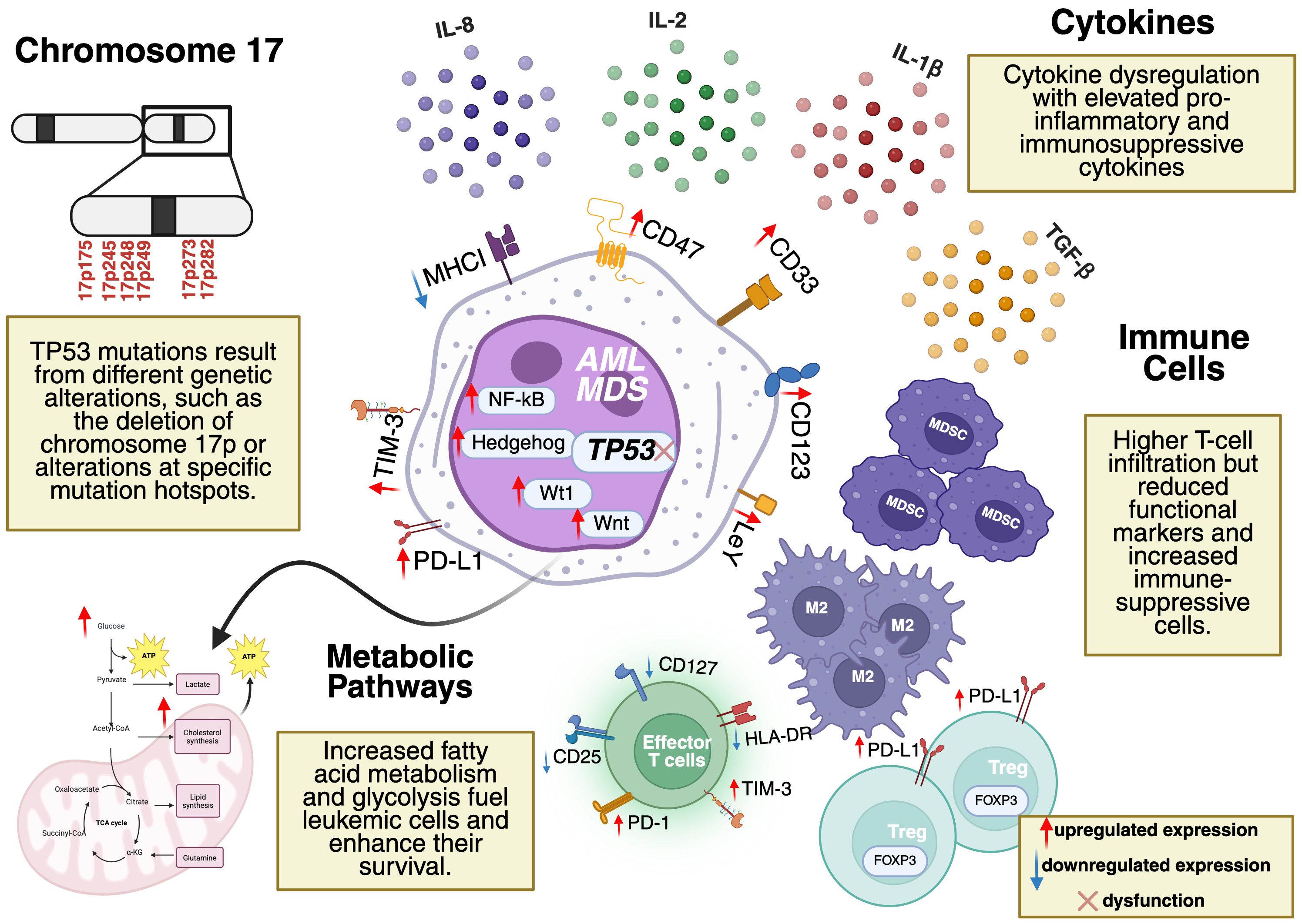
Figure 1. Tumor microenvironment (TME) in TP53-mutated AML and MDS. Key features include cytokine dysregulation (elevated IL-1β, IL-8, TGF-β), immune suppression (Tregs, MDSCs, reduced effector T-cell functionality), and metabolic reprogramming (enhanced glycolysis, fatty acid metabolism). Mutation hotspots in TP53 (codons 175, 245, 248, 249, 273, 282) are located on Chromosome 17p. Dysregulated pathways (e.g., NF-κB, Hedgehog, Wnt) and overexpressed markers (e.g., CD123, CD33, CD47) drive leukemic progression and represent potential therapeutic targets. Figure created by the author using BioRender.
3.1 Cytokine dysregulation and oncogenic signaling
In TP53-mutated MDS and AML, cytokine dysregulation is key in shaping the immunosuppressive TME. Elevated levels of pro-inflammatory and immunosuppressive cytokines such as IL-1β, IL-2, IL-8, and TGF-β contribute to leukemic cell survival and immune dysfunction (39). These cytokines interact with oncogenic signaling pathways, including Hedgehog, Wnt, and NF-κB, which further promote immune evasion.
3.1.1 Hedgehog signaling in immune evasion
The Hedgehog (Hh) pathway is critical for HSC regulation and differentiation but becomes dysregulated in TP53-mutated AML and MDS, leading to self-renewal of malignant stem cells and suppression of anti-tumor immunity (40). Aberrant Hh signaling promotes
● Accumulation of immunosuppressive M2 macrophages, which suppress anti-tumor immunity.
● PD-L1 overexpression, leading to T-cell exhaustion and immune evasion.
● Inhibition of effector T-cell function, further reducing immune-mediated tumor elimination (41, 42).
3.1.2 Wnt signaling and immunosuppressive microenvironment
Wnt signaling is fundamental to stem cell proliferation and differentiation but is often hyperactivated in TP53-mutated malignancies, promoting leukemia stem cell expansion and therapy resistance (43). Additionally, Wnt signaling contributes to immune evasion by
● Inducing the accumulation of Tregs and tumor-associated macrophages, which suppress anti-tumor immunity (44, 45).
● Driving T-cell exhaustion, leading to reduced CTL activity against leukemic cells (44, 45).
Currently, no clinical trials have specifically targeted Wnt inhibitors in hematologic malignancies, highlighting a gap in therapeutic development.
3.1.3 NF-κB signaling and pro-tumor immune regulation
Aberrant activation of NF-κB signaling in TP53-mutated AML and MDS contributes to immune evasion through
● Upregulation of anti-apoptotic proteins (e.g., BCL-XL, MCL-1) that sustain malignant clones.
● Increased production of IL-6 and TNF-α, which promote leukemia progression and chemotherapy resistance.
● Expansion of immunosuppressive cell populations, including Tregs and MDSCs, which inhibit CTLs and natural killer (NK) cells (46).
Due to the role of NF-κB in leukemic survival and immune suppression, its inhibitors combined with immune-modulating agents may benefit TP53-mutated AML/MDS.
3.2 Immune checkpoints and T-cell dysfunction
TP53-mutated AML and MDS are associated with dysfunctional T-cell immunity despite increased immune cell infiltration in the bone marrow. Studies have shown that TP53-mutated AML patients exhibit
● Reduced functional memory T-cell markers (e.g., CD127, CD25, HLA-DR) (39).
● Increased regulatory T-cell (Treg) populations, which suppress anti-tumor immunity (47, 48).
● Upregulated metabolic pathways (e.g., glycolysis, fatty acid metabolism, and oxidative phosphorylation) that enhance Treg-mediated immune suppression (49).
Additionally, CD8+ CTLs in TP53-mutated AML show hallmarks of exhaustion, including increased expression of mucin-domain containing-3 (TIM3) and PD-1 (47). The upregulation of immune senescence markers, such as PD-L1 and T-cell immunoreceptors with Ig and ITIM domains (TIGIT), further promotes immune evasion by preventing effective T-cell activation (39)
A bone marrow biopsy study evaluating T-cell infiltration in relapsed TP53-mutated AML found that PD-L1 expression was significantly higher in CD8+ T cells from patients with multiple relapses compared to newly diagnosed cases (47). This suggests that immune checkpoint dysregulation plays a progressive role in disease evolution and resistance to therapy.
4 Therapeutic approaches
For over 3 decades, chemotherapy has remained the standard treatment for AML and MDS. However, patients with TP53-mutated AML and MDS exhibit significantly lower response rates to conventional therapies, including cytotoxic chemotherapy, hypomethylating agents (HMAs) such as azacitidine and decitabine, and venetoclax-based regimens. Compared to patients with wild-type TP53, those harboring TP53 mutations have a markedly worse prognosis, with a median overall survival (OS) of only 5–10 months (50).
Currently, allogeneic HSCT (allo-HSCT) remains the only potentially curative option for TP53-mutated MDS and AML patients eligible for transplantation. However, outcomes remain suboptimal, as TP53 mutations are associated with higher relapse rates and poor long-term survival following transplantation (51). HMAs are frequently used as cytoreductive therapy before allo-HSCT and as monotherapy in patients ineligible for transplant due to advanced age or comorbidities (52).
Despite these treatment strategies, effective therapies for TP53-mutated disease remain an urgent unmet need. The development of novel targeted therapies and immunotherapies has the potential to overcome treatment resistance and improve survival outcomes. Emerging approaches, including agents that restore TP53 function, immune-based therapies, and metabolic pathway inhibitors, represent promising avenues for improving disease management and enhancing survival and quality of life in these high-risk patients.
Table 1 summarizes current and investigational therapeutic strategies, including their mechanisms of action and clinical progress.
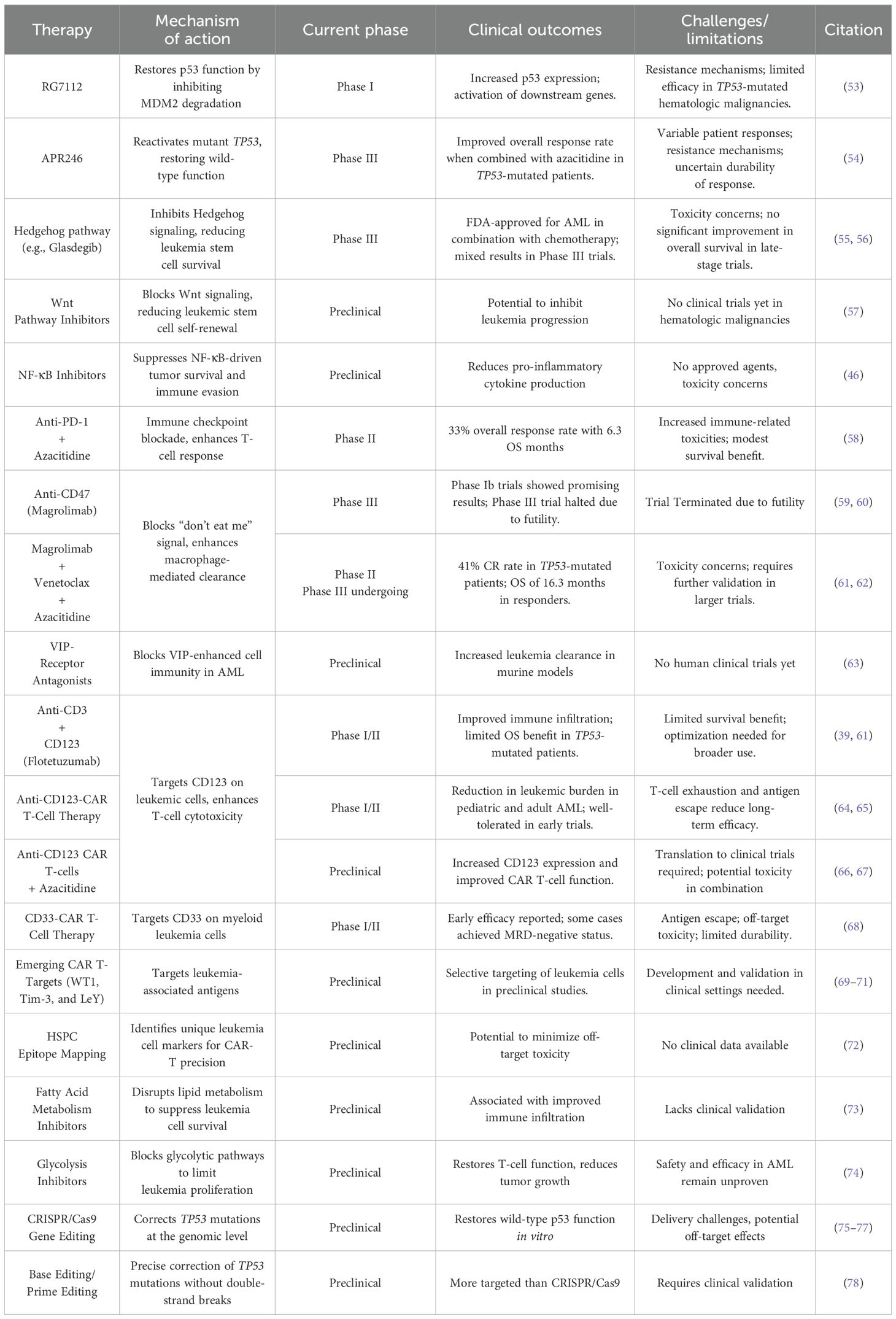
Table 1. Summary of therapeutic strategies for TP53-Mutated MDS and AML.
4.1 Targeting TP53 mutations directly
Directly targeting TP53 mutations represents a promising therapeutic strategy, aiming to restore or enhance p53 function in TP53-mutated AML and MDS. Several small-molecule agents are currently in clinical trials, focusing on either stabilizing p53 expression or reactivating mutant p53 to restore its tumor-suppressive functions.
MDM2 inhibitors, such as RG7112 and idasanutlin, are designed to prevent p53 degradation by inhibiting MDM2, a negative regulator of p53. Blocking MDM2-p53 interactions enables p53 accumulation and apoptotic pathway activation. MDM2 inhibitors have shown preclinical efficacy. However, early-phase clinical trials in leukemia patients have reported limited success, as their effectiveness depends on the presence of wild-type TP53, which is frequently absent in TP53-mutated AML and MDS (53).
APR246 (eprenetapopt) reactivates mutant p53, restoring apoptosis and cell cycle arrest. In multiple clinical trials, APR246 combined with azacitidine has demonstrated enhanced anti-tumor activity, leading to higher response rates in TP53-mutated AML and MDS compared to azacitidine alone (79–81). APR246 is now in Phase III trials evaluating response rates and durability (54).
Given the complexity of TP53 mutations and resistance mechanisms, combining TP53-reactivating agents with immunotherapies or other targeted treatments is being actively investigated. These combinations may enhance disease control and survival in patients with TP53 mutations (82).
4.2 Pathway-targeted therapies
Targeting dysregulated signaling pathways represents a promising therapeutic approach in TP53-mutated AML and MDS, particularly given their role in leukemia stem cell survival, immune evasion, and therapy resistance. Several pathways, including Hh, Wnt, and NF-κB, have been implicated in disease progression, and efforts to develop targeted inhibitors are ongoing.
4.2.1 Hedgehog pathway inhibition
The Hh signaling pathway, involving key components such as SMO, SHH, and GLI3, is critical in stem cell maintenance and differentiation. Aberrant activation of this pathway has been associated with resistance to HMAs in AML and MDS, promoting leukemia stem cell survival and self-renewal (83).
Preclinical studies have shown that combining Hh inhibitors with 5-azacytidine exhibit synergistic cytotoxic effects in AML models (84, 85). Among these inhibitors, glasdegib, an SMO inhibitor, has shown promising activity in preclinical models and early-phase clinical trials, including in patients with TP53-mutated AML (86–88). In the Phase II BRIGHT AML 1003 trial, glasdegib combined with low-dose cytarabine (LDAC) improved complete remission (CR) rates, leading to FDA approval (87).
However, the Phase III BRIGHT AML 1019 trial failed to demonstrate an improvement in OS, limiting glasdegib’s clinical impact in a broader population (55, 56). Differences in trial design, treatment intensity, or patient stratification including, co-mutation profiles, karyotype complexity, TP53 variant allele frequency (VAF), and functional status, may have influenced therapeutic outcomes. The Phase III enrolled a larger population and combined glasdegib with intensive chemotherapy compared to the smaller Phase II that used low-dose chemotherapy. These findings underscore the need for future studies to incorporate comprehensive molecular and clinical stratification to evaluate glasdegib and other Hh inhibitors in combination with targeted therapies for TP53-mutated AML and MDS.
4.3 Immune checkpoint inhibitors and immunotherapy
The use of immune checkpoint inhibitors targeting PD-1 (programmed cell death protein 1) and PD-L1 (programmed cell death ligand 1) has revolutionized cancer treatment, particularly in solid tumors. However, their efficacy in AML and MDS, particularly in TP53-mutated cases, has been limited. While PD-1/PD-L1 inhibitors combined with HMAs such as azacitidine have shown enhanced immune responses, overall clinical outcomes remain modest (89, 90).
4.3.1 PD-1/PD-L1 inhibitors in TP53-mutated AML and MDS
A Phase II trial evaluating nivolumab (PD-1 inhibitor) combined with azacitidine in relapsed/refractory AML reported a 33% overall response rate and a median OS of 6.3 months but with limited long-term benefit (58). Similarly, in higher-risk MDS, combination therapies involving PD-1/PD-L1 inhibitors with HMAs have not demonstrated significantly improved survival compared to HMAs alone (91). Additionally, these therapies have been associated with increased immune-related adverse events, including cytopenias and infections, particularly in older and frail patients, further complicating their clinical application.
The limited efficacy of immune checkpoint blockade in TP53-mutated AML and MDS may be attributed to several factors, including
● Intrinsic resistance mechanisms such as mutations in PD-1/PD-L1 signaling pathways.
● Defective antigen presentation due to MHC dysfunction, reducing T-cell activation.
● An immunosuppressive bone marrow TME suppressing T-cell infiltration and function (92).
Given these challenges, combination strategies are being explored to overcome immune resistance. For instance, integrating PD-1 inhibitors with chimeric antigen receptor T-cell therapy has shown promise in other hematologic malignancies, such as diffuse large B-cell lymphoma with TP53 alterations (93).
4.3.2 Targeting CD47: “Don’t Eat Me” signal blockade
Beyond PD-1/PD-L1 inhibitors, other immune-modulating agents are being investigated. One such approach targets CD47, a key regulator of the “don’t eat me” immune evasion mechanism.
Magrolimab, a CD47-blocking antibody, enhances macrophage-mediated phagocytosis, promoting immune-mediated leukemia clearance. In a Phase Ib trial in MDS patients, magrolimab combined with azacitidine achieved a 40% CR rate and a median OS of 16.3 months, with a 2-year OS of 77% in patients undergoing allogeneic stem cell transplantation (59). However, despite these promising early-phase results, a Phase III trial evaluating magrolimab with azacitidine was discontinued due to futility (60).
Conversely, a Phase II study examining a triple regimen of venetoclax, azacitidine, and magrolimab in newly diagnosed and relapsed/refractory AML demonstrated encouraging response rates with manageable toxicity. Among patients with TP53 mutations, the CR rate was 41%, with a 1-year OS of 53%, compared to 83% in patients with wild-type TP53. A Phase III trial is currently underway to further evaluate this triplet combination in newly diagnosed AML (61, 62).
4.3.3 Emerging checkpoint targets: vasoactive intestinal polypeptide signaling
In 2024, a study published in Blood identified VIP signaling as a potential immune checkpoint in TP53-mutated AML. VIP was found to be overexpressed in CD34-high and TP53-mutated AML cells, contributing to immune suppression by interacting with VPAC1 receptors on myeloid cells and VPAC2 receptors on lymphoid cells (94).
Interestingly, in non-mutated AML, VIP signaling was correlated with enhanced opsonization, suggesting that VIP may influence the efficacy of antibody-based therapies (94). Preclinical models of murine AML treated with a VIP receptor antagonist showed immune-cell-mediated leukemia eradication, with long-term survival rates of 40% in VIP-negative cases and 75% in VIP-positive models (63).
These findings highlight VIP signaling as a novel immunotherapeutic target in TP53-mutated AML, warranting further exploration in clinical trials.
4.4 CAR T-cell and NK-cell therapies
Adoptive cell therapies, including chimeric antigen receptor (CAR) T-cell therapy and NK-cell therapy, have emerged as promising therapeutic strategies in TP53-mutated AML and MDS. These therapies harness the immune system to selectively target tumor-associated antigens overexpressed in leukemic cells, enabling immune-mediated elimination. Among the most well-characterized targets in AML and MDS are CD123 and CD33. Both are highly expressed in leukemic stem cells and blasts, making them ideal candidates for CAR-based therapies.
Despite promising preclinical data, the clinical translation of CAR T-cell therapies in AML and MDS remains challenging due to factors such as antigen heterogeneity, TME-induced immunosuppression, and therapy resistance (95). Strategies to improve efficacy, including dual-targeting approaches, CAR modifications, and combination therapies, are currently being explored.
4.4.1 CD123-CAR T-cell therapy
CD123, the alpha chain of the interleukin-3 receptor, is widely overexpressed in AML and MDS, particularly on leukemia stem cells, making it a key target for immunotherapies (96). Several therapeutic approaches have been developed to exploit CD123, including bispecific antibodies and CAR T-cell therapies.
4.4.1.1 Bispecific CD123/CD3 therapy
Flotetuzumab, a dual-affinity retargeting antibody, simultaneously binds CD3 on T cells and CD123 on leukemia cells, enabling T-cell-mediated leukemia cell destruction. A clinical study evaluating flotetuzumab in 35 patients with relapsed/refractory AML, including 14 with TP53 mutations, demonstrated increased immune infiltration in patients with TP53 mutations. However, these patients had a median OS of only 4.5 months, compared to 18.5 months in TP53-wild-type cases, underscoring the persistent poor prognosis associated with TP53 mutations despite immunotherapy (39, 61).
4.4.1.2 CD123-CAR T-cell therapy
CD123-CAR T-cell therapy employs genetically modified T cells engineered to recognize and eliminate CD123-expressing leukemia cells. In a Phase I trial of pediatric relapsed/refractory AML, CD123-CAR T-cell therapy demonstrated significant anti-tumor activity with a favorable safety profile, as no Grade 2 or higher cytokine release syndrome (CRS) or neurotoxicity was observed (64). In adult patients, the therapy led to a reduction in tumor burden at dose level 2, but sustained responses were limited due to poor CAR T-cell persistence (65). Consequently, ongoing clinical trials are incorporating FCA lymphodepletion regimens and prophylactic tocilizumab to mitigate CRS and improve CAR T-cell durability.
4.4.1.3 Combination strategies to enhance CD123-CAR T-cell efficacy
Combining CD123-CAR T-cell therapy with HMAs, such as azacitidine, is being explored to further enhance efficacy (66, 67); this approach is hypothesized to increase CD123 expression in leukemia cells, improving CAR T-cell recognition and cytotoxicity while also enhancing T-cell activation to strengthen anti-leukemic effects. These findings support the continued development of combination strategies integrating CAR T-cell therapy with epigenetic modulators to improve outcomes in TP53-mutated AML (59).
4.4.2 CD33-CAR T-cell therapy
CD33 is another widely expressed myeloid antigen in AML and MDS, making it a valuable target for CAR T-cell therapy. In a Phase I/II clinical trial, CD33-CAR T-cell therapy demonstrated early efficacy, with some patients achieving CR with minimal residual disease negativity (68).
However, significant challenges remain, including manufacturing delays associated with autologous CAR T-cell production and rapid disease progression, which can outpace CAR T-cell expansion and activity (97). To address these limitations, ongoing research is focusing on optimizing CAR constructs to enhance persistence and reduce T-cell exhaustion while also exploring allogeneic “off-the-shelf” CAR T-cell therapies to mitigate manufacturing delays and improve accessibility. Despite these obstacles, CD33-targeted CAR T-cell therapy remains an area of active investigation, with efforts directed toward refining its clinical applicability in high-risk TP53-mutated AML and MDS.
4.4.3 Emerging targets and challenges
Beyond CD123 and CD33, additional tumor-associated antigens are being explored for CAR T-cell therapy in TP53-mutated AML and MDS. Notable targets include WT1, a transcription factor overexpressed in leukemia stem cells; Tim-3, a checkpoint receptor involved in immune evasion; and LeY, an antigen found in a subset of myeloid leukemias. These are thus promising candidates for immunotherapy (69–71).
Despite their potential, translating CAR T-cell therapy into clinical use faces challenges such as antigen escape, off-target toxicity, and T-cell exhaustion. Leukemia cells can downregulate target antigen expression, leading to relapse, while shared antigen expression with normal hematopoietic cells increases the risk of prolonged myelosuppression. Additionally, T-cell exhaustion due to prolonged activation remains a barrier to achieving durable responses (98).
To overcome these limitations, researchers are exploring multiplex CAR T-cell designs to target multiple antigens simultaneously, reducing antigen escape. Hematopoietic stem and progenitor cell (HSPC) epitope mapping is being investigated to help differentiate leukemia cells from healthy cells, minimizing off-target toxicity (72). Additionally, combining CAR T-cell therapy with immune checkpoint blockades, such as PD-1 inhibitors, may enhance T-cell function and persistence, potentially leading to more sustained anti-leukemic responses.
4.5 Modulating the TME
The TME is crucial in the progression and treatment resistance of TP53-mutated AML and MDS. In these malignancies, the TME is highly immunosuppressive, driven by pro-tumorigenic cytokines, metabolic reprogramming, and immune evasion mechanisms. A key factor is IL-8, which promotes leukemic cell proliferation and therapy resistance (99, 100). Preclinical studies suggest that neutralizing IL-8 with specific antibodies can reduce AML cell growth and enhance sensitivity to chemotherapy, highlighting its potential as a therapeutic target (100).
Beyond cytokine dysregulation, metabolic alterations within the TME further contribute to immune evasion and disease persistence. Dysregulated lipid metabolism and glycolysis provide crucial energy sources for leukemic cells while supporting the survival of immunosuppressive populations. Strategies that disrupt these metabolic dependencies may help reshape the TME, enhancing anti-tumor immune responses and improving treatment outcomes for TP53-mutated AML and MDS.
4.5.1 Lipid metabolism
Lipid metabolism alterations are increasingly recognized as key contributors to leukemic progression and therapy resistance in TP53-mutated AML. A 2024 multi-omic analysis published in Blood found that specific TP53 hotspot mutations (R175H, R273H) were associated with enhanced oxidative phosphorylation and a higher prevalence of primitive leukemia stem and progenitor states (101). Conversely, TP53-wild-type and R248Q-mutated AML cells displayed enrichment in cellular senescence, heme metabolism, and fatty acid metabolism pathways, suggesting that TP53 mutations may drive metabolic reprogramming, promoting stemness and aggressive disease behavior (101).
In addition to its role in leukemia cell survival, lipid metabolism influences immune cell function within the TME. A 2022 study in Lipid Insights identified a fatty acid metabolism-related signature correlating with immune cell infiltration and clinical outcomes in AML, indicating its prognostic value (102). This suggests that targeting fatty acid metabolism could not only disrupt leukemic cell survival but also enhance anti-tumor immunity.
The cholesterol pathway has also been implicated in AML resistance mechanisms. A recent Blood study found that TP53-mutated AML cells exhibited increased cholesterol metabolism, contributing to immune evasion and reduced CAR T-cell efficacy (103). Notably, simvastatin-mediated cholesterol pathway inhibition improved CAR T-cell function, reducing T-cell exhaustion and enhancing tumor clearance (73). These findings suggest that combining CAR T-cell therapy with lipid metabolism inhibitors may improve responses in TP53-mutated AML and MDS, warranting further clinical investigation.
4.5.2 Glycolysis
Glycolytic reprogramming is another hallmark of TP53-mutated AML, enabling leukemic cells to sustain rapid proliferation and evade immune surveillance. A 2024 study published in Frontiers in Pharmacology examined the metabolic and physiological hallmarks of TP53-mutated AML, highlighting an upregulated glycolysis pathway as a key driver of chemoresistance and immune evasion (104, 105).
The reliance on glycolysis in TP53-mutated AML suggests that targeting this metabolic pathway may enhance therapeutic efficacy. Preclinical models indicate that inhibiting glycolysis can reduce leukemia cell proliferation, sensitize AML cells to standard chemotherapy, and restore T-cell function within the TME (74). However, clinical validation is still required to determine the safety and efficacy of glycolysis inhibitors in TP53-mutated AML and MDS.
4.6 Gene-editing technologies
Advances in gene-editing technologies, particularly CRISPR/Cas9, represent a promising frontier in the treatment of TP53-mutated MDS and AML. These tools aim to correct TP53 mutations at the genomic level, potentially restoring normal p53 function and immune regulation. Preclinical studies have demonstrated success in repairing TP53 mutations, reinstating wild-type p53 activity, and selectively eliminating p53-deficient cells while sparing normal ones (75–77). Additionally, computational tools such as the Computational CRISPR Strategy improve precision, while emerging techniques such as base and prime editing enhance mutation correction (78).
Despite these advances, several challenges remain, particularly in safe and efficient delivery to HSPCs. Current methods, including viral vectors and lipid nanoparticles, pose risks of toxicity, immunogenicity, and off-target effects, potentially leading to genomic instability or the emergence of new oncogenic drivers (69). Timely and scalable gene-editing methods are necessary given the rapid progression of TP53-mutated AML.
Ongoing research is focused on improving guide RNA design, developing high-fidelity Cas9 variants, and refining targeting specificity to minimize off-target effects. While gene editing holds long-term therapeutic potential, its clinical success will depend on advances in delivery methods, safety validation, and successful clinical translation.
5 Conclusion
Treating TP53-mutated MDS and AML remains challenging due to poor prognoses, resistance to standard therapies, and limited treatment options. However, recent advances in understanding the role of TP53 mutations in immune dysregulation and tumor progression have opened new avenues for therapeutic innovation. These approaches, including targeted therapies, immunotherapies, metabolic interventions, and gene editing, are emerging as promising alternatives. Enhancing combination therapies, improving immune modulating, and advancing gene-editing delivery will be essential for improving patient outcomes.
Author contributions
MMA: Conceptualization, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Acknowledgments
The author sincerely appreciates Dr. Hammad Tashkandi for his valuable feedback and insights. Special thanks to BioRender.com for providing the platform used to create the diagram; Albakri, M. (2025) https://BioRender.com/i62r794.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Language support was provided by ChatGPT to assist with grammar refinement and clarity during manuscript preparation. Reference management was conducted using EndNote to organize and format citations. Figure design was assisted by BioRender to generate a professional schematic summarizing key concepts.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kastenhuber ER and Lowe SW. Putting p53 in context. Cell. (2017) 170:1062–78. doi: 10.1016/j.cell.2017.08.028
2. Bykov VJN, Eriksson SE, Bianchi J, and Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. (2018) 18:89–102. doi: 10.1038/nrc.2017.109
3. Sullivan KD, Galbraith MD, Andrysik Z, and Espinosa JM. Mechanisms of transcriptional regulation by p53. Cell Death Differ. (2018) 25:133–43. doi: 10.1038/cdd.2017.174
4. Carlsen L, Zhang S, Tian X, de la Cruz A, George A, Arnoff TE, et al. The role of p53 in anti-tumor immunity and response to immunotherapy. Front Mol Biosci. (2023) 10:1148389. doi: 10.3389/fmolb.2023.1148389
5. Cooks T, Harris CC, and Oren M. Caught in the cross fire: p53 in inflammation. Carcinogenesis. (2014) 35:1680–90. doi: 10.1093/carcin/bgu134
6. Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. (2016) 37:865–76. doi: 10.1002/humu.23035
7. Vousden KH and Prives C. Blinded by the light: the growing complexity of p53. Cell. (2009) 137:413–31. doi: 10.1016/j.cell.2009.04.037
9. Riley T, Sontag E, Chen P, and Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. (2008) 9:402–12. doi: 10.1038/nrm2395
10. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. (1997) 88:323–31. doi: 10.1016/s0092-8674(00)81871-1
11. Alvarado-Ortiz E, de la Cruz-Lopez KG, Becerril-Rico J, Sarabia-Sanchez MA, Ortiz-Sanchez E, and Garcia-Carranca A. Mutant p53 gain-of-function: role in cancer development, progression, and therapeutic approaches. Front Cell Dev Biol. (2020) 8:607670. doi: 10.3389/fcell.2020.607670
12. Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood. (2005) 106:3150–9. doi: 10.1182/blood-2005-02-0553
13. Olivier M, Hollstein M, and Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. (2010) 2:1. doi: 10.1101/cshperspect.a001008
14. Leroy B, Anderson M, and Soussi T. TP 53 mutations in human cancer: database reassessment and prospects for the next decade. Hum mutation. (2014) 35:672–88. doi: 10.1002/humu.22552
15. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. (2013) 502:333–9. doi: 10.1038/nature12634
16. Prochazka KT, Pregartner G, Rucker FG, Heitzer E, Pabst G, Wolfler A, et al. Clinical implications of subclonal TP53 mutations in acute myeloid leukemia. Haematologica. (2019) 104:516–23. doi: 10.3324/haematol.2018.205013
17. Chen S, Wang Q, Yu H, Capitano ML, Vemula S, Nabinger SC, et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat Commun. (2019) 10:5649. doi: 10.1038/s41467-019-13542-2
18. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. (2015) 126:9–16. doi: 10.1182/blood-2015-03-631747
19. Sill H, Zebisch A, and Haase D. Acute myeloid leukemia and myelodysplastic syndromes with TP53 aberrations - A distinct stem cell disorder. Clin Cancer Res. (2020) 26:5304–9. doi: 10.1158/1078-0432.CCR-20-2272
20. van Kampen F, Clark A, Soul J, Kanhere A, Glenn MA, Pettitt AR, et al. Deletion of 17p in cancers: Guilt by (p53) association. Oncogene. (2025) 44:637–51. doi: 10.1038/s41388-025-03300-8
21. Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. (2012) 119:2114–21. doi: 10.1182/blood-2011-08-375758
22. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. (2022) 36:1703–19. doi: 10.1038/s41375-022-01613-1
23. Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. (2022) 140:1200–28. doi: 10.1182/blood.2022015850
24. Weinberg OK, Siddon A, Madanat YF, Gagan J, Arber DA, Dal Cin P, et al. TP53 mutation defines a unique subgroup within complex karyotype de novo and therapy-related MDS/AML. Blood advances. (2022) 6:2847–53. doi: 10.1182/bloodadvances.2021006239
25. Bernard E, Nannya Y, Hasserjian RP, Devlin SM, Tuechler H, Medina-Martinez JS, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. (2020) 26:1549–56. doi: 10.1038/s41591-020-1008-z
26. Bernard E, Tuechler H, Greenberg PL, Hasserjian RP, Arango Ossa JE, Nannya Y, et al. Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM evidence. (2022) 1:EVIDoa2200008. doi: 10.1056/EVIDoa2200008
27. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. New Engl J Med. (2016) 374:2209–21. doi: 10.1056/NEJMoa1516192
28. Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood J Am Soc Hematology. (2022) 140:1345–77. doi: 10.1182/blood.2022016867
29. Greenberg PL, Stone RM, Abaza Y, Al-Kali A, Anand S, Ball B, et al. NCCN guidelines® Insights: myelodysplastic syndromes, version 2.2025: featured updates to the NCCN guidelines. J Natl Compr Cancer Network. (2025) 23:66–75. doi: 10.6004/jnccn.2025.0013
30. Short NJ, Montalban-Bravo G, Hwang H, Ning J, Franquiz MJ, Kanagal-Shamanna R, et al. Prognostic and therapeutic impacts of mutant TP53 variant allelic frequency in newly diagnosed acute myeloid leukemia. Blood advances. (2020) 4:5681–9. doi: 10.1182/bloodadvances.2020003120
31. Ciurea SO, Chilkulwar A, Saliba RM, Chen J, Rondon G, Patel KP, et al. Prognostic factors influencing survival after allogeneic transplantation for AML/MDS patients with TP53 mutations. Blood J Am Soc Hematology. (2018) 131:2989–92. doi: 10.1182/blood-2018-02-832360
32. Shahzad M, Iqbal Q, Tariq E, Ammad-Ud-Din M, Butt A, Mushtaq AH, et al. Outcomes with allogeneic hematopoietic stem cell transplantation in TP53-mutated myelodysplastic syndrome: A systematic review and meta-analysis. Crit Rev oncology/hematology. (2024) 196:104310. doi: 10.1016/j.critrevonc.2024.104310
33. Xiao W, Du N, Huang T, Guo J, Mo X, Yuan T, et al. TP53 mutation as potential negative predictor for response of anti-CTLA-4 therapy in metastatic melanoma. EBioMedicine. (2018) 32:119–24. doi: 10.1016/j.ebiom.2018.05.019
34. Lyu H, Li M, Jiang Z, Liu Z, and Wang X. Correlate the TP53 mutation and the HRAS mutation with immune signatures in head and neck squamous cell cancer. Comput Struct Biotechnol J. (2019) 17:1020–30. doi: 10.1016/j.csbj.2019.07.009
35. Liu Z, Jiang Z, Gao Y, Wang L, Chen C, and Wang X. TP53 mutations promote immunogenic activity in breast cancer. J Oncol. (2019) 2019:5952836. doi: 10.1155/2019/5952836
36. Wang X and Sun Q. TP53 mutations, expression and interaction networks in human cancers. Oncotarget. (2017) 8:624–43. doi: 10.18632/oncotarget.13483
37. Wen XM, Xu ZJ, Jin Y, Xia PH, Ma JC, Qian W, et al. Association analyses of TP53 mutation with prognosis, tumor mutational burden, and immunological features in acute myeloid leukemia. Front Immunol. (2021) 12:717527. doi: 10.3389/fimmu.2021.717527
38. Wang B, Niu D, Lai L, and Ren EC. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat Commun. (2013) 4:2359. doi: 10.1038/ncomms3359
39. Vadakekolathu J, Lai C, Reeder S, Church SE, Hood T, Lourdusamy A, et al. TP53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy in AML. Blood Adv. (2020) 4:5011–24. doi: 10.1182/bloodadvances.2020002512
40. Jing J, Wu Z, Wang J, Luo G, Lin H, Fan Y, et al. Hedgehog signaling in tissue homeostasis, cancers, and targeted therapies. Signal Transduct Target Ther. (2023) 8:315. doi: 10.1038/s41392-023-01559-5
41. Petty AJ, Li A, Wang X, Dai R, Heyman B, Hsu D, et al. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J Clin Invest. (2019) 129:5151–62. doi: 10.1172/JCI128644
42. Petty AJ, Dai R, Lapalombella R, Baiocchi RA, Benson DM, Li Z, et al. Hedgehog-induced PD-L1 on tumor-associated macrophages is critical for suppression of tumor-infiltrating CD8+ T cell function. JCI Insight. (2021) 6:6–6. doi: 10.1172/jci.insight.146707
43. Xiao Q, Werner J, Venkatachalam N, Boonekamp KE, Ebert MP, and Zhan T. Cross-talk between p53 and Wnt signaling in cancer. Biomolecules. (2022) 12:453. doi: 10.3390/biom12030453
44. Gruszka AM, Valli D, and Alcalay M. Wnt signalling in acute myeloid leukaemia. Cells. (2019) 8:1403. doi: 10.3390/cells8111403
45. Yuan Y, Wu D, Hou Y, Zhang Y, Tan C, Nie X, et al. Wnt signaling: Modulating tumor-associated macrophages and related immunotherapeutic insights. Biochem Pharmacol. (2024) 223:116154. doi: 10.1016/j.bcp.2024.116154
46. Blagih J, Buck MD, and Vousden KH. p53, cancer and the immune response. J Cell Sci. (2020) 133:jcs237453. doi: 10.1242/jcs.237453
47. Williams P, Basu S, Garcia-Manero G, Hourigan CS, Oetjen KA, Cortes JE, et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer. (2019) 125:1470–81. doi: 10.1002/cncr.31896
48. Vadakekolathu J, Altmann H, Wobus M, von Bonin M, Schmitz M, Baretton GB, et al. Spatially resolved transcriptomics unveils unique T-cell dysfunctional states and prognostic gene expression signatures in TP53-mutated acute myeloid leukemia. Blood. (2023) 142:291. doi: 10.1182/blood-2023-174871
49. Abolhalaj M, Sincic V, Lilljebjorn H, Sanden C, Aab A, Hagerbrand K, et al. Transcriptional profiling demonstrates altered characteristics of CD8(+) cytotoxic T-cells and regulatory T-cells in TP53-mutated acute myeloid leukemia. Cancer Med. (2022) 11:3023–32. doi: 10.1002/cam4.4661
50. Daver NG, Maiti A, Kadia TM, Vyas P, Majeti R, Wei AH, et al. TP53-mutated myelodysplastic syndrome and acute myeloid leukemia: biology, current therapy, and future directions. Cancer Discov. (2022) 12:2516–29. doi: 10.1158/2159-8290.CD-22-0332
51. Steiner N, Klyuchnikov E, Badbaran A, Massoud R, Zeck G, Nachbaur D, et al. Impact of TP53 mutation on outcome after allogeneic stem cell transplantation for patients with MDS/AML not in complete remission. Blood. (2024) 144:3571. doi: 10.1182/blood-2024-205849
52. Scalzulli E, Pepe S, Colafigli G, and Breccia M. Therapeutic strategies in low and high-risk MDS: What does the future have to offer? Blood Rev. (2021) 45:100689. doi: 10.1016/j.blre.2020.100689
53. Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, et al. Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res. (2016) 22:868–76. doi: 10.1158/1078-0432.CCR-15-0481
54. Therapeutics A. A phase III Multicenter, Randomized, Open Label Study of APR-246 in Combination with Azacitidine Versus Azacitidine Alone for the Treatment of (Tumor Protein) TP53 Mutant Myelodysplastic Syndromes. Doylestown, Pennsylvania, USA: Aprea Therapeutics (2021).
55. Jaramillo S, Krisam J, Le Cornet L, Kratzmann M, Baumann L, Eissymont O, et al. Randomized phase III GnG study on two schedules of gemtuzumab ozogamicin as adjunct to intensive induction therapy and double-blinded intensive post-remission therapy with or without glasdegib in patients with newly diagnosed acute myeloid leukemia. Haematologica. (2024) 109:1973. doi: 10.3324/haematol.2023.284346
56. Sekeres MA, Montesinos P, Novak J, Wang J, Jeyakumar D, Tomlinson B, et al. Glasdegib plus intensive or non-intensive chemotherapy for untreated acute myeloid leukemia: results from the randomized, phase 3 BRIGHT AML 1019 trial. Leukemia. (2023) 37:2017–26. doi: 10.1038/s41375-023-02001-z
57. Pepe F, Bill M, Papaioannou D, Karunasiri M, Walker A, Naumann E, et al. Targeting Wnt signaling in acute myeloid leukemia stem cells. Haematologica. (2021) 107:307. doi: 10.3324/haematol.2020.266155
58. Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discov. (2019) 9:370–83. doi: 10.1158/2159-8290.CD-18-0774
59. Sallman DA, Al Malki MM, Asch AS, Wang ES, Jurcic JG, Bradley TJ, et al. Magrolimab in combination with azacitidine in patients with higher-risk myelodysplastic syndromes: final results of a phase Ib study. J Clin Oncol. (2023) 41:2815–26. doi: 10.1200/JCO.22.01794
60. ClinicalTrials.gov. Study NCT04313881: Clinical Trial Information (2025). Available online at: https://www.clinicaltrials.gov/study/NCT04313881 (Accessed January 11, 2025).
61. Daver N, Konopleva M, Maiti A, Kadia TM, DiNardo CD, Loghavi S, et al. Phase I/II study of azacitidine (AZA) with venetoclax (VEN) and magrolimab (Magro) in patients (pts) with newly diagnosed older/unfit or high-risk acute myeloid leukemia (AML) and relapsed/refractory (R/R) AML. Blood. (2021) 138:371. doi: 10.1182/blood-2021-153638
62. Daver NG, Liu K, Kuwahara SB, Caldwell K, and Vyas P. AML-577 A phase III, randomized trial of magrolimab in combination with venetoclax and azacitidine in previously untreated patients with acute myeloid leukemia who are ineligible for intensive chemotherapy (ENHANCE-3). Clin Lymphoma Myeloma Leukemia. (2023) 23:S313–4. doi: 10.1016/S2152-2650(23)01083-2
63. Petersen CT, Li JM, and Waller EK. Administration of a vasoactive intestinal peptide antagonist enhances the autologous anti-leukemia T cell response in murine models of acute leukemia. Oncoimmunology. (2017) 6:e1304336. doi: 10.1080/2162402X.2017.1304336
64. Naik S, Madden RM, Lipsitt A, Lockey T, Bran J, Rubnitz JE, et al. Safety and anti-leukemic activity of CD123-CAR T cells in pediatric patients with AML: preliminary results from a phase 1 trial. Blood. (2022) 140:4584–5. doi: 10.1182/blood-2022-170201
65. Sallman DA, DeAngelo DJ, Pemmaraju N, Dinner S, Gill S, Olin RL, et al. Ameli-01: a phase I trial of UCART123v1. 2, an anti-CD123 allogeneic CAR-T cell product, in adult patients with relapsed or refractory (R/R) CD123+ acute myeloid leukemia (AML). Blood. (2022) 140:2371–3. doi: 10.1182/blood-2022-169928
66. El Khawanky N, Hughes A, Yu W, Myburgh R, Matschulla T, Taromi S, et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat Commun. (2021) 12:6436. doi: 10.1038/s41467-021-26683-0
67. Cummins KD, Frey N, Nelson AM, Schmidt A, Luger S, Isaacs RE, et al. Treating relapsed/refractory (RR) AML with biodegradable anti-CD123 CAR modified T cells. Blood. (2017) 130:1359. doi: 10.1182/blood.V130.Suppl_1.1359.1359
68. Shah NN, Tasian SK, Kohler ME, Hsieh EM, Baumeister SH, Summers C, et al. CD33 CAR T-cells (CD33CART) for children and young adults with relapsed/refractory AML: dose-escalation results from a phase I/II multicenter trial. Blood. (2023) 142:771–1. doi: 10.1182/blood-2023-179667
69. He X, Feng Z, Ma J, Ling S, Cao Y, Gurung B, et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood. (2020) 135:713–23. doi: 10.1182/blood.2019002779
70. Rafiq S, Purdon TJ, Daniyan AF, Koneru M, Dao T, Liu C, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia. (2017) 31:1788–97. doi: 10.1038/leu.2016.373
71. Brenner MK. CAR T cells for acute myeloid leukemia: the LeY of the land. Mol Ther. (2013) 21:1983–4. doi: 10.1038/mt.2013.234
72. Casirati G, Cosentino A, Mucci A, Salah Mahmoud M, Ugarte Zabala I, Zeng J, et al. Epitope editing enables targeted immunotherapy of acute myeloid leukaemia. Nature. (2023) 621:404–14. doi: 10.1038/s41586-023-06496-5
73. Zaky MY, Fan C, Zhang H, and Sun XF. Unraveling the anticancer potential of statins: mechanisms and clinical significance. Cancers (Basel). (2023) 15:4787. doi: 10.3390/cancers15194787
74. Yang Y, Pu J, and Yang Y. Glycolysis and chemoresistance in acute myeloid leukemia. Heliyon. (2024) 10:e35721. doi: 10.1016/j.heliyon.2024.e35721
75. Enache OM, Rendo V, Abdusamad M, Lam D, Davison D, Pal S, et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat Genet. (2020) 52:662–8. doi: 10.1038/s41588-020-0623-4
76. Chira S, Gulei D, Hajitou A, and Berindan-Neagoe I. Restoring the p53 ‘Guardian’phenotype in p53-deficient tumor cells with CRISPR/Cas9. Trends Biotechnol. (2018) 36:653–60. doi: 10.1016/j.tibtech.2018.01.014
77. Zhan H, Xie H, Zhou Q, Liu Y, and Huang W. Synthesizing a genetic sensor based on CRISPR-cas9 for specifically killing p53-deficient cancer cells. ACS Synth Biol. (2018) 7:1798–807. doi: 10.1021/acssynbio.8b00202
78. Niu X, Deng K, Liu L, Yang K, and Hu X. A statistical framework for predicting critical regions of p53-dependent enhancers. Brief Bioinform. (2021) 22:bbaa053. doi: 10.1093/bib/bbaa053
79. Mishra A, Tamari R, DeZern AE, Byrne MT, Gooptu M, Chen YB, et al. Eprenetapopt plus azacitidine after allogeneic hematopoietic stem-cell transplantation for TP53-mutant acute myeloid leukemia and myelodysplastic syndromes. J Clin Oncol. (2022) 40:3985–93. doi: 10.1200/JCO.22.00181
80. Cluzeau T, Sebert M, Rahme R, Cuzzubbo S, Lehmann-Che J, Madelaine I, et al. Eprenetapopt plus azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia: A phase II study by the groupe francophone des myelodysplasies (GFM). J Clin Oncol. (2021) 39:1575–83. doi: 10.1200/JCO.20.02342
81. Sallman DA, DeZern AE, Garcia-Manero G, Steensma DP, Roboz GJ, Sekeres MA, et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J Clin Oncol. (2021) 39:1584–94. doi: 10.1200/JCO.20.02341
82. Zhang Q, Bykov VJN, Wiman KG, and Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. (2018) 9:439. doi: 10.1038/s41419-018-0463-7
83. Abraham A and Matsui W. Hedgehog signaling in myeloid Malignancies. Cancers (Basel). (2021) 13:4888. doi: 10.3390/cancers13194888
84. Huang K, Sun Z, Ding B, Jiang X, Wang Z, Zhu Y, et al. Suppressing Hedgehog signaling reverses drug resistance of refractory acute myeloid leukemia. Onco Targets Ther. (2019) 12:7477–88. doi: 10.2147/OTT.S216628
85. Chaudhry P, Singh M, Triche TJ, Guzman M, and Merchant AA. GLI3 repressor determines Hedgehog pathway activation and is required for response to SMO antagonist glasdegib in AML. Blood J Am Soc Hematology. (2017) 129:3465–75. doi: 10.1182/blood-2016-05-718585
86. Savona MR, Pollyea DA, Stock W, Oehler VG, Schroeder MA, Lancet J, et al. Phase Ib study of glasdegib, a hedgehog pathway inhibitor, in combination with standard chemotherapy in patients with AML or high-risk MDS. Clin Cancer Res. (2018) 24:2294–303. doi: 10.1158/1078-0432.CCR-17-2824
87. Cortes JE, Heidel FH, Hellmann A, Fiedler W, Smith BD, Robak T, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. (2019) 33:379–89. doi: 10.1038/s41375-018-0312-9
88. Lemos T and Merchant A. The hedgehog pathway in hematopoiesis and hematological Malignancy. Front Oncol. (2022) 12:960943. doi: 10.3389/fonc.2022.960943
89. Zeidan AM, Boddu P, Wood BL, Zelterman D, Little RF, Ivy SP, et al. Blast MRD AML-2: blockade of PD-1 added to standard therapy to target measurable residual disease (MDR) in acute myeloid leukemia (AML) 2-a randomized phase 2 study of the venetoclax, azacitidine, and pembrolizumab versus venetoclax and azacitidine as first line therapy in older patients with AML who are ineligible or who refuse intensive chemotherapy. Blood. (2020) 136:11–2. doi: 10.1182/blood-2020-139752
90. Zeidan AM, Boss I, Beach C, Copeland WB, Thompson E, Fox BA, et al. A randomized phase 2 trial of azacitidine with or without durvalumab as first-line therapy for older patients with AML. Blood Advances. (2022) 6:2219–29. doi: 10.1182/bloodadvances.2021006138
91. Zeidan AM, Boss I, Beach C, Copeland WB, Thompson E, Fox BA, et al. A randomized phase 2 trial of azacitidine with or without durvalumab as first-line therapy for higher-risk myelodysplastic syndromes. Blood Advances. (2022) 6:2207–18. doi: 10.1182/bloodadvances.2021005487
92. Marei HE, Hasan A, Pozzoli G, and Cenciarelli C. Cancer immunotherapy with immune checkpoint inhibitors (ICIs): potential, mechanisms of resistance, and strategies for reinvigorating T cell responsiveness when resistance is acquired. Cancer Cell Int. (2023) 23:64. doi: 10.1186/s12935-023-02902-0
93. Xue B, Luo X, Liu Y, Ye S, Zhou L, Li S, et al. CAR T-cell therapy combined with PD-1 inhibitors significantly improve the efficacy and prognosis of r/r DLBCL with TP53 alterations. Blood. (2023) 142:3515. doi: 10.1182/blood-2023-180500
94. Zeng F, Chaudagar K, Wang Y, Li J-M, and Waller EK. VIP expression drives an immunosuppressive tumor microenvironment in TP53-mutated AML. Blood. (2024) 144:4299. doi: 10.1182/blood-2024-208170
95. Liu Z, Lei W, Wang H, Liu X, and Fu R. Challenges and strategies associated with CAR-T cell therapy in blood Malignancies. Exp Hematol Oncol. (2024) 13:22. doi: 10.1186/s40164-024-00490-x
96. Testa U, Pelosi E, and Castelli G. CD123 as a therapeutic target in the treatment of hematological Malignancies. Cancers. (2019) 11:1358. doi: 10.3390/cancers11091358
97. Tambaro FP, Singh H, Jones E, Rytting M, Mahadeo KM, Thompson P, et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia. (2021) 35:3282–6. doi: 10.1038/s41375-021-01232-2
98. Isidori A, Cerchione C, Daver N, DiNardo C, Garcia-Manero G, Konopleva M, et al. Immunotherapy in acute myeloid leukemia: where we stand. Front Oncol. (2021) 11:656218. doi: 10.3389/fonc.2021.656218
99. Ryningen A, Wergeland L, Glenjen N, Gjertsen BT, and Bruserud Ø. In vitro crosstalk between fibroblasts and native human acute myelogenous leukemia (AML) blasts via local cytokine networks results in increased proliferation and decreased apoptosis of AML cells as well as increased levels of proangiogenic Interleukin 8. Leukemia Res. (2005) 29:185–96. doi: 10.1016/j.leukres.2004.06.008
100. Vijay V, Miller R, Vue GS, Pezeshkian MB, Maywood M, Ast AM, et al. Interleukin-8 blockade prevents activated endothelial cell mediated proliferation and chemoresistance of acute myeloid leukemia. Leuk Res. (2019) 84:106180. doi: 10.1016/j.leukres.2019.106180
101. Vadakekolathu J, Skuli S, Boocock DJ, Coveney C, Ikpo EG, Wang B, et al. Multi-omic analyses of TP53-mutated acute myeloid leukemia identify prognostic metabolic signatures. Blood. (2024) 144:2911. doi: 10.1182/blood-2024-201311
102. Zhang HB, Sun ZK, Zhong FM, Yao FY, Liu J, Zhang J, et al. A novel fatty acid metabolism-related signature identifies features of the tumor microenvironment and predicts clinical outcome in acute myeloid leukemia. Lipids Health Dis. (2022) 21:79. doi: 10.1186/s12944-022-01687-x
103. Mueller J, Schimmer R, Koch C, Schneiter F, Fullin J, Lysenko V, et al. TP53 deficiency in AML confers resistance to CAR T-cells that can be overcome by targeting the cholesterol or Wnt pathways. Blood. (2023) 142:1019. doi: 10.1182/blood-2023-185567
104. Motlagh AV, Mahdevar M, Mirzaei S, Entezari M, Hashemi M, Hushmandi K, et al. Introduction of mutant TP53 related genes in metabolic pathways and evaluation their correlation with immune cells, drug resistance and sensitivity. Life Sci. (2022) 303:120650. doi: 10.1016/j.lfs.2022.120650
Keywords: TP53 mutation, myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), immune evasion, tumor microenvironment, targeted therapies
Citation: Albakri MM (2025) TP53-mutated MDS and AML: immune dysregulation, tumor microenvironment, and emerging therapeutic strategies. Front. Oncol. 15:1655486. doi: 10.3389/fonc.2025.1655486
Received: 28 June 2025; Accepted: 30 July 2025;
Published: 20 August 2025.
Edited by:
Teresa de Souza Fernandez, National Cancer Institute (INCA), BrazilReviewed by:
Dinesh Pendharkar, Sarvodaya Hospital and Research Centre, IndiaNathalia Lopez Duarte, Rio de Janeiro Municipal Health Secretariat (SMS-RJ), Brazil
Copyright © 2025 Albakri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marwah M. Albakri, bWFsYmFrcmlAdGFpYmFodS5lZHUuc2E=