 Jingyu Feng1
Jingyu Feng1 Jun Yang1,2*
Jun Yang1,2*- 1Department of Neurosurgery, Peking University Third Hospital, Peking University, Beijing, China
- 2Center for Precision Neurosurgery and Oncology of Peking University Health Science Center, Peking University, Beijing, China
The development of gliomas is linked to neuroplasticity. Neurons, which are largely nonregenerative in adulthood, rely on axons and synapses to rebuild the neural network in response to experience and injury. Neural stem cells and immune cells coordinate “creation” (e.g., neurogenesis) and “clearance” (e.g., synaptic pruning), guided by signals from neural circuits. This review summarizes neuroplasticity mechanisms and explores their connection to gliomas, revealing that glioma cells hijack neural network derived signals to promote growth, migration, and stem-like properties, while simultaneously disrupting normal neural conduction. Similar to oligodendrocyte precursor cells (OPCs), gliomas exploit neural network regulation but are prone to uncontrolled proliferation. Moreover, glioma induced neural hyperexcitability disrupts circuit homeostasis, creating a permissive microenvironment for glioma progression. Consequently, neuroplasticity will contribute to the study of glioma related mechanisms and the development of more targeted strategies for prevention and control.
Graphical Abstract. A schematic that visually defines the structure of this review.
1 Introduction
Gliomas, which originate from central nervous system (CNS) cells and account for 75% of malignant primary brain tumors in adults, are the most common type of primary brain tumor (1–5). Classically, tumor cell proliferation was regarded as an “autonomous” process driven by genetic defects, with neural signaling interactions considered secondary (6). However, recent evidence indicates glioma cells are active participants, expressing neuron-like ion channels and neurotransmitter receptors to decode neural signals and regulate invasion, metabolism, and drug resistance (7, 8).
In the normal brain, neurons form a complex signaling network through electrical activity and neurotransmitter release (e.g., glutamate, γ-aminobutyric acid (GABA)) to regulate cognition and movement, a process termed neural plasticity (9, 10). Histopathologic and lineage analyses confirm that neural stem cells (NSCs), glial progenitors (e.g., OPCs), and astrocytes are potential origins of gliomas (11–13). These cells are all involved in regulating nervous system plasticity in the brain (14). It is widely recognized that cancer arises from the dysregulation of homeostatic mechanisms governing tissue repair and stem cell self-renewal (15). In the adult brain, NSCs and glial progenitor cells exhibit characteristics associated with central nervous system cancers, including a strong proliferative potential and diversity (16). Meanwhile, NSCs are regulated by the same cellular pathways that are active in brain tumors, such as the Notch, Wnt, and NF-κB signaling pathways (17–19). In the stem cells of the adult brain, OPCs constitute a major proliferative population, uniformly distributed throughout the adult rodent brain (20, 21). Numerous studies have shown that OPC or earlier pre-OPC cells are present in various forms of gliomas (22, 23). Dysregulation of myelin plasticity promotes glioma cell proliferation in primary brain cancer (24). Synaptosomal-associated protein 25 (SNAP25), a synaptic plasticity protein, is significantly correlated with the progression of glioma (25, 26). In summary, neuroplasticity is closely linked to the initiation and progression of gliomas, particularly in myelin plasticity. Aberrant plastic repair mechanisms may drive the development of gliomas, while post-glioma repair processes can further promote glioma progression.
This review summarizes current knowledge on glioma-neuron interactions from the perspective of neuroplasticity, dissecting the intricate mechanisms and structural alterations underlying neuroplasticity. Previous studies have discussed the interrelationship between myelin plasticity and glioma-neuron interactions, proposing that gliomas hijack myelin growth signals to promote self-proliferation (27, 28). Based on these findings, we analyze the relationship between normal neuroplasticity and abnormal glioma behavior in terms of proliferation, migration, stem-like properties, and immune interactions. We further explore the impacts of local neural signals, remote neural signals, and external signals on gliomas (Figure 1). We found that glioma-neuron interactions closely resemble the mechanisms of neuroplasticity, but disrupt the homeostatic balance inherent to normal neuroplasticity. The objective of this review is to delineate the correlations between neuroplasticity and glioma-neuron interactions, offering promising future directions for research.
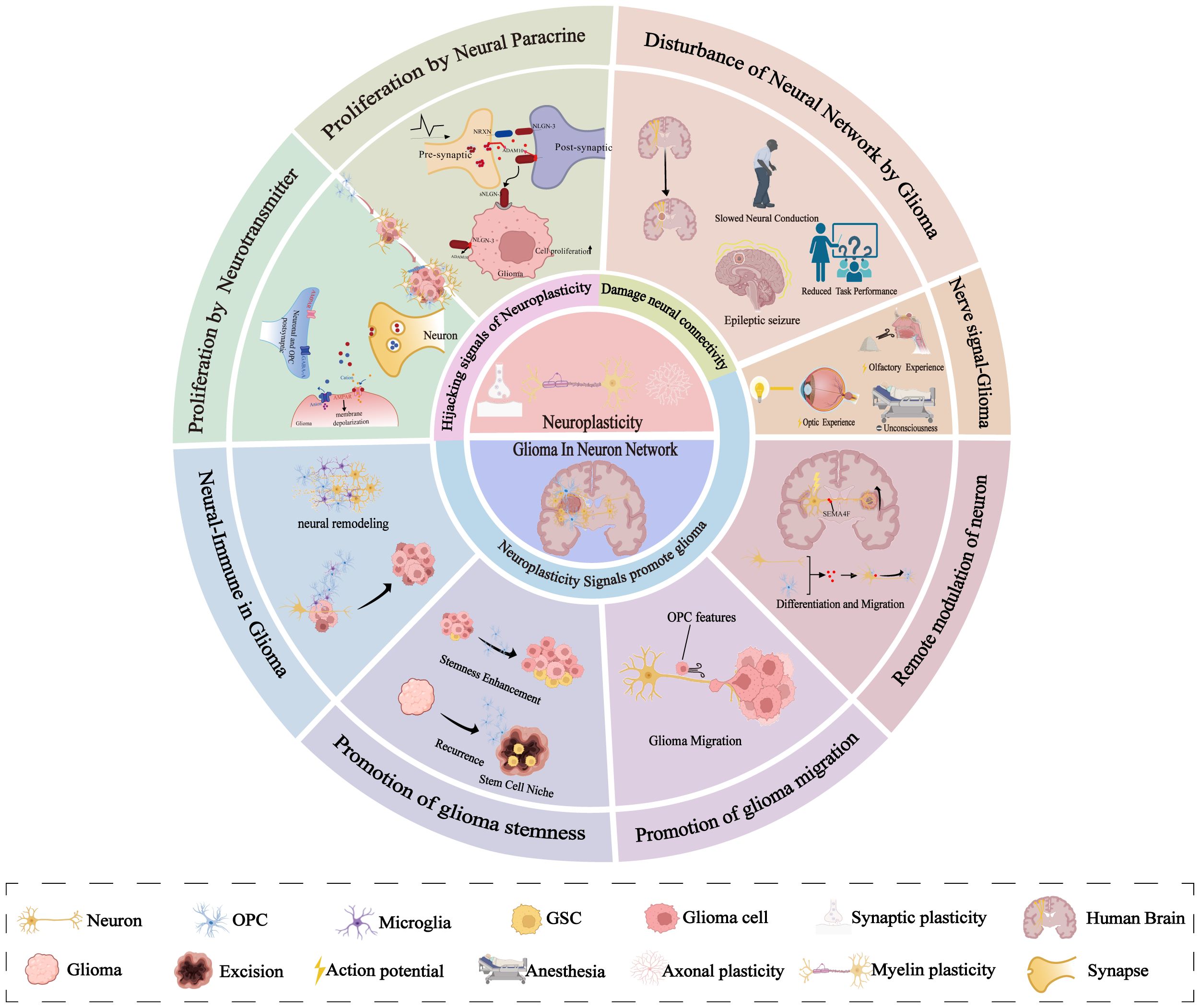
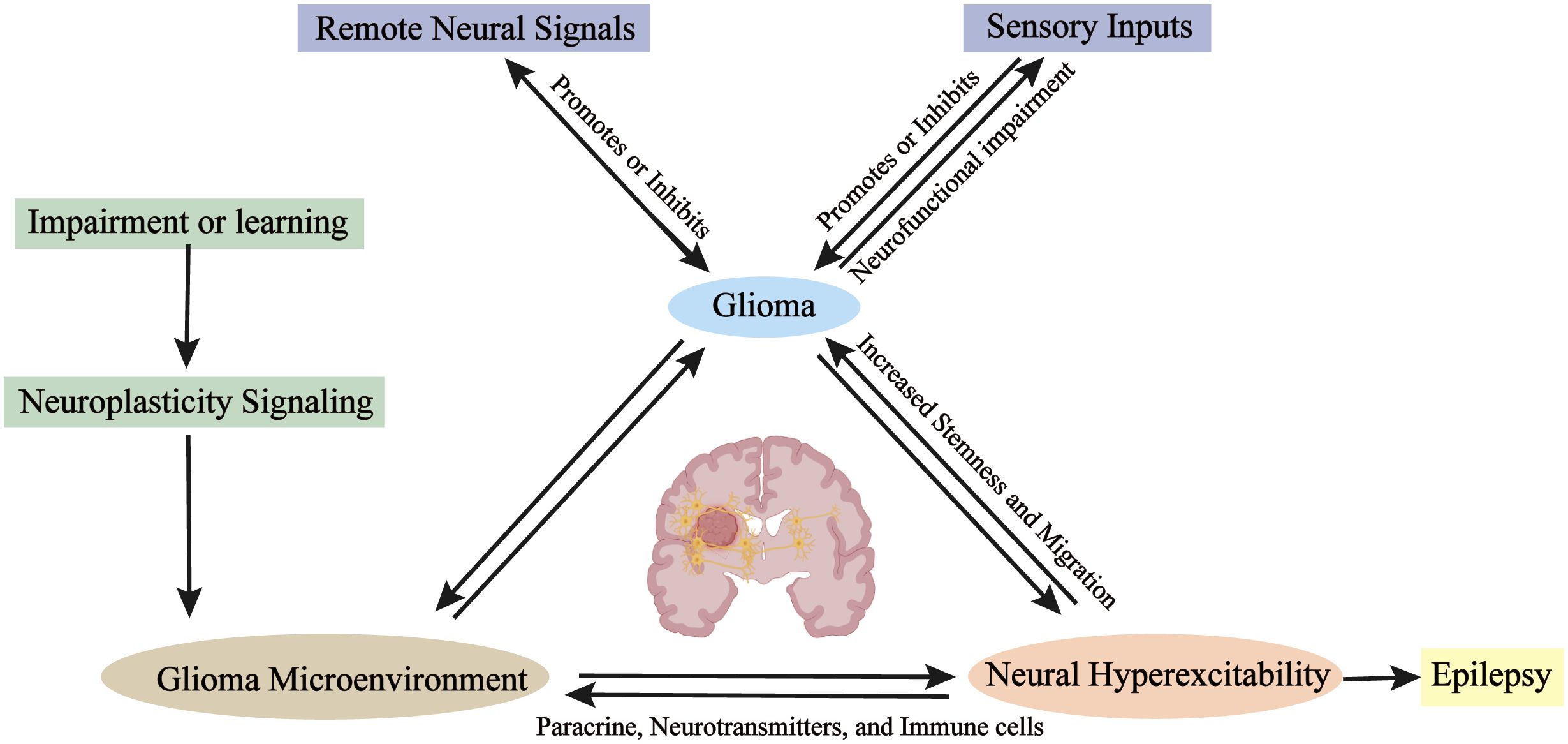
Figure 1. Specific aspects of glioma-neuron interactions and neural plasticity. Neural plasticity governs myelin, axon, and synaptic remodeling via neuronal signaling, with gliomas preferentially arising in highly plastic regions. Gliomas hijack neural stem cell repair mechanisms—forming synapses to receive neurotransmitters and paracrine factors—while neural inputs (e.g., visual/olfactory stimuli, anesthesia) significantly impact glioma growth. Bidirectional glioma-neuron communication drives epileptogenesis and impairs functions. Distant neurons participate via neural networks. Neuronal signals enhance glioma cell migration and the acquisition of stem-like properties. Neurons also indirectly regulate gliomas through immune cell crosstalk.
2 Neuroplasticity
Neuroplasticity refers to the brain’s capacity to reorganize its structure, function, or connectivity in response to intrinsic or extrinsic stimuli, a process that elicits both functional and morphological alterations. This dynamic process allows us to adapt to different environments and plays an important role in learning, memory, and injury recovery (29). It is well known that neurons in the adult brain are not regenerative upon death (30). Consequently, the remodeling following injury and learning primarily relies on the regrowth and reinnervation of axons (31). Axonal growth forms or strengthens more synaptic connections. Synaptic connections are highly plastic, with the number and strength of synapses changing significantly during development or in response to training (32).
Glia cells act as active regulators of neuroplasticity through their interactions with neurons and exhibit structural plasticity during learning (33, 34). The glia-neuronal crosstalk differs in physiological conditions and various brain disorders (35). Axonal growth relies on the regeneration of myelin sheaths. During the development of the brain, Oligodendrocytes (OLs) are the myelinating cells of the CNS that are generated from OPCs (36), which proliferate and differentiate during embryonic development. OPCs originate in the subventricular zone (SVZ) (37). However, to promote brain repair, OPCs are typically distributed throughout the gray and white matter, where they exhibit strong proliferative and migratory capabilities, as well as the constant capacity for surveillance (20, 38). During the development, myelin sheaths are “optimally distributed” throughout the nervous system. After developmental maturation, OPCs generate OLs involved in adaptive myelination (39). Neuronal activity, learning, and socialization influence myelin formation, this dynamic change in turn regulates signaling in neural circuits and is associated with emotional and cognitive functions, known as “myelin adaptation” (40) (Figure 2). Consequently, neuroplasticity can be summarized as three interrelated aspects of axonal plasticity myelin plasticity, and synaptic plasticity, which together form the basis of brain adaptation and plasticity.
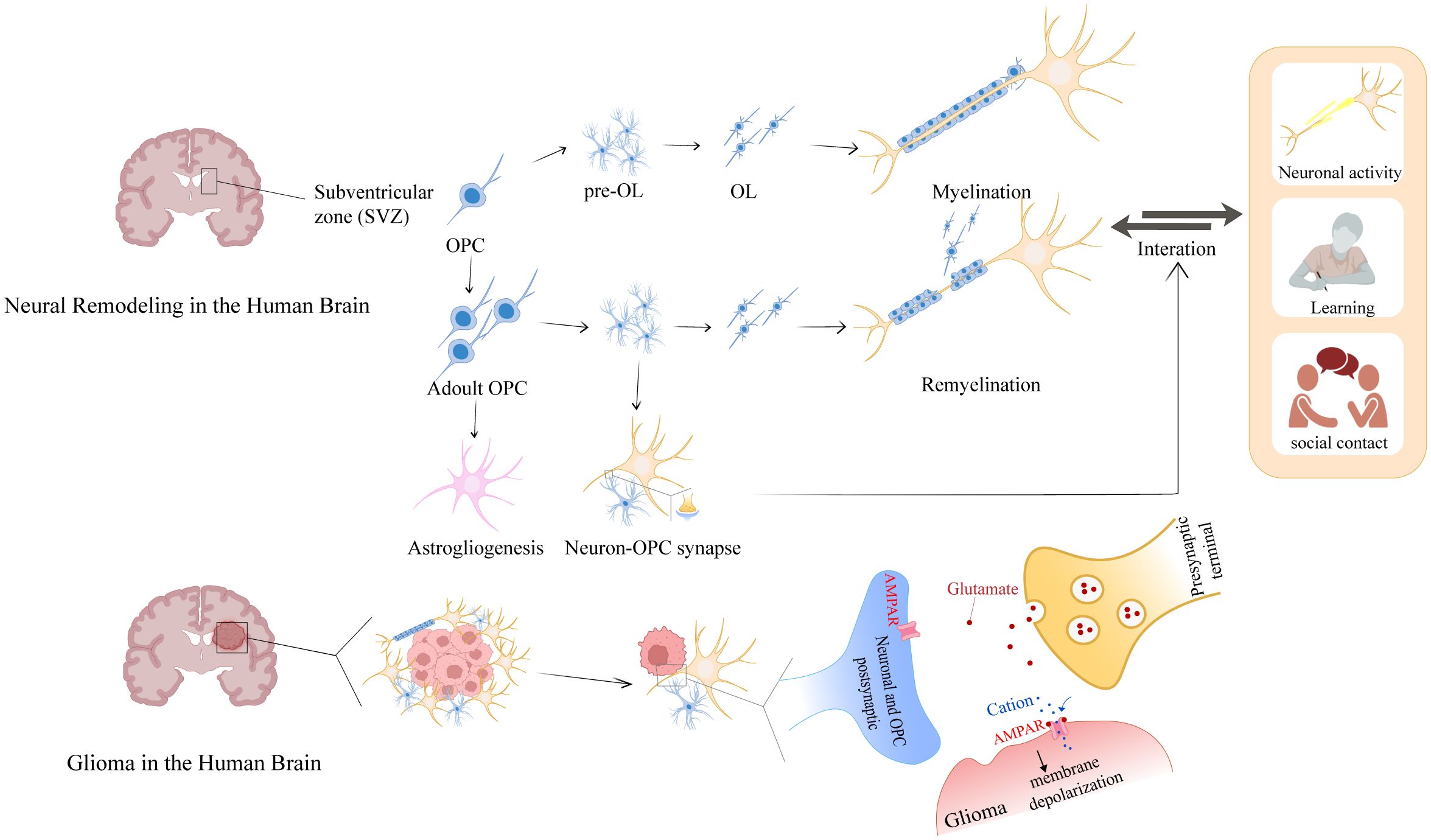
Figure 2. Glioma-neuron interactions of myelin plasticity and glioma intervention. During development, the SVZ serves as the primary source of OPCs. In the adult brain, OPCs that are distributed and reserved throughout the brain establish synaptic connections with neurons to receive repair and remodeling signals. These signals drive OPC proliferation and differentiation into mature OLs, which myelinate axons to facilitate neural network formation. Notably, glioma harbors OPC-like glioma cells that usurp this neuroregulatory pathway: these cells aberrantly repurpose neuronal-derived signals—originally dedicated to myelin repair—for autonomous growth, thereby illuminating a pivotal crosstalk between myelin plasticity and oncogenic mechanisms.
3 Neural signals influencing glioma growth
Studies have shown that neuronal activity can drive glioma progression via synaptic connections (7, 8), with early neuronal activity found to promote OPC proliferation and differentiation (41). A mouse model in which general anesthesia was used to reduce neuronal activity has demonstrated that low neural signals inhibited the growth and invasion of patient-derived glioblastoma (7). In normal physiological conditions, neural signals from external sensory stimuli can directly impact glioma development, and manipulating olfactory receptor neuron activity influences glioma progression (42). Additionally, stimulation of optic nerve activity promotes optic nerve glioma growth, while reducing visual input inhibits tumor formation and maintenance (43). Surprisingly, radiotherapy—a common treatment modality—accelerates tumor growth by enhancing neuronal activity (44). Collectively, these results indicate that neural signals promote glioma proliferation and differentiation, with such signals being moderately associated with learning and remodeling of the nervous system.
3.1 Synaptic transmission
Myelin plasticity homeostasis is important for the prevention of gliomas. The structural basis of myelin adaptation lies in OPCs’ ability to form true synapses with glutamatergic and GABAergic neurons, suggesting neuronal electrical activity regulates OPC proliferation and differentiation (45). In OPCs, GABA positively stimulates signaling cascades, which promote myelination as well as neural recovery (46, 47). Recent studies have further revealed that the OPC can receive inputs from multiple brain regions, illustrating that OPCs have strikingly comprehensive synaptic access to brain-wide projection networks (48). OPC postsynaptic molecules gradually lose the ability to be modulated by neurons during differentiation. As the unique glial cell to forms synapses with neurons, OPCs can accurately predict the location of future myelin production (49).
In recent years, Michelle Monje has illustrated the formation of structural synapses between glioma cells and neurons in the tumor microenvironment through electrophysiological and ultrastructural observations in two works from 2019 (7, 8), which opens up new directions for researchers. Interestingly, it has been revealed through single-cell transcriptomics that glioma cells forming synaptic structures predominantly belong to an OPC-like subpopulation (8). Spontaneous glutamatergic postsynaptic currents are present in such cells. It has been demonstrated that neuronal action potentials induce spontaneous inward currents (SIC) in GB, thereby promoting cancer development (7).
Neuron-glioma signal transmission occurs via calcium permeable AMPA receptors. Glutamate released from presynaptic membranes triggers depolarization by activating AMPARs on glioma cell membranes, with receptor inhibitors significantly impeding synaptic communication (7). Similarly, neuronal activity that promotes OPC myelination also involves AMPA glutamate receptors (50). However, in addition to glutamatergic synapses, GABAergic synapses have recently been discovered between gliomas and neurons, with both types able to coexist on a single glioma cell (51). Similar to OPCs in the early developmental stage, the Na-K-2Cl cotransporter (NKCC1) elevates Cl− levels in glioma cells (52), which tends to an efflux of Cl− upon activation of GABAARs. Therefore, GABAergic neuron-to-OPC and GABAergic neuron-to-glioma cell synapses cause depolarization (53). Notably, mature OPCs receive GABA-A-mediated inhibition through upregulated K+-Cl- cotransporter 2 (KCC2), while early developmental OPCs respond to promotive signals (47), but gliomas show only promotive effects (Figure 3).
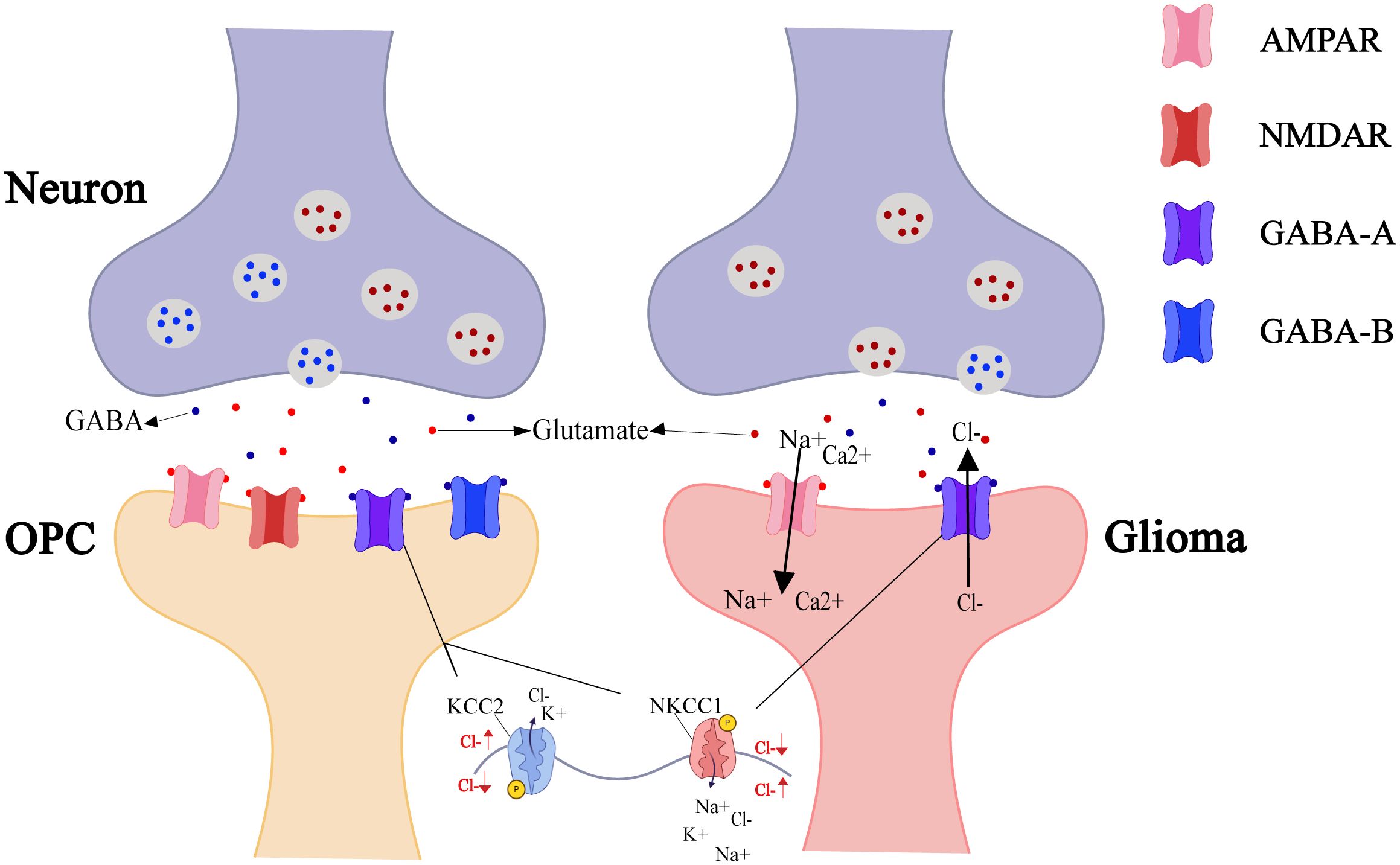
Figure 3. Neuro-OPC vs. neuro-glioma synaptic transmission. As the only glial cells forming synapses with neurons, OPCs undergo neural remodeling and repair regulated by neuronal-released neurotransmitters (e.g., GABA, glutamate). OPC postsynaptic membranes express AMPAR, NMDAR, GABA-A, and GABA-B receptors to integrate excitatory/inhibitory signals from the neurons. In contrast, certain glioma cells also express AMPAR and GABA-A receptors to promote self-growth. AMPAR activation opens Na+/Ca²+ channels, with cation influx inducing membrane depolarization. GABA-A activation opens Cl- channels, causing efflux of intracellularly accumulated Cl- (due to NKCC1 transporter activity) and inducing depolarization.
Glioma cells exhibit cellular properties similar to those of OPCs, suggesting that interactions between these cancer cells and neurons may be informed by the known neuronal regulation of their putative cellular origins (27).
3.2 Neural paracrine NLGN-3
In addition to neuronal regulation of synaptic transmission, Glioma cells appear to have also learned to respond to neuronal signals by the paracrine signaling of neural plasticity. Neurexins (NRXNs) and Neuroligins (NLGNs) are synaptic cell adhesion molecules that mediate presynaptic-postsynaptic neuronal connections and play critical roles in synapse development and signaling (54, 55). Neuroligin-3 (NLGN3) is predominantly distributed within the postsynaptic membranes of neurons and OPCs (56). This protein is released from these membranes in an activity-dependent manner, with secreted NLGN3(s-NLGN3) being cleaved from neurons and oligodendrocyte progenitor cells (OPCs) by A Disintegrin And Metalloproteinase10 (ADAM10) (57). As postsynaptic regulators of synaptic plasticity (58), s-NLGN3 critically mediates neuromodulation in gliomas by binding to glioma cell membranes (59). Gαi1/3 is activated by s-NLGN3 induction and mediates downstream oncogenic signaling pathways (60). Additionally, s-NLGN3 activates the PI3K-mTOR pathway, thereby promoting the proliferation and migration of glioma cells (61, 62). Unexpectedly, P13K upregulates NLGN-3 gene expression in glioma cells, generating more sNLGN-3 in the glioma microenvironment (57, 61) (Figure 4).
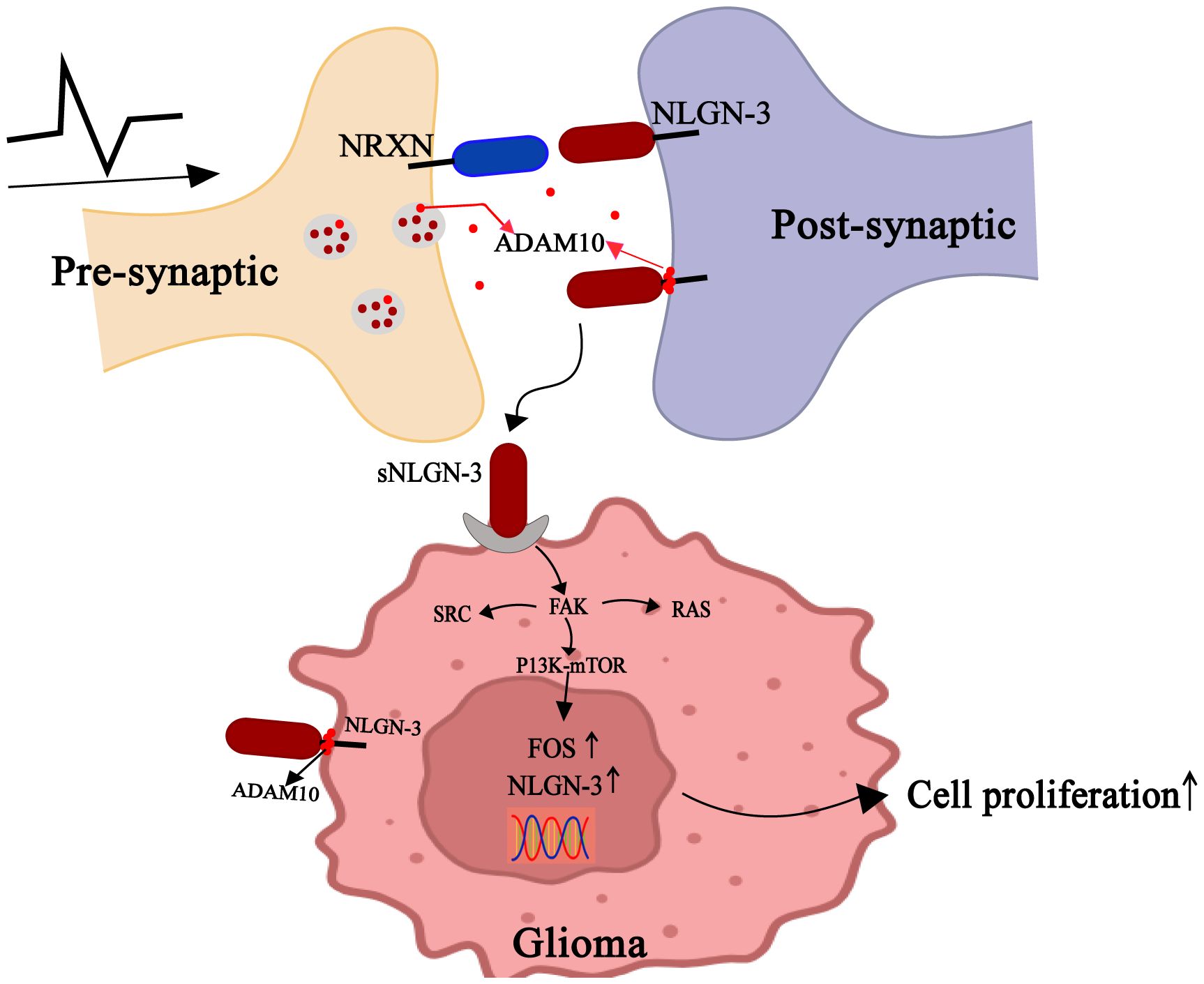
Figure 4. Gliomas exploit paracrine signals of neural plasticity to promote self-development. Neurons release ADAM10 from synaptic vesicles in an activity-dependent manner, which cleaves NLGN3 on neuronal or OPC membranes to shed s-NLGN3. Glioma cells competitively combine s-NLGN3 to activate multiple oncogenic pathways, including PI3K-mTOR, SRC kinase, and the SHC-RAS-RAF-MEK-ERK cascade. Concurrently, ADAM10-mediated cleavage sheds s-NLGN3 from glioma cell membranes, establishing autocrine positive feedback.
ADAM10 is highly enriched in synaptic vesicles (63). Reportedly, treatment with ADAM10 inhibitors suppresses the growth of adult and pediatric glioblastoma cells, an effect mediated by blocking ADAM10-dependent release of NLGN3 from neurons (64). s-NLGN3 shedding in the tumor microenvironment also drives optic pathway glioma (OPG) formation and growth. Mutations in the tumor suppressor gene NF1 (neurofibromatosis 1) in retinal neurons and increased optic nerve activity were both associated with increased NLGN3 shedding (43). Notably, NLGN3 is not the exclusive regulator of activity-dependent glioma growth, as NLGN3 deficiency only partially attenuates glioma cell mitogenic potential rather than completely abolishing it.
3.3 BDNF
Brain-derived neurotrophic factor (BDNF) is a survival factor for certain neurons during development (65). Signals via two different types of receptors: myosin-related kinase (Trk) B and p75 kilodalton neurotrophic receptor (p75). BDNF exerts divergent roles in distinct cell types and microenvironments, potentially exhibiting either oncogenic or tumor-suppressive effects (66). Overexpression of BDNF and/or Trk-B has been reported in multiple cancer types (67). However, in the healthy brain, BDNF functions as a paracrine trophic factor to promote adaptive synaptic plasticity (68). During cognitive activities like thinking and learning, neuronal activity orchestrates BDNF gene transcription, mRNA trafficking to dendrites, and BDNF protein secretion (69–72). BDNF has been shown to promote proliferation and differentiation of NSCs—particularly into the oligodendroglial lineage—in a dose-dependent fashion, a process that promotes myelin remodeling and is regulated by insulin (73, 74). Interestingly, insulin can also promote the proliferation and survival of glioblastomas (75). BDNF regulates malignant synapse-like connections between neurons and glioma cells in malignant gliomas. BDNF signaling via the tropomyosin-related kinase B (Trk-B) receptor promotes trafficking of AMPA receptors to glioma cell membranes, thereby modulating the amplitude of postsynaptic currents (28). Consequently, neurons are potently driven to promote malignant tumor proliferation via synaptic-like connections.
3.4 Neural network remote regulation of glioma growth
Neuromodulation in gliomas is not regional, with gliomas forming a close connection with neural networks (76, 77). The magnitude of regulation of gliomas growing in different brain regions varies, which is influenced by the surrounding neural network environment and conduction. Research finds that gliomas are more frequent in cortical regions that inherently have higher activity levels (78). Furthermore, the type of neurons that form synaptic connections with gliomas varies depending on the location in the brain region, with mostly long-distance glutamatergic neurons in the cortex and short-distance GABAergic neurons in the striatum (79). This is similar to the synaptic connections between OPC and neurons (80).
Gliomas have been shown to integrate into the brain’s network structure, which encompasses connections among tumor-tumor, neuron-tumor, and tumor-other cell-type interactions (81). In the network structure, cancer cells are interlinked through specialized membranous conduits, called tumor microtubules (TM) (76). However, this interconnection is not ubiquitous. The cellular network formed by TMs contributes to an enhancement in the stemness characteristics and drug resistance of the tumor (82). Based on the network structure, neuronal projections from brain regions remote from the primary tumor contribute to tumorigenesis. Activation of neurons contralateral to gliomas using chemical genetics was revealed to promote not only glioma proliferation but also early infiltration. Surprisingly, severing the interhemispheric connections inhibits the activity-dependent acceleration of infiltration observed in intact controls, while mechanistic investigations identify Semaphorin 4F (SEMA4F) as a key mediator linking remote neuronal activity to glioma progression (83). SEMA4F is expressed by neurons and OPCs, and it stimulates OPC differentiation (84). In the migration of OPCs, Sema4F contributes to the correct migration of OPCs along the nerve, thereby preventing cell dispersion and intermingling (85). Diffuse midline glioma (DMG), a malignant pediatric tumor originating in the midline of the brain (86), arises from and closely resembles oligodendroglial lineage precursors regardless of its specific anatomical location (23). Recent studies reveal that mesencephalic cholinergic nuclei drive proliferation of both healthy OPCs and DMG cells in their projection targets via a circuit-dependent mechanism, providing the first evidence for distance regulation by cholinergic neuronal activity (87).
This phenomenon highlights the requirement for global neural activity in glioma development, which is strongly correlated with the migration of OPCs during repair and remodeling processes. Disruption of this neural pathway could potentially impede glioma progression.
4 Neural signals influencing glioma migration
It is well established that the dissemination of glioma cells contributes to their incurability (88), yet this process differs fundamentally from the metastasis of other solid cancers, which typically do not spread to distant organs (89). There are several possible reasons: First, although glioma cells bind to blood vessels, they may not be able to break through the basement membrane into the vasculature system (90). Second, extra-neural tissues may lack an appropriate growth microenvironment to support glioma proliferation. Clinical investigations have shown that following complete resection, postoperative recurrence predominantly occurs in the local white matter (91). The neuronal soma resides in the gray matter, while axons—including those forming the corpus callosum, the largest interhemispheric commissure—occupy the white matter, which is composed of a broad array of neural fibers (92, 93). Gliomas that spread along the white matter bundles of the corpus callosum are called butterfly gliomas (94). The prognosis for these patients is often poor, and severing the corpus callosum can largely prevent the spread of gliomas (83). The axonal architecture in the white matter creates a more permissive microenvironment for the dissemination of glioma cells.
White matter, situated beneath the gray matter cortex, consists of myelinated neuronal fibers that facilitate rapid signal transmission within the brain (95). Myelin plays a critical role in tumor spread. It serves as a highly permissive substrate for glioma cell adhesion and migration (96). The microenvironment of the CNS inherently exhibits resistance to glioma cell infiltration. Inhibitory molecules in CNS myelin (e.g., Nogo/Semaphorins/Ephrins, etc.) also suppress glioma cell migration and proliferation (97). This is based on another crucial function of myelin: to prevent excessive axonal regeneration, sprouting, and cellular infiltration into the brain parenchyma (98). Neuronal activity induces adaptive changes in myelin structure and function. Correspondingly, this activity significantly influences the invasive behavior of glioblastoma cells, including the formation, growth, and movement of TMs (99). Neurons paradoxically exhibit tumor-promoting effects on gliomas, though emerging studies reveal that adult post-mitotic neurons can induce apoptosis in both murine and human glioma cells (100). Moreover, in vitro co-culture reveals that the migratory ability of glioblastoma cells is inhibited by contact with neurons (101). The underlying mechanism may involve neuron-regulated glioma cells exhibiting characteristics resembling those of OPCs (8). Overall, neurons normally regulate OPCs to promote myelination repair, but may pathologically facilitate glioma migration along axons by misidentifying tumor cells as OPCs.
5 Neural networks enhance glioma cell stemness
The cellular composition of glioma is not homogeneous (102). Glioma cells with stemness, called glioma stem cells (GSCs), promote heterogeneity and drug resistance in gliomas (103). As normal stem and progenitor cells participate in tissue development and repair, these developmental programs re-emerge in CSCs to support the development and progressive growth of glioma (104). Current research suggests that GSC may be derived from NSCs residing in the SVZ in adults, as they share many common features (105, 106). The researchers believe that if GSCs are the glioma cells responsible for generating the tumors, then the developmentally analogous relationship is the NSC-OPC axis (107). In fully developed individuals, NSCs can differentiate into OPCs as the primary source of myelination contribution (108). Olig2 is highly expressed in OPCs as well as in GSCs (109). Culturing glioma cells with conditioned medium from OPCs, which contains secreted factors, indicates that soluble factors secreted by OPCs enhance the stem-like properties of glioma cells, thereby contributing to tumorigenesis, therapeutic resistance, and recurrence (110). OPCs and macrophages/microglia form a distinct microenvironment for glioma cells at the tumor boundary, with particularly prominent aggregation in recurrent lesions. In this microenvironment, OPCs may drive the acquisition of stemness in glioma cells (91, 110, 111). Neuronal activity enhances the stemness of glioma cells. Studies have shown that exosomes derived from active neurons promote glioma progression and radioresistance by inducing phenotypic and metabolic transformation of GSCs (112). In summary, we suggest that the aggregation of OPCs at the tumor may misregulate the enhancement of glioma stemness, and this regulation can be potentiated by electrical activity stimulation.
6 Neural-immune interplay in glioma
In addition to directly mediating tumor growth, neurons can promote the tumor by modulating immune cell function. Astrocytes perform supporting functions for neurons and oligodendrocytes (113). Microglia are recognized as mononuclear phagocytic cells that play a significant role in immune response and homeostasis within the CNS (114). They contribute to the formation, maintenance, and reshaping of neuronal circuits by clearing dead cells and participating in neural repair through pruning (115, 116). In neuroplasticity, complex interactions between neurons, T cells, and microglia (117). Neurons play a crucial role in regulating microglia activation, as neurons secrete factors such as CD200 (118), SEMA3A (119), and CX3CL1 (120) can modulate microglial cell properties to different degrees. Whereas this regulation promotes the process of neuronal repair and remodeling in the normal brain, in the glioma setting, neurons produce reduced mid-term to activate T cells, which in turn leads to an increase in T cell Ccl4 secretion and microglial cell secretion of Ccl5 to sustain glioma cell growth (117).
Overall, while glioma growth stimulates immune cell repair and participates in neural remodeling, signaling impulses from neurons can, in turn, facilitate this process. However, this process seems to be exploited by the glioma cells for the use of self-growth.
7 Disturbance of neural network by glioma
Neuron–glioma interactions are bidirectional. Based on subdural electrocorticography, sampling of normal and glioma-infiltrated cortex during speech showed that glioma infiltration affected the brain’s ability to encode information during nuanced tasks (121). Recent studies have revealed that tumor-associated cortical networks exhibit hyperexcitability (8). Tumor-induced disruption of synaptic network activity in the peritumoral region leads to alterations in network excitability (122). Although neuronal over-excitation maintains task-specific neuronal responses, the tumor-affected cerebral cortex loses the ability to decode complex words (123).
In addition to impairing brain function, epilepsy is diagnosed in 70–90% of patients with glioma (124). However, further investigation has revealed that the abnormal enhancement of peritumoral neuronal network activity and the prevalent epileptiform activity were closely associated with the formation of new synapses, with glioma cells forming these new synapses remaining as OPC-like cells (125). In the glioma-surrounding tissue, extracellular glutamate levels were found to be 100 times higher than in the unaffected brain (126). Glutamate secretion from gliomas stimulates peritumoral neuronal receptors, leading to neuronal hyperexcitability and epileptic seizures (127).
8 Conclusion and future directions
There is growing evidence that different types of cancers originate from distinct “progenitor cells”, which undergo the first or multiple genetic hits leading to the onset of cancer (128). Therefore, the origin and progression of cancer cells in different locations depend on the surrounding environment and cell type. Gliomas, the most prevalent primary malignant tumors in the adult CNS, are likely triggered by the daily remodeling and repair processes of glial cells, during which multiple factors induce malignant changes. OPCs, the most active stem cells in the brain and responsible for myelin plasticity, are also found aggregating around gliomas.
The underlying mechanism of this OPC aggregation—whether driven by reparative recruitment or malignant transformation during the initial repair process—remains inconclusive. Mosaic Analysis with Double Markers (MADM)-based lineage tracing revealed significant abnormal growth prior to malignancy only in OPCs (129). Notably, accumulating evidence has established that OPC aggregation significantly accelerates glioma progression. Stem cells not only have the mission of proliferation and differentiation but also require multiple factors (e.g., neuroregulatory signals, paracrine factors) to promote or inhibit the function (130). Similarly, in gliomas, the factors that regulate OPC also regulate the glioma cells and even form similar synaptic connections. It seems that brain cancer learns the mechanisms of neural plasticity.
However, these regulators also exhibit bidirectionality. As a key modulator of synaptic plasticity (72), BDNF contributes to physiological synaptic regulation through neuronal activity and drives tumor progression through BDNF-TrkB-mediated malignant synapse enhancement (28). Its effects are not unilaterally protumorigenic: mature BDNF/TrkB signaling drives glioma growth, migration, and anti-apoptotic effects, while proBDNF/p75NTR activation inhibits these processes (131). Additionally, lncRNA BDNF-AS suppresses malignancy by targeting RAX2 (132). This functional difference depends on the type of cells involved, the selective binding of receptor subtypes, and microenvironmental characteristics (66). GABA shows more pronounced bidirectionality (133). In DMG, NKCC1-mediated high intracellular Cl- converts the action of GABA to membrane depolarization, promoting proliferation (51). Additionally, GABA maintains GSC quiescence for post-surgical recurrence (134). Conversely, GABAaR activation inhibits proliferation in low-grade gliomas via enhanced inhibitory signaling, although a mechanism potentially weakened by GABAaR downregulation in glioblastoma (135). In vitro experiment, neuronal GABAaR activation directly suppresses glioma growth (136). This bidirectionality resembles the functional differences of GABAergic signaling in OPC regulation (47). This suggests that targeted modulation of Cl- currents in glioma cells may provide a novel therapeutic approach to halt tumor progression (137).
Current experimental models of tumor-neuron interactions predominantly rely on in vitro cell co-culture (138) or xenografts in immunodeficient mice (8, 61). Though they partially reflect the interactions between gliomas and neurons, they cannot replicate the 3D structure of in vivo neural circuits, neurotransmitter microenvironment, or brain region-specific neuroplasticity. However, related studies have made progress. An in vitro 3D model constructed using 3D bioprinting technology, consisting of an outer hemisphere containing neurons and an inner hemisphere containing glioma cells (139). Modeling glioblastoma invasion using human brain organoids (140). Co-culture system using patient-derived GBM organoids and human induced pluripotent stem cells (hiPSCs) (141). Despite these advancements, further optimization is still needed to more realistically simulate the physiological environment of tumor-neuron interactions in vivo. Another important consideration is that there are significant differences between pediatric and adult gliomas in terms of genetic background, site of origin, and clinical behavior (142). Adult gliomas often originate in the supratentorial region and are often accompanied by neuroplastic compensatory mechanisms. In contrast, pediatric gliomas predominantly occur in brain regions with active neurogenesis, including the brainstem and thalamus. The progression may be more closely linked to the active neuroplasticity during the brain development stage (143). Therefore, it is necessary to study the differences between the contributions of “developmental neuroplasticity” and “pathological neuroplasticity” in childhood and adult gliomas.
Molecules related to neuroplasticity may serve as potential targets for glioma treatment, but the specific mechanisms remain unclear. ADAM10 is highly expressed in gliomas; however, the mechanism by which ADAM10 balances neuroplasticity and glioma phenotypes through cleaving different substrates remains elusive (64). AMPAR is the core subtype of glutamate receptors. Pharmacological inhibition of AMPAR activity using Talampanel has demonstrated potential in the clinical management of newly diagnosed glioblastoma (144), but the impact of long-term AMPAR inhibition on normal neurological function has not yet been systematically validated. Cav3, as a T-type calcium channel in synaptic plasticity, can be utilized in inhibiting glioma development through disconnecting nerve cell and OPC-like glioma cell interaction (145, 146). Rabies-mediated genetic ablation of neurons halts glioblastoma progression (44). Unexpectedly, some commonly used drugs have been found to have tumor-promoting effects, such as Lorazepam (51). Currently, there is growing evidence that multiple neuroplasticity signals are exploited to influence the progression of gliomas, suggesting that learning and remodeling are closely related to the initiation and progression of gliomas. We believe that broader plasticity regulatory mechanisms can inspire the study of abnormal tumor proliferation. Meanwhile, based on the study and modulation of neuroplasticity, more effective treatments for controlling the progression of gliomas will be discovered.
Author contributions
JF: Writing – original draft. JY: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was funded by the National Natural Science Foundation of China, grant number 82272675.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ADAM10, A Disintegrin and Metalloproteinase10; AMPAR, α-Amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors; BDNF, Brain Derived Neurotrophic Factor; CCL4, CC chemokine 4; CCL5, CC chemokine 5; CD200, Cluster of Differentiation 200; CNS, Central Nervous System; CX3CL1, Chemokine (C-X3-C Motif) Ligand 1; DMG, Diffuse Midline Glioma; GABA, γ-aminobutyric acid; GSCs, Glioma Stem Cells; hiPSCs, Human induced Pluripotent Stem Cells; KCC2, K+-Cl- cotransporter 2; MADM, Mosaic Analysis with Double Markers; NF1, Neurofibromatosis type 1; NLGNs, Neuroligins; NLGN3, Neuroligin-3; NSCs, Neural Stem Cells; NKCC1, Na-K-2Cl cotransporter; NRXNs, Neurexins; OLs, Oligodendrocytes; OPG, Optic Pathway Glioma; OPCs, Oligodendrocyte Precursor Cells; RAX2, Retina and anterior neural fold homeobox 2; SEMA4F, Semaphorin 4F; SIC, Spontaneous Inward Currents; SNAP25, Synaptosomal-associated protein 25; SVZ, Subventricular Zone; s-NLGN3, secreted NLGN3; TM, Tumor Microtubules; SEMA3A, Semaphorin 3A.
References
1. Lapointe S, Perry A, and Butowski NA. Primary brain tumours in adults. Lancet. (2018) 392:432–46. doi: 10.1016/S0140-6736(18)30990-5
2. Behin A, Hoang-Xuan K, Carpentier AF, and Delattre J-Y. Primary brain tumours in adults. Lancet. (2003) 361:323–31. doi: 10.1016/S0140-6736(03)12328-8
3. Germano I, Swiss V, and Casaccia P. Primary brain tumors, neural stem cell, and brain tumor cancer cells: where is the link? Neuropharmacology. (2010) 58:903–10. doi: 10.1016/j.neuropharm.2009.12.019
4. Zheng S, Alfaro-Munoz K, Wei W, Wang X, Wang F, Eterovic AK, et al. Prospective clinical sequencing of adult glioma. Mol Cancer Ther. (2019) 18:991–1000. doi: 10.1158/1535-7163.MCT-18-1122
5. Yang K, Wu Z, Zhang H, Zhang N, Wu W, Wang Z, et al. Glioma targeted therapy: insight into future of molecular approaches. Mol Cancer. (2022) 21:39. doi: 10.1186/s12943-022-01513-z
6. Evan GI and Vousden KH. Proliferation, cell cycle and apoptosis in cancer. nature. (2001) 411:342–8. doi: 10.1038/35077213
7. Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, Kessler T, et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature. (2019) 573:532–8. doi: 10.1038/s41586-019-1564-x
8. Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M, et al. Electrical and synaptic integration of glioma into neural circuits. Nature. (2019) 573:539–45. doi: 10.1038/s41586-019-1563-y
9. Teleanu RI, Niculescu A-G, Roza E, Vladâcenco O, Grumezescu AM, and Teleanu DM. Neurotransmitters—Key factors in neurological and neurodegenerative disorders of the central nervous system. Int J Mol Sci. (2022) 23:5954. doi: 10.3390/ijms23115954
10. Dhuriya YK and Sharma D. Neuronal plasticity: neuronal organization is associated with neurological disorders. J Mol Neurosci. (2020) 70:1684–701. doi: 10.1007/s12031-020-01555-2
11. Pojo M and Costa BM. Molecular hallmarks of gliomas. In: Garami M, editor. Molecular Targets of Cns Tumors. Rijeka: IntechOpen (2011).
12. Wesseling P and Capper D. Who 2016 classification of gliomas. Neuropathol Appl Neurobiol. (2018) 44:139–50. doi: 10.1111/nan.12432
13. Zong H, Verhaak RG, and Canoll P. The cellular origin for Malignant glioma and prospects for clinical advancements. Expert Rev Mol diagnostics. (2012) 12:383–94. doi: 10.1586/erm.12.30
14. Zhang Y, Liao Q, Wen X, Fan J, Yuan T, Tong X, et al. Hijacking of the nervous system in cancer: mechanism and therapeutic targets. Mol Cancer. (2025) 24:44. doi: 10.1186/s12943-025-02246-5
15. Beachy PA, Karhadkar SS, and Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. (2004) 432:324–31. doi: 10.1038/nature03100
16. Reya T, Morrison SJ, Clarke MF, and Weissman IL. Stem cells, cancer, and cancer stem cells. nature. (2001) 414:105–11. doi: 10.1038/35102167
17. Guo R, Han D, Song X, Gao Y, Li Z, Li X, et al. Context-dependent regulation of notch signaling in glial development and tumorigenesis. Sci Adv. (2023) 9:eadi2167. doi: 10.1126/sciadv.adi2167
18. Widera D, Kaus A, Kaltschmidt C, and Kaltschmidt B. Neural stem cells, inflammation and nf-κb: basic principle of maintenance and repair or origin of brain tumours? J Cell Mol Med. (2008) 12:459–70. doi: 10.1111/j.1582-4934.2007.00208.x
19. Li X, Kim HJ, Yoo J, Lee Y, Nam CH, Park J, et al. Distant origin of glioblastoma recurrence: neural stem cells in the subventricular zone serve as a source of tumor reconstruction after primary resection. Mol Cancer. (2025) 24:64. doi: 10.1186/s12943-025-02273-2
20. Maki T. Novel roles of oligodendrocyte precursor cells in the developing and damaged brain. Clin Exp Neuroimmunol. (2017) 8:33–42. doi: 10.1111/cen3.12358
21. Geha S, Pallud J, Junier MP, Devaux B, Leonard N, Chassoux F, et al. Ng2+/olig2+ Cells are the major cycle-related cell population of the adult human normal brain. Brain Pathol. (2010) 20:399–411. doi: 10.1111/j.1750-3639.2009.00295.x
22. Larjavaara S, Mäntylä R, Salminen T, Haapasalo H, Raitanen J, Jääskeläinen J, et al. Incidence of gliomas by anatomic location. Neuro-oncology. (2007) 9:319–25. doi: 10.1215/15228517-2007-016
23. Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci. (2011) 108:4453–8. doi: 10.1073/pnas.1101657108
24. Mancusi R and Monje M. The neuroscience of cancer. Nature. (2023) 618:467–79. doi: 10.1038/s41586-023-05968-y
25. Huang Q, Lian C, Dong Y, Zeng H, Liu B, Xu N, et al. Snap25 inhibits glioma progression by regulating synapse plasticity via gls-mediated glutaminolysis. Front Oncol. (2021) 11:698835. doi: 10.3389/fonc.2021.698835
26. Noor A and Zahid S. A review of the role of synaptosomal-associated protein 25 (Snap-25) in neurological disorders. Int J Neurosci. (2017) 127:805–11. doi: 10.1080/00207454.2016.1248240
27. Pan Y and Monje M. Neuron–glial interactions in health and brain cancer. Advanced Biol. (2022) 6:2200122. doi: 10.1002/adbi.202200122
28. Taylor KR, Barron T, Hui A, Spitzer A, Yalçin B, Ivec AE, et al. Glioma synapses recruit mechanisms of adaptive plasticity. Nature. (2023) 623:366–74. doi: 10.1038/s41586-023-06678-1
29. Voss P, Thomas ME, Cisneros-Franco JM, and de Villers-Sidani É. Dynamic brains and the changing rules of neuroplasticity: implications for learning and recovery. Front Psychol. (2017) 8:274878. doi: 10.3389/fpsyg.2017.01657
30. Sorrells SF, Paredes MF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature. (2018) 555:377–81. doi: 10.1038/nature25975
31. Horner PJ and Gage FH. Regenerating the damaged central nervous system. Nature. (2000) 407:963–70. doi: 10.1038/35039559
32. Turrigiano GG and Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. (2004) 5:97–107. doi: 10.1038/nrn1327
33. Rangel-Gomez M, Alberini CM, Deneen B, Drummond GT, Manninen T, Sur M, et al. Neuron–glial interactions: implications for plasticity, behavior, and cognition. J Neurosci. (2024) 44. doi: 10.1523/JNEUROSCI.1231-24.2024
34. Laming PR, Kimelberg H, Robinson S, Salm A, Hawrylak N, Müller C, et al. Neuronal–glial interactions and behaviour. Neurosci Biobehav Rev. (2000) 24:295–340. doi: 10.1016/S0149-7634(99)00080-9
35. Adamczyk A. Glial–neuronal interactions in neurological disorders: molecular mechanisms and potential points for intervention. Int J Mol Sci.. (2023) 24(7):6274. doi: 10.3390/ijms24076274
36. Auguste YSS, Ferro A, Kahng JA, Xavier AM, Dixon JR, Vrudhula U, et al. Oligodendrocyte precursor cells engulf synapses during circuit remodeling in mice. Nat Neurosci. (2022) 25:1273–8. doi: 10.1038/s41593-022-01170-x
37. Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, and Alvarez-Buylla A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci. (2006) 26:7907–18. doi: 10.1523/jneurosci.1299-06.2006
38. Bergles DE and Richardson WD. Oligodendrocyte development and plasticity. Cold Spring Harbor Perspect Biol. (2016) 8:a020453. doi: 10.1101/cshperspect.a020453
39. Ma Z, Zhang W, Wang C, Su Y, Yi C, and Niu J. A new acquaintance of oligodendrocyte precursor cells in the central nervous system. Neurosci Bull. (2024) 40:1573–89. doi: 10.1007/s12264-024-01261-8
40. Mount CW and Monje M. Wrapped to adapt: experience-dependent myelination. Neuron. (2017) 95:743–56. doi: 10.1016/j.neuron.2017.07.009
41. Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science. (2014) 344:1252304. doi: 10.1126/science.1252304
42. Chen P, Wang W, Liu R, Lyu J, Zhang L, Li B, et al. Olfactory sensory experience regulates gliomagenesis via neuronal igf1. Nature. (2022) 606:550–6. doi: 10.1038/s41586-022-04719-9
43. Pan Y, Hysinger JD, Barron T, Schindler NF, Cobb O, Guo X, et al. Nf1 mutation drives neuronal activity-dependent initiation of optic glioma. Nature. (2021) 594:277–82. doi: 10.1038/s41586-021-03580-6
44. Tetzlaff SK, Reyhan E, Layer N, Bengtson CP, Heuer A, Schroers J, et al. Characterizing and targeting glioblastoma neuron-tumor networks with retrograde tracing. Cell. (2025) 188:390–411.e36. doi: 10.1016/j.cell.2024.11.002
45. Káradóttir R, Cavelier P, Bergersen LH, and Attwell D. Nmda receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. (2005) 438:1162–6. doi: 10.1038/nature04302
46. Lin S-c and Bergles DE. Synaptic signaling between gabaergic interneurons and oligodendrocyte precursor cells in the hippocampus. Nat Neurosci. (2004) 7:24–32. doi: 10.1038/nn1162
47. Bai X, Kirchhoff F, and Scheller A. Oligodendroglial gabaergic signaling: more than inhibition! Neurosci Bull. (2021) 37:1039–50. doi: 10.1007/s12264-021-00693-w
48. Mount CW, Yalçın B, Cunliffe-Koehler K, Sundaresh S, and Monje M. Monosynaptic tracing maps brain-wide afferent oligodendrocyte precursor cell connectivity. Elife. (2019) 8:e49291. doi: 10.7554/eLife.49291
49. Li J, Miramontes TG, Czopka T, and Monk KR. Synaptic input and ca2+ Activity in zebrafish oligodendrocyte precursor cells contribute to myelin sheath formation. Nat Neurosci. (2024) 27:219–31. doi: 10.1038/s41593-023-01553-8
50. Fannon J, Tarmier W, and Fulton D. Neuronal activity and ampa-type glutamate receptor activation regulates the morphological development of oligodendrocyte precursor cells. Glia. (2015) 63:1021–35. doi: 10.1002/glia.22799
51. Barron T, Yalçın B, Su M, Byun YG, Gavish A, Shamardani K, et al. Gabaergic neuron-to-glioma synapses in diffuse midline gliomas. Nature. (2025) 639(8056):1060–8. doi: 10.1038/s41586-024-08579-3
52. Kurki SN, Uvarov P, Pospelov AS, Trontti K, Hübner AK, Srinivasan R, et al. Expression patterns of nkcc1 in neurons and non-neuronal cells during cortico-hippocampal development. Cereb Cortex. (2023) 33:5906–23. doi: 10.1093/cercor/bhac470
53. Zonouzi M, Scafidi J, Li P, McEllin B, Edwards J, Dupree JL, et al. Gabaergic regulation of cerebellar ng2 cell development is altered in perinatal white matter injury. Nat Neurosci. (2015) 18:674–82. doi: 10.1038/nn.3990
54. Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. (2008) 455:903–11. doi: 10.1038/nature07456
55. Craig AM and Kang Y. Neurexin–neuroligin signaling in synapse development. Curr Opin Neurobiol. (2007) 17:43–52. doi: 10.1016/j.conb.2007.01.011
56. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An rna-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. (2014) 34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
57. Venkatesh HS, Tam LT, Woo PJ, Lennon J, Nagaraja S, Gillespie SM, et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature. (2017) 549:533–7. doi: 10.1038/nature24014
58. Connor SA and Siddiqui TJ. Synapse organizers as molecular codes for synaptic plasticity. Trends Neurosci. (2023) 46:971–85. doi: 10.1016/j.tins.2023.08.001
59. Liu R, Qin XP, Zhuang Y, Zhang Y, Liao HB, Tang JC, et al. Glioblastoma recurrence correlates with nlgn 3 levels. Cancer Med. (2018) 7:2848–59. doi: 10.1002/cam4.1538
60. Wang Y, Y-y L, Chen M-b, Cheng K-W, Qi L-n, Zhang Z-q, et al. Neuronal-driven glioma growth requires Gαi1 and Gαi3. Theranostics. (2021) 11:8535. doi: 10.7150/thno.61452
61. Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell. (2015) 161:803–16. doi: 10.1016/j.cell.2015.04.012
62. Li X, Wu C, Chen N, Gu H, Yen A, Cao L, et al. Pi3k/akt/mtor signaling pathway and targeted therapy for glioblastoma. Oncotarget. (2016) 7:33440. doi: 10.18632/oncotarget.7961
63. Lundgren JL, Ahmed S, Schedin-Weiss S, Gouras GK, Winblad B, Tjernberg LO, et al. Adam10 and bace1 are localized to synaptic vesicles. J neurochem. (2015) 135:606–15. doi: 10.1111/jnc.13287
64. Smith TM Jr., Tharakan A, and Martin RK. Targeting adam10 in cancer and autoimmunity. Front Immunol. (2020) 11:499. doi: 10.3389/fimmu.2020.00499
65. Carroll P, Lewin GR, Koltzenburg M, Toyka KV, and Thoenen H. A role for bdnf in mechanosensation. Nat Neurosci. (1998) 1:42–6. doi: 10.1038/242
66. Colucci-D’Amato L, Speranza L, and Volpicelli F. Neurotrophic factor bdnf, physiological functions and therapeutic potential in depression, neurodegeneration and brain cancer. Int J Mol Sci. (2020) 21:7777. doi: 10.3390/ijms21207777
67. Xiong J, Zhou L, Lim Y, Yang M, Zhu Y-H, Li Z-W, et al. Mature brain-derived neurotrophic factor and its receptor trkb are upregulated in human glioma tissues. Oncol Lett. (2015) 10:223–7. doi: 10.3892/ol.2015.3181
68. Caldeira MV, Melo CV, Pereira DB, Carvalho R, Correia SS, Backos DS, et al. Brain-derived neurotrophic factor regulates the expression and synaptic delivery ofα-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem. (2007) 282:12619–28. doi: 10.1074/jbc.M700607200
69. Lu B. Bdnf and activity-dependent synaptic modulation. Learn Memory. (2003) 10:86–98. doi: 10.1101/lm.54603
70. Lu B, Nagappan G, and Lu Y. Bdnf and synaptic plasticity, cognitive function, and dysfunction. Handb. Exp. Pharmacol. (2014) 220:223–50. doi: 10.1007/978-3-642-45106-5_9
71. Bramham CR and Messaoudi E. Bdnf function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. (2005) 76:99–125. doi: 10.1016/j.pneurobio.2005.06.003
72. Kowiański P, Lietzau G, Czuba E, Waśkow M, Steliga A, and Moryś J. Bdnf: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol Neurobiol. (2018) 38:579–93. doi: 10.1007/s10571-017-0510-4
73. Langhnoja J, Buch L, and Pillai P. Potential role of ngf, bdnf, and their receptors in oligodendrocytes differentiation from neural stem cell: an in vitro study. Cell Biol Int. (2021) 45:432–46. doi: 10.1002/cbin.11500
74. Langhnoja J, Buch L, Chruvattil R, Gupta S, and Pillai P. Insulin receptor regulates neurotrophin and neurotrophin receptor expression in the differentiation of neural stem cells: in vitro study. J Biochem Mol Toxicol. (2025) 39:e70198. doi: 10.1002/jbt.70198
75. Gong Y, Ma Y, Sinyuk M, Loganathan S, Thompson RC, Sarkaria JN, et al. Insulin-mediated signaling promotes proliferation and survival of glioblastoma through akt activation. Neuro-oncology. (2015) 18:48–57. doi: 10.1093/neuonc/nov096
76. Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. (2015) 528:93–8. doi: 10.1038/nature16071
77. Jung E, Osswald M, Blaes J, Wiestler B, Sahm F, Schmenger T, et al. Tweety-homolog 1 drives brain colonization of gliomas. J Neurosci. (2017) 37:6837–50. doi: 10.1523/jneurosci.3532-16.2017
78. Numan T, Breedt LC, Maciel B, Kulik SD, Derks J, Schoonheim MM, et al. Regional healthy brain activity, glioma occurrence and symptomatology. Brain. (2022) 145:3654–65. doi: 10.1093/brain/awac180
79. Hsieh AL, Ganesh S, Kula T, Irshad M, Ferenczi EA, Wang W, et al. Widespread neuroanatomical integration and distinct electrophysiological properties of glioma-innervating neurons. Proc Natl Acad Sci. (2024) 121:e2417420121. doi: 10.1073/pnas.2417420121
80. Habermacher C, Angulo MC, and Benamer N. Glutamate versus gaba in neuron-oligodendroglia communication. Glia. (2019) 67:2092–106. doi: 10.1002/glia.23618
81. Venkataramani V, Schneider M, Giordano FA, Kuner T, Wick W, Herrlinger U, et al. Disconnecting multicellular networks in brain tumours. Nat Rev Cancer. (2022) 22:481–91. doi: 10.1038/s41568-022-00475-0
82. Xie R, Kessler T, Grosch J, Hai L, Venkataramani V, Huang L, et al. Tumor cell network integration in glioma represents a stemness feature. Neuro-oncology. (2021) 23:757–69. doi: 10.1093/neuonc/noaa275
83. Huang-Hobbs E, Cheng Y-T, Ko Y, Luna-Figueroa E, Lozzi B, Taylor KR, et al. Remote neuronal activity drives glioma progression through sema4f. Nature. (2023) 619:844–50. doi: 10.1038/s41586-023-06267-2
84. Armendáriz BG, Bribian A, Pérez-Martínez E, Martínez A, de Castro F, Soriano E, et al. Expression of semaphorin 4f in neurons and brain oligodendrocytes and the regulation of oligodendrocyte precursor migration in the optic nerve. Mol Cell Neurosci. (2012) 49:54–67. doi: 10.1016/j.mcn.2011.09.003
85. Carulli D, de Winter F, and Verhaagen J. Semaphorins in adult nervous system plasticity and disease. Front Synaptic Neurosci. (2021) 13. doi: 10.3389/fnsyn.2021.672891
86. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
87. Drexler R, Drinnenberg A, Gavish A, Yalçin B, Shamardani K, Rogers AE, et al. Cholinergic neuronal activity promotes diffuse midline glioma growth through muscarinic signaling. Cell. (2025) 188(17):4640–57.e30. doi: 10.1101/2024.09.21.614235
88. Seker-Polat F, Pinarbasi Degirmenci N, Solaroglu I, and Bagci-Onder T. Tumor cell infiltration into the brain in glioblastoma: from mechanisms to clinical perspectives. Cancers. (2022) 14:443. doi: 10.3390/cancers14020443
89. Cuddapah VA, Robel S, Watkins S, and Sontheimer H. A neurocentric perspective on glioma invasion. Nat Rev Neurosci. (2014) 15:455–65. doi: 10.1038/nrn3765
90. Bernstein JJ and Woodard CA. Glioblastoma cells do not intravasate into blood vessels: 124. Neurosurgery. (1995) 36:124–32. doi: 10.1227/00006123-199501000-00016
91. Hide T and Komohara Y. Oligodendrocyte progenitor cells in the tumor microenvironment. Tumor Microenvironment: Non-Hematopoietic Cells. (2020) 1234:107–22. doi: 10.1007/978-3-030-37184-5_8
93. Aboitiz F, Scheibel AB, Fisher RS, and Zaidel E. Fiber composition of the human corpus callosum. Brain Res. (1992) 598:143–53. doi: 10.1016/0006-8993(92)90178-C
94. Dadario NB, Zaman A, Pandya M, Dlouhy BJ, Gunawardena MP, Sughrue ME, et al. Endoscopic-assisted surgical approach for butterfly glioma surgery. J neuro-oncol. (2022) 156:635–44. doi: 10.1007/s11060-022-03945-5
95. Fields RD. White matter in learning, cognition and psychiatric disorders. Trends Neurosci. (2008) 31:361–70. doi: 10.1016/j.tins.2008.04.001
96. Giese A, Kluwe L, Laube B, Meissner H, Berens ME, and Westphal M. Migration of human glioma cells on myelin. Neurosurgery. (1996) 38:755–64. doi: 10.1227/00006123-199604000-00026
97. Iwadate Y, Fukuda K, Matsutani T, and Saeki N. Intrinsic protective mechanisms of the neuron-glia network against glioma invasion. J Clin Neurosci. (2016) 26:19–25. doi: 10.1016/j.jocn.2015.07.024
98. Sandvig A, Berry M, Barrett LB, Butt A, and Logan A. Myelin-, reactive glia-, and scar-derived cns axon growth inhibitors: expression, receptor signaling, and correlation with axon regeneration. Glia. (2004) 46:225–51. doi: 10.1002/glia.10315
99. Venkataramani V, Yang Y, Schubert MC, Reyhan E, Tetzlaff SK, Wißmann N, et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell. (2022) 185:2899–917. doi: 10.1016/j.cell.2022.06.054
100. Liu Y, Carlsson R, Ambjørn M, Hasan M, Badn W, Darabi A, et al. Pd-L1 expression by neurons nearby tumors indicates better prognosis in glioblastoma patients. J Neurosci. (2013) 33:14231–45. doi: 10.1523/jneurosci.5812-12.2013
101. Romão LF, Mendes FA, Feitosa NM, Faria JC, Coelho-Aguiar JM, de Souza JM, et al. Connective tissue growth factor (Ctgf/ccn2) is negatively regulated during neuron-glioblastoma interaction. PloS One. (2013) 8:e55605. doi: 10.1371/journal.pone.0055605
102. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. nature. (2004) 432:396–401. doi: 10.1038/nature03128
103. Biserova K, Jakovlevs A, Uljanovs R, and Strumfa I. Cancer stem cells: significance in origin, pathogenesis and treatment of glioblastoma. Cells. (2021) 10:621. doi: 10.3390/cells10030621
104. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, and Rich JN. Cancer stem cells in glioblastoma. Genes Dev. (2015) 29:1203–17. doi: 10.1101/gad.261982.115
105. Sanai N, Tramontin AD, Quinones-Hinojosa A, Barbaro NM, Gupta N, Kunwar S, et al. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature. (2004) 427:740–4. doi: 10.1038/nature02301
106. Llaguno SA, Chen J, Kwon C-H, Jackson EL, Li Y, Burns DK, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. (2009) 15:45–56. doi: 10.1016/j.ccr.2008.12.006
107. Laug D, Glasgow SM, and Deneen B. A glial blueprint for gliomagenesis. . Nat Rev Neurosci. (2018) 19:393–403. doi: 10.1038/s41583-018-0014-3
108. Moyon S, Holloman M, and Salzer JL. Neural stem cells and oligodendrocyte progenitor cells compete for remyelination in the corpus callosum. Front Cell Neurosci. (2023) 17:1114781. doi: 10.3389/fncel.2023.1114781
109. Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and Malignant glioma. Neuron. (2007) 53:503–17. doi: 10.1016/j.neuron.2007.01.009
110. Hide T, Komohara Y, Miyasato Y, Nakamura H, Makino K, Takeya M, et al. Oligodendrocyte progenitor cells and macrophages/microglia produce glioma stem cell niches at the tumor border. eBioMedicine. (2018) 30:94–104. doi: 10.1016/j.ebiom.2018.02.024
111. Hide T, Shibahara I, and Kumabe T. Novel concept of the border niche: glioblastoma cells use oligodendrocytes progenitor cells (Gaos) and microglia to acquire stem cell-like features. Brain tumor Pathol. (2019) 36:63–73. doi: 10.1007/s10014-019-00341-2
112. Guo X, Qiu W, Wang C, Qi Y, Li B, Wang S, et al. Neuronal activity promotes glioma progression by inducing proneural-to-mesenchymal transition in glioma stem cells. Cancer Res. (2024) 84:372–87. doi: 10.1158/0008-5472.CAN-23-0609
113. Lipi B, Jaldeep L, and Prakash P. Role of astrocytic mecp2 in regulation of cns myelination by affecting oligodendrocyte and neuronal physiology and axo–glial interactions. Exp Brain Res. (2018) 236:3015–27. doi: 10.1007/s00221-018-5363-7
114. Borst K, Dumas AA, and Prinz M. Microglia: immune and non-immune functions. Immunity. (2021) 54:2194–208. doi: 10.1016/j.immuni.2021.09.014
115. Kalafatakis I and Karagogeos D. Oligodendrocytes and microglia: key players in myelin development, damage and repair. Biomolecules. (2021) 11:1058. doi: 10.3390/biom11071058
116. Guedes JR, Ferreira PA, Costa JM, Cardoso AL, and Peça J. Microglia-dependent remodeling of neuronal circuits. J Neurochem. (2022) 163:74–93. doi: 10.1111/jnc.15689
117. Guo X, Pan Y, Xiong M, Sanapala S, Anastasaki C, Cobb O, et al. Midkine activation of cd8+ T cells establishes a neuron–immune–cancer axis responsible for low-grade glioma growth. Nat Commun. (2020) 11:2177. doi: 10.1038/s41467-020-15770-3
118. Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, et al. Down-regulation of the macrophage lineage through interaction with ox2 (Cd200). Science. (2000) 290:1768–71. doi: 10.1126/science.290.5497.1768
119. Majed HH, Chandran S, Niclou SP, Nicholas RS, Wilkins A, Wing MG, et al. A novel role for sema3a in neuroprotection from injury mediated by activated microglia. J Neurosci. (2006) 26:1730–8. doi: 10.1523/JNEUROSCI.0702-05.2006
120. Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and cx3cr1-expressing microglia. Proc Natl Acad Sci. (1998) 95:10896–901. doi: 10.1073/pnas.95.18.10896
121. Aabedi AA, Lipkin B, Kaur J, Kakaizada S, Valdivia C, Reihl S, et al. Functional alterations in cortical processing of speech in glioma-infiltrated cortex. Proc Natl Acad Sci U.S.A. (2021) 118. doi: 10.1073/pnas.2108959118
122. Campbell SL, Buckingham SC, and Sontheimer H. Human glioma cells induce hyperexcitability in cortical networks. Epilepsia. (2012) 53:1360–70. doi: 10.1111/j.1528-1167.2012.03557.x
123. Krishna S, Choudhury A, Keough MB, Seo K, Ni L, Kakaizada S, et al. Glioblastoma remodelling of human neural circuits decreases survival. Nature. (2023) 617:599–607. doi: 10.1038/s41586-023-06036-1
124. van Breemen MS, Wilms EB, and Vecht CJ. Epilepsy in patients with brain tumours: epidemiology, mechanisms, and management. Lancet Neurol. (2007) 6:421–30. doi: 10.1016/S1474-4422(07)70103-5
125. Zhang Y, Duan W, Chen L, Chen J, Xu W, Fan Q, et al. Potassium ion channel modulation at cancer-neural interface enhances neuronal excitability in epileptogenic glioblastoma multiforme. Neuron. (2024) 113(2):225–43.e10. doi: 10.1016/j.neuron.2024.10.016
126. Roslin M, Henriksson R, Bergström P, Ungerstedt U, and Tommy Bergenheim A. Baseline levels of glucose metabolites, glutamate and glycerol in Malignant glioma assessed by stereotactic microdialysis. J neuro-oncol. (2003) 61:151–60. doi: 10.1023/A:1022106910017
127. Buckingham SC, Campbell SL, Haas BR, Montana V, Robel S, Ogunrinu T, et al. Glutamate release by primary brain tumors induces epileptic activity. Nat Med. (2011) 17:1269–74. doi: 10.1038/nm.2453
129. Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. (2011) 146:209–21. doi: 10.1016/j.cell.2011.06.014
130. Fuchs E and Segre JA. Stem cells: A new lease on life. Cell. (2000) 100:143–55. doi: 10.1016/s0092-8674(00)81691-8
131. Xiong J, Zhou L, Lim Y, Yang M, Zhu Y-H, Li Z-W, et al. Mature bdnf promotes the growth of glioma cells in vitro. Oncol Rep. (2013) 30:2719–24. doi: 10.3892/or.2013.2746
132. Su R, Ma J, Zheng J, Liu X, Liu Y, Ruan X, et al. Pabpc1-induced stabilization of bdnf-as inhibits Malignant progression of glioblastoma cells through stau1-mediated decay. Cell Death Dis. (2020) 11:81. doi: 10.1038/s41419-020-2267-9
133. Huang Q, Chen L, Liang J, Huang Q, and Sun H. Neurotransmitters: potential targets in glioblastoma. Cancers. (2022) 14:3970. doi: 10.3390/cancers14163970
134. Blanchart A, Fernando R, Häring M, Assaife-Lopes N, Romanov RA, Andäng M, et al. Endogenous gabaa receptor activity suppresses glioma growth. Oncogene. (2017) 36:777–86. doi: 10.1038/onc.2016.245
135. D'Urso PI, D'Urso OF, Storelli C, Mallardo M, Gianfreda CD, Montinaro A, et al. Mir-155 is up-regulated in primary and secondary glioblastoma and promotes tumour growth by inhibiting gaba receptors. Int J Oncol. (2012) 41:228–34. doi: 10.3892/ijo.2012.1420
136. Parmigiani E, Scalera M, Mori E, Tantillo E, and Vannini E. Old stars and new players in the brain tumor microenvironment. Front Cell Neurosci. (2021) 15:709917. doi: 10.3389/fncel.2021.709917
137. Hua T, Shi H, Zhu M, Chen C, Su Y, Wen S, et al. Glioma−Neuronal interactions in tumor progression: mechanism, therapeutic strategies and perspectives (Review). Int J Oncol. (2022) 61:104. doi: 10.3892/ijo.2022.5394
138. Niu X, Zhang Y, and Wang Y. Co-culture models for investigating cellular crosstalk in the glioma microenvironment. Cancer Pathogenesis Ther. (2024) 2:219–30. doi: 10.1016/j.cpt.2023.11.002
139. Bai L, Hao Z, Wang S, Zhou J, Yao S, Pei N, et al. Biomimetic three-dimensional glioma model printed in vitro for the studies of glioma cells and neurons interactions. Int J Bioprinting. (2023) 9:715. doi: 10.18063/ijb.715
140. Krieger TG, Tirier SM, Park J, Jechow K, Eisemann T, Peterziel H, et al. Modeling glioblastoma invasion using human brain organoids and single-cell transcriptomics. Neuro-oncology. (2020) 22:1138–49. doi: 10.1093/neuonc/noaa091
141. Sun Y, Wang X, Zhang Z, Park KH, Wu Y, Dong W, et al. Cholinergic neuron-to-glioblastoma synapses in a human ipsc-derived co-culture model. Stem Cell Rep. 20(7):102534 doi: 10.1016/j.stemcr.2025.102534
142. Rodriguez FJ, Vizcaino MA, and Lin M-T. Recent advances on the molecular pathology of glial neoplasms in children and adults. J Mol diagnostics. (2016) 18:620–34. doi: 10.1016/j.jmoldx.2016.05.005
143. Ostrom QT, Adel Fahmideh M, Cote DJ, Muskens IS, Schraw JM, Scheurer ME, et al. Risk factors for childhood and adult primary brain tumors. Neuro-oncology. (2019) 21:1357–75. doi: 10.1093/neuonc/noz123
144. Radin DP. Ampa receptor modulation in the treatment of high-grade glioma: translating good science into better outcomes. Pharmaceuticals. (2025) 18:384. doi: 10.3390/ph18030384
145. Leresche N and Lambert RC. T-type calcium channels in synaptic plasticity. Channels (Austin). (2017) 11:121–39. doi: 10.1080/19336950.2016.1238992
Keywords: glioma, neural mechanisms, neuroplasticity, neural networks, migration, glioma stem cell, tumorigenesis, tumor microenvironment
Citation: Feng J and Yang J (2025) Glioma-neuron interactions: insights from neural plasticity. Front. Oncol. 15:1661897. doi: 10.3389/fonc.2025.1661897
Received: 08 July 2025; Accepted: 29 August 2025;
Published: 11 September 2025.
Edited by:
Soma Sengupta, University of North Carolina at Chapel Hill, United StatesReviewed by:
Joshua John Breunig, Cedars Sinai Medical Center, United StatesJaldeep Langhnoja, University of Cincinnati, United States
Copyright © 2025 Feng and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Yang, MTM5MDEyOTEyMTFAMTYzLmNvbQ==