 Xiaoyang Ma
Xiaoyang Ma Xiaolin Yu1,2
Xiaolin Yu1,2- 1Department of Clinical Laboratory, Zigong Fourth People’s Hospital, Zigong, China
- 2Sichuan Vocational College of Health and Rehabilitation, Zigong, China
In tumors, extrachromosomal DNA (ecDNA) is an important driver of oncogene expression, genomic instability, the evolution of drug resistance, and poor patient prognosis. ecDNA is present in various tumors but is rarely found in normal cells. Here, we provide a detailed review of the structure, genetics, occurrence, outcomes, and functions of ecDNA, offering further reference for research on ecDNA.
1 Introduction
In 1965, Cox et al. (1) discovered a large number of small double chromatin bodies outside the chromosomes in five cases of pediatric embryonal tumors and one case of a rare type of adult bronchial carcinoma. In 1967, Radloff et al. (2) isolated and detected circular DNA of varying lengths in the HeLa cervical cancer cell line. The double chromatin bodies reported by Cox et al. (1) and the circular DNA reported by Radloff et al. (2) are both located extrachromosomally and are now collectively referred to as ecDNA. In 2024, Bailey et al. (3) analyzed whole-genome sequencing data from 14,778 tumor samples of 39 types and found that 17.1% of tumor samples contained ecDNA. The high detection rate of ecDNA in tumors has prompted extensive research to elucidate the role of ecDNA in tumorigenesis and progression. This article will comprehensively discuss the formation mechanisms, biological structure, genetic patterns, and functions of ecDNA in tumors, as well as its potential clinical applications, providing guidance for further research on ecDNA.
2 Structural and inheritance dynamics of ecDNA
2.1 Physical and functional architecture
Approximately 30% of ecDNA exist in pairs within the nucleus, and thus, for a long time, they were referred to as double minutes (4). ecDNA has a complex structure, lacks centromeres, and can originate from multiple chromosomes (5). The frequency, copy number, and size of ecDNA vary greatly among different tumors (3). The prevalence of ecDNA across various cancers is shown in Figure 1. Typically, ecDNA exists in a circular form (4). However, ecDNA is distinct from extrachromosomal circular DNA (eccDNA). eccDNA has a small molecular weight (<1 kb), does not undergo amplification, does not contain complete gene sequences or regulatory elements, does not carry mutated genes, and can appear in normal tissues (5–11). ecDNA has a large molecular weight (>100 kb), undergoes clonal selection, possesses self-replication and amplification capabilities, can contain oncogenes, regulatory elements, recombinant genes, and mutated genes, and is rare in normal tissues (5, 6, 9–12).
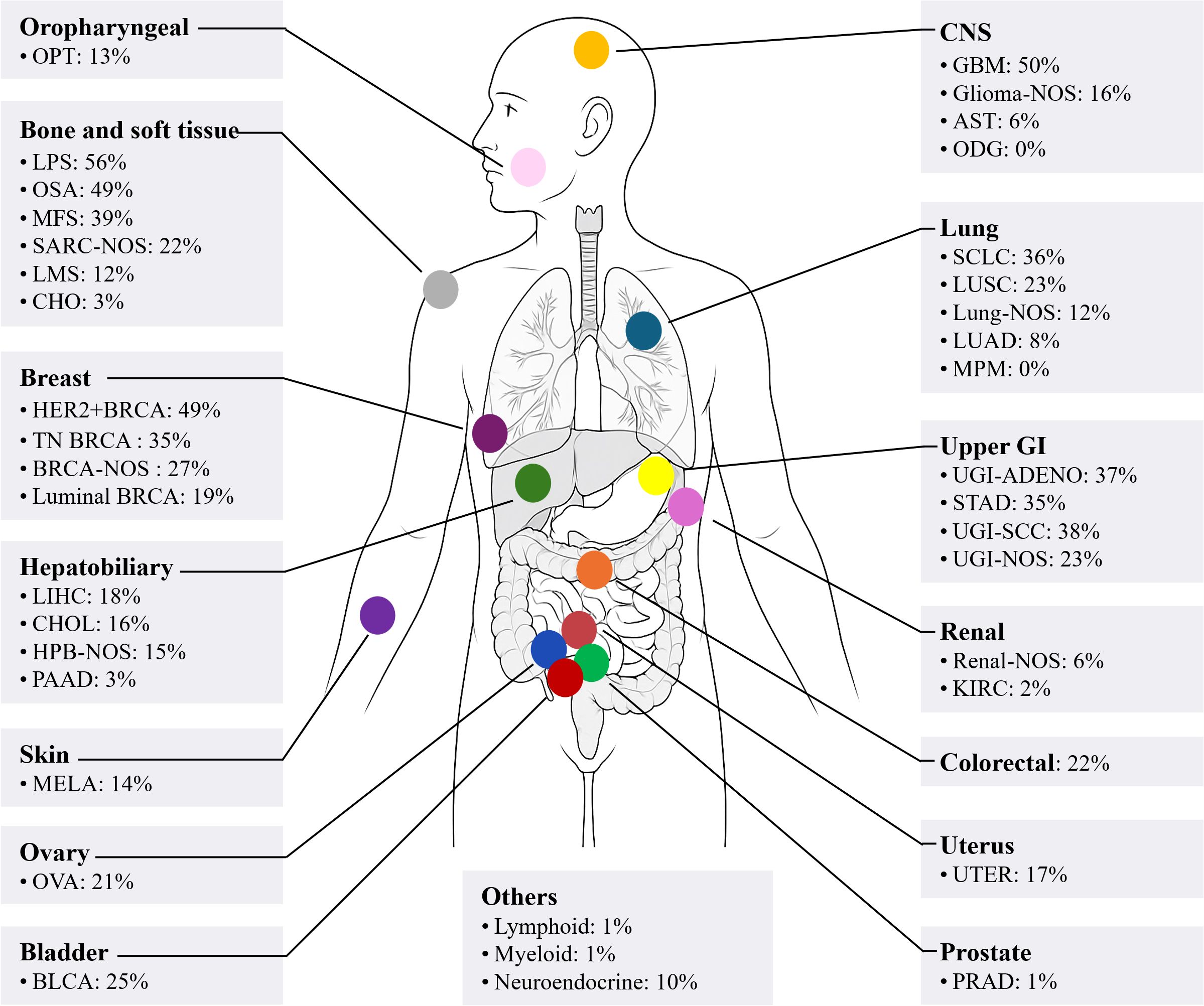
Figure 1. A body map of the prevalence of ecDNA in various cancers. OPT, oropharyngeal tumour; LPS, liposarcoma; OSA, primary conventional osteosarcoma; MFS, myxofibrosarcoma; SARC, sarcoma; NOS, not-otherwise specified; LMS, leiomyosarcoma; CHO, chordoma; BRCA, breast cancer; TN, triple negative; LIHC, liver hepatocellular carcinoma; CHOL, cholangiocarcinoma; HPB, hepatopancreatobiliary cancer; PAAD, pancreatic adenocarcinoma; MELA, malignant melanoma; OVA, ovarian cancer; BLCA, bladder cancer; CNS, central nervous system; GBM, glioblastoma; AST, astrocytoma; ODG, oligodendroglioma; SCLC, small cell lung cancer; LUSC, lung squamous cell carcinoma; LUAD, lung adenocarcinoma; MPM, malignant pleural mesothelioma; GI, gastrointestinal; UGI, upper gastrointestinal; ADENO, adenocarcinoma; STAD, stomach adenocarcinoma; SCC, squamous cell carcinoma; KIRC, clear cell renal cell carcinoma; UTER, endometrial cancer; PRAD, prostate adenocarcinoma. The data were extracted using Getdata Graph Digitizer (https://getdata-graph-digitizer.com/) from Bailey et al. (3), 2024, on June 6, 2025.
ecDNA can carry a variety of common oncogenes, such as MYC, MYCN, Jun, KRAS, MYCL, MDM2, epidermal growth factor receptor (EGFR), fibroblast growth factor receptor 2 (FGFR2), platelet derived growth factor receptor alpha (PDGFRA), erb-b2 receptor tyrosine kinase 2 (ERBB2), and cyclin-dependent kinase 4 (CDK4), among others (13, 14). Even when carrying the same oncogene, ecDNA can exhibit substantial variability in both size and sequence composition (15). ecDNA with distinct sequence architectures are referred to as ecDNA species. ecDNA can simultaneously carry multiple oncogenes (13), and share adjacent regulatory regions (16). In addition, there exists a class of ecDNA that does not carry oncogenes but only carries promoters, enhancers, or long noncoding RNA (lncRNA) regulatory elements, referred to as regulatory ecDNA (3). Compared to ecDNA carrying oncogenes, regulatory ecDNA have a simpler structure, smaller size, and lower copy number (3). Whole-genome sequencing of human papillomavirus mediated oropharyngeal cancer (HPVOPC) revealed that HPVOPC contains ecDNA composed of host genome and HPV-host genome hybrids, and both types of ecDNA can carry multiple oncogenes (17). Additionally, ecDNA can also carry immune regulatory genes and inflammation-related genes (3, 13). Gene function enrichment analysis confirmed that genes carried by ecDNA are often upregulated in biological processes such as cell cycle, cell division, and DNA damage, and downregulated in processes related to the immune system (18).
2.2 Non-Mendelian segregation mechanisms
During mitosis, the spindle apparatus pulls the centromeres of chromosomes, guiding their alignment and equal segregation to ensure that daughter cells have identical chromosomal DNA. Multiple studies have shown that the copy number of ecDNA among tumor cells exhibits significant heterogeneity (19, 20). ecDNA does not appear to follow Mendelian inheritance during cell division, differing from chromosomal inheritance patterns (21). FISH-based methods combined with unbiased image analysis have shown that after mitosis in multiple tumor cell lines, the number of ecDNA in daughter cells follows a Gaussian distribution, and the segregation process is independent of tumor type and ecDNA species (22). Subsequently, CRISPR-based ecDNA tagging with live-cell imaging was used to dynamically track ecDNA during the cell cycle, further confirming that ecDNA undergoes random segregation during cell division (22, 23). Analysis of The Cancer Genome Atlas Program (TCGA) database showed that more than 25% of ecDNA-containing (ecDNA+) tumors contain more than two types of ecDNA, and different ecDNA species coexist at copy numbers much higher than expected by chance (14). Therefore, when cells contain multiple ecDNA species, their segregation may not be completely independent and random. Recent studies have shown that when tumor cells contain multiple ecDNA species, cooperative ecDNA species are coordinately inherited through mitotic co-segregation (14). In summary, during cell division, a single type of ecDNA segregates into daughter cells in a binomial random manner, while multiple coexisting ecDNA species can be co-segregated into daughter cells (Figure 2).
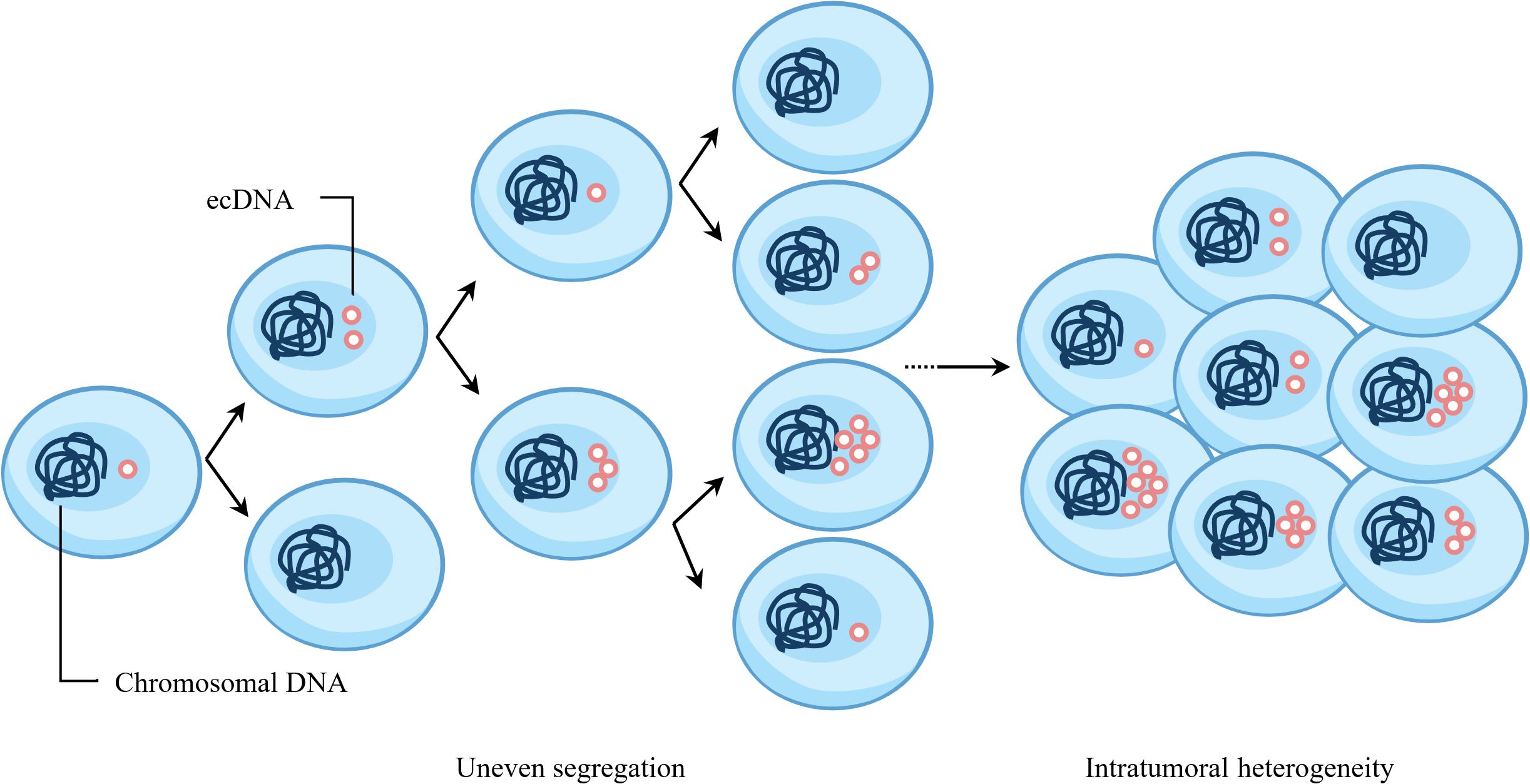
Figure 2. Random segregation of ecDNA promotes intratumoral heterogeneity of cancer. ecDNA does not follow Mendelian inheritance and undergoes random segregation during cell division. After multiple rounds of cell division, the copy number of ecDNA in cells will exhibit significant heterogeneity.
3 Formation of ecDNA
Since the discovery of ecDNA, researchers have been committed to exploring its origin and formation mechanisms. Analysis of single nucleotide variant (SNV) frequency has shown that ecDNA and chromosomal DNA are haplotypically distinct, providing evidence for the hypothesis that ecDNA originates from chromosomes (24). Currently, several models have been proposed to explain the formation of ecDNA, such as the excisional model, breakage-fusion-bridge (BFB) cycle, translocation-bridge amplification (TB amplification), and chromothripsis. Below, we discuss these models in detail (Figure 3).
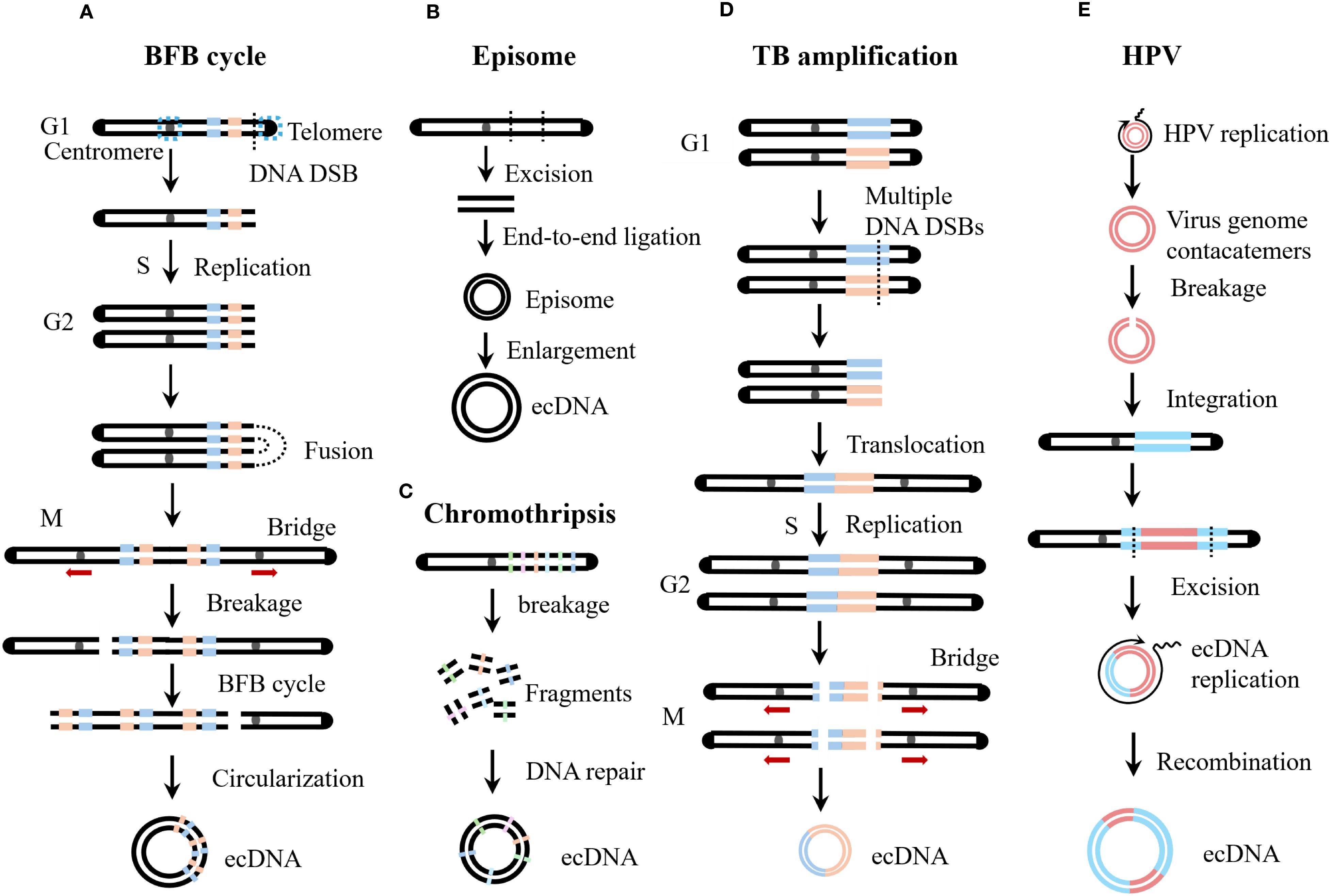
Figure 3. Biogenesis of ecDNA. (A) Breakage-Fusion-Bridge (BFB) cycles start during a bridge formation (usually between sister chromatids) as a stabilizing repair intermediate for DNA double-strand break (DSB). Unequal mitotic separation and breakage of the bridged chromosomes creates an inverted duplication on one chromosome, and a deletion on the other. The broken end result in continued BFB cycles until the telomere is re-capped. (B) The episome model is also known as the excisional model. Episomes are derived from excisied DNA fragments, and they can enlarge to form ecDNA by over-replication or recombination. The excisional model is divided into the scarring excisional model and the scarless excisional model. (C) Chromothripsis generates multiple chromosomal fragments via a single catastrophic event. These fragments may undergo misrepair and illegitimate reassembly, driving either massive chromosomal rearrangements or ecDNA formation. (D) Translocation-bridge (TB) amplification occurs in the G1 phase. Inter-chromosomal translocation directly creates the dicentric chromosome. During mitosis, dicentric chromosomes separate to form chromosomal bridges, which break and circularize to form ecDNA. (E) Human papillomavirus (HPV) integration drives host genome amplification and structural rearrangement, resulting in virus-host tandem DNA formation. Subsequent excision of viral DNA by host cells generates hybrid virus-host ecDNA.
3.1 Excisional model
The excisional model, also known as the episome model, is a simple hypothesis regarding the origin of ecDNA. The excisional model posits that ecDNA originates from double-stranded DNA breaks on chromosomes, so the resulting ecDNA usually have simple structures and low diversity (25). As early as 1988, researchers confirmed that episomes can be directly formed from deleted chromosomal fragments (26). Subsequently, a case of acute myeloid leukemia was reported in which the leukemic cells contained double minutes carrying the MYC gene, and a chromosome 8 with a deletion in the MYC region was also present, suggesting that the MYC in the double minutes originated from chromosome 8 (27). Through next-generation sequencing, single nucleotide polymorphism array, fluorescent in situ hybridization, and polymerase chain reaction-based techniques, the genomic structure and evolutionary mechanisms of seven MYC ecDNA+ tumor cell lines were analyzed, revealing that ecDNA are gradually formed through multiple steps such as amplification, recombination, and deletion of ancestral episomes from a single chromosome (28). Chromosome conformation capture (Hi-C) analysis showed that spatial proximity is not required for the generation of ecDNA, and long-distance double-stranded breaks can still efficiently form ecDNA (29). In addition, CRISPR-C technology can be used to artificially cut specific genes to construct ecDNA+ cell models (12, 22).
According to whether the chromosome can be accurately repaired after DNA fragment excision, the excisional model is divided into the scarring excisional model and the scarless excisional model. In the scarring excisional model, DNA fragment excision occurs before DNA replication, and the organism connects the chromosomal break ends through nonhomologous end joining (NHEJ) (25). In the scarless excisional model, DNA fragments are usually formed by replication fork breakage, and the missing part of the chromosome is precisely repaired through a homologous recombination-dependent DNA replication process using the normal sister chromatid as a template (25). Deep sequencing of the junctions between ecDNA and chromosomal excision scars revealed that the formation of ecDNA and chromosomal scars is independent, with NHEJ predominantly repairing chromosomal scars, while microhomology-mediated end joining (MMEJ) is more common in ecDNA circularization (29). However, some studies have shown that the breakpoints of circular amplicons often have no sequence homology or only minimal sequence homology (<5 bp), suggesting that NHEJ is the main mechanism for ecDNA formation (30).
3.2 Breakage-fusion-bridge cycle
BFB cycles were originally described by Barbara McClintock in 1939 for the fate of a dicentric chromosome during meiotic mitosis and endosperm development in maize (31). BFB events are common in tumors and are a frequent cause of increased oncogene copy number (32, 33). The BFB cycle begins with telomere loss, and chromosomes lacking telomeres or sister chromatids of telomere-deficient chromosomes after replication fuse to form dicentric chromosomes (34). During cell division, dicentric chromosomes are pulled in opposite directions by spindle fibers, forming chromosomal bridges (35). Chromosomal bridges break under mechanical tension during cell division, generating new telomere-deficient chromosomes and broken chromosomal fragments (34). In the absence of telomeres, BFB will continue to occur in subsequent generations of cells until telomeres are restored (32). Studies have shown that there is a strong overlap between oncogenes amplified by BFB cycles and those amplified by ecDNA, suggesting that chromosomal bridge fragments generated by the BFB cycle can circularize to form ecDNA (32).
3.3 Translocation-bridge amplification
Chromosomal translocations can also form dicentric chromosomes. During cell division, dicentric chromosomes separate to form chromosomal bridges, which break and circularize to form ecDNA (36). TB amplification elucidates the amplification mechanism of key oncogenes ERBB2 and cyclin D1 (CCND1) in breast cancer (36). Chromosomal translocation is the most common cause of chromosomal bridge formation in tumors, and tumors with TB amplification often exhibit loss of heterozygosity (LOH) on the bridge arm (36). In the TB amplification model, dual-LOH occurs on two chromosomal arms, so chromosomal translocation in TB amplification occurs in the G1 phase (36).
3.4 Chromothripsis
Chromothripsis refers to the occurrence of a large number of random breaks in one or several chromosomes within a short period (37). Chromosomal shattering does not occur through the accumulation of gene mutations but is a single catastrophic genomic event (38). Fanconi anaemia (FA), a model syndrome of genome instability, is caused by a deficiency in DNA interstrand crosslink repair resulting in chromosome breakage (39). Studies have shown that the FA pathway is a driving factor for chromothripsis, with the core FA complex monoubiquitinating and activating FANCI-FANCD2, which recruits the SLX4-XPF-ERCC1 endonuclease to cleave micronuclear chromosomes, triggering large-scale chromosomal shattering (40). The shattered chromosomal fragments undergo erroneous joining and assembly, leading to extensive chromosomal rearrangement or the formation of ecDNA (41). Chromothripsis is widespread in tumors, with an incidence exceeding 40% in glioblastoma, lung adenocarcinoma, osteosarcoma, and liposarcoma (42). Additionally, approximately 50% of cases with circular amplicons exhibit chromothripsis (30, 43). Some researchers also believe that chromothripsis may arise through TREX1-mediated fragmentation of dicentric chromosomes formed in telomere crisis (44). BFB cycles and chromothripsis are hallmarks of telomere crisis (45). Therefore, chromothripsis and BFB cycles may share a common origin and coexist and promote each other in the formation of ecDNA (30, 43).
4 Viral integration and ecDNA biogenesis
HPV is a small, non-enveloped virus with a circular double-stranded DNA genome, and more than 200 genotypes have been identified to date (46, 47). According to its carcinogenicity, HPV is classified into low-risk and high-risk types (48). High-risk HPV infection can lead to malignant tumors such as cervical cancer, vaginal cancer, penile cancer, anal cancer, oropharyngeal cancer, and head and neck cancer (49). After HPV infection, integration of its genome into the host chromatin is a characteristic step in cellular carcinogenesis, ensuring constitutive expression of the E6/E7 oncogenes (50, 51). As mentioned above, HPVOPC contains virus-host hybrid ecDNA (17, 52). Does HPV play a specific role in the formation of virus-host hybrid ecDNA? Studies have shown that HPV integration can mediate amplification and rearrangement of the host genome, altering the local chromosomal structure and forming virus-host tandem DNA (53–55). Subsequently, researchers found that this tandem DNA sequence in various HPV-related tumors exhibits repetitive, diverse, and interrelated structural features, which were named “heterocateny” (56). Heterocateny is driven by the HPV genome, exists both intrachromosomally and extrachromosomally, and its formation process is as follows (1): HPV replication forms unstable viral genome concatemers (2); viral genome concatemers integrate into host DNA (3); host cells excise viral DNA, forming virus-host hybrid ecDNA (4); virus-host ecDNA undergo replication, amplification, and rearrangement, forming diverse ecDNA (5); virus-host ecDNA can recombine into chromosomes again, and undergo further excision, circularization, replication, amplification, and rearrangement, resulting in even more diverse ecDNA (56). In summary, HPV integration into the host genome can drive the formation of virus-host ecDNA, which are variable in size and structurally diverse (57).
5 ecDNA promotes oncogene expression
Pan-cancer analysis shows that oncogenes encoded by ecDNA have the highest expression levels in tumor transcriptomes (15). In fact, the expression levels of EGFR, MYC, CDK4, and MDM2 genes commonly carried by ecDNA rank in the top 1% of tumor genomes (15). Increased gene expression usually involves increased gene copy number and altered transcriptional regulation. Below, we discuss in detail the mechanisms by which ecDNA promotes increased oncogene expression (Figure 4).
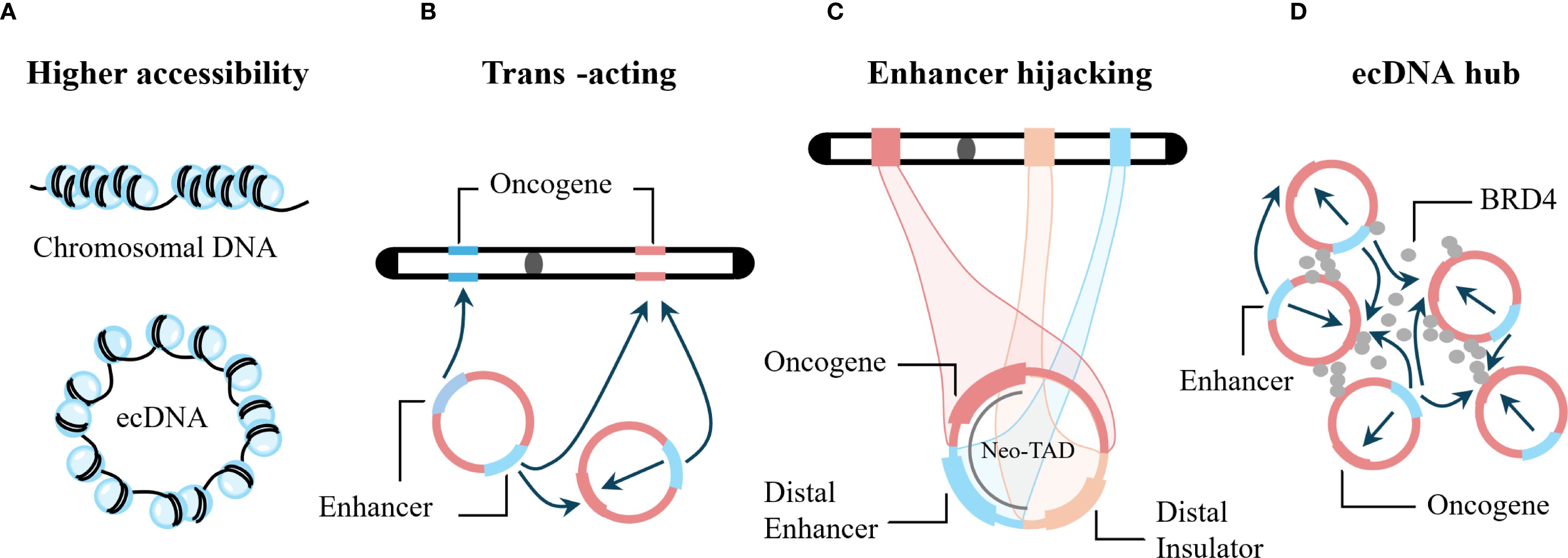
Figure 4. Examples of how ecDNA can promote oncogene expression. (A) The nucleosome structure of ecDNA has higher chromatin accessibility, enabling increased transcription. (B) ecDNA functions as a trans-acting mobile element that enhances gene expression from both chromosomal loci and other ecDNA molecules. (C) ecDNA hijacks both local and distal chromosomal enhancers to elevate oncogene expression. (D) ecDNAs assemble into transcriptional hubs that amplify gene transcription across proximal ecDNA molecules through shared regulatory elements, including enhancers.
5.1 Amplification of ecDNA copy number
In tumors, gene amplification is a measure to antagonize anti-cancer treatments by directly increasing the dosage of target proteins (58), or by activating an alternative cell-proliferation pathway (59). More commonly, genes promoting tumorigenesis and progression employ gene amplification to increase their protein level (60). Gene amplification occurs in two forms: linear amplification and circular amplification, manifested as homogeneously staining regions (HSR) and ecDNA, respectively (61). Analysis of 8,068 circularly amplified genes and 6,247 linearly amplified genes in 77 tumor samples showed that the copy number of circularly amplified genes was significantly higher than that of linearly amplified genes (15). Therefore, increased copy number of oncogenes carried by ecDNA is one of the main mechanisms for increased gene expression. In addition, engineered ecDNA can also spontaneously accumulate in primary cells and promote cell proliferation, transformation, immortalization, and drive tumor formation (62).
5.2 ecDNA exhibits high transcriptional activity
ecDNA can carry multiple regulatory regions, resulting in a non-linear relationship between transcriptional output and gene copy number. Transcription of ecDNA is very common (63). After normalizing the copy number of oncogenes carried by ecDNA and chromosomal oncogenes, oncogenes on ecDNA still produce more transcripts, significantly higher than those on chromosomes (15, 30). Studies have shown that ecDNA can promote oncogene transcription through non-copy number-dependent mechanisms such as increased chromatin accessibility and enhancer hijacking (64). In addition, as the copy number of ecDNA increases, the transcription level of genes carried by ecDNA also increases accordingly (20). Below, we discuss in detail the mechanisms underlying the high transcriptional activity of ecDNA.
5.2.1 High chromatin accessibility of ecDNA
Chromatin accessibility refers to the physical contact permissibility of nuclear macromolecules with chromatinized DNA, which is mainly determined by the distribution and occupancy of nucleosomes, as well as other DNA-binding factors (65, 66). Accessible chromatin is a hallmark of active DNA regulatory elements (67). The accessible regions comprise only ~2-3% of the whole genome, and more than 90% of these regions are yet to be captured by transcription factors (68). ecDNA is also composed of nucleosome units and possesses chromatin structural features (15). Assay for transposase-accessible chromatin using sequencing (ATAC-seq) and transposase-accessible chromatin with visualization (ATAC-see) experiments have shown that the nucleosome structure of ecDNA is more loosely assembled, lacks higher-order compaction, and has higher chromatin accessibility (15, 64).
5.2.2 ecDNA as mobile enhancers
Gene expression is regulated by genomic enhancers that recruit transcription factors and cofactors to activate transcription from target core-promoters (69). In the HMS001 human papillomavirus-associated oropharyngeal cancer cell line, HPV can integrate into host enhancer regions, forming ecDNA containing enhancer-E6/E7 promoter complexes, and CRISPR interference with this enhancer can reduce E6/E7 expression (52). Therefore, ecDNA can enhance the expression of its own carried oncogenes. Does ecDNA also affect genes at other loci? Studies using artificial enhancer ecDNA to transfect the PC3 prostate cancer cell line (ecDNA−) showed that this ecDNA triggered genome-wide chromosomal gene transcriptional activation in PC3 cells (70). In addition, in cervical cancer cell lines containing HPV virus-host ecDNA with super-enhancers, these super-enhancers can promote strong and extensive intra- and inter-chromosomal interactions (71). The above studies indicate that ecDNA can act as mobile enhancers, facilitating extensive internal interactions and genome-wide chromosomal interactions, thereby broadly promoting gene expression (15, 70).
5.2.3 Enhancer hijacking
Genes can hijack distal enhancers to compensate for the loss of local gene regulatory elements, thereby enhancing gene expression, a phenomenon known as enhancer hijacking (16). Enhancer hijacking can form new topological domains and is an effective mechanism for driving oncogene expression (72, 73). Studies have shown that in the CHP-212 neuroblastoma cell line, MYCN on ecDNA can hijack the enhancer element of the tribbles pseudokinase 2 (TRIB2) gene (16), CDX2 on ecDNA in COLO320-DM cells can hijack the MYC enhancer (74), and in SNU16 cells, MYC, FGFR2, CD44, and pyruvate dehydrogenase complex component X (PDHX) on ecDNA can mutually hijack enhancers (74). In addition, in hematological tumors, ecDNA can also promote oncogene transcription through enhancer hijacking (64). The above studies indicate that enhancer hijacking is common on ecDNA and is an important reason for oncogene overexpression promoted by ecDNA.
5.2.4 Promoter hijacking
ecDNA in COLO320-DM cells contains multiple copies of the long noncoding RNA gene PVT1, and PVT1 often fuses with the MYC gene, constituting more than 70% of MYC gene transcripts (75). The PVT1-MYC fusion gene is formed by the fusion of the PVT1 promoter and exon 1 with exons 2 and 3 of MYC, that is, the PVT1 promoter and exon 1 replace the MYC gene promoter and exon 1 (75). CRISPR interference experiments inhibiting the PVT1 gene promoter showed that the total amount of MYC gene transcripts decreased (75). High-throughput conformation capture with chromatin immunoprecipitation (HiChIP) experiments showed that multiple enhancers can significantly interact with the PVT1-MYC promoter, and its H3K27ac signal is higher than that of the classic MYC gene promoter (75). In addition, studies of multiple small cell lung cancer (SCLC) cell lines found that the MYCL gene on ecDNA can significantly increase MYCL expression by hijacking the RLF promoter (76). In summary, ecDNA can increase the expression of related genes by hijacking promoters, facilitating broader contact with enhancers.
5.2.5 ecDNA hub
FISH technology has confirmed that ecDNA in PC3 cells, COLO320-DM cells, SNU16 cells, and HK359 glioma cell lines all exhibit a significant tendency to aggregate in the nucleus (75). Aggregated ecDNA are referred to as ecDNA hubs (75). In addition, FGFR2 ecDNA and MYC ecDNA in SNU6 cells are intertwined in the same hub (75). Bromodomain containing 4 (BRD4) is a member of the bromodomain and extra-terminal domain (BET) family, which also includes BRD1, BRD3, and bromodomain testis associated (BRDT) (77). Live-cell imaging has shown that BRD4 protein is highly enriched in the ecDNA hub of COLO320-DM cells (75). JQ1 is a broad-spectrum BET inhibitor targeting all four BET proteins (77). JQ1 can disperse the ecDNA hub, causing ecDNA to be distributed diffusely, suggesting that BRD4 is a key mediator for the formation and maintenance of the ecDNA hub (75, 78).
Immunofluorescence staining has confirmed that the ecDNA hub in glioblastoma-derived neurosphere cell lines is co-localized with RNA polymerase II (RNAPII), suggesting that the ecDNA hub can promote the aggregation and recruitment of functional transcriptional machinery (23). Transfection of COLO320-DM cells with a PVT1 promoter-NanoLuc luciferase (PVT1p-nLuc) plasmid confirmed the presence of inter-molecular enhancer-promoter activation in the ecDNA hub and determined that PVT1p can be trans-activated in the ecDNA hub (75). In SNU6 cells, the enhancer on FGFR2 ecDNA can trans-activate MYC gene expression (75). Therefore, co-localization of FGFR2 ecDNA and MYC ecDNA can further promote MYC expression. In COLO320-DM cells, aggregation of MYC ecDNA predicts MYC pre-mRNA expression levels better than MYC copy number (78). In summary, the ecDNA hub increases the spatial proximity between regulatory elements and oncogenes, leading to increased oncogene expression. In addition, after JQ1 disperses the ecDNA hub in COLO320-DM cells, the expression of the MYC gene carried by ecDNA also decreases, further confirming that the ecDNA hub can promote oncogene expression (75).
It is worth noting that not all ecDNA in all cells can form ecDNA hubs or exhibit high transcriptional activity. Studies have shown that in glioblastoma stem cells, there is no aggregation or close interaction between ecDNA carrying EGFR, MYC, and PDGFR, nor with transcriptional condensates, and the increase in transcriptional products is due to increased copy number (79). In summary, increased oncogene expression on ecDNA involves multiple mechanisms, which may coexist or exist independently.
6 ecDNA promotes tumor heterogeneity, evolution, drug resistance, and poor prognosis
Tumor cells continuously evolve into populations with intratumoral heterogeneity (80). Tumor heterogeneity is caused by genetic, epigenetic, transcriptomic, and phenotypic heterogeneity, and natural selection and Darwinian evolution drive tumor progression and drug resistance on this basis (80, 81). Therefore, tumor heterogeneity is a key factor leading to drug resistance, treatment failure, and death in patients (82). Studies have shown that ecDNA plays an important role in promoting tumor heterogeneity, evolution, and drug resistance (83, 84).
6.1 ecDNA copy number, epigenetic, and genetic heterogeneity
As mentioned above, ecDNA does not follow Mendelian inheritance and undergoes random segregation during cell division. Therefore, after multiple rounds of cell division, the copy number of ecDNA in cells will exhibit significant heterogeneity (Figure 2) (21, 22). When the cellular microenvironment changes, natural selection enables cells to rapidly accumulate ecDNA carrying oncogenes to cope with adverse environments (22). ecDNA+ tumors adapt faster, exhibit more pronounced intratumoral heterogeneity, and develop drug resistance earlier than BFB-amplified tumors (32). DNA methylation is an important epigenetic modification regulating gene expression (85). In multiple SCLC cell lines, the DNA methylation level in ecDNA+ cells is slightly lower than that in ecDNA− cells (76). Using nanopore sequencing technology, it was found that the methylation level of the EGFR gene promoter region on ecDNA in GBM39 cells is significantly lower than that of the same region on chromosomes (24). Therefore, ecDNA can regulate gene methylation status to achieve higher transcriptional activity.
In glioblastoma, exons 2–7 of EGFR are often deleted, resulting in the constitutively active mutant EGFRvIII (86). EGFRvIII in GBM39 cells is mainly located on ecDNA, while chromosomes usually contain full-length wild-type EGFR (24). Therefore, EGFRvIII ecDNA in glioblastoma can provide a unique selective advantage for tumor evolution (14). Studies have shown that the expression of apolipoprotein B mRNA editing enzyme catalytic polypeptide 3 (APOBEC3) is significantly higher in ecDNA+ tumors than in ecDNA− tumors, and 31% of samples containing ecDNA exhibit kyklonic events (APOBEC3 kataegis and ecDNA occurring simultaneously) (87). Among all kyklonic events, 41% overlap with known tumor driver genes, resulting in mutations in tumor driver genes (87). Thus, APOBEC3 plays an important role in ecDNA mutation and tumor evolution. In addition, HPV-host ecDNA can undergo multiple rounds of amplification and recombination, forming highly heterogeneous virus-host ecDNA, thereby promoting heterogeneity and clonal evolution in HPV-related tumors (56). The above studies indicate that ecDNA exhibits copy number heterogeneity and species heterogeneity among cells, as well as genetic and epigenetic heterogeneity with chromosomal DNA.
6.2 ecDNA promotes selection and drug resistance
Studies have shown that the detection rate of ecDNA is significantly increased in tumor patients receiving chemotherapy and targeted therapy, suggesting that ecDNA may be an adaptive mechanism for tumor cells to cope with treatment pressure (3). DHFR gene amplification is the main cause of acquired methotrexate resistance (88). The amplification of the DHFR gene in the methotrexate-resistant HT29 human colon cancer cell line undergoes three stages: pre-amplification, HSR, and ecDNA, with ecDNA being the main driver of resistance (88). After methotrexate treatment of HAP1 cells containing DHFR ecDNA, the copy number of ecDNA increased in a strongly dose-dependent manner (22). In urothelial cancer, the CCND1 gene undergoes amplification through ecDNA-mediated structural variants (SVs), driving cell cycle progression and enhancing cellular adaptability under selective therapeutic pressure (89). Treatment of GBM39-EC (EGFRvIII located on ecDNA) and GBM39-HSR (EGFRvIII located on HSR) cells with the EGFR tyrosine kinase inhibitor (TKI) erlotinib showed that GBM39-EC cells resist erlotinib by reducing the copy number of EGFRvIII ecDNA, while the copy number of EGFRvIII in GBM39-HSR cells remains unchanged, and these cells remain sensitive to erlotinib (22). In glioblastoma patients receiving EGFR TKI targeted therapy, tumor cells acquire EGFR TKI resistance by eliminating EGFRvIII ecDNA, and EGFRvIII ecDNA reappear after drug withdrawal (90). Pancreatic ductal adenocarcinoma (PDAC) is usually difficult to survive in a WNT-deficient environment, and acquired WNT independence can promote PDAC progression (19). In PDAC organoid cultures under WNT-deficient conditions, MYC ecDNA+ cells can be selected to adapt to the WNT-deficient environment (19). In addition, GBM39-HSR cells are extremely sensitive to glucose deprivation, while GBM39-EC cells show no significant changes (22). The above studies indicate that ecDNA+ tumors have a stronger selective advantage and can rapidly adapt to changes in the microenvironment when facing metabolic stress or drug treatment, leading to rapid tumor progression and drug resistance.
6.3 ecDNA leads to poor prognosis
Studies have shown that patients with ecDNA+ medulloblastoma were more than twice as likely to relapse and three times as likely to die within 5 years of diagnosis (20). After adjusting for tumor type, stage, age, sex, and genomic instability in 14,778 tumor patients (39 types), it was found that ecDNA detection was associated with tumor stage, metastasis, and shorter overall survival (3). Research indicates that reduced expression of MHC class I molecules in ecDNA+ urothelial carcinoma cells enables immune evasion from T cell attack, thereby contributing to poor prognosis in ecDNA+ urothelial cancer patients (91). Moreover, ecDNA can harbor genes regulating immune and inflammatory responses, which is associated with reduced T cell infiltration in cancer patients (3). Therefore, ecDNA may affect tumor progression and prognosis by influencing the expression of immune-related genes, such as inhibiting immune clearance of tumor cells or promoting immune evasion (3, 92). The p53 tumor suppressor protein is a transcription factor that inhibits cell division or survival in response to various stresses (93). TP53 mutations are associated with enhanced chromosomal instability, including increased amplification of oncogenes and deep deletion of tumor suppressor genes (94, 95). Studies have shown that ecDNA is the main driver of the progression of high-grade dysplasia Barrett’s esophagus (HGD) to esophageal adenocarcinoma (EAC), and TP53 mutations drive the formation of ecDNA (13). TP53 mutations are significantly enriched in ecDNA+ endometrial cancer, renal cancer, breast cancer, and medulloblastoma (3, 12, 20). Therefore, some researchers believe that the impact of ecDNA on patient survival is due to TP53 mutations (20). The overall survival rate of neuroblastoma patients with ecDNA-derived rearrangements is significantly lower than that of patients without ecDNA-derived rearrangements, and the overall survival rate of patients with MYCN ecDNA-derived rearrangements is also significantly lower than that of patients without MYCN ecDNA-derived rearrangements (96). Therefore, genomic instability caused by the integration of ecDNA into chromosomes may be another mechanism by which it affects patient survival outcomes (97). In addition, in SCLC, ecDNA is the main source of the RLF-MYCL oncogenic fusion (76). RLF-MYCL, the most common oncogenic fusion in small cell lung cancer, can accelerate transformation and proliferation of murine SCLC and increase metastatic dissemination and the diversity of metastatic sites (98). In summary, multiple studies have confirmed that ecDNA can lead to poor patient prognosis, making it another indicator for prognosis prediction. However, the biological mechanisms by which ecDNA drives tumor progression and metastasis require further study to provide more evidence for the development of future targeted therapies.
7 Fate of ecDNA
ecDNA plays an important role in tumorigenesis and progression. Can ecDNA persist long-term after abnormal formation in cells? Next, we discuss the fate of ecDNA in cells (Figure 5).
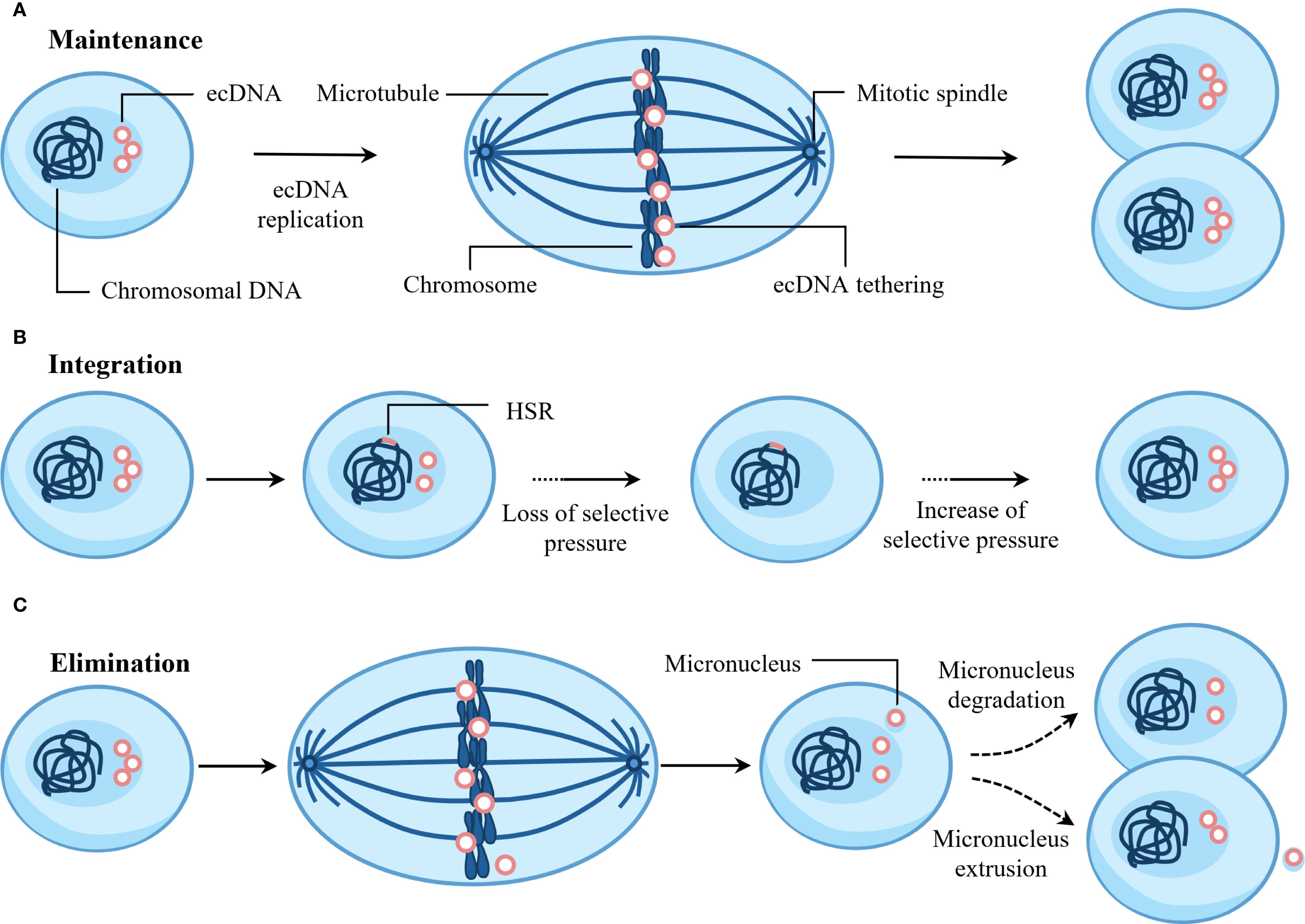
Figure 5. Fate of ecDNA. (A) ecDNA is maintained via active replication and mitotic tethering to chromosomes during cell division. (B) Random integration of ecDNA into chromosomes generates homogeneously staining regions (HSRs). This integration event is positively selected upon loss of selective pressure. Functioning as ecDNA reservoirs, HSRs can regenerate new ecDNAs when favorable selection pressures reappear. (C) ecDNA may undergo elimination through micronucleus formation, followed by extrusion or degradation.
7.1 Maintenance
ecDNA exhibits autonomous replication and undergoes replication only once during the S phase of the cell cycle (99). ecDNA replication activates the ataxia telangiectasia mutated (ATM)-mediated DNA damage response (DDR) pathway (12). This DDR pathway is essential for ecDNA maintenance, and its inhibition would disrupt ecDNA circularization (12). Furthermore, during mitosis, ecDNA achieves segregation to daughter cell nuclei by attaching to chromosomes – a phenomenon termed “hitchhiking” or “tethering” (100). Research indicates that ecDNA achieves efficient nuclear segregation by tethering to mitotic chromosome ends, and this tethering is essential for ecDNA maintenance (101). In addition, ecDNA can form new, more complex structures through replication, amplification, and rearrangement (41, 102).
7.2 Integration
As early as 1985, research revealed that MYC amplification in human colonic carcinoma cell lines evolved from ecDNA to HSR on an X chromosome (103). Studies have shown that focal amplification of BRAF in the M249 human melanoma cell line resistant to vemurafenib and selumetinib initially exists as ecDNA and can subsequently integrate into chromosomes to form homogeneously staining regions (HSR) (104). In neuroblastoma, most genomic structural rearrangements are caused by the integration of ecDNA into chromosomes (96). For example, after chromosome 2 breaks, ecDNA containing MYCN, NBAS, and rs13028343 are formed, and the part of ecDNA containing NBAS and rs13028343 can integrate into chromosome 13, causing the doublecortin like kinase 1 (DCLK1) gene to break (96). In addition, JQ1 can induce the integration of ecDNA into chromosomes, leading to the elimination of ecDNA in cells (102).
7.3 Elimination
ecDNA usually carries oncogenes, so eliminating ecDNA can induce cell differentiation and reverse the tumor phenotype. Studies have shown that COLO320-DM cells contain ecDNA micronuclei, and low concentrations of hydroxyurea can further induce their formation (105). Hydroxyurea induces DNA double-strand breaks, causing ecDNA to aggregate and lag behind mitotic chromosomes, eventually forming micronuclei (106). Subsequently, CRISPR Cas9 was employed to induce precise double-strand breaks in ecDNA. This approach confirmed that damaged ecDNA is prone to aggregation, and that aggregated ecDNA subsequently detaches from mitotic chromosomes, forming micronuclei (107). In addition, gemcitabine also promotes micronucleus formation of ecDNA in human ovarian cancer cells (108). The MDC1-TOPBP1-CIP2A complex mediates the tethering of chromosomal fragments, allowing them to be transmitted as a whole to daughter cells (109). Therefore, the MDC1-TOPBP1-CIP2A complex may also explain why broken ecDNA are more likely to aggregate, but this requires further research. Moreover, recent studies demonstrate that BRD4 plays a significant role in the nuclear segregation of ecDNA, and inhibition of BRD4 impairs ecDNA clustering during mitotic segregation, ultimately leading to micronucleation (101). Subsequently, ecDNA micronuclei are degraded in cells through autophagy or apoptosis-like processes, or extruded from cells through exocytosis-like mechanisms, becoming the main pathways for ecDNA elimination (105).
8 ecDNA as a potential therapeutic target
ecDNA promotes massive transcription of oncogenes and rapid genomic evolution in tumor patients, leading to drug resistance and reduced survival rates. Therefore, ecDNA is an important potential therapeutic target in tumors. Under normal conditions, replication forks on ecDNA exhibit slightly reduced speed and an elevated stalling rate, indicating that they persistently operate under a certain degree of replication stress (110). The reduced fork speed on ecDNA may be associated with increased replication pressure resulting from high-copy gene amplification (111). Studies demonstrate that hydroxyurea can further induce replication stress on ecDNA, exacerbating replication impairment and reducing fork speed, ultimately depleting ecDNA within cells (110). Furthermore, ecDNA undergoes extensive transcription, leading to significantly increased levels of transcription-replication conflicts (63). Such conflicts can cause replication fork reversal and DNA breakage (112). Transcription-replication conflict, replication stress, and DNA damage can drive activation of the S-phase checkpoint (63). The S-phase checkpoint involving checkpoint kinase 1 (CHK1) is essential for fork stability in response to fork stalling (113). The CHK1 protein kinase is essential to ensure genome integrity and cell survival (114). Tang et al. (63) employed a CHK1 inhibitor and CRISPR knockout assays, demonstrating that ecDNA+ tumor cells exhibit heightened sensitivity to CHK1 inhibition, and CHK1 knockout was shown to induce ecDNA damage and subsequent cell death. Subsequently, the oral CHK1 inhibitor BBI-2779 was further applied in a mouse gastric cancer model containing FGFR2 ecDNA, confirming that BBI-2779 can inhibit gastric cancer growth and cause sustained tumor regression in mice (63). Similar to BBI-2779, BBI-355 is also an oral, potent, selective CHK1 small molecule inhibitor in development as an ecDNA-directed therapy (ecDTx). BBI-355 is also an oral, potent, selective CHK1 small molecule inhibitor in development as an ecDNA directed therapy (ecDTx). BBI-825 is an oral, potent, selective ribonucleotide reductase (RNR) small molecule inhibitor. The combination regimen of BBI-355 and BBI-825 has entered phase 1/2 clinical development for the treatment of patients with proto-oncogene-amplified cancers (NCT05827614). Furthermore, leveraging the unique structural features of ecDNA, we have summarized potential ecDNA-specific therapeutic strategies in Table 1.
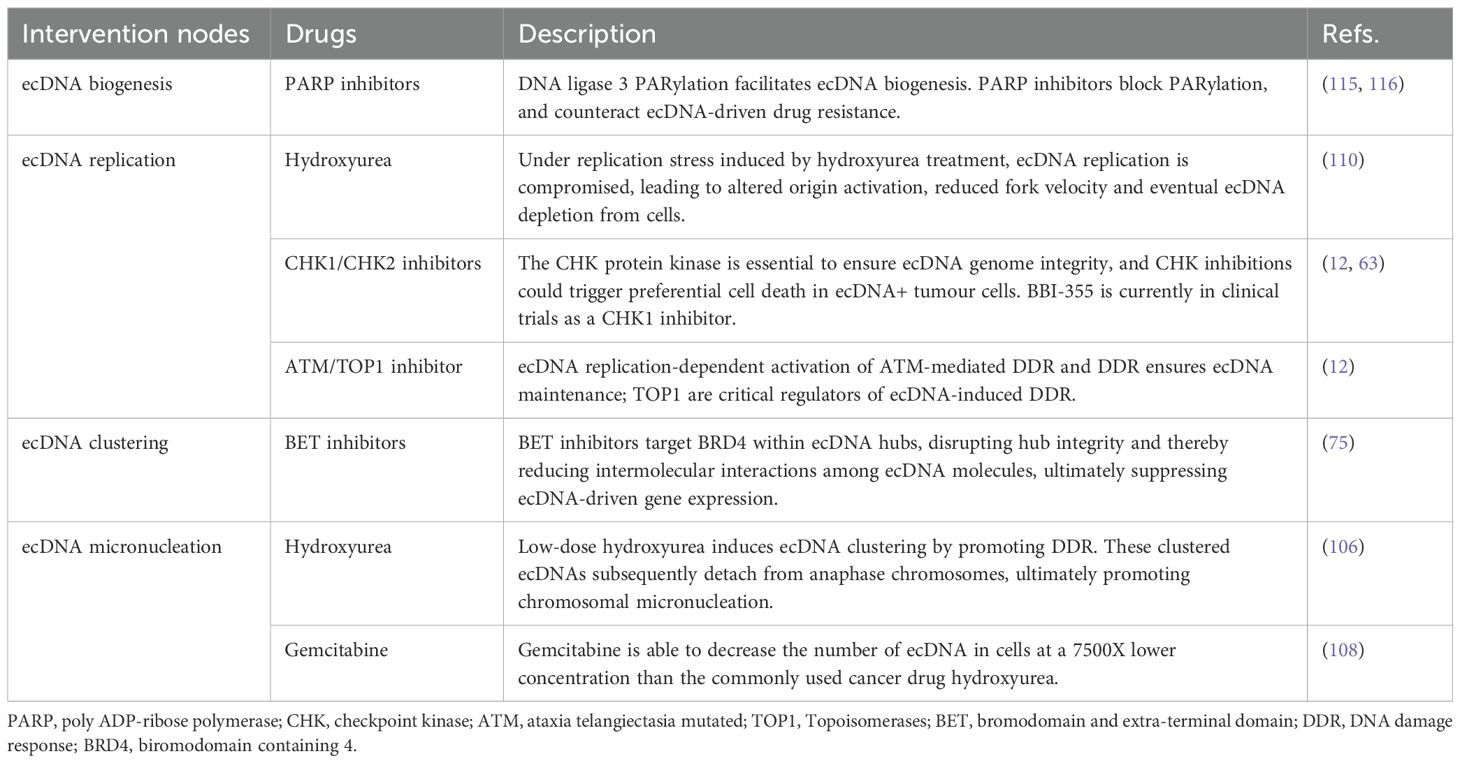
Table 1. Potential targeted therapeutic strategies for ecDNA.
9 Conclusions and perspectives
Currently, the detection of ecDNA primarily relies on two complementary approaches: DNA sequencing and imaging-based technologies. High-throughput sequencing enables comprehensive characterization of ecDNA sequence composition and dynamic alterations, yet it remains limited in resolving spatial organization and intercellular variability. In contrast, advanced imaging techniques allow direct visualization and real-time tracking of ecDNA dynamics but lack the capacity to provide precise sequence-level information. Furthermore, ecDNA detection rates vary substantially across cancer types, and the vast diversity of ecDNA-associated oncogenes introduces significant complexity for the development of ecDNA-targeted therapeutics. Adding to these challenges, the profound intratumoral heterogeneity driven by ecDNA through non-Mendelian inheritance mechanisms substantially reduces the diagnostic reliability of single-time tissue biopsies or liquid biopsies.
Future advances are likely to emerge from integrative strategies that combine high-resolution imaging modalities—such as three-dimensional (3D) reconstruction and live-cell imaging—with next-generation and single-molecule sequencing platforms. Such multimodal approaches are expected to provide a systematic understanding of the spatiotemporal dynamics of ecDNA and its influence on tumor evolution, clonal selection, and treatment response. In parallel, therapeutic strategies may increasingly focus on key regulatory nodes that govern ecDNA biogenesis and maintenance, offering potential broad-spectrum targets across multiple tumor types. Additionally, multi-region sampling and single-cell sequencing are anticipated to mitigate false-negative results associated with intratumoral heterogeneity, thereby improving the sensitivity and reliability of ecDNA detection.
In summary, although ecDNA was first described over six decades ago, its critical roles in oncogenesis and cancer progression have only recently gained widespread recognition. A growing body of evidence demonstrates that ecDNA serves as a major driver of oncogene amplification, genomic instability, intratumoral heterogeneity, and therapeutic resistance. Ongoing studies are progressively elucidating the biological processes underlying ecDNA formation, maintenance, clustering, and clearance, laying a theoretical foundation for the development of ecDNA-targeted interventions, with several candidate compounds currently advancing into clinical trials. Nonetheless, key questions remain unresolved regarding the cellular origins, transcriptional regulation, and three-dimensional spatial architecture of ecDNA, highlighting the urgent need for further mechanistic investigations and technological innovations.
Author contributions
XM: Methodology, Resources, Visualization, Writing – original draft, Writing – review & editing. XY: Data curation, Investigation, Writing – review & editing. CW: Supervision, Writing – review & editing. LS: Conceptualization, Methodology, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was funded by the key project of Sichuan Vocational College of Health and Rehabilitation (CWKY-2022Z-04).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cox D, Yuncken C, and Sprigges AI. Minute chromatin bodies in Malignant tumours of childhood. Lancet. (1965) 1:55–8. doi: 10.1016/S0140-6736(65)90131-5
2. Radloff R, Bauer W, and Vinograd J. A dye-buoyant-density method for the detection and isolation of closed circular duplex DNA: the closed circular DNA in HeLa cells. Proc Natl Acad Sci U S A. (1967) 57:1514–21. doi: 10.1073/pnas.57.5.1514
3. Bailey C, Pich O, Thol K, Watkins TBK, Luebeck J, Rowan A, et al. Origins and impact of extrachromosomal DNA. Nature. (2024) 635:193–200. doi: 10.1038/s41586-024-08107-3
4. Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature. (2017) 543:122–5. doi: 10.1038/nature21356
5. Lv W, Pan X, Han P, Wu S, Zeng Y, Wang Q, et al. Extrachromosomal circular DNA orchestrates genome heterogeneity in urothelial bladder carcinoma. Theranostics. (2024) 14:5102–22. doi: 10.7150/thno.99563
6. Jiang X, Pan X, Li W, Han P, Yu J, Li J, et al. Genome-wide characterization of extrachromosomal circular DNA in gastric cancer and its potential role in carcinogenesis and cancer progression. Cell Mol Life Sci. (2023) 80:191. doi: 10.1007/s00018-023-04838-0
7. Henriksen RA, Jenjaroenpun P, Sjostrom IB, Jensen KR, Prada-Luengo I, Wongsurawat T, et al. Circular DNA in the human germline and its association with recombination. Mol Cell. (2022) 82:209–17 e7. doi: 10.1016/j.molcel.2021.11.027
8. Moller HD, Mohiyuddin M, Prada-Luengo I, Sailani MR, Halling JF, Plomgaard P, et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat Commun. (2018) 9:1069. doi: 10.1038/s41467-018-03369-8
9. Wu N, Wei L, Zhu Z, Liu Q, Li K, Mao F, et al. Innovative insights into extrachromosomal circular DNAs in gynecologic tumors and reproduction. Protein Cell. (2024) 15:6–20. doi: 10.1093/procel/pwad032
10. Hung KL, Mischel PS, and Chang HY. Gene regulation on extrachromosomal DNA. Nat Struct Mol Biol. (2022) 29:736–44. doi: 10.1038/s41594-022-00806-7
11. Chamorro Gonzalez R, Conrad T, Stober MC, Xu R, Giurgiu M, Rodriguez-Fos E, et al. Parallel sequencing of extrachromosomal circular DNAs and transcriptomes in single cancer cells. Nat Genet. (2023) 55:880–90. doi: 10.1038/s41588-023-01386-y
12. Kang X, Li X, Zhou J, Zhang Y, Qiu L, Tian C, et al. Extrachromosomal DNA replication and maintenance couple with DNA damage pathway in tumors. Cell. (2025) 188:1–17. doi: 10.1016/j.cell.2025.04.012
13. Luebeck J, Ng AWT, Galipeau PC, Li X, Sanchez CA, Katz-Summercorn AC, et al. Extrachromosomal DNA in the cancerous transformation of Barrett’s oesophagus. Nature. (2023) 616:798–805. doi: 10.1038/s41586-023-05937-5
14. Hung KL, Jones MG, Wong IT, Curtis EJ, Lange JT, He BJ, et al. Coordinated inheritance of extrachromosomal DNAs in cancer cells. Nature. (2024) 635:201–9. doi: 10.1038/s41586-024-07861-8
15. Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature. (2019) 575:699–703. doi: 10.1038/s41586-019-1763-5
16. Helmsauer K, Valieva ME, Ali S, Chamorro Gonzalez R, Schopflin R, Roefzaad C, et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat Commun. (2020) 11:5823. doi: 10.1038/s41467-020-19452-y
17. Pang J, Nguyen N, Luebeck J, Ball L, Finegersh A, Ren S, et al. Extrachromosomal DNA in HPV-mediated oropharyngeal cancer drives diverse oncogene transcription. Clin Cancer Res. (2021) 27:6772–86. doi: 10.1158/1078-0432.CCR-21-2484
18. Lin MS, Jo S-Y, Luebeck J, Chang HY, Wu S, Mischel PS, et al. Transcriptional immune suppression and up-regulation of double-stranded DNA damage and repair repertoires in ecDNA-containing tumors. eLife. (2024) 12:RP88895. doi: 10.7554/eLife.88895
19. Fiorini E, Malinova A, Schreyer D, Pasini D, Bevere M, Alessio G, et al. MYC ecDNA promotes intratumour heterogeneity and plasticity in PDAC. Nature. (2025) 640:811–20. doi: 10.1038/s41586-025-08721-9
20. Chapman OS, Luebeck J, Sridhar S, Wong IT, Dixit D, Wang S, et al. Circular extrachromosomal DNA promotes tumor heterogeneity in high-risk medulloblastoma. Nat Genet. (2023) 55:2189–99. doi: 10.1038/s41588-023-01551-3
21. deCarvalho AC, Kim H, Poisson LM, Winn ME, Mueller C, Cherba D, et al. Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat Genet. (2018) 50:708–17. doi: 10.1038/s41588-018-0105-0
22. Lange JT, Rose JC, Chen CY, Pichugin Y, Xie L, Tang J, et al. The evolutionary dynamics of extrachromosomal DNA in human cancers. Nat Genet. (2022) 54:1527–33. doi: 10.1038/s41588-022-01177-x
23. Yi E, Gujar AD, Guthrie M, Kim H, Zhao D, Johnson KC, et al. Live-cell imaging shows uneven segregation of extrachromosomal DNA elements and transcriptionally active extrachromosomal DNA hubs in cancer. Cancer Discov. (2022) 12:468–83. doi: 10.1158/2159-8290.CD-21-1376
24. Hung KL, Luebeck J, Dehkordi SR, Colon CI, Li R, Wong IT, et al. Targeted profiling of human extrachromosomal DNA by CRISPR-CATCH. Nat Genet. (2022) 54:1746–54. doi: 10.1038/s41588-022-01190-0
25. Vogt N, Lefèvre S-H, Apiou F, Dutrillaux A-M, Cör A, Leuraud P, et al. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc Natl Acad Sci U S A. (2004) 101:11368–73. doi: 10.1073/pnas.0402979101
26. Carroll SM, Derose ML, Gaudray P, Moore CM, Needham-Vandevanter DR, Hoff DDV, et al. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Mol Cell Biol. (1988) 8:1225–533. doi: 10.1128/mcb.8.4.1525-1533.1988
27. Poddighe PJ, Wessels H, Merle P, Westers M, Bhola S, Loonen A, et al. Genomic amplification of MYC as double minutes in a patient with APL-like leukemia. Mol Cytogenetics. (2014) 7:67. doi: 10.1186/s13039-014-0067-6
28. L’Abbate A, Macchia G, D’Addabbo P, Lonoce A, Tolomeo D, Trombetta D, et al. Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res. (2014) 42:9131–45. doi: 10.1093/nar/gku590
29. Rose JC, Belk JA, Wong IT, Luebeck J, Horn HT, Daniel B, et al. Disparate pathways for extrachromosomal DNA biogenesis and genomic DNA repair. Cancer Discov. (2025) 15:69–82. doi: 10.1158/2159-8290.CD-23-1117
30. Kim H, Nguyen NP, Turner K, Wu S, Gujar AD, Luebeck J, et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet. (2020) 52:891–7. doi: 10.1038/s41588-020-0678-2
31. McClintock B. The stability of broken ends of chromosomes in zea mays. Genetics. (1941) 26:234–82. doi: 10.1093/genetics/26.2.234
32. Raeisi Dehkordi S, Wong IT, Ni J, Luebeck J, Zhu K, Prasad G, et al. Breakage fusion bridge cycles drive high oncogene number with moderate intratumoural heterogeneity. Nat Commun. (2025) 16:1497. doi: 10.1038/s41467-025-56670-8
33. Tanaka H and Watanabe T. Mechanisms underlying recurrent genomic amplification in human cancers. Trends Cancer. (2020) 6:462–77. doi: 10.1016/j.trecan.2020.02.019
34. Umbreit NT, Zhang CZ, Lynch LD, Blaine LJ, Cheng AM, Tourdot R, et al. Mechanisms generating cancer genome complexity from a single cell division error. Science. (2020) 368:6488. doi: 10.1126/science.aba0712
35. Singh M, Raseley K, Perez AM, MacKenzie D, Kosiyatrakul ST, Desai S, et al. Elucidation of the molecular mechanism of the breakage-fusion-bridge (BFB) cycle using a CRISPR-dCas9 cellular model. Nucleic Acids Res. (2024) 52:11689–703. doi: 10.1093/nar/gkae747
36. Lee JJ, Jung YL, Cheong TC, Espejo Valle-Inclan J, Chu C, Gulhan DC, et al. ERα-associated translocations underlie oncogene amplifications in breast cancer. Nature. (2023) 618:1024–32. doi: 10.1038/s41586-023-06057-w
37. Simovic-Lorenz M and Ernst A. Chromothripsis in cancer. Nat Rev Cancer. (2025) 25:79–92. doi: 10.1038/s41568-024-00769-5
38. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. (2011) 144:27–40. doi: 10.1016/j.cell.2010.11.055
39. Webster ALH, Sanders MA, Patel K, Dietrich R, Noonan RJ, Lach FP, et al. Genomic signature of Fanconi anaemia DNA repair pathway deficiency in cancer. Nature. (2022) 612:495–502. doi: 10.1038/s41586-022-05253-4
40. Engel JL, Zhang X, Wu M, Wang Y, Espejo Valle-Inclan J, Hu Q, et al. The Fanconi anemia pathway induces chromothripsis and ecDNA-driven cancer drug resistance. Cell. (2024) 187:6055–70.e22. doi: 10.1016/j.cell.2024.08.001
41. Rosswog C, Bartenhagen C, Welte A, Kahlert Y, Hemstedt N, Lorenz W, et al. Chromothripsis followed by circular recombination drives oncogene amplification in human cancer. Nat Genet. (2021) 53:1673–85. doi: 10.1038/s41588-021-00951-7
42. Cortes-Ciriano I, Lee JJ, Xi R, Jain D, Jung YL, Yang L, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet. (2020) 52:331–41. doi: 10.1038/s41588-019-0576-7
43. Shoshani O, Brunner SF, Yaeger R, Ly P, Nechemia-Arbely Y, Kim DH, et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature. (2021) 591:137–41. doi: 10.1038/s41586-020-03064-z
44. Maciejowski J, Li Y, Bosco N, Campbell PJ, and de Lange T. Chromothripsis and kataegis induced by telomere crisis. Cell. (2015) 163:1641–54. doi: 10.1016/j.cell.2015.11.054
45. Dewhurst SM, Yao X, Rosiene J, Tian H, Behr J, Bosco N, et al. Structural variant evolution after telomere crisis. Nat Commun. (2021) 12:2093. doi: 10.1038/s41467-021-21933-7
46. Williamson A-L. Recent developments in human papillomavirus (HPV) vaccinology. Viruses. (2023) 15:1440. doi: 10.3390/v15071440
47. Wei F, Georges D, Man I, Baussano I, and Clifford GM. Causal attribution of human papillomavirus genotypes to invasive cervical cancer worldwide: a systematic analysis of the global literature. Lancet. (2024) 404:435–44. doi: 10.1016/S0140-6736(24)01097-3
48. Wang R, Pan W, Jin L, Huang W, Li Y, Wu D, et al. Human papillomavirus vaccine against cervical cancer: Opportunity and challenge. Cancer Lett. (2020) 471:88–102. doi: 10.1016/j.canlet.2019.11.039
49. Oyouni AAA. Human papillomavirus in cancer: Infection, disease transmission, and progress in vaccines. J Infect Public Health. (2023) 16:626–31. doi: 10.1016/j.jiph.2023.02.014
50. Schmitz M, Driesch C, Beer-Grondke K, Jansen L, Runnebaum IB, and Durst M. Loss of gene function as a consequence of human papillomavirus DNA integration. Int J Cancer. (2012) 131:E593–602. doi: 10.1002/ijc.27433
51. Bodelon C, Untereiner ME, Machiela MJ, Vinokurova S, and Wentzensen N. Genomic characterization of viral integration sites in HPV-related cancers. Int J Cancer. (2016) 139:2001–11. doi: 10.1002/ijc.30243
52. Nakagawa T, Luebeck J, Zhu K, Lange JT, Sasik R, Phillips C, et al. Inhibition of human-HPV hybrid ecDNA enhancers reduces oncogene expression and tumor growth in oropharyngeal cancer. Nat Commun. (2025) 16:2964. doi: 10.1038/s41467-025-57447-9
53. Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. (2014) 24:185–99. doi: 10.1101/gr.164806.113
54. Peter M, Stransky N, Couturier J, Hupe P, Barillot E, de Cremoux P, et al. Frequent genomic structural alterations at HPV insertion sites in cervical carcinoma. J Pathol. (2010) 221:320–30. doi: 10.1002/path.2713
55. Cao C, Hong P, Huang X, Lin D, Cao G, Wang L, et al. HPV-CCDC106 integration alters local chromosome architecture and hijacks an enhancer by three-dimensional genome structure remodeling in cervical cancer. J Genet Genomics. (2020) 47:437–50. doi: 10.1016/j.jgg.2020.05.006
56. Akagi K, Symer DE, Mahmoud M, Jiang B, Goodwin S, Wangsa D, et al. Intratumoral heterogeneity and clonal evolution induced by HPV integration. Cancer Discov. (2023) 13:910–27. doi: 10.1158/2159-8290.CD-22-0900
57. Deshpande V, Luebeck J, Nguyen ND, Bakhtiari M, Turner KM, Schwab R, et al. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat Commun. (2019) 10:392. doi: 10.1038/s41467-018-08200-y
58. Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. (2001) 293:876–80. doi: 10.1126/science.1062538
59. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. (2007) 316:1039–43. doi: 10.1126/science.1141478
60. Zhu K, Jones MG, Luebeck J, Bu X, Yi H, Hung KL, et al. CoRAL accurately resolves extrachromosomal DNA genome structures with long-read sequencing. Genome Res. (2024) 34:1344–54. doi: 10.1101/gr.279131.124
61. Kadali VN and Shoshani O. Aberrant nuclei with amplified DNA in cancer. Trends Cancer. (2025) 11:9–11. doi: 10.1016/j.trecan.2024.09.005
62. Pradella D, Zhang M, Gao R, Yao MA, Gluchowska KM, Cendon-Florez Y, et al. Engineered extrachromosomal oncogene amplifications promote tumorigenesis. Nature. (2025) 637:955–64. doi: 10.1038/s41586-024-08318-8
63. Tang J, Weiser NE, Wang G, Chowdhry S, Curtis EJ, Zhao Y, et al. Enhancing transcription-replication conflict targets ecDNA-positive cancers. Nature. (2024) 635:210–8. doi: 10.1038/s41586-024-07802-5
64. Zhang H, Liu B, Cheng J, Li Z, Jia M, Li M, et al. Characterization and integrated analysis of extrachromosomal DNA amplification in hematological Malignancies. Neoplasia. (2024) 56:101025. doi: 10.1016/j.neo.2024.101025
65. Fyodorov DV, Zhou BR, Skoultchi AI, and Bai Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol. (2018) 19:192–206. doi: 10.1038/nrm.2017.94
66. Routh A, Sandin S, and Rhodes D. Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure. Proc Natl Acad Sci U S A. (2008) 105:8872–7. doi: 10.1073/pnas.0802336105
67. Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, et al. The chromatin accessibility landscape of primary human cancers. Science. (2018) 362:6413. doi: 10.1126/science.aav1898
68. Chen Y, Liang R, Li Y, Jiang L, Ma D, Luo Q, et al. Chromatin accessibility: biological functions, molecular mechanisms and therapeutic application. Signal Transduct Target Ther. (2024) 9:340. doi: 10.1038/s41392-024-02030-9
69. Zabidi MA and Stark A. Regulatory enhancer-core-promoter communication via transcription factors and cofactors. Trends Genet. (2016) 32:801–14. doi: 10.1016/j.tig.2016.10.003
70. Zhu Y, Gujar AD, Wong CH, Tjong H, Ngan CY, Gong L, et al. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell. (2021) 39:694–707.e7. doi: 10.1016/j.ccell.2021.03.006
71. Tian R, Huang Z, Li L, Yuan J, Zhang Q, Meng L, et al. HPV integration generates a cellular super-enhancer which functions as ecDNA to regulate genome-wide transcription. Nucleic Acids Res. (2023) 51:4237–51. doi: 10.1093/nar/gkad105
72. Liu T, Wang J, Yang H, Jin Q, Wang X, Fu Y, et al. Enhancer coamplification and Hijacking promote oncogene expression in liposarcoma. Cancer Res. (2023) 83:1517–30. doi: 10.1158/0008-5472.CAN-22-1858
73. Wang X and Yue F. Hijacked enhancer-promoter and silencer-promoter loops in cancer. Curr Opin Genet Dev. (2024) 86:102199. doi: 10.1016/j.gde.2024.102199
74. Mortenson KL, Dawes C, Wilson ER, Patchen NE, Johnson HE, Gertz J, et al. 3D genomic analysis reveals novel enhancer-hijacking caused by complex structural alterations that drive oncogene overexpression. Nat Commun. (2024) 15:6130. doi: 10.1038/s41467-024-50387-w
75. Hung KL, Yost KE, Xie L, Shi Q, Helmsauer K, Luebeck J, et al. ecDNA hubs drive cooperative intermolecular oncogene expression. Nature. (2021) 600:731–6. doi: 10.1038/s41586-021-04116-8
76. Pongor LS, Schultz CW, Rinaldi L, Wangsa D, Redon CE, Takahashi N, et al. Extrachromosomal DNA amplification contributes to small cell lung cancer heterogeneity and is associated with worse outcomes. Cancer Discov. (2023) 13:928–49. doi: 10.1158/2159-8290.CD-22-0796
77. Wang ZQ, Zhang ZC, Wu YY, Pi YN, Lou SH, Liu TB, et al. Bromodomain and extraterminal (BET) proteins: biological functions, diseases, and targeted therapy. Signal Transduct Target Ther. (2023) 8:420. doi: 10.1038/s41392-023-01647-6
78. Weiser NE, Hung KL, and Chang HY. Oncogene convergence in extrachromosomal DNA hubs. Cancer Discov. (2022) 12:1195–8. doi: 10.1158/2159-8290.CD-22-0076
79. Purshouse K, Friman ET, Boyle S, Dewari PS, Grant V, Hamdan A, et al. Oncogene expression from extrachromosomal DNA is driven by copy number amplification and does not require spatial clustering in glioblastoma stem cells. Elife. (2022) 11:e80207. doi: 10.7554/eLife.80207
80. Dagogo-Jack I and Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. (2018) 15:81–94. doi: 10.1038/nrclinonc.2017.166
81. McGranahan N and Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. (2017) 168:613–28. doi: 10.1016/j.cell.2017.01.018
82. Greaves M. Evolutionary determinants of cancer. Cancer Discov. (2015) 5:806–20. doi: 10.1158/2159-8290.CD-15-0439
83. Kanayama K, Imai H, Hashizume R, Matsuda C, Usugi E, Hirokawa YS, et al. Extrachromosomal DNA dynamics contribute to intratumoural receptor tyrosine kinase genetic heterogeneity and drug resistance in gastric cancer. Mol Cancer Res. (2025) 23:503–14. doi: 10.1158/1541-7786.MCR-24-0741
84. Pal Choudhuri S, Girard L, Lim JYS, Wise JF, Freitas B, Yang D, et al. Acquired cross-resistance in small cell lung cancer due to extrachromosomal DNA amplification of MYC paralogs. Cancer Discov. (2024) 14:804–27. doi: 10.1158/2159-8290.CD-23-0656
85. Martisova A, Holcakova J, Izadi N, Sebuyoya R, Hrstka R, and Bartosik M. DNA methylation in solid tumors: functions and methods of detection. Int J Mol Sci. (2021) 22:4247 doi: 10.3390/ijms22084247
86. Hoogstrate Y, Ghisai SA, de Wit M, de Heer I, Draaisma K, van Riet J, et al. The EGFRvIII transcriptome in glioblastoma: A meta-omics analysis. Neuro Oncol. (2022) 24:429–41. doi: 10.1093/neuonc/noab231
87. Bergstrom EN, Luebeck J, Petljak M, Khandekar A, Barnes M, Zhang T, et al. Mapping clustered mutations in cancer reveals APOBEC3 mutagenesis of ecDNA. Nature. (2022) 602:510–7. doi: 10.1038/s41586-022-04398-6
88. Meng XN, Ma JF, Liu YH, Li SQ, Wang X, Zhu J, et al. Dynamic genomic changes in methotrexate-resistant human cancer cell lines beyond DHFR amplification suggest potential new targets for preventing drug resistance. Br J Cancer. (2024) 130:1819–27. doi: 10.1038/s41416-024-02664-0
89. Nguyen DD, Hooper WF, Liu W, Chu TR, Geiger H, Shelton JM, et al. The interplay of mutagenesis and ecDNA shapes urothelial cancer evolution. Nature. (2024) 635:219–28. doi: 10.1038/s41586-024-07955-3
90. Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. (2014) 343:72–6. doi: 10.1126/science.1241328
91. Lv W, Zeng Y, Li C, Liang Y, Tao H, Zhu Y, et al. Spatial–temporal diversity of extrachromosomal DNA shapes urothelial carcinoma evolution and the tumor immune microenvironment. Cancer Discovery. (2025) 15:1225–46. doi: 10.1158/2159-8290.Cd-24-1532
92. Rui R, Zhou L, and He S. Cancer immunotherapies: advances and bottlenecks. Front Immunol. (2023) 14:1212476. doi: 10.3389/fimmu.2023.1212476
93. Kastenhuber ER and Lowe SW. Putting p53 in context. Cell. (2017) 170:1062–78. doi: 10.1016/j.cell.2017.08.028
94. Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M, et al. Integrated analysis of TP53 gene and pathway alterations in The Cancer Genome Atlas. Cell Rep. (2019) 28:1370–84.e5. doi: 10.1016/j.celrep.2019.07.001
95. Danovi S. TP53-dependent genomic instability. Nat Genet. (2022) 54:1584. doi: 10.1038/s41588-022-01216-7
96. Koche RP, Rodriguez-Fos E, Helmsauer K, Burkert M, MacArthur IC, Maag J, et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat Genet. (2020) 52:29–34. doi: 10.1038/s41588-019-0547-z
97. Requesens M, Foijer F, Nijman HW, and de Bruyn M. Genomic instability as a driver and suppressor of anti-tumor immunity. Front Immunol. (2024) 15:1462496. doi: 10.3389/fimmu.2024.1462496
98. Ciampricotti M, Karakousi T, Richards AL, Quintanal-Villalonga A, Karatza A, Caeser R, et al. Rlf-Mycl gene fusion drives tumorigenesis and metastasis in a mouse model of small cell lung cancer. Cancer Discov. (2021) 11:3214–29. doi: 10.1158/2159-8290.CD-21-0441
99. Barker PE, Drwinga HL, Hittelman WN, and Maddox AM. Double minutes replicate once during S phase of the cell cycle. Exp Cell Res. (1980) 130:353–60. doi: 10.1016/0014-4827(80)90012-9
100. Kanda T, Otter M, and Wahl GM. Mitotic segregation of viral and cellular acentric extrachromosomal molecules by chromosome tethering. J Cell Sci. (2001) 114:49–58. doi: 10.1242/jcs.114.1.49
101. Nichols A, Choi Y, Norman RX, Chen Y, Striepen J, Salataj E, et al. Chromosomal tethering and mitotic transcription promote ecDNA nuclear inheritance. Mol Cell. (2025) 85:2839–53. doi: 10.1016/j.molcel.2025.06.013
102. Goble K, Mehta A, Guilbaud D, Fessler J, Chen J, Nenad W, et al. Leveraging AI to automate detection and quantification of extrachromosomal DNA to decode drug responses. Front Pharmacol. (2024) 15:1516621. doi: 10.3389/fphar.2024.1516621
103. Lin CC, Alitalo K, Schwab M, George D, Varmus HE, and Bishop JM. Evolution of karyotypic abnormalities and C-MYC oncogene amplification in human colonic carcinoma cell lines. Chromosoma. (1985) 92:11–5. doi: 10.1007/BF00327240
104. Song K, Minami JK, Huang A, Dehkordi SR, Lomeli SH, Luebeck J, et al. Plasticity of extrachromosomal and intrachromosomal BRAF amplifications in overcoming targeted therapy dosage challenges. Cancer Discov. (2022) 12:1046–69. doi: 10.1158/2159-8290.CD-20-0936
105. Shimizu N, Shimura T, and Tanaka T. Selective elimination of acentric double minutes from cancer cells through the extrusion of micronuclei. Mutat Res. (2000) 448:81–90. doi: 10.1016/S0027-5107(00)00003-8
106. Shimizu N, Misaka N, and Utani K. Nonselective DNA damage induced by a replication inhibitor results in the selective elimination of extrachromosomal double minutes from human cancer cells. Genes Chromosomes Cancer. (2007) 46:865–74. doi: 10.1002/gcc.20473
107. Oobatake Y and Shimizu N. Double-strand breakage in the extrachromosomal double minutes triggers their aggregation in the nucleus, micronucleation, and morphological transformation. Genes Chromosomes Cancer. (2020) 59:133–43. doi: 10.1002/gcc.22810
108. Yu L, Zhao Y, Quan C, Ji W, Zhu J, Huang Y, et al. Gemcitabine eliminates double minute chromosomes from human ovarian cancer cells. PloS One. (2013) 8:e71988. doi: 10.1371/journal.pone.0071988
109. Trivedi P, Steele CD, Au FKC, Alexandrov LB, and Cleveland DW. Mitotic tethering enables inheritance of shattered micronuclear chromosomes. Nature. (2023) 618:1049–56. doi: 10.1038/s41586-023-06216-z
110. Jaworski JJ, Pfuderer PL, Czyz P, Petris G, Boemo MA, and Sale JE. ecDNA replication is disorganized and vulnerable to replication stress. Nucleic Acids Res. (2025) 53:gkaf711. doi: 10.1093/nar/gkaf711
111. Chowdhry S, Garcia S, Nguyen N-P, Celeste A, Tse E, Milutinovic S, et al. Replication stress and the inability to repair damaged DNA, the potential “Achilles’ heel” of ecDNA+ tumor cells. Cancer Res. (2022) 82:1520. doi: 10.1158/1538-7445.AM2022-1520
112. Browning KR and Merrikh H. Replication-transcription conflicts: A perpetual war on the chromosome. Annu Rev Biochem. (2024) 93:21–46. doi: 10.1146/annurev-biochem-030222-115809
113. Gong Y, Wang Z, Zong W, Shi R, Sun W, Wang S, et al. PARP1 UFMylation ensures the stability of stalled replication forks. Proc Natl Acad Sci U S A. (2024) 121:e2322520121. doi: 10.1073/pnas.2322520121
114. Michelena J, Gatti M, Teloni F, Imhof R, and Altmeyer M. Basal CHK1 activity safeguards its stability to maintain intrinsic S-phase checkpoint functions. J Cell Biol. (2019) 218:2865–75. doi: 10.1083/jcb.201902085
115. Qin L-N, Wu T, Zhen X-T, Zhao Y-L, Zhou J-S, Cheng S-B, et al. Extrachromosomal DNA biogenesis is dependent on DNA looping and religation by YY1-Lig3-PARylation complex. Mol Cell. (2025) 85:1–18. doi: 10.1016/j.molcel.2025.07.007
Keywords: ecDNA, tumorigenesis, oncogene, heterogeneity, cancer progression
Citation: Ma X, Yu X, Wu C and Song L (2025) The role of extrachromosomal DNA in tumorigenesis and progression. Front. Oncol. 15:1665024. doi: 10.3389/fonc.2025.1665024
Received: 14 July 2025; Accepted: 29 August 2025;
Published: 15 September 2025.
Edited by:
Gunnar Boysen, University of Arkansas for Medical Sciences, United StatesReviewed by:
Xiongbin Kang, Bielefeld University, GermanyDavid W. Ussery, Oklahoma State University, United States
Copyright © 2025 Ma, Yu, Wu and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyang Ma, MTg0MDgyNzUwMzRAMTYzLmNvbQ==