 José Ignacio Erices1
José Ignacio Erices1 Evelin González2
Evelin González2 Marcela Salgado1,3Carol Barahona-Ponce4‡Matías Freire5
Marcela Salgado1,3Carol Barahona-Ponce4‡Matías Freire5 Gonzalo Sepúlveda-Hermosilla5Diego Ampuero5Alejandro Blanco2Valentina Gárate-Calderón1,4
Gonzalo Sepúlveda-Hermosilla5Diego Ampuero5Alejandro Blanco2Valentina Gárate-Calderón1,4 Pablo Báez-Benavides6
Pablo Báez-Benavides6 Camilo Tapia-Valladares1
Camilo Tapia-Valladares1 Jessica Toro1,7Iván Gallegos1,5,6,8
Jessica Toro1,7Iván Gallegos1,5,6,8 Olga Barajas1,7,9Mónica Ahumada1,7,9Verónica Sanhueza10Loreto Spencer11Gonzalo De Toro12Erik Morales13,14Lorena Gutiérrez15Fernanda Morales1Arnaldo Marin1
Olga Barajas1,7,9Mónica Ahumada1,7,9Verónica Sanhueza10Loreto Spencer11Gonzalo De Toro12Erik Morales13,14Lorena Gutiérrez15Fernanda Morales1Arnaldo Marin1 Nelson M. Varela1
Nelson M. Varela1 Justo Lorenzo Bermejo4
Justo Lorenzo Bermejo4 Ricardo Armisén2
Ricardo Armisén2 Katherine Marcelain1,7*
Katherine Marcelain1,7*- 1Departamento de Oncología Básico Clínico, Facultad de Medicina, Universidad de Chile, Santiago, Chile
- 2Centro de Genética y Genómica, Instituto de Ciencias e Innovación en Medicina, Facultad de Medicina Clínica Alemana, Universidad del Desarrollo, Santiago, Chile
- 3Departamento de Tecnología Médica, Facultad de Medicina, Universidad de Chile, Santiago, Chile
- 4Statistical Genetics Research Group, Institute of Medical Biometry, Heidelberg University, Heidelberg, Germany
- 5Corporación de Fomento de la Producción (CORFO) Center of Excellence in Precision Medicine, Pfizer Chile, Santiago, Chile
- 6Centro de Informática Médica y Telemedicina, Facultad de Medicina, Universidad de Chile, Santiago, Chile
- 7Centro para la Prevención y el Control del Cáncer, Centro para la Prevención y el Control del Cáncer (CECAN), Universidad de Chile, Santiago, Chile
- 8Departamento de Anatomía Patológica, Hospital Clínico de la Universidad de Chile, Santiago, Chile
- 9Departamento de Medicina Interna, Hospital Clínico de la Universidad de Chile, Santiago, Chile
- 10Department of Pathology, Hospital Padre Hurtado, Santiago, Chile
- 11Department of Pathology, Hospital Clínico Regional Guillermo Grant Benavente, Concepción, Chile
- 12School of Medical Technology, Universidad Austral de Chile at Puerto Montt, Puerto Montt, Chile
- 13Department of Pathology, Hospital Regional de Talca, Talca, Chile
- 14Department of Preclinical Sciences, Faculty of Medicine, Universidad Católica del Maule, Talca, Chile
- 15Department of Pathology, Hospital San Juan de Dios, Santiago, Chile
Introduction: Gallbladder cancer (GBC) is a highly aggressive malignancy with one of the highest incidence rates reported in Chile. Despite its clinical impact, molecular characterization of GBC in Latin American populations remains limited, and the absence of effective targeted therapies underscores the urgent need for new therapeutic strategies.
Methods: We collected 118 tumor samples, of which 56 passed sequencing quality control using the Oncomine™ Comprehensive Assay v1. Somatic variants were identified with ANNOVAR and Cancer Genome Interpreter, and ancestry was inferred using ADMIXTURE and PCA with ancestry-informative markers. Comparative analyses were performed with Japanese, Singaporean, and U.S. cohorts.
Results: A total of 535 somatic mutations were detected in 43 genes, with TP53 (30%), TSC2 (29%), and NOTCH1 (27%) being the most frequently mutated. We identified 121 clinically actionable variants in ATM, BRCA1/2, EGFR, ERBB2, and other genes. Exploratory analysis suggested an association between higher Mapuche ancestry and TP53 mutations. Comparative analyses revealed distinct mutational patterns in the Chilean cohort relative to Asian and U.S. datasets.
Conclusion: This ancestry-informed genomic analysis provides the first comprehensive landscape of Chilean GBC, identifying actionable alterations with potential therapeutic relevance and supporting the development of population-specific precision oncology strategies.
1 Introduction
Gallbladder cancer (GBC) is the most frequent, aggressive and lethal malignancy of the biliary tract neoplasms. It presents a poor prognosis and low five-year survival rates, due to late diagnosis and limited treatment option. The regions with the highest incidence and mortality rates include Bolivia, India, China, Japan, Bangladesh, and Chile (1, 2). These differences in incidence have been related to both lifestyle and genetic factors, but some risk factors for GBC have also been identified, such as cholelithiasis, chronic inflammation, advanced age and female sex (2, 3). However, the lack of specific symptoms of GBC makes early diagnosis difficult, which decreases the efficacy of available treatments and the potential for curative interventions (4). In fact, it is estimated that only 20% of GBC cases are detected at stages responsive to curative surgical resection (cholecystectomy) (5). While chemotherapy, utilizing gemcitabine and cisplatin, is an option for patients with unresectable GBC, not all patients respond favorably to this treatment (4, 5). Consequently, developing novel strategies to improve the treatment of GBC and facilitate early diagnosis of the pathology is vital to improving the life expectancy of affected patients.
The advent of next-generation sequencing (NGS) has revolutionized precision medicine, enabled detailed genomic characterization of cancers and identified actionable mutations that guide the development of targeted therapies. In GBC, genomic studies have revealed recurrent somatic mutations in genes like TP53, KRAS, SMAD4, and ERBB2, many of which represent promising therapeutic targets due to their roles in oncogenic signaling pathways (6). Large-scale genomic analyses, particularly from regions with high GBC incidence like China, confirm a high prevalence of alterations in genes such as TP53, KRAS, and ATM, often with substantial geographic variation, underscoring the influence of population-specific factors on GBC’s mutational landscape (7). These findings emphasize the imperative of integrating genomic profiling into clinical practice and investigating regional molecular patterns to advance precision medicine in GBC.
The Chilean population is admixed, consisting mainly of European and Amerindian ancestry. The main Amerindian groups in Chile are the Aymaras, located in the north of the country, and the Mapuches, located in the south (8). Despite the high incidence and mortality of GBC in Chile, particularly among women with high Amerindian ancestry, genomic studies in this population remain scarce (2, 8). Those that exist are often limited by small sample sizes and do not adequately consider the unique genetic admixture of the Chilean population, which may influence tumor biology and treatment response. These limitations underscore the pressing need for comprehensive genomic investigations that incorporate ancestry-informed analyses, thereby improving our understanding of GBC pathogenesis and supporting the development of more effective, tailored therapeutic strategies for underserved populations.
To bridge this knowledge gap and identify potential therapeutic targets, our study characterized the genomic landscape of a cohort of Chilean GBC patients. We identified 535 somatic variants across 43 of 56 GBC samples. Notably, 22% of these alterations are reported as predictive markers in clinical guidelines for various cancers. Furthermore, our analysis suggests a possible association between higher Mapuche ancestry and the frequency of TP53 mutations. These findings are crucial for informing the design of new treatment options and prevention strategies, grounded in the specific genetic background of the Chilean population.
2 Materials and methods
2.1 Patients and samples
Samples were collected in several sites along the country: Clínica Indisa, Biobanco de Tejidos y Fluidos de la Universidad de Chile, Hospital Padre Hurtado, Hospital Regional de Concepción, Hospital Regional de Talca, Hospital de Puerto Montt, Hospital San Juan de Dios, Instituto Nacional del Cáncer, Hospital Regional de Coquimbo, and Hospital Regional de Arica. Hematoxylin and eosin-stained tissue sections, derived from Formalin-Fixed Paraffin-Embedded (FFPE), underwent a thorough review by two pathologists. The tumor region was delineated. Only specimens with a percentage greater than 10% of tumor content were eligible for inclusion in this study. Areas characterized by significant necrosis and intra-tumoral fibrosis were excluded from the analysis. Unstained tissue sections (5 µm-thick) were prepared, deparaffinized, and the tumor tissue specimen was carefully collected for subsequent DNA extraction.
2.2 Library preparation and sequencing
DNA was extracted from paraffin-embedded samples using the RecoverAll™ Total Nucleic Acid Isolation Kit (Thermo Fisher Scientific). A 20 ng DNA input was used for library preparation with the Oncomine™ Comprehensive Assay V1 (OCAv1, Thermo Fisher Scientific), following the manufacturer’s protocol. Library preparation was performed using the Ion Chef™ System, with a final library concentration of 100 pM. Purified DNA was quantified using the Qubit™ dsDNA HS Assay and the Quant-iT™ PicoGreen® dsDNA Reagent Kit (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA). DNA purity was evaluated by measuring the absorbance ratio at 260/280 nm. The DNA integrity and fragmentation status were assessed using the High Sensitivity Genomic DNA Analysis Kit (DNF-488) on a Fragment Analyzer system (Agilent Technologies, formerly Advanced Analytical). Sequencing was conducted on an Ion PGM™ System in single-read mode using the Ion 550 kit-CHEF (Thermo Fisher Scientific). The list of genes analyzed and the sequencing quality metrics for all analyzed samples, including total reads, coverage, and uniformity, are summarized in Supplementary Tables S1 and 2. Of the 118 FFPE tumor samples collected, 66 (55.9%) yielded sufficient quantity and quality of DNA to proceed to sequencing. The remaining 52 samples (44.1%) failed pre-sequencing QC due to low DNA yield, high degradation. Many of the samples that failed due to low DNA yield or high degradation were obtained from various pathology centers. In these cases, inconsistencies in pre-analytical processes, such as prolonged fixation times and the use of non-buffered formalin, may have significantly contributed to DNA degradation. Ten samples were excluded during post-sequencing quality control for failing to meet predefined variant calling thresholds (≥3 million total reads and ≥90% on-target rate). The final genomic analysis was therefore performed on the remaining 56 patient samples (Supplementary Figure S1).
2.3 Bioinformatic sequencing data analysis
The data preprocessing and processing were carried out using the OCAv1 v5.18 DNA workflow, using default parameters of the GRCh37/hg19 reference genome. For alignment and variant calling, stringent parameters were defined. Single Nucleotide Variants (SNV) and Indels required a minimum allele frequency of 0.05 and 0.07. The minimum coverage for a variant to be considered was set at 10x for SNV and Indels. Additionally, the minimum coverage for the variant location was set at 200x. Variant annotation was performed using ANNOVAR (9), including RefGene, GnomAD v2.1.1, ESP6500, 1000 Genomes phase 3, CADD v1.3, dbSNP v150, COSMIC v94, CLINVAR 2021, ICGC28, ABraOM, and Snp138NonFlaged. To enhance the filtering of germline variants in tumor samples, large and local population germline variant databases were interrogated:CSVS (10), GnomAD (overall and population specific); BIPMed (Brazilian Initiative on Precision Medicine); and a Chilean database (3, 11) (variants imputed from genotyping two arrays with 1,313 and 2,249 samples from Chilean individuals, and whole exome sequencing (WES) data from 87 individuals (NCBI dbSNP database under accession code 1062069) (12).
2.4 Public databases
Mutation data of GBC samples were extracted from the Memorial Sloan Kattering Clinical Sequencing cohorts (6) through cBioportal (https://www.cbioportal.org, accessed on march 1st 2024). In addition, GBC mutation data from Japanese and Singaporean samples were extracted from the International Cancer Genome Consortium (ICGC) website (downloaded on march 1st 2024). Only somatic protein-affecting variants found in primary tumors were analyzed in all datasets.
2.5 Analysis of actionable and driver variants
The mutational landscape of GBC samples was characterized using the R/maftools package (13). To identify actionable and driver variants within the cancer samples, the Cancer Genome Interpreter platform (https://www.cancergenomeinterpreter.org/home) was employed. Variants were labeled according to the level of evidence (A, B, C, and D) obtained from VICC integrated knowledge base, following the AMP/ASCO/CAP guidelines. OncodriveMUT and BoostDM algorithms were used for prediction of driver variants (14). Only predicted drivers that were also annotated as “oncogenic or likely-oncogenic” in OncoKB (oncokb.org) were considered.
2.6 Ancestry analysis in patients with gallbladder cancer
Genetic ancestry was evaluated using genomic DNA extracted from peripheral blood samples of 37 individuals. The ancestry analysis was performed using the ADMIXTURE software (15) and Genetic PCA with the Eigenstrat function (16). This approach allows for supervised estimation of individual ancestry components, including African, European, Native American Mapuche and Aymara, using a large number of Single Nucleotide Polymorphism (SNP) markers previously defined as Ancestry informative marker (AIM) (15). The panel for the preselection of ancestry-informative markers and the estimation of individual ancestry proportions in genetically admixed Chileans, which included 63 Aymara individuals (17) and 28 Mapuche individuals (17, 18). Additionally, it comprised 206 Europeans (99 Utah residents with Northern and Western European ancestry [CEU] and 107 Iberians from Spain [IBS]) and 108 African Yoruba from Ibadan, Nigeria (YRI) from the 1000 Genomes Project (17).
2.7 Statistical analysis
A Chi-square test was performed to compare mutation frequencies between the Chilean GBC cohort and those from Japan (GBC-JPN), Singapore (GBC-SGN), and the MSK2022 cohort. p-values were adjusted using the Benjamini–Hochberg method to control the false discovery rate (FDR), given the large number of genes tested simultaneously. Pairwise Chi-square tests with Yates’ continuity correction were performed as post hoc comparisons in cases where the adjusted p-value (FDR) was below 0.05. Multivariate logistic regression models were adjusted to evaluate the association between ancestry and the presence of variants in target genes. Sex, age at the time of collection, history of cholelithiasis, and GBC histology were included as covariates using the following model:
In this model, each target gene status (mutated/non-mutated) was modeled as a function of each ancestry, grouped as high/low, along with the effect of other covariates mentioned. A significance level of 0.05 was defined as statistically significant. Patients were categorized into two groups using the median of each estimated genetic ancestry (European, Mapuche, Aymara, and African). All statistical analyses were conducted using R 4.2.1 software.
3 Results
3.1 Clinical characteristics of gallbladder cancer patients
We collected a total of 118 FFPE biopsies from GBC. To account for the high heterogeneity of the Chilean population, samples were gathered from several medical centers throughout the country. Of these, 94 (76.6%) were from female patients and 24 (23.4%) from male patients, respectively, with an average age of 62 years at the time of collection. Seventy-eight patients (66.1%) had a history of cholelithiasis in their clinical history (Table 1).
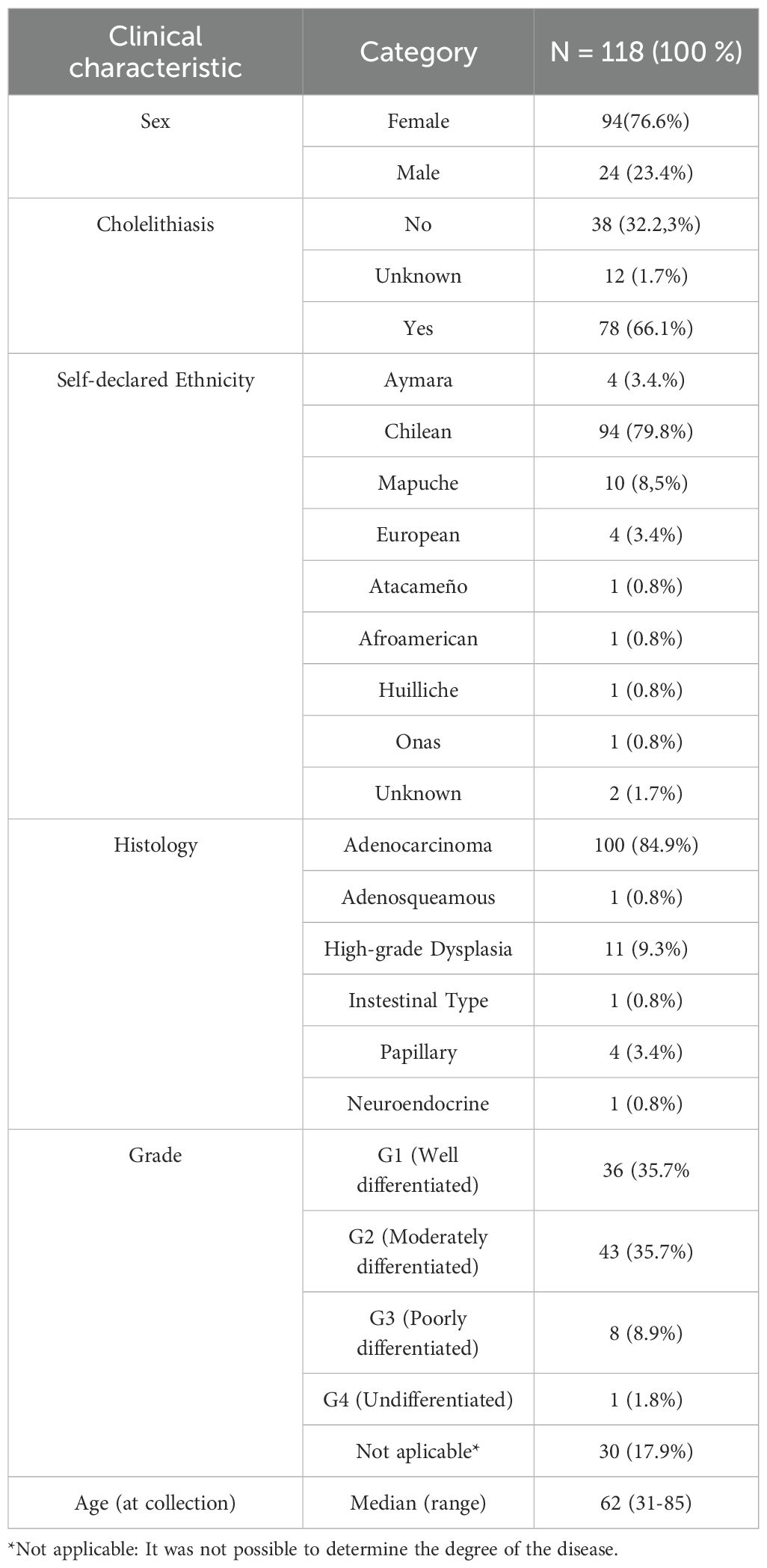
Table 1. Clinical characteristics of patients included in the study.
In terms of ethnicity, most patients self-declared Chilean (Admixed) (79.8%), followed by Mapuche (8.5%), Aymara (3.4%) and European (3.4%). Genetic ancestry distribution at individual level is provided in Supplementary Figure S3. The specific genetic ancestry As for the histological diagnosis of the samples, a varied distribution in this type of cancer was revealed, with adenocarcinoma being the most frequent type (84.9%), while squamous cell carcinomas and high-grade dysplasia were less common. Most patients were diagnosed at G1 and G2 stages at the time of surgery (Table 1).
3.2 Somatic mutation profile of gallbladder cancer
Just 56 of the 118 samples met all quality control criteria for DNA, library, and sequencing data (Supplementary Figure S1). This finding underscores the significant influence of tissue processing and other pre-analytical factors on the successful execution and quality of molecular analyses. We identified 535 somatic mutations across 43 tumor samples. The majority of these somatic variants, 469 (87.66%), were missense mutations, followed by 34 (6.36%) nonsense mutations, 12 (2.24%) splice site mutations, and 5 (0.93%) frameshift deletion mutations. (Figure 1A), with the majority falling under the Single Nucleotide Variants (SNV) category (Figure 1B). The C > T nucleotide variation was the most frequent substitution among the detected somatic variants. Additionally, transitions (Ti) are more prevalent than transversions (Tv) among the SNVs identified in all genes with mutations (Figures 1C, D).
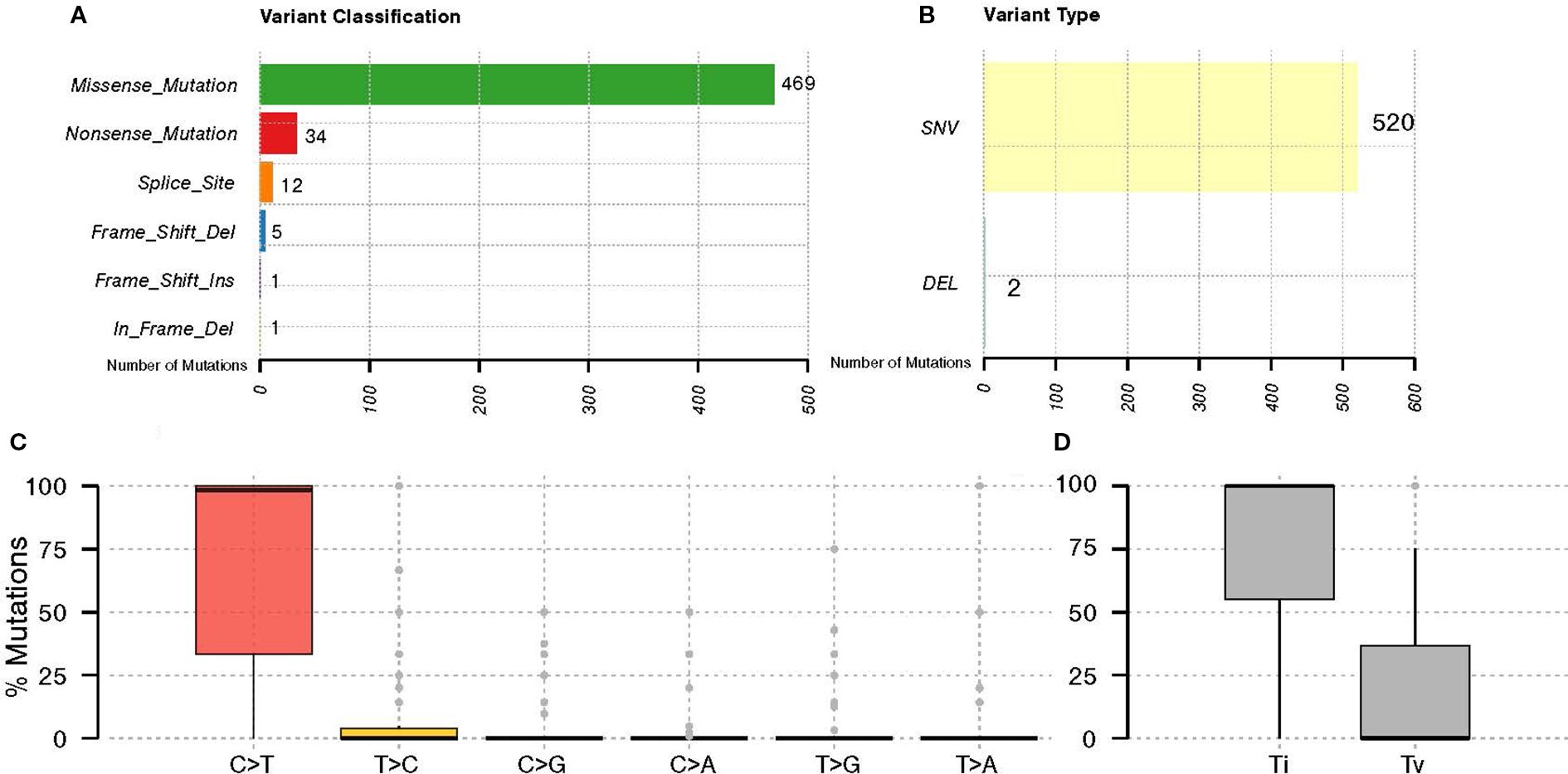
Figure 1. General somatic variant’s classification in GBC. The graph displays the distribution of (A) variant classifications, (B) types of variants identified in GBC patients, (C) nucleotide substitutions of the identified variants, and (D) their classification as transitions (Ti) and transversions (Tv).
Somatic mutations were identified in 41 out of the 161 genes analyzed. The most frequently mutated genes included TP53 (30%), TSC2 (29%), NOTCH1 (27%), BRCA2 (16%), KRAS (16%), and PTCH1 (16%) (Figure 2). The customized oncoplot integrates both gene-level and pathway-level alterations, grouping genes according to their corresponding oncogenic signaling pathways. In this analysis, 2/6 (33.3%) genes in the TP53 pathway and 15/85 (17.7%) genes in the RTK–RAS pathway were altered. Notably, the RTK–RAS pathway was affected in 50% of GBC samples, followed by the TP53 pathway (38%), NOTCH (37%), and PI3K (32%) (Figure 2). Among the 10 most mutated genes, several genes show a significant co-occurrence, such as MLH1/TSC2, NOTCH1/MLH1, and NOTCH1/TSC2, among others. Mutations in TP53 and KRAS seem to be mutually exclusive, although this trend did not reach statistical significance (Figure 3).
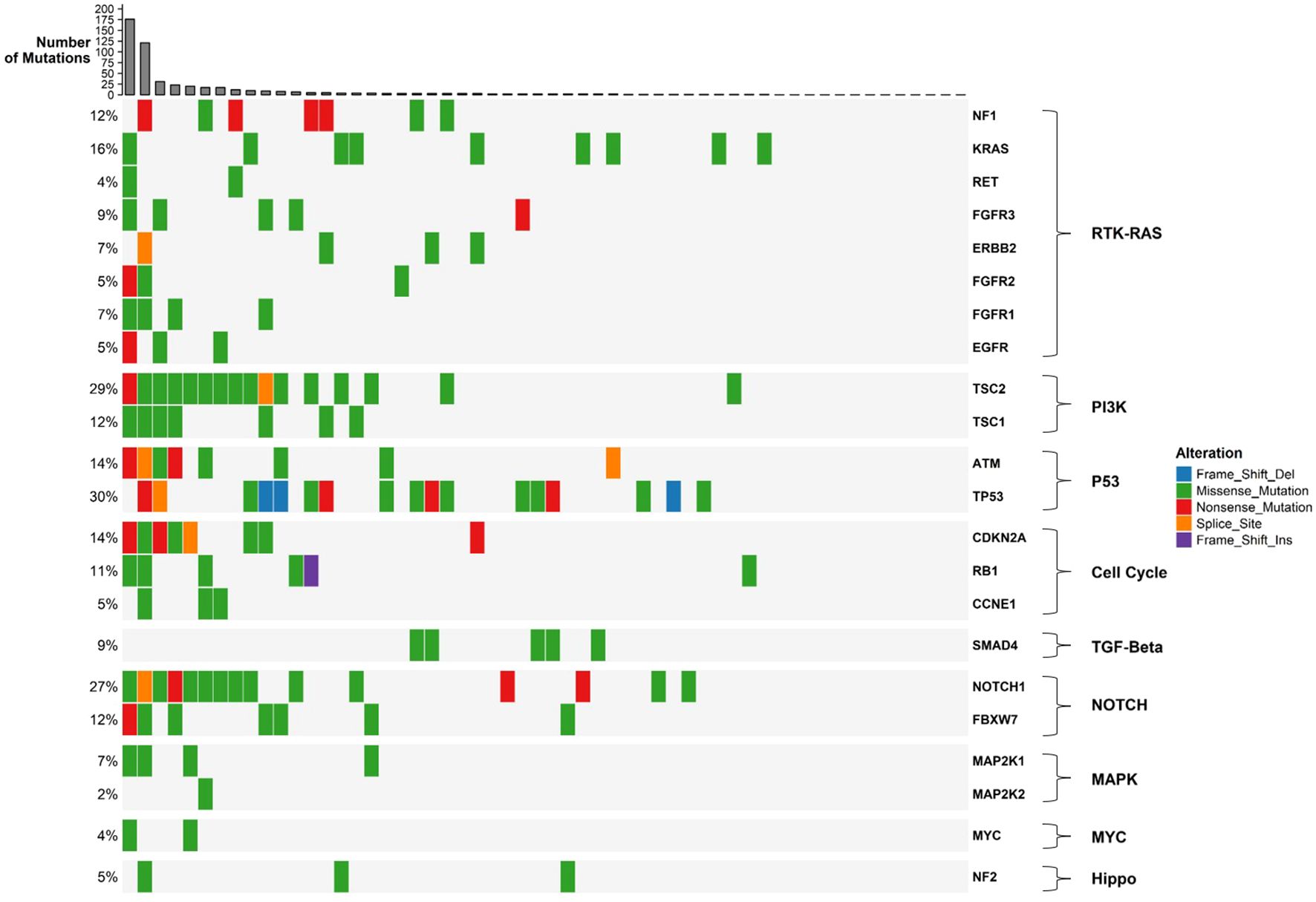
Figure 2. Oncoplot of gallbladder cancer samples grouped by oncogenic signaling pathways. The plot shows somatic mutations in 56 gallbladder cancer samples, with genes grouped according to their associated oncogenic pathways as defined by the TCGA framework. Each colored square represents a mutation type (see legend). Barplots on the top show the number of mutations per sample, while barplots on the right indicate the number and percentage of samples affected for each gene or pathway.
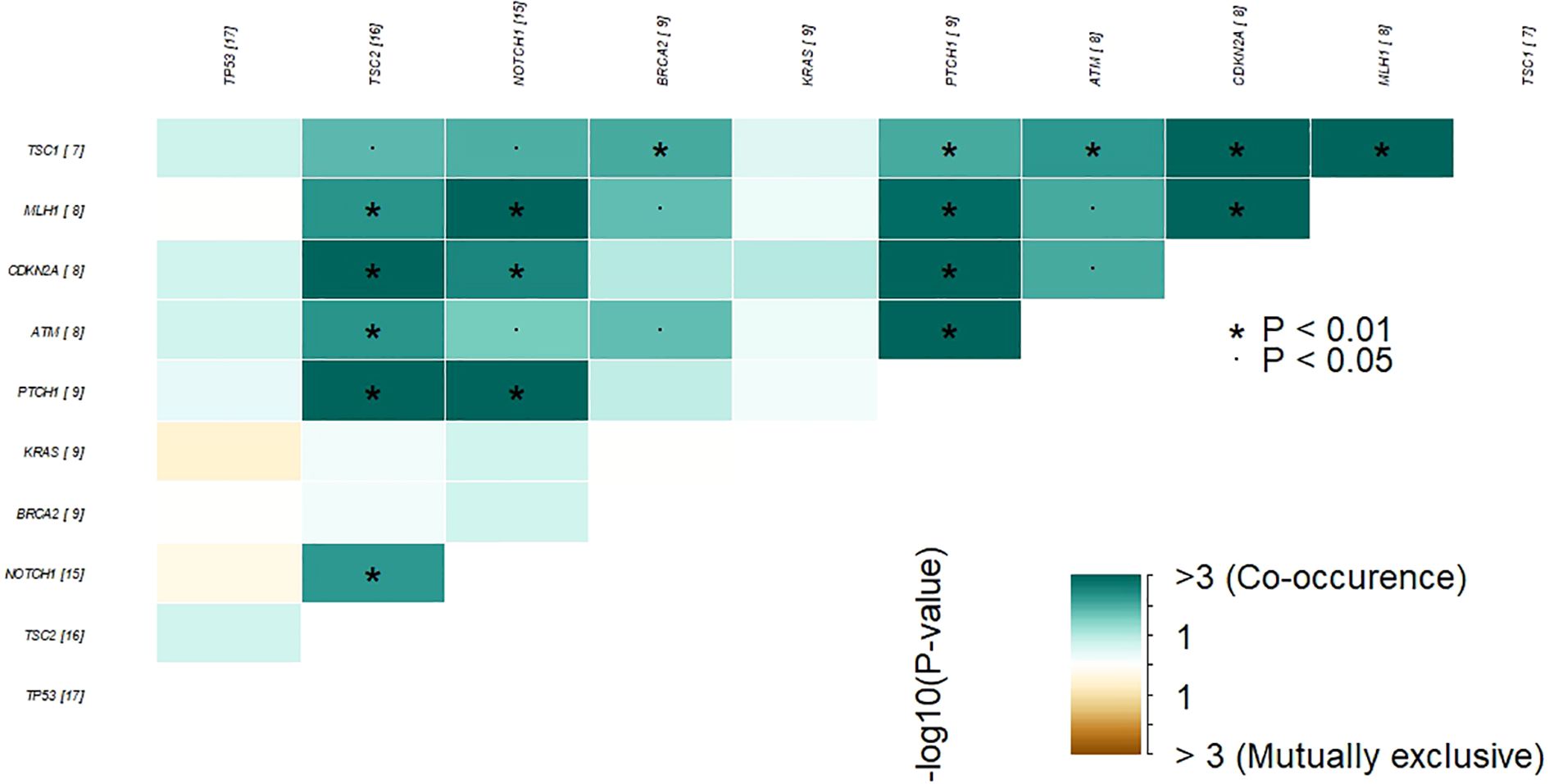
Figure 3. Co-occurrence and mutual exclusivity of the most frequently altered genes in GBC tumor samples. Heatmap showing statistically significant patterns of co-occurrence and mutual exclusivity among the top recurrently mutated genes in our cohort. Color intensity represents the –log10(p-value) for each gene pair. Green tones indicate co-occurrence, while brown tones indicate mutual exclusivity. Asterisks denote statistical significance (P < 0.05; P < 0.01). The numbers in brackets next to each gene indicate the number of patients in our cohort harboring mutations in that gene.
3.3 Identification of somatic variants with actionable potential
Given the limited therapeutic options for patients with GBC, identifying targets for therapies that have already demonstrated clinical benefit in other tumor types may enable the inclusion of GBC patients in ongoing or future trials. In our cohort, all 44 patients with somatic variants harbored one or more actionable alteration with supporting Level A evidence. The most frequently altered genes were KRAS wildtype (n=35, predictive of response to anti-EGFR therapies such as panitumumab and cetuximab in colorectal cancer), TSC2 (n=16, associated with everolimus in renal angiomyolipoma and giant cell astrocytoma), and BRCA2 (n=9, associated with PARP inhibitors such as rucaparib in ovarian, prostate, and pancreas cancers). Additional Level A alterations were identified in ATM, BRCA1, EGFR, ERBB2, FGFR2, FGFR3, PTCH1, RET, and TSC1, all of which predict responsiveness to specific targeted therapies. Importantly, we detected KRAS mutations (G12D/V/C, G13D, Q61H) associated with resistance to EGFR inhibitors such as cetuximab and panitumumab in colorectal and lung cancer, and responsiveness to KRAS inhibitors sotorasib and adagrasib in colorectal, NSCLC, pancreatic, ampullary, and hepatobiliary cancer (Table 2).
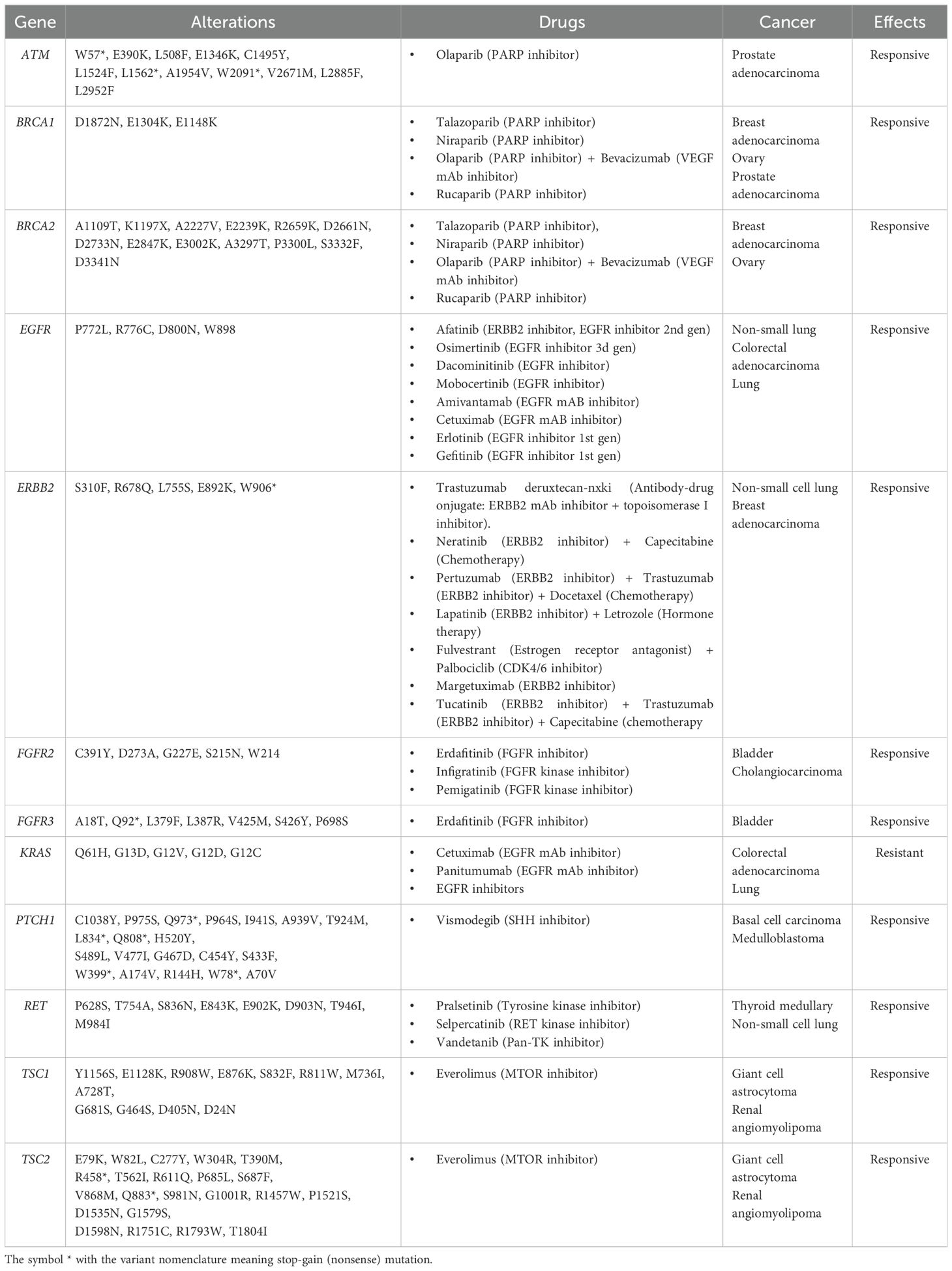
Table 2. Predictive biomarkers found in GBC samples from Chilean patients and supported by level A evidence in various cancers (according to AMP/ASCO/CAP guidelines).
3.4 Comparative analysis of somatic mutation frequencies and oncogenic variants across diverse gallbladder cancer cohorts
Although genomic data on GBC are available in public repositories, this data is still scarce and has a limited representation of Latin American patients. Given the marked geographical disparities in GBC incidence and mortality, it is plausible to hypothesize that underlying environmental and genomic background may favor the activation of distinct molecular oncogenic mechanisms in high incidence regions. To explore this possibility, first we compared the mutation frequencies in the 41 genes identified in the Chilean cohort with those reported in previously published datasets MSK-IMPACT 2022 cohort, as well as two GBC cohorts from Japan and Singapore (obtained from the International Cancer Genome Consortium (ICGC) (https://dcc.icgc.org/). Despite the limited sample size, we found significant differences in the mutation frequencies of all genes but ERBB2 across cohorts (Table 3).
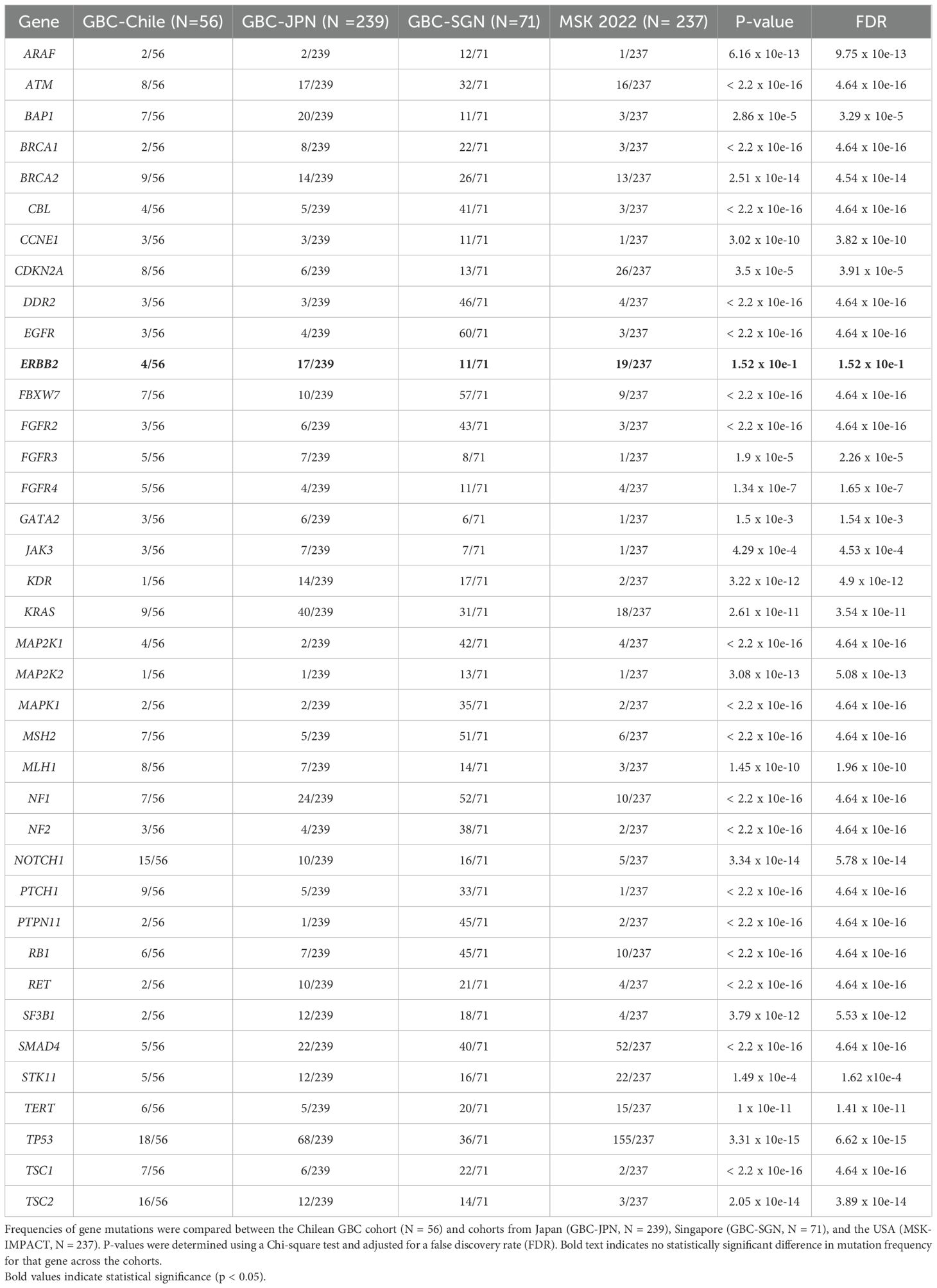
Table 3. Differential somatic mutation frequencies in gallbladder cancer across geographic cohorts.
Next, we identified variants predicted and annotated as oncogenic and likely-oncogenic in 23 (41.07%) out of the 56 Chilean samples analyzed. These mutations are distributed in 12 genes. TP53 and KRAS exhibit the highest frequencies of oncogenic/likely-oncogenic variants in 18% and 9% of the studied samples, respectively, followed by ERBB2 (5%) (Supplementary Figure S2). TP53 variants are predominantly missense mutations situated within the p53 DNA-binding domain (Figure 4A). KRAS recurrent oncogenic G12D/C/V, G13D, and Q61H mutations were found (Figure 4B). In ERBB2 gene, activating S310F, R678Q, and L755S were identified at Furin-like, TMD, and Kinase domains (Figure 4C).
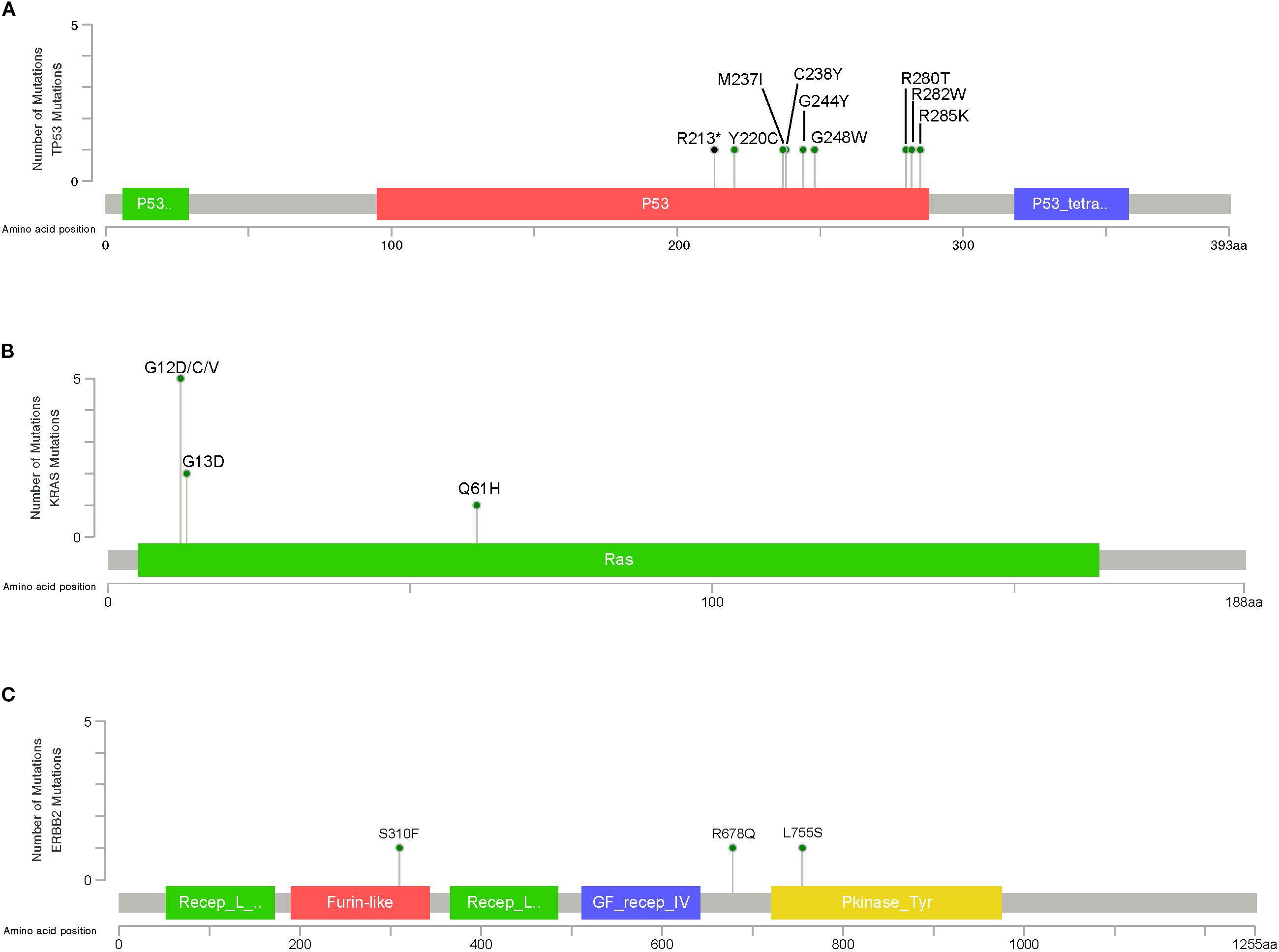
Figure 4. Lollipop plot illustrating the distribution and number of driver mutations in (A) TP53, (B) KRAS, and (C) ERBB2, identified in GBC patients. The gray bar represents the entire protein with the different amino acid positions (aa). The colored boxes are specific functional domains. The vertical axis represents the number of variants per sample. Green and black circles represent missense and nonsense variants, respectively.
To further explore whether the overall differences detected in gene mutation frequencies between GBC from mostly White (MSK.,2022), Asian (GBC-JPN, GBC-SGNAs), and Chilean patients involved key driver mutations, oncogenic variants in the 12 driver genes were identified and compared in all cohorts. Most oncogenic mutations were also found in one or more studies, especially hotspot mutations in CDKN2A, ERBB2, SMAD4, FBXW7, KRAS, and TP53. Nevertheless, differences are also evident, in tumor suppressor genes ATM and TP53. Notably, in Chilean samples, likely oncogenic EGFR R776C, BRCA2 E3002K, and MLH1 W666* and Q562*, were found. No oncogenic mutations were identified in these genes in the referenced studies (Table 4).
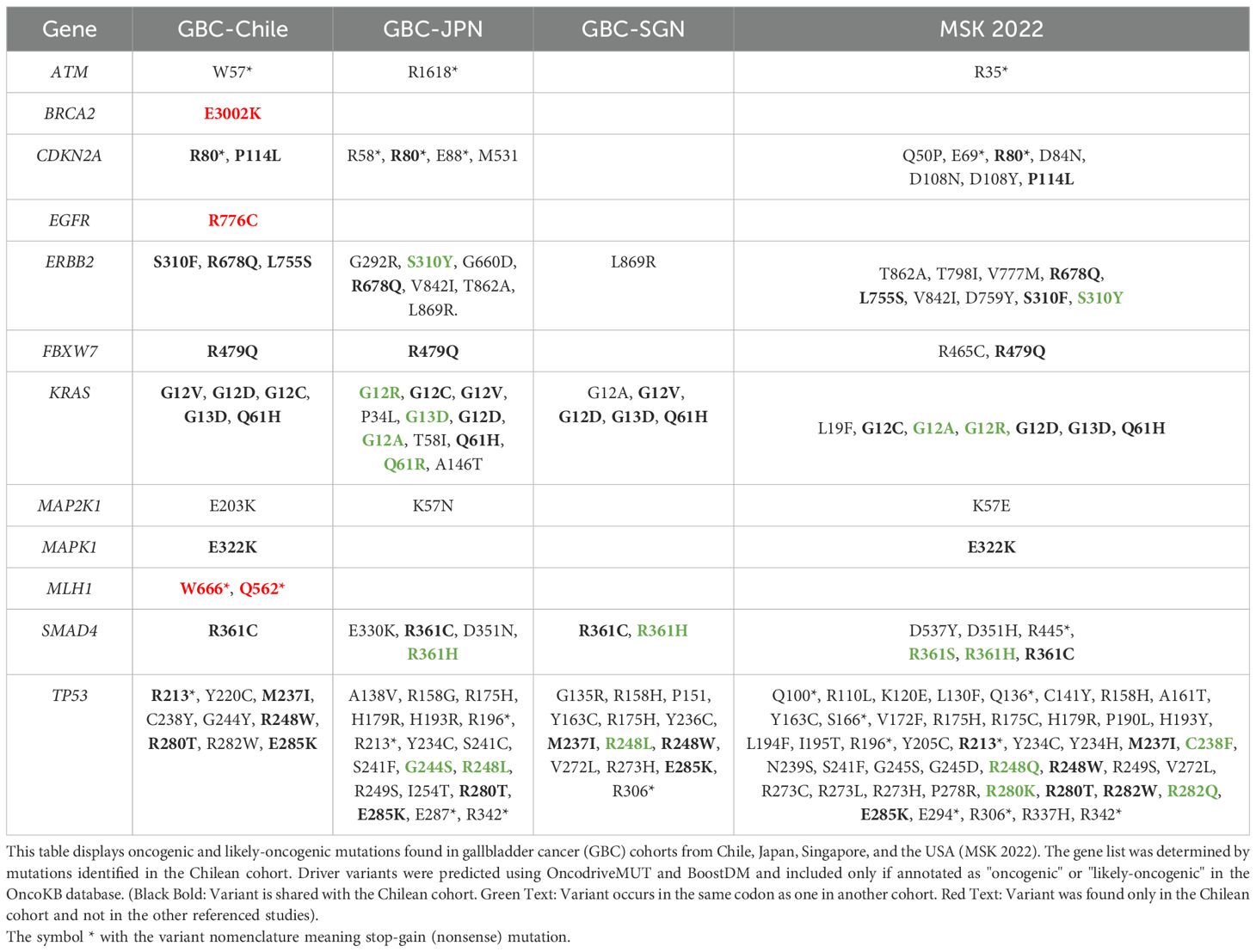
Table 4. Shared and unique oncogenic driver mutations in Chilean GBC compared to other populations.
3.5 Exploring the relationship between ancestry, somatic mutations, and TP53 in Chilean GB
Genomic studies have provided evidence supporting that Chilean Amerindian genetic ancestry is a risk factor for GBC (3). In fact, a correlation has been established indicating that Chileans with a higher proportion of South Amerindian genetic ancestry (Mapuche, MAP) have a higher risk for GBC (3, 8). With the aim to assess whether this genetic-ancestry-related risk for GBC is also associated with the risk for a specific molecular oncogenic mechanism of disease, we used a panel of Ancestry informative marker (AIM) to estimate the proportion of ancestral genetic backgrounds for 37 out of 43 patients with somatic variants. As expected, European and Mapuche genetic ancestries are predominant in the analyzed individuals (0.493, sd: 0.145; and 0.34, sd: 0,129, respectively), followed by Chilean main North Amerindian Aymara (0.137; sd: 0,221), and African (0.019; sd: 0.16) ancestries (Supplementary Figure S3).
A general overview of the somatic mutation´s data suggest that higher proportion of Mapuche ancestry would associate with a lower number of somatic mutations (Supplementary Figures S4, S5). Although these results are preliminary, this led us to investigate the potential association between Mapuche ancestry and specific gene mutation frequency in TP53 (the most frequently mutated gene in our restricted cohort). Higher proportion of Mapuche ancestry may be positively associated with mutations in TP53; however, the result is not significant (OR = 6.73, 95% CI: 1.14, 59.6, p = 0.053). In addition, poorly differentiate (G3) tumors showed positive associations regarding the mutational status of TP53 (p= 0.006) in a multivariate analysis (Table 5).
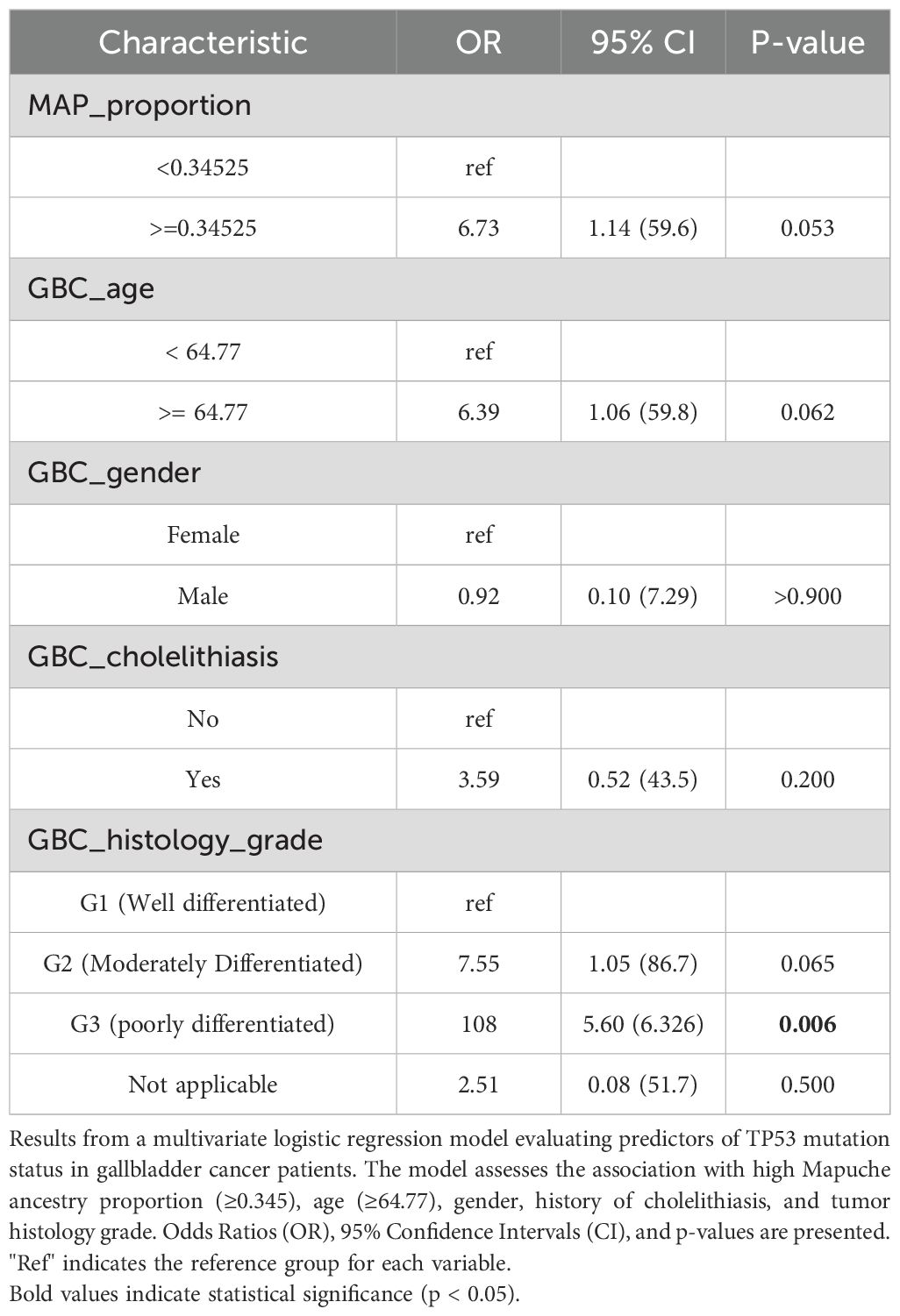
Table 5. Multivariate analysis of factors associated with TP53 mutation in GBC.
4 Discussion
Gallbladder cancer (GBC) is a highly aggressive biliary tract malignancy, consistently associated with poor overall survival (1–3). Despite its relatively low global incidence, GBC exhibits profound geographical disparities. Notably, Chile reports among the highest incidence and mortality rates worldwide, trailing only Bolivia and Bangladesh (1). The absence of specific targeted therapies for GBC underscores the critical need for advanced approaches. Next-generation sequencing (NGS) offers a promising avenue by enabling the identification of actionable and driver genetic variants that could inform treatment strategies.
In recent years, several studies have emphasized the need to characterize the genomic landscape of solid tumors, particularly those that exhibit marked geographic disparities in incidence and mortality, and for which no effective treatment currently exists—as is the case for GBC (1–3). Using molecular records from different geographical regions is essential for understanding population-specific disease characteristics, enabling the development of tailored prevention and management strategies for high-incidence areas like Latin America (19).
In this context, our study investigated the molecular landscape of GBC in a cohort of Chilean patients using a targeted gene panel. Our findings reveal that TP53 mutations are the most common alterations (30%), followed by mutations in TSC2, NOTCH1, BRCA2, KRAS, and PTCH1. This pattern aligns with previous research; for instance, Narayan et al. (20) demonstrated that Chilean GBC patients exhibited the highest frequency of TP53 mutations when compared to cohorts from Japan and the USA. This observation was further supported by the same group’s subsequent targeted sequencing of 233 GBC patients, where TP53 remained the most frequently mutated gene (6). Similarly, Nepal and colleagues (21) reported TP53 as the most mutated gene in a GBC exome analysis that included Chilean individuals. These findings resonate with Yu et al.’s study (7) on 117 Chinese GBC patients, which also identified TP53 as the most frequently mutated gene. Their work suggested that TP53 mutation frequencies in Chinese patients did not differ significantly from Western cohorts, implying a common pathogenic event in GBC development across diverse populations.
4.1 Global and local genomic insights in GBC
Beyond our findings, studies in other high-incidence regions, such as by Mishra et al. in India, similarly show a high frequency of TP53 mutations (90.90%), alongside other common alterations like SMAD4, NOTCH1, and ERBB2 (22). This shared high frequency of TP53 mutations between Chilean and Indian GBC patients may stem from common risk factors like gender, bowel habits, and cholelithiasis (23). Chronic inflammation induced by cholelithiasis is a plausible mechanism, potentially triggering TP53 mutations that contribute to GBC development (24). While a common variant in the ABCB4 gene (rs4148808) linked to gallstone disease might play a role in both populations, further studies are needed for verification (25). It is crucial to note that differences in variant allele frequency (VAF) thresholds (e.g., >1% in Mishra et al. vs. >5% in our study) could influence observed mutation frequencies and interpretations of somatic variants.
We also observed a high number of non-synonymous somatic mutations in two patients, both carrying variants in DNA repair genes such as MLH1 and MSH2. Alterations in these genes have been associated with increased mutational load and may suggest potential sensitivity to immune checkpoint inhibitor (ICI) therapies (26). Furthermore, the predominance of C > T substitutions in our mutation pattern indicates a possible APOBEC-mediated mutational signature (27), a finding also reported in a whole exome sequencing study including Chilean GBC patients (21). This opens avenues for signature-based therapies.
Our cohort showed a female-to-male ratio of 4:1, consistent with the well-established female predominance in GBC incidence reported in Chile and other high-incidence regions (1–3). This disparity is thought to be largely driven by the higher prevalence of gallstones and chronic cholecystitis among women, potentially influenced by hormonal and metabolic factors (1–3). While sex-related differences in mutational patterns were not statistically evaluated in our series due to sample size constraints, previous studies have suggested that certain genomic alterations in GBC may vary by sex, including recent evidence describing distinct clinical and molecular profiles between men and women (28). However, these differences are often confounded by underlying risk factor distributions rather than direct sex-linked pathogenic mechanisms. Nonetheless, the integration of sex as a biological variable in future genomic studies of GBC could help clarify its role in tumor development and progression.
4.2 Actionable targets and therapeutic implications
Given the limited approved targeted therapies for GBC and the reliance on systemic chemotherapy, we explored the actionable potential of genetic alterations in our Chilean cohort. Using the CGI platform, we identified clinically actionable alterations with drug recommendations in genes such as ATM, BRCA1/2, EGFR, ERBB2, FGFR3, KRAS, PTCH1, RET, and TSC1/2. While many of these drugs are established in other solid tumors, their specific utility in GBC is emerging. For instance, panitumumab combined with gemcitabine and oxaliplatin showed promising results in KRAS wild-type biliary tract cancer (29), and the SGNTUC-019 study demonstrated the efficacy of Tucatinib and Trastuzumab in HER2-positive metastatic biliary tract cancer (30). The recent FDA approval of sotorasib, a KRAS G12C inhibitor, further highlights the potential for targeted therapies (31).
Notably, ATM, BRCA1, and BRCA2 mutations are highly significant due to their involvement in the DNA damage response (DDR) pathway. PARP inhibitors (e.g., olaparib, niraparib) have shown efficacy in BRCA-mutated cancers (32), with recent evidence suggesting benefit in GBC patients with BRCA2 alterations (33). Although ATM alterations are less studied in GBC, their reported presence in biliary tract tumors suggests they could serve as predictive biomarkers (34, 35). These findings collectively underscore the potential for implementing personalized therapies for Chilean GBC patients by repurposing existing drugs based on specific genomic alterations.
The recurrent involvement of RTK-RAS, PI3K, and TP53 pathways in our cohort underscores their central role in the biology of BTC. Alterations in the RTK-RAS pathway, including KRAS mutations, are known to drive tumor proliferation and survival, and may influence sensitivity to targeted agents or MEK inhibitors (36). Similarly, PI3K pathway activation, through mutations in PIK3CA or upstream receptor tyrosine kinases, has been implicated in resistance to standard chemotherapy, and PI3K/AKT/mTOR inhibitors are currently being evaluated in BTC clinical trials (37). TP53 inactivation, the most frequent event in our cohort, is not only associated with loss of cell cycle control and genomic instability but has also been linked to reduced sensitivity to gemcitabine-based chemotherapy in BTC. This highlights p53 as a potential therapeutic target, with different strategies depending on mutational status: in TP53-mutated BTC, direct reactivation of mutant p53 or synthetic lethality approaches targeting cell cycle checkpoint proteins (e.g., Chk1, ATR, Wee1) may be promising; in wild-type p53 BTC, inhibition of its negative regulators (e.g., MDM2, WIP1) could enhance p53 activity (38). Such pathway-level characterization in GBC not only improves our understanding of tumor biology but also informs the rational design of targeted and combination therapies, particularly in the context of overcoming chemoresistance.
4.3 Shared genomic drivers and regional differences
Using computational algorithms, we identified driver variants in TP53, KRAS, and ERBB2, genes frequently mutated in other biliary tract tumors (39). TP53 variants, often influenced by external mutagens, include R280 (associated with aristolochic acid exposure) (40, 41) and R248 (linked to tobacco smoke in lung cancer) (42). The recurrence of R248W and R282W variants may stem from increased CpG site methylation during inflammation, a process contributing to GBC tumorigenesis (43, 44).
Our comparison with international cohorts revealed that ERBB2 was the only gene without significant mutation frequency differences across our Chilean cohort and those from the USA, Singapore, and Japan (45, 46). This consistent frequency, also reported by Nepal et al. (21) and Bitter et al. (47), and, importantly, the frequency of ERBB2 alterations was also found to be comparable between Chilean and U.S. cohorts (48). It has been reported that GBC patients harboring ERBB2 alterations exhibit improved overall survival. Thus, ERBB2 may represent a broadly applicable prognostic and therapeutic biomarker across diverse populations (48).
4.4 Ancestry, mutational signatures, and therapeutic opportunities
Beyond common drivers, our study uncovered distinct patterns. Two patients exhibited hypermutated tumors, potentially linked to variants in DNA repair genes (MLH1, MSH2) and suggesting responsiveness to immune checkpoint inhibitor therapies (26). The predominance of C > T substitutions in our cohort also points to an APOBEC-mediated mutational signature (27), consistent with previous findings in Chilean GBC patients (21), opening avenues for signature-based therapeutic strategies.
Furthermore, we explored the association between Mapuche ancestry and TP53 mutation likelihood. While not statistically significant, our exploratory analysis suggested a possible association between higher Mapuche ancestry and TP53 mutation status (OR = 6.73, p = 0.053). While this trend did not reach statistical significance, it may reflect underlying biological differences warranting further investigation. This observation should be considered hypothesis-generating, and validation in larger, independent GBC cohorts will be necessary to confirm or refute this potential ancestry-related effect. This aligns with prior research linking Mapuche ancestry to GBC risk variants (e.g., in ABCG8, TRAF3, ABCB1, ABCB4 genes) and increased susceptibility to gallstones, a known inducer of TP53 mutations via chronic inflammation (24, 25, 49, 50). Such observations underscore the influence of genetic ancestry on tumor profiles, as seen in pan-cancer analyses (51).
These results suggest that there may be tumor profiles associated with genetic ancestry. Although the results are preliminary, they indicate that patients with greater Mapuche ancestry may have a lower mutation rate, which could reflect biological differences in the tumorigenesis of this type of malignant tumor, supporting the need to incorporate genomic studies in GBC.
Recently, Zhu et al., reported overall similar mutation profiles in a Chilean vs US GBC cohorts. However, the immune profiles were highly distinct, characterized by higher densities of T cells and PD-1, but lower macrophages in tumors from Chilean patients (52). Although these clinically relevant differences may reflect lifestyle and/or environmental factor dissimilarities between the 2 countries, their findings reinforce the idea that the genetic origin of patients could modulate cancer biology and response to treatment, thus supporting the development of precision medicine strategies that incorporate ancestry, especially in underrepresented populations such as Chileans.
4.5 Actionable targets and future directions
Given the scarcity of approved GBC therapies, identification of actionable alterations (e.g., ATM, BRCA1/2, EGFR, ERBB2, FGFR3, KRAS, PTCH1, RET, TSC1/2) in Chilean patients provide a rationale for personalized treatment, particularly as specific therapies for KRAS and HER2-positive BTC are emerging (29–31). Mutations in ATM and BRCA1/2, crucial for DNA damage response, highlight the potential for PARP inhibitors, with promising results already seen in a GBC patient with a BRCA2 mutation (32, 33).
Despite these insights, our study has limitations. For instance, the NGS assay used (Oncomine Comprehensive Assay v1 (OCAv1), although it covers a panel of 161 clinically relevant cancer genes, it does not detect alterations in, for example, ARID1A, which is frequently mutated in GBC, including in Chileans (21, 52, 53).
In addition, although this assay is designed to detect a certain number of structural variants, only a few samples passed QC for RNA sequencing. Same with CNV. This could lead to underestimation of clinically relevant events such as ERBB2 amplification (49). The future application of broader sequencing approaches, such as whole-exome sequencing (WES) or whole-genome sequencing (WGS), in Chilean GBC cohorts would allow for a more comprehensive characterization of the mutational spectrum, the discovery of novel prognostic or therapeutic biomarkers, and the strengthening of analyses by integrating the ancestral component.
Nevertheless, challenges with FFPE sample quality have to be urgently addressed to further advance in a comprehensive and reliable genomic characterization of GBC in the country. Optimizing biopsy handling and preanalytical conditions are crucial to minimize sample attrition and enhance genomic profiling (54–56). Future work should also consider liquid biopsies (cfDNA) as a complementary strategy, given their concordance with tissue mutations and potential for non-invasive detection of actionable variants (57).
In conclusion, we identified actionable and driver genetic variants that may inform tumor response in Chilean GBC patients, laying the groundwork for personalized GBC therapies. Future research should leverage comprehensive genomic methodologies, such as whole-exome sequencing (WES), whole-genome sequencing (WGS), and liquid biopsies, to overcome current limitations and further elucidate GBC’s complex genetic and environmental interactions.
Data availability statement
The data presented in the study are deposited in the Zenodo repository, accession number DOI: https://doi.org/10.5281/zenodo.17051169.
Ethics statement
This study was conducted in compliance with relevant guidelines and regulations, following approval by “Comité ética de la investigación en seres humanos (CEISH) de la Facultad de Medicina, Universidad de Chile”, “Comité de ética científico del Servicio de Salud Metropolitano Oriente” and “Comité de ético científico del Hospital Clínico de la Universidad de Chile”. Informed consent was obtained from all patients included in this study. The study was conducted in accordance with the Declaration of Helsinki, and approved by the “Scientific or Research Ethics Committee of the Hospital Clínico Universidad de Chile (HCUCH)” (X9001083 protocol, Acta No. 17/2017). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
JE: Writing – review & editing, Formal Analysis, Writing – original draft, Methodology, Investigation, Conceptualization. EG: Formal Analysis, Investigation, Data curation, Writing – review & editing, Methodology. MS: Writing – original draft, Formal Analysis, Methodology, Writing – review & editing, Investigation. CB-P: Resources, Project administration, Methodology, Writing – review & editing, Investigation. MF: Supervision, Formal Analysis, Writing – review & editing, Methodology, Investigation. GS-H: Formal Analysis, Investigation, Writing – review & editing. DA: Writing – review & editing, Formal Analysis, Methodology, Investigation. AB: Formal Analysis, Investigation, Methodology, Writing – review & editing. VG: Project administration, Writing – review & editing, Investigation. PB-B: Writing – review & editing, Investigation, Formal Analysis. CT: Writing – review & editing, Investigation, Formal Analysis. JT: Methodology, Investigation, Formal Analysis, Writing – review & editing. IG: Writing – review & editing, Investigation, Resources. OB: Investigation, Resources, Writing – review & editing. MA: Resources, Investigation, Writing – review & editing. VS: Investigation, Resources, Writing – review & editing. LS: Investigation, Resources, Writing – review & editing. Gd: Writing – review & editing, Investigation, Resources. EM: Resources, Investigation, Writing – review & editing. LG: Writing – review & editing, Resources, Investigation. FM: Investigation, Writing – review & editing. AM: Writing – review & editing, Investigation. NV: Writing – review & editing, Investigation, Formal Analysis. JL: Conceptualization, Methodology, Writing – review & editing, Funding acquisition. RA: Resources, Conceptualization, Writing – review & editing, Funding acquisition, Supervision. KM: Writing – review & editing, Data curation, Supervision, Writing – original draft, Investigation, Funding acquisition, Conceptualization, Resources.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was funded by CORFO International Center of Excellence Program #13CEE2-21602, ANID Grants No. ACT210079, FONDEF IT16I10051, FONDECYT 3250470, FONDAP 152220002 (CECAN) & the European Union’s Horizon 2020 research and innovation program (grant 825741).
Acknowledgments
We thank Alicia Colombo and the staff from the “Biobanco de Fluidos y Tejidos de la Universidad de Chile” (BTUCH), Daniela Diez, and Vania Montecinos for sample processing and clinical data collection.
Conflict of interest
GS, AB, RA, and MF were Pfizer Chile employees.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1658528/full#supplementary-material
References
1. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
2. Roa JC, García P, Kapoor VK, Maithel SK, Javle M, and Koshiol J. Gallbladder cancer. Nat Rev Dis Primers. (2022) 8:69. doi: 10.1038/s41572-022-00398-y
3. Zollner L, Boekstegers F, Barahona Ponce C, Scherer D, Marcelain K, Gárate-Calderón V, et al. Gallbladder cancer risk and indigenous South American Mapuche ancestry: instrumental variable analysis using ancestry-informative markers. Cancers (Basel). (2023) 15:4033. doi: 10.3390/cancers15164033
4. Okumura K, Gogna S, Gachabayov M, Felsenreich DM, McGuirk M, Rojas A, et al. Gallbladder cancer: Historical treatment and new management options. World J Gastrointest Oncol. (2021) 13:1317–35. doi: 10.4251/wjgo.v13.i10.1317
5. Muszynska C, Lundgren L, Lindell G, Andersson R, Nilsson J, Sandström P, et al. Predictors of incidental gallbladder cancer in patients undergoing cholecystectomy for benign gallbladder disease: Results from a population-based gallstone surgery registry. Surgery. (2017) 162:256–63. doi: 10.1016/j.surg.2017.02.009
6. Giraldo NA, Drill E, Satravada BA, Dika IE, Brannon AR, Dermawan J, et al. Comprehensive molecular characterization of gallbladder carcinoma and potential targets for intervention. Clin Cancer Res. (2022) 28:5359–67. doi: 10.1158/1078-0432.CCR-22-1954
7. Yu H, Xu Y, Gao W, Li M, He J, Deng X, et al. Comprehensive germline and somatic genomic profiles of Chinese patients with biliary tract cancer. Front Oncol. (2022) 12:930611. doi: 10.3389/fonc.2022.930611
8. Lorenzo Bermejo J, Boekstegers F, González Silos R, Marcelain K, Baez Benavides P, Barahona Ponce C, et al. Subtypes of Native American ancestry and leading causes of death: Mapuche ancestry-specific associations with gallbladder cancer risk in Chile. PloS Genet. (2017) 13:e1006756. doi: 10.1371/journal.pgen.1006756
9. Wang K, Li M, and Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
10. Peña-Chilet M, Roldán G, Perez-Florido J, Ortuño FM, Carmona R, Aquino V, et al. CSVS, a crowdsourcing database of the Spanish population genetic variability. Nucleic Acids Res. (2021) 49:1130–7. doi: 10.1093/nar/gkaa794
11. Eyheramendy S, Martinez FI, Manevy F, Vial C, and Repetto GM. Genetic structure characterization of Chileans reflects historical immigration patterns. Nat Commun. (2015) 6:6472. doi: 10.1038/ncomms7472
12. Poli MC, Rebolledo-Jaramillo B, Lagos C, Orellana J, Moreno G, Martín LM, et al. Decoding complex inherited phenotypes in rare disorders: the DECIPHERD initiative for rare undiagnosed diseases in Chile. Eur J Hum Genet. (2024) 32:1227–37. doi: 10.1038/s41431-023-01523-5
13. Mayakonda A, Lin DC, Assenov Y, Plass C, and Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. (2018) 28:1747–56. doi: 10.1101/gr.239244.118
14. Tamborero D, Rubio-Perez C, Deu-Pons J, Schroeder MP, Vivancos A, Rovira A, et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. (2018) 10:25. doi: 10.1186/s13073-018-0531-8
15. Alexander DH, Novembre J, and Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. (2009) 19:1655–64. doi: 10.1101/gr.094052.109
16. Patterson N, Price AL, and Reich D. Population structure and eigenanalysis. PloS Genet. (2006) 2:e190. doi: 10.1371/journal.pgen.0020190
17. Reich D, Patterson N, Campbell D, Tandon A, Mazieres S, Ray N, et al. Reconstructing Native American population history. Nature. (2012) 488:370–4. doi: 10.1038/nature11258
18. Lindo J, Haas R, Hofman C, Apata M, Moraga M, Verdugo RA, et al. The genetic prehistory of the Andean highlands 7000 years BP though European contact. Sci Adv. (2018) 4:eaau4921. doi: 10.1126/sciadv.aau4921
19. Piñeros M, Vignat J, Colombet M, Laversanne M, Ferreccio C, Heise K, et al. Global variations in gallbladder cancer incidence: What do recorded data and national estimates tell us? Int J Cancer. (2025) 156:1358–68. doi: 10.1002/ijc.35232
20. Narayan RR, Creasy JM, Goldman DA, Gönen M, Kandoth C, Kundra R, et al. Regional differences in gallbladder cancer pathogenesis: Insights from a multi-institutional comparison of tumor mutations. Cancer. (2019) 125:575–85. doi: 10.1002/cncr.31850
21. Nepal C, Zhu B, O’Rourke CJ, Liu T, Li Q, Zhang W, et al. Integrative molecular characterisation of gallbladder cancer reveals micro-environment-associated subtypes. J Hepatol. (2021) 74:1132–44. doi: 10.1016/j.jhep.2020.11.033
22. Mishra S, Kumari S, Srivastava P, Pandey A, Shukla S, and Husain N. Genomic profiling of gallbladder carcinoma: Targetable mutations and pathways involved. Pathol Res Pract. (2022) 232:153806. doi: 10.1016/j.prp.2022.153806
23. Mishra K, Behari A, Shukla P, Tsuchiya Y, Endoh K, Asai T, et al. Risk factors for gallbladder cancer development in northern India: A gallstones-matched, case-control study. Indian J Med Res. (2021) 154:699–706. doi: 10.4103/ijmr.IJMR_201_19
24. Rodriguez-Meira A, Norfo R, Wen S, Chédeville AL, Rahman H, O’Sullivan J, et al. Single-cell multi-omics identifies chronic inflammation as a driver of TP53-mutant leukemic evolution. Nat Genet. (2023) 55:1531–41. doi: 10.1038/s41588-023-01480-1
25. Boekstegers F, Marcelain K, Barahona Ponce C, Baez Benavides PF, Müller B, de Toro G, et al. ABCB1/4 gallbladder cancer risk variants identified in India also show strong effects in Chileans. Cancer Epidemiol. (2020) 65:101643. doi: 10.1016/j.canep.2019.101643
26. Salem ME, Bodor JN, Puccini A, Xiu J, Goldberg RM, Grothey A, et al. Relationship between MLH1, PMS2, MSH2 and MSH6 gene-specific alterations and tumor mutational burden in 1057 microsatellite instability-high solid tumors. Int J Cancer. (2020) 147:2948–56. doi: 10.1002/ijc.33115
27. Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. (2023) 614:E41. doi: 10.1038/s41586-020-1943-3
28. Bernardez B, Higuera O, Martinez-Callejo V, Cardeña-Gutiérrez A, Marcos Rodríguez JA, Santaballa Bertrán A, et al. Sex and gender differences in cancer pathogenesis and pharmacology. Clin Transl Oncol. (2025). doi: 10.1007/s12094-025-03894-1
29. Hezel AF, Noel MS, Allen JN, Abrams TA, Yurgelun M, Faris JE, et al. Phase II study of gemcitabine, oxaliplatin in combination with panitumumab in KRAS wild-type unresectable or metastatic biliary tract and gallbladder cancer. Br J Cancer. (2014) 111:430–36. doi: 10.1038/bjc.2014.343
30. Nakamura Y, Mizuno N, Sunakawa Y, Canon JL, Galsky MD, Hamilton E, et al. Tucatinib and trastuzumab for previously treated human epidermal growth factor receptor 2-positive metastatic biliary tract cancer (SGNTUC-019): A phase II basket study. J Clin Oncol. (2023) 41:5569–78. doi: 10.1200/JCO.23.00606
31. Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J Med Chem. (2022) 65:3123–33. doi: 10.1021/acs.jmedchem.1c01688
32. Muzzana M, Broggini M, and Damia G. The landscape of PARP inhibitors in solid cancers. Onco Targets Ther. (2025) 18:297–317. doi: 10.2147/OTT.S499226
33. Chen Y, Fan X, Lu R, Zeng S, and Gan P. PARP inhibitor and immune checkpoint inhibitor have synergism efficacy in gallbladder cancer. Genes Immun. (2024) 25:307–16. doi: 10.1038/s41435-024-00280-9
34. Lin J, Dong K, Bai Y, Zhao S, Dong Y, Shi J, et al. Precision oncology for gallbladder cancer: insights from genetic alterations and clinical practice. Ann Transl Med. (2019) 7:467. doi: 10.21037/atm.2019.08.67
35. De Paolis E, Urbani A, Salvatore L, Foca L, Tortora G, Minucci A, et al. A novel ATM pathogenic variant in an Italian woman with gallbladder cancer. Genes (Basel). (2021) 12:313. doi: 10.3390/genes1202031
36. Moffat GT, Hu ZI, Meric-Bernstam F, Kong EK, Pavlick D, Ross JS, et al. KRAS allelic variants in biliary tract cancers. JAMA Netw Open. (2024) 7:e249840. doi: 10.1001/jamanetworkopen.2024.9840
37. Corti F, Nichetti F, Raimondi A, Niger M, Prinzi N, Torchio M, et al. Targeting the PI3K/AKT/mTOR pathway in biliary tract cancers: A review of current evidences and future perspectives. Cancer Treat Rev. (2019) 72:45–55. doi: 10.1016/j.ctrv.2018.11.001
38. Wu CE, Pan YR, Yeh CN, and Lunec J. Targeting P53 as a future strategy to overcome gemcitabine resistance in biliary tract cancers. Biomolecules. (2020) 10:1474. doi: 10.3390/biom10111474
39. Javle M, Bekaii-Saab T, Jain A, Wang Y, Kelley RK, Wang K, et al. Biliary cancer: Utility of next-generation sequencing for clinical management. Cancer. (2016) 122:3838–47. doi: 10.1002/cncr.30254
40. Koshiol J, Gao YT, Dean M, Egner P, Nepal C, Jones K, et al. Association of aflatoxin and gallbladder cancer. Gastroenterology. (2017) 153:488–94.e1. doi: 10.1053/j.gastro.2017.04.005
41. Hollstein M, Moriya M, Grollman AP, and Olivier M. Analysis of TP53 mutation spectra reveals the fingerprint of the potent environmental carcinogen, aristolochic acid. Mutat Res. (2013) 753:41–9. doi: 10.1016/j.mrrev.2013.02.003
42. Hainaut P and Pfeifer GP. Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb Perspect Med. (2016) 6:a026179. doi: 10.1101/cshperspect.a026179
43. Baugh EH, Ke H, Levine AJ, Bonneau RA, and Chan CS. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. (2018) 25:154–60. doi: 10.1038/cdd.2017.180
44. Brägelmann J, Barahona Ponce C, Marcelain K, Roessler S, Goeppert B, Gallegos I, et al. Epigenome-wide analysis of methylation changes in the sequence of gallstone disease, dysplasia, and gallbladder cancer. Hepatology. (2021) 73:2293–310. doi: 10.1002/hep.31585
45. Kuipers H, de Bitter TJJ, de Boer MT, van der Post RS, Nijkamp MW, de Reuver PR, et al. Gallbladder cancer: current insights in genetic alterations and their possible therapeutic implications. Cancers (Basel). (2021) 13:5257. doi: 10.3390/cancers13215257
46. Mishra SK, Kumari N, and Krishnani N. Molecular pathogenesis of gallbladder cancer: An update. Mutat Res. (2019) 816-18:111674. doi: 10.1016/j.mrfmmm.2019.111674
47. de Bitter TJJ, de Reuver PR, de Savornin Lohman EAJ, Kroeze LI, Vink-Börger ME, van Vliet S, et al. Comprehensive clinicopathological and genomic profiling of gallbladder cancer reveals actionable targets in half of patients. NPJ Precis Oncol. (2022) 6:83. doi: 10.1038/s41698-022-00327-y
48. Mondaca S, Walch H, Sepúlveda S, Schultz N, Muñoz G, Yaqubie A, et al. Clinical and genomic characterization of ERBB2-altered gallbladder cancer: exploring differences between an american and a Chilean cohort. JCO Glob Oncol. (2024) 10:e2400090. doi: 10.1200/GO.24.00090
49. Bustos BI, Pérez-Palma E, Buch S, Azócar L, Riveras E, Ugarte GD, et al. Variants in ABCG8 and TRAF3 genes confer risk for gallstone disease in admixed Latinos with Mapuche Native American ancestry. Sci Rep. (2019) 9:772. doi: 10.1038/s41598-018-35852-z
50. Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, Oddson A, Gylfason A, et al. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet. (2015) 47:435–44. doi: 10.1038/ng.3247
51. Yuan J, Hu Z, Mahal BA, Zhao SD, Kensler KH, Pi J, et al. Integrated analysis of genetic ancestry and genomic alterations across cancers. Cancer Cell. (2018) 34:549–60.e9. doi: 10.1016/j.ccell.2018.08.019
52. Zhu Y, Solis Soto LM, Pant S, Vega EA, Vinuela E, Bozorgui B, et al. Population-specific immunogenomic alterations in gallbladder cancer and prognostic significance. Mod Pathol. (2025) 38:100824. doi: 10.1016/j.modpat.2025.100824
53. Salvo M, González-Feliú E, Toro J, Gallegos I, Maureira I, Miranda-González N, et al. Validation of an NGS panel designed for detection of actionable mutations in tumors common in Latin America. J Pers Med. (2021) 11:899. doi: 10.3390/jpm11090899
54. Zhang C, Wang Y, Hu X, Qin L, Yin T, Fu W, et al. An improved NGS library construction approach using DNA isolated from human cancer formalin-fixed paraffin-embedded samples. Anat Rec (Hoboken). (2019) 302:941–46. doi: 10.1002/ar.24002
55. Steiert TA, Parra G, Gut M, Arnold N, Trotta JR, Tonda R, et al. A critical spotlight on the paradigms of FFPE-DNA sequencing. Nucleic Acids Res. (2023) 51:7143–62. doi: 10.1093/nar/gkad519
56. Oba U, Kohashi K, Sangatsuda Y, Oda Y, Sonoda KH, Ohga S, et al. An efficient procedure for the recovery of DNA from formalin-fixed paraffin-embedded tissue sections. Biol Methods Protoc. (2022) 7:bpac014. doi: 10.1093/biomethods/bpac014
Keywords: gallbladder cancer, next-generation sequencing (NGS), driver mutations, genetic ancestry, personalized therapies
Citation: Erices JI, González E, Salgado M, Barahona-Ponce C, Freire M, Sepúlveda-Hermosilla G, Ampuero D, Blanco A, Gárate-Calderón V, Báez-Benavides P, Tapia-Valladares C, Toro J, Gallegos I, Barajas O, Ahumada M, Sanhueza V, Spencer L, De Toro G, Morales E, Gutiérrez L, Morales F, Marin A, Varela NM, Lorenzo Bermejo J, Armisén R and Marcelain K (2025) The mutational landscape and actionable targets of gallbladder cancer: an ancestry-informed and comparative analysis of a Chilean population. Front. Oncol. 15:1658528. doi: 10.3389/fonc.2025.1658528
Received: 02 July 2025; Accepted: 08 September 2025;
Published: 03 October 2025.
Edited by:
Daniel P. Bezerra, Oswaldo Cruz Foudantion (FIOCRUZ), BrazilReviewed by:
Antonio Sandoval-Cabrera, Universidad Autónoma del Estado de México, MexicoShelly Pathak, Novogene Bioinformatics Technology Co., Ltd, China
Uttara Saran, University of Texas MD Anderson Cancer Center, United States
Copyright © 2025 Erices, González, Salgado, Barahona-Ponce, Freire, Sepúlveda-Hermosilla, Ampuero, Blanco, Gárate-Calderón, Báez-Benavides, Tapia-Valladares, Toro, Gallegos, Barajas, Ahumada, Sanhueza, Spencer, De Toro, Morales, Gutiérrez, Morales, Marin, Varela, Lorenzo Bermejo, Armisén and Marcelain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katherine Marcelain, a21hcmNlbGFpbkB1Y2hpbGUuY2w=
‡ORCID: Carol Barahona-Ponce, orcid.org/0000-0001-9505-4965